Embed Size (px)
Citation preview

Am J Clin Pathol 2013;139:491-514 491491 DOI: 10.1309/AJCP83AOQTMLOJTM 491
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
Non–Mycosis Fungoides Cutaneous T-Cell Lymphomas
Report of the 2011 Society for Hematopathology/European Association for Haematopathology Workshop
Leticia Quintanilla-Martinez, MD,1 Patty M. Jansen, MD, PhD,2 Marsha C. Kinney, MD,3 Steven H. Swerdlow, MD,4 and Rein Willemze, MD, PhD5
Key Words: Skin; T-cell lymphomas; CD30+ T-cell lymphoproliferations
DOI: 10.1309/AJCP83AOQTMLOJTM
A b s t r a c t
Primary cutaneous T-cell lymphomas (CTCL) excluding mycosis fungoides (MF) were discussed in 2 sessions of the 2011 Society for Hematopathology/European Association of Haematopathology Workshop, Los Angeles, CA. Session 2 focused on primary cutaneous CD30+ T-cell lymphoproliferative disorders and their differential diagnosis, including systemic CD30+ T-cell lymphoma secondarily infiltrating the skin. Interesting features like special morphologic variants and atypical phenotypes were presented. In addition, the possibility of rare ALK+ primary cutaneous lymphomas was discussed. Session 3 examined other more uncommon non-MF CTCLs, including subcutaneous panniculitis-like T-cell lymphoma, extranodal NK/T-cell lymphoma, hydroa vacciniforme–like T-cell lymphoma, and rare subtypes of primary cutaneous peripheral T-cell lymphoma, not otherwise specified. In addition, systemic T-cell lymphomas involving the skin secondarily, such as angioimmunoblastic T-cell lymphoma, were included in this session. In this report, novel findings, areas of special interest, and diagnostic challenges emerging from the cases submitted to the workshop will be highlighted. The necessity to integrate histologic, immunophenotypical, genetic, and in particular, clinical data to arrive at the correct diagnosis, and subsequently provide adequate treatment, is emphasized.
The term primary cutaneous T-cell lymphoma (CTCL) refers to cutaneous T-cell lymphomas that manifest in the skin with no evidence of extracutaneous disease at the time of diagnosis. CTCL accounts for approximately 70% of all primary cutaneous lymphomas ❚Table 1❚, with the vast majority being mycosis fungoides (MF) and its variants (summarized by Song et al1 in this issue of the Journal). Two sessions of the 2011 Society for Hematopathology/European Association for Haematopathology Workshop were devoted to discussion of CTCL other than MF. The 2008 World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissue together with the European Organisation for Research and Treatment of Cancer (EORTC) recognize as CTCL other than MF the group of primary cutaneous CD30+ T-cell lymphoprolif-erative disorders (LPDs) and a group of more uncommon non-MF CTCLs including primary CTCL, rare subtypes.2,3
Session 2 dealt with the topic of primary cutaneous CD30+ T-cell LPDs. Submitted cases included in this ses-sion illustrated the morphologic and immunophenotypical spectrum of primary and secondary cutaneous CD30+ T-cell LPD. This session was divided into cases representing pri-mary cutaneous anaplastic large cell lymphoma (C-ALCL), ALK+ C-ALCL, lymphomatoid papulosis (LyP), borderline cases, and secondary cutaneous CD30+ ALCL.
Session 3 dealt with more uncommon CTCL subtypes including subcutaneous panniculitis-like T-cell lymphoma (SPTCL), primary cutaneous g/d T-cell lymphoma (PCGD-TCL), extranodal NK/T-cell lymphoma, hydroa vaccin-iforme-like T-cell lymphoma, and (rare subtypes of) primary cutaneous peripheral T-cell lymphoma, not otherwise speci-fied (PTCL-NOS). In addition, systemic T-cell lymphomas
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

492 Am J Clin Pathol 2013;139:491-514492 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
involving the skin secondarily, such as angioimmunoblastic T-cell lymphoma, were included in this session. This review summarizes the discussions and conclusions of sessions 2 and 3 on CTCL other than MF. It is not intended to provide a com-prehensive review of CTCL, but to highlight novel findings, areas of special interest, and diagnostic challenges illustrated by the spectrum of interesting and sometimes unusual cases submitted to the workshop.
Primary Cutaneous CD30+ T-Cell Lymphoproliferative Disorders
Primary cutaneous CD30+ T-cell lymphoproliferative disorders are the second most common group of CTCLs, accounting for approximately 30% of the cases.2 Three types are recognized in the 2008 WHO classification: C-ALCL, lymphomatoid papulosis (LyP), and borderline lesions.4 These entities seem to represent a spectrum of related condi-tions that show overlapping clinical and histologic features and may coexist in individual patients ❚Table 2❚. The disease must be distinguished from systemic ALCL with cutaneous involvement, which represents a separate disease with differ-ent clinical features, treatment, and outcome.
❚Table 1❚WHO-EORTC Classification of Cutaneous T-Cell Lymphomas
Frequency, Primary Cutaneous T-Cell and NK-Cell Lymphoma %a
Indolent clinical behavior Mycosis fungoides 44 Mycosis fungoides variants and subtypes 6 Subcutaneous panniculitis-like T-cell lymphoma 1 Primary cutaneous CD4+ small/medium 2 T-cell lymphoma Hydroa-vacciniforme–like lymphoma <1Primary cutaneous CD30+ lymphoproliferative disorders Primary anaplastic large cell lymphoma 8 Lymphomatoid papulosis 12Aggressive clinical behavior Sézary syndrome 3 Primary cutaneous NK/T-cell lymphoma, nasal type <1 Primary cutaneous CD8+ aggressive epidermotropic <1 cytotoxic T-cell lymphoma Primary cutaneous g/d T-cell lymphoma <1 Primary cutaneous peripheral T-cell 2 lymphoma, unspecifiedSecondary T-cell neoplasms ALK+ anaplastic large cell lymphoma — ALK– anaplastic large cell lymphoma — Angioimmunoblastic T-cell lymphoma — Adult T-cell leukemia/lymphoma —
ALK, anaplastic lymphoma kinase; NK, natural killer; WHO-EORTC, World Health Organization/European Organisation for Research and Treatment of Cancer.
a The frequency given represents the percentage of all B- and T-cell primary cutaneous lymphomas.
❚Table 2❚Differential Diagnosis Between C-ALCL and LyP
C-ALCL LyP
Clinical criteria Solitary, grouped or multifocal (~20%) nodular lesions Recurrent self-healing grouped or disseminated No clinical evidence of other CTCL papulonodular skin lesionsa
No extracutaneous involvement Concurrent with MF, C-ALCL, cHL, or systemic ALCL (20%Histology Dense nodular infiltrate composed of large pleomorphic, Type A: Wedge-shaped infiltrate with scattered CD30+ cells, anaplastic, immunoblastic, or RS-like cells intermingled with inflammatory cells Clusters of small reactive lymphocytes and eosinophils Type B: Epidermotropic infiltrate of small atypical CD30+ may be found or CD30– cells resembling MF Epidermotropism is not observed Type C: Dense infiltrate of cohesive CD30+ large atypical cells Morphologic variants mimicking C-ALCL Neutrophil-rich (pyogenic) Type D: Epidermotropic infiltrate of small- to medium-sized Pseudoepitheliomatous atypical CD8+ and CD30+ lymphoid cells resembling primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphomaPhenotype CD30+ in at least 75% of tumor cells; CD3+, CD4+ (~95%) CD30+ lymphoid cells mostly CD4+ CD8+ (~5%), variable loss of pan-T-cell antigens (CD2, CD3, CD8+ (~5%) mostly pediatric population, and type D; variable CD5); cytotoxic proteins+ loss of pan-T-cell antigens (CD2, CD3, CD5) EMA– (–/+) CLA+ Genotype TCR genes clonally rearranged TCR genes clonally rearranged in ∼ 60% IRF4/MUM1 translocation (∼ 26%) 5-Year survival, % 95 100Treatment Solitary lesions: Surgical resection or radiotherapy Topical steroids, PUVA, low-dose MTX Multiple lesions: low-dose MTXb Extracutaneous spread: Multiagent chemotherapy
