Embed Size (px)
Citation preview

Molecular Microbiology (2006)
59
(3), 779–794 doi:10.1111/j.1365-2958.2005.04983.xFirst published online 30 November 2005
© 2005 Blackwell Publishing LtdNo claim to original Australian government works
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382X© 2005 Blackwell Publishing Ltd; No claim to original Australian government works
? 2005
59
3779794
Original Article
Proteins at the malaria host–parasite interfaceT.
Spielmann
et al.
Accepted 31 October, 2005. *For correspondence. [email protected]; Tel. (
+
61) 73 3620 420; Fax (
+
61)73 3620 104.
Organization of ETRAMPs and EXP-1 at the parasite–host cell interface of malaria parasites
Tobias Spielmann,
1
* Donald L. Gardiner,
1
Hans-Peter Beck,
2
Katharine R. Trenholme
1
and David J. Kemp
1
1
Infectious Diseases and Immunology Division, Queensland Institute of Medical Research, PO Royal Brisbane Hospital, QLD 4029, Australia.
2
Department of Medical Parasitology and Infection Biology, Swiss Tropical Institute, Basel CH 4002, Switzerland.
Summary
The parasite–host cell interface is a key compartmentof vacuolated intracellular pathogens but little isknown about its molecular composition and architec-ture. We used
in vivo
cross-linking to analyse theparasite–host cell interface of asexual stages of themost virulent human malaria parasite
Plasmodiumfalciparum
. We show that the integral membrane pro-tein members of the early transcribed membrane pro-tein (ETRAMP) family and exported protein 1 (EXP-1),which are components of the parasite–host cell inter-face, form complexes of oligomeric arrays in thiscompartment. The most notable feature is that eachETRAMP member and EXP-1 define separate arrays,demonstrating that the protein distribution in thismembrane is non-random. Each of three recombinantETRAMPs readily oligomerized in bacterial mem-branes, confirming that these arrays can form inde-pendently of other
Plasmodium
proteins. We proposethat the malaria parasite–host cell interface containspatches of integral membrane proteins forming amosaic of different microdomains in this membrane.
Introduction
Malaria, caused by infection with the apicomplexan para-sites of the genus
Plasmodium
, remains a global healthproblem. Many intracellular pathogens, including
Plasmo-dium falciparum
, the most virulent of the malaria para-sites, develop inside a vacuole that separates them fromthe host cell cytosol (Moulder, 1985; Garcia-del Portillo
and Finlay, 1995). The only mode of contact with the hostcell is via the membrane of this vacuole, termed the par-asitophorous vacuole membrane (PVM) in apicomplexanparasites. This structure is formed during invasion oruptake of the pathogen into the host cell and as theinterface between parasite and host cell, plays a key rolein survival and propagation of intracellular pathogens. Atthe same time it has to be both a barrier and a gate thatallows passage of nutrients, waste products and signals.
While most intracellular pathogens face an active hostcell, imposing on the PVM the additional task of avoidinglysosomal fusion or acidification of the vacuole (Méresse
et al
., 1999; Hackstadt, 2000), asexual stages of humanmalaria parasites develop inside red blood cells (RBCs),which are devoid of organelles or a protein traffickingsystem. Malaria parasites are therefore forced to refurbishtheir host cell, requiring protein export and induction ofmembranous structures in the RBC (Cooke
et al
., 2001).All these modifications have to be established beyond theparasite’s cellular boundaries and across the PVM.
Despite the importance of the vacuolar membrane formany intracellular parasites, little is known about its archi-tecture, its molecular components and how they mediatethe functions of this compartment. In
Plasmodium
a non-specific solute pore has been described in the PVM (Desai
et al
., 1993). Protein export across the PVM requires ATP(Ansorge
et al
., 1996) and is mediated by a specific signalpresent in a large proportion of exported proteins (Hiller
et al
., 2004; Marti
et al
., 2004). Certain areas of the PVMinvaginate together with the parasite plasma membrane,forming a cytostome to engulf haemoglobin of the host(Slomianny
et al
., 1985). Other areas extend into the hostcell cytoplasm to become a tubovesicular network (TVN)(Elmendorf and Haldar, 1993). To date the moleculesinvolved in these processes are unknown and it is unclearhow different regions in the PVM differentiate into struc-tures as diverse as the TVN or the cytostome.
To date exported protein 1 (EXP-1) is the only welldocumented integral PVM protein in malaria parasites(Simmons
et al
., 1987; Kara
et al
., 1988) but its functionremains unknown. We recently described a new genefamily coding for highly charged integral PVM proteinstermed early transcribed membrane proteins (ETRAMPs)(Spielmann and Beck, 2000; Spielmann
et al
., 2003)found in all
Plasmodium
species for which sequence dataare available (Carlton
et al
., 2002; Birago
et al
., 2003;

780
T. Spielmann
et al.
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
Spielmann
et al
., 2003; our unpublished data). ETRAMPsare small single pass transmembrane proteins orientedsuch that the C-terminus, the most divergent region of themolecule, faces the RBC cytosol (Spielmann
et al
., 2003).Remarkably for integral membrane proteins, some
etramps
are among the 10 most highly transcribed genesin asexual stages of
P. falciparum
(Le Roch
et al
., 2003),a fact that is also reflected
in vivo
(Daily
et al
., 2004).ETRAMPs are developmentally regulated with six of the13 family members found exclusively in early stage para-sites (ring stage). These are replaced by a second set ofETRAMPs, concomitant with transition of parasites to thetrophozoite stage. Transcripts of another three
etramp
sare not detectable in asexual blood stages (Spielmann
et al
., 2003). One of these (ETRAMP13) has an ortho-logue in rodent malarias that is induced in salivary glandsporozoites and is essential for liver stage development(Kaiser
et al
., 2004; Mueller
et al
., 2005a). Hence differentETRAMPs are required in different development stages.
It has become increasingly clear that not only are sol-uble proteins often organized into higher order structures(Alberts, 1998; Gavin
et al
., 2002), but also that the spatialorganization of many membrane proteins is highly sophis-ticated. This is most evident in membranes at interfacesinvolved in exchanging signals with the environment orother cells. Examples of this are oligomeric arrays of bac-terial chemotaxis receptors (Maddock and Shapiro, 1993),the clustering of ion channels and receptors in synapses(Froehner, 1993), the organization of proteins in the immu-nological synapse in T cells (Delon and Germain, 2000)and B cell receptor complexes (Matsuuchi and Gold,2001).
Here we report the use of
in vivo
cross-linking to anal-yse the molecular architecture of the malaria PVM. Weshow that the interface between
P. falciparum
parasitesand their host cell contains ETRAMPs and EXP-1 orga-nized into oligomeric arrays. Furthermore we provide evi-dence that different ETRAMPs are present in separatearrays, leading to a mosaic distribution of clustered inte-gral PVM proteins. We also show by expression in
Escher-ichia coli
that ETRAMPs can form oligomers independentof other
Plasmodium
proteins or lipid rafts, indicating thatETRAMPs alone are sufficient to create different micro-domains in a biological membrane.
Results
Evidence for ETRAMP protein complexes by cross-linking of whole infected RBCs
In order to analyse the organization of ETRAMPs in thePVM, we developed an
in vivo
cross-linking procedure for
Plasmodium
-infected RBCs that enables the situation inlive parasites to be frozen and captured. This protocol isbased on formaldehyde, a widely used protein–protein or
DNA–protein cross-linking agent that penetrates intactcells and cross-links amino groups that lie within 2 Å ofone another (reviewed in Orlando
et al
., 1997). Cross-linked intact infected RBCs (IRBCs) were then quenched,hypotonically lysed and extracted proteins analysed byWestern blotting. This avoids detection of possible non-specific protein aggregation, which may occur after lysis.
We had previously raised two antisera, one specific forthe ring stage-specific ETRAMP2, the second specific forthe constitutively expressed ETRAMP4 (Spielmann
et al
.,2003). In Western blots of non-cross-linked parasiteextracts, the ETRAMP4 serum detected a single band of17 kDa and the ETRAMP2 serum detected a band of13 kDa (Spielmann
et al
., 2003; Fig. 1A and B). However,in Western blots of 3D7 or D10 parasites cross-linked asdescribed above, the ETRAMP4 serum reacted with addi-tional bands that migrated above the ETRAMP4 mono-mer, resulting in a ladder of bands (Fig. 2A). This suggeststhat ETRAMP4 is part of a protein complex. In contrast,after cross-linking the ETRAMP2 band was no longerdetected in Western blots with the ETRAMP2 serumand no additional bands were apparent (Fig. 2A). Thisindicates that cross-linking masked the epitopes inETRAMP2, possibly by reacting with lysine residues in theepitopes or by covalently linking an interacting protein thatthen covers the epitopes.
Generation of parasites expressing C-terminally tagged ETRAMPs
To overcome the problem of masking and to verify ourinitial results for ETRAMP4 with an antibody of indepen-dent specificity, we generated transgenic
P. falciparum
(D10) parasites stably expressing either ETRAMP2 orETRAMP4 with a C-terminal myc tag (parasitesD10E2myc and D10E4myc). Both transfected culturesexpressed the transgene from an episomal plasmid underthe constitutive
hsp
86 promoter. Western blots showedthat both the tagged and the endogenous ETRAMPs wereexpressed in the transgenic parasites (Fig. 1A, B, E andF). Immunofluorescence analysis with an anti-myc serumshowed a circular staining pattern in acetone-fixedD10E2myc and D10E4myc parasites (Fig. 1C and D),which is typical of the ETRAMP staining pattern seen withanti-ETRAMP serum in untransfected parasites. This sug-gests that the tagged ETRAMPs were faithfully exported.This was further supported by their colocalization inimmunofluorescence assays (IFAs) with the respectiveendogenous ETRAMPs (Fig. 1C and D). Although thesera specific for the endogenous ETRAMPs recognizesboth the tagged and the untagged ETRAMPs, a distinctlocalization of the tagged ETRAMP would be expected tohave resulted in areas stained with the specific ETRAMPsera but not the anti-myc serum.

