Embed Size (px)
Citation preview

Synthetic Metals, 10 (1985) 181 - 191 181
OPTICAL ABSORPTION AND LUMINESCENCE IN POLY(4,4'-DIPHENYLENEDIPHENYLVINYLENE)
W. J. FEAST and I. S. MILLICHAMP
Department of Chemistry, University of Durham, South Road, Durham DH1 3LE (U.K.)
R. H. FRIEND and M. E. HORTON
Cavendish Laboratory, Madingley Road, Cambridge CB3 0HE (U.K.)
D. PHILLIPS, S. D. D. V. RUGHOOPUTH and G. RUMBLES
The Royal Institution, 21 Albermarle Street, London WIX 4BS (U.K.)
(Received August 14, 1984}
Abstract
Poly(4,4'-diphenylenediphenylvinylene), PDPV, is a soluble conjugated polymer that shows a degree of conjugation similar to that in poly-(para- phenylene). The optical properties of thin films exposed to AsF s show the appearance of features below the ~-~r* gap at 3 eV that can be interpreted in a model of dopant-induced polaron and bipolaron defects. When excited above the 7r-~* gap, PDPV shows a strong luminescence peaked at 2.4 eV. The Stokes' shift of 1 eV can be accounted for by radiative decay from a photogenerated polaron-exci ton defect.
1. Introduct ion
The transport properties of conjugated polymers have been much studied since the discovery of the Pennsylvannia group [1] that semiconducting polyacetylene prepared as films by the Shirakawa route [2] could be chem- ically doped to metallic levels of conductivity of up to 1000 (~ cm) -1. Extensive theoretical and experimental work [3, 4] has shown that the carriers introduced on to the polymer chain through the doping reaction are not present as free electrons or holes (as for conventional semiconductors) but are intrinsically trapped on to localized defects produced by a reorganiza- t ion of bond alternation along the chain. The defect that has been postulated for the trans isomer of polyacetylene is a kink, or bond-alternation defect at which the sense of bond alternation is reversed; it possesses one non-bonding Pz orbital, singly occupied (spin 1/2) for the uncharged state [5, 6]. The defect is calculated, in the absence of pinning potentials, to move as a particle along chains, and has been called a soliton defect. It may be charged, either by the addition or removal of an electron, to become non-magnetic. The
0379-6779/85/$3.30 © Elsevier Sequoia/Printed in The Netherlands

182
separation of charge from spin is an unusual property of this defect and there is experimental evidence to suggest that high conductivity can be achieved in a material exhibiting no magnetic behaviour [ 7 ].
Other conjugated polymers that have been studied do not possess the symmetry of trans-polyacetylene and for these systems the bond alternation defect just discussed separates distinct phases on the chain that are not degenerate. These polymers are still dopable and still exhibit very high levels of conductivity; for example poly(paraphenylene) can be doped with AsF s to give a conductivity of 500 (~2 cm) -1 [8]. For this polymer a bond alterna- tion defect converts the lower energy, benzenoid structure into the higher energy, quinonoid structure. However, these defects may still occur in bound pairs, with a short quinonoid structure region between them. If this bound pair is singly charged, it still possesses spin, and can be termed a polaron; if it is doubly charged it becomes spinless, and can be termed a bipolaron. A schematic illustration of the polaron defect is shown in Fig. 1. There have recently been extensive calculations of these polaron and bipolaron defect states [9 - 15]. Two energy levels are pulled out of the valence and conduc- tion bands to give 'bonding' and 'antibonding' polaron levels in the gap. These are shown schematically in Fig. 2 for different charge configurations.
conduction / / '7/ / / / /" / / / / / / / / / / / / / / / / ' / I band
-f-~ spin 1/2 - - ~' ~ spin 0 - - ~' spin 0 i charge *e II '1 charge*2e i charge 0
/ / 1 1 / / / / / ~ 7"/-/-/-/-/75 valence bond
polaron bipoLoron poIoron-exciten
Fig. 1. Schematic illustration of a polaron defect on a polyparaphenylene chain, showing the quinonoid character of the defect.