ALCL, anaplastic large cell lymphoma ; C-ALCL, cutaneous ALCL; cHL, classic Hodgkin lymphoma; CTCL, cutaneous T-cell lymphoma; EMA, epithelial membrane antigen; LyP, lymphomatoid papulosis; MF, mycosis fungoides; MTX, methotrexate; PUVA, psoralen and UVA light therapy; RS, Reed-Sternberg; TCR, T-cell receptor.
a Self-healing is defined as spontaneous regression of individual lesions within weeks or months, regardless of whether new lesions appear.b MTX therapy has been proposed,15 but more studies are necessary to confirm the benefit of this treatment.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

Am J Clin Pathol 2013;139:491-514 493493 DOI: 10.1309/AJCP83AOQTMLOJTM 493
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
Primary C-ALCLC-ALCL is defined as a CTCL composed of large ana-
plastic, pleomorphic cells with expression of CD30 in more than 75% of tumor cells. It occurs predominantly in the adult/elderly population, is rare in children, and shows a male pre-dominance (2:1).4 Clinically, most patients have solitary or localized nodules, tumors, or papules that frequently become ulcerated. Partial or complete regression may occur, but skin relapses are often observed. Multifocal lesions are reported in approximately 20% of the patients, as was demonstrated in 3 of the submitted cases (cases 13, 266, and 299). Regional lymph nodes may be involved in 10% of the patients (see the end of this section and the section on lymphomatoid papulosis).
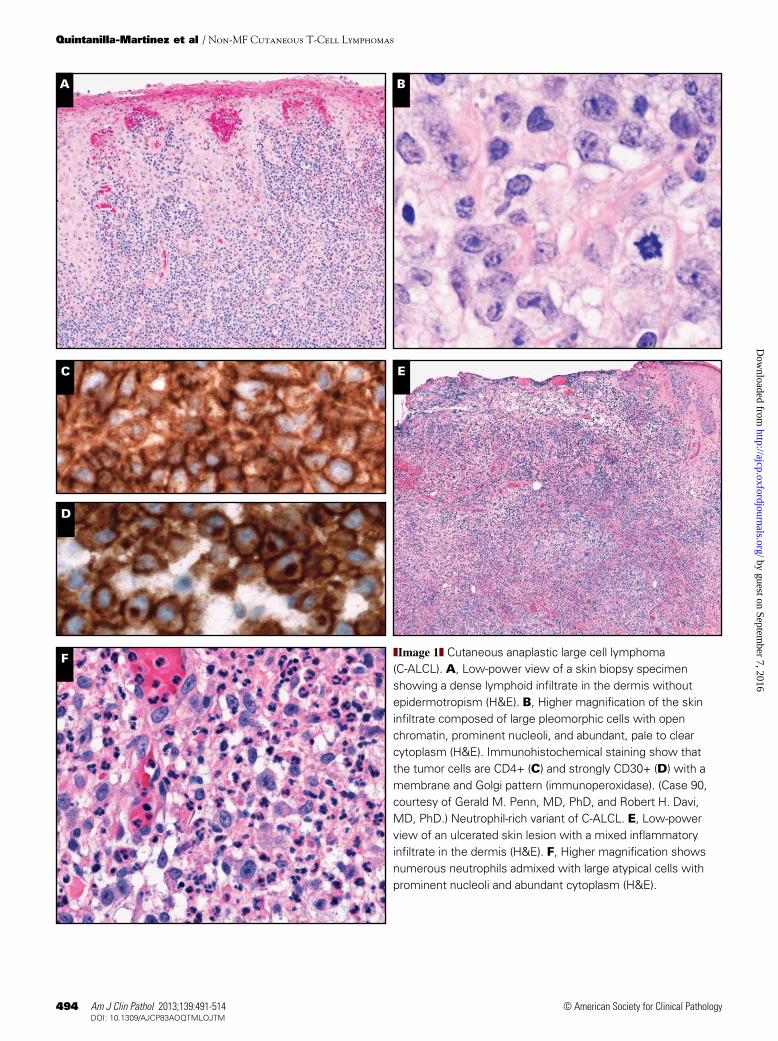
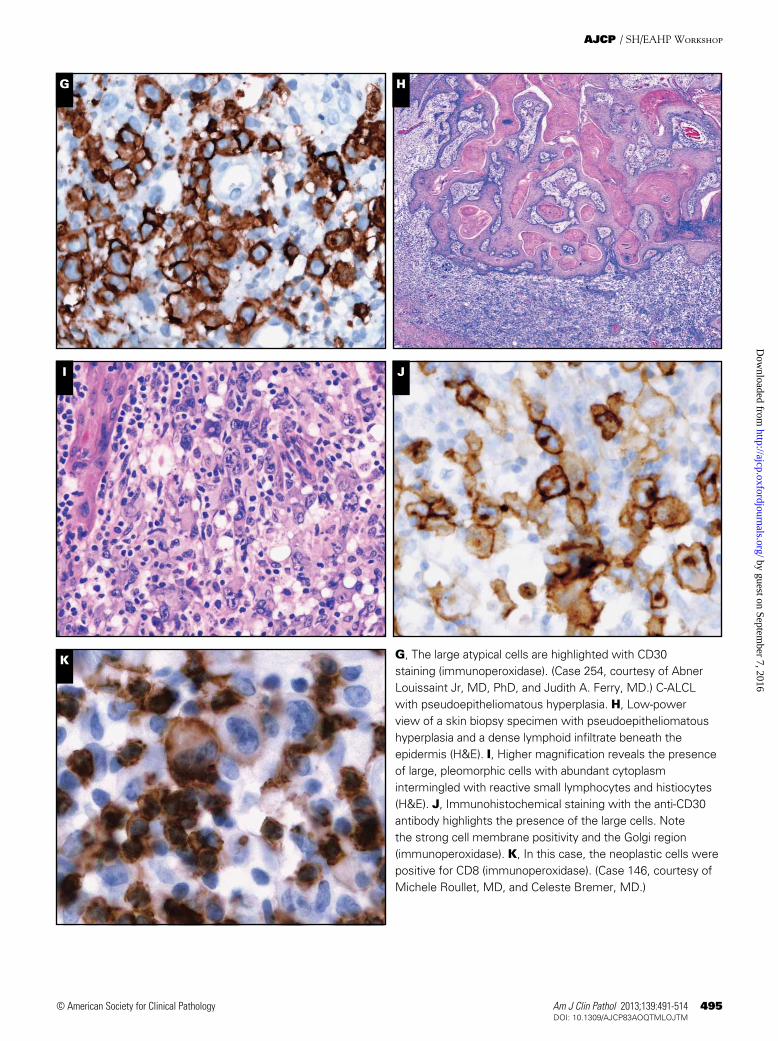
Morphologically, the infiltrate is usually diffuse and involves the dermis and subcutaneous tissue characterized by cohesive sheets of large, pleomorphic, and sometimes multi-nucleated giant cells resembling Reed-Sternberg cells ❚Image 1A❚, ❚Image 1B❚, ❚Image 1C❚, and ❚Image 1D❚. Although epidermal invasion may occur, epidermotropism is rare. A discrete inflammatory background may be present; however, when the inflammatory background is abundant, a differen-tial diagnosis with LyP or even reactive conditions may be considered. Two cases submitted illustrated this differential diagnosis. In case 13, the extensive granulomatous reaction delayed the diagnosis of C-ALCL for several months. In case 254, the ulcerated lesion with numerous admixed neutrophils (neutrophil-rich or pyogenic variant) masked the neoplastic lymphoid population, mimicking an inflammatory condition such as pyoderma ❚Image 1E❚ and ❚Image 1F❚.5 In both cases CD30 staining highlighted the atypical cells in the background and together with the clinical presentation helped to render the correct diagnosis ❚Image 1G❚. Some cases, in addition to an abundant inflammatory infiltrate, are also accompanied by prominent epidermal hyperplasia mimicking squamous cell carcinoma. Case 146 was an example of the so-called pseudo-epitheliomatous hyperplasia variant of C-ALCL ❚Image 1H❚, ❚Image 1I❚, and ❚Image 1J❚. Most patients with pseudoepi-theliomatous hyperplasia have 1 or several lesions that show spontaneous regression and even complete resolution within a few months of initial diagnosis. Because these cases seem to follow a relatively benign clinical course, a conservative watch and wait approach is recommended to avoid unneces-sary surgery and/or radiation.6
The immunophenotype of C-ALCL is characterized by a uniform membrane and Golgi expression of CD30 in most tumor cells (Image 1D, Image 1G, and Image 1J). The neo-plastic cells show an activated CD4+ T-cell phenotype (Image 1C) with variable loss of CD2, CD5, CD7, and/or CD3, and expression of cytotoxic granule–associated proteins gran-zyme B, perforin, and/or TIA-1. Three cases submitted to the workshop represented examples of the classic phenotype of
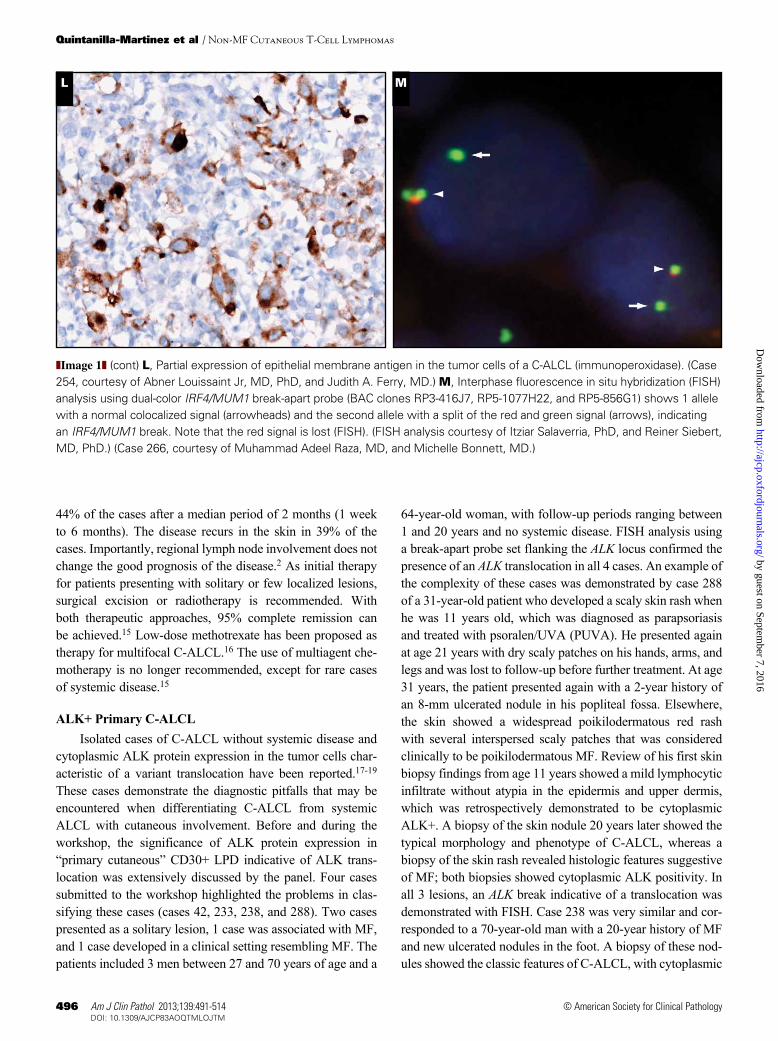
C-ALCL (cases 13, 90, and 299). Fewer than 5% of the cases have a cytotoxic CD8+ T-cell phenotype, as was illustrated in case 146 ❚Image 1K❚. Interestingly, although CD4/CD8 double negative phenotype is rare, 5 cases submitted to the workshop revealed this phenotype (cases 30, 174, 220, 254, and 266). Of note, 3 of these cases developed under special conditions such as seroma associated in a patient with derma-tomyositis (case 30), in an HIV-positive patient (case 174), and in a post-transplant setting (case 220). Although several reports describe C-ALCL in transplant recipients7,8 and HIV+ patients,5,9 neither conditions are recognized as an associated or predisposing clinical feature of conventional C-ALCL. Interestingly, a relatively high frequency of the neutrophil-rich variant of C-ALCL has been reported in patients with compromised immune systems.10 Unlike systemic CD30+ lymphomas, most C-ALCL lesions express the cutaneous lymphocyte antigen (CLA), but are negative for the epithelial membrane antigen (EMA). Nevertheless, weak and focal het-erogeneous expression of EMA may be observed and does not preclude the diagnosis of C-ALCL ❚Image 1L❚.11 Staining for the anaplastic lymphoma kinase (ALK) protein, indicative of the t(2;5) chromosomal translocation or its variants is negative (exceptions described later in this article).
Genetically, most cases show clonal rearrangement of T-cell receptor (TCR) genes. Recently, a recurrent transloca-tion involving IRF4/MUM1 oncogene locus at chromosome 6p25.3 was initially described to occur in 57% of C-ALCLs, as opposed to systemic ALCL or other peripheral T-cell lymphomas, not otherwise specified, where this translocation was rarely observed.12 Two further studies corroborated the presence of IRF4/MUM1 translocation almost exclusively in C-ALCL; however, the frequency was lower—20% in one study and 26% in the second study.13,14 The panel per-formed fluorescence in situ hybridization (FISH) analysis in 8 C-ALCL submitted cases (cases 13, 30, 90, 146, 220, 254, 266, and 299) using a break-apart IRF4 probe (BAC clones RP3-416J7, RP5-1077H22, and RP5-856G1). One case was positive (case 266) ❚Image 1M❚, accounting for 13% of the cases analyzed. Taken together, all the cases reported in the literature12-14 and the cases analyzed in this workshop, the frequency of IRF4 translocation in C-ALCL is estimated to be around 26% (24/90 cases). The expression of IRF4/MUM1 protein does not correlate with the presence of the transloca-tion and cannot be used as surrogate marker. Interestingly, despite the absence of IRF4/MUM1 translocation in systemic ALCL cases, the vast majority of cases show a strong expres-sion of IRF4/MUM1 protein, suggesting that the expression of this protein might be important for the pathogenesis of cutaneous and systemic ALCL cases.
The prognosis of C-ALCL is favorable, with 5-year survival rates between 76% and 96%.15 Spontaneous tumor regression, either partial or complete, is observed in 23% to
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
494 Am J Clin Pathol 2013;139:491-514494 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
A B
E
F
D
C
❚Image 1❚ Cutaneous anaplastic large cell lymphoma (C-ALCL). A, Low-power view of a skin biopsy specimen showing a dense lymphoid infiltrate in the dermis without epidermotropism (H&E). B, Higher magnification of the skin infiltrate composed of large pleomorphic cells with open chromatin, prominent nucleoli, and abundant, pale to clear cytoplasm (H&E). Immunohistochemical staining show that the tumor cells are CD4+ (C) and strongly CD30+ (D) with a membrane and Golgi pattern (immunoperoxidase). (Case 90, courtesy of Gerald M. Penn, MD, PhD, and Robert H. Davi, MD, PhD.) Neutrophil-rich variant of C-ALCL. E, Low-power view of an ulcerated skin lesion with a mixed inflammatory infiltrate in the dermis (H&E). F, Higher magnification shows numerous neutrophils admixed with large atypical cells with prominent nucleoli and abundant cytoplasm (H&E).
