Embed Size (px)
Citation preview

American Mineralogist, Volume 97, pages 1849–1857, 2012
0003-004X/12/1112–1849$05.00/DOI: http://dx.doi.org/10.2138/am.2012.4164 1849
New structural features of the high-pressure synthetic sheet-disilicate Phase-X, K(2–x)Mg2Si2O7Hx
Mark D. Welch,1,* Jürgen konzett,2 luca BinDi,3 SiMon c. kohn,4 anD Daniel J. FroSt5
1Department of Mineralogy, The Natural History Museum, Cromwell Road, London SW7 5BD, U.K.2Institute of Mineralogy and Petrology, University of Innsbruck, Innrain 52, A-6020 Innsbruck, Austria
3Dipartimento di Scienze della Terra, Università di Firenze, Via La Pira 4, I-50121 Firenze, Italy4School of Earth Sciences, University of Bristol, Queens Road, Bristol BS8 1RJ, U.K.
5Bavarian Research Institute of Experimental Geochemistry and Geophysics, University of Bayreuth, D-95440 Bayreuth, Germany
aBStract
The structure of the synthetic high-pressure sheet-disilicate Phase-X (PhX), a possible host of H2O and K in the mantle, has been determined for a crystal synthesized at 16 GPa/1300 °C/23 h. The composition of the sample is close to K1.5Mg2Si2O7H0.5, which is 50% PhX/50% Anhydrous-PhX and has 25% of interlayer K sites vacant. The structures of four crystals were determined by single-crystal X-ray diffraction and had very similar diffraction characteristics and structural results; the structure of one of the larger crystals is reported here. Reflection intensity statistics strongly indicate that PhX is centrosymmetric, space group P63/mcm, in contrast to other studies that have reported non-centrosymmetric space group P63cm. While it was possible to obtain good agreement indices for refinements in P63cm, there were strong correlations between atoms that are equivalent in P63/mcm, suggesting that the correct structure is centrosymmetric. Full anisotropic refinement in space group P63/mcm gave R1 = 0.036, wR2 = 0.079, GoF = 1.467. As with all previous studies of PhX, the H atom was not located. Difference-Fourier maps of the residual electron density indicated that the K atom is displaced from the 4c site lying on the sixfold axis on to three split 12j sites 0.2 Å away, each hav-ing ¼ occupancy, giving a total of 3 K atoms per unit cell and corresponding to 1.5 K apfu, in good agreement with the content derived from electron microprobe analysis. Diffraction patterns of all four crystals examined, reconstructed from the full-intensity data collection, consistently show the presence of a large hexagonal superstructure with dimensions 8asub × 8asub × csub, having Z = 128, compared with Z = 2 for the two-layer subcell. Complex arrays of superlattice reflections occur in layers with l = 2n, but are absent from l = 2n + 1 layers.
Unpolarized infrared spectra of single crystals of PhX were obtained that are similar to those reported previously in the literature. Spectra in the OH-stretching region consist of a major absorp-tion band at 3595 cm–1 and three much weaker bands at 3690, 3560, and 3405 cm–1. Bond-valence analysis of PhX indicates that O1 is very over-bonded, whereas O2 is slightly under-bonded and a possible site for protonation. We present geometrical and crystal-chemical arguments that exclude O1 as a candidate for protonation, whereas a much better case can be made for O2. In PhX structures, H must be located at a partially occupied site with a multiplicity 4 ≤ m ≤ 24 in P63/mcm or 4 ≤ m ≤ 12 in P63cm. Such low occupancies for H sites are the likely reason for their invisibility to diffraction. We outline a model for the incorporation of H into PhX of composition K1.5Mg2Si2O7H0.5 that suggests a mechanism for ordering based upon avoidance of H and K, coupled with K-site vacancies. Such behavior may also be the origin of the superstructure. The P63/mcm structure and the presence of an underlying superstructure may well be characteristic of ordered intermediate compositions at or near PhX50/Anhydrous-PhX50. Identification of a new space group and recognition of a previously unob-served superstructure point to new possibilities for PhX and its derivatives that may bear significantly upon their stability at mantle conditions.
Keywords: Phase-X, structure, X-ray diffraction, superlattice, infrared spectroscopy
* E-mail: [email protected]
introDuction
High-pressure disilicates having the general stoichiometry (K,Na)2–xMg2Si2O7Hx (0 ≤ x ≤ 1) and layered structures have been synthesized at high pressure in model alkali-bearing peridotites (Luth 1997; Gasparik and Litvin 1997; Inoue et al. 1998; Konzett
and Fei 2000). Phase-X (PhX) is stable over 1150–1400 °C and 9–17 GPa, and its high thermal stability allows this phase to store and transport water and potassium not only in subduction settings but also in convective mantle.
The two K-bearing end-members of the series have com-positions KMg2Si2O7H (“PhX” sensu stricto) and K2Mg2Si2O7 (“anhydrous-PhX”), and intermediate compositions have also been synthesized. Although charge-balance in this series requires

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X1850
protonation via the substitution HK–1, the H atom has never been located, despite having a clear spectroscopic signature as structural OH (Yang et al. 2001; Mancini et al. 2002; Bindi et al. 2007). The interlayer alkali atom site can be fully or partially occupied by K or Na. Interlayer vacancies (denoted by the symbol ¨ in this paper) are a common feature of the crystal chemistry of the PhX group. Interlayer vacancies also occur in variants containing octahedrally coordinated Fe3+, e.g., (K1.31Na0.02¨0.67) (Mg1.50Fe3+
0.37Al0.05)Si2O7H0.36 (Bindi et al. 2007), implying an ideal anhydrous end-member stoichiometry (K¨)(MgFe3+)Si2O7.