Fig. 2. Energy level diagram for positively charged polaron and bipolaron defects and the neutral polaron-exciton defect. Possible optical transitions are indicated.
The neutral defect, shown as a singiet in the Figure, has two electrons, one on each level. As discussed in Section 4, its formation is expected following photoexcitat ion and its characteristics can explain photoluminescence in some polymers. The positive polaron and bipolaron levels are also shown. Possible optical transitions between these levels and the band states are in- dicated. Fesser et al. [ 11] have calculated oscillator strengths for such tran- sitions on defects on a polyacetylene chain, and find that all the transitions shown are expected to have large oscillator strengths. The energies of forma- t ion for the polaron and bipolaron defects have been calculated [12, 13], and it is found that a polaron state is expected for a single charge on the polymer chain, but that a bipolaron is energetically preferred to two polarons for a doubly-charged polymer chain.
Experimental work on those polymers that are expected to exhibit polaron and bipolaron defects has been quite limited because most of these polymers are available only as infusible, insoluble powders. This is true of poly(paraphenylene) (PPP), which is usually prepared in a powder form with

183
a relatively low degree of polymerization, with typically no more than 12 phenyl groups in a chain. It has a larger band gap than polyacetylene, of 3 - 3.5 eV, but is still dopable with strong acceptors to give high conductivities [8]. Polypyrrole can be prepared as oxidized thin films at the anode of an electrochemical cell, and the IBM group has worked extensively on these materials [16]. Poly(phenylenevinylene) (PPV) has been prepared in much the same powder form as PPP, and shows similar properties [17, 18]. Two of the present authors have recently reported the synthesis of a related polymer, poly(4,4'-diphenylenediphenylvinylene), (PDPV), the structure of which is shown in Fig. 3 [19]. This polymer is soluble in common solvents such as chloroform, and the feasibility of forming films by evaporation from solu- tion has greatly extended the range of possible experiments on this polymer. Presumably the solubility is related to the presence of the two phenyl groups on the two vinylene carbons. We report in this paper a s tudy of some of the electronic properties of this polymer.
n
Fig. 3. Chemical structure of PDPV, poly(4,4'-diphenylenediphenylvinylene).
2. Polymer synthesis and characterization
PDPV was prepared by a condensation polymerization of 4,4'-dibenzoyl- biphenyl using TiCI3/LiA1H4 as the coupling reagent, as described in ref. 19. The product was purified by reprecipitation twice from chloroform or THF solution into methanol. Films were cast by evaporation from chloroform solution.
Various analyses confirm that the product is a genuine polymer, with a substantial degree of polymerization. Gel permeation chromatography analyses give 'polystyrene equivalent' values of Mn and Mw of typically 36 000 and 74 000. These values will overestimate the molecular weight of this rigid, conjugated polymer, and vapour pressure osmometry estimates for the molecular weight give a lower limit of ~ 4200. This gives, as a minimuM, ~ 13 repeat units in the polymer chain, or 26 phenyl rings plus 13 vinylene units along the conjugated chain. These values are considerably higher than those reported for other phenylene-containing conjugated polymers [20]. Infrared spectrometry showed no trace of either carbonyl or hydroxyl groups, which are the likely polymer chain termini.
The cis-trans distribution of the vinylene units is not unambiguously known at present. From the method of preparation, both cis and trans double bonds would be expected. The 13C n.m.r, spectrum of PDPV is shown in Fig. 4, together with the off-resonance spectrum.

184
:,,~,'~,~',,~,
ir I ] 1 2 3 ~ I i~l!i I
r I
~ V'."*'¢~%(."
I I I I I I
150 1~.5 lz, O 135 130 125
Fig. 4. 13C n.m.r, spectra for PDPV recorded at 90.56 MHz in CDCI3, using a Bruker WH360 spectrometer (SERC/Edinburgh University Facility). Lower trace is the broad band proton-decoupled spectrum. Chemical shifts are in ppm downfield from the internal TMS standard.
