Embed Size (px)
Citation preview

doi:10.1016/j.jmb.2009.09.045 J. Mol. Biol. (2009) 394, 576–586
Available online at www.sciencedirect.com
PhhR Binds to Target Sequences at DifferentDistances with Respect to RNA Polymerase inOrder to Activate Transcription
M.CarmenHerrera1, TinoKrell1, XiaodongZhang2 and Juan-LuisRamos1⁎
1Department of EnvironmentalProtection, EstaciónExperimental del Zaidín, CSIC,C/Profesor Albareda 1, E-18001Granada, Spain2Imperial College,SW7 2AZ London, UKReceived 28 April 2009;received in revised form10 September 2009;accepted 17 September 2009Available online23 September 2009
*Corresponding author. E-mail [email protected] used: tsp, transcrip
integration host factor; ITC, isothermcalorimetry.
0022-2836/$ - see front matter © 2009 E
The NtrC-family PhhR protein of Pseudomonas putida is involved in thecontrol of themetabolism of aromatic amino acids, and it is a dual regulatoryprotein. When PhhR acts as an activator, it stimulates transcription from itscognate promoters with RNA polymerase/σ70 rather than with σ54, as is thecase for most members of the family. The target binding sites in repressedand activated promoters are defined by the 5′-TGTAAAN6TTTACA-3′consensus sequence. PhhR binds to target sites as a dimer with affinity in therange of 0.03 to 6.6 μM, as shown by isothermal titration calorimetry. PhhRactivates transcription from both the PP2827 and PP2078 promotersregardless of the absence or presence of aromatic amino acids, whereasPhhR stimulates transcription from certain positively regulated promoters(PphhA, PPP3122, PPP3434, and Phmg ) only in the presence of phenylalanine andtyrosine or their corresponding keto acids (i.e., phenylpyruvate and p-hydroxyphenylpyruvate). A surprising feature of PhhR-mediated transcrip-tional activation is that PhhR may bind to one or two upstream targetsequences that are located at different distances from the RNA polymerasebinding site. This allows PhhR to function as a class I regulator (target sites at−66/−83), a class II regulator (target sites around −40), as well as anenhancer protein (target sites N−128). When functioning as an enhancerprotein, PhhR-mediated transcription is modulated by the integration hostfactor protein. PhhR represses transcription from its own promoter and thepromoter of the paaY gene by steric hindrance.
© 2009 Elsevier Ltd. All rights reserved.
Edited by M. Gottesman
Keywords: amino acid; NtrC family; σ70; DNA binding protein; enhancerIntroduction
In prokaryotes, the activity of RNA polymerasecan be modulated by a number of transcriptionalregulatory proteins known as activators and repres-sors. In a recent survey of regulators, it was foundthat about 80% of described regulators are exclu-sively activators or exclusively repressors, with theremaining being dual regulators.1The interactions between RNA polymerase and
activators are strongly influenced by the location atwhich they bind to the promoter relative to thetranscription start point (tsp), and a wide range of
ress:
tion start point; IHF,al titration
lsevier Ltd. All rights reserve
locations and interactions between regulators andRNA polymerase have been described.2 The mostfrequently observed regulator binding sites arefound close to the −35 region of the promoter.2–4
In other promoters, known as class I promoters, theactivator binding site is close to the −60 position andoften involves interactions between the regulatorand the extended C-terminal end of α subunit ofRNA polymerase.2 Other upstream activatorsequences have also been found more than 100 bpaway from the tsp, a feature often associated withregulators that act with RNA polymerase/σ54,5,6
although exceptions exist. Examples of such excep-tions are TyrR of Escherichia coli and NtrC-like ofRhodopseudomonas palustris, both of which functionwith RNA polymerase/σ70.7,8 Less frequentlyoccurring activator binding sites are located at the−10/−35 regions of promoters, which are utilised bythe MerR family of transcriptional regulators.Structural studies have shown that when MerR
d.
577PhhR, RNA Polymerase, and Transcription
family members bind to −10/−35 motifs, which arenot present on the same DNA face, they are ableactivate expression in these promoters by distortingthe DNA so that the RNA polymerase binding sitesbecome aligned for correct recognition.9
Repression of transcription occurs through vari-ous mechanisms that inhibit the key processes oftranscription initiation. The most often describedrepression mechanism involves binding of therepressor protein to the promoter in a way thatimpedes the binding of RNA polymerase, althoughit should be mentioned that steric hindrance is justone of the several mechanisms used by repressors toachieve their function.10
