Embed Size (px)
Citation preview

J. Parallel Distrib. Comput. 66 (2006) 1489–1502www.elsevier.com/locate/jpdc
Parallel evolution strategy on grids for the protein threading problem�
Alioune Ngom∗
School of Computer Science, 5115 Lambton Tower, University of Windsor, 401 Sunset Avenue, N9B 3P4, Windsor, Ont., Canada
Received 8 October 2005; received in revised form 12 June 2006; accepted 15 August 2006Available online 16 October 2006
Abstract
The protein threading problem is the problem of determining the three-dimensional structure of a given but arbitrary protein sequence froma set of known structures of other proteins. This problem is known to be NP-hard and current computational approaches to threading areunrealistic for long proteins and/or large template data sets. In this paper, we propose an evolution strategy for the solution of the proteinthreading problem. We also propose three parallel methods for fast threading. Our experiments produced encouraging preliminary results interm of threading energy as well as significant reduction in threading time.© 2006 Elsevier Inc. All rights reserved.
Keywords: Protein; Threading; Three-dimensional structure; Fold; Energy; Alignment; Prediction; Evolution
1. Introduction
The Human Genome Project identified thousands of genesand proteins. To understand the biological functions and func-tional mechanisms of these proteins, the knowledge of theirthree-dimensional structures is required. The Structural Ge-nomic Initiatives, launched by NIH in 1999, intends to deter-mine these protein-structures within a decade [4].
The laboratory methods that are currently available for deter-mining the three-dimensional structure of a protein are X-raycrystallography or nuclear magnetic resonance (NMR) spec-troscopy. These methods are expensive, very time-consuming(ranging from weeks to months) and are quite difficult for high-throughput production. The Structural Genomic Initiatives tookthe strategy to determine protein structures using experimentalmethods only for a small fraction of all known proteins and toemploy computational techniques to predict the correct struc-tures for the rest of the proteins [4]. The basic premises behindthis idea are that there is a limited number of unique folds innature and different proteins share significant structural simi-larity. Thus, determining the unique structural folds of a few
� This work was supported by NSERC under Grant no. RGPIN228117-06.∗ Fax: +1 519 973 7093.
E-mail address: [email protected]: http://www.cs.uwindsor.ca/∼angom.
0743-7315/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.jpdc.2006.08.005
proteins in laboratory may allow to predict the structures of thevast majority of proteins. Protein threading represents one ofthe most promising such techniques according to the CASP5[11] and CAFASP3 [6] reports.
2. Protein threading
In protein threading (see Fig. 1), one matches a protein se-quence (called “query”) to a set of known protein structures(called “templates”). The template that best matches the queryis the predicted structure. The process of matching a query toa template is called “threading” and it involves aligning thequery to the template, and then determining the best alignmentamong all possible alignments between the query and the tem-plate. The quality of an alignment is determined by an objectivefunction. There are many defined objective functions in litera-ture for protein threading and are all based on computing theminimum free energy of the query sequence when it assumesthe fold (or three-dimensional structure) of the template.
A query is a sequence of amino acids of a given a protein.A template is also a sequence of amino acids but includes thethree-dimensional (3D) coordinates of all atoms for each aminoacids in the sequence. In three-dimension, a template is a seriesof cores (such as �-helix, �-sheet), loops, links and turns. Cores,loops, links and turns are the basic folds that subsequences of asequence will take, and the sequence of amino acids determines

1490 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
Fig. 1. The protein threading process.
how a protein folds in 3D. Threading a query against a templateis to determine which basic folds the amino acids of the querybelong to (this is done by aligning the query to the template)and then compute the free energy of the query when it assumesthe full fold of the template.
Numerous algorithms have been proposed for protein thread-ing [13]. They can be grouped into two categories based onthe energy models. To summarize, during a threading process,the amino acids of a query are assigned to the 3D atomic po-sitions of a template and then compute the energy of the cor-responding fold to determine the query’s compatibility to thetemplate’s structure. The common energy functions are basedon statistical methods and are biologically meaningful.
In the first category of threading approaches, the energy func-tion is based on the individual 3D atomic positions of the aminoacids. Interaction between the amino acids are not taken intoaccount. A simple dynamic programming method, such as Gen-THREADER [9], is employed to optimize the energy function.The prediction speed is fast but the accuracy is not always good.In the second category, the energy function is more complexas it takes into account the pairwise interactions between pairsof amino acids of the query sequence, beside considering thesingleton interactions of individual amino acids as in the firstcategory. Threading using such energy functions yields moreaccurate predictions than those in the first category, but withmore computation. It is known that protein threading problem isNP-hard when variable gaps and pair-wise interactions are con-sidered simultaneously [12]. Some approximation algorithmsare used to optimize the energy function [13].
Akutsu et al. [1] has proved that the protein threading prob-lem is MAX-SNP-hard, which means that it cannot be approxi-mated to an arbitrary accuracy in polynomial time. To overcomethe computational difficulty, several approaches have been pro-posed. One approach is to employ frozen approximation, thatis, when calculating a pairwise contact potential for a residueit is assumed that its contacting residues are the same in thetemplate and in the query structure. Such assumption reducesthe complexity of the general protein threading problem but at
the expense of threading accuracy [24]. Another approach at-tacks the problem through statistical sampling [5]. This typeof approach does not guarantee to find the globally optimalthreading, though it works better than frozen approximation ap-proach. Also, most current threading algorithms limit the align-ments to core regions only, defined either as regions of helicesand sheets or as regions buried inside the protein. This limita-tion is mainly introduced for computational efficiency and isonly partly justified by the data. Beside core restrictions, otherlimitations include gap restrictions (they are restricted only toloop regions in most methods), limited number of representa-tive templates, and so on.
In this paper, we propose evolution strategy and parallel evo-lution strategies for protein threading. Our methods use distancedependent pair-wise interactions and hydrophobic matrices forindividual amino acids as well as variable length gaps in thealignments [3] without predefined core elements. and core andgap restrictions.
3. Related studies
In Yadgari et al. [19,20], genetic algorithms (GA), an effi-cient method [8] based on the principle of natural selectionand evolution, was used to perform sequence to structure align-ments. To the best of our knowledge, this is the only paper inliterature that discusses GA for the protein threading problem.They discussed an effective method for representing such align-ments and such that they can be processed by GA using ap-propriate mutation and crossover operators. Threading by GAdoes not include any built-in core region limitations, since itis powerful enough to find good alignments without relying onsuch limitations. They tested the performance of their GA onsix sequence-structure pairs that were taken from a database ofstructural alignments [7] where the sequence of one protein wasthreaded through the structure of the other. In these cases thestructures of both proteins are known, so the reference align-ment, which is based on a structural super-positions of the twostructures, is quite reliable. Their goal was not to design or testenergy functions. Rather, they used an available energy func-tion to explore the performance of the GA, and use the struc-tural alignments as a reference point to be compared with theresults of the GA threading. Their experiments showed that GAconsistently scored much better than the structural alignments,and that the threading alignments obtained by GA were qual-itatively similar to the structural alignments. They concludedthat the ability of the GA to find better than the “correct” struc-tural alignments was due to its ability to take advantage of thepeculiarities of the energy function they used.
Yanev et al. [21,22] introduced a parallel divide and conquerapproach for protein threading. As far as we know, there is noother paper in literature that discusses parallelization methodfor the protein threading problem. First, they formulated theprotein threading problem as a generalized shortest path prob-lem (GSPP), then use a mathematical programming approachto solve the threading problem via the GSPP. In particular,the threading solutions are computed as shortest paths usingmixed integer programming (MIP). Next, they proposed a di-

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1491
vide and conquer strategy for the formulated GSPP problem inorder to parallelize their approach. In such a strategy, an ini-tial problem is split into r sub-problems processed by p pro-cessors, and then the solution of the original problem is com-puted as the minimum of all solutions of the sub-problems.Their parallel algorithm is based on centralized dynamic loadbalancing in which each slave processor applies MIP to solveits given sub-problems and then return the results to the mas-ter processor. Their parallel algorithm produced a super-linearspeedup and did suffer from high communication overheads.Their algorithms were tested on small as well as large proteinsusing up to eight processors and compared them to the corre-sponding sequential methods. Their computational experimentsshowed significant reduction in total running time as well asa good prediction accuracy when compared to a branch-and-bound method [14] for protein threading. As with Yadgari etal. [20], Yanev et al. [22] were not interested in testing energyfunctions. The function they used considered only the pair-wiseinteractions and the residue hydrophobicities. Also, their for-mal model of the protein threading problem included core andgap restrictions.
