Embed Size (px)
Citation preview

P R I N C I P L E S O F
Toxicology Testing
F R A N K A B A R I L E
S E C O N D E D I T I O N

Principles of Toxicology Testing


Principles of Toxicology Testing
Second Edition
Frank A. Barile, PhDDepartment of Pharmaceutical Sciences, St. John’s University,
College of Pharmacy and Health Sciences, Queens, New York, USA
Boca Raton London New York
CRC Press is an imprint of theTaylor & Francis Group, an informa business

CRC Press
Taylor & Francis Group
6000 Broken Sound Parkway NW, Suite 300
Boca Raton, FL 33487-2742
© 2013 by Taylor & Francis Group, LLC
CRC Press is an imprint of Taylor & Francis Group, an Informa business
No claim to original U.S. Government works
Version Date: 20130225
International Standard Book Number-13: 978-1-84214-529-6 (eBook - PDF)
This book contains information obtained from authentic and highly regarded sources. While all reasonable efforts have
been made to publish reliable data and information, neither the author[s] nor the publisher can accept any legal respon-
sibility or liability for any errors or omissions that may be made. The publishers wish to make clear that any views or
opinions expressed in this book by individual editors, authors or contributors are personal to them and do not neces-
sarily reflect the views/opinions of the publishers. The information or guidance contained in this book is intended for
use by medical, scientific or health-care professionals and is provided strictly as a supplement to the medical or other
professional’s own judgement, their knowledge of the patient’s medical history, relevant manufacturer’s instructions and
the appropriate best practice guidelines. Because of the rapid advances in medical science, any information or advice
on dosages, procedures or diagnoses should be independently verified. The reader is strongly urged to consult the drug
companies’ printed instructions, and their websites, before administering any of the drugs recommended in this book.
This book does not indicate whether a particular treatment is appropriate or suitable for a particular individual. Ulti-
mately it is the sole responsibility of the medical professional to make his or her own professional judgements, so as to
advise and treat patients appropriately. The authors and publishers have also attempted to trace the copyright holders of
all material reproduced in this publication and apologize to copyright holders if permission to publish in this form has
not been obtained. If any copyright material has not been acknowledged please write and let us know so we may rectify
in any future reprint.
Except as permitted under U.S. Copyright Law, no part of this book may be reprinted, reproduced, transmitted, or uti-
lized in any form by any electronic, mechanical, or other means, now known or hereafter invented, including photocopy-
ing, microfilming, and recording, or in any information storage or retrieval system, without written permission from the
publishers.
For permission to photocopy or use material electronically from this work, please access www.copyright.com (http://
www.copyright.com/) or contact the Copyright Clearance Center, Inc. (CCC), 222 Rosewood Drive, Danvers, MA 01923,
978-750-8400. CCC is a not-for-profit organization that provides licenses and registration for a variety of users. For
organizations that have been granted a photocopy license by the CCC, a separate system of payment has been arranged.
Trademark Notice: Product or corporate names may be trademarks or registered trademarks, and are used only for
identification and explanation without intent to infringe.
Visit the Taylor & Francis Web site athttp://www.taylorandfrancis.com
and the CRC Press Web site athttp://www.crcpress.com

Dedication
To Pauline


vii
Contents
Preface viiiAcknowledgments xBiography xi
SECTION I BASIC CONCEPTS IN TOXICOLOGY TESTING
Chapter 1. Introduction to principles of toxicology 1
Chapter 2. Effects of chemicals 8
Chapter 3. Toxicokinetics 23
Chapter 4. Risk assessment and regulatory toxicology 44
Chapter 5. Descriptive animal toxicology tests 58
SECTION II TOXICOLOGY TESTING IN VIVO
Chapter 6. Acute toxicology testing 74
Chapter 7. Subchronic and chronic toxicology testing 93
Chapter 8. Acute dermal toxicity testing 106
Chapter 9. Acute ocular toxicity testing 116
Chapter 10. Toxicity testing for fertility and reproduction 122
Chapter 11. Carcinogenicity and mutagenicity testing in vivo 142
SECTION III TOXICOLOGY TESTING IN VITRO
Chapter 12. Introduction to in vitro toxicology testing 157
Chapter 13. Cell culture methodology 163
Chapter 14. Cell culture methods for acute toxicology testing 187
Chapter 15. Acute local toxicology testing in vitro 206
Chapter 16. Toxicokinetic studies in vitro 224
Chapter 17. Mutagenicity and carcinogenicity testing in vitro 229
Chapter 18. Reproductive and teratogenicity studies in vitro 256
Chapter 19. High-throughput screening and microarray analysis 264
Chapter 20. Toxicogenomics and epigenetic testing in vitro 276
Chapter 21. Experimental design and statistics 289
Chapter 22. Standardization and validation of alternative methods 302
Chapter 23. Applications of alternative models for toxicology testing 317
Index 331

viii
Preface
In the fi rst edition, I commented in the Preface that the science of toxicology testing has evolved in the last few decades from an applied and supportive science to its own refi ned and technical discipline. I also mentioned that the development and maturity of the discipline has been sporadic. In the last 5 years since the fi rst edition, however, the latter description has not held true. In fact, the fi eld has soared in its applications of the techniques and principles that defi ne the fi eld. Its progress has been prompted not by public health initiatives and needs but by the discovery of new disciplines, such as epigenetics, toxicogenetics, and toxicodynamics (for which new chapters have been added in the second edition). Thus, new roles have emerged for the application of both in vivo and in vitro techniques. These new roles have allowed for unique investigations in toxicology testing, followed by introduction of exciting principles to the fi eld. No sooner have the burgeoning advances of biotechnology allowed for the development of corresponding in vitro systems that complement traditional animal toxi-cology testing methods, but the new disciplines have been fi tted as well to important applica-tions within the toxicology arena.
As in the fi rst edition, the book begins with an introduction into the fundamentals of toxicology (Section I) to prepare students for the subsequent topics and continues through with a discussion of toxicokinetics and human risk assessment. This introductory material is useful in understanding the applications of toxicology testing.
Section II describes the fundamental principles of toxicology testing in animals in greater detail. This section describes acute toxicity studies as well as subchronic and chronic studies performed in animals. Special emphasis is placed on study design and determination of classical indicators for acute and chronic testing, such as the LD50. Other short- and long-term animal toxicity testing methodologies including dermal, ocular, and reproductive toxicity testing are discussed. Mutagenicity and carcinogenicity studies are also discussed in separate chapters.
Section III introduces and discusses in vitro alternatives to animal toxicology tests. This section emphasizes cell culture methodology and cellular methods for acute systemic toxicity, target organ toxicity, and local toxicity. The advantages and disadvantages of alter-native methods are presented. Special features of this section describe the use of high-throughput screening and its applications, the concepts of standardization and validation of in vitro techniques, especially large, organized validation efforts currently supported by US and EU regulatory agencies, and the theories supporting the development of in vitro meth-odologies. Undergraduate and graduate toxicology students and industrial and academic research laboratories will fi nd the text useful for the entry level students in the discipline or for establishing a toxicology testing laboratory, respectively.
The juxtaposition of the principles of animal toxicology testing in the same text as in vitro alternative methods highlights the importance of both fi elds for interpretation of the signifi cance and relevance of the other. Thus, the discussions continuously refer to the cor-responding methods available and the potential results from complementary designs of studies. In fact, both animal and in vitro toxicology testing methods are currently employed,

PREFACE ix
often together, in toxicological analysis, derivation of mechanisms of toxicity, mutagenic-ity testing, and preclinical drug development.
Several excellent texts are available in the fi eld concerning the details of individual protocols. Consequently, although some procedures are outlined in detail, the emphasis is on the principles of the disciplines rather than on the particular steps of the techniques. In fact, the title, Principles of Toxicology (rather than Toxicity) Testing, emphasizes the uni-versal application of the fi eld as a scientifi c discipline as opposed to amplifi cation of labo-ratory techniques. In addition, the book highlights contemporary issues in toxicology testing including the various means of possible exposure to chemicals, high-throughput screening of chemicals for preclinical drug development, and an overview of applications. Overall, the reader is challenged to interpret the signifi cance of toxicology testing results and to construct a logical approach toward the ultimate purpose of testing. Thus, the infor-mation contained herein is presented with great enthusiasm, particularly for the students prepared to dedicate their careers to this intriguing and fascinating scientifi c discipline.

x
Acknowledgments
I would like to extend my appreciation to the editorial staff at Informa Healthcare, Inc., for their interest, professionalism, and commitment to the project, in particular to Ms. Amber Thomas, Ms. Claire Bonnett, and Mr. Oscar Heini, as well as the staff at Exeter Premedia Services. My colleagues and staff at the College of Pharmacy, Department of Pharmaceutical Sciences, afforded invaluable comments, suggestions, and support throughout the project. Finally, I have had the pleasure to know and been fortunate to guide undergraduate and graduate toxicology and pharmacy students during the writing of this text, especially Angela Aliberti, Sanket Gadhia, Tak Lee, and Sanjay Dholakiya. This work would not have progressed fl uently or competently without their continuous feedback.

xi
Biography
Frank A. Barile, Ph.D., is a full professor in the Toxicology Division of the Department of Pharmaceutical Sciences at St. John’s University College of Pharmacy and Health Sciences, New York.
Dr. Barile received his B.S. in Pharmacy (1977), M.S. in Pharmacology (1980), and Ph.D. in Toxicology (1982) at St. John’s University. After a postdoctoral fellowship in Pulmo-nary Pediatrics at the Albert Einstein College of Medicine, Bronx, New York, he moved to the Department of Pathology, Columbia University, St. Luke’s Roosevelt Hospital, New York, as a research associate. In these positions, he investigated the role of pulmonary toxicants on collagen metabolism in cultured lung cells. In 1984, he was appointed as an assistant profes-sor in the Department of Health Sciences at City University of New York. Sixteen years later, he rejoined St. John’s University in the Department of Pharmaceutical Sciences and became an instrumental part of the toxicology program in the College of Pharmacy.
Dr. Barile holds memberships in several professional associations, including the US Society of Toxicology, American Association of University Professors, American Associa-tion for the Advancement of Science, American Society of Hospital Pharmacists, New York City Pharmacists Society, New York Academy of Sciences, and New York State Council of Health System Pharmacists. He has been appointed as a consultant scientist with several professional groups, including the Department of Pediatrics, Schneider’s Children’s Hos-pital, Long Island Jewish/Cornell Medical Centers, New York, as a committee member of SACATM, ICCVAM/NICEATM, and NIEHS, and as President and Past-President of the In Vitro and Alternative Methods Specialty Section, US Society of Toxicology. He is also the recipient of the Public Health Service Medallion from the Director of the NIEHS, Dr. Linda Birnbaum (2009). He was recently appointed as Editor-In-Chief (Rest of the World) for Toxicology In Vitro.
Dr. Barile has been the recipient of Public Health Service research grants from the National Institutes of Health (NIGMS), including awards from the Minority Biomedical Research Support program, the Minority High School Student Research Apprentice pro-gram, and AREA program.
Dr. Barile has authored and coauthored approximately 75 papers and abstracts in peer-reviewed biomedical and toxicology journals as well as three books and one contrib-uted chapter. He contributed original in vitro toxicology data to the international Multi-center Evaluation for Cytotoxicity program, along with distinguished international investigators. He lectures regularly to toxicology and pharmacy undergraduate and gradu-ate students in the toxicological and pharmaceutical sciences (he was awarded Professor of the Year for the College of Pharmacy by the University Student Government Associa-tion in 2003). Dr. Barile continues to perform fundamental research on the cytotoxic effects of environmental chemicals and therapeutic drugs on cultured human and mam-malian stem cells.


1
1 Introduction to principles of toxicology
INTRODUCTION
From its inception in the medieval era to its maturity as a distinct and separate discipline in the twenty fi rst century, toxicology was taught primarily as an applied science. The fi eld incorporated the various approaches from a variety of disciplines, not limited to the broad sciences of chemistry and biology. In particular, toxicology evolved from applications of analytical and clinical chemists whose job defi nitions included chemical identifi cation and analysis of body fl uids.
The fi rst modern toxicologists were chemists who had specialized training in inorganic separation methods including chromatographic techniques. Analytical chemists later employed thin layer and gas chromatography. The development of methods for forensic analysis greatly promoted the fi eld. These advances were particularly important for accep-tance within the legal community and in jurisprudence arenas. Furthermore, as the instru-mentation evolved and the technology became more exacting, high-performance liquid chromatography was incorporated into the analytical arsenal in order to isolate minute quantities of compounds from complex mixtures of toxicological importance. Eventually, biological applications exerted their infl uence, incorporating such specialties as microbiol-ogy, genetics, and cell culture methodology. Today, the fi eld has burgeoned into areas of specialization, some of which are hardly identifi able with their ancestral origins.
TYPES OF TOXICOLOGY
GENERAL TOXICOLOGY
General toxicology involves studies of exposure to chemical, biological, or physical agents and the untoward consequences that affect biological systems. The term, however, has been replaced by descriptions of areas that more closely refl ect the specialized fi elds of study within the discipline. The development of advanced methodologies in biotechnology, the requirement for increased training, and the involvement of toxicology in legal applications have made it necessary to more accurately label the discipline according to the expanding body of specialties. As a result, a variety of descriptions further defi ne the fi eld of toxicology.
MECHANISTIC TOXICOLOGY
Mechanistic toxicology involves the identifi cation of the cause, pathway, reaction, and cel-lular modifi cation associated with the toxicity of a chemical at the level of the cell, tissue, or organ. The classifi cation of toxicity of a chemical, therefore, may be expressed in terms of its mechanism of toxicity, its site of action, target area, or the organ most affected by the toxic insult. A similar expression, mechanism of action, is universally applied in the study of

2 PRINCIPLES OF TOXICOLOGY TESTING
pharmacology. Thus, mechanistic toxicology seeks to determine the biochemical, physio-logical, or organic basis of a toxic agent’s effects on biological systems.
REGULATORY TOXICOLOGY
Regulatory toxicology relates to the administrative dogma associated with the potential exposure to toxic agents encountered in the environment, in occupational settings, and in the home. Regulatory toxicology defi nes, directs, and dictates the rate at which an individual may encounter a synthetic or naturally occurring toxin and establishes guidelines for its maintenance in the environment, for risks due to possible exposure within the community and within the remedial market. The guidelines are generally promulgated by agencies whose jurisdiction and regulations are established by federal, state, and local authorities.
DESCRIPTIVE TOXICOLOGY
Descriptive toxicology is a subjective attempt to explain toxic agents and their applications. The list of descriptive areas developed principally as a method for bridging the vacuum between science and the public’s understanding of the fi eld, especially when it became nec-essary for nonscientifi c sectors to comprehend and interpret the importance of toxicology. This is especially true for public sector interpretation so that they could translate the infor-mation for the development of regulations and guidelines. For instance, the study of metals in the environment (metal toxicology) has become a popular discipline for toxicologists interested in examining the roles of heavy or trace metals in the environment.
FORENSIC TOXICOLOGY
From its inception in the medieval era to its maturity as a distinct and separate discipline in the 1950s, toxicology was taught primarily as an applied science. More recently, toxicology has evolved from applications of analytical and clinical chemists whose job defi nition was the chemical identifi cation and analysis of body fl uids. Thus the fi rst modern toxicologists were chemists with specialized training in inorganic separation methods, including chro-matographic techniques. Eventual evolution toward liquid chromatographic methods allowed analysis and isolation of minute quantities of compounds from complex mixtures of toxicological importance. Forensic toxicology thus integrates these techniques to identify compounds of sometimes unrelated poisons from biological specimens as a result of inci-dental or deliberate exposure. Initially, forensic sciences profi ted from the application of the principles of chemical separation methods for the identifi cation of controlled substances in body fl uids. Later, forensic toxicology applied biological principles of antigen–antibody interaction for paternity testing. By using the principles of blood grouping and “exclusion” of the potential outcomes of paternal contribution to offspring phenotype, it was feasible to eliminate the possibility of a male as the father of a child. Antigen–antibody interactions also became the basis for enzyme-linked immunosorbent assays (ELISA) currently used for spe-cifi c and sensitive identifi cation of drugs in biological fl uids. Radioimmunoassays (RIAs) utilize similar antigen–antibody reactions while incorporating radiolabeled ligands as indi-cators. DNA separation and sequencing techniques have now almost totally replaced tradi-tional paternity exclusion testing. These methods are also the basis for inclusion or exclusions of evidence in criminal and civil cases.
CLINICAL TOXICOLOGY
Clinical toxicology is also considered a descriptive category. However, toxic agents with clinical applications are also characterized in other toxicological fi elds. Clinical toxicology has evolved and branched from its counterpart, forensic toxicology, to include identifi cation,

INTRODUCTION TO PRINCIPLES OF TOXICOLOGY 3
diagnosis, and treatment of a condition, pathology, or disease resulting from environmental, therapeutic, or illicit exposure to chemicals or drugs. Treatment may involve amelioration of the signs and symptoms or controlling the underlying pathology. In clinical toxicology, expo-sure is commonly understood to include individual risk of contact with a toxin, either deliber-ate, accidental, or intentional. Exposure may further be defi ned to include population risk.1 Table 1.1 defi nes other descriptive fi elds of toxicology. More recently, the fi eld has blos-somed into more broadly defi ned areas including the study of apoptosis, receptor- mediated signal transduction, gene expression, proteomics, oxidative stress, and toxicogenomics, among others.
COMMON TERMS AND NOMENCLATURE
The classic defi nition of toxicology has traditionally been understood as the study of xeno-biotics, the science of poisons, and refers particularly to the interactions of exogenous agents with biological systems. For purposes of organizing the nomenclature, chemicals, compounds, and drugs are often referred to as agents or substances. Because such agents induce undesirable effects, they are usually alluded to as toxins. Consequently, toxicology involves internal and external physiological exposures to toxins and their interactions with organisms. Gradually, the term has evolved to include many chemically or physically unre-lated classes of agents. What transforms a chemical into a toxin, therefore, depends more on the duration of exposure, dose (or concentration), or route of exposure and less on the chemical structure, product formulation, or intended use of the material. As a result, almost any agent has the potential for toxicity and thus falls within the broad defi nition of toxicology.
1Risk to groups of persons from exposure to radiation, pollutants, and chemical or biological threats, which neces-sitates identifi cation, diagnosis, and treatment.
TABLE 1.1Other Descriptive Fields of Toxicology
Descriptive Field Defi nition
Genetic toxicology Incorporates molecular biology principles in applications of toxicological sciences, as applied to toxic agents that interfere with normal physiological function
Occupational toxicology Examines hazards associated with toxic exposure in the workplace, including industrial, agricultural, and communal sectors
In vitro toxicology Development of cell culture and biochemical techniques as alternatives to animal toxicity testing; a current defi nition of this fi eld includes alternative methods to animal toxicology, which more accurately describes the applications of in vitro methods
Analytical toxicology Chemical and biochemical procedures and methods associated with identifi cation, analysis, reactions, and detection of toxic substances in specimens
Developmental toxicology Study of toxic substances and their potential effects on biological reproduction, mating, fetal development, and embryogenesis
Immunotoxicology Study of toxic substances and their potential effects on immunity and resistanceNeurotoxicology Study of toxic substances and their potential effects on the function and activity of
the nervous systems

4 PRINCIPLES OF TOXICOLOGY TESTING
APPLICATIONS OF TOXICOLOGY
RESEARCH
Academic Applications
In the academic arena, research toxicologists examine the broad issues of toxicology in the laboratory setting. Academic concerns include all the public health areas in which progress in understanding toxicological sciences is necessary and include the elucidation of mechanistic, clinical, and descriptive toxicological theories. Research methods are modifi ed to answer specifi c questions that arise from toxicological concerns that affect public health.
Industrial Applications
Research toxicologists employed in toxicity testing in the biotechnology and pharmaceutical industries perform toxicity testing—the screening of chemicals and drugs that have toxic potential—before they are marketed. Preclinical testing in the pharmaceutical industry involves the conduct of phase I trials to test the toxicities of candidate agents that have been chemically and biochemically screened as potentially useful therapeutic drugs. The toxicity testing procedures include both in vitro and animal protocols.
REGULATORY TOXICOLOGY
Regulatory toxicologists are employed primarily in government administrative agencies, as consultants to government and industry or as representatives of industrial concerns. In this role, they sanction, approve, and monitor the uses of chemicals by establishing rules and guidelines. The guiding principles are promulgated through laws enacted by appropriate federal, state, and local jurisdictions that grant regulatory agencies their authority. Thus, through these regulations, an agency determines who is accountable and responsible for manufacturing, procurement, distribution, marketing, and, ultimately, release and dispens-ing of chemical substances to the public.
FORENSIC TOXICOLOGY
Forensic toxicologists integrate appropriate techniques to identify compounds arising from mixtures of sometimes unrelated poisons as a result of incidental or deliberate exposure. Initially, forensic sciences profi ted from the application of the principles of chemical separa-tion methods for the identifi cation of controlled substances in body fl uids. Later, forensic toxicologists applied biological principles of antigen–antibody interaction for paternity test-ing. By using the principles of blood grouping and the exclusion of the possible outcomes of paternal contributions to offspring phenotypes, it became possible to eliminate a male as a possible father of a child.
Antigen–antibody interactions also became the basis for ELISA and enzyme multi-plied immunoassay technique, which are currently used for specifi c and sensitive identi-fi cation of drugs in biological fl uids. RIAs utilize similar antigen–antibody reactions while incorporating radiolabeled ligands as indicators. DNA separation and sequencing techniques have now almost totally replaced traditional paternity exclusion testing. These methods are also the basis for inclusion or exclusion of evidence in criminal and civil cases.

INTRODUCTION TO PRINCIPLES OF TOXICOLOGY 5
CLINICAL TOXICOLOGY
Clinical toxicologists have evolved and branched away from their corresponding forensic applications. The clinical toxicologist is interested in the identifi cation, diagnosis, and treat-ment of a condition, pathology, or disease resulting from an environmental, therapeutic, or illicit exposure to chemicals or drugs. Exposure is commonly understood to include the individual risk of contact with a toxin.
CLASSIFICATION OF TOXIC AGENTS
Classifi cation of toxic agents is a daunting task, considering the vast numbers and com-plexities of chemical compounds in the public domain. The availability of the variety of chemicals, drugs, and physical agents, along with their varied toxicological and pharmaco-logical effects, means that a single agent may be listed in several different categories. Even compounds with similar structures or toxicological actions may be alternatively grouped according to their activities or physical states. The following outline of the classifi cation system is generally accepted for demonstrating the complex nature of toxins.
CLASSIFICATION ACCORDING TO USE
Pesticides
The U.S. Environmental Protection Agency defi nes a pesticide as a substance or a mixture of substances intended to prevent, destroy, repel, or mitigate a pest. In general, pesticides are classifi ed according to their biological targets. The four major classes of pesticides are insec-ticides, herbicides, rodenticides, and fungicides. Table 1.2 lists these categories and their general subclasses. Because of the physiological and biochemical similarities of target spe-cies and mammalian organisms, an inherent toxicity is associated with pesticides in mam-malian organisms. In addition, within each classifi cation, compounds are identifi ed according to mechanism of action, chemical structure, or semisynthetic source. For instance, although many fungicide categories exist, fungicidal toxicity in humans is mostly low order, with the exception of therapeutic antifungal agents, principally because of their specifi c mechanisms of action. Similarly, fumigants range from carbon tetrachloride to ethylene oxide and are used to kill insects, roundworms, and fungi in soil, stored grain, fruits, and vegetables. Their toxicity, however, is limited to occasional occupational exposure.
Food and Industrial Additives
Direct food and color additives are intentionally incorporated in foods and food-processing operations for purposes of changing, enhancing, or masking color. They are also used for a variety of functionalities ranging from anticaking agents to stabilizers, thickeners, and tex-turizers. Food and industrial additives fall into the fi eld of food toxicology and readers are referred to review articles listed at the end of this chapter for information concerning food ingredients and contaminants.
Therapeutic Drugs
Toxicological classifi cation of therapeutic agents follows their pharmacological mechanisms of action or their principal target organs of toxicity. Several important references address clinical toxicologies of therapeutic drugs as extensions of their adverse reactions and direct effects resulting from their excessive use.

6 PRINCIPLES OF TOXICOLOGY TESTING
SOURCES OF TOXINS
BOTANICAL
Contact dermatitis caused by poison ivy is a well-characterized syndrome of acute infl amma-tion. Today, many compounds of botanical origin are classifi ed as herbal supplements, imply-ing that their origins are botanical. Their importance in maintaining health is also related to their natural derivations. The toxicities of these agents, however, are poorly understood.
ENVIRONMENTAL
As a result of industrialization, many chemicals are associated with and classifi ed according to their continuous presence in the environment, that is, water, land, and soil. The phenom-enon is not limited to developed Western nations and is becoming a problem among develop-ing South American, Asian, and African countries.
Environmental toxicology is a distinct discipline encompassing the areas of air pollu-tion and ecotoxicology. A discussion of air pollution necessarily includes outdoor and indoor air pollution, presence of atmospheric sulfuric acid, airborne particulate matter, interaction of photochemicals with the environment, and chemicals found in smog. Ecotoxicology is the branch of environmental toxicology that investigates the effects of environmental chemicals on the ecosystems in question. Readers are referred to the review articles at the end of this chapter for further discussion.
SUGGESTED READINGS
Abraham J. The science and politics of medicines control. Drug Saf 2003; 26: 135.Bois FY. Applications of population approaches in toxicology. Toxicol Lett 2001; 120: 385.Eaton DL, Klassen CD. Principles of toxicology. In: Klassen CD, ed. Casarett and Doull’s Toxicology:
The Basic Science of Poisons, 7th edn. Chapter 2 New York: McGraw-Hill, 2007.
TABLE 1.2Classification of Pesticides
Pesticide Class Classifi cation According to Target
Insecticide Organophosphorus estersOrganochlorine compoundsCarbamate estersPyrethroid estersBotanical derivatives
Herbicide Chlorophenoxy compoundsBipyridyl derivativesChloroacetanilidesPhosphonomethyl amino acids
Rodenticide Anticoagulantsα-Napthyl thioureaMiscellaneous metals, inorganic, natural products
Fungicide General areas of agricultural, domestic, and therapeutic antifungal agents

INTRODUCTION TO PRINCIPLES OF TOXICOLOGY 7
Ettlin RA, Dybing E, Eistrup C, et al. Careers in toxicology in Europe: options and requirements. Report of a workshop presented at the EUROTOX Congress in London (September 17–20, 2000). Arch Toxicol 2001; 75: 251.
Gennings C. On testing for drug/chemical interactions: defi nitions and inference. J Biopharm Stat 2000; 10: 457.
Greenberg G. Internet resources for occupational and environmental health professionals. Toxicology 2002; 178: 263.
Guzelian PS, Victoroff MS, Halmes NC, James RC, Guzelian CP. Evidence-based toxicology: a comprehensive framework for causation. Hum Exp Toxicol 2005; 24: 161.
Kaiser J. Toxicology: tying genetics to the risk of environmental diseases. Science 2003; 300: 563.Meyer O. Testing and assessment strategies including alternative and new approaches. Toxicol Lett
2003; 140–141: 21–30.Tennant RW. The National Center for Toxicogenomics: using new technologies to inform mechanis-
tic toxicology. Environ Health Perspect 2002; 110: A8.
REVIEW ARTICLES
Aardema MJ, MacGregor JT. Toxicology and genetic toxicology in the new era of “toxicogenomics”: impact of “-omics” technologies. Mutat Res 2002; 499: 13.
Abraham J, Reed T. Progress, innovation and regulatory science in drug development: the politics of international standard setting. Soc Stud Sci 2002; 32: 337.
Barnard RC. Some regulatory defi nitions of risk: interaction of scientifi c and legal principles. Regul Toxicol Pharmacol 1990; 11: 201.
Collins TF. History and evolution of reproductive and developmental toxicology guidelines. Curr Pharm Des 2006; 12: 1449.
Eason C, O’Halloran K. Biomarkers in toxicology versus ecological risk assessment. Toxicology 2002; 181: 517.
Fostel J, Choi D, Zwickl C, et al. Chemical effects in biological systems data dictionary (CEBS-DD): a compendium of terms for the capture and integration of biological study design descrip-tion, conventional phenotypes, and “omics” data. Toxicol Sci 2005; 88: 585.
Harris SB, Fan AM. Hot topics in toxicology. Int J Toxicol 2002; 21: 383.Johnson DE, Wolfgang GH. Assessing the potential toxicity of new pharmaceuticals. Curr Top Med
Chem 2001; 1: 233.Lewis RW, Billington R, Debryune E, et al. Recognition of adverse and nonadverse effects in toxicity
studies. Toxicol Pathol 2002; 30: 66.Reynolds VL. Applications of emerging technologies in toxicology and safety assessment. Int J Toxicol
2005; 24: 135.Rietjens IM, Alink GM. Future of toxicology: low dose toxicology and risk–benefi t analysis. Chem
Res Toxicol 2006; 19: 977.Schrenk D. Regulatory toxicology: objectives and tasks defi ned by the working group of the
German society of experimental and clinical pharmacology and toxicology. Toxicol Lett 2002; 126: 167.
Sexton K, Hattis D. Assessing cumulative health risks from exposure to environmental mixtures - three fundamental questions. Environ Health Perspect 2007; 115: 825.
Thong HY, Maibach HI. Hormesis [biological effects of low-level exposure (B.E.L.L.E.)] and derma-tology. Cutan Ocul Toxicol 2007; 26: 329.
Valerio LG Jr. In silico toxicology for the pharmaceutical sciences. Toxicol Appl Pharmacol 2009; 241: 356.
Waddell WJ. The science of toxicology and its relevance to MCS. Regul Toxicol Pharmacol 1993; 18: 13.
Wolfgang GH, Johnson DE. Web resources for drug toxicity. Toxicology 2002; 173: 67.

8
2 Effects of chemicals
TOXICOLOGICAL EFFECTS
GENERAL CLASSIFICATION
As noted in chapter 1, chemical and physical agents are categorized in part according to several indicators of toxicity that render them amenable for understanding their potential toxic effects. Prediction of toxicity of a chemical lies in the ability to recognize its potential adverse effects based on factors not necessarily related to physicochemical properties. This chapter explores a variety of local and systemic reactions elicited by chemical exposure and the physiological and immunological basis of such effects.
CHEMICAL ALLERGIES
The four types of immunological hypersensitivity (allergic) reactions include the following:
1. Type I antibody-mediated reactions.2. Type II antibody-mediated cytotoxic reactions.3. Type III immune complex reactions.4. Type IV delayed-type hypersensitivity cell-mediated immunity.
Type I antibody-mediated reactions occur in three phases. The initial or sensitization phase is triggered by contact with a previously unrecognized antigen. This reaction entails binding of the antigen to the immunoglobulin E (IgE) present on the surfaces of mast cells and basophils. The second or activation phase follows after an additional dermal or muco-sal challenge with the same antigen. This phase is characterized by degranulation of mast cells and basophils, with a subsequent release of histamine and other soluble mediators. The third stage, the effector phase, is characterized by accumulation of preformed and newly synthesized chemical mediators that precipitate local and systemic effects. Degranu-lation of neutrophils and eosinophils completes the late-phase cellular response.
Antigens involved in type I reactions are generally airborne pollens including mold spores and ragweeds as well as food ingredients. Ambient factors such as heat and cold, drugs (opioids, antibiotics), and metals (silver, gold) precipitate chemical allergies of the type I nature. Because of their small molecular weights, the majority of chemicals and drugs, as single entities, generally circulate undetected by immune surveillance systems. Consequently, in order to initiate the sensitization phase of an antigenic response, chemicals are immunologically handled as haptens.1 Examples of type I hypersensitivity syndromes are described in Table 2.1 and typical effects of chemical allergies are listed in Table 2.2.
1 Haptens are small molecular weight chemical entities (<1000 Da) that nonspecifi cally bind to larger circulating polypeptides or glycoproteins. Binding induces a conformational change in the original larger complex. The chemical entity bound to the larger molecule is no longer recognized as part of the host (self) system, rendering it susceptible to immune attack.

EFFECTS OF CHEMICALS 9
The effects of chemical allergies are usually acute and appear immediately when compared with other hypersensitivity reactions described below.
Type II antibody-mediated cytotoxic reactions differ from type I in the nature of anti-gen, the cytotoxic character of the antigen–antibody reaction, and the type of antibody formed (IgM, IgG). In general, antibodies are induced against target antigens that are altered cell membrane determinants. Type II reactions include complement–mediated (CM) reactions, antibody-dependent cell-mediated cytotoxicity, antibody-mediated cellu-lar dysfunction, transfusion reactions, Rh incompatibility reactions, autoimmune reactions, and chemical-induced reactions, the last of which is of greater interest in experimental toxicology.
TABLE 2.1Examples of Type I Hypersensitivity Syndromes, Causes, and Effects
Syndrome Causes Effects
Allergic rhinitis (hay fever) Pollen, mold spores ↑ Capillary permeability in nasal and frontal sinuses and mucosal membranes, ↑ vasodilation
Food allergies Lectins and proteins present in nuts, eggs, shellfi sh, dairy products
↑ Capillary permeability, ↑ vasodila-tion, ↑ smooth muscle contraction
Atopic (allergic) dermatitis Localized exposure to drugs and chemicals
Initial local mast cell release of cytokines, followed by activation of neutrophils and eosinophils
Asthma Chemicals, environmental, behavioral Chronic obstructive reaction of LRT involving airway hyperactivity and cytokine release
Abbreviation: LRT, lower respiratory tract.
TABLE 2.2Descriptive Inflammatory Effects of Chemical Allergies
Effect Description
Bullous Large dermal blistersErythema Redness or infl ammation of skin due to dilation and congestion of superfi cial capillaries
(e.g., sunburn)Flare Reddish, diffuse blushing of the skinHyperemia Increased blood fl ow to tissue or organInduration Raised, hardened, thickened skin lesionMacule Flat red spot on skin due to increased blood fl owPapule Raised red spot on skin due to increased blood fl ow and antibody localizationPetechial Small, pinpoint dermal hemorrhagic spotsPruritus ItchingPurulent Suppuration; production of pus containing necrotic tissue, bacteria, infl ammatory cellsUrticaria Pruritic skin eruption characterized by wheal formationVesicular Small dermal blistersWheal Raised skin lesion due to accumulation interstitial fl uid

10 PRINCIPLES OF TOXICOLOGY TESTING
As with type I reactions, chemical-induced type II cytotoxicity requires that an agent behave as a hapten. The chemical binds to the target cell membrane and proceeds to operate as an altered cell membrane determinant. This determinant changes the conformational appearance of a component of the cell surface, not unlike the effect described above for a hapten. Thus, it induces a series of responses that terminate in antibody induction. The determinant attracts a variety of immune surveillance reactions including a CM cytotoxic reaction, recruitment of granulocytes, or deposit of immune complexes within the cell membrane. A variety of therapeutic drugs are known to induce type II reactions, particu-larly with continuous administration. Examples include antibiotics and cardiovascular agents, among others.
Type III immune complex reactions are localized responses mediated by antigen– antibody immune complexes. Type III reactions are stimulated by microorganisms and involve activation of complement. Systemic (serum sickness) and localized (Arthus reaction) immune complex disease, infection-associated immune complex disease (rheumatic fever), and occupational diseases (opportunistic pulmonary fungal infections) induce complement–antibody–antigen complexes that trigger the release of cytokines and recruitment of granulo-cytes, resulting in increased vascular permeability and tissue necrosis.
Type IV (delayed-type) hypersensitivity cell-mediated immunity involves antigen- specifi c T-cell activation. The reaction starts with an intradermal or mucosal challenge (sensitization stage). CD4+ T cells then recognize MHC-II (major histocompatibility class-II) antigens on antigen-presenting cells (such as Langerhans cells) and differentiate to T
H1
cells. This sensitization stage requires a prolonged local contact with the agent, from sev-eral days to weeks. A subsequent repeat challenge stage induces differentiated T
H1 (mem-
ory) cells to release cytokines, further stimulating the attraction of phagocytic monocytes and granulocytes. The release of lysosomal enzymes from the phagocytes results in local tissue necrosis. Contact hypersensitivity resulting from a prolonged exposure to natural products and metals, for example, is caused by the lipophilicity of the chemicals in skin secretions, thus acting as a hapten.
IDIOSYNCRATIC REACTIONS
Idiosyncratic reactions are abnormal responses to chemicals or drugs generally resulting from uncommon genetic predisposition. An exaggerated response to the skeletal muscle relaxant properties of succinylcholine, a depolarizing neuromuscular blocker, classifi es as a typical idiosyncratic reaction. In some subjects, a congenital defi ciency in plasma cholines-terase results in a reduction in the rate of succinylcholine deactivation. As succinylcholine accumulates, respiration fails to return to normal during the postoperative period. Similarly, the cardiotoxic action of cocaine is exaggerated in cases of congenital defi ciency of plasma esterases, which are necessary for metabolism of the drug. A paucity of circulating enzymes allows for uncontrolled, sympathetically mediated effects in experimental animals and humans.
IMMEDIATE VS. DELAYED HYPERSENSITIVITY
In contrast to immune hypersensitivity reactions, some chemical effects are immediate or delayed, depending on the mechanism of toxicity. The acute effects of sedative hypnotics are of immediate consequence—high concentrations in animals or humans result in death from respiratory depression. The effects of carcinogens, however, may not be demonstrated for generations in humans or only after several years of exposure in rodents. An important exam-ple of this has been demonstrated with the link between diethylstilbestrol administration to

EFFECTS OF CHEMICALS 11
child-bearing women in the 1950s and the subsequent development of clear cell adenocarci-noma vaginal cancers in the offspring.
REVERSIBLE VERSUS IRREVERSIBLE EFFECTS
In general, the effects of most chemicals or drugs are reversible until a critical point is reached, that is, when a vital function is compromised or a mutagenic, teratogenic, or carcinogenic effect develops. In fact, the carcinogenic effects of chemicals such as those present in tobacco smoke may be delayed for decades until irreversible cellular transfor-mation occurs. Reversibility of the acute effects of chemicals is achieved through the administration of antagonists, enhancement of metabolism or elimination, delay of absorption, intervention of another toxicological procedure that decreases toxic blood concentrations, or termination of the exposure.
LOCAL VERSUS SYSTEMIC EFFECTS
As discussed below, local or systemic effects of a compound depend on the site of exposure. The integument (skin) and lungs are targets of chemical exposure because they frequently serve as the fi rst sites of contact with environmental chemicals. Oral exposure requires absorption and distribution of an agent prior to the development of systemic effects. Hyper-sensitivity reaction types I and IV are precipitated by local activation of immune responses following a sensitization phase, while chemical-induced type II reactions are elicited through oral or parenteral administration.
MUTAGENIC AND CARCINOGENIC EFFECTS
Mutagenic and carcinogenic effects of chemical exposure are discussed in detail in chapter 11.
BIOCHEMICAL PROPERTIES
CHEMICAL STRUCTURE
Important components of a toxicity testing protocol include the identifi cation, categoriza-tion, synthesis, and toxicity screening of chemicals according to their classifi cations. Because chemical and drug development depends upon the availability of parent compounds, the synthesis of structurally related compounds is an important process. Thus, understanding the chemical structure of a compound allows for reasonable prediction of many of its anticipated and adverse effects. Some examples of agents that are categorized according to their molec-ular structures include organophosphorus insecticides, heavy metals, benzodiazepines (seda-tive hypnotics), and imidazolines (tranquilizers).
MECHANISM OF ACTION
Similarly, it is convenient to organize toxic agents according to their physiological or bio-chemical targets. Examples include mutagens, hepatotoxic compounds, methemoglobin-producing agents, and acetylcholinesterase inhibitors.
EXPOSURE
In the presence of specifi c circumstances, any chemical has the potential for toxicity, that is, the same dose of a chemical may be harmless if limited to oral exposure but toxic if

