Embed Size (px)
Citation preview

MINIREVIEWS
On the Origins of a Vibrio Species
Tammi Vesth & Trudy M. Wassenaar & Peter F. Hallin &
Lars Snipen & Karin Lagesen & David W. Ussery
Received: 3 July 2009 /Accepted: 17 September 2009 /Published online: 15 October 2009# The Author(s) 2009. This article is published with open access at Springerlink.com
Abstract Thirty-two genome sequences of various Vibrio-naceae members are compared, with emphasis on whatmakes V. cholerae unique. As few as 1,000 gene familiesare conserved across all the Vibrionaceae genomes ana-lysed; this fraction roughly doubles for gene familiesconserved within the species V. cholerae. Of these,approximately 200 gene families that cluster on variouslocations of the genome are not found in other sequencedVibrionaceae; these are possibly unique to the V. choleraespecies. By comparing gene family content of the analysedgenomes, the relatedness to a particular species is identifiedfor two unspeciated genomes. Conversely, two genomes
presumably belonging to the same species have suspicious-ly dissimilar gene family content. We are able to identify anumber of genes that are conserved in, and unique to, V.cholerae. Some of these genes may be crucial to the nicheadaptation of this species.
Introduction
The species concept for bacteria has long been under siegefrom several angles, and now with thousands of bacterialgenomes being sequenced, the disputes have intensified [8].One frequently used definition of a bacterial species is “acategory that circumscribes a (preferably) genomicallycoherent group of individual isolates/strains sharing a highdegree of similarity in (many) independent features,comparatively tested under highly standardized conditions”[12]. Such independent features are usually phenotypes thatcan easily be tested. For a new species to be defined,amongst other criteria, inter-species DNA–DNA hybrid-isation has to be below 70%, although this rule is notwithout its limitations [18]. In the late 1970s and 1980s, the16S rRNA gene sequence was introduced as a molecularclock that could be used to infer phylogenetic relationships[50]. Ideally, isolates belonging to the same species haveidentical or nearly identical 16S rRNA genes, and thesediffer from isolates belonging to different species [32, 44].In practice, this is not always the case. Examples exist ofdifferent species sharing identical rRNA genes (forinstance, E. coli and Shigella [37] that are even placed indifferent genera); in addition, isolates of one species canhave different rRNA genes beyond the 97% that isconsidered to demarcate species [4]. Lateral transfer ofgenetic material (to which ribosomal genes are believed tobe resistant) destroys the phylogenetic relationship, so that
T. Vesth : T. M. Wassenaar : P. F. Hallin : L. Snipen :K. Lagesen :D. W. Ussery (*)Center for Biological Sequence Analysis,Department of Systems Biology,The Technical University of Denmark,Building 208,2800 Kgs. Lyngby, Denmarke-mail: [email protected]
T. M. WassenaarMolecular Microbiology and Genomics Consultants,Zotzenheim, Germany
P. F. HallinNovozymes A/S,Krogshøjvej 36,2880 Bagsværd, Denmark
L. SnipenBiostatistics, Department of Chemistry, Biotechnology,and Food Sciences, Norwegian University of Life Sciences,Ås, Norway
K. LagesenCentre for Molecular Biology and Neuroscience and Instituteof Medical Microbiology, University of Oslo,Oslo, Norway
Microb Ecol (2010) 59:1–13DOI 10.1007/s00248-009-9596-7

phylogenies based on alternative housekeeping genes candiffer from a 16S rRNA tree and frequently are not even inaccordance to each other. Such observations question thevalidity of a phylogenetic tree as the most suitable modelfor bacterial ancestry, when multiple genetic transferswould produce a network-like evolutionary structure [6].On the other hand, it is observed that lateral gene transfer ismost frequent between genetically related members sharinga similar base content and occupying the same ecologicalniche [29]. Nevertheless, a core of genes can be recognisedthat produce coherent phylogenetic trees, though these maynot represent the species’ complete evolutionary history asthey comprise only a minor fraction of the genetic contentof the organism [35].
Whether a tree or a network is more accurate to describephylogeny, in either case bacterial species may be consid-ered as a cloud of isolates having a higher level of geneticsimilarity to each other than to organisms belonging to adifferent species. When such clouds have fuzzy andoverlapping borders, the species concept falls apart but thatwill only apply to certain cases [7]. Since 16S rRNA genesare not informative on the level of diversity within aspecies, the 'density' of a cloud of isolates making up aspecies cannot be determined by this gene. Those genesshared by all isolates belonging to one species comprise thecore genome of that species [39], and the degree ofdiversity in the remaining non-core genes determines thedensity of the species cloud.
We hypothesised that certain genes can be recognised asspecific to a particular species, to be conserved in thatspecies but not present in related species. We tested ourhypothesis with complete genome sequences of the bacte-rial family Vibrionaceae, which belong to the γ-Proteobacteria and comprises eight genera. Most availablegenome sequences belong to the genus Vibrio. This genuscontains 51 recognised species [10, 46] which are mainlyfound in marine environments, frequently living in associ-ation with marine organisms such as corals, fish, squid orzooplankton. Most of them are symbionts and only a feware human pathogens, notably particular serotypes of V.cholerae producing cholera, Vibrio parahaemolyticus(causing gastroenteritis) and Vi vulnificus (causing woundinfections) [46]. Other Vibrionaceae, including V. vulnifi-cus, Aliivibrio salmonicida and V. harveyi, are fish orshellfish pathogens and have major economic impact.Photobacterium profundum, representing another genuswithin the Vibrionaceae, was also included.
The gene content of 32 available sequenced Vibrionaceaegenomes was compared and the results were analysed invarious ways. The data allowed us to identify possible V.cholerae-specific genes, since this species was representedby 18 genomes that was a sufficient number to testconservation both within the species and across species.
We found that a two-component signal transduction pathwayis uniquely conserved in V. cholerae but is not found outsidethis species. Our findings further indicated that possibly arelatively small set of genes could confer niche specialisationallowing V. cholerae to be adopted to a unique environment,so that over time V. cholerae have become a distinct species.
Materials and Methods
Genomes and Gene Annotations Used
Publicly available genome sequences of Vibrionaceae wereselected that were provided in less than 300 contigs and inwhich full-length 16S rRNA sequence could be found usingthe rRNA gene finder RNAmmer [19]. The 32 genomesequences included are shown in Table 1.
The gene annotations as provided in GenBank wereused, except for those genomes marked “Easygene” inTable 1 where protein annotation was not available in theRefSeq file at the time of analysis, and we used EasyGene[20] to identify the genes. As a control, an availableGenBank annotation was compared to a generated Easy-gene annotation to confirm that the number of identifiedgenes was comparable.
Ribosomal RNA Analysis
RNAmmer [19] was used to identify 16S rRNA sequenceswithin the 32 genomes. Sequences were considered reliableif they were between 1,400 and 1,700 nucleotides long andhad an RNAmmer score above 1,800. In cases where theprogram found multiple and variable 16S sequences withina genome, one of these (with satisfactory RNAmmerscores) was arbitrarily chosen. The sequences were alignedusing PRANK [23, 24], and the program MEGA4 was usedto elucidate a phylogenetic tree [45]. Within MEGA4, thetree was created using the Neighbor-Joining method withthe uniform rate Jukes–Cantor distance measure and thecomplete-delete option. Five hundred resamplings weredone to find the bootstrap values.
Pan-Genome Family Clustering
Clustering based on shared gene families from the Vibriopan-genome was constructed, based on BLASTP similarityusing default settings. A BLASTP hit was consideredsignificant if the alignment produced at least 50% identityfor at least 50% of the length of the longest gene (eitherquery or subject). Using this criterion, each pair of genesproducing a significant reciprocal best hit was scored asbelonging to the same gene family. A genome matrix wasconstructed, containing one row for each genome and one
2 T. Vesth et al.

column for each gene family. Cell (i, j) in this matrix is 1 ifgenome i has a member in gene family j, 0 otherwise. Ahierarchical clustering, with average linkage based on theManhattan distance between genomes was then performed.Two trees were made, one with more weight given to genefamilies present in most (90%, or between 27 and 30)Vibrio genomes (“stabilome”), and the other with moreweight given to gene families present in only a few (two,three, or four) genomes (“mobilome”). Thus, the originalBoolean matrix is now scaled differently, depending on thenumber of genomes in each gene family [44]. For both
trees, singletons (families which are only found in onegenome) have been excluded.
Pan- and Core Genome Analysis
The results of the BLAST analysis were also used toconstruct a pan- and core genome plot as follows. Based onclusterings from the pan-genome family tree, an ordered setof genomes was constructed with V. cholerae genomes atthe start. For the first chosen genome, all BLAST hits foundin the second genome were recorded and the accumulative
Table 1 Vibrionaceae genomes used in this analysis
GPID Organism Contigs Accession/GenBank Status No. of genes Ref.
