Embed Size (px)
Citation preview

ORIGINAL ARTICLE
p53 suppresses structural chromosome instability after mitotic arrest
in human cells
WB Dalton1,2, B Yu1,2 and VW Yang1,2,3
1Division of Digestive Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, GA, USA; 2Biochemistry,Cell and Developmental Biology Graduate Program, Emory University School of Medicine, Atlanta, GA, USA and 3Departmentof Hematology and Medical Oncology, Emory University School of Medicine, Atlanta, GA, USA
The p53 tumor suppressor inhibits the proliferation of cellsthat undergo prolonged activation of the mitotic checkpoint.However, the function of this antiproliferative response isnot well defined. Here, we report that p53 suppressesstructural chromosome instability after mitotic arrest inhuman cells. In both HCT116 colon cancer cells and normalhuman fibroblasts, DNA breaks occurred during mitoticarrest in a p53-independent manner, but p53 was required tosuppress the proliferation and structural chromosomeinstability of the resulting polyploid cells. In contrast, cellsmade polyploid without mitotic arrest exhibited neithersignificant structural chromosome instability nor p53-dependent cell cycle arrest. We also observed that p53suppressed both the frequency and structural chromosomeinstability of spontaneous polyploids in HCT116 cells.Furthermore, time-lapse videomicroscopy revealed thatpolyploidization of p53�/� HCT116 cells is frequentlyaccompanied by mitotic arrest. These data suggest that afunction of the p53-dependent postmitotic response is theprevention of structural chromosome instability afterprolonged activation of the mitotic checkpoint. Accordingly,our study suggests a novel mechanism of tumor suppressionfor p53, as well as a potential function for p53 in theoutcome of antimitotic chemotherapy.Oncogene (2010) 29, 1929–1940; doi:10.1038/onc.2009.477;published online 11 January 2010
Keywords: p53; cell cycle arrest; chromosomal instabil-ity; DNA damage; mitotic checkpoint; polypoidization
Introduction
The p53 tumor suppressor represents a central defenseagainst human cancer (Vousden and Lane, 2007). Itsinactivation is one of the most common alterations inhuman tumors, and numerous studies have establishedthe tumor suppressing properties of p53 (Toledo andWahl, 2006). A principal mechanism of this tumor
suppression is the induction of growth arrest and/orapoptosis in cells that suffer DNA damage (Vousden andLu, 2002). In this way, p53 inhibits the propagation ofcells that harbor potentially oncogenic DNA alterations.In addition, other forms of stress have been shown toactivate p53-dependent responses (Vousden and Lane,2007). One example is prolonged activation of the mitoticcheckpoint, which elicits a p53-dependent cell cycle arrest(Ganem and Pellman, 2007). This ‘postmitotic’ response,so named because growth arrest is actually imposed oncells that have exited from prolonged mitosis, has beenobserved in numerous cell systems (Cross et al., 1995;Minn et al., 1996; Di Leonardo et al., 1997; Lanni andJacks, 1998; Andreassen et al., 2001; Rajagopalan et al.,2004; Chan et al., 2008). Despite the ubiquity of thepostmitotic response, its mechanism is not well defined(Stukenberg, 2004; Ganem and Pellman, 2007).
One clue to the function of the postmitotic responsemay be that prolonged activation of the mitoticcheckpoint has been causally implicated in tumorigen-esis (Dalton and Yang, 2009). Indeed, mitosis isfrequently prolonged in cancer cells, and several geneticand epigenetic defects, which cause mitotic arrest, cancontribute to cancer (Dalton and Yang, 2009). For someof these defects, such as inactivation of Rb and hCDC4,oncogenic activation of c-Myc, and the presenceof supernumerary chromosomes and/or centrosomes,prolonged mitosis is one of many cellular effects,which may or may not be oncogenic (Hernando et al.,2004; Rajagopalan et al., 2004; Fujiwara et al., 2005;Yang et al., 2008). However, mitotic arrest and canceralso develop in mice overexpressing Mad2, a proteinprincipally involved in mitotic checkpoint signaling,providing strong evidence that prolonged mitoticcheckpoint activation can directly promote tumorigen-esis (Sotillo et al., 2007). Accordingly, the p53-depen-dent postmitotic response may serve to inhibit thepropagation of cells, which acquire oncogenic propertiesduring prolonged activation of the mitotic checkpoint.
What aspects of mitotic arrest might be oncogenic?Certainly, one candidate is aneuploidy and/or tetra-ploidy resulting from the chromosome missegregationand/or cytokinesis failure, which can follow prolongedactivation of the mitotic checkpoint (Ganem andPellman, 2007). Indeed, in some contexts, aneuploidyand tetraploidy have themselves been causallyimplicated in tumorigenesis (Fujiwara et al., 2005;
Received 10 July 2009; revised 7 November 2009; accepted 22 November2009; published online 11 January 2010
Correspondence: Dr VW Yang, Division of Digestive Diseases,Department of Medicine, Emory University School of Medicine,201 Whitehead Research Building, 615 Michael Street, Atlanta,GA 30322, USA.E-mail: [email protected]
Oncogene (2010) 29, 1929–1940& 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00
www.nature.com/onc

Weaver et al., 2007). At the same time, we and othersrecently found that mitotic arrest can induce structuralchromosome changes resulting from double-strandedDNA breaks (Dalton et al., 2007; Quignon et al., 2007;Stevens et al., 2007). Given the role of structuralchromosome aberrations in tumorigenesis, these obser-vations suggest that one way prolonged mitosis couldpromote cancer is through introduction of DNA breaks.By extension, one function of the p53-dependentpostmitotic response may be to prevent this structuralchromosome instability. To investigate this possibility,we have measured structural chromosome instabilityresulting from mitotic arrest in human colon cancer cellsand normal fibroblasts, which differ only in their p53status. Our results show that, by imposing growth arrestand/or apoptosis in cells whose DNA is damaged duringmitotic arrest, p53 suppresses structural chromosomeinstability after prolonged mitotic checkpoint activationin human cells.
Results
Both p53þ /þ and p53�/� cells acquire g-H2AX foci duringmitotic arrestTo examine the function of p53 in structural chromo-some instability after prolonged mitosis, we first askedwhether p53 influences the acquisition of DNA damageduring pharmacologic induction of mitotic arrest inisogenic p53þ /þ and p53�/� HCT116 cells (Bunz et al.,1998). On treatment with the microtubule-depolymeriz-ing agent nocodazole, both p53þ /þ and p53�/� cellsexhibited a transient rise in mitotic index, which peakedat 12–18 h (Figure 1a). This was followed by ‘mitoticslippage,’ a process whereby mitotically arrested cellsreturn to interphase without undergoing cell division(Figure 1a) (Rieder and Maiato, 2004). To determine theextent of DNA damage acquired during this arrest, wemeasured the formation of g-H2AX, the phosphorylatedform of histone H2AX, which forms around sites ofDNA breaks (Rogakou et al., 1999). Similar to ourearlier findings (Dalton et al., 2007), nocodazole-arrested prometaphase p53þ /þ cells showed an increasein g-H2AX foci, as compared with prometaphasecontrols (Figure 1b). Increased g-H2AX foci were alsoobserved in p53�/� cells (Figure 1b), and flow cytometricanalysis showed that the magnitude of this increase wascomparable with p53þ /þ cells (Figure 1c). To determinewhether increased g-H2AX was indeed the result ofmitotic arrest, and not some other effects of nocodazole,we performed the assay after siRNA-mediated knock-down of the Eg5 mitotic kinesin protein, whoseinactivation prevents centrosome separation, producesa monopolar spindle, and thereby induces mitotic arrest(Koller et al., 2006). Indeed, knockdown of Eg5produced an elevated mitotic index, monopolar spin-dles, and increased g-H2AX foci in mitotic p53þ /þ andp53�/� cells (Figures 1d–f). Thus, both pharmacologicand genetic manipulations, which provoke mitotic arrestthrough distinct mechanisms, produce evidence of DNAbreaks in p53þ /þ and p53�/� HCT116 cells.