12 PRINCIPLES OF TOXICOLOGY TESTING
inhaled or administered parenterally. Thus the route and site of exposure exert signifi cant infl uence in determining the toxicity of a substance. More frequently, a therapeutic dose for an adult may be toxic for an infant or a child. Similarly, a substance may not exert adverse effects until a critical threshold is achieved. Thus, in order to induce toxicity, it is necessary for the chemical to accumulate in a physiological compartment at a concentra-tion suffi ciently high to reach the threshold value. Finally, repeated administration over a specifi ed period of time also determines the potential for toxicity. The following discussion details the circumstances for exposure and dose of a chemical that favors or deters the potential for toxicity.
ROUTE
Oral Administration
Undoubtedly, oral exposure to toxins is the most frequently encountered route of contact. Oral administration involves the presence of several physiological barriers that must be penetrated or circumvented if adequate blood concentrations of a compound is achieved (Fig. 2.1). The mucosal layers of the oral cavity, pharynx, and esophagus consist of strat-ifi ed squamous epithelium that serves to protect the upper gastrointestinal (GI) lining from the effects of contact with physical and chemical agents. Simple columnar epithe-lium lines the stomach and villi of the intestinal tract that function in digestion, secretion, and absorption. Immediately underlying the epithelium is the lamina propria, a mucosal layer rich in blood vessels and nerves. Mucosa-associated lymphoid tissue is layered within this level, where prominent lymphatic nodules sustain the presence of phagocytic macrophages and granulocytes. Salivary and intestinal glands contribute to the digestive process by secreting saliva and digestive juices. The submucosa, muscularis, and serosa complete the strata that form the anatomical envelope of the GI tract. Enteroendocrine and exocrine cells in the GI tract secrete hormones and, in the stomach, secrete acid and gastric lipase.
The primary function of the stomach is mechanical and chemical digestion of food, while absorption is secondary. Several factors infl uence the transit and stability of a chem-ical in the stomach, thereby infl uencing gastric emptying time (GET). The presence of food delays absorption and dilutes the contents of the stomach, thus reducing subsequent chem-ical transit. An increase in the relative pH of the stomach causes a negative feedback inhibi-tion of stomach churning and motility, which also delays the gastric emptying process. Any factor that slows down stomach motility will increase the amount of time that a chemical stays in the stomach, prolonging the GET. Thus, the longer the GET, the greater the dura-tion of a chemical’s presence within the stomach, and the more susceptibility to gastric enzyme degradation and acid hydrolysis, in spite of any increase in pH. In addition, a prolonged GET delays passage to and subsequent absorption in the intestinal tract.
Intranasal Administration
Intranasal insuffl ation is a popular method for therapeutic administration of corticosteroids and sympathomimetic amines and for the illicit use of drugs of abuse such as cocaine and opioids. The dosage form in therapeutic use is usually aerosolized in a metered nasal inhaler. In the case of illicit use, crude drugs are inhaled (snorted) through the nares as fi ne or coarse powders. In both cases, absorption is rapid due to the extensive network of capillaries in the lamina propria of the mucosal lining within the nasopharynx. Thus the absorption rate rivals that of pulmonary inhalation.
EFFECTS OF CHEMICALS 13
Inhalation
The vast surface area of the upper and lower respiratory tracts allows for wide and immedi-ate distribution of inhaled powders, particulates, aerosols, and gases. Figure 2.2 illustrates the thin alveolar wall that separates airborne particulates from access to capillary mem-branes. Once a drug is ventilated to the alveoli, it is transported across the alveolar epithelial lining to the capillaries, resulting in rapid absorption.
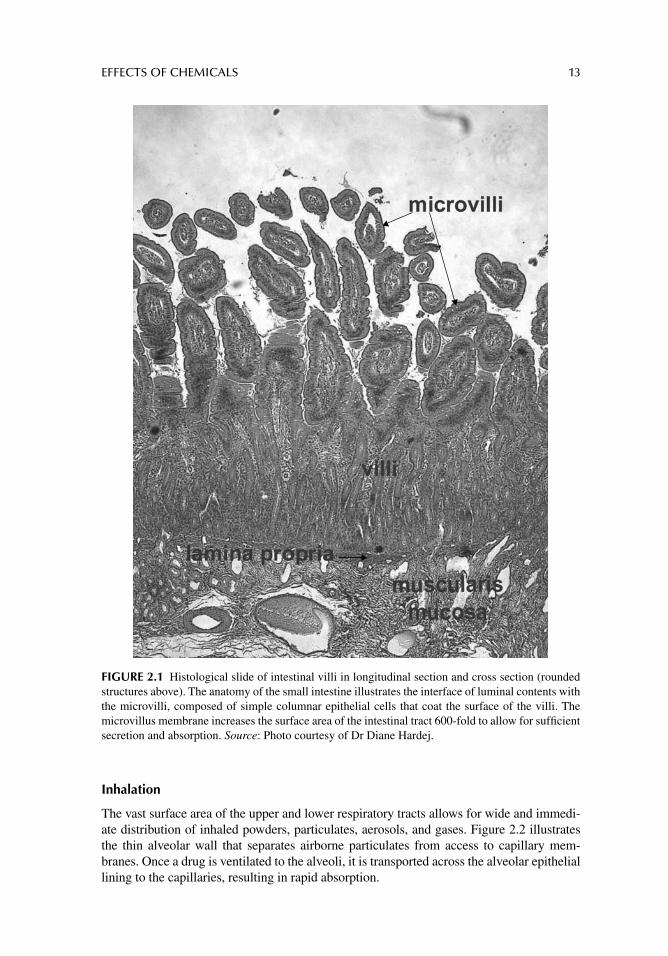
FIGURE 2.1 Histological slide of intestinal villi in longitudinal section and cross section (rounded structures above). The anatomy of the small intestine illustrates the interface of luminal contents with the microvilli, composed of simple columnar epithelial cells that coat the surface of the villi. The microvillus membrane increases the surface area of the intestinal tract 600-fold to allow for suffi cient secretion and absorption. Source: Photo courtesy of Dr Diane Hardej.

14 PRINCIPLES OF TOXICOLOGY TESTING
Dermal Routes
As an organ, the integumentary system (skin) allows for the greatest surface area for physi-ological contact with the external environment. The skin affords a wide range of therapeutic interventions through application of chemicals and drugs. Topical administration is the most common form of dermal application, yet also include epidermal, intradermal, and transder-mal injections—upper dermal injections are still considered topical applications.
Figure 2.3 illustrates the layers of the skin: the epidermis, the outermost layer, and the underlying dermis and subcutaneous (hypodermis) layers. Epidermal and upper dermal injections have the poorest absorption capabilities of the parenteral routes primarily because of limited circulation. Consequently, intradermal and epidermal injections are limited to eliciting reactions that might occur on the skin surface, such as testing for immunological reactions.2 Yet surface application of fat-soluble substances has a propen-sity for dermal absorption, principally through the oil-rich content of the dermal layers. Thus lipid-soluble chemicals are slowly but readily absorbed by non-ionic passage through the epidermal layers. For instance, oil-rich creams, lotions, and ointments are formulated to profi t from this type of pharmacokinetic model. Similarly, newer delivery methods,
2 The Tine test for tuberculosis and skin allergy testing are examples of immunological diagnostic therapeutic injections for determination of intradermal reactivity.
Alveolus Capillary
FIGURE 2.2 Pulmonary alveolus showing thin capillary–air interface. Source: From Barile, Frank A., Clinical Toxicology: Principles and Mechanisms, 2nd edition, CRC Press, 2010.
Epidermis
Dermis
Hair follicleFat
Blood vessel
Sensorynerve
Fibroblasts
Sweat gland
FIGURE 2.3 Cross section of dermal epithelium and underlying basal lamina structures. Epidermis, dermis, and underlying (subcutaneous) layers of skin. Source: From Artifi cial Skin, News Features and Articles, NIH, NIGMS, 2003.

EFFECTS OF CHEMICALS 15
such as topical application systems (patches) are formulated to deliver precise quantities of lipid-soluble chemicals and drugs via the dermal routes. Thus, prolonged contact, con-trolled release from the drug delivery unit, and increased surface area combine to allow the transfer of precise amounts of a compound through the epidermal barriers to the underly-ing capillary circulation.
Parenteral Routes
Parenteral administration includes subcutaneous, intramuscular, intraperitoneal, and intrave-nous (IV) injections. Parenteral routes, in general, are subject to minimal initial enzymatic degradation or chemical neutralization, thus bypassing the hindrances associated with pas-sage through epithelial barriers. The deeper dermal and subcutaneous layers provide entrance to a richer supply of venules and arterioles. A subcutaneous injection forms a depot within the residing adipose tissue with subsequent leakage into the systemic circulation, or the majority of the injection can enter the arterioles and venules. Intramuscular injections ensure access to a more extensive vascular network within skeletal muscle, accounting for more rapid exposure than subcutaneous injections. IV injection is the most rapid method of chem-ical exposure because access to the circulation is direct and immediate.3 Thus, the route of exposure contributes as much to toxicity as the dose.
DURATION AND FREQUENCY
Acute Exposure
In general, any exposure shorter than or equal to 24-hour is regarded as acute. Exposure to most toxic gases (carbon monoxide, hydrogen cyanide) requires less than 24 hours for toxic-ity. In most experimental settings, however, 72 hours may still be considered acute exposure, such as in continuous low-dose exposure to hepatotoxic agents. In addition, a single IV injection of a chemical is certainly classifi ed as an acute exposure. Subacute exposure gener-ally refers to continuous or repeated exposure to a chemical for more than 72 hours but less than a month.
Chronic Exposure
Chronic exposure is any relative period for which continuous or repeated exposure beyond the acute phase is required for the same chemical to induce a toxic response. Subchronic exposure is understood to involve a duration between acute and chronic. The traditional time of subchronic exposure is accepted as 1–3 months. In experimental and clinical toxicity, however, a subchronic exposure may include repeated exposure for a period longer than 3 months. Thus, the terms are fl exible adaptations to defi ne the onset of chemical intoxica-tion. In addition, considerable overlap in judgment is involved when labels are assigned to exposure periods.
Frequency of administration involves repeated doses of a drug or toxin during the exposure period. Repeated administration of the same dose of a chemical within a period defi ned as acute or chronic generally establishes a greater potential for adverse effects. Similarly, continuous repeated exposure to a toxin, especially during an acute period, has a greater toxic potential.
3 Although the inhalation route (see above) rivals IV administration by virtue of the extensive surface area and capillary exposure in the vast regions of the pulmonary alveoli.

16 PRINCIPLES OF TOXICOLOGY TESTING
ACCUMULATION
Dose, duration, frequency, and route of exposure contribute to chemical toxicity in part through accumulation of a compound in physiological compartments. A normal dosage schedule is determined according to a chemical’s half-life (t
1/2) in plasma and its intended
response, that is, the time required for plasma levels to decrease to one-half of the measured or estimated concentration. Thus, if the frequency of administration occurs often within the t1/2
of a chemical, its concentration in a compartment is likely to increase beyond the desir-able level. Accumulation results from overloading of a chemical within this compartment.
According to Physiological Compartment
Biological systems in their entirety are considered one-compartment models. Ideally, a chemical capable of distributing uniformly throughout the body would maintain steady-state levels for the exposure period. Blood levels4 of a compound may also decrease uniformly, assuming a constant rate of elimination. Physiologically, however, the body is not a homo-geneous chamber. A chemical, once absorbed, can distribute and/or bind to one or more of many physiological sites. The distribution of a chemical depends largely on its physico-chemical characteristics (see chap. 3, for detailed descriptions of absorption, distribution, metabolism, and elimination).
The compartments include whole blood, serum and serum proteins, plasma and plasma proteins, adipose tissue, interstitial and extracellular fl uids, alveolar air space, and bone marrow. In addition, a chemical may preferentially accumulate in any tissue or organ, thus acting as a discrete compartment. For instance, many therapeutic drugs such as warfarin (a vitamin K antagonist anticoagulant) nonspecifi cally bind to circulating plasma proteins, resulting in apparently lower blood concentrations than anticipated.5 Heavy metals prefer-entially accumulate in adipose tissue, kidney, and bone. Consequently their toxicity may be experienced for prolonged periods as the compounds are slowly released from this com-partment years after exposure has ceased. Accumulation, therefore, is predicted based on a chemical’s apparent volume of distribution (V
d), which is estimated as the total dose of
chemical administered divided by the concentration of chemical in the plasma for a given period. In general, the greater the V
d, the greater the potential for accumulation in some
physiological compartment.6
According to Chemical Structure
Accumulation is also determined by a chemical’s structure and its interaction within the physiological compartment. This phenomenon is guided by the chemical’s predominant state of existence in a physiological fl uid, that is, it remains in the fl uid compartment as an ionic or non-ionic species. In general, at physiological pH, lipid-soluble compounds will preferen-tially remain in their non-ionic states, to bind to and accumulate in membranes of tissues and organs. Conversely, water-soluble compounds remain as ionic species at the pH of blood. Thus, because they are less prone to tissue binding, the ions are readily available for renal secretion and elimination (see chap. 3, for a complete discussion).
4 The blood level and plasma level terms are often used interchangeably, although blood and plasma are distinct anatomical compartments. 5 Drugs or chemicals that bind to circulating plasma proteins are generally not pharmacologically active, are structurally incapable of binding to receptors, and are preferentially excluded from fi ltering through the renal glomeruli. 6 A standard measure for the accumulated internal dose of a chemical is body burden , which refers to the amount of chemical stored in one or several physiological compartments or in the body as a whole.

EFFECTS OF CHEMICALS 17
CHEMICAL INTERACTIONS
POTENTIATION
Potentiation of toxicity occurs when the toxic effect of one chemical is enhanced in the pres-ence of a toxicologically unrelated agent. The situation can be described numerically as 0 + 2 > 2, where a relatively nontoxic chemical alone has little or no effect (0) on a target organ, but may enhance the toxicity of another coadministered chemical (2). The hepatotoxicity of carbon tetrachloride, for instance, is greatly enhanced in the presence of isopropanol.
ADDITIVE EFFECTS
Two or more chemicals whose combined effects are equal to the sum of the individual effects is described as an additive interaction. This is the case with the additive effects of a combina-tion of sedative hypnotics and ethanol (drowsiness, respiratory depression). Numerically this is summarized as “2 + 2 = 4.”
SYNERGISTIC EFFECTS
By defi nition, a synergistic effect is indistinguishable from potentiation except that, in some references, both chemicals must have some cytotoxic activity. Numerically, synergism occurs when the sum of the effects of two chemicals is greater than the additive effects, such as the effect experienced with a combination of ethanol and antihistamines (1 + 2 > 3). The synergism and potentiation are terms often used synonymously.
ANTAGONISTIC EFFECTS
The opposing actions of two or more chemical agents, not necessarily administered simultane-ously, are considered antagonistic interactions. Different types of antagonism are as follows:
Functional antagonism: The opposing physiological effects of chemicals, such as with central nervous system stimulants versus depressants.
Chemical antagonism: Drugs or chemicals that bind to, inactivate, or neutralize target compounds such as the actions of chelators in metal poisoning.
Dispositional antagonism: The interference of one agent with the absorption, distri-bution, metabolism, or excretion of another; examples of agents that interfere with absorption, metabolism, and excretion include activated charcoal, phenobarbital, and diuretics, respectively.
Receptor antagonism: The occupation of toxicological or pharmacological receptors by competitive or noncompetitive agents, such as the use of tamoxifen in the preven-tion of estrogen-induced breast cancer.
DOSE–RESPONSE RELATIONSHIP
GENERAL ASSUMPTIONS
A discussion of the effects of a chemical as a result of exposure to a particular dose is neces-sarily followed by an explanation of the pathway by which that dose elicited the response. This relationship has traditionally been known as the dose–response relationship.7 The result
7 In certain fi elds of toxicology, particularly mechanistic and in vitro systems, the term dose-response is more accurately referred to as concentration-effect. This change in terminology makes note of the specifi c effect on a measurable parameter that corresponds in vivo to a precise plasma concentration.
18 PRINCIPLES OF TOXICOLOGY TESTING
of exposure to the dose is any measurable, quantifi able, or observable indicator. The response depends on the quantity and route of chemical exposure or administration within a given period. Two types of dose–response relationships exist, depending on the number of subjects and doses tested.
Graded Dose–Response
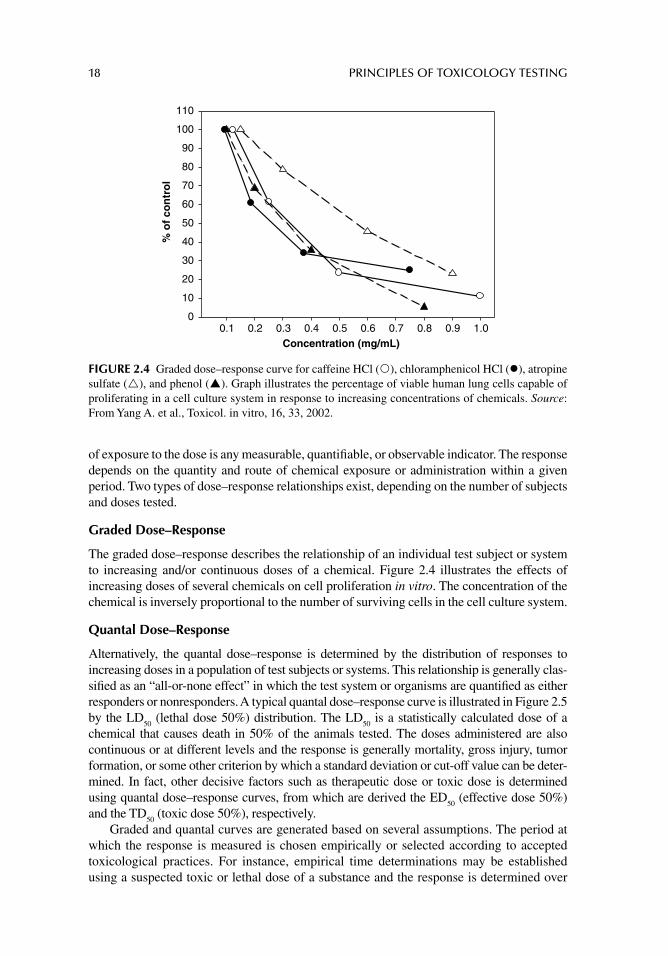
The graded dose–response describes the relationship of an individual test subject or system to increasing and/or continuous doses of a chemical. Figure 2.4 illustrates the effects of increasing doses of several chemicals on cell proliferation in vitro. The concentration of the chemical is inversely proportional to the number of surviving cells in the cell culture system.
Quantal Dose–Response
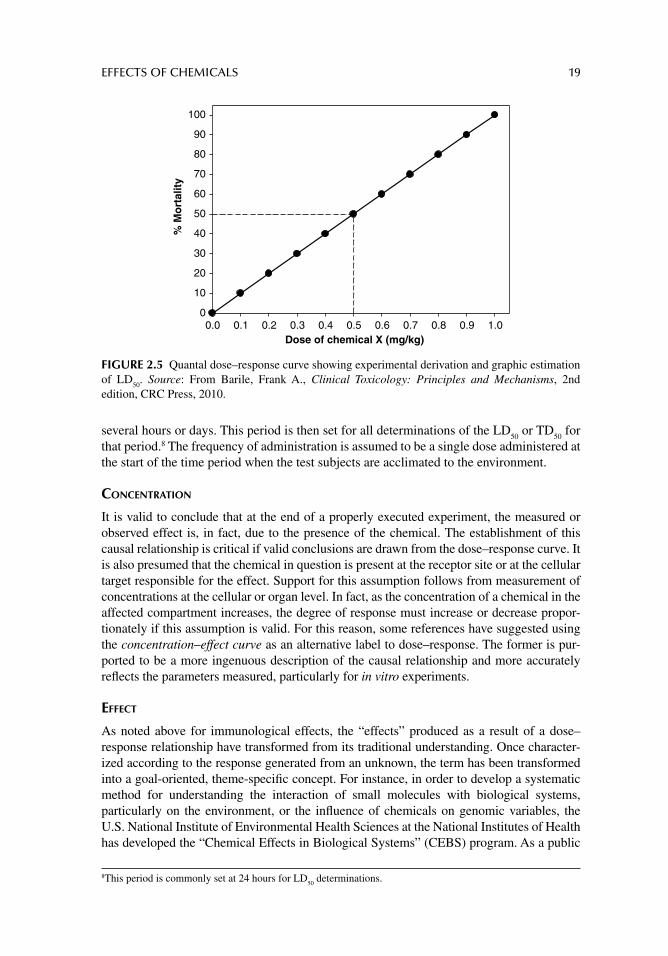
Alternatively, the quantal dose–response is determined by the distribution of responses to increasing doses in a population of test subjects or systems. This relationship is generally clas-sifi ed as an “all-or-none effect” in which the test system or organisms are quantifi ed as either responders or nonresponders. A typical quantal dose–response curve is illustrated in Figure 2.5 by the LD
50 (lethal dose 50%) distribution. The LD
50 is a statistically calculated dose of a
chemical that causes death in 50% of the animals tested. The doses administered are also continuous or at different levels and the response is generally mortality, gross injury, tumor formation, or some other criterion by which a standard deviation or cut-off value can be deter-mined. In fact, other decisive factors such as therapeutic dose or toxic dose is determined using quantal dose–response curves, from which are derived the ED
50 (effective dose 50%)
and the TD50
(toxic dose 50%), respectively.Graded and quantal curves are generated based on several assumptions. The period at
which the response is measured is chosen empirically or selected according to accepted toxicological practices. For instance, empirical time determinations may be established using a suspected toxic or lethal dose of a substance and the response is determined over
Concentration (mg/mL)0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
% o
f co
ntr
ol
0
10
20
30
40
50
60
70
80
90
100
110
FIGURE 2.4 Graded dose–response curve for caffeine HCl ( ), chloramphenicol HCl ( ), atropine sulfate ( ), and phenol ( ). Graph illustrates the percentage of viable human lung cells capable of proliferating in a cell culture system in response to increasing concentrations of chemicals. Source: From Yang A. et al., Toxicol. in vitro, 16, 33, 2002.
EFFECTS OF CHEMICALS 19
several hours or days. This period is then set for all determinations of the LD50
or TD50
for that period.8 The frequency of administration is assumed to be a single dose administered at the start of the time period when the test subjects are acclimated to the environment.
CONCENTRATION
It is valid to conclude that at the end of a properly executed experiment, the measured or observed effect is, in fact, due to the presence of the chemical. The establishment of this causal relationship is critical if valid conclusions are drawn from the dose–response curve. It is also presumed that the chemical in question is present at the receptor site or at the cellular target responsible for the effect. Support for this assumption follows from measurement of concentrations at the cellular or organ level. In fact, as the concentration of a chemical in the affected compartment increases, the degree of response must increase or decrease propor-tionately if this assumption is valid. For this reason, some references have suggested using the concentration–effect curve as an alternative label to dose–response. The former is pur-ported to be a more ingenuous description of the causal relationship and more accurately refl ects the parameters measured, particularly for in vitro experiments.
EFFECT
As noted above for immunological effects, the “effects” produced as a result of a dose–response relationship have transformed from its traditional understanding. Once character-ized according to the response generated from an unknown, the term has been transformed into a goal-oriented, theme-specifi c concept. For instance, in order to develop a systematic method for understanding the interaction of small molecules with biological systems, particularly on the environment, or the infl uence of chemicals on genomic variables, the U.S. National Institute of Environmental Health Sciences at the National Institutes of Health has developed the “Chemical Effects in Biological Systems” (CEBS) program. As a public
8 This period is commonly set at 24 hours for LD 50
determinations.
Dose of chemical X (mg/kg)0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
% M
ort
alit
y
0
10
20
30
40
50
60
70
80
90
100
FIGURE 2.5 Quantal dose–response curve showing experimental derivation and graphic estimation of LD
50. Source: From Barile, Frank A., Clinical Toxicology: Principles and Mechanisms, 2nd
edition, CRC Press, 2010.

20 PRINCIPLES OF TOXICOLOGY TESTING
resource, the CEBS database houses data of interest for environmental health scientists and other toxicologists interested in toxicogenomics.9 CEBS has received depositions of data from academic, industrial, and governmental laboratories. It is designed to display data in the context of biology and study design and to permit data integration across studies for novel meta-analysis.
The CEBS-Data Dictionary (CEBS-DD) is designed to standardize nomenclature needed to describe a toxicogenomics study in a structured yet intuitive format. The Diction-ary facilitates the capture and display of toxicogenomics data in a biological context in CEBS and associates molecular events with the toxicology/pathology phenotype, as observed in the individual experiments. CEBS-DD is developed with a focus on acute tox-icity studies, but with a design that will permit it to be extended to other areas of toxicology and biology with the addition of domain-specifi c terms.
The destination of a program such as CEBS, therefore, is to create a reference toxi-cogenomics information system of studies on environmental chemicals or stressors and their effects. In addition, the database has generated relational and descriptive data com-pendia on toxicologically important genes, groups of genes, single nucleotide polymor-phisms, and mutant and knock-out phenotypes in animal models, which are relevant to human diseases and health. More specifi cally, observing effects through the CEBS data-base has enabled hypothesis-driven and discovery research in environmental toxicology.
RECEPTORS AND RECEPTOR SITES
Pharmacodynamics and toxicodynamics10 refer to the study of the biochemical and physiolog-ical effects of drugs and toxins and their mechanisms of action or toxicity, respectively. The objectives of this study, therefore, is to understand the biochemical or physical interactions between the chemical agent and the target cell and to characterize the sequence, scope, time course, and explanation of its effect. Thus a complete analysis provides the basis for rational therapeutic drug use, understanding the mechanisms of action or toxicity, describing the ratio-nale for therapeutic intervention in poisoning, and predicting the complex interactions between drugs and drugs, food, the environment, and the underlying pathological basis of a disease.
A receptor is traditionally defi ned as a cellular macromolecule to which a chemical agent may bind in order to initiate, maintain, continue, block, or prevent an effect. A recep-tor is characterized according to its macromolecular structure, site of action, regulatory role, functional quality, structure–activity relationship, and quantitative analysis. Agents requiring the interaction with receptors may also be distinguished from actions of chemi-cals not mediated by receptors. Several informative suggested readings and review articles are noted at the end of the chapter, which discuss drug receptor interactions and their phar-macological and toxicological basis for understanding dose–response relationships.
CRITERIA FOR MEASUREMENT
Except for lethality, the selection of a measurable or observable end point is crucial. A desir-able biomarker accurately refl ects the presence of a chemical at a cellular or molecular site or suggests that a toxic effect originates from its action at the target organ. Selection of a measurable end point thus depends on the suspected mechanism of toxicity, if known, or on
9 The study of the response of a genome to environmental stressors and toxicants. Toxicogenomics combines genetics, genome-scale mRNA expression (transcriptomics), cell and tissue-wide protein expression (proteomics), metabolite profi ling (metabonomics), and bioinformatics with conventional toxicology in an effort to understand the role of gene–environment interactions in disease. 10 For convenience, the terms are used interchangeably throughout the text.

EFFECTS OF CHEMICALS 21
empirical determinations based on the chemical formula. Some biomarkers are also subjec-tive, such as reliance on histological grading, calculation of the degree of anesthesia, pain, motor activity, or behavioral change. Thus the standards for quantifying the end point are determined and established prior to the experimental setup.
SUGGESTED READINGS
Acquavella J, Doe J, Tomenson J, et al. Epidemiologic studies of occupational pesticide exposure and cancer: regulatory risk assessments and biologic plausibility. Ann Epidemiol 2003; 13: 1.
Ashby J. The leading role and responsibility of the international scientifi c community in test develop-ment. Toxicol Lett 2003; 140: 37.
Carnevali O, Maradonna F. Exposure to xenobiotic compounds: looking for new biomarkers. Gen Comp Endocrinol 2003; 131: 203.
Eaton DL, Gilbert SG. Principles of toxicology. In: Klaassen CD, ed. Casarett and Doull’s Toxicol-ogy: The Basic Science of Poisons, 7th edn. Ch. 2 New York: McGraw-Hill, 2007.
Fostel J, Choi D, Zwickl C, et al. Chemical effects in biological systems–data dictionary (CEBS-DD): a compendium of terms for the capture and integration of biological study design descrip-tion, conventional phenotypes, and ‘omics data. Toxicol Sci 2005; 88: 585.
Olsen CM, Meussen-Elholm ET, Hongslo JK, Stenersen J, Tollefsen KE. Estrogenic effects of envi-ronmental chemicals: an interspecies comparison. Comp Biochem Physiol C Toxicol Phar-macol 2005; 141: 267.
U.S. National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), Chemical Effects in Biological Systems (CEBS). [Available from: http://www.niehs.nih.gov/research/resources/databases/cebs/index.cfm] [Last accessed May 2012].
Yang A, Cardona DL, Barile FA. Subacute cytotoxicity testing with cultured human lung cells.
Toxicol In Vitro 2002; 16: 33.
REVIEW ARTICLES
Afshari CA, Hamadeh HK, Bushel PR. The evolution of bioinformatics in toxicology: advancing toxicogenomics. Toxicol Sci 2011; 120: S225.
Basketter DA, Evans P, Gerberick GF, Kimber IA. Factors affecting thresholds in allergic contact dermatitis: safety and regulatory considerations. Contact Dermat 2002; 47: 1.
Bolt HM, Kiesswetter E. Is multiple chemical sensitivity a clinically defi ned entity? Toxicol Lett 2002; 128: 99.
Collings FB, Vaidya VS. Novel technologies for the discovery and quantitation of biomarkers of toxicity. Toxicology 2008; 245: 167.
Dewhurst IC. Toxicological assessment of biological pesticides. Toxicol Lett 2001; 120: 67.Edler L, Kopp-Schneider A. Statistical models for low dose exposure. Mutat Res 1998; 405: 227.Efroymson RA, Murphy DL. Ecological risk assessment of multimedia hazardous air pollutants:
estimating exposure and effects. Sci Total Environ 2001; 274: 219.Feron VJ, Cassee FR, Groten JP, et al. International issues on human health effects of exposure to
chemical mixtures. Environ Health Perspect 2002; 110: 893.Foster JR. The functions of cytokines and their uses in toxicology. Int J Exp Pathol 2001; 82: 171.Furtaw EJ Jr. An overview of human exposure modeling activities at the U.S. EPA’s National Expo-
sure Research Laboratory. Toxicol Ind Health 2001; 17: 302.Gaylor DW, Kodell RL. Dose–response trend tests for tumorigenesis adjusted for differences in sur-
vival and body weight across doses. Toxicol Sci 2001; 59: 219.Goetz AK, Singh BP, Battalora M, et al. Current and future use of genomics data in toxicology:
opportunities and challenges for regulatory applications. Regul Toxicol Pharmacol 2011; 61: 141.
Goldman LR. Epidemiology in the regulatory arena. Am J Epidemiol 2001; 154: 18.

22 PRINCIPLES OF TOXICOLOGY TESTING
Green S, Goldberg A, Zurlo J. TestSmart high production volume chemicals: an approach to imple-menting alternatives into regulatory toxicology. Toxicol Sci 2001; 63: 6.
Hakkinen PJ, Green DK. Alternatives to animal testing: information resources via the Internet and World Wide Web. Toxicology 2002; 173: 3.
Hanson ML, Solomon KR. New technique for estimating thresholds of toxicity in ecological risk assessment. Environ Sci Technol 2002; 36: 3257.
Hastings KL. Implications of the new FDA/CDER immunotoxicology guidance for drugs. Int Immu-nopharmacol 2002; 2: 1613.
Hattan DG, Kahl LS. Current developments in food additive toxicology in the USA. Toxicology 2002; 181: 417.
Herbarth O, Fritz G, Krumbiegel P, et al. Effect of sulfur dioxide and particulate pollutants on bron-chitis in children: a risk analysis. Environ Toxicol 2001; 16: 269.
Isbister GK. Data collection in clinical toxinology: debunking myths and developing diagnostic algo-rithms. J Toxicol Clin Toxicol 2002; 40: 231.
Jarup L. Health and environment information systems for exposure and disease mapping, and risk assessment. Environ Health Perspect 2004; 112: 995.
Kenna LA, Labbe L, Barrett JS, Pfi ster M. Modeling and simulation of adherence: approaches and applications in therapeutics. AAPS J 2005; 7: E390.
Kimber I, Gerberick GF, Basketter DA. Thresholds in contact sensitization: theoretical and practical considerations. Food Chem Toxicol 1999; 37: 553.
Konsoula Z. Chemical and drug receptor interactions. Chapter 11 In: Barile FA, ed. Clinical Toxicol-ogy: Principles and Mechanisms, 2nd edn. New York, London: Informa HealthCare Pub-lishers, 2010.
Krishnan K, Johanson G. Physiologically-based pharmacokinetic and toxicokinetic models in cancer risk assessment. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2005; 23: 31.
Maurer T. Skin as a target organ of immunotoxicity reactions. Dev Toxicol Environ Sci 1986; 12: 147.McCarty LS. Issues at the interface between ecology and toxicology. Toxicology 2002; 181: 497.McCurdy T, Glen G, Smith L, Lakkadi Y. The national exposure research laboratory’s consolidated
human activity database. J Expo Anal Environ Epidemiol 2000; 10: 566.Melnick RL, Kohn MC. Dose-response analyses of experimental cancer data. Drug Metab Rev 2000;
32: 193.Moschandreas DJ, Saksena S. Modeling exposure to particulate matter. Chemosphere 2002; 49:
1137.Moya J, Phillips L. Overview of the use of the U.S. EPA exposure factors handbook. Int J Hyg Envi-
ron Health 2002; 205: 155.Mueller SO. Xenoestrogens: mechanisms of action and detection methods. Anal Bioanal Chem 2004;
378: 582.Palmer JR, Hatch EE, Rosenberg CL, et al. Risk of breast cancer in women exposed to diethylstilbes-
trol in utero: preliminary results (U.S.). Cancer Causes Control 2002; 13: 753.Pennington D, Crettaz P, Tauxe A, et al. Assessing human health response in life cycle assessment
using ED10s and DALYs: part 2, Noncancer effects. Risk Anal 2002; 22: 947.Schneider T, Vermeulen R, Brouwer DH, et al. Conceptual model for assessment of dermal exposure.
Occup Environ Med 1999; 56: 765.Strickland JA, Foureman GL. U.S. EPA’s acute reference exposure methodology for acute inhalation
exposures. Sci Total Environ 2002; 288: 51.Tanaka E. Toxicological interactions between alcohol and benzodiazepines. J Toxicol Clin Toxicol
2002; 40: 69.Waters M, Stasiewicz S, Merrick BA, et al. CEBS–Chemical Effects in Biological Systems: a public
data repository integrating study design and toxicity data with microarray and proteomics data. Nucleic Acids Res 2008; 36: D892–900. [Available from: http://www.ncbi.nlm.nih.gov/pubmed/17962311]
Wexler P, Phillips S. Tools for clinical toxicology on the World Wide Web: review and scenario. J Toxicol Clin Toxicol 2002; 40: 893.

23
3 Toxicokinetics
INTRODUCTION
RELATIONSHIP TO PHARMACOKINETICS
The study of drug disposition in physiological compartments is traditionally referred to as pharmacokinetics. Toxicokinetics includes the more appropriate study of exogenous com-pounds (xenobiotics), the adverse effects associated with their concentrations, routes through the physiological compartments, and the ultimate metabolic fate. Compartmental toxicoki-netics entails administration or exposure to a chemical, rapid equilibration to the central compartments (plasma and tissues), followed by distribution to peripheral compartments. Consequently, the principles of pharmacokinetics and toxicokinetics historically have been used interchangeably.
ONE-COMPARTMENT MODEL
A one-compartment model is the simplest representation of a system that describes the fi rst-order extravascular absorption of a xenobiotic. The absorption rate is constant (k
a) and allows
for entry into central compartments (plasma and tissues). A constant fi rst-order elimination (k
e1) is explained in the next paragraph. The reaction is illustrated below. Few compounds
follow one-compartment modeling; agents such as creatinine equilibrate rapidly, distribute uniformly throughout blood, plasma, and tissues, and are eliminated quickly.
Reaction 1: Chemical One-compartment Excretedka kel
Drug or toxin elimination processes generally follow zero-order or fi rst-order kinetics. First-order elimination and fi rst-order kinetics are terms that describe the rate of elimination of a drug in proportion to the drug concentration: an increase in the concentration of the chemical results in an increase in the rate of elimination. This usually occurs at low drug concentrations. Alternatively, zero-order elimination refers to the removal of a fi xed amount of drug independent of the concentration: increasing plasma concentration of the chemical does not proceed with a corresponding increase in elimination. Zero-order kinetics usually occurs when biotransforming enzymes are saturated. Clinically, this situation is the most common cause of drug accumulation and toxicity. Michaelis–Menton kinetics occurs when a compound is metabolized according to both processes. For instance, at low concentrations, ethanol is metabolized by fi rst-order kinetics (dose dependent). As the blood alcohol concen-tration increases, the process switches to zero-order (dose independent) elimination.
TWO-COMPARTMENT MODEL
In a two-compartment model, a xenobiotic chemical requires a longer time for the concentra-tion to equilibrate with tissues and plasma. Essentially, molecules enter the central

24 PRINCIPLES OF TOXICOLOGY TESTING
compartment at a constant rate (ka) and may also be eliminated through a constant fi rst-order
elimination (kel) process. However, a parallel reaction (Reaction 2) occurs when the chemical
enters and exits peripheral compartments through fi rst-order rate constants for distribution (k
d1 and k
d2, respectively). The distribution phases are variable but rapid and relative to elimi-
nation. Most drugs and xenobiotics undergo two- or multicompartment modeling behavior.
Reaction 2: Chemical First-compartment
Second compartment
Excreted
kd1
ka kel
kd2
MULTI-COMPARTMENT MODEL
It is useful to conceptualize the distribution of a chemical substance into a limited number of compartments, dictated in part by the volume of distribution of the agent and the physico-chemical infl uences of the major plasma volume. However, physiologically, the body does not render itself to simple models and, thus, requires construction of complex systems to explain drug and chemical distribution. For drugs whose target organ is also the end organ for pharmacological (clinical) response, the two-compartment model suffi ces. However, for a majority of drugs and agents predominantly administered by other than the intravenous route, the location of the target organ is either distant from the plasma or is separated by fac-tors that ultimately infl uence response at the end organ. Thus, the multicompartment model accounts for chemicals whose target organ response is separated by a factor of time, or intensity of effect, from the site of absorption. Reaction 3 displays the diagram for a typical multicompartment toxicodynamic system. Behavior of a chemical in the intestinal tract, for instance, is regulated by the physiological force of absorption, which necessarily becomes the fi rst compartment. However, this is not necessarily the rate-limiting step, but the chemi-cal’s distribution is so infl uenced. Thus, for orally administered drugs, the end organ response is suffi ciently prolonged due to the signifi cantly slower rate of absorption. Consequently, rate of absorption through the intestinal tract contributes to subsequent distribution through the intermediate compartment.
ka kel
Reaction 31: Chemical
kd1 kd2
Intermediate compartments
ExcretedMulti-compartment
APPLICATIONS TO TOXICOLOGY TESTING
Clinical applications of kinetics and the phenomena of absorption, distribution, metabolism, and elimination (ADME) are useful in the monitoring of toxicological behavior of chemi-cals. Assigning a compound to a compartmental system, particularly a two-compartment model or the more complex multicompartment model, allows for prediction of adverse
1Illustration of a toxicokinetic multi-compartment model; ka is the constant for rate of absorption, k
el is the constant
rate for elimination, kd1
and kd2
represent constants for rates of distribution toward and away from the intermediate compartments, respectively.

