Embed Size (px)
Citation preview

Current Pharmaceutical Design, 2003, 9, 599-625 599
1381-6128/03 $41.00+.00 © 2003 Bentham Science Publishers Ltd.
Neuronal High-Affinity Sodium-Dependent Glutamate Transporters(EAATs): Targets for the Development of Novel Therapeutics AgainstNeurodegenerative Diseases
Giuseppe Campiani*,1, Caterina Fattorusso2, Meri De Angelis1,Bruno Catalanotti2,Stefania Butini1, Roberto Fattorusso3 , Isabella Fiorini1, Vito Nacci1 and EttoreNovellino4
1Dipartimento Farmaco Chimico Tecnologico (DFCT), Universita’ degli Studi di Siena, via AldoMoro, 53100 Siena, Italy. 2Dipartimento di Chimica delle Sostanze Naturali (DCSN), Universita’degli Studi di Napoli “Federico II”, via D. Montesano 49, 80131 Napoli, Italy.3Dipartimento diScienze Ambientali, Universita’ degli Studi di Napoli II, Via Vivaldi 43, Caserta, Italy.4Dipartimento di Chimica Farmaceutica e Tossicologica (DCFT), Universita’ degli Studi diNapoli “Federico II”, via D. Montesano 49, 80131 Napoli, Italy
Abstract: L-Glutamate is the major excitatory neurotransmitter in mammalian central nervoussystem, and excitatory amino acid transporters (EAATs) are essential for terminating synapticexcitation and for maintaining extracellular glutamate concentration below toxic levels. Althoughthe structure of these channel-like proteins has not been yet reported, their membrane topology has been hypothesisedbased on biochemical and protein sequence analyses. In the case of an inadequate clearance from synaptic cleft and fromthe extrasynaptic space, glutamate behaves as a potent neurotoxin, and it may be related to several neurodegenerativepathologies including epilepsy, ischemia, amyotrophic lateral sclerosis, and Alzheimer disease. The recent boom ofglutamate is demonstrated by the enormous amount of publications dealing with the function of glutamate, with its role onmodulation of synaptic transmission throughout the brain, mainly focusing: i) on the structure of its receptors, ii) onmolecular biology and pharmacology of Glu transporters, and iii) on the role of glutamate uptake and reversal uptake inseveral neuropathologies. This review will deal with the recent and most interesting published results on Glu transportersmembrane topology, Glu transporters physiopathological role and Glu transporters medicinal chemistry, highlighting theguidelines for the development of potential neuroprotective agents targeting neuronal high-affinity sodium-dependentglutamate transporters.
1. INTRODUCTION
L-Glutamate (1, Glu) is an important nutrient involved inseveral biochemical pathways such as gluconeogenesis andammonia detoxification. In the mammalian Central NervousSystem (CNS) Glu plays an even more important role as anexcitatory neurotransmitter modulating the functions of mostneuronal circuits [1]. Glu is involved in most aspects ofnormal brain functioning including cognition, memory andlearning [2], its role in the CNS has been the object ofextensive investigation in the modern neuroscience andliterature on this topic is rapidly growing [3]. In neuronsglutamate is stored in specialized presynaptic vesicles(loaded by specific vesicular transporters) [4] and is releasedinto the synaptic cleft, upon different stimuli, by fusion ofthe vesicles with the plasma membrane. Once in the synapticcleft, glutamate activates a variety of proteins includingreceptors (ionotropic iGluRs and metabotropic mGluRs),electrogenic transport systems (excitatory amino acid
*Address correspondence to this author at the Dipartimento FarmacoChimico Tecnologico, Universita’ degli Studi di Siena, via Aldo Moro,53100 Siena, Italy; Tel: 0039-0577-234172; Fax: 0039-0577-234333;E-mail: [email protected].
transporters, EAATs), and enzymes, situated on the pre- andpostsynaptic membrane, and also on astroglia [5-8]. Thedegree of the postsynaptic response is influenced by anumber of factors that affect the time dependence of theglutamate concentration in the synapse, such as the rate ofpresynaptic release, the rate of passive diffusion in theextrasynaptic space and, most importantly, the rate of activeglutamate transport.
The glutamatergic neurotrasmission is terminated byclearance of Glu from the cleft by an uptake mechanism,which uses an electrochemical gradient as driving force, andis operated by neuronal and astroglial sodium-dependenthigh affinity transporter proteins [9]. An imbalance inglutamatergic neurotrasmission has been related to manypathophysiological conditions in the CNS. In fact, Glu is apotential neurotoxin (up to 1-3 µM) and the transporter-mediated uptake is believed to play an important role inmediating the balance between the physiological andpathological actions of this excitatory transmitter [10].Excitotoxicity would be mostly mediated by iGluRsactivation, but at the physiological level and in normal brainsthe desensitization process of iGluRs shapes synapticresponses and provides a critical mechanism of neuropro-tection at central synapses.

600 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
This high affinity transport system, [11] in addition tomaintaining glutamate concentrations below desensitizingand excitotoxic levels (high nanomolar range), contributes tothe termination of the excitatory signal and to the recyclingof the transmitter into the metabolic pool (the glutamate-glutamine cycle) [9]. The link between impairment oftransporter functions and excitotoxic concentrations of Glusuggests that transporter dysfunction could be a mechanismof neurodegeneration and cell death [12]. Of consequence,targeting excitatory amino acids transporters could lead tothe development of new therapeutic strategies against avariety of pathological conditions, including epilepsy, ische-mia, amyotrophic lateral sclerosis, and Alzheimer’s disease.
In the last decade, despite the enormous amount ofpublications on structure, molecular biology and pharmaco-logy of Glu transporters, few ligands that specifically interactwith these transport systems have been developed. Thoughmolecular pharmacology aspects of EAATs have beenrecently reviewed by different authors, [2,3,6,11,13-15] theaim of this review is to provide the reader with an overviewof recent medicinal chemistry advances in the developmentof EAAT ligands, providing a survey of the present status ofthe most important structural and functional aspects ofEAATs, exploring the potential of this family of channel-likeproteins as targets for drug design.
2. GLUTAMATE TRANSPORTERS: FUNCTIONALFEATURES
Amino acid (AA) transporters across the plasmamembrane of eukaryotic and bacterial cells mediate andregulate the supply of AA to cells for cellular nutrition butalso they fulfill specialized cellular functions. All known andputative transport proteins have been grouped into familiesbased on sequence similarity. To date 174 transporterfamilies have been identified of which the secondarytransporters represent the largest functional category [16].Many of these secondary transporter families belong to thesame structural class, [17,18] while, based on computationalanalyses of the aminoacid sequences and hydropathyprofiles, glutamate transporters form a unique structural classof membrane proteins [16], as will be discussed in section 4.The glutamate transporters family includes transporters forneutral and acidic amino acids. EAATs are responsible forthe translocation of L-glutamate, D- and L-aspartate andsome other acidic amino acids such as cysteate, although,with low affinity, Glu is also transported by ASC neutralamino acid transporters (ASCTs) [19]. The uptake process,as well as the potential consequences of decreases intransport capacity, is particularly significant in view of theexcitotoxic properties of L-glutamate. The rapid removal ofglutamate, from the synaptic cleft by high-affinity uptake isthought to influence the kinetics of Glu receptor activation,and to contribute to (i) the termination of the excitatorysignal, (ii) the maintenance of extracellular glutamate levelsbelow those that could induce excitotoxic damage, butmaintaining synaptic communication, and (iii) the recyclingof the transmitter via the glutamine cycle. Anyway, EAATsremove Glu not only from the cleft, but also from theextracellular space because iGluRs and mGluRs are locatedin the cell body (nerve terminals, dendrites) [9]. Since
enzymes that could extrasynaptically metabolize the excessof Glu have never been identified, the cellular uptake is themost rapid mechanism of clearance, and it is the mechanismresponsible for the long-term maintenance of low extracellu-lar concentrations of Glu. To date, five different mammaliansubtypes of transporters have been cloned (EAAT1(GLAST), EAAT2 (GLT1), EAAT3 (EAAC1), EAAT4 andEAAT5) [2,20,21]. Cloning of the different transporters hasrevealed a highly specific distribution pattern of theindividual transporter molecules in different CNS regions.The process of Glu uptake is driven by the free energy storedin electrochemical gradients of sodium and potassium ionsover the membrane [22-24]. Numerous functional studies ofnatively and heterologously expressed Glu transporterssubtypes showed that the transport process is electrogenicand it is coupled to the co-transport of two/three sodiumions, one proton, and a counter-transport of one potassiumion, per transported Glu. Additionally, some transportersfunction as glutamate-gated chloride channels [21,25-27].While the thermodynamically uncoupled chloride conduc-tance has been associated with the activation of transporters,this anion flux is not linked stoichiometrically to Glutransport. Each Glu transporter subtype differs in the relativeproportion of the current generated by the charge transfers(Glu and ions). Transporters GLT-1, EAAC1 and GLASTshow a weak chloride flux, while in EAAT4 and EAAT5 thechloride current dominates the current elicited by ion-coupled co-transport. It has been proposed that chloride fluxdetermines a negative potential that induce an increase ofGlu reuptake.
2a. Mechanism of the Glutamate Uptake Process andRegulation
2a.1. Glu Translocation and Ion-Cotransport
Functional properties of EAAs transport have beenestablished in a number of in vivo and recombinantexpression systems through pharmacological techniques suchas reconstitution of purified transporters in artificialmembranes, and electrophysiological methods [28]. Thetransporters present ligand-gated channel properties[27,29,30] and the translocation of Glu is ion-dependent[31]. Accordingly, these proteins contain structural features,like water-filled pores and pore loops that may be related tochannel-like properties [27,29,30], and a pentamericorganization of the transporters similar to that of otherchannel proteins has been recently reported. The transporterscatalyze the uptake, efflux and exchange of Glu, with theuptake and efflux that show a transport cycle in oppositedirections. The process of transport is electrogenic and isstimulated by the negative membrane potential. The co-transport of sodium ions provides the driving force for theuptake of Glu. Two or three sodium ions and a proton arecotransported per Glu molecule, and a potassium ion, [32]translocated in distinct steps, is only required to reorient theempty transporter. In 1997 Kanner and co-workers [32]demonstrated in GLT-1 that E404 is important for carryingout the potassium transporting limb of the cycle, suggestingthat this AA, surrounded by histidine 326, aspartates 398 and470, may be intimately associated with ion permeationpathway (the cation binding site). Mutagenesis studies on ratEAAT2 indicate that E404 not only affect the interaction of

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 601
potassium, but also of sodium and glutamate selectivity. Themutant E404D strongly prefers aspartate over glutamate as asubstrate. The binding sequence of Na+, H+ and Glu has notyet been established and it is under debate [31a,33]. Severalstudies indicate that the Glu and sodium binding sitesintimately interact and this is in agreement with recentmutagenesis studies. The coupling stoichiometry is a criticaldeterminant of the steady state distribution ofneurotransmitter across the membrane [34-38]. It has beenrecently demonstrated that, by measuring the transportcurrent reversal potential in EAAT3 as a function of theextracellular concentration of Glu, Na+, H+ and K+, thestoichiometry of the transport cycle was three sodium ionsper Glu, with a proton co transported [31a,33]. On the otherhand, Kanai and co-workers [31b] reported that two sodiumions were co transported with each Glu molecule by EAAC1.In the human EAAT3 the stoichiometry of proton-substratesymport was found to depend on the charge of thetransported substrate. Thus, transport of Glu and cysteate,anionic at physiological pH, is associated with the transportof one proton; some evidences suggest that the co-transportof a proton could be explained assuming that the proton isnecessary for bridging the substrate to a residue in thesubstrate binding site. This mechanism implies that thesubstrate binding site could exist in a protonated andunprotonated form. On GLT-1 Kanner and co-workers [28]speculated that the transport cycle could be summarized asfollows: on the outside three sodium ions bind a specificdomain of the transporters (ion binding site). Then a Glumolecule binds to the receptor, followed by translocation,debinding the Glu and sodium inside the cell. Potassiumcontributes to reorient the transporter; it binds from insideand is released outside (the various Glu transporter subtypesmay have different transport cycles). For EAATs an anionconductance (passive chloride flux), thermodynamicallyindependent of the transport process, has been shown [3,21].The transporters GLT-1, EAAC1 and GLAST showrelatively small chloride flux, while in EAAT4 and EAAT5the anion current is prominent, and the chloride currentsdominate the currents elicited by ion-coupled co-transport. Infact, Amara and co-workers [27,39] reported that, althoughthis associated chloride conductance varies among the clonedsubtypes, the currents generated in EAAT4 are almostentirely due to the flux of chloride ions. Furthermore Amaraalso described cloning and characterization of the humanretinal Glu transporter EAAT5, [21] in which Glu uptake issodium and voltage-dependent and chloride independent, buttransporter current is largely carried by chloride ions. Ingeneral, the transport rate does not depend on anionconcentration, and the transport could take place in absenceof chloride. At present, there are not structural evidences tosuggest whether Glu and chloride anions permeate the samepore or pass through different pathways in the transporterproteins. These anions are not completely inert and in fact,Attwell [40] suggested that the chlorides inside the channel-like proteins slow down the Glu transport by altering the rateconstants for the transition in and out of the conducting state.
2a.2. Mechanisms of High-Affinity Glu TransportersRegulation
Recent studies suggest that EAATs can be rapidlyregulated by a variety of mechanisms [41-43]. Regulation of
Glu transport may occur as a result of changes in the steadystate levels of these transporters that are probably related toaltered rates of transcription and translation [44,45]. Inaddition there are other mechanisms of modulation of Glutransport. Rapid regulation correlates with changes inphosphorylation rate of the proteins and this may relate tothe efficiency of the transporters. Furthermore, as found fortransporters of other neurotransmitters, activation of proteinkinases can affect the expression of EAATs (in tumor celllines the cell surface expression of EAAC1 appears to beregulated by pathways mediated by both protein kinase Cand phosphatidylinositol-3-OH kinase). Phosphorylation byprotein kinase C (PKC) increases the activity of EAAT2, andactivation of PKC increases EAAT3 activity (this occurs, atleast in part, through an increase in cell-surface expression)[46,47]. Up-regulation of Glu transport by GLT-1 andGLAST, and not by EAAC1, may be induced by substratessuch as Glu and Asp [44]. The increase of Glu transportstimulated by Glu itself may be an endogenous mechanismto increase the clearance of Glu from the extracellularenvironment. It has been very recently demonstrated byRothstein and co-workers [48] that expression of EAAC1may be affected by an endogenous, peptidic, inhibitorymodulator, namely GTRAP3-18. This regulatory protein isexpressed in several tissues, it localizes to the cell membraneand cytoplasm and specifically interact with the C-terminaldomain where the Glu binding site of EAAC1 is located. Anincrease of the expression of GTRAP3-18 determines areduction of EAAC1-mediated Glu transport, by loweringsubstrate affinity. Of consequence, GTRAP3-18 may have akey role in the regulation of the neurotransmitter andmetabolic functions of EAAC1. The same authors alsodescribed the modulation of EAAT4 by two otherendogenous proteins, [49] GTRAP41 and GTRAP48. Theseproteins modulate Glu transport activity by interacting withthe C-terminus of EAAT4 proteins. GLT-1, GLAST andEAAT4 are probably regulated via different signallingmechanisms. In fact, as recently demonstrated, neuronalsoluble factors may determine the induction of GLT-1 inastroglia, where they have little effect on the expression ofGLAST, but induce the supporting machinery for regulationof GLAST expression by the astroglial metabotropicreceptors mGluR3 and mGluR5 [41]. Down-regulation ofGLAST in astrocytes is induced by long-term treatment witha group I mGluR agonist DHPG, while DCG-IV, a group IIagonist showed an opposite effect. In general, Schousboedemonstrated that co-culturing of the astrocytes with neuronsinduces the expression of GLT-1 and that neuron-inducedup-regulation of GLT-1 does not require direct contactbetween neurons and astrocytes [41]. Thus soluble factorsmay be involved in this regulatory process [41]. Arachidonicacid (ArA) is a polyunsaturated fatty acid currentlyrecognized as a diffusible signalling molecule capable ofmodulating several cellular and intercellular processes. ArAis able to physiologically modulate the biochemicalproperties of various membrane proteins such as channels,receptors and transporters [50,51]. Studies on isolatedsalamander retina glial cells demonstrated that ArA caninterfere with electrogenic Glu uptake in a concentrationdependent manner. ArA directly acts on the proteininhibiting or enhancing the transport currents, depending ontransporter subtype.

