Embed Size (px)
Citation preview

lable at ScienceDirect
Biomaterials 31 (2010) 345–357
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomateria ls
Prediction of the solubility of cucurbitacin drugs in self-associatingpoly(ethylene oxide)-b-poly(a-benzyl carboxylate 3-caprolactone)block copolymer with different tacticities usingmolecular dynamics simulation
Sarthak K. Patel a, Afsaneh Lavasanifar a,b, Phillip Choi a,*
a Department of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 2V4b Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada T6G 2N8
a r t i c l e i n f o
Article history:Received 26 August 2009Accepted 13 September 2009Available online 30 September 2009
Keywords:Molecular dynamics simulationHydrophobic drugsDi-block copolymerTacticitySolubilityInteraction parameter
* Corresponding author.E-mail address: [email protected] (P. Choi).
0142-9612/$ – see front matter � 2009 Elsevier Ltd.doi:10.1016/j.biomaterials.2009.09.051
a b s t r a c t
Molecular dynamics (MD) simulation was used to investigate the solubility of two hydrophobic drugsCucurbitacin B (CuB) and Cucurbitacin I (CuI) in poly(ethylene oxide)-b-poly(a-benzyl carboxylate3-caprolactone) (PEO-b-PBCL) block copolymers with different tacticities. In particular, di-blockcopolymer with three different tacticities viz. PEO-b-iPBCL, PEO-b-sPBCL, and PEO-b-aPBCL were used.The solubility was quantified by calculating the corresponding Flory–Huggins interaction parameters (c)using random binary mixture models with 10 wt% of drug. The tacticity of the di-block copolymer wasfound to influence significantly the solubility of two drugs in it. In particular, based on MD simulationresults, only PEO-b-sPBCL exhibited solubility while the other two did not. Given the fact that the drugswere shown to be soluble in PEO-b-PBCL experimentally, it is predicted that the tacticity of the di-blockcopolymer synthesized in experiment is syndiotactic. This predication matches well with the dominantring opening polymerization of cyclic lactones to syndiotactic polymers by stannous octoate as catalystused to prepare PEO-b-PBCL block copolymers in our previous experiments. The simulation resultsshowed that the solubility of the drugs in PEO-b-sPBCL is attributed to the favorable intra-molecularinteraction of the di-block copolymer and favorable intermolecular interaction between the di-blockcopolymer and the drugs. Radial distribution function analysis provides useful insights into the natureand type of the intermolecular interactions.
� 2009 Elsevier Ltd. All rights reserved.
1. Introduction
One key concept used by many researchers to increase theloading capacity of a self-associating block copolymer for a givenhydrophobic drug is to introduce certain chemical moieties intothe backbone of the block copolymer that are capable of inter-acting with the drug, with specific interactions of the typesleading to negative heat of mixing [1–3]. In such studies, theblock copolymer structure suitable for the solubilization ofa given drug is usually selected based on predictions of Flory–Huggins interaction parameters calculated by the group contri-bution method (GCM) [4–7]. However, the stereochemistry andtacticity of polymer are not taken into account in predicting thesolubility of drugs in block copolymers this way. The objective of
All rights reserved.
this study was to assess whether application of moleculardynamics (MD) simulation for the determination of drug/blockcopolymer Flory–Huggins interaction parameter can address theshortcoming of GCM in this aspect and provide a reliable meansto predict the solubilisation of drugs in block copolymers havingdifferent tacticities.
Recently, Mahmud et al. [8] have reported successful synthesis ofself-associating tailor-made carriers having pendant aromatic and/orother reactive functional groups on the PCL block of the PEO-b-PCLblock copolymers (Fig. 1(A)). These core-functionalized micellesespecially PEO-b-poly(a-benzyl carboxylate 3-caprolactone) (PEO-b-PBCL) (Fig. 1(B)) have shown great potential in improving loadingcapacity of two anti-cancer drugs CuB and CuI (Fig. 2) [9]. Thepresence of pendant benzyl carboxylate groups on the PCL block mayinduce the formation of additional inter or intra-molecular specificinteractions (e.g., p–p interactions, hydrogen bonds, etc.) whichwould eventually lead to the improvement of the drug loadingcapacity of the di-block copolymer.

H3 C
H
H
C
H
H
O C
O
H
m n
A
*
C C C C
H
H
H
H
H
H
H
H
H
H
H3
C O
C O C
H
H
C
H
H
O C
O
C O
C O H
m n
B
C C C C
H
C
H
H
H
H
H
H
H
HO
O CH2
Fig. 1. Chemical structures of (A) PEO-b-PCL; and (B) PEO-b-PBCL.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357346
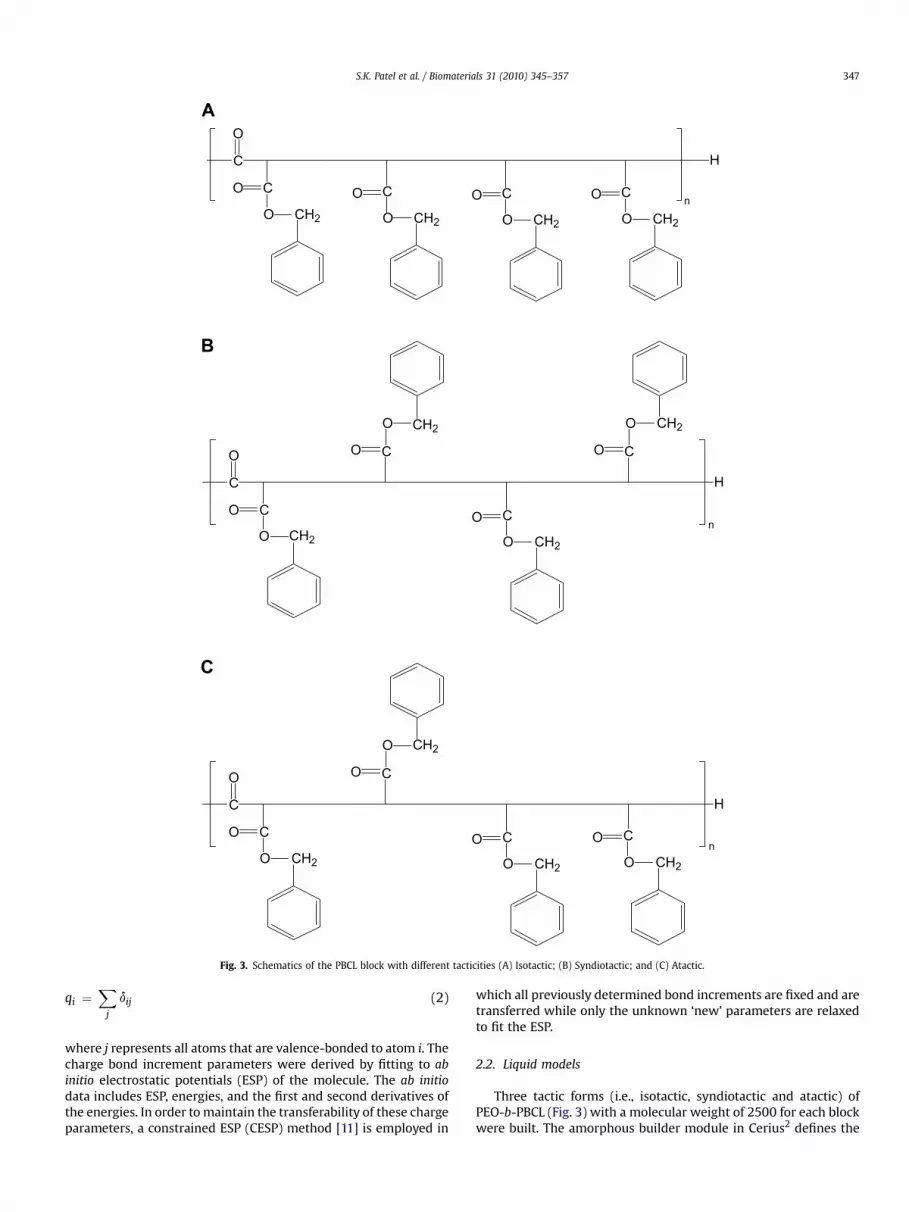