TOXICOKINETICS 25
effects of agents based on distribution and equilibration within the system. It also allows for establishing therapeutic interventions based on kinetic factors due to chemical poisoning. Examples of ADME factors that infl uence the fate of a chemical, and how these substances are handled toxicologically and therapeutically, are discussed next.
ABSORPTION
IONIC AND NON-IONIC PRINCIPLES
How does the ionic or non-ionic species of a chemical relate to absorption? The eukaryotic cell membrane is characterized as a fl exible, semipermeable barrier that maintains a homeo-static environment for normal cellular functions by preventing chemicals, ions, and water from traversing through it easily. Figure 3.1 illustrates the structure of a eukaryotic cell membrane. The membrane consists of phospholipid polar heads, glycolipids, integral pro-teins organized toward the periphery of the membrane (hydrophilic region), and nonpolar tails and cholesterol directed inward (hydrophobic region). The rest of the membrane is interspersed with transmembrane channel proteins and other intrinsic proteins. The fl uidity of the membrane is accounted for by the presence of cholesterol and integral proteins. The
Outside of cell
Inside of cell
Hydrophilicregion
Hydrophilicregion
Hydrophobicregion
Glycoprotein Cell surface
Phospholipid Proteins
Cytoplasm
FIGURE 3.1 Fluid mosaic model of the normal eukaryotic cell membrane showing polar heads (hydrophilic regions) and nonpolar tails (hydrophobic region). Other structural features, such as transmembrane proteins and surface glycoproteins, contribute to the fl uidity and selective permeabil-ity of the membrane. Source: Photo courtesy of Mr Trevor Gallant, http://kvhs.nbed.nb.ca/gallant/biology/biology.html.

26 PRINCIPLES OF TOXICOLOGY TESTING
phospholipid polar groups and nonpolar tails are derived from triglycerides. Thus, the lipid bilayer structure of the cell membrane owes its functional activity in part to the polar envi-ronment contributed by the phosphate and carboxyl moieties (heads) and the relatively nonpolar saturated long carbon chains (tails).
Because of the selectively permeable arrangement of the cell membrane, the ability of a chemical to bind to or penetrate intact membranes is determined by its polarity, that is, its water or lipid solubility. In turn, the relative contribution of a chemical to its polarity is infl uenced by the acidic or basic nature of the chemical in solution, which is determined by the proportion of the chemical’s propensity to dissociate or associate, respectively. Accord-ing to the Brönsted–Lowry theory of acids and bases, an acidic compound dissociates (donates a proton); a basic molecule associates (accepts a proton). The dissociation con-stant for an acid can be summarized according to the following formula:
+ −
a[H ][A ]K =
[HA]
where Ka is the dissociation constant for an acid; [H+], [A–], and [HA] represent the hydro-
gen ion, conjugate acid, and undissociated acid concentrations, respectively. The pKa derived
from the negative logarithm of the acid dissociation constant is calculated as follows:
pKa = −log K
a
The pKa of a drug, therefore, is defi ned as the pH at which 50% of the compound is
ionized. Similarly, pKb is the negative logarithm of the base dissociation constant (K
b). In
general, acids and bases are classifi ed according to their abilities to dissociate relative to the pH of the solution. Independent of the pH, however, the lower the pK
a of an acid, the
stronger the acid. Similarly, the lower the pKa (or higher the pK
b) of a base, the stronger
the base. Unlike most chemicals that display a wide variety of dissociative abilities in solu-tion, therapeutic drugs are usually weak acids or bases, neutral, or amphoteric compounds.
In general, strong acidic and basic characteristics of chemicals are determined by the relative presence of carboxyl anions and amine groups in solution, respectively. For instance, benzoic acid, with a pK
a of 4, readily dissociates, leaving the carboxyl anion
after dissociation. Its low pKa calculates a high dissociation constant (K
a) equal to 1 × 10–4,
resulting in a compound possessing strong acidic properties. Similarly, aniline is a highly protonated species with a pK
b of 10 and corresponding K
b of 1 × 10–10 (pK
a + pK
b = 14;
thus, although it is a base possessing an amine group, the pKa of aniline can also be
expressed as 4). The low dissociation constant of the base indicates that the H+ ions are closely held to the nitrogen of the amine and contribute to its strong basic nature.
HENDERSON–HASSELBACH EQUATION AND DEGREE OF IONIZATION
The relationship of the ionization of a weakly acidic drug, its pKa, and the pH of a solution
is adequately demonstrated by the exquisite application of the Henderson–Hasselbach equa-tion. The equation allows for the prediction of the non-ionic and ionic states of a compound at a given pH. Following are the formulas:
Acid: pH = pKa + log ([A−]/[HA])
Base: pH = pKa + log ([HA]/[A−])

TOXICOKINETICS 27
The equations are derived from the logarithmic expression of the dissociation con-stant formula above. Small changes in pH near the pK
a of a weakly acidic or basic drug,
markedly affect its degree of ionization. This is more clearly shown with rearrangement of the Henderson–Hasselbach equations, such that:
For an Acid: pKa − pH = log ([HA]/[A−])
For a Base: pKa − pH = log ([A−]/[HA])
When an acidic compound enters the stomach, and before the establishment of equilib-rium (e.g., pK
a = 4 in an average pH of the stomach = 2), the Henderson–Hasselbach equa-
tion predicts that the relative ratio of non-ionized to ionized species of the acidic compounds will be calculated at 100:1. This transforms the compound predominantly to the non-ionic form within the acidic environment of the stomach, rendering it more lipophilic. Lipophilic compounds have a greater tendency for absorption within that compartment.2
In the proximal small intestine, in the areas of the duodenum and jejunum where the pH is approximately 8, the same compound will be transformed to exist predominantly in the ionized state. The relative ratio of non-ionized to ionized species is reversed (1:104). Thus, within the weakly basic environment of the proximal intestine, a strongly acidic drug is less lipophilic, more ionized, and slower to be absorbed.
Conversely, for a strong basic compound with a pKa = 4 (pK
b = 10) in the stomach, the
Henderson–Hasselbach equation computes the ratio of ionized to non-ionized species at 100:1. Thus, a basic compound is more ionized, less lipophilic, and slower to be absorbed in the stomach. In the basic environment of the proximal intestine (pH = 8), however, the ionized to non-ionized species ratio is 1:104, rendering it more lipophilic and imparting a greater propensity for absorption.
The knowledge of the pKa of a chemical is useful in predicting its absorption in these
compartments. In addition, as explained below, this information is helpful in determining distribution and elimination functions. Tables 3.1 and 3.2 summarize the chemical proper-ties and behaviors of acidic, neutral, and basic chemicals and their ionization potential in the stomach and small intestine. Based on the extremely acidic environment of an empty stomach, acids or bases are either completely non-ionized and highly lipophilic or ionized and hydrophilic. In the basic environment of the small intestine, although some ionization is present, absorption of a neutral compound (weak acid) is favored over that of an acid.
2 Note that the absorption rate of the stomach is limited and is secondary to its digestion and churning functions.
TABLE 3.1Summary of Chemical Properties and Behavior in the Stomach Environment (pH = 2) for Acidic, Neutral, and Basic Compounds
Compound Properties Acidic Neutral (Weak Acid) Basic
pKa 3 7 10Non-ionic:ionic ratio 10:1 105:1 1:108
Acidic/basic nature Free acid, highly lipophilic
Non-ionic, lipophilic Protonated, hydrophilic, very low lipophilicity
Absorption Favorable Favorable Not favorable

28 PRINCIPLES OF TOXICOLOGY TESTING
Note that some toxicokinetic principles that govern chemical absorption are not always signifi cant factors in the determination of toxic effects. This is primarily due to the circumstances surrounding toxic chemical exposure. Consequently, some of the circum-stances that infl uence chemical absorption in a therapeutic setting are considered in the following text. Some of these factors include route of administration, presence of food in the intestinal tract, and gastric emptying time. Other circumstances such as product for-mulation, chemical and physical characteristics of a drug product, drug interactions, con-current presence of gastrointestinal diseases, individual categories of toxic exposure, and special circumstances that alter their effects are considered in later chapters.
ROUTE OF ADMINISTRATION
As outlined in chapter 2, the oral route of administration creates the most signifi cant barrier to absorption. The primary function of the stomach is mechanical and chemical digestion of food, whereas absorption is secondary. Consequently, several factors infl uence the transit and stability of a chemical in the stomach, thereby infl uencing the length of time the substance remains in the stomach environment. Naturally, this time factor precipitates a dynamic inter-action with, and reaction from, the stomach, in order to adjust to the changing contents. For instance, the time required for the newly presented contents to pass through the stomach on its transit to the duodenum is known as the gastric emptying time (GET). Thus the presence of food slows down the peristaltic movement of stomach activity, activates churning and digestion, and dilutes the contents of the stomach, all in an attempt to increase contact with the foodstuff. Ultimately, this delays chemical transit and increases GET—the net effect is a reduction in the ultimate absorption in the latter stages of the intestinal tract. In addition, the relative pH of the stomach increases and causes a compounding negative feedback inhibition of stomach churning and motility, which also results in the delay of gastric emptying. Thus, the longer the GET, the greater the duration of a chemical presence within the stomach, and the more susceptibility to gastric enzyme degradation and acid hydrolysis. In addition, pro-longed GET delays passage to and subsequent absorption in the intestinal tract.
ABSORPTION IN NASAL AND RESPIRATORY MUCOSA
Nasal Mucosa
The vestibule of the nasal cavity and nasopharynx is covered by olfactory epithelium. This mucous membrane lining consists of an extensive network of capillaries and pseudostratifi ed columnar epithelium interspersed with goblet cells. Inspired air enters the nasal conchae and
TABLE 3.2Summary of Chemical Properties and Behavior in the Small Intestine (pH = 8–10) Environment for Acidic, Neutral, and Basic Compounds
Compound Properties Acidic Neutral (Weak Acid) Basic
pKa 3 7 10Non-ionic:ionic ratio 10–5:1 to 10–7:1 10–1:1 to 10–3:1 10–2:1 to 1:1Acidic/basic nature Proton donor very low
lipophilicityWeak acid some ionization
low lipophilicitySome ionization
lipophilicAbsorption Not favorable Some absorption over
length of intestinal tractFavorable

TOXICOKINETICS 29
is warmed by circulating blood in the capillaries, while mucus secreted by the goblet cells moistens the air and traps airborne particles. Thus, several factors facilitate rapid availability and absorption from chemical exposure through the nasal route:
1. Vast surface areas provided by capillary circulation and epithelial cells in mucous membranes
2. Ability of the nasal mucosa to trap dissolved and particulate substances3. Phospholipid secretions of the nasal mucosa (resulting in a relatively neutral pH)
Non-ionized drugs are more rapidly absorbed in this compartment relative to their ion-ized counterparts.
Respiratory Mucosa
The upper and lower respiratory tracts extend from the nasal cavity to the lungs. The con-ducting portion responsible for ventilation consists of the pharynx, larynx, trachea, bronchi, bronchioles, and terminal bronchioles. The respiratory portion that serves as the main site for gas exchange consists of the respiratory bronchioles, alveolar ducts, alveolar sacs, and alve-oli. Most of the upper and lower tracts are covered by ciliated secretory epithelial cells.
Aside from the nasal epithelium, however, signifi cant absorption of airborne substances does not occur above the level of the respiratory bronchioles. As respiratory bronchioles penetrate deeply into the lungs, the epithelial lining changes from simple cuboidal to simple squamous, enhancing the surface absorptive capability of the mucosa. In addition, the thin 0.5-μm respiratory membrane is composed of 300 million alveoli and further supported by an extensive network of capillary endothelium (see Fig. 2.2). This anatomy allows rapid diffusion of chemicals released as gases within aerosols or carried by humidifi ed air.
ABSORPTION AND TRANSPORT THROUGH OCULAR, PLACENTAL, VAGINAL, AND RECTAL ROUTES
Specialized compartments and membranes, such as the ocular and placental membranes, and the vaginal and rectal routes, follow the same principles of ionic/non-ionic distribution in order to penetrate the biomembranes. For instance, the placenta is fully developed by the third month of pregnancy and is formed by the embryonic chorion (syncytial trophoblast) and the maternal endometrium (decidua basalis). Gases, vitamins, amino acids, small sugars, ions, as well as waste products diffuse across or are stored in the placental membrane. In addition, viruses and organic chemicals also access the fetus by virtue of their lipid-soluble properties. Thus, the same principles of ionization that govern absorption of chemicals infl uence absorp-tion, distribution, and passage through these barriers. Non-ionized, lipid-soluble substances gain access more easily than ionized, water-soluble materials. Interestingly, passage through to the fetal membranes does not depend on the number of placental layers formed as the fetus matures, but is a consequence of the physicochemical characteristics. In addition, among these tissues, biotransformation properties of the placenta are unique to this organ and contribute additional properties of detoxifi cation (see the section, “Biotransformation”).
TRANSPORT OF MOLECULES
Absorption of chemicals requires basic mechanisms of transporting molecules across cell membranes and fl uid barriers. These transport vehicles include diffusion, active transport, facilitated diffusion, and fi ltration.

30 PRINCIPLES OF TOXICOLOGY TESTING
The simplest mechanism for transportation of molecules involves diffusion, defi ned as the transport of molecules across a semipermeable membrane. The most common pathway of diffusion is passive transport by which molecules are transported from an area of high- to low-solute concentration, down the concentration gradient. This is not an energy- dependent process and no electrical gradient is generated. Lipophilic molecules, small ions, and elec-trolytes generally gain access through membrane compartments by passive diffusion because they are not repelled by the phospholipid bilayers of cell membranes. Most passive diffusion processes, however, are not molecularly selective.
In contrast to passive transport, polar substances such as amino acids, nucleic acids, and carbohydrates are moved by an active transport process. This mechanism shuttles ionic substances from areas of low-solute concentration to high-solute concentration, against a concentration gradient. Active transport is an energy-dependent process and requires mac-romolecular carriers. These two requirements render this process susceptible to metabolic inhibition and consequently a site for toxicological or pharmacological infl uence. In addi-tion, active transport processes are saturable systems, implying that they have a limited number of carriers, making them competitive and selective.
Facilitated diffusion resembles active transport with the exception that substrate move-ment is not compulsory against a concentration gradient and it is not energy-dependent. For example, macromolecular carriers augment the transfer of glucose across gastrointestinal epithelium through to the underlying capillary circulation.
Filtration allows for the movement of solutes across epithelial membrane barriers along with water movement. The motive force here is hydrostatic pressure. In general, the molecular weight cutoff for solutes transported along with water is about 100 to 200 mw units. Under normal circumstances, several membranes throughout the body with pores or fenestrations averaging 4 Å in diameter are designed to allow entry for larger molecules. These special membranes permit coarse fi ltration of lipophilic or hydrophilic substances. Such fi ltration processes are described for the renal nephron. The fi ltration slits formed by the interdigitating podocytes in the glomerular apparatus and Bowman’s capsule prevent the passage of cells and solutes above 45 kD. Under normal circumstances, most of the smaller molecules pass through the fi lter, only to be reabsorbed in the proximal and distal convoluted tubules and collecting tubules.
Many organs exhibit passive fi ltration as a fundamental process for the passage of water. Intestinal absorption of water involves a transport method by which water and dis-solved nutrients are coarsely fi ltered and absorbed through the intestinal lumen. At the tis-sue level, the inotropic action of the cardiac system is the driving force for capillary perfusion. The thin endothelial and epithelial membranes that form the walls of the pulmo-nary alveoli are the only barriers for the diffusion of gases into the alveolar spaces.
DISTRIBUTION
FLUID COMPARTMENTS
Anatomically, the body is not a homogeneous entity. Water occupies approximately 70% of total body weight—the physiological water compartments, therefore, play signifi cant roles in chemical distribution and in the chemical’s ultimate fate. Table 3.3 illustrates the composi-tion and contribution of water-based compartments relative to body weight. The ability of a compound to gain access to these compartments is determined by factors that infl uence the compound’s subsequent fate. The distribution of the substance to the target organ and the receptor site infl uences metabolic fate and excretion, such that the latter are reasonably

TOXICOKINETICS 31
predicted by such factors. These dynamic features include (1) physicochemical properties of a substance, such as lipid solubility and ionic or non-ionic principles; (2) binding to macro-molecules; and (3) physiological forces of blood fl ow and capillary perfusion. The coopera-tion of theses forces determines the relative bioavailability of a compound. These concepts are further discussed next.
IONIC AND NON-IONIC PRINCIPLES
The same mechanisms that govern absorption of molecules across semipermeable mem-branes also determine the bioavailability of a compound. Thus a compound with a propen-sity for a non-ionic existence is more likely to be lipophilic and thus has greater ability to penetrate phospholipid membranes. Thus the substance has greater access and more favor-able distribution to liver, kidney, muscle, adipose, and other membrane-bound visceral organs. Conversely, molecules that are highly polarized in plasma have diminished access to organs and tissues, because of their limited ability to penetrate charged membranes. As with absorption, pH and pK
a have similar infl uences over ionization, or the lack of ioniza-
tion, at the tissue level. Similarly, as an indicator of distribution to organs, the apparent volume of distribution (V
d) is used as a measure of the amount of chemical in the entire
body (mg/kg) relative to that in the plasma (mg/L) over a specifi ed period. The following formula defi nes the relationship as:
Vd (L/kg) = dose (mg/kg)/plasma concentration (mg/L)
Thus, Vd is useful in estimating the extent of distribution of a substance in the body.
Table 3.4 shows human Vd values, in increasing order, for 25 chemicals whose toxicity is
well established. Examination of these numbers indicates that a compound with a large Vd
(e.g., 19.0, D-propoxyphene HCl) is highly lipophilic and distributes throughout tissue compartments. This kinetic property explains why D-propoxyphene HCl has a special affi nity for the central nervous system. Thus, a compound with a high V
d necessarily
expresses a low-plasma concentration relative to dose. Conversely, a compound with a low V
d (e.g. 0.4 for caffeine) is highly bound to circulating plasma proteins and thus has a
plasma concentration 2.5-fold of the mean total body dose.Another criterion for estimating bioavailability is the calculation of the area under the
plasma concentration versus time curve (shaded area, AUC, Fig. 3.2). Dividing AUC, follow-ing an oral dose, by AUC, following an intravenous dose, and multiplying by 100 yields
TABLE 3.3Compartments and Their Relative Percentages of Total Body Weight
Compartment DescriptionRelative %
of Body Weight
Plasma water (PL) Noncellular water component of blood 10%Interstitial water (IS) Fluids relating to the interstices (spaces) of tissue 15%Extracellular water (EC) PL + IS 25%Intracellular water (IC) Confi ned to cytosol (within cells) 45%Total body water (TBW) EC + IC 70%Solids Tissue binding capacity 30%Total body weight Solids + TBW 100%
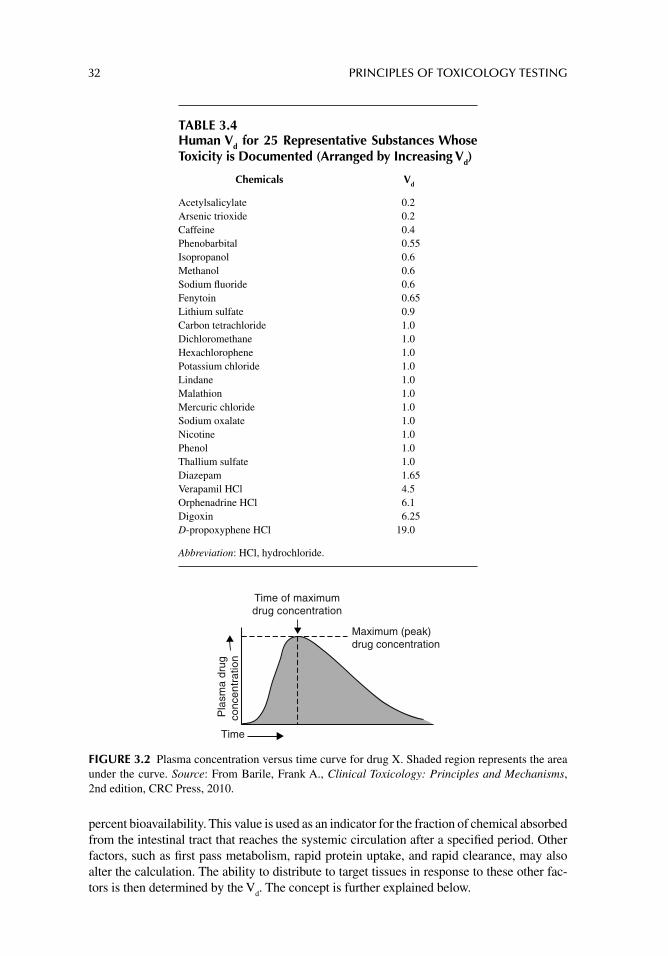
32 PRINCIPLES OF TOXICOLOGY TESTING
percent bioavailability. This value is used as an indicator for the fraction of chemical absorbed from the intestinal tract that reaches the systemic circulation after a specifi ed period. Other factors, such as fi rst pass metabolism, rapid protein uptake, and rapid clearance, may also alter the calculation. The ability to distribute to target tissues in response to these other fac-tors is then determined by the V
d. The concept is further explained below.
TABLE 3.4Human Vd for 25 Representative Substances Whose Toxicity is Documented (Arranged by Increasing Vd)
Chemicals Vd
Acetylsalicylate 0.2Arsenic trioxide 0.2Caffeine 0.4Phenobarbital 0.55Isopropanol 0.6Methanol 0.6Sodium fl uoride 0.6Fenytoin 0.65Lithium sulfate 0.9Carbon tetrachloride 1.0Dichloromethane 1.0Hexachlorophene 1.0Potassium chloride 1.0Lindane 1.0Malathion 1.0Mercuric chloride 1.0Sodium oxalate 1.0Nicotine 1.0Phenol 1.0Thallium sulfate 1.0Diazepam 1.65Verapamil HCl 4.5Orphenadrine HCl 6.1Digoxin 6.25D-propoxyphene HCl 19.0
Abbreviation: HCl, hydrochloride.
Time of maximumdrug concentration
Maximum (peak)drug concentration
Pla
sma
drug
conc
entr
atio
n
Time
FIGURE 3.2 Plasma concentration versus time curve for drug X. Shaded region represents the area under the curve. Source: From Barile, Frank A., Clinical Toxicology: Principles and Mechanisms, 2nd edition, CRC Press, 2010.
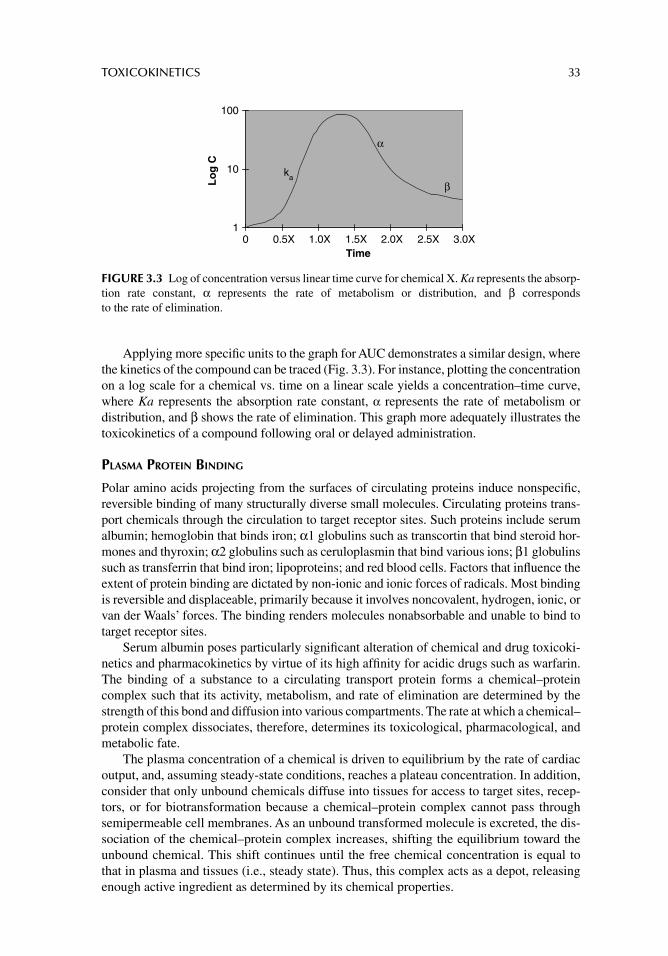
TOXICOKINETICS 33
Applying more specifi c units to the graph for AUC demonstrates a similar design, where the kinetics of the compound can be traced (Fig. 3.3). For instance, plotting the concentration on a log scale for a chemical vs. time on a linear scale yields a concentration–time curve, where Ka represents the absorption rate constant, α represents the rate of metabolism or distribution, and β shows the rate of elimination. This graph more adequately illustrates the toxicokinetics of a compound following oral or delayed administration.
PLASMA PROTEIN BINDING
Polar amino acids projecting from the surfaces of circulating proteins induce nonspecifi c, reversible binding of many structurally diverse small molecules. Circulating proteins trans-port chemicals through the circulation to target receptor sites. Such proteins include serum albumin; hemoglobin that binds iron; α1 globulins such as transcortin that bind steroid hor-mones and thyroxin; α2 globulins such as ceruloplasmin that bind various ions; β1 globulins such as transferrin that bind iron; lipoproteins; and red blood cells. Factors that infl uence the extent of protein binding are dictated by non-ionic and ionic forces of radicals. Most binding is reversible and displaceable, primarily because it involves noncovalent, hydrogen, ionic, or van der Waals’ forces. The binding renders molecules nonabsorbable and unable to bind to target receptor sites.
Serum albumin poses particularly signifi cant alteration of chemical and drug toxicoki-netics and pharmacokinetics by virtue of its high affi nity for acidic drugs such as warfarin. The binding of a substance to a circulating transport protein forms a chemical–protein complex such that its activity, metabolism, and rate of elimination are determined by the strength of this bond and diffusion into various compartments. The rate at which a chemical–protein complex dissociates, therefore, determines its toxicological, pharmacological, and metabolic fate.
The plasma concentration of a chemical is driven to equilibrium by the rate of cardiac output, and, assuming steady-state conditions, reaches a plateau concentration. In addition, consider that only unbound chemicals diffuse into tissues for access to target sites, recep-tors, or for biotransformation because a chemical–protein complex cannot pass through semipermeable cell membranes. As an unbound transformed molecule is excreted, the dis-sociation of the chemical–protein complex increases, shifting the equilibrium toward the unbound chemical. This shift continues until the free chemical concentration is equal to that in plasma and tissues (i.e., steady state). Thus, this complex acts as a depot, releasing enough active ingredient as determined by its chemical properties.
1
10
100
0 0.5X 1.0X 1.5X 2.0X 2.5X 3.0XTime
α
kaβL
og
C
FIGURE 3.3 Log of concentration versus linear time curve for chemical X. Ka represents the absorp-tion rate constant, α represents the rate of metabolism or distribution, and β corresponds to the rate of elimination.

34 PRINCIPLES OF TOXICOLOGY TESTING
Chemicals that are extensively bound to circulating or tissue proteins pose signifi cant toxicological risks. Toxic effects of such chemicals extend well beyond acute exposures. Chronic and subchronic exposure to lead, for instance, has signifi cant long-term neuro-logical consequences by virtue of its affi nity for depositing in bone. Therapeutically, steady-state concentrations of warfarin are easily disrupted when another acidic drug such as acetylsalicylic acid (aspirin) displaces it from circulating albumin binding sites. Since warfarin is extensively protein bound (97%), a displacement of as little as 1% can double the amount of warfarin available for interaction with target organs, resulting in signifi cant adverse hemodynamic events. Consequently, displacement of protein-bound chemicals results in increased unbound chemical concentration in plasma and increased toxicological and pharmacological effects, followed by enhanced metabolism.3
LIPIDS
Distribution of chemicals is also determined in part by their lipid/water partition coeffi cients. Two chemicals with similar pK
a values such as amphetamine (9.8) and chlorpromazine (9.3)
have different lipid affi nities in their non-ionized forms. At basic pH (8 to 9), both chemicals are completely non-ionized. However, at pH 10 or above, the partition coeffi cient of chlor-promazine in an organic solvent (K
d = 106.3) is 103.5 times greater than that of amphetamine
(Kd = 101.8), allowing for greater absorption of the former in the contents of the intestinal tract.
Similarly, the polychlorinated biphenyls and the contact insecticides such as DDT have high lipid solubilities, dissolve in neutral fats, and thus readily accumulate in adipose tissue.
LIVER AND KIDNEY
The high capacities of the liver and kidney for binding and concentrating toxicants, coupled with their extensive circulation and metabolic activities, render these organs remarkably susceptible to chemical attack. Further discussion of the metabolic roles that the liver and kidney play in chemical detoxifi cation are addressed below.
BLOOD–BRAIN BARRIER
Formed by the tight junctions of capillary endothelial cells and surrounded by basement membrane and an additional layer of neuroglial cells, the blood–brain barrier (BBB) imposes a formidable hindrance to the passage of molecules. Although often misconstrued as an anatomical structure, it is primarily a physiologically functional entity, selectively allowing passage of a few water-soluble molecules, ions, and glucose (by active transport). It is suf-fi ciently impermeable to most compounds including high-molecular-weight proteins and chemicals. Lipid-soluble elements such as oxygen, carbon dioxide, alcohol, anesthetics, and the opioid analgesics enjoy relatively easy access to the central nervous system via the intact BBB. When the BBB loses its barrier function, as occurs with bacterial or viral encephalitis, the compromised membrane is then susceptible to unregulated entry.
PLACENTA
The placenta is fully developed by the third month of pregnancy and is formed by the embry-onic chorion (syncytial trophoblast) and the maternal endometrium (decidua basalis). Although gases, vitamins, amino acids, small sugars, ions, and waste products diffuse across
3 It is important to note that the bioavailability of a chemical at a receptor site is infl uenced more by rate of diffu-sion (V
d ) than by rate of dissociation. This is especially true when the displacement of an acidic chemical with a
small apparent V d displays toxicologically important consequences.

TOXICOKINETICS 35
or are stored in the placental membrane, viruses and lipid-soluble chemicals have relative easy access to the fetus. Thus, the same principles of ionization that govern absorption of chemicals infl uence distribution and passage through the placenta. Non-ionized lipid-soluble substances penetrate more easily than ionized water-soluble materials. In addition, passage through the fetal membranes does not depend on the number of placental layers formed as a fetus matures, but is directed by the complexity of the circulatory pathways formed between maternal and fetal systems. Biotransformation properties of the placenta are addressed next.
OTHER FACTORS THAT AFFECT DISTRIBUTION
Physiological distribution of a drug or chemical is infl uenced by a variety of dynamic fea-tures interacting to achieve equilibrium. Regardless of concentration, when a drug meets the two minimum conditions for distribution, that is, relatively small molecular weight and high water solubility, then the main requirements for favorable absorption and distribution are satisfi ed. If molecular size and solubility were the only important criteria, however, then most drugs would traverse biomembranes without the prerequisite of carrier proteins or active transport. In addition, the presence of charges on drug molecules affects ionization, association, and dissociation, eventually resulting in equivocal passage through membranes. Consequently, other factors that shape distribution of chemicals into physiological compart-ments are listed and briefl y described in Table 3.5. These include the pH partition principle,
TABLE 3.5Other Factors That Affect Distribution
Factors Description Consequence
pH partition principle Difference in pH between two compartments infl uences non-ionic diffusion of weakly acidic or basic drugs
Concentration of non-ionized form only is equally distributed between two compartments, whereas concentration of ionized moiety depends on pH differential
Electrochemical potential
Positively charged outer membrane and negatively charged inner cell membrane affect migration of an ionized substance
Higher concentration of cations, for example, once inside the cell, remain inside, and thus are moving along the electrochemical gradient (electrochemical potential)
Donnan distribution (equilibrium)
Presence of an impermeable charged macromolecule on one side of a cell membrane will alter permeability of other ionized particles
Molecules with the same charge as the impermeable macromolecule are committed to the opposite side of the membrane—due to mutual ionic repulsion (or Donnan equilibrium)
Binding and storage Drugs bound to intracellular or circulating macromolecules cannot negotiate partitioning among physiological compartments
Partitioning is determined by binding capacity and binding constant (K), as long as the concentration of the drug is not increased beyond K
Non-equilibrium and redistribution
True equilibrium among compartments is physiologically nonexistent; equilibrium between plasma and other compartments (e.g., fat, liver, muscle) does not occur at equal rates
This non-equilibrium exists because of redistribution to the rate-limiting compartment (e.g., fat) and is affected by biotransformation, elimination, and the practicality of dosing intervals

36 PRINCIPLES OF TOXICOLOGY TESTING
electrochemical and Donnan distribution effect, binding and storage, and non-equilibrium and redistribution. These principles are not functionally independent of other pressures but contribute to the understanding of distribution kinetics. A detailed discussion of these factors is found in the suggested readings and review articles.
BIOTRANSFORMATION
PRINCIPLES OF DETOXIFICATION
Biotransformation is defi ned as the modifi cation of an endogenous or exogenous chemical (xenobiotic) in a biological system.4 Xenobiotic transformation plays a crucial role in main-taining homeostasis during chemical exposure. This is accomplished primarily by converting lipid-soluble (nonpolar) nonexcretable xenobiotics to polar water-soluble compounds, ren-dering them accessible for elimination in the bile and/or urine. The outcomes of biotransfor-mation include facilitation of excretion, conversion of toxic parent compounds to nontoxic metabolites (detoxifi cation), and conversion of nontoxic or nonreactive parent compounds to toxic or reactive metabolites (bioactivation).
Detoxifi cation of substances by combination with the bile is part of the fi rst pass phe-nomenon. This process renders the exogenous chemical inactive by conjugation with bile acids. The conjugated chemicals are then selectively excreted into the gall bladder. Table 3.6 lists agents that are selectively captured, conjugated, and excreted in the bile. In general, oral exposure to compounds with higher bile-to-plasma ratio (Class B, Table 3.6) have a greater propensity to distribute to the bile acid compartment.
PHASE I REACTIONS
Phase I reactions involve hydrolysis, reduction, and oxidation of chemicals to more hydro-philic, usually smaller, entities. The most important enzymes utilized in hydrolysis phase I reactions include carboxylesterase, azo- and nitroreductases, and quinone reductases. Carbo-xylesterases cleave carboxylic acid esters, amide, or thioesters. Azo- and nitro-reduction reactions incorporate two enzymes: azoreductases (—N=N—) from intestinal fl ora, and liver cytochrome P450 reductases. Quinone reduction reactions involve quinone reductases and NADPH cytochrome P450 reductases. The former are involved in two-electron reduction reactions (detoxifi cation) and the latter in one-electron reduction reactions (bioactivation).
Oxidative enzymes include dehydrogenases and cytochrome P450 oxidases. These ubiquitous enzymes are responsible for detoxifi cation of a variety of exogenous substances. For example, alcohol dehydrogenase (ADH) and aldehyde dehydrogenases convert alco-hols to the corresponding aldehydes and acids, respectively.
4 Exogenous chemicals are often referred to as xenobiotics when foreign substances access biological systems.
TABLE 3.6Bile-to-Plasma Ratio of Representative Xenobiotics
Classifi cationBile-to-Plasma
Ratio Representative Compounds
Class A 1:1 → 10:1 Na+, K+, glucoseClass B 10:1 → 1000:1 Arsenic, bile acids, lidocaine, phenobarbitalClass C Less than 1:1 Albumin, metals (zinc, iron, gold, chromium)

TOXICOKINETICS 37
Among phase I biotransformation enzymes, the cytochrome P450 (designated CYP) superfamily of enzymes is the most versatile and ubiquitous, accounting for 60% of drug and chemical biotransformation. The enzyme system is predominantly composed of mono-oxygenase, heme-containing proteins, and are associated mostly with the hepatic micro-somal (liver homogenate fraction) of the smooth muscle endoplasmic reticulum, as well as with mitochondria. The enzymes are classifi ed into families, subfamilies, or subtypes of subfamilies, according to their amino acid sequences. They catalyze the incorporation of one atom of oxygen, or facilitate the removal of one atom of hydrogen, into or from the substrate, respectively. Other types of enzymes involved in the P450 system are hydroxy-lases, dehydrogenases, esterases, N-dealkylation and oxygen transferases. Examples of phase I reactions are noted in Table 3.7.
PHASE II REACTIONS
In general, phase II reactions usually follow phase I reactions with glucuronidation, sulfa-tion, acetylation, methylation, and conjugation of the molecule. These reactions usually occur by interacting amino acids with the phase I metabolites. Phase II enzymes act on hydroxyl, sulfhydril, amino, and carboxyl functional groups present on either the parent compound or the phase I metabolite. Therefore, if a functional group is already present on the parent compound, phase I reactions are circumvented.5
5 For instance, because of the presence of hydroxyl groups, morphine undergoes direct phase II conversion to morphine-3-glucuronide, whereas codeine must endure transformation with both phases I and II .
TABLE 3.7Predominant Biotransformation Pathways and Their Substrates
Biotransformation Pathway Reactions Drug or Chemical Substrates
Phase I Reactions Oxidation Amitriptyline; erythromycin; dextromethorphan; indomethacin; barbiturates; cyclosporine; chlorpheniramine; APAP; amphetamines
Dealkylation, deamination, N-oxidation
Diazepam; tamoxifen; theophylline
Reduction ChloramphenicolHydrolysis ACE inhibitors; local anesthetics
(e.g., cocaine, lidocaine, procaine)Dehalogenation Halogenated general anesthetics;
aromatic hydrocarbon gases (e.g., lacrimating agents)
Phase II Reactions Glucuronidation Sulfonamide antibiotics; APAP; morphine; THC; chloramphenicol
Sulfation Estrogens; methyldopa; APAP
Acetylation Sulfonamide antibiotics; procainamide; sulfonamides; isoniazid; clonazepam
With glutathione APAP; ROS/RNS
Abbreviations: ACE, angiotensin-converting enzyme; APAP, acetyl para-amino phenol (acetaminophen); RNS, reactive nitrogen species; ROS, reactive oxygen species; THC, tetrahydrocanabinol.