Signals 1, 2, 3 and 4 are associated with carbons not bearing hydrogen. Of these, the highest field signal is broader {partially resolved in the off- resonance spectrum), which might possibly be due to vinyl carbons in cis and trans environments. Examination of the repeat unit for the polymer with a trans vinylene unit (Fig. 3) implies that if there is free rotation about the carbon-carbon single bonds there are only five environments for carbons bearing hydrogen. It is clear from the spectrum that there are indeed five main signals (5 - 9) but two of these (7 and 9) are resolved into doublets. This observation cannot be unambiguously interpreted, but is consistent with doubling of environments by restricted rotation and/or the presence of c/s and trans isomeric repeat units. Provisional assignments of the observed signals are shown in Fig. 5.
o
0 O; I I
• d
o = 126 .4 b= 125.8,125.9 c = 131.3 or 131.? d = 131.7 or 131.3 e = 127.6,127.7
Fig. 5. ]3C n.m.r, chemical shift assignments for PDPV.
The assignments are in general agreement with those proposed by Horhold et al. [21] although the spectra available to these workers were very poorly resolved.
The distribution of cis and trans vinylene units is expected to have a marked effect on the crystallinity of the polymer. A structural investigation using electron microscopy is currently in progress.

185
3. Results
D.c. conductivity with AsF s doping The room temperature d.c. conductivity was measured as a function of
AsF s uptake. Free-standing films of thickness 15 pm exposed to AsF s at a pressure of 30 mbar showed a slow uptake and a slow rise in conductivity, with a value of o ~ 5 × 10 -4 (~2 cm) -1 achieved after 24 hours' exposure, and 5% by weight uptake of AsF5. This slow doping rate is consistent with the doping of a fully-dense polymer film, and contrasts with the much more rapid reaction observed with PPP powder. The value of conductivity achieved is still very low, but it is similar to the values reported by Wnek et al. [17] for PPV at similar concentrations of AsF s. At levels of up to 40% by weight, however, they report that the conductivity of compressed pellets reaches a value of I (~2 cm) -1.
Optical absorption with AsFs doping Optical absorption spectra of thin films of PDPV were recorded over
the energy range 0.5 - 5.5 eV, as a function of exposure to AsFs. Figure 6 shows the spectra for an undoped film, and the same film after exposure to 30 mbar of AsF s for 4 hours and again after I month , in the energy range up to 4 eV. The undoped film shows the onset of the band-band absorption edge at 3 eV, with a peak in absorption at 3.5 eV. This gap is very similar to that observed in PPP [22], and is presumed to be the ~r-lr* gap, as discussed later. Absorption spectra from solutions of PDPV in chloroform axe similar to that for the undoped film in Fig. 6. The extinction coefficient measured
PDPV I pristine 2 exposed to AsR 3 for 4hours
3 exposed to AsF 5 for t month
I :
Fig. 6. Optical absorption spectra of PDPV before and after exposure to AsFs.

186
PDPV ~ ' ~ | pristine / 2 exposed to.AsF 5 followed /
J c
Fig. 7. Optical absorption spectra of a PDPV film before doping and after prolonged exposure to AsF s followed by exposure to moist air.
at the absorption peak at 3.5 eV is 1.6 X 104 mole -] cm - ] , (using the molec- ular weight of the PDPV repeat unit).
On exposure to AsFs there is a general increase in absorption below this gap, with structure apparent. For curve 2, measured after 4 hours ' exposure to AsF s, a well-resolved peak at 0.8 eV is observed, together with a broad band extending from 1.7 eV to 2.5 eV. There is little change in the absorption above the gap. For curve 3, measured after 1 month ' s exposure, the absorp- t ion features below the gap are stronger, and the feature at higher energy is sharper, now showing an edge at 1.7 eV and a well-defined peak at 2.3 eV. In addition, there has been some significant alteration to the u.v. spectrum, and the band-edge is now less well defined.
After exposing this doped film to moist air, the features below 3 eV dis- appeared, indicating a reversible doping reaction. However, the changes in the u.v. spectra observed after 1 month 's exposure to AsFs remained. Figure 7 shows this spectrum up to 5.5 eV, and also the spectrum measured for the pristine film. The u.v. spectra are very different, and indicate that some im- por tan t irreversible reactions have taken place.