PEO-b-PBCL di-block copolymers are synthesized by the ringopening polymerization of a-benzyl carboxylate-3-caprolactoneusing methoxy-PEO as an initiator and stannous octoate as a catalyst[8]. It is worth noting that benzyl carboxylate-3-caprolactonemonomer possesses one asymmetric carbon and is used in thepolymerization reaction as a racemic mixture. Upon polymerization,the hydrophobic block of the di-block copolymer can potentiallycontain similar or different stereo-isomers leading to different tac-ticities viz. isotactic, syndiotactic and atactic forms. The site labelledC* in Fig. 1(B), is termed pseudo-asymmetric or chiral carbon atomcenter in the PBCL repeating unit. The stereo chemical configurations
HO
O
H
O
OH
OH O
OAc
A
HO
O
H
O
OH
OH O
OH
B
Fig. 2. Chemical structures of (A) Cucurbitacin B (CuB); and (B) Cucurbitacin I (CuI).
of successive chiral carbon atoms in the backbone of the di-blockcopolymer will define its tacticity. Fig. 3 shows a schematic diagramof the three possible stereo-isomers of the PBCL block. If configura-tions of all the successive chiral carbon atoms are the same (i.e., thesubstituent branches lie on the same side of the reference plane), thePBCL block is termed isotactic (Fig. 3(A)). If configurations ofsuccessive chiral carbon atoms differ (i.e., the substituent branchesappear alternatively above and below the reference plane), the PBCLblock is termed syndiotactic (Fig. 3(B)). Alternatively, when config-urations at the chiral centers are more or less random then the PBCLblock is termed atactic (Fig. 3(C)). Since specific interactions dependon the tacticity of a polymer [10], tacticity of the di-block copolymeris expected to affect the solubility of a drug in the micelle formed bythe di-block copolymer. And it is expected that the differences inintermolecular interactions between drug and the di-block copol-ymer could be captured by the corresponding Flory–Huggins inter-action parameter (c). Therefore, in this work, we applied thetechnique of molecular dynamics simulation to determine thecompatibility between the two anti-cancer drugs (CuB and CuI) usedby Molavi et al. [9] and PEO-b-PBCL with different tacticities. Inparticular, Flory–Huggins interaction parameters (c) of variousbinary PEO-b-PBCL/drug pairs were calculated and were used toinfer the drug solubility. To this end, the stereo configuration of PEO-b-PBCL that would yield higher solubility can be determined. Suchmolecular level understanding can also aid in the process of chem-ical tailoring of the di-block copolymer to obtain drug carriers withoptimized functional properties.
2. Simulation methodology
2.1. Software and force field
All MD simulations reported here were performed usinga commercially available software package Materials Studio (MSModeling version 4.2, Accelrys) run on a Silicon Graphics (SGI)workstation cluster. The initial liquid state models of all di-blockcopolymers were generated based on the rotational isomeric state(RIS) theory using the amorphous builder module available inanother commercial software Cerius2 from the same vendor. Theinter-atomic interactions were modeled with the COMPASS(condensed-phase optimized molecular potentials for atomisticsimulation studies) force field [11] which is an all-atom force fieldoptimized to predict structural, conformational and thermophys-ical condensed phase properties for the most common organic/inorganic molecules, including polymers. In this force field, thetotal potential energy of the system is represented by the sum ofbonded and non-bonded interactions expressed as:
E ¼ Eb þ Eq þ Ef þ Ec þ Ecross þ Evdw þ EQ (1)
The first five valence terms represent internal coordinates ofbond (b), angle (q), torsion angle (f), Wilson out-of-plane angle (c),and the cross-coupling terms (Ecross). The cross-coupling termsinclude combinations of two or more internal coordinates (e.g.,bond–bond, bond–angle and bond–torsion) that would predictvibration frequencies and structural variations associated withconformational changes. The last two terms of Equation (1)represent non-bonded interactions consisting of Lennard Jones (LJ)9–6 function for the dispersive interactions and Coulombic functionfor electrostatic interactions. In the COMPASS force field, the partialatomic charges of a molecule are calculated from the charge bondincrement, dij, which represents the charge separation between twovalence-bonded atoms i and j [11]. Thus, the net partial atomiccharge qi on an atom i is considered to be the summation of allcharge bond increments.
C
O
CO
O CH2
CO CO CO
H
n
A
C
O
CO CO
H
CO CO
n
B
C
O
CO CO
H
CO
O CH2
n
C
CO
O CH2 O CH2 O CH2
O CH2 O CH2
O CH2 O CH2
O CH2 O CH2 O CH2
Fig. 3. Schematics of the PBCL block with different tacticities (A) Isotactic; (B) Syndiotactic; and (C) Atactic.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357 347
qi ¼X
dij (2)
jwhere j represents all atoms that are valence-bonded to atom i. Thecharge bond increment parameters were derived by fitting to abinitio electrostatic potentials (ESP) of the molecule. The ab initiodata includes ESP, energies, and the first and second derivatives ofthe energies. In order to maintain the transferability of these chargeparameters, a constrained ESP (CESP) method [11] is employed in
which all previously determined bond increments are fixed and aretransferred while only the unknown ‘new’ parameters are relaxedto fit the ESP.