Proteins at the malaria host–parasite interface
781
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
In accordance with the activity of the
hsp86
promoter,ETRAMP4myc was constitutively expressed in asexualblood stages (Fig. 1E), similar to previously observedendogenous ETRAMP4 expression (Spielmann
et al
.,2003). Although under the same promoter (and in theidentical vector differing only in the ETRAMP codingsequence), ETRAMP2myc was absent in late-stage par-asites in IFA and Western experiments (Fig. 1F and datanot shown), matching expression of the genomic copy ofETRAMP2. These results might indicate that ringETRAMPs not only are regulated at the transcriptionallevel, but are also actively removed in a stage-specificmanner. To exclude possible alterations in the transfectedparasites or a strain-specific effect, 3D7 parasitesexpressing ETRAMP2myc and ETRAMP4myc were gen-erated to obtain parasites 3D7E2myc and 3D7E4myc.Like D10E2myc, ETRAMP2myc was again absent from
late stages in 3D7E2myc parasites as judged from IFAand Western experiments (data not shown).
In vivo
cross-linking of parasites expressing C-terminally tagged ETRAMPs confirms that ETRAMPs are part of protein complexes
To corroborate our initial cross-linking results obtainedwith ETRAMP4 and ETRAMP2, the
in vivo
cross-linkingexperiments were repeated using the transgenic para-sites. When parasites D10E2myc and D10E4myc werecross-linked and analysed in immunoblots, the anti-mycsera reacted with a ladder of bands for both transgenicETRAMPs (Fig. 2A). The band sizes of the ladders dif-fered between E2myc and E4myc. No ladder wasdetected with the anti-myc serum in Western blots usinguntransfected parasites. The sizes of the ETRAMP4myc
C
A
GAPDH
E2myc
α-ETRAMP2α-myc α-ETRAMP4α-myc
D10 E2myc
D10 E2myc
D10 E4myc
D10 E4myc
E4myc
GAPDH
α-ETRAMP2 α-myc merge + nuclei
E
0-10
10-2
016
-2624
-3434
-44
2-12
12-2
218
-2826
-3636
-2
F
B
Dα-ETRAMP4 α-myc merge + nuclei
hours post invasion hours post invasion
Fig. 1.
Transgenic expression of myc-tagged ETRAMP2 and ETRAMP4.A and B. The transgenic parasites express the tagged ETRAMP as demonstrated by Western analysis using specific sera to ETRAMP2 in D10E2myc parasites (A) or sera specific for ETRAMP4 in D10E4myc parasites (B). The tagged ETRAMPs are visible as an additional band of slightly larger size (open arrow) above the endogenous ETRAMP (solid arrow). This band is absent in untransfected D10 parasites.C and D. Immunofluorescence analysis in ace-tone-fixed IRBCs: the tagged transgenes detected via the myc tag colocalize with the respective chromosomally derived ETRAMPs in a typical peripheral staining in the transfected parasites D10E2myc (C) and D10E4myc (D). C shows two IRBCs, each with a double infection of ring-stage parasites and D shows a double infection of trophozoites. Hoechst was used to stain the nuclei. No signal was seen with anti-myc sera in untransfected parasites (not shown).E. Western blot with stage-specific D10E2myc parasite extracts showing ETRAMP2myc (E2myc) expression in early, mid and late ring stages only (lanes 1–3; hours post invasion are indicated above each lane) but not in trophozo-ites and schizonts (lanes 4 and 5).F. Western blot with stage-specific D10E4myc parasite extracts showing constitutive expres-sion of the transgene in accordance with the
hsp86
promoter used. Tagged ETRAMPs were detected with an anti-myc antibody.Filters shown in E and F were reprobed using PfGAPDH to demonstrate equal loading.

782
T. Spielmann
et al.
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
bands were slightly larger than those observed withETRAMP4. This is consistent with an increased molecularweight due to the myc tag. Similar results were obtainedwith 3D7E2myc and 3D7E4myc (data not shown). Henceboth ETRAMPs appear to be part of a protein complexpresent in live parasites.
The formaldehyde-induced cross-linking could bereversed by boiling extracts in SDS-PAGE sample buffer.This converted the ladder of bands back to the monomericETRAMP band normally seen in Western blots with non-cross-linked extracts. Reversal of the cross-linking alsoled to reappearance of the ETRAMP2 band in untrans-fected parasites when probed with the specific ETRAMP2serum (Fig. 2A).
To verify these results, we also used two alternativecross-linkers [Dimethyl 3,3
′
-dithiobispropionimidate·2HCl(DTBP) and Bis(sulphosuccinimidyl)suberate (BS
3
)] oncrude membrane preparations of IRBCs. These cross-linkers have a different reaction chemistry and spacerlength to formaldehyde. No difference in the Western blotbanding pattern of ETRAMP2myc was observed withthese alternative cross-linkers in D10E2myc parasiteswhen compared with extracts derived from formaldehydecross-linked parasites (data not shown). For ETRAMP4and ETRAMP4myc, each cross-linker caused a slightmobility shift in all bands (data not shown and Fig. 3). Thismobility shift was minimal and also seen with the mono-meric band. This indicates that it was most likely due tocross-linker-induced changes in charge or conformationof the ETRAMP rather than cross-linking of different com-plex components.
EXP-1 is also part of a protein complex
To date the only well documented integral PVM protein isEXP-1. Like ETRAMPs, EXP-1 is a highly charged, singlepass membrane protein with a classical signal peptide. Itshows no more than 22% amino acid identity to anyETRAMP (data not shown). Hence, while EXP-1 andETRAMPs seem to be structurally related, they share littleor no significant sequence similarity. The structural simi-larity might indicate that EXP-1 has similar properties toETRAMPs. We therefore tested whether EXP-1 is alsopart of a protein complex.
Western analysis of
in vivo
cross-linked parasiteextracts probed with specific EXP-1 antisera also showeda ladder of bands above the EXP-1 monomer (Fig. 2A).This indicates that EXP-1 is also part of a protein complexpresent in live parasites, analogous to what is seen withETRAMPs. Similar results were obtained using the alter-native cross-linkers DTBP and BS
3
except for a singleband that showed a different mobility when cross-linkedwith BS
3
(data not shown and Fig. 3).
Different ETRAMPs and EXP-1 are not part of the same protein complex
The clear bands representing the dimer (first band abovethe monomer) and higher order oligomers in Westernblots with our cross-linked extracts indicated that eachstep in the ladder was represented by the specific inter-action of the corresponding ETRAMP with a single spe-cies of protein only. If there had been non-specific
Fig. 2.
ETRAMPs and EXP-1 are part of protein complexes.A.
In vivo
formaldehyde cross-linking reveals ETRAMP and EXP-1 complexes that can be reversed by boiling. Western blots of extracts of
in vivo
cross-linked IRBCs are shown. Parasite cultures used are indicated on top, sera used for detection are shown below each filter. Cross-linking was reverted by boiling the sample. The asterisk denotes a band picked up by the anti-myc antibodies upon prolonged exposure that was not related to the transgene, as it was also present in untransfected parasites (data not shown).B. Comparison of individual ETRAMPs and EXP-1 on the same gel. Filters were cut if different sera were used. Note that the lowest band of ETRAMP2myc in its comparison with EXP-1 corresponds to the second band in the ladder usually seen with ETRAMP2myc. The ETRAMP2myc monomer was allowed to migrate out of the gel to obtain a better resolution. ETRAMP4myc and ETRAMP2myc were detected via the tag, EXP-1 and ETRAMP4 with specific sera.
**
Formaldehydeboiled
+ + - + + - + + -
- + - - + - - + -
α-mycα-ETRAMP4
3D7 D10E4myc D10E2myc+ + -
- + -
3D7
α-ETRAMP2
66
56
97
43
35
27
20
116
158212
66
35
20
212
66
35
20
212
14
66
35
20
14
181
64
26
15
+ + -
- + -
3D7
α-EXP-1
66
35
20
212
181
64
26
64
26
15
116ETRAMP4m
yc
ETRAMP2m
yc
EXP-1
ETRAMP2m
yc
ETRAMP4
EXP-1
A B

Proteins at the malaria host–parasite interface
783
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
interactions of individual ETRAMP monomers with otherproteins such as different ETRAMPs (which show differentmobility in SDS-PAGE) this would have led to a numberof dimeric bands. As this was not the case, it seemedlikely that the nearest neighbour(s) of the monomers of agiven ETRAMP were all identical.
To test if these could be specific heterologous interac-tions between different ETRAMPs or EXP-1, we ranextracts of cross-linked parasites on the same Westerngel. None of the dimer bands co-migrated (Fig. 2B). Sucha co-migration would be expected if a direct heteropoly-meric interaction was occurring between any of the pro-teins analysed here. In fact, none of the bands of aparticular complex showed identical migration to any ofthe bands of another complex (Fig. 2B). This suggeststhat each ladder represents a different protein complex.Analysis of band sizes revealed that the ladder incrementswere roughly similar for each complex and that the aver-
age increment was close to the theoretical monomer-sizeof each protein analysed (see Supplementary Table S1).These results show that ETRAMP2, ETRAMP4 and EXP-1 interact with either themselves or other proteins of asimilar molecular weight and that they are part of distinctprotein complexes.
ETRAMP and EXP-1 complexes are found outside of the parasite cell
While we had previously localized ETRAMPs to thePVM (Spielmann
et al
., 2003), it was not clear whatproportion of total ETRAMP proteins are found insidethe parasite cell at any given time, possibly denselypacked in export organelles. To demonstrate that theETRAMP and EXP-1 complexes are present in thePVM, and do not originate from unexported proteinwithin the parasite itself, the membrane-impermeable
Fig. 3.
Selective permeabilization and subsequent cross-linking demonstrates that ETRAMP complexes are present in the PVM. Cultured parasite were subdivided into three fractions that were either kept in PBS (PBS), released by saponin treatment (saponin) or hypotonically lysed with 5 mM HEPES to obtain crude membranes (HEPES). Each of these fractions were mock-treated (–), treated with BS
3
(BS
3
) or with formaldehyde (F). Anti-myc probed Western blots of D10E4myc and D10E2myc parasites are shown in A and B respectively. The asterisk denotes a band picked up by the anti-myc antibodies upon prolonged exposure required to detect ETRAMP2myc. This band was not related to the transgene, as it was also present in untransfected parasites (data not shown). C shows EXP-1 and D shows ETRAMP4 both detected in D10. The arrow (C) indicates a band changing mobility with different cross-linkers. Integrity of the saponin-released parasites was demonstrated by minimal release of
P. falciparum
GAPDH in the supernatant of each treatment (shown for A).
PBS saponin HEPESPBS saponin HEPESBS3PBS
sapo
nin
HEPES
GAPDH in SN
F- BS3 F- BS3 F- BS3 F- BS3 F- BS3 F-
A B
*
14
19
25
36
4760
19
25
36
47
60
81
114
PBS saponin HEPESBS3 F- BS3 F- BS3 F-
CPBS saponin HEPESBS3 F- BS3 F- BS3 F-
D
19
25
36
47
6081
114
19
25
36
47
6081
114
ETRAMP4myc ETRAMP2myc
EXP-1 ETRAMP4