p53 inhibits the polyploidization and survivalof postmitotic cellsHaving observed that p53 does not influence theacquisition of DNA damage during mitotic arrest, wenext investigated whether the cellular consequences ofthis damage are dependent on p53. We first determinedthe fates of p53þ /þ and p53�/� cells after nocodazoletreatment. Similar to results from earlier studies (Kimet al., 2004; Vogel et al., 2004; Castedo et al., 2006),p53�/� cells exhibited greater polyploidization andreduced apoptosis when compared with p53þ /þ cellsduring 48 h nocodazole (Figures 2a–e; SupplementaryFigure S1). As the specificity of gene targeting in cancercells is not always certain (Pfleghaar et al., 2005; Matobaet al., 2006), we independently confirmed the p53dependence of these phenotypes using miRNA (Supple-mentary Figure S2). Moreover, because short-term ratesof apoptosis do not always correspond to long-termrates of survival (Bunz et al., 1999), we also determinedthe clonogenicity of these nocodazole-treated cells. Thisanalysis revealed that colony survival of p53�/� cellsexposed to 48 h nocodazole, while low overall, wasfivefold of that of p53þ /þ cells (Figure 2f). Collectively,these data indicate that p53 inhibits polyploidization,promotes apoptosis, and suppresses clonogenicitiy afterpharmacologic induction of mitotic arrest in HCT116cells.
p53 inhibits structural chromosome instabilityin postmitotic cellsBy inducing growth arrest and apoptosis in cells, whichhave undergone prolonged mitotic arrest, p53 could actto suppress the structural chromosome changes, whichresult from DNA damage acquired during prolongedmitosis. If true, p53�/� cells made polyploid afterprolonged mitosis might be expected to exhibit not onlygreater survival, but also more structural chromosomeaberrations than their p53þ /þ counterparts. To test thishypothesis, we examined p53þ /þ and p53�/� HCT116cells for the presence of chromosome aberrations after48 h of nocodazole. Consistent with our earlier findings(Dalton et al., 2007), nocodazole-induced polyploidHCT116 cells exhibited multiple types of structuralchromosome aberrations (Figure 3). Notably, nocoda-zole-treated p53�/� cells exhibited a 50% higher burdenof aberrations than p53þ /þ cells (Po0.0001) (Figure 3).This result suggests that p53 preferentially inhibits thepolyploidization and survival of those cells that sufferthe greatest DNA damage during mitotic arrest. In thisway, p53 suppresses structural chromosome instabilityafter mitotic arrest in HCT116 cells.
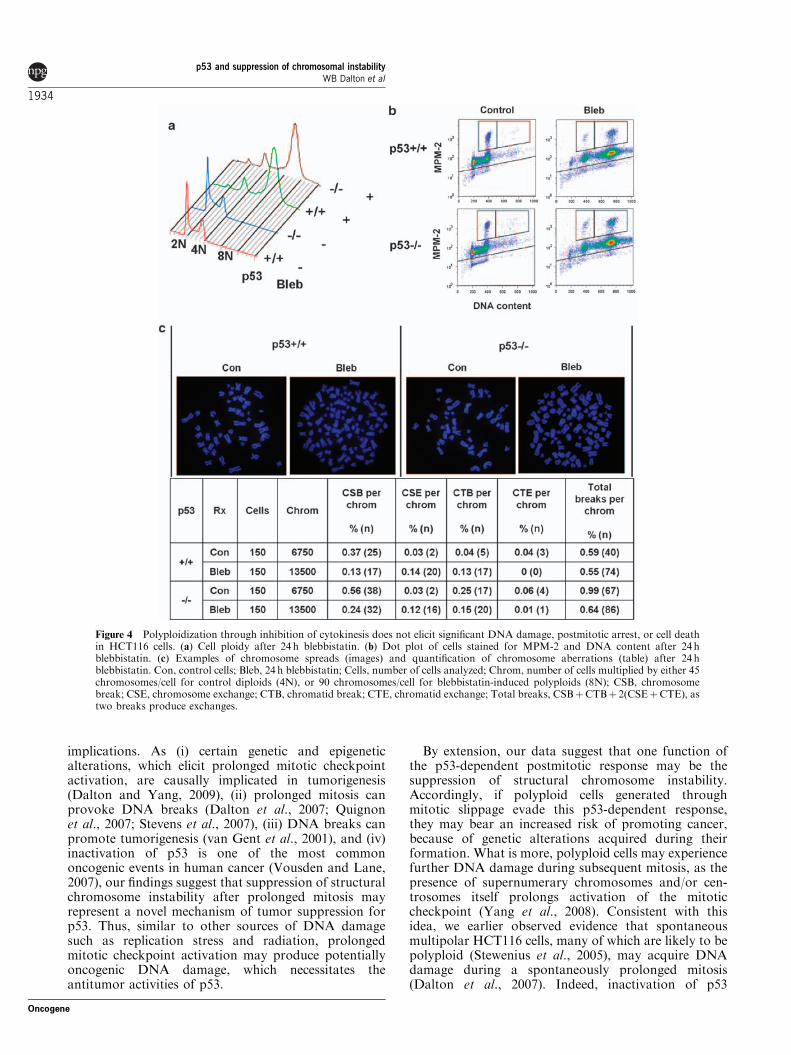
Polyploidy is not responsible for structural chromosomeinstability after mitotic arrestTo address the possibility that the polyploid state itselfmight elicit structural chromosome changes (Ganemet al., 2007), we determined the cell fate and genomicstability of p53þ /þ and p53�/� HCT116 cells treated withthe myosin inhibitor blebbistatin, which creates poly-ploid cells through inhibition of cytokinesis without
p53 and suppression of chromosomal instabilityWB Dalton et al
1930
Oncogene

provoking mitotic arrest (Wong and Stearns, 2005).In contrast to nocodazole, blebbistatin was capable ofproducing polyploidy without significant cell death, cellcycle arrest, or increased chromosome aberrations(Figures 4a–c). Moreover, no difference in theseparameters was observed between p53þ /þ and p53�/�
cells (Figures 4a–c). This result indicates that it isprolonged mitotic arrest, and not polyploidy per se, thatelicits significant DNA damage and p53-dependentsuppression of structural chromosome instability innocodazole-treated HCT116 cells. Interestingly,although the total number of breaks per chromosomewas not increased by blebbistatin treatment, thefrequencies of certain aberrations did change(Figure 4c). This pattern was also observed aftercytokinesis inhibition using dihydrocytochalasin B(unpublished observations). Thus, although polyploidi-zation itself does not account for the observed structuralchromosome instability after prolonged mitotic arrest, itmay be that polyploidization, or pharmacologic inhibi-tion of cytokinesis, may induce some degree of DNAdamage.