36 V. cholerae N16961a 2 AE003852.1 Fully sequenced 3,828 [15]
15667 V. cholerae O395 TIGRa 2 CP000626.1 Fully sequenced 3,875 [11]
32853 V. cholerae O395 TEDAa 2 CP001235.1 Fully sequenced 3,934 [49]
33555 V. cholerae MJ-1236a 2 CP001485.1 Fully sequenced 3,774 [31]
15666 V. cholerae MO10a 153 NZ_AAKF00000000 Unfinished (Easygene) 3,421 [5]
15670 V. cholerae V52a 268 NZ_AAKJ00000000 Unfinished (NCBI) 3,815 [16]
33559 V. cholerae BX330286a 8 NZ_ACIA00000000 Unfinished (NCBI) 3,632 [31]
33557 V. cholerae B33a 17 NZ_ACHZ00000000 Unfinished (NCBI) 3,748 [31]
33553 V. cholerae RC9a 11 NZ_ACHX00000000 Unfinished (NCBI) 3,811 [31]
32851 V. cholerae M66-2 2 CP001233.1 Fully sequenced 3,693 [49]
18495 V. cholerae MZO-2 162 NZ_AAWF00000000 Unfinished (NCBI) 3,425 [16]
18265 V. cholerae 1587 254 NZ_AAUR00000000 Unfinished (NCBI) 3,758 [16]
18253 V. cholerae 2740-80 257 NZ_AAUT00000000 Unfinished (NCBI) 3,771 [16]
17723 V. cholerae AM-19226 154 NZ_AATY00000000 Unfinished (Easygene) 3,407 [33]
33561 V. cholerae 12129 12 NZ_ACFQ00000000 Unfinished (NCBI) 3,574 [31]
33549 V. cholerae VL426 5 NZ_ACHV00000000 Unfinished (NCBI) 3,461 [31]
33579 V. cholerae TM 11079-80 35 NZ_ACHW00000000 Unfinished (NCBI) 3,621 [31]
33551 V. cholerae TMA 21 20 NZ_ACHY00000000 Unfinished (NCBI) 3,600 [31]
13564 V. campbellii AND4 143 NZ_ABGR00000000 Unfinished (NCBI) 3,935 [13]
19857 V. harveyi BAA-1116 3 CP000789.1 Fully sequenced 6,064 [1]
349 V. vulnificus CMCP6 2 AE016795.2 Fully sequenced 4,538 [38]
1430 V. vulnificus YJ016 3 BA000037.2 Fully sequenced 5,028 [3]
19397 V. shilonii AK1 158 NZ_ABCH00000000 Unfinished (NCBI) 5,360 [41]
15693 Vibrio sp. Ex25 222 NZ_AAKK00000000 Unfinished (Easygene) 4,004 [16]
13616 Vibrio sp. MED222 99 NZ_AAND00000000 Unfinished (NCBI) 4,590 [36]
32815 V. splendidus LGP32 2 FM954973.1 Fully sequenced 4,434 [27]
19395 V. parahaemolyticus 16 78 NZ_ACCV00000000 Unfinished (Easygene) 3,780 [9]
360 V. parahaemolyticus 2210633 2 BA000031.2 Fully sequenced 4,832 [25]
12986 A. fischeri ES114 3 CP000020.1 Fully sequenced 3,823 [42]
19393 A. fischeri MJ11 3 CP001133.1 Fully sequenced 4,039 [26]
30703 A. salmonicida LFI1238 6 FM178379.1 Fully sequenced 4,284 [17]
13128 P. profundum SS9 3 CR354531.1 Fully sequenced 5,480 [48]
GPID genome project identifier at NCBI. Contigs the number of contiguous sequences, which for a completely sequenced genome is at least two(for two chromosomes) and can be up to six when plasmids are present. Unfinished sequences are represented by multiple contigs perchromosomea Strains containing the genes encoding the cholera enterotoxin subunits are indicated
Origins of V. cholerae 3

number of gene families (as defined above) now recognised intotal was plotted for the pan-genome. The number of genefamilies with at least one representative gene in both genomeswas plotted for the core genome. A running total is plotted forthe pan-genome which increases as more genomes are added,whilst the core genome representing conserved gene familiesslowly decreases with the addition of more genomes.
Whole-Genome BLAST Analysis and Constructionof a BLAST Matrix
The predicted genes of every genome (annotated or foundby Easygene) were translated and every gene was com-pared, by BLASTP against every other genome and its owngenome. In the latter case, the hit to self was ignored. The50/50 rule for BLAST hits as described above was used. Ifthese requirements were met, genes were combined in agene family. The BLAST results were visualised in aBLAST matrix [2], which summarises the results ofgenomic pairwise comparisons and reports, both as per-centage and as absolute numbers, the number of reciprocalBLAST hits as a fraction of the total number of genefamilies found in the two genomes. For easier visualinspection, the cells in the matrix are coloured darker as
the fraction of similarity increases. Hits identified within agenome are differently coloured.
BLAST Atlas
BLAST results were also visualised in a BLAST atlas, thistime visualising, for all genes in the reference genome Vcholerae N16961, their best hit in all other genomes, againwith a threshold of 50% identity over at least 50% of thelength of the query protein. The atlas displays the hits as theyare located in the reference strain [14]. The BLAST scoresobtained for each queried gene is plotted, so that conservedand variable regions are located with respect to the referencegenome. Note that genes absent in the reference genome arenot shown in the lanes of the query genomes.
Results
Ribosomal RNA Analysis
A phylogenetic tree based on the 16S rRNA gene extractedfrom the 32 analysed Vibrionaceae genomes is shown inFig. 1. The 18 V. cholerae genomes build a tight subcluster,
45
65
56
88
55
86
AA
Vibrio sp. MED222
Vibrio sp. Ex25
Figure 1 Phylogenetic tree ofthe 16S rRNA gene extractedfrom 32 sequenced Vibriogenomes listed in Table 1. En-vironmental V. cholerae lackingthe cholera enterotoxin genesare highlighted in bright green,whilst pathogenic V. choleraegenomes are in dark green.Further colouring was used forspecies for which two genomesare represented
4 T. Vesth et al.
quite distanced from the other species. Above this in thefigure, another subcluster comprising eight genomes repre-senting at least six species is recognised, and within thiscluster the two V. parahaemolyticus genes are not found onthe same branch. A third cluster, a bit further removed,includes Aliivibrio fischeri and A. almonidica as well as V.splendidus and Vibrio species MED 222; the gene ofPhotobacterium profundum is the most distant.
Pan-Genome Family Trees
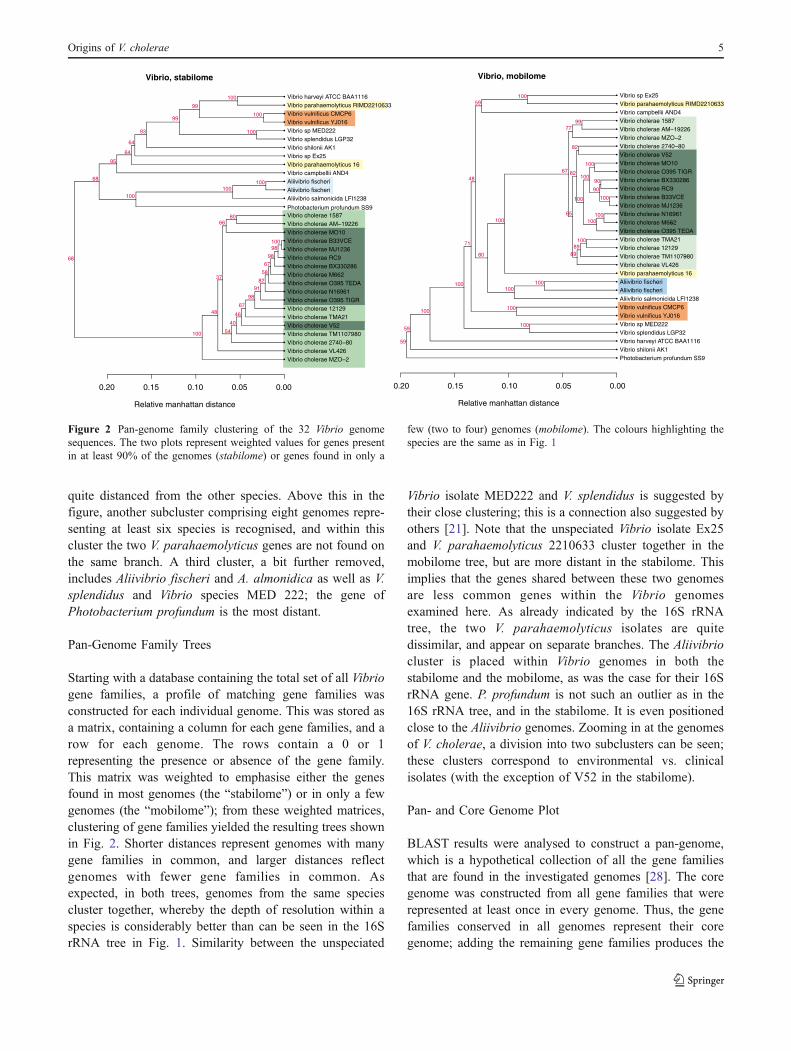
Starting with a database containing the total set of all Vibriogene families, a profile of matching gene families wasconstructed for each individual genome. This was stored asa matrix, containing a column for each gene families, and arow for each genome. The rows contain a 0 or 1representing the presence or absence of the gene family.This matrix was weighted to emphasise either the genesfound in most genomes (the “stabilome”) or in only a fewgenomes (the “mobilome”); from these weighted matrices,clustering of gene families yielded the resulting trees shownin Fig. 2. Shorter distances represent genomes with manygene families in common, and larger distances reflectgenomes with fewer gene families in common. Asexpected, in both trees, genomes from the same speciescluster together, whereby the depth of resolution within aspecies is considerably better than can be seen in the 16SrRNA tree in Fig. 1. Similarity between the unspeciated
Vibrio isolate MED222 and V. splendidus is suggested bytheir close clustering; this is a connection also suggested byothers [21]. Note that the unspeciated Vibrio isolate Ex25and V. parahaemolyticus 2210633 cluster together in themobilome tree, but are more distant in the stabilome. Thisimplies that the genes shared between these two genomesare less common genes within the Vibrio genomesexamined here. As already indicated by the 16S rRNAtree, the two V. parahaemolyticus isolates are quitedissimilar, and appear on separate branches. The Aliivibriocluster is placed within Vibrio genomes in both thestabilome and the mobilome, as was the case for their 16SrRNA gene. P. profundum is not such an outlier as in the16S rRNA tree, and in the stabilome. It is even positionedclose to the Aliivibrio genomes. Zooming in at the genomesof V. cholerae, a division into two subclusters can be seen;these clusters correspond to environmental vs. clinicalisolates (with the exception of V52 in the stabilome).