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 495495 DOI: 10.1309/AJCP83AOQTMLOJTM 495
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
G H
I J
K G, The large atypical cells are highlighted with CD30 staining (immunoperoxidase). (Case 254, courtesy of Abner Louissaint Jr, MD, PhD, and Judith A. Ferry, MD.) C-ALCL with pseudoepitheliomatous hyperplasia. H, Low-power view of a skin biopsy specimen with pseudoepitheliomatous hyperplasia and a dense lymphoid infiltrate beneath the epidermis (H&E). I, Higher magnification reveals the presence of large, pleomorphic cells with abundant cytoplasm intermingled with reactive small lymphocytes and histiocytes (H&E). J, Immunohistochemical staining with the anti-CD30 antibody highlights the presence of the large cells. Note the strong cell membrane positivity and the Golgi region (immunoperoxidase). K, In this case, the neoplastic cells were positive for CD8 (immunoperoxidase). (Case 146, courtesy of Michele Roullet, MD, and Celeste Bremer, MD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
496 Am J Clin Pathol 2013;139:491-514496 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
44% of the cases after a median period of 2 months (1 week to 6 months). The disease recurs in the skin in 39% of the cases. Importantly, regional lymph node involvement does not change the good prognosis of the disease.2 As initial therapy for patients presenting with solitary or few localized lesions, surgical excision or radiotherapy is recommended. With both therapeutic approaches, 95% complete remission can be achieved.15 Low-dose methotrexate has been proposed as therapy for multifocal C-ALCL.16 The use of multiagent che-motherapy is no longer recommended, except for rare cases of systemic disease.15
ALK+ Primary C-ALCLIsolated cases of C-ALCL without systemic disease and
cytoplasmic ALK protein expression in the tumor cells char-acteristic of a variant translocation have been reported.17-19 These cases demonstrate the diagnostic pitfalls that may be encountered when differentiating C-ALCL from systemic ALCL with cutaneous involvement. Before and during the workshop, the significance of ALK protein expression in “primary cutaneous” CD30+ LPD indicative of ALK trans-location was extensively discussed by the panel. Four cases submitted to the workshop highlighted the problems in clas-sifying these cases (cases 42, 233, 238, and 288). Two cases presented as a solitary lesion, 1 case was associated with MF, and 1 case developed in a clinical setting resembling MF. The patients included 3 men between 27 and 70 years of age and a
64-year-old woman, with follow-up periods ranging between 1 and 20 years and no systemic disease. FISH analysis using a break-apart probe set flanking the ALK locus confirmed the presence of an ALK translocation in all 4 cases. An example of the complexity of these cases was demonstrated by case 288 of a 31-year-old patient who developed a scaly skin rash when he was 11 years old, which was diagnosed as parapsoriasis and treated with psoralen/UVA (PUVA). He presented again at age 21 years with dry scaly patches on his hands, arms, and legs and was lost to follow-up before further treatment. At age 31 years, the patient presented again with a 2-year history of an 8-mm ulcerated nodule in his popliteal fossa. Elsewhere, the skin showed a widespread poikilodermatous red rash with several interspersed scaly patches that was considered clinically to be poikilodermatous MF. Review of his first skin biopsy findings from age 11 years showed a mild lymphocytic infiltrate without atypia in the epidermis and upper dermis, which was retrospectively demonstrated to be cytoplasmic ALK+. A biopsy of the skin nodule 20 years later showed the typical morphology and phenotype of C-ALCL, whereas a biopsy of the skin rash revealed histologic features suggestive of MF; both biopsies showed cytoplasmic ALK positivity. In all 3 lesions, an ALK break indicative of a translocation was demonstrated with FISH. Case 238 was very similar and cor-responded to a 70-year-old man with a 20-year history of MF and new ulcerated nodules in the foot. A biopsy of these nod-ules showed the classic features of C-ALCL, with cytoplasmic
L M
❚Image 1❚ (cont) L, Partial expression of epithelial membrane antigen in the tumor cells of a C-ALCL (immunoperoxidase). (Case 254, courtesy of Abner Louissaint Jr, MD, PhD, and Judith A. Ferry, MD.) M, Interphase fluorescence in situ hybridization (FISH) analysis using dual-color IRF4/MUM1 break-apart probe (BAC clones RP3-416J7, RP5-1077H22, and RP5-856G1) shows 1 allele with a normal colocalized signal (arrowheads) and the second allele with a split of the red and green signal (arrows), indicating an IRF4/MUM1 break. Note that the red signal is lost (FISH). (FISH analysis courtesy of Itziar Salaverria, PhD, and Reiner Siebert, MD, PhD.) (Case 266, courtesy of Muhammad Adeel Raza, MD, and Michelle Bonnett, MD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

Am J Clin Pathol 2013;139:491-514 497497 DOI: 10.1309/AJCP83AOQTMLOJTM 497
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
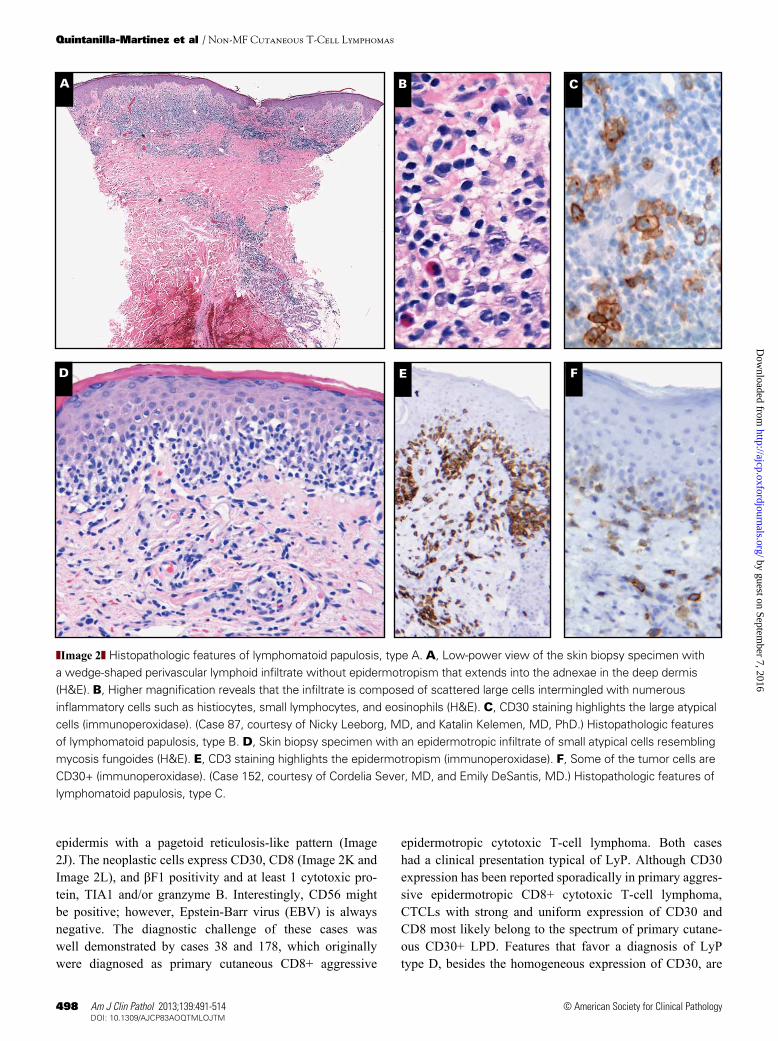
subtypes represent a spectrum of overlapping features that in part correlates with the age of the skin lesion biopsy speci-men, and may be present in individual patients at the same time, as was demonstrated in case 135. Immunophenotypi-cally, the CD30+ large atypical cells are CD4+ in most cases, but CD8+ cases, especially in children, have been reported and represent about 5% of LyP types A-C.22 Although clon-ally rearranged TCR genes have been detected in 60% of the lesions, from a clinical perspective LyP is not considered a malignant disorder. Ten cases were submitted to the workshop with the diagnosis of LyP (cases 69, 87, 128, 135, 152, 162, 178, 222, 256, and 273) representing the wide morphologic spectrum of the disease. LyP type A lesions are characterized by a wedge-shaped infiltrate with scattered or small clusters of large, sometimes multinucleated or Reed-Sternberg–like, CD30-positive cells intermingled with numerous inflamma-tory cells, such as histiocytes, small lymphocytes, neutrophils, and/or eosinophils (Image 2A, Image 2B, and Image 2C). LyP type A is the most common histologic presentation (cases 87, 135, and 256). LyP type B is uncommon (<10%) and is characterized by an epidermotropic infiltrate of small atypical cells with cerebriform nuclei that histologically resembles MF (Image 2D and Image 2E). Although originally it was reported that the tumor cells in LyP type B are CD30 negative,2,3 further experience has shown that many cases are CD30 posi-tive,15 as demonstrated by case 152 (Image 2F). LyP type C lesions are composed of a monotonous population of cohesive sheets of CD30+ large anaplastic lymphoid cells with only few admixed reactive inflammatory cells mimicking C-ALCL (Image 2G). The differential diagnosis with C-ALCL is not always easy and requires a careful clinicopathologic correla-tion. This was seen in case 69, a 68-year-old man who had papules on the right foot that resolved over 8 weeks. The lesions recurred 2 months later in the legs and earlobes and regressed after 3 to 4 weeks. Morphologically, the lesions were characterized by a diffuse, monotonous infiltration of large, cohesive CD30+, CD3+, CD4–, CD8– cells involving mainly the dermis (Image 2H and Image 2I). Clonal analysis revealed that the different lesions were clonally related. Inter-estingly, FISH analysis revealed a break in the IRF4/MUM1 locus indicative of a 6p25.3 translocation characteristic of C-ALCL.13 Over a period of several years the patient has continued with the same clinical findings. Despite the striking morphology and presence of the IRF4/MUM1 break, the panel agreed that because of the waxing and waning clinical picture, this case was better classified as LyP type C.
Recently, cases with clinical features of LyP but with histopathologic changes mimicking primary aggressive epi-dermotropic CD8+ cytotoxic T-cell lymphoma have been recognized and the term LyP type D has been proposed for this unusual variant.23 This variant is characterized by medium-sized, pleomorphic lymphocytes infiltrating the
ALK positivity and ALK break demonstrated with FISH. The patient experienced a recurrence of similar lesions 7 years later on the back and buttocks, which were clonally related to the skin nodules in the foot, as demonstrated by identical clonal product size peaks in the TCR gene rearrangement studies. Unfortunately, the clonal relatedness of the MF lesion and the ALK+ C-ALCL could not be demonstrated, leaving open the question of whether these represented 2 independent lesions or if the ALK+ C-ALCL was the result of transformed MF. However, neither of these patients developed a systemic disease. In case 233, a 27-year-old man with a solitary lesion, the ALK staining was nuclear and cytoplasmic, characteristic of a t(2;5) chromosomal translocation. This patient had been followed up for only 1 year, and the possibility that he might develop a systemic disease cannot be excluded.
After extensive discussions, the panel arrived at the con-clusion that cases with ALK positivity, such as the 4 cases submitted to the workshop, should be diagnosed as ALK+ C-ALCL until data from more cases can show whether these cases represent true ALK+ C-ALCL cases or whether some cases represent transformed MF with secondary ALK trans-location. Further proof that primary cutaneous ALK+ ALCLs exist was provided by a recent series of pediatric ALK+ ALCL, in which 6 children presented with a single skin lesion without developing systemic disease after a median follow-up of 7 years (ratio, 1-8 years).20 Interestingly, in contrast to the cases in the workshop where 3 of 4 cases expressed cytoplas-mic ALK protein indicative of a variant translocation, in the pediatric population 5 of 6 cases showed nuclear-cytoplasmic ALK staining characteristic of the t(2;5) chromosomal trans-location. Indeed, the question regarding how these patients should be treated still remains. In the reported cases, as well as in the cases in the workshop, complete resection of the lesion with or without local radiation resulted in complete remis-sions. Conservative therapy seems justified after systemic disease is thoroughly excluded in these cases.
Lymphomatoid Papulosis LyP is defined as a chronic, recurrent, self-healing skin
disease composed of large anaplastic, immunoblastic, or Hodgkin-like cells in a marked inflammatory background. LyP occurs mainly in adults (median age, 45 years) but chil-dren may also be affected.4,21 A male predominance has been found, with a male-to-female ratio of 2 to 3:1. Clinically, LyP is characterized by the presence of papular, papulonecrotic, and/or nodular skin lesions at different stages of develop-ment affecting predominantly the trunk and extremities. Skin lesions regress spontaneously within 3 to 12 weeks, and may leave superficial, hyperpigmented scars. The duration of the disease may vary from a couple of months up to many years.4
The 2008 WHO classification recognizes 3 histologic subtypes of LyP, types A, B, and C ❚Image 2❚. These histologic
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
498 Am J Clin Pathol 2013;139:491-514498 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
epidermotropic cytotoxic T-cell lymphoma. Both cases had a clinical presentation typical of LyP. Although CD30 expression has been reported sporadically in primary aggres-sive epidermotropic CD8+ cytotoxic T-cell lymphoma, CTCLs with strong and uniform expression of CD30 and CD8 most likely belong to the spectrum of primary cutane-ous CD30+ LPD. Features that favor a diagnosis of LyP type D, besides the homogeneous expression of CD30, are
epidermis with a pagetoid reticulosis-like pattern (Image 2J). The neoplastic cells express CD30, CD8 (Image 2K and Image 2L), and βF1 positivity and at least 1 cytotoxic pro-tein, TIA1 and/or granzyme B. Interestingly, CD56 might be positive; however, Epstein-Barr virus (EBV) is always negative. The diagnostic challenge of these cases was well demonstrated by cases 38 and 178, which originally were diagnosed as primary cutaneous CD8+ aggressive
A
D
CB
E F
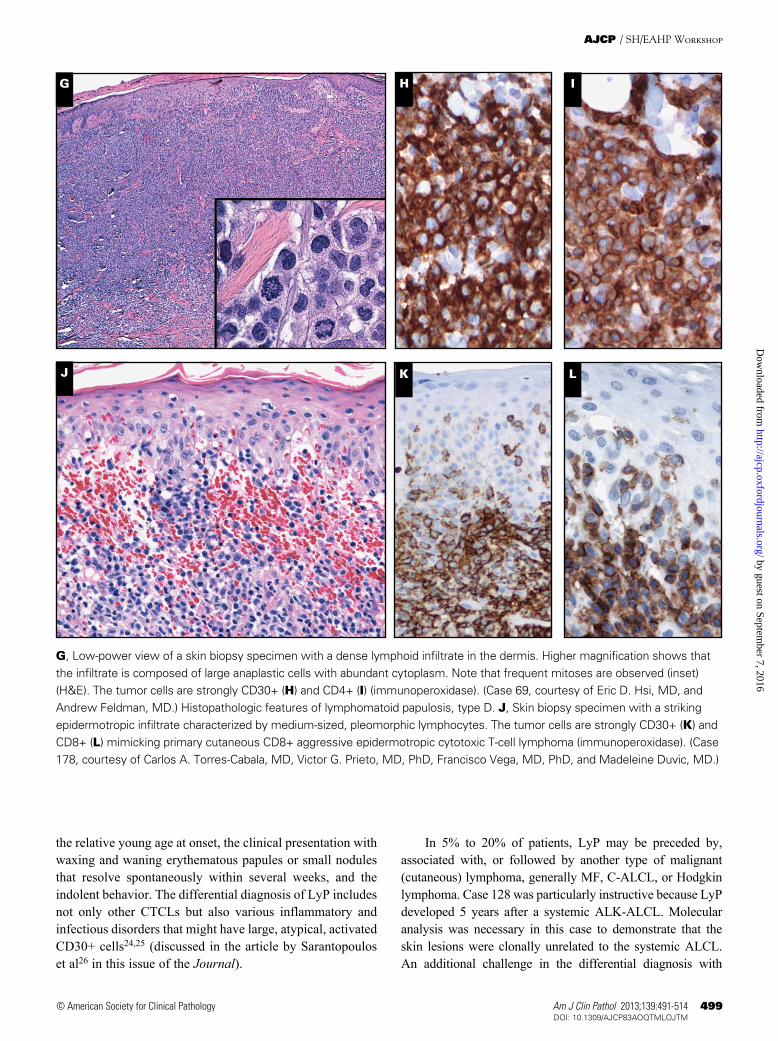
❚Image 2❚ Histopathologic features of lymphomatoid papulosis, type A. A, Low-power view of the skin biopsy specimen with a wedge-shaped perivascular lymphoid infiltrate without epidermotropism that extends into the adnexae in the deep dermis (H&E). B, Higher magnification reveals that the infiltrate is composed of scattered large cells intermingled with numerous inflammatory cells such as histiocytes, small lymphocytes, and eosinophils (H&E). C, CD30 staining highlights the large atypical cells (immunoperoxidase). (Case 87, courtesy of Nicky Leeborg, MD, and Katalin Kelemen, MD, PhD.) Histopathologic features of lymphomatoid papulosis, type B. D, Skin biopsy specimen with an epidermotropic infiltrate of small atypical cells resembling mycosis fungoides (H&E). E, CD3 staining highlights the epidermotropism (immunoperoxidase). F, Some of the tumor cells are CD30+ (immunoperoxidase). (Case 152, courtesy of Cordelia Sever, MD, and Emily DeSantis, MD.) Histopathologic features of lymphomatoid papulosis, type C.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 499499 DOI: 10.1309/AJCP83AOQTMLOJTM 499
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
In 5% to 20% of patients, LyP may be preceded by, associated with, or followed by another type of malignant (cutaneous) lymphoma, generally MF, C-ALCL, or Hodgkin lymphoma. Case 128 was particularly instructive because LyP developed 5 years after a systemic ALK-ALCL. Molecular analysis was necessary in this case to demonstrate that the skin lesions were clonally unrelated to the systemic ALCL. An additional challenge in the differential diagnosis with
the relative young age at onset, the clinical presentation with waxing and waning erythematous papules or small nodules that resolve spontaneously within several weeks, and the indolent behavior. The differential diagnosis of LyP includes not only other CTCLs but also various inflammatory and infectious disorders that might have large, atypical, activated CD30+ cells24,25 (discussed in the article by Sarantopoulos et al26 in this issue of the Journal).