p53 suppresses structural chromosome instability aftermitotic arrest in normal human fibroblastsTo test whether p53 suppresses structural chromosomeinstability after mitotic arrest in untransformed human
cells, we examined the response to nocodazole of humandiploid fibroblasts (HDFs) in which p53 was silencedthrough lentivirus-mediated expression of miRNAs(Figure 5a). In HDFs expressing either p53 or controlmiRNAs, nocodazole produced an increase in g-H2AXfoci during mitotic arrest (Figure 5b). Thus, as inHCT116 cells, p53 does not influence the acquisition ofDNA damage during mitotic arrest in normal humancells. Also similar to HCT116 cells, HDFs expressingp53 miRNA exhibited an increase in polyploidization(3.5- to 7.5-fold, Po0.05) and structural chromosomeinstability (2.6- to 2.8-fold, Po0.01) after nocodazoletreatment, as compared with control miRNA (Figures5c–e). In addition to harboring chromosome aberrationssuch as fragments and dicentrics, a minority of HDFsalso exhibited evidence of chromosome pulverization,which can occur when micronuclei enter mitosis prema-turely (Supplementary Figure S3) (Kato and Sandberg,1967; Ikeuchi et al., 1972). Nocodazole-induced celldeath, however, did not seem to be influenced by p53knockdown in HDFs (Supplementary Figure S4),showing that the function of p53 on apoptosis afterprolonged mitotic arrest is cell-type dependent, asdiscussed earlier. These data, therefore, show that, byinducing postmitotic cell cycle arrest, p53 suppressesstructural chromosome instability after mitotic arrest inuntransformed, as well as transformed, human cells.
Figure 1 Both p53þ /þ and p53�/� HCT116 cells acquire DNA damage during mitotic arrest. (a) Diploid mitotic index of HCT116 cellsduring 48h nocodazole, as determined by MPM-2 flow cytometry. Means and s.e.m.s are from 2–3 independent experiments.(b) Images of prometaphase cells stained for g-H2AX and a-tubulin. Nuclei were counterstained with DAPI. Noc, 18h nocodazole;g-irr, 30min after 2Gy g-irradiation. (c) Flow cytometric analysis of mitotic g-H2AX. Cells were treated with or without 18hnocodazole and stained for CC-3 and g-H2AX. CC-3-positive cells, which are mitotic (Thibodeau and Vincent, 1991), were gated, andg-H2AX signals of the gated cells are shown. Data are representative of two independent experiments. (d) Western blot analysis of Eg5 inHCT116 cells 24 h after transfection with Eg5-specific, or control, siRNA. Actin was used as a loading control. (e) Diploid mitotic indexof HCT116 cells (blue bars, p53þ /þ ; maroon bars, p53�/�) 24 h after transfection with Eg5-specific, or control, siRNA, as determinedby MPM-2 flow cytometry. Means and s.e.m.s are from two independent experiments. *, Po0.05, for t-tests, as compared with control.(f) Images of prometaphase cells stained for g-H2AX and a-tubulin 24h after transfection with Eg5-specific, or control, siRNA.
p53 and suppression of chromosomal instabilityWB Dalton et al
1931
Oncogene

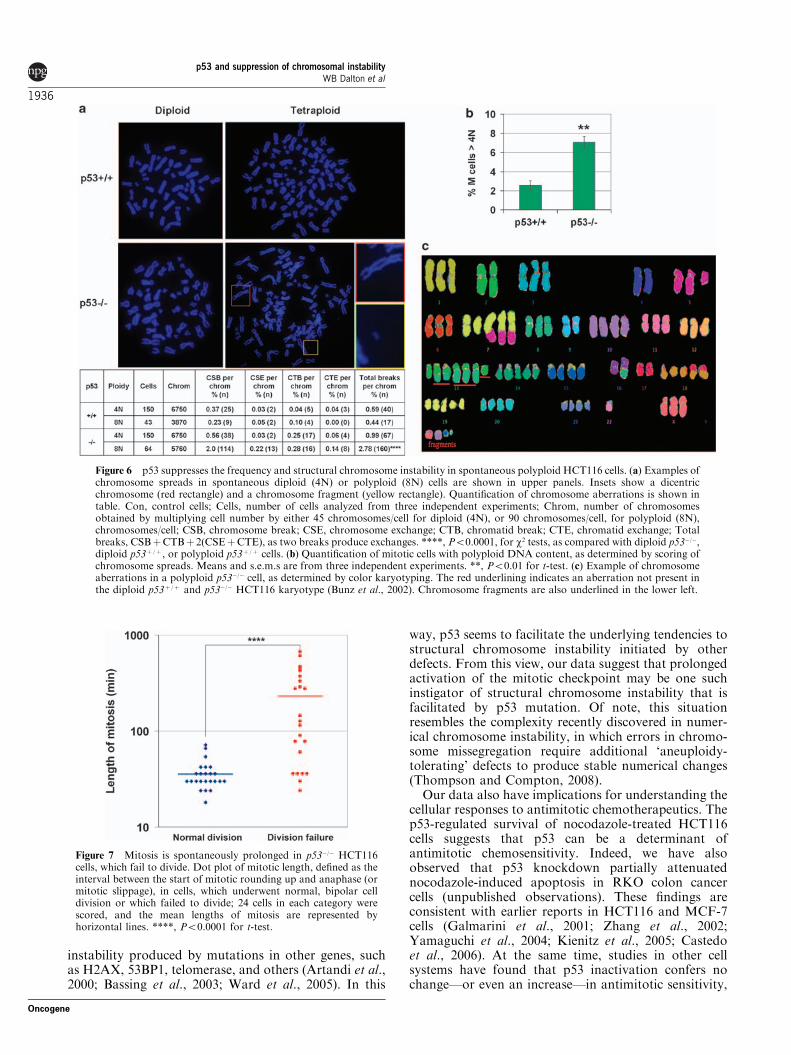
p53 suppresses structural chromosome instabilityin spontaneous polyploidsInterestingly, while performing cytogenetic analysis ofHCT116 cells, we noticed that many spontaneouspolyploid p53�/� cells contained chromosome aberra-tions, including chromosome fragments, dicentrics, andchromatid breaks (Figure 6a). Indeed, quantificationrevealed a 2.8-fold increase in the number of breaksper chromosome in polyploid (8N) vs diploid (4N)
p53�/� cells (Po0.0001) (Figure 6a). Color karyotyping ofthese spontaneous p53�/� polyploids (44N) confirmedthe presence of cells with chromosome fragments andchromosome rearrangements (Figure 6c). In contrast,spontaneous p53þ /þ polyploids did not exhibit anelevated frequency of chromosome aberrations, whencompared with p53þ /þ diploid cells (Figure 6a). More-over, we found that, while of low overall abundance,spontaneous polyploid mitotic cells were threefold more
Figure 2 p53 inhibits the polyploidization and survival of HCT116 cells after prolonged mitotic arrest. (a) Example of cell ploidy (leftpanel) and quantification of polyploidy (right panel) in nocodazole-treated cells. Means and s.e.m.s are from 2–3 independentexperiments. *, Po0.05; **, Po0.01 for t-tests. (b) Dot plots of cells stained for MPM-2 and DNA content after 24 and 48 hnocodazole treatment. (c) Quantification of polyploid (44N) mitotic index (upper right gate in (b)) after 48 h nocodazole. Means ands.e.m.s are from 2–3 independent experiments. *, Po0.05, **, Po0.01 for t-tests. (d) Quantification of cell death (lower gate in (b))during 48 h nocodazole treatment. Cells that were entirely negative for the MPM-2 phosphoepitope were found, through fluorescentmicroscopy, to be apoptotic (Supplementary Figure S1). Means and s.e.m.s are from 2–3 independent experiments. *, Po0.05, **,Po0.01 for t-tests. (e) Western blot analysis of p53 and cleaved PARP levels during 48 h nocodazole. Actin was used as a loadingcontrol. (f) Example of colony survival after 48 h nocodazole (left panel) and its quantification (right panel). % survival is the numberof nocodazole-treated colonies divided by the number of control colonies, although more sparse control flasks were used forquantification (see Materials and methods). Means and s.e.m.s are from three independent experiments. **, Po0.01 for t-test.
p53 and suppression of chromosomal instabilityWB Dalton et al
1932
Oncogene

frequent in p53�/�, as compared with p53þ /þ , cells(Figure 6b), consistent with earlier studies (Bunz et al.,2002, Pantic et al., 2006). These findings thus indicatethat, in HCT116 cells, p53 suppresses both thefrequency and the structural chromosome instability ofspontaneous polyploids.