38 PRINCIPLES OF TOXICOLOGY TESTING
Phase II biotransformation enzymes include UDP-glucuronosyltransferases, sulfo-transferases, acetyltransferases, methyltransferases, and glutathione S-transferases. Except for acetylation and methylation reactions, phase II reactions, along with enzyme cofactors, interact with different functional groups to increase hydrophilic properties. Glucuronida-tion is the main pathway for phase II reactions. It converts nonpolar, lipid-soluble com-pounds to polar, water-soluble metabolites. Sulfation reactions involve the transfer of sulfate groups from PAPS6 to the xenobiotic, yielding sulfuric acid esters that are excreted into urine.
Glutathione (GSH) conjugation is responsible for detoxifi cation (or bioactivation) of xenobiotics. GSH conjugates are excreted intact in bile, or converted to mercapturic acid in kidney and excreted in urine.
Table 3.7 lists representative drugs and chemicals involved primarily with phase I and/or phase II biotransformation reactions. Table 3.8 lists other frequently cited therapeutic and nontherapeutic agents that are predominantly metabolized via the cytochrome P450 enzyme system.
ENZYME SYSTEMS
P450 Enzyme Pathways
Among phase I biotransformation enzymes, the cytochrome P450 system is resourceful and adaptable. The enzymes are predominantly mono-oxygenase, heme-containing proteins, located primarily in liver microsomes and mitochondria. The superfamily system is catego-rized according to its amino acid sequence. The enzymes catalyze the incorporation of one atom of oxygen or facilitate the removal of one atom of hydrogen within the interaction with the substrate. Other types of enzymes involved in the P450 system are hydroxylases, dehy-drogenases, esterases, N-dealkylation and oxygen transferases.
Phase II biotransformation enzymes include UDP-glucuronosyltransferase, sulfotrans-ferases, acetyltransferases, methyltransferases, and glutathione S-transferases. Except for acetylation and methylation reactions, phase II reactions incorporate enzyme cofactors to facilitate interaction with different functional groups, thereby increasing hydrophilic prop-erties. Glucuronidation is the main pathway for phase II reactions; it is responsible for converting nonpolar lipid-soluble compounds to polar water-soluble metabolites. Sulfation reactions transfer sulfate groups from PAPS to the xenobiotic, yielding water-soluble, urine-bound sulfuric acid esters.
Glutathione Conjugation and Other Oxidative–Reductive Reaction Systems
GSH conjugation is responsible for detoxifi cation (or bioactivation) of xenobiotics. GSH conjugates are excreted intact in bile or converted to mercapturic acid in the kidneys and eliminated in the urine.
Other enzymes used in hydrolysis phase I reactions include carboxylesterase, azo- and nitroreductases, and quinone reductases. Carboxylesterases cleave carboxylic acid esters, amides, and thioesters. Azo- and nitroreduction reactions incorporate two enzymes: azore-ductases (–N=N–) from intestinal fl ora and liver cytochrome P450 reductases. Quinone reduction reactions involve quinone reductases and NADPH cytochrome P450 reductases. The former are involved in two-electron reduction reactions (detoxifi cation) and the latter in one-electron reduction reactions (bioactivation).
6 3-phosphoadenosine-5-phosphosulfate

TOXICOKINETICS 39
TABLE 3.8Examples of Chemical Changes That Occur with Phase I Reactions
Type of Reaction Process Substrate Metabolite (s)
Oxidation Aromatic hydroxylation R R HO
Aliphatic hydroxylation CH3R H2CR OH
Epoxidation HCR
HC R'
H2 H2CR O C R'
N-hydroxylation NH2HN OH
O-dealkylation OR CH3 OHH
+R HC O
N-dealkylationR CH3
HN
NH2H
+R HC O
S-dealkylation R CH3S SHR HC OH
+
DeaminatonR CH3
HC
NH2
CR NH3+O
CH3
S-oxidation SR R' SR R'
O
Dechlorination CCl4 CHCl3
Amine oxidation H2CR NH2
HCR NH3+O
Dehydrogenation H2CR OH
CR H++O
H
Reduction Azo- NR N R' R 'R+NH2 NH2
Nitro- NO2R NH2R
Carbonyl- R R'C
O
HCR R'
OH
Hydrolysis Ester- C OR
O
R' CR 'R+O
OHOH
Amide CR NH2
O
CR +O
OHNH3
R = aromatic, alkyl, allyl, or other long-chain hydrocarbon.

40 PRINCIPLES OF TOXICOLOGY TESTING
Oxidative enzymes include ADHs and cytochrome P450 oxidases. For example, ADH and aldehyde dehydrogenases convert alcohols to corresponding aldehydes and acids, respectively.
ELIMINATION
URINARY EXCRETION
Urinary excretion represents the most important route of chemical, drug, and waste elimina-tion in the body. Normal glomerular fi ltration rate (GFR, ~110–125 mL/min) is determined by the endothelial fenestrations (pores) formed by the visceral layers of the specialized endothelial cells of the glomeruli. The pores are coupled with the overlying glomerular base-ment membranes and the fi ltration slits of the pedicels. This three-layer membrane permits the passage of noncellular materials, with a molecular weight cutoff of ~45–60K. The fi ltrate is then collected in Bowman’s capsule. Reabsorption follows in the proximal convoluted tubules, loop of Henle, and distal convoluted tubules. The collecting ducts have limited reab-sorption and secretion capabilities. Control of the transfer of molecules back to the capillary circulation or elimination in the fi nal fi ltrate depends predominantly on reabsorption and secretion processes.
As with absorption through biological membranes and distribution in physiological compartments, urinary elimination of toxicants is guided by ionic and non-ionic principles. While Na+ and glucose are actively transported or facilitated by symporters across the proximal tubules, other larger polar compounds are prevented from reabsorption to the capillary circulation and are eventually eliminated in the urine. Non-ionic nonpolar com-pounds are transported by virtue of their ability to move across phospholipid membranes. Clinically, these principles are somewhat susceptible to manipulation for reducing chemi-cal toxicity. For instance, it has long been proposed that treatment of overdoses of barbitu-rates (pKα values between 7.2 and 7.5) may be effectively treated with sodium bicarbonate. The mechanism of this therapeutic intervention relies, in theory, on the alkalinization of the urine, rendering the acidic drug ionic, water soluble, and more amenable to elimination. In practice, however, the buffering capacity of the plasma coupled with a large dose of the sedative or hypnotic makes this a treatment modality of last resort.
Other organic ions undergo active tubular secretion. Organic anion transporter (oat) proteins (organic acid transporters) and organic cation transporter (oct) proteins (organic base transporters) both actively secrete polar molecules from capillaries to the proximal convoluted tubule lumen. Some classic examples include the active tubular secretion of para-aminohippuric acid by oat proteins as well as sulfonamide antibiotics and methotrex-ate that compete for the same secretory pathways. Probenecid, a uricosuric agent used in the treatment of gout, competes for oat proteins as part of its therapeutic mechanism. While small doses of the drug actually depress the excretion of uric acid by interfering with tubu-lar secretion but not reabsorption, larger doses depress the reabsorption of uric acid and lead to increased excretion and decreases in serum levels. Similarly, small doses of proben-ecid decrease the renal excretion of penicillins that compete with the same oat proteins, necessitating adjustment of antibiotic dosage regimens.
FECAL ELIMINATION
With the exception of cholestyramine, a cholesterol-binding drug, and the herbicide para-quat, the presence of nonabsorbable, nontransformed xenobiotics in the feces is rare. This

TOXICOKINETICS 41
is because many metabolites of compounds formed in the liver are excreted into the intes-tinal tract via the bile. Furthermore, organic anions and cations are biotransformed and actively transported into bile by carrier mechanisms similar to those in the renal tubules.
Chemicals ingested via oral exposure enter the hepatic portal system and liver sinusoids, only to be extensively cleared by the liver. This toxicokinetic phenomenon is known as hepatic fi rst pass elimination. According to the principles of elimination, compounds may be classifi ed based on their bile-to-plasma (B:P) ratios. Table 3.5 outlines the classifi cations of representative chemicals based on this ratio. Substances with high B:P ratios are readily eliminated in bile. Drugs such as phenobarbital are extensively removed by the liver, which explains why the oral bioavailability of these drugs is low despite complete absorption. Other compounds, such as lidocaine, a local anesthetic, are extensively eliminated by fi rst pass elimination and thus oral administration is precluded. Compounds with low B:P values are not subject to this phenomenon and maintain relatively higher plasma and other compartmen-tal concentrations.
PULMONARY ELIMINATION
Most gases, aerosolized molecules, and volatile liquids are eliminated by simple diffusion via exhalation. The rate of pulmonary elimination is inversely proportional to the blood solubility of a compound. Thus, compounds with low blood solubility, such as ethylene dioxide, have high vapor pressure levels and are readily volatilized and eliminated as pulmo-nary effl uents. Conversely, compounds with high blood solubility levels such as ethanol, carbon tetrachloride, and chloroform have low vapor pressures. Their elimination through the lungs is more a function of concentration than of solubility. Similarly, lipid-soluble gen-eral anesthetic agents have long half-lives in plasma because of their low vapor pressures and high plasma solubilities.
MAMMARY GLAND SECRETION
The mammary glands are modifi ed sudoriferous glands that produce a lipid-laden secre-tion (breast milk) that is excreted during lactation. The lobules of the glands consist of alveoli surrounded by one or two layers of epithelial and myoepithelial cells anchored to well-developed basement membranes. Unlike the BBB and placenta, these cells do not interpose a signifi cant barrier to transport of molecules. In addition, breast milk is a watery secretion with a signifi cant lipid component. Consequently, both water- and lipid-soluble substances are transferred through the breast milk. In particular, basic chemicals readily accumulate in the weakly acidic or alkaline environment of breast milk (pH 6.5–7.5). Accordingly, nursing infants are exposed to almost any substance present in the maternal circulation.
OTHER SECRETIONS
Elimination of chemicals through saliva, tears, hair follicles, and sudoriferous (watery) or sebaceous (oily) sweat glands is a function of plasma concentration. Elimination is also infl uenced by liver metabolism, passage through the intestinal tract and pulmonary system, and by limited access to local glandular secretions. Thus, secretion of com-pounds through mucous, ophthalmic, and dermal glands is not signifi cant. In forensic toxicology, however, certain chemicals may be detected in minute concentrations in hair follicles and nails when plasma levels seem to be nonexistent. Chronic arsenic and lead poisoning have been detected by measuring the metals bound to hair follicles and hair shafts.

42 PRINCIPLES OF TOXICOLOGY TESTING
SUGGESTED READINGS
Kauffman FC. Sulfonation in pharmacology and toxicology Drug Metab Rev. 2004; 36: 823.Klaassen CD, Slitt AL. Regulation of hepatic transporters by xenobiotic receptors. Curr Drug Metab
2005; 6: 309.Medinsky MA, Valentine JL. Principles of toxicology. In: Klaassen CD, ed. Casarett and Doull’s
Toxicology: The Basic Science of Poisons, 6th edn. Ch. 7 New York: McGraw-Hill, 2001.Norinder U, Bergstrom CA. Prediction of ADMET properties. Chem Med Chem 2006; 1: 920.Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer.
Oncogene 2006; 25: 1679.Strolin-Benedetti M, Whomsley R, Baltes EL. Differences in absorption, distribution, metabolism
and excretion of xenobiotics between the paediatric and adult populations. Expert Opin Drug Metab Toxicol 2005; 1: 447.
Urso R, Blardi P, Giorgi G. A short introduction to pharmacokinetics. Eur Rev Med Pharmacol Sci 2002; 6: 33.
Wijnand HP. Pharmacokinetic model equations for the one- and two-compartment models with fi rst-order processes in which the absorption and exponential elimination or distribution rate
constants are equal. J Pharmacokinet Biopharm 1988; 16: 109.
REVIEW ARTICLES
Barile FA, Dierickx PJ, Kristen U. In vitro cytotoxicity testing for prediction of acute human toxicity. Cell Biol Toxicol 1994; 10: 155.
Bell LC, Wang J. Probe ADME and test hypotheses: a PATH beyond clearance in vitro-in vivo cor-relations in early drug discovery. Expert Opin Drug Metab Toxicol 2012; 8: 1131–55.
Bodo A, Bakos E, Szeri F, Váradi A, Sarkadi B. The role of multidrug transporters in drug availability, metabolism and toxicity. Toxicol Lett 2003; 140: 133.
Buchanan JR, Burka LT, Melnick RL. Purpose and guidelines for toxicokinetic studies within the National Toxicology Program. Environ Health Perspect 1997; 105: 468.
Cascorbi I. Genetic basis of toxic reactions to drugs and chemicals. Toxicol Lett 2005; 162: 16.Castellino S, Groseclose MR, Wagner D. MALDI imaging mass spectrometry: Bridging biology and
chemistry in drug development. Bioanalysis 2011; 3: 2427.Dalvie D. Recent advances in the applications of radioisotopes in drug metabolism, toxicology and
pharmacokinetics. Curr Pharm Des 2000; 6: 1009.El-Tahtawy AA, Tozer TN, Harrison F, Lesko L, Williams R. Evaluation of bioequivalence of highly
variable drugs using clinical trial simulations II. Comparison of single and multiple-dose trials using AUC and Cmax. Pharm Res 1998; 15: 98.
Gamage N, Barnett A, Hempel N, et al. Human sulfotransferases and their role in chemical metabo-lism. Toxicol Sci 2005; 90: 5.
Henchoz Y, Bard B, Guillarme D, et al. Analytical tools for the physicochemical profi ling of drug candidates to predict absorption/distribution. Anal Bioanal Chem 2009; 394: 707.
Hsieh Y, Chen J, Korfmacher WA. Mapping pharmaceuticals in tissues using MALDI imaging mass spectrometry. J Pharmacol Toxicol Methods 2007; 55: 193.
Ikeda M. Application of biological monitoring to the diagnosis of poisoning. J Toxicol Clin Toxicol 1995; 33: 617.
Ioannides C, Lewis DF. Cytochromes P450 in the bioactivation of chemicals. Curr Top Med Chem 2004; 4: 1767.
Ishii K, Katayama Y, Itai S, Ito Y, Hayashi H. A new pharmacokinetic model including in vivo dis-solution and gastrointestinal transit parameters. Biol Pharm Bull 1995; 18: 882.
Krzyzanski W, Jusko WJ. Mathematical formalism for the properties of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm 1997; 25: 107.
Lacey LF, Keene ON, Duquesnoy C, Bye A. Evaluation of different indirect measures of rate of drug absorption in comparative pharmacokinetic studies. J Pharm Sci 1994; 83: 212.

TOXICOKINETICS 43
Lin JH. Species similarities and differences in pharmacokinetics. Drug Metab Dispos 1995; 23: 1008.Lock EA, Smith LL. The role of mode of action studies in extrapolating to human risks in toxicology.
Toxicol Lett 2003; 140: 317.Luo Y, Liu C, Qu Y, Fang N. Towards single-cell analysis for pharmacokinetics. Bioanalysis 2012;
4: 453.Mahmood I, Miller R. Comparison of the Bayesian approach and a limited sampling model for the
estimation of AUC and Cmax: a computer simulation analysis. Int J Clin Pharmacol Ther 1999; 37: 439.
Matthies M. Exposure assessment of environmental organic chemicals at contaminated sites: a mul-ticompartment modelling approach. Toxicol Lett 2003; 140: 367.
Meineke I, Gleiter CH. Assessment of drug accumulation in the evaluation of pharmacokinetic data. J Clin Pharmacol 1998; 38: 680.
Myllynen P, Pasanen M, Pelkonen O. Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta 2005; 26: 361.
Perri D, Ito S, Rowsell V, Shear NH. The kidney: the body’s playground for drugs: an overview of renal drug handling with selected clinical correlates. Can J Clin Pharmacol 2003; 10: 17.
Plusquellec Y, Courbon F, Nogarede S. Consequence of equal absorption, distribution and/or elimina-tion rate constants. Eur J Drug Metab Pharmacokinet 1999; 24: 197.
Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 2002; 91: 1358.
Ramanathan M. Pharmacokinetic variability and therapeutic drug monitoring actions at steady state. Pharm Res 2000; 17: 589.
Repetto MR, Repetto M. Concentrations in human fl uids: 101 drugs affecting the digestive system and metabolism. J Toxicol Clin Toxicol 1999; 37: 1.
Rogers JF, Nafziger AN, Bertino JS Jr. Pharmacogenetics affects dosing, effi cacy, and toxicity of cytochrome P450-metabolized drugs. Am J Med 2002; 113: 746.
Sugiura Y, Setou M. Imaging mass spectrometry for visualization of drug and endogenous metabo-lite distribution: toward in situ pharmacometabolomes. J Neuroimmune Pharmacol 2010; 5: 31.
Trim PJ, Francese S, Clench MR. Imaging mass spectrometry for the assessment of drugs and metab-olites in tissue. Bioanalysis 2009; 1: 309.
van den Berg H, van den Anker JN, Beijnen JH. Cytostatic drugs in infants: a review on pharmacoki-netic data in infants. Cancer Treat Rev 2012; 38: 3.

44
4 Risk assessment and regulatory toxicology
INTRODUCTION
The U.S. Food and Drug Administration (U.S.FDA) is responsible for the approval of thera-peutic drugs only after they are determined to be safe for use in the conditions for which they have been tested. The conditions (indications) are part of the effectiveness requirements out-lined on the labeling (“package insert”) marketed with the drug. Other regulatory agencies, such as the Consumer Products Safety Commission (CPSC) and Environmental Protection Agency (EPA) are responsible for safety, production, marketing, and overall exposure to non-therapeutic chemicals, consumer products, as well as monitoring of environmental and occu-pational standards associated with these commodities, respectively. Consequently, modern concepts of risk management are based on the foundation that the pharmaceutical and chemi-cal industries, health care professionals, regulatory agencies, as well as patients and consum-ers, have a responsibility to minimize the risks associated with these activities. Risk management, therefore, depends on the nature of the product as established historically. In general, for pharmaceutical products manufactured in the United States, the risk management process is categorized into four key elements: (1) risk assessment (RA), (2) risk quantifi cation, (3) development and implementation of risk management tools, and (4) evaluation of the effec-tiveness of these tools. This chapter will concentrate on RA principally because this broad topic has applications to the safety and effi cacy of most other consumer products, particularly those that are relevant to toxicological risk evaluation. In addition, RA is involved with maintenance of environmental and occupational regulations. Accordingly, the following sections categorize and discuss RA with particular respect to understanding (1) hazard identifi cation, (2) dose–response assessment, (3) exposure assessment, and (4) risk characterization.
RISK ASSESSMENT
RA is a complex process aimed at determining, evaluating, and predicting the potential for harm to a defi ned community from exposure to chemicals and their by-products. The toler-ance of chemicals in the public domain is initially determined by the need for such chemi-cals, by the availability of alternative methods for obtaining the desired qualities of the chemicals, and by the economic impact of the presence or absence of the chemical agents on the economic status of the region.
For instance, over the last few decades, the amount and frequency of use of pesticides and herbicides have increased exponentially, especially in the agricultural and commer-cial industries. Use of these chemicals has resulted in signifi cant benefi ts for commercial, household, and industrial concerns, especially in relation to greater yields of agricultural products as it curtailed the growth of wild-type plants and attack of pests. Indeed, food products are available in abundance and more sustainable produce has been achieved.

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 45
However, the balance between safe, effective use of such chemicals and the risk to the population constantly undergoes reevaluationto determine whether pesticides and herbi-cides are associated with developmental, carcinogenic, endocrine, or other toxicological abnormalities.
Quantitative risk assessment is an estimation of toxic exposure concentrations or doses that, within a defi ned probability level, lead to specifi c increases in lifetime incidence rates for an undesirable consequence associated with the toxic substance. Factors used to esti-mate prediction models depend largely on accumulated retrospective and prospective data including the applicability of the scientifi c knowledge. In actuality, RA is based on a lack of available data for a substance with potential for human or animal toxicity and the calcu-lation of an estimate of potential toxicity on the data that are available. Consequently, gathering reliable and valid toxicity data for the prediction of human and animal health risks relies on a systematic approach in the quantitative RA evaluation process. The follow-ing steps form the analytical approach:
1. Hazard identifi cation2. Dose–response assessment3. Exposure assessment (potential for exposure)4. Risk characterization (characterization of risk)
HAZARD IDENTIFICATION
In an effort to determine whether a health risk exists, the process of hazard identifi cation uses qualitative methods to evaluate the potential for adverse health effects of chemical or physical substances. The information incorporated into the database is obtained from rele-vant retrospective epidemiological studies and case studies. In addition, controlled animal and in vitro studies (including teratogenicity, mutagenicity, and carcinogenicity studies) are also conducted to contribute to RA evaluation.
The data gathered from these experimental studies contribute to understanding levels and routes of exposure, time of administration, multiple target organ toxicity, and combina-tions of chemicals. The relevance of the toxicity data and extrapolation for human risk assessment, however, require careful interpretation of the information in order to construct valid conclusions as to health risk. As discussed in later chapters on animal toxicology test-ing (Section II), organization of animal studies is particularly important in order to extrap-olate data for human RA.
DOSE–RESPONSE ASSESSMENT
As the relevant studies are examined for assessment of potential toxicity, the second objec-tive of RA is the determination of the relationship between the dose administered and the expected/observed response (dose–response assessment). Factors used to estimate the dose or doses associated with the appearance of adverse effects are recognized according to the determination of the following statistical computations:
Quantal relationship: Number of test subjects or individuals in a population that dem-onstrate an effect; varies with level of exposure (see Fig. 2.5)
Graded relationship: Severity of lesions exhibited by the test subjects or individuals; graded relationship is proportional to dose
Continuous relationship: Alteration of a physiological or biological indicator (e.g., body weight) with respect to dosage regimen

46 PRINCIPLES OF TOXICOLOGY TESTING
Accordingly, defi nitive proof of a dose–response relationship is understood as a statistically positive correlation between the intensity and frequency of the effect with increasing dose. An important factor that infl uences interpretation of the relationship involves the recognition of experimental thresholds for chemicals in animal studies and demonstration of similar thresholds in human populations. Thresholds defi ne acceptable exposure levels within human populations. Consequently, how is the dose–response rela-tionship estimated for prediction of human or animal RA? The following criteria have historically been judged as invaluable for forecasting risks to the community: selection of an appropriate animal species; determination of a suffi cient number of animals for an experimental setup; incorporation of a variety of chemical exposure levels; estimation of an adequate duration and frequency of administration of the agent that mimics lifelike exposures; and, use of suitable in vitro screening techniques1 to calculate threshold esti-mates for subsequent in vivo experimentation.
Many model systems are currently available that provide primarily bibliographical information or summary factual data from toxicological studies. Relatively few contain adequate information to allow application of dose–response models. The EPA Gene-Tox database, the EPA/IARC Genetic Activity Profi le database, and the Toxicological Activity Profi le database are examples of prototype profi les derived from a variety of test systems based on thousands of chemicals. They are used to support hazard classifi cation of poten-tial toxicants. Waters and Jackson (2008) describe the contents of several databases that use dose–response evaluation in hazard identifi cation and RA. Woodall (2008) describes the Exposure–Response database that allows entry of detailed toxicological results from exper-imental studies in support of dose–response assessment. These models permit a high level of automation in performance of various types of dose–response analyses.
EXPOSURE ASSESSMENT
Exposure assessment is an estimation of the intensity, frequency, and duration of human exposure or the potential of exposure to a toxic substance present in the environment. A variety of exposure possibilities that describe the magnitude, duration, schedule, and route of exposure to a substance are determined. Exposure assessment also entails determination of the extent of exposure and the circumstances surrounding the type of chemical release. Available toxicity data, current limits of environmental pollution, and estimations of current or projected intakes are included in the parameters. Information gathered in this manner help address the uncertainties associated with the conclusions about exposure assessment in par-ticular and RA in general.
Assessment Modeling. Figures 4.1 through 4.4 illustrate modeling scenarios that track movement of contaminants through environmental paradigms. For instance, groundwater models quantify the movement of subsurface water and provide inputs to subsurface contaminant transport models (Fig. 4.1). The simulation provides insight into groundwater and contaminant behavior and allows for quantitative assessments for environmental decision making. Surface water models (Fig. 4.2) simulates contaminant movement and concentration in lakes, streams, estuaries, and marine environments. This model allows regulators to understand how exposure to contaminants affects aquatic environments. Similarly, models that emulate possible contamination in the food chain can aid in tracing contaminated aquatic and terrestrial environments that
1 In vitro methods, using test tubes or cell cultures, involve alternative methods to animal experimentation. These systems are used as preliminary screening tests for estimating toxic exposures, which avoids sacrifi ce of a large numbers of animals from the beginning.

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 47
typically result in the bioaccumulation of chemicals within all trophic levels of an eco-system and for estimating chemical impacts on exposed plant, prokaryotic and eukary-otic kingdoms (Fig. 4.3). Finally, multimedia models track contaminants that travel through the atmosphere, soil, surface water, and cohabitants (Fig. 4.4). An advantage of the multimedia computer simulation is its ability to monitor and quantify the impacts of exposure as the contaminants navigate concurrently and sequentially throughout these environments.
RISK CHARACTERIZATION
Risk characterization involves the accumulation of data used in estimating the probable incidence of an adverse health effect o humans under various conditions of exposure. This component of RA incorporates the most reliable human databases and animal studies that
Ground waterpenetration
Aquifer layer
FIGURE 4.1 Ground Water Model. Source: Adapted from U.S. EPA public website, http://www.epa.gov/ceampubl.
Contaminant movement
FIGURE 4.2 Surface Water Model. Source: Adapted from U.S. EPA public website, http://www.epa.gov/ceampubl.

48 PRINCIPLES OF TOXICOLOGY TESTING
have been thoroughly reviewed. In addition, EPA describes risk characterization as the risk associated with particular exposures in situations considered for regulation. Risk character-ization has since evolved from the initial descriptions of the management of RA through to the decision-making process. Presentation and display of information, exposure times, potency (unit risk), nature and weight of evidence, uncertainty of the component parts, and distribution of population risk, calculate in the fi nal decision-making process. Overall, risk characterization translates the strengths and weaknesses of the assessment by purposefully synthesizing risk considerations into an incorporated, reasonable analysis. Thus, risk char-acterization is the fi nal, integrative step of RA and conveys the risk assessor’s judgment as to the nature and existence of human health or ecological risks. Together with hazard iden-tifi cation, dose–response relationship, and exposure assessment, risk characterization enables the formation of model systems and their integration in RA prediction.
Water masses Land masses
Contaminantmovement
FIGURE 4.3 Food Chain Model. Source: Adapted from U.S. EPA public website, http://www.epa.gov/ceampubl.
Contaminantmovement
Aquifer
River bed
FIGURE 4.4 Multimedia Model. Source: Adapted from U.S. EPA public website, http://www.epa.gov/ceampubl.

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 49
THRESHOLD RELATIONSHIPS
Estimation of low dose exposure levels and determining their effects in human populations are particularly diffi cult processes. Threshold values represent a range of the lowest possible exposures that are tolerated by humans in the environment or in an occupational setting without risk of adverse health effect. The dose–response relationship however does not always calculate with good correlations, particularly at lower concentrations. This is scien-tifi cally frustrating for the most part because experimental situations that attempt to mimic low-dose exposures, as those encountered in the environment, are not necessarily linear at low levels. Consequently, data obtained from chronic in vivo and valid in vitro studies must be carefully examined before the data are used to support the estimation of threshold values. Table 4.1 summarizes threshold relationships and associated toxic classifi cation types used in applications of RA.
REGULATORY TOXICOLOGY
REGULATORY SYSTEMS
Regulatory systems, particularly in the biopharmaceutical industry, have evolved from its origins in national affairs to include an interactive global organization. Global regulatory affairs organizations within an industry provide strategic leadership and direction of the government regulatory requirements for product research and development. These systems use regulatory knowledge to ensure compliance and create opportunities in highly regulated environments. The main goal of these systems is to develop functional roles for the presenta-tion of a chemical or drug within the biopharmaceutical industry. Briefl y, these phases include the following:
1. Product development: This group helps formulate strategy, coordinates policy with regulatory agencies, and reviews regulatory submissions. In addition, product development coordinates manufacturing, registration and maintenance of the product in the market.
2. Chemistry, manufacturing and control: This group is involved in the develop-ment of strategy for manufacturing plans and processes, facility inspections, and prepares conformance and compliance documents with current good manufac-turing processes.
TABLE 4.1Definition of Threshold Values and Associated Toxic Classification Types
Classifi cation Type Description ApplicationsMethod of Calculation
Threshold Non-zero threshold Adverse health effects are unlikely to occur below this level
Systemic organ toxicity; germ-cell mutations; developmental, reproductive toxicity
Safety (uncertainty) factor method
Non-threshold Zero threshold Adverse health effects are possible at any dosage level
Mutagenic and carcinogenic effects
Risk analysis method

50 PRINCIPLES OF TOXICOLOGY TESTING
3. Policy and regulatory intelligence: Monitors emerging trends, policies, regulations and guidelines for strategic purposes, and is responsible for regulatory intelligence, and external participation.
4. Regulatory submission: This is responsible for publishing and compilation of data, technology management, assembly of submissions, and document standards for dispatching to the regulatory agencies.
5. Product labeling: Develops labeling associated with the marketing and professional presentation, ensures compliance and accuracy of label, and is responsible for the package insert and literature, container labels and packaging, updating the product information, and the label content reviews.
6. Promotion and advertising: Generates competitive and compliant promotional materials, regulates review of promotional copies and publications, and is the pri-mary liaison with regulatory agencies on promotional and advertising materials.
In the United States, several administrative regulatory agencies are responsible for application, enforcement, and establishment of rules and regulations associated with safe and effective chemical and drug use. Table 4.2 lists the relevant regulatory agencies and the regulatory acts under their jurisdiction.
NUCLEAR REGULATORY COMMISSION
Headed by a fi ve-member commission, the Nuclear Regulatory Commission (NRC) is an independent agency established by the Energy Reorganization Act of 1974 to regulate civil-ian use of nuclear materials.2 The NRC’s primary mission is to protect public health and safety and the environment from the effects of radiation from nuclear reactors, materials, and waste facilities. In addition, it monitors national defense and promotes security from radio-logical threat. NRC carries out its mission through the enactment of policy; protection of workers from radiation hazards; development of standards; inspection of facilities; investi-gation of cases; research and development; licensing the procurement, storage, and use of radiation materials; and adjudication.
ENVIRONMENTAL PROTECTION AGENCY
Headed by an administrator who is appointed by the President, the mission of the EPA is to protect human health and safeguard the natural environment. The agency employs over 18,000 engineers, scientists, environmental protection specialists, and staff in program offi ces, regional offi ces, and laboratories.
EPA’s mandate arises from federal laws protecting human health and the environment. It oversees natural resources, human health, economic growth, energy, transportation, agri-culture, industry, and international trade in establishing environmental policy. EPA also provides leadership in environmental science, research, education and assessment efforts, and works closely with other federal agencies, state and local governments, and Indian tribes to develop and enforce regulations. EPA sets national standards and delegates
2 The Atomic Energy Act of 1954 established a single agency known as the Atomic Energy Commission that was responsible for the development and production of nuclear weapons as well as for the development and safety regulation of civilian uses of nuclear materials. The Act of 1974 split these functions and assigned the responsibil-ity for the development and production of nuclear weapons and other energy-related work to the Department of Energy, while delegating the regulatory work to the NRC.
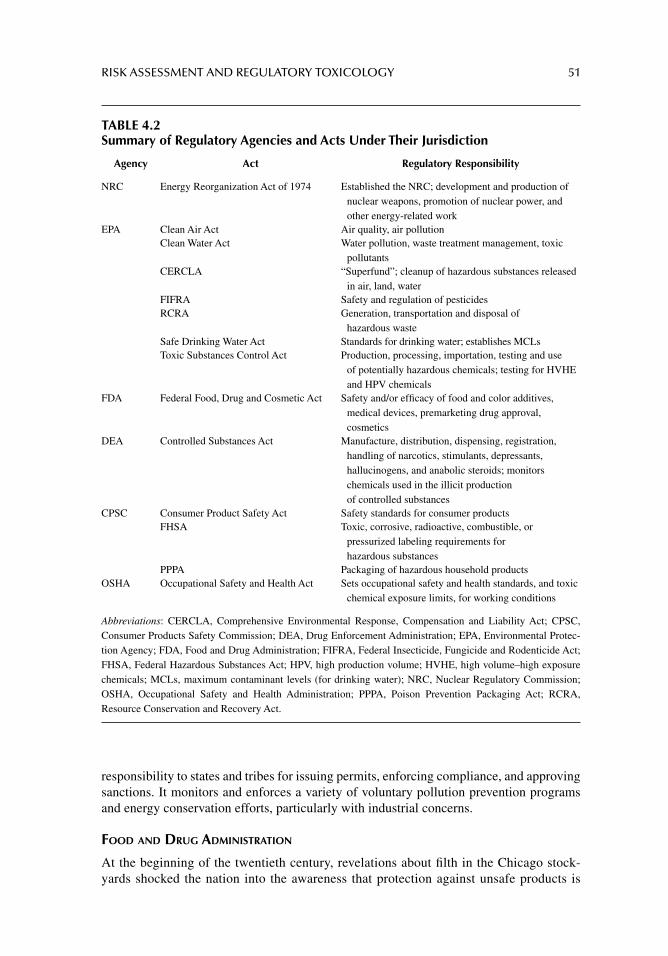
RISK ASSESSMENT AND REGULATORY TOXICOLOGY 51
responsibility to states and tribes for issuing permits, enforcing compliance, and approving sanctions. It monitors and enforces a variety of voluntary pollution prevention programs and energy conservation efforts, particularly with industrial concerns.
FOOD AND DRUG ADMINISTRATION
At the beginning of the twentieth century, revelations about fi lth in the Chicago stock-yards shocked the nation into the awareness that protection against unsafe products is
TABLE 4.2Summary of Regulatory Agencies and Acts Under Their Jurisdiction
Agency Act Regulatory Responsibility
NRC Energy Reorganization Act of 1974 Established the NRC; development and production of nuclear weapons, promotion of nuclear power, and other energy-related work
EPA Clean Air Act Air quality, air pollutionClean Water Act Water pollution, waste treatment management, toxic
pollutantsCERCLA “Superfund”; cleanup of hazardous substances released
in air, land, waterFIFRA Safety and regulation of pesticidesRCRA Generation, transportation and disposal of
hazardous wasteSafe Drinking Water Act Standards for drinking water; establishes MCLsToxic Substances Control Act Production, processing, importation, testing and use
of potentially hazardous chemicals; testing for HVHE and HPV chemicals
FDA Federal Food, Drug and Cosmetic Act Safety and/or effi cacy of food and color additives, medical devices, premarketing drug approval, cosmetics
DEA Controlled Substances Act Manufacture, distribution, dispensing, registration, handling of narcotics, stimulants, depressants, hallucinogens, and anabolic steroids; monitors chemicals used in the illicit production of controlled substances
CPSC Consumer Product Safety Act Safety standards for consumer productsFHSA Toxic, corrosive, radioactive, combustible, or
pressurized labeling requirements for hazardous substances
PPPA Packaging of hazardous household productsOSHA Occupational Safety and Health Act Sets occupational safety and health standards, and toxic
chemical exposure limits, for working conditions
Abbreviations: CERCLA, Comprehensive Environmental Response, Compensation and Liability Act; CPSC, Consumer Products Safety Commission; DEA, Drug Enforcement Administration; EPA, Environmental Protec-tion Agency; FDA, Food and Drug Administration; FIFRA, Federal Insecticide, Fungicide and Rodenticide Act; FHSA, Federal Hazardous Substances Act; HPV, high production volume; HVHE, high volume–high exposure chemicals; MCLs, maximum contaminant levels (for drinking water); NRC, Nuclear Regulatory Commission; OSHA, Occupational Safety and Health Administration; PPPA, Poison Prevention Packaging Act; RCRA, Resource Conservation and Recovery Act.