Pan- and Core Genome Plot
BLAST results were analysed to construct a pan-genome,which is a hypothetical collection of all the gene familiesthat are found in the investigated genomes [28]. The coregenome was constructed from all gene families that wererepresented at least once in every genome. Thus, the genefamilies conserved in all genomes represent their coregenome; adding the remaining gene families produces the
0.20 0.15 0.10 0.05 0.00
Vibrio, stabilome
Relative manhattan distance
Vibrio cholerae MZO 2Vibrio cholerae VL426Vibrio cholerae 2740 80Vibrio cholerae TM1107980Vibrio cholerae V52Vibrio cholerae TMA21Vibrio cholerae 12129Vibrio cholerae O395 TIGRVibrio cholerae N16961Vibrio cholerae O395 TEDAVibrio cholerae M662Vibrio cholerae BX330286Vibrio cholerae RC9Vibrio cholerae MJ1236Vibrio cholerae B33VCEVibrio cholerae MO10Vibrio cholerae AM 19226Vibrio cholerae 1587Photobacterium profundum SS9
Aliivibrio salmonicida LFI1238Aliivibrio fischeriAliivibrio fischeriVibrio campbellii AND4Vibrio parahaemolyticus 16Vibrio sp Ex25Vibrio shilonii AK1Vibrio splendidus LGP32Vibrio sp MED222Vibrio vulnificus YJ016Vibrio vulnificus CMCP6Vibrio parahaemolyticus RIMD2210633Vibrio harveyi ATCC BAA1116
10098
98
67
58
100
82
100
91
100
98
67
46
100
80
40
100
54
66
37
48
100
99
99
93
100
64
64
95
68
68
0.20 0.15 0.10 0.05 0.00
Vibrio, mobilome
Relative manhattan distance
Photobacterium profundum SS9Vibrio shilonii AK1Vibrio harveyi ATCC BAA1116Vibrio splendidus LGP32Vibrio sp MED222Vibrio vulnificus YJ016Vibrio vulnificus CMCP6Aliivibrio salmonicida LFI1238Aliivibrio fischeriAliivibrio fischeriVibrio parahaemolyticus 16Vibrio cholerae VL426Vibrio cholerae TM1107980Vibrio cholerae 12129Vibrio cholerae TMA21Vibrio cholerae O395 TEDAVibrio cholerae M662Vibrio cholerae N16961Vibrio cholerae MJ1236Vibrio cholerae B33VCEVibrio cholerae RC9Vibrio cholerae BX330286Vibrio cholerae O395 TIGRVibrio cholerae MO10Vibrio cholerae V52Vibrio cholerae 2740 80Vibrio cholerae MZO 2Vibrio cholerae AM 19226Vibrio cholerae 1587Vibrio campbellii AND4Vibrio parahaemolyticus RIMD2210633Vibrio sp Ex25
100
100
90
90
100
100
100
100
100
99
89
82
89
82
65
77
67
100
100
100
100
100
100
80
59
48
71
100
100
59
59
Figure 2 Pan-genome family clustering of the 32 Vibrio genomesequences. The two plots represent weighted values for genes presentin at least 90% of the genomes (stabilome) or genes found in only a
few (two to four) genomes (mobilome). The colours highlighting thespecies are the same as in Fig. 1
Origins of V. cholerae 5

pan-genome. The resulting pan- and core genome plot isshown in Fig. 3. The genomes start with the documentedclinical isolates of V. cholerae and then follow the ordersuggested by the pan-genome family clustering (Fig. 2),although genomes from the same species were kepttogether (the two V. parahaemolyticus genomes were splitin the trees). As more genomes are added in the plot, thenumber of gene families in the pan-genome (blue line)increases, and the number of conserved gene families (redline) in the core genome decreases, albeit at a lower rate.This is because every genome can add many novel (andfrequently different) genes to the pan-genome but onlydecreases the core genome with a few genes that are absent
in that particular strain but that were conserved in thepreviously analysed genomes. The pan-genome curveincreases with a relative steep slope when a novel speciesis added, as is obvious when a V. parahaemolyticus genomeis added after the last V. cholerae. A stable plateau can beseen for the pan-genome of V. cholerae around 6,500 genes.Nevertheless, a small increase occurs when adding V.cholerae 11587; this is caused by the difference betweenthe two subclusters of V. cholerae seen in Fig. 2. V.cholerae strain 2740-80 behaves atypical in all the figuresshown; although documented as an environmental isolate, itappears closer to the clinical isolates, in terms of overallgenomic properties.
Pan genome
Core genome
New gene families
V. cholerae N
16961
V. cholerae M
66-2 V
. cholerae O395 T
ED
A
V. cholerae O
395 TIG
R
V. cholerae M
O10
V. cholerae B
X330286
V. cholerae R
C9
V. cholerae M
J1236 V
. cholerae B33V
CE
V. cholerae 2740-80
V. cholerae 1587
V. cholerae A
M-19226
V. cholerae M
ZO
-2
V. cholerae 12129
V. cholerae T
MA
21 V
. cholerae TM
11079-80
V. cholerae V
L426
V. cholerae V
52
Vibrio. sp M
ED
222
V.splendidus LG
B2
A. fisheri E
S114
A. fisheri M
J11
A.salm
onicida LFI1238
Vibrio sp E
x25
V.cam
pbelliiV
.harveyi BA
A-1116
V.shilonii A
K1
P.profundum
SS
9
V. parahaem
. 2210633V
. parahaem. 16
V. vulnificus C
MC
P6
V. vulnificus Y
J016
5000
25000
20000
10000
0
15000
Figure 3 Pan- and core genome plot of the 32 Vibrionaceae genomes. The colours highlighting species are the same as in Fig. 1
6 T. Vesth et al.

3.0
%11
0 / 3
,665
88.1
%3,
495
/ 3,9
66
80.4
%3,
303
/ 4,1
09
77.1
%3,
186
/ 4,1
34
92.9
%3,
489
/ 3,7
54
79.6
%3,
139
/ 3,9
44
85.8
%3,
291
/ 3,8
37
83.2
%3,
325
/ 3,9
95
82.4
%3,
302
/ 4,0
09
83.4
%3,
315
/ 3,9
73
76.9
%3,
165
/ 4,1
17
64.7
%2,
888
/ 4,4
63
70.4
%2,
922
/ 4,1
53
68.7
%2,
874
/ 4,1
81
75.9
%3,
094
/ 4,0
77
71.6
%3,
010
/ 4,2
05
73.6
%3,
045
/ 4,1
35
70.3
%2,
933
/ 4,1
74
40.0
%2,
421
/ 6,0
55
42.2
%2,
215
/ 5,2
54
44.3
%2,
515
/ 5,6
83
41.7
%2,
552
/ 6,1
16
38.4
%2,
291
/ 5,9
71
40.3
%2,
326
/ 5,7
71
35.0
%1,
949
/ 5,5
61
34.0
%1,
963
/ 5,7
71
32.1
%1,
846
/ 5,7
47
38.7
%2,
143
/ 5,5
36
35.8
%2,
018
/ 5,6
37
32.5
%2,
385
/ 7,3
36
31.2
%2,
143
/ 6,8
62
27.2
%1,
946
/ 7,1
65
4.2
%15
5 / 3
,729
88.8
%3,
489
/ 3,9
27
74.9
%3,
187
/ 4,2
53
89.7
%3,
485
/ 3,8
84
78.1
%3,
147
/ 4,0
32
82.5
%3,
278
/ 3,9
71
80.7
%3,
321
/ 4,1
17
80.8
%3,
319
/ 4,1
06
81.3
%3,
320
/ 4,0
85
76.7
%3,
195
/ 4,1
67
64.9
%2,
940
/ 4,5
33
70.3
%2,
965
/ 4,2
17
67.2
%2,
880
/ 4,2
88
77.2
%3,
155
/ 4,0
88
72.6
%3,
068
/ 4,2
26
74.9
%3,
101
/ 4,1
42
69.2
%2,
953
/ 4,2
67
39.6
%2,
438
/ 6,1