2.2. Liquid models
Three tactic forms (i.e., isotactic, syndiotactic and atactic) ofPEO-b-PBCL (Fig. 3) with a molecular weight of 2500 for each blockwere built. The amorphous builder module in Cerius2 defines the

S.K. Patel et al. / Biomaterials 31 (2010) 345–357348
tacticity of polymer using meso-diad ratio, which is generallydefined as the relative proportion of isotactic monomer pairs ina given polymer. Thus, a polymer with a meso-diad ratio of 0.0 isa syndiotactic polymer and a polymer with a ratio of 1.0 is anisotactic polymer. A meso-diad ratio of 0.5 was used to build anatactic polymer. Throughout the manuscript, the di-block copoly-mer with isotactic, syndiotactic, and atactic stereo configurations ofPBCL repeating unit will be denoted as PEO-b-iPBCL, PEO-b-sPBCL,and PEO-b-aPBCL, respectively. The method of Theodorou and Suter[12] was employed in the amorphous builder module of Cerius2 tobuild the bulk amorphous states of pure di-block copolymers, drugsand their mixtures subjected to periodic boundary conditions anddensity constraint. The detailed procedure for constructing suchinitial structures is described elsewhere [13]. The reason for usingamorphous structures for the purpose of computing Flory–Hugginsinteraction parameters has been justified in our previous work[13,14].
In order to acquire the density values of the pure di-blockcopolymers and drugs at the simulation temperature of 473 K, wecarried out MD simulation in isobaric–isothermal (NPT) statisticalensemble (P¼ 1 atm; T¼ 473 K) using the density values from theGCM as the initial values. Here, the temperature was chosen toensure that both the drug molecules and the di-block copolymersare in the liquid (amorphous) state. The simulated density can beobtained from the knowledge of equilibrated unit cell volume,which is allowed to fluctuate during the NPT MD simulation. Thepressure and temperature of the systems were controlled usingAndersen barostat [15] and Nose thermostat [16] algorithms,respectively. It is worth noting that NPT MD simulation was usedto determine the density of the pure drugs and pure PEO-b-PBCLdi-block copolymers. The density values of the mixtures werethen calculated from the pure density values of constituentsubstances by assuming that the volume change on mixing isnegligible. These mixture and pure substance density values wereused in the subsequent canonical (NVT) MD simulations todetermine the Flory–Huggins interaction parameters (c). Therationale behind using NVT rather than NPT MD simulation was toreduce the computational time.
MD simulations for systems with large number of atoms typi-cally require days or weeks to perform even with the use of currenthigh end workstations. Consequently, only hundred or thousand ofpicoseconds (ps) simulation regime is accessible by this technique.
H3C
H
H
H
H
O
O C C O C C
m
H
CO
O
φ1φ2 φ4 φ6
φ6
φ4
Fig. 4. Schematics of all the torsion
Unfortunately, this is insufficient time for systems such as polymersin the amorphous state to undergo very drastic reorientation andrelaxation. Thus, it is crucial that the initial state of system ofdi-block copolymers be a representative of the equilibrated state aspossible. Unlike torsion angles, conformational features like bondlengths and bond angles, initially set to the equilibrium values,generally adjust very quickly during the course of the MD simula-tion. Thus, the most important constraint imposed during modelconstruction was the specification of the distribution of the torsionangles of the skeletal bonds in the di-block copolymers. Thesedistributions were determined using the rotational isomeric state(RIS) theory [17,18]. It is worth noting that the RIS theory can onlybe used for polymers in their amorphous state. The distribution ofthe torsion angles was determined by applying the Boltzmannweighting factor to the energies of the RIS minima to determine thedistribution of torsion angles. The initial distribution of torsionangles for PEO block of the PEO-b-PBCL di-block copolymerremains the same as it was in PEO-b-PCL di-block copolymer. Thedetailed method can be found in our previous work [13]. Weidentified a total of nine torsion angles for PEO-b-PBCL and they aredepicted in Fig. 4. In Table 1, we list the values of RIS minima andthe respective tolerances for all nine torsion angles that havesignificant influence to the conformation of the di-block copolymer.Once the RIS state distribution was determined, the RIS stateswere populated allowing for the angle tolerances stated in Table 1.It is worth noting that the initial conformations of the di-blockcopolymers used in the NPT MD simulations were also subjected tothe RIS constraints.
2.3. Molecular dynamics simulation
All initial amorphous structures are in relatively high energystate and hence before performing MD simulations, they weresubjected to energy minimization step using the conjugate gradientmethod in order to remove strong van der Waals overlaps.Canonical (NVT) MD simulations were carried out at 473 K usingthe Nose thermostat [16]. The velocity Verlet method, with a timestep of 0.001 ps, was used as an integrator in all simulations. Thenon-bonded dispersive interactions were evaluated using atombased cut-off distance of 9.50 Å with a spline width of 1 Å, while thelong-range electrostatic interactions especially important in 3Dperiodic systems were evaluated using the well-known Ewald
O H
n
C C C C
H
H
H
H
H
H
H
H
CH2
φ5 φ7
φ8
φ9
φ3
angles identified in PEO-b-PBCL.

Table 1Rotational isomeric states for all identified unique torsion angles.
Skeletal bond State 1a State 2a State 3a State 4a
F1 170� 20 300� 10 330� 10 N/AF2 40� 10 60� 10 280� 10 350� 10F3 270� 10 290� 10 330� 10 N/AF4 130� 10 300� 10 N/A N/AF5 50� 10 90� 10 120� 10 N/AF6 110� 10 N/A N/A N/AF7 70� 15 180� 10 230� 5 N/AF8 70� 10 N/A N/A N/AF9 140� 10 160� 10 330� 10 N/A
a Data are torsion angle� tolerance.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357 349
summation method [19]. Long-range tail corrections were appliedto the non-bonded interactions during MD simulations. Simula-tions were carried out until the total energy of the system wasstabilized. Each simulation was carried out for a total of 2,000 ps.The properties of interest (e.g., the total energy, radial distributionfunction, etc.) were calculated by averaging over the last 500 ps ofthe corresponding trajectory file. Here, we adopted the MD simu-lation approach in which the internal energy changes on mixingwere calculated and then used to obtain c parameters for the drug/di-block copolymer pairs [13].