Two alternative structures for anhydrous-PhX, K2Mg2Si2O7, have been reported: a one-layer structure having the centrosym-metric space group P31m (Matsuzaki et al. 2010), and a two-layer structure having the non-centrosymmetric space group P63cm (Yang et al. 2001; Mancini et al. 2002). Until the study of Matsuzaki et al. (2010), the trigonal structure had only been found for anhydrous sodic-PhX, ideally Na2Mg2Si2O7 (Yang et al. 2001).
Raman and infrared spectra of PhX in the OH-stretching region 3000–4000 cm–1 are generally similar despite their dif-ferent compositions. They consist of an intense narrow band at ∼3600 cm–1 which has an associated broader minor band at ∼3570 cm–1. The spectrum of end-member PhX reported by Mancini et al. (2002, Fig . 3) shows a wider spectral range in which additional weak, broad bands centered at 3410 and 4506 cm–1 occur. Mancini et al. (2002) suggest that the 4506 cm–1 band may be due to silanol groups, although independent evidence for such groups was lacking. The spectra of Fe3+-bearing PhX (Bindi et al. 2007) and K1.54Mg1.93Si1.89O7H1.04 (Yang et al. 2001), which are only given for the 3500–3700 cm–1 range are almost identical. No systematic study of the OH vibrational spectra of PhX structures has been made, and assignments of bands to specific OH environments have not been made. Here we report the results of a single-crystal XRD and IR study of PhX, which indicate new and intriguing aspects of the crystal chemistry of this group of high-pressure silicates.
experiMental MethoDS
Sample synthesisPhX was synthesized from K2CO3, Mg(OH)2, and SiO2 mixed in ratio 1:4:4
(corresponding to 2KMg2Si2O7H + 3H2O + CO2) at the Bavarian Research Institute of Experimental Geochemistry and Geophysics using a 1000 t multi-anvil press. Run conditions were 16 GPa and 1300 °C with a run duration of 23 h. The product was a mixture of PhX with clinoenstatite and a quench phase (Fig. 1). The composition of PhX was determined by electron microprobe analysis using a JEOL JXA 8100 Superprobe operated at 15 kV, 5nA (to minimize K volatilization) with a 0.01 × 0.01 mm2 raster and counting for 20 s on peak and 10 s on background. Element standards were quartz (Si), periclase (Mg), and orthoclase (K). Uncertainties (2σ) on elements for the analysis conditions used correspond to SiO2 1.0% relative, MgO 1.3% relative and K2O 1.5% relative. Twenty microprobe analyses were col-lected (Table 1) and analytical totals ranged from 97.61–100.34%. The empirical chemical formula calculated to 2 Si apfu is K1.55(7)Mg2.05(3)Si2O7H0.35(9), for which the H content was calculated for charge balance to 14 positive charges, as required by the general stoichiometry of PhX structures, all of which have seven O atoms per formula. An analogous calculation normalizing to 2 Mg apfu (Table 1) gives an average formula K1.51(6)Mg2Si1.95(3)O7H0.61(7). Within one or two standard devia-tions of the K, Mg, and Si contents, the average formula allows for a composition K1.5Mg2Si2O7H0.5, corresponding to PhX50Anhydrous-PhX50. It was not possible to determine the H content directly, due to the very small quantities of material available. As we shall see, structure refinement indicates that the samples studied have compositions very close to the 1:1 intermediate.
X-ray diffractionFour crystals of PhX were examined by single-crystal XRD using an Agilent
Technologies XcaliburE four-circle diffractometer at The Natural History Museum (London) operating with graphite-monochromated MoKα radiation (λ = 0.71073 Å) at 50 kV/45mA, and equipped with an Eos CCD-type area-detector. All four crystals had sharp diffraction maxima with no reflection streaking parallel to c*, indica-tive of a well-ordered structure lacking layer stacking disorder. A whole sphere of reflections was collected to 32 °θ with 100% completeness to 30 °θ. Omega scans of 1° frame-width and a 40 s frame-time were used. Further information relating to data collection is given in Table 2. Raw intensities were corrected for Lorentz, polarization, and absorption effects and converted to structure factors using the CrysalisPro program (Agilent Technologies).
Infrared spectroscopyUnpolarized infrared spectra were collected for two crystals (PhX4 and PhX5)
for which almost identical structures had been determined by X-ray diffraction. The crystals were already mounted on 0.01 mm diameter carbon fibers that themselves were glued to glass fibers. It was, therefore, possible to suspend the samples in the instrument without having to place them on a KBr disk. The samples were oriented so that the crystallographic z-axis (normal to the crystal flake) was approximately parallel to the propagation direction of the beam. Spectra were collected over the range 4000–450 cm–1 using a Thermo-Nicolet iN10MX infrared microscope. The presence of epoxy glue in some areas of the sample surface required careful posi-tioning of the infrared beam. For one of the crystals a grid of spectra with a 0.01 mm spacing was acquired using a 0.02 × 0.02 mm2 beam. By comparing with a spectrum of the glue, an area of crystal PhX4 that was essentially uncontaminated by glue was found. For the PhX5 crystal a 0.04 × 0.04 mm2 beam was used. A spectral resolution of either 2 cm–1 (PhX4) or 4 cm–1 (PhX5) was used.
reSultS
X-ray diffractionAll four crystals displayed the same diffraction characteristics
and gave very similar results for structure determination. We report structural data for one of the larger and better crystals. Information relevant to data reduction and structure refinement is given in Table 2.
Reconstructed diffraction patterns for the four crystals ob-tained using the Unwarp facility in Crysalis show many superlat-tice reflections for hkl with l = 2n that define a 8asub metrically hexagonal supersheet (40.5 × 40.5 Å). These superlattice reflec-
Figure 1. Polished mount of the experimental run capsule of the PhX synthesis. Cen = clinoenstatite, Q = quench phase. The crystals studied were extracted from the PhX-rich central region.