G IH
J LK
G, Low-power view of a skin biopsy specimen with a dense lymphoid infiltrate in the dermis. Higher magnification shows that the infiltrate is composed of large anaplastic cells with abundant cytoplasm. Note that frequent mitoses are observed (inset) (H&E). The tumor cells are strongly CD30+ (H) and CD4+ (I) (immunoperoxidase). (Case 69, courtesy of Eric D. Hsi, MD, and Andrew Feldman, MD.) Histopathologic features of lymphomatoid papulosis, type D. J, Skin biopsy specimen with a striking epidermotropic infiltrate characterized by medium-sized, pleomorphic lymphocytes. The tumor cells are strongly CD30+ (K) and CD8+ (L) mimicking primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (immunoperoxidase). (Case 178, courtesy of Carlos A. Torres-Cabala, MD, Victor G. Prieto, MD, PhD, Francisco Vega, MD, PhD, and Madeleine Duvic, MD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

500 Am J Clin Pathol 2013;139:491-514500 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
Secondary Cutaneous CD30+ Anaplastic Large T-Cell Lymphoma
The differential diagnosis of a cutaneous CD30+ LPD includes MF with transformed CD30+ large cells and skin localization of a systemic CD30+ ALCL. Both systemic ALK+ and ALK– ALCLs frequently involve lymph nodes and extranodal sites including skin, soft tissues, and bone. One of the major problems in the differential diagnosis is that there are no reliable histologic criteria to differentiate between a primary and secondary cutaneous CD30+ ALCL. It has been suggested that to exclude a secondary cutane-ous CD30+ ALCL, adequate staging in all patients with a cutaneous CD30+ ALCL should be done.16 Seven cases of systemic ALCL with skin involvement were submitted to the workshop. In 2 of 3 systemic ALK– ALCLs submitted (cases 103 and 107) the skin was the initial manifestation of the disease. In general, systemic involvement occurs in less than 6 months, as occurred in these 2 cases (2 and 4 months, respectively). More importantly, patients with secondary cuta-neous CD30+ ALCL have a significant difference in survival, with a survival of 25% at 5 years vs more than 90% in patients with primary CD30+ C-ALCL.16 Three of the 4 ALK+ ALCL cases (20, 73, and 167) were typical examples of the small cell variant with CD8+ phenotype. The small cell variant of ALK+ ALCL is frequently associated with widespread dis-seminated disease and the initial biopsy may come from skin or other extranodal sites, contributing to the difficulty in mak-ing this diagnosis.28 This variant represents approximately 10% of ALK+ ALCL, and the skin is involved in 78% of these cases. The patients are often young, as demonstrated in cases 20 and 73, and morphologically, the dermis is infiltrated by numerous small lymphocytes with irregular nuclei, which are often CD30–; only a minor population of large cells are CD30+.29 Although a particularly useful diagnostic feature in the small cell variant is the tendency of ALK+, CD30+, large cells to be distributed in a perivascular pattern rather than ran-domly scattered, in the cases submitted to the workshop only randomly scattered cells were CD30+ and ALK+.
Uncommon Types of CTCL Other Than MF
Session 3 focused on relatively uncommon types of CTCLs that have been identified as distinct clinical enti-ties with characteristic clinical and pathologic features based on the WHO-EORTC classification for cutaneous lymphomas and the 2008 WHO classification.2,3 SPTCL and PCGD-TCLs are now considered 2 different entities involving mainly the subcutaneous tissue with very dif-ferent clinical prognoses ❚Table 3❚.30 PCGD-TCL, primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma, and primary cutaneous CD4+ small/medium
systemic ALCL is the fact that 5% to 10% of LyP cases may involve regional lymph nodes,16 which does not make it a sys-temic disease, as was shown in cases 222 and 273. Useful in the differential diagnosis is the fact that the nodal lesions are in the lymphatic drainage of the dominant cutaneous lesion. Without clinical information, it is not possible, based on morphology alone, to distinguish between lymphatic drainage or nodal involvement by systemic ALCL. Both might show prominent subcapsular and sinusoidal infiltration. To compli-cate things more, recently it was reported that nodal involve-ment by C-ALCL may mimic classic Hodgkin lymphoma, not only morphologically but also phenotypically with coex-pression of CD30 and CD15 by the tumor cells that may lead to a mistaken diagnosis of classic Hodgkin lymphoma. In these cases molecular analysis is necessary to demonstrate a common T-cell clone.27 Of note, in some cases the regional lymphadenopathy might regress spontaneously. Patients with regional lymph node involvement have a similar prognosis to those who only have skin lesions.16 LyP has an excellent prognosis; only 2% of the patients died of systemic disease after a median follow-up duration of 77 months.16
Borderline CasesAccording to the 2008 WHO classification, the term
“borderline” refers to cases in which, despite careful clinico-pathologic correlation, a definite distinction between C-ALCL and LyP cannot be made. However, follow-up will generally disclose whether such patients have C-ALCL or LyP. It is important to stress that the borderline diagnosis should only be used at the time of first diagnosis and not for individual lesions in the follow-up of the disease. Case 209 exemplified the difficulties that might be encountered in daily practice. An 11-year-old girl presented with a 6-week history of a 1.5-cm nodule in the right proximal forearm and a large indurated mass in the right arm. Biopsies of both lesions were per-formed. The nodule had the typical morphology of C-ALCL with sheets of cohesive large, pleomorphic CD30+ cells and pseudoepitheliomatous hyperplasia of the skin. The indurated mass had the morphology of LyP with scattered large CD30+ cells in an inflammatory background. One lesion was surgi-cally removed and the other underwent spontaneous resolu-tion after 4 months. A diagnosis of C-ALCL was proposed because the large, rapidly growing lesion suggested an aggres-sive lymphoma. However, the panel felt that because of the age of the patient and the morphologic discordance between the 2 lesions, this case was better classified as “borderline.” Nevertheless, despite the clinical presentation, children and young patients (under 20 years) usually have LyP.16 In these cases, a watch and wait period is recommended to avoid unnecessary treatment. In most cases, the larger skin lesions disappeared completely within 2 to 4 months, as was well illustrated in this case.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

Am J Clin Pathol 2013;139:491-514 501501 DOI: 10.1309/AJCP83AOQTMLOJTM 501
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
and dermis are typically uninvolved. Rimming of individual fat cells by neoplastic T cells is a helpful, though nonspecific diagnostic feature. Necrosis, karyorrhexis, and cytophagocy-tosis are common findings. In the early stages, the neoplastic infiltrates may lack significant atypia and a heavy inflamma-tory infiltrate may predominate. The neoplastic cells have an α/β+, CD3+, CD4–, CD8+ T-cell phenotype ❚Image 3C❚ and ❚Image 3D❚, with expression of cytotoxic proteins. CD30 and CD56 are rarely, if ever, expressed. Positive staining for βF1, preferably in combination with negative staining for TCR g/d, is extremely helpful in differentiating SPTCL from PCGD-TCL. If these stains cannot be performed, or in case of negative staining for βF1, other criteria listed in Table 3 may be helpful, as illustrated by 1 of the 7 cases of SPTCL submitted to the workshop (case 161). This concerned a pediatric patient who first had a localized subcutaneous nodular lesion on the back, and 7 years later developed new lesions on the right arm. Because the neoplastic T-cells were negative for βF1 and only partly positive for CD8, a diagnosis of PCGD-TCL was considered. However, the strictly subcutaneous infiltrates without dermal or epidermal involvement, negative CD56 staining, and prolonged sur-vival argued against this diagnosis, so a definite diagnosis of SPTCL was made.
Approximately 20% of patients with SPTCL may have associated autoimmune diseases, most commonly systemic lupus erythematous. Furthermore, lupus profundus panniculi-tis and SPTCL may have overlapping clinical and histologic features, making this differential diagnosis rather difficult (discussed in the article by Sarantopoulos et al26 in this issue of the Journal).
PCGD-TCLPCGD-TCL is a lymphoma composed of a clonal pro-
liferation of mature, activated g/d T cells with a cytotoxic phenotype.2,3,33 This group includes cases previously known as SPTCL with a g/d T-cell phenotype. PCGD-TCLs gener-ally present with disseminated plaques and/or ulceronecrotic nodules or tumors. Mucosal and other extranodal sites are
T-cell lymphoma remain provisional entities; more informa-tion is needed before they can be considered distinct enti-ties. EBV-positive natural killer (NK)/T-cell LPDs can also infiltrate primarily the skin including the new recognized entity hydroa vacciniforme (HV)–like lymphoma (HVLL) and extranodal NK/T-cell lymphoma, nasal type.
SPTCLSPTCL was originally defined as a cytotoxic T-cell
lymphoma, which preferentially infiltrated the subcutaneous tissue, was often complicated by a hemophagocytic syndrome (HPS), had an aggressive clinical course, and should there-fore be treated with aggressive multiagent chemotherapy.31,32 More recent studies showed clinical, histologic, and immuno-phenotypical differences between SPTCL with an α/β T-cell phenotype compared with those with a γ/δ T-cell phenotype, thus suggesting different entities (Table 3).30 In recent clas-sifications, the term SPTCL is only used for cases with an α/β T-cell phenotype, whereas cases expressing the γ/δ TCR are reclassified as PCGD-TCL.2,3,33
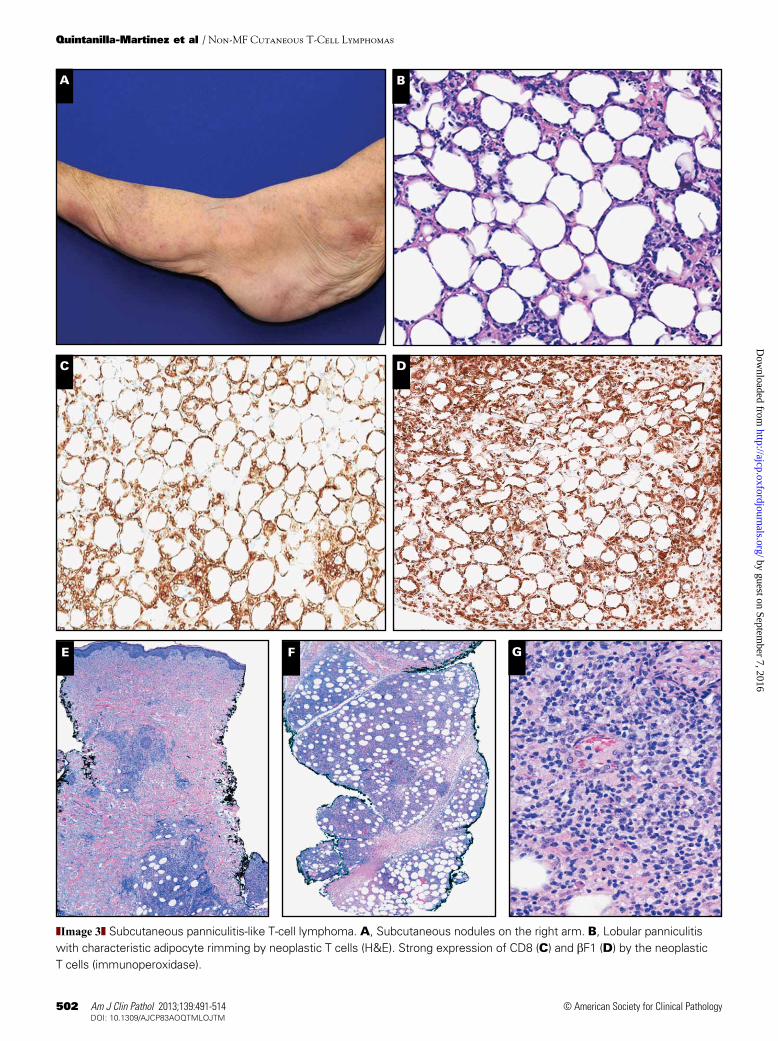
SPTCLs occur in adults as well as young children. Patients generally have solitary or multiple nodules or deeply seated plaques, which mainly involve the legs, arms, and trunk ❚Image 3A❚. Ulceration is uncommon. Systemic symp-toms such as fever, fatigue, and weight loss, and laboratory abnormalities, including cytopenias and elevated liver func-tion test results, are common, but a frank HPS is observed in only 15% of patients. Dissemination to extracutaneous sites is rare. SPTCLs have an excellent prognosis in particular if not associated with an HPS. For SPTCLs without associated HPS, systemic steroids or other immunosuppressive agents are recommended, whereas in cases of solitary skin lesions, radio-therapy is advised. Only in cases with progressive disease not responding to immunosuppressive therapy and in cases with HPS should multiagent chemotherapy be considered.