784
T. Spielmann
et al.
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
cross-linker BS
3
was used on selectively permeabilizedIRBCs.
Infected RBCs were treated with saponin, whichreleases the parasite from the RBC but leaves the parasiteplasma membrane intact. These cells were then treatedwith BS
3
and analysed by Western blotting. ForETRAMP4, ETRAMP4myc, ETRAMP2myc and EXP-1,BS
3
treatment resulted in similar ladders of bands to thoseseen with formaldehyde (which is cell-permeable). Treat-ment of intact IRBCs however, resulted in a ladder ofbands with formaldehyde only, but not with BS
3
(Fig. 3).Crude membrane preparations, used as a positive control,resulted in the same pattern of bands observed with sapo-nin-treated IRBCs (Fig. 3). The integrity of the parasitesreleased by saponin was confirmed by the minimalrelease of
P. falciparum
glyceraldehyde-3-phosphatedehydrogenase (GAPDH) (Fig. 3).
These results confirm that although BS
3
has noaccess to the parasite interior, ETRAMPs and EXP-1 stillbecame cross-linked in saponin-treated cells. This dem-onstrates that these complexes were present outside ofthe parasite’s boundary. No cross-linking was observedusing intact IRBCs and BS
3
, in accordance with theknown membrane impermeability of this compound.Thus ETRAMP and EXP-1 complexes are present in thePVM after export of these proteins out of the parasitecell. Furthermore, the similar cross-linking efficiencybetween BS
3
and formaldehyde indicates that mostETRAMP proteins are present outside of the parasitecell.
A membrane environment is crucial for the stability of ETRAMP and EXP-1 complexes
In order to study the properties of the ETRAMP and EXP-1 interaction partners, crude membranes of IRBCs wereprepared and treated with different reagents designed toremove components of the complex depending on theirbinding properties. After treatment, the membranes werewashed, cross-linked and analysed on Western blots toidentify whether a given treatment either disrupted thecomplex or removed components.
Treatment of crude membrane preparations with 1 MKCl did not result in disruption or removal of componentsof ETRAMP4myc and EXP-1 complexes. This was evi-dent from a banding pattern in Western blots that wascomparable in relative intensity to the pattern in extractsobtained by
in vivo
cross-linking or untreated crudemembranes (Fig. 4). Doubling the KCl concentration orusing a different salt (NaCl) also had no effect (Fig. 4).Similarly, low-salt treatment (crude membranes wereprepared by hypotonic lysis of IRBCs in low salt) ortreatment with 50 mM EDTA had no effect (Fig. 4). Theseresults demonstrate that ETRAMP and EXP-1 complexes
were not affected by high- or low-salt treatment. Transientdenaturing with 2 M urea or removal of cholesterol withmethyl-
β
-cyclodextrin (MBC), also did not alter the com-plex composition of the bands seen on Western blots(Fig. 4).
These findings indicate that the interaction partnerswere neither soluble proteins nor peripheral membraneproteins, as these would have been removed by thesetreatments. It therefore seemed likely that the complexconsisted entirely of integral membrane proteins. To con-firm this, the membranes were disrupted with 0.2% TritonX-100 prior to cross-linking (Fig. 4). This led to completedisruption of the complex. A control sample cross-linkedin a similar volume without Triton X-100 left the complexintact. Results for ETRAMP2myc were similar toETRAMP4myc (data not shown). This indicates that amembrane environment is critical for complex stability andthat complexes containing ETRAMPs are arrays of inte-gral membrane proteins. A less profound effect wasobserved with EXP-1, where Triton X-100 only partiallyprevented cross-linking (Fig. 4).
Fig. 4.
ETRAMP and EXP-1 complexes consist of integral membrane proteins. Crude membranes were prepared from IRBCs, treated with the reagents indicated on top of each lane prior to cross-linking and then compared with
in vivo
cross-linked samples by immunoblotting (see
Experimental procedures
).A. ETRAMP4myc detected in D10E4myc parasites via the myc tag.B. EXP-1 detected in D10.TX100, Triton X-100.
B
inviv
o
1MKCl
2MKCl
2MNaC
l
50m
MEDTA
20m
MM
BC
2MUre
a
0.2%
TX100
TX100
cont
rol
A
182
63
37
20
182
63
37
20
E4myc
EXP-1
Proteins at the malaria host–parasite interface
785
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
59
, 779–794No claim to original Australian government
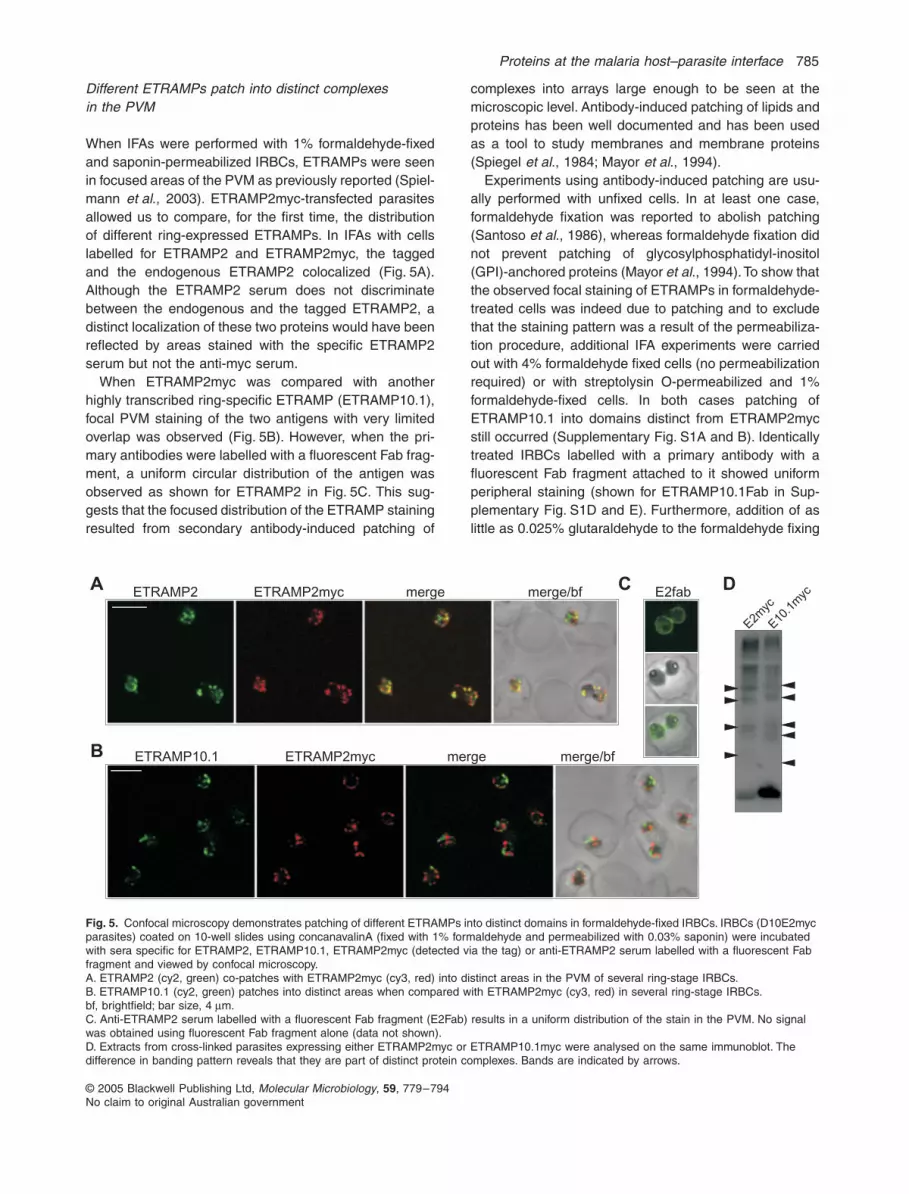
Different ETRAMPs patch into distinct complexes in the PVM
When IFAs were performed with 1% formaldehyde-fixedand saponin-permeabilized IRBCs, ETRAMPs were seenin focused areas of the PVM as previously reported (Spiel-mann
et al
., 2003). ETRAMP2myc-transfected parasitesallowed us to compare, for the first time, the distributionof different ring-expressed ETRAMPs. In IFAs with cellslabelled for ETRAMP2 and ETRAMP2myc, the taggedand the endogenous ETRAMP2 colocalized (Fig. 5A).Although the ETRAMP2 serum does not discriminatebetween the endogenous and the tagged ETRAMP2, adistinct localization of these two proteins would have beenreflected by areas stained with the specific ETRAMP2serum but not the anti-myc serum.
When ETRAMP2myc was compared with anotherhighly transcribed ring-specific ETRAMP (ETRAMP10.1),focal PVM staining of the two antigens with very limitedoverlap was observed (Fig. 5B). However, when the pri-mary antibodies were labelled with a fluorescent Fab frag-ment, a uniform circular distribution of the antigen wasobserved as shown for ETRAMP2 in Fig. 5C. This sug-gests that the focused distribution of the ETRAMP stainingresulted from secondary antibody-induced patching of
complexes into arrays large enough to be seen at themicroscopic level. Antibody-induced patching of lipids andproteins has been well documented and has been usedas a tool to study membranes and membrane proteins(Spiegel
et al
., 1984; Mayor
et al
., 1994).Experiments using antibody-induced patching are usu-
ally performed with unfixed cells. In at least one case,formaldehyde fixation was reported to abolish patching(Santoso
et al
., 1986), whereas formaldehyde fixation didnot prevent patching of glycosylphosphatidyl-inositol(GPI)-anchored proteins (Mayor
et al
., 1994). To show thatthe observed focal staining of ETRAMPs in formaldehyde-treated cells was indeed due to patching and to excludethat the staining pattern was a result of the permeabiliza-tion procedure, additional IFA experiments were carriedout with 4% formaldehyde fixed cells (no permeabilizationrequired) or with streptolysin O-permeabilized and 1%formaldehyde-fixed cells. In both cases patching ofETRAMP10.1 into domains distinct from ETRAMP2mycstill occurred (Supplementary Fig. S1A and B). Identicallytreated IRBCs labelled with a primary antibody with afluorescent Fab fragment attached to it showed uniformperipheral staining (shown for ETRAMP10.1Fab in Sup-plementary Fig. S1D and E). Furthermore, addition of aslittle as 0.025% glutaraldehyde to the formaldehyde fixing
Fig. 5.
Confocal microscopy demonstrates patching of different ETRAMPs into distinct domains in formaldehyde-fixed IRBCs. IRBCs (D10E2myc parasites) coated on 10-well slides using concanavalinA (fixed with 1% formaldehyde and permeabilized with 0.03% saponin) were incubated with sera specific for ETRAMP2, ETRAMP10.1, ETRAMP2myc (detected via the tag) or anti-ETRAMP2 serum labelled with a fluorescent Fab fragment and viewed by confocal microscopy.A. ETRAMP2 (cy2, green) co-patches with ETRAMP2myc (cy3, red) into distinct areas in the PVM of several ring-stage IRBCs.B. ETRAMP10.1 (cy2, green) patches into distinct areas when compared with ETRAMP2myc (cy3, red) in several ring-stage IRBCs.bf, brightfield; bar size, 4
µm.C. Anti-ETRAMP2 serum labelled with a fluorescent Fab fragment (E2Fab) results in a uniform distribution of the stain in the PVM. No signal was obtained using fluorescent Fab fragment alone (data not shown).D. Extracts from cross-linked parasites expressing either ETRAMP2myc or ETRAMP10.1myc were analysed on the same immunoblot. The difference in banding pattern reveals that they are part of distinct protein complexes. Bands are indicated by arrows.
ETRAMP2 ETRAMP2myc merge merge/bf E2fabA
B
C
ETRAMP10.1 ETRAMP2myc merge merge/bf
D
E2myc
E10.1
myc