Mitotic slippage after prolonged mitosis occursspontaneously in p53�/� HCT116 cellsAlthough the origin of spontaneously damaged p53�/�
polyploids is unknown, one possibility is that they arisefrom cells that undergo mitotic slippage after mitoticarrest, for example because of the appearance ofspontaneous spindle defects, which we earlier observedat low frequency in HCT116 cells (Dalton et al., 2007).To examine this possibility, we examined mitoticprogression using time-lapse videomicroscopy. Thisanalysis revealed that 4% (24/592) of mitoses resultedin spontaneous cell division failure in p53�/� HCT116
cells. Moreover, the average length of mitosis in thesecells was significantly longer than that of cells thatcompleted a normal, bipolar mitosis (229 vs 36min,Po0.0001) (Figure 7). Indeed, some cells spent up to10 h in spontaneous mitotic arrest before undergoingmitotic slippage (Supplementary Videos S1–S3). Thesedata thus indicate that mitotic slippage after prolongedmitosis can occur spontaneously and are consistent withthe possibility that this mechanism of polyploidizationmay be responsible for the increased structural chromo-some instability in polyploid p53�/� HCT116 cells.
Discussion
Our study shows that in both human colon cancer cellsand normal human fibroblasts, p53 suppresses structuralchromosome instability after prolonged activation of themitotic checkpoint. This finding has several important
Figure 3 p53 suppresses structural chromosome instability after mitotic arrest in HCT116 cells. Examples of chromosome spreads incells treated with or without nocodazole (Noc) for 48 h are shown in upper panels. Insets show chromosome fragments (upper left,lower left, lower right), a chromatid break (lower left), a ring chromosome (upper right), and dicentric chromosomes (upper-middleright, lower-middle right). Quantification of chromosome aberrations is shown in table. Con, control cells; Noc, 48 h nocodazole;Cells, number of cells analyzed from three independent experiments; Chrom, number of chromosomes obtained by multiplying cellnumber by either 45 chromosomes/cell for control diploids (4N), or 90 chromosome/cell for nocodazole-induced polyploids (8N);CSB, chromosome break; CSE, chromosome exchange; CTB, chromatid break; CTE, chromatid exchange; Total breaks,CSBþCTBþ 2(CSEþCTE), as two breaks produce exchanges; ****, Po0.0001, for w2 test, as compared with nocodazole-treatedp53þ /þ cells.
p53 and suppression of chromosomal instabilityWB Dalton et al
1933
Oncogene
implications. As (i) certain genetic and epigeneticalterations, which elicit prolonged mitotic checkpointactivation, are causally implicated in tumorigenesis(Dalton and Yang, 2009), (ii) prolonged mitosis canprovoke DNA breaks (Dalton et al., 2007; Quignonet al., 2007; Stevens et al., 2007), (iii) DNA breaks canpromote tumorigenesis (van Gent et al., 2001), and (iv)inactivation of p53 is one of the most commononcogenic events in human cancer (Vousden and Lane,2007), our findings suggest that suppression of structuralchromosome instability after prolonged mitosis mayrepresent a novel mechanism of tumor suppression forp53. Thus, similar to other sources of DNA damagesuch as replication stress and radiation, prolongedmitotic checkpoint activation may produce potentiallyoncogenic DNA damage, which necessitates theantitumor activities of p53.
By extension, our data suggest that one function ofthe p53-dependent postmitotic response may be thesuppression of structural chromosome instability.Accordingly, if polyploid cells generated throughmitotic slippage evade this p53-dependent response,they may bear an increased risk of promoting cancer,because of genetic alterations acquired during theirformation. What is more, polyploid cells may experiencefurther DNA damage during subsequent mitosis, as thepresence of supernumerary chromosomes and/or cen-trosomes itself prolongs activation of the mitoticcheckpoint (Yang et al., 2008). Consistent with thisidea, we earlier observed evidence that spontaneousmultipolar HCT116 cells, many of which are likely to bepolyploid (Stewenius et al., 2005), may acquire DNAdamage during a spontaneously prolonged mitosis(Dalton et al., 2007). Indeed, inactivation of p53
Figure 4 Polyploidization through inhibition of cytokinesis does not elicit significant DNA damage, postmitotic arrest, or cell deathin HCT116 cells. (a) Cell ploidy after 24 h blebbistatin. (b) Dot plot of cells stained for MPM-2 and DNA content after 24 hblebbistatin. (c) Examples of chromosome spreads (images) and quantification of chromosome aberrations (table) after 24 hblebbistatin. Con, control cells; Bleb, 24 h blebbistatin; Cells, number of cells analyzed; Chrom, number of cells multiplied by either 45chromosomes/cell for control diploids (4N), or 90 chromosomes/cell for blebbistatin-induced polyploids (8N); CSB, chromosomebreak; CSE, chromosome exchange; CTB, chromatid break; CTE, chromatid exchange; Total breaks, CSBþCTBþ 2(CSEþCTE), astwo breaks produce exchanges.
p53 and suppression of chromosomal instabilityWB Dalton et al
1934
Oncogene

increased the frequency of these damaged multipolarcells (data not shown). Thus, p53-dependent postmitoticarrest may suppress a ‘vicious cycle’ of structuralchromosome instability occurring during the forma-tion—and propagation—of polyploid cells. This mayhelp explain why polyploid cells created throughcytokinesis inhibition—and thus without prior mitoticarrest—have increased tumorigenicity in a p53-deficientbackground (Fujiwara et al., 2005).