602 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Several studies have also demonstrated that Glu transportmay be affected by amyloid-β protein, the major constituentof the insoluble and cytotoxic amyloid plaques [52-54]. Itoperates an indirect modulation by stimulating an increase ofintracellular calcium ion levels through activation of iGluRs,with a subsequent production of nitric oxide (NO), the latterof which is capable of inhibiting the Glu transporters. SinceNO is produced in response to the stimulation of NMDA-type receptors, this diffusible messenger could be added tothe list of potential mediator of the Glu-sensitive regulationof Glu transport [3]. Furthermore, β-amyloid may decreaseGlu transport [55] via amyloid-β-dependent inactivation ofglutamine synthetase, the astroglial enzyme that convertstranslocated Glu into glutamine. Modulation of Glu uptakecan be obtained also through redox reactions of the SH-groups [56]. In fact, thiol-oxidazing agents such as DTNBmay turn down Glu transport through GLAST and GLT-1,suggesting that Glu transporters may have a SH-based(cysteine residues) redox regulatory mechanism [57,58].Furthermore, there is also an interaction between EAATs andmGluRs. In the cerebellum Glu transporters and mGluRshave an overlapping postsynaptic distribution, and so Gluuptake may serve as a general mechanism for regulatingmGluR-initiated synaptic depression [59].
3. LOCALIZATION AND EXPRESSION OF GLUTRANSPORTERS
Cloning of the different transporters has revealed a highlyspecific distribution pattern of the individual transportermolecules in different CNS regions and in the periphery[60]. Northern blotting analyses [61] have localized humanand rat Glu transporters in the central nervous system and inperipheral tissues. EAAT1, EAAT2 and EAAT4 mRNAs areexpressed in brain, EAAT5 mRNA is prominent in retina,and EAAT3 mRNA is found throughout the brain andperipheral tissues (kidney) [3,23,25,62-70]. In brain EAAT3are found in somas and dendrites of small and largepyramidal neurons. At the cellular level, EAAT2 (GLT-1)are present in astrocytes and to a lesser extent into neurons.EAAT3 (EAAC1) and EAAT1 (GLAST) are present inneurons and glia, and EAAT4 are mostly restricted todendrites of Purkinje cells (cerebellum). EAAT5 are presentin both neurons and glia in retina and may participate invisual processes. Neuronal EAAT3 and EAAT4 are locatedoutside the synaptic cleft and contribute less significantly tothe Glu uptake in the brain than the two astroglial transporterEAAT1 and EAAT2. EAAT3 are also localized topresynaptic GABA-containing terminals and may have ametabolic role in providing Glu for GABA metabolism. Lossof brain EAAT3 expression interfaces with GABA synthesisand results in epilepsy. Peripherally in kidneys EAAT3 maycontribute to renal acidic amino acid reabsorption, acid/basebalance and amino acid metabolism. At the tissue level inbrain [71], EAAT2 are abundantly expressed in thehippocampus, cerebral cortex and striatum, with lowconcentration in the cerebellum, while EAAT1 are highlyexpressed in this latter region. EAAT3 are present inhippocampus, caudate-putamen, and cortex [71].Furthermore, different carriers are localised in the emato-encefalic barrier, and are also responsible of the uptake ofGlu in the synaptic vesicles [72,4]. In fact, sodium dependent
Glu transport was described in the abluminal membrane ofthe blood-brain barrier, and this translocation is mediated byEAAT1, EAAT2, and EAAT3; these transporters may assistin maintaining low Glu levels in the extracellular fluid. Insynaptic vesicles, prior to its depolarization-triggered,calcium dependent release from neuron terminals, glutamateis transported into synaptic vesicles in an ATP-dependentmanner by a specific uptake system. The protein thatmediates this uptake, referred to as VGLUT1, is unrelated toother neurotransmitter transporters and was originallycharacterized as a putative inorganic phosphate transporter.In contrast to the sodium-dependent EAATs, vesicularuptake is coupled to an electrochemical proton gradientgenerated by a vacuolar type ATPase (V-ATPase). In rats,GLAST, GLT-1 and EAAC1 are also expressed in theolfactory bulb. EAAC1 are expressed in neurons, whileGLAST and GLT-1 were found in glial cells throughout theolfactory bulb. In a recent article Utsumi and co-workers,[63] using double staining immunohistochemistry,demonstrated that GLAST and GLT-1 were expressed inastrocytes and that GLAST were intensely expressed in thesubpendymal layer where precursor cells exist. Thisobservation indicates that Glu transporters play their uniquerole not only in neurotransmission but also in celldifferentiation.
4. STRUCTURAL ANALYSIS MEMBRANE TOPO-LOGY
Glu transporters belong to a large and widespread familyof transport proteins with members in eukaryotes, bacteriaand archaea [14], related, either directly or via a commonthird member of the family, by amino acid sequenceidentities of at least 30%. Since the three-dimensionalstructure of proteins is far better conserved than amino acidsequences, all transporters in the glutamate transporterfamily should share the same global structure, including theirmembrane topology. Secondary transporters, such asEAATs, use the free energy stored in ions and/or solutegradient over the membrane to drive transport. In the case ofGlu transporters, the electrochemical gradient of Na+ and K +
across the plasma membrane is used as driving force for theuptake [14,40]. Intriguingly, the transporters also appear tofunction as chloride channels [27, 30,40] indicating thatthese proteins combine both transport and channel features.
Since the three-dimensional structures of secondarytransporters have not been solved, topological models havebeen proposed on the basis of biochemical data andcomputational analysis of their amino acid sequences. Atypical secondary transporter consists of a single polypeptidechain that forms a bundle of membrane-spanning alpha-helices connected by loops of various sizes and there isevidence that EAAT membrane topology would consist ofeight membrane spanning segments (TM1-8) with the aminoand carboxyl terminals located intracellularly [30]. All theproposed models agree in predicting that the N-terminal halfof Glu transporters includes six hydrophobic segments,organized in transmembrane helices (TM1-6) [3,14],experimentally confirmed in the glutamate transporterEAAT2 of rat [73]. On the contrary, the C-terminal half ofthe protein does not clearly show alternating regions of high

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 603
and low hydrophobicity and its membrane topology is still amatter of debate. It includes a stretch of 150 residues (340-490, GLT-1 numbering) which is well conserved not only inglutamate transporters, but also in small neutral amino acidtransporters [74-77], as well as in bacterial glutamate anddicarboxylic acid transporters [78]. This part of the proteincontains several sequence motifs, which are involved inrecognition and/or translocation of glutamate and cotrans-ported cations [74,78]. Surprisingly, analysis of hydropathyprofiles, as well as biochemical and mutagenesis studies,suggest that EAAT C-terminal half presents structuralfeatures that are unique among secondary transporters.
Topological models of GLT-1 and GltT (bacterial GluTransporter from Bacillus Stearothermophilus) [79,80] agreein predicting that TM6 is followed by an intracellular loopthat reenters the membrane (first reentrant loop, RL1) [73-75,77-81], a seventh membrane-spanning helix (TM7) [81],and another reentrant loop entering the membrane from theextracellular side (RL2) [73]. The presence of reentrant loopshas never been reported for secondary transporters and, in arecent work, Brocke and co-workers [79] demonstrated theproximity of the two oppositely oriented reentrant loops inGLT-1, resembling the structure of reentrant loops present inwater-filled pores found in channel proteins [82].
The second reentrant loop is followed by the C-terminusportion of the transporters ending in the cytoplasm [73,74],which inevitably means that there must be an eighthmembrane-spanning segment (TM8) [81]. The structuralarrangement of this portion is still controversial because ofthe hydropathy profile of the region. It has been reported thatthe hydrophilic residues of amphipathic helix 8 (G374-A393GltT numbering) face an aqueous pore in the interior of theprotein while its hydrophobic side is likely to face the lipidbilayer [80,81]. Moreover, on the basis of GLT-1biotinylation studies [73], Grunewald and co-workerssuggested the presence of another highly amphipathicoutward facing segment (S443-L465 GLT-1 numbering)connecting the second reentrant loop and TM8 (linker).
On the other hand, studies performed using thiol-modifying reagents on the C-terminal half of EAAT1[76,83,84] led to a model in which TM6 is followed by: i)the membrane-spanning domain 7, ii) three functionaldomains (8, 9, 10) highly accessible extracellularly, formedby at least one reentrant loop (domain 8) and a waterexposed α-helix (domain 10), and, finally, iii) anothertransmembrane helix named domain 11, ending with the C-terminus located in the cytoplasm.
Freeze-fracture electron microscopy studies revealed thatthe human neuronal glutamate transporter EAAT3, expressedin Xenopus laevis oocytes, assembled into the membrane byassuming a pentameric structure, thus offering new insightsinto its function as both glutamate transporter and glutamate-gated ion channel [85].
4a. Functionally Important Aminoacid Residues
The recent cloning and expression of recombinantglutamate transporter proteins in heterologous expression
systems, allowed a number of questions concerning themolecular basis of transporter function to be addressed. Inparticular, an important aspect of such studies has been theidentification of domains involved in substrate and ionrecognition and translocation through the membrane.
Glutamate transporters contain a well conserved serineand threonine rich stretch (T267-T272, T360-T366 GltT andGLT-1 numbering, respectively) which would reside in theproposed RL1 portion of human EAAT2 homologuestopological models. This stretch plays an important role inthe transporter functionality, as demonstrated by theevidence that N-ethylmaleimide (NEM) modifications onsingle cysteine mutants introduced in the GltT T267-T272motif, blocked the transporter activity [74]. Interestingly,three of these mutants (S269C, S270C, E271C) wereaccessible to cysteine-modifying reagents from both sides ofthe membrane. In the same study it has been demonstratedthat glutamate protected S269C, S270C and E271C mutantsfrom modification with permeable reagents, suggesting thatthese residues are spatially related to the glutamate bindingsite. The above mentioned results highlighted that, in thecatalytic cycle of the transporter, the substrate binding sitecould be exposed alternatively to the extra and theintracellular face of the membrane. This hypothesis seems tobe confirmed by cysteine mutagenesis and chemicallymodifying reagents accessibility studies on GLT-1 firstreentrant loop (R340-P390) [75]. These experiments provedthat the GLT-1 intracellular loop RL1 contains at least oneresidue (A364), located in the threonine/serine conservedstretch, that is exposed to the extracellular environment andwhose accessibility to modifying reagent is restricted in thepresence of glutamate and DHK.
A second domain that was proved to be involved inglutamate transport is located in the region modelled as amembrane spanning helix (TM7) in the EAAT2 topologicalmodel, and as a reentrant loop in the EAAT1 model. TM7segment of GLT-1 transporter (V391-I411) has beenextensively investigated by Kanner and co-workers[32,77,78,86]. Cysteine scanning mutagenesis studies provedthat replacement of the aminoacids N396-T400 led to theloss of transporter function [78]. Also E404 and D398 arerequired for intrinsic activity of GLT-1, since their mutationto asparagine afforded a transporter which exhibitedimpaired glutamate transport. Furthermore, a full activity isstill not retained in E404D and D398E mutants, showing thatit is not sufficient to merely have a negative charge in thisposition [86]. Although the E404D mutant exhibited anunimpaired substrate dependent anion conductance and anunaffected affinity to sodium, it lost the ability to interactwith potassium [32]. Nevertheless, the proximity of sodiumand potassium binding sites is strongly supported by theevidence that the GLT-1 Y403P mutant locks the transporterin the exchange mode by losing the ability to interact withpotassium, and, at the same time, affects sodium selectivity[77]. It has to be emphasized that Y403 can be alternatelyaccessible from either side of the membrane (depending onthe conformational state of the protein), consistent with itsrole as structural determinant of the potassium binding site[78]. It is noteworthy that another aminoacid residue foundto be involved in potassium binding (A407), would be

604 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
positioned just one turn away from Y403 and E404 whenTM7 is modelled as an α-helix [77].
The segment supposed to form a transmembrane helix inGLT-1 and GltT models, corresponds to a reentrant loop inthe EAAT1 topological model. Seal and Amara investigatedthe entire segment, by mutating each of the 24 highlyconserved residues (P392-Q415, EAAT1 numbering) tocysteine [83]. In this study, they reported that EAAT1substrates and blockers prevent the reaction between thetransporter and impermeant sulfhydryl reagents with A395Cmutant, while sodium slows the reaction of membranepermeant reagents with Y405C and E406C. These results arein agreement with those obtained for the GLT-1 transporter,despite the difference between EAAT1 and GLT-1 models.
Although the GLT-1 RL2 portion has been observed tobe largely extracellular, there is at least one residue (A431)which is accessible from both sides of the membrane. Thisobservation, together with those reported for the firstputative reentrant loop and the demonstration of the spatialproximity of RL1 and RL2 [79], suggests that residuesalternatively exposed to either cellular surfaces could drivethe transport of the substrate and co-transported cationsthrough the membrane. Two other serine residues have beenfound to be involved in substrate and co-transported cationsrecognition and translocation; experiments performed on theGLT-1 transporter revealed that these residues (S440, S443)lay in the second reentrant loop [87]. In particular, S440Cmutant reaction with sulfhydryl reagents is prevented by thepresence of DHK and substrates, while the S440G GLT-1mutant shows a seven fold decreased affinity only for DHK.Interestingly, serine clusters were found in the ligand bindingsites of both ionotropic and metabotropic Glu receptors [88,89]. Moreover, when S440 and S443 are mutated to aglycine and an asparagine, respectively, despite transporteraffinity for K+ was not affected but substrate transportbecame almost equally efficient with Li+ or with Na+,indicating that these residues could be also involved indetermining Na+ selectivity [87]. Another evidence of theinvolvement of RL2 in substrate interaction, is thatsulfhydryl modification of I421C mutant inhibits the uptakeof radiolabeled substrate and affects the anion coupled butnot the uncoupled fluxes [90]. Interestingly, similar resultswere obtained on EAAT1 transporter. In fact, sulfhydrylmodification of V449C led to the inactivation of substratetransport but it did not affect substrate gated anionconductance [91].
An elegant study, carried out by Mitrovic and co-workers. using EAAT1/EAAT2 chimeric transporters andselective ligands, showed that substrate sensitivity resides ina stretch of 57 residues (G442-R499, EAAT1 numbering,included in domain 10 and 11 according to the topologicalmodel), of which only 17 are not conserved between EAAT1and EAAT2 [92].
These findings were followed by an insightful analysis ofthe segment A446-G459 of EAAT1 (domain 10) [84], inwhich all residues were scanned by single cysteine mutation,and the resulting mutants’ transporter functionality, as wellas their reactivity with sulfhydryl agents, were evaluated.Several of the cysteine substitution mutants displayed altered
kinetic properties for substrate transport (L448C, M451C,V458C) and/or for binding of a non-transported substrateanalogue (L448C, L455C, S457C, I453C). Moreover, ineight mutants, the reaction with sulfhydryl reagents wasprevented by EAAT1 substrates and inhibitors, suggestingthat this domain may form part of permeation pathway forsubstrates.
The putative amphipathic membrane spanning helix 8may also constitute part of the substrate binding site ortranslocation pore, too [92]. Mutagenesis studies in the Glutransporters GLAST-1 and rat EAAT3 confirmed that helix 8is involved in substrate recognition [93,94]. Indeed, a pivotalrole in substrate interactions is thought to be played by R447(EAAC1 numbering), since its mutation to neutral ornegatively charged amino acid residues completely abolishestransport of L-Glutamate and D- and L-Aspartate, withoutimpairing cysteine transport . Also the carboxyl-terminalmembrane spanning segment 8 of the glutamate transporterGltT (G374-Q404) was studied by cysteine-scanningmutagenesis [80]; of the resulting mutants four were inactive(D380C, R383C, T384C, and N87C), and two showedseverely reduced glutamate transport activity (R337C,M381C).
It can be concluded that despite some controversy on themembrane topology of the glutamate transporters, there isgeneral agreement about the localization of glutamate andco-transported ions binding sites, as well as theirtranslocation pathway, in the C-terminal half of the protein.
5. THE ROLE OF GLUTAMATE TRANSPORTERS INNEUROPATHOLOGIC CONDITIONS: NEW THERA-PEUTIC TARGETS
Although there is a wealth of evidence to show that Gluand its receptors are involved in learning, memory and otherplastic changes in the CNS, paradoxically, excessivestimulation of glutamate receptors appears to underlie anumber of human neurological disorders andneurodegenerative diseases [3,95-97]. Excessiveextracellular Glu has been considered as a pathogenicelement of neuronal damage and death [98-100]. InterstitialGlu clearance depends on the activity of sodium-dependentGlu transporters, and the linkage between impairedtransporter function and excitotoxic concentration of Glusuggest their role in acute conditions such as stroke, CNSischemia, [101-104] and seizure, as well as in chronicneurodegenerative diseases such as the Alzheimer’s disease(AD), [105] Huntington’s disease, [106] and amyotrophiclateral sclerosis (ALS) [107].
5a. Uptake and Reversal Uptake: Implications in SpecificPathologies
5a.1. EAATs and Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis is a chronic, progressivedisease characterized by selective degeneration of upper andlower motor neurons. The progression of the degeneration israpid, leading to extensive paralysis and usually to deathfrom respiratory complications [108,109]. The neurodegene-