786 T. Spielmann et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
solution markedly reduced patching (SupplementaryFig. S1C). This is in agreement with the results reportedfor patching of GPI-anchored proteins (Mayor et al.,1994).
These results strongly suggest that the focused distri-bution of ETRAMPs is due to patching. ThereforeETRAMPs (and ETRAMP complexes that will be chemi-cally cross-linked by formaldehyde and can be regardedas a single patchable unit) are diffusable in the membraneeven after formaldehyde treatment.
Antibody-mediated patching can be used to determinethe association between membrane proteins (see e.g.Constantinescu et al., 2001). It is thought that membraneproteins that interact do co-patch, whereas non-interact-ing proteins segregate into different foci. As ETRAMP2and ETRAMP2myc co-patch but ETRAMP2myc andETRAMP10.1 do not, this indicates that ETRAMP2 andETRAMP10.1 are present in different cross-linked com-plexes. This can be concluded because a mixedETRAMP2myc and ETRAMP10.1 array would have bind-ing sites for both primary antibodies (and thus for bothsecondary antibodies), leading to co-patching. However,this was not observed.
To verify the finding that different ring stage-specificETRAMPs form distinct complexes, 3D7 parasites weretransfected with a construct mediating expression ofETRAMP10.1myc. ETRAMP10.1myc was also found inthe PVM in IFA (data not shown) and gave rise to a slightlydifferent ladder of bands after cross-linking, when com-pared with ETRAMP2myc in Western blot experiments(Fig. 5D).
Individual ETRAMPs form homo-oligomers in the E. coli inner membrane independent of other Plasmodium proteins
The cross-linking and patching experiments indicated thatdifferent ETRAMPs and EXP-1 form separate complexes.As the size increments of the ladder of bands was relatedto the size of the monomer of the particular ETRAMPanalysed and the interaction partners also had the prop-erties of integral membrane proteins, this indicated thatETRAMP arrays may consist of homo-oligomers. Immu-noprecipitation experiments to establish if ETRAMPs con-sist of homo-oligomers were inconclusive because of lowefficiency of the immunoprecipitation and poor stability ofpurified ETRAMPs during reversal of the cross-linking. Itwould also be difficult to rule out the presence of otherproteins using this technique. Detection of, for example,ETRAMP2 in ETRAMP2myc complexes immunoprecipi-tated via the myc tag, would not exclude the presence ofother components. Even if it were possible to isolate suf-ficient complex to analyse by mass spectrometry, theabsence of spectra to other peptides would not definitely
exclude the presence of other proteins. We therefore usedthe alternative approach of expressing ETRAMPs inE. coli to analyse whether individual ETRAMPs were ableto oligomerize in the absence of other P. falciparumproteins.
Initially ETRAMP2myc (recE2myc) was expressed inE. coli. This recombinant ETRAMP was inserted into theE. coli inner membrane with the C-terminus facing theperiplasm, as determined by immunofluorescence andproteinase K spheroblast protection assays (Fig. 6A andB). Cross-linking of bacteria after induction of recombinantprotein expression resulted in a ladder of bands detectedin Western blots with an anti-myc antibody. The extent ofoligomerization appeared not to be dependent on levelsof induction (Fig. 6C). This indicates a wide ranging con-centration independence for complex formation. ThusETRAMP2 is capable of forming homo-oligomers in thebacterial inner membrane, independent of other Plasmo-dium proteins.
Next we tested if this was also the case for otherETRAMPs and whether the tag had any influence oncomplex formation. Myc-tagged ETRAMP10.1(recE10.1myc) or FLAG-tagged ETRAMP4 andETRAMP2 (recE2FLAG, recE4FLAG) as well as an N-terminally truncated version of ETRAMP4 (recNtrE4myc)were expressed in E. coli (Supplementary Fig. S2 andFig. 6). RecE2myc was larger than parasite-derivedETRAMP2myc and recE10.1myc even slightly larger thanrecE2myc. Trace amounts of a smaller band of identicalsize to the parasite-derived tagged ETRAMP wereobserved below the dominant band (Fig. 6 and data notshown). The most likely explanation is that the signalpeptide was not efficiently cleaved in E. coli. Interestingly,recE2FLAG showed predominantly the smaller band, indi-cating efficient cleavage of the signal peptide. Further-more, Western analysis showed a faint larger band in allbacteria expressing recombinant ETRAMPs, indicatingthe formation of SDS-resistant ETRAMP dimers at thesehigh protein concentrations (asterisks in Fig. 6D). Higherorder oligomers could be seen in overexposed filters (datanot shown). After cross-linking of intact bacteria, a ladderof bands was seen in Western blots with all recombinantETRAMPs (Fig. 6E; see Supplementary Table S1 for bandsizes). This indicates that ETRAMP10.1 and ETRAMP4can also oligomerize independently of other P. falciparumproteins. This was independent of the tag used, as FLAG-tagged ETRAMPs also formed oligomers.
It was only possible to show incorporation into theE. coli inner membrane of recE2myc, as intact sphero-blasts could not be obtained of the bacteria expressingthe other recombinant ETRAMPs due to filamentousgrowth. However, peripheral staining in IFA stronglysuggested membrane insertion for all recombinantETRAMPs, although recE4FLAG and recE10.1myc

Proteins at the malaria host–parasite interface 787
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
1 2 3 α-E2 brightfieldA
E2myc
control
B
α-mycα-FLAG
E2FLA
G
E2myc
E10.1
myc
NtrE4m
yc
E4FLA
G
****
*
E2FLA
G
E2myc
E10.1
myc
NtrE4m
yc
E4FLA
G
α-FLAG α-FLAGα-myc
C
0.00
2
arabinose
D
α-myc brightfield
E F
665643
35
27
20
212
20
15
182
63
20
14
3D7NtrE
4myc
α-E4
0.00
002
182
63
20
26
37
49
82116
15
Fig. 6. ETRAMPs oligomerize in the E. coli inner membrane independent of other P. falciparum proteins.A. Immunoblot with spheroblasts of bacteria expressing recE2myc that were treated with proteinaseK and 1% Triton X-100 (lane 1), pro-teinaseK alone (lane2) or mock-treated (lane3). A cytoplasmic control protein was not digested, demonstrating integrity of spheroblasts, whereas recE2myc (detected by anti-myc anti-bodies) was digested in lane 2, demonstrating insertion of this protein into the E. coli inner membrane with the C-terminus facing the peri-plasmic space. Coomassie-stained gels of the same sample also confirmed integrity of spher-oblast (not shown).B. Anti-ETRAMP2 and anti-myc antibodies show a peripheral stain in bacteria expressing recE2myc in confocal microscopy. Bar size, 4 µm.C. Bacteria expressing recE2myc treated with formaldehyde and analysed by Western blotting with anti-myc antibodies show efficient cross-linking of recE2myc independent of induction levels.D. Expression of myc and FLAG-tagged ETRAMPs in E. coli detected by Western anal-ysis of total bacterial extracts. Asterisks show SDS-resistant dimer bands. Note the faint band visible below recE2myc probably indicating inefficient cleavage of the signal peptide, whereas recE2FLAG is cleaved more readily.E. Efficient oligomerization independent of other P. falciparum proteins can be detected in all recombinant ETRAMPs upon formaldehyde cross-linking of total bacteria and subsequent Western analysis.F. The ladder of bands of cross-linked recombi-nant NtrE4myc is indistinguishable from the one seen with ETRAMP4 in 3D7 parasites in immunoblots of 12% PAGE gels probed with anti-ETRAMP4 serum.