PhhR is the ultimate regulator of L-phenylalaninecatabolism in a number of pathogenic and non-pathogenic Pseudomonas strains.11–14 Additionally,based on microarray assays and a wide series ofpromoter fusions, we have recently shown that PhhRis a dual regulator.15 In fact, the PhhR regulon in-cludes 21 genes/operons whose gene productsinclude proteins that are involved in L-phenylalaninecatabolism via two parallel pathways, efflux pumpsthat might prevent phenylalanine accumulation inthe cells as well as proteins of unknown function.15
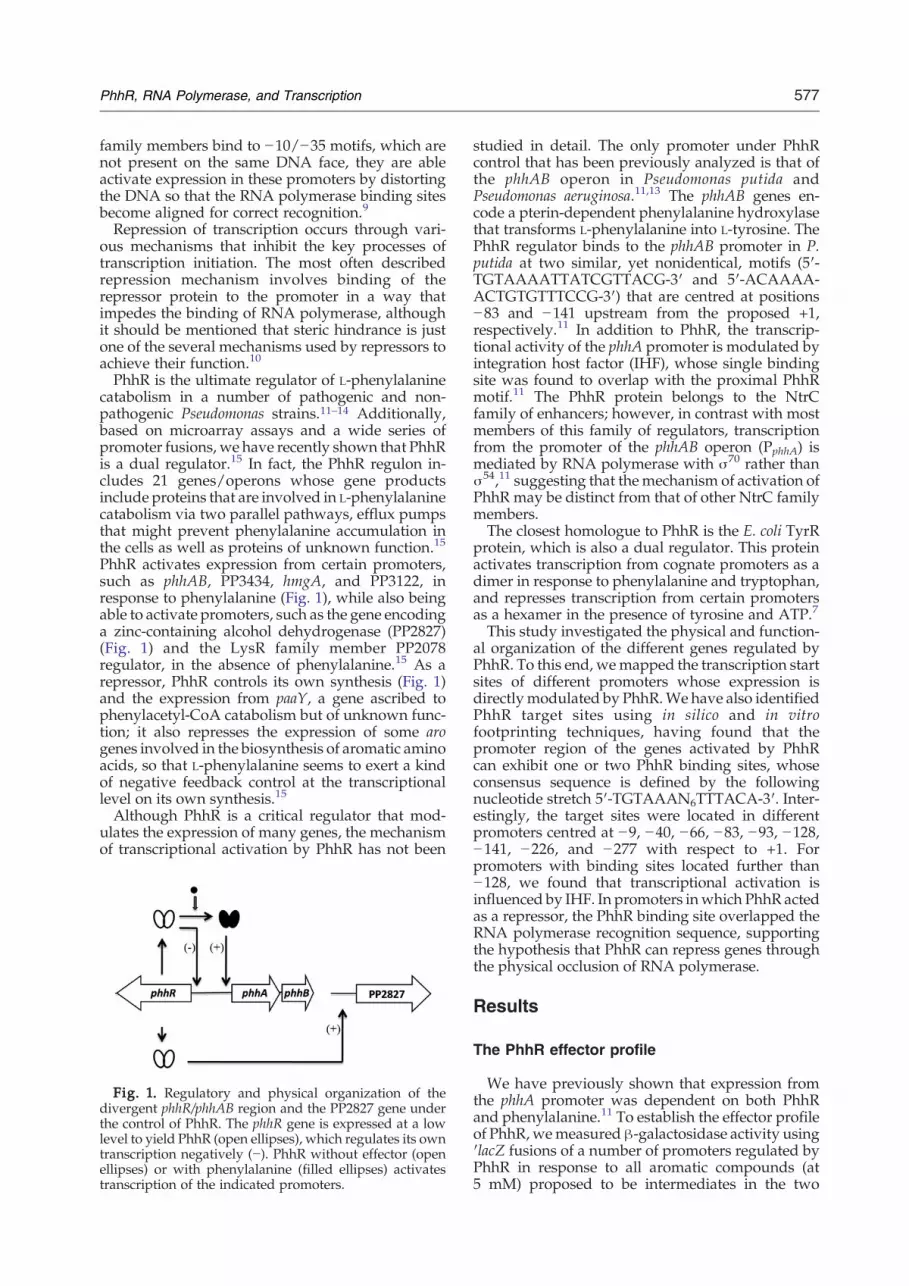
PhhR activates expression from certain promoters,such as phhAB, PP3434, hmgA, and PP3122, inresponse to phenylalanine (Fig. 1), while also beingable to activate promoters, such as the gene encodinga zinc-containing alcohol dehydrogenase (PP2827)(Fig. 1) and the LysR family member PP2078regulator, in the absence of phenylalanine.15 As arepressor, PhhR controls its own synthesis (Fig. 1)and the expression from paaY, a gene ascribed tophenylacetyl-CoA catabolism but of unknown func-tion; it also represses the expression of some arogenes involved in the biosynthesis of aromatic aminoacids, so that L-phenylalanine seems to exert a kindof negative feedback control at the transcriptionallevel on its own synthesis.15
Although PhhR is a critical regulator that mod-ulates the expression of many genes, the mechanismof transcriptional activation by PhhR has not been
Fig. 1. Regulatory and physical organization of thedivergent phhR/phhAB region and the PP2827 gene underthe control of PhhR. The phhR gene is expressed at a lowlevel to yield PhhR (open ellipses), which regulates its owntranscription negatively (−). PhhR without effector (openellipses) or with phenylalanine (filled ellipses) activatestranscription of the indicated promoters.
studied in detail. The only promoter under PhhRcontrol that has been previously analyzed is that ofthe phhAB operon in Pseudomonas putida andPseudomonas aeruginosa.11,13 The phhAB genes en-code a pterin-dependent phenylalanine hydroxylasethat transforms L-phenylalanine into L-tyrosine. ThePhhR regulator binds to the phhAB promoter in P.putida at two similar, yet nonidentical, motifs (5′-TGTAAAATTATCGTTACG-3′ and 5′-ACAAAA-ACTGTGTTTCCG-3′) that are centred at positions−83 and −141 upstream from the proposed +1,respectively.11 In addition to PhhR, the transcrip-tional activity of the phhA promoter is modulated byintegration host factor (IHF), whose single bindingsite was found to overlap with the proximal PhhRmotif.11 The PhhR protein belongs to the NtrCfamily of enhancers; however, in contrast with mostmembers of this family of regulators, transcriptionfrom the promoter of the phhAB operon (PphhA) ismediated by RNA polymerase with σ70 rather thanσ54,11 suggesting that themechanism of activation ofPhhR may be distinct from that of other NtrC familymembers.The closest homologue to PhhR is the E. coli TyrR
protein, which is also a dual regulator. This proteinactivates transcription from cognate promoters as adimer in response to phenylalanine and tryptophan,and represses transcription from certain promotersas a hexamer in the presence of tyrosine and ATP.7
This study investigated the physical and function-al organization of the different genes regulated byPhhR. To this end, wemapped the transcription startsites of different promoters whose expression isdirectlymodulated by PhhR.We have also identifiedPhhR target sites using in silico and in vitrofootprinting techniques, having found that thepromoter region of the genes activated by PhhRcan exhibit one or two PhhR binding sites, whoseconsensus sequence is defined by the followingnucleotide stretch 5′-TGTAAAN6TTTACA-3′. Inter-estingly, the target sites were located in differentpromoters centred at −9, −40, −66, −83, −93, −128,−141, −226, and −277 with respect to +1. Forpromoters with binding sites located further than−128, we found that transcriptional activation isinfluenced by IHF. In promoters inwhich PhhR actedas a repressor, the PhhR binding site overlapped theRNA polymerase recognition sequence, supportingthe hypothesis that PhhR can repress genes throughthe physical occlusion of RNA polymerase.
Results
The PhhR effector profile
We have previously shown that expression fromthe phhA promoter was dependent on both PhhRand phenylalanine.11 To establish the effector profileof PhhR,wemeasuredβ-galactosidase activity using′lacZ fusions of a number of promoters regulated byPhhR in response to all aromatic compounds (at5 mM) proposed to be intermediates in the two

Table 1. Plasmids and strains used in this study
Strain Genotype Ref
P. putida KT2440 CmR; ApR 16MCH4 (phhR:aphA3) CmR; KmR 11P. putida KT2440-IHF3 ihfA∷Km 17E. coli DH5α F′/hsdR17, recA1, gyrA 18E. coli BL21 (DE3) F−, ompI, hsdSB
(r−B m−B), gal, dcm, met
Novagen
PlasmidspMP220 ′lacZ; IncP; TcR 19pMCR1 PphhR∷lacZ; TcR 11pMCA1 PphhA∷lacZ; TcR 11pMChmg Phmg∷lacZ; TcR 15pMC3122 PPP3122∷lacZ; TcR 15pMC3434 PPP3434∷lacZ; TcR This studypMC2827 PPP2827∷lacZ; TcR 15pMC3285 PPPaaY∷lacZ; TcR 15pMC2078 PPP2078∷lacZ; TcR This studypMC3122P ‘orfPP3122’∷pGEMT, ApR This study
ApR, CmR, KmR, and TcR stand for resistance to ampicillin,chloramphenicol, kanamycin, and tetracycline.