3. Results and discussion
3.1. Molecular dynamics simulation
Table 2 summarizes the computed density values for the di-block copolymers, drugs and their mixtures (10 wt% drug) ata pressure of 1 atm and a temperature of 473 K. It is worth notingthat the density values for all three tactic forms viz. PEO-b-iPBCL,PEO-b-sPBCL and PEO-b-aPBCL were assumed to be the same.Therefore, only the density of PEO-b-iPBCL was calculated.Generally, density values of stereo-isomers of polymers differ onlyin the crystalline state due to their different packing characteris-tics while their density values are essentially the same in theamorphous state. For example, the densities of i-polypropyleneand s-polypropylene are identical in the liquid (amorphous) statewhile differ significantly in their solid state [20]. In the presentwork, since the amorphous state of the di-block copolymer was ofinterest, we feel justified to use the same density value for allthree tactic forms of PEO-b-PBCL. The density values of the binarymixtures were calculated using those of the pure components
Table 2Computed densities of CuB, CuI, all tactic forms of PEO(2500)-b-PBCL(2500) di-blockcopolymer and their mixtures (10 wt% drug) in the liquid state from NPT MDsimulations at 1 atm and 473 K along with the number of drug molecules involved.
Drug/block copolymer/mixture Density(g/cm3)
No. of drugmolecules
No. of blockcopolymerchains
Cucurbitacin B (CuB) 1.15 – –
Cucurbitacin I (CuI) 1.12 – –
PEO-b-iPBCL (P1)PEO-b-sPBCL (P2)PEO-b-aPBCL (P3)
1.18 – 1
CuB & P1
CuB & P2
CuB & P3
1.18 1 1
CuI & P1
CuI & P2
CuI & P3
1.18 1 1
obtained from MD simulation, not directly from the MD simula-tion of the mixture. We have demonstrated in our previous work[13,14] that the density values for di-block copolymers andhydrophobic drugs computed using the COMPASS force field arereproducible and reliable. The details of computing Flory–Hugginsinteraction parameter (note that c¼DHm/(RTf1f2) where fi is thevolume fraction of component i and internal energy change onmixing (DEm¼DHm as DPV w 0) values obtained from the NVT MDsimulation can be found in reference 13). The mean potentialenergies of the bulk states of the pure di-block copolymers, drugsand their mixtures were calculated by using time average of thepotential energy over the last 500 ps of the correspondingtrajectories of 2000 ps at equal interval of 2 ps. The velocityautocorrelation functions of the drug molecules, not shown here,indicated that the drug molecules de-correlated in less than 2 ps.The analysis of mean values and standard errors were performedusing the Sigma Plot version 11.0 (Systat Software, Inc.).
The computed c values are plotted in Fig. 5(A). We note that theerror bars shown in the figure are ensemble fluctuations and weredetermined using the data of last 500 ps of the corresponding MDtrajectories. According to Flory–Huggins solution theory, lower c
values indicate higher affinity between the components and hencebetter solubility of one component into the other. Since the solu-bility prediction from simulation is solely based on the qualitativetrend in c values of different systems, we have compared the c
values of the current systems viz. PEO(2500)-b-PBCL(2500) withthose of PEO(2500)-b-PCL(2500) [14].
Fig. 5(A) shows negative c values only for binary mixtures con-taining syndiotactic version of di-block copolymer (i.e., PEO-b-sPBCL).Comparing these c values with those of non-functionalized mixturesystems containing PEO-b-PCL, these c values decreased by aboutthree times. Comparing this observation with the experimental drugloading data (mole drug/mole di-block copolymer) (Fig. 5(B)) of CuBand CuI in PEO(5000)-b-PCL(5000) and PEO(5000)-b-PBCL(4700), ourMD results on PEO-b-sPBCL are consistent with experiment. It shouldbe pointed out that the tacticity of the PEO-b-PBCL was unknown inthe experimental case. However, the simulation data strongly suggestthat the tacticity of the experimentally synthesized PEO-b-PBCLshould be syndiotactic. Various mechanistic and NMR studies alongwith predictions from Monte Carlo calculations performed by Kri-cheldorf et al. [21,22] and Thakur et al. [23,24] demonstrate thatstannous octoate, an achiral catalyst, commonly used in the ringopening polymerization of lactides shows a clear preference for syn-diotactic addition. Since our synthesis process of PEO-b-PBCL usesstannous octoate catalyst for the ring opening polymerization ofa-benzyl carboxylate-3-caprolactone, it is likely that stereo form of thedi-block copolymer is syndiotactic. However, further NMR experi-ment is needed to verify the tacticity of the di-block copolymer.
The next step is to identify the factors that contribute to thefavourable interactions in the mixture containing PEO-b-sPBCL andthe drugs. Functionalization of PCL core of PEO-b-PCL with aromaticbenzyl carboxylate group is expected to affect the intra-molecularinteractions to a certain extent in the pure polymer state due to thepresence of aromatic rings in the di-block copolymer structure. Thestrength of these intra-molecular interactions indirectly affectsthe strength of intermolecular interactions between the di-blockcopolymer and drug molecules in the mixture environmentbecause these intermolecular associations would have to overcomeexisting intra-molecular associations in pure polymer.
3.2. Intra-molecular non-bonded energy
In order to assess the intra-molecular specific interactions,a preliminary study on the electrostatic and dispersive contribu-tions to the intra-molecular non-bonded energy values for pure

PEO-b-PCL
PEO-b-iPBCL
PEO-b-sPBCL
PEO-b-aPBCL
Flo
ry
-H
ug
gin
s In
te
ra
ctio
n P
ara
me
te
r (χ)
-1.6-1.2-0.8-0.40.00.40.81.21.62.02.42.8
CuBCuI
A
PEO(5000)-b-PCL(5000)
PEO(5000)-b-PBCL(4700)
Dru
g L
oad
in
g(m
ole d
ru
g/m
ole p
olym
er)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8B
CuBCuI
Fig. 5. (A) Plot of the computed Flory–Huggins interaction parameters (c) of binarymixtures of two cucurbitacins and three tactic forms of PEO-b-PBCL using moleculardynamics simulation along with the data for PEO-b-PCL from our previous work [14].(Data are means. Bars are standard errors). (Note: the red arrow indicates decrease in cvalues). (B) Comparison plot of experimentally measured drug loading capacity (moledrug/mole polymer) of PEO-b-PCL and PEO-b-PBCL micelles for cucurbitacins asreported by Molavi et al. [9]. (Data are means. Bars are standard deviations).
PEO-b-PCL
PEO-b-iPBCL
PEO-b-sPBCL
PEO-b-aPBCL
En
erg
y (K
ca
l/m
ol)
-300
-250
-200
-150
-100
-50
0
50
100
150
200Electrostatic EnergyDispersive Energy
Fig. 6. Comparison of electrostatic and dispersive energy values for all the tactic formsof PEO-b-PBCL and PEO-b-PCL in their pure state. (Note: the red arrow indicates thedrastic decrease in electrostatic energy value).