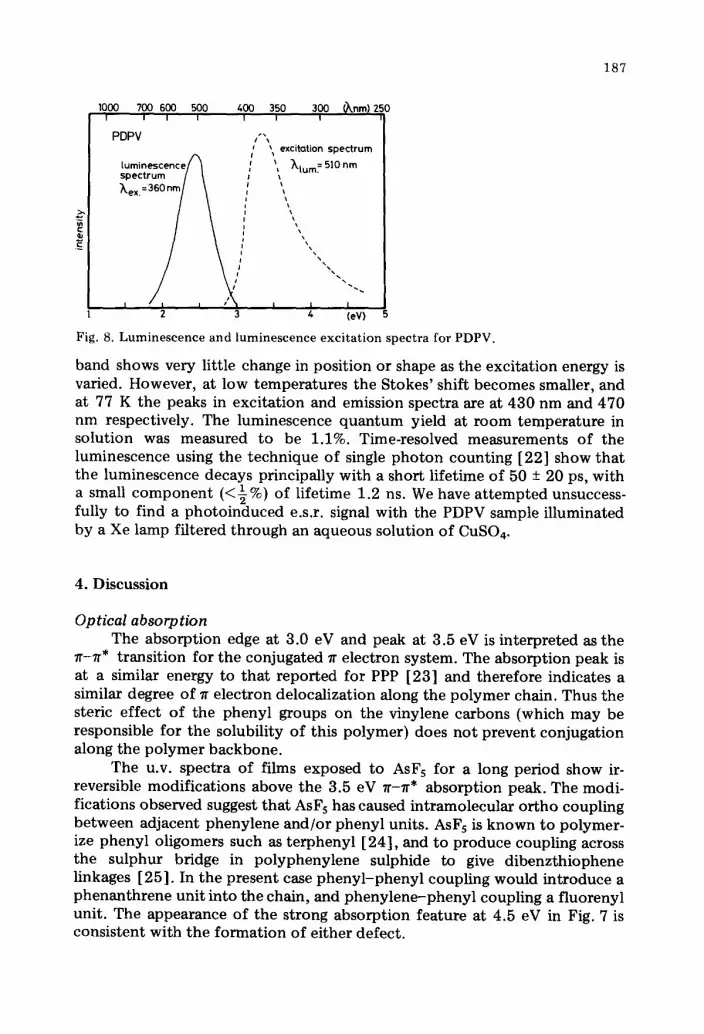
Luminescence PDPV both as a film and in solution shows a strong green fluorescence
when illuminated with u.v. radiation. The fluorescence and excitat ion spectra were measured using a Xe lamp light source, and results at room temperature are shown in Fig. 8. The spectra for the films and solutions are similar. The luminescence appears in a broad band (of width ~ 0.75 eV), with a peak at 2.4 eV. The intensity of this luminescence as a funct ion of the excitation energy shows an onset at 3 eV and a peak at 3.3 eV. This matches very closely the optical absorption spectrum shown in Figs. 6 and 7. The luminescence
187
1000 ?00 600 500 | I I I I I I
PDPV ~" "~ i ~ excitation spec t rum
'
luminescenc ~ ~ "~kium = 51Ohm spec t rum / ~ , '~
Xex.=360.m/ \ : ',,
I I I I Z 3 4 (eV)
400 350 300 Qknm) 250
Fig. 8. Luminescence and luminescence excitation spectra for PDPV.
band shows very little change in position or shape as the excitation energy is varied. However, at low temperatures the Stokes' shift becomes smaller, and at 77 K the peaks in excitation and emission spectra are at 430 nm and 470 nm respectively. The luminescence quantum yield at room temperature in solution was measured to be 1.1%. Time-resolved measurements of the luminescence using the technique of single photon counting [22] show that the luminescence decays principally with a short lifetime of 50 + 20 ps, with a small component (<½%) of lifetime 1.2 ns. We have a t tempted unsuccess- fully to find a photoinduced e.s.r, signal with the PDPV sample illuminated by a Xe lamp filtered through an aqueous solution of CuSO4.