578 PhhR, RNA Polymerase, and Transcription
parallel pathways for L-phenylalanine degradation,namely, tyrosine, p-hydroxyphenylpyruvate, phe-nylpyruvate, and homogentisate. We also includedtryptophan in this series of assays because it is alsoan aromatic amino acid. We used the low-copy-number pMP220 vector for all the following ′lacZfusions. For positively regulated promoters, we usedthe previously available PphhA∷′lacZ fusion andconstructed a fusion of the PP3122 promoter regionto ′lacZ (Table 1). We found that phenylalanine,tyrosine, p-hydroxyphenylpyruvate, and phenylpyr-uvate were good effectors, whereas homogentisatewas a very weak effector and tryptophan showed noeffect at all on expression from the constructs(Table 2). Further support of the positive effect ofthese amino acids and keto acids was obtainedwhena fusion of other PhhR-regulated promoters fused to′lacZ was used (Supplementary Table S1). To testfor effectors that influence repression by PhhR, weused a fusion of the paaY promoter to ′lacZ, since wehave also shown that expression of paaY is repressedby PhhR.15 Upon measuring expression from thePpaaY∷′lacZ construct in response to the above effec-tors, we found that again, L-phenylalanine, L-tyrosine,p-hydroxyphenylpyruvate, and phenylpyruvatewere effectors, whereas tryptophan and homogenti-sate showed no effect on expression from thisconstruct (Table 2).
Table 2. Profile of PhhR effectors measured with positively a
Promoterfusion
β-Gal
— Phe Tyr
PphhA∷′lacZ 5±1 2780±150 630±30PPP3122∷′lacZ 20±4 1445±41 5460±145PpaaY∷′lacZ 40±6 345±65 500±15
P. putidaKT2440, bearing the indicated promoter∷′lacZ fusion in the loM9 minimal medium with glucose as a carbon source and 5 mM ofindependent determinations done in duplicate, and values are the aveOther conditions are given in Materials andMethods. Phe, Tyr, Trp, 4Otryptophan, 4-hydroxyphenylpyruvate, phenylpyruvate, and homoge
Oligomeric form of PhhR
To determine the oligomeric form of PhhR, wepurified the protein to homogeneity, as describedpreviously,11 and carried out gel filtration assaysunder different conditions. We found that close to85% of the PhhR protein existed as a dimer when theassay was carried out in the absence of aromaticamino acids (Fig. 2a and b), and that the remainingprotein eluted as a peak corresponding to thehexameric form of the protein. We collected frac-tions corresponding to both peaks and analyzedthem on SDS-PAGE and verified that the proteinwas, in fact, PhhR (Fig. 2c). In order to test whetherthe presence of aromatic amino acids influences theoligomeric state of PhhR, we preincubated theprotein with 5 mM of L-phenylalanine or L-tyrosineand eluted the protein with buffers containing 5–10 mM of the same aromatic amino acid. The elutionprofile of the protein under these conditions wasidentical with that seen in the absence of aromaticamino acids (data not shown).A major difference between NtrC family members
and PhhR is that PhhR does not hydrolyse ATP. Inspite of this, we tested if the oligomeric state of PhhRwas affected by the presence of ADP or ATP, but nochange in the conformational state was observed(not shown). We found no change in the oligomericstate when incubation was performed in thepresence of 1 to 10 mM of each aromatic aminoacid and 100 μM–10 mM of ATP (see Fig. 2 for arepresentative result using 1 mM tyrosine and100 μM ATP). The results showed that the dimer/hexamer ratio remained 6.7±0.2 regardless of theprotein concentration and regardless of the presenceof nucleotides.Although the set of chromatographic results
supported that PhhR is a dimer both in the absenceand in the presence of L-phenylalanine or L-tyrosine,interactions with L-phenylalanine and L-tyrosinemay have an influence on the conformational stateof PhhR as determined during the establishment ofoptimal conditions for crystallization of this protein(data not shown). We found that the presence of thearomatic amino acids favours the crystallization ofthe protein and that a set of thin needles areproduced only when the aromatic amino acid waspresent (not shown). Further optimization of thecrystallisation conditions is being carried out asthese crystals are extremely fragile and are not
nd negatively regulated promoters
actosidase (Miller units)
Trp 4-Ohphe Phenylp Hmg
5±1 150±25 2260±50 30±220±4 1220±90 605±12 110±12050±13 485±60 1650±50 45±10
w-copy-number, wide-host-range pMP220 vector,18 was grown onthe indicated aromatic compound. Data are the average of fourrage of all determinations and the corresponding standard errors.H-phe, Phenyl, and Hmg stand for L-phenylalanine, L-tyrosine, L-ntisate, respectively.

Fig. 2. Determination of the oligomeric state of PhhR by analytical gel filtration. (a) Elution profile from the analysis of86 μM purified PhhR. Continuous track, protein eluted in the absence of effectors; dotted line, protein eluted in thepresence of 1 mM tyrosine and 100 μM ATP. (b) Calibration curve using protein standards: lactalbumin (A; 14 kDa),carbonic anhydrase (29 kDa), chicken egg albumin (45 kDa), bovine serum albumin monomer (D; 66 kDa), and bovineserum albumin dimer (E; 132 kDa). Shown is a liner regression of the resulting five elution volumes. The elution volumesand the corresponding molecular mass of PhhR are indicated. (c) SDS-PAGE of fractions eluted as 11 ml (lanes 1 and 2)and 13 ml (lanes 3 and 4); lane M is a molecular weight marker.
579PhhR, RNA Polymerase, and Transcription
suitable for the resolution of three-dimensionalstructure.We also explored the possibility that the oligo-
meric state of PhhR may be influenced by binding tospecific DNA target sequences. Electrophoreticmobility shift assays were carried out using a 460-bp fragment of PP3122 (from +100 to −360) withincreasing concentrations of PhhR in the absenceand in the presence of 10 mM L-phenylalanine orL-tyrosine. Figure 3 shows the results in the absence(Fig. 3a) and in the presence of phenylalanine
Fig. 3. Electrophoretic mobility shift assay of PhhRwith the PP3122 intergenic region in the absence (a) and inthe presence (b) of phenylalanine. Binding reactions werecarried out with 0.5 nM DNA and increasing concentra-tions of PhhR (from left to right, 0–2.5 μM) for 30 min andsubjected to electrophoresis as indicated in Materials andMethods.
(Fig. 3b). We found that the retarded band wasobserved at concentrations above 1 μM PhhR andthat the size of the retarded band was not influencedby the presence of amino acids (Fig. 3), whichsuggests that the oligomeric state of PhhR whenbound to its target sites is not influenced by thepresence of amino acids. We determined the molec-ularmass of theDNA/PhhR complex and found thatit agrees with the size of one dimer of PhhR boundper target site. These data agree with our earlierstudies that showed that PhhR binds to both phhApromoter binding sites as a dimer.
Determination of the transcription start point ofpromoters activated and repressed by PhhR andidentification of PhhR binding sites by footprinting
As a first step in the determination of the potentialbinding sites for PhhR-regulated promoters, wedecided to define the tsp of these promoters bycarrying out primer extension assays. In a previousstudy, we determined the tsp of phhA and phhR. Inthe current study, we identified the tsp of theremaining known activated promoters (i.e., PP3122,PP3434, PP2078, PP2827, as well as the recentlyidentified PhhR-repressed paaY promoter; Fig. 4 andSupplementary Fig. S1).The tsp of the activated promoters (PP3122,
PP3434, PP2078, and PP2827) were determinedusing RNA prepared from cells in the exponentialgrowth phase in the presence of phenylalanine. Forthe repressed promoter, the tsp was determinedusing RNA from cells devoid of PhhR regulator inorder to facilitate a high yield of the signal. Figure 4

Fig. 4. Transcription start point of PP3122 (a) andPP3285 (b). Cells were grown inM9minimal mediumwithglucose as the sole carbon source in the presence of 5 mMphenylalanine. RNA was prepared from the cells in themid-exponential growth phase, as described in Materialsand Methods. Lanes 1 to 4 are sequencing ladders used todetermine the exact site of the analyzed cDNAs. Lane 5corresponds to the sample, and M is a single-strand sizemarker. On the right is the size of the cDNA extendedproduct, and at the bottom is the deduced hexamers at −10and −35 for the corresponding promoters.