788 T. Spielmann et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
showed some concentration at the bacterial poles in addi-tion to the peripheral staining (Supplementary Fig. S2).These results indicate that recombinant ETRAMPs areable to homo-oligomerize in a heterologous membraneenvironment and that these results are not due to non-specific cytoplasmic aggregation in E. coli.
RecNtrE4myc showed similar migration in SDS-PAGEto parasite-derived ETRAMP4. To show that the ladderobserved in the parasite could be derived from ETRAMP4alone, cross-linked recNtrE4myc extracts and cross-linkedparasite extracts were compared in Western blots probedwith anti-ETRAMP4 antisera. Migration of ETRAMP4bands from the parasite extract was identical to therecNtrE4myc bands in immunoblots of 12% PAGE gels(Fig. 6F). However, ETRAMP2myc, which had a similarmonomeric size to recE2FLAG, did not show exactly thesame spacing as the ladder of the recombinant protein(data not shown). This may indicate an interaction withanother protein, although our results clearly demonstratethat it can efficiently homo-oligomerize in bacteria and thatETRAMP2 defines a different complex to any of the othermolecules analysed in this study.
ETRAMPs are a major component of the Plasmodium PVM
The high transcription levels of etramps demonstrated bytranscriptome analysis (Le Roch et al., 2003) and ourNorthern blot results (Spielmann and Beck, 2000; Spiel-mann et al., 2003) may not necessarily translate into highabundance of ETRAMP protein given our observationabove of post-translational processing. Therefore thenumber of ETRAMP molecules per parasite cell wasestimated, using dilutions of recombinantly expressedETRAMPs as a standard (Supplementary Fig. S3). Usingseveral different cultures, we obtained values of 300 000–470 000 ETRAMP4 molecules per single 3D7 parasiteand between 140 000 and 550 000 ETRAMP4 moleculesper D10 parasite cell. Cultures containing predominantlytrophozoite stage parasites contained higher copy num-bers of ETRAMP4, possibly indicating an adjustment ofETRAMP4 copy number to the larger size of this stage.For ETRAMP2 we obtained copy numbers of 450 000–810 000 per 3D7 ring-stage parasite and 500 000–710 000 in D10.
To put these figures into context, the resulting proteindensity in the PVM was estimated (see supplementarydata). A young trophozoite-stage parasite with a diameterof 2.5 µm containing 300 000 ETRAMP4 molecules wouldhave approximately one ETRAMP4 molecule per ∼65 nm2
of PVM. A ring-stage parasite of 1 µm radius would haveup to one ETRAMP2 molecule per ∼15 nm2 PVM giventhe estimate of up to 810 000 ETRAMP2 molecules perring-stage parasite. The most abundant erythrocyte mem-
brane protein, glycophorin A, is present at about 1 × 106
copies per cell. This corresponds to a density of oneglycophorin A molecule per 135 nm2 membrane. ThusETRAMPs are present at higher densities than glycoph-orin A.
Discussion
Intracellular pathogens cause some of the most seriousdiseases of humans and animals. The most intimate con-tact between host and vacuolated intracellular pathogensis the membrane of the vacuole (Garcia-del Portillo andFinlay, 1995; Méresse et al., 1999). Despite the impor-tance of this compartment, our knowledge particularlyabout the pathogen-derived factors that shape its proper-ties, is still limited. Many of these factors may well beunique to the pathogen. Unravelling novel pathogen-specific processes could direct research towards noveltargets for chemotherapeutic intervention.
In this work the organization of ETRAMPs and EXP-1at the PVM of intact RBC, infected with asexual stages ofhuman malaria parasites was analysed. This provides forthe first time insights into the molecular architecture of thiscompartment. ETRAMP2, ETRAMP10.1, ETRAMP4 andEXP-1 were demonstrated to be components of separateoligomeric arrays in the PVM. Despite their abundance inthe same membrane, they do not randomly interact witheach other or other proteins. Such interactions would haveled to more than one band that corresponds to the dimerand an increasing number of bands for higher order oli-gomers in Western blots using extracts derived fromcross-linked IRBCs. This was not the case, demonstratingthat a given monomer interacts with only a single type ofprotein. Furthermore interactions were highly specific witheach complex appearing as a ladder of distinctive bandswith average size increments close to the theoreticalmolecular weight of the respective monomer.
It is important to note, that the approach of cross-linkingintact IRBCs, followed by subsequent cell lysis, excludesinteractions due to protein aggregation after lysis. There-fore our results can only be explained by close proximityof ETRAMPs to each other in the PVM of live parasites.This is further supported by the comparable resultsobtained using three different cross-linking agents, irre-spective of whether live intact parasites, saponin-releasedparasites or crude membrane preparations were used.
Although all ETRAMPs show different mobility in SDS-PAGE, they are of a similar molecular weight. The ladderof bands seen in cross-linked extracts could theoreticallybe due to specific interaction between a monomer of oneETRAMP with that of a different ETRAMP. However, mostcritically, there was no overlap within the linear separationrange between any of the ladders derived from differentETRAMPs or EXP-1. Therefore each defines an entirely

Proteins at the malaria host–parasite interface 789
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
distinct complex. Even if they were hetero-oligomers, thisexcludes mixed ETRAMP arrays. These data, togetherwith the ability of recombinant ETRAMPs to homo-oligomerize in E. coli, suggest that ETRAMP and EXP-1complexes are largely homo-oligomeric in vivo. In accor-dance with this hypothesis, both recombinant NtrE4mycand parasite-derived ETRAMP4, which showed identicalmigration as monomers, gave rise to identical laddersupon cross-linking.
Despite the evidence for ETRAMP homo-oligomers, thesize increments between the bands in the ladder from agiven ETRAMP was not exactly identical. This was mostlikely due to cross-linker-induced change in charge orconformation of the complex, affecting its migration duringelectrophoresis. In accordance with this interpretation,this phenomenon was also seen with complexes derivedfrom recombinant ETRAMPs.
Absence of mixed ETRAMP oligomers was alsosupported in IFA experiments, as ETRAMP2 andETRAMP10.1 patched into different domains. We previ-ously observed distinct localization for EXP-1 when com-pared with ETRAMP2 and ETRAMP10.1, although at thattime we were not aware that this did not reflect the truesituation in vivo but was induced by patching with thesecondary antibody and that these molecules were partof complexes cross-linked by the fixative (Spielmannet al., 2003). Therefore, although our data strongly sup-port oligomeric arrays in the PVM defined by differentETRAMPs, these arrays are either too dynamic or toosmall to be resolved as visible domains by fluorescentmicroscopy.
It has been shown that additional proteins can stabilizeoligomers of integral membrane proteins or influence theirspatial distribution. For example CheA and CheW do soby interacting with bacterial chemotaxis receptors (Mad-dock and Shapiro, 1993). Such proteins might be difficultto detect, especially if they were present at low stoichiom-etry or if they interacted only transiently. However, no suchprotein seems to be required for the formation of ETRAMPcomplexes. This was evident by efficient oligomerizationof ETRAMPs in bacterial membranes, unless this functionwas replaced by a bacterial protein. However, this isunlikely to be an abundant protein interacting directly withmonomers, which would have been detected as an irreg-ularity in the migration of the ladder of cross-linked recE-TRAMPs. Nevertheless, recE10.1myc and recE4FLAGshowed some tendency to localize near the bacterialpoles. This might indicate some interaction with E. coliproteins in this region. Also the filamentous growth of thebacteria expressing the recombinant ETRAMPs may bedue to interference with bacterial proteins associated withthe inner membrane that are involved in bacterial celldivision, as has previously been observed (Ferguson andShaw, 2004).
Early transcribed membrane protein complexes consistof patches of integral membrane proteins associated in alipid bilayer-dependent manner and therefore have to beseen as arrays of oligomeric proteins suspended in themembrane. This complicates analysis of size and compo-sition, as cross-linking is indispensable before they canbe isolated from the membrane. The size estimation ofcomplexes of such integral membrane proteins can befurther confounded because of detergent interferencewith sedimentation and gel filtration analysis. The highabundance of ETRAMPs in the PVM could indicate thatthey are present as confluent areas of distinct oligomericarrays. Individual arrays could contain far more ETRAMPmolecules than the 5–11 bands that have been discernedin Western blots with cross-linked extracts of parasites orETRAMP-expressing bacteria. Indeed there was a con-siderable amount of unresolved high-molecular-weightmaterial that was not seen in non-cross-linked extracts(Figs 2–4 and 6 and data not shown). Furthermore,unless cross-linking is 100% efficient, a most unlikely sit-uation, Western blots must give an underestimate of thearray sizes. Thus no estimate of the upper size is possibleand it may prove difficult to test this, as our IFA patchingresults indicate relatively unrestricted movement forcross-linked complexes. This could reflect dynamicassociation/disassociation and movement of arraycomponents.
It has been previously shown that green fluorescentprotein-fusions of soluble proteins exported to the parasi-tophorous vacuole of ring stages often appear as a beadof strings in live parasites (Waller et al., 2000) and thatthe diffusion of parasitophorous vacuole soluble proteinswas partially restricted in these cells (Adisa et al., 2003).The patching results show that ETRAMPs are diffusablein the PVM even after treatment with formaldehyde. Thisindicates that ETRAMPs are not strongly attached to afixed scaffold or otherwise permanently restricted withintheir movement in the PVM. This could be comparable tothe situation with GPI-anchored proteins on the surface ofmammalian cells. These proteins still can be patched afterformaldehyde fixation (Mayor et al., 1994), possiblybecause they have no moiety interacting with the cytosk-eleton underlying the plasma membrane.
It is noteworthy that other protein complexes may alsobe present in the PVM. The high-molecular-mass rhoptrycomplex appears to be delivered to the parasitophorousvacuole or the PVM upon RBC invasion (Ling et al., 2004).Furthermore, a P. falciparum stomatin homologue wasproposed to be involved in the nucleation of proteinscaffolds in lipid rafts in the PVM (Hiller et al., 2003),although the complex state of this protein at the PVMremains to be experimentally demonstrated. Any inter-action between ETRAMPs and these molecules has notyet been established.