54
41.6
%2,
225
/ 5,3
54
43.7
%2,
527
/ 5,7
81
41.2
%2,
564
/ 6,2
24
38.0
%2,
307
/ 6,0
67
39.8
%2,
339
/ 5,8
73
34.8
%1,
967
/ 5,6
47
33.7
%1,
977
/ 5,8
65
32.1
%1,
873
/ 5,8
28
38.3
%2,
156
/ 5,6
31
35.9
%2,
049
/ 5,7
13
32.6
%2,
405
/ 7,3
80
31.1
%2,
163
/ 6,9
48
27.1
%1,
964
/ 7,2
45
4.3
%15
7 / 3
,665
80.2
%3,
271
/ 4,0
79
81.1
%3,
280
/ 4,0
46
80.2
%3,
169
/ 3,9
53
83.4
%3,
267
/ 3,9
19
81.4
%3,
309
/ 4,0
67
81.6
%3,
311
/ 4,0
57
81.9
%3,
311
/ 4,0
41
81.6
%3,
264
/ 4,0
00
67.6
%2,
983
/ 4,4
13
72.2
%2,
986
/ 4,1
36
69.7
%2,
916
/ 4,1
83
78.0
%3,
149
/ 4,0
38
73.5
%3,
065
/ 4,1
72
75.5
%3,
089
/ 4,0
92
69.7
%2,
944
/ 4,2
21
40.0
%2,
440
/ 6,0
94
42.3
%2,
236
/ 5,2
83
44.5
%2,
539
/ 5,7
07
41.9
%2,
575
/ 6,1
51
38.5
%2,
311
/ 6,0
04
40.4
%2,
345
/ 5,8
08
35.3
%1,
972
/ 5,5
81
34.2
%1,
983
/ 5,7
97
32.5
%1,
873
/ 5,7
69
38.8
%2,
162
/ 5,5
66
36.4
%2,
055
/ 5,6
47
33.1
%2,
415
/ 7,2
99
31.5
%2,
169
/ 6,8
84
27.5
%1,
971
/ 7,1
79
3.3
%12
0 / 3
,599
77.3
%3,
164
/ 4,0
93
75.1
%3,
024
/ 4,0
28
79.4
%3,
143
/ 3,9
56
76.0
%3,
147
/ 4,1
41
75.3
%3,
136
/ 4,1
62
76.3
%3,
142
/ 4,1
20
77.5
%3,
153
/ 4,0
66
65.1
%2,
880
/ 4,4
23
68.5
%2,
869
/ 4,1
91
69.0
%2,
860
/ 4,1
45
71.5
%2,
986
/ 4,1
75
68.5
%2,
914
/ 4,2
56
69.8
%2,
942
/ 4,2
17
66.3
%2,
833
/ 4,2
71
38.4
%2,
348
/ 6,1
20
41.3
%2,
181
/ 5,2
77
42.8
%2,
449
/ 5,7
18
40.0
%2,
473
/ 6,1
85
37.0
%2,
227
/ 6,0
26
38.6
%2,
251
/ 5,8
39
33.6
%1,
884
/ 5,6
12
32.5
%1,
896
/ 5,8
27
30.6
%1,
777
/ 5,8
04
37.9
%2,
110
/ 5,5
60
34.7
%1,
968
/ 5,6
77
31.7
%2,
323
/ 7,3
37
30.4
%2,
098
/ 6,8
93
26.3
%1,
893
/ 7,2
08
2.8
%99
/ 3,
560
80.7
%3,
108
/ 3,8
53
87.0
%3,
272
/ 3,7
62
82.9
%3,
277
/ 3,9
54
82.6
%3,
267
/ 3,9
54
83.7
%3,
275
/ 3,9
15
78.4
%3,
157
/ 4,0
29
67.3
%2,
909
/ 4,3
20
73.7
%2,
947
/ 4,0
01
71.8
%2,
896
/ 4,0
36
79.5
%3,
125
/ 3,9
32
74.1
%3,
024
/ 4,0
83
76.0
%3,
059
/ 4,0
25
73.2
%2,
952
/ 4,0
34
41.4
%2,
445
/ 5,9
05
43.8
%2,
234
/ 5,1
01
45.9
%2,
535
/ 5,5
26
42.9
%2,
564
/ 5,9
77
39.7
%2,
312
/ 5,8
25
41.7
%2,
346
/ 5,6
26
36.3
%1,
965
/ 5,4
13
35.3
%1,
981
/ 5,6
19
33.3
%1,
863
/ 5,5
93
40.3
%2,
167
/ 5,3
78
37.3
%2,
045
/ 5,4
77
33.6
%2,
410
/ 7,1
81
32.3
%2,
164
/ 6,7
06
28.0
%1,
962
/ 7,0
16
1.8
%59
/ 3,
353
83.0
%3,
126
/ 3,7
68
83.2
%3,
208
/ 3,8
55
85.6
%3,
244
/ 3,7
90
86.6
%3,
253
/ 3,7
57
74.3
%2,
987
/ 4,0
18
67.8
%2,
836
/ 4,1
84
81.0
%2,
989
/ 3,6
88
71.5
%2,
801
/ 3,9
15
77.9
%3,
002
/ 3,8
56
73.0
%2,
908
/ 3,9
86
75.2
%2,
954
/ 3,9
28
73.5
%2,
863
/ 3,8
97
42.4
%2,
408
/ 5,6
83
45.9
%2,
223
/ 4,8
42
47.1
%2,
503
/ 5,3
10
44.1
%2,
533
/ 5,7
43
40.6
%2,
270
/ 5,5
92
42.9
%2,
314
/ 5,3
88
37.7
%1,
947
/ 5,1
65
36.6
%1,
964
/ 5,3
71
34.4
%1,
846
/ 5,3
60
41.6
%2,
140
/ 5,1
39
38.7
%2,
021
/ 5,2
25
34.3
%2,
377
/ 6,9
32
33.0
%2,
137
/ 6,4
67
28.7
%1,
944
/ 6,7
66
2.9
%10
0 / 3
,429
91.4
%3,
373
/ 3,6
89
90.1
%3,
346
/ 3,7
15
91.2
%3,
355
/ 3,6
79
78.6
%3,
113
/ 3,9
62
68.1
%2,
876
/ 4,2
26
74.9
%2,
944
/ 3,9
32
72.2
%2,
861
/ 3,9
60
83.0
%3,
135
/ 3,7
77
78.5
%3,
061
/ 3,9
01
80.4
%3,
080
/ 3,8
31
76.4
%2,
970
/ 3,8
87
41.8
%2,
434
/ 5,8
18
44.3
%2,
232
/ 5,0
38
46.4
%2,
534
/ 5,4
64
43.3
%2,
559
/ 5,9
16
39.9
%2,
299
/ 5,7
68
41.9
%2,
334
/ 5,5
71
36.6
%1,
961
/ 5,3
54
35.5
%1,
974
/ 5,5
63
33.4
%1,
852
/ 5,5
47
40.6
%2,
159
/ 5,3
23
37.4
%2,
032
/ 5,4
28
33.8
%2,
403
/ 7,1
16
32.4
%2,
155
/ 6,6
49
28.2
%1,
960
/ 6,9
57
2.8
%10
2 / 3
,619
90.4
%3,
439
/ 3,8
05
91.7
%3,
455
/ 3,7
66
75.5
%3,
125
/ 4,1
41
66.4
%2,
917
/ 4,3
93
73.3
%2,
983
/ 4,0
71
69.0
%2,
868
/ 4,1
58
79.4
%3,
144
/ 3,9
61
75.8
%3,
073
/ 4,0
56
77.3
%3,
098
/ 4,0
09
73.1
%2,
977
/ 4,0
72
40.8
%2,
445
/ 5,9
93
43.0
%2,
240
/ 5,2
12
45.2
%2,
546
/ 5,6
33
42.3
%2,
572
/ 6,0
75
38.9
%2,
309
/ 5,9
32
40.9
%2,
343
/ 5,7
33
35.7
%1,
969
/ 5,5
22
34.6
%1,
982
/ 5,7
32
32.7
%1,
868
/ 5,7
05
39.5
%2,
169
/ 5,4
93
36.7
%2,
048
/ 5,5
85
33.3
%2,
418
/ 7,2
52
31.8
%2,
169
/ 6,8
17
27.6
%1,
965
/ 7,1
22
3.0
%10
9 / 3
,575
96.0
%3,
531
/ 3,6
78
74.6
%3,
103
/ 4,1
60
66.3
%2,
908
/ 4,3
86
73.6
%2,
979
/ 4,0
45
69.1
%2,
864
/ 4,1
45
79.3
%3,
138
/ 3,9
58
75.1
%3,
061
/ 4,0
77
77.1
%3,
088
/ 4,0
07
73.4
%2,
971
/ 4,0
50
41.1
%2,
450
/ 5,9
57
43.4
%2,
242
/ 5,1
71
45.5
%2,
548
/ 5,5
99
42.7
%2,
578
/ 6,0
32
39.3
%2,
314
/ 5,8
92
41.2
%2,
346
/ 5,6
97
35.9
%1,
971
/ 5,4
85
34.8
%1,
984
/ 5,6
94
33.0
%1,
872
/ 5,6
67
39.7
%2,
168
/ 5,4
59
37.0
%2,
051
/ 5,5
45
33.5
%2,
420
/ 7,2
25
32.1
%2,
173
/ 6,7
78
27.7
%1,
965
/ 7,0
93
2.6
%92
/ 3,
593
75.4
%3,
111
/ 4,1
24
67.0
%2,
915
/ 4,3
48
74.3
%2,
982
/ 4,0
14
70.0
%2,
873
/ 4,1
02
80.3
%3,
147
/ 3,9
18
76.3
%3,
076
/ 4,0
29
78.0
%3,
097
/ 3,9
71
73.8
%2,
975
/ 4,0
30
41.3
%2,
449
/ 5,9
36
43.6
%2,
244
/ 5,1
43
45.8
%2,
552
/ 5,5
68
42.9
%2,
579
/ 6,0
05
39.4
%2,
314
/ 5,8
68
41.4
%2,
346
/ 5,6
70
36.1
%1,
971
/ 5,4
58
35.0
%1,
985
/ 5,6
67
33.2
%1,
872
/ 5,6
41
40.0
%2,
171
/ 5,4
28
37.2
%2,
052
/ 5,5
16
33.6
%2,
420
/ 7,1
93
32.2
%2,
173
/ 6,7
52
27.8
%1,
967
/ 7,0
64
3.5
%12
5 / 3
,567
64.7
%2,
847
/ 4,4
03
68.5
%2,
844
/ 4,1
50
68.3
%2,
820
/ 4,1
26
73.1
%3,
000
/ 4,1
03
69.2
%2,
925
/ 4,2
26
71.3
%2,
953
/ 4,1
42
65.6
%2,
791
/ 4,2
56
37.9
%2,
313
/ 6,0
99
41.1
%2,
153
/ 5,2
42
42.2
%2,
409
/ 5,7
11
39.4
%2,
432
/ 6,1
72
36.9
%2,
208
/ 5,9
89
38.0
%2,
213
/ 5,8
23
32.8
%1,
843
/ 5,6
11
31.9
%1,
857
/ 5,8
17
30.2
%1,
747
/ 5,7
86
38.2
%2,
104
/ 5,5
04
34.4
%1,
944
/ 5,6
45
31.0
%2,
282
/ 7,3
62
30.3
%2,
079
/ 6,8
56
25.7
%1,
850
/ 7,1
98
2.8
%99
/ 3,
586
70.6
%2,
886
/ 4,0
87
68.0
%2,
806
/ 4,1
26
69.5
%2,
908
/ 4,1
85
64.3
%2,
805
/ 4,3
65
70.2
%2,
930
/ 4,1
72
65.2
%2,
768
/ 4,2
46
38.2
%2,
320
/ 6,0
80
41.2
%2,
152
/ 5,2
28
41.4
%2,
372
/ 5,7
35
39.1
%2,
413
/ 6,1
76
36.4
%2,
186
/ 6,0
03
38.0
%2,
209
/ 5,8
11
33.1
%1,
848
/ 5,5
85
32.1
%1,
861
/ 5,7
95
30.0
%1,
736
/ 5,7
91
37.3
%2,
064
/ 5,5
37
34.8
%1,
952
/ 5,6
06
30.8
%2,
270
/ 7,3
79
29.7
%2,
044
/ 6,8
87
25.6
%1,
841
/ 7,1
94
2.5
%84
/ 3,