580 PhhR, RNA Polymerase, and Transcription
shows the results for the positively regulated PP3122promoter and the repressed paaY promoter. The tspsequences of the positively regulated promoterswere aligned according to the +1 position, revealingthat the most conserved region (−16 to −11)corresponds to a 5′-TAWART-3′ consensus. Thissequence resembles the proposed GATATT hexamerthat has been proposed to be recognized by σ70 inPseudomonas.20 Upon determining the tsp for thenegatively regulated paaY promoter, we found thesequence 5′-ATTAAT-3′ (−10), which also showssimilarity to the sequence recognized by RNApolymerase/σ70. No sequence similar to the σ54-recognized consensus GGN9TGCA in the −24/−12stretch was found.6
Since PhhR recognizes two target sites in phhA,which is the only PhhR-regulated promoter con-firmedwith in vitro and in vivo data,11 we aligned thesequences of the two sites and derived a consensus
sequence (5′-NNNAAAANTNTNNTTNCG-3′) thatwas used to screen from positions +1 to −500 of eachpromoters in order to identify potential PhhRbinding sites. Using the derived consensus, wewere able to locate the two PhhR boxes present inthe phhA promoter, centred at −141 and −83 (Fig. 5).Upon screening the other promoters, two bindingsites were found for PP3122 centred at −93 and −40;two were also found for PP3434 centred at −277 and−226; a single binding site was found for PP2078 at−128; for PP2827 at −66; and, surprisingly, at −9 forthe hmgA promoter. For the repressed promoters, asingle potential binding site was found centred at+10 for PP3285 (paaY), and two sites at −41 and +16were found for phhR (Fig. 5).The sites that we found using the derived
consensus for phhA and phhR were the same asthose that have been previously characterized,confirming the efficacy of the screen.11 These insilico derived binding sites were then validated forthe PP3122 promoter using footprint assays (Fig. 6).In this series of assays, increasing concentrations ofPhhR (0 to 15 μM) were incubated with a 466-bpDNA fragment spanning −366 to +100 in the PP3122promoter. We found that two regions were pro-tected, and that these regions corresponded to thedistal and proximal sites that were identified above,confirming and validating our in silico results.
Thermodynamic characterization of theinteractions of PhhR with its target sequences
To shed further light on the binding processbetween PhhR and its target sequences, we deter-mined the thermodynamic parameters of interac-tions between a homogeneous preparation of PhhRand 26-mer double-stranded oligonucleotides (at3 μM) corresponding to each of the PhhR bindingsites that were identified above. Figure 7 showstitration results for the two PhhR target sites of thePP3434 promoter as a representative example (othertitration curves are given in Supplementary Fig. S2).Binding experiments gave rise in all cases toexothermic heat changes indicative of favourableenthalpy changes, which were accompanied byeither favourable or unfavourable entropy changes(Table 3). In the case of the PP3434 sites, we foundthat enthalpy changes and the affinity of PhhR werehigher for the site corresponding to −226 (725 nM)than for the more distant site centred at −277(6.6 μM). We also inferred from the stoichiometry ofbinding that one PhhR dimer binds per site,suggesting that each PhhR monomer recognizeshalf of each site. Isothermal titration calorimetry(ITC) data derived for the remaining targetsequences are presented in Table 3. It should benoted that affinity of PhhR for these binding sitesvaries between 30 nM for the high-affinity site of thephhA promoter11 and 6.6 μM for the low-affinity siteof the PP3434 promoter. For most target sequences,the affinities were in the range of 200 to 660 nM. TheITC data revealed no significant difference inbinding affinity according to the position of the

Fig. 5. PhhR and IHF binding sites in the mapped promoters. Schematic representation of the fusion of differentpromoters to ′lacZ. The lacZ gene is represented by a thick arrow. Green boxes represent PhhR binding sites, and thenumbers above indicate the centre of the target site. Blue ellipses indicate IHF sites; the centre of the site is also indicted. β-Galactosidase activity was determined for cells in the exponential phase in the parental strain KT2440, its isogenic IHFmutant, and its isogenic PhhR mutant, in the absence (−) or in the presence of phenylalanine (+phe). Data are the averageof at least three independent assays done in duplicate. Standard error was below 10% of the given number.
581PhhR, RNA Polymerase, and Transcription
site with respect to +1 or between the repressed andthe activated promoters.
IHF influences transcription from some of thePhhR-regulated promoters
We have previously shown that a single IHFbinding site is present in the phhA promoter, whichoverlaps with the proximal PhhR binding site(Fig. 5). Furthermore, using an IHF-null back-ground,21 we showed that the presence of IHFreduced expression from the phhA promoter.11 IHFbinding sites were found by visually searching thesequences of the promoter regions of PP3434(centred at −150) and PP2078 (centred at −76), aswell as the two repressed promoters of phhR andpaaY (centred at −41). Similar searches showed thatno IHF sites were present within the PP3122 or hmgpromoters. To fully assess the potential influence ofIHF on expression from these promoters, we con-structed appropriate fusions to ′lacZ and measuredβ-galactosidase activity in the IHF-proficient andIHF-deficient backgrounds. Similarly to what wefound for phhA, expression from phhR and paaY inthe IHF-deficient background was higher than thatin the parental background. This was particularlyevident in the presence of phenylalanine (Fig. 5).More strikingly, PP2078 and PP3434 expression wasfound to be completely IHF dependent, as they werenot expressed at all under an IHF-null background.In a PhhR-deficient background, transcription frompositively regulated promoters failed to occur, as
expected (Fig. 5).We can therefore conclude that IHFmodulates PhhR-mediated transcription, and that itseffect may be influenced by the location of the IHFsite with respect to the tsp, although PhhR is the keyregulator. Promoters with no IHF binding sites(PP3122 and hmg) exhibited no changes in theexpression pattern between IHF-null and IHF-proficient cells, as expected (Fig. 5).