4. Problem statement
The protein threading problem is to determine the three-dimensional structure of a given but arbitrary protein sequence,called query, from a set of known structures of other proteins,called templates. Yadgari et al. [19,20], discussed a genetic al-gorithm approach for threading. However, their method threadsa single query against a single template only. They also reportresults only on queries/templates of length 300 amino acids atmost.
As far as we know, the current threading algorithms onlythread a single query against a single template. To thread againstmany templates, a given algorithm is applied sequentially manytimes, each time on a different template. Large queries cannotbe threaded that way against a template database within rea-sonable time bounds, particularly for large templates and largedatabases. In matter of fact, vast number of proteins have neverbeen attempted due to their sizes [21,22].
With an appropriate parallelization, there is a possibility tothread (large) queries against a (large) set of (large) templateswithin reasonable time bounds. There is even a possibility tothread multiple queries against multiple templates within rea-sonable time bounds.
In the following two sections, we first discuss a novel thread-ing method based on evolution strategy (ES) and then presenttwo parallelization of our methods for fast protein threading.
5. Evolution strategy-based threading
ES was first introduced by Rechenberg [16] and is anotherclass of evolutionary approaches. Unlike GA, the original ESevolved only a single solution at a time, used only mutation asgenetic operator, and real vector coded problem representation.The original ES worked as follows: starting from an originalrandom solution, mutation operation is applied to the current
unique solution, then the mutant replaces the parent only if ithas better fitness value, and this is repeated many times untilwe reach a satisfactory solution with respect to an objectivefunction. Although the original ES is simpler and faster thanGA it does, however, have less ability than GA to escape fromlocal optima. Current ES algorithms improve the original ESin many ways. There are two common variation of ES and theyare (�, �)-ES and (� + �)-ES. In both variations, each parentproduces � offsprings through mutation and there are � parents.In (�+�)-ES, � best individuals from all the (�+�) individualsare selected to form the next generation. In (�, �)-ES, � bestindividuals from the set of � offsprings are selected as parentsfor the next generation.
5.1. Problem representation
Our problem representation is same as in Yadgari et al. [20].Given a query Q and a template T, we seek to find the bestalignment between Q and T. Given a template database D =T1, T2, . . . , Tt , we seek to find the best alignment from the setof all alignments between Q and all Ti , 1� i� t . The templatethat aligns best with Q is therefore the solution we are lookingfor. The search space is the space of all possible alignmentsbetween Q and all Ti . In order to apply ES to the proteinthreading problem, we therefore, need to represent an alignmentappropriately for ES to evolve it and find the best solution.
An alignment between Q and given T is represented as afixed length string of integers S = S1, S2, S3, . . . , S|T | where
0�Si � |Q|, 1� i� |T |, and∑i=|T |
i=1 Si = |Q|.Fig. 2 shows the correspondence between a query-template
alignment and an integer string representation of the alignment.Si = 0 represents a structure deletion; Si = 1 represents amatch between a given amino acid in the query and a templateposition (a match means that the amino acid is assigned to thatposition in the template’s structure); Si > 1 represents gaps inthe sequence when aligned to the template.
The template’s structure is represented by the continuousbold trace through the circles that represent the structural po-sitions (12 in the example). The query sequence on the topis threaded through this structure and the associated encoding111100111311 is shown in the bottom. In this threading, thefirst four amino acids S, W, F, I of the query are matched to thefirst four structural positions of the template (that is S, W, F, Iassume the positions 1, 2, 3 and 4). Next in the threading, theamino acids G, N, A assume the positions 7, 8, and 9, whichare then set to values 1 in the encoding. Position 5 and 6 of thetemplate are not matched to the query (this is represented bythe dash line) and thus specify the structural deletions in thealignment (thus, corresponding positions in the encoding areset to 0). The following amino acid in the query, L takes po-sition 10 of the template, but its corresponding position in theencoding is set to 3 (not 1) to signify that after this structuralposition, the next 2 amino acids of the query are not matched toany structural position (this is an example of sequence deletionwhere the deleted letters are G and A). Thus a position Si = v
(v > 1) in the encoding specifies that the next v − 1 letters of

1492 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
Sequence - SWFIGNALGATS
SWFI - - GNALTS
StructureDeletion
SequenceDeletion
1 1 1 1 1 1 1 1 130 0
S
A
GL
A
N
GI
F
W
S
Structuralposition
4
3
2
1
12
11
10
9
8
7
56
T
Fig. 2. Representation of a query-template alignment (adopted from Yadgariet al. [20]).
the query are not matched to any structural position while thecurrent amino acid takes position i. Last query letters, T and Sare matched to the last positions and corresponding positionsin the encoding take 1 each. Below, we show the alignmentbetween Q and T, and the corresponding encoding E.
T: 1 2 3 4 5 6 7 8 9 10 11 12Q: S W F I G N A L G A T SE: 1 1 1 1 0 0 1 1 1 3 1 1
In this paper, we use (�+�)-ES method. Initially, we generate arandom population of � parents, that is each parent is initially arandom integer vector S = S1, S2, . . . , S|T |, where 0�Si � |Q|,1� i� |T | and such that the constraint
∑i=|T |i=1 Si = |Q| is
satisfied. The random integer vectors are drawn uniformly.
5.2. Objective function
Besides the problem representation, the design of an ap-propriate objective function is also fundamental. The objectivefunction is needed to assess how good or bad a candidate so-lution is. Candidates are selected according to their objectivevalues and the best � candidates among the current candidatesare always selected as the new parents for the next genera-tion. In a protein molecule, the bonds between the atoms of itsamino acids determine the three dimensional conformation ofthe molecule in space. Thus the three-dimensional structure ofthe protein is decided by the linear sequence of its amino acids.A protein assumes a fold at low energy state. Therefore, an en-
ergy function can be used to determine, if a query protein foldsat low energy state. The energy state of the template’s fold isknown as well as its atomic coordinates in three dimension.The query’s actual fold is unknown but we want to predict it.When the query is aligned to the template and folded accord-ingly, we can then compute the energy of the fold. If the actualunknown fold of the query is the same as the template’s fold,then it will have the energy of the template’s fold, assumingthat the fold is attained from the best alignment between thequery and the template.
Many energy functions are defined in literature. In this paper,we use the energy function discussed in Yadgari et al. [20]which is itself based on the energy function studied in Bryantet al. [3]. Given an alignment, the energy of its associated foldis given by
Etotal = Epair + Esingle + Egap.
Epair reflects the interactions between all pairs of amino acidsin the given query, and is computed from the pairwise potentialenergy matrix of Bryant et al. [3]. Esingle describes how wellthe individual amino acids of the query match their assignedstructural positions of the template, and is computed from thehydrophobic potential energy matrix of Bryant et al. [3]. Egap isthe alignment gap penalty function which is set to three energyunits per each gap. We refer the reader to paper Bryant et al.[3] for discussion of how Etotal is computed from the energymatrices.
The best solution vectors are those whose associated foldshave the lowest energy. Since our ES maximizes an objectivefunction, we transform the energy function appropriately for ESto maximize. The transformed energy function is the objectivefunction that ES will use in order to select the best solution.We will use the term ‘fitness function’ as the objective value isnormalized such that it is between 0 and 1.
Given a query Q, a template T and a solution vector S =S1, S2, . . . , S|T |, our fitness function is defined as
F(S) = 1− E(S)− Emin
Emax − Eminor F(S) = Emax − E(S)
Emax − Emin,
where E(S) is the actual energy of the fold associated with thealignment corresponding to S, Emin (respectively, Emax) is thelowest (respectively, highest) bound on the energy value and canbe obtained by assigning to each column of the alignment, theminimum (respectively, maximum) possible energy value fromthe energy matrices. Thus, we always have Emin �E(S)�Emax.The closer E(S) is to Emin then the lower is the energy of thefold, and thus the larger is the fitness of S.