Earlier studies have shown that the function of p53 inthe regulation of spontaneous structural chromosomeintegrity is context dependent. For example, increasedstructural aberrations have been observed in bonemarrow cells of p53�/� mice (Bouffler et al., 1995) andfibroblasts from patients with germline p53 mutations(Bischoff et al., 1990), but not in lymphoblastoid cellsfrom patients with germline p53 mutations (Lalle et al.,1995) or isogenic human cancer cell lines (Bunz et al.,2002) (see Supplementary Information for a discussion
of the last example). Similar context dependence hasbeen observed in cytogenetic and comparative genomichybridization studies of human tumor tissue, as p53mutations correlate with structural chromosome in-stability in several case series (De Angelis et al., 1999;Jong et al., 2004; Kleivi et al., 2005), but not in others(Eshleman et al., 1998; Curtis et al., 2000; Westra et al.,2005). Interestingly, some of this context dependencemay be due to differences in the types of p53 mutationsthat occur, as there is evidence that gain-of-function p53mutations are more likely than null p53 mutations topromote structural chromosome instability (Song et al.,2007). Conversely, much of the complexity may resultfrom the different genetic backgrounds of tissue typesand individual tumors in which p53 is mutated. Indeed,several studies have shown that while p53 mutationalone may have minimal effects on structural chromo-some instability in certain mouse models, its mutationcan greatly exacerbate structural chromosome
Figure 5 p53 suppresses structural chromosome instability after mitotic arrest in IMR90 human diploid fibroblasts (HDFs).(a) Western blot analysis of p53 after 72 h nocodazole in cells stably expressing either control or two independent p53 miRNAs. Actinwas used as a loading control. (b) Images of prometaphase cells stained for g-H2AX and a-tubulin. Con, control; Noc, 18 hnocodazole. (c) Quantification of polyploid mitotic index in miRNA-expressing cells (blue bars, miRNA-Con; maroon bars, miRNA-p53-1; yellow bars, miRNA-p53-2) after 72 h nocodazole, as determined by MPM-2 flow cytometry. Means and s.e.m.s are from twoindependent experiments. *, Po0.05 for t-tests, as compared with miRNA-Con cells treated with 48 h nocodazole. (d) Examples ofchromosome spreads in cells treated with or without 72 h nocodazole. Untreated (upper) or nocodazole-treated (lower) cells expressingeither control (left) or p53-1 (right) miRNAs. Insets show dicentric chromosomes. (e) Quantification of chromosome aberrations. Con,control cells; Noc, 72 h nocodazole; Cells, number of cells analyzed from two independent experiments; Chrom, number ofchromosomes obtained by multiplying cell number by either 46 chromosomes/cell for control diploids (2N), or 92 chromosomes/cellfor nocodazole-induced polyploids (8N); CSB, chromosome break; CSE, chromosome exchange; CTB, chromatid break; CTE,chromatid exchange; Total breaks, CSBþCTBþ 2(CSEþCTE), as two breaks produce exchanges; **, Po0.01; ***, Po0.001, for w2
tests, as compared with nocodazole-treated cells expressing control miRNA.
p53 and suppression of chromosomal instabilityWB Dalton et al
1935
Oncogene
instability produced by mutations in other genes, suchas H2AX, 53BP1, telomerase, and others (Artandi et al.,2000; Bassing et al., 2003; Ward et al., 2005). In this
way, p53 seems to facilitate the underlying tendencies tostructural chromosome instability initiated by otherdefects. From this view, our data suggest that prolongedactivation of the mitotic checkpoint may be one suchinstigator of structural chromosome instability that isfacilitated by p53 mutation. Of note, this situationresembles the complexity recently discovered in numer-ical chromosome instability, in which errors in chromo-some missegregation require additional ‘aneuploidy-tolerating’ defects to produce stable numerical changes(Thompson and Compton, 2008).
Our data also have implications for understanding thecellular responses to antimitotic chemotherapeutics. Thep53-regulated survival of nocodazole-treated HCT116cells suggests that p53 can be a determinant ofantimitotic chemosensitivity. Indeed, we have alsoobserved that p53 knockdown partially attenuatednocodazole-induced apoptosis in RKO colon cancercells (unpublished observations). These findings areconsistent with earlier reports in HCT116 and MCF-7cells (Galmarini et al., 2001; Zhang et al., 2002;Yamaguchi et al., 2004; Kienitz et al., 2005; Castedoet al., 2006). At the same time, studies in other cellsystems have found that p53 inactivation confers nochange—or even an increase—in antimitotic sensitivity,
Figure 6 p53 suppresses the frequency and structural chromosome instability in spontaneous polyploid HCT116 cells. (a) Examples ofchromosome spreads in spontaneous diploid (4N) or polyploid (8N) cells are shown in upper panels. Insets show a dicentricchromosome (red rectangle) and a chromosome fragment (yellow rectangle). Quantification of chromosome aberrations is shown intable. Con, control cells; Cells, number of cells analyzed from three independent experiments; Chrom, number of chromosomesobtained by multiplying cell number by either 45 chromosomes/cell for diploid (4N), or 90 chromosomes/cell, for polyploid (8N),chromosomes/cell; CSB, chromosome break; CSE, chromosome exchange; CTB, chromatid break; CTE, chromatid exchange; Totalbreaks, CSBþCTBþ 2(CSEþCTE), as two breaks produce exchanges. ****, Po0.0001, for w2 tests, as compared with diploid p53�/�,diploid p53þ /þ , or polyploid p53þ /þ cells. (b) Quantification of mitotic cells with polyploid DNA content, as determined by scoring ofchromosome spreads. Means and s.e.m.s are from three independent experiments. **, Po0.01 for t-test. (c) Example of chromosomeaberrations in a polyploid p53�/� cell, as determined by color karyotyping. The red underlining indicates an aberration not present inthe diploid p53þ /þ and p53�/� HCT116 karyotype (Bunz et al., 2002). Chromosome fragments are also underlined in the lower left.
Figure 7 Mitosis is spontaneously prolonged in p53�/� HCT116cells, which fail to divide. Dot plot of mitotic length, defined as theinterval between the start of mitotic rounding up and anaphase (ormitotic slippage), in cells, which underwent normal, bipolar celldivision or which failed to divide; 24 cells in each category werescored, and the mean lengths of mitosis are represented byhorizontal lines. ****, Po0.0001 for t-test.
p53 and suppression of chromosomal instabilityWB Dalton et al
1936
Oncogene

showing the cell-type specificity of this effect (Woodset al., 1995; Minn et al., 1996; Wahl et al., 1996; Taoet al., 2007). Our own observation that p53 knockdowndid not significantly affect nocodazole-induced apopto-sis in HDFs is consistent with this idea. This contextdependence is likely influenced by the same factorsenumerated above for the function of p53 in structuralchromosome instability, as tissue of origin and p53mutation type have earlier been implicated in p53-dependent sensitivity to other chemotherapeutics (Luand El-Deiry, 2009). Thus, a conservative interpretationof these findings is that p53 may be a determinant ofantimitotic sensitivity in a subset of human tumors,which also share other modulators of drug sensitivity.Identification of such modulators will be an interestingarea for future work, and might include investigationsinto DNA repair pathways (Swanton et al., 2009),as well as other p53 family members, such as p63 andp73, both of which are known to influence chemosensi-tivity (Muller et al., 2006). Finally, with the ongoingdevelopment of pharmaceuticals, which partially restorep53 activity in cancer cells (Lu and El-Deiry, 2009), itmay be worthwhile to determine whether such agentsmight work in synergy with antimitotics by restoringp53-dependent apoptotic and growth arrest signalingafter prolonged mitotic arrest.
An important but unanswered question is whattriggers p53 during this process (Stukenberg, 2004;Ganem and Pellman, 2007; Chan et al., 2008). Althoughan initial proposal was that p53 is activated by amechanism, which senses, or counts, the presence of atetraploid genome (Andreassen et al., 2001), subsequentstudies showed that tetraploid cells produced in theabsence of prolonged mitosis do not necessarily undergop53-dependent growth arrest (Uetake and Sluder, 2004,2007; Wong and Stearns, 2005). Indeed, the results ofour blebbistatin experiment (Figure 5) support theconclusions of these later studies. Another proposalhas been that the transcriptional repression, whichoccurs during mitosis leads to inhibition of p53degradation, which in turn leads to progressive accu-mulation of p53 protein during prolonged mitotic arrest(Blagosklonny, 2006). However, this ‘mitotic timer’model for p53 is not supported by studies showing thatconditions that dramatically shorten mitotic arrest donot diminish the accumulation of p53 (Vogel et al.,2004; Chan et al., 2008), nor by data showing that p53accumulation occurs after, and not during, mitoticarrest (Minn et al., 1996).