Discussion
The P. putida PhhR transcriptional regulator is adual regulator in that it can act both as an activatorand as a repressor of transcription. In the currentstudy, we present data in support of PhhR function-ing as a dual regulator through at least three differentmodes of action: (1) activation in response toaromatic amino acids, (2) activation regardless ofthe presence of aromatic amino acids, and (3)repression by competing with RNA polymerase.This is supported by data in Table 2, Fig. 5, andSupplementary Table S2.As an activator, PhhR exhibits a number of
distinct characteristics. Of particular interest is thatPhhR is able to activate promoters that contain twoPhhR binding sites (phhAB, PP3122, and PP3434) aswell as those that contain a single binding site(PP2078, hmg, and PP2827)—a feature that is sharedwith TyrR,22,23 the closest homologue of PhhR. Italso of note that for the first set of promoters—thosewith two binding sites—their expression is both
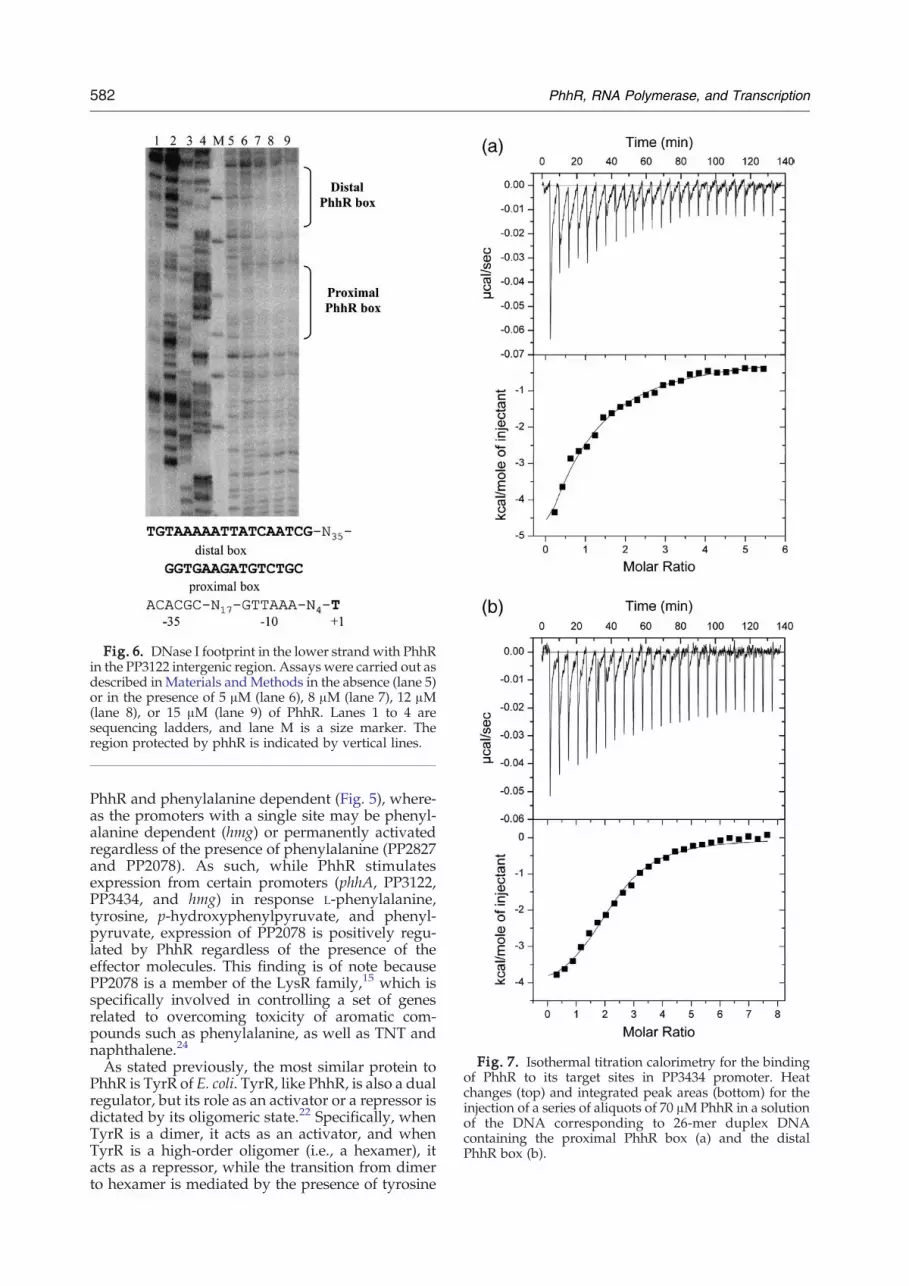
Fig. 6. DNase I footprint in the lower strand with PhhRin the PP3122 intergenic region. Assays were carried out asdescribed in Materials andMethods in the absence (lane 5)or in the presence of 5 μM (lane 6), 8 μM (lane 7), 12 μM(lane 8), or 15 μM (lane 9) of PhhR. Lanes 1 to 4 aresequencing ladders, and lane M is a size marker. Theregion protected by phhR is indicated by vertical lines.
Fig. 7. Isothermal titration calorimetry for the bindingof PhhR to its target sites in PP3434 promoter. Heatchanges (top) and integrated peak areas (bottom) for theinjection of a series of aliquots of 70 μMPhhR in a solutionof the DNA corresponding to 26-mer duplex DNAcontaining the proximal PhhR box (a) and the distalPhhR box (b).
582 PhhR, RNA Polymerase, and Transcription
PhhR and phenylalanine dependent (Fig. 5), where-as the promoters with a single site may be phenyl-alanine dependent (hmg) or permanently activatedregardless of the presence of phenylalanine (PP2827and PP2078). As such, while PhhR stimulatesexpression from certain promoters (phhA, PP3122,PP3434, and hmg) in response L-phenylalanine,tyrosine, p-hydroxyphenylpyruvate, and phenyl-pyruvate, expression of PP2078 is positively regu-lated by PhhR regardless of the presence of theeffector molecules. This finding is of note becausePP2078 is a member of the LysR family,15 which isspecifically involved in controlling a set of genesrelated to overcoming toxicity of aromatic com-pounds such as phenylalanine, as well as TNT andnaphthalene.24
As stated previously, the most similar protein toPhhR is TyrR of E. coli. TyrR, like PhhR, is also a dualregulator, but its role as an activator or a repressor isdictated by its oligomeric state.22 Specifically, whenTyrR is a dimer, it acts as an activator, and whenTyrR is a high-order oligomer (i.e., a hexamer), itacts as a repressor, while the transition from dimerto hexamer is mediated by the presence of tyrosine

Table 3. Affinity of PhhR for its binding sites as determined from ITC titrations
Boxes PhhR KD (μM) ΔH (kcal/mol)TΔS
(kcal/mol)
Activated genesphhAB (PP4490)a GCTGACAAAAACTGTGTTTCCGCCAG 2.03±0.25 −2.59 5.18phhAB (PP4490)a GTTTTGTAAAATTATCGTTACGAAAA 0.03±0.01 −14.7 −4.47PP3122 GTTTTGTAAAAATTATCAATCGATGA 1.71±0.13 −9.59 −2.20PP3122 TTGCGGTGAAGATGTCTGCACACGCT 2.2±0.2 −1.80 5.81PP3434 AGGTTGTCGAGGATGTGTTCGACGTG 0.72±0.1 −4.43 3.81PP3434 TGGCTGCTGGCCTGCATTTCACGTTC 6.6±1 −1.33 5.96Hmg (PP4621) GCGGAGCAAATTACGTTATTCGTAAT 2.12±0.38 −7.51 0.2PP2078 GTGCGGTCATGGTGGCGTTACTCGTA 0.27±0.06 −11.3 −2.29PP2827 CAGTGGTAATTAGAGGTTCACAAAGG 2.47±0.21 −2.37 5.27
Repressed genespaaY (PP3285) CACAAGTGATACACGATTGACGACCA 0.85±0.1 −5.02 3.09phhR (PP4489)a GTTTTGTAAAATTATCGTTACGAAAA 0.03±0.01 −14.7 −4.47phhR (PP4489)a GCTGACAAAAACTGTGTTTCCGCCAG 2.03±0.25 −2.59 5.18
ITC assays were carried out as described in Materials and Methods.a Data from Herrera and Ramos.11
583PhhR, RNA Polymerase, and Transcription
and ATP.22,23 Furthermore, as a repressor, thehexameric form of TyrR binds to high- and low-affinity sites, creating loops that prevent the bindingof RNA polymerase to target promoters.7 The datathat we have obtained in this study suggest thatPhhR differs from TyrR in that it is preferentially adimer regardless of the presence of aromatic aminoacids or ATP. It is therefore not surprising that themechanism of PhhR-mediated repression seems todiffer from that of TyrR in that PhhR binding sitesoverlap with RNA polymerase binding sites, as isthe case in phhR and paaY promoters, suggestingthat steric hindrance rather than the oligomeric stateof PhhR is important for PhhR-mediated repression.This is of interest because there are few NtrC-likeregulators that act as repressors.6,21
Modulation of some of the genes in the PhhRregulon involves cooperative action with IHF; in thisline is our observation that in an IHF-deficientbackground, expression from the two repressedpromoters (phhR and paaY) is higher than that in theisogenic-proficient strain. In terms of PhhR positive-ly regulated promoters that had IHF binding sites(phhA, PP3434, and PP2078), we also found thatsteric factors are important in determining whetherIHF exerts a positive or a negative effect. For phhA,IHF exerts a negative effect on expression due to thecompetition between IHF and PhhR for the proximalactivation site; while in the other two promoters, inwhich PhhR binding sites are located N−128 bp (i.e.,PP3434 and PP2078) and steric hindrance does notoccur, we found that IHF enhances expression,which suggests that DNA bending21,24–26 is neededto facilitate interactions between the regulatorbound at upstream activator sequences and theRNA polymerase bound to the −10/−35 elements.Another difference between PhhR and other