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X 1851
tions are absent from l = 2n + 1 layers. Representative Unwarp diffraction patterns of the crystal for which data are reported are shown in Figure 2. CrysalisPro finds the metrically hexagonal supercell easily in a unit-cell search. We made a second full intensity data collection at 100 K using a 0.5° frame-width and a 100 s frame-time in the hope of increasing the I/σ(I) of these relatively weak superlattice reflections. The unconstrained unit-cell parameters for this 100 K supercell (Z = 128) are: a = 40.338(2), b = 40.369(2), c = 13.2015(5) Å, α = 90.022(3)°, β = 89.998(3)°, γ = 119.915(5)°, and V = 18633(1) Å3 obtained from 4557 reflections; the corresponding constrained hexagonal cell has a = 40.378(2), c = 13.1966(6) Å, and V = 18633(1) Å3. However, it was not possible to integrate reflection intensities satisfactorily. Unwarp images show that overall the intensities of superlattice decrease markedly beyond 9 °θ (a resolution of ∼2.3 Å) and are very weak beyond 16 °θ (∼1.3 Å). This rapid in-tensity die-off results in a prohibitively low data:parameter ratio and precluded determination of the superstructure. However, we discuss the possible origin of this supercell later.
As it was not possible to determine the superstructure, we focus on the substructure. In addition to the large supercell mentioned above, Unwarp images show a strong c/2 (6.6 Å) sublattice. However, there are numerous hkl with l = 2n + 1, but many are weak. For example, the reconstructed hk1 diffraction pattern, a representative part of which is shown in Figure 2b,
consists only of sublattice reflections, most of which are weaker than their counterparts in l = 2n layers. Hence, the PhX crystal discussed has a two-layer 13.2 Å repeat.
Structure solution and refinement were carried out using the program SHELX (Sheldrick 2008). E-statistics strongly indicate that PhX is centrosymmetric (|E2 – 1| = 1.006 and a probability of 86%). Although it was possible to solve and refine the structure in non-centrosymmetric space group P63cm, there are strong correlations between atoms that are equivalent in the centrosym-metric space group P63/mcm, e.g., z(O2,O3) = 0.97, z(Si1,Si2) = 0.86, x(O2,O3) = 0.73. Furthermore, the Flack parameter for refinements in P63cm is close to 0.5, but with a large standard deviation (0.48 ± 0.16). This result also suggests the likelihood that the correct structure is centrosymmetric. Consequently, the structure was refined in space group P63/mcm (no.193). Approximate positions of K, Mg, and Si atoms were obtained from structure solution, and the two oxygen atoms appeared in a difference-Fourier map after the first few cycles of refinement. Atom coordinates and displacement parameters are given in Table 3. Interatomic distances are given in Table 4, calculated bond-valences are given in Table 5, and a list of structure factors is given in Table 61. (CIF1 also available.)
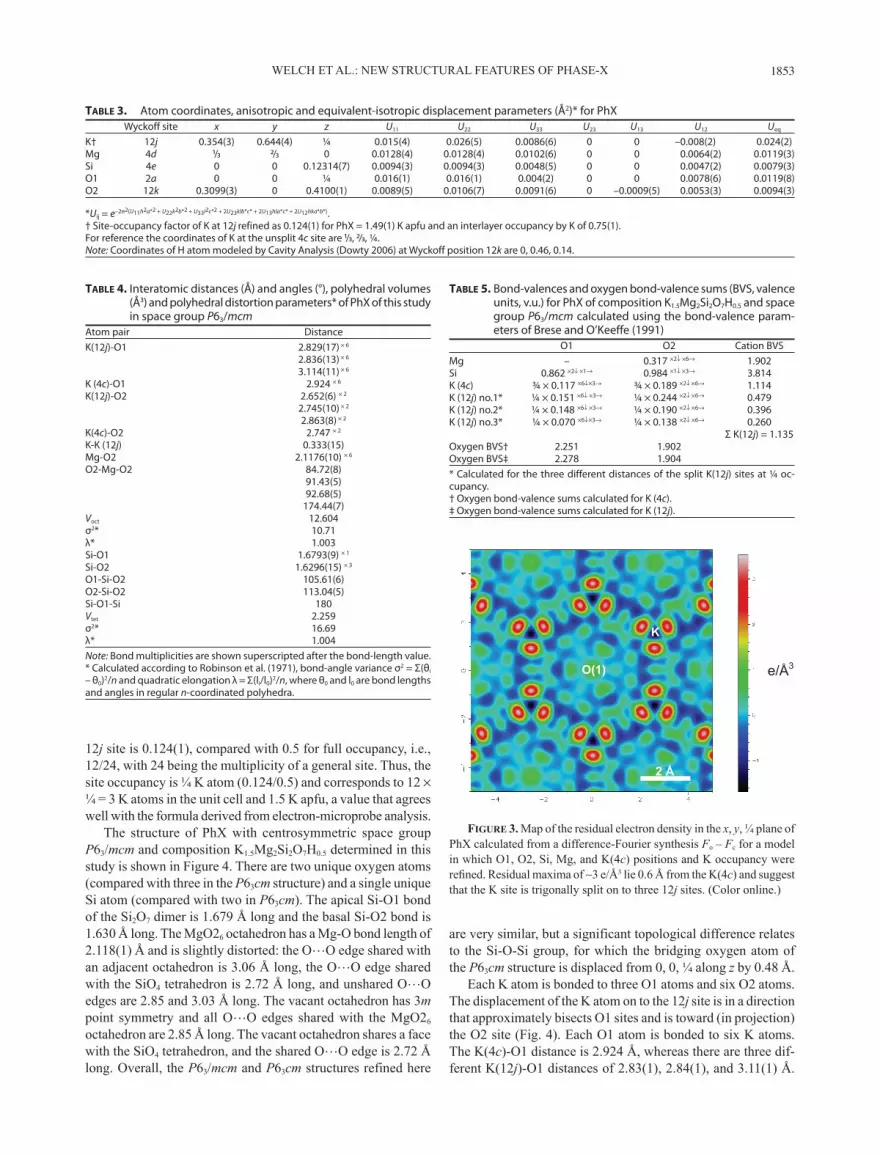
The K atom was initially located at special position 4c (⅓ ⅔ ¼) lying at the inversion center on the sixfold axis and its site occupancy refined to 0.75(1). However, after all atoms except H had been located and their coordinates and isotropic displace-ment parameters refined to R1 = 0.098, a difference-Fourier map revealed significant residual electron-density maxima (3.5 e/Å3) ∼0.6 Å away from the 4c site, suggesting that K is located slightly off the sixfold axis and is split into three equivalent 12j sub-sites (x y ¼) around the 4c position. A difference-electron-density map derived from difference-Fourier synthesis is shown in Figure 3, in which the sets of three residual 12j sub-site maxima are evident. The coordinates, occupancy level and anisotropic displacement parameters of the K atom were refined successfully at 0.354(3) 0.644(4) ¼ (12j), which lies 0.2 Å away from the 4c site at (⅓, ⅔, ¼). A very similar displacement (0.18 Å) of K from the threefold axis (at a 4b site) of the non-centrosymmetric P63cm structure was reported by Mancini et al. (2002). The distance between adjacent K sub-sites is 0.335(15) Å. The refined value of the occupancy factor (as defined in SHELX, Sheldrick 2008) of the