As DNA damage is a well-established activator of p53(Vousden and Lu, 2002), it is tempting to speculate thatpostmitotic activation of p53 is induced, or influenced,by DNA damage acquired during mitotic arrest. Indeed,our finding that loss of p53 increases the burden ofchromosome aberrations in nocodazole-induced poly-ploids—but does not affect the initial acquisition ofDNA damage during mitotic arrest—suggests that thep53-dependent postmitotic response preferentially in-hibits the most damaged cells. This, in turn, suggeststhat the extent of DNA damage acquired duringprolonged mitosis influences cell fate. In this way, the
postmitotic activation of p53 by DNA damage would beanalogous to p53 activation after blockage of nucleotidebiosynthesis, which, while initially believed to be anon-genotoxic inducer of p53 (Linke et al., 1996), hasrecently been shown, with more sensitive assays, tocause DNA damage (Hastak et al., 2008).
At the same time, it remains possible that DNAdamage is one, but not the only, determinant of p53activation after prolonged mitotic arrest (Ganem andPellman, 2007). Indeed, we earlier observed significantcell-type variation in nocodazole-induced g-H2AX foci(Dalton et al., 2007), which raises the question ofwhether DNA damage could account for p53 activationafter mitotic arrest in all cell types. Along these lines, alack of increased g-H2AX in nocodazole-treated U20Scells has been reported (Aylon et al., 2006; Chan et al.,2008). However, multiple studies have now reportedevidence that DNA damage accompanies treatments,which induce mitotic arrest in a variety of cell lines(Tighe et al., 2004; Wong and Stearns, 2005; Daltonet al., 2007; Quignon et al., 2007; Stevens et al., 2007; Shiet al., 2008). Furthermore, it is important to note that alack of increased g-H2AX does not rule out the presenceof DNA damage, as there are DNA lesions, such assingle-stranded breaks and base alterations, which donot induce g-H2AX (Rogakou et al., 1998). In fact,some of the chromatid-type aberrations we observedafter prolonged mitotic arrest could result from lesionsother than double-stranded breaks (Zhuanzi et al.,2007). Clearly, future studies into the mechanismsresponsible for DNA damage during mitotic arrest willbe needed to determine its function in postmitotic p53activation. Nonetheless, we believe our data show that p53functions to inhibit the potentially dangerous conse-quences of DNA damage acquired during mitotic arrest.
Materials and methods
Cell lines and treatmentsIMR90 HDFs were obtained from the ATCC. p53þ /þ andp53�/� HCT116 cells were kindly provided by B Vogelstein(Johns Hopkins Medical Institution, Baltimore, MD, USA).HCT116 cells were cultured in McCoy’s and seeded at adensity of 3� 104 cells/cm2 onto fibronectin-coated dishes orslides 24 h before experiments. IMR90 cells were cultured inDulbecco0s Modified Eagle Medium and also seeded at adensity of 3� 104 cells/cm2 24 h before experiments. Nocoda-zole and blebbistatin (Sigma-Aldrich, St Louis, MO, USA)were used at 200 nM and 150 mM, respectively, the minimumconcentrations, which completely inhibited cell division inHCT116 cells (data not shown). g-irradiation was performedwith a Cs-137 Gammacell. Stealth Select siRNAs targeted toEg5, control siRNA, and Lipofectamine RNAiMax wereobtained from, and used according to the instructions of,Invitrogen (Carlsbad, CA, USA). miRNAs targeting p53, or anon-specific control, were also obtained from Invitrogen,initially as DNA oligos. These oligos were then cloned intothe pLenti6-GW/EmGFP-miR lentiviral expression vector(Invitrogen), and these vectors were transfected into 293FTcells along with the pLP/VSVG, pLP1, and pLP2 plasmids(Invitrogen) to produce miRNA-containing lentivirus.
p53 and suppression of chromosomal instabilityWB Dalton et al
1937
Oncogene

HCT116 and IMR90 were transduced with high-titer virus,and pools of hundreds of blasticidin-resistant colonies wereexpanded to produce stable knockdown cell lines.
ImmunodetectionFor immunocytochemistry, cells were fixed with 2% formal-dehyde/PBS and permeabilized with 0.2% Triton-X 100.Antibody incubations were 1 h at room temperature, andDNA was counterstained with DAPI. Images were acquiredwith a Zeiss Axioskop 2 Plus microscope. For MPM-2 flowcytometry, cells were harvested by trypsinization and fixedovernight at �20 1C in 70% ethanol. Antibody incubationswere 1 h at room temperature, and DNA was counterstainedwith propidium iodide. For g-H2AX/CC-3 flow cytometry,cells were harvested by trypsinization, fixed with 2% for-maldehyde/PBS, permeabilized with methanol, and incubatedovernight at 4 1C with primary antibodies, followed the nextday by 1 h incubations with secondary antibodies. Similar toMPM-2, CC-3 is an antibody, which specifically stains mitoticcells, but is an IgG2a allotype, and can thus be usedsimultaneously with IgG1 antibodies, such as g-H2AX(Thibodeau and Vincent, 1991). Data were acquired using anFACSCalibur (Becton-Dickinson, San Jose, CA, USA) andanalyzed with Flowjo. Immunoblotting was performed asdescribed earlier (Yoon et al., 2005).The following antibodies were used: mouse anti-g-H2AX
(Upstate, Billerica, MA, USA), rat anti-a-tubulin (Chemicon,Temecula, CA, USA), mouse MPM-2 (Upstate), mouse CC-3(a gift from M. Vincent, Universite Laval, Quebec, Qc, Canada),rabbit anti-Eg5 (Abcam, Cambridge, MA, USA), goat anti-p53(Santa Cruz, Santa Cruz, CA, USA), rabbit anti-cleaved-PARP(Cell Signaling, Danvers, MA, USA), and mouse anti-actin(Sigma-Aldrich). All fluorescent secondary antibodies wereAlexa-conjugates (Molecular Probes, Carlsbad, CA, USA).
Cytogenetic analysesChromosome spreads were prepared using standard cytoge-netic techniques, DNA was stained with DAPI, and imageswere obtained using a Zeiss Axioskop 2 Plus microscope.Scoring of chromosome aberrations was performed accordingto the classification system of Savage (Savage, 1976).Furthermore, all scoring was performed, in which possible,
in a blinded manner. For color karyotyping of spontaneouspolyploid p53�/� cells, analysis was performed by ChrombiosGmbH (Munich, Germany).
Colony survivalAfter 48 h nocodazole treatment, HCT116 cells were harvestedby trypsinization and seeded into T75 flasks. After 9 days,colonies were stained with methylene blue, photographs of theflasks were taken, and colony number was quantified usingMetamorph imaging software. For quantification of untreatedcontrol colonies, 1/100 the number of cells used in nocodazole-treated samples were seeded into T75 flasks and colonynumber was normalized accordingly, as use of the samenumber of cells as nocodazole-treated samples producedcolonies, which were too dense to be quantified (Figure 2f).
Time-lapse imagingPhase-contrast images of p53�/� HCT116 cells grown inside a37 1C, 5% CO2 chamber were automatically obtained at 6minintervals in multiple locations over 48 h using an OlympusIX81 microscope. All images were analyzed with Slidebook.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank D Pallas, P Doetsch, D Jones, A Corbett, G Davis,and M Wiltenburg for discussion and support, and BVogelstein for providing cell lines. This work was supportedin part by grants from the National Institutes of Healthto VWY (DK52230, DK64399, and CA84197) and toWBD (5T32GM008367-18). This work was supported in partby grants from the National Institutes of Health toVWY (DK52230, DK64399, and CA84197) and to WBD(5T32GM008367).