SPTCL reveals subcutaneous histologic infiltrates simu-lating a panniculitis with small, medium, or sometimes large pleomorphic T cells with hyperchromatic nuclei and often many macrophages ❚Image 3B❚. The overlying epidermis
❚Table 3❚Differential Diagnosis Between SPTCL and PCGD-TCL
SPTCL PCGD-TCL
Phenotype TCR-β1+, CD4–, CD8+, CD56– TCR-g1+, CD4–, CD8–, CD56+Architecture Subcutaneous Subcutaneous and/or epidermal/dermalHPS Rare (17%) Common (45%)5-Year survival 91% (without HPS) vs 46% (with HPS) 11%Treatment Systemic steroids Systemic chemotherapy
HPS, hemophagocytic syndrome; PCGD-TCL, primary cutaneous g/d T-cell lymphoma; SPTCL, subcutaneous panniculitis-like T-cell lymphoma.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
502 Am J Clin Pathol 2013;139:491-514502 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
A B
C D
GFE
❚Image 3❚ Subcutaneous panniculitis-like T-cell lymphoma. A, Subcutaneous nodules on the right arm. B, Lobular panniculitis with characteristic adipocyte rimming by neoplastic T cells (H&E). Strong expression of CD8 (C) and bF1 (D) by the neoplastic T cells (immunoperoxidase).
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 503503 DOI: 10.1309/AJCP83AOQTMLOJTM 503
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
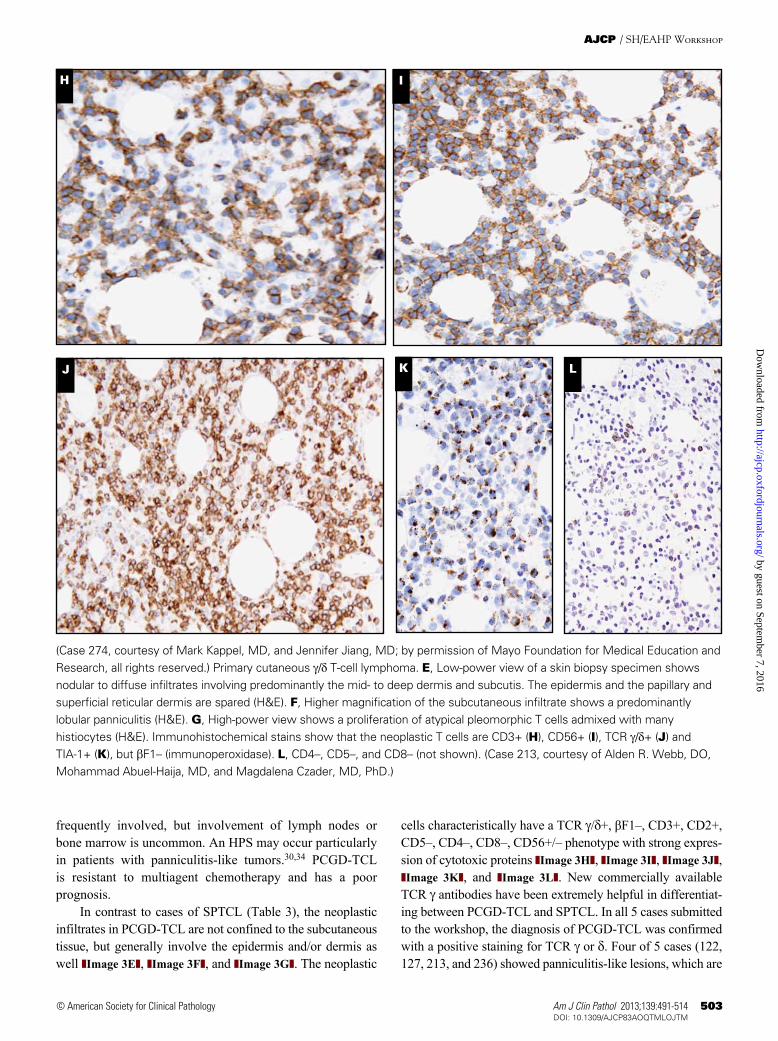
cells characteristically have a TCR g/d+, βF1–, CD3+, CD2+, CD5–, CD4–, CD8–, CD56+/– phenotype with strong expres-sion of cytotoxic proteins ❚Image 3H❚, ❚Image 3I❚, ❚Image 3J❚, ❚Image 3K❚, and ❚Image 3L❚. New commercially available TCR g antibodies have been extremely helpful in differentiat-ing between PCGD-TCL and SPTCL. In all 5 cases submitted to the workshop, the diagnosis of PCGD-TCL was confirmed with a positive staining for TCR g or d. Four of 5 cases (122, 127, 213, and 236) showed panniculitis-like lesions, which are
frequently involved, but involvement of lymph nodes or bone marrow is uncommon. An HPS may occur particularly in patients with panniculitis-like tumors.30,34 PCGD-TCL is resistant to multiagent chemotherapy and has a poor prognosis.
In contrast to cases of SPTCL (Table 3), the neoplastic infiltrates in PCGD-TCL are not confined to the subcutaneous tissue, but generally involve the epidermis and/or dermis as well ❚Image 3E❚, ❚Image 3F❚, and ❚Image 3G❚. The neoplastic
H I
J LK
(Case 274, courtesy of Mark Kappel, MD, and Jennifer Jiang, MD; by permission of Mayo Foundation for Medical Education and Research, all rights reserved.) Primary cutaneous g/d T-cell lymphoma. E, Low-power view of a skin biopsy specimen shows nodular to diffuse infiltrates involving predominantly the mid- to deep dermis and subcutis. The epidermis and the papillary and superficial reticular dermis are spared (H&E). F, Higher magnification of the subcutaneous infiltrate shows a predominantly lobular panniculitis (H&E). G, High-power view shows a proliferation of atypical pleomorphic T cells admixed with many histiocytes (H&E). Immunohistochemical stains show that the neoplastic T cells are CD3+ (H), CD56+ (I), TCR g/d+ (J) and TIA-1+ (K), but bF1– (immunoperoxidase). L, CD4–, CD5–, and CD8– (not shown). (Case 213, courtesy of Alden R. Webb, DO, Mohammad Abuel-Haija, MD, and Magdalena Czader, MD, PhD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

504 Am J Clin Pathol 2013;139:491-514504 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
mycosis fungoides, respectively. Most cases have a genuine NK-cell origin with no CD3 surface antigen and no TCR expression, together with a germline state of the TCR gene. However, there is evidence of a g/d T-cell origin based on immunophenotype or clonal TCR rearrangement in some cases. Current evidence does not suggest any differences in biology, clinical behavior, or prognosis between “T” and “NK” cases.37
Thirteen cases of extranodal NK/T-cell lymphoma in the skin were submitted to the workshop. Concurrent nasal and skin localization was noted in 1 case (case 3). Dissemination to extracutaneous sites was described in 5 cases. A CD8+, CD56– T-cell phenotype was described in 1 case (case 132); in addition, a clonal rearrangement of the g-TCR gene was noted in case 78. One case of extranodal NK-cell lymphoma was observed in the context of renal transplantation, and should hence be classified as monomorphic NK/T cell post-transplant lymphoproliferative disease.
HV and HVLLHVLL is one of the EBV+ lymphoproliferative disorders
of childhood that has been recognized by the 2008 WHO classification.38 Its name arises from the clinical similarities to HV, which is a rare condition of childhood associated with hypersensitivity to ultraviolet light that leads to a necrotic papulovesicular eruption with scarring on sun-exposed areas, typically the face and the dorsum of the hands. No ethnic or racial association is seen in HV cases, as illustrated in case 56, a white 8-year-old girl with HV. In contrast, HVLL is a lymphoproliferative disorder of most often CD8+ T-cell but rarely NK-cell derivation seen mainly in children from Asia, Mexico, or Central and South America.39,40 Although the skin lesions are not exclusively limited to sun-exposed areas and do not only appear after sun-exposure, like HV skin lesions, a seasonal increase occurs during the summer.41 Recurrent lesions may be seen for up to 15 years before progression to systemic involvement, which is accompanied by symptoms of fever, wasting, lymphadenopathy, and hepatosplenomegaly. In some patients, episodes of the disease and HV-like symp-toms are precipitated by a hypersensitivity to mosquito bites
HV, hypersensitivity to mosquito bites, and HVLL appear to represent different cutaneous manifestations of chronic active EBV infection and produce similar histologic findings, characterized by NK-cell or T-cell infiltrates that may extend from the epidermis to the subcutis, showing necrosis, angio-centricity, and angioinvasion. CD56 is positive only in the NK cell–derived lesions, mainly patients with hypersensitivity to mosquito bites. As part of the disease definition, EBV must be demonstrated on EBER in situ hybridization in all cases. Cur-rently a clear morphologic division is not seen between HV and HVLL.42 Both disorders are EBV associated, and it has been proposed that the most useful criterion to separate these
considered to have a more aggressive clinical behavior than cases with only dermal or epidermal involvement. Interest-ingly, in 2 cases (122 and 213) the infiltrates were confined to the subcutis and deep dermis, whereas the overlying skin was not involved. In 2 other cases (127 and 136), dermis and epidermis were not present in the biopsy specimens. Case 213 had a 10-year history of recurrent self-remitting subcutaneous nodules clinically diagnosed as erythema nodosum before a diagnosis of PCGD-TCL with predominantly panniculitis-like involvement of subcutis and deep dermis was made. Unfortunately, biopsy specimens from these preceding skin lesions were not available. Similarly, in case 127, presenting with multiple subcutaneous nodules, histologically consistent with a diagnosis of PCGD-TCL, subcutaneous nodules had developed 3 years before the diagnosis of PCGD-TCL had been made and 2 years after successful cyclophosphamide, hydroxydaunorubicin, vincristine (Oncovin), prednisone (CHOP) therapy. Biopsies of these lesions were reportedly nondiagnostic or the lesions were diagnosed as lipoma. In both cases a relationship between these nonspecific or benign lesions and PCGD-TCL could not be established. Neverthe-less, the presence of such nonspecific subcutaneous nodules may precede or follow a diagnosis of lymphoma (cases 127 and 213), and the strictly subcutaneous infiltrates (cases 122 and 213) and the apparently indolent course in some of these cases are more consistent with a diagnosis of SPTCL; these findings highlight the overlapping features between PCGD-TCL and SPTCL in these cases.