52 PRINCIPLES OF TOXICOLOGY TESTING
beyond any individual’s means in an industrial economy. The U.S. Congress responded to Upton Sinclair’s best-selling book titled The Jungle by passing the Food and Drug Act of 1906 that prohibited interstate commerce of misbranded and adulterated foods and drugs.
Enforcement of the law was entrusted to the U.S. Department of Agriculture’s Bureau of Chemistry, which later became the the Food and Drug Administration (FDA). The 1906 act was the fi rst of more than 200 laws, some of which are discussed here, that constitute a comprehensive and effective network of public health and consumer protections. FDA’s mission, therefore, is to promote and protect public health by ensuring the safety and effi -cacy of products in the market and monitoring them for continued safety.
Overall, the FDA regulates $1 trillion worth of products a year. It is under the jurisdiction of the Department of Health and Human Services (HHS) and consists of nine centers: the Center for Biologics Evaluation and Research, the Center for Devices and Radiological Health, the Center for Drug Evaluation and Research, the Center for Food Safety and Applied Nutrition, the Center for Veterinary Medicine, the National Center for Toxicological Research, the Office of the Commissioner, and the Office of Regulatory Affairs.
The Code of Federal Regulations (CFR) is a codifi cation of the rules published in the Federal Register by executive departments and agencies of the U.S. government. The CFR is divided into 50 titles (volumes) that represent broad areas subject to federal regu-lation, with environmental regulations contained mainly in Title 40. Products regulated by the FDA through the CFR include food and food additives (except for meat, poultry, and certain egg products); medical and surgical devices; therapeutic drugs; biological products (including blood, vaccines, and tissues for transplantation); animal drugs and feed; and radiation-emitting consumer and medical products. The FDA also acts to pre-vent the willful contamination of all regulated products and improve the availability of medications to prevent or treat injuries caused by biological, chemical, and nuclear agents.
The federal Food, Drug, and Cosmetic Act (FD&C) of 1938 was passed after a legally marketed toxic elixir killed 107 children and adults. The FD&C Act authorized the FDA to demand evidence of safety for new drugs, issue standards for foods, and conduct factory inspections.
The Kefauver–Harris Amendments of 1962 were inspired by the thalidomide tragedy in Europe. The amendments strengthened the rules for drug safety and required manufac-turers to prove drug effectiveness.3
The Medical Device Amendments of 1976 followed a U.S. Senate fi nding that faulty medical devices had caused 10,000 injuries, including 731 deaths. These amendments established guidelines for safety and effectiveness of new medical devices.
DRUG ENFORCEMENT ADMINISTRATION
The Drug Enforcement Administration (DEA) is under the policy guidance of the Sec-retary of State and HHS and is headed by an administrator. Its mission is to enforce controlled substances laws and regulations and prosecute individuals and organizations involved in the growing, manufacture, or distribution of controlled substances appear-ing in or destined for illicit traffi c. It also recommends and supports non-enforcement
3 Prompted by the FDA’s vigilance and monitoring of clinical cases, thalidomide was prevented from entering the U.S. market.

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 53
programs aimed at reducing the availability of illicit controlled substances in domestic and international markets. The DEA’s primary responsibilities include the following:
1. Investigation and preparation for the prosecution of major violators of controlled substance laws operating at interstate and international levels
2. Prosecution of criminals and drug gangs3. Management of a national drug intelligence program in cooperation with federal,
state, local, and foreign offi cials to collect, analyze, and disseminate strategic and operational drug intelligence information
4. Seizure of assets derived from, traceable to, or intended to be used for illicit drug traffi cking
5. Enforcement of provisions of the Controlled Substances Act (see the next para) as they pertain to the manufacture, distribution, and dispensing of legally produced controlled substances
6. Coordination and cooperation with federal, state, and local law enforcement offi -cials on mutual drug enforcement efforts including the reduction of the availability of illicit abuse-type drugs, crop eradication, and training of foreign offi cials
The Controlled Substances Act (CSA), Title II of the Comprehensive Drug Abuse Prevention and Control Act of 1970, is a consolidation of numerous laws. It regulates the manufacture and distribution of narcotics, stimulants, depressants, hallucinogens, ana-bolic steroids, and chemicals used in the illicit sale or production of controlled substances. The CSA places all regulated substances onto one of fi ve schedules, based on therapeutic importance, toxicity, and potential for abuse or addiction. Table 4.3 describes the sched-ules and lists examples of drugs within these classifi cations. Regulatory proceedings initi-ated by the DEA or HHS may add, delete, or change the schedule of a drug or other substance. Other interested parties including drug manufacturers, medical and pharmacy societies or associations, public interest groups concerned with drug abuse, state and local government agencies, and individuals may also petition the DEA to modify a schedule.
The CSA also creates a closed system of distribution for those authorized to handle controlled substances. The cornerstone of this system is the registration of all those autho-rized by the DEA to handle such substances. All registered individuals and fi rms are required to maintain complete and accurate inventories and records of all transactions involving con-trolled substances and provide security for the storage of controlled substances.
CONSUMER PRODUCTS SAFETY COMMISSION
The CPSC is an independent federal regulatory agency created by Congress in 1972 under the Consumer Product Safety Act.4 Its mission is to protect the public from unreasonable risks of injuries and deaths associated with some 15,000 types of consumer products by informing the public about product hazards through local and national media coverage, pub-lication of booklets and product alerts, a web site, a telephone hotline, the National Injury Information Clearinghouse, its Public Information Center, and responses to the Freedom of Information Act requests.
4 The CPSC is headed by three presidential nominees who serve as commissioners (one is the chair person). Com-missioners are confi rmed by the Senate for staggered 7-year terms. The commissioners set policy. The CPSC is not part of any other governmental department or agency. The Congressional Affairs, Equal Employment and Minority Enterprise, General Counsel, Inspector General, Secretary, and Executive Director groups report directly to the chairman.

54 PRINCIPLES OF TOXICOLOGY TESTING
TABLE 4.3Controlled Substances Schedules and Description
Schedules Description of Regulated Substances Some Examples of Listed Substances
I Drug or substance has a high potential for abuse, has no currently accepted medical use in treatment in the USA, and there is a lack of accepted safety for its use under medical supervision
Benzylmorphine, etorphine, heroin, DMT, marijuana, LSD, mescaline, peyote, psilocybin, cocaine, THC
II The drug or substance has a high potential for abuse, has a currently accepted medical use in treatment in the USA with or without severe restrictions, and may lead to severe psychological or physical dependence
Fentanyl, levorphanol methadone, opium and derivatives
III The drug or substance has a potential for abuse less than those in schedules I and II, has a currently accepted medical use in treatment in the USA, and may lead to moderate or low physical dependence or high psychological dependence
Amphetamine, phenmetrazine methylphenidate, phencyclidine, nalorphine, anabolic steroids
IV The drug or substance has a low potential for abuse relative to those drugs or substances in schedule III, has a currently accepted medical use in treatment in the USA, and may lead to limited physical dependence or psychological dependence
Barbital, chloral hydrate, meprobamate, phenobarbital
V The drug or substance has a low potential for abuse, has a currently accepted medical use in treatment in the USA, and may lead to limited physical dependence or psychological dependence
Not >200 mg of codeine/100 ml; not >2.5 mg of diphenoxylate and not <25 mcg of atropine sulfate per dosage unit (Lomotil®)a
aEach tablet or 5 ml of the liquid formulation contain 2.5 mg diphenoxylate and 0.025 mg atropine.Abbreviations: DMT, dimethyl tryptamine; LSD, lysergic acid diethylamide; THC, tetrahydrocannabinols.
The CPSC fulfi lls its mission through the development of voluntary standards for industry and by issuing and enforcing mandatory standards or banning consumer products if no feasible standard adequately protects the public. The CPSC obtains recalls of products or arranges for product modifi cations. It also conducts research on potential product haz-ards, informs and educates consumers, and responds to consumer inquiries. Table 4.4 lists some of the product guidelines under CPSC jurisdiction.
CPSC administers the Poison Prevention Packaging Act (PPPA) of 1970. The PPPA requires child-resistant packaging of hazardous household products. The number of reported deaths from ingestions of toxic household substances by children since the incep-tion of this act has declined signifi cantly. However, it is estimated that more than one mil-lion calls to poison control centers are still registered following unintentional exposure to medicines and household chemicals by children under 5 years of age. More than 85,000 children are examined in emergency departments and almost 50 child deaths result from this type of exposure each year.5
5 According to the National Electronic Injury Surveillance System (a CPSC database tracking emergency room visits) and the American Association of Poison Control Centers (2003).

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 55
Some of the reasons accounting for continuing ingestions include the availability of non–child-resistant packaging on request for prescription medications and generally for over-the-counter medications; inadequate quality control by manufacturers leading to defective child-resistant closures; misuse of child-resistant packaging in the home (failure to replace or secure caps, transferring contents to non–child-resistant packages); and viola-tions by health professionals. Consequently, the CPSC has designed a textbook to educate health professionals, particularly pharmacists and physicians, about the child-resistant packaging program. It is intended to be incorporated into medical and pharmacy school curricula to bring greater awareness of their legal responsibilities. Table 4.5 lists some of the substances covered by the PPPA regulations.
OCCUPATIONAL SAFETY AND HEALTH ADMINISTRATION
Created by the Occupational Safety and Health Act of 1970, Occupational Safety and Health Administration (OSHA) assures safe and healthful working conditions. It
TABLE 4.4Examples of Some Consumer Products with Established Safety and Monitoring Guidelines Under CPSC Jurisdiction
All Terrain Vehicles Safety Home Heating EquipmentArt and Crafts Safety Household Products SafetyBicycle Safety Indoor Air QualityChild Safety Older Consumers’ SafetyChildren’s Furniture Outdoor Power Equipment SafetyClothing Safety Playground SafetyConsumer Product Safety Review Poison PreventionCrib Safety and SIDS Reduction Pool and Spa SafetyElectrical Safety Public Use ProductsFire Safety Recreational and Sports SafetyGeneral Information ReportsHoliday Safety Toy Safety
Abbreviation: SIDS, sudden infant death syndrome.
TABLE 4.5Substances Covered by the PPPA of 1970
Therapeutic Category Examples of Drugs and Substances
Analgesics Aspirin, acetaminophen, methyl salicylateControlled substances Opioids, S/H, stimulantsVitamins and dietary supplements Iron-containing drugs and other dietary supplementsLocal anesthetics Lidocaine, dibucaine, and minoxidilNonprescription NSAIDs Ibuprofen, naproxen, and ketoprofenNonprescription antihistamines Diphenhydramine (Benadryl®)a
Nonprescription antidiarrheal products Loperamide (Imodium®)General Human oral prescription drugs (with some
exceptions and exemptions)
a Each tablet or capsule contains 25 mg of diphenhydramine; each dosage form contains 2 mg of loperamide.
Abbreviations: NSAIDs, nonsteroidal anti-infl ammatory drugs; S/H, sedative/hypnotics.

56 PRINCIPLES OF TOXICOLOGY TESTING
accomplishes this task through inspectors and staff personnel. OSHA authorizes the enforcement of the standards developed under the act in cooperation with 26 states. In addition, it provides for research, information, education, and training in occupational safety and health. As a testament to the impact of the agency and the act, workplace fatalities and occupational injury and illness rates have been reduced by 40–50% while U.S. employment doubled from 56 to 111 million workers at 7 million sites since 1971.
The agency fulfi lls its mandate by setting standards established by regulations. For example, the OSHA Lead Standards for General Industry and Construction require employers to provide biological monitoring for workers exposed to airborne lead above the action level. Employers must provide monitoring of lead and zinc protoporphyrin (or free erythrocyte protoporphyrin) levels in blood. The analyses must be performed by labo-ratories that meet accuracy requirements specifi ed by OSHA. The OSHA List of Labora-tories Approved for Blood Lead Analysis is designed to serve as a source to locate laboratories that OSHA has determined meet the requirements of the accuracy provisions of the lead standards. Laboratories voluntarily provide profi ciency test data to OSHA for evaluation.
SUGGESTED READINGS
Clarkson TW. Principles of risk assessment. Adv Dent Res 1992; 6: 22.Collins TF. History and evolution of reproductive and developmental toxicology guidelines. Curr
Pharm Des 2006; 12: 1449.Cross J, Lee H, Westelinck A, et al. Postmarketing drug dosage changes of 499 FDA-approved new
molecular entities. Pharmacoepidemiol Drug Saf 2002; 11: 439.Dietz FK, Ramsey JC, Watanabe PG. Relevance of experimental studies to human risk. Environ
Health Perspect 1983; 52: 9.Hays SM, Becker RA, Leung HW, et al. Biomonitoring equivalents: a screening approach for inter-
preting biomonitoring results from a public health risk perspective. Regul Toxicol Pharma-col 2007; 47: 96.
Luft J, Bode G. Integration of safety pharmacology endpoints into toxicology studies. Fundam Clin Pharmacol 2002; 16: 91.
Nicoll A, Murray V. Health protection: strategy and a national agency. Publ Health 2002; 116: 129.Patterson J, Hakkinen PJ, Wullenweber AE. Human health risk assessment: selected Internet and
world wide web resources. Toxicology 2002; 173: 123.Ukwu HN. Global Regulatory Systems: A Strategic Primer for Biopharmaceutical Product Develop-
ment and Registration. CenterWatch, 2011. [Available from: http://store.centerwatch.com/p-287-global-regulatory-systems-a-strategic-primer-for-biopharmaceutical-product-
development-and-registration.aspx]
REVIEW ARTICLES
Acquavella J, Doe J, Tomenson J, et al. Epidemiologic studies of occupational pesticide exposure and cancer: regulatory risk assessments and biologic plausibility. Ann Epidemiol 2003; 13: 1.
Baht RV, Moy GG. Monitoring and assessment of dietary exposure to chemical contaminants. World Health Stat Q 1997; 50: 132.
Barlow SM, Greig JB, Bridges JW, et al. Hazard identifi cation by methods of animal-based toxicol-ogy. Food Chem Toxicol 2002; 40: 145.
Bernauer U, Oberemm A, Madle S, Gundert-Remy U. The use of in vitro data in risk assessment. Basic Clin Pharmacol Toxicol 2005; 96: 176.
Boyes WK, Dourson ML, Patterson J, et al. EPA’s neurotoxicity risk assessment guidelines. Fundam Appl Toxicol 1997; 40: 175.

RISK ASSESSMENT AND REGULATORY TOXICOLOGY 57
Buchanan JR, Burka LT, Melnick RL. Purpose and guidelines for toxicokinetic studies within the National Toxicology Program. Environ Health Perspect 1997; 105: 468.
Bull RJ, Conolly RB, De Marini DM, et al. Incorporating biologically based models into assessments of risk from chemical contaminants. J Am Water Works Assoc 1993; 85: 49.
Carere A, Benigni R. Strategies and governmental regulations. Teratog Carcinog Mutagen 1990; 10: 199.Casciano DA. FDA: a science-based agency. FDA Consum 2002; 36: 40.Cimino MC. Comparative overview of current international strategies and guidelines for genetic
toxicology testing for regulatory purposes. Environ Mol Mutagen 2006; 47: 362.Clewell HJ 3rd, Andersen ME, Barton HA. A consistent approach for the application of pharmacokinetic
modeling in cancer and noncancer risk assessment. Environ Health Perspect 2002; 110: 85.Conolly RB. The use of biologically based modeling in risk assessment. Toxicology 2002; 181: 275.Cornfi eld J, Rai K, Van Ryzin J. Procedures for assessing risk at low levels of exposure. Arch Toxicol
Suppl 1980; 3: 295.Creton S, Billington R, Davies W, et al. Application of toxicokinetics to improve chemical risk
assessment: Implications for the use of animals. Regul Toxicol Pharmacol 2009; 55: 291.Dorato MA, Engelhardt JA. The no-observed-adverse-effect level in drug safety evaluations: Use,
issues, and defi nition(s). Regul Toxicol Pharmacol 2005; 42: 265.Eason C, O’Halloran K. Biomarkers in toxicology versus ecological risk assessment. Toxicology
2002; 181: 517.Indans I. The use and interpretation of in vitro data in regulatory toxicology: cosmetics, toiletries and
household products. Toxicol Lett 2002; 127: 177.Jensen J, Pedersen MB. Ecological risk assessment of contaminated soil. Rev Environ Contam Toxi-
col 2006; 186: 73.Kowalski L, Denne J, Dyer R, et al. Overview of EPA Superfund human health research program. Int
J Hyg Environ Health 2002; 205: 143.Lathers CM. Risk assessment in regulatory policy making for human and veterinary public health. J
Clin Pharmacol 2002; 42: 846.Hansson SO, Ruden C. Evaluating the risk decision process. Toxicology 2006; 218: 100.Hayashi Y. Designing in vitro assay systems for hazard characterization: basic strategies and related
technical issues. Exp Toxicol Pathol 2005; 57: 227.Liebsch M, Spielmann H. Currently available in vitro methods used in the regulatory toxicology.
Toxicol Lett 2002; 127: 127.Lorber M. Indirect exposure assessment at the United States Environmental Protection Agency. Tox-
icol Ind Health 2001; 17: 145.Lu FC, Sielken RL Jr. Assessment of safety/risk of chemicals: inception and evolution of the ADI and
dose-response modeling procedures. Toxicol Lett 1991; 59: 5.Pirkle JL, Sampson EJ, Needham LL, et al. Using biological monitoring to assess human exposure to
priority toxicants. Environ Health Perspect 1995; 103: 45.Temple R. Policy developments in regulatory approval. Stat Med 2002; 21: 2939.U.S. Environmental Protection Agency, Environmental Management, Risk Assessment, Exposure
Assessment Programs. 2012. [Available from: http://www.epa.gov/ebtpages/enviriskassessmentexposureassessment.html]
U.S. Environmental Protection Agency, Risk Assessment and Management: Framework for Decision-Making. 1984.
U.S. Environmental Protection Agency, Risk Characterization Handbook, Science Policy Council, Publication No. EPA 100-B-00-002 (2003); 2012. [Available from: http://www.epa.gov/OSA/spc/pdfs/rchandbk.pdf]
Van Ryzin J. Quantitative risk assessment. J Occup Med 1980; 22: 321.Weed DL. Weight of evidence: a review of concept and methods. Risk Anal 2005; 25: 1545.Waters M, Jackson M. Databases applicable to quantitative hazard/risk assessment-Towards a predic-
tive systems toxicology. Toxicol Appl Pharmacol 2008; 233: 34.Woodall GM. An exposure-response database for detailed toxicity data. Toxicol Appl Pharmacol
2008; 233: 14.

58
5 Descriptive animal toxicology tests
CORRELATION WITH HUMAN EXPOSURE
HUMAN RISK ASSESSMENT
The information derived from descriptive animal toxicology tests is useful in determining the potential toxicity of a compound to humans as well as animals. The objective of these tests is to identify a toxic chemical at an early stage of its development, marketing, or distri-bution, especially if the substance is already commercially available. Together with in vitro tests, animal tests are applied as biological markers of chemically induced risks, whether the substances are synthetic or occur naturally.
These validated batteries of tests are used by regulatory agencies to screen for or predict human toxic effects in an attempt to establish a signifi cant frame of reference or standard for monitoring environmental chemical threats, therapeutic adverse reactions, and com-mercial and occupational toxicity. Detailed discussions of risk assessment and its applica-tion for estimating toxic potential for humans are presented in chapter 4.
PREDICTIVE TOXICOLOGY AND EXTRAPOLATION TO HUMAN TOXICITY
The ability to predict toxicity in humans and provide a responsible level of safety is probably the most daunting task arising from animal testing. Correlation of results of toxicology test-ing described in animals with human exposure requires careful consideration of the param-eters of the animal tests, including the selection of species with similar physiologies as humans. The initial selection of the appropriate chemical substance, dose, frequency, dura-tion of exposure, and route of administration dictates the subsequent setup of the project. Ostensibly minor, but not less signifi cant, is the consideration of animal housing, care, and maintenance of the animals; cost-effectiveness; veterinary aspects of the study; and selection of a laboratory diet. These factors also calculate into the toxicology testing equation. The variety of tests available to achieve valid conclusions with a particular chemical must be systematically chosen according to the guidelines of the regulatory agencies.
ANIMAL WELFARE AND U.S.ANIMAL WELFARE ACT
The U.S. Animal Welfare Act (originally enacted in 1966) is promulgated under the Code of Federal Regulations (Title 7, Sections 2131–2156). In general, the act authorizes the Secretary of Agriculture to regulate transport, sale, and handling of animals such as dogs, cats, nonhuman primates, guinea pigs, hamsters, and rabbits intended to be used in research or “for other purposes.” Several amendments have been enacted, including Public Law 91–579 (1970) that expanded the list of animals covered by the act to include all warm-blooded animals used or intended for use in experimentation or exhibition.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 59
Research facilities are defi ned according to the standards set by the act. Regulations have been developed regarding record keeping and humane care and treatment of animals in or during commerce, exhibition, experimentation, and transport. The regulations also mention inspections and appropriate use of anesthetics, analgesics, and tranquilizers. A further amendment to the act (Public Law 99–198, Food Security Act of 1985, Subtitle F: Animal Welfare) is referred to as the Improved Standards for Laboratory Animals Act. The amendment clarifi es what is meant by “humane care” and details specifi cs such as sanitation, housing, and ventilation. It directs the Secretary of Agriculture to institute regulations providing exercise for dogs and mandates provisions for establishing an ade-quate physical environment promoting the psychological health of nonhuman primates. It specifi es that pain and distress must be minimized in experimental procedures and requires that principal investigators consider alternatives to such procedures. It also defi nes experimental practices that are considered painful.
The 1985 Act also establishes a requirement for formation of and describes the role and composition of institutional animal care and use committees (IACUCs) and defi nes the responsibilities of the Animal and Plant Health Inspection Service. Also included is the formation of an information service at the National Agricultural Library to assist the stake-holders regulated by the act in preventing unintended duplication of research, providing employee training, searching for ways to reduce or replace animal testing, and obtaining information on how to decrease pain and distress.
The current version of the regulations, as developed by the U.S. Department of Agriculture, specifi es how to comply with the Animal Welfare Act and its amendments. It is divided into four sections: Defi nitions, Regulations, Standards, and Rules of Prac-tice Governing Proceedings under the Animal Welfare Act. The Defi nitions section describes the terms used in the legislation. For example, the term “animal” specifi cally excludes rats of the genus Rattus and mice of the genus Mus used in research. The Regulations section includes subparts pertaining to licensing, registration, inspection of research facilities, responsibilities of attending veterinarians, and regulations governing adequate veterinary care. It also promulgates guidelines associated with stolen animals, records, compliance with standards and holding periods, and miscellaneous topics, such as confi scation and destruction of animals, and access to and inspection of records and property.
The bulk of the third section provides the standards for specifi c species. Included are sections covering cats and dogs, guinea pigs, hamsters, rabbits, nonhuman primates, marine mammals, and the general category of other warm-blooded animals. Standards include those for facilities and operations, health and husbandry systems, and transporta-tion. The fi nal section sets forth the rules of practice applicable to adjudicating administra-tive proceedings under Section 19 of the Animal Welfare Act (see Suggested Readings and Review Articles listed at the end of this chapter).
The remaining parts of this chapter defi ne, describe, and compare the general approach with in vivo toxicology tests. It also outlines the traditional animal experimentation through which most toxicologists and biomedical researchers gained their fundamental under-standing of the fi eld. Section II addresses animal toxicology tests, including the basic principles, techniques and methodologies associated with designing, executing, and com-pleting these studies. This section describes animal experimentation techniques in detail and presents the foundation for future projects. A thorough understanding of these chapters reinforces the importance of preventing and avoiding misuse, overuse, and unnecessary waste of animals. Finally, Section III introduces in vitro methodologies as alternative methods to animal toxicology testing. This text, therefore, is valuable as an aid for the

60 PRINCIPLES OF TOXICOLOGY TESTING
graduate student, junior investigator, as well as the seasoned toxicologist, to explore the options of a variety of different experimental pathways to ascertain the answers to important toxicological questions.
CHEMICALS
SELECTION OF CHEMICALS
The selection of appropriate chemicals for investigation and the aptness of the application of these chemicals for fulfi lling the objectives of a study establish the foundation for an entire project. Starting a project requires a thorough knowledge of the chemicals under consider-ation including, but not limited to, the physicochemical properties of the test agents, the suspected or known mechanisms of toxicity, and the applications and uses of the agents in the public sector.
Solubility
Physicochemical properties involve knowledge of the physical states of chemicals. This aspect is important not only to demonstrate the commercial availability of a chemical, but also to properly manipulate the agent in a laboratory. For instance, the aqueous or lipid solubility of a chemical determines the choice of a suitable vehicle. Thus, a sub-stance with greater water solubility is more easily dissolved in aqueous media including buffers, electrolytes, or drinking water. Poor water-soluble chemicals require additional measures in order to deliver the appropriate concentrations to test subjects. Therefore a chemical with poor water solubility precludes its inclusion in water vehicles because combining a chemical with an aqueous solvent leads to inaccurate vehicle concentra-tions. Thus, water-insoluble chemicals are diffi cult to administer parenterally or via drinking water.
Lipid-soluble chemicals require dissolution in organic solvents either as stock solu-tions or for whole vehicle delivery. Dissolving a test agent in an organic solvent to formu-late a stock solution renders the agent temporarily soluble, after which the extent of solubility in the subsequent water vehicle for in vivo administration is effectively deter-mined. The selected organic vehicle is necessarily inert and becomes part of the vehicle control group. Alternatively, insoluble agents in aqueous media necessitate preparing an emulsion or suspension, thus requiring additional manipulations, such as shaking, separate storage conditions, observance of shelf life, or homogenization. In any event, whether an agent is dissolved in an organic vehicle or is formulated as part of an emulsion or suspension, the preparation must be acceptable for administration via an oral, parenteral, or inhalation route.
Vehicle
Limited or poor aqueous solubility necessitates formulation of an emulsion, suspension, or solution in vegetable oils or organic solvents to form a homogeneous preparation to deliver the correct dose. The use of vegetable oils such as corn, olive, peanut, sunfl ower, and soy bean oils as delivery vehicles is not without precautions. Oils generally enhance absorption of orally administered agents more than equivalent concentrations delivered in aqueous media. Thus, chemicals orally administered in oils risk greater target organ toxicity by virtue of their experimentally enhanced absorption. In addition, as the vehicle and test substance are metabolized in vivo, some infl uence on chemical interactions is possible.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 61
Several studies with pesticides, herbicides, polyethylene glycols, and alcohols revealed unanticipated results when administered orally or parenterally in oil-based vehicles (see Suggested Readings and Review Articles).
Similarly, chemicals dissolved in oil-based vehicles are well suited for dermal appli-cations. Preparation of solutions, suspensions, or emulsions in oil or organic vehicles ensures effi cient penetration of the skin and enhanced dermal absorption. This technique of dermal administration is limited by the method of application to the animal. Several models developed for dermal testing in animals secure proper dermal delivery of test agents (see chap. 8).
As with gastrointestinal or parenteral administration, proper dissolution in an appropriate vehicle is necessary for nasal or respiratory inhalation of toxicants. The vehicle and dissolved agent are administered through any of several methods appropriate for inhalation studies including exposure of animals to high pressure aerosolization or release of vapors in contained, enclosed chambers. Consequently, with agents that have poor water solubility, the interaction of the vehicle with pulmonary tissue may infl uence mucous membrane interaction or toxicity. For instance, oil-based vehicles cause local upper respiratory irritation or infl ammation of lower pulmonary airways, resulting in respiratory infl ammatory reactions. Infl ammation unre-lated to the test agent thus must be distinguished from reactions caused by the chemical. In addition, insoluble solid particles present in a suspension or emulsion confound interpretation of results and require additional testing. A degradation product may interact with a test chemi-cal or may be erroneously considered as a metabolite formed in vivo.
ROUTE OF ADMINISTRATION
Selection of the appropriate route of administration is important in toxicological analysis and evaluation of the degree of toxicity. In general, as the objective of a toxicology study is to mimic human or animal risk exposure, the route of administration of a chemical to a test species should correlate with normal circumstances encountered for human exposure. For example, studies that attempt to understand toxicant exposure for human occupational toxicology require development of toxicity testing protocols that offer oral ingestion, dermal contact, and inhalation as important routes of possible contact.
Alternatively, domestic environmental exposure studies involving pesticides, herbicides, or fungicides should use an inhalation or dermal exposure animal model. Hence, develop-ment of animal models for toxicology testing should consider incorporating a parallel route of administration that imitates human exposure.
SELECTION OF APPROPRIATE ANIMAL SPECIES
As with the selection of an appropriate route of administration in designing toxicological studies, it is important to consider several criteria before selecting a suitable animal species. The criteria include the following:
1. Classifi cation of the toxic agent according to human exposure (see chap. 2).2. Anatomic, physiological, and metabolic similarity of the animal species to humans.3. Age, life expectancy, and sex of the animals.4. Objectives of the experiment.5. Previous experience with the species, convenience in handling the animal, housing
requirements, and daily care for the species.6. Cost effi ciency of the study.

62 PRINCIPLES OF TOXICOLOGY TESTING
It should be emphasized that animal models are selected based on their ability to substitute for the effects seen with chemical exposure to humans. The classifi cation of the chemical determines the route of administration, duration, and dosage, and subse-quently infl uences species selection. For acute toxicity studies, the rodent model is most appropriate based on the versatility of administration and the short duration correspond-ing to the physiological parameters of rodents. Dosage in any animal species must be adjusted based on metabolic rate and historical toxicological data available for the rodent species.
Although no perfect surrogate for humans exists, each species contributes distinct fea-tures that underscore biological and physiological differences among the various groups. It is generally accepted that differences in absorption, distribution, biotransformation, and elimination of chemicals exist among animal species and from humans, thus necessitating careful interpretation of toxicological data. Table 5.1 lists some of the major characteristics of mammalian species commonly used in toxicological investigations. Considerations affecting the selection and incorporation of animals in toxicology testing studies are also listed.
It is common knowledge that no single species accurately refl ects the complex char-acteristics of human physiology and metabolic activity. Interspecies variability is expected. In fact, this variability exists even within the same species, especially when the parame-ters of a study have been adequately controlled. Careful attention to criteria used in repeating studies (time of day when animals are handled, sexes, and numbers of animals used) will minimize species variation. The goal of using animal models in toxicological assessment, therefore, is to produce results that can be extrapolated and interpreted for human risk assessment and refl ect the most signifi cant correlations with the objectives of a study. As will be shown in later chapters, if the objective of a study is the demonstration of risk, for example with a suspected human carcinogen, then the interpretation of the results is based on the understanding of the differences between the animal models and humans.
Interspecies variability is also minimized by selecting animals bred under controlled environments. In order to decrease experimental variability, it is important to select animals with the following uniform characteristics:
1. Use of inbred (homogeneous) or outbred (heterogeneous) strains of the animal.1
2. Uniform mean body weights.3. Dependable yearly survival rates.4. Similar incidence of age-related diseases.5. Similar occurrence of spontaneous tumor formation.6. Typical responses to standard diet.
Knowledge and assurances by the breeders that these parameters are strictly monitored decrease unsuspected interspecies variability.
1 Outbred strains of rodents are closed colonies propagated through random mating in an established breeding system, avoiding the mating of close relatives. While a colony of animals is reasonably uniform in characteristics, each individual is genetically distinct. Outbred strains show genetic variabilities among colonies from different sources even though they may bear a generic name (e.g., Wistar, Spragu–Dawley, or Swiss albino). They also vary phenotypically. Even within a single colony, genetic characteristics change with time and breeding.
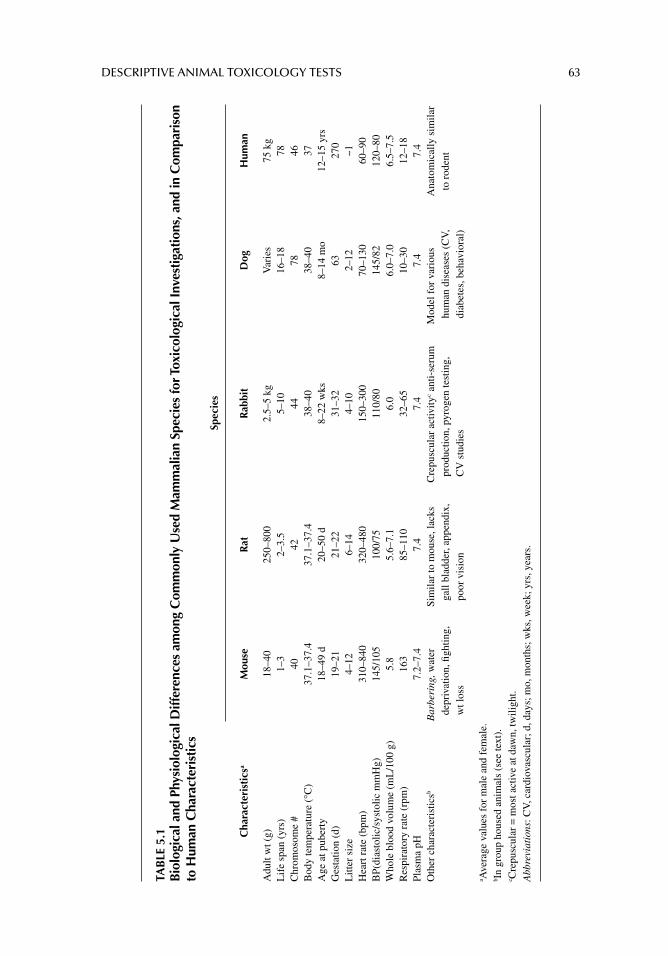
DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 63
TAB
LE 5
.1B
iolo
gica
l and
Phy
siol
ogic
al D
iffer
ence
s am
ong
Com
mon
ly U
sed
Mam
mal
ian
Spec
ies
for T
oxic
olog
ical
Inve
stig
atio
ns, a
nd in
Com
pari
son
to H
uman
Cha
ract
eris
tics
Spec
ies
Cha
ract
eris
tics
aM
ouse
Rat
Rab
bit
Dog
Hum
an
Adu
lt w
t (g)
18–4
025
0–80
02.
5–5
kgV
arie
s75
kg
Lif
e sp
an (
yrs)
1–3
2–3.
55–
1016
–18
78C
hrom
osom
e #
4042
4478
46B
ody
tem
pera
ture
(°C
)37
.1–3
7.4
37.1
–37.
438
–40
38–4
037
Age
at p
uber
ty18
–49
d20
–50
d8–
22 w
ks8–
14 m
o12
–15
yrs
Ges
tatio
n (d
)19
–21
21–2
231
–32
6327
0L
itter
siz
e4–
126–
144–
102–
12~1
Hea
rt r
ate
(bpm
)31
0–84
032
0–48
015
0–30
070
–130
60–9
0B
P(di
asto
lic/s
ysto
lic m
mH
g)14
5/10
510
0/75
110/
8014
5/82
120–
80W
hole
blo
od v
olum
e (m
L/1
00 g
)5.
85.
6–7.
16.
06.
0–7.
06.
5–7.
5R
espi
rato
ry r
ate
(rpm
)16
385
–110
32–6
510
–30
12–1
8Pl
asm
a pH
7.2–
7.4
7.4
7.4
7.4
7.4
Oth
er c
hara
cter
istic
sbB
arbe
ring
, wat
er
depr
ivat
ion,
fi gh
ting,
w
t los
s
Sim
ilar
to m
ouse
, lac
ks
gall
blad
der,
appe
ndix
, po
or v
isio
n
Cre
pusc
ular
act
ivity
c ant
i-se
rum
pr
oduc
tion,
pyr
ogen
test
ing,
C
V s
tudi
es
Mod
el f
or v
ario
us
hum
an d
isea
ses
(CV
, di
abet
es, b
ehav
iora
l)
Ana
tom
ical
ly s
imila
r to
rod
ent
a Ave
rage
val
ues
for
mal
e an
d fe
mal
e.b I
n gr
oup
hous
ed a
nim
als
(see
text
).c C
repu
scul
ar =
mos
t act
ive
at d
awn,
twili
ght.
Abb
revi
atio
ns: C
V, c
ardi
ovas
cula
r; d
, day
s; m
o, m
onth
s; w
ks, w
eek;
yrs
, yea
rs.

64 PRINCIPLES OF TOXICOLOGY TESTING
COST-EFFECTIVENESS
One of the most important criteria for selecting an appropriate animal species is cost. Animal testing is more expensive than in vitro methods. A budget for animal testing must account for the following:
1. Procurement of animals, housing, and maintenance within the animal care facility. 2. Daily requirements for food, water, and bedding.3. Employment and training of animal-care staff.4. On-staff or on-call veterinarian.5. Adherence to proper procedures for removal and disposal of specimens and waste.
The decision to institute, maintain, and develop animal toxicology studies involves a variety of expected and unexpected costs. These expenses are outlined in Table 5.2, and include the costs of purchasing and shipping, providing and maintaining housing accom-modations, animal feed and supplies, technical care, and utilities. The overhead costs associated with establishing and maintaining an animal care facility may be prohibitive, especially for smaller research industries or academic institutions. Interestingly, the bud-gets required for animal toxicology testing have, in part, prompted the development of in vitro alternative methods with comparatively less expensive requirements.
Accordingly, rodents are the most commonly used models for toxicity testing. This is principally based on the predicted costs of acute and chronic studies, ease of handling, convenient housing requirements, and the vast comparative databases available for most chemicals tested. Consequently, the decision to use rodents is based more on cost than on their biological similarities to humans. Based on these criteria, rodents are generally appro-priate models for toxicology testing. However, it is important to note that cost is not an adequate substitute for extensive experience with the biological parameters of animal mod-els, target organ toxicity, classifi cation of chemicals, and toxicokinetics. Selection of rodents as test models, therefore, must include a comparative analysis of the advantages and disadvantages of other animal species.
The number of animals used in a test project necessarily contributes to the cost of a project. According to the data from a survey (U.S. Public Health Service, 2006) more than 50,000,000 animals were used in toxicology testing studies in the United States in 2005.
TABLE 5.2Cost-Effective Considerations in Animal Toxicity Studies
Cost-Effectiveness of Animal Toxicity Studies Description
Animals Procurement, housing, maintenanceDaily animal requirements Food, water, beddingHousing expenses Utilities, animal careTechnical staff Technical and auxiliary animal care, training, employment expensesMedical care Veterinarian servicesWaste disposal Contract company for disposal of specimens and wasteIACUC Indirect and direct costs associated with a viable committee
Abbreviation: IACUC, Institutional Animal Care and Use Committee.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 65
While this conglomerate number represents an unwieldy statistic, the decision to incorpo-rate a specifi c number of animals per treatment group depends on the factors mentioned earlier. Among those factors not listed previously is the probability of occurrence of a toxic insult.
The probability of a toxic effect occurring in a human is inversely proportional to the number of animals required to demonstrate the effect. That is, as the probability of an occurrence in humans decreases, the greater the number of test animals will be required to demonstrate a parallel risk of occurrence. For human exposure to most environmental pol-lutants and commercially available chemicals, the probability of occurrence of toxic effects is less than 0.01%. Consequently, this 1:10,000 rate requires several thousands of animals to demonstrate a comparable causal relationship. Since this number of animals in practice is prohibitive, 10–25 animals per treatment group (minimum fi ve dosage groups per chemical plus controls) are generally used in acute or chronic toxicity studies, respec-tively. Understandably, the risk of not detecting all the potential adverse effects (false negatives) as a consequence of toxic insult within the dosage ranges used increases with fewer animals.
ANIMAL HOUSING
As noted above, animal housing rules and regulations and other animal care requirements are specifi ed in the U.S. Animal Welfare Act contained in the Code of Federal Regulations (as amended, 1990). The act outlines the conditions required for animal housing in approved animal care facilities. Among the regulations that apply to housing requirements, the rules dictate the physical environment, lighting, number of animals housed per square foot based on animal and cage size, and job specifi cations for animal care personnel.
DIET
It has been consistently shown that long-term calorie restriction (CR) is the only intervention that slows down the rate of aging and increases mean and maximum life spans in short-lived species. CR extends life spans and retards age-related chronic diseases in a variety of species including rats, mice, fi sh, fl ies, worms, yeasts, and, by circumstantial evidence, in humans. Although the mechanism for this occurrence is unclear, it is well understood that CR expands the effi ciency of metabolic rates, reduces oxidative stress, improves insulin sensitivity, and enhances neuroendocrine and sympathetic nervous system functions. Rodent studies dem-onstrate that CR opposes the development of many age-associated pathophysiological changes including detrimental alterations in brain function, learning, and behavior. Long-term CR in monkeys has shown benefi cial effects similar to those seen in rodents. Implica-tions associated with the benefi ts of long-term CR in longer-lived species such as primates and humans are materializing.
It is now generally accepted that rodents fed 8–20% less than the ad libitum intake exhibit weight loss, have increased longevity, and are less susceptible to a variety of dis-eases and age-related changes in organ function. In addition, decreased incidence of spon-taneous and chemically induced tumor formation appears to correlate with reduction in total caloric content, rather than the elimination of any single micronutrient or macronutri-ent. Thus, the conditions under which laboratory animals are maintained can signifi cantly infl uence the results of toxicology studies used for risk assessment. In fact, the results of numerous chronic toxicology studies have now been questioned because of the original dietary parameters under which the projects were conducted. Consequently, it is reasonable to ask: Are animals maintained on CR diets more resistant to chronic toxicity effects?