References
Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. (2001).Tetraploid state induces p53-dependent arrest of nontransformedmammalian cells in G. Mol Biol Cell 12: 1315–1328.
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L et al.(2000). Telomere dysfunction promotes non-reciprocal transloca-tions and epithelial cancers in mice. Nature 406: 641–645.
Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M.(2006). A positive feedback loop between the p53 and Lats2tumor suppressors prevents tetraploidization. Genes Dev 20:2687–2700.
Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff Met al. (2003). Histone H2AX: a dosage-dependent suppressor ofoncogenic translocations and tumors. Cell 114: 359–370.
Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, GiovanellaBC et al. (1990). Spontaneous abnormalities in normal fibroblastsfrom patients with Li-Fraumeni cancer syndrome: aneuploidy andimmortalization. Cancer Res 50: 7979–7984.
Blagosklonny MV. (2006). Prolonged mitosis versus tetraploidcheckpoint: how p53 measures the duration of mitosis. Cell Cycle
5: 971–975.
Bouffler SD, Kemp CJ, Balmain A, Cox R. (1995). Spontaneousand ionizing radiation-induced chromosomal abnormalities inp53-deficient mice. Cancer Res 55: 3883–3889.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JPet al. (1998). Requirement for p53 and p21 to sustain G2 arrest afterDNA damage. Science 282: 1497–1501.
Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KWet al. (2002). Targeted inactivation of p53 in human cells does notresult in aneuploidy. Cancer Res 62: 1129–1133.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay Let al. (1999). Disruption of p53 in human cancer cells alters theresponses to therapeutic agents. J Clin Invest 104: 263–269.
Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen Pet al. (2006). Apoptosis regulation in tetraploid cancer cells. EMBO
J 25: 2584–2595.Chan YW, On KF, Chan WM, Wong W, Siu HO, Hau PM et al.
(2008). The kinetics of p53 activation versus cyclin E accumulationunderlies the relationship between the spindle-assemblycheckpoint and the postmitotic checkpoint. J Biol Chem 283:15716–15723.
p53 and suppression of chromosomal instabilityWB Dalton et al
1938
Oncogene

Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, IdzerdaRL et al. (1995). A p53-dependent mouse spindle checkpoint.Science 267: 1353–1356.
Curtis LJ, Georgiades IB, White S, Bird CC, Harrison DJ, Wyllie AH.(2000). Specific patterns of chromosomal abnormalities areassociated with RER status in sporadic colorectal cancer. J Pathol
192: 440–445.Dalton WB, Nandan MO, Moore RT, Yang VW. (2007). Human
cancer cells commonly acquire DNA damage during mitotic arrest.Cancer Res 67: 11487–11492.
Dalton WB, Yang VW. (2009). The role of prolonged mitoticcheckpoint activation in the formation and treatment of cancer.Future Oncol 5: 1363–1370.
De Angelis PM, Clausen OP, Schjolberg A, Stokke T. (1999).Chromosomal gains and losses in primary colorectal carcinomasdetected by CGH and their associations with tumour DNA ploidy,genotypes and phenotypes. Br J Cancer 80: 526–535.
Di Leonardo A, Khan SH, Linke SP, Greco V, Seidita G, Wahl GM.(1997). DNA rereplication in the presence of mitotic spindleinhibitors in human and mouse fibroblasts lacking either p53 orpRb function. Cancer Res 57: 1013–1019.
Eshleman JR, Casey G, Kochera ME, Sedwick WD, Swinler SE, VeiglML et al. (1998). Chromosome number and structure both aremarkedly stable in RER colorectal cancers and are not destabilizedby mutation of p53. Oncogene 17: 719–725.
Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D.(2005). Cytokinesis failure generating tetraploids promotestumorigenesis in p53-null cells. Nature 437: 1043–1047.
Galmarini CM, Falette N, Tabone E, Levrat C, Britten R,Voorzanger-Rousselot N et al. (2001). Inactivation of wild-typep53 by a dominant negative mutant renders MCF-7 cells resistant totubulin-binding agent cytotoxicity. Br J Cancer 85: 902–908.
Ganem NJ, Pellman D. (2007). Limiting the proliferation of polyploidcells. Cell 131: 437–440.
Ganem NJ, Storchova Z, Pellman D. (2007). Tetraploidy, aneuploidyand cancer. Curr Opin Genet Dev 17: 157–162.
Hastak K, Paul RK, Agarwal MK, Thakur VS, Amin AR, Agrawal Set al. (2008). DNA synthesis from unbalanced nucleotide poolscauses limited DNA damage that triggers ATR-CHK1-dependentp53 activation. Proc Natl Acad Sci USA 105: 6314–6319.
Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M,Hemann M et al. (2004). Rb inactivation promotes genomicinstability by uncoupling cell cycle progression from mitotic control.Nature 430: 797–802.
Ikeuchi T, Weinfeld H, Sandberg AA. (1972). Chromosome pulveriza-tion in micronuclei induced by tritiated thymidine. J Cell Biol 52:97–104.
Jong YJ, Li LH, Tsou MH, Chen YJ, Cheng SH, Wang-Wuu S et al.(2004). Chromosomal comparative genomic hybridization abnorm-alities in early- and late-onset human breast cancers: correlationwith disease progression and TP53 mutations. Cancer Genet
Cytogenet 148: 55–65.Kato H, Sandberg AA. (1967). Chromosome pulverization in human
binucleate cells following colcemid treatment. J Cell Biol 34: 35–45.Kienitz A, Vogel C, Morales I, Muller R, Bastians H. (2005). Partial
downregulation of MAD1 causes spindle checkpoint inactivationand aneuploidy, but does not confer resistance towards taxol.Oncogene 24: 4301–4310.
Kim KT, Ongusaha PP, Hong YK, Kurdistani SK, Nakamura M, LuKP et al. (2004). Function of Drg1/Rit42 in p53-dependent mitoticspindle checkpoint. J Biol Chem 279: 38597–38602.
Kleivi K, Diep CB, Pandis N, Heim S, Teixeira MR, Lothe RA.(2005). TP53 mutations are associated with a particular pattern ofgenomic imbalances in breast carcinomas. J Pathol 207: 14–19.
Koller E, Propp S, Zhang H, Zhao C, Xiao X, Chang M et al. (2006).Use of a chemically modified antisense oligonucleotide library toidentify and validate Eg5 (kinesin-like 1) as a target for anti-neoplastic drug development. Cancer Res 66: 2059–2066.
Lalle P, Moyret-Lalle C, Wang Q, Vialle JM, Navarro C, Bressac-dePaillerets B et al. (1995). Genomic stability and wild-type p53
function of lymphoblastoid cells with germ-line p53 mutation.Oncogene 10: 2447–2454.
Lanni JS, Jacks T. (1998). Characterization of the p53-dependentpostmitotic checkpoint following spindle disruption. Mol Cell Biol
18: 1055–1064.Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. (1996). A
reversible, p53-dependent G0/G1 cell cycle arrest induced byribonucleotide depletion in the absence of detectable DNA damage.Genes Dev 10: 934–947.
Lu C, El-Deiry WS. (2009). Targeting p53 for enhanced radio- andchemo-sensitivity. Apoptosis 14: 597–606.
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova Oet al. (2006). p53 regulates mitochondrial respiration. Science 312:1650–1653.