305
73.1
%2,
818
/ 3,8
54
76.7
%2,
961
/ 3,8
61
71.6
%2,
868
/ 4,0
06
77.2
%2,
983
/ 3,8
66
71.6
%2,
802
/ 3,9
15
42.3
%2,
399
/ 5,6
75
46.0
%2,
220
/ 4,8
21
46.3
%2,
464
/ 5,3
26
43.0
%2,
483
/ 5,7
81
39.9
%2,
238
/ 5,6
09
41.8
%2,
264
/ 5,4
18
36.7
%1,
906
/ 5,1
92
35.6
%1,
922
/ 5,3
98
33.6
%1,
805
/ 5,3
77
41.6
%2,
134
/ 5,1
35
38.1
%1,
994
/ 5,2
28
33.3
%2,
327
/ 6,9
84
32.4
%2,
098
/ 6,4
81
28.1
%1,
904
/ 6,7
82
2.2
%73
/ 3,
311
71.8
%2,
849
/ 3,9
68
67.9
%2,
780
/ 4,0
92
71.8
%2,
855
/ 3,9
74
68.6
%2,
743
/ 4,0
01
39.7
%2,
303
/ 5,7
96
42.7
%2,
113
/ 4,9
53
43.5
%2,
367
/ 5,4
37
40.1
%2,
373
/ 5,9
19
38.0
%2,
171
/ 5,7
07
39.9
%2,
202
/ 5,5
14
34.9
%1,
843
/ 5,2
82
34.2
%1,
872
/ 5,4
73
31.9
%1,
743
/ 5,4
69
39.1
%2,
048
/ 5,2
44
36.4
%1,
935
/ 5,3
17
32.1
%2,
268
/ 7,0
62
31.2
%2,
045
/ 6,5
65
26.9
%1,
851
/ 6,8
69
2.1
%72
/ 3,
454
78.6
%3,
059
/ 3,8
94
82.5
%3,
117
/ 3,7
80
77.1
%2,
975
/ 3,8
61
42.9
%2,
463
/ 5,7
45
44.3
%2,
213
/ 5,0
01
45.9
%2,
501
/ 5,4
51
43.5
%2,
550
/ 5,8
59
40.1
%2,
293
/ 5,7
19
42.0
%2,
320
/ 5,5
30
36.8
%1,
951
/ 5,3
07
35.7
%1,
970
/ 5,5
13
33.5
%1,
845
/ 5,5
01
40.4
%2,
139
/ 5,2
93
37.7
%2,
026
/ 5,3
67
34.2
%2,
394
/ 7,0
02
32.6
%2,
153
/ 6,6
00
28.2
%1,
949
/ 6,9
15
2.4
%83
/ 3,
442
78.0
%3,
045
/ 3,9
06
73.0
%2,
911
/ 3,9
89
40.8
%2,
400
/ 5,8
76
43.5
%2,
200
/ 5,0
58
46.1
%2,
513
/ 5,4
55
42.2
%2,
506
/ 5,9
41
39.2
%2,
272
/ 5,7
96
41.1
%2,
303
/ 5,6
03
36.1
%1,
940
/ 5,3
73
34.9
%1,
952
/ 5,5
86
32.9
%1,
824
/ 5,5
45
39.4
%2,
118
/ 5,3
70
37.3
%2,
022
/ 5,4
18
33.4
%2,
359
/ 7,0
60
31.8
%2,
123
/ 6,6
82
27.9
%1,
942
/ 6,9
69
2.9
%99
/ 3,
427
74.7
%2,
922
/ 3,9
14
42.4
%2,
451
/ 5,7
81
44.9
%2,
242
/ 4,9
98
45.7
%2,
506
/ 5,4
80
42.9
%2,
536
/ 5,9
06
39.8
%2,
292
/ 5,7
56
41.6
%2,
314
/ 5,5
69
36.2
%1,
940
/ 5,3
52
35.2
%1,
954
/ 5,5
58
33.0
%1,
831
/ 5,5
49
40.3
%2,
145
/ 5,3
20
37.3
%2,
019
/ 5,4
07
33.4
%2,
375
/ 7,1
15
32.0
%2,
130
/ 6,6
56
27.9
%1,
941
/ 6,9
54
1.9
%62
/ 3,
316
40.9
%2,
360
/ 5,7
69
43.5
%2,
155
/ 4,9
58
45.1
%2,
444
/ 5,4
15
42.3
%2,
475
/ 5,8
45
39.2
%2,
230
/ 5,6
84
41.5
%2,
272
/ 5,4
79
36.3
%1,
906
/ 5,2
50
35.3
%1,
926
/ 5,4
55
33.1
%1,
804
/ 5,4
48
39.8
%2,
086
/ 5,2
45
37.8
%2,
001
/ 5,2
88
33.0
%2,
325
/ 7,0
56
32.0
%2,
097
/ 6,5
49
27.9
%1,
909
/ 6,8
51
3.2
%14
7 / 4
,662
43.5
%2,
547
/ 5,8
58
48.9
%2,
994
/ 6,1
28
46.4
%3,
042
/ 6,5
50
41.4
%2,
698
/ 6,5
23
43.9
%2,
762
/ 6,2
93
35.9
%2,
233
/ 6,2
19
35.5
%2,
270
/ 6,4
00
31.9
%2,
074
/ 6,4
94
64.9
%3,
384
/ 5,2
14
47.0
%2,
741
/ 5,8
27
46.4
%3,
371
/ 7,2
66
35.2
%2,
581
/ 7,3
33
29.6
%2,
295
/ 7,7
53
2.1
%79
/ 3,
683
47.2
%2,
608
/ 5,5
24
43.2
%2,
597
/ 6,0
13
43.7
%2,
492
/ 5,7
05
45.4
%2,
507
/ 5,5
23
36.2
%1,
976
/ 5,4
64
34.5
%1,
965
/ 5,6
96
32.3
%1,
842
/ 5,7
05
45.0
%2,
357
/ 5,2
32
37.8
%2,
081
/ 5,5
03
34.9
%2,
496
/ 7,1
60
34.3
%2,
276
/ 6,6
34
27.9
%1,
972
/ 7,0
61
2.8
%12
1 / 4
,337
67.5
%3,
741
/ 5,5
40
41.9
%2,
637
/ 6,3
01
43.7
%2,
670
/ 6,1
12
36.3
%2,
179
/ 6,0
05
35.3
%2,
191
/ 6,2
11
32.6
%2,
040
/ 6,2
50
46.1
%2,
626
/ 5,6
97
39.9
%2,
372
/ 5,9
42
38.7
%2,
880
/ 7,4
39
34.4
%2,
472
/ 7,1
84
29.4
%2,
212
/ 7,5
34
3.1
%15
0 / 4
,773
39.7
%2,
672
/ 6,7
28
40.8
%2,
680
/ 6,5
65
34.2
%2,
205
/ 6,4
48
33.1
%2,
209
/ 6,6
72
30.5
%2,
050
/ 6,7
15
43.2
%2,
655
/ 6,1
43
37.5
%2,
396
/ 6,3
87
37.0
%2,
900
/ 7,8
32
33.0
%2,
516
/ 7,6
15
27.8
%2,
222
/ 7,9
79
2.3
%10
3 / 4
,463
72.3
%3,
688
/ 5,1
01
36.6
%2,
201
/ 6,0
16
35.7
%2,
219
/ 6,2
13
33.1
%2,
074
/ 6,2
69
40.2
%2,
413
/ 5,9
99
36.9
%2,
259
/ 6,1
24
34.6
%2,
682
/ 7,7
62
36.5
%2,
593
/ 7,1
05
28.1
%2,
155
/ 7,6
67
2.8
%11
8 / 4
,277
38.7
%2,
246
/ 5,8
08
38.1
%2,
277
/ 5,9
79
34.9
%2,
114
/ 6,0
65
42.3
%2,
451
/ 5,7
95
38.6
%2,
289
/ 5,9
31
36.7
%2,
759
/ 7,5
16
36.7
%2,
562
/ 6,9
82
29.5
%2,
198
/ 7,4
56
2.6
%96
/ 3,
691
75.0
%3,
261
/ 4,3
46
55.5
%2,
683
/ 4,8
38
34.5
%1,
963
/ 5,6
92
33.6
%1,
915
/ 5,6
95
29.6
%2,
214
/ 7,4
78
30.4
%2,
085
/ 6,8
66
30.3
%2,
110
/ 6,9
68
2.9
%11
2 / 3
,894
52.4
%2,
666
/ 5,0
85
33.9
%1,
991
/ 5,8
74
32.5
%1,
919
/ 5,9
03
29.3
%2,
244
/ 7,6
65
29.4
%2,
083
/ 7,0
82
29.7
%2,
127
/ 7,1
69
3.3
%11
1 / 3
,378
30.5
%1,
813
/ 5,9
39
30.3
%1,
795
/ 5,9
23
28.3
%2,
095
/ 7,4
06
26.7
%1,
916
/ 7,1
68
28.3
%1,
980
/ 6,9
89
2.7
%10
3 / 3
,886
46.2
%2,
452
/ 5,3
07
43.4
%2,
981
/ 6,8
75
34.5
%2,
335
/ 6,7
62
28.0
%2,
022
/ 7,2
22
2.3
%88
/ 3,
822
45.0
%3,
018
/ 6,7
02
30.9
%2,
144
/ 6,9
48
25.5
%1,
872
/ 7,3
39
3.9
%20
1 / 5
,117
30.1
%2,
581
/ 8,5
74
26.1
%2,
254
/ 8,6
24
3.9
%20
0 / 5
,078
25.9
%2,
170
/ 8,3
70
5.0
%24
3 / 4
,897
V.ch
olera
e
N1696
1
V.ch
olera
e03
95 T
EDA
V.ch
olera
e03
95 T
IGR
V.ch
olera
eV52
V.ch
olera
eM
66-2
MO10
V.ch
olera
eBX33
0286
V.ch
olera
eRC9
V.ch
olera
eM
J123
6
V.ch
olera
eB33
VCE
V.ch
olera
e27
40-8
0
V.ch
olera
e
V.ch
olera
e15
87
V.ch
olera
eAM
-192
26
V.ch
olera
eM
ZO-2
V.ch
olera
e
1212
9
V.ch
olera
e
TM11
079-
80
V.ch
olera
eTM
A21
V.ch
olera
eVL4
26
V.pa
raha
emoly
ticus
2210
633
V.pa
raha
emoly
ticus
16
V.vu
lnific
usCM
CP6
V.vu
lnific
usYJ0
16
V.sp
ecies
MED22
2
V.sp
lendid
usLG
P32
A.fisch
eri ES11
4
A.fisch
eri M
J11
A.salm
onici
daLF
I123
8
V.sp
ecies
Ex25
V.ca
mpb
ellii
AND4
V.ha
rvey
i BAA1116
V.sh
ilonii
AK1
P.pro
fund
umSS9
V.ch
olera
eN16
961
V.ch
olera
e03
95 T
EDA
V.ch
olera
e03
95 T
IGR
V.ch
olera
eV52
V.ch
olera
eM
66-2
V.ch
olera
eM
O10
V.ch
olera
eBX33
0286
V.ch
olera
eRC9
V.ch
olera
eM
J123
6
V.ch
olera
eB33
VCE
V.ch
olera
e27
40-8
0
V.ch
olera
e15
87
V.ch
olera
eAM
-192
26
V.ch
olera
eM
ZO-2
V.ch
olera
e12
129
V.ch
olera
eTM
1107
9-80
V.ch
olera
eTM
A21
V.ch
olera
eVL4
26
V.pa
raha
emoly
ticus
221
0633
V.pa
raha
emoly
ticus
16
V.vu
lnific
usCM
CP6
V.vu
lnific
usYJ0
16
V.sp
ecies
MED22
2
V.sp
lendid
usLG
P32
A.fisch
eriES11
4
A.fisch
eriM
J11
A.salm
onici
daLF
I123
8
V.sp
ecies
Ex25
V.ca
mpb
ellii
AND4
V.ha
rvey
iBAA1116
V.sh
ilonii
AK1
P.pro
fund
umSS9
Hom
olog
y w
ithin
pro
teom
es
6.0
%0.