790 T. Spielmann et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
It has previously been speculated that ETRAMPs maybe involved in invasion (Birago et al., 2003). However, wehave been unable to localize ETRAMPs in late schizontsor merozoites (data not shown) and our Western blot dataconfirmed exclusive ring-stage expression for ETRAMP2and ETRAMP10.1 (Spielmann et al., 2003). Semiquanti-tative proteomics data confirm that five of the sixETRAMPs we previously classified as ring stage-specificwere found almost exclusively in ring stages (Le Rochet al., 2004). Together with the high abundance ofETRAMPs in the PVM, invasion as the function ofETRAMPs now seems unlikely. Rather the strong invest-ment in the ring stage indicates that the main function ofETRAMPs lies with the PVM and the initiation of parasitedevelopment in the new host cell as has been previouslyhypothesized (Spielmann et al., 2003). In accordancewith this, the rodent malaria orthologue of PfETRAMP13is upregulated in infective sporozoites (Kaiser et al.,2004) and essential for liver stage development (Muelleret al., 2005a). Parasites lacking this gene were able toinvade liver cells but failed to develop to liver schizonts, afact elegantly used to obtain a protective experimentallive vaccine in mice (Mueller et al., 2005a). Furthermore,another rodent malaria sporozoite gene coding for asmall integral membrane protein was also essential forliver stage development (Mueller et al., 2005b). Althoughlacking some of the typical features of ETRAMPs, thisgene was considered a possible orthologue ofPfetramp10.3 (PF10_0164). This accords with its local-ization in the liver stage PVM (Mueller et al., 2005b).Therefore it seems that ETRAMPs are essential in theearly development of different malaria intracellular lifecycle stages. This may explain our failure to obtain knock-outs for asexual stage etramps to date (our unpubl. data).Moreover, EXP-1 and its Plasmodium yoelii orthologueare expressed in the PVM of liver stages (Doolan et al.,1996), which indicates that the liver stage PVM couldcontain a similar organization of EXP-1 and liver stage-specific ETRAMPs.
ETRAMP2, ETRAMP10.1, ETRAMP4 and EXP-1 areall expressed in ring-stage parasites, giving rise to fourseparate complexes. By analogy, it can be inferred thatother ETRAMPs share these properties, although thisremains to be determined experimentally. With the fourother remaining ring stage-specific ETRAMPs, this wouldlead to a patchwork of eight kinds of distinct array typesin the ring-stage PVM. The copy number estimation forETRAMP2 and ETRAMP4 indicated that they are presentin densities higher than glycophorin A, the most abundantRBC membrane protein. Taking into account that besidesetramp2 also etramp11.1, etramp11.2 and etramp10.1belong to the most highly transcribed genes in asexualstage parasites and that exp-1 shows higher transcriptionthan etramp4 (Le Roch et al., 2003), it seems possible
that ETRAMPs and EXP-1 represent the dominant proteincomponent of the PVM.
Each of the proteins analysed appears to define a sep-arate array, leading to a mosaic of domains that accordingto the patching results may be dynamic in size and posi-tion of the individual arrays. The observation thatETRAMPs are capable of oligomerizing in a heterologoussystem, shows that their presence in the PVM is sufficientto spontaneously create microdomains consisting of dif-ferent ETRAMP arrays. This would be a simple and ele-gant way for the cell to create polarity in the host–parasiteinterface. Similar to lipid rafts (Simons and Toomre, 2000;Edidin, 2003), these arrays of short integral membraneproteins could provide platforms to compartmentalize dif-ferent processes or serve as seeds to initiate differentstructures such as TVN and cytostome in the PVM. Anal-ogous to proteins clustered in a lipid raft-dependent man-ner, ETRAMPs can be clustered to microscopically visibledomains by antibody-mediated patching, and are detectedas oligomers upon in vivo cross-linking. ETRAMP oligo-merization was not itself lipid raft-dependent, as choles-terol depletion with MBC or saponin treatment had noeffect on complex formation and oligomerization was alsoobserved in E. coli, which does not contain cholesterol.This does not preclude them being present in lipid rafts,which in fact has been demonstrated for EXP-1 (Laueret al., 2000). However, it indicates that rafts are not thebasis for the observed oligomerization.
Small, self-assembling integral membrane proteins maybe a general way to achieve local polarization in biologicalmembranes. The organization of the malaria PVM may bereminiscent of the organization of compartments found incomplex membrane systems such as the endocytic path-way (Gruenberg, 2001). Finally, the presence of proteinarrays raises the exciting possibility of conformationalspread, a phenomenon allowing cross-talk between com-plex components by allosteric interaction (Bray and Duke,2004).
Experimental procedures
Plasmid construction
For P. falciparum transfection constructs, etramp2 andetramp4 were polymerase chain reaction (PCR)-amplifiedfrom 3D7 gDNA using primers E2Xfw (5′-ACCGCTCGAGCAAAATGAAACTCTCCAAAATC-3′; stop and start codonsare in bold, restriction sites underlined in italics) and E4Xfw(5′-ACCGCTCGAGCAAAAAATGAAATTGAGCAAC-3′) withreverse primers E2mycrv (5′-TGGCCTCGAGTTAtaaatcttcttcgcttatgagtttttgttcAGGCCTGGCTCTGTACATGGTACTTG-3′;myc and FLAG tag coding sequences are shown in lowercase) and E4mycrv (5′-TGGCCTCGAGTTAtaaatcttcttcgcttatgagtttttgttcAGGCCTAACAGTAGTGGGTACTGCTG-3′)appending the coding sequence for the myc tag followedby a stop codon and cloned into vector pHHC*/DR0.28

Proteins at the malaria host–parasite interface 791
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
(O’Donnell et al., 2002) replacing the CAT gene that was cutout using XhoI. This resulted in vectors DR0.28E2myc andDR0.28E4myc. Cloning of etramp10.1 into the same site ofvector pHHC*/DR0.28 to obtain DR0.28E10.1myc will bedescribed elsewhere.
For expression of ETRAMPs in bacteria, the plasmidpBAD/Thio-TOPO (Invitrogen) was cleaved with NcoI andPmeI to release the thioredoxin gene. etramp2myc was PCR-amplified from DR0.28E2myc using primers E2Xfw andmycstoprv (5′-TTAtaaatcttcttcgcttatgag-3′) and cleaved withXhoI. The XhoI and NcoI overhangs of insert and vectorwere blunted and ligated to obtain pNoT-BAD/E2myc. ForNtrE4myc expression, etramp4 was PCR-amplified with prim-ers ETR4Xfw and mycstoprv from DR0.28E4myc, cleavedwith BspHI (cleaves internally in codon for Met 38) andligated into pBAD/Thio-TOPO cleaved with NcoI and PmeIto obtain pNoT-BAD/NtrE4myc. For recE10.1myc, plasmidpRSFDuet-1 (Novagen) was cleaved with BamHI, blunted,then cleaved with NcoI and ligated with a BspHI-cleavedinsert PCR-amplified from DR0.28E10.1myc with primersE10.1Bfw (5′-ACCGCATATGAAACTCTCCAAAATCTTATATTTC-3′) and mycstoprv to obtain pRSF-E10.1myc. ForrecE2FLAG and recE4FLAG, pRSFDuet-1 and inserts PCR-amplified with primers E2Nfw (5′-ACCGCATATGAAACTCTCCAAAATCTTATATTTC-3′) and E2FLAGrv (5′-CATATCCGTCGACTTA cttgtcatcatcgtctttataatcAGGCCT GGCTCTGTACATGGTACTTG-3′) or E4Nfw (5′-ACCGCATATGAAATTGAGCAACTTATTTTAC-3′) and E4dFLAGrv (5′-CATATCCGTCGACTTActtgtcatcatcgtctttataatccttatcatcgtcatccttgtagtcAGGCCTAACAGTAGTGGGTACTGCTG-3′) were NdeI-cleaved andligated to obtain pRSF-E2FLAG and pRSF-E4FLAG. Allconstructs were checked by sequencing.
Parasite culture and transfections
Parasites clones 3D7 and D10 were cultured in RPMI con-taining 10% human serum according to standard methods(Trager and Jensen, 1976). Synchronization was performedusing sorbitol (Lambros and Vanderberg, 1979). Transfec-tions were performed according to standard methods (Wuet al., 1996).
Preparation of parasite extracts
For saponin lysis, parasites were washed in PBS, releasedfrom IRBCs by incubation with 0.03% saponin/PBS on ice for10 min, centrifuged at 16 000 g for 5 min and washed threetimes in ice-cold PBS. For hypotonic lysis, an equivalent of10 ml parasite culture (5% haematocrit, 5–10% parasi-taemia) was washed in PBS and incubated in 10 ml ice-cold5 mM HEPES for 1 h on ice or frozen at −20°C. Broken cells(verified microscopically and by complete release ofPfGAPDH) were centrifuged at 5000 g for 20 min at 4°C andwashed three times in 1.5 ml of ice-cold PBS using centrifu-gations at 16 000 g. The resulting pellet depleted of solubleprotein is referred to as ‘crude membranes’.
Cross-linking
For in vivo cross-linking, an equivalent of 10 ml P. falciparumculture (5% haematocrit, 5–10% parasitaemia) was washed
in PBS, resuspended in 5 ml of PBS and total IRBCs werecross-linked by adding formaldehyde to 1% and incubated at37°C for 30 min at 37°C. TrisHCl pH 8.0 was added to 30 mMto quench the reaction. The cells were spun down at 3000 gand the pellet resuspended in 10 ml of 10 mM Tris HCl pH 8.0to lyse the cells. This suspension was either stored at −20°Covernight or incubated on ice for 1 h, spun at 5000 g for15 min and the pellet washed three times in 1.5 ml ice-coldPBS with intermittent centrifugations at 16 000 g. The layeron top of the pellet representing erythrocyte ghost mem-branes was removed. Proteins were extracted by resuspend-ing the final pellet in two volumes of 4% SDS/1% T-X-100 in0.5× PBS and stored at −80°C. Before loading, the samplewas warmed to room temperature and centrifuged at16 000 g for 5 min and the supernatant used for SDS-PAGE.
For cross-linking of crude membrane fractions, saponin-released parasites or selectively permeabilized IRBCs, anequivalent of 2 ml of thus treated parasite culture (5% hae-matocrit, 5–10% parasitaemia) was washed and incubated ineither 720 µl of 1% formaldehyde in PBS for 15 min at 37°C,10 mg ml−1 DTBP (Pierce) in 500 µl of triethanolamine pH 8.0or in 500 µl PBS containing 0.25 mg ml−1 BS3 (SIGMA).DTBP and BS3 incubations were performed at room temper-ature for 20 min. Reactions were quenched by adding TrisHClpH 8.0 to 30 mM, incubated at room temperature for 10 minand then centrifuged at 16 000 g for 5 min. The pellet waswashed once in PBS/30 mM TrisHCl pH 8.0, resuspended in4%SDS/1%T-X-100 in 0.5× PBS and stored at −80°C untilSDS-PAGE. Reversal of cross-linking was performed by boil-ing the sample for 30 min at 98°C in 1× Laemmli buffer.
Total bacteria were cross-linked in 1% formaldehyde inPBS for 15 min at 37°C. TrisHCl pH 8.0 was added to 50 mM,the cells washed in PBS and directly resuspended in Laemmlisample buffer.
Solubility properties of ETRAMP complex components
Crude membranes (complete cell lysis was confirmed micro-scopically and by complete release of PfGAPDH) derivedfrom 40 ml of culture (5% haematocrit, 5–10% parasitaemia)were divided equally and treated for 30 min with 1 ml of 1 MKCl/PBS, 2 M KCl/PBS, 2 M NaCl/PBS, 50 mM EDTA pH 8.0,on ice or 1 ml 20 mM MBC in RPMI at 37°C or 1 ml 2 M Urea/50 mM triethanolamine pH 8.0 at room temperature respec-tively. Treated membranes were washed in 1 ml ice-cold PBSand cross-linked with formaldehyde as described undercross-linking. For testing the effect of detergents, equalamounts of crude membranes were divided into two tubesand brought to either 720 µl of PBS or 720 µl of PBS/0.2%Triton-X-100 respectively. Formaldehyde was added to 1% inboth aliquots and the samples incubated at 37°C for 15 min.Then TrisHCl pH 8.0 was added to 30 mM, incubated at roomtemperature for 10 min and the sample without Triton waswashed as described for cross-linking. The sample contain-ing Triton was precipitated using TCA.
Western analysis
Proteins resolved on reducing SDS-PAGE gels were trans-ferred to Porablot 0.2 µm PVDF membranes (Macherey-