4. Discussion
Optical absorption The absorption edge at 3.0 eV and peak at 3.5 eV is interpreted as the
7r-Tr* transition for the conjugated ~r electron system. The absorption peak is at a similar energy to that reported for PPP [23] and therefore indicates a similar degree of Ir electron delocalization along the polymer chain. Thus the steric effect of the phenyl groups on the vinylene carbons (which may be responsible for the solubility of this polymer) does not prevent conjugation along the polymer backbone.
The u.v. spectra of films exposed to AsFs for a long period show ir- reversible modifications above the 3.5 eV 7r-lr* absorption peak. The modi- fications observed suggest that AsFs has caused intramolecular or tho coupling between adjacent phenylene and/or phenyl units. AsFs is known to polymer- ize phenyl oligomers such as terphenyl [ 24], and to produce coupling across the sulphur bridge in polyphenylene sulphide to give dibenzthiophene linkages [ 25]. In the present case phenyl -phenyl coupling would introduce a phenanthrene unit into the chain, and phenylene-phenyl coupling a fluorenyl unit. The appearance of the strong absorption feature at 4.5 eV in Fig. 7 is consistent with the formation of either defect.

188
The electronic properties of doped PPP and related polymers such as PDPV can be accounted for by the creation of polaron and bipolaron defects. It has been known for some time that although PPP and polypyrrole can be prepared in highly conducting forms, they exhibit no Pauli paramagnetic susceptibility [26], and in the case of polypyrrole, an e.s.r, line which arises from spins that are not involved in the transport process [27]. The presence of non-magnetic bipolarons accounts for these observations.
The appearance of absorption features below the r-Tr* gap is consistent with optical transitions between band states and the bonding and antibonding polaron/bipolaron levels in the gap. Optical absorption measurements on highly oxidized samples of PPP and polypyrrole have shown strong absorption bands at ~ 1 eV, well below the 7r-u* gap [8, 16]. This is also evident in in- elastic electron scattering experiments on AsFs-doped PPP [ 28]. More specific information about the positions of polaron and bipolaron levels is obtained with lightly-doped polymer, and a detailed study of lightly-oxidized poly- pyrrole has recently been reported by the IBM group [29].
The features observed in lightly<loped PDPV, shown in Fig. 6, are also well explained by the polaron/bipolaron defect model. The three optical transitions involving a polaron defect, calculated to have a large oscillator strength [11] are those between the valence (conduction) band and each of the two polaron levels, and between the two levels for a positive (negative) polaron. For the bipolaron, the occupation of levels is such that only the two band to defect-level transitions are possible, as indicated in Fig. 2. The bipolaron defect is expected at high dopant concentrations, and the spectrum for the more heavily doped sample in Fig. 6 shows peaks at 0.8 and 2.3 eV. These can be associated with the valence band to lower bipolaron level and valence band to upper bipolaron level transitions, with the two levels 0.8 eV away from the band edges. The more lightly<loped sample shows the 0.8 eV absorption and a broader absorption feature from 1.7 to 2.5 eV. This may arise from a band to upper level transition at 2.5 eV and the lower level to upper level transition at 1.7 eV for a polaron defect, which should be present at lower dopant concentrations.
Luminescence The results shown in Fig. 8 indicate that the strong luminescence centred
at 2.4 eV has an excitation spectrum that coincides with the lr-lr* absorption band, extending from 3 to 4 eV and peaking at 3.3 eV. This indicates that the luminescence arises from the conjugated ~ electron system along the polymer chain. A very important feature of the luminescence is the large Stokes' shift of the emitted photons, with a difference of 0.9 eV between the peak in the excitation spectrum and the peak in the luminescence spectrum. This loss of energy requires that the optically excited states can lose energy non-radiatively to the lattice before the radiative decay. The polaron defect model that we have used to account for the dopant-induced absorption features in the ~-~* gap of PDPV can also provide a natural ex- planation for the Stokes' shift, as discussed below.