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 605
ration in ALS may be the result of a complex interaction ofseveral factors [110-112]. Although an abnormal metabolismof Glu has been suggested to have a role in thepathophysiological process of ALS, oxidative stress or analteration of the metabolism of zinc may in such a waycontribute to degeneration of motor neurons [113]. It hasbeen recently proven that a defect on zinc/copper superoxidedismutase (SOD) can be causative of ALS [112,114]. In factit was demonstrated by Rothstein and co-workers [107] thattransgenic mice that overexpressed mutant human Cu/ZnSOD developed ALS-like symptoms similar to familial andsporadic ALS. Cell injury by oxidative processes may beinvolved in most ALS cases. Furthermore, loss of zinc fromALS-mutant SOD renders the enzymes cytotoxic.
Abnormal Glu uptake may be also involved in ALS[115,116]. It has been found that in patients with ALS thereis a marked decrease in the maximal velocity of transport forEAATs in synaptosomes from spinal cord, brain cortex andbrain stem, while transport was unaffected in hippocampusand striatum. Alteration of Glu transport has been suggestedto be an important factor in progressive neurodegenerationand loss of motor neurons from specific brain regions. Themechanism by which Glu may induce neurodegeneration areas follows: i) a reduced uptake may generate high synapticlevels of the neurotransmitter with consequent overactivationof iGluRs; ii) hyperactivation of EAA receptors causes alarge increase in the intracellular free calcium concentrationthat appears to be associated with excitotoxic neuronal celldeath. In general there is a reduced uptake of Glu. Thedefective Glu uptake found in autopsy material taken fromALS patients seems to be mainly due to a selective loss ofGLT-1, and not of GLAST and other transporters, in spinalcord. In fact, several studies revealed that up to 60% to 70%of patients with sporadic ALS have a 30% to 90% loss of theEAAT2 protein, in both motor cortex and spinal cord. Sincein ALS there is no significant astroglial loss, this loss ofEAAT2 seems not to be secondary to neuron death [95].Thus, oxidative injury (free radical formation) andexcitotoxic mechanisms to ALS may be linked. Trotti andco-workers reported in 1999 that the C-terminal half of GLT-1 is the target of mutated SOD enzymes.
5a.2. Glutamate Uptake, Amyloid-β (Aβ) Peptide, andAlzheimer’s Disease (AD)
AD is a chronic and progressive neurodegenerativedisorder and the most common cause of dementia all aroundthe world [117]. The hallmarks of the disease are plaques(Ab) and neurofibrillary tangles [118]. Several studies havedemonstrated that defects on Glu transporters capacity couldbe implicated in the neurodegeneration observed in AD[119-121]. Although there is evidence that Ab may interferewith Glu translocation [55] (reactive oxygen species aremediators of Ab toxic effects so it is possible that Abproduces Glu transporters oxidation and dysfunction [122]),it is debatable whether aberrant Glu metabolism is asignificant factor in AD. Since Glu transporters cruciallycontribute to the detoxification of extracellular Glu, theirdysfunction may increase susceptibility to Glu toxicity,contributing to neuronal cell death in AD. Li and co-workersin 1997, and later Masliah and co-workers [123] reported asignificant reduction in EAAT2 protein expression levels in
the midfrontal cortex of Alzheimer diseased brains and in theneocortex of transgenic mouse models. Since Glutransporters crucially contribute to the detoxification ofextracellular Glu, interference with these proteins mayaggravate progressive neurodegenerations. Very recentlyScott and co-workers [105] reported that EAAT1 wasstrongly expressed in a subset of cortical pyramidal neuronsin AD brains, and that this abnormal expression wasassociated to tau protein deposition resulting inneurofibrillary tangles. The presence of abnormal amount oftau proteins was also found in those neurons highlyexpressing EAAT1, suggesting that EAAT1 changes may berelated to tau expression in dementia cases showingAlzheimer-type pathologies.
Recent studies have also demonstrated an inhibition ofGlu uptake by Ab in the brain. These findings were furtherconfirmed in amyloid precursor protein (APP) transgenicanimals. Transgenic mice expressing human APP showed asignificant decrease in Vmax and Km for Glu uptake and adecrease in protein expression of GLT-1 and GLAST [53].Glu transporters are also sensitive to oxidizing agents. β-Amyloid in complex with copper may generate hydrogenperoxide and O2
- ion is involved in neurodegeneration andprotein denaturation [124-126]. In fact, oxidative damage iselevated in AD brain, and Ab is thought to mediate thisoxidative damage. Lipid oxidation by Ab can result in theformation of 4-hydroxy-2-nonenal (HNE) which has beensuggested to play a major role in the toxicity of Ab_(highHNE levels have been found in AD brains) [52]. Ab or HNEdetermine an inhibition of Glu transporters when added toneurons in vitro. Thus the reduced EAATs activity in ADbrain may be related to the covalent modification of theseproteins by HNE. In fact, since EAATs are regulated by athiol-based redox mechanism, HNE reacting with cysteines,may disrupt the function of EAATs [56]. Furthermore, in ADthe altered APP processing not only causes an increase ofAb, but also results in a decreased secretion of APPa , thephysiological role of which is to protect againstexcitotoxicity, to stimulate Glu transport and to regulateintracellular calcium levels [123,127,128].
Although direct pathogenetic implications of EAATs inAD have not yet been proven, the alterations of EAATsfunction in patients with Alzheimer-type pathologies may berelated to different factors (for example, changes inexpression of EAATs occur before neurofibrillarypathology). Nonetheless an increasing body of evidenceindicates Glu-mediated toxicity (and abnormal Glutransporter function) might specifically contribute to AD.
5a.3. Glutamate Uptake and Epilepsy
As an excitatory neurotransmitter Glu plays a role in theinitiation and/or spreading in the seizure activity, and adefect of uptake may be responsible of the increase in Gluconcentration noted in the brain of patients with variousforms of epilepsy [95,129,130]. Hyperactivation of AMPAand NMDA receptors may then be responsible for the higherconductance. Although epilepsy is a cluster of disordershaving complex etiology, (most of the studies have beenconducted in animal models [131]), a general decrease inGlu transporter expression may be involved in the disease.

606 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Valproate, [132] an antiepileptic drug, increases the levels ofGLAST and GABA transporters after several days oftreatment.
5a.4. Glutamate Reversed Uptake in CNS Ischaemia
CNS ischemia and traumatic brain injuries are leadingcauses of death or severe mental and physical disabilitiesworldwide, and effective clinical treatment regimens are stilllacking [114,118,130,133,134,135]. Cell death after CNSischemia is mainly mediated by a massive release ofexcitatory amino acids, an increase of intracellular calciumload and the formation of free radicals. One of the majorevents during CNS ischemia is cerebral energy failure whichimpairs the ability of cells to maintain their electrochemicalgradients. Weakening of gradients may be the cause ofalteration of Glu homeostasis. In parallel, during ischemiathe brain parenchyma undergoes changes in oxygen tensionranging from anoxia (ischemic core) to ipoxia (penumbrazone). As a result the redox equilibria are completelydisrupted and, as for other redox-sensitive proteins, thefunction of Glu transporters is profoundly modified [57].Taking into consideration the different mechanisms thatcould lead to excessive Glu release in the synaptic space,Attwell and co-workers [103] demonstrated in CA1pyramidal neurons that, after the start of ischaemic insult,Glu release was mainly operated by reversal uptake of Glutransporters. Transporter-mediated Glu homeostasis failsdramatically in ischemia: instead of removing extracellularGlu to protect neurons, transporters act as a site of efflux ofGlu, triggering neuronal death. This reversal of uptake maybe prevented by non-transportable inhibitors blockade of Glutransporters [2].
5b. Glutamate Transporters as a Target for the Develop-ment of Novel Neuroprotecting Agents
It is now clear that glutamate transporters, and EAAT2 inparticular, are crucial for Glu homeostasis, and the EAATvulnerability to oxidants adds new evidence that excitotoxicand oxidative-stress mechanisms may act synergistically tocause neuronal damage. Consequently this fact has importanttherapeutic implications since it states that the use of anti-excitotoxicity and anti-oxidants compounds may serve asneuroprotective agents and that Glu transporters mayrepresent the major pharmacological targets to modulate Gluaccumulation in the intersynaptic space in a number ofneurodegenerative diseases. Glu has been implicated inchronic progressive neurodegenerations such as Alzheimer’sdisease and Amyotrophic Lateral Sclerosis (ALS) (a chronicsuppression of Glu transport has been proposed to be criticalin ALS), as well as in acute pathological conditionsoccurring in epilepsy and in the period of ischemia thatfollows a stroke [109,107,136]. Since Glu transportersubtypes display different physiological roles in specificbrain regions, the design of selective ligands (to modulatethe uptake or to interfere with the Glu efflux targeting aspecific EAAT subtype) may be useful to treat specificneuropathologies. Excitotoxicity is mainly due to abnormalactivation of iGluRs. Overstimulation of iGluRs can, in fact,promote an increase in intracellular calcium load, triggeringcell death. Glutamate transporters may be the major source
of extracellular Glu by acting as a site of efflux of glutamatefrom the intracellular compartment, and thus increasing theGlu concentration available to iGluRs. This latter mechanismhas been implicated during acute brain ischemia andepileptic seizure [103,104,137,138]. Therefore the design ofselective inhibitors of reverse transport (non transportableinhibitors or blockers), with very low affinity for iGluRs,represents a new strategy for the development ofneuroprotective agents to reduce the acute damage ofspecific regions of the brain during the ischemic insult Fig.(1) [2].
Another new approach to the therapy of chronicneurodegenerative diseases involving glutamate toxicity, isthe design of selective and competitive EAAT substrateshaving negligible GluR affinity. Such agents could betransported across the blood brain barrier (BBB) and intopresynaptic vesicles by specific transporters and thenreleased in combination with Glu into the synaptic cleft, Fig.(2).
6. EAAT LIGANDS: TARGET SPECIFICITY ANDGUIDELINES TO SELECTIVE COMPETITIVESUBSTRATES INHIBITORS AND BLOCKERS
In the last decade a huge number of Glu analogues havebeen synthesised and tested for their ability to discriminateamong iGluRs, mGluRs and EAATs. Despite the potential ofEAATs as a target for the development of new drugs againstneurodegenerative pathologies, few glutamate relatedcompounds have been described as selective EAATcompetitive substrates and inhibitors, and an even muchsmaller number of ligands, that can discriminate among thedifferent EAAT subtypes has been reported until to date. L-Glutamic acid (1), the endogenous EAAT ligand, is a highlyflexible molecule with several different low energy bioactiveconformations that can account for Glu interaction withvarious receptors characterised by different binding sites[15,139]. Although both Glu (1) and Asp (2), theendogenous substrates of EAATs, can be translocated by thesame transporters, these aminoacids show a differentenantioselective EAAT interaction. In fact, while Glu istranslocated and its enantiomer (3) shows low affinity forEAATs, but not for other glutamate receptors, L-aspartateand D-aspartate (D-Asp, 4) are apparently translocated witha similar rate. In depth studies, performed on aspartateenantiomers, showed an enantioselective EAAT interactionfor D- and L-aspartate, depending on the brain region andcell culture taken into consideration [140]. In fact, Mitrovicand co-workers [141], Gordon and co-workers [142], andvery recently Takamoto and co-workers [140], throughbinding studies performed by using 2 and 4 to displace[3H]L-aspartate and [3H]D-aspartate from cerebralneocortex, hippocampus and cerebellar cortex, demonstrateda non-stereoselective interaction with the binding sites of thetransporters in the forebrain, while a higher affinity for Asp(2) was found in the cerebral cortex (data reported in Table1).
While in forebrain GLT-1 are highly expressed, theGLAST subtype is particularly abundant in cerebral cortex.This unprecedently described aspartate enantioselective

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 607
Fig. (1). A Possible Neuroprotection Mechanism Based on EAAT Inhibitors Blocking the Glu Reversed Uptake.
Fig. (2). A Possible Neuroprotection Pathway Through EAAT Selective Substrates.

608 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Table 1. Inhibition of [3H]L-and [ 3H]D-Aspartate Binding inSections of Frozen Rat Brain by D and L-Aspartatea
D-Asp (4) L-Asp (2)
IC50 (nM) n IC50 (nM) n
(a) [3H]L-aspartate
Cerebral Neocortex 340 45 375 46
Hippocampus 285 44 444 46
Striatum 320 47 410 46
Septal nuclei 500 47 640 45
Cerebellar cortex 1.3b 43 440 43
(b) [3H]D-aspartate
Cerebral Neocortex 510 43 370 43
Hippocampus 340 43 630 42
Striatum 340 43 410 42
Septal nuclei 580 43 335 41
Cerebellar cortex 1.03b 47 290 48
aData from Ref. [140]The values are means ± S.E.M. calculated from inhibitions measured at eight inhibitorconcentrations from 0.02 to 5 µM, up to six points per concentration, n is the totalnumber of experimental points.bDifferent from other regions at least at P< 0.01 by ANOVA (Tukey-Kramer test)
interaction was found to be similar to that seen for a Glutransporter recently cloned from an insect nervous system[143]. D-Aspartate has been widely used to study Glutransporters since it is inert to metabolism. Starting from thestructures of the endogenous substrates Glu and Asp, in thelast years several analogues have been synthesized andtested.
These related compounds are mostly characterised bymodification of the distal carboxyl group or by aconformationally restricted structure (cyclic or heterocyclicskeletons) to identify and adequately mimic the variousbioactive conformations of Glu, responsible for interactionwith the different Glu receptor systems. Depending onspecific structural features, some compounds behave ascompetitive substrates being themselves translocated by thetransporter, inducing release of excitatory aminoacid fromthe cytoplasm by means of heteroexchange or exchange withan intracellular transporter substrate (named substrate- ortransported-inhibitors); others behave as non substrateinhibitors since they prevent the molecular transportmechanism from working (non transported inhibitors or theso-called uptake blockers) [20,144,145] (Table 2 ).
Substrate inhibitors can increase extracellular Glu levelsby provoking (i.e. L-trans-2,4-PDC [146]) Glu release viaheteroexchange, or by greatly enhancing reversal uptakeinduced by a metabolic insult [144]. On the contrary, nontransportable inhibitors attenuate EAAT reversal uptake (Fig.(1)), providing useful tools to further examine the
contribution of reversed Glu transport to the increased Glulevels in ischemic brains.
6a. Substrate Inhibitors and Uptake Blockers
6a.1. Substrate-Based EAAT Ligands: Alkyl vs CycloalkylGlutamate Analogues
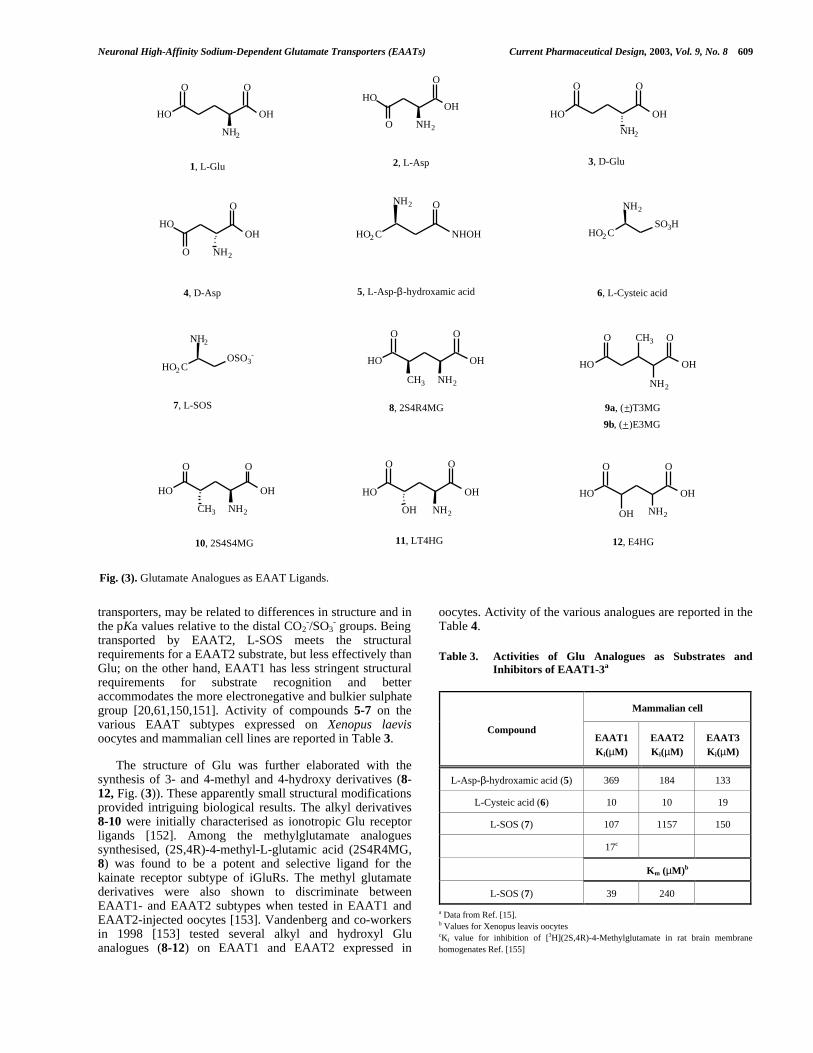
Initial synthetic and biochemical studies, focusing on themodification of the structure of the endogenous substratesnamely Asp and Glu, led to the identification of potentsubstrate inhibitors and uptake blockers. Most of the firstgeneration of Asp and Glu analogues presentedmodifications or bioisosteric replacements of the distalcarboxyl group, and were tested on synaptosomes andastrocytes taken from cortical tissues. Accordingly, L-aspartate-β-hydroxamate (5), cysteic acid (6), L-serine-O-sulphate (L-SOS, 7) and several esters of Glu and Asp wereprepared (Fig. (3)).
These compounds retained the substrate activity of theendogenous substrates towards EAATs, although showingdifferent properties and selectivity [20,147-149]. As an Aspanalogue, the hydroxamic derivative 5 is a substrateinhibitor. Anyway, it inhibits the Glu transport mainlythroughout the EAAT3 subtype with respect to EAAT1 andEAAT2, while L-SOS (7), another Asp analogue, inhibits theEAAT1 more efficiently than the EAAT2 and EAAT3transport. These differences in the ways in which compound7, Glu and D-Asp (4) interact with excitatory amino acid
Table 2. Heteroexchange Induced by Glutammate TransportInhibitors in Cultured Neurons and Astrocytes over5 or 30 mina
Percentage of baseline D-[14C]aspartate release
Astrocytes NeuronsInhibitor (100 µM)
5 min 30 min 5 min 30 min
None 100 100 100 100
L-Glu (1) 290b 215b NA NA
D-Asp (4) 260b 154b 184b 487b
L-t-2,4-PDC (32) 280b 305b 213b 590b
L-β-THA (19) 242b 298b 185b 501b
cis-4-Me-L-trans-2,4-PDC (37) 190c 120 150b 325b
L-3,4-MPDC (34) 230b 225b 154b 452b
DL-TBOA 135 170b 98 128
DHK (31) NA NA 105 119
L-t-2,3-PDC (33) NA NA 109 152c
aData from Ref. [144]Data are means ± SEM of between four and six determinations. bP< 0.01, cP< 0.05compared with control [14C]D-aspartate using one-way ANOVA followed byDunnett’s multiple comparison test.
Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 609
transporters, may be related to differences in structure and inthe pKa values relative to the distal CO2
-/SO3- groups. Being
transported by EAAT2, L-SOS meets the structuralrequirements for a EAAT2 substrate, but less effectively thanGlu; on the other hand, EAAT1 has less stringent structuralrequirements for substrate recognition and betteraccommodates the more electronegative and bulkier sulphategroup [20,61,150,151]. Activity of compounds 5-7 on thevarious EAAT subtypes expressed on Xenopus laevisoocytes and mammalian cell lines are reported in Table 3.
The structure of Glu was further elaborated with thesynthesis of 3- and 4-methyl and 4-hydroxy derivatives (8-12, Fig. (3)). These apparently small structural modificationsprovided intriguing biological results. The alkyl derivatives8-10 were initially characterised as ionotropic Glu receptorligands [152]. Among the methylglutamate analoguessynthesised, (2S,4R)-4-methyl-L-glutamic acid (2S4R4MG,8) was found to be a potent and selective ligand for thekainate receptor subtype of iGluRs. The methyl glutamatederivatives were also shown to discriminate betweenEAAT1- and EAAT2 subtypes when tested in EAAT1 andEAAT2-injected oocytes [153]. Vandenberg and co-workersin 1998 [153] tested several alkyl and hydroxyl Gluanalogues (8-12) on EAAT1 and EAAT2 expressed in
oocytes. Activity of the various analogues are reported in theTable 4.
Table 3. Activities of Glu Analogues as Substrates andInhibitors of EAAT1-3a
Mammalian cell
CompoundEAAT1Ki(µM)
EAAT2Ki(µM)
EAAT3Ki(µM)
L-Asp-β-hydroxamic acid (5) 369 184 133
L-Cysteic acid (6) 10 10 19
L-SOS (7) 107 1157 150
17c
Km (µM)b
L-SOS (7) 39 240
a Data from Ref. [15].b Values for Xenopus leavis oocytescKi value for inhibition of [3H](2S,4R)-4-Methylglutamate in rat brain membranehomogenates Ref. [155]
Fig. (3). Glutamate Analogues as EAAT Ligands.
HO OH
O O
NH2
HO OH
O O
NH2
HOOH
O
NH2O
HOOH
O
NH2O
HO2 C NHOH
NH2
HO2 C
NH2
SO3H
HO2 C
NH2
OSO3-
HO OH
O O
HO OH
O O
NH2CH3
HO OH
O O
CH3
HO OH
O O
OH
HO OH
O O
OH
CH3
NH2
O
NH2NH2NH2
1, L-Glu 3, D-Glu2, L-Asp
4, D-Asp 6, L-Cysteic acid
7, L-SOS
5, L-Asp-β-hydroxamic acid
8, 2S4R4MG
10, 2S4S4MG 11, LT4HG 12, E4HG
9a, (+)T3MG
9b, (+)E3MG