Table 1. Electron microprobe analyses (n = 20) of PhXwt% oxides
SiO2 43.66 43.16 42.69 43.12 42.80 43.90 43.13 43.39 43.24 42.42 42.95 42.95 42.91 42.56 43.30 43.28 42.89 43.33 42.18 44.13MgO 29.70 29.94 29.36 31.04 30.24 29.33 29.22 30.02 30.17 28.95 29.38 30.23 29.15 29.21 29.95 30.09 30.19 29.84 28.78 30.13K2O 24.88 26.52 28.29 25.09 25.61 25.92 26.55 25.73 24.99 26.24 26.90 25.55 27.77 27.66 26.58 25.63 25.40 25.84 28.17 25.08 98.24 99.62 100.34 99.25 98.65 99.15 98.90 99.14 98.40 97.61 99.23 98.73 99.83 99.43 99.83 99.00 98.48 99.01 99.13 99.34
Cations apfu (ΣSi = 2)Mg 2.03 2.05 2.07 2.05 2.07 2.02 2.05 2.04 2.05 2.09 2.06 2.06 2.06 2.08 2.04 2.05 2.06 2.04 2.10 2.01K 1.45 1.56 1.69 1.48 1.52 1.50 1.57 1.51 1.47 1.57 1.59 1.51 1.65 1.65 1.56 1.51 1.51 1.52 1.70 1.45
Cations apfu (ΣMg = 2)Si 1.97 1.95 1.93 1.95 1.93 1.98 1.95 1.96 1.95 1.92 1.94 1.94 1.94 1.92 1.96 1.96 1.94 1.96 1.91 1.99K 1.43 1.53 1.63 1.44 1.47 1.49 1.53 1.48 1.44 1.51 1.55 1.47 1.60 1.59 1.53 1.47 1.46 1.49 1.62 1.44Notes: In one case, cation contents are normalized to 2 Si apfu and in the other case to 2 Mg apfu. Similar average formulae are obtained for the two normalization schemes.
Table 2. Information relating to data collection and structure refine-ment of Phase-X
Phase-XCell contents 2[K1.5Mg2Si2O7H0.5]Crystal system hexagonalSpace group P63/mcm (no.193)Unit cell parameters (Å) 5.0646(2), 13.2379(5)Volume (Å3) 294.06(2)Reflections used for cell refinement, I >7σ(I) 1462Calculated density (g/cm3) 3.116Crystal dimensions (mm) 0.141 × 0.104 × 0.030Theta (°) and indices ranges for data collection 3.07–32.45, ±7, ±7, ±19Absorption correction (µ = 1.87 mm–1) GaussianTransmission max, min 0.95, 0.85Reflections collected 4977Average I/σ(I) 9.47Rint (6/mmm) 0.034Independent reflections 215Independent reflections with I > 2σ(I) 201Data completeness to 30 °θ (%) 100Data/restraints/parameters 215/0/21Goodness-of-fit on F2 1.467R1, wR2 indices [I > 2σ(I)] 0.032, 0.078R1, wR2 indices (all data) 0.036, 0.079Max shift/e.s.d. 0.000Largest diff. peak and hole (e Å–3) 0.26, –0.34
1 Deposit item AM-12-093, Table 6 and CIFs. Deposit items are available two ways: For a paper copy contact the Business Office of the Mineralogical Society of America (see inside front cover of recent issue) for price information. For an electronic copy visit the MSA web site at http://www.minsocam.org, go to the American Mineralogist Contents, find the table of contents for the specific vol-ume/issue wanted, and then click on the deposit link there.

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X1852
Figure 2. (a, b, and c) Representative parts of reconstructed hk0, hk1, and hk2 diffraction patterns of PhX created using Unwarp. Many weak superlattice reflections are evident in the hk0 and hk2 patterns, whereas they are absent from the hk1 pattern. In fact all l = 2n layers have superlattice reflections, whereas l = 2n + 1 layers do not. (d and e) Schematics of the arrays of superlattice reflections in a and c. The intensity arrays are complex and consist of chevrons and trapezia each of five reflections. In the hk2 pattern (e), additional triangles of very weak superlattice reflections also occur. These three arrays can be viewed as triangles of six points in which one point is missing (chevron and trapezium) or three are missing (triangle). (f) An enlargement of the hk2 diffraction pattern highlighted in e showing indexed superlattice reflections (italics) and four sublattice reflections.
WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X 1853
12j site is 0.124(1), compared with 0.5 for full occupancy, i.e., 12/24, with 24 being the multiplicity of a general site. Thus, the site occupancy is ¼ K atom (0.124/0.5) and corresponds to 12 × ¼ = 3 K atoms in the unit cell and 1.5 K apfu, a value that agrees well with the formula derived from electron-microprobe analysis.
The structure of PhX with centrosymmetric space group P63/mcm and composition K1.5Mg2Si2O7H0.5 determined in this study is shown in Figure 4. There are two unique oxygen atoms (compared with three in the P63cm structure) and a single unique Si atom (compared with two in P63cm). The apical Si-O1 bond of the Si2O7 dimer is 1.679 Å long and the basal Si-O2 bond is 1.630 Å long. The MgO26 octahedron has a Mg-O bond length of 2.118(1) Å and is slightly distorted: the O…O edge shared with an adjacent octahedron is 3.06 Å long, the O…O edge shared with the SiO4 tetrahedron is 2.72 Å long, and unshared O…O edges are 2.85 and 3.03 Å long. The vacant octahedron has 3m point symmetry and all O…O edges shared with the MgO26 octahedron are 2.85 Å long. The vacant octahedron shares a face with the SiO4 tetrahedron, and the shared O…O edge is 2.72 Å long. Overall, the P63/mcm and P63cm structures refined here
are very similar, but a significant topological difference relates to the Si-O-Si group, for which the bridging oxygen atom of the P63cm structure is displaced from 0, 0, ¼ along z by 0.48 Å.
Each K atom is bonded to three O1 atoms and six O2 atoms. The displacement of the K atom on to the 12j site is in a direction that approximately bisects O1 sites and is toward (in projection) the O2 site (Fig. 4). Each O1 atom is bonded to six K atoms. The K(4c)-O1 distance is 2.924 Å, whereas there are three dif-ferent K(12j)-O1 distances of 2.83(1), 2.84(1), and 3.11(1) Å.
Table 3. Atom coordinates, anisotropic and equivalent-isotropic displacement parameters (Å2)* for PhX Wyckoff site x y z U11 U22 U33 U23 U13 U12 Ueq
K† 12j 0.354(3) 0.644(4) ¼ 0.015(4) 0.026(5) 0.0086(6) 0 0 –0.008(2) 0.024(2)Mg 4d ¹/3 ²/₃ 0 0.0128(4) 0.0128(4) 0.0102(6) 0 0 0.0064(2) 0.0119(3)Si 4e 0 0 0.12314(7) 0.0094(3) 0.0094(3) 0.0048(5) 0 0 0.0047(2) 0.0079(3)O1 2a 0 0 ¼ 0.016(1) 0.016(1) 0.004(2) 0 0 0.0078(6) 0.0119(8)O2 12k 0.3099(3) 0 0.4100(1) 0.0089(5) 0.0106(7) 0.0091(6) 0 –0.0009(5) 0.0053(3) 0.0094(3)
*Uij = e–2π2(U11h2a*2 + U22k2b*2 + U33l2c*2 + 2U23klb*c* + 2U13hla*c* + 2U12hka*b*). † Site-occupancy factor of K at 12j refined as 0.124(1) for PhX = 1.49(1) K apfu and an interlayer occupancy by K of 0.75(1).For reference the coordinates of K at the unsplit 4c site are ¹/3, ²/₃, ¼.Note: Coordinates of H atom modeled by Cavity Analysis (Dowty 2006) at Wyckoff position 12k are 0, 0.46, 0.14.
Table 4. Interatomic distances (Å) and angles (°), polyhedral volumes (Å3) and polyhedral distortion parameters* of PhX of this study in space group P63/mcm
Atom pair DistanceK(12j)-O1 2.829(17) × 6
2.836(13) × 6
3.114(11) × 6
K (4c)-O1 2.924 × 6
K(12j)-O2 2.652(6) × 2
2.745(10) × 2
2.863(8) × 2
K(4c)-O2 2.747 × 2
K-K (12j) 0.333(15)Mg-O2 2.1176(10) × 6
O2-Mg-O2 84.72(8) 91.43(5) 92.68(5) 174.44(7)Voct 12.604σ2* 10.71λ* 1.003Si-O1 1.6793(9) × 1
Si-O2 1.6296(15) × 3
O1-Si-O2 105.61(6)O2-Si-O2 113.04(5)Si-O1-Si 180Vtet 2.259σ2* 16.69λ* 1.004Note: Bond multiplicities are shown superscripted after the bond-length value.* Calculated according to Robinson et al. (1971), bond-angle variance σ2 = Σ(θi – θ0)2/n and quadratic elongation λ = Σ(li/l0)2/n, where θ0 and l0 are bond lengths and angles in regular n-coordinated polyhedra.
Table 5. Bond-valences and oxygen bond-valence sums (BVS, valence units, v.u.) for PhX of composition K1.5Mg2Si2O7H0.5 and space group P63/mcm calculated using the bond-valence param-eters of Brese and O’Keeffe (1991)
O1 O2 Cation BVSMg – 0.317 ×2↓ ×6→ 1.902Si 0.862 ×2↓ ×1→ 0.984 ×1↓ ×3→ 3.814K (4c) ¾ × 0.117 ×6↓×3→ ¾ × 0.189 ×2↓ ×6→ 1.114K (12j) no.1* ¼ × 0.151 ×6↓ ×3→ ¼ × 0.244 ×2↓ ×6→ 0.479K (12j) no.2* ¼ × 0.148 ×6↓ ×3→ ¼ × 0.190 ×2↓ ×6→ 0.396K (12j) no.3* ¼ × 0.070 ×6↓×3→ ¼ × 0.138 ×2↓ ×6→ 0.260 Σ K(12j) = 1.135Oxygen BVS† 2.251 1.902 Oxygen BVS‡ 2.278 1.904 * Calculated for the three different distances of the split K(12j) sites at ¼ oc-cupancy.† Oxygen bond-valence sums calculated for K (4c).‡ Oxygen bond-valence sums calculated for K (12j).
e/Å3
K
O(1)
2 Å
Figure 3. Map of the residual electron density in the x, y, ¼ plane of PhX calculated from a difference-Fourier synthesis Fo – Fc for a model in which O1, O2, Si, Mg, and K(4c) positions and K occupancy were refined. Residual maxima of ∼3 e/Å3 lie 0.6 Å from the K(4c) and suggest that the K site is trigonally split on to three 12j sites. (Color online.)