189
The quantum yield of 1.1% indicactes that the principal recombination mechanism is a non-radiative process. This is evident also in the short lifetime measured for the luminescence decay, of 50 + 20 ps. The expected radiative lifetime, rR, can be estimated from the extinction coefficient, e at the absorp- tion peak at 3.5 eV through the approximate relation ra -1 = 104 e (with rR in s, e in mole- ' cm- 1). This gives a value of 6.4 ns. If the decay is dominated by a faster, non-radiative process giving a decay time r, the quantum yield 7? is simply related to r and ra by r = ~rR. ~?rR takes the value 70 ps, in agreement with the measured value of 50 -+ 20 ps. Non-radiative relaxation in unsaturated systems is known to be very rapid. In simple olefins, the excited single and ground states are connected by a twisting motion around the carbon-carbon bond that gives a non-bonding configuration for the two Pz orbitals, and allows depopulation of the excited state by a non-radiative route [30, 31]. Similar twisting movements are clearly possible within the repeat unit of PDPV.
The evolution with time of the photoexcited 7r electron system on a conjugated polymer has been modelled by Su and Schrieffer [32] for the case of trans-polyacetylene. They find that within a period of the order of the inverse optic phonon frequency (~ 10 -13 s) the excited electron and hole relax to form two soliton states which lie at mid-gap. For the other conju- gated polymers, in which soliton defects are not favoured, it is expected that the soliton states remain bound, as a polaron-exciton [9]. For this bound defect, rapid radiative recombination is expected. The contrast between trans-polyacetylene which is a photoconductor , but exhibits no luminescence and cis-polyacetylene, which shows no photoconductivi ty but some lumi- nescence is in broad agreement with this model [33]. However, the lumi- nescence in cis-polyacetylene is extremely rapid, with a lifetime of less than 10 ps [34, 35], has a very low quantum yield (~10 -s) and is Stokes' shifted by only 0.15 eV. This suggests that there is an alternative very rapid non- radiative decay channel [34], and that the polaron-exciton is not fully relaxed before recombination.
For PDPV the fully relaxed polaron-exciton defect is indicated schemat- ically in Fig. 2. The two polaron levels are singly occupied so that the defect remains neutral. The defect is indicated as being in the spin-singlet configura- tion, consistent with the absence of a photo-induced e.s.r, signal. Radiative decay of this defect involves the transition of the electron in the upper polaron levels to the lower level, as indicated in the Figure. This is followed by relaxation of the lattice to its undistorted configuration, so that the doubly-occupied lower level falls back to the valence band, and the un- occupied upper level is reabsorbed into the conduction band. The Stokes' shift of 0.9 eV measured thus places the two polaron levels about 0.45 eV away from the band edges. This is smaller than the 0.8 eV separation inferred earlier for the positively charged polaron and bipolaron defects, and indicates that the degree of bond reorganization from benzenoid to quinonoid charac- ter is weaker for the neutral defect. This trend is in agreement with the calculations of Bredas et al. for polaron and bipolaron defects on poly(para- phenylene) [13]. The reduction in Stokes' shift measured at 77 K must

190
indicate that the polymer chain has not been able to fully relax to give the polaron-exciton defect before recombination of the electron-hole pair.
Since the polaron and bipolaron defects on polyparaphenylene are calculated to extend over only some four or five phenylene rings [13, 14], this polaron-exciton defect should also be formed in phenylene oligomers under photoexcitat ion. These materials luminescence with a high quantum efficiency and are known to show a large Stokes' shift [36]. For example, paraterphenyl shows a peak in emission at 3.0 eV for both photoexcitat ion [36] and electron beam excitation [37], but the lowest-energy peak in optical absorption is at 4.5 eV [24]. The shorter chain oligomers show con- siderable structure in the emission spectrum with well-resolved peaks sepa- rated by typically 0.1 to 0.15 eV, and total emission widths of ~ 1/2 eV. These peaks arise from transitions to the excited vibrational states of the electronic ground state. For longer chain oligomers the structure is less clearly resolved, and we can expect for a polymeric material such as PDPV, which is at most only partially crystalline, that the structure is no longer resolved. However, the width of the luminescence emission spectrum for PDPV is comparable to those observed in these oligomers.