S.K. Patel et al. / Biomaterials 31 (2010) 345–357350
PBCL was carried out and compared with the values for pure PCL.Fig. 6 shows the time average values of the electrostatic anddispersive energy values for pure states of PEO-b-iPBCL, PEO-b-sPBCL, and PEO-b-aPBCL along with the values for pure state PEO-b-PCL for the sake of comparison. The energy values were averagedover the last 500 ps of the corresponding trajectory of 2000 ps. It is
clear from the Fig. 6 that dispersive energy became unfavourable inPBCL while electrostatic energy became favourable. In fact, due tothe functionalization, the electrostatic energy decreased substan-tially from positive to negative values, indicating stronger intra-molecular interactions in the cases of PEO-b-PBCL as compared toPEO-b-PCL. This is also consistent with our experimental observa-tion showing a higher rigidity of the PBCL compared to the PCL corestructures estimated by a fluorescence probe study [7]. An increasein the attractive interactions can be attributed to the increase in theattractive forces due to more atoms carrying opposite charges andthe number of hydrogen bonds formed. Here, it should be pointedout that the electrostatic energy contribution originates fromColumbic interactions and specific interactions like intra-molecularhydrogen bonds, p–p interactions, etc. Since the commercial soft-ware package Materials Studio was unable to display the hydrogenbonds formed with aromatic rings, we could not carry out thecounting of hydrogen bonds in this case. Instead, we examined theradial distribution functions (RDF) among different intra-molecularand intermolecular segments in the pure and mixture states of thesystems. Hence, we will be defining a particular pair to be inter-acting only based on RDF plots showing the proximity of differentsegments. But before discussing RDF calculations, we brieflydescribe different types of specific interactions probable betweendifferent segments of the di-block copolymers and between thosewith the drug molecules in the following section.
3.3. Specific interactions
In systems involving long chain di-block copolymers, specificinteractions play a vital role in inducing miscibility by achievingnegative heats of mixing. Fig. 7 shows all probable intra-molecularand intermolecular specific interactions between di-block copoly-mers and drug molecules. The numbering of all these interactions islisted in Table 3.
Aromatic–aromatic/p–p interactions: Benzene rings are gener-ally considered to be non-polar but their electron distribution isa complex multi-pole with no net dipole moment. This multi-polardistribution makes benzene have electron rich faces and partialpositive charges on hydrogen atoms around the edges. Hence,

π --- π interactions
O
H
n
H
C
H
H
H
H
H
H
H
HO
O CH2
Drug
C
O
H
nC C OC C C C
C OC C C C
H
C
H
H
H
H
H
H
H
HO
O C
Drug
Drug --- C=O(Chain)Drug --- H(Chain)
C
O
C O H
n
H
C
H
H
H
H
H
H
H
HO
O CH2
Drug
C=O or O-H groups
Drug---π interactions
O
H
n
C C C CC C OC C C C
H
C
H
H
H
H
H
H
H
HO
O CH2Drug
Drug ---C=O(Branch)
Fig. 7. Different types of intra-molecular and intermolecular interactions probable in binary mixtures of PEO-b-PBCL di-block copolymer and drug molecules.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357 351
when benzene rings centroid comes between 4.5 and 7 Å separa-tion distance [25], the positively charged edge interacts with thenegatively charged faces. Such type of interaction is termed as p–pinteractions. In a study by Barlow et al. [26], this type of interactionbetween aromatic rings of polystyrene and tetramethyl bisphenol-A-polycarbonate polymers were found to be responsible forinducing miscibility in their corresponding blends. In the presentwork, we have aromatic rings on substituent branches of di-block
copolymers and hence intra-molecular p–p interactions would beimportant.
Drug–H(Chain): The H(Chain) are hydrogen atoms attached toactivated carbons on the backbone of the di-block copolymer. Thecarbonyl groups on drug molecules favourably interact and formmultiple hydrogen bonds with these hydrogen atoms. This type ofconventional hydrogen bonding has already been discussed in ourprevious work [14].

Table 3Numbering of intra-molecular and intermolecular interactions shown in Fig. 7.
Interactionnumber
Interacting pair Type of interaction
1 Aromatic–aromatic/p–p Intra-molecular2 Drug–H(Chain) Intermolecular3 Drug–C]O(Chain) Intermolecular4 Drug–C]O(Branch) Intermolecular5 Drug–p (C]O or –OH
groups of drug)Intermolecular
A
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL
sPBCL
aPBCL
B
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL/CuB
sPBCL/CuB
aPBCL/CuB
iPBCL/CuI
sPBCL/CuI
aPBCL/CuI
Fig. 8. RDF plots for Interaction No. 1 (listed in Table 3) in (A) pure di-block copolymersystem, and (B) mixture system of drug/di-block copolymer.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357352
Drug–C]O(Chain): The hydroxyl (–OH) groups present in thedrug molecules interact and form hydrogen bonds with thecarbonyl (–C]O) groups present in backbone of the di-blockcopolymer. For details, refer our previous work [14].
Drug–C]O (Branch): This type of interaction is similar to theprevious one except that drug molecules interact with the carbonyl(–C]O) groups present in the substituent branches.
Drug–p interactions: Drug molecules contain various carbonyland hydroxyl groups. These groups could interact with the aromaticrings either by n–p/C]O–p or p–hydrogen bonding interactions(i.e. OH–p interactions). The miscibility is induced in polyester/PCblend through the formation of n–p complex between estercarbonyl and PC aromatic rings [27]. In another example, p–hydrogen bonding interactions between p electrons of PC aromaticgroups and –OH groups of styrene-co-4-vinyl phenol copolymerwas responsible for inducing miscibility in their correspondingpolymer blends [28].
3.4. Radial distribution function (RDF)
The spatial correlation between any two molecules or segmentsof molecules can be described using radial distribution function(RDF). The value of RDF is a relative measure rather than an abso-lute one. The RDF gAB(r) between two selected groups A and B canbe calculated using following general expression:
gAB ¼NABðrÞ � V
ðNANB � NABÞ4pr2dr(3)
where NA and NB are the numbers of atoms in groups A and B,respectively, NAB is the number of atoms common to both groups Aand B, and V is the unit cell volume.