610 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Table 4. Transport Parameters for Various GlutamateDerivativesa
EAAT1 EAAT2
CompoundKm
(µM)Imax
Km/Kb
(µM)Imax
L-Glu (1) 20 1 18 1
L-KA (30) > 3000 - 17 0
L-t-2,4-PDC (32) 26 0.8 2.3 0.2
(±)-T3MG (9a) NA - 18 0
(±)-E3MG (9b) NA - NA
2S4R4MG (8) 54 0.8 3.4 0
2S4S4MG (10) NA - NA
LT4HG (11) 60 0.8 48 0.9
E4HG (12) > 1000 ∼1.0 > 1000 ∼1.0
aData from Ref. [153]Imax values are relative to the maximal current genereted by glutamate. T3MG,2S4RMG, and Kainate blocked glutamate transport by EAAT2. The number of celltested for each compound on both EAAT1 and EAAT2 was between three and eight.
These authors calculated the dose-dependent inwardcurrent generated by exposure to the selected compounds ofEAAT1 and EAAT2, expressed in Xenopus laevis oocytes,and screened the series of analogues at 100 µM. (±)-threo-3-Methylglutamic acid (T3MG, 9a) caused a block of theEAAT2 current induced by 30 µM Glu, and Schild analysisof the effect induced by increasing doses of 9a indicated thatthis Glu analogue was a competitive inhibitor of EAAT2transport with a negligible effect on EAAT1, showingpharmacological properties similar to kainate anddihydrokainate. Its erythro isomer E3MG (9b) did not showany inhibitory effect on either transporter. Recently, theinvestigation of T3MG (9a) has been also extended tooocytes expressing EAAT3 and EAAT4 and to mammaliancell lines (MDCK cells) expressing the EAAT1-3 subtypes[154]. It was found that 9a inhibits EAAT4-mediatedcurrents (IC50 = 109 µM) with a potency similar to thattowards EAAT2 (IC 50 = 90 µM). T3MG did not inhibit bothEAAT1 and EAAT3 at higher concentrations (less than 50%inhibition at 1 mM) and by itself did not elicit currents inEAAT1, EAAT2 and EAAT3. In addition to examining Glu-elicited currents, the same authors measured the effect ofT3MG on uptake of [3H]L-glutamate (or aspartate) in amammalian system. Uptake of [3H]L-glutamate and [3H]D-aspartate was completely inhibited in a dose-dependentfashion, as in oocytes, and T3MG was found selective forEAAT2 (inhibition of [3H]L-glutamate, IC50 = 82.3 µM[154] over EAAT1 and EAAT3 (50% of inhibition found at300 µM-1 mM concentration range)). T3MG (9a) producedan equipotent inhibition of [3H]D-aspartate uptake byEAAT2 and EAAT4 in both cortical and cerebellarsynaptosomes (in both subtypes the IC50s were about 90 µMand 105 µM in both regions, respectively) [154]. Thesestudies further indicated that T3MG (9a) was also capable of
eliciting anion currents in oocyte EAAT4, with a currentsimilar to that elicited by Glu. The investigation performedon 9a indicated that this glutamate analogue might serve as auseful pharmacological tool to determine EAAT subtypeexpression. If the methyl substituent is placed at position 4,close to the distal carboxyl group, the isomer 2S4R4MG (8)showed a pharmacological profile similar to that of T3MG(9a). Compound 8 was found to be a competitive non-transported blocker on EAAT2, binding to the samerecognition site as do kainate and 9a. Its tritiated form [3H]8has been also employed for the characterization of Glureceptors and recently it has been used as a novel ligand forthe investigation of Glu transporters [155] On the contrary,exposure of EAAT1 to 2S4R4MG highlighted a dosedependent inward transport current, indicating that atEAAT1 this compound behaves as a competitive substrate.Its isomer, (2S,4S)-4-methylglutamic acid (2S4S4MG, 10),did not show any affinity for both EAAT1 and EAAT2.These data account for a stereoselective interaction atEAAT1,2 for alkylglutamate analogues, and the differentintrinsic pharmacological properties and selectivity seen forthe active compounds may be rationalised in terms ofdifficulties to optimally orient the distal carboxyl function inpresence of a substituent at position 3 and 4. Replacement ofthe methyl group at position 4 by a hydroxy function, led toL-threo-4-hydroxyglutamic acid (LT4HG, 11) and (±)-erythro-4-hydroxyglutamic acid (E4HG, 12). Thesecompounds were investigated by Vandenberg and co-workers on EAAT1 and EAAT2 on oocytes. As shown inTable 4, LT4HG (10) is a potent EAAT1- and EAAT2-substrate (Kms = 61 µM and 48 µM. respectively), allowingtransport to occur and generating current amplitudes similarto those elicited by Glu (1), while its erythro analogue 12was found less potent at the same transporter subtypes [153].
The structure of glutamate was further elaborated withthe synthesis of 2-(2-carboxycyclopropyl)glycine derivatives(L-CCGs), conformationally restricted analogues ofglutamate [156]. These compounds, shown in Fig.(4),constrained the Glu conformations to an extended or foldedform, with consequently different mutual orientation of thetwo carboxylic functionalities.
Among the four isomer of CCGs, (2S,1'S,2'S)-L-CCG(L-CCG-I, 13), that resembles an extended conformation ofGlu, was characterized as a potent and selective mGluRsagonist, while (2S,1'R,2'S)-L-CCG (L-CCG-IV, 16),mimicking a folded Glu conformation, was found to beactive at NMDA receptors. (2S,1'R,2'R)-L-CCG (L-CCG-II,14) and (2S,1'S,2'R)-L-CCG (L-CCG-III, 15) were notsignificantly active at iGluRs and mGluRs, but wereinhibitors of Glu transport systems [157-160]. Thesecompounds confirmed that a specific conformation isrequired for a selective interaction with the various Glureceptors. Compound 15 is the most potent substrateinhibitor of [3H]glutamate uptake of the series, with Kis inthe range of 1-5 µM (cerebellar and cortical synaptosomes)[161] and has been also tested at individual transportersubtypes [162]. In COS-7 cells expressing EAAT2 15inhibits the [3H]glutamate uptake with a IC50 of 0.29 µM,while in COS-1 cells expressing EAAT1 , it showed a Ki forinhibition of Glu uptake of 7.5 µM [163]. The compoundalso behaved as competitive inhibitor in oocytes expressing

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 611
EAAC1 (Ki of about 10 µM). While 15 is the most potentcompetitive inhibitor, the isomeric compound L- CCG-IV(16), although unselective for EAATs, was also found tomodulate Glu uptake. In fact, in cerebellar and corticalsynaptosomes this compound was able to block the [3H]L-glutamate uptake with IC50 of 3.8 _M and 24 _M, respectively. In EAAT2-
injected COS-7 cells, 16 was reported to be a competitive inhibitor with a IC50 of 1.1 _M.
Furthermore, 14 was also found to inhibit the uptake, although to a lesser extent than 15 and
16 [162]. In a recent paper [164] the pharmacological profile of 15 was compared to
2S4R4MG (8), L-threo-3-hydroxyaspartate (L-_-THA, 19, Fig. (6)), L-SOS (7), and L-
anti,endo-3,4-methanopyrro-lidine dicarboxylate (L-3,4-MPDC, 34, Fig. (7)), using fresh-frozen sections of rat brain(Table 5).
Table 5. Inhibition of 20 nM [3H]L-Aspartate Binding inThaw-Mounted Horizontal Sections of Fresh-FrozenRat Braina,b
CerebralNeocortex
Hippocampus CerebellarCortex
Compound
IC50(µM) IC50(µM) IC50(µM)
2S4R4MG (8) (0.2-80.0 µM) 3 2 5
L-3,4-MPDC (34) (1-100 µM) 36 51 96
L-SOS (7) (5-100 µM) 11 15 10
L-THA (19) (0.1-10.0 µM) 1 1 2
L-CCG III (15) (0.1-80.0 µM) 1 1 2
L-CCG IV (16) 100 µM 59 55 78
L-CCG I (13) 100 µM 82 85 85
aData from Ref. [164]bThe valeus are means ± SEM. Typical approximate values of controls were 190-200fmol/mg tissue (cerebral neocortex), 260-270 fmol/mg tissue (hippocampus) and 790-850 fmol/mg (cerebellar cortex)
The strongest inhibitor of [3H]L-aspartate uptake was 15.In contrast 34 and the other L-CCG isomers, were an orderof magnitude weaker while the conformationally constrainedGlu analogues ((R,S)-1-aminoindan-1,5-dicarboxylic acid(AIDA), trans-1-aminocyclobutane-1,3-dicarboxylic acid
(trans-ACBD, 18, Fig. (5)), (R,S)-3,5-dihydroxyphenyl-glycine (DHPG) and (1S,3S)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD)) were found inactive at 100 µMconcentrations [164]. In general, for the active compounds,only subtle regional variations in the characteristic ofinhibitors of [3H]L-aspartate binding were reported [164]. Inparticular, 8 was reinvestigated, and although previousstudies reported its ability to discriminate between GLT-1and GLAST subtypes when these proteins are expressed inXenopus laevis oocytes, this compound does not stronglydifferentiate between forebrain and cerebellum [3H]L-aspartate binding [164].
F ig. ( 5) . A minoc yc lobutane D ic a rboxylic A cid ( ACBD ) D er iva tives.
Fletcher and coworkers [165] reported that while scarcelyactive on iGluRs, cis-1-aminocyclobutane-1,3-dicarboxylicacid (cis-ACBD) (17) (Fig. (5)) was a potent inhibitor of Gluuptake (K i = 8 µM). Its isomer trans-ACBD (18) is inactiveas uptake inhibitor, but binds with high affinity to NMDAreceptors (Ki = 0.05 µM) [165].
Griffiths and co-workers [166] established that 17 is asubstrate inhibitor since it induces an efflux of [3H]D-aspartate from cerebellar granule cells in a manner consistentwith transporter-mediated heteroexchange. In a further seriesof experiments performed using COS-7 cells expressingEAAT1, EAAT2 and EAAT3 17 showed Ki values of 120µM, 600 µM, and 240 µM, respectively [20].
6a.2. Hydroxy-, Acyl and O-Alkyl Analogues of Aspartate
Another series of Asp analogues is characterized by ahydroxy group at position 3, bearing either acyl or alkylsubsituents. The most important compounds of this series arelisted in Fig. (6).
Fig. (4). 2-(2-Carboxycyclopropyl)-Glycine (CCG) Derivatives.
HO2 C CO2H
NH2 HO2 C
CO2H
NH2
H
NH2
CO2H H
NH2
CO2H
H H
HCO2H
NH2
CO2HH
HHO2 C
CO2H
NH2
CO2H
H HHO2 C H
H H
13,(2S,1'S,2S) L- CCG-I 14,(2S,1'R,2'R) L-CCG-II 15,(2S,1'S,2'R) L-CCG-III 16,(2S,1'R,2'S) L-CCG-IV
Extended form Folded form
CO2HH
NH2HO2 C
CO2HHO2 C
NH2H
17, cis-ACBD 18, trans-ACBD