5.3. Mutation
Mutation helps an evolutionary process to explore new areasof the search space by generating totally new solution vectors.It also helps to maintain the diversity of a population, whichin turn helps to escape local optimum traps. Mutation opera-tion randomly alters certain positions of a given solution vector.With our representation, mutation should be designed in such

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1493
a way that it always produces valid solutions, that is the con-straints
∑i=|T |i=1 Si = |Q| and 0�Si � |Q| must be satisfied on
resulting solutions. Our mutation operation gives valid vectorsand is as follows.
We randomly select a solution to mutate then randomly gen-erate an integer n ∈ [0,
|T |� ] and randomly select two positions
p1, p2 ∈ [1, |T |] to alter (n is the number of times that pairsof positions will be altered and � ∈ {1, 2, . . . , |T |} controls themagnitude of n). We then increase SP1 and decrease SP2 by asame small random amount m ∈ [0,
|Q|� ] (� ∈ {1, 2, . . . , |Q|}
controls the magnitude of m), given a parent vector S to pro-duce a mutant vector. The offset m must be selected such that0�Spi
±m� |Q|, to ensure the validity of the mutant. This pro-cess of altering a pair of positions is repeated n times. For ex-ample, if parent S = 1110401031, n = 1, p1 = 4, p2 = 9 andm = 2, then after mutation, the mutant will be 1112401011. Itshould be noted that mutation operation is applied � times onthe current set of parents to yield � mutants.
Mutation is a disruptive operation, and the degree of disrup-tion depends on the value of �; the smaller is �, the larger themaximum value of n will be, and therefore the greater the levelof disruption will be. The magnitude of m also induces a certainlevel of “local disruption” in an alignment. That is, a large valueof m implies a large “block” of amino acid deletions in eitherthe query or the template, and the resulting alignment will befar from the original alignment; the larger is �, the smaller willbe the maximum magnitude of m, and thus the lower will bethe degree of local disruption. Another source of disruption isthe distance between the positions p1 and p2; since larger dis-tance will disrupt the parent too much. One could constrain thedistance between p1 and p2 such that the resulting alignmentis closer to the parent alignment. Through trial-and-errors, wefound that value 4 for both control parameters � and � gave thebest results, and thus we set � = � = 4 in all our experiments.
InYadgari et al. [20], mutation is performed by altering a sin-gle position in the string, then a compensating change is donein other position/s. For example, if 1102114101 is changed to1104114101 (here m = 2), then the two additional insertionsis compensated by two deletions in at most two random posi-tions elsewhere in the string, e.g. 1104004101 or 1104113011.In Yadgari et al. [20], the amount of change is m < 5, and thenumber of positions altered by this single mutation is at mostm+ 1 whereas our single mutation changes only two positionsexactly and thus it is locally less disruptive. Yadgari et al. [20]also designed a mutation operator that does not require a com-pensation process. This is done by selecting a small substringand reversing it. For example, 1102114101 to 1141120101. Itis clear that this mutation method is not very explorative sincegood strings with value, say 3 at certain position, will never begenerated.
5.4. Recombination
We also applied a recombination operator on randomly se-lected pairs of parents. Recombination aims to combine geneticmaterials from two parents and to pass them on to the next gen-
eration, depending on the fitness of the parents. First, we gen-erate a random integer n ∈ [0,
�� ] (� = {1, 2, . . . , �} controls
the magnitude of n) as the number of recombination operationsto apply on the current population of � parents. We then repeatthe process of recombination n times, as follows.
Randomly select two parents S1 and S2, randomly generatea binary mask M and construct the offsprings C1 and C2 in thefollowing manner: at position p, C1p ← S1p and C2p ← S2p
if Mp = 01, else C1p ← S2p and C2p ← S1p if Mp = 1. Thefollowing example shows the recombination process.
S1 = 1 1 0 2 0 1 0 3S2 = 2 0 1 0 3 1 0 1
M = 1 0 0 1 0 1 1 0
C1 = 1 0 1 2 3 1 0 1C2 = 2 1 0 0 0 1 0 3
We may need to correct a child Ci , if it is not valid, so asto satisfy the constraints discussed earlier. In the example, C1and C2 are invalid and thus will be corrected to make themvalid. Our correction method is as follows. For a child S, wefirst compute the sum s = ∑i=|T |
i=1 Si and let d = s − |Q|.We then adjust some values Si according to the sign and/orthe magnitude of d so as to satisfy the constraints. In all ourexperiments, we used � = 4 since this gave best results.
Yadgari et al. [20] used single-point crossover as recombi-nation operator, which also suffers from the same correctionproblem as ours. We did experiments, using ES, to compare ourmutation and recombination operators against those in Yadgariet al. [20] and obtained better results with our genetic opera-tors. However, this does not mean that our operators are bet-ter at all (since we did not perform experiments with GA tocompare the operators). GA and ES use different evolutionaryprinciples and, at least for the problem at hand, use differentcontrol parameters. If we could compare the operators under“both” GA and ES with “equivalent” sets of values for theirrespective control parameters, then we could say which amongthe operators are the best. The control parameters of our ES are�, �, �, �, �, number of generations and number of local op-timizations (see below). The number of control parameters ofthe GA is m, populations size, mutation rate and crossover rate(and possibly more). Determining equivalent sets of values forthe control parameters of ES and GA is difficult. For example,our ES does not have the problem of “early convergence” as inGA and therefore GA needs good values for its parameters tomaintain the diversity of its population.
5.5. ES-Based algorithm
Fig. 3 illustrates our ES approach for protein threading, calledEST algorithm. Starting from a random initial population Uof � candidate solutions, we create subsequent generations byrecombining members of U and then create a set M of � mutantsfrom the set U, and finally, the next � parents are selectedfrom the best in R ∪M . We also keep track of the global bestsolution and preserve it across generations. The inner while-

1494 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
Fig. 3. EST algorithm.
loop locally optimizes the actual best solution in R∪M in orderto escape a local optimum trap, and the global best solutionis updated only if its fitness is worse than the fitness of thelocal best solution found in the current population R ∪M . Ourmethod is elitist since the local best solution in the currentgeneration and the global best solution are passed onto the nextgeneration.
6. Parallel EST for protein threading
6.1. Single query single template threading
The single query single template parallel EST (SQST-PEST)method threads one query against one template, in parallel. InSQST-PEST (see Fig. 4) the master processor creates a popu-lation from a query Q and a template T, and then continuouslyapplies recombination and mutation operators on the currentpopulation for a certain number of generations. Each slave pro-cessor evaluates only the fitness of each solution in its givensub-population and then searches and returns its local best so-lution to the master.
The computation of the fitness values is the most costlyprocess in EST, particularly for large values of � + � andfor large templates T. SQST-PEST thus parallelizes EST byevaluating the current population concurrently on p slaveprocessors. Each slave can, in parallel, evaluate a distinctsubset of the population, search for good solutions andthen return its local best solution to the master. In this ap-proach, the master interacts with each slave only twice ineach generation: one interaction to send a subpopulation toa slave, and another to receive a solution from the slave.The total communication complexity between the masterand a slave within a generation is TComm = �+�
pO(|T |),
and the computation complexity of the slave is TComp =�+�p
O(|T |2) (see Section 7.2). The ratio TCommTComp
= 1O(|T |)
implies that the communication overhead increases with p.
Fig. 4. SQST-PEST algorithm.
However, if SQST-PEST is used on large templates, the cost ofthis overhead will be small compared with the task’s granularitywhich increases with |T |. Therefore, SQST-PEST is best usedwith large templates.
The partitioning of R∪M into p slaves proceeds as follows.Since (�+�)�p, each slave will receive exactly �+�
psolutions
first. If there are still solutions remaining to be distributed, theneach of the first [(� + �) modp] slaves will receive one moresolution. The same method is also applied in our next parallelEST. Also for a better performance in both parallel methods, themaster can process one subset while waiting to receive resultsfrom the slaves. This technique has not been implemented inthis research but it is being undertaken.