3.4.1. Intra-molecular p–p interactionsFig. 8(A) and (B) shows RDF plots for intra-molecular p–p
interactions in PEO-b-PBCL and the binary mixtures formed by thedi-block copolymer and the drugs, respectively. It is worth notingthat throughout the plots of RDF analysis, the iPBCL is usedinterchangeably for PEO-b-iPBCL and similarly sPBCL for PEO-b-sPBCL and aPBCL for PEO-b-aPBCL. It is obvious from Fig. 8(A) thatp–p correlation is highest in case of iPBCL while lowest in case ofsPBCL indicating favourable p–p interactions in the case of iPBCL.In order to establish favourable intermolecular interactions/contacts between di-block copolymer and drug molecules, theexisting intra-molecular interactions in the di-block copolymerneeds to be overcome. Thus, due to lowest p–p correlation in thecase of sPBCL, we expect to observe maximum intermolecularassociations between this di-block copolymer and drug mole-cules. In Fig. 8(B) we compare p–p correlation of different di-blockcopolymers in a mixture environment of both drugs. In the PEO-b-PBCL/CuB mixtures, the intra-molecular p–p correlation is stillstrong in the isotactic form of the copolymer while in the mixturecontaining CuI, the intra-molecular p–p correlation decreasesdrastically for all tactic forms, which we believe is due to strong
correlation of CuI drug with the aromatic rings. In a later section,we will show that the intermolecular OH–p interactions betweenthe CuI drug and the aromatic ring has weakened the intra-molecular p–p interactions. It is obvious that the local packing ofthe substituent branches will be different in different stereo-isomers and effect of this local packing is visible on the strength ofthe p–p interactions.
3.4.2. Intermolecular specific interactions for CuBRDF plots showing correlations for interaction no. 2, 3, 4, and 5
listed in Table 3 are shown in Fig. 9(A), (B), (C), and (D), respectively,for all the tactic forms of di-block copolymer and CuB mixture.Since the c values of PEO-b-sPBCL has decreased (almost threetimes) as compared to that of PEO-b-PCL, we try to compare thecorrelations for interactions no. 2 and 3 between PEO-b-PBCL andPEO-b-PCL. From Fig. 9(A) and (B), it is clear that the correlation ofCuB with H(Chain) and C]O(Chain) in PEO-b-PBCL is similar tothose in PEO-b-PCL. While correlation for isotactic form is found tobe slightly higher than other forms of di-block copolymer. Fig. 9(C)shows higher correlation between CuB and C]O(Branch) for PEO-b-sPBCL and PEO-b-aPBCL. The probable reason behind this

A
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL
sPBCL
aPBCL
PCL
B
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL
sPBCL
aPBCL
PCL
C
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL
sPBCL
aPBCL
D
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
iPBCL
sPBCL
aPBCL
Fig. 9. RDF plots of CuB drug in di-block copolymer for (A) Interaction No. 2, (B) Interaction No. 3, (C) Interaction No. 4, and (D) Interaction No. 5 listed in Table 3.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357 353
observation is more free space available for interaction with thecarbonyl groups of the substituent branches in such copolymers.From Fig. 9(D), it is clear that for PEO-b-sPBCL, the correlation ofCuB with the aromatic ring is a lot higher than other di-blockcopolymers. There are two probable reasons: (i) due to the alter-nating arrangement of the branches in the syndiotactic version ofthe di-block copolymer, the exposure of the CuB drug is more withthe aromatic rings; (ii) CuB drug is not able to pierce through thestrong intra-molecular p–p interactions present in isotactic andatatic forms of the di-block copolymer. Here, p–p interactions arestrong in mixtures with CuB as compared to with CuI, as shown inFig. 8(B).
3.4.3. Comparison between CuB and CuISince the difference between c values of CuB and CuI is negli-
gible in the case of PEO-b-iPBCL as compared to PEO-b-sPBCL andPEO-b-aPBCL. We compare correlations for CuB and CuI drugs forthe latter two cases only.
(a) PEO-b-sPBCL
Fig. 10 shows RDF plots comparing correlations for CuB andCuI in PEO-b-sPBCL. Compared to CuI, CuB shows more correla-tions with the backbone atoms like H(Chain) and C]O(Chain)indicating its favourable interactions with them. Since thecarbonyl groups present on drug molecules are responsible for
favourable interactions (through hydrogen bonding) with the Hatoms attached to the activated carbon atoms, CuB with morecarbonyl groups, it shows more correlation with the backbone Hatoms. Both drugs show almost similar correlations/interactionswith the carbonyl group and aromatic ring of substituentbranches (Fig. 10(C) and (D)).
(b) PEO-b-aPBCL
Fig. 11 compares the RDF plots for CuB and CuI in PEO-b-aPBCL.In this case, both drugs show almost similar correlations with theH(Chain), while for the correlation with C]O(Chain) (Fig. 11(B)),the first coordination shell signified by the first peak (at w1.96 Å inthe present case) for CuB is absent in CuI. This first coordinationshell is an indication of favourable interactions (mainly specificinteractions like hydrogen bonding). Thus, Fig. 11(B) implies thatCuB has strong favourable interactions with C]O(Chain) but notCuI. The correlations of drug molecules with C]O(Branch) issimilar for both drugs (Fig. 11(C)). The surprising result was foundfor the correlation between drug molecules and the aromatic ringsfor the present atactic form of copolymer. The correlations of CuIwith the aromatic ring was much higher than that found in the CuBcase (Fig. 11(D)). The probable reasons behind this surprising resultwill be discussed later.
In short, the above comparison can be summarized asfollows: for the PEO-b-sPBCL/drug binary mixture case, there is

A
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9
0 1 2 3 4 5 6 7 8 9
r(Å)
g(r)
g(r)
CuB
CuI
B
0
0.2
0.4
0.6
0.8
1
1.2
r(Å)
C
D
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1 2 3 4 5 6 7 8 9 r(Å)
g(r)
g(r)
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9r(Å)
CuB
CuI
CuB
CuI
CuB
CuI
Fig. 10. RDF plots of comparison of CuB and CuI drugs in PEO-b-sPBCL copolymer for (A) Interaction No. 2, (B) Interaction No. 3, (C) Interaction No. 4, and (D) Interaction No. 5 listedin Table 3.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357354
almost equal correlations/interactions between the drugs andgroups of substituent branches i.e., carbonyl group andaromatic rings. CuB shows more interactions with the backboneatoms (H(Chain) and C]O(Chain)) while CuI interacts mainlywith the substituent branch atoms (C]O and aromatic ring).This is because CuI has 4 hydroxyl groups as compared to 3 inthe CuB case.