members of the NtrC family of transcriptionalregulators is that in contrast to the majoritymembers of this family, which function with σ54,
PhhR has been shown to transcribe the phhApromoter using RNA polymerase/σ70.11 We furtherexplored this difference considering promoters thathave been characterized in the current study. Upondetermining the tsp of the promoters under PhhRcontrol, we searched for potential target sequencessimilar to those recognized by σ70 for σ54. In all ofthe analyzed promoters, we found −10/−35 regionsrather than −12/−24 regions, further indicating thatin contrast to most of NtrC family members,transcription is mediated by RNA polymerase/σ70.Another relevant insight gained by the current
study are the locations of the PhhR binding siteswith respect to the +1 tsp, which, in the promotersinvestigated, vary from −9 to −277. This suggeststhat the different interactions that occur betweenPhhR and its target sequences are important indetermining how PhhR activity is modulated. Assuch, for the PP3122 promoter, which has a PhhRbox at −40 and another at −93, we propose that it isa class II promoter (Fig. 8). Furthermore, we proposethat PhhR bound to the high-affinity −40 site mayestablish direct interactions with σ70. On the otherhand, the PP2827 promoter has a PhhR box centredaround −66, making it a class I promoter, such thatPhhR would activate transcription by interactingwith the α subunit of the RNA polymerase.28 In thisregard, this diverse binding ability places PhhR inthe same category as the CAP protein of E. coli,which is able to function both as a class I and as aclass II activator.27
The hmgA promoter requires some specific atten-tion, since this gene is the first gene in the hmgABCoperon and is involved in homogentisate meta-bolism.12 Homogentisate catabolic genes are knownto be induced when P. putida cells are grown onhomogentisate, and early studies show that theexpression of this operon is mediated by the hmgRgene product.12 HmgR is known to bind the Phmgpromoter spanning positions −16 to +29with respect

Fig. 8. PhhR as a class I, class II, and enhancer-like activator. The scheme is based on Busby and Ebright's presentationof regulator binding sites with respect to RNA polymerase binding sites.27 Current research in our lab is directed toestablish interactions between RNA polymerase and PhhR, and the depicted interaction should be considered as aworking model. (a–c) Physical organization of the promoters of PP2827, PP3122, and PP2078.
584 PhhR, RNA Polymerase, and Transcription
to the Phmg transcription start site, at a perfectlypalindromic 17-bp motif (5′-TCGTAATCTGA-TTACGA-3′).12 An unexpected finding of ourstudy is that the induction of the hmgABCD operonwas also PhhR- and L-phenylalanine dependent.15 Itcould be argued that this is due to the synthesis ofhomogentisate from L-phenylalanine, but the factthat hmgA expression occurred with L-phenylalaninein an hpdmutant background suggests that the effectcannot be attributed to the internal synthesis ofhomogentisate from phenylalanine. With regard tothe PhhR-mediated activation of PhmgA in thepresence of phenylalanine, it should be noted thatthe PhhR box identified in this study spans positions−18 to −1, partially overlapping with the sequencerecognized by the HmgR.We hypothesize that PhhRsterically inhibits the ability of HmgR to bind to itstarget site, thereby facilitating the recruitment ofRNA polymerase to the promoter, although furtherexperiments are required to confirm this.In summary, we have shown that PhhR is a dual
regulator that modulates activation of transcriptionby binding to the PhhR box(es) and establishingpotential interactions with the RNA polymerase/σ70. As a repressor, PhhRmay occlude the binding ofRNA polymerase to its target sites. As an activator, itseems to work as a class I, class II, and enhancer-likeregulator. This is dictated by the location of the PhhRbox upstream of −35, which may, in turn, lead to
differential regulator–RNA polymerase σ70 interac-tions. The specifics of these interactions betweenPhhR and RNA polymerase require further researchin order to explain the novel and interesting modesof activation and repression mediated by PhhR.
Materials and Methods
Bacterial strains, plasmids, and culture media
The bacterial strains, cloning vectors, and plasmids usedin this study are shown in Table 1. P. putidaKT2440 and theΔphhRmutant strain were grown in M9 minimal mediumwith glucose [0.5 (wt/vol)] as the carbon source.16 Whenindicated, 5 mM phenylalanine was added and cultureswere incubated at 30 °C and shaken on an orbital platformoperating at 200 strokes per minute. E. coli strains weregrown at 37 °C in LB medium with shaking. E. coli DH5αwas used for gene cloning, and E. coli BL21 (DE3) was usedfor protein expression. When required, antibiotics wereused at the following final concentrations in microgramsper millilitre: ampicillin, 100; chloramphenicol, 30; kana-mycin, 50; and tetracycline, 20.