6.2. Single query multiple template threading
The single query multiple templates parallel EST (SQMT-PEST) method threads one query against a set of templates, inparallel. In SQMT-PEST (see Fig. 5) the master distributes sub-sets of templates to the slaves, each slave then serially appliesEST algorithm on each template it receives and then returns itslocal best solution to the master.
Unlike SQST-PEST, SQMT-PEST parallelizes EST by hav-ing each slave perform EST concurrently on distinct subsetsof templates. The slaves are much slower than those in SQST-PEST. Here, the master interacts with each slave only twicewithin its lifetime; first to send templates to a slave, and sec-ond to receive results from the slave. The total communicationcomplexity between the master and a slave is O(
|T |p
t), and the
computation complexity of the slave is (� + �)O(|T |2p
t) (see
Section 7.3). The ratio TCommTComp
= 1(�+�)O(|T |) implies that the

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1495
Fig. 5. SQMT-PEST algorithm.
gain from parallelization increases with p, since there is nocommunication overhead.
7. Performance issues
For the parallel ESTs, we must distinguish between compu-tation time (of master and each slave) and communication time(between processors), and the sum of these gives their totaltime complexities. Also, to efficiently use a parallel system, abalanced workload among the processors is required. To com-pare the workload balancing effectiveness of a parallel method,we define the percentage of load imbalance (PLI) by PLI =LargestLoadSmallestLoad
× 100, that is PLI is the percentage of the overallprocessing time that the first finished processor must wait forthe last processor to finish. This number also affects the degreeof parallelism (i.e. speedup). If PLI is less than 1%, we achieveover 99% degree of parallelism. Therefore, a parallel methodwith a lower PLI is more efficient than another with a higherPLI. The workload is perfectly balanced if PLI = 0. To bal-ance the workload in our parallel ESTs, the master processorpartitions its data set into p portions, where p is the number ofslave processors allocated. To achieve ideal workload balance,where PLI = 0, the size of each portion must be equal to thesize of the data set divided by p.
Let {T1, T2, . . . , Tt } be a set of t templates and p be the num-ber of slaves. Obtaining the ideal size for each portion T j inSQMT-PEST is not easily attained since the templates in T j
may have greatly varying lengths. The last template assigned toeach portion does not always result in the perfect size for thatportion, with the template being either too short or too long.Therefore in this case, some slaves will run longer than otherslaves, and the slaves that receive the largest portions will runthe longest. Perfect balance is achieved when the sum of the
template lengths in each portion T j is equal to∑i=t
i=1 |Ti |p
. WithSQMT-PEST, we can use the load balancing algorithm of Yapet al. [23], for distributing the templates to each slave Sj as fol-lows. First, the templates are sorted in decreasing length order.Then starting from the longest one, each template is placed intothe portion T j that has the current smallest sum of templatelengths. In the case of a tie, the smallest numbered portion isselected. Finally, each portion T j is sent to its correspondingslave Sj . Load balancing process is not included in SQMT-PEST algorithm shown in Fig. 5; here, each portion T j has thesame number t
pof templates but not necessarily the same size,
and we must have (t mod p) = 0 to achieve best performance.In SQST-PEST, each generation consists of (� + �) solutionsof equal length. Here, ideal workload balance is attained onlywhen [(� + �) mod p] = 0, otherwise some slaves will havemore solutions than others and thus will run the longest.
Let us denote by TSeq the time of the fastest algorithm forsolving the protein threading problem on one processor, and byTPar the time of our parallel EST to solve the problem. As usual,the speedup is given as S(p) = TSeq
TPar, given p processors. For
TSeq we will choose the time of EST since we do not know thebest protein threading algorithm; alternatively, we can choosethe time of parallel EST executed on one slave only.
7.1. Complexity of EST
The calculation of the fitness value is usually the most costlyoperation in evolutionary computation, as it involves decodinga chromosome into an alignment and then obtaining the fitnessvalue from the alignment. Given a query Q, a template T and acandidate solution vector S, the time complexity of the energyE(S) is O(|T |2); since the computation of the Epair term is themost expensive and that the entry for each pair of positions (oramino acids) in T is searched for in the two dimensional energymatrix of Bryant et al. [3]. Both our mutation and recombinationalgorithms run in O(|T |) time each. In all our experiments, weused � < � (where � = b�, b > 1) and � = a|T | (a > 0). Thishelps to define good upper-bounds on our algorithm.
In a given generation, the evaluation of solutions and thelocal optimization contribute the most to the complexity ofEST. After observing EST and summing up the complexitiesof all main operations, we obtain a time complexity of (� +�)O(|T |2) + �O(� + �) = a+ab
bO(|T |3) since � = b� =
a|T |, (a > 0, b > 1).
7.2. Complexity of SQST-PEST
In a given generation, we must distinguish between com-putation time and communication time. Given p slaves, themaster sends �+�
p+ 1 solutions to a slave and receives one
solution from the slave. Since the master communicates witha slave through a queue under MPI and that it takes O(T )
time to send/receive a solution, therefore, the total communi-cation complexity is (�+�)
pO(|T |). The master’s computation
includes recombination, mutation, and selecting the best so-lution and the next parents: this gives a time complexity of(�+ �)O(|T |)+ �O(�)+ �O(p). A slave’s computation onlyincludes evaluation and local optimization: therefore, a slaveruns in time �+�
pO(|T |2).
Taking the sum of communication and all computationtimes, SQST-PEST runs in time �+�
pO(|T |)2 + �O(�+ p) =
a+abb
O(|T |3p
) + abO(|T |p), in a given generation. The second
term comes from the last statement in the repeat loop of themaster. In that statement, we apply “multiple elitist strategy”,that is the best solutions from all slaves are added to the currentpopulation and the best � current solutions will be used as next
1496 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
parents. Multiple-elitism adds a time overhead as p increases,however, such overhead can be avoided by not applying mul-tiple elitism. We can randomly replace a current solution byGlobal_best and select � best solutions out of � + � currentsolutions instead of � + � + p solutions. Somehow, SQST-PEST can improve EST in search strategy by taking advantageof parallelism with the cost of having an extra time overheadthat is linear on p. If we disallow multiple elitism, then SQST-PEST will be about p times faster than EST, given p slaves.SQST-PEST has a speedup of O(p).
7.3. Complexity of SQMT-PEST
Unlike in SQST-PEST, the slaves in SQMT-PEST do all thework. Each slave calls EST sequentially on Q and t
ptemplates
(t �p) and returns its best solution to the master. Therefore,a slave’s computation time is t
p(� + �)O(|T |2) + t
p�O(� +
�) = a+abb
O(|T |3p
t). The master’s computation time is O(t)+O(p) + O(|T |); its only task is to send/receive data to/fromslaves and find the best solution out of p results from slaves.The communication time is O(
|T |p
t).The time complexity of SQMT-PEST is therefore
a+abb
O(|T |3p
t) + O(p). Again, SQMT-PEST is p times faster
than EST. Here the master does not iterate, so the overhead ofO(p) time due to its last statement occurs only once. AlthoughSQMT-PEST is cubic on |T |, it is also linear on t (the numberof templates). SQMT-PEST also has a speedup of O(p).
8. Computational experiments, results and discussions
Unfortunately due to infrastructure update and other prob-lems at our university node, we have had difficulties to accessthe thousands of processors available in Ontario through theUniversity of Windsor. We could use only up to eight processorsin total. Certain problems have been solved only very recentlybut some still remains. We are planning to perform more exper-iments with more processors once our node is fully working.
Our experiments aim to determine the performance of ourmethods. We used Grid-enabled environments on SHARCNET(http://www.sharcnet.ca/) for our parallel algorithms. We ob-tained our protein information from the protein data bank (PDBat http://www.rcsb.org/pdb/). We compared our EST algorithmwith the genetic algorithm threading (GAT) method of Yadgariet al. [20] on the same proteins in Yadgari et al. [20]. But wefound that some of the protein data of Yadgari et al. [20] havebeen updated in PDB, and that those data retrieved from PDBdo not match with the length of some of the proteins used inYadgari et al. [20] specified. We used the latest version of PDBdata set to perform the experiments using the same energy func-tion of Bryant et al. [3] as used in Yadgari et al. [20].