612 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Fig. (6). Hydroxy, Acyl and O-Alkyl Analogues of Aspartic Acid.
To this subset of potent EAAT ligands belongs (±)-threo-β-hydroxyaspartic acid (D,L-THA), a compound that hasbeen extensively used to characterise Glu transporters. Itsenantiomers (L- threo- and D-threo-, 19 and 20 respectively)and the racemic form have been found to markedly decreaseGlu uptake with no enantioselectivity of interaction[20,21,61,64,167]. Superfusion of 19, 20 and D,L-THA (at100 µM conc.) in Xenopus laevis oocytes, injected withcRNA encoding EAAT1, elicited transport current; the effectof 19 was similar to that of Glu, while the effects of the D,L-form and 20 were somewhat weaker[167]. In general, D,L-THA interacts with all EAAT subtypes, behaving as asubstrate inhibitor at EAAT1-4 (with Kms in the range of 1-40 µM), while acting as non-transportable inhibitor atEAAT5 (Ki = 1 µM).
Arriza and co-workers [21] reported the cloning and thefunctional characterisation of the human Glu transporterEAAT5. Application of Glu to oocytes expressing EAAT5,generated a voltage- and concentration-dependent currentthat was similar to that observed with D- and L-aspartate, butsuperior to that observed with D-glutamate indicating astereoselective interaction for Glu towards EAAT5. D,L-THA, and, to a slightly lesser extent, L-trans-pyrrolidine-2,4-dicarboxylic acid (L-t-2,4-PDC, 32, Fig. (7)) were foundpotent inhibitors of Glu uptake and Glu elicited currents inEAAT5-expressed oocytes, while the potent EAAT2 blockerkainic acid had a negligible effect. Both compounds D,L-THA and 32 reduced the EAAT5 leak current.
Due to the importance of D,L-THA in thecharacterisation of EAATs, several analogues of THA havebeen synthesized in order to explore the effect of varioussubstituents on potency and EAAT subtype selectivity. Thefirst series of compounds was the acyl derivatives of THA inwhich the hydroxy group was replaced by a aliphatic oraromatic ester moiety. Most of the new analogues weresynthesized in racemic form, due to the lowenantioselectivity observed with D,L-THA isomers. In 1997LeBrun and co-workers [167] designed a series of racemicanalogues of D,L-THA characterised by an ester function atposition 3. As reported in Fig. (6), the new compoundspresented an acetoxy function (D,L-threo-β-acetoxyasparticacid, TAcOAsp, 21), a propionyloxy (D,L-threo-β-propionyloxyaspartic acid, TPnOAsp, 22), a benzoyl groupat C-3 (D,L-threo-β-benzoyloxyaspartic acid, TBzOAsp,23), a 1-naphthoyl (D,L-threo-β-(1-naphthoyl)oxyasparticacid, T1NpOAsp, 24) or a 2-naphthoyl system (D,L-threo-β-(2-naphthoyl)oxyaspartic acid, T2NpOAsp, 25). Thesecompounds were initially tested on oocytes injected withcRNA expressing bovine EAAT1. Addition of compounds21-25 resulted in a significant decrease in [14C]glutamateuptake. Although all these new analogues were less potentthan D,L-THA (Km = 9 µM) in inhibiting Glu uptake,compounds 21 and 22 were found to be substrate inhibitorswith Km of 40 and 64 µM (EAAT1), respectively. Incontrast, compounds 23-25 gave results consistent with ablock of glutamate-induced currents. In fact, compounds 23and 24 were non-transported inhibitors and their Ki values,calculated by Schild analysis, were 17 and 52 µM (EAAT1),respectively. More recently, Shimamoto and co-workersreported the development of a series of new THA-relatedanalogues characterised by an ethereal methoxy (L-threo-β-methoxyaspartic acid, L-TMOA, 26), benzyloxy (L-threo-β-benzyloxyaspartic acid, L-TBOA, 27), 1-naphthylmethyloxy(L-threo-β-(1-naphthyl)methyloxyaspartic acid, L-TNOA1,28), and 2-naphthylmethyloxy (L-threo-β-(2-naphthyl)methyloxyaspartic acid, L-TNOA2, 29) function at C-3[168]. These compounds were designed to bypass thestability problems inherent to the ester analogues 21-25. Infact, these THA analogues undergo a facile acyl transferreaction giving inactive amides. Initially the racemic form ofTBOA was pharmacologically characterised [164] and laterthe same authors developed a synthetic strategy to theoptically pure form of these ether derivatives [168]. Forthese ether derivatives, inhibition of Glu uptake wasmeasured in COS-1 cells expressing EAAT1-3 (Table 6) andonly D,L-TBOA and L-TMOA (26), were tested on Xenopuslaevis oocytes expressing EAAT4,5 (Table 7) [169]. Amongthe THA analogues, L-TBOA (27), L-TNOA1 (28) and L-TNOA2 (29) were the most potent blockers for humanEAAT1-3. Threo isomers were more potent as blockers thanthe erythro counterparts (L-EBOA and D-EBOA), and L-isomers were more efficacious than the D-enantiomers(Table 6).
Electrophysiological analysis on Xenopus laevis oocytesexpressing EAAT1-3 was performed in order to assess thesubstrate or blocking properties of the new analogues. Whilethe arylmethyl analogues 27-29 as non-transported blockersdid not elicited detectable current in EAAT1-3, L-TMOA(26) proved to be a substrate for EAAT1 and EAAT3 and ablocker for EAAT2. To characterise the action of D,L-
OH
OH
HO
O
OH
OH
NH2
HO
O
HO
OH
NH2
HO
O OCH2R'
NH2
O R
OH
O
NH2 OO
O
O
O
Me, TAcOAsp (21)Et, TPnOAsp (22)Ph, TBzOAsp (23) 1-Naphthyl, T1NpOAsp (24) 2-Naphthyl, T2NpOAsp (25)
R =
H, L-TMOA (26)Ph, L-TBOA (27)1-Naphthyl, L-TNOA1 (28)2-Naphthyl, L-TNOA2 (29)
R' =
19, L-β-THA 20, D-β-THA

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 613
TBOA and L-TMOA, and to investigate the function ofEAAT4 and EAAT5, the same research group performedelectrophysiological analyses in EAAT4 and EAAT5expressed in Xenopus laevis oocytes [169]. As THA, D,L-TBOA and L-TMOA (26) strongly reduced Glu-inducedcurrents generated by EAAT5, with L-TMOA (26) being themost potent inhibitor, blocking the EAAT5 leak current. InEAAT4, D,L-TBOA behaved as a non transportable blocker,while D,L-THA and L-TMOA (26) acted as substrateinhibitors (Table 7).
D,L-THA and L- t-2,4-PDC are transported into cells andmust compete with intracellular Glu and Asp for access tothe transport site. These two compounds have been used[170] to obtain evidences that transporter reversal plays arole in ischemia-evoked increase of extracellular Glu levels.
The recent development of D,L-TBOA provided anadditional opportunity to investigate the contribution ofreversed Glu transport to Glu levels, in vitro, in astrocytesand glutamatergic neurons [171], and in vivo in ischemic ratcerebral cortex [172]. In a recent work Waagepetersen andco-workers [171] investigated the effect of D,L-TBOA,compared to L-t-2,4-PDC, on uptake and release incerebellar astrocytes and granule neurons, using [3H]D-aspartate. These compounds inhibited, in both cell types, theuptake of [3H]D-aspartate (IC50 values 10-100 µM), althoughD,L-TBOA was found slightly more potent. In neurons andastrocytes, release of preloaded [3H]D-aspartate wasstimulated by L-t-2,4-PDC while D,L-TBOA had no effect.The data obtained by these authors confirmed that D,L-TBOA is a non transported blocker, while L-t-2,4-PDC is asubstrate inhibitor that stimulates heteroexchange with
Table 6. IC50 Values of THA Derivatives for Blocking [14C]L-Glutamate Uptake in COS-1 Cells Expressing EAAT1-3 and Potencyof the Hydroxyaspartate Derivatives to Induce a Transport Current in the Absence or Presence of L-Glu (100 µM) inOocytes Expresing EAAT1-3a,b
EAAT1 I/I(cont)(%) EAAT2 I/I(cont)(%) EAAT3 I/I(cont)(%)
CompoundCompd
(100µM)+Glu
(100µM)IC50
(µM)Compd
(100µM)+Glu
(100µM)IC50
(µM)Compd
(100µM)+Glu
(100µM)IC50
(µM)
DL-TBOA 0 30 48 0 0 7 0 13 8
L-TBOA (27) 0 14.5 23 0 0 4 0 8 7
D-TBOA 0 85 640 0 0 64 0 28 21
L-EBOA 0 77 820 0 0 164 0 64 134
D-EBOA 0 100 >1000 0 61 220 0 83 161
L-TMOA (26) 4.7 33 70 0 0 2 6 25 8
L-TNOA1 (28) 0 10 15 0 0 1 0 13 5
L-TNOA2 (29) 0 18 16 0 0 2 0 30 6.5
aDa from Ref. [168]bThe results are expressed relative to the current obtained with 100 µM L-Glu in the same cells (mean ± S.D., n=3).
Table 7. Pharmacological Properties of THA Derivatives on EAAT4 and EAAT5a
EAAT4 EAAT5
Compound
Km(µM) Ki(µM) Imax Km(µM) Ki(µM) Imax
L-Glu(1) 1 0.4 72 1.0
L-Asp (2) 1 1.0 13 d 0.7
D,L-THA 0.6 0.3 2.5 b,c
L-TMOA (26) 1.2 0.3 1.0 b,c
D,L-TBOA 4.4b 0 3.2 b,c
aData from Ref. [169]bKi values determined by Schild analysis.cThese THA derivatives also block the leakage of currents.dValue previosly reported by Arrizza et al.[21]

614 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
[3H]D-aspartate. Other authors [172] investigated thecontribution of reversed Glu transport in vivo in ischemic ratcerebral cortex. The 50% reductions in ischemia-evoked Aspand Glu efflux seen in D,L-TBOA (100 µM)-treated animals,similar to that observed with KA (30) and DHK (31)(selective blockers of EAAT2), added further support to thehypothesis of reversed uptake by EAATs in depolarizedcells. Furthermore, the similarity of the magnitude of theresponses to both blockers (D,L-TBOA and DHK) stronglysuggests that glial EAAT2 transporters are the primaryagents mediating reversed transport [173].
6a.3. Pyrrolidine Derivatives: PDCs, MPDCs, Kainic Acid(KA) and Dihydrokainic Acid (DHK)
Among the pyrrolidine-based Glu analogue, the naturalproduct kainic acid (KA, 30) has been extremely importantfor the characterisation of Glu receptors. Kainic acid is aneurotoxin that specifically interact with ionotropic AMPAand KA receptors. Furthermore, KA is a non transportedinhibitor of EAAT2. This natural product is characterized byan isopropenyl chain at C-4 of the pyrrolidine ring. Thisside-chain is strategic for iGluRs and EAATs interaction andselectivity. Hydrogenation of the KA double bond providedin fact dihydrokainate (DHK, 31) a Glu analogue character-ized by improved EAAT inhibitory activity (in synaptosomalpreparation KiKA= 60 µM vs KiDHK = 30 µM), and amarkedly reduced affinity for iGluRs. Removal of the C-4side-chain highly increased the affinity for iGluRs, reducing[3H]D-aspartate uptake [174]. Subsequently, on oocytesexpressing EAAT subtypes, DHK (31) was defined as acompetitive inhibitor of EAAT2 with negligible activity onother subtypes and by Arriza and co-workers [20] wasfurther characterized as a non-transportable inhibitor of theEAAT2 subtype. The structure of the natural product KA(30) was further elaborated to provide pyrrolidine analoguescharacterized by improved EAAT specificity and subtypeselectivity.
Pyrrolidine dicarboxylates were design by Bridges andco-workers [175] as constrained analogues of Glu,
mimicking specific Glu conformations with pharmacophoricgroups in well-defined positions. L/D-trans- and L/D-cis-2,4-Pyrrolidine dicarboxylic acids (32 and isomers),(Fig.(7)), were prepared and tested for their ability to inhibittransport of [3H]L-glutamate into synaptosomes, and toinhibit Glu binding to AMPA, KA, and NMDA receptors.
All the isomers at 100 µM concentration showed asimilar weak affinity for iGluRs while, against Glu uptake,L-t-2,4-PDC (32) proved to be a potent competitive inhibitorof Glu transport (Km = 16.6 µM, Ki = 5.06 µM). The D-trans- and the two cis isomers were found much less activethan 32. After an initial use of these compounds toinvestigate the pharmacology of Glu uptake in physiologicalpreparations, they have been more recently employed inexpression systems and to probe the specificity of the clonedtransporters. Thus, Koch and co-workers [176] characterizeda number of pyrrolidine analogues on rat cortical synapto-somal preparations and on oocytes expressing EAAT1-3,measuring the transport inhibition properties and determin-ing the substrate activity evaluating the heteroexchange-mediated release of [3H]D-aspartate (Table 8).
L-t-2,4-PDC (32) was shown to be a more potentcompetitive inhibitor of [3H]D-aspartate uptake than Glu intorat forebrain synaptosomes with a Ki of 1.5 µM, while for itscis isomer a K i of 66 µM (Table 8) was calculated. Althoughthis latter compound initially binds the substrate site withlower affinity, once it occupies the recognition site, it can betranslocated as effectively as the substrate L-aspartate.Furthermore, 32 can stimulate the efflux of [3H]D-aspartatefrom granule cells by heteroexchange. In oocytes expressingEAAT1-4 [20,27] 32 was characterized as a substrateinhibitor with Km values in the range of 2.6-28 µM. LikeD,L-THA, 32 proved to be a non transportable blocker ofEAAT5. At present this compound represents a perfectpharmacological tool to investigate functioning of all fiveEAAT subtypes. Its cis-4-methyl analogue 33 has also beensynthesized and, tested in astrocyte cultures showed a Ki of43 µM [144].
Fig. (7). Pyrrolidine Derivatives: Kainic (KA), and Dihydrokainic (DHK) Acids, PDCs, MPDCs.
NH
CO2H
CO2H
CH2H3 C
NH
CO2H
HO2 C
NH
CO2H
NH
CO2H
CO2H
CH3H3 C
CO2H
NH
CO2H
HH
CO2H
NH
HO2 C
CO2HNH
CO2H
CO2H
H3 C
NH
CO2H
H3 C CO2H
30, KA 32, L-t-2,4-PDC31, DHK
34, L-t-2,3-PDC 36, L-3,4-MPDC 37, L-2,4-MPDC35, cis-5-methyl- L-t-2,3-PDC
33, cis-4-methyl- L-t-2,4-PDC

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 615
Regioisomers of 2,4-PDC, L-t-2,3-PDC (34) and its(2S,3S,5S)-5-methyl analogue (35) were also developed byBridges and co-workers [177,178]. Positioning of a carboxylfunction at position 3 of the pyrrolidine ring provided acompound that mimics the aspartate bioactive conformation.With respect to 32, shifting the carboxyl function to C-3significantly altered the pharmacological profile of thesepyrrolidine derivatives. In fact 34 showed high affinity foriGluRs and this compound and its cis-5-methyl analogue 35showed a lower ability to inhibit the [3H]D-aspartate uptakeinto synaptosomes than did L-t-2,4-PDC (Ki = 33 µM and 37µM respectively [176,179]). In this preparation bothcompounds proved to be competitive inhibitors, but did notproduce levels of efflux different from those observed inabsence of inhibitors [176]. At 300 µM 34 failed to stimulateheteroexchange, in presence or absence of Glu. Furthermore,in oocytes expressing the human transporter clones EAAT1,EAAT2 or EAAT3, application of 30 µM of 34 did not elicita current, proving its non-transportable inhibition propertiesat EAAT2, with little activity at EAAT1 and EAAT3 [176].In 1994 and later in 1998 Esslinger and Bridges [145, 180]described the development of other two novel competitiveinhibitors, starting from 32 as a model: L-anti-endo-3,4-
methanopyrrolidine dicarboxylate (L-3,4-MPDC, 36) and2,4-methanopyrrolidine 2,4-dicarboxylate (L-2,4-MPDC,37). The inhibitory properties of these compounds ontransporter function were assessed on synaptosomes[176,180], and only L-3,4-MPDC blocked the uptake of[3H]D-aspartate, proving to be a competitive inhibitor of[3H]D-aspartate uptake with little activity as substrate (Ki =4.9 µM, Table 9). In oocytes expressing EAAT2 [145], thiscompound was identified as a potent non-transportableinhibitor, with a Ki of 1.6 µM.
The other conformationally locked analogue of 32 is 2,4-MPDC (37)[145]. In synaptosomes from rat forebrain thiscompound at 250 µM concentration produced a completeinhibition of [3H]D-aspartate uptake similar to that shown by36 and 32. When 37 was applied to voltage-clamped oocytesexpressing EAAT2, it induced currents in a dose-dependentmanner with a Km of 45 µM. In synaptosomes from ratforebrain 37 was a substrate of EAAT2, and, to a lesserextent, of EAAT1 and EAAT3. A correlation betweensynaptosomal and cloned EAAT1-3 pharmacology of 37 isshown in Table 10.
Table 8. Ki Values and Exchange Rates for Some Pyrrolidine Derivativesa,b
CompoundKi
(µM)Concentration
(µM)Exchange rate
(pmol/mg protein/2 min)
L-Glu (1) 5 1005010
362344316
L-Asp (2) 2 10020
278266
L-2,4-MPDC (37) 7 10075
282280
L-cis-2,4-PDC 66 100500
206260
L-trans-2,4-PDC (32) 1.5 10015
250208
L-3,4-MPDC (36) 5 10050
120120
KA (30) 59 100600
00
Cis-5-Methyl-L-t-2,3-PDC (35) 37 100350
30
DHK (31) 28 100300
011
DHK+ (31)
L-Glu (1)L-t-2,3-PDC (34) 33
300
10100300
60
319
L-trans-2,3-PDC+ (34)L-Glu (1)
30010
76
a Data from Ref. [176]