5. Conclusion
PDPV is a soluble conjugated polymer that shows properties charac- teristic of simpler phenylene chain polymers such as poly(paraphenylene). In view of its processibility, it may be considered as an important example of this type of conjugated polymer, in which polaron and bipolaron defects have been proposed to explain the transport and optical properties. Mea- surements of optical absorption on AsFs~Joped films show the appearance of absorption features below the 7r-Tr* gap that are consistent with a polaron or bipolaron defect model. Similarly the Stokes' shifted luminescence at 2.4 eV can be explained by radiative decay from a polaron-exciton excited state.
Further studies of structure, electrical, magnetic and optical properties are in progress.
Acknowledgements
We thanks Drs. D. C. Bott, C. K. Chai, A. D. Yoffe and Prof. D. Bloor for manyhelpful discussions, Dr. I. Sadler for recording the C ~3 n.m.r, spec- tra, and Dr. S. R. Elliot for providing facilities for the photoinduced e.s.r. experiments.
References
1 c. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 39 (1977) 1098.

191
2 T. Ito, H. Shirakawa and S. Ikeda, J. Poly. Sci. Polym. Chem. Ed., 12 (1974) 11. 3 Proceedings o f the Int. Conf. on Low-Dimensional Conductors, Boulder, Colorado,
U.S.A., Aug. 1981, Mol. Cryst. Liq. Cryst., 77 and 83 (1982). 4 Proceedings o f the Int. Conf. on Conducting Polymers, Les Arcs, France, Dec. 1982,
J. Phys. (Paris) Colloq., 44 (1983) C3. 5 M.J. Rice, Phys. Lett. , 71A (1979) 152. 6 W. P. Su, J. R. Schrieffer and A. J. Heeger, Phys. Rev. Lett., 42 (1979) 1698;Phys.
Rev. B, 22 (1980) 2099. 7 S. Ikehata, J. Kaufer, T. Woerner, A, Pron, M. A. Druy, A. Sivak, A. J. Heeger and A.
G. MacDiarmid, Phys. Rev. Lett. , 45 (1980) 1123. 8 L. Shacklette, R. R. Chance, D. M. Ivory, G. G. Miller and R. H. Baughman, Synth.
Met., 1 (1980)307. 9 S. A. Brazovskii and N. N. Kirova, Pis'ma Zh. Eksp. Teor. Fiz., 33 (1981) 4 [Sov.
Phys. JETP Lett. , 33 (1980) 4. 10 D. K. Campbell and A. R. Bishop, Phys. Rev. B, 24 (1981) 4859. 11 K. Fesser, A. R. Bishop and D. K. Campbell, Phys. Rev. B, 27 (1983)4804. 12 J. P. Albert and C. Jouanin, Phys. Rev. B, 26 (1982) 955. 13 J. L. Bredas, R. R. Chance and R. Silbey, Phys. Rev. B, 26 (1982) 5843. 14 J. L. Bredas, B. Themans and J. M. Andre, Phys. Rev. B, 26 (1982) 6000. 15 J. L. Bredas, B. Themans and J. M. Andre, Phys. Rev. B, 27 (1983) 7827. 16 K. K. Kanazawa, A. F. Diaz, W. D. Gill, P. M. Grant, G. B. Street, G. P. Gardine and
J. F. Kwak, Synth. Met., 1 (1980) 329. 17 G. E. Wnek, J. C. W. Chien, F. E. Karasz and C. P. Lillya, Polymer, 20 (1979) 1441. 18 J. R. Reynolds, F. E. Karasz, J. C. W. Chien, K. D. Gourley and C. P. Lillya, J. Phys.
(Paris) Colloq., 44 (1983) C3 - 693. 19 W.J. Feast and I. S. Millichamp, Polymer Commun., 24 (1983) 102. 20 We note, however, that there is a recent report of polyparaphenylene prepared with a
high degree of polymerization. D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, J. Chem. Soc., Chem. Commun., (1983) 954.
21 B. Schroter, H. H. Horhold and D. Raabe, Mahromol., 182 (1981} 3185. 22 R. A. Lampert, L. A. Chewter, D. Phillips, D. V. O'Connor, A. J. Roberts and S. R.