8.1. Experiments with parameters � and �
We did experiments to find best values for � and �.Through trials-and-errors, we obtained best results when
200
150
100
50
0
-50
-100
-150
-200
-250
-300
Ene
rgy
Tim
e (m
in)
0 0.5 |T| 1 |T| 1.5 |T| 2
λ
Energy (EST)
Energy (SQST)
Time (EST)
Time (SQST)
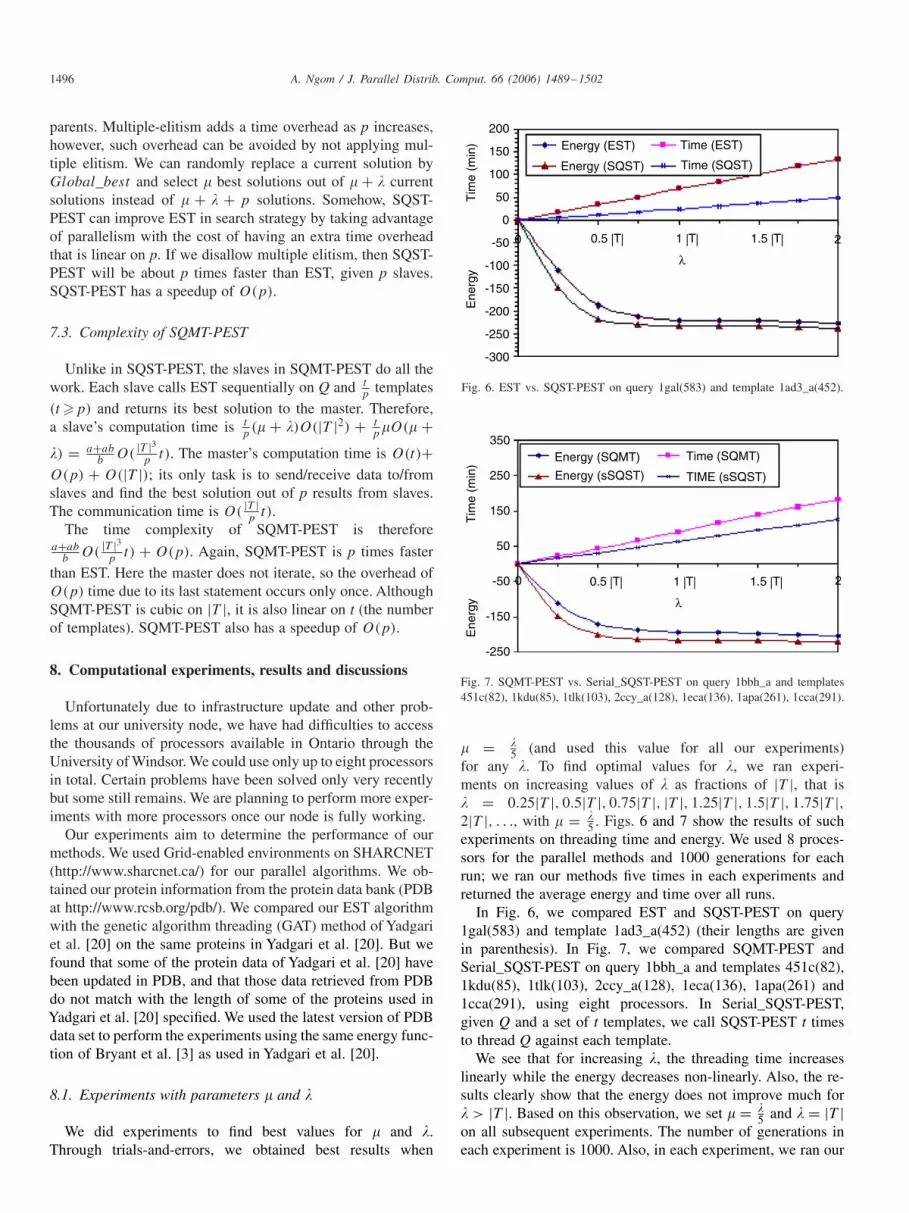
Fig. 6. EST vs. SQST-PEST on query 1gal(583) and template 1ad3_a(452).
350
250
150
50
-50
-150
-250
Ene
rgy
Tim
e (m
in)
0 0.5 |T| 1 |T| 1.5 |T| 2
λ
Energy (SQMT)
Energy (sSQST)
Time (SQMT)
TIME (sSQST)
Fig. 7. SQMT-PEST vs. Serial_SQST-PEST on query 1bbh_a and templates451c(82), 1kdu(85), 1tlk(103), 2ccy_a(128), 1eca(136), 1apa(261), 1cca(291).
� = �5 (and used this value for all our experiments)
for any �. To find optimal values for �, we ran experi-ments on increasing values of � as fractions of |T |, that is� = 0.25|T |, 0.5|T |, 0.75|T |, |T |, 1.25|T |, 1.5|T |, 1.75|T |,2|T |, . . ., with � = �
5 . Figs. 6 and 7 show the results of suchexperiments on threading time and energy. We used 8 proces-sors for the parallel methods and 1000 generations for eachrun; we ran our methods five times in each experiments andreturned the average energy and time over all runs.
In Fig. 6, we compared EST and SQST-PEST on query1gal(583) and template 1ad3_a(452) (their lengths are givenin parenthesis). In Fig. 7, we compared SQMT-PEST andSerial_SQST-PEST on query 1bbh_a and templates 451c(82),1kdu(85), 1tlk(103), 2ccy_a(128), 1eca(136), 1apa(261) and1cca(291), using eight processors. In Serial_SQST-PEST,given Q and a set of t templates, we call SQST-PEST t timesto thread Q against each template.
We see that for increasing �, the threading time increaseslinearly while the energy decreases non-linearly. Also, the re-sults clearly show that the energy does not improve much for� > |T |. Based on this observation, we set � = �
5 and � = |T |on all subsequent experiments. The number of generations ineach experiment is 1000. Also, in each experiment, we ran our

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1497
Table 1EST vs. GAT (Yadgari et al. [20])
Q (Length) T (Length) GAT EST w/o. Rec.
Ethr Time Ethr Imp (%) Time Imp (%)
1bbh(131) 2ccy(128) −175 29 −173.1± 4.3 −0.6 18.3± 3.1 36.91ash(147) 1cca(291) −111 33 −121.2± 4.1 9.2 32.2± 3.3 2.42pfl(156) 1kdu(85) −94 13 −91.2± 4.9 −3.0 9.2± 2.5 29.22rhe(114) 1tlk(103) −122 20 −109.5± 4.4 −10.2 11.1± 2.5 44.51ccr(112) 451c(82) −37 14 −39.5± 4.6 6.8 8.1± 1.9 42.11rtc(268) 1apa(261) −491 120 −479.5± 5.1 −2.3 77.5± 4.1 35.4
EST w. Rec.
Ethr Imp (%) Time Imp (%)
1bbh(131) 2ccy(128) −175 29 −191.5± 3.8 9.1 22.5± 2.9 22.411ash(147) 1cca(291) −111 33 −165.2± 4.1 48.8 36.1± 3.3 −9.42pfl(156) 1kdu(85) −94 13 −121.4± 3.9 29.1 11.3± 2.7 1.32rhe(114) 1tlk(103) −122 20 −141.5± 4.1 15.98 14.3± 2.5 28.51ccr(112) 451c(82) −37 14 −56.1± 4.1 51.6 10.3± 2.1 26.41rtc(268) 1apa(261) −491 120 −513.2± 5.1 4.48 83.5± 4.2 30.4
methods 5 times and give the average result of 5 runs as wellas the standard deviation. All times are reported in minute andEthr in the tables below is the energy value.
8.2. EST versus GAT comparison
In Table 1, we compare EST with the GAT method ofYadgariet al. [20] on their data set. Yadgari et al. [20] used 1000 gener-ations and a population size of 300. The improvements (undercolumn Imp) of EST over GAT are computed as
Imp = VEST − VGAT
VGAT× 100,
where V{GAT or EST} is either energy or time; a positive valuemeans EST is Imp% better than GAT. The improvements wereaveraged over five runs.