For PEO-b-aPBCL/drug binary mixture case, the intra-molec-ular p–p interactions between aromatic rings are strongcompared to the previous case and hence both drugs showalmost equal and reduced interactions with backbone atoms andthe carbonyl group of branches. As stated earlier, the onlysurprising part was the large difference between the correlations/interactions of the drugs with the aromatic rings. CuI has muchhigher correlation as compared to CuB. It is evident in p–pinteractions graph for the binary mixtures of di-block copolymersand drugs (Fig. 8(B)) that CuI reduces intra-molecular p–pinteractions drastically. To find the possible reasons behind thesetwo inter related observations, we further examined the corre-lation functions of specific groups (especially hydroxyl groups) onthe drug molecules with the aromatic ring. Here, the drugmolecules could interact with the aromatic ring either by OH–pinteractions or C]O–p interactions. Since the focus was partic-ularly on the intermolecular interactions between drug moleculesand aromatic ring of di-block copolymer, we examined the PEO-b-sPBCL case, which has minimal intra-molecular p–pinteractions.
3.4.4. OH–p interactionsFig. 12 shows RDF plot for the correlation between all hydroxyl
(–OH) groups of each drug and the aromatic rings on the di-blockcopolymer. The hydroxyl groups of CuI, having higher correlation,are in fact closer to the aromatic rings as compared to CuB. We alsoexamined such correlations of individual –OH groups on the drugmolecules. Here, we adopted a numbering scheme shown inFig. 13(A) and (B) to identify different groups present on the drugmolecules. Fig. 14 shows the RDF plots for the correlations ofindividual hydroxyl groups of CuI and compared them with the–OH group (1) of CuB. CuIH(1) and CuIH(4) are closer to aromaticrings as compared to other –OH groups. CuIH(4), being attached tothe free end of the molecule, is relatively flexible compared to otherhydroxyl groups and hence it can easily adjust itself to havemaximum interactions with the aromatic ring. This particular –OHgroup is absent in CuB. The maximum correlation was observed forCuIH(1). This correlation was much higher compared to that ofCuBH(1). This observation was very surprising. However, it may beattributed to the differences in the partial atomic charges on thedrug molecules. Fig. 15(A) and (B) show structures of CuB and CuI,respectively, along with the partial atomic charges displayed on the–OH groups of interest. Owing to the presence of double bond nearthe –OH group (1) in CuI, the partial atomic charge of the oxygenatom in the –OH group is less negative than that of CuB. Due to this,the electro negativity disparity of –OH(1) is different in both casesas seen in Fig. 15. The –OH(1) group is more polar in CuB ascompared to the one in CuI. The more polar –OH bond forms strong

C
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
0 1 2 3 4 5 6 7 8 9r(Å)r(Å)
0
0.2
0.4
0.6
0.8
1
1.2
g(r)
D
A
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9
g(r)
B
CuB
CuI
CuB
CuI
CuB
CuI
CuB
CuI
Fig. 11. RDF plots of comparison of CuB and CuI drugs in PEO-b-aPBCL copolymer for (A) Interaction No. 2, (B) Interaction No. 3, (C) Interaction No. 4, and (D) Interaction No. 5 listedin Table 3.
HOH
O
OH
OH O
O
1
2
2 3
3
A
S.K. Patel et al. / Biomaterials 31 (2010) 345–357 355
dipole and has tendency to get attracted to other strong dipoles likecarbonyl groups (–C]O) rather than engaging themselves with therelatively weak OH–p interactions. Thus, the less polar OH(1) groupof CuI will have favourable interactions with the aromatic ringwhile OH(1) group of CuB will have favourable interactions withthe carbonyl groups. All the above factors contribute towardshigher correlation of CuIH with the aromatic ring. Moreover, CuI
00.20.40.60.8
11.21.41.61.8
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
CuB -OH
CuI -OH
Fig. 12. RDF plot showing comparison of correlation between –OH groups of CuB andCuI and aromatic ring of di-block copolymer.
O
O4
1
HO
O
H
O
OH
OH O
OH
1
2
2 3
4
3
1
B
Fig. 13. Numbering scheme of H and O atoms of Hydroxyl and carbonyl groups in(A) CuB and (B) CuI.

0
0.5
1
1.5
2
2.5
3
0 1 2 3 4 5 6 7 8 9r(Å)
g(r)
CuIH(1)
CuIH(2)
CuIH(3)
CuIH(4)
CuBH(1)
Fig. 14. RDF plot showing correlations between all –OH groups of CuI, –OH(1) group ofCuB and aromatic ring of di-block copolymer.
S.K. Patel et al. / Biomaterials 31 (2010) 345–357356
interacts mostly through –OH groups with the p electrons of thearomatic ring while CuB interacts through carbonyl groups –C]Owith the edges (–CH) of the aromatic ring.
In general, from the above RDF plots, we found that for drugs likeCuB and CuI, with multitude of hydroxyl and carbonyl groups, to bemiscible in functionalized block copolymers, the drugs need tointeract not only with the substituent branches but also with thebackbone atoms (in the form of multiple hydrogen bonding withbackbone atoms). These intermolecular associations in the mixture ofdrug/copolymer largely depends on the strength of intra-molecularp–p interactions between aromatic rings of pure di-block copolymer.MD simulation is a powerful tool to predict these interplay of intraand intermolecular interactions.
Fig. 15. Partial atomic charges on –OH(1) group on (A) CuB and (B) CuI.
4. Conclusions
A molecular dynamics simulation approach was used tocompute the Flory–Huggins interaction parameters for binary di-block copolymer/drug mixtures formed by two hydrophobic drugsCuB and CuI and PEO-b-PBCL with three different tacticities viz.PEO-b-iPBCL, PEO-b-sPBCL, and PEO-b-aPBCL. It was found thatonly the c values for the binary mixtures containing PEO-b-sPBCLagree with experiment. In particular, the c values of the two otherstereo-isomers of PEO-b-PBCL are even much higher than those ofthe mixtures containing PEO-b-PCL. The results suggest that syn-diotactic configuration is the ideal stereo-isomer for inducingmiscibility between the drugs and PEO-b-PBCL. The strength of theintra-molecular p–p interactions was found to be dependent on thestereo configurations of the di-block copolymers. In order to inducemiscibility in the drug/copolymer mixture, intermolecular associ-ations/interactions need to be established through specific inter-actions which aid in achieving negative values of heats of mixingand hence negative c values. The formation of such intermolecularspecific interactions is largely hindered by the presence of intra-molecular p–p interactions. Hence, the syndiotactic version of di-block copolymer which exhibits minimal p–p interactionssucceeds in establishing favourable intermolecular specific inter-actions between the drug molecules and the di-block copolymer.
Acknowledgements
The authors would like to thank the Natural Sciences andEngineering Research Council of Canada (NSERC) for supportingthis work financially. This research has been enabled by the use ofWestGrid computing resources, which are funded in part by theCanada Foundation for Innovation, Alberta Innovation andScience, BC Advanced Education, and the participating researchinstitutions. WestGrid equipment is provided by IBM, HewlettPackard and SGI.