Meech, Anal. Chem., 55 (1983) 68. 23 B. Tieke, Ch. Bubeck and G. Lieser, J. Phys. (Paris) Colloq. 44 (1983) C3 - 753;
G. Froyer, J. Y. Goblot, J. L. Guilbert, F. Maurice and Y. Pelous, J. Phys. (Paris) Colloq., 44 (1983) C3 - 745.
24 L. W. Shacklette, H. Eckhardt, R. R. Chance, G. G. Miller, D. M. Ivory and R. H. Baughman, J. Chem. Phys., 73 (1980) 4098.
25 R. L. Elsenbaumer, L. W. Shacklette, J. W. Sourn and R. H. Baughman, Mol. Cryst. Liq. Cryst., 83 (1982) 229.
26 M. Peo, S. Roth, K. Dransfeld, B. Tieke, J. Hocker, H. Gross, A. Grupp and H. Sixl, Solid State Commun., 35 (1980) 119.
27 J. C. Scott, P. Pfluger, M. T. Krounbe and G. B. Street, Phys. Rev. B, 28 (1983) 2140. 28 G. Crecelius, M. Stamm, J. Fink and J. J. Ritsko, Phys. Rev. Lett., 50 (1983) 1498. 29 K. Yakushi, L. J. Lauchlan, T. C. Clarke and G. B. Street, J. Chem. Phys., 79 (1983)
4774. 30 R. M. Hochstrasser, Pure Appl. Chem., 52 (1980) 2683. 31 S. D. D. V. Rughooputh, D. Phillips, D. Bloor and D. J. Ando, Chem. Phys. Lett. ,
106 (1984) 247. 32 W. P. Su and J. R. Schrieffer, Proc. Natl. Aead. Sci. U.S.A. 77 (1980) 5626. 33 L. Lauchlan, S. Etemad, T. C. Chung, A. J. Heeger and A. G. MacDiarmid, Phys. Rev.
B, 24 (1981) 3701. 34 W. Hayes, C. N. Ironside, J. F. Ryan, R. P. Steele and R. A. Taylor, J. Phys. C, 16
(1983) L729. 35 J. R. Andrews, T. E. Orlowski, H. Gibson, M. L. Slade, W. Knox and B. Wittsmerhaus,
Phys. Rev. B, 27 (1983) 6545. 36 R. C. Sangster and J. W. Irvine, J. Chem. Phys., 24 (1956) 670. 37 M. De Mets, K. J. Howlett and A. D. Yoffe, J. Microsc., 102 (1974) 125.

![Synthesis and characterization of novel polyurethanes based on 4,4′-[1,4-phenylenedi-diazene-2,1-diyl]bis(2-carboxyphenol) and 4,4′-[1,4-phenylenedi-diazene-2,1-diyl]bis(2-chlorophenol)](https://img.pdfslide.net/doc/110x75/635f4751bf72c5cede076411/synthesis-and-characterization-of-novel-polyurethanes-based-on-44-14-phenylenedi-diazene-21-diylbis2-carboxyphenol.jpg)


![Bis{μ-4,4′-dimethoxy-2,2′-[propane-1,2-diylbis(nitrilomethylidyne)]diphenolato}bis({4,4′-dimethoxy-2,2′-[propane-1,2-diylbis(nitrilomethylidyne)]diphenol}manganese(III)) bis(hexafluoridophosphate](https://img.pdfslide.net/doc/110x75/6320013e069357aa4506000b/bism-44-dimethoxy-22-propane-12-diylbisnitrilomethylidynediphenolatobis44-dimethoxy-22-propane-12-diylbisnitrilomethylidynediphenolmanganeseiii.jpg)



















![Poly[[diaquabis(μ 2 -4,4′-bipyridyl)cobalt(II)] dinitrate tetrahydrate]](https://img.pdfslide.net/doc/110x75/63559cabb4909beae3007236/polydiaquabism-2-44-bipyridylcobaltii-dinitrate-tetrahydrate.jpg)