As expected, EST gives faster threading time than GAT.On average, EST is 31.75% faster without recombination, and16.6% faster with recombination. Without recombination, ESTyields generally worse energies than GAT with a maximum im-provement of 9.2% only and an average improvement of only−0.02%. Thus both methods are comparable at least on thisdata set. With recombination, EST gives much better energiesthan GAT with improvements up to 51.6% and an average en-ergy improvement of 26.51%.
It should be noted that Yadgari et al. [20] used both recom-bination and mutation operators in his GAT threading exper-iments, with 1000 generations and a population size of 300solutions, and the energy and time results averaged over fiveruns for each experiment. Our EST experiments used a pop-ulation size of (� + �) = 6 |T |5 , where |T | is the size of thegiven template input T. In Table 1, the length of the templatesranges from 82 to 291, hence the population size ranges from 98to 349 solutions. The energy performance of our EST without
recombination compares well with that of GAT even with asmall population size (see rows 3 and 5). There are many rea-sons for this. First, our EST algorithm applies a local searchstrategy in order to escape a local optimum trap. Second, ourEST is multiple-elitist since it always selects the best � solu-tions, including the local and global best solutions, of the cur-rent population as parents for the next generation. Third, ESTis able to maintain a diverse population at all times by gener-ating � mutants in each generation (which is another way toescape local optimum trap) and thus avoids premature conver-gence; consequently, the subsequent set of � parents will con-tain “distinct” good solutions. EST explores the search spacemuch more efficiently than GAT even with a smaller popu-lation size or without recombination. Without recombinationhowever, EST has less (if not none) exploitation ability thanGAT. That is, other than the fitness value, EST will not makeuse other informations already present in a good solution in or-der to better guide his rearch. That is the reason why EST with-out recombination did not perform better than GAT in general.With recombination though, EST clearly outperforms GAT dueto its augmented ability to exploit good solutions.
8.3. Self-threading experiments
Given a good energy function and considering the resultsabove, we assume that our methods are capable of determiningthe optimal threading, in most cases. We tested EST and SQST-PEST to see whether they can find the native fold of a queryQ when Q is threaded against its own template T; that is, T isQ with its structure information. In self-threading, the energyof T is called native energy, Enat, and can be obtained fromthe energy matrices of Bryant et al. [3] given Q. We did self-threading experiments with EST and SQST-PEST and Table 2,shows the native energy of proteins with the energy found by

1498 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
Table 2Self-threading with EST and SQST-PEST
Q (Length) Enat EST w/o. Rec. EST w. Rec.
Ethr Acc (%) Time Ethr Acc (%) Time
1kdu(85) −14.9 −14.9 100 8.7 −14.9 100 10.11tlk(122) −139.5 −139.5 100 11.8 −139.5 100 13.62ccyA(128) −79.2 −79.2 100 18.1 −79.2 100 21.11apa(261) −122.2 −122.2 100 73.2 −122.2 100 78.31cca(292) −119.9 −124.5 96.2 79.5 −124.5 96.2 86.31cem(363) −199.2 −193.5 97.1 100.1 −199.2 100 110.21gpl(432) −211.3 −203.8 96.4 116.6 −211.3 100 129.53grs(478) −176.1 −167.1 94.8 123.5 −173.4 98.5 139.1
SQST-PEST w/o. Rec. SQST-PEST w. Rec.
Ethr Acc (%) Time Ethr Acc (%) Time
1cem(363) −199.2 −199.2 100 27.8 −199.2 100 31.41gpl(432) −211.3 −211.3 100 33.6 −211.3 100 38.53grs(478) −176.1 −176.1 100 37.5 −176.1 100 41.11gal(583) −231.1 −231.1 100 47.1 −231.1 100 52.61w63_A(618) −273.2 −273.2 100 51.6 −273.2 100 58.1101g_A(722) −366.8 −359.6 98.1 61.1 −366.8 100 68.31mvw_A(840) −311.9 −291.6 93.49 69.7 −298.7 95.7 81.2
self-threading. In the table,
Acc =(
1−∣∣∣∣Ethr − Enative
Enative
∣∣∣∣)× 100
is the accuracy of self-threading with EST or SQST-PEST.EST gives an accuracy of > 94%. Without recombination,
EST found the exact answer in 50% of the tests, whereas withrecombination, the exact answer is found in 75% of the tests.Thus most energies are 100% accurate. Also, in particular forruns without recombination, the accuracy degrades as the lengthof the proteins increases. Clearly however, EST is able to findthe optimal threading given enough time and/or larger values of�. The effect of recombination can be seen on proteins, 1cem,1gpl and 3grs, where we see greater accuracy than withoutrecombination. Last, the entry for protein 1cca shows Ethr tobe lower than Enative. Yadgari et al. [18] observed a similar casein their self-threading experiments. This case was examined byPanchenko [15] and they told us that it can happen for someproteins that the energy of optimized alignment (the alignmentwhich we find with EST) is lower than the energy of the selfalignment, but that it should be rather an exception than therule. In general, the energy of a self-alignment should be thelowest but not necessarily the case, since proteins are not in their“global” energy minimum. One should be more concerned onhow well a threading algorithm distinguishes native templatesfrom non-native ones.
We used eight processors for self-threading with SQST-PEST. By taking advantage of parallelism, SQST-PEST appliesmultiple-elitism by inserting the best local solutions from allslaves, as well as the best solution produced so far, in the nextgeneration. Moreover, SQST-PEST has a better explorationcapability than EST since each slave processor explores dis-
tinct area of the search space to find its best solution. ThusSQST-PEST has a better search strategy than EST. The con-sequence of this strategy can be seen when we self-threadthe following three proteins, 1cem, 1gpl and 3grs, using ESTand SQST-PEST. Clearly, SQST-PEST improves the result ofEST on these three proteins; it was able to find their nativeenergies, with or without recombination and in faster time.In general, for 1000 generations, all the reported threadingtimes for SQST-PEST are very fast compared to those of ESTand are consistent with our complexity analysis of Section 7.The accuracies of SQST-PEST are > 93% and degrade asthe length of the proteins increases, however, given enoughtime and/or larger values of �, SQST-PEST is able to find theoptimal solutions.
In the sections below, we reported only the results of ourmethods with recombination. All our methods produced besttime results but with worse energy results, when recombina-tion is not added; thus we found it unnecessary to report suchexperiments.
8.4. SQST-PEST versus PDCT comparison
We compared SQST-PEST with the parallel divide-and-conquer threading (PDCT) approach of Yanev et al. [21,22].Yanev et al. [21] used large proteins but a different and simplerenergy function than SQST-PEST. Also, their model of proteinthreading problem is not the same as ours. In their model, in-sertions and deletions (i.e. gaps) are not allowed within the coresegments of a template during alignment. This restriction onthe core segments simplifies the general problem of threadingand thus yields a smaller search space than ours. In addition,their parallel algorithm is based on centralized dynamic load

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1499
Table 3SQST-PEST vs. PDCT (Yanev et al. [21])
Q (Length) T (Length) p PDCT time SQST w. Rec.
Ethr Time Imp (%)
2bmh(455) 1cem(363) 3 11.63 −323.5± 3.3 23.2± 1.8 −1003min(522) 1gpl(432) 3 13.33 −419.1± 3.8 29.5± 2.1 −121.82cyp(294) 3grs(478) 4 30.50 −271.3± 3.5 26.3± 3.1 23.11gal(583) 1ad3_a(452) 4 43.20 −228.2± 3.4 31.4± 2.9 27.3
balancing, where the master gives work on demand to idleslaves in a dynamic way whereas our parallel method is basedon static load balancing, where the workload is assigned priorto execution. Yanev et al. [21] used a different problem rep-resentation for their candidate solutions as well as a differentoptimization approach such that the work load is not knownbeforehand and this lead to frequent communications betweenmaster and slaves. In PDCT the master partitions the thread-ing problem into r distinct smaller subproblems that are thendistributed to p slaves for processing, whereas in SQST-PESTthe slaves solve the same problem (not smaller subproblems)by exploring different areas of the search space. The experi-ments in Yanev et al. [22] showed that the gain from the PDCTparallelism increases with the problem size, and that thereis an optimal value for pair (r, p) such that PDCT performsoptimally. Table 3 shows the results of SQST-PEST on theproteins used in Yanev et al. [21].