66 PRINCIPLES OF TOXICOLOGY TESTING
Nutrition is of importance in toxicological bioassays and research because diet composi-tion and the conditions of administration affect the metabolism and activities of xenobiotic test substances and alter the results and reproducibility of long-term studies. Ad libitum-fed animals are not well-controlled subjects for experimental studies and this type of feeding leads to considerable interlaboratory variability. In fact, the U.S. Food and Drug Administra-tion addressed the problems associated with uncontrolled food consumption and suggested levels of dietary control that are appropriate to achieve standardized growth curves.
The evidence suggests that the primary adaptation appears to be a rhythmic hypercorti-cism in the absence of elevated adrenocorticotropic hormone levels. This characteristic hypercorticism evokes a spectrum of responses such as (1) decreased glucose uptake and metabolism by peripheral tissues; (2) decreased mitogenic response; (3) reduced infl amma-tory response; (4) reduced oxidative damage to proteins and DNA; (5) reduced reproduc-tive capacity; (6) altered drug metabolizing enzyme expression.
Mechanistic data from dietary restriction studies suggest that obese animals are more susceptible to chemically induced toxicity, thus presenting problems in comparability of information. This is especially important in comparing data generated today from histori-cal databases. Several studies testing this paradigm have demonstrated that dietary control increases animal survival in 2-year studies and bioassay sensitivity. Consequently, the method of devising experimental toxicology protocols that may be infl uenced by dietary components, especially in young animals, has a direct impact on a variety of metabolic factors.
Metabolically, CR reduces the hyperinsulinemia characteristically seen with ad libitum-fed rats, subsequently decreasing the regulatory expression of metabolizing enzymes. CR markedly decreases the acute toxicities of several agents, the mechanism of which is mediated through enhanced immunosuppression, reduced rate of metabo-lism of test chemicals, or prevention of biotransformation. In addition, suspected or known mammalian carcinogens show signifi cantly lower incidence of cancer induc-tion in CR animals. CR also interferes with normal circadian rhythms, as rodents appear to consume their allotted food rapidly when on reduced diets. Interestingly, CR and changes in feeding behavior alter the circadian cycles of serum hormones such as insulin, thyroid hormone, and corticosterone. Overall, CR suppresses xenobiotic metabolism in young animals and exerts a moderate effect to maintain these enzymes in older animals.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE
The U.S. Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals was promulgated in 1986 and implemented the Health Research Extension Act of 1985 (Public Law 99–158, entitled “Animals in Research”). The Offi ce of Laboratory Ani-mal Welfare (OLAW) at the U.S. National Institutes of Health, which has the responsibility for the general administration and coordination of policy on behalf of the PHS, provides specifi c guidance, instruction, and materials to institutions on the utilization and care of vertebrate animals used in testing, research, and training. The PHS policy requires institu-tions to establish and maintain proper measures to ensure the appropriate care and incorpo-ration of all animals involved in research, research training, and biological testing activities conducted or supported by the PHS. In addition, any activity involving animals must include a written assurance acceptable to the PHS setting forth compliance with this policy. Assurances are submitted to OLAW at the U.S. National Institutes of Health, and are eval-uated by OLAW to determine the adequacy of an institution’s proposed program for the

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 67
care and use of animals in PHS-conducted or - supported activities. In summary, the pro-gram assurance must include the following information:
1. A list of all branches and major components of the institution.2. Lines of authority and responsibilities for administering the program.3. The qualifi cations, authorities, and responsibilities of veterinarians who participate
in the program.4. The membership list of the IACUC established in accordance with the requirements. 5. The procedures that the IACUC follows to fulfi ll the requirements.6. The health program for personnel who work in laboratory animal facilities or have
frequent contacts with animals.7. A synopsis of the training or instruction in the humane practice of animal care and
use. Training or instruction in research or testing methods that minimize the num-ber of animals required to obtain valid results and minimize animal distress must be offered to scientists, animal technicians, and other personnel involved in animal care, treatment, or use.
8. The gross square footage of each animal facility, the species housed, an average daily inventory of animals in each facility, and any other pertinent information requested by OLAW.
Other functions of the IACUC include periodic reviews of the institution’s program, inspection of facilities, preparation of reports, and submission of suggestions and recom-mendations on improving the program.
METHODOLOGIES
The following discussion presents an introduction to the methods, techniques, and protocols involved in establishing an animal model for toxicology testing. Subsequent chapters will highlight the models in greater detail. The purpose of this section, therefore, is to systemati-cally organize the subject matter for the investigator, that is, by understanding the material in an organizational context, the reader can prepare and design a study more thoroughly in a variety of contexts. For some readers, organization of the topics may suffi ce, while for others, the information may require a more detailed study.
A BRIEF HISTORY OF ANIMAL TOXICOLOGY TESTING PROTOCOLS
Systemic toxicity refers to the untoward consequences of xenobiotic exposure observed in an organism after absorption and distribution of the agent. The effects are generally moni-tored distally from the initial route of entry. Acute toxicity is seen after administration of either a single dose or several doses of a chemical within 24–72 hours (Klaasen 2008). The substance is administered orally, dermally, or via inhalation. Acute systemic toxicity testing, therefore, estimates the potential of a substance to cause generalized toxic effects after acute exposure.
Acute systemic toxicity testing is traditionally undertaken with the use of animals of the mammalian species, and the most common form of administration is oral. Historically, assessment of toxicity was based solely on the calculation of the LD
50 or median lethal dose;
that is, the estimated dose that causes death in 50% of the population of the species exposed under the defi ned conditions of the test (Barile 2008). First introduced in 1927 (Trevan

68 PRINCIPLES OF TOXICOLOGY TESTING
1927), the LD50
served as the end point for testing if the dose was administered orally.2 Since then, the test has progressed through numerous standardized protocols by the primary inter-national regulatory agencies, including the Organization for Economic Cooperation and Development (OECD; Guideline 401)3 and the Environmental Protection Agency (EPA; Guideline 798.1175). The guidelines established the choice of animals for testing which varied according to the forms of administration. The rodent is still the preferred species for oral and inhalation testing whereas the rat or rabbit is preferred for dermal testing. The num-ber of animals used to determine the LD
50 values ranged from 100 to 200 per study, depend-
ing on the test substance (Galson 2000). In 2001, OECD 401 Guideline was rescinded and replaced by more humane OECD protocols for in vivo toxicity testing.
Benign, Restructured OECD protocols: Fixed Dose Procedure (FDP), Acute Toxic Class Method (ATCM), Up and Down Procedure (UDP), OPPTS 870.1100 (EPA; Acute Oral Toxicity).
There was signifi cant concern regarding the number of animals consumed in toxicity testing studies using the lethal dose approach. In an attempt to reduce, refi ne and/or replace the number of animals used in in vivo studies, several methods were developed in combina-tion with animal tests, as alternative methods for animal toxicity testing.
In 1992, the fi xed-dose procedure (FDP; OECD Guideline 420: Acute Oral Toxicity) was adopted by the OECD. In contrast to the lethal dose method, the toxicity end points for the FDP was marked by the observation of ostensible signs of toxicity, that is “evident toxicity” (OECD 2001). The method incorporates a range-fi nding phase with single ani-mals prior to dosing of fi ve animals. The fi xed doses include 5, 50, 300, or 2000 mg/kg. However, this procedure renders the technique more subjective; it estimates the LD
50 range
while generating results with less confi dence. Soon after, the acute toxic class method (ATCM) was adopted as an alternative to the FDP.
This procedure incorporated biometric calculations using physicochemical data prior to animal experimental testing, to arrive at a starting point for in vivo toxicology testing (OECD, 1996). Essentially, the ATCM uses sequential testing parameters with three animals per sex at each step of the protocol. In FDP, subsequent doses are adjusted accordingly based on the results of the previous step. Alternatively, no further testing is needed if the previous lower dose demon-strates signifi cant toxicity (OECD 2001a). Interestingly, the dose levels are designed to allow for optimal integration of the results with international classifi cation systems, such as the glob-ally harmonized classifi cation system, so as to properly classify chemicals.
An up-and-down procedure (UDP) was adopted in 1998 as an alternative to lethal dose testing, the basis of which is to expose animals separately and individually. Depending on the outcome observed from one fi xed starting dose in one animal, subsequent doses are adjusted (increased or decreased) in the next animal. The procedure is repeated and contin-ued until toxic doses are computed (OECD 1998). The UDP appears to be the most accept-able method for achieving a reliable point estimate for the LD
50 when regulatory
requirements so mandate. Thus the UDP forms the basis for the EPA acute oral toxicity testing guideline (OPPTS 870.1100). The latter requires a point estimate of the LD
50 with
confi dence intervals for regulation of chemicals, particularly pesticides.Eventually, the FDP, ATCM, and the UDP were evaluated and were determined to be
statistically valid as acute toxicology testing methods. Although the procedures reduce the
2 The Lethal Concentration or LC 50
is the endpoint determined when the agent is administered by inhalation, as well as to estimate serum or organ concentrations (Gribaldo et al . 2005). 3 An international trade organization that includes the United States, Japan, and several member states of the European Union.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 69
number of animals used in toxicology tests and refi ne the pain and suffering associated with animal experimentation, further in vitro methods have since been developed to assess acute systemic toxicology testing as methods to further replace some of the in vivo proce-dures currently mandated by regulatory agencies (see Section III of this book, for an exten-sive discussion of the development of in vitro methodologies).
ROUTES OF EXPOSURE, DURATION, AND FREQUENCY OF DOSING
Establishment of routes of exposure, duration, and frequency of dosing in experimental pro-tocols requires individual attention to the objectives of the experiment. Thus, a further dis-cussion on experimental setup involving dosing and administration is presented in subsequent chapters.
TOXICITY INDICATORS
The biological disposition and effect of a chemical administered to an animal test subject are determined by the toxicokinetics of the interaction between the animal and the chemical. Figure 5.1 illustrates an overview of toxicokinetic (or pharmacokinetic) disposition of a chemical in vivo and its purported metabolic fate.
At any time after administration, the interaction can be identifi ed within any compart-ment. In addition, depending on the infl uence of the chemical on the compartment, the reactions may be concurrent and/or sequential. Also, the alteration of the compartment by a toxicant infl uences subsequent interactions and the detection of an effect at a particular time is shaped by the possibility of numerous events occurring between the chemical and physiological targets. The ability to detect the effect must be responsive enough to monitor the toxicological events at that moment. Therefore, in order to apply quantitative monitors to toxicological studies, sensitive toxicity indicators within a reliable dosage range are to be instituted. Proper experimental planning requires anticipation of the toxic effects and the most suitable toxicity endpoints to monitor that effect.
As described for in vitro techniques (see Section III of this book), a battery of pre-defi ned, standardized, and validated tests are also essential for animal testing throughout the
BIOTRANSFORMATION
LOCUS OF ACTION“RECEPTORS”
bound free free bound
Free drug
MetabolitesBound drug
TISSUERESERVOIRS
ABSORPTION EXCRETION
SYSTEMICCIRCULATION
FIGURE 5.1 Overview of toxicokinetic or pharmacokinetic disposition of a chemical in vivo and its purported metabolic fate.

70 PRINCIPLES OF TOXICOLOGY TESTING
experiment. A variety of end points, dosages (or concentrations), and responses (effects) allow construction of a dose–response relationship that accurately refl ects the events. Accordingly, a reasonable cost-effective approach to selecting appropriate toxicity indica-tors and inclusion of a proper number of animals will generate productive results. Data obtained from several indicators will help to construct a valid explanation of toxicological activity.
REQUIRED LD50 AND TWO ROUTES IN ACUTE SYSTEMIC EXPOSURE
Probably the most creative aspect of designing a toxicology study is the construction of the initial experimental protocol. For instance, initial decisions for the experimental setup must include (1) the species of animal, (2) number of animals, (3) route of administration, (4) estimation of initial doses (concentrations), and (5) length of the study (albeit an acute study).
Species of Animal
As previously mentioned above (“Selection of Appropriate Animal Species”) various strains of the laboratory rodent are routinely used in the determination of the acute LD
50.
Notwithstanding the physiological and anatomical similarity of the rodent to the human, and the abundant toxicological data that have already been accumulated with rodents, sev-eral persistent factors render this species the more preferable experimental subject. For instance, rodents are conveniently cost-effective, readily available internationally, have minimal individual variations within the species, and are technically easy to maintain and handle. Consequently, it is not fortuitous that the rodent is the preferred experimental ani-mal for toxicology studies.
Number of Animals
A suffi cient number of animals must be anticipated to accommodate any contingencies throughout the length of the study. Enough animals are included to assess predicted and unanticipated toxicity associated with the chemical agent. Unsuspected toxicity or prema-ture morbidity or mortality may precipitate an insuffi cient number of animals to complete the study. Thus, by the end of the project, a shortage of test subjects will render the computa-tion of the statistics invalid.
Route of Administration
The experimental route of administration should mimic the route of exposure most fre-quently encountered in a natural setting. For instance, the study of oral or injectable thera-peutic drugs will suggest an oral or parenteral route of experimental exposure, respectively. Similarly, gaseous toxicants encountered in the environment or occupational setting will favor an inhalation or dermal route of laboratory exposure. When a route is decided upon, there may be factors associated with the animal and/or chemical which will favor a specifi c methodology to accommodate the route of exposure. For example, oral administration may take the form of incorporating the chemical into solid food, solubilizing the agent in a liquid or water, or administration of the substance using other oral devices, such as oral feeding syringes or tubes. Injectable agents can be administered intravenously, intramuscularly, or subcutaneously, depending on the study objective, drug application, or protocol conve-nience. Ultimately, the selection depends on the details of the setup.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 71
Estimation of Initial Doses
Initial doses or target concentrations are usually decided with range-fi nding tests. In general, the larger the number of animals used per group dose, the more likely the LD
50 will be deter-
mined more accurately. However, as the LD50
is not required to be very accurate and because there is an ethical need to minimizing the number of animals sacrifi ced for the accuracy of the LD
50, there is no urgency to sacrifi ce many animals for the sake of calculating a defi nitive
LD50
. Consequently, there is a balance therefore between fi nding an approximate starting dose for subsequent toxicity tests and the needless slaughter of large numbers of live ani-mals. The methodologies discussed earlier (section, “Methodologies”) outline the humane approach to arriving at a screening dose, such as the LD
50, for subsequent testing.
Length of the Study
The length of a toxicology study, although acute, is a relative phenomenon. A 24-hour, 7-day, or 30-day test may all be considered acute relative to more lengthy periods. However, the U.S. EPA requires that a study must incorporate a 24-hour interval for an acute LD
50
study, regardless of the route of exposure. In comparison to humans, the 24-hour exposure correlates to an emergency situation. For instance, considering that the rodent’s life expec-tancy is short, an experimental exposure of 24 hours occupies a greater percentage of its life span than the equivalent time in humans or many other mammalian species. It is reasonable to request, therefore, that the time of exposure in rodents be proportionate to the duration of exposure in humans. In comparison to other species, and assuming that the rodent life span is about 2 years, 24 hours represents approximately 0.14% of relative life time exposure. The equivalent percent time of exposure in humans, with an average life span of 78 years, calculates to 40 days. Consequently, a 24-hour rodent LD
50 requires more than a month of
exposure to the same chemical in humans, assuming the same parameters are maintained constant. This relative computation is often not a consideration in human or animal risk assessment when considering acute LD
50s performed in rodents. Nevertheless, 24 hours has
been traditionally used as the cutoff for LD50
determinations in acute studies, in spite of the differences in species or route of exposure. Hence, the LD
50 is a gross estimate of the relative
toxicity of a chemical agent, whose interpretation is of limited value, whose calculation is approximate at best, and unfortunately, whose time in this advanced biomedical setting has passed, yet whose utility is still regarded as valuable in the regulatory hierarchy.
CHRONIC AND SUBCHRONIC EXPOSURE
Besides the acute time frame, several exposure periods are incorporated into chronic and sub-chronic studies. Subchronic exposure usually involves a 90-day exposure period, with or with-out repeated dosing intervals. Chronic studies are more diffi cult to design and execute, more cumbersome to organize, and expensive to conduct. Most of these studies are reserved for carcinogenicity and mutagenicity testing or toxicokinetic studies whose interest is ultimately in understanding the accumulation phenomenon. The rodent is the animal of choice in chronic toxicology testing. Other parameters are also of interest and may be of value in chronic tests, including hematology analysis, clinical chemistry, and histological examination.
SUMMARY OF TYPES OF TESTS
Table 5.3 outlines additional animal toxicology testing methods that are often performed along with LD
50 protocols or as specifi c target tests. These include methods or models for the
determination of ocular irritation, local irritation, teratogenicity, developmental toxicity,

72 PRINCIPLES OF TOXICOLOGY TESTING
perinatal and postnatal toxicity, mutagenicity, immunotoxicity, and toxicokinetics. Although they are mentioned briefl y in this and previous sections, more extensive discussions of the techniques and other toxicity model indicators designed for specifi c project objectives are detailed in subsequent chapters (Section II).
SUGGESTED READINGS
Bayne KA. Environmental enrichment of nonhuman primates, dogs and rabbits used in toxicology studies. Toxicol Pathol 2003; 31: 132.
Dirks AJ, Leeuwenburgh C. Caloric restriction in humans: potential pitfalls and health concerns. Mech Ageing Dev 2006; 127: 1.
Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr 2003; 78: 361.
Keenan KP, Laroque P, Dixit R. Need for dietary control by caloric restriction in rodent toxicology and carcinogenicity studies. J Toxicol Environ Health B Crit Rev 1998; 1: 135.
U.S. Department of Agriculture, National Agricultural Library, Animal Welfare Information Center.
[Available from: http://www.nal.usda.gov] [Last accessed June 2012].
REVIEW ARTICLES
Arts JH. Inhalation toxicity studies: OECD guidelines in relation to REACH and scientifi c develop-ments. Exp Toxicol Pathol 2008; 60: 125.
Bakand S, Hayes A. Troubleshooting methods for toxicity testing of airborne chemicals in vitro. J Pharmacol Toxicol Methods 2010; 661: 76.
Barrow PC. Reproductive toxicology studies and immunotherapeutics. Toxicology 2003; 185: 205.Borgert CJ. Evaluation of EPA’s Tier 1 Endocrine Screening Battery and recommendations for
improving the interpretation of screening results. Regul Toxicol Pharmacol 2011; 59: 397.
TABLE 5.3Other Types of Descriptive Animal Techniques Used as Specific Target Organ or Objective Tests
Methods Descriptive Protocol Preferred Species
Dermal testing Local irritancy Guinea pigDraize method Ocular irritancy RabbitFertility and reproductive tests Developmental toxicity Rabbit and rodent littersImmunotoxicity studies Toxicity related to immune
functionRodent
Mammalian mutagenesis Altered foci inductiona; skin neoplasm, pulmonary neoplasm breast cancer induction
Rodent
Mammalian teratology, whole embryo culture
Teratogenicity Rabbit, rodent
Perinatal and postnatal studies Developmental toxicity Rabbit, rodent Toxicokinetic studies Infl uence on A,D,M,Ea Rodent and larger mammals
aIn rodent liver.Abbreviations: A,D,M,E, absorption; distribution; metabolism; elimination.

DESCRIPTIVE ANIMAL TOXICOLOGY TESTS 73
Bucher JR. The National Toxicology Program Rodent Bioassay: designs, interpretations, and scien-tifi c contributions. Ann NY Acad Sci 2002; 982: 198.
Cooper RL. Current developments in reproductive toxicity testing of pesticides. Reprod Toxicol 2009; 28: 180.
Faqi AS. A critical evaluation of developmental and reproductive toxicology in nonhuman primates. Syst Biol Reprod Med 2012; 58: 23.
Faulks SC, Turner N, Else PL, Hulbert AJ. Calorie restriction in mice: effects on body composition, daily activity, metabolic rate, mitochondrial reactive oxygen species production, and mem-brane fatty acid composition. J Gerontol A Biol Sci Med Sci 2006; 61: 781.
Harper JM, Salmon AB, Chang Y, et al. Stress resistance and aging: infl uence of genes and nutrition. Mech Ageing Dev 2006; 127: 687.
Hayes A, Bakand S. Inhalation toxicology. EXS 2010; 100: 461.James ML. IACUC training: from new-member orientation to continuing education. Lab Anim (NY)
2002; 31: 26.Leakey JEA, Seng JE, Allaben WT. Infl uence of body weight, diet, and stress on aging, survival and
pathological endpoints in rodents: implications for toxicity testing and risk assessment. Reg Res Perspect 2004; 4: 1.
Lilienblum W, Dekant W, Foth H, et al. Alternative methods to safety studies in experimental ani-mals: role in the risk assessment of chemicals under the new European Chemicals Legisla-tion (REACH). Arch Toxicol 2008; 82: 211.
Merry BJ. Oxidative stress and mitochondrial function with aging the effects of calorie restriction. Aging Cell 2004; 3: 7.
Meyer O. Testing and assessment strategies, including alternative and new approaches. Toxicol Lett 2003; 140: 21.
Michael B, Yano B, Sellers RS, et al. Evaluation of organ weights for rodent and non-rodent toxicity studies: A review of regulatory guidelines and a survey of current practices. Toxicol Pathol 2007; 35: 742.
OECD Detailed Review Paper (DRP) number 31. “Cell transformation assays for detection of chem-ical Carcinogens”: main results and conclusions. Mutat Res 2012; 744: 8.
Pauluhn J. Overview of testing methods used in inhalation toxicity: from facts to artifacts. Toxicol Lett 2003; 140: 183.
Pitts M. A guide to the new ARENA/OLAW/IACUC guidebook. Lab Anim (NY) 2002; 31: 40.Rogers JV, McDougal JN. Improved method for in vitro assessment of dermal toxicity for volatile
organic chemicals. Toxicol Lett 2002; 135: 125.Silverman J. Animal breeding and research protocols: the missing link. Lab Anim (NY) 2002; 31: 19.Smialowicz RJ. The rat as a model in developmental immunotoxicology. Hum Exp Toxicol 2002;
21: 513.Spielmann H, Grune B, Liebsch M, et al. Successful validation of in vitro methods in toxicology by
ZEBET, the National Centre for Alternatives in Germany at the BfR (Federal Institute for Risk Assessment). Exp Toxicol Pathol 2008; 60: 225.
Steneck NH. Role of the institutional animal care and use committee in monitoring research. Ethics Behav 1997; 7: 173.
Vasseur P, Lasne C. OECD Detailed Review Paper (DRP) number 31 on “Cell Transformation Assays for Detection of Chemical Carcinogens”: main results and conclusions. Mutat Res 2012; 744: 8.
Warheit DB, Donner EM. Rationale of genotoxicity testing of nanomaterials: Regulatory require-ments and appropriateness of available OECD test guidelines. Nanotoxicology 2010; 4: 409.
Whitney RA. Animal care and use committees: history and current policies in the United States. Lab Anim Sci 1987; 37: 18.

74
6 Acute toxicology testing
OBJECTIVES OF ACUTE TOXICOLOGY TESTS
The information derived from acute animal toxicology studies is essential for determining the potential toxicity of a chemical to humans and other life forms. The objective of acute studies is to identify the potential toxicology of chemicals, whether commercially available or in development. The underlying premise of using animals in acute testing is that the effects of administration of a synthetic agent on an animal mimic the possible outcomes that the agent may produce in humans or other mammals. The system also implies that short-term tests in animals parallel acute exposure in the human population.
In general, the time of exposure, dose of chemical, selection of animal species, and biological parameters used in acute studies are fl exible. To qualify as an acute test, a study must be organized for comparison with human situations. The substance is administered orally, locally, parenterally, or via inhalation, and toxic effects should correlate with increasing doses. In fact, acute toxicology studies are designed to express the potency of a toxicant as a correlative dose–response relationship.
LD50 AND ACUTE TOXICOLOGY TESTS
In accordance with U.S. Food and Drug Administration regulations that establish current chemical and drug testing protocols using animal experiments, the LD
50 (median lethal dose
50%) of a substance must be established. The LD50
represents the estimated dose that causes death in 50% of the population of the species exposed under the defi ned conditions of the test. Each LD
50 test performed for a chemical must include at least two routes of exposure,
usually oral and parenteral routes. Depending on the nature of a substance, the routes can be modifi ed to include testing for inhalation, dermal, or other selective exposure. For inhalation or aquatic studies, toxicology is presented as the median lethal concentration (LC
50) the
estimated concentration of environmental exposure resulting in 50% mortality of the popula-tion of experimental animals.
LD50
data represent lethality and do not refl ect the acute toxic properties of a compound, nor does the value suggest enough information to adequately categorize a compound. The LD
50 also does not correlate well with information on different mechanisms of action of
toxic agents, especially when the agents are in different toxicological categories. Compari-sons are signifi cant only when agents are homologous and have the same or similar mecha-nisms of action. In fact, analysis of LD
50 data simply translates a value into a comparative
indicator of immediate toxicology for a specifi c agent in a particular strain, age group, and sex of a species of animal.
The concept of the LD50
developed in the early years of the twentieth century when many medicinal agents were available as impure and frequently toxic mixtures, extracts, or tinctures of biologically derived materials. The test was used to standardize the potencies of such biologically derived medicinal agents. Calculation of the lethal potency of a substance

ACUTE TOXICOLOGY TESTING 75
could theoretically aid in assessing its therapeutic potency. Consequently, LD50
methodol-ogy developed to yield dose–response relationships to standardize biological preparations according to lethal potency.
In general, assessment of the lethal properties of a chemical (and hence the determina-tion of its LD
50) has lost its integral value to the acute toxicology phase of the safety evalu-
ation process. The value and signifi cance of the LD50
determination for any agent environmental, commercial, or clinical yield little information toward explaining its mech-anism of toxicity or action. The reasons for performing LD
50 protocols are no longer valid,
considering the current state of advanced biotechnology. Knowledge of the lethal range of a chemical in a particular species under specifi c test guidelines does not necessarily imply protective safeguards in any other species, including humans.
It is important to know the subtle nuances of chemical injury resulting from diverse exposure routes, injurious doses, and duration of exposure. In addition, information about gross adverse reactions and wide ranging doses of a chemical needed to induce such reac-tions is ascertained from structure–activity relationships when known. Proper protective and security measures are still based almost entirely on other mechanistic toxicology tests in order to ensure safe handling, transport, and treatment upon exposure. More valu-able information on the pathophysiology of toxic exposure, therefore, can be obtained from conducting other animal studies and alternative acute toxicology tests as screening models.
Consequently, it is understandable that controversy has arisen over the use of the LD50
as an antiquated measure of toxicology. The test originated when other indicators of toxic-ity were not available or were not very sensitive, and the fl edgling biotechnology was not supportive of needs. In addition, the number of animals required for validation of the pro-tocol is large and it has been suggested that the method needlessly destroys large numbers of animals. Some suggestions for updating the test include (1) the use of far fewer animals in estimating starting doses for further testing (see chap. 5), (2) consideration of fewer routes of exposure, and (3) obtaining additional information during a study that adds to the body of knowledge available for the chemical. In addition, the tremendous amount of acute, toxicological data already in existence about currently available chemicals and chemical classes does not support the repeated generation of LD
50 data with the introduction of a new
formulation of the same compound, a new derivative of the same chemical class, or a dif-ferent combination of known chemicals. In fact, complete elimination of the test has also been suggested.
The European Union has mandated that the LD50
must be phased out for all chemicals by the year 2009 (Directive OECD 2001). In addition, as animal tests are further refi ned and alternative tests are developed, it is anticipated that LD
50 testing will be only of his-
torical interest.
ORGANIZATION OF STUDIES
Historically, the selection of laboratory mice or rats for study depends more on practical considerations than on biological imperatives or applicability to humans. Rodent species are more economic to use than other animals, have fewer interspecies variations, are readily available, and are technically easier to handle. In addition, housing and caring for rodents are more feasible and practical than they are for other species.
Non-rodent LD50
data are necessary when the mouse and rat LD50
values are signifi cantly different. Although this suggests species differences in the absorption, distribution, biotrans-formation, or elimination of a toxicant, this information is obtained from mechanistic studies

76 PRINCIPLES OF TOXICOLOGY TESTING
rather than through the performance of additional LD50
tests. The disadvantages associated with non-rodent species are precisely the factors that discourage use of other non-rodent animal species: cost, handling, and technical measures.
Protocols for acquiring and acclimating animals are important to ensure study success. Animals should be purchased from dependable breeders and placed in the quarantine area of an animal care facility for 7–14 days prior to the initiation of an experiment. They are examined at appropriate stages for gross pathology and general health. A protocol includes the euthanasia of a randomly selected subpopulation of animals for blood chemistry tests and gross and histopathological examinations of organs. Extra animals are usually pur-chased because the screening process necessitates elimination of some animals. Acclima-tion to the animal care facility is important to reduce the stress associated with transport and adaptation to the new environment.
Table 6.1 summarizes other organizational considerations in toxicology studies. These include the plans associated with scheduling and performance before, during, and after the study. In general, intra- or interlaboratory reproducibility of data depends on proper control of factors before initiation of the protocol (Table 6.1), as much as on the technical perfor-mance during an experiment.
TABLE 6.1Necessary Steps for Procuring and Handling Animals Prior to Initiation of Experiment
Organization Consideration Description
Prior to purchasing Selection of species, numbers, sexes, and ages of animals
Biological and economical calculations, applicability to human risk assessment, different or same sex, housing requirements, extra animals
Selection of commercial animal source
Reputation of breeder, cost, availability of animals, technical support
Type of study Acute, chronic, systemic, or target organ toxicology
Field of study Basic or applied toxicology testingAnimal care center Screen center for adequate availability of:
suffi cient technical services; suffi cient housing; laboratory environment; backup generators; off-hour coverage; proper disposal services; veterinary care
Immediately after purchasing and prior to initiation of experiment
Quarantine and screening Acclimation of animals to new environment (animal care center); observe for general health or pathology; reduce animal stress
Physical examination Gross observations; blood chemistry and histopathological analysis
Preparations Procurement of supplies and technical help; ensure housing requirements
Immediately before initiation of experiment
Preparations Preparation of solutions, special diets, perishable supplies; proper allocation of time
Physical examination Removal of unhealthy animalsDuring experiment Maintain constant
experimental conditionsEnsure adequate supplies and technical services;
help; maintain housing requirements; maintain consistent and constant experimental protocols

ACUTE TOXICOLOGY TESTING 77
OECD TEST GUIDELINES FOR IN VIVO TESTING
The Organization for Economic Cooperation and Development (OECD) test guidelines are a collection of relevant test methods used by government, industry and academic laborato-ries to determine the safety of chemicals and chemical preparations. The methods have been designed by international cooperation and include tests for physicochemical properties of chemicals, human health effects, environmental effects, and degradation and accumulation in the environment. This section outlines test guidelines promulgated for animal testing, including tests for acute, chronic, and local applications. Table 6.2 also lists and summarizes OECD in vivo animal test guidelines (OECD Guidelines for the Testing of Chemicals). Table 6.3 presents working documents and review papers related to the development of the OECD Guidelines for Testing of Chemicals while Table 6.4 lists Guidance Documents and Detailed Review Documents for Animal Testing of Chemicals (Extended Titles).
OECD TEST GUIDELINES FOR ACUTE TOXICITY TESTING: RANGE-FINDING TESTS
Determination of LD50
values is not inherently precise. The original procedure fi rst described by Trevan in 1927 often used 50 animals or more and was a benchmark for comparing the toxicity of chemicals. The nature of the LD
50 is that a certain percentage of animals survive a
particular dose; the next higher dose also shows a percentage of animals surviving, albeit lower. The incongruity arises with the survival of animals that should not necessarily survive after they receive subsequently higher doses. Thus, the possibility exists of killing one animal with a lower dose and failing to kill another animal with the next higher dose. To avoid such anomalies, dosages must be reasonably close to demonstrate precise lethal concentrations.
Alternatively, arbitrary selection of an appropriate concentration of toxicant is admin-istered to few animals, followed by careful observation after 24 hours. Subsequent groups of animals receive 0.75- to 1.25-fold of the original dose, depending on whether the con-centration was lethal or well tolerated, respectively. Administration is further adjusted as the experiment progresses and can minimize the number of animals sacrifi ced. A variety of established techniques have been traditionally used for LD
50 determinations with the inten-
tion of minimizing the number of animals. Listed below are the frequently used methods:
1. Up-and-down procedure (UDP) or staircase method (OECD 425)2. Fixed-dose approach (FDP, British Toxicology Society, OECD 420)3. Acute toxic class method (ATCM, OECD 436)
UP-AND-DOWN PROCEDURE
The revised UDP test method (ICCVAM, NIH Publication 02-4501) has three components:
1. A primary test providing an improved estimate of acute oral toxicology with a reduction in the number of animals used when compared with classical methods
2. A limit test for substances anticipated to have minimal toxicological properties3. A supplemental test to determine slope and confi dence interval (CI) for the dose–
response curve
In the revised UDP primary test, an appropriate dose is administered orally to one ani-mal (usually 175 mg/kg as the default starting dose) and the animal is observed up to 14 days.