Extranodal NK/T-Cell LymphomaExtranodal NK/T-cell lymphoma, nasal type, is an EBV+
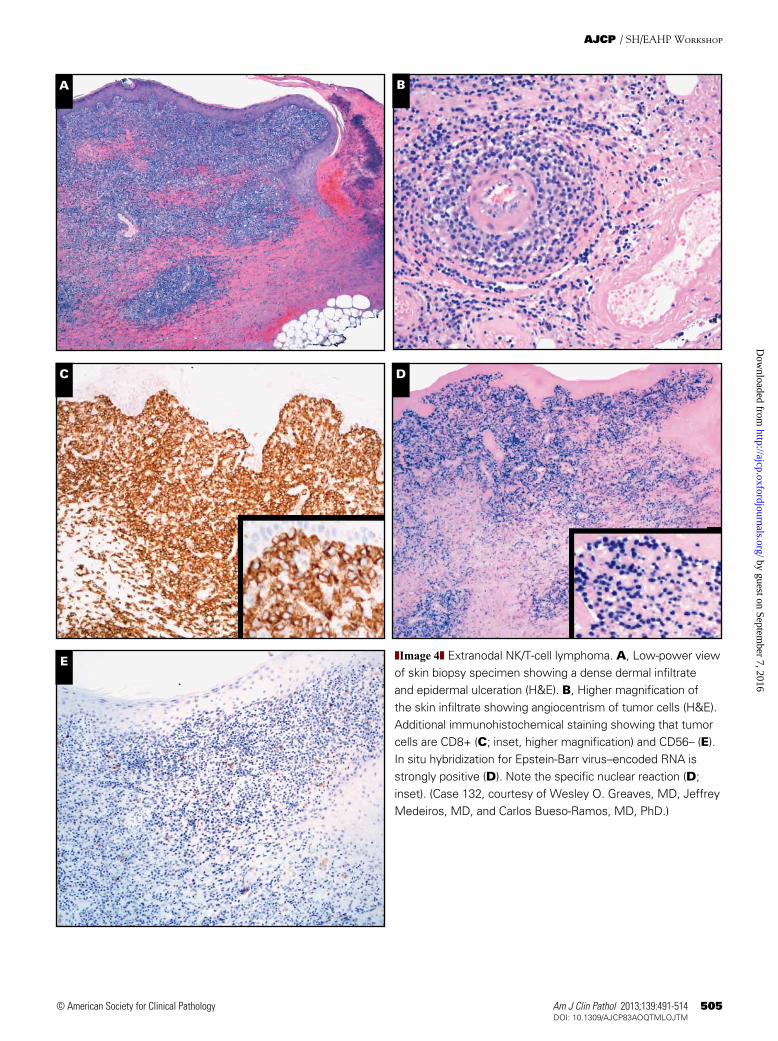
lymphoma of small, medium, or large cells usually with an NK-cell, or more rarely a cytotoxic T-cell, phenotype. The skin is the second most common site of involvement after the nasal cavity/nasopharynx, and skin involvement may be a primary or secondary manifestation of the disease.35 Although the prognosis of nasal forms is better than that of extranasal forms, the overall prognosis remains poor.36 Diagnosis requires a biopsy showing a diffuse proliferation of lymphoid cells harboring a surface CD3–, cytoplasmic CD3e+, CD56+/– phenotype, and a cytotoxic profile (posi-tive for TIA-1, perforin, or granzyme B) ❚Image 4A❚, ❚Image 4B❚, ❚Image 4C❚, ❚Image 4D❚, and ❚Image 4E❚. Demonstra-tion of EBV in the biopsy specimen (with EBV-encoded RNA [EBER] in situ hybridization or immunohistochemistry for latent membrane protein 1) is mandatory (Image 4D). Infiltration of the tumor cells is frequently angiocentric and angiodestructive, with frequent zonal necrosis (Image 4B). On H&E sections, subcutaneous localization with some rim-ming around adipocytes (cases 169 and 221), or extensive epidermotropism with Pautrier microabscess formation (cases 176 and 218), rendered a differential diagnosis of SPTCL and
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 505505 DOI: 10.1309/AJCP83AOQTMLOJTM 505
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
A B
C D
E ❚Image 4❚ Extranodal NK/T-cell lymphoma. A, Low-power view of skin biopsy specimen showing a dense dermal infiltrate and epidermal ulceration (H&E). B, Higher magnification of the skin infiltrate showing angiocentrism of tumor cells (H&E). Additional immunohistochemical staining showing that tumor cells are CD8+ (C; inset, higher magnification) and CD56– (E). In situ hybridization for Epstein-Barr virus–encoded RNA is strongly positive (D). Note the specific nuclear reaction (D; inset). (Case 132, courtesy of Wesley O. Greaves, MD, Jeffrey Medeiros, MD, and Carlos Bueso-Ramos, MD, PhD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

506 Am J Clin Pathol 2013;139:491-514506 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
Primary Cutaneous CD8+ Aggressive Epidermotropic Cytotoxic T-Cell Lymphoma
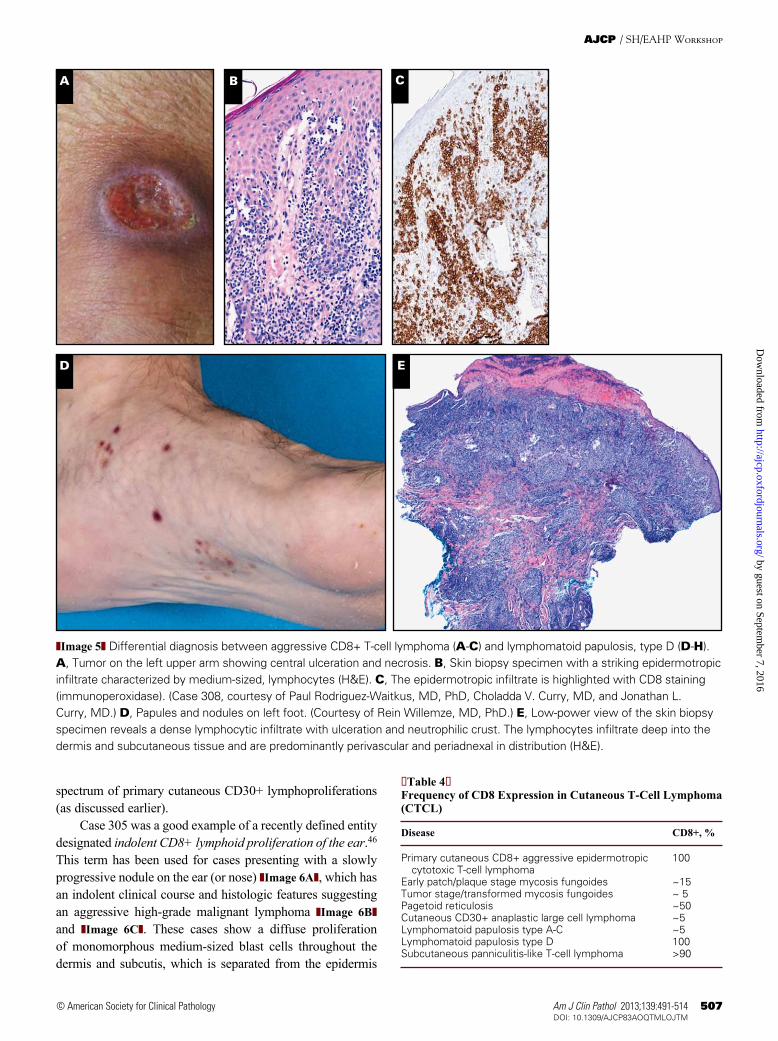
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphomas are characterized by a prolifera-tion of epidermotropic CD8+ cytotoxic T cells, an aggressive clinical behavior, and poor prognosis.45 Clinically, these lymphomas have localized or disseminated eruptive papules, nodules, and tumors with central ulceration and necrosis or superficial, hyperkeratotic patches and plaques ❚Image 5A❚. These lymphomas tend to disseminate to mucosal and other extranodal sites (lung, testis, central nervous system, and oral mucosa), but lymph nodes are often spared. Involvement of the oral mucosa and/or tongue was reported in 2 of 10 cases submitted to the workshop (cases 131 and 157). The histologic appearance is variable, ranging from a lichenoid pattern with marked, pagetoid epidermotropism in early patch-like lesions to diffuse dermal infiltrates in nodular and tumorous lesions, in which marked epidermotropism and folliculotropism are generally maintained ❚Image 5B❚ and ❚Image 5C❚. Epidermal necrosis and ulceration, as well as invasion and destruction of adnexal skin structures, are commonly found. Angiocentricity and angioinvasion may be present. Tumor cells may be small-medium or medium-large with pleomorphic or blastic nuclei. They have a βF1+, CD3+, CD8+, granzyme B+, perforin+, TIA-1+, CD45RA+, CD45RO–, CD2–, CD4–, CD5–, CD7–/+ T-cell phenotype. Uncommonly, the neoplastic T cells may express CD30, as seen in one of the submitted cases (case 85). The proliferation fraction is generally high.
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphomas should be differentiated from other types of CTCLs or cutaneous LPDs expressing a CD8+ cytotoxic T-cell phenotype ❚Table 4❚, including LyP type D (as discussed earlier) ❚Image 5D❚, ❚Image 5E❚, ❚Image 5F❚, ❚Image 5G❚, and ❚Image 5H❚. Differentiation from SPTCL (subcutaneous plaques and nodules), early-stage MF (eczematous patches and plaques), and pagetoid reticulosis (solitary slowly expanding plaque) is generally not difficult, in particular when the clinical features are taken into account. Differentiation from rare cases of CD8+ tumor stage or trans-formed MF, which may also show ulcerating tumors, may be much more difficult. Clinical information and histologic examination of preceding or concurrent patches or plaques are essential to make a correct diagnosis. Occasionally, patients with classic CD4+ early-stage MF may develop (ulcerating) tumors with a cytotoxic CD8+ T-cell phenotype, as illustrated in case 279. In this patient both the small neoplastic CD4+ T cells in early patches and plaques and the medium-sized CD8+ neoplastic T cells in a subsequent tumor showed loss of CD2 and CD5 as well as the same T-cell clone. Although pri-mary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphomas may occasionally express CD30, CTCL showing coexpression of CD8 and CD30 usually belong to the
2 entities is clonal analysis of the TCR genes. If the skin lesion shows a clonal TCR rearrangement, a diagnosis of HVLL should be rendered. Remarkably, the 2 HVLL cases submit-ted to the workshop were both adult patients with a history of recurrent skin lesions since childhood. Case 177 concerned a 31-year old Peruvian man with a 21-year history of blistering and scarring eruptions on the face, neck, chest, and extremi-ties, as well as a hypersensitivity to mosquito bites, before developing mild systemic symptoms. A progressive disease course was noted in case 269, which concerned a 36-year-old Peruvian patient with facial edema, disseminated skin rash, and inguinal lymphadenopathy, who died of lymphoma 2 years later. Although HVLL is characterized by a monoclo-nal proliferation of T cells, its treatment remains uncertain. Chemotherapy has been used in many patients, but the effect is usually transient and does not induce sustained remission in most cases. A conservative approach is recommended as first-line therapy.43
PTCL-NOS
PTCL-NOS is a heterogeneous group of nodal and extranodal mature T-cell lymphomas, and primary presenta-tion in the skin is uncommon. The diagnosis can only be made by excluding other specifically defined categories of mature T-cell lymphoma in the current WHO classification. Complete clinical examination and an accurate clinical his-tory are critical to particularly rule out (transformed) MF or 1 of the 3 subtypes of primary cutaneous PTCL-NOS that have been recognized in recent classifications, ie, PCGD-TCL, primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma, and primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma (PCSM-TCL), which will be discussed separately. The overall survival rates of PTCL-NOS presenting in the skin are poor and inde-pendent of the presence or absence of extracutaneous disease at the time of diagnosis, cell size, or expression of CD4+ or CD8+ phenotype.44
The panel considered PTCL-NOS to be the preferred diagnosis in 19 cases submitted to the workshop. In 11 of 19 cases (cases 52, 86, 112, 160, 166, 244, 249, 278, 283, 289, and 293) skin involvement was secondary to primary involve-ment of extracutaneous sites. In 8 of 19 cases, the skin was the primary site of involvement (cases 26, 29, 57, 183, 199, 203, 272, and 282) and subsequently extracutaneous spread was reported in 2 cases (203 and 272). In most of these cases, it was impossible to definitively exclude late-stage MF from the differential diagnosis, because detailed clinical data or pic-tures of the lesions were missing. In cases 52, 57, and 183, the submitted diagnosis of a PCGD-TCL was rendered unlikely because of a lack of demonstrable TCR g/d+ phenotype.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 507507 DOI: 10.1309/AJCP83AOQTMLOJTM 507
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
❚Table 4❚Frequency of CD8 Expression in Cutaneous T-Cell Lymphoma (CTCL)
Disease CD8+, %
Primary cutaneous CD8+ aggressive epidermotropic 100 cytotoxic T-cell lymphomaEarly patch/plaque stage mycosis fungoides ~15Tumor stage/transformed mycosis fungoides ~ 5Pagetoid reticulosis ~50Cutaneous CD30+ anaplastic large cell lymphoma ~5Lymphomatoid papulosis type A-C ~5Lymphomatoid papulosis type D 100Subcutaneous panniculitis-like T-cell lymphoma >90
D
CBA
E
❚Image 5❚ Differential diagnosis between aggressive CD8+ T-cell lymphoma (A-C) and lymphomatoid papulosis, type D (D-H). A, Tumor on the left upper arm showing central ulceration and necrosis. B, Skin biopsy specimen with a striking epidermotropic infiltrate characterized by medium-sized, lymphocytes (H&E). C, The epidermotropic infiltrate is highlighted with CD8 staining (immunoperoxidase). (Case 308, courtesy of Paul Rodriguez-Waitkus, MD, PhD, Choladda V. Curry, MD, and Jonathan L. Curry, MD.) D, Papules and nodules on left foot. (Courtesy of Rein Willemze, MD, PhD.) E, Low-power view of the skin biopsy specimen reveals a dense lymphocytic infiltrate with ulceration and neutrophilic crust. The lymphocytes infiltrate deep into the dermis and subcutaneous tissue and are predominantly perivascular and periadnexal in distribution (H&E).
spectrum of primary cutaneous CD30+ lymphoproliferations (as discussed earlier).