For a given fixed number of slaves p, Yanev et al. [21]reported results for different values of parameter pairs (r, p),however in Table 3 we wrote down only the best time obtainedby PDCT among all r values for a given value of p and com-pared it to the results obtained by SQST-PEST. We do not havea fair comparison since both methods are very different in theirprotein threading models and representations as well as theiroptimization approaches. We report results for p = 3 and 4only, on four threading cases. SQST-PEST was much slowerthan PDCT in two of the proteins. Notice that SQST-PEST gavefaster times than PDCT on its two hardest tasks (see last tworows of the table). Also in the table, we reported only the re-sults of SQST-PEST with recombination. We did experimentswithout recombination and obtained faster times but worse en-ergies than with recombination; we also found that even with-out recombination, SQST-PEST performed worse than PDCTin half of the tests.
8.5. Efficiency of SQST-PEST
To study the effect of the number of slave processors, or inother words, the effect of multiple-elitism on the energy, weapplied SQST-PEST on the hardest pair of query-template usedin Yanev et al. [21], with increasing number of slaves. Table 4reports the results of such experiments.
With respect to the energy, the gain from SQST-PEST paral-lelization increases with the number of slaves p. As more slavesare added, a larger and more distinct area of the search space
1 2 3 4 5 6 71
1.2
1.4
1.6
1.8
2
2.2
2.4
2.6
2.8
3
Number of Slaves
Spee
dup
Fig. 8. Speedup produced by SQST-PEST.
is explored by each slave. Furthermore, SQST-PEST becomesincreasingly multiple-elitist as more slaves implies more “bestlocal solutions” are passed on to the next generation for ex-ploitation. With respect to time though, the efficiency of SQST-PEST parallelism decreases with p, due to the increasing com-munication overhead. This is illustrated in Fig. 8 where we seea decrease in the speedup obtained by SQST-PEST for largerp. The amount of communication overhead is proportional tothe number of generations g as well as to the number of slaves.Each slave interacts with the master twice in each genera-tion, so the total communication overhead increases with eitherg or p.
8.6. Serial_SQST-PEST versus SQMT-PEST comparison
We can thread a query against t templates by callingSQST-PEST t times serially, once for each template. Wetested Serial_SQST-PEST and SQMT-PEST with the query

1500 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
Table 4SQST-PEST on query 1gal(583) and template 1ad3_a(452)
SQST w. Rec.
p 1 2 3 4 5 6 7
Ethr −216.8± 3.8 −220.1± 4.1 −224.2± 3.9 −228.2± 3.4 −232.6± 4.1 −233.3± 3.3 −234.8± 3.4Time 70.5± 1.9 48.5± 2.1 36.4± 2.9 31.4± 2.7 28.5± 1.9 25.4± 1.8 23.6± 1.6
Table 5Serial_SQST-PEST vs. SQMT-PEST on query 1bbh_a and templates 451c(82), 1kdu(85), 1tlk(103), 2ccy_a(128), 1eca(136), 1apa(261), 1cca(291)
p SQMT w. Rec. Serial_SQST w. Rec.
T : Ethr Time T : Ethr Imp (%) Time
1 2ccy_a : −191.5± 3.5 273.2± 2.8 2ccy_a : −191.6± 3.8 0.05 267.3± 1.62 2ccy_a : −192.1± 3.7 197.8± 3.1 2ccy_a : −198.1± 4.1 3.12 174.9± 2.13 2ccy_a : −192.4± 4.1 144.7± 2.6 2ccy_a : −204.3± 3.3 6.2 121.1± 1.84 2ccy_a : −191.5± 4.5 112.7± 2.9 2ccy_a : −210.1± 3.1 9.7 91.5± 2.75 2ccy_a : −192.8± 4.1 100.2± 2.1 2ccy_a : −212.6± 4.1 10.3 72.9± 2.66 2ccy_a : −193.2± 4.1 96.5± 2.9 2ccy_a : −214.1± 3.6 10.8 66.4± 2.17 2ccy_a : −193.5± 3.6 91.7± 3.5 2ccy_a : −215.3± 3.6 11.2 63.9± 3.1
1bbh_a and the seven different templates 451c(82), 1kdu(85),1tlk(103), 2ccy_a(128), 1eca(136), 1apa(261) and 1cca(291),with increasing number of slaves. In Table 5, T is the templatepredicted by the algorithm, and Imp is the energy improvementof Serial_SQST-PEST over SQMT-PEST.
Both parallel methods produced the same prediction forany p. Both approaches gave better energy for larger p, withSerial_SQST-PEST achieving greater energy reduction as seenon the Imp column. Again, this is due to the better searchstrategy of SQST-PEST over EST (see Section 8.3) and to theincreased multiple-elitism and exploration ability of SQST-PEST when p is large (see Section 8.5); recall that each slaveof SQMT-PEST calls EST on each template it receives fromthe master. With respect to time, Serial_SQST-PEST paral-lelism is much more efficient than SQMT-PEST parallelismalso, although the efficiency decreases with p, as seen in Fig.9. SQMT-PEST suffers from a very high load imbalance as thetemplates have very diverse lengths and this, in turn, greatlyaffects its speedup. At this point, we did not apply any loadbalancing algorithm on SQMT-PEST; we only required thatthe slaves receive roughly equal number of templates. In thiscase, to greatly reduce the load imbalance in SQMT-PEST,it is necessary that all templates be roughly of equal lengthsand that (t mod p) = 0 for an almost perfect load balancing.Serial_SQST-PEST yields an almost perfect load balancingsince the slaves process sub-populations of roughly equalsizes for given templates; perfect balance is obtained onlywhen [(� + �) mod p] = 0. The decrease in the efficiency ofSerial_SQST-PEST parallelism is due only to its increasingcommunication overhead for larger p. We are planning to addload balancing method in SQMT-PEST and to perform moreexperiments on both parallel ESTs to study the effects of loadbalancing and other parameters on their speedup.
1 2 3 4 5 6 71
1.5
2
2.5
3
3.5
4
4.5
Number of Slaves
Spee
dup
SQMT
Serial_SQST
Fig. 9. Speedups produced by SQMT-PEST and Serial_SQST-PEST.
8.7. Preliminary results on a combined parallel EST
In the SQMT-PEST algorithm of Fig. 5, we can substituteEST by SQST-PEST to obtain a new parallel EST we callComb-PEST. Comb-PEST will be faster than SQMT-PESTsince each EST process is now parallelized using SQST-PEST.

A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502 1501
Table 6SQMT-PEST vs. Serial_SQST-PEST vs. Comb-PEST
Q (Length) T (Length) SQMT w. Rec. Serial_SQST w. Rec. Comb w. Rec.
Ethr Time Ethr Time Ethr Time
1bbh_a(131) 2ccy_a(128) −193.5± 4.1 91.7± 3.5 −215.3± 3.6 63.9± 3.1 −213.2± 3.6 52.7± 2.91apa(261) −193.2± 3.2 −194.2± 3.1 −191.8± 3.41cca(291) −164.4± 3.8 −172.1± 3.6 −170.8± 3.11kdu(85) −134.1± 3.4 −145.3± 3.8 −145.2± 3.41tlk(103) −131.2± 3.3 −136.5± 3.3 −133.7± 3.2451c(82) −129.1± 3.2 −137.2± 4.1 −137.2± 3.71eca(136) −116.6± 3.1 −122.1± 4.1 −121.9± 4.1
The energy result of Comb-PEST will also be better than thatof SQMT-PEST since SQST-PEST has a better search strategythan EST. We ran Comb-PEST with recombination on the samedata set as above, using seven slaves. The preliminary resultsare shown in Table 6.