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X1854
Figure 4. Structural aspects of PhX with the P63/mcm structure and composition K1.5Mg2Si2O7H0.5. (a) Polyhedral representation projected on to (110) showing reversal of the direction of octahedral sheet leading to a doubled c repeat, relative to (b) the one-layer structure of the trigonal P31m polytype of anhydrous-PhX (Matsuzaki et al. 2010). (c) A view of the environment of the vacant octahedron projected on to (001) showing the H position found by cavity analysis. (d) The same structural fragment as in c viewed along the x axis to show the out-of-plane O-H vector orientation. (e and f) The array of O1, O2, and K(12j) sites projected on to (001), with O1 and K(12j) interlayer sites being coplanar and O2 atoms lying below the interlayer. In e, the anisotropic displacement ellipsoids of O1, O2, and K(12j) are shown; in f, positions and K(12j)-O2 distances are shown. See text for discussion.

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X 1855
The K(4c)-O2 distance is 2.75 Å; the three different K(12j)-O2 distances are 2.652(6), 2.75(1), and 2.863(8) Å. Each O2 atom is bonded to one Si [1.630(2) Å], two Mg atoms [2.118(1) Å], and two K atoms (see above). The anisotropic displacement el-lipsoid of K(12j) is cigar-shaped with U33 >> U22 > U11, and U33 lies within the x, y, ¼ plane.
Infrared spectroscopyUnpolarized infrared absorption spectra in the OH-stretching
region 3750–3200 cm–1 of two different crystals of PhX for which almost identical structures were determined are shown in Figure 5. The two spectra are very similar and comprise an intense nar-row band at 3595 cm–1 with a minor shoulder at 3560 cm–1, and weak broad absorption features centered at 3690 and 3405 cm–1. The spectra are similar to the unpolarized IR spectrum of PhX reported by Mancini et al. (2002), which has an intense narrow band at 3602 cm–1 and a much weaker band at 3410 cm–1. The minor shoulder we observe at 3560 cm–1 is near the position of the minor peak at 3575 cm–1 in the Raman spectrum of Yang et al. (2001). Thus, the IR spectra in the OH-stretching region of our PhX crystals are comparable to spectra reported in the literature.
DiScuSSion
Two new features of the crystal-chemistry of PhX structures have been discovered in the present study: (1) a new space group P63/mcm, and (2) a large supercell. Potassium contents very close to 1.5 apfu for all four crystals studied suggest that there is a sig-nificant crystal-chemical control operating that may stabilize this composition at high P and T. Three key observations concerning the infrared spectra in the OH-stretching region of the two PhX crystals reported here are: (1) they are essentially the same as each other; (2) they are very similar to published Raman and
infrared spectra of various PhX structures in the OH-stretching region; (3) they are dominated by a relatively narrow peak at 3595 cm–1, with a shoulder at 3560 cm–1, with other absorptions at 3690 and 3405 cm–1.
We now provide an analysis of possible H locations in PhX using the bond-valence method (Brown 1981), cavity analysis (e.g. Dowty 2006), and correlations between O-H vibrational frequencies and O…O distances (Libowitzky 1999) to suggest a plausible mechanism for the development of superstructures in PhX. It should, however, be borne in mind that bond-valence analysis relates to the average (sub-)structure of PhX determined here, and so may not allow definitive identification of oxygen sites for protonation. Furthermore, due to the wide variation of K coordination polyhedra, the bond-valence constant R0 for K (2.13 v.u.; Brese and O’Keeffe 1991) is not well constrained, and this lack of constraint can result in considerable deviations from unity for K bond-valence sums. Even for structurally re-lated minerals, K bond-valence sums can be very different. For example, K bond-valence sums for the K-micas muscovite and phlogopite are 1.16 and 0.84 v.u., respectively. In view of these limitations of the bond-valence method in the present case, we also use geometrical and crystal-chemical arguments to evaluate O1 and O2 as sites of protonation.
Bond-valence analysisIn calculating bond-valence sums, the occupancies of 4c and
12j sites must be considered. Potassium at 4c has an occupancy of ¾, a K(4c)-O1 bond-length of 2.924 Å, and a bond valence of 0.117 v.u., which reduces to 0.117 × ¾ = 0.088 v.u. Bond-valence analysis (Table 5) indicates that the O1 atom is over-bonded to the same extent for K at 4c and 12j positions. Six K(4c)-O1 bonds contribute 0.088 × 6 = 0.528 v.u. to the O1 bond-valence sum. Adding the contribution from the two Si-O1 bond valences (0.862 × 2) gives a bond-valence sum for O1 of 2.252 v.u. The corresponding calculation for K(12j), which has an occupancy of ¼, must consider three different K(12j)-O1 distances at 2.829, 2.836, and 3.114 Å, with bond-valences of 0.151, 0.148, and 0.070 v.u., respectively. The average bond-valence of a K(12j)-O1 bond is ¼(0.369) = 0.092 v.u. The total contribution from six of these bonds is 0.552 v.u., giving an O1 bond-valence sum (adding two Si-O) of 2.278 v.u.