792 T. Spielmann et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
Nagel) in a tankblot device (Bio-Rad) using 10 mM CAPSpH 11.2 without methanol at 4°C for at least 12 h. Blockingand antibody incubations were carried out in 3.5% fat-freemilk/PBS, washing was performed in PBS and detection wasperformed using ECL (Amersham). Rabbit anti-myc (CellSignaling Technologies) was used 1/1000; mouse mono-clonal anti-myc (SIGMA) 1/4000; M2 mouse monoclonalanti-FLAG (SIGMA) was diluted to 2.5 µg µl−1 and used1/8000; rabbit anti-maltose binding protein 1/2500 (NewEngland Bioloabs). Anti-ETRAMP sera raised againstETRAMP C-termini have been described (Spielmann et al.,2003) and were used at 1/500 for anti-ETRAMP2, 1/650 foranti-ETRAMP4. The anti-EXP-1 serum (raised against theC-terminus) was a kind gift of Dr Lingelbach (Philipps-University Marburg, Germany) and was used 1/650. Mousemonoclonal anti-GAPDH antibodies were a kind gift ofDr Daubenberger (Swiss Tropical Institute) and were used1/2000. Sheep horseradish peroxidase (HRP)-conjugatedsecondary antibodies (Chemicon) were used 1/5000.
Immunofluorescence assays
Immunofluorescence assays were carried out essentiallyas described (Spielmann et al., 2003). Parasites orbacteria were fixed in 100% acetone at room temperaturefor 10 min or 100% methanol at −20°C for 20 min (forbacteria expressing recE2myc, recE10.1myc andrecE4FLAG). For patching experiments, 10-well slideswere precoated with concanavalinA (0.5 mg ml−1 in dH2O)for 30 min at 37°C, then washed parasites were added,incubated at room temperature for 15 min and unboundcells were removed by washing with PBS. Wells coatedwith IRBC were treated with either (i) 1% formaldehydeand 0.03% saponin in PBS for 10 min at 4°C; (ii) 4% form-aldehyde in PBS for 15 min at 4°C; (iii) 12 units activatedstreptolysin (Sigma) in 20 µl of PBS at 37°C for 15 min,washed in PBS and fixed with 1% formaldehyde or 1%formaldehyde/0.025% glutaraldehyde for 15 min at 4°C.Antibody incubations were in 10 mg ml−1 BSA in PBS,washing in PBS. Antibody dilutions used were: mouseanti-ETRAMP2 (1/240), anti-ETRAMP4 (1/400), anti-ETRAMP10.1 (1/300), rabbit anti-myc (Cell SignalingTechnologies) was used 1/150; M2 mouse monoclonalanti-FLAG (SIGMA) was diluted to 2.5 µg µl−1 and used1/2000. Secondary antibodies used were cy2-, cy3- andTexasRed-conjugated Goat AffiniPure antibodies (JacksonImmunoResearch).
For IFAs with fluorescent Fab fragment labelled antibodies,1 µl of crude mouse serum (containing 50% glycerol) or 1 µgof mouse monoclonal anti-myc antibody (SIGMA) was usedwith 5 µl of Alexa Fluor 488 or Alexa Fluor 594 Zenon mouseIgG1 labelling reagent (Molecular Probes) according to theZenon labelling protocol.
Samples were analysed on a Zeiss upright fluorescentmicroscope using cy2 and TexasRed. Confocal microscopywas performed using a Leica TCS SP2 microscope, cy2(excitation source used was a 20 mW Argon Laser at488 nm) and cy3 (excitation source used was a 1.2 mWHeNe Laser at 543 nm) labelling was viewed sequentially. Nobleed through was observed when cells stained with a singlefluorophore were viewed.
Expression of recombinant ETRAMPs and spheroblast proteinaseK protection assay
Overnight starter cultures of bacteria were diluted 1/10 intofresh LB containing 10–100 µM IPTG (Duet vectors) or0.00002–0.02% arabinose (pBAD vectors). Recombinantprotein expression was carried out for 90 min at 37°C andbacteria were immediately used for IFA, cross-linking orWestern analysis. Spheroblasts proteinaseK protectionassay and testing of spheroblast integrity using Coomassiestaining were performed as described (Mendrola et al.,2002). Integrity of spheroblasts was also confirmed by rep-robing the Western blot with serum against maltose bindingprotein (New England Biolabs), which cross-reacts to a cyto-plasmic E. coli protein.
Estimation of ETRAMP2 and ETRAMP4 copy number
In order to determine the concentration of recombinantETRAMPs, five dilutions of partially purified recNtrE4myc andrecE2myc (see Supplementary Fig. S2) were resolved bySDS-PAGE alongside serial dilutions of either a BSAstandard (Pierce) or chicken egg lysozyme (Sigma, concen-tration confirmed by measuring absorbance at OD280). Non-saturated images of Coomassie-stained gels were collectedusing GeneScan software (SynGene) and densitometricallyanalysed by the GeneTools software (SynGene). Quantitycalibrations were performed using a quadratic standard curvegenerated with the dilutions of either BSA or lysozyme. Gene-Tools software then determined the amount of the respectiverecombinant ETRAMP. The average of the values obtainedwith the two standards was used as the final concentration.
The concentrations determined for the recombinantETRAMPs were then used to determine the concentration ofETRAMP2 and ETRAMP4 in parasite cultures as follows:the extracts containing the recombinant ETRAMPs wereresolved by SDS-PAGE and blotted together with parasiteextracts derived from several different P. falciparum culturesharvested at different times over several weeks. Five dilu-tions of recNtrE4myc or recE2myc and three dilutions ofeach parasite culture were analysed. For each culture used,amount of packed RBCs and parasitaemia was recorded(number of RBCs per volume of packed cells was deter-mined by the Queensland Health Pathology and ScientificServices) to determine the exact number of parasites loadedper lane. Blots were reacted with sera specific for the C-terminus of ETRAMP2 or ETRAMP4. Non-saturated imagesof X-rays were collected using GeneScan software (Syn-Gene) and densitometrically analysed by the GeneToolssoftware (SynGene). The concentration of ETRAMP2 andETRAMP4 in each parasite extract was estimated by theGeneTools software using a quadratic standard curve gener-ated from dilutions of either recNtrE4myc or recE2myc. Theestimated amounts for each parasite sample were in goodagreement with the dilution loaded onto the gel. The valueobtained was then used to calculate the number of ETRAMPmolecules in each extract using the theoretical mass of therecombinant protein in the standard (4.65 × 1010 copies pernanogram recE2myc; 4.93 × 1010 copies per nanogramrecNtrE4myc). The number of ETRAMP molecules per lanewas divided by the number of parasites loaded to obtain