As expected, Comb-PEST is much faster than SQMT-PEST. Its energy results are just slightly worse than those ofSerial_SQST-PEST but still much better than those of SQMT-PEST. Although it is not clear why at this stage, Comb-PESTwas faster than SQST-PEST in this example and more in-vestigation is needed in order to characterize the efficiencyof Comb-PEST with respect to SQST-PEST. More work isneeded to study and improve the performance of Comb-PEST,but these preliminary results are at least very encouraging atthis early stage of the research.
9. Conclusion and future research directions
In this paper we described a novel ES-based approach to pro-tein threading. With recombination, our EST algorithm givesmuch better results, both in energy and threading time, thanan existing GA-based method. Without recombination, EST iscomparable to the GA-based approach but much faster. Wediscussed parallel EST methods for threading a single queryagainst a single template or against a set of templates. Weimplemented the parallel ESTs on Grid-enabled platforms forHigh-Performance Computing. Due to its multiple-elitism andits greater exploration capability, Serial_SQST-PEST approachproduced better energy results than the other parallel EST meth-ods we described. We plan to test our methods on longer pro-teins as well as larger set of proteins and larger number ofslaves processors. More study is also required for Comb-PEST.It would be of interest to know what the statistical distributionof vector S = S1, S2, . . . , S|T | is. It seems that values 0 and1 should have high probability and values like |Q| should bevery infrequent. It also seems that values S1 and S|T | might besignificantly larger than interior values, since this would be aform of local alignment if Q contained T plus additional aminoacids on either end; thus, if S1 and S|T | are allowed to mutatefaster than the rest, then perfect subsequence matches wouldbe more easily found. Therefore, we plan to study the statisti-cal distribution of S and define better mutation operators. We
also intent to define recombination operators that do not yieldinvalid offsprings. In this paper, we were only interested in de-termining the best alignment between a query and a templategiven an energy function. We plan to use better energy func-tion than the one discussed in the paper. Also, a threading scorebetween a query and a template may not provide enough infor-mation about whether the template is the “correct” fold. Thatis, from the threading scores between a query and a pool oftemplates, we generally cannot tell if the query’s correct foldtemplate is in the pool, nor can we always tell which is the cor-rect fold even if it is there. A criterion is needed for estimatingthe confidence level of the predicted structure. Therefore as fu-ture research, we intent to study and include criterion, such asthe P-value scheme of Jones [9] and Karlin et al. [10], to assigna meaning to a threading score.
References
[1] T. Akutsu, S. Miyano, On the approximation of protein threading,Theoret. Comput. Sci. 210 (1999) 264–275.
[2] S.H. Bryant, Evaluation of threading specificity and accuracy, Proteins:Structure, Function and Genetics 26 (1996) 172–185.
[3] S.H. Bryant, C.E. Lawrence, An empirical energy function for threadingprotein sequence through folding motif, Proteins 16 (1993) 92–112.
[4] S.K. Burley, S.C. Almo, J.B. Bonanno, M. Capel, M.R. Chance, T.Gaasterland, D. Lin, A. Sali, F.W. Studier, S. Swaminathan, Structuralgenomics: beyond the Human Genome Project, Nature Genetics 23(1999) 151–157.
[5] O.H. Crawford, A fast, stochastic algorithm for protein threading,Bioinformatics 15 (1999) 66–71.
[6] D. Fischer, L. Rychlewski, R.L. Dunbrack, A.R. Ortiz, A. Elofsson,CAFASP3: the third critical assessment of fully automated structureprediction methods, Proteins 53 (2003) 503–516.
[7] A. Godzik, 〈http://cape6.scripps.edu/adam/service/alignbase.html〉, 1998.[8] D.E. Goldberg, Genetic Algorithms in Search, Optimization, and
Machine Learning, Addison-Wesley, Reading, MA, 1989.[9] D.T. Jones, GenTHREADER: an efficient and reliable protein fold
recognition method for genomic sequences, J. Mol. Biol. 287 (1999)797–815.
[10] S. Karlin, S.F. Altschul, Methods for assessing the statistical significanceof molecular sequence features by using general scoring schemes, Proc.Natl. Acad. Sci. USA, 87, 2264–2268.
[11] L.N. Kinch, J.O. Wrabl, S.S. Krishna, I. Majumdar, R.I. Sadreyev, Y. Qi,J. Pei, H. Cheng, N.V. Grishin, CASP5 assessment of fold recognitiontarget predictions, Proteins: Structure, Function and Genetics, FifthMeeting on the Critical Assessment of Techniques for Protein StructurePrediction (CASP5), vol. 53(S6), 2003, pp. 385–409.

1502 A. Ngom / J. Parallel Distrib. Comput. 66 (2006) 1489–1502
[12] R. Lathrop, The protein threading problem with sequence amino acidinteraction preferences is NP-complete, Protein Engineering 7 (1994)1059–1068.
[13] R. Lathrop, R. Rogers, J. Bienkowska, B. Bryant, L. Butorovic, C.Gaitatzes, R. Nambudripad, J. White, T. Smith, Analysis and algorithmsfor protein sequence-structure alignment, in: S.L. Salzberg, D.B. Searls,S. Kasif (Eds.), Computer Methods in Molecular Biology, Elsevier,Amsterdam, 1998, pp. 227–283.
[14] R. Lathrop, T. Smith, Global optimum threading with gapped alignmentand empirical pairs core functions, J. Mol. Biol. 255 (1996) 641–665.
[15] A.R. Panchenko, A. Marchler-Bauer, S.H. Bryant, Combination ofthreading potentials and sequence profiles improves fold recognition,J. Mol. Biol. 296 (2000) 1319–1331.
[16] I. Rechenberg, Evolutionsstrategie: Optimierung technischer systemenach prinzipien der biologischen evolution, Frommann-Holzboog Verlag,Stuttgart, 1973.
[17] J. Xu, M. Li, G. Lin, D. Kim, Y. Xu, Protein Threading bylinear programming, Pacific Symposium in Biocomputing (PSB), WorldScientific, Singapore, 2003 pp. 264–275.
[18] J. Yadgari, A. Amir, R. Unger, Representation and data structureof genetic algorithms for protein threading, 〈http://citeseer.ist.psu.edu/yadgari97representation.html〉, 1997.
[19] J. Yadgari, A. Amir, R. Unger, Genetic Algorithms for Protein Threading,ISMB, 1998, pp. 193–202.
[20] J. Yadgari, A. Amir, R. Unger, Genetic Threading, Constraints, vol. 6,Kluwer Academic Publishers, Dordrecht, 2001, pp. 271–292.
[21] N. Yanev, R. Andonov, Solving protein threading problem in parallel,Workshop on High Performance Computational Biology, in: Proceedings
of the 17th International Parallel and Distributed Processing Symposium,Nice, France, April, IEEE Computer Society Press, Los Alamitos, CA,2003.
[22] N. Yanev, R. Andonov, Parallel divide and conquer approach for theprotein threading problem, Concurrency and Computation: Practice andExperience, vol. 16, 2004, pp. 961–974.
[23] T.K. Yap, O. Frieder, R.L. Shapiro, Parallel computation in biologicalsequence analysis, IEEE Trans. Parallel Distrib. Systems 9 (3) (1998)283–294.
[24] B. Zhang, L. Jaroszewski, L. Rychlewski, A. Godzik, Similaritiesand differences between nonhomologous proteins with similar folds:evaluation of threading strategies, Fold Des. 2 (1997) 307–317.
Dr. Alioune Ngom received the B.Sc. degree incomputer science in 1992 from the Universityof Québéc at Trois-Rivières, Québéc, and theM.Sc. and Ph.D. degrees in computer sciencein 1995 and 1998, respectively, from the Uni-versity of Ottawa, Ontario. From 1998 to 2000he was an Assistant Professor in the depart-ment of Computer Science at Lakehead Univer-sity, Thunder Bay, Ontario. In Summer 2000,he joined the faculty in the School of ComputerScience at the University of Windsor, Ontario,Canada, where he currently holds the positionof Associate Professor. His current research and
teaching interests include multiple-valued logic and algebra, discrete neu-ral computation, applications of evolutionary computation, wireless mobilecomputing, bioinformatics and computational molecular biology.





