Minn AJ, Boise LH, Thompson CB. (1996). Expression of Bcl-xL andloss of p53 can cooperate to overcome a cell cycle checkpointinduced by mitotic spindle damage. Genes Dev 10: 2621–2631.
Muller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH,Schilling T. (2006). One, two, three–p53, p63, p73 and chemosensi-tivity. Drug Resist Updat 9: 288–306.
Pantic M, Zimmermann S, Daly E, Opitz OG, Popp S, Boukamp Pet al. (2006). Telomere dysfunction and loss of p53 cooperatein defective mitotic segregation of chromosomes in cancer cells.Oncogene 25: 4413–4420.
Pfleghaar K, Heubes S, Cox J, Stemmann O, Speicher MR. (2005).Securin is not required for chromosomal stability in human cells.PLoS Biol 3: e416.
Quignon F, Rozier L, Lachages AM, Bieth A, Simili M, Debatisse M.(2007). Sustained mitotic block elicits DNA breaks: one-stepalteration of ploidy and chromosome integrity in mammalian cells.Oncogene 26: 165–172.
Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW,Vogelstein B et al. (2004). Inactivation of hCDC4 can causechromosomal instability. Nature 428: 77–81.
Rieder CL, Maiato H. (2004). Stuck in division or passing through:what happens when cells cannot satisfy the spindle assemblycheckpoint. Dev Cell 7: 637–651.
Rogakou EP, Boon C, Redon C, Bonner WM. (1999). Megabasechromatin domains involved in DNA double-strand breaks in vivo.J Cell Biol 146: 905–916.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. (1998).DNA double-stranded breaks induce histone H2AX phosphoryla-tion on serine 139. J Biol Chem 273: 5858–5868.
Savage JR. (1976). Classification and relationships of inducedchromosomal structual changes. J Med Genet 13: 103–122.
Shi J, Orth JD, Mitchison T. (2008). Cell type variation in responses toantimitotic drugs that target microtubules and kinesin-5. Cancer Res
68: 3269–3276.Song H, Hollstein M, Xu Y. (2007). p53 gain-of-function cancer
mutants induce genetic instability by inactivating ATM. Nat Cell
Biol 9: 573–580.Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-
Cardo C, Lowe SW et al. (2007). Mad2 overexpression promotesaneuploidy and tumorigenesis in mice. Cancer Cell 11: 9–23.
Stevens JB, Liu G, Bremer SW, Ye KJ, Xu W, Xu J et al. (2007). Mitoticcell death by chromosome fragmentation. Cancer Res 67: 7686–7694.
Stewenius Y, Gorunova L, Jonson T, Larsson N, Hoglund M,Mandahl N et al. (2005). Structural and numerical chromosomechanges in colon cancer develop through telomere-mediatedanaphase bridges, not through mitotic multipolarity. Proc Natl
Acad Sci USA 102: 5541–5546.Stukenberg PT. (2004). Triggering p53 after cytokinesis failure. J Cell
Biol 165: 607–608.Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q et al. (2009).
Chromosomal instability determines taxane response. Proc Natl
Acad Sci USA 106: 8671–8676.Tao W, South VJ, Diehl RE, Davide JP, Sepp-Lorenzino L, Fraley
ME et al. (2007). An inhibitor of the kinesin spindle proteinactivates the intrinsic apoptotic pathway independently of p53 andde novo protein synthesis. Mol Cell Biol 27: 689–698.
p53 and suppression of chromosomal instabilityWB Dalton et al
1939
Oncogene

Thibodeau A, Vincent M. (1991). Monoclonal antibody CC-3recognizes phosphoproteins in interphase and mitotic cells. Exp
Cell Res 195: 145–153.Thompson SL, Compton DA. (2008). Examining the link between
chromosomal instability and aneuploidy in human cells. J Cell Biol
180: 665–672.Tighe A, Johnson VL, Taylor SS. (2004). Truncating APC mutations
have dominant effects on proliferation, spindle checkpoint control,survival and chromosome stability. J Cell Sci 117: 6339–6353.
Toledo F, Wahl GM. (2006). Regulating the p53 pathway: in vitrohypotheses, in vivo veritas. Nat Rev Cancer 6: 909–923.
Uetake Y, Sluder G. (2004). Cell cycle progression after cleavagefailure: mammalian somatic cells do not possess a ‘tetraploidycheckpoint’. J Cell Biol 165: 609–615.
Uetake Y, Sluder G. (2007). Cell-cycle progression without an intactmicrotuble cytoskeleton. Curr Biol 17: 2081–2086.
van Gent DC, Hoeijmakers JH, Kanaar R. (2001). Chromosomalstability and the DNA double-stranded break connection. Nat Rev
Genet 2: 196–206.Vogel C, Kienitz A, Hofmann I, Muller R, Bastians H. (2004).
Crosstalk of the mitotic spindle assembly checkpoint with p53 toprevent polyploidy. Oncogene 23: 6845–6853.
Vousden KH, Lane DP. (2007). p53 in health and disease. Nat Rev
Mol Cell Biol 8: 275–283.Vousden KH, Lu X. (2002). Live or let die: the cell’s response to p53.
Nat Rev Cancer 2: 594–604.Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW
et al. (1996). Loss of normal p53 function confers sensitization to Taxolby increasing G2/M arrest and apoptosis. Nat Med 2: 72–79.
Ward IM, Difilippantonio S, Minn K, Mueller MD, Molina JR, Yu Xet al. (2005). 53BP1 cooperates with p53 and functions as a haplo-insufficient tumor suppressor in mice. Mol Cell Biol 25: 10079–10086.
Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW.(2007). Aneuploidy acts both oncogenically and as a tumorsuppressor. Cancer Cell 11: 25–36.
Westra JL, Boven LG, van der Vlies P, Faber H, Sikkema B,Schaapveld M et al. (2005). A substantial proportion of micro-satellite-unstable colon tumors carry TP53 mutations while notshowing chromosomal instability. Genes Chromosomes Cancer 43:194–201.
Wong C, Stearns T. (2005). Mammalian cells lack checkpoints fortetraploidy, aberrant centrosome number, and cytokinesis failure.BMC Cell Biol 6: 6.
Woods CM, Zhu J, McQueney PA, Bollag D, Lazarides E. (1995).Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol Med 1: 506–526.
Yamaguchi H, Chen J, Bhalla K, Wang HG. (2004). Regulation of Baxactivation and apoptotic response to microtubule-damaging agentsby p53 transcription-dependent and -independent pathways. J Biol
Chem 279: 39431–39437.Yang Z, Loncarek J, Khodjakov A, Rieder CL. (2008). Extra
centrosomes and/or chromosomes prolong mitosis in human cells.Nat Cell Biol 10: 748–751.
Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB,Yang VW. (2005). Kruppel-like factor 4 prevents centrosomeamplification following gamma-irradiation-induced DNA damage.Oncogene 24: 4017–4025.
Zhang H, Shi X, Zhang QJ, HampongM, Paddon H, Wahyuningsih Det al. (2002). Nocodazole-induced p53-dependent c-Jun N-terminalkinase activation reduces apoptosis in human colon carcinomaHCT116 cells. J Biol Chem 277: 43648–43658.
Zhuanzi W, Wenjian L, Dejuan Z, Wei W, Xigang J, Qingxiang G.(2007). Chromatid-type aberrations following irradiation in G0lymphocytes with heavy ions. Mutat Res 617: 98–103.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
p53 and suppression of chromosomal instabilityWB Dalton et al
1940
Oncogene





