616 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Table 9. Effects of Glutamate Analogues on GlutamateTransport in Astrocyte Culturesa
Compound IC50 (µM) Ki (µM)Imax (%
Inhibitors)
L-THA (19) 41 17 93
DL-TBOA 41 17 86
L-t-2,4-PDC (32) 74 31 97
L-3,4-MPDC (36) 87.5 37 92
cis-4-Methyl-L-t-2,4-PDC (33) 102 43 81
aData from Ref. [144]
6a.4. Novel Azole-Based Bioisosteres of Glutamic andAspartic Acid
Recently, two new classes of EAAT ligands werereported [181-183] (Fig. (8)).
Krogsgaard-Larsen and co-workers [181] have long beeninvolved in the synthesis of glutamate analogues, bindingmainly AMPA receptors. Recently they reported thepharmacological evaluation of a new selective EAAT2inhibitor (±)-, (+)-, and (-)-TDPA, based on a 3-hydroxy-1,2,5-thiadiazole skeleton (38a,b). These compounds weredeveloped with the objective of investigating the effect ofdifferent heterocyclic bioisosteric groups of the distal acidicfunction of Glu. These compounds were tested on COS-7cells transiently transfected with EAAT1, EAAT2 andEAAT3. While (R)-TDPA (38b) was found to be inactive asan inhibitor of the three transporters, (S)-TDPA (38a) was aselective inhibitor of EAAT2 subtype, but lacked specificitysince it interacted also with iGluRs. Although this unspecificinteraction limited its use as a pharmacological tool, 38arepresents one of the few examples of selective inhibitors ofa EAAT subtype, and prompted the Vedso research group[182] to search for other bioisosteric azoles mimicking thedistal carboxyl group of (S)-glutamic acid. Accordingly, anew series of 1-hydroxyazoles was synthesized and testedagainst ionotropic Glu receptors and EAAT1-3. The different
Table 10. Correlation Between Synaptosomal and Cloned EAAT1-3a,b
Synaptosomes EAAT1 EAAT2 EAAT3Compound
Ki efflux Km Imax Km or Ki* Imax Km Imax
µM % of Glu µM % of Glu µM % of Glu µM % of Glu
L-Glu (1) 5 100 20 100 18 100 28 100
DHK (31) 28 3 n.d. n.d. 9* 0 n.d. n.d.
L-t-2,3-PDC (34) 33 5 n.d. n.d. 12* 0 n.d. n.d
L-2,4-MPDC (37) 7 82 87 40 45 115 85 54
aData from Ref. [176]bAnalogs were evaluated as blockers and substrates inhibitors in oocytes expressing EAAT1, EAAT2, and EAAT3. In the instances where compounds exhibited no substrate activity(*), K i values were calculated by Schild analysis. In the heteroexchange studies, analogs were tested at concentation ≈10 x greater than the Ki values for inhibition of D-aspartateuptake. Values indicating substrates activity are reported as a percentage of the activity of L-glutamate at 37°C.
Fig. (8). Novel Azole-Based Bioisosters of Glutamic and Aspartic Acid.
NN
N
HO
O
H2N
NS
N
HO
O
38a, (S)-TDPA
R
O N
CO2H
CO2H
NH2
O N
CO2H
CO2HH2N
N O
CO2HHO2 C
N O
CO2HHO2 C
38b, (R)-TDPA
H2N
ON
HO
O
H2NOH OH OH
39a, R= H39b, R= CH3
41 42 43 44
40
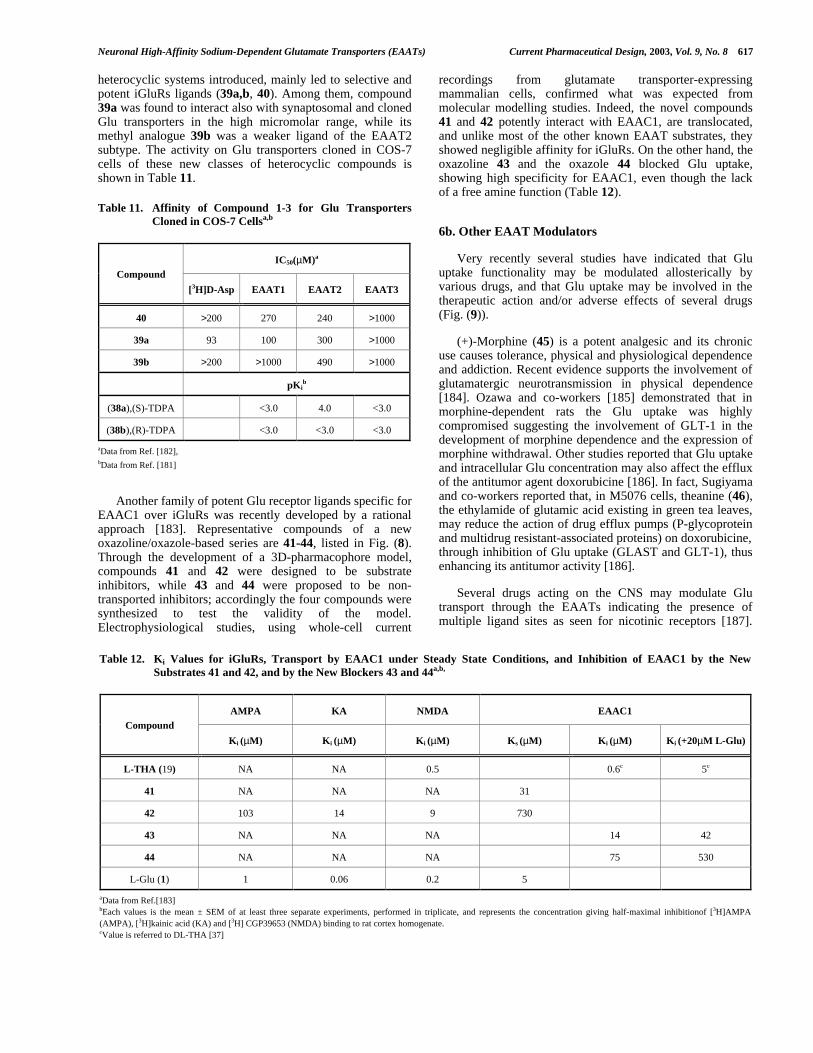
Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 617
heterocyclic systems introduced, mainly led to selective andpotent iGluRs ligands (39a,b, 40). Among them, compound39a was found to interact also with synaptosomal and clonedGlu transporters in the high micromolar range, while itsmethyl analogue 39b was a weaker ligand of the EAAT2subtype. The activity on Glu transporters cloned in COS-7cells of these new classes of heterocyclic compounds isshown in Table 11.
Table 11. Affinity of Compound 1-3 for Glu TransportersCloned in COS-7 Cellsa,b
IC50(µM)a
Compound
[3H]D-Asp EAAT1 EAAT2 EAAT3
40 >200 270 240 >1000
39a 93 100 300 >1000
39b >200 >1000 490 >1000
pKib
(38a),(S)-TDPA <3.0 4.0 <3.0
(38b),(R)-TDPA <3.0 <3.0 <3.0
aData from Ref. [182],bData from Ref. [181]
Another family of potent Glu receptor ligands specific forEAAC1 over iGluRs was recently developed by a rationalapproach [183]. Representative compounds of a newoxazoline/oxazole-based series are 41-44, listed in Fig. (8).Through the development of a 3D-pharmacophore model,compounds 41 and 42 were designed to be substrateinhibitors, while 43 and 44 were proposed to be non-transported inhibitors; accordingly the four compounds weresynthesized to test the validity of the model.Electrophysiological studies, using whole-cell current
recordings from glutamate transporter-expressingmammalian cells, confirmed what was expected frommolecular modelling studies. Indeed, the novel compounds41 and 42 potently interact with EAAC1, are translocated,and unlike most of the other known EAAT substrates, theyshowed negligible affinity for iGluRs. On the other hand, theoxazoline 43 and the oxazole 44 blocked Glu uptake,showing high specificity for EAAC1, even though the lackof a free amine function (Table 12).
6b. Other EAAT Modulators
Very recently several studies have indicated that Gluuptake functionality may be modulated allosterically byvarious drugs, and that Glu uptake may be involved in thetherapeutic action and/or adverse effects of several drugs(Fig. (9)).
(+)-Morphine (45) is a potent analgesic and its chronicuse causes tolerance, physical and physiological dependenceand addiction. Recent evidence supports the involvement ofglutamatergic neurotransmission in physical dependence[184]. Ozawa and co-workers [185] demonstrated that inmorphine-dependent rats the Glu uptake was highlycompromised suggesting the involvement of GLT-1 in thedevelopment of morphine dependence and the expression ofmorphine withdrawal. Other studies reported that Glu uptakeand intracellular Glu concentration may also affect the effluxof the antitumor agent doxorubicine [186]. In fact, Sugiyamaand co-workers reported that, in M5076 cells, theanine (46),the ethylamide of glutamic acid existing in green tea leaves,may reduce the action of drug efflux pumps (P-glycoproteinand multidrug resistant-associated proteins) on doxorubicine,through inhibition of Glu uptake (GLAST and GLT-1), thusenhancing its antitumor activity [186].
Several drugs acting on the CNS may modulate Glutransport through the EAATs indicating the presence ofmultiple ligand sites as seen for nicotinic receptors [187].
Table 12. Ki Values for iGluRs, Transport by EAAC1 under Steady State Conditions, and Inhibition of EAAC1 by the NewSubstrates 41 and 42, and by the New Blockers 43 and 44a,b,
AMPA KA NMDA EAAC1Compound
Ki (µM) Ki (µM) Ki (µM) Ks (µM) Ki (µM) Ki (+20µM L-Glu)
L-THA (19) NA NA 0.5 0.6c 5c
41 NA NA NA 31
42 103 14 9 730
43 NA NA NA 14 42
44 NA NA NA 75 530
L-Glu (1) 1 0.06 0.2 5
aData from Ref.[183]bEach values is the mean ± SEM of at least three separate experiments, performed in triplicate, and represents the concentration giving half-maximal inhibitionof [3H]AMPA(AMPA), [3H]kainic acid (KA) and [3H] CGP39653 (NMDA) binding to rat cortex homogenate.cValue is referred to DL-THA [37]

618 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
Neuroleptics and benzodiazepines (Bzs) interfere withEAAT functionality. Bzs represent a well known class ofpotent anxiolytic and sedative agents whose effects in theCNS are mediated by different mechanisms. For example,Bzs not only modulate GABA-gated chloride channels, butcan antagonize AMPA receptors, reducing the excitotoxiceffect of Glu [188]. Furthermore, it has been recentlydescribed that, in CHO cells and Xenopus laevis oocytesexpressing EAAC1, low concentrations of medazepam,oxazepam (47), lorazepam and diazepam (48) may stimulatethe transport activity of EAAC1, whereas highconcentrations inhibit it [189]. Palmeda and co-workers[189] suggested that the neuronal Glu transporter EAAC1may also be a target of Bzs. The effect of Bzs on EAATssuggest a direct interaction with EAAC1 and these authorsproposed the presence of two different sites for Bzs onEAAC1, one stimulatory and another one inhibitory. Morerecently, the same authors [190] investigated the effect ofBzs on EAAT1 and EAAT2 expressed on CHO cells anddemonstrated that not only neuronal but also glialtransporters may be targeted by Bzs. Diazepam (48) wasfound to be the most potent modulator, and oxazepam (47)was found to be an activator or inhibitor at low and high
concentrations, respectively. In general, a potential role ofneuronal and glial transporters in the action of Bzs wasdemonstrated and such modulation of EAAT functionality byBzs suggests an additional mechanism for the anxiolyticeffect of this class of drugs.
Activity and expression of Glu transporters may bemodulated also by second messengers and protein kinases.Recent evidence suggests that activation of protein kinase C(PKC), but not A (PKA), efficiently, but indirectly, regulatesthe EAAT activity. Application of phorbol 12 myristate-13-acetate (PMA, a PKC agonist) to oocytes expressingEAAC1, determined a decrease in the Glu-evoked uptakecurrent [191].
Niflumic acid (49), a non-steroidal anti inflammatorydrug (NSAID), may modulate Glu uptake by EAAT4expressed in Xenopus laevis oocytes [192] (Table 13).
Table 13. Kinetic Constant for L-Glu and L-Asp- ElicitedCurrents in the Absence and Presence of 300 µMa
0 µM NFA (49) 300 µM NFA (49)
Substrate K0.5 (µM) Imax K0.5 (µM) Imax
L-Glu 1.1 1 1 5
L-Asp 2.5 1 1.3 2.5
aData from Ref. [192]Values presented are mean ± S.E.M. from 4 cells and 6 cells for L-Asp. Imax isnormalized to the Imax without niflumic acid (NFA). K0.5 values for either L-Glu or L-Asp were not significantly different in the absence and presence of niflumic acid ( L-Glu, P= 0.85; L-Asp, P= 0.21), but Imax values are significantly different (L-Glu, P<0.001; L-Asp, P= 0.047)
This NSAID significantly enhanced the substrate-gatedcurrents in EAAT4 without affecting the affinity of theendogenous substrate towards EAAT4. Although its activitywas inhibited by the Glu transporter blocker D,L-TBOA, itwas demonstrated that niflumic acid (49) could beconsidered an allosteric modulator by acting at a differentrecognition site, representing a lead compound for thedevelopment of novel EAAT modulators.
(+)-Amphetamine is a drug that can modulate Glu uptake[193]. (+)-Amphetamine (50), in fact, induces an increase ofdelayed nature in Glu efflux in the ventral tegmental area(VTA), and this effect may be involved in (+)-amphetamineneurotoxicity (associated with the production of reactiveoxygen species (ROS)). The mechanism underlying (+)-amphetamine-induced Glu efflux has been recentlycharacterized [193]. Repeated elevation of Glu in VTA leadsto the induction of behavioral sensitization. This effect ismediated by Glu transporters, is inhibited by DHK, and mayinvolve ROS. The inhibitory activity of DHK (31) indicatesthat the (+)-amphetamine-induced Glu efflux is due toreversal of the Glu transporter. This mechanism is mainlydue to the disruption of ionic gradients necessary for Glu-inward transport, induced by the drug. Use of α-phenyl-N-tert-butyl nitrone, which reacts with free radicals to prevent
Fig.(9). Other EAAT Modulators.
NH2
CH3
O
NH
H N OH
O
H
O
NH2
N
HN O
OHCl N
NO
OH
Cl
HO
HO
CH3
Et
CH3
NH
N
CO2H
CF3
50, D-(+)-Amphetamine
45, (+)-Morphine 46, Theanine
47, Oxazepam 48, Diazepam
49, Niflumic Acid

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 619
neuronal damage, and D-phenylalanine, a trapping reagentfor hydroxyl radicals, attenuate amphetamine toxicity andthe development of behavioural sensitization, indicating adirect role of ROS in the activity/toxicity of 50.
7. STRUCTURAL AND CONFORMATIONALREQUIREMENTS FOR BINDING TO EAATS ANDTRANSLOCATION
Due to the lack of high resolution structures of EAATs,only ligand-based approaches have been used to elucidatethe structural and conformational features required forEAATs binding and translocation. The small number ofEAATs specific ligands so far developed, makes acomprehensive structure-activity relationship analysisdifficult to formulate. Most of the known ligands bear an α-aminoacid moiety, separated by 1-to-3 methylenes from adistal acidic group. These three functional groups (aprotonatable nitrogen and two acidic functions) can beassumed to be the transporter interacting points, and herewill be referred to as pharmacophoric points or groups. Sincevarious glutamate analogues exhibited selectivity towardsthe different glutamate interacting proteins, the first attemptto find the conformational requirements for binding toEAATs was based on the assumption that Glu binds to itsreceptors by different conformations. Starting from thishypothesis, Ohfune and co-workers synthesized through astereoselective pathway L-2-(2-carboxycyclopropyl)glycines(L-CCGs, 13-16). These compounds, which mimic differentGlu conformations, displayed selective affinity for differentglutamate receptors; among them, L-CCG-III (15) was foundto be a potent uptake inhibitor [160]. When superimposedupon L-t-2,4-PDC (32) by fitting the three pharmacophoricpoints, the cyclopropane ring system of L-CCG-III did notlie in the same area occupied by the cyclopropane ringsystems of the inactive L-CCG-I (13) and L-CCG-IV (16)isomers; furthermore the α-carboxyl group of 15 and thecorresponding carboxyl group of 32 showed a differentorientation and position with respect to the α-carboxylicfunctions of 13 and 16 [164]. These data further supportedthe hypothesis of different Glu conformations for selectivebinding to the various Glu receptors.
Furthermore, in order to define a three dimensionalpharmacophoric model for EAAT ligands, several otherspeculations have been made about the Glu bindingconformation. High affinity sodium dependent glutamatetransporters bind either D- and L-Aspartate while being ableto discriminate between glutamate enantiomers since D-glutamate (which potently activates ionotropic receptors) hasvery low affinity for the transport systems. Cooper and co-workers [194] gave a possible explanation for thisexperimental observation by proposing that the aspartatebinding (extended) conformation allows both Aspenantiomers to bind to the active site as “mirror images”; onthe other hand, comparing the putative (partially folded) L-glutamate binding conformation with that of D-glutamate, itis not possible to conciliate the same orientation of D- and L-glutamate pharmacophoric groups, when they bind as mirrorimages, with the same orientation of the carbon backbone[184]. In fact, according to this model, the steric hindrance ofthe D-glutamate carbon chain buckles in a different area
from that occupied by the chain of L-Glu, thus possiblyprecluding the interaction with the binding site. Vandenbergand co-workers, proposed a different Glu binding mode forEAAT1 and EAAT2, comparing constrained Glu analoguesacting on both transporters with kainate (selective forEAAT2) [153]. These authors suggested that Glu bindseither EAAT1 and EAAT2 in the extended conformation,while the partially folded conformation is adopted byEAAT2 blockers. The authors also rationalised the samemechanism of action on EAAT2 of T3MG (9a), 2S4R4MG,(8), and kainate (30), by the observation that, when theirpharmacophoric groups are superimposed, the methyl groupof T3MG aligns with the ring structure of kainate at thecorresponding position, and the methyl group of 2S4R4MGlays in the same region of the kainate isopropene side chainand pyrrolidine ring. Since 9a was found inactive towardsEAAT1, while 8 is a substrate inhibitor, it seems that onlythe methyl at position 4 is tolerated for the binding to thistransporter subtype.
Through the synthesis and the pharmacologicalevaluation of conformationally restricted analogues of Glu,such as 2,4-pyrrolidine dicarboxylates isomers, Bridges andco-workers proposed that Glu binds EAATs in a partiallyfolded conformation, so that the distance between the twocarboxyl groups is 4.2 ±0.3 Å and the distance between thedistal carboxyl function and the nitrogen is 3.4 ±0.3 Å [175].
In general, modification on the Asp backbone are muchmore well tolerated, leading to potent EAAT ligands. Indeed,a number of L-THA (19) derivatives, bearing differenthydrophobic substituents on the hydroxyl function (21-29),have been synthesised and tested [163,167]. Although mostof the resulting compounds were able to bind to thetransporters, they behaved as substrate inhibitors or blockersdepending on the nature of the substituents. Furthermore, itwas observed that, when the pharmacophoric groups of L-TBOA (27) and DHK (31) are superimposed, their bulkysubstituents largely overlap [168].
An attempt to rationalise the molecular basis of thedifferentiation between the intrinsic pharmacologicalproperties of substrate inhibitors and blockers was reportedby Bridges and co-workers, who hypothesised that therewere at least two pharmacophores for EAAT interaction: onefor substrate inhibitors and the other for non-transportableinhibitors [145]. These authors proposed that the positioningof the functional groups of the non-substrates is exemplifiedby the folded conformation of L-t-2,4-PDC (32), resembledby L-3,4-MPDC (36). On the other hand, the more extendedconformer of L-t-2,4-PDC (32), resembled by L-2,4-MPDC(37), lock the structure in a position suitable for a substrate-like activity, typifying good substrates. The role of the sterichindrance in differentiation of substrate and non-transportable EAAT inhibitors has been highlighted by Kochand co-workers [176]. By the superimposition of thepharmacophoric points of several EAAT ligands, it wasobserved that both, substrate and non-transportableinhibitors, exhibited a high degree of overlap with L-glutamate, while steric excess in specific positions representsa determining, or at least predicting, factor in whether or notthe analogues can be translocated. More recently, asystematic analysis of the different structural determinants