78 PRINCIPLES OF TOXICOLOGY TESTING
TAB
LE 6
.2O
ECD
Gui
delin
es: I
n V
ivo
Gui
delin
e fo
r A
nim
al T
esti
ng o
f Che
mic
als
(Acc
ordi
ng t
o th
e Ye
ar S
ubm
itte
d)
Test
Gui
delin
e N
umbe
rD
ate
subm
itte
d (Y
ear)
Titl
eD
escr
ipti
on
411
1981
Subc
hron
ic D
erm
al T
oxic
ity: 9
0-da
y St
udy
Der
mal
ana
lysi
s of
hea
lth h
azar
ds o
f so
lid o
r liq
uid
test
sub
stan
ce41
019
81R
epea
ted
Dos
e, D
erm
al T
oxic
ity:
21/
28-d
ay S
tudy
Der
mal
ana
lysi
s of
hea
lth h
azar
ds o
f so
lid o
r liq
uid
test
sub
stan
ce41
519
83O
ne-G
ener
atio
n R
epro
duct
ion
Toxi
city
Stu
dyG
ener
al in
form
atio
n co
ncer
ning
the
effe
cts
of a
test
sub
stan
ce o
n m
ale
and
fem
ale
repr
oduc
tive
perf
orm
ance
478
1984
Gen
etic
Tox
icol
ogy:
Rod
ent D
omin
ant L
etha
l Tes
tIn
duct
ion
of a
dom
inan
t let
hal e
vent
aft
er e
xpos
ure
to a
test
sub
stan
ce, w
hich
cau
ses
embr
yoni
c or
fet
al d
eath
402
1987
Acu
te D
erm
al T
oxic
ityH
ealth
haz
ard
likel
y to
ari
se f
rom
sho
rt-t
erm
exp
osur
e to
a s
olid
or
liqui
d te
st
subs
tanc
e vi
a a
derm
al r
oute
406
1992
Skin
Sen
sitiz
atio
nH
ealth
haz
ard
likel
y to
ari
se f
rom
exp
osur
e to
a te
st s
ubst
ance
via
intr
ader
mal
in
ject
ion
and/
or e
pide
rmal
app
licat
ion
421
1995
Rep
rodu
ctio
n/ D
evel
opm
enta
l Tox
icity
Scr
eeni
ng T
est
Test
sub
stan
ce is
adm
inis
tere
d in
gra
duat
ed d
oses
to s
ever
al g
roup
s of
mal
es
and
fem
ales
419
1995
Del
ayed
Neu
roto
xici
ty o
f O
rgan
opho
spho
rus
Subs
tanc
es: 2
8-da
y R
epea
ted
Dos
e St
udy
Ass
essm
ent a
nd e
valu
atio
n of
the
toxi
c ef
fect
s of
org
anop
hosp
orus
sub
stan
ces
486
1997
Uns
ched
uled
DN
A S
ynth
esis
Tes
t with
Mam
mal
ian
Liv
er C
ells
in v
ivo
Iden
tifi c
atio
n of
sub
stan
ces
that
indu
ce D
NA
rep
air
afte
r ex
cisi
on a
nd r
emov
al
of D
NA
reg
ions
of
dam
age
indu
ced
by c
hem
ical
sub
stan
ces
408
1998
Rep
eate
d D
ose
90-D
ay O
ral T
oxic
ity S
tudy
in R
oden
tsId
entifi
cat
ion
of h
ealth
haz
ards
like
ly to
ari
se f
rom
exp
osur
e to
a te
st s
ubst
ance
via
or
al a
dmin
istr
atio
n40
919
98R
epea
ted
Dos
e 90
-Day
Ora
l Tox
icity
Stu
dy in
N
on-R
oden
tsId
entifi
cat
ion
of h
ealth
haz
ards
like
ly to
ari
se f
rom
sho
rt-t
erm
exp
osur
e to
a te
st s
ubst
ance
via
ora
l adm
inis
trat
ion
414
2001
Pren
atal
Dev
elop
men
t Tox
icity
Stu
dyD
evel
opm
enta
l tox
icity
test
des
igne
d to
pro
vide
gen
eral
info
rmat
ion
on th
e ef
fect
s of
pr
enat
al e
xpos
ure
on p
regn
ant t
est a
nim
als
and
on th
e de
velo
ping
org
anis
m41
620
01Tw
o-G
ener
atio
n R
epro
duct
ion
Toxi
city
Two-
gene
ratio
n re
prod
uctio
n te
st d
esig
ned
to p
rovi
de g
ener
al in
form
atio
n on
the
effe
cts
of a
test
sub
stan
ce o
n th
e in
tegr
ity a
nd p
erfo
rman
ce o
f th
e m
ale
and
fem
ale
repr
oduc
tive
syst
ems

ACUTE TOXICOLOGY TESTING 79
420
2002
Acu
te O
ral T
oxic
ity -
Fix
ed-D
ose
Proc
edur
e (s
ee te
xt)
Use
of
only
mod
erat
ely
toxi
c do
ses
are
used
in th
e m
ain
stud
y; a
dmin
istr
atio
n of
do
ses
that
are
exp
ecte
d to
be
leth
al s
houl
d be
avo
ided
404
2002
Acu
te D
erm
al I
rrita
tion/
Cor
rosi
onId
entifi
cat
ion
of h
ealth
haz
ards
like
ly to
ari
se f
rom
exp
osur
e to
a li
quid
or
solid
test
su
bsta
nce
by d
erm
al a
pplic
atio
n40
520
02A
cute
Eye
Irr
itatio
n/C
orro
sion
Iden
tifi c
atio
n of
hea
lth h
azar
ds li
kely
to a
rise
fro
m e
xpos
ure
to a
test
sub
stan
ce
(liq
uids
, sol
ids,
and
aer
osol
s) b
y ap
plic
atio
n on
the
eye
427
2004
Skin
Abs
orpt
ion:
In
Vivo
Met
hod
An
in v
ivo
perc
utan
eous
abs
orpt
ion
stud
y; p
rovi
des
a lin
k ne
cess
ary
to e
xtra
pola
te
from
ora
l stu
dies
fol
low
ing
derm
al e
xpos
ure
426
2007
Dev
elop
men
tal N
euro
toxi
city
Stu
dyA
dev
elop
men
tal n
euro
toxi
city
stu
dy; i
dent
ifi ca
tion
of e
ffec
ts a
fter
rep
eate
d ex
posu
re to
a s
ubst
ance
dur
ing
in u
tero
and
ear
ly p
ostn
atal
dev
elop
men
t44
020
07U
tero
trop
hic
Bio
assa
y in
Rod
ents
An
in v
ivo
shor
t-te
rm s
cree
ning
test
bas
ed o
n th
e in
crea
se in
ute
rine
wei
ght o
r ut
erot
roph
ic r
espo
nse
407
2008
Rep
eate
d D
ose
28-d
ay O
ral T
oxic
ity S
tudy
in
Rod
ents
Iden
tifi c
atio
n of
hea
lth h
azar
ds li
kely
to a
rise
fro
m e
xpos
ure
to a
test
sub
stan
ce v
ia
oral
adm
inis
trat
ion
425
2008
Acu
te O
ral T
oxic
ity: U
p-an
d-D
own
Proc
edur
e (s
ee te
xt)
Est
imat
ion
of a
n L
D50
with
a c
onfi d
ence
inte
rval
; allo
ws
for
clas
sifi c
atio
n of
a
subs
tanc
e fo
r ac
ute
toxi
city
acc
ordi
ng to
the
Glo
bally
Har
mon
ized
Sys
tem
of
clas
sifi c
atio
n an
d la
belin
g40
320
09A
cute
Inh
alat
ion
Toxi
city
Iden
tifi c
atio
n of
hea
lth h
azar
ds li
kely
to a
rise
fro
m s
hort
-ter
m e
xpos
ure
to a
test
ar
ticle
(ga
s, v
apor
, or
aero
sol/p
artic
ulat
e te
st a
rtic
le)
by in
hala
tion
452
2009
Chr
onic
Tox
icity
Stu
dies
Cha
ract
eriz
atio
n of
a te
st s
ubst
ance
in a
mam
mal
ian
spec
ies
(pri
mar
ily r
oden
ts)
follo
win
g pr
olon
ged
and
repe
ated
exp
osur
e45
320
09C
ombi
ned
Chr
onic
Tox
icity
/Car
cino
geni
city
Stu
dies
Iden
tify
the
maj
ority
of
chro
nic
and
carc
inog
enic
eff
ects
and
det
erm
ine
dose
–res
pons
e re
latio
nshi
ps f
ollo
win
g pr
olon
ged
and
repe
ated
exp
osur
e44
120
09H
ersh
berg
er B
ioas
say
in R
ats
An
in v
ivo
shor
t–te
rm s
cree
ning
test
to e
valu
ate
the
abili
ty o
f a
chem
ical
to e
licit
biol
ogic
al a
ctiv
ities
con
sist
ent w
ith a
ndro
gen
agon
ists
, ant
agon
ists
or
5 á-
redu
ctas
e in
hibi
tors
(Con
tinu
ed)
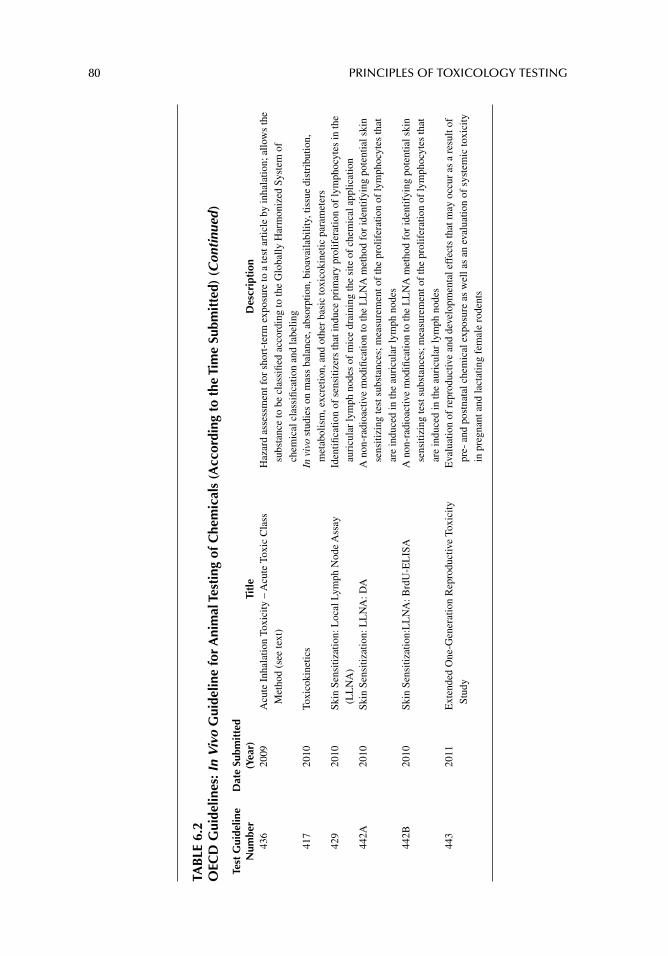
80 PRINCIPLES OF TOXICOLOGY TESTING
TAB
LE 6
.2O
ECD
Gui
delin
es: I
n V
ivo
Gui
delin
e fo
r A
nim
al T
esti
ng o
f Che
mic
als
(Acc
ordi
ng t
o th
e Ti
me
Subm
itte
d) (
Con
tinu
ed )
Test
Gui
delin
e N
umbe
rD
ate
Subm
itte
d (Y
ear)
Titl
eD
escr
ipti
on43
620
09A
cute
Inh
alat
ion
Toxi
city
– A
cute
Tox
ic C
lass
M
etho
d (s
ee te
xt)
Haz
ard
asse
ssm
ent f
or s
hort
-ter
m e
xpos
ure
to a
test
art
icle
by
inha
latio
n; a
llow
s th
e su
bsta
nce
to b
e cl
assi
fi ed
acco
rdin
g to
the
Glo
bally
Har
mon
ized
Sys
tem
of
chem
ical
cla
ssifi
catio
n an
d la
belin
g41
720
10To
xico
kine
tics
In v
ivo
stud
ies
on m
ass
bala
nce,
abs
orpt
ion,
bio
avai
labi
lity,
tiss
ue d
istr
ibut
ion,
m
etab
olis
m, e
xcre
tion,
and
oth
er b
asic
toxi
coki
netic
par
amet
ers
429
2010
Skin
Sen
sitiz
atio
n: L
ocal
Lym
ph N
ode
Ass
ay
(LL
NA
)Id
entifi
cat
ion
of s
ensi
tizer
s th
at in
duce
pri
mar
y pr
olif
erat
ion
of ly
mph
ocyt
es in
the
auri
cula
r ly
mph
nod
es o
f m
ice
drai
ning
the
site
of
chem
ical
app
licat
ion
442A
2010
Skin
Sen
sitiz
atio
n: L
LN
A: D
AA
non
-rad
ioac
tive
mod
ifi ca
tion
to th
e L
LN
A m
etho
d fo
r id
entif
ying
pot
entia
l ski
n se
nsiti
zing
test
sub
stan
ces;
mea
sure
men
t of
the
prol
ifer
atio
n of
lym
phoc
ytes
that
ar
e in
duce
d in
the
auri
cula
r ly
mph
nod
es44
2B20
10Sk
in S
ensi
tizat
ion:
LL
NA
: Brd
U-E
LIS
AA
non
-rad
ioac
tive
mod
ifi ca
tion
to th
e L
LN
A m
etho
d fo
r id
entif
ying
pot
entia
l ski
n se
nsiti
zing
test
sub
stan
ces;
mea
sure
men
t of
the
prol
ifer
atio
n of
lym
phoc
ytes
that
ar
e in
duce
d in
the
auri
cula
r ly
mph
nod
es44
320
11E
xten
ded
One
-Gen
erat
ion
Rep
rodu
ctiv
e To
xici
ty
Stud
yE
valu
atio
n of
rep
rodu
ctiv
e an
d de
velo
pmen
tal e
ffec
ts th
at m
ay o
ccur
as
a re
sult
of
pre-
and
pos
tnat
al c
hem
ical
exp
osur
e as
wel
l as
an e
valu
atio
n of
sys
tem
ic to
xici
ty
in p
regn
ant a
nd la
ctat
ing
fem
ale
rode
nts

ACUTE TOXICOLOGY TESTING 81
TABLE 6.3List of OECD Guidance Documents and Detailed Review Papers for Animal Testing of Chemicals
Test Guideline Number
Date Submitted
(Year) Title Description
47 2004 DRP on Fish Screening Assays for the Detection of Endocrine Active Substances
Fish screening assays for chemicals active at the endocrine level on the reproductive system of test animals
46 2004 DRP on Amphibian Metamorphosis Assay for the Detection of Thyroid Active Substances
Amphibian metamorphosis as a model for the detection of chemicals affecting the thyroid axis
45 2004 GD on Use of Multimedia Models for Estimating Overall Environmental Persistence and Long-Range Transport
Use of multimedia models in risk assessment and management of chemicals: POPs; PBTs
44 2004 Descriptions of Selected Key Generic Terms used in Chemical Hazard/Risk Assessment
Internationally harmonized generic and technical terms used in chemical hazard/risk assessment; help facilitate the mutual use and acceptance of the assessment of chemicals
36 2002 Report of the OECD/UNEP Workshop on the Use of Multimedia Models for Estimating Overall Environmental Persistence and Long-Range Transport for PBTS/POPS Assessment
Report from the OECD/UNEP Workshop on the Use of Multimedia Models for Estimating Overall Persistence and Long-range Transport in the Context of PBTs/POPs Assessment, October 2001
35 2002 Guidance Notes for Analysis and Evaluation of Chronic Toxicity and Carcinogenicity Studies
Outlines core concepts to obviate the need to consult large numbers of text books, while still providing sources of more detailed or specifi c information
32 2002 Guidance Notes for Analysis and Evaluation of Repeat-Dose Toxicity Studies
General guide to the analysis and evaluation of data from studies involving repeated exposures of toxicity test species to pesticides and other chemicals
7 2002 GD on Direct Phototransformation of Chemicals in Water
Presents basic concepts of photochemistry applicable to aqueous photolysis, as well as the existing national guidelines and standards
5 2002 Report of the SETAC/OECD Workshop on Avian Toxicity Testing
Presents a report on a Workshop on Avian Toxicity Testing held in Pensacola, Florida, USA, in 1994
4 2002 Report of the OECD Workshop on Environmental Hazard/Risk Assessment
Environment Monograph report of the OECD Workshop on Environmental Hazard/Risk Assessment, London 1994
2 2002 DRP on Biodegradability Testing Reviews biodegradability testing to identify whether there is a need to revise existing OECD test guidelines or develop new guidelines
1 2002 GD for the Development of OECD Guidelines for Testing of Chemicals
Describes the process of Test Guideline development, including the structure of the Test Guidelines Program, the responsibilities of those involved and the procedures that should be followed
Abbreviations: DRD, Detailed Review Document; DRP, Detailed Review Paper; GD, Guidance Document; GHS, globally harmonized system; OECD, Organization for Economic Co-operation and Development; PBTs, persistent and bio-accumulating toxins; POPs, persistent organic pollutants.

82 PRINCIPLES OF TOXICOLOGY TESTING
TABLE 6.4List of OECD Guidance Documents and Detailed Review Documents for Animal Testing of Chemicals (Extended Titles)
Test Guideline Number
Date (Year)
Submitted Extended Titles
30 2002 DRD on Hazard Classifi cation Systems for Mixtures33 2002 DRD on Hazard Classifi cation Systems for Specifi c Target Organ Systemic Toxicity42 2004 GD on Reporting Summary Information on Environmental, Occupational, and
Consumer Exposure41 2004 DRD on Classifi cation in OECD Member Countries of Substances and Mixtures
which come in contact with water release toxic gasesDRD on Classifi cation in OECD Member Countries of Substances and Mixtures
which cause Respiratory Tract Irritation and Corrosion38 2004 Detailed Background Review of the Uterotrophic Bioassay37 2002 DRD on Classifi cation Systems for Substances which Pose an Aspiration Hazard33 2002 Harmonized Integrated Classifi cation System for Human Health and Environmental
Hazards of Chemical Substances and Mixtures30 2002 DRD on Hazard Classifi cation Systems for Mixtures29 2002 GD on Transformation/ Dissolution of Metals and Metal Compounds
in Aqueous Media28 2004 GD for the Conduct of Skin Absorption Studies27 2002 GD on the Use of GHS for Classifi cation of Chemicals which are Hazardous for
Aquatic Environments26 2002 Revised Analysis of Responses Received from Member Countries to the
Questionnaire on Data Requirements for Acute Oral Toxicity25 2002 DRD on Hazard Classifi cation Systems for Specifi c Target Organ Systemic Toxicity
Following Single or Repeated Exposure in OECD Member Countries24 2002 GD on Acute Oral Toxicity Testing23 2002 GD on Aquatic Toxicity Testing of Diffi cult Substances and Mixtures21 2002 DRP Appraisal of Test Methods for Sex Hormone Disrupting Chemicals19 2002 GD on the Recognition, Assessment, and Use of Clinical Signs as Human Endpoints
for Experimental Animals Used in Safety Evaluation18 2002 Report of the OECD Workshop on Improving the Use of Monitoring Data in the
Exposure Assessment of Industrial Chemicals17 2002 Environmental Exposure Assessment Strategies for Existing Industrial Chemicals in
OECD Member Countries16 2002 DRD on Classifi cation Systems for Skin Irritation/Corrosion in OECD
Member Countries15 2002 DRD on Classifi cation Systems for Reproductive Toxicity in OECD Member Countries14 2002 DRD on Classifi cation Systems for Eye Irritation/Corrosion in OECD Member
Countries13 2002 DRD on Classifi cation Systems for Sensitizing Substances in OECD Member Countries12 2002 DRD on Classifi cation Systems for Germ-Cell Mutagenicity in OECD Member Countries11 2002 DRP on Aquatic Testing Methods for Pesticides and Industrial Chemicals10 2002 Report of the OECD Workshop on Statistical Analysis of Aquatic Toxicity Data 9 2002 GD for the Conduct of Studies of Occupational Exposure to Pesticides During
Agricultural Application 8 2002 Report of the OECD Workshop on Sharing Information about New Industrial
Chemicals Assessment
Abbreviations: DRD, Detailed Review Document; DRP, Detailed Review Paper; GD, Guidance Document; GHS, Globally Harmonized System; OECD, Organization for Economic Cooperation and Development.

ACUTE TOXICOLOGY TESTING 83
If it survives 48 hours after treatment, a preset higher dose (0.5 log spacing) is administered orally to a second animal. If the fi rst animal dies, the second animal is dosed at a preset lower dose. Dosing stops when one of three criteria is satisfi ed,1 with as few as six but not more than 15 animals used per test.
In the revised UDP limit test, one animal receives a limit dose (2.0 or 5.0 g/kg). If the animal dies, the UDP primary test is conducted. If the animal lives, two more animals are dosed concurrently at the limit dose. If both of these animals live (i.e., three animals sur-vive), the UDP limit test is stopped. If one or both of the two animals die, additional ani-mals are dosed sequentially at the limit dose until either three animals survive or three animals die, with the maximum number of animals tested set at fi ve. If three animals sur-vive, the LD
50 is above the limit dose. Conversely, if three animals die, the LD
50 is below
the limit dose level. In the UDP supplemental test for determining slope and CI, three treatment schedules
at increasing dose levels are initiated at 10- to 30-fold below the estimated LD50
obtained in the primary test. Dosing continues in each sequence until an animal dies. All data, including those obtained in the primary test are then considered in a statistical model that estimates the slope and CI for calculating the LD
50.
FIXED-DOSE PROCEDURE
The FDP was fi rst proposed in 1984 by the British Toxicology Society as an alternative to the conventional LD
50 test (OECD Test Guideline 401) for determining acute oral toxicity. The
FDP used fewer animals and caused less suffering than the LD50
test and provided informa-tion on acute toxicology that allowed substances to be classifi ed according to the European Union hazard classifi cation system. In 1992, the FDP was introduced as OECD Test Guide-line 420. In 1999, as part of an initiative to phase out Test Guideline 401, a review of the FDP was undertaken, the aim of which was to provide further reductions and refi nements, and classifi cation according to the criteria of the United Nations’ Globally Harmonized System (GHS) of classifi cation and labeling of chemicals.
ACUTE TOXIC CLASS METHOD
Traditional methods for assessing acute toxicology use deaths of tested animals as intended endpoints. The ATCM (OECD Test Guideline 423) avoids this criterion as an exclusive indicator by incorporating evident clinical signs of toxicity at one of a series of fi xed dose levels on which to base classifi cation of the test material. Refi nements to this protocol have been introduced in order to minimize the suffering and distress of test animals. The oral ATCM uses three animals of one sex per step, thus incorporating considerably fewer ani-mals. The method provides information on the hazardous properties of a chemical and allows it to be ranked and classifi ed according to the GHS classifi cations of chemicals that cause acute toxicology. Another specifi c ATCM procedure, known as the inhalation ATC method is based on the specifi c requirements for inhalation toxicity testing.
The ATCM is based on a stepwise procedure, such that suffi cient information is obtained on the acute toxicology of a test substance to enable its classifi cation at any of the defi ned concentrations. The test requires simultaneous testing at two steps using three ani-mals of one sex per step. If evidence demonstrates that one gender is more susceptible, the
1Criteria include these points: Three consecutive animals survive at the upper bound dose, or fi ve reversals occur in any six consecutive animals tested, or at least four animals follow the fi rst reversal with the likelihood ratios exceeding the statistical critical value.

84 PRINCIPLES OF TOXICOLOGY TESTING
test is continued with the most susceptible gender. The absence or presence of compound-related mortality of the animals exposed at one step will determine the next step:
1. No further testing needed2. Testing of the most susceptible gender only3. Testing of an additional three male and three female animals at the next higher or
next lower concentration level
When there are indications that a test material is likely to be nontoxic, a limit test is performed as described earlier (see section, “Up-and-Down Procedure”). In addition, the ATCM further outlines standards and recommendations for the selection of animal species, housing and feeding conditions, preparation of animals, and mode of exposure, among other proposals.
CLASSICAL LD50
A variety of factors should be considered when planning and organizing a study for the determination of LD
50, not all of which are associated with the calculation of 95% confi -
dence limits. Most preparations among the different routes of administration are similar although some changes are required as shown below.
ORAL LD50
Table 6.5 summarizes some of the parameters and factors that are monitored as a classical oral LD
50 study commences. Such factors include but are not limited to (1) randomization of
animals, (2) maintenance of a narrow range of body weights, (3) appropriate number of animals per group, (4) identifi cation of individual test subjects, (5) fasting, and (6) availabil-ity of water. Dosage range is determined according to techniques outlined above. Prelimi-nary range-fi nding experiments are initiated to minimize the extent of no lethality or 100% lethality, thus reducing the number of groups in the total study. This method also improves the precision of the LD
50 determination.
DERMAL LD50
Dermal LD50
studies are conducted on toxicants if the probable exposure route is through skin absorption. As with the oral LD
50, lethality is generally assessed in two species, one of
which is non-rodent. Also, the test substance is applied to shaved skin in increasing doses to
TABLE 6.5Considerations During Preparation for Oral LD50 Study
Parameter Factors for Consideration
Randomization of animals Unbiased distribution into groupsNarrow range of body weights Uniform distribution of similar sized animalsNumber of animals For classical LD
50, average of 10 per treatment group
Identifi cation of individual animals Ensures individual observation and monitoring; allows for group housingFasting (16–24 hrs) Optimal GI absorptionWater ad libitum Prevention of dehydration

ACUTE TOXICOLOGY TESTING 85
several groups of experimental animals, one dose per group. The parameters involved with the dermal LD
50 determination are the same as those described for the oral LD
50 (Table 6.5).
Some of the factors that are unique to dermal studies are summarized in Table 6.6.Most of the variability in dermal LD
50 studies arises from these parameters. Incomplete
absorption of a toxicant due to poor vehicle solubility, its inability to penetrate intact skin, and lack of uniformity in the application method are largely responsible for the inconsis-tencies seen with dermal LD
50 studies. Because of known or suspected inadequate absorp-
tion, an LC50
(median lethal concentration 50%) is determined after absorption and may substitute for the lack of a reliable dosage determination. In fact, cutoff values of 1 g/kg doses constitute an upper limit. Also, animals are prepared at least 24 hours prior to initia-tion of the experiment, and the application area is prepared to encompass about 10% of total body surface area. Application procedure and volume of material are consistent and uniform—planned so as to minimize technical variability. Occlusive devices or mechanical restraints are avoided if possible.
INHALATION LD50
Air-borne toxic materials that are transported via gases, aerosols, smoke, or ventilation necessitate the determination of acute inhalation LD
50. As described for the classical LD
50,
rodents and a non-rodent species are exposed for 4–24 hours to a test substance in increas-ing concentrations (one concentration per group, at least four doses plus a control group). Well-controlled inhalation studies incorporate a negative pressure, dynamic inhalation sys-tem with programmable airfl ow settings. Currently used systems, such as that shown in Figure 6.1, are capable of delivering precise test material concentrations, continuously monitoring toxicant in the exposure chambers, and include exhaust ducts capable of shunt-ing chamber air through ductal fume vents. Some systems accommodate either whole–body exposure or oral–nasal exposure only. The range of doses is capable of producing a corre-sponding series of toxic effects and mortality rates to facilitate assessment of acute toxicity for LD
50 or LC
50 determination.
Table 6.7 presents some of the diffi culties unique to inhalation studies. The concentra-tion of toxicant is calculated as the total amount of test substance delivered through the inhalation system with respect to the air pressure and volume of air, as monitored by head-space gas chromatography. Concentrations are calculated, monitored, and adjusted as nec-essary. However, net toxic concentrations delivered to the animals are infl uenced by airfl ow rates, particle size distribution, and air temperature and humidity, all of which can be mon-itored via serial programmable detectors.
TABLE 6.6Considerations During Preparation for Dermal LD50 Study
Parameter Factors for Consideration
Formulation Solids dissolved in water or inert oil-based vehicle (dimethyl sulfoxide, propylene glycol); liquid, paste, ointment, patch
Application to skin Shave fur 24 hrs prior to test; uniform application; 2–3 cm2 area for smaller, 3–5 cm2 for larger animals (10%); maintain consistency; application volume = 1–2 mL/kg
Absorption of toxicant Depends on water-soluble or lipid-soluble properties (latter is more rapid)Solubility Lipid-soluble liquid more rapidly absorbed; water-soluble liquid requires more time;
use occlusive dressing if necessaryVariability of results Dermal studies associated with a high degree of variability; determine LC
50

86 PRINCIPLES OF TOXICOLOGY TESTING
TABLE 6.7Parameters for Consideration: Special Emphasis on Inhalation LD50 Studies
Parameter Factors for Consideration
Concentration of a delivered test agent
Airfl ow rate into chamber; air temperature and humidity; monitoring of concentration in chamber; check integrity of exposure chamber
Particle size Particle size of particulates determines distribution to target organ (lower or upper respiratory tract); size infl uences the solubility of a toxicant in air
Respiration rate Light and dark cycles affect respiration rate; contact with irritant induces behavioral, secretory, and infl ammatory responses altering exposure rate
Control groups Infl uence of co-solvents, additives, air pressure
FIGURE 6.1 Photo of a rodent exposure chamber for inhalation (smoking) toxicology studies. Shown is the TE-10 smoking machine with its hood unit, air compressor, rat exposure chambers, meters, and controls. Source: Courtesy of Teague Enterprises, Davis, California, USA.
OTHER CONSIDERATIONS WITH LD50 DETERMINATIONS
Adequate planning of an experimental protocol minimizes errors and unanticipated results. Consideration of several factors, as discussed previously, reduces experimental error, opti-mizes result analysis, and maximizes resource utilization. In addition, laboratories are now encouraged to consider using currently available in vitro toxicology screening protocols and assays in addition to in vivo studies. In vitro studies are particularly benefi cial as preliminary screening tests prior to animal toxicity studies. They are also used to supplement, reduce, or refi ne animal testing as well as yield mechanistic data in support of LD
50 determinations.
ROUTE OF ADMINISTRATION
A toxic agent is administered according to the environmental, occupational, or clinical expo-sure expected in humans or animals. Although most exposures are via the oral or local route (and not limited to ocular, dermal, or pulmonary routes) as encountered with occupational exposure, some agents such as therapeutic drugs and biologicals may require testing by

ACUTE TOXICOLOGY TESTING 87
parenteral routes. Oral administration may also be refl ected in animal testing using several methods. For instance, acute oral administration is accomplished through
1. adding a chemical to drinking water. This requires controlled distribution of the volume of water to deliver the appropriate concentration of test chemical.
2. adding a test chemical to solid food. This also necessitates the monitoring of food volume intake and includes the risk of reduced absorption of chemical in the pres-ence of stomach contents.
3. administering via gastric gavage. The agent is dissolved in a solvent vehicle and administered through a round tipped gastric needle and syringe. By maintaining constant volumes (2–5 mL/kg total volume per oral injection), precise dosages (mg/kg body weight) are allotted.
4. diet supplement. Animals have natural aversions to otherwise miscible liquid chem-icals such as alcohols and bitter, irritating, organic liquids that are added directly to drinking water, to the extent that the animals will dehydrate before consuming enough of the test substance. In this situation, a complete liquid diet is prepared from commercially available fortifi ed liquid diets or reconstituted powder formula-tions. The liquid diet is then supplemented with the active ingredient by volume (% w/v), allowing for well-controlled delivery of test material. After initial acclima-tion and weight loss, the combination of hunger and thirst drives overcomes the natural aversion to the added test ingredient.
Dermal application of toxic substances is also subject to solubility considerations such as dissolution of the active ingredient in various forms: a solution, powder, ointment, cream, and paste. The objective is to ensure adequate penetration of the vehicle and active ingredient through dermal barriers. This may necessitate covering the treated area with occlusive wrapping. The substance remains in contact for the specifi ed exposure time, after which the area is rinsed clean of material.
With inhalation exposure of chemicals in the form of gases, vapors, particulate matter, or aerosols, the principal consideration is the delivery of the correct concentration. In addi-tion, inhalation studies require placing adequate numbers of animal in a toxicant-infused chamber at different times to accommodate the range of concentrations. Exposure times vary from 2 hours up to 96 hours, after which the animals are removed from the chamber.
DURATION
Although LD50
studies are generally performed during the fi rst 24-hour exposure period, needless suffering is avoided by removing and euthanizing prostrate, ill, or dead animals from treatment groups. Suffi cient results are also obtained by determining the time of mor-bidity or mortality, as well as employing any toxicological analyses.
GENERAL APPEARANCE OF ANIMALS
Although mortality is the major goal of an LD50
study, data gathering may be optimized by performing other toxicological analyses. Close observation of treated animals throughout a study period should include the following:
1. General appearance (compared to control animals) 2. Onset, intensity, and duration of toxic effects

88 PRINCIPLES OF TOXICOLOGY TESTING
3. Changes in behavior, activity, respiration, appetite, fl uid intake, or food retention 4. Monitoring of body weight, food intake, water consumption, and skin or fur turgor
SPECIMEN COLLECTION AND GROSS PATHOLOGY
Depending on the animal species, blood samples are obtained during the course of an exper-iment and at its conclusion, from both rodent and non-rodent species. Microliter blood sam-ples are acquired using puncture methods or small skin incisions. Milliliter blood samples are secured using tail or ocular venipuncture in rodents or ear venipuncture in larger mam-mals. In addition, urinalysis and fecal analysis are readily accomplished on samples col-lected from animals placed overnight in appropriate metabolic cages. Non-invasive neurological assessment is performed with behavioral testing apparatus (rotorod, maze, and learning reinforcement activities). In addition, the study is designed to conduct gross patho-logical examination at any time, including histological examination of tissue specimens and hematological analysis. Selected organs are quickly and precisely dissected into 1- to 2-cm sections and immersed in an appropriate fi xative for further histological processing.
BIOLOGICAL VARIATION
Intra- and interspecies differences exist in the metabolism and toxicology of chemicals and within a population of animals randomly selected for a study. Variation of response is observed in heterogeneous, randomly bred, strains of animals, posing the possibility of encountering substantial errors in estimating LD
50 values. In addition, signifi cant skewing of
the dose–response relationship may be encountered in inbred animals—animals whose homogeneous genetic pool origins suggest similar responses.
In addition, extrapolations of LD50
data to human toxicology and carcinogenicity are not necessarily linear because humans may be more susceptible than monkeys, rats, and mice to toxicological effects in general and carcinogenic chemicals in particular. Striking interspecies variation in susceptibility to several carcinogens is known; for example, rats are the most sensitive species to afl atoxin B1. Effi cient conjugation with glutathione, how-ever, confers afl atoxin B1 resistance to mice. Extremely large interspecies differences in herbicide and pesticide-induced toxicity are known among guinea pigs (more susceptible) and hamsters (most resistant). One of the most complex challenges, therefore, is awareness of the biological variations present within species and extrapolation of this information from laboratory animals to humans.
DETERMINATION OF ACUTE LETHALITY
As noted in chapter 3, the path by which a dose elicits a particular response is known as the dose–response (or concentration effect) relationship. The observed response is a calculated observation, assuming that the response is a result of exposure to a chemical and is measured and quantifi ed. The response also depends on the quantity of chemical exposure and admin-istration within a given period. Two types of dose–response relationships exist, depending on the number of subjects and doses tested. The graded dose–response (Fig. 6.2) describes the relationship of test subjects to logarithmic increases in the dose or concentration of a chem-ical. The concentration of the chemical is proportional to the number of surviving subjects in the experimental system or any other parameter of morbidity.
The quantal dose–response (Fig. 6.3) is determined by the distribution of responses to increasing doses in a population of test subjects. This relationship is generally classifi ed as an all-or-none effect and the animals are quantifi ed as either responders or nonresponders. Figure 6.4 shows a typical graph for comparing ED
50 (median effective dose 50%) to LD
50.
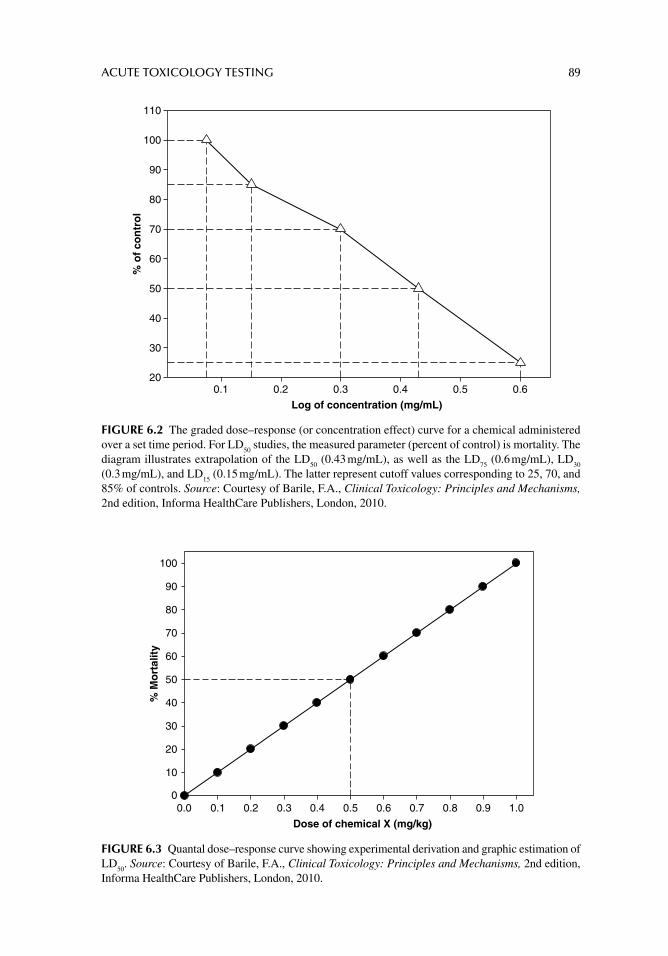
ACUTE TOXICOLOGY TESTING 89
Log of concentration (mg/mL)0.1 0.2 0.3 0.4 0.5 0.6
% o
f co
ntr
ol
20
30
40
50
60
70
80
90
100
110
FIGURE 6.2 The graded dose–response (or concentration effect) curve for a chemical administered over a set time period. For LD
50 studies, the measured parameter (percent of control) is mortality. The
diagram illustrates extrapolation of the LD50
(0.43 mg/mL), as well as the LD75
(0.6 mg/mL), LD30
(0.3 mg/mL), and LD
15 (0.15 mg/mL). The latter represent cutoff values corresponding to 25, 70, and
85% of controls. Source: Courtesy of Barile, F.A., Clinical Toxicology: Principles and Mechanisms, 2nd edition, Informa HealthCare Publishers, London, 2010.
Dose of chemical X (mg/kg)
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
% M
ort
alit
y
0
10
20
30
40
50
60
70
80
90
100
FIGURE 6.3 Quantal dose–response curve showing experimental derivation and graphic estimation of LD
50. Source: Courtesy of Barile, F.A., Clinical Toxicology: Principles and Mechanisms, 2nd edition,
Informa HealthCare Publishers, London, 2010.
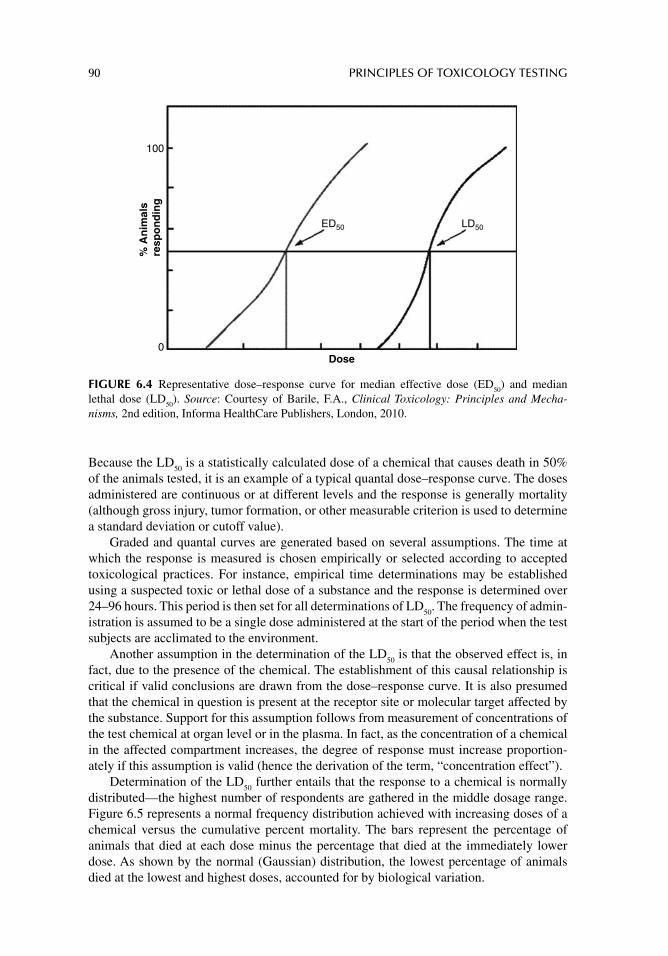
90 PRINCIPLES OF TOXICOLOGY TESTING
Because the LD50
is a statistically calculated dose of a chemical that causes death in 50% of the animals tested, it is an example of a typical quantal dose–response curve. The doses administered are continuous or at different levels and the response is generally mortality (although gross injury, tumor formation, or other measurable criterion is used to determine a standard deviation or cutoff value).
Graded and quantal curves are generated based on several assumptions. The time at which the response is measured is chosen empirically or selected according to accepted toxicological practices. For instance, empirical time determinations may be established using a suspected toxic or lethal dose of a substance and the response is determined over 24–96 hours. This period is then set for all determinations of LD
50. The frequency of admin-
istration is assumed to be a single dose administered at the start of the period when the test subjects are acclimated to the environment.
Another assumption in the determination of the LD50
is that the observed effect is, in fact, due to the presence of the chemical. The establishment of this causal relationship is critical if valid conclusions are drawn from the dose–response curve. It is also presumed that the chemical in question is present at the receptor site or molecular target affected by the substance. Support for this assumption follows from measurement of concentrations of the test chemical at organ level or in the plasma. In fact, as the concentration of a chemical in the affected compartment increases, the degree of response must increase proportion-ately if this assumption is valid (hence the derivation of the term, “concentration effect”).
Determination of the LD50
further entails that the response to a chemical is normally distributed—the highest number of respondents are gathered in the middle dosage range. Figure 6.5 represents a normal frequency distribution achieved with increasing doses of a chemical versus the cumulative percent mortality. The bars represent the percentage of animals that died at each dose minus the percentage that died at the immediately lower dose. As shown by the normal (Gaussian) distribution, the lowest percentage of animals died at the lowest and highest doses, accounted for by biological variation.
100%
An
imal
sre
spo
nd
ing
Dose
ED50 LD50
0
FIGURE 6.4 Representative dose–response curve for median effective dose (ED50
) and median lethal dose (LD
50). Source: Courtesy of Barile, F.A., Clinical Toxicology: Principles and Mecha-
nisms, 2nd edition, Informa HealthCare Publishers, London, 2010.
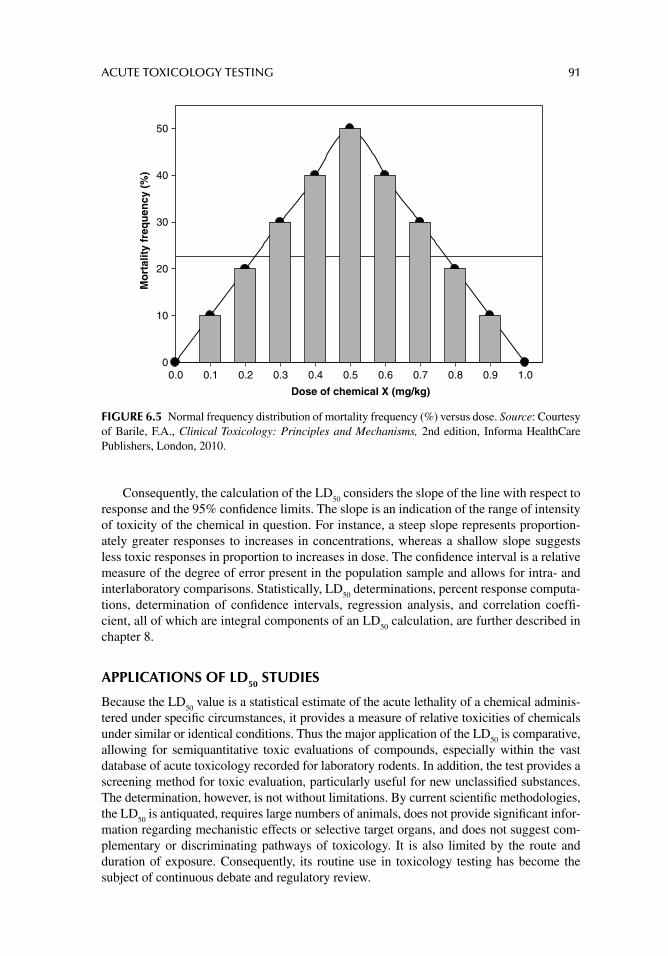
ACUTE TOXICOLOGY TESTING 91
Dose of chemical X (mg/kg)
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Mo
rtal
ity
freq
uen
cy (
%)
0
10
20
30
40
50
FIGURE 6.5 Normal frequency distribution of mortality frequency (%) versus dose. Source: Courtesy of Barile, F.A., Clinical Toxicology: Principles and Mechanisms, 2nd edition, Informa HealthCare Publishers, London, 2010.
Consequently, the calculation of the LD50
considers the slope of the line with respect to response and the 95% confi dence limits. The slope is an indication of the range of intensity of toxicity of the chemical in question. For instance, a steep slope represents proportion-ately greater responses to increases in concentrations, whereas a shallow slope suggests less toxic responses in proportion to increases in dose. The confi dence interval is a relative measure of the degree of error present in the population sample and allows for intra- and interlaboratory comparisons. Statistically, LD
50 determinations, percent response computa-
tions, determination of confi dence intervals, regression analysis, and correlation coeffi -cient, all of which are integral components of an LD
50 calculation, are further described in
chapter 8.
APPLICATIONS OF LD50 STUDIES
Because the LD50
value is a statistical estimate of the acute lethality of a chemical adminis-tered under specifi c circumstances, it provides a measure of relative toxicities of chemicals under similar or identical conditions. Thus the major application of the LD
50 is comparative,
allowing for semiquantitative toxic evaluations of compounds, especially within the vast database of acute toxicology recorded for laboratory rodents. In addition, the test provides a screening method for toxic evaluation, particularly useful for new unclassifi ed substances. The determination, however, is not without limitations. By current scientifi c methodologies, the LD
50 is antiquated, requires large numbers of animals, does not provide signifi cant infor-
mation regarding mechanistic effects or selective target organs, and does not suggest com-plementary or discriminating pathways of toxicology. It is also limited by the route and duration of exposure. Consequently, its routine use in toxicology testing has become the subject of continuous debate and regulatory review.