The O2 atom is bonded to one Si (1.630 Å, 0.984 v.u.), two Mg (2.118 Å, 0.317 v.u.) and two K atoms. For ¾ K at 4c (2.747 Å), the bond-valence is 0.189 × ¾ = 0.142 v.u., and the bond-valence sum of O2 for K at 4c is 0.984 + 0.317 × 2 + 0.142 × 2 = 1.902 v.u. A similar calculation for K(12j), considering the three different K(12j)-O2 distances and ¼ K occupancy gives 1.904 v.u. Thus, the bond-valence sums of O2 for K(4c) and K(12j) are the essentially same and indicate under-bonding of this oxygen atom. Compared with the highly over-bonded O1 atom, bond-valence analysis suggests that O2 is a more likely candidate for protonation.
Hydrogen location in PhXEnd-member PhX, KMg2Si2O7H with the two-layer structure,
has two H per unit cell (Z = 2). Space groups P63/mcm and P63cm have two Wyckoff sites of multiplicity m = 2: P63/mcm has 2a at the 62m inversion center 0, 0, ¼ ( 0, 0, ¾) that is occupied
Figure 5. Unpolarized infrared spectra of single crystals of PhX (crystals PhX4 and PhX5). See text for discussion.
320033003400350036003700
34053690
3560
3595
PhX4
PhX5
Wavenumber (cm-1)

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X1856
by O1, and 2b at the 3.m inversion center at the center of the vacant octahedron at 0, 0, 0 (0, 0, ½). Neither of these sites is a plausible location for H because 2a is occupied by O1 and 2b is 1.97 Å away from O2 and H would need to be bonded to six oxygen atoms. Consequently, H in end-member PhX must be located on a partially occupied site with m ≥ 4, i.e., m = 4 with ½ occupancy, m = 6 with ⅓ occupancy, m = 8 with ¼ occupancy, m = 12 with 1⁄6 occupancy, or m = 24 with 1⁄12 occupancy. For the case of PhX studied here which is close to K1.5Mg2Si2O7H0.5 and has one H per unit cell, these H occupancies are halved. A similar argument exists for end-member PhX in space group P63cm (but with a maximum m = 12). Such partial occupancies lead to H being extremely difficult or impossible to locate by diffraction methods, while having a distinctive spectroscopic signature. For space group P63/mcm, Wyckoff positions with multiplicities of 4 (4c, 4d, 4e), 6 (6f, 6g), and 8 (8h) can be discounted as possible H sites on the grounds that they are already occupied, or are too close to Mg, or imply implausible O-H distances. This evaluation also holds for the P63cm structure.
The correlation between d(Odonor…Oacceptor) and OH-stretching frequency was calibrated by Libowitzky (1999, Fig. 1 therein) and is useful for exploring possible hydrogen-bonded O-H…O linkages. For PhX, the main absorption band at 3595 cm–1 lies within the region of weakly hydrogen-bonded O-H, with d(O…O) > 3 Å. The minor broad band centered at 3405 cm–1 cor-relates with a d(O…O) of ∼2.85 Å, and is indicative of a stronger hydrogen bond. This distance correlates well with the shorter edge-length of the vacant octahedron (2.85 Å), and so may point to the presence of an H site lying inside the vacancy and defined by an O-H…O linkage associated with this O2…O2 edge.
In an attempt to find a plausible H site in PhX, we have carried out a bond-valence analysis of all the previous structure models by transforming them to the current space group P63/mcm. Such a procedure yielded quite different bond-valence sums for the two oxygen atoms of 2.19–2.27 and 1.87–1.91 v.u. for O1 and O2, respectively. On this basis, the O2 position could, therefore, be a site for protonation. For example, using the cavity analysis routine of the program ATOMS 6.3 (Dowty 2006), and maximiz-ing the distance of H (for a typical O-H bond length of 0.95 Å) from the fully occupied atom positions, gives one possible set of atomic coordinates for the H atom at 0, 0.46, 0.14 (Fig. 4). These atom coordinates correspond to a 12k Wyckoff position lying 2.34 Å from Si. For ½H apfu and one H per unit-cell (for PhX studied here), such a site has an occupancy of only 8.33% (1/12), representing a small contribution to the bond-valence sum of O2 that avoids over-bonding it. The O-H vector bisects two K positions and is directed toward O1. The H…O1 distance is long at 3.10 Å, which is consistent with a OH vibrational frequency of >3550 cm–1 (Libowitzky 1999). We provisionally assign the major 3595 cm–1 band to this O2H group, which has a dipole vector oriented 39° to the di-octahedral sheet. The H atom of this OH group lies ∼2.1 Å from the K(12j) sites. This distance is quite short and suggests the possibility of H…K repulsion and H-K avoidance. Such avoidance of H and K, involving vacancies at K sites coupled with H occupancy, is a plausible mechanism for generating superstructures in PhX.
Some further comments should be made regarding O1 as a potential site of protonation. Laying aside the highly over-
bonded nature of this oxygen atom that on its own would seem to preclude protonation, we use geometrical and crystal-chemical arguments to show that protonation of O1 is very unlikely. The longest d(Si…H) occurs when H lies in the x, y, ¼ plane. For a typical hydroxyl bond-length of 0.98 Å and a Si-O of 1.65 Å (this study), Pythagoras’ theorem gives d(Si…H) of 1.93 Å, compared with 2.34 Å for H bonded to O2. The shortest d(Si…H) we have found in the mineralogical literature is 2.06 Å for a non-bridging OH in the pyroxenoid serandite (Jacobsen et al. 2000). Hence, the d(Si…H) associated with O1H in PhX is exceptionally short. A further problem for a O1-H bond is the formation of a suitable hydrogen bond, for which O2 at the base of the SiO4 tetrahedron is the only available acceptor. To achieve a O1-H…O2 hydrogen bond, H has to leave the x, y, ¼ plane and move toward O2, thereby shortening d(Si…H) even more.