620 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
required for a EAAT ligand to behave as a substrate inhibitoror a blocker, led to the rational design of new EAATtransported and non-transportable inhibitors [183]. The novelcompounds are characterised by a oxazole/oxazolineskeleton bearing two carboxylic functions which aredifferently spaced and orientated. The lack of a protonatablenitrogen in these oxazole based compounds indicates that thepositive charge on the amine function is not essential forEAAT binding. According to this 3D-pharmacophoricmodel, all analysed EAAT ligands can share the sameorientation of the acidic functions, even if the distancebetween the carboxylic carbons can vary from 3.7 to 4.9 Å.The distance between the two carboxylic functions is notable to discriminate between substrate inhibitors andblockers, but between glutamate and aspartate-relatedderivatives, which are present in both classes, while thedifference in activity depends on the differences in thevolume distribution of the rest of the molecule with respectto the axis connecting the two carboxylic groups (Fig. (10)).From the superimposition reported in Fig. (10) it can beobserved that a constrained folded glutamate-like
conformation (i.e. L-3,4-MPDC (white)) forces the volumeexcess to occupy the edge opposite to the one wherecarboxylic groups are located (blockers), while a constrainedunfolded glutamate-like conformation (i.e. L-t-2,4-PDC(magenta)) allows a quite uniform volume distribution onboth sides of the backbone (substrate inhibitors).
8. PERSPECTIVES
Although our understanding of Glu uptake is stillincomplete, the Glu transporters appear to possess morerefined functions than Glu removal, thus playing a key role
in the regulation of synaptic transmission. Their density andlocalization in specific brain regions may have criticalimplication in brain (and peripheral) functioning. Byconsequence, abnormalities in Glu transport may be at leastpartially responsible for several neurological diseases.Recently the number of selective substrates and inhibitors tobe used as probes of structure and function definition of thedifferent EAAT subtypes is slowly increasing. The resultingstructure-activity relationship data will allow refinement ofthe pharmacological properties of the new ligands and willlead to increasingly detailed pharmacophore models of thedifferent EAAT substrate binding domains for a futuredesign of subtype selective ligands. Furthermore, veryrecently a number of proteins capable of modulating Glutransporter subtype functioning have been identified. Futuremanipulation of these proteins may also provide noveltherapeutic means to regulate Glu transport. In conclusion,such compounds with suitable pharmacokinetic propertiesand the ability to modulate glutamate uptake in differentbrain regions, will have intriguing clinical implications,especially on neurodegenerative disorders.
REFERENCES
References 195-197 are related articles recently published inCurrent Pharmaceutical Design.
[1] Headley, P. M., Grillner, S. Trends Pharmacol. Sci., 1990,11, 205.
[2] Seal, R. P., Amara, S.G. Annu. Rev. Pharmacol. Toxicol.,1999, 39, 431. b) Amara, S.G., Fontana, A.C.K.,Neurochem. Int., 2002, in press.
[3] Danbolt, N. C. Prog. Neurobiol., 2001, 65, 1.
Fig. (10). Superimposition of known blockers and substrate inhibitors within the exceeding volume for blockers, with respect to substrateinhibitors (red framework), and for substrate inhibitors with respect to blockers (cyan framework). (A) Selective substrate inhibitors: L-t-2,4-PDC (32, magenta), L-CCG III (15, green), L-THA (19, orange). (B) Selective blockers: L-3,4-MPDC (36, white), L-TBOA (27, yellow), 44(green). Superimposition was obtained by fitting the protonatable nitrogen (blue) and the two carboxylic carbons (oxygens in red).Hydrogens are omitted for clarity. Adapted from ref. [183].

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 621
[4] Takamori, S., Rhee, J. S., Rosenmund, C., Jahn, R. Nature,2000, 407, 189.
[5] Bergles, D. E., Roberts, J. D., Somogyi, P., Jhar, C.E.Nature, 2000, 405, 187.
[6] Gegelashvili, G., Schousboe, A. Mol. Pharm., 1997, 52, 6.
[7] Michaelis, E. K. Prog. Neurobiol., 1998, 54, 369.
[8] Cotman, C, W., Kahle, J. S., Miller, S. E., Bridges, R. J.,Psychopharmacology: The Fourth Generation of Progress,Bloom, F.E. and Kupfers, D. J., Eds; Raven Press: NewYork, 1997, pp. 75-85.
[9] Bergles, D.E., Diamond, J. S., Jahr, C. E. Curr. Opin.Neurobiol., 1999, 9, 293.
[10] Takahashi, M., Billups, B., Rossi, D., Sorentis, M., Hamann,M., Attwell, D. J. Exp. Biol., 1997, 200, 401.
[11] Armer, R. C. Curr. Medicinal Chem., 2000, 7, 199.
[12] Tanaka, K. Neurosci. Res., 2000, 37, 15.
[13] Palacín, M., Estévez, R., Bertran, J., Zorzano, A.Physiological Rev., 1998, 78, 969.
[14] Slotboom, D. J., Konings, W. N., Lolkema, J. S. Microbiol.Mol. Biol. Rev., 1999, 63, 293.
[15] Bridges, R. J., Kavanaugh, M. P., Chamberlin, A. R. Curr.Pharm. Design, 1999, 5, 363.
[16] Saier, M. H., Jr. http://www-biology.ucsd.edu/-msaier/transport/. 1998
[17] Lolkema, J. S., Slotboom, D. J. Mol. Membr. Biol., 1998,15, 33.
[18] Lolkema, J. S., Slotboom, D. J. FEMS Microbiol. Rev.,1998, 22, 305.
[19] Kanai, Y. Curr. Opin. Cell Biol., 1997, 9, 565.
[20] Arriza, J. L., Fairman, W. A., Wadiche, J. I., Murdoch, G.H., Kavanaugh, M. P., Amara, S. G. J. Neurosci., 1994, 14,5559.
[21] Arriza, J. L., Eliasof, S., Kavanaugh, M. P., Amara, S. G.Proc. Natl. Acad. Sci. USA, 1997, 94, 4155.
[22] Donly, B. C., Richman, A., Hawkins, E., McLean, H.Caveney, S. Eur. J. Biochem., 1997, 248, 535.
[23] Levy, L. M., Warr, O., Attwell, D. J. Neurosci., 1998, 18,9620.
[24] Zerangue, N., Kavanaugh, M. P. Nature, 1996, 383, 634.
[25] Eliasof, S., Arriza, J. L., Leighton, B. H., Kavanaugh, M. P.,Amara, S. G. J. Neurosci., 1998, 18, 698.
[26] Billups, B. D., Rossi, D., Attwell, D. J. Neurosci., 1996, 16,6722.
[27] Fairman, W. A., Vandenberg, R. J., Arriza, J. L.,Kavanaugh, M. P., Amara, S. G. Nature, 1995, 375, 599.
[28] Kanner, B. I., Kavanaugh, M. P., Bendahan, A. Biochem.Soc. Trans., 2001, 29, 707.
[29] Slotboom, D. J., Konings, W. N., Lolkema, J. S. FEBS Lett.,2001, 492, 183.
[30] Slotboom, D. J., Konings, W. N., Lolkema, J. S. TrendsBiochem. Sci., 2001, 26, 534.
[31] Kanner, B. I., Bendahan, A., Biochemisty, 1982, 21, 6327.b) Kanai Y., Nussberger, S., Romero, M. F., Boron, W. F.,Hebert, S.C., Hediger, M. A. J. Biol. Chem., 1995, 270,16561.
[32] Kavanaugh, M. P., Bendahan, A., Zerangue, N., Zhang, Y.,Kanner, B. I. J. Biol. Chem., 1997, 272, 1703.
[33] Pinnes, G., Kanner, B. I. Biochemisty, 1990, 29, 11209.
[34] Otis, T. S., Jahr, C.E. J. Neurosci.,1998,18, 7099.
[35] Auger, C., Attwell, D. Neuron, 2000, 28, 547.
[36] Wadiche, J. I., Kavanaugh, M. P. J. Neurosci., 1998, 18,7650.
[37] Grewer, C. F., Watzke, N., Wiessner, M., Rauen, T. Proc.Natl. Acad. Sci. USA, 2000, 97, 9706.
[38] Otis, T. S., Kavanaugh, M. P. J. Neurosci., 2000,20, 2749.
[39] Wadiche, J. L., Amara, S. G., Kavanaugh, M. P. Neuron,1995, 15, 721.
[40] Attwell, D., Laughlin, S.B. J. Physiol., 2000, 512, 61P.
[41] Gegelashvili, G., Dehnes, Y., Danbolt, N. C., Schousboe, A.Neurochem. Int., 2000, 37, 163.
[42] Liu, Y., Krantz, D. E., Waites, C., Edwards, R. H. Tr. CellBiol., 1999, 9, 356.
[43] Gegelashvili, G., Schousboe, A. Brain Res. Bull., 1998, 45,233.
[44] Munir, M., Correale, D. M., Robinson, M. B. Neurochem.Int., 2000, 37, 147.
[45] Conradt, P. J., Stoffel, W. J. Neurochem., 1997, 68, 1244.
[46] Davis, K. E., Straff, J. D., Weinstein, E. A., Bannerman, P.G., Correale, D. M. Rothstein, J. D., Robinson, M. B. J.Neurosci., 1998,18, 2475.
[47] Casado, M., Bendahan, A., Zafra, F., Danbolt, N. C.,Gimenez, C., Kanner, B. I. J. Biol. Chem., 1993, 268,27313.
[48] Lin, C. G., Orlov, I., Ruggiero, A. M., Dykes-Hoberg, M.,Lee, A., Jackson, M., Rothstein, J. D. Nature, 2001, 410, 84.
[49] Jackson, M., Song, W., Liu, M., Jin, L., Dykes-Hoberg, M.,Lin, C. G., Bowers, W. J., Federoff, H. J., Sternweis, P. C.,Rothstein, J. D. Nature, 2001, 410, 89.
[50] Zerangue, N., Arriza, J. L., Amara, S. G., Kavanaugh, M. P.J. Biol. Chem., 1995, 270, 6433.
[51] Trotti, D., Volterra, A., Lehere, K. P. Rossi, D., Gjesdal, O.,Recagni, G., Danbolt, N. C. J. Biol. Chem., 1995, 270, 9890.
[52] Lauderback, C. M., Hackett, J. M., Huang, F., Keller, J. N.,Szweda, L. I., Markesbery, W. R., Butterfield, D. A. J.Neurochem., 2001, 78, 413.
[53] Liang, Z., Valla, J., Sefidvash-Hockeley, S., Rogers, J., Li,R. J. Neurochem., 2002, 80, 807.

622 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
[54] Yatin, S., Varadarajan, S., Butterfield, D. A. J. Alzheimer’sDis., 2000, 2, 123.
[55] Lauderback, C. M., Harris-White, M. E., Pedigo, N., Wang,Y., Varney, J. M., Butterfield, D. A. Life Sci., 1999, 65,1977.
[56] Trotti. D., Rizzini, B., Rossi, D., Haugeto, O., Racagni, G.,Danbolt, N. C., Volterra A. Eur. J. Nuerosci., 1997, 9, 1236.
[57] Trotti, D., Danbolt, N. C., Volterra, A. Trends Pharmacol.Sci., 1998, 19, 328.
[58] Reis, H. J., Gomez, M. V., Kalapothakis, E., Diniz, C. R.,Cordeiro, M. N., Prado, M. A. M., Romano-Silva, M. A.Neuroreport., 2000, 11, 2191.
[59] Brasnjo G., Otis, T. S. Neuron, 2001, 31, 607.
[60] Hertz, L. Prog. Neurobiol., 1979, 13, 277.
[61] Kanai, Y., Hediger, M. A. Nature, 1992, 360, 467.
[62] Schmitt, A., Asan, E., Lesch, K. P., Kugler, P. Neurosci.,2002, 109, 45.
[63] Utsumi, M., Ohno, K., Onchi, H., Sato, K., Tohyama, M.,Mol. Brain Res., 2001, 92, 1.
[64] Pines, G., Danbolt, N. C., Bjøras, M., Zhang, Y., Bendahan,A., Eide, L., Koepsell, H., Storm-Mathisen, J., Seeberg, E.,Kanner, B. I. Nature, 1992, 360, 464.
[65] Storck, T., Schultz, S., Hofmann, K., Stoffel, W. Proc. Natl.Acad. Sci., USA, 1992, 89, 10955.
[66] Tanaka, K. Neurosci. Res., 1993, 16, 149.
[67] Zelenaia, O., Schlang, B. D., Gochenauer, G. E., Ganel, R.,Song, W., Beesley, J. S., Grinspan, J. B., Rothstein, J. D.,Robinson, M. B. Mol. Pharm., 2000, 57, 667.
[68] Sims, K. D., Robinson, M. B., Grit. Rev. Neurobiol., 1999,13, 169.
[69] Nakayama, T., Kawakami, H., Tanaka, K., Nakamura, S.Mol. Brain Res., 1996, 36, 189.
[70] Hediger, M. A. J. Physiol., 1999, 277, F487-F492.
[71] Banner, S. J., Fray, A. E., Ince, P.G., Steward, M., Cookson,M. R., Shaw, P. J. Neuroscience, 2002, 109, 27.
[72] O’Kane, R. L., Martínez-López, I., DeJoseph, M. R., Viña,J. R., Hawkins R. A. J. Biol. Chem., 1999, 274, 31891.
[73] Grunewald, M., Bendahan, A., Kanner, B. I. Neuron, 1998,21, 623.
[74] Slotboom, D. J., Sobczak, I., Konings, W. N., Lolkema, J. S.Proc. Natl. Acad. Sci. USA, 1999, 95, 14282.
[75] Grunewald, M., and Kanner, B. I. J. Biol. Chem., 2000, 275,9684.
[76] Seal, R. P., Leighton, B. H., Amara, S. G. Neuron, 2000, 25,695.
[77] Zhang, Y., Bendahan, A., Zarbiv, R., Kavanaugh, M. P.,Kanner, B. I. Proc. Natl. Acad. Sci. USA, 1998 95, 751.
[78] Zarbiv, R., Grunewald, M., Kavanaugh, M. P., Kanner, B. I.J. Biol. Chem., 1998, 273, 14231.
[79] Brocke, L., Bendahan, A., Grunewald, M., Kanner, B. I. J.Biol. Chem., 2002, 277, 3985.
[80] Slotboom, D. J., Konings, W. N., Lolkema, J. S. J. Biol.Chem., 2001, 276(14), 10775.
[81] Slotboom, D. J., Lolkema, J. S., Konings, W. N. J. Biol.Chem., 1996, 271(49), 31317.
[82] Murata, K., Mitsuoka, K., Hirai, T., Walz, T., Agre, P.,Heymann, J. B., Engel, A., Fujiyoshi, Y. Nature, 2000, 407,599.
[83] Seal, R. P. and Amara, S. G. Neuron, 1998, 6, 1487.
[84] Leighton, B. H., Seal, R. P., Shimamoto, K., Amara, S. G. J.Biol. Chem. 2002, avaliable on www.jbc.org , JBC Papers inPress Manuscript M202508200.
[85] Eskandari, S., Kreman, M., Kavanaugh, M. P., Wright, E.M., Zampighi, G. A. Proc. Natl. Acad. Sci. USA, 2000, 97,8641.
[86] Pines, G., Zhang, Y., Kanner, B. I. J. Biol. Chem., 1995,270(29),17093.
[87] Zhang, Y., and Kanner, B. I. Proc. Natl. Acad. Sci. USA,1999, 96, 1710.
[88] Kunishima, N., Shimada, Y., .Tsuji, Y., Sato, T.,Yamamoto, M., Kumasaka, T., Nakanishi, H.J., Morikawa,K. Nature, 2000 407, 971.
[89] Armstrong, N., Sun, Y., Chen, G.Q., Gouaux, E. Nature,1998 395, 913.
[90] Borre, L., Kavanaugh, M.P., Kanner, B.I. J. Biol. Chem.,2002, 277(16), 13501.
[91] Seal, R. P., Shigeri, Y., Eliasof, S., Leighton, B. H., Amara,S. G. Proc. Natl. Acad. Sci. USA, 2001, 98, 15324.
[92] Mitrovic, A. D., Amara, S. G., Johnstone, G. A. R.,Vandenberg, R. J. Biol. Chem., 1998, 273(24), 14698.
[93] Bendahan, A., Armon, A., Madani, N., Kavanaugh, M.P.,Kanner, B.I. J. Biol. Chem., 2000, 275, 37436.
[94] Conradt, M., and Stoffel, W. J. Biol. Chem., 1995, 270,25207.
[95] Maragakis, N. J., Jeffrey, M. D., Rothstein, M. D. Arch.Neurol., 2001, 58, 365.
[96] Tanaka, K., Watase, K., Mabe, T., Yamada, K., Watanabe,M., Takahashi, K., Iwama, T., Ichihara, N., Kikuchi, T.,Okuyama, S., Kawashima, N., Hori, S., Takimoto, M.,Wada, K. Science, 1997, 276, 1699.
[97] Peghini, P., Janzen, J., Stoffel, W. EMBO J., 1997, 16,3822.
[98] Kato, S., Negishi, K., Mawatari, K., Kuo, C. H.Neuroscience, 1992, 48, 903.
[99] Yasui, Y. Mawatari, K., Higuchi, Y., Tanii, H., Kato, S. In:Free Radicals in Brain Physiology and Disorders. Packer,L., Hiramatsu, L., Yoshikawa, T.: Academic Press, SanDiego, CA, 1996, pp. 51-67.
[100] Garthwaite, G., Williams, G.D., Garthwaite, J. Eur. J.Neurosci., 1992, 4, 353.