Analytical gel filtration chromatography
Analytical gel filtration chromatography using an ÅktaFPLC system (Amersham Bioscience) was carried out todetermine the oligomeric state of PhhR in the absence or in

585PhhR, RNA Polymerase, and Transcription
the presence of effector. Purified PhhR protein at aconcentration of 86 μM was loaded onto a Superdex-20010/300 GL column (Amersham Bioscience) and equilibrat-ed in buffer [100 mM phosphate (pH 7.2), supplementedwith 1 mM ethylenediaminetetraacetic acid (EDTA),10 mM β-mercaptoethanol, 100 mM NaCl, 300 mMimidazole]. Protein was eluted at a constant flow rate of1 ml/min, and the absorbance of the eluate wasmonitoredat 280 nm. The column was calibrated with α-lactalbumin(14.2 kDa), carbonic anhydrase (29 kDa), chicken eggalbumin (45 kDa), and bovine serum albumin (66 and132 kDa) (Sigma). Aliquots of the peak fractions wereanalyzed by SDS-PAGE according to standard protocols.The molecular mass of PhhR was inferred by plotting theelution volume of PhhR and marker proteins against theLn molecular mass of marker proteins, according to theinstructions provided by Amersham Bioscience.
β-Galactosidase assays
We used fusions of the PhhR target promoters of genesto a promoterless ′lacZ gene in the low-copy-numberpMP220 vector. The corresponding promoter regions wereamplified by PCR with primers incorporating restrictionsites (3′ end: EcoRI site for PP3434 promoter and BamHIsite for the PP2078 promoter; 5′ end: PstI site for allpromoters) in order to facilitate the fusion of the promotersto ′lacZ. Upon amplification, DNA was digested withEcoRI or BamHI and PstI and ligated to EcoRI- or BamHI –PstI-digested pMP220. The fusion constructs were con-firmed by DNA sequencing. The plasmids were electro-porated into the wild-type P. putida KT2440 strain and theΔihfmutant strain. The corresponding transformants weregrown overnight in M9 medium with glucose plustetracycline. The cultures were then diluted 100-fold inthe same medium and grown to a turbidity of about 0.6 at660 nm. Aliquots were prepared and incubated in theabsence or presence of 5 mM effector at 30 °C for 2 h withshaking. β-Galactosidase activity was assayed in permea-bilized whole cells according to Miller's method.29 Assayswere run in triplicate and were repeated for at least threeindependent experiments.
Preparation of RNA and primer extension analysis
P. putida KT2440 and the phhR-deficient regulatorknockout mutant were grown overnight in M9 minimalmedium with 25 mM glucose as the sole carbon source.Cells were then diluted 100-fold in fresh medium until theculture reached a turbidity of about 0.6 at 660 nm, andaliquots were incubated in the absence and in the presenceof 5 mM phenylalanine at 30 °C for 15 min. Cells (10 ml)were harvested by centrifugation (9000g for 15 min) indisposable plastic tubes pre-cooled in liquid nitrogen andwere kept at−80 °C until use. RNAwas extracted using theTRI Reagent (ref. 9738, Ambion, Austin, TX) and modifiedas follows. The lysis step was carried out at 60 °C, and afinal digestion stepwith RNase-free DNasewas carried outat the end of the process followed by purification usingRNeasy columns (cat. no. 74104, Qiagen, Hilden). TheRNA concentration was determined spectrophotometri-cally at 260 nm and its integrity was assessed by agarosegel eletrophoresis.For primer extension analysis, primers were labeled at
their 5′ ends with [γ-32P]ATP and T4 polynucleotidekinase.30 About 105 cpm of the labeled primers washybridized to 30 μg of total RNA, and extension wascarried out using avian myeloblastosis virus reverse
transcriptase. Electrophoresis of cDNA products wasdone using a urea–polyacrylamide sequencing gel toseparate the reaction products, and gels were exposed to aphosphor screen (Fuji Photo Film Co., Ltd.) for 24–48 h.Phosphor screens were scanned using a phosphorimaginginstrument (Molecular Imager FX, Bio-Rad).
DNase I footprinting
A466-bp PCR fragmentwas used in these assays. For thetop strand of the PP3122/PP3123 operon, we amplifiedDNAusing primers 5′-TGAATTCGGCAGTCAGGCTG-3′(end-labeled with [γ-32P]ATP as described above) and 5′-CGCGCCGCTTGATTTCGCTG-3′. For the bottom strand,the same primers were used but the latter primer was end-labeled. Solutions containing 5 nM (104 cpm) of eachlabeled probe were incubated without or with His6-taggedPhhR at 5, 8, 12, or 15 μM for 30 min at 30 °C in 10-μlreaction mixtures. The solutions were then treated with40 μl of DNase I (final concentration, 1.4×10−4 U/μl)diluted in 10 mM Tris–HCl (pH 7) supplemented with2.55 mM MgCl2, 1 mM CaCl2, 0.1 mM EDTA, and 50 mMKCl. After 4 min at 30 °C, reactions were stopped byadding 2 μl of 0.5 mM EDTA. The solutions were thenextracted with phenol, and the DNA was precipitatedusing 2 volumes of ethanol and then resuspended in 5 μl ofTE [10 mM Tris–0.1 mM EDTA (pH 8.0)] and 2.5 μl ofloading dye. Equal amounts of DNA (5,000–6,000 cpm)were heated to 90 °C for 3 min and electrophoresedthrough a 6.5% (wt/vol) denaturing polyacrylamide gel.Sequencing ladders were generated with the corre-sponding labeled primer, using a T7 DNA polymerasesequencing kit (USB-Amersham) and the pMC3122Pplasmid.
Isothermal titration calorimetry
Measurements were done on a VP-Microcalorimeterat 25 °C. The protein was thoroughly dialyzed against100 mM phosphate buffer, 250 mM NaCl, 10 mMmagnesium acetate, 2 mM DTT, and 1 mM EDTA(pH 7.2). For these DNA binding studies, oligonucleo-tides corresponding to both strands of the PhhR targetsequences were synthesized. Annealing was carried outby mixing equimolar amounts (at a concentration of60 μM) of each complementary oligonucleotide in50 mM phosphate buffer (pH 7.0), 5 mM EDTA, and750 mM NaCl. The mixture was incubated at 95 °C for25 min and then chilled on ice and dialyzed in thebuffer used for ITC studies. The titrations involvedinjections of 70 μM PhhR into a solution containing3 μM of the 26-mer duplex DNA. Titration curves werefitted by a nonlinear least squares method (ORIGINsoftware) to a function for the binding of a ligand to amacromolecule.31,32
Acknowledgements
Work at Ramos's lab was supported by projectsPSYSMO GEN2006-27750-C5-5-E/SYS and CONSO-LIDER-C BIO2006-05668. We thank M. Mar Fandilaand Carmen Lorente for secretarial assistance andBenjamin Pakuts for checking the language.

586 PhhR, RNA Polymerase, and Transcription
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2009.09.045
References
1. de Lorenzo, V., Silva-Rocha, R., Carbajosa, G., Galvão,T. C. & Cases, I. (2009). Sensing xenobiotic com-pounds: lessons from bacteria that face pollutants inthe environment. In Sensory Mechanisms in Bacteria(Spiro, S. & Dixon, R., eds), Horizon Scientific Press,Norwich, UK.
2. Busby, S. & Ebright, R. H. (1994). Promoter structure,promoter recognition, and transcription activation inprokaryotes. Cell, 79, 743–746.