Additional information on the characteristic geometry of silanol groups involving protonated Si-O-Si bridging oxygen atoms is provided by Geisinger and Gibbs (1981) and Gibbs (1982) who showed conclusively from quantum-mechanical calculations that three-coordinated Si-O-Si bridging oxygen atoms, including Si-O(-H)-Si groups, are associated with highly bent Si-O-Si angles (120–140°). Hence, a protonated Si-O(-H)-Si group should be markedly bent. For the PhX structure deter-mined here, the maximum (100%) limit of the O1 displacement ellipsoid allows for a displacement from 0, 0, ¼ of only 0.24 Å, corresponding to ∠Si-O1-Si of 164°. A ∠Si-O1-Si of, say, 140° that is more consistent with the results of Geisinger and Gibbs (1981) implies an O1 displacement from 0, 0, ¼ of 0.67 Å, a distance that amounts to a split O1 site that could be detected by diffraction methods, but for which there is no empirical evidence in this or any published studies of PhX. From these various geo-metrical, crystal-chemical, and non-empirical considerations, we conclude that O1 is not a likely candidate for protonation, and that a much better case can be made for protonation at O2, as we have described above.
The nature of the superstructureAbove, we have given an evaluation of plausible H locations
in PhX and suggested that H…K repulsion, coupled to K-site vacancies, provides a means of ordering and the development of superstructures. As the substructure of PhX examined here has space group P63/mcm, we examined group-supergroup relationships to determine the possible symmetry of the super-structure. The maximal klassengleiche subgroup of P63/mcm with an enlarged cell (isomorphic) with pa, pb, c, where p = 8, is P63/mcm, implying that the metrically hexagonal super-sheet motif has 128 interlayer alkali sites. For 1.5 K apfu, this corresponds to 32 vacancies per unit interlayer motif. The task then becomes one of arranging 32 vacancies over 128 sites to get the required 8asub hexagonal motif. To evaluate the wider oc-currence and significance of superstructures in PhX and related phases, a systematic study by diffraction and spectroscopy of the KMg2Si2O7H–K2Mg2Si2O7 series would seem to be in order.
acknoWleDgMentSWe thank reviewers Andrew Locock and Franco Mancini for their insightful
and constructive comments on this manuscript, and Associate Editor Ian Swainson for his efficient editorial handling. M.D.W. acknowledges The Natural History Museum (London) for support of his research on hydrous minerals.

WELCH ET AL.: NEW STRUCTURAL FEATURES OF PHASE-X 1857
reFerenceS citeDBindi, L., Bobrov, A., and Litvin, Y.A. (2007) Incorporation of Fe3+ in phase-X,
A2–xM2Si2O7Hx, a potential high-pressure K-rich hydrous silicate in the mantle. Mineralogical Magazine, 71, 265–272.
Brese, N.E. and O’Keeffe, M. (1991) Bond-valence parameters for solids. Acta Crystallographica, B47, 192–197.
Brown, I.D. (1981) The bond-valence method: an empirical approach to chemical structure and bonding. In M. O’Keeffe and A. Navrotsky, Eds., Structure, and Bonding in Crystals II, p. 1–30. Academic Press, New York.
Dowty, E. (2006) ATOMS for Windows. Version 6.3. Shape Software, Kingsport, Tennessee.
Gasparik, T. and Litvin, Y.A. (1997) Stability of Na2Si2O7 and melting relations on the forsterite-jadeite join at pressure up to 22 GPa. European Journal of Mineralogy, 9, 311–326.
Geisinger, K.L. and Gibbs, G.V. (1981) SiSSi and SiOSi bonds in molecules and solids: A comparison. Physics and Chemistry of Minerals, 7, 204–210.
Gibbs, G.V. (1982) Molecules as models for bonding in crystals. American Min-eralogist, 67, 421–450.
Inoue, T., Irifune, T., Yurimoto, H., and Miyagi, I. (1998) Decomposition of K-amphibole at high pressure: implications for subduction zone volcanism. Physics of the Earth and Planetary Interiors, 107, 221–231.
Jacobsen, S.D., Smyth, J.R., Swope, R.J., and Sheldon, R.I. (2000) Two proton positions in the very strong hydrogen bond of serandite, NaMn2[Si3O8(OH)]. American Mineralogist, 85, 745–752.
Konzett, J. and Fei, Y. (2000) Transport and storage of potassium in the Earth’s upper mantle and transition zone: an experimental study to 23 GPa in simplified
and natural bulk compositions. Journal of Petrology, 41, 583–603.Libowitzky, E. (1999) Correlation of O-H stretching frequencies and O-H…O hy-
drogen bond lengths in minerals. Monatshefte für Chemie, 130, 1047–1059.Luth, R.W. (1997) Experimental study of the system phlogopite-diopside from 3.5
to 17 GPa. American Mineralogist, 82, 1198–1209.Mancini, F., Harlow, G.E., and Cahill, C.L. (2002) The crystal structure and cation
ordering of phase-X-(K1–x–n)2(Mg1–n[Al,Cr]n)2Si2O7H2x: A potential K- and H-bearing phase in the mantle. American Mineralogist, 87, 302–306.
Matsuzaki, T., Hagiya, K., Shatskiy, A., Katsura, T., and Matsui, M. (2010) Crystal structure of anhydrous Phase-X, K1.93(Mg2.02Cr0.02)Si2.00O7. Journal of Mineral-ogical and Petrological Sciences, 105, 303–308.
Robinson, D.K., Gibbs, G.V., and Ribbe, P.H. (1971) Quadratic elongation: a quantitative of distortion in coordination polyhedra. Science, 172, 567–570.
Sheldrick, G.M. (2008) A short history of SHELX. Acta Crystallographica, A64, 112–122.
Yang, H., Konzett, J., and Prewitt, C.W. (2001) Crystal structure of Phase-X, a high pressure alkali-rich hydrous silicate and its anhydrous equivalent. American Mineralogist, 86, 1483–1488.
Manuscript received March 10, 2012Manuscript accepted July 16, 2012 Manuscript handled by ian swainson




