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 623
[101] Tao, F., Lu, S. D., Zhang, L. M., Huang, Y. L., Sun, F. Y.Neuroscience, 2001, 102, 503.
[102] Fukamachi, S., Furuta, A., Ikeda, T., Ikenoue, T., Kaneoka,T., Rothstein, J. D. Iwaki, T. Develop. Brain Res., 2001,132, 131.
[103] Rossi, D. J., Oshima, T., Attwell, D. Nature, 2000, 403, 316.
[104] Koch, H. P., Chamberlin, A. R., Bridges, R. J. Mol. Pharm.,1999, 55, 1044.
[105] Scott, H. L., Pow, D. V., Tannenberg, A. E. G., Dodd, P. R.J. Neurosci., 2002, 22, RC206
[106] Liévens, J. C., Woodman, B., Mahal, A., Spasic-Boscovic,O., Samuel, D., Kerkerian-LeGoff, L., Bates, G. P.Neurobiol. Disease, 2001, 8, 807.
[107] Rothstein, J. D., Martin L. J., Kuncl, R. W. N. Engl. J. Med.,1992, 326, 1464.
[108] Rothstein, J. D., Tsai, G., Kuncl, R.W., Clawson, L.,Cornbalth, D., Drachman, D., Pestronk, A., Stauch, B.,Coyle J.T. Ann. Neurol., 1990, 28, 18.
[109] Plaitakis, A., Constantakakis, E., Smith, J. Ann. Neurol.,1988, 24, 446.
[110] Brown, J. A., Nijar, M. S. Mol. Cell Biochem., 1995, 151,49.
[111] Cookson, M. R., Shaw, P.J. Brain Pathol., 1999, 9, 165.
[112] Morrison, B. M., Morrison, J. H. Brain Res. Rev. , 1999, 29,121.
[113] Robberecht, W. J. Neurol., 2000, 247, 1.
[114] Ripps, M.E., Huntley, G.W., Hof, P.R., Morrison, J. H.,Gordon, J. W. Proc. Natl. Acad. Sci., USA, 1995, 92, 689.
[115] Shaw, P. J. Ann. Neurol., 1999, 46, 803.
[116] Spencer, P.S. Drug Metab. Rev., 1999, 31, 561.
[117] Alloul, K.,Sauriol, L., Kennedy, W., Laurier, C., Tessier, G.,Novosel, S., Contandriopoulos, A. Arch. Gerontol. Geriatr.,1998, 27, 189.
[118] Kalra, S., Arnold, D. L., Cashman, N. R. J. Neurol. Sci.,1999, 165(suppl 1): S27-S32.
[119] Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A.,Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, P.,Deng, H., Rahmani, Z., Krizus, A., McKenna-Yasek, D.,Cayabyab, A., Gaston, S. M., Berger, R., Tanzi, R., E.,Halperin, J., J., Herzfeldt, B., Van den Bergh, R., Hung, W.,Bird, T., Deng, G., Mulder, D., W., Smyth, C., Laing, N.,G., Soriano, E., Pericak-Vance, ,M., A., Haines, J., Rouleau,G., A., Gusella, J., S., Horvitz, H. R., Brown, R. H., jrNature, 1993, 362, 59.
[120] Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C.,Polchow, C. Y., Alexander, D. D., Caliendo, J., Hentati, A.,Kwon, Y. W., Deng, H., Chen, W., Zhai, P., Sufit, R. L.,Siddique, T. Science, 1994, 264, 1772.
[121] Masliah, E. Alford, M., De Teresa, R., Mallory, M., Hansen,L. Ann. Neurol., 1996, 40, 759.
[122] Markesbery, W.R., Carney, J.M. Brain Pathol., 1999, 9,133.
[123] Masliah, E., Alford, M., Mallory, M., Rockenstein, E.,Moechars, D., Vanleuven, F. Exp. Neurol., 2000, 163, 381.
[124] Lauderback, C. M., Hackett, J., Keller, J. N., Varadarajan,S., Szweda, L., Kindy, M. S., Butterfield, D. A.Biochemistry, 2001, 40, 2548.
[125] Varadarajan, S., Yatin, S., Aksenova, M., Butterfield, D. A.J. Struct. Biol., 2000, 130, 184.
[126] Varadarajan, S., Kanski, J., Aksenova, M., Lauderback, C.M., Butterfield, D. A. J. Am. Chem. Soc., 2001, 123, 5625.
[127] Mattson, M. P., Guo, Z. H., Geiger, J. D. J. Neurochem.,1999, 73, 532.
[128] Masliah, E., Westland, C. E., Rockenstein, E., Abraham, C.R., Mallory, M., Veinberg, I., Sheldon, E., Mucke, L.Neuroscience, 1997, 78, 135.
[129] Tessler, S. Danbolt, N. C., Faull, R. L. M., Storm-Mathisen,J., Emson, P. C., Neuroscience, 1999, 88, 1083.
[130] Niebroj-Dodosz, I., Janik, P. Acta Neurol. Scand., 1999,100, 6.
[131] Akbar, M. T., Torp, R., Danbolt, N. C., Levy, L. M.,Meldrum, B. S., Ottersen, O. P. Neuroscience, 1997, 78,351.
[132] Hassel, B., Iversen, E. G., Gjerstad, L., Taubøll, E. J.Neurochem., 2001, 77, 1285
[133] Billups, B., Attwell, D. Nature, 1996, 379, 171.
[134] Szatkowski, M., Barbour, B., Attwell, D. Nature, 1990, 348,443.
[135] Storm-Mathisen, J. Danbolt, N. C., Rothe, F. Prog. BrainRes., 1992, 94, 225.
[136] Choi, D. W. Brain Res., 1994, 100, 47.
[137] Drejer, D. J., Benveniste, H., Diemer, N. H., Schousboe, A.J. Neurochem., 1985, 45, 145.
[138] Choi, D. W., Rothman, S. M. Annu. Rev. Neurosci., 1990,13, 171.
[139] Bräuner-Osborne, H., Egebjerg, J., Nielsen, E.Ø., Madsen,U., Krogsgaard-Larsen, P. J. Med. Chem., 2000, 43, 2609.
[140] Takamoto, A., Quiggin, L. B., Lieb, I., Shave, E., Balcar, V.J., Yoneda, Y. Life Sci., 2002, 70, 991.
[141] Mitrovic, A. D., Jhonston, G. A. R. Neurochem. Int., 1994 ,24, 583.
[142] Gordon, R. D., Balazs, R. J. Neurochem., 1983, 40, 1090
[143] Donly, C., Jevnikar, J., McLean, H., Caveney, S. Insect.Biochem. Molec. Biol., 2000, 30, 369.
[144] Anderson, C. M., Bridges, R. J., Chamberlin, A. R.,Shimamoto, K., Yasuda-Kamatani, Y., Swanson, R. A. J.Neurochem., 2001, 79, 1207.
[145] Esslinger, C. S., Koch, H. P., Kavanaugh, M. P., Philips, D.P., Chamberlin, A. R., Thompson, C. M., Bridges, R. J.Bioorg. Med. Chem. Lett., 1998, 8, 3101.
[146] Volterra, A., Bezzi, P., Rizzini, B.L., Trotti, D. Eur. J.Neurosci., 1996, 8, 2019.

624 Current Pharmaceutical Design, 2003, Vol. 9, No. 8 Campiani et al.
[147] Balcar, V. J., Johnston, G. A. R. J. Neuorochem., 1972, 19,2657.
[148] Roberts, P. J., Watkins, J. C. Brain Res., 1975, 85, 120.
[149] Griffiths, R., Grieve, A., Dunlop, J., Damgaard, I., Fosmark,H., Schousboe, A. Neurochem. Res., 1989, 14, 333.
[150] Vandenberg, R. J., Mitrovic, A. D., Jhonston, G. A. R. Br. J.Pharmacol., 1998, 123, 1593
[151] Vandenberg, R. J. Clin. Expt. Pharmacol. Physiol., 1998,25, 393
[152] Gu, Z. Q., Hesson, D.P., Pelletier, J. C., Maccecchini, M. L.,Zhou, L. M. Skolnick, P. J. Med. Chem., 1995, 38, 2518.
[153] Vandenberg, R. J., Mitrovic, A. D., Chebib, M., Balcar, J.,Jhonston, G. A. R. Mol. Pharm., 1997, 51, 809.
[154] Eliasof, S., McIlvain, H. B., Petroski, R. E., Foster, A. C.,Dunlop, J. J. Neurochem., 2001, 77, 550.
[155] Apricó, K., Beart, P. M., Lawrence, A. J., Crawford, D.,O’Shea, R. D. J. Neurochem., 2001, 77, 1218.
[156] Yamanoi, K., Ohfune, Y., Watanabe, K., Li, P.-N.,Takeuchi, H. Tetrahedron Lett., 1988, 29, 1181.
[157] Shinoazaki, H., Ishida, M., Shimamoto, K., Ohfune, Y. Br.J. Pharmacol., 1989, 98, 1213.
[158] Kawai, M., Horikawa, Y., Ishihara, T., Shimamoto, K.,Ohfune, Y. Eur. J. Pharmacol., 1992, 211, 195.
[159] Robinson, M. B., Sinor, J.D., Dowd, L. A., Kerwin, J. F., Jr.J. Neurochem., 1993, 60, 167.
[160] Shimamoto, K., Ohfune, Y. J. Med. Chem., 1996, 39, 407.
[161] Garlin, A., Sinor, A., Lee, S., Grinspan, J., Robinson, M. J.Neurochem., 1995, 64, 2572.
[162] Yamashita, H., Kawakami, H., Zhang, H., Hagiwara, T.,Tanaka, K., Nakamura, S. Eur. J. Pharmacol., 1995, 289,387.
[163] Shimamoto, K., LeBrun, B., Yasuda-Kamatani, Y.,Sakaitani, M., Shigeri, Y., Yumoto, N., Nakajima, T. Mol.Pharmacol., 1998, 53, 195.
[164] Lieb, I., Chebib, M., Cooper, B., Dias, L. S., Balcar, V. J.Neurochem. Int., 2000, 36, 319.
[165] Fletcher, E. J., Mewett, K. N., Drew, C. A., Allan, R.D.,Jhonston, G. A. R. Neurosci. Lett., 1991,121,133.
[166] Griffiths, R., Dunlop, J., Gorman, A., Senior, J., Grieve, A.Biochem. Pharmacol., 1994, 47, 267.
[167] LeBrun, B., Sakaitani, M., Shimamoto, K., Yasuda-Kamatani, Y., Nakajima, T. J. Biol. Chem., 1997, 272,20336.
[168] Shimamoto, K., Shigeri, Y., Yasuda-Kamatani, Y., LeBrun,B., Yumoto, N., Nakajima, T. Bioorg. Med. Chem. Lett.,2000, 10, 2407.
[169] Shigeri, Y., Shimamoto, K., Yasuda-Kamatani, Y., Seal, R.P., Yumoto, N., Nakajima, T., Amara, S.G. J. Neurochem .,2001, 79, 297.
[170] Longuemare, M.C., Swanson, R. A. J. Neurosci. Res., 1995,40, 379.
[171] Waagepetersen, H.S., Shimamoto, K., Schousboe, A.Neurochem. Res., 2001, 26, 661.
[172] Phillis, J.W., O’Regan, M.H. Brain Res., 2000, 868, 105.
[173] Ogata, T., Nakamura, Y., Shibata, T., Kataoka, K. J.Neurochem., 1992, 58, 1957.
[174] Sonnenberg, J. D., Koch, H.P., Willis, C. L., Bradbury, F.,Dauenhauer, D., Bridges, R. J., Chamberlin, A. R. Bioorg.Med. Chem. Lett., 1996, 6, 1607.
[175] Bridges, R. J., Stanley, M. S., Anderson, M. W., Cotman, C.W., Chamberlin, A. R. J. Med. Chem., 1991, 34, 717
[176] Koch, H. P., Kavanaugh, M. P., Esslinger, C. S., Zerangue,N., Humphrey, J. M., Amara, S. G., Chamberlin, A. R.,Bridges, R. J. Mol. Pharm., 1999, 56, 1095.
[177] Humphrey, J. M., Bridges, R. J, Hart, J. A., Chamberlin, A.R. J. Org. Chem., 1994, 59, 2467.
[178] Willis, C. L., Dauenhauer, D., Humphrey, J. M.,Chamberlin, A. R, Buller, A., Monaghan, D. T., Bridges, R.J. Toxicol. Appl. Pharmacol., 1997, 144, 45.
[179] Willis, C. L., Humpherey, J.M., Koch, H. P., Hart, J.A.,Blakely, T., Ralston, L., Baker, C.A., Shim, S., Kadri, M.,Chamberlin, A. R. Bridges, R. J. J. Neuropharmacol., 1996,35, 531.
[180] Bridges, R. J., Lovering, F. E., Koch, H. P., Cotman, C. W.,Chamberlin, A. R. Neuroci. Lett., 1994, 174, 193.
[181] Bräuner-Osborne, H., Hermit, M. B., Nielsen, B.,Krogsgaard-Larsen, P., Johansen, T. N. Eur. J. Pharmacol.,2000, 406, 41.
[182] Stensbøl, T. B., Uhlmann, P., Morel, S., Eriksen, B. L.,Felding, J., Kromann, H., Hermit, M. B., Greenwood, J. R.,Bräuner-Osborne, H, Madsen, U., Junager, F., Krogsgaard-Larsen, P., Begtrup, M., Vedsø, P. J. Med. Chem., 2002, 45,19.
[183] Campiani, G., De Angelis, M., Armaroli, S., Fattorusso, C.,Catalanotti, B., Ramunno, A., Nacci, V., Novellino, E.,Grewer, C., Ionescu, D., Rauen, T., Griffiths, R., Sinclair,C., Fumagalli, E., Mennini, T. J. Med. Chem., 2001, 44,2507.
[184] Zhu, H. Rockhold, R.W., Ho, I. K. Jpn. J. Pharmacol.,1998, 76, 1.
[185] Ozawa, T., Nakagawa, T., Shige, K., Minami, M., Satoh, M.Brain Res., 2001, 905, 254.
[186] Sugiyama, T., Sadzuka, Y., Tanaka, K., Sonobe, T. Toxicol.Lett., 2001, 121, 89.
[187] Albuquerque, E. X., Pereira, E. F. R., Alkondon M.,Schrattenholz, A., Maelicke, A. J. Recept. Signal Transduct.Res., 1997, 17, 243.
[188] May, P.C., Robinson, P. M., Fuson, K. S. Neuroci. Lett.,1999, 262, 219.
[189] Palmada, M., Böhmer, C., Centelles, J. J., Kinne, R. K. H. J.Neurochem., 1999, 73, 2389.

Neuronal High-Affinity Sodium-Dependent Glutamate Transporters (EAATs) Current Pharmaceutical Design, 2003, Vol. 9, No. 8 625
[190] Palmada, M., Kinne-Saffran, E., Centelles, J. J., Kinne, R.K. H. Neurochem. Int., 2002, 40, 321.
[191] Trotti, D., Peng, J., Dunlop, J., Hediger, M. A. Brain Res .,2001, 914, 196.
[192] Poulsen, M. V., Vandenberg, R. J. J. Physiol., 2001, 534.1,159.
[193] Wolf. M. E., Xue, C., Li, Y., Wavak, D. J. Neurochem.,2000, 75, 1634.
[194] Copper B., Chebib, M., Shen, J., King, N.J.C., Darvey, I.G.,Kuchel, P.W., Rothstein, J.D., Balcar, V.J. Arch. Biochem.Biophys., 1998, 353, 356.
[195] Danysz, W., Parsons, C. G., Jirgensons, A., Kauss, V. andTillner, J. Curr. Pharm. Des., 2002, 8(10), 835-43.
[196] Skerry, T. M. and Taylor, A. F. Curr. Pharm. Des., 2001,7(8), 737-50.
[197] Szekely, J. I., Torok, K. and Mate, G. Curr. Pharm. Des.,2002, 8(10), 887-912.





