0 %H
omol
ogy
betw
een
prot
eom
es 90.0
%30
.0 %
Figure 4 BLAST matrix ofthe 32 Vibrionaceae genomes.The colours highlighting thespecies are the same as in Fig. 1.Since the reciprocal similarity(reported as percent) is notreadable at this resolution, everymatrix cell is coloured using thescales as indicated. The bottomrow identifies hits (other thanhits-to-self) found within a ge-nome. Four matrix cells report-ing high pairwise similarities areoutlined; their numbers arespecified in the text
Origins of V. cholerae 7

0M
0.5M1M
1.5M
2M2.
5MV. cholerae 01 El Tor N16961chromosome 12,961,149 bp
Gap B
Gap A
Gap C
Gap D
Gap G
Gap E
Gap F
0k125k
25
0k
375k
500k625k75
0k
875
k
1000k
V. cholerae 01 El Tor N16961chromosome 21,072,310 bp
Superintegron
P.profundum SS9
V.shilonii AK1
V.harveyi BAA-116
V.campebellii AND4
V.parahaemolyticus 16
V.parahaemolyticus 2210633
Vibrio spp. Ex25
A.salmonicida LF11238
A.fischeri MJ11
A.fischeri ES114
V.splendidus LGP32
V.species MED222
V.vulnificus YJ016
V.vulnificus CMCP6
V.cholerae VL426
V.cholerae 12129
V.cholerae TMA21
V.cholerae TM11079-80
V.cholerae 1587
V.cholerae AM-19226
V.cholerae MZO-2
V.cholerae 2740-80
V.cholerae BX330286
V.cholerae B33VCE
V.cholerae RC9
V.cholerae MJ1236
V.cholerae M66-2
V.cholerae V52
V.cholerae MO10
V.cholerae O395 TEDA
V.cholerae 0395 TIGR
V.cholerae N16961
Stacking energy
Position preference
Global direct repeats
GC skew
genes positive strandgenes negatve strand
Outer circle
Inner circle
8 T. Vesth et al.

When the first genome of A. fischeri is added, which isnot a member of the Vibrio genus, it does not addsignificantly more novel genes to the pan-genome thanVibrio genomes did. This contrasts with P. profundumwhich produces a sharp increase in the pan-genome, asdoes, interestingly, V. shilonii. Note that there are approx-imately 20,200 total gene families within the 32 sequencedVibrionaceae genomes, whereas the core genome decreasesto approximately 1,000 gene families.
BLAST Comparison Visualised in a BLAST Matrix
A BLAST matrix provides a visual overview of reciprocalpairwise whole-genome comparisons, as shown in Fig. 4.The stronger a matrix cell is coloured, the more similaritywas detected between the gene content of two genomes. Ascan be seen in the lower right triangle, all V. choleraegenomes are highly similar, with similarity ranging between64% and 93% for any given pair of genomes. No statisticaldifference was observed when comparing clinical isolatesto environmental isolates. The two A. fischeri and the twoV. vulnificus genomes also share a high degree of identitywithin their species (75% and 67%, respectively), visible atthe bottom of the matrix. In contrast, the two V. para-haemolyticus genomes only share 35% identity, which isnot higher than the similarity detected between genomes ofdifferent species. With 72% similarity, isolate MED222most closely matches V. splendidus and with 65% isolateEX25 again shares most similarity with V. parahaemolyti-cus 2210633.
BLAST Atlas
A BLAST atlas was constructed using V. cholerae N16961(O1, El Tor) as the reference genome, shown in Fig. 5. Thebest blast hits identified in the query genomes areplotted in the lanes around the reference genome, withdifferent colours for different species. In general,chromosome 1 is more strongly conserved than chro-mosome 2. A large part of chromosome 2 of N16961displays very little conservation in the other genomes;this area represents a super integron [40] that containsthe V. cholerae-specific repeat (VCR) sequences, as well
as a high number of gene cassettes. The repeat sequencesare visible as black boxes in the repeat lane of thereference genome (second inner lane). Although all V.cholerae genomes contain a superintegron, its genes arevery diverse between isolates [34] which explains the lackof blast hits in this region.
Several regions of the atlas have been highlighted. GapsB, C, D and F on chromosome 1 (indicated in green)contain genes that are conserved in the representedgenomes of V. cholerae but not in the other Vibrionaceae.The gaps marked A, E and G indicate regions that arespecific to the toxigenic, clinical isolates only. Annotated,V. cholerae-specific genes present in all these regions arelisted in Table 2 (hypothetical genes are excluded). Genesspecific for toxinogenic V. cholerae identified in gap Ainclude, amongst others, biosynthesis genes for the toxinco-regulated pilus (which is required for transmission of theprophage CTXΦ carrying the enterotoxin genes), as well asgenes encoding citrate lyase. Note that the genes in gap Aare also found in the environmental isolate V. cholerae2740-80.
Gap B contains a number of outer membrane proteingenes involved in sugar modification that are found in all V.cholerae genomes. Genes from gap C encoding a histidinekinase two-component signal transduction regulatory sys-tem are also conserved within the species, as genes in gapsD and F, involved in chemotaxis and possible multidrugresistance.
Gap E, containing genes conserved in toxigenic strainsonly, holds the prophage CTXΦ that contains the genesencoding cholera enterotoxin subunits A and B; thisenterotoxin is responsible for the excessive, watery diar-rhoea typical for cholera. Upon binding to target cell GM1gangliosides, enterotoxin enters the cell and stimulatesadenylate cyclase by ADP ribosylation. The resultantincreased cyclic AMP levels induce excessive electrolytemovement and sodium plus water secretion [43]. StrainM66-2 is believed to be a precursor of the seventhpandemic V. cholerae that lacks the prophage CTXΦ andthe enterotoxin genes [11]. Gap E bears the RTX toxinoperon, which encodes a pore-forming cytotoxin [22]. AnRTX toxin is also present in environmental isolate 2740-80and in V. vulnificus.