Appendix
Figures with essential colour discrimination. Figures in thisarticle may be difficult to interpret in black and white. The fullcolour images can be found in the on-line version, at doi:10.1016/j.biomaterials.2009.09.051.
References
[1] Lavasanifar A, Samuel J, Sattari S, Kwon GS. Block copolymer micelles forthe encapsulation and delivery of amphotericin B. Pharm Res 2002;19:418–22.
[2] Yokoyama M, Satoh A, Sakurai Y, Okano T, Matsumura Y, Kakizoe T, et al.Incorporation of water-insoluble anticancer drug into polymeric micelles andcontrol of their particle size. J Control Release 1998;55:219–29.
[3] Lee J, Cho EC, Cho K. Incorporation and release behavior of hydrophobic drugin functionalized poly(D,L-lactide)-block-poly(ethylene oxide) micelles.J Control Release 2004;94:323–35.
[4] Liu J, Xiao Y, Allen C. Polymer–drug compatibility: a guide to the developmentof delivery systems for the anticancer agent, ellipticine. J Pharm Sci 2004;93:132–43.
[5] Forrest ML, Zhao A, Won CY, Malick AW, Kwon GS. Lipophilic prodrugs ofHsp90 inhibitor geldanamycin for nanoencapsulation in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J Control Release 2006;116:139–49.
[6] Latere Dwan’Isa JP, Rouxhet L, Preat V, Brewster ME, Arien A. Prediction ofdrug solubility in amphiphilic di-block copolymer micelles: the role of poly-mer–drug compatibility. Pharmazie 2007;62:499–504.
[7] Mahmud A, Patel S, Molavi O, Choi P, Samuel J, Lavasanifar A. Self-associatingpoly(ethylene oxide)-b-poly(a-cholesteryl carboxylate-3-caprolactone) blockcopolymer for the solubilization of STAT-3 inhibitor cucurbitacin I. Bio-macromolecules 2009;9:471–8.
[8] Mahmud A, Xiong X, Lavasanifar A. Novel self-associating poly(ethylene oxi-de)-block-poly(3-caprolactone) block copolymers with functional side groupson the polyester block for drug delivery. Macromolecules 2006;39:9419–28.

S.K. Patel et al. / Biomaterials 31 (2010) 345–357 357
[9] Molavi O, Ma Z, Mahmud A, Alshamsan A, Samuel J, Lai R, et al. Polymericmicelles for the solubilization and delivery of STAT3 inhibitor cucurbitacins insolid tumors. Int J Pharm 2008;347:118–27.
[10] Flory PJ. Statistical mechanics of chain molecules. New York: Hanser; 1989.[11] Sun H. COMPASS: an ab initio force-field optimized for condensed-phase
applications – overview with details on alkane and benzene compounds.J Phys Chem B 1998;102:7338–64.
[12] Theodorou DN, Suter UW. Atomistic modeling of mechanical properties ofpolymeric glasses. Macromolecules 1986;19:139–54.
[13] Patel S, Lavasanifar A, Choi P. Application of molecular dynamics simulationto predict the compatibility between water-insoluble drugs and self-asso-ciating poly(ethylene oxide)-b-poly(3-caprolactone) block copolymers. Bio-macromolecules 2008;9:3014–23.
[14] Patel SK, Lavasanifar A, Choi P. Roles of nonpolar and polar intermolecularinteractions in the improvement of the drug loading capacity of PEO-b-PCL withincreasing PCL content for two hydrophobic cucurbitacin drugs. Bio-macromolecules 2009;10:2584–91.
[15] Andersen HC. Molecular dynamics simulations at constant pressure and/ortemperature. J Chem Phys 1980;72:2384–93.
[16] Nose S. A unified formulation of the constant-temperature moleculardynamics methods. J Chem Phys 1984;81:511–9.
[17] Flory PJ. Foundations of rotational isomeric state theory and general methodsfor generating configurational averages. Macromolecules 1974;7:381–92.
[18] Mattice WL, Suter UW. Conformational theory of large molecules: the rota-tional isomeric state model in macromolecular systems. New York: J. Wiley;1994.
[19] Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu Rev Biophys Biomol Struct 1999;28:155–79.
[20] Bai F, Li F, Calhoun BH, Quirk RP, Cheng S. Physical constant of poly(propylene).In: Brandrup J, Immergut EH, Grulke EA, editors. Polymer handbook. 4th ed.New York: A Wiley Interscience Publications; 1999. p. V/21–30.
[21] Kricheldorf HR, Boettcher C, Tonnes K. Polylactones: 23. Polymerization ofracemic and meso D, L-lactide with various organotin catalysts – stereo-chemical aspects. Polymer 1992;33:2817–24.
[22] Kricheldorf HR, Kreiser-Saunders I, Boettcher C. Polylactones: 31. Sn(II)oc-toate-initiated polymerization of L-lactide: a mechanistic study. Polymer1995;36:1253–9.
[23] Thakur KAM, Kean RT, Hall ES, Kolstad JJ, Lindgren TA, Doscotch MA, et al.High-resolution 13C and 1H solution NMR study of poly(lactide). Macromole-cules 1997;30:2422–8.
[24] Thakur KAM, Kean RT, Hall ES, Kolstad JJ, Munson EJ. Stereochemical aspects oflactide stereo-copolymerization investigated by 1H NMR: a case of changingstereospecificity. Macromolecules 1998;31:1487–94.
[25] Burley SK, Petsko GA. Aromatic–aromatic interaction: a mechanism of proteinstructure stabilization. Science 1985;229:23–8.
[26] Barlow JW, Paul DR. Polymer blends and alloys – a review of selectedconsiderations. Polym Eng Sci 1981;21:985–96.
[27] Cruz CA, Barlow JW, Paul DR. The basis for miscibility in polyester–poly-carbonate blends. Macromolecules 1979;12:726–31.
[28] Li G, Cowie JMG, Arrighi V. Miscibility of polymer blends of poly(styrene-co-4-hydroxystyrene) with bisphenol-A polycarbonate. J Appl Polym Sci 1999;74:639–46.






![Nanostructured poly(urethane)s and poly(urethane-urea)s from reactive solutions of poly[styrene-b-butadiene-b-(methyl methacrylate)]-triblock copolymers](https://img.pdfslide.net/doc/110x75/63494541b88fb0854f02e452/nanostructured-polyurethanes-and-polyurethane-ureas-from-reactive-solutions.jpg)






