Proteins at the malaria host–parasite interface 793
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
copies per parasite. This value was adjusted for the lossbetween determination of parasite number and loading ontothe gel. Parasites used for the quantification experimentswere released from RBCs by saponin treatment and washedthree times in PBS before loading. Quantification ofP. falciparum GAPDH as well as counting of thus treatedparasites compared with total IRBCs revealed a consistentloss of 50% of parasites (data not shown). Therefore,ETRAMP copy number was adjusted accordingly by multi-plying the value obtained by two to get the final copy numberper parasite. Estimation of ETRAMP and glycophorin A den-sities in the membrane are described in supplementarydata.
Acknowledgements
We thank Drs K. Lingelbach and C. Daubenberger for anti-bodies. T.S. gratefully acknowledges the Swiss National Sci-ence Foundation, the Novartis Foundation, the FreiwilligeAkademische Gesellschaft Basel for support during thiswork. D.K., D.G. and K.T. gratefully acknowledge supportfrom Australian NHMRC (Program grant 290208) and MarkNicholson, Alice Hill and The Tudor Foundation.
References
Adisa, A., Rug, M., Klonis, N., Foley, M., Cowman, A.F., andTilley, L. (2003) The signal sequence of exported protein-1 directs the green fluorescent protein to the parasito-phorous vacuole of transfected malaria parasites. J BiolChem 278: 6532–6542.
Alberts, B. (1998) The cell as a collection of proteinmachines: preparing the next generation of molecular biol-ogists. Cell 92: 291–294.
Ansorge, I., Benting, J., Bhakdi, S., and Lingelbach, K. (1996)Protein sorting in Plasmodium falciparum-infected redblood cells permeabilized with the pore-forming proteinstreptolysin O. Biochem J 315: 307–314.
Birago, C., Albanesi, V., Silvestrini, F., Picci, L., Pizzi, E.,Alano, P., et al. (2003) A gene-family encoding smallexported proteins is conserved across Plasmodium genus.Mol Biochem Parasitol 126: 209–218.
Bray, D., and Duke, T. (2004) Conformational spread: thepropagation of allosteric states in large multiprotein com-plexes. Annu Rev Biophys Biomol Struct 33: 53–73.
Carlton, J.M., Angiuoli, S.V., Suh, B.B., Kooij, T.W., Pertea,M., Silva, J.C., et al. (2002) Genome sequence and com-parative analysis of the model rodent malaria parasitePlasmodium yoelii yoelii. Nature 419: 512–519.
Constantinescu, S.N., Keren, T., Socolovsky, M., Nam, H.,Henis, Y.I., and Lodish, H.F. (2001) Ligand-independentoligomerization of cell-surface erythropoietin receptor ismediated by the transmembrane domain. Proc Natl AcadSci USA 98: 4379–4384.
Cooke, B.M., Mohandas, N., and Coppel, R.L. (2001) Themalaria-infected red blood cell: structural and functionalchanges. Adv Parasitol 50: 1–86.
Daily, J.P., Le Roch, K.G., Sarr, O., Fang, X., Zhou, Y., Ndir,O., et al. (2004) In vivo transcriptional profiling of Plasmo-dium falciparum. Malar J 3: 30.
Delon, J., and Germain, R.N. (2000) Information transfer atthe immunological synapse. Curr Biol 10: R923–R933.
Desai, S.A., Krogstad, D.J., and McCleskey, E.W. (1993)A nutrient-permeable channel on the intraerythrocyticmalaria parasite. Nature 362: 643–646.
Doolan, D.L., Hedstrom, R.C., Rogers, W.O., Charoenvit, Y.,Rogers, M., de la Vega, P., and Hoffman, S.L. (1996)Identification and characterization of the protective hepa-tocyte erythrocyte protein 17 kDa gene of Plasmodiumyoelii, homolog of Plasmodium falciparum exported protein1. J Biol Chem 271: 17861–17868.
Edidin, M. (2003) The state of lipid rafts: from modelmembranes to cells. Annu Rev Biophys Biomol Struct 32:257–283.
Elmendorf, H.G., and Haldar, K. (1993) Secretory transportin Plasmodium. Parasitol Today 9: 98–102.
Ferguson, P.L., and Shaw, G.S. (2004) Human S100Bprotein interacts with the Escherichia coli division proteinFtsZ in a calcium-sensitive manner. J Biol Chem 279:18806–18813.
Froehner, S.C. (1993) Regulation of ion channel distributionat synapses. Annu Rev Neurosci 16: 347–368.
Garcia-del Portillo, F., and Finlay, B.B. (1995) The variedlifestyles of intracellular pathogens within eukaryotic vacu-olar compartments. Trends Microbiol 3: 373–380.
Gavin, A.C., Bosche, M., Krause, R., Grandi, P., Marzioch,M., Bauer, A., et al. (2002) Functional organization of theyeast proteome by systematic analysis of protein com-plexes. Nature 415: 141–147.
Gruenberg, J. (2001) The endocytic pathway: a mosaic ofdomains. Nat Rev Mol Cell Biol 2: 721–730.
Hackstadt, T. (2000) Redirection of host vesicle traffickingpathways by intracellular parasites. Traffic 1: 93–99.
Hiller, N.L., Akompong, T., Morrow, J.S., Holder, A.A., andHaldar, K. (2003) Identification of a stomatin orthologuein vacuoles induced in human erythrocytes by malariaparasites: a role for microbial raft proteins in apicom-plexan vacuole biogenesis. J Biol Chem 278: 48413–48421.
Hiller, N.L., Bhattacharjee, S., van Ooij, C., Liolios, K.,Harrison, T., Lopez-Estrano, C., and Haldar, K. (2004) Ahost-targeting signal in virulence proteins reveals a secre-tome in malarial infection. Science 306: 1934–1937.
Kaiser, K., Matuschewski, K., Camargo, N., Ross, J., andKappe, S.H. (2004) Differential transcriptome profilingidentifies Plasmodium genes encoding pre-erythrocyticstage-specific proteins. Mol Microbiol 51: 1221–1232.
Kara, U.A., Stenzel, D.J., Ingram, L.T., Bushell, G.R., Lopez,J.A., and Kidson, C. (1988) Inhibitory monoclonal antibodyagainst a (myristylated) small-molecular-weight antigenfrom Plasmodium falciparum associated with the parasito-phorous vacuole membrane. Infect Immun 56: 903–909.
Lambros, C., and Vanderberg, J.P. (1979) Synchronizationof Plasmodium falciparum erythrocytic stages in culture. JParasitol 65: 418–420.
Lauer, S., VanWye, J., Harrison, T., McManus, H., Samuel,B.U., Hiller, N.L., et al. (2000) Vacuolar uptake of hostcomponents, and a role for cholesterol and sphingomyelinin malarial infection. EMBO J 19: 3556–3564.
Le Roch, K.G., Zhou, Y., Blair, P.L., Grainger, M., Moch, J.K.,Haynes, J.D., et al. (2003) Discovery of gene function by

794 T. Spielmann et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 59, 779–794No claim to original Australian government
expression profiling of the malaria parasite life cycle. Sci-ence 301: 1503–1508.
Le Roch, K.G., Johnson, J.R., Florens, L., Zhou, Y.,Santrosyan, A., Grainger, M., et al. (2004) Global analysisof transcript and protein levels across the Plasmodiumfalciparum life cycle. Genome Res 14: 2308–2318.
Ling, I.T., Florens, L., Dluzewski, A.R., Kaneko, O., Grainger,M., Yim Lim, B.Y., et al. (2004) The Plasmodium falciparumclag9 gene encodes a rhoptry protein that is transferred tothe host erythrocyte upon invasion. Mol Microbiol 52: 107–118.
Maddock, J.R., and Shapiro, L. (1993) Polar location of thechemoreceptor complex in the Escherichia coli cell. Sci-ence 259: 1717–1723.
Marti, M., Good, R.T., Rug, M., Knuepfer, E., and Cowman,A.F. (2004) Targeting malaria virulence and remodelingproteins to the host erythrocyte. Science 306: 1930–1933.
Matsuuchi, L., and Gold, M.R. (2001) New views of BCRstructure and organization. Curr Opin Immunol 13: 270–277.
Mayor, S., Rothberg, K.G., and Maxfield, F.R. (1994)Sequestration of GPI-anchored proteins in caveolae trig-gered by cross-linking. Science 264: 1948–1951.
Mendrola, J.M., Berger, M.B., King, M.C., and Lemmon, M.A.(2002) The single transmembrane domains of ErbB recep-tors self-associate in cell membranes. J Biol Chem 277:4704–4712.
Méresse, S., Steele-Mortimer, O., Moreno, E., Desjardins,M., Finlay, B., and Gorvel, J.P. (1999) Controlling the mat-uration of pathogen-containing vacuoles: a matter of lifeand death. Nat Cell Biol 1: E183–E188.
Moulder, J.W. (1985) Comparative biology of intracellularparasitism. Microbiol Rev 49: 298–337.
Mueller, A.K., Labaied, M., Kappe, S.H., and Matuschewski,K. (2005a) Genetically modified Plasmodium parasites asa protective experimental malaria vaccine. Nature 433:164–167.
Mueller, A.K., Camargo, N., Kaiser, K., Andorfer, C., Frevert,U., Matuschewski, K., and Kappe, S.H. (2005b) Plasmo-dium liver stage developmental arrest by depletion of aprotein at the parasite–host interface. Proc Natl Acad SciUSA 102: 3022–3027.
O’Donnell, R.A., Freitas-Junior, L.H., Preiser, P.R.,Williamson, D.H., Duraisingh, M., McElwain, T.F., et al.(2002) A genetic screen for improved plasmid segregationreveals a role for Rep20 in the interaction of Plasmodiumfalciparum chromosomes. EMBO J 21: 1231–1239.
Orlando, V., Strutt, H., and Paro, R. (1997) Analysis ofchromatin structure by in vivo formaldehyde cross-linking.Methods 11: 205–214.
Santoso, S., Zimmermann, U., Neppert, J., and Mueller-Eckhardt, C. (1986) Receptor patching and capping of
platelet membranes induced by monoclonal antibodies.Blood 67: 343–349.
Simmons, D., Woollett, G., Bergin-Cartwright, M., Kay, D.,and Scaife, J. (1987) A malaria protein exported into a newcompartment within the host erythrocyte. EMBO J 6: 485–491.
Simons, K., and Toomre, D. (2000) Lipid rafts and signaltransduction. Nat Rev Mol Cell Biol 1: 31–39.
Slomianny, C., Prensier, G., and Charet, P. (1985) Ingestionof erythrocytic stroma by Plasmodium chabaudi trophozo-ites: ultrastructural study by serial sectioning and 3-dimen-sional reconstruction. Parasitology 90: 579–588.
Spiegel, S., Kassis, S., Wilchek, M., and Fishman, P.H.(1984) Direct visualization of redistribution and capping offluorescent gangliosides on lymphocytes. J Cell Biol 99:1575–1581.
Spielmann, T., and Beck, H.P. (2000) Analysis of stage-specific transcription in Plasmodium falciparum reveals aset of genes exclusively transcribed in ring stage parasites.Mol Biochem Parasitol 111: 453–458.
Spielmann, T., Fergusen, D.J., and Beck, H.P. (2003)etramps, a new Plasmodium falciparum gene family codingfor developmentally regulated and highly charged mem-brane proteins located at the parasite–host cell interface.Mol Biol Cell 14: 1529–1544.
Trager, W., and Jensen, J.B. (1976) Human malaria parasitesin continuous culture. Science 193: 673–675.
Waller, R.F., Reed, M.B., Cowman, A.F., and McFadden, G.I.(2000) Protein trafficking to the plastid of Plasmodiumfalciparum is via the secretory pathway. EMBO J 19: 1794–1802.
Wu, Y., Kirkman, L.A., and Wellems, T.E. (1996) Transforma-tion of Plasmodium falciparum malaria parasites by homol-ogous integration of plasmids that confer resistance topyrimethamine. Proc Natl Acad Sci USA 93: 1130–1134.
Supplementary material
The following supplementary material is available for thisarticle online:Appendix S1. Estimation of ETRAMP and glycophorin Adensities in the membrane.Fig. S1. Patching of ETRAMPs occurs under different per-meabilisation conditions of IRBCs but is reduced upon glut-araldehyde treatment.Fig. S2. Expression of recombinant ETRAMPs in E. coli.Fig. S3. Example of a Western blot used for estimation ofETRAMP4 and ETRAMP2 copy numbers per parasite cell.Table S1. Band sizes of ETRAMP complexes.
This material is available as part of the online article fromhttp://www.blackwell-synergy.com





