Gap G on chromosome 2 consists of a set of five genes,all in the same orientation, in a putative operon, flanked bygenes on the complimentary strand. This appears to be aremnant of a mobile element, as these genes are flanked bya transposase gene on the 3′ end, and there is a small globalrepeat on the 5′ end. Only the first two of the five genes havean assigned function, with the first gene being a GMPreductase, and the second a putative DNA methyltransferase.The remaining three genes are hypothetical, but theirstrikingly strong conservation in all pathogenic strains and
�Figure 5 BLAST atlas with V. cholerae strain N16961 as a referencestrain, showing chromosomes 1 (top) and 2 (bottom). The bestBLAST hits identified with genes from N16961 in the other V.cholerae genomes are represented in dark red, for the location as itappears in N16961. Blast hits in the other genomes are shown invarious colours as indicated to the right. Major areas conserved in V.cholerae but not in other Vibrionaceae are identified as gap B, gap C,gap D and gap F in green; areas that are found in toxigenic V. choleraeonly are marked black as gap A, gap E and gap G. The superintegronon chromosome 2 of V. cholerae is also indicated
Origins of V. cholerae 9

Table 2 A selection of genes located in the gaps marked in Fig. 5
Gap A (850000–913000)
852903–851557 Citrate/sodium symporter
853165–854235 Citrate (pro-3S)-lyase ligase
854287–854583 Citrate lyase subunit gamma
854565–855455 Citrate lyase, beta subunit
855391–856995 Citrate lyase, alpha subunit
856992–857528 citX protein
857506–858447 citG protein
869812–866873 Helicase-related protein
870391–869813 Tellurite resistance protein-related
871298–870819 Transcriptional regulator, putative
873242–874225 Transposase, putative
876974–880015 ToxR-activated gene A protein
881390–884728 Inner membrane protein, putative
885773–886267 tagD protein
888405–886543 Toxin co-regulated pilus biosynthesis
888846–889511 Toxin co-regulated pilus biosynthesis
889496–889906 Toxin co-regulated pilus biosynthesis
890449–891123 Toxin co-regulated pilin
891203–892495 Toxin co-regulated pilus biosynthesis
892495–892947 Toxin co-regulated pilus biosynthesis
892950–894419 Toxin co-regulated pilus biosynthesis
894412–894867 Toxin co-regulated pilus biosynthesis
894855–895691 Toxin co-regulated pilus biosynthesis
895707–896165 Toxin co-regulated pilus biosynthesis
896155–897666 Toxin co-regulated pilus biosynthesis
897641–898663 Toxin co-regulated pilus biosynthesis
898673–899689 Toxin co-regulated pilus biosynthesis
899896–900726 TCP pilus virulence regulatory protein
900726–901487 Leader peptidase TcpJ
901494–903374 Accessory colonization factor AcfB
903380–904150 Accessory colonization factor AcfC
904648–905556 tagE protein
906206–905559 Accessory colonization factor AcfA
914124–912856 Phage family integrase
Gap B (975000–1010000)
978644–979144 Phosphotyrosine protein phosphatase
981833–982387 Serine acetyltransferase-related protein
982384–983532 Exopolysacch. biosynth protein EpsF
983529–984938 Polysacch. export protein, putative (gfcE)
986166–986597 Serine acetyltransferase-related protein
986597–987937 capK protein, putative
987913–989010 Polysaccharide biosynthesis protein, putative
1001910–1002437 Polysaccharide export-related protein (gfcE)
1002462–1004675 Putative exopolysacch. biosynth protein
Gap C (1130000–1160000)
1139646–1142912 Chitinase, putative
1147856–1148998 Response regulator
1149033–1149398 Response regulator
1149990–1151309 Sensory box sensor histidine kinase
1151321–1152625 Sensor histidine kinase
1152625–1154235 Response regulator
1154252–1155595 Response regulator
1157228–1155624 Sensor histidine kinase
1158044–1157232 Periplasmic binding protein-related
Gap D (1478000–1520000)
2086826–2087584 CDP-diacylglycerol-glyc.-3-phosph-3-phosphatidyltransferase
2087587–2088519 Phosphatidate cytidylyltransferase
2094741–2095604 PvcB protein
2098112–2097183 LysR family transcriptional regulator
2098432–2100258 pvcA protein
2117923–2119977 Methyl-accepting chemotaxis protein
2120575–2120030 Transcriptional regulator
2120663–2121826 Benzoate transport protein
Gap E (1537000–1587500)
1541452–1543170 Sensor histidine kinase/response regulator
1545396–1543231 Toxin secretion transporter, putative
1546802–1545399 RTX toxin transporter
1548919–1546757 RTX toxin transporter
1549662–1550123 RTX toxin activating protein
1550108–1563784 RTX toxin RtxA
1564376–1564152 RstC protein
1564844–1564470 RstB1 protein
1565901–1564822 RstA1 protein
1566027–1566365 Transcriptional repressor RstR
1567341–1566967 Cholera enterotoxin, B subunit
1568114–1567338 Cholera enterotoxin, A subunit
1569412–1568213 Zona occludens toxin
1569702–1569409 Accessory cholera enterotoxin
1571241–1570993 Colonization factor
1571760–1571377 RstB2 protein
1572817–1571738 RstA1 protein
1572943–1573281 Transcriptional repressor RstR
1577272–1575704 Phage replication protein Cri
1582123–1580555 Phage replication protein Cri
1583160–1583513 Transposase OrfAB, subunit A
1583510–1584382 Transposase OrfAB, subunit B
Gap F (1896000–1956000)
1896092–1897327 Phage family integrase
1900831–1898009 Helicase, putative
1903632–1902898 Chemotaxis protein MotB-related
1908858–1905790 Type I restriction enzyme HsdR
1916009–1913628 DNA methylase HsdM, putative
1933231–1935654 Neuraminidase
1936007–1935801 Transcriptional regulator
1936121–1936597 DNA repair protein RadC, putative
1938391–1937519 Transposase OrfAB, subunit B
1938732–1938388 Transposase OrfAB, subunit A
1941671–1941351 Transcriptional regulator, putative
Table 2 (continued)
10 T. Vesth et al.

complete absence of homologues in the other Vibrio genomesstrongly point towards a potential biological significance.
Discussion
The recent availability of many Vibrionaceae genomes,including a substantial number of V. cholerae genomes,allows the possibility to take a closer look at the similaritiesand differences of species within the genus Vibrio. This canexamine, on a genome scale, what distinguishes V. choleraefrom the other Vibrio species. Since not all V. choleraeisolates are pathogenic, the presence of the prophage-bearing cholera enterotoxin, the main virulence factor forcholera, is not a suitable marker for this species. Weattempted to identify a set of V. cholerae-specific genes,and also explored the internal diversity within the V.cholerae genomes that have been sequenced to date.
On a phylogenetic tree based on the 16S ribosomal RNAgene, those isolates that do not belong to the genus Vibriowere positioned as outliers, as expected. This tree furtherindicated the closest resembling 16S rRNA sequence forthe two sequenced Vibrio strains that are currently notassigned to a species. It was observed that the twosequenced V. parahaemolyticus strains were not placedtogether. The complete gene content of each genome wasnext compared by BLAST and the results were pooled intogene families which were subjected to cluster analysis. Thisprovided evidence that the 18 V. cholerae genomes fall intotwo subclusters, one mainly containing clinical isolates andthe other environmental isolates.
The gene family clustering, subsequent pan-genomeanalysis and the pairwise BLAST results, as summarisedin the BLAST matrix, all supported the relatedness ofVibrio species Ex25 to V. parahaemolyticus 2210633 butnot to V. parahaemolyticus 16. This latter genome was quitedifferent from V. parahaemolyticus 2210633 in all analyses.Although it is possible that the species V. parahaemolyticusis far more genetically diverse than V. cholerae, A. fischerior V. vulnificus, an alternative explanation is that one of the
sequenced isolates is perhaps incorrectly named as V.parahaemolyticus. The similarity between Vibrio speciesMED222 and V. splendidus based on gene families is inagreement with their related 16S rRNA genes and pub-lished data [21]. However, in contrast to what the ribosomalgene suggests, our whole-genome comparison indicates thatthe three Aliivibrio genomes (A. salmonicida and two A.fischeri) are not so different from Vibrio after all. Theirrecent placement in the genus Aliivibrio, a decision basedon five genes (the 16S rRNA gene and four housekeepinggenes) and phenotypical characteristics [47], appears not tobe reflective of the whole genome picture presented here.
The BLAST results were graphically summarised in aBLAST atlas, which visualised V. cholerae-specific geneclusters. These coded for polysaccharide biosynthesisenzymes, response regulators and chemotaxis proteins,amongst others. In addition, a V. cholerae-specific, histidinekinase two-component signal transduction regulatory sys-tem was identified. The two-component signal transductionpathway is a powerful regulating system for bacteria toadapt to a particular ecological niche. There is a precedentfor this claim, as the introduction of a single regulatoryprotein in Vibrio fischeri strain MJ11 has been shown tospecifically enable colonization of the squid Euprymnascolopes [26].
As expected, the main differences observed between V.cholerae clinical isolates and the environmental strains aredue to genes related to virulence. Two exceptions are thepresence of a number of virulence genes in the environ-mental strain V. cholerae 2740-80 and the absence ofenterotoxin genes in clinical isolate M66-2. It has alreadybeen suggested that M66-2 might be a predecessor ofpandemic, enterotoxic V. cholerae [11]. From sequencecomparison of four housekeeping genes, it was concludedthat V. cholerae 2740-80 is intermediary between toxigenicand non-toxigenic isolates [30]. This view is confirmed bythe data presented here, although we propose to considerthe possibility that the isolate arose from a pandemic clonethat has lost the CTXΦ prophage, rather than being aprecursor of a pathogen.
In conclusion, several different methods of genomecomparisons have yielded a picture of V. cholerae genomesas forming a distinct cluster, compared to related species,and a relatively small number of genes might be responsiblefor environmental niche adaptation and hence for genera-tion of this distinct species. Likely candidates includemultiple two-component signal transduction regulatoryproteins as well as chemotaxis proteins.
Acknowledgements We would like to thank Tim Binnewies forearly work on this project, and also to the Danish Research Councilsand the DTU Globalization funds for financial support.
1942032–1941658 Middle operon regulator-related
1944457–1943306 eha protein
Gap G (chromosome II, 21300–223000)
213207–214250 GMP reductase
214574–215725 DNA methyltransferase
220262–219825 IS1004 transposase
All gene annotations are taken from the reference genome V. choleraestrain N16961. Hypothetical proteins were excluded. Gaps A, E and Gare conserved in pathogenic strains, whereas gaps B, C, D and F areconserved in all V. cholerae genomes analysed (Figure 1)
Table 2 (continued)
Origins of V. cholerae 11

Open Access This article is distributed under the terms of theCreative Commons Attribution Noncommercial License which per-mits any noncommercial use, distribution, and reproduction in anymedium, provided the original author(s) and source are credited.