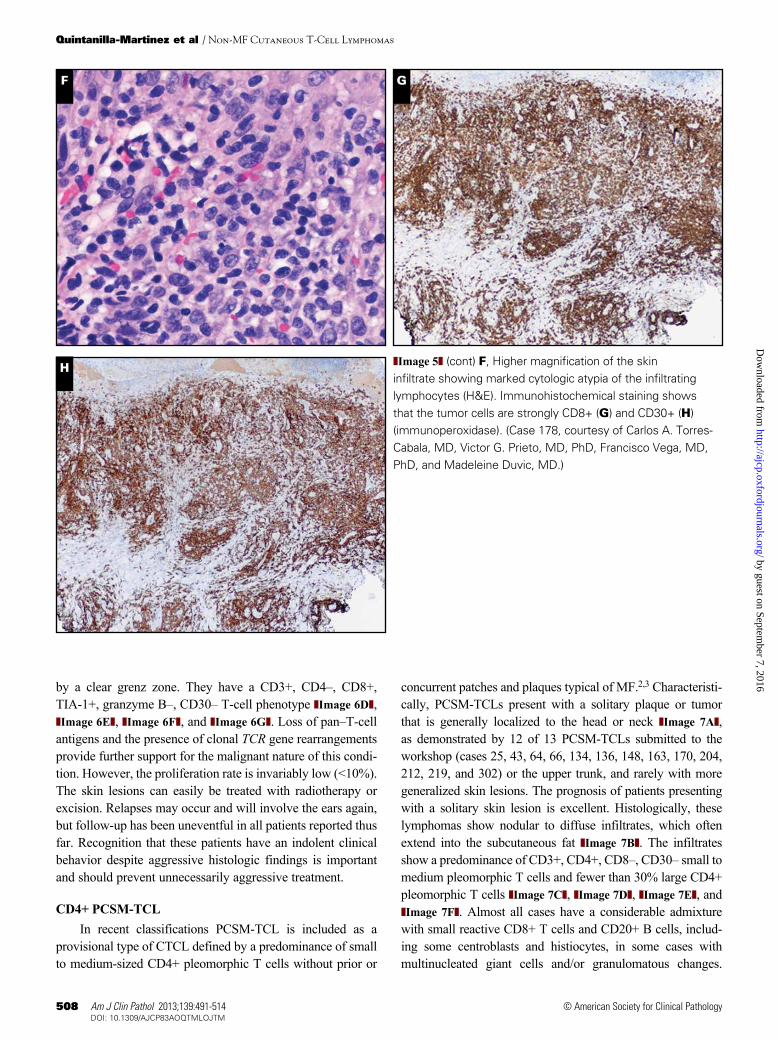
Case 305 was a good example of a recently defined entity designated indolent CD8+ lymphoid proliferation of the ear.46 This term has been used for cases presenting with a slowly progressive nodule on the ear (or nose) ❚Image 6A❚, which has an indolent clinical course and histologic features suggesting an aggressive high-grade malignant lymphoma ❚Image 6B❚ and ❚Image 6C❚. These cases show a diffuse proliferation of monomorphous medium-sized blast cells throughout the dermis and subcutis, which is separated from the epidermis
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
508 Am J Clin Pathol 2013;139:491-514508 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
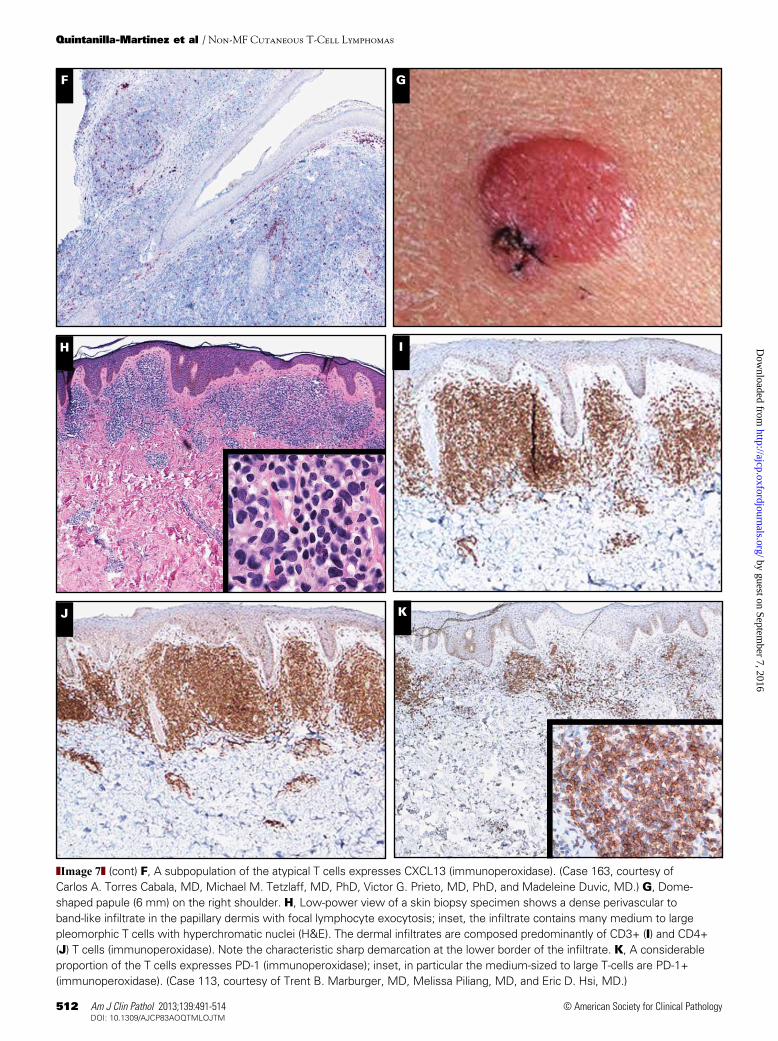
concurrent patches and plaques typical of MF.2,3 Characteristi-cally, PCSM-TCLs present with a solitary plaque or tumor that is generally localized to the head or neck ❚Image 7A❚, as demonstrated by 12 of 13 PCSM-TCLs submitted to the workshop (cases 25, 43, 64, 66, 134, 136, 148, 163, 170, 204, 212, 219, and 302) or the upper trunk, and rarely with more generalized skin lesions. The prognosis of patients presenting with a solitary skin lesion is excellent. Histologically, these lymphomas show nodular to diffuse infiltrates, which often extend into the subcutaneous fat ❚Image 7B❚. The infiltrates show a predominance of CD3+, CD4+, CD8–, CD30– small to medium pleomorphic T cells and fewer than 30% large CD4+ pleomorphic T cells ❚Image 7C❚, ❚Image 7D❚, ❚Image 7E❚, and ❚Image 7F❚. Almost all cases have a considerable admixture with small reactive CD8+ T cells and CD20+ B cells, includ-ing some centroblasts and histiocytes, in some cases with multinucleated giant cells and/or granulomatous changes.
by a clear grenz zone. They have a CD3+, CD4–, CD8+, TIA-1+, granzyme B–, CD30– T-cell phenotype ❚Image 6D❚, ❚Image 6E❚, ❚Image 6F❚, and ❚Image 6G❚. Loss of pan–T-cell antigens and the presence of clonal TCR gene rearrangements provide further support for the malignant nature of this condi-tion. However, the proliferation rate is invariably low (<10%). The skin lesions can easily be treated with radiotherapy or excision. Relapses may occur and will involve the ears again, but follow-up has been uneventful in all patients reported thus far. Recognition that these patients have an indolent clinical behavior despite aggressive histologic findings is important and should prevent unnecessarily aggressive treatment.
CD4+ PCSM-TCLIn recent classifications PCSM-TCL is included as a
provisional type of CTCL defined by a predominance of small to medium-sized CD4+ pleomorphic T cells without prior or
F G
❚Image 5❚ (cont) F, Higher magnification of the skin infiltrate showing marked cytologic atypia of the infiltrating lymphocytes (H&E). Immunohistochemical staining shows that the tumor cells are strongly CD8+ (G) and CD30+ (H) (immunoperoxidase). (Case 178, courtesy of Carlos A. Torres-Cabala, MD, Victor G. Prieto, MD, PhD, Francisco Vega, MD, PhD, and Madeleine Duvic, MD.)
H
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 509509 DOI: 10.1309/AJCP83AOQTMLOJTM 509
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
A
C
B
❚Image 6❚ Indolent CD8+ proliferation of the ear. A, Infiltrated plaque on the left ear. B, Low-power view of a skin biopsy specimen showing a diffuse nonepidermotropic infiltrate throughout the entire dermis (H&E). C, Higher magnification shows a monotonous proliferation of medium to large pleomorphic T cells with prominent nucleoli. Considerable karyorrhexis can be noted (H&E).
Plasma cells are generally present, in some cases even in large numbers, which may initially suggest a diagnosis of cutaneous marginal zone B-cell lymphoma, as illustrated by case 64 in this workshop. Eosinophils are generally few or absent. The proliferation rate is low, varying between less than 5% and at most 20%. Consistent with recent studies, the medium to large atypical T cells expressed PD-1, BCL6, and CXCL13, but not CD10 in all 13 submitted cases.47,48
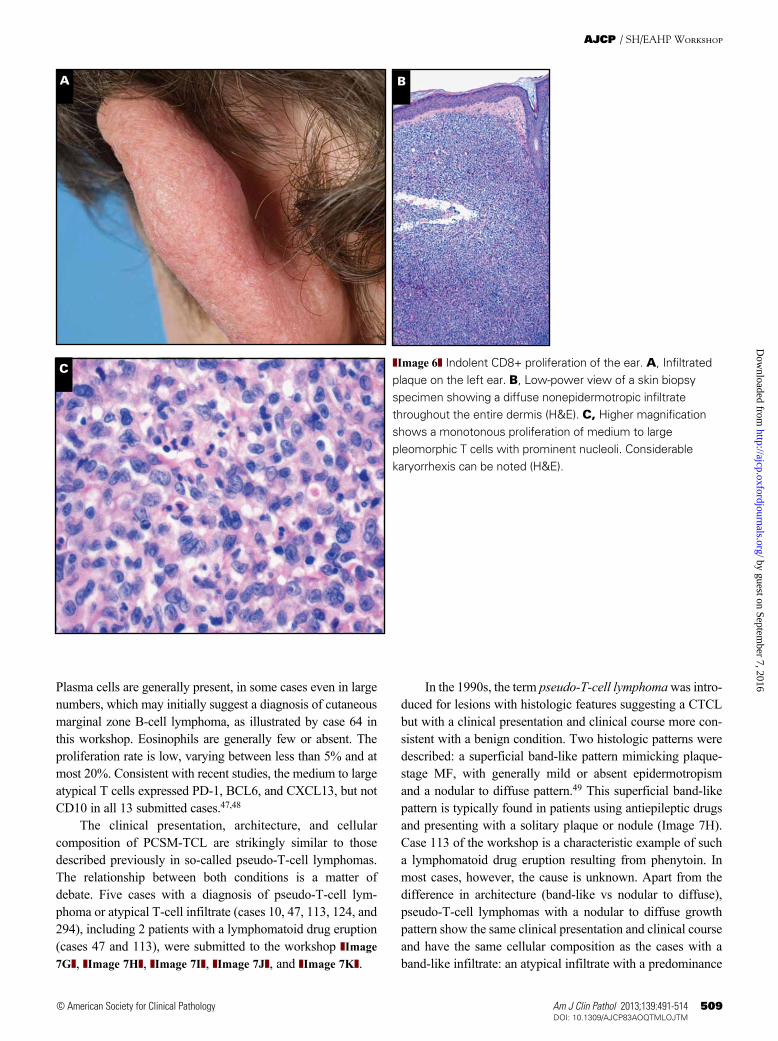
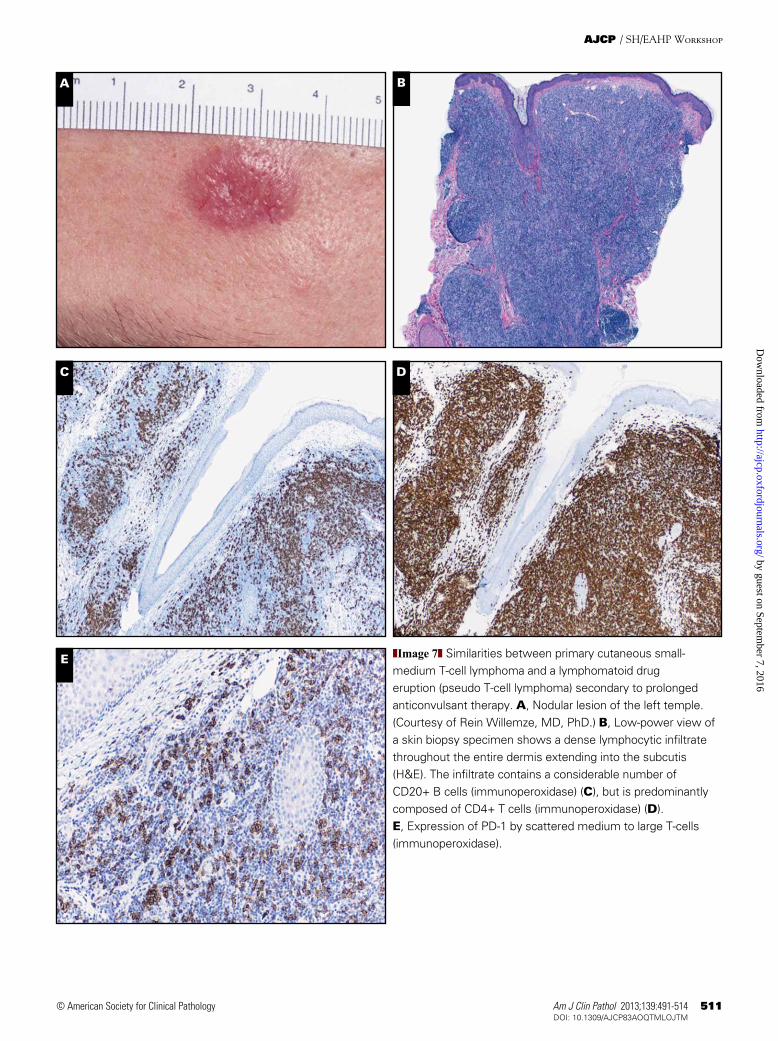
The clinical presentation, architecture, and cellular composition of PCSM-TCL are strikingly similar to those described previously in so-called pseudo-T-cell lymphomas. The relationship between both conditions is a matter of debate. Five cases with a diagnosis of pseudo-T-cell lym-phoma or atypical T-cell infiltrate (cases 10, 47, 113, 124, and 294), including 2 patients with a lymphomatoid drug eruption (cases 47 and 113), were submitted to the workshop ❚Image 7G❚, ❚Image 7H❚, ❚Image 7I❚, ❚Image 7J❚, and ❚Image 7K❚.
In the 1990s, the term pseudo-T-cell lymphoma was intro-duced for lesions with histologic features suggesting a CTCL but with a clinical presentation and clinical course more con-sistent with a benign condition. Two histologic patterns were described: a superficial band-like pattern mimicking plaque-stage MF, with generally mild or absent epidermotropism and a nodular to diffuse pattern.49 This superficial band-like pattern is typically found in patients using antiepileptic drugs and presenting with a solitary plaque or nodule (Image 7H). Case 113 of the workshop is a characteristic example of such a lymphomatoid drug eruption resulting from phenytoin. In most cases, however, the cause is unknown. Apart from the difference in architecture (band-like vs nodular to diffuse), pseudo-T-cell lymphomas with a nodular to diffuse growth pattern show the same clinical presentation and clinical course and have the same cellular composition as the cases with a band-like infiltrate: an atypical infiltrate with a predominance
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

510 Am J Clin Pathol 2013;139:491-514510 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
that most cutaneous pseudo-T-cell lymphomas, including both cases with an MF-like pattern (including lymphomatoid drug eruptions) and cases with a nodular pattern, contain clonal T-cells. Among the cases submitted to the workshop, 12 of 13 PCSM-TCL and all 5 cases with a pseudo-T-cell lymphoma, including the 2 cases with a lymphomatoid drug eruption, showed clonal TCR gene rearrangements.