3. Gallegos, M. T., Schleif, R., Bairoch, A., Hofmann, K. &Ramos, J. L. (1997). Arac/XylS family of transcrip-tional regulators. Microbiol Mol Biol. Rev. 61, 393–410.
4. Ishihama, A. (1986). Transcription signals and factorsin Escherichia coli. Adv. Biophys. 21, 163–173.
5. Pérez-Martín, J. & de Lorenzo, V. (1997). Coactivationin vitro of the σ54-dependent promoter Pu of the TOLplasmid of Pseudomonas putida by HU and themammalian HMG-protein. Annu. Rev. Microbiol. 51,593–628.
6. Buck, M., Gallegos, M. T., Studholme, D. H., Guo, Y. &Gralla, J. D. (2000). The bacterial enhancer-dependentσ54 (σN) transcription factor. J. Bacteriol. 182, 4129.
7. Pittard, J., Camakaris, H. & Yang, J. (2005). The TyrRregulon. Mol. Microbiol. 55, 16–26.
8. Davies, K. M., Skamnaki, V., Johnson, L. N. & Vénien-Bryan, C. (2006). Structural and functional studies ofthe response regulator HupR. J. Mol. Biol. 359,276–288.
9. Brown, N. L., Stoyanov, J. V., Kidd, S. P. & Hobman,J. L. (2003). Formation of aromatic amino acid pools inEscherichia coli K-12. FEMS Microbiol. Rev. 27, 145–163.
10. Rojo, F. (1999). Repression of transcription initiation inbacteria. J. Bacteriol. 181, 2987–2991.
11. Herrera, M. C. & Ramos, J. L. (2007). Catabolism ofphenylalanine by Pseudomonas putida: The NtrC-family PhhR regulator binds to two sites upstreamfrom the phhA gene and stimulates transcription withσ70. J. Mol. Biol. 366, 1374–1386.
12. Arias-Barrau, E., Olivera, E. R., Luengo, J. M.,Fernández, C., Galán, B., García, J. L. et al. (2004). Thehomogentisate pathway: a central catabolic pathwayinvolved in the degradation of L-phenylalanine, L-tyrosine, and 3-hydroxyphenylacetate in Pseudomonasputida. J. Bacteriol. 186, 5062–5077.
13. Song, J. & Jensen, R. A. (1996). PhhR, a divergentlytranscribed activator of the phenylalanine hydroxilasegene cluster of Pseudomonas aeruginosa. Mol. Microbiol.22, 497–507.
14. Zhao, G., Xia, T., Song, J. & Jensen, R. A. (1994).Pseudomonas aeruginosa possesses homologues ofmammalian phenylalanine hydroxylase and 4α carbi-nolamine dehydratase/DCoH as part of a three-component gene cluster. Proc. Natl Acad. Sci. USA,91, 1366–1370.
15. Herrera, M. C., Duque, E., Rodríguez-Herva, J. J.,Fernández-Escamilla, A. & Ramos, J. L. (2009).Identification and characterization of the PhhR reg-ulon in Pseudomonas putida. Environ. Microbiol.submitted.
16. Abril, M. A., Michán, C., Timmis, K. N. & Ramos, J. L.(1989). Regulator and enzyme specificities of the TOLplasmid-encoded upper pathway for degradation ofaromatic hydrocarbons and expansion of the substraterange of the pathway. J. Bacteriol. 171, 6782–6790.
17. Marqués, S., Gallegos, M. T., Manzanera, M., Holtel,A., Timmis, K. N. & Ramos, J. L. (1998). Activation andrepression of transcription at the double tandemdivergent promoters for the xylR and xylS genes ofthe TOL plasmid of Pseudomonas putida. J. Bacteriol.180, 2889–2894.
18. Gobert, A. P., McGee, D. J., Akhtar, M., Mendz, G. L.,Newton, J. C. & Cheng, Y. (2001). Helicobacter pyloriarginase inhibits nitric oxide production by eukaryoticcells: a strategy for bacterial survival. Proc. Natl Acad.Sci. USA, 98, 13844–13849.
19. Spaink, H. P., Okker, R. J. H., Wijffelman, C. A., Pees,E. & Lugtenberg, B. (1987). Promoters in the nodula-tion region of the Rhizobium leguminosarum Symplasmid pRL1JI. Plant Mol. Biol. 9, 27–39.
20. Domínguez-Cuevas, P. & Marqués, S. (2004). InPseudomonas: Virulence and Gene Regulation(Ramos, J. L., ed.), vol. II, pp. 319–343. KluwerAcademic/Plenum Publishers, New York, NY.
21. Pérez-Martín, J., Timmis, K. N. & de Lorenzo, V.(1994). Co-regulation by bent DNA. J. Biol. Chem. 269,22657–22662.
22. Wilson, T. J., Maroudas, P., Howlett, G. J. & Davidson,B. E. (1994). Ligand-induced self-association of theEscherichia coli regulatory protein TyrR. J. Mol. Biol.238, 309–318.
23. Lawley, B., Fujita, N., Ishihama, A. & Pittard, A. J.(1995). The TyrR protein of Escherichia coli is a class Itranscription factor. J. Bacteriol. 177, 238–241.
24. Fernández, M., Duque, E., Pizarro-Tobías, P., vanDillewijn, P., Wittich, R. M. & Ramos, J. L. (2009).Microbial responses to xenobiotic compounds. Iden-tification of genes that allow Pseudomonas putidaKT2440 to cope with 2,4,6-trinitrotoluene.. MicrobialBiotechnol. 2, 287–294.
25. Bertoni, G., Noboyuki, F., Ishihama, A. & de Lorenzo,V. (1998). Active recruitment of σ54-RNA polymeraseto the Pu promoter of Pseudomonas putida: role of IHFand α-CTD. EMBO J. 17, 5120–5128.
26. Austin, S., Buck, M., Cannon, W., Eydmann, T. &Dixon, R. (1994). Purification and in vitro activities ofthe native nitrogen fixation control proteins NifA andNifL. J. Bacteriol. 176, 3460–3465.
27. Busby, S. & Ebright, R. H. (1999). Transcriptionactivation by catabolite activator protein (CAP). J.Mol. Biol. 293, 199–213.
28. Jovanovic, G., Lloyd, L. J., Stumpf, P. H.,Mayhew,A. J.& Buck,M. (2006). Induction and function of the phageshock protein extracytoplasmic stress response inEscherichia coli. J. Biol. Chem. 281, 21147–21161.
29. Miller, U. H. (1972). Experiments in Molecular Genetics.Cold Spring Harbor Laboratory, New York.
30. Marqués, S., Ramos, J. L. & Timmis, K. N. (1993).Analysis of the mRNA structure of the Pseudomonasputida TOL meta-fission pathway operon. Biochim.Biophys. Acta, 1216, 227–236.
31. Lacal, J., Guazzaroni, M. E., Busch, A., Krell, T. &Ramos, J. L. (2008). Hierarchical binding of the TodTresponse regulator to its multiple recognition sites atthe tod pathway operon promoter. J. Mol. Biol. 376,325–337.
32. Krell, T. (2008). Microcalorimetry: a response tochallenges in modern biotechnology. MicrobialBiotechnol. 1, 126–136.





