References
1. Bassler B et al. (2007) CP000789.1: Direct submission toGenBank
2. Binnewies TT, Hallin PF, Staerfeldt HH, Ussery DW (2005)Genome update: proteome comparisons. Microbiol 151:1–4
3. Chen CY, Wu KM, Chang YC, Chang CH, Tsai HC, Liao TL, LiuYM, Chen HJ, Shen AB, Li JC, Su TL, Shao CP, Lee CT, Hor LI,Tsai SF (2003) Comparative genome analysis of Vibrio vulnificus,a marine pathogen. Genome Res 13:2577–2587
4. Clayton RA, Sutton G, Hinkle PS, Bult C, Fields C (1995)Intraspecific variation in small-subunit rRNA sequences inGenBank: why single sequences may not adequately representprokaryotic taxa. Int J Syst Bacteriol 45:595–599
5. Colwell R, Grim CJ, Young S, Jaffe D, Gnerre S, Berlin A,Heiman D, Hepburn T, Shea T, Sykes S, Alvarado L, Kodira C,Heidelberg J, Lander E, Galagan J, Nusbaum C, Birren B (2008)NZ_AAKF00000000: Direct submission to GenBank
6. Doolittle WF (1995) Phylogenetic classification and the universaltree. Science 284:2124–2129
7. Doolittle WF, Papke RT (2006) Genomics and the bacterialspecies problem. Genome Biol 7:116
8. Doolittle WF, Zhaxybayeva O (2009) On the origin of prokaryoticspecies. Genome Res 19:744–756
9. Edwards R, Ferriera S, Johnson J, Kravitz S, Beeson K, Sutton G,Rogers Y-H, Friedman R, Frazier M, Venter JC (2008)NZ_ACCV00000000: Direct submission to GenBank
10. Farmer JJ, Janda JM (2005) Vibrionaceae. In: Bergey’smanual of systematic bacteriology, 2nd edn, vol 2 part B.Springer, New York, pp 491–546
11. Feng L, Reeves PR, Lan R, Ren Y, Gao C, Zhou Z, Ren Y, ChengJ, Wang W, Wang J, Qian W, Li D, Wang L (2008) A recalibratedmolecular clock and independent origins for the cholera pandemicclones. PLoS ONE 3:e4053
12. Gevers D, Cohan FM, Lawrence JG, Sprat BG, Coeyne T, Feil EJ,Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL,Swings J (2005) Re-evaluating prokaryotic species. Nat RevMicrobiol 3:733–739
13. Hagstrom A, Ferriera S, Johnson J, Kravitz S, Beeson K, SuttonG, Rogers Y-H, Friedman R, Frazier M, Venter JC (2007)NZ_ABGR00000000: Direct submission to GenBank
14. Hallin PF, Binnewies TT, Ussery DW (2008) The genomeBLASTatlas—a GeneWiz extension for visualization of whole-genome homology. Mol Biosyst 4:363–371
15. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML,Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, GillSR, Nelson KE, Read TD, Tettelin H, Richardson D, ErmolaevaMD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonaldL, Utterback T, Fleishmann RD, Nierman WC, White O, SalzbergSL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM(2000) DNA sequence of both chromosomes of the cholerapathogen Vibrio cholerae. Nature 406:477–483
16. Heidelberg J, SebastianY.NZ_AAKJ00000000,NZ_AAUT00000000,NZ_AAKK00000000, NZ_AAUR00000000, NZ_AAWF00000000:Direct submission to GenBank
17. Hjerde E, Lorentzen MS, Holden MT, Seeger K, Paulsen S, BasonN, Churcher C, Harris D, Norbertczak H, Quail MA, Sanders S,Thurston S, Parkhill J, Willassen NP, Thomson NR (2008) Thegenome sequence of the fish pathogen Aliivibrio salmonicida
strain LFI1238 shows extensive evidence of gene decay. BMCGenomics 9:616
18. Konstantinidis T, Ramette A, Tiedje JA (2006) The bacterialspecies definition in the genomic era. Phil Trans R Soc B361:1929–1940
19. Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T,Ussery DW (2007) RNAmmer: consistent and rapid annotation ofribosomal RNA genes. Nucleic Acids Res 35:3100–3108
20. Larsen TS, Krogh A (2003) EasyGene—a prokaryotic gene finderthat ranks ORFs by statistical significance. BMC Bioinformatics4:29
21. Le Roux F, Zouine M, Chakroun N, Binesse J, Saulnier D,Bouchier C, Zidane N, Ma L, Rusniok C, Lajus A, Buchrieser C,Médigue C, Polz MF, Mazel D (2009) Genome sequence of Vibriosplendidus: an abundant planctonic marine species with a largegenotypic diversity. Environ Microbiol 11:1959–1970
22. Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE,Calderwood SB, Fraser C, Mekalanos JJ (1999) Identification ofa Vibrio cholerae RTX toxin gene cluster that is tightly linked tothe cholera toxin prophage. Proc Natl Acad Sci U S A 96:1071–1076
23. Loytynoja A, Goldman N (2005) An algorithm for progressivemultiple alignment of sequences with insertions. Proc Natl AcadSci U S A 102:10557–10562
24. Loytynoja A, Goldman N (2008) Phylogeny-aware gap placementprevents errors in sequence alignment and evolutionary analysis.Science 320:1632–1635
25. Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T,Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A,Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, HattoriM, Iida T (2003) Genome sequence of Vibrio parahaemolyticus: apathogenic mechanism distinct from that of V. cholerae. Lancet361:743–749
26. Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG(2009) A single regulatory gene is sufficient to alter bacterial hostrange. Nature 458:215–218
27. Mazel D, Le Roux F (2008) FM954973.1: Direct submission toGenBank
28. Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R(2005) The microbial pan-genome. Curr Opin Genet Dev15:589–594
29. Medrano-Soto A, Moreno-Hagelsieb G, Vinuesa P, Christen JA,Collado-Vides J (2001) Succesful lateral transfer requires codonusage compatibility between foreign genes and recipient genomes.Mol Biol Evol 21:1884–1894
30. Mohapatra SS, Ramachandran D, Mantri CK, Colwell RR, SinghDV (2009) Determination of relationships among non-toxigenicVibrio cholerae O1 biotype El Tor strains from housekeepinggene sequences and ribotype patterns. Res Microbiol 160:57–62
31. Munk A, Tapia R, Green L, Rogers Y, Detter JC, Bruce D, Brettin TS,Colwell R, Grim C, Vonstein V, Bartels D. CP001485.1,NZ_ACHV00000000, NZ_ACHY00000000, NZ_ACHW00000000,NZ_ACHX00000000, NZ_ACHZ00000000, NZ_ACIA00000000,NZ_ACFQ00000000: Direct submission to GenBank
32. Murray RG, Stackebrandt E (1995) Taxonomic note: implemen-tation of the provisional status Candidatus for incompletelydescribed procaryotes. Int J Syst Bacteriol 45:186–187
33. Nierman WC (2006) NZ_AATY00000000: Direct submission toGenBank
34. Pang B, Yan M, Cui Z, Ye X, Diao B, Ren Y, Gao S, Zhang L,Kan B (2007) Genetic diversity of toxigenic and nontoxigenicVibrio cholerae serogroups O1 and O139 revealed by array-basedcomparative genomic hybridization. J Bacteriol 189:4837–4879
35. Philippe H, Douady CJ (2003) Horizontal gene transfer andphylogenetics. Curr Opin Microbiol 6:498–505
12 T. Vesth et al.

36. Pinhassi J, Pedros-Alio C, Ferriera S, Johnson J, Kravitz S,Halpern A, Remington K, Beeson K, Tran B, Rogers Y-H,Friedman R, Venter JC (2006) NZ_AAND00000000: Directsubmission to GenBank
37. Pupo GM, Lan R, Reeves PR (2000) Multiple independent originsof Shigella clones of Escherichia coli and convergent evolution ofmany of their characteristics. Proc Natl Acad Sci U S A97:10567–10572
38. Rhee JH, Kim SY, Chung SS, Lee SE, Choy HE (2002)AE016795.2: Direct submission to GenBank
39. Riley MA, Lizotte-Waniewski M (2009) Population genomics andthe bacterial species concept. Methods Mol Biol 532:367–377
40. Rowe-Magnus DA, Guérout AM, Mazel D (1999) Super-integrons. Res Microbiol 150:641–651
41. Rosenberg E, Ferriera S, Johnson J, Kravitz S, Beeson K, SuttonG, Rogers Y-H, Friedman R, Frazier M. Venter JC (2006)NZ_ABCH00000000: Direct submission to GenBank
42. 3Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, GunsalusR, Lostroh P, Lupp C, McCann J, Millikan D, Schaefer A, Stabb E,Stevens A, Visick K, Whistler C, Greenberg EP (2005) Completegenome sequence of Vibrio fischeri: a symbiotic bacterium withpathogenic congeners. Proc Natl Acad Sci U S A 102:3004–3009
43. Sánchez J, Holmgren J (2005) Virulence factors, pathogenesis andvaccine protection in cholera and ETEC diarrhoea. Curr OpinImmunol 17:388–398
44. Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA,Kämpfer P, Maiden MC, Nesme X, Rosselló-Mora R, Swings J,Trüper HG, Vauterin L, Ward AC, Whitman WB (2002) Report ofthe ad hoc committee for the re-evaluation of the species definitionin bacteriology. Int J Syst Evol Microbiol 52:1043–1047
45. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: MolecularEvolutionary Genetics Analysis (MEGA) software version 4.0.Mol Biol Evol 24:1596–1599
46. Thompson FL, Iida T, Swings J (2004) Biodiversity of vibrios.Microbiol Mol Biol Rev 68:403–431
47. Urbanczyk H, Ast JC, Higgins MJ, Carson J, Dunlap PV (2007)Reclassification of Vibrio fischeri, Vibrio logei, Vibrio salmoni-cida and Vibrio wodanis as Aliivibrio fischeri gen. nov., comb.nov., Aliivibrio logei comb. nov., Aliivibrio salmonicida comb.nov. and Aliivibrio wodanis comb. nov. Int J Syst Evol Microbiol57:2823–2829
48. Vezzi A, Campanaro S, D'Angelo M, Simonato F, Vitulo N, LauroFM, Cestaro A, Malacrida G, Simionati B, Cannata N, RomualdiC, Bartlett DH, Valle G (2005) Life at depth: Photobacteriumprofundum genome sequence and expression analysis. Science30:1459–1461
49. Wang L, Feng L, Reeves P, Lan R, Ren Y, Gao C, Zhou Z, Ren Y,Wang W (2008) CP001233.1. CP001235.1: Direct submission toGenBank
50. Woese CR (1987) Bacterial evolution. Microbial Rev 51:221–271
Origins of V. cholerae 13





