During the workshop, the overlapping features between PCSM-TCL and pseudo-T-cell lymphomas in patients with a solitary lesion were widely recognized. Because differentiat-ing criteria are lacking, cutaneous CD4+ small-medium T-cell lymphoproliferation was suggested as a unifying term for this condition. Alternatively, Beltraminelli et al50 suggested the term “small- to medium-sized pleomorphic T-cell nodules of undetermined significance,” and emphasized that these cases
of small to medium-sized lymphocytes with variable num-bers, but always less than 30% medium to large CD3+, CD4+, CD8– T cells; a considerable admixture with CD8+ T cells, CD20+ B cells, and histiocytes; a low proportion of prolifer-ating cells; and no or only focal infiltration of the epidermis or follicular epithelium. Consistent with a recent study, in all 5 cases submitted to the workshop the atypical CD4+ T cells expressed PD-1, BCL6, and CXCL13 (Image 7I, Image 7J, and Image 7K). Obviously these characteristics are identical to those of the PCSM-TCL described above.
Demonstration of a T-cell clone and loss of pan-T-cell antigens (except from CD7) are currently used as criteria to differentiate PCSM-TCL from pseudo-T-cell lymphomas. However, with the introduction of more sensitive techniques to demonstrate clonality (Biomed-2 protocols), it now appears
GF
D E
❚Image 6❚ (cont) The atypical T-cells strongly express CD3 (D), CD8 (F) and TIA-1 (G), but are completely negative for CD5 (E). (Case 305, courtesy of Patty M. Jansen, MD, PhD, and Rein Willemze, MD, PhD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
Am J Clin Pathol 2013;139:491-514 511511 DOI: 10.1309/AJCP83AOQTMLOJTM 511
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
A B
C D
E ❚Image 7❚ Similarities between primary cutaneous small-medium T-cell lymphoma and a lymphomatoid drug eruption (pseudo T-cell lymphoma) secondary to prolonged anticonvulsant therapy. A, Nodular lesion of the left temple. (Courtesy of Rein Willemze, MD, PhD.) B, Low-power view of a skin biopsy specimen shows a dense lymphocytic infiltrate throughout the entire dermis extending into the subcutis (H&E). The infiltrate contains a considerable number of CD20+ B cells (immunoperoxidase) (C), but is predominantly composed of CD4+ T cells (immunoperoxidase) (D). E, Expression of PD-1 by scattered medium to large T-cells (immunoperoxidase).
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from
512 Am J Clin Pathol 2013;139:491-514512 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
J K
H I
GF
❚Image 7❚ (cont) F, A subpopulation of the atypical T cells expresses CXCL13 (immunoperoxidase). (Case 163, courtesy of Carlos A. Torres Cabala, MD, Michael M. Tetzlaff, MD, PhD, Victor G. Prieto, MD, PhD, and Madeleine Duvic, MD.) G, Dome-shaped papule (6 mm) on the right shoulder. H, Low-power view of a skin biopsy specimen shows a dense perivascular to band-like infiltrate in the papillary dermis with focal lymphocyte exocytosis; inset, the infiltrate contains many medium to large pleomorphic T cells with hyperchromatic nuclei (H&E). The dermal infiltrates are composed predominantly of CD3+ (I) and CD4+ (J) T cells (immunoperoxidase). Note the characteristic sharp demarcation at the lower border of the infiltrate. K, A considerable proportion of the T cells expresses PD-1 (immunoperoxidase); inset, in particular the medium-sized to large T-cells are PD-1+ (immunoperoxidase). (Case 113, courtesy of Trent B. Marburger, MD, Melissa Piliang, MD, and Eric D. Hsi, MD.)
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

Am J Clin Pathol 2013;139:491-514 513513 DOI: 10.1309/AJCP83AOQTMLOJTM 513
© American Society for Clinical Pathology
AJCP / SH/EAHP Workshop
6. Zayour M, Gilmore E, Heald P, et al. A distinct entity in the spectrum of the CD30+ cutaneous lymphoproliferative diseases: oligolesional nodules with pseudoepitheliomatous hyperplasia followed by spontaneous resolution. Am J Dermatopathol. 2009;31:37-43.
7. Salama S. Primary “cutaneous” T-cell anaplastic large cell lymphoma, CD30+, neutrophil-rich variant with subcutaneous panniculitic lesions, in a post-renal transplant patient: report of unusual case and literature review. Am J Dermatopathol. 2005;27:217-223.
8. Ravat FE, Spittle MF, Russell-Jones R. Primary cutaneous T-cell lymphoma occurring after organ transplantation. J Am Acad Dermatol. 2006;54:668-675.
9. Jhala DN, Medeiros LJ, Lopez-Terrada D, et al. Neutrophil-rich anaplastic large cell lymphoma of T-cell lineage: a report of two cases arising in HIV-positive patients. Am J Clin Pathol. 2000;114:478-482.
10. Papalas JA, Van Mater D, Wang E. Pyogenic variant of primary cutaneous anaplastic large-cell lymphoma: a lymphoproliferative disorder with a predilection for the immunocompromised and the young. Am J Dermatopathol. 2010;32:821-827.
11. Vergier B, Beylot-Barry M, Pulford K, et al. Statistical evaluation of diagnostic and prognostic features of CD30+ cutaneous lymphoproliferative disorders: a clinicopathologic study of 65 cases. Am J Surg Pathol. 1998;22:1192-1202.
12. Feldman AL, Law M, Remstein ED, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23:574-580.
13. Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
14. Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
15. Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
16. Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
17. Kadin ME, Pinkus JL, Pinkus GS, et al. Primary cutaneous ALCL with phosphorylated/activated cytoplasmic ALK and novel phenotype: EMA/MUC1+, cutaneous lymphocyte antigen negative. Am J Surg Pathol. 2008;32:1421-1426.
18. Su LD, Schnitzer B, Ross CW, et al. The t(2;5)-associated p80 NPM/ALK fusion protein in nodal and cutaneous CD30+ lymphoproliferative disorders. J Cutan Pathol. 1997;24:597-603.
19. Sasaki K, Sugaya M, Fujita H, et al. A case of primary cutaneous anaplastic large cell lymphoma with variant anaplastic lymphoma kinase translocation. Br J Dermatol. 2004;150:1202-1207.
20. Oschlies I, Lisfeld J, Lamant L, et al. ALK-positive anaplastic large cell lymphoma limited to the skin: clinical, histopathological and molecular analysis of 6 pediatric cases: a report from the ALCL99 study [published online ahead of print July 6]. Haematologica. 2012;98:50-56.
should not be treated aggressively. Whatever term one pre-fers, there is consensus that patients presenting with a solitary lesion and the characteristic histologic and immunopheno-typical features described herein have an excellent prognosis. A recent study suggested that staging procedures are not required in these patients, and that if the skin lesions do not resolve spontaneously after biopsy, they should be treated primarily with intralesional steroids or surgical excision, and only in exceptional circumstances, with radiotherapy. PCSM-TCLs that do not meet the criteria of the cases described herein are rare and should be fully staged. A recent study has suggested that PCSM-TCL with rapidly growing bulky tumors, a low percentage of admixed CD8+ T cells, and/or a high proliferative fraction are at risk to develop progressive disease; these findings await further confirmation.51
From the 1Institute of Pathology, and Comprehensive Cancer Center (CCC), Eberhard-Karls-University of Tübingen, Tübingen, Germany; 2Departments of Pathology and 5Dermatology, Leiden University Medical Center, Leiden, The Netherlands; 3Department of Pathology, University of Texas Health Science Center at San Antonio; and 4Division of Hematopathology, Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA.
Address reprint requests to Dr Quintanilla-Martínez: Institute of Pathology and Comprehensive Cancer Center (CCC), University of Tübingen, Liebermeisterstr 8, D-72076 Tübingen, Germany; [email protected].
Acknowledgments: The authors thank Dr Itziar Salaverria and Dr Reiner Siebert from the Institute of Human Genetics, Christian-Albrechts University, Campus Kiel, Germany, for performing the fluorescence in situ hybridization analysis using an IRF4/MUM1 break-apart probe in the cutaneous anaplastic large cell lymphoma cases submitted to the workshop. We also thank the workshop participants for their case submissions and for use of their images.
References 1. Song SX, Willemze R, Swerdlow SH, et al. Mycosis
fungoides: report of the 2011 Society for Hematopathology/European Association for Haematopathology Workshop. Am J Clin Pathol. 2013;139:466-490.
2. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
3. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
4. Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008:300-301.
5. Burg G, Kempf W, Kazakov DV, et al. Pyogenic lymphoma of the skin: a peculiar variant of primary cutaneous neutrophil-rich CD30+ anaplastic large-cell lymphoma: clinicopathological study of four cases and review of the literature. Br J Dermatol. 2003;148:580-586.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

514 Am J Clin Pathol 2013;139:491-514514 DOI: 10.1309/AJCP83AOQTMLOJTM
© American Society for Clinical Pathology
Quintanilla-Martinez et al / Non-MF Cutaneous T-Cell Lymphomas
38. Quintanilla-Martinez L, Kimura H, Jaffe ES. EBV-positive T-cell lymphoproliferative disorders of childhood. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
39. Barrionuevo C, Anderson VM, Zevallos-Giampietri E, et al. Hydroa-like cutaneous T-cell lymphoma: a clinicopathologic and molecular genetic study of 16 pediatric cases from Peru. Appl Immunohistochem Mol Morphol. 2002;10:7-14.
40. Nava VE, Jaffe ES. The pathology of NK-cell lymphomas and leukemias. Adv Anat Pathol. 2005;12:27-34.
41. Magana M, Sangueza P, Gil-Beristain J, et al. Angiocentric cutaneous T-cell lymphoma of childhood (hydroa-like lymphoma): a distinctive type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 1998;38:574-579.
42. Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
43. Kimura H, Ito Y, Kawabe S, et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood. 2012;119:673-686.
44. Bekkenk MW, Vermeer MH, Jansen PM, et al. Peripheral T-cell lymphomas unspecified presenting in the skin: analysis of prognostic factors in a group of 82 patients. Blood. 2003;102:2213-2219.
45. Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas: a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
46. Petrella T, Maubec E, Cornillet-Lefebvre P, et al. Indolent CD8-positive lymphoid proliferation of the ear: a distinct primary cutaneous T-cell lymphoma? Am J Surg Pathol. 2007;31:1887-1892.
47. Rodriguez Pinilla SM, Roncador G, Rodriguez-Peralto JL, et al. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma expresses follicular T-cell markers. Am J Surg Pathol. 2009;33:81-90.
48. Cetinozman F, Jansen PM, Willemze R. Expression of programmed death-1 in primary cutaneous CD4-positive small/medium-sized pleomorphic T-cell lymphoma, cutaneous pseudo-T-cell lymphoma, and other types of cutaneous T-cell lymphoma. Am J Surg Pathol. 2012;36:109-116.
49. Rijlaarsdam JU, Scheffer E, Meijer CJ, et al. Cutaneous pseudo-T-cell lymphomas: a clinicopathologic study of 20 patients. Cancer. 1992;69:717-724.
50. Beltraminelli H, Leinweber B, Kerl H, et al. Primary cutaneous CD4+ small-/medium-sized pleomorphic T-cell lymphoma: a cutaneous nodular proliferation of pleomorphic T lymphocytes of undetermined significance? A study of 136 cases. Am J Dermatopathol. 2009;31:317-322.
51. Garcia-Herrera A, Colomo L, Camos M, et al. Primary cutaneous small/medium CD4+ T-cell lymphomas: a heterogeneous group of tumors with different clinicopathologic features and outcome. J Clin Oncol. 2008;26:3364-3371.
21. Nijsten T, Curiel-Lewandrowski C, Kadin ME. Lymphomatoid papulosis in children: a retrospective cohort study of 35 cases. Arch Dermatol. 2004;140:306-312.
22. de Souza A, Camilleri MJ, Wada DA, et al. Clinical, histopathologic, and immunophenotypic features of lymphomatoid papulosis with CD8 predominance in 14 pediatric patients. J Am Acad Dermatol. 2009;61:993-1000.
23. Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. Description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
24. Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
25. Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1177.
26. Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology Workshop. Am J Clin Pathol. 2013;139:536-551.
27. Eberle FC, Song JY, Xi L, et al. Nodal involvement by cutaneous CD30-positive T-cell lymphoma mimicking classical Hodgkin lymphoma. Am J Surg Pathol. 2012;36:716-725.
28. Kinney MC, Higgins RA, Medina EA. Anaplastic large cell lymphoma: twenty-five years of discovery. Arch Pathol Lab Med. 2011;135:19-43.
29. Kinney MC, Collins RD, Greer JP, et al. A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. Am J Surg Pathol. 1993;17:859-868.
30. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111:838-845.
31. Gonzalez CL, Medeiros LJ, Braziel RM, et al. T-cell lymphoma involving subcutaneous tissue: a clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15:17-27.
32. Jaffe ES, Ralfkiaer E. Subcutaneous panniculitis-like T-cell lymphoma. In: Jaffe ES, Harris NL, Stein H, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001:212-213.
33. Gaulard P, Berti E, Willemze R, et al. Primary cutaneous peripheral T-cell lymphomas, rare subtypes. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008:302-303.
34. Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
35. Chan JKC, Quintanilla-Martinez L, Ferry JA, et al. Extranodal NK/T-cell lymphoma, nasal type. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008:285-288.
36. International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124-4130.
37. Huang Y, de Reynies A, de Leval L, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood. 2010;115:1226-1237.
by guest on September 7, 2016
http://ajcp.oxfordjournals.org/D
ownloaded from

