92 PRINCIPLES OF TOXICOLOGY TESTING
SUGGESTED READINGS
Arts JH, Muijser H, Jonker D, et al. Inhalation toxicity studies: OECD guidelines in relation to REACH and scientifi c developments. Exp Toxicol Pathol 2008; 60: 125.
Clode SA. Assessment of in vivo assays for endocrine disruption. Best Pract Res Clin Endocrinol Metab 2006; 20: 35.
Huxley A. Testing is necessary on animals as well as in vitro. Nature 2006; 439: 138.Lamb JC, Brown SM. Chemical testing strategies for predicting health hazards to children. Reprod
Toxicol 2000; 14: 83.Travis KZ, Pate I, Welsh ZK. The role of the benchmark dose in a regulatory context. Regul Toxicol
Pharmacol 2005; 43: 280.
REVIEW ARTICLES
Botham PA. Acute systemic toxicity: prospects for tiered testing strategies. Toxicol In Vitro 2004; 18: 227.
Brent RL. Utilization of animal studies to determine the effects and human risks of environmental toxicants (drugs, chemicals, and physical agents). Pediatrics 2004; 113: 984.
Gelbke HP, Kayser M, Poole A. OECD test strategies and methods for endocrine disruptors. Toxicol-ogy 2004; 205: 17.
Hayes A, Bakand S. Inhalation toxicology. EXS 2010; 100: 461.Kim JS, Song KS, Sung JH, et al. Genotoxicity, acute oral and dermal toxicity, eye and dermal irrita-
tion and corrosion and skin sensitisation evaluation of silver nanoparticles. Nanotoxicology 2012; Epub ahead of print.
Krishna G, Urda G, Theiss J. Principles and practices of integrating genotoxicity evaluation into routine toxicology studies: a pharmaceutical industry perspective. Environ Mol Mutagen 1998; 32: 115.
Landsiedel R, Ma-Hock L, Haussmann HJ, et al. Inhalation studies for the safety assessment of nano-materials: Status quo and the way forward. Wiley Interdiscip Rev Nanomed Nanobiotech-nol 2012; 4: 399.
Meyer O. Testing and assessment strategies, including alternative and new approaches. Toxicol Lett 2003; 140: 21.
Organisation for Economic Co-operation and Development (OECD) Guidelines for the Testing of Chemicals. [Available from: http://www.oecd.org/daf/internationalinvestment/guidelinesformultinationalenterprises/] [last accessed December 2012].
Pauluhn J. Overview of inhalation exposure techniques: strengths and weaknesses. Exp Toxicol Pathol 2005; 57: 111.
Rispin A, Farrar D, Margosches E, et al. Alternative methods for the median lethal dose (LD50
) test: the up-and-down procedure for acute oral toxicity. ILAR J 2002; 43: 233.
Rosenkranz HS, Cunningham AR. Lack of predictivity of the rat lethality (LD50
) test for ecological and human health effects. Altern Lab Anim 2005; 33: 9.
Rosolen SG, Rigaudiere F, Le Gargasson JF, Brigell MG. Recommendations for a toxicological screening ERG procedure in laboratory animals. Doc Ophthalmol 2005; 110: 57.
Schlede E, Genschow E, Spielmann H, et al. Oral acute toxic class method: a successful alternative to the oral LD
50 test. Regul Toxicol Pharmacol 2005; 42: 15.
Stallard N, Whitehead A. A statistical evaluation of the fi xed dose procedure. Altern Lab Anim 2004; 32: 73–80.
The Revised Up-and-Down Procedure: A Test Method for Determining the Acute Oral Toxicology of Chemicals, 2001, NIH Publication 02-4501. [Available from: http://iccvam.niehs.nih.gov/ docs/docs.htm#udp] [last accessed December 2012].
Walker DK. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Br J Clin Pharmacol 2004; 58: 601.
Warheit DB, Donner EM. Rationale of genotoxicity testing of nanomaterials: regulatory requirements and appropriateness of available OECD test guidelines. Nanotoxicology 2010; 4: 409.

93
7 Subchronic and chronic toxicology testing
INTRODUCTION
As discussed in chapter 6 covering acute toxicity studies, the shorter duration of exposure in acute studies allows monitoring of the effects of a chemical at high doses in a brief period. Such acute exposures occur in a variety of circumstances, particularly in emergency, occupational, environmental, and domestic settings. More frequently, clinical toxicology has shown that acute exposures to drugs of abuse and therapeutic drugs are of major inter-national concern and treatment of accidental or intentional overdoses poses a signifi cant challenge. For purposes of comparison, however, as much toxicity occurs with extended interactions with lower nonfatal concentrations of chemicals as that occurs with short intense exposures.
In addition, much regulatory emphasis is placed on long-term human and animal expo-sures to environmental toxins, occupational exposure to hazardous substances, and chronic effects of food and dietary supplements. Consequently, there exists an important incentive for the scientifi c toxicology sector to develop test methods for predicting relative chronic hazards and risks of exposure to chemicals in daily life. As a result, simulation of these interactions requires the development of several types of studies in order to mimic the human situation.
TYPES OF SUBCHRONIC AND CHRONIC TOXICITY TESTS
OBJECTIVES AND DEFINITIONS
In general, in animal and human risk assessment, acute and chronic durations of exposure are relative terms intended for comparative purposes. Any exposure up to 24 hours is regarded as acute. Exposure to many toxic gases (e.g., carbon monoxide, hydrogen cyanide) requires less than 24 hours to produce toxicity. Also, 72 hours may still constitute an acute exposure, such as in continuous low-dose exposure of children to acetaminophen and exposure of house pets to herbicides and insecticides. Subacute exposure is a term that has fallen into disuse. It formerly referred to repeated exposure of more than 72 hours but less than a month.
Chronic exposure is any relative period for which continuous or repeated exposure beyond the acute phase is required for the same chemical to induce a toxic response. For convenience, the term chronic also applies to the subchronic period. Subchronic expo-sure is also understood to involve a duration that is between acute and chronic. The traditional time of subchronic exposure is understood to be a period of 1–3 months. Thus, all these terms are fl exible adaptations for defi ning the onset of chemical intoxica-tion and a considerable overlap in judgment may occur in assigning labels to exposure

94 PRINCIPLES OF TOXICOLOGY TESTING
periods. In contrast to acute studies, the objectives of subchronic and chronic studies are as follows:
1. Determine the toxicological effects of repeated administration of test chemicals on potential target organs at subchronic or chronic dosages
2. Establish the dose–response (or concentration effect) relationships using various indicators over the selected dosage ranges and durations of exposure
3. Experimentally verify a maximum dosage level that does not promote ostensible toxicity with repeated exposures
4. Propose a mechanism of toxicity that complements or contrasts with acute studies of the chemical or class of agents
The following discussion describes the information necessary for addressing these objectives and explains how the experimental approach differs from the design of acute toxicology testing studies. It is important to note that comparability of results depends upon the similarities of the test criteria.
FACTORS ASSOCIATED WITH CHRONIC TOXICITY
Frequency of Exposure
It is important to emphasize that frequency of exposure involves repeated doses of a toxin during a certain period. Thus, continuous repeated exposure to a toxin, especially during a subchronic or chronic period, has greater toxic potential. In addition, dose, duration, frequency, and route of exposure contribute to chemical toxicity in part through accumulation of a compound in physiological compartments. A chemical’s toxic profi le is determined according to its half-life (t
1/2) in plasma and its effect, that is, the time
required for plasma levels to decrease to one-half of the measured or estimated concen-trations. If the frequency of exposure exceeds the t
1/2 of a chemical, its concentration in a
compartment is likely to increase beyond the desirable level. Consequently, in long-term interactions with chemicals, accumulation results from the overloading of an agent within this compartment.
Accumulation and Distribution
Once absorbed, a chemical distributes and/or binds to one or more of many physiological sites. The distribution of a chemical depends largely on its physiochemical characteristics. The compartments include whole blood, serum and serum proteins; plasma and plasma proteins; adipose tissue; interstitial and extracellular fl uids; alveolar air space; and bone marrow. In addition, any tissue or organ may preferentially accumulate a chemical, thus act-ing as a discrete compartment, as with the preferential accumulation of heavy metals in adipose tissue.
Consequently, toxicity may be experienced for a prolonged period as a compound is slowly released from this compartment, years after exposure has ceased. Accumulation, therefore, is predicted based on a chemical’s apparent volume of distribution (V
d), which is
estimated as the total dose of drug in the body divided by the concentration of drug in the plasma for a given period. In general, the greater the V
d, the greater is the potential for
accumulation in some physiological compartments. A standard measure for accumulated internal quantity of a chemical is the body burden, defi ned as the amount of chemical stored in one or several physiological compartments or in the body as a whole.

SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 95
Infl uence of Chemical Structure
Accumulation and its infl uence on chronic toxicity are also determined by a chemical’s structure and its interactions within physiological compartments. This phenomenon is guided by the chemical’s predominant ionic or non-ionic state of existence in a physiological fl uid. In general, at physiological pH, lipid-soluble compounds preferentially remain in a non-ionic state, preferring to bind to, penetrate, and accumulate in membranes of tissues and organs. Conversely, water-soluble compounds remain as ionic species at the pH of the blood. Thus, because they are less prone to tissue binding, the ions are readily available for renal secretion and elimination.
OBJECTIVES OF CHRONIC TOXICITY STUDIES
The goals of conducting chronic toxicity studies are similar to those of acute studies, with a few important differences. The objectives that overlap with acute studies include (1) determination of the lethal and toxic concentrations of a chemical and its effect on organs and tissues, (2) identifi cation of the causal relationship between the administered dose and the altered physiological, biochemical, and morphological changes, and (3) monitoring of animal species variations in response to an agent. Major differences between chronic studies and acute experiments, however, rely on frequency, accumula-tion, and length of exposure to a toxic agent. For instance, chronic studies are generally conducted to
1. measure or assess the toxic effects of lower, more frequently administered doses of a chemical, thus analyzing for repeated cumulative exposure.
2. determine cumulative effects of repeated exposure. 3. examine the toxicological effects of increasing doses of chemicals over extended
periods. 4. identify recovery of subjects after removal of the source of exposure. 5. predict long-term adverse health effects in the species arising from intermittent,
repeated, or continuous exposure.
Finally, chronic studies generally complement acute studies, assuming that the condi-tions are structured to enhance the results obtained in acute experiments.
EXPERIMENTAL DESIGN
As noted earlier, chronic and subchronic are often relative terms, especially in relation to the species. Historically, 2 years is a typical period for conducting chronic studies in rodents. However, 2 years does not represent a signifi cant portion of the total life span of other spe-cies such as dogs (average life expectancy of 10–15 years) or rabbits (5–6 years). Even in rodents sustaining calorie-restricted diets, the 2-year exposure period is signifi cantly less representative of their healthier, longer life span.
In general, as the life expectancy of the mammalian species increases, the exposure period must be adjusted to mimic chronic exposure. Thus it becomes more diffi cult to assign and extrapolate the chronic period in a particular species to adequately correlate the results of such a study to human risk assessment. Consequently, the lengths of chronic stud-ies (6 months to 3 years) are variable and fl exible and are determined according to the appropriateness of the objectives of a project.

96 PRINCIPLES OF TOXICOLOGY TESTING
OECD GUIDELINES FOR THE TESTING OF CHEMICALS
The Organization for Economic Cooperation and Development (OECD) Guidelines for the testing of chemicals are available on the website: http://www.oecd.org/chemicalsafety/testingofchemicals/chemicalstestingdraftoecdguidelinesforthetestingofchemicals-sections1-5.htm. Since the original publication in 1981, 18 addenda have been published. The guidelines are divided into fi ve sections; each section lists adopted guidelines with links to the draft propos-als. These are the sections:
Section 1: Physical Chemical Properties Section 2: Effects on Biotic Systems Section 3: Degradation and Accumulation Section 4: Health EffectsSection 5: Other Test Guidelines
The site also lists the Monograph Series on Testing and Assessment [i.e., Guidance documents (GDs) and Review documents (RDs)].
Section 1, Physical Chemical Properties, outlines monographs detailing procedures associated with determination of pH, acidity and alkalinity; density of liquids and solids; and, viscosity of liquids. Section 2, Effects on Biotic Systems, outlines a draft proposal for a new test guideline and offers revised monographs on several existing test guidelines, including the Fish embryo toxicity (FET) test, the Fish early life-stage test [test guideline (TG) 210], and the Daphnia magna reproduction test (TG 211). Section 3, Degradation and Accumulation, summarizes test guidelines for Bioaccumulation in Fish: Aqueous and Dietary Exposure (Draft TG 305), and Draft Validation Report of a ring test for the dietary exposure in fi sh. Section 4, Health Effects, offers an extensive list of guidance documents and monographs on the health effects of a variety of chemicals and toxins. The GDs, their numbers, and a brief description are summarized in Table 7.1.
SELECTION OF DOSAGE LEVELS
Dosage levels for chronic toxicology studies are selected based on existing information avail-able about a chemical from acute studies, known toxicological effects, animal and human epidemiological data, knowledge of the species reaction to chemicals of similar classes, and known toxic concentrations of chemicals from similar classes. In addition, in vitro data are also recommended as a method for screening chemicals for in vivo studies. Thus, familiarity and awareness of doseresponse curves, LD
50, EC
50, and IC
50 (in vitro inhibitory concentration
50% or concentration that causes 50% of measured response from a control in an in vitro system) values for the chemical aid an investigator in identifying the proper doses for a chronic in vivo study.
Understanding the details of the target end point is also essential for determining the dosage levels. As will be seen in later chapters, the dosages established in studies whose objectives involve the evaluation of histological or pathological monitoring for toxicity may be different from carcinogenic projects. As with acute studies, an investigator has some fl exibility in the selection of dosage levels, usually numbering up to fi ve groups plus a vehicle and/or blank control. In addition, preliminary experiments with small numbers of animals with repeated dosages are necessary to establish lowest and highest effector groups.
Doses are then selected based on preliminary range-fi nding pilot projects. Finally, recognition of the pharmacokinetics of the chemical, if available, contributes to the pre-diction of its behavior in physiological compartments during the course of a study. Con-sequently, a pilot study on limited numbers of animals for several weeks supplies the
SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 97
TAB
LE 7
.1O
ECD
Gui
danc
e D
ocum
ents
and
Mon
ogra
phs
on t
he H
ealt
h Ef
fect
s of
Che
mic
als
and
Toxi
ns—
In V
ivo
Gui
delin
es f
or A
nim
al T
esti
ng
of C
hem
ical
s Se
ctio
n 4.
Hea
lth
Effe
ctsa
TGN
umbe
rTi
tle
Obj
ecti
veD
escr
ipti
on
474
Mam
mal
ian
Ery
thro
cyte
Mic
ronu
cleu
s Te
stD
etec
t dam
age
to c
hrom
osom
es o
r m
itotic
app
arat
us
of e
ryth
robl
asts
indu
ced
by a
test
sub
stan
ce
Eva
luat
es th
e fo
rmat
ion
of m
icro
nucl
ei in
ery
thro
blas
ts
475
Mam
mal
ian
Bon
e M
arro
w C
hrom
osom
al
Abe
rrat
ion
Test
Det
ect s
truc
tura
l chr
omos
omal
abe
rrat
ions
Det
ects
chr
omos
omal
abe
rrat
ions
in b
one
mar
row
cel
ls o
f an
imal
s in
duce
d by
test
com
poun
ds
Dra
ftIn
Vitr
o C
arci
noge
nici
ty: S
yria
n H
amst
er
Em
bryo
Cel
l Tra
nsfo
rmat
ion
Ass
ayId
entifi
es
test
sub
stan
ces
that
hav
e ca
rcin
ogen
ic
pote
ntia
l in
vivo
Iden
tifi e
s in
duct
ion
of p
heno
typi
c al
tera
tions
in c
ells
that
are
ch
arac
teri
stic
of
tum
orig
enic
cel
ls45
7B
G1L
uc E
stro
gen
Rec
epto
r (E
R)
Tra
nsac
tivat
ion
In V
itro
Ass
ayPr
ovid
es a
qua
litat
ive
asse
ssm
ent o
f in
vitr
o an
ti-es
trog
enic
act
ivity
Use
s an
ER
-res
pons
ive
repo
rter
gen
e (l
uc)
in h
uman
ova
rian
ad
enoc
arci
nom
a ce
ll lin
e (B
G1)
to d
etec
t sub
stan
ces
with
in
vitr
o E
R a
ntag
onis
t act
ivity
430
In V
itro
Skin
Cor
rosi
on: T
rans
cuta
neou
s E
lect
rica
l Res
ista
nce
Test
Met
hod
Scre
enin
g fo
r no
n-co
rros
ive
and
corr
osiv
e su
bsta
nces
an
d m
ixtu
res
Iden
tifi e
s co
rros
ives
by
thei
r ab
ility
to p
rodu
ce a
loss
of
norm
al s
trat
um c
orne
um in
tegr
ity a
nd b
arri
er f
unct
ion
431
In V
itro
Skin
Cor
rosi
on: R
econ
stru
cted
H
uman
Epi
derm
is (
RhE
) Te
st M
etho
dSc
reen
ing
for
non-
corr
osiv
e an
d co
rros
ive
subs
tanc
es
and
mix
ture
sId
entif
y co
rros
ive
subs
tanc
es b
y th
eir
abili
ty to
pen
etra
te th
e st
ratu
m c
orne
um; c
ytot
oxic
ity is
mea
sure
d us
ing
the
MT
T
(3-(
4, 5
-dim
ethy
lthia
zoly
l-2)
-2, 5
-dip
heny
ltetr
azol
ium
br
omid
e) a
ssay
473
In v
itro
Mam
mal
ian
Chr
omos
ome
Abe
rrat
ion
Test
Iden
tify
agen
ts th
at c
ause
str
uctu
ral c
hrom
osom
e ab
erra
tions
in c
ultu
red
mam
mal
ian
cells
Cel
ls a
re a
naly
zed
mic
rosc
opic
ally
for
chr
omos
ome
aber
ratio
ns a
fter
exp
osur
e to
the
test
sub
stan
ce
487
In v
itro
Mam
mal
ian
Cel
l M
icro
nucl
eus
Test
Gen
otox
icity
test
for
the
dete
ctio
n of
mic
ronu
clei
in
the
cyto
plas
m o
f in
terp
hase
cel
lsD
etec
ts th
e ac
tivity
of
clas
toge
nic
and
aneu
geni
c ch
emic
als
in c
ells
that
hav
e un
derg
one
cell
divi
sion
dur
ing
or a
fter
ex
posu
re to
the
test
sub
stan
ceD
PSSt
ably
Tra
nsfe
cted
Tra
nsac
tivat
ion
In V
itro
Ass
ays
Det
ect e
stro
gen
agon
ists
Acc
ompa
nies
TG
455
, ER
TA
b
draf
t TG
Stab
ly T
rans
fect
ed T
rans
activ
atio
n In
Vitr
o A
ssay
sD
etec
t est
roge
n re
cept
or a
goni
sts
Acc
ompa
nies
TG
455
, ER
TA
(Con
tinu
ed)

98 PRINCIPLES OF TOXICOLOGY TESTING
TAB
LE 7
.1O
ECD
Gui
danc
e D
ocum
ents
and
Mon
ogra
phs
on t
he H
ealt
h Ef
fect
s of
Che
mic
als
and
Toxi
ns—
In V
ivo
Gui
delin
es f
or A
nim
al T
esti
ng
of C
hem
ical
s Se
ctio
n 4.
Hea
lth
Effe
ctsa (
Con
tinu
ed )
TG
Num
ber
Titl
eO
bjec
tive
Des
crip
tion
draf
t TG
BG
1 L
uc E
stro
gen
Rec
epto
r T
rans
crip
tiona
l Act
ivat
ion
Test
Met
hod
Iden
tifi e
s es
trog
en r
ecep
tor
agon
ists
and
ant
agon
ists
In v
itro
assa
y pr
ovid
ing
data
abo
ut s
elec
ted
endo
crin
e m
echa
nism
s-m
amm
alia
n an
d no
n-m
amm
alia
n m
etho
dsdr
aft T
GFl
uore
scei
n L
eaka
ge T
est M
etho
dIn
vitr
o te
st m
etho
d to
cla
ssif
y ch
emic
als
as o
cula
r co
rros
ives
and
sev
ere
irri
tant
sR
ecom
men
ded
as a
n in
itial
ste
p to
iden
tify
ocul
ar c
orro
sive
s an
d se
vere
irri
tant
sup
date
d 40
5 A
cute
Eye
Irr
itatio
n/C
orro
sion
In v
ivo
acut
e oc
ular
irri
tatio
n an
d/or
cor
rosi
on in
or
der
to r
educ
e an
imal
pai
n an
d di
stre
ssD
eter
min
e th
e de
gree
of
eye
irri
tatio
n/co
rros
ion
by s
cori
ng
lesi
ons
of c
onju
nctiv
a, c
orne
a, a
nd ir
is, a
t spe
cifi c
inte
rval
s.dr
aft T
GSt
ably
Tra
nsfe
cted
Tra
nscr
iptio
nal
Act
ivat
ion
In V
itro
Ass
ayId
entif
y es
trog
en a
goni
st a
ctiv
ity o
f ch
emic
als
Acc
ompa
nies
TG
455
, ER
TA
draf
t TG
BG
1Luc
ER
TA
Tes
t Met
hod
Iden
tify
ER
ago
nist
s an
d an
tago
nist
sSc
reen
ing
in v
itro
TA a
ssay
s ba
sed
upon
the
prod
uctio
n of
E
R r
epor
ter
gene
pro
duct
indu
ced
by a
che
mic
aldr
aft T
GT
he C
ytos
enso
r M
icro
phys
iom
eter
Tes
t M
etho
dId
entif
y ch
emic
als
clas
sifi e
d as
ocu
lar
corr
osiv
e,
seve
re ir
rita
nt, a
nd n
on-i
rrita
nt
Haz
ard
clas
sifi c
atio
n an
d la
belin
g of
che
mic
als
for
eye
corr
osio
n/ir
rita
tion,
acc
ordi
ng to
UN
GH
S dr
aft T
G (
4xx)
H29
5R S
tero
idog
enes
is A
ssay
Det
ect s
ubst
ance
s th
at a
ffec
t and
roge
n or
est
roge
n st
eroi
d ho
rmon
e pr
oduc
tion
Iden
tify
chem
ical
s th
at in
duce
or
inhi
bit t
he p
rodu
ctio
n of
T
and
E2
draf
t TG
(X
XX
)T
GR
In
Viv
o G
ene
Mut
atio
n A
ssay
s U
se o
f re
port
er g
enes
for
the
dete
ctio
n of
gen
e m
utat
ions
indu
ced
in v
ivo
by te
st s
ubst
ance
sU
se tr
ansg
enic
rod
ents
that
con
tain
mul
tiple
cop
ies
of
chro
mos
omal
ly in
tegr
ated
pla
smid
s or
vec
tors
All
docu
men
ts a
re c
onta
ined
in S
ecti
on 4
, Hea
lth
Effe
cts,
OE
CD
Gui
deli
nes
for
the
Test
ing
of C
hem
ical
s; s
ubm
itted
201
1–20
12.
a Ref
ers
to C
hem
ical
s Te
stin
g: D
raft
OE
CD
Gui
deli
nes
for
the
Test
ing
of C
hem
ical
s –
Sect
ions
1–5
.b I
n vi
tro
TA a
ssay
s ar
e ba
sed
upon
the
prod
uctio
n of
a re
port
er g
ene
prod
uct i
nduc
ed b
y a
chem
ical
, fol
low
ing
bind
ing
of th
e ch
emic
al to
a s
peci
fi c re
cept
or a
nd s
ubse
quen
t dow
nstr
eam
tr
ansa
ctiv
atio
n.A
bbre
viat
ions
: DPS
, Dra
ft P
erfo
rman
ce S
tand
ards
, acc
ompa
ny T
est G
uide
lines
(T
G);
E2,
17β
–est
radi
ol; E
R, e
stro
gen
rece
ptor
, T, t
esto
ster
one;
TA
, tra
nsac
tivat
ion
assa
y; T
GR
, tra
ns-
geni
c ro
dent
; UN
GH
S, U
nite
d N
atio
ns G
loba
lly H
arm
oniz
ed S
yste
m o
f C
lass
ifi ca
tion
and
Lab
elin
g of
Che
mic
als.

SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 99
necessary information to commence a full study. It also reduces the chances of altering the parameters during the course of the project, such as adjustment of doses, signifi cant changes in the number of animals, or modifi cation of the study parameters to accommo-date for excessive mortality or lack of effects.
SELECTION OF ANIMALS
Selection of the number and species of animal relies on the objectives of a chronic study, the parameters established in the experimental setup, and the ability to monitor and adjust to unanticipated and untoward results. In general, the selection of animals is as important in preparing for chronic studies as it is in acute experiments. Table 7.2 outlines some of these parameters that bear similarities to those established for acute studies (see Table 6.1). How-ever, considering that chronic experiments are longer in duration, attention is drawn to understanding the toxicological indicators and effects that may be unpredicted in such experiments.
In addition, maintenance of constant experimental conditions during the course of a 2-year study is more daunting than maintaining conditions for 24- to 96-hour acute experi-ments. Other factors to be monitored during the conduct of a chronic toxicity testing proj-ect are outlined in Table 7.3. Some of the circumstances that require premature sacrifi cing of animals are included. It is important that the decision to terminate the animal’s inclusion in the experiment is documented and that enough analyses of specimens are performed to determine a causal relationship, that is, whether the state of the animal is induced by exper-imental conditions or by factors unrelated to the test chemical.
TABLE 7.2Steps Necessary for Procuring and Handling Animals Prior to Initiation of Chronic Experiments
Organization Consideration Description
Prior to purchasing Selection of species, number, sex, and age of animals
Biological and economical calculations; applicability to human risk assessment; usually same sex; long-term arrangement for housing requirements; young animals
Selection of commercial animal source
Same as with acute studies: reputation of breeder, cost, availability of animals, technical support
Type of study Toxicological assessment; for carcinogenicity studyImmediately after
purchasing and prior to initiation of experiment
Quarantine and screening Same as with acute studies: acclimation of animals to new environment (animal care center); observe for general health or pathology; reduce stress
Physical examination Gross observations; histopathological and blood chemistry analysis
Prepare for experiment Procure supplies, technical help, ensure housing requirement for duration of study
Immediately before initiation of experiment
Prepare for experiment Continuous preparation of solutions, special diets, perishable supplies
Physical examination Removal of unhealthy animalsDuring experiment Maintain constant
experimental conditionsEssential for chronic studies: ensure adequate
supplies, technical help, maintain housing requirements, maintain consistent and constant experimental protocols

100 PRINCIPLES OF TOXICOLOGY TESTING
Other considerations during the course of a chronic study include (1) the importance of acclimating the animals to the housing and laboratory environment prior to initiation of the study as with acute studies, (2) maintaining the animals free of infection, and (3) pur-chasing extra animals for smaller parallel experiments that provide a set of baseline values for physiological, biochemical, and morphological endpoints during the treatment and post-treatment periods and during parallel acute range-fi nding studies. Finally, as with acute studies, the animals are randomly assigned to treatment groups and cages, labeled for identifi cation, repositioned weekly in the quarters, and examined using physiological and toxicological indicators.
MONITORING DAILY CRITERIA OF CHRONIC TOXICOLOGY STUDIES
Chronic studies require persistence, diligence, and endurance on the part of the investiga-tor and personnel, principally because of the need for daily observation and handling of animals. Table 7.4 lists the criteria for monitoring the daily routine aspects of a chronic study. Attention to these factors aids in analyzing the daily progress of a study, ensures humane treatment of animals and strengthens the reliability of results. In addition, daily observations aid staff in noticing subtle underlying toxic phenomena or target organ pathology.
Daily observation of animals also involves minding routine biological parameters as indicators of health status. Table 7.5 outlines biological indicators that are employed during selected intrastudy intervals to ascertain biological condition. As with the rou-tine observations, physiological and morphological parameters are designed to assess appearance and time sequence of toxicity, especially in the detection of target organ toxicity. In addition, general criteria used to determine the daily appearance of the ani-mals may overlap with monitoring the same criteria, for example, observing when and how an animal approaches the food and water versus measuring the amount of nutrition consumed (Tables 7.4 and 7.5). This is particularly important if the chemical additive interferes with appeal for the solid or liquid nutrition. Consequently, careful observa-tion of dietary habits avoids the confusion between toxicity due to chemical agent ver-sus the incorrect conclusion of the development of a pathology due to inadequate dietary intake.
Blood, urine, and fecal specimen collection is performed at selected intervals without the need to destroy animals prior to termination of the study. Urine and fecal collection
TABLE 7.3Circumstances Requiring Consideration or Modification During Chronic Toxicity Study
Circumstance Factors for Consideration
Euthanasia Impaired health, anorexia, debility, pain, weight lossNumber of treatment groups According to the number of doses (low, intermediate, high) plus controlNumber of animals per groups 5–10; criteria based on effi cient use of data and avoiding unnecessary
wasting of animalsLength of study Range of 90 days to 3 yrsRepresentative periods
for analysis during studySubgroups of animals analyzed at selected points
during course of project; at least 4 intervalsRecovery study Performed at the termination of chronic study with surviving animals

SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 101
devices are non-invasive and are available as attachments to cages. Blood sample collection requires training but is performed either by venipuncture via the tail vein method or through capillary puncture of the inner canthus of the eye. In both situations, an improper technique or a repeated use of the same animal presents risk of infection. Non-invasive procedures are also used to assess neurological, behavioral, and learning parameters. These include tech-niques such as measuring motor rod balance, maze performance, tail-fl ick procedure for pain measurement, and reward stimuli for learned behaviors.
Urinalysis, blood clinical chemistry, and hematology measurements are routine indi-cators of toxicity and ensure identifi cation of toxicological events. These indicators are also suggestive of toxicity mechanisms and lead to the understanding of target organ effects. In particular, plasma levels of specifi c enzymes are often refl ective of target organ damage and are used to identify interactions occurring within other organs. Similarly, routine urinalysis provides evidence of toxicant-induced changes and is suggestive of
TABLE 7.5Monitoring Biological Parameters During Chronic Studies
Indicator Biological Parameters Measured
Observation for moribund or dead animals Necropsy, organ/body weight ratio, fi xation and processing of specimens for histological analysis
Time Documenting appearance of pathology, sampling intervals, specimen collection, time of death
Aging Differentiating age-related changes from agent-induced toxicityEuthanasia Interval sampling of selected groups of animals for intrastudy
monitoringNormal physiological functions Food and water intake, pair feedinga, urine output, fecal
excretionPhysiological parameters Body weights, measurement of daily food and water
consumption, blood and urine clinical chemistries
aPair-feeding refers to matching groups of treated and untreated animals; both groups receive the same quantities of nutrition. Assuming that the chemical treatment alters adequate food and liquid intake, the treated group thus becomes the rate-limiting nutrition control while the control group serves as an indicator for differentiating the effect of the toxicant from that of inadequate nutrition.
TABLE 7.4Monitoring Routine Features of Chronic Studies
Routine Feature Observations
Appearance Condition of fur, general cleanlinessBehavior Grooming, alertness, aggressiveness, irritability, irascibility (ease of excitement),
food and water consumption, sedationCages Removal of waste, absence of foul odors, fresh beddingQuarters Adequate lighting, proper day-and-night cycles, temperature and humidity
control, weekend and holiday schedulesAnimal care personnel Adherence to schedule, suffi cient coverage for off-hours, training

102 PRINCIPLES OF TOXICOLOGY TESTING
metabolic as well as target organ toxicity. Considerable information including evidence of biotransformation products of administered chemicals can be obtained from these measurements.
DURATION OF STUDIES
During the course of chronic and subchronic studies, it is important to understand that selec-tion of a termination time should prevent interpretation of age-related changes and patholo-gies as changes due to chemical induction. Subchronic studies are generally 21–90 days in duration, depending upon the route of administration and toxicological end points of interest. The duration of chronic studies, however, has not been clearly defi ned.
Traditionally, based on the approximate life spans of rodents, chronic experiments have been designated to continue for 2 years. The FDA recently suggested that these studies should be reduced to 6–18 months, depending on the animal species. The basis for this argument relies on recent evidence suggesting that toxicity observed in the second year of a chronic study is similar to that seen during the fi rst year. In fact, since age-related mortal-ity and morbidity are uncommon in the early months of chronic experiments, most toxici-ties observed during this time are causal and dose dependent.
This approach however may not adequately mimic the level of lifetime exposure for a rodent. In addition, the susceptibility of an animal to chemical insult increases with age, thus obviating the pathological consequences of chemical administration in older animal populations. Consequently, it may be necessary to design separate projects whose objec-tives focus on detecting an age-dependent, toxicant-induced pathology. These studies may need to highlight different starting ages for the animals, continuing with exposure for 6–12 months. A primary concern, therefore, is that chronic studies are designed according to the expected life spans of the species involved.
RECOVERY EXPERIMENTS
A recovery experiment or the reversibility of toxicant-induced injury after prolonged expo-sure is important for understanding cumulative toxicity, reorganization of physiological parameters, regeneration of normal biological status in an organism, and repair mechanisms. The proper design of subchronic or chronic studies should include effi cient use of resources to incorporate recovery from toxicity. The inclusion of such studies in a project helps iden-tify the progression, regression, or amelioration of a toxicant-induced injury after termina-tion of exposure. A posttreatment recovery period of 1–3 months is usually adequate to address these objectives and is accomplished by a random selection of representative surviv-ing animals at various intervals.
ANALYSIS OF RESULTS
Chronic and subchronic studies rely on normal distribution of effects based on the selec-tion of appropriate dosage ranges. Normal distribution requires a linear or logarithmic dose–response effect using corresponding dosage groups, that is, linear or logarithmic increases of dosages, respectively. Consequently, valid dose–effect responses depend on the appearance of expected effects at the highest and lowest doses. These responses are based on predicted levels of parameters based on dosage groups and include the frank effect level, lowest observed adverse effect level, no observed adverse effect level, and no observed effect level. Table 7.6 describes these parameters and their signifi cance in evalu-ating the results of chronic and subchronic toxicology tests. Further descriptions of hazard assessment are addressed in chapter 4.

SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 103
SUGGESTED READINGS
Alden CL, Lynn A, Bourdeau A, et al. A critical review of the effectiveness of rodent pharmaceutical carcinogenesis testing in predicting for human risk. Vet Pathol 2011; 48: 772–84.
Allaben WT, Turturro A, Leakey JE, Seng JE, Hart RW. FDA points-to-consider documents: the need for dietary control for the reduction of experimental variability within animal assays and the use of dietary restriction to achieve dietary control. Toxicol Pathol 1996; 24: 776–81.
Barlow SM. Agricultural chemicals and endocrine-mediated chronic toxicity or carcinogenicity. Scand J Work Environ Health 2005; 31: 141–5.
Brausch JM, Connors KA, Brooks BW, Rand GM. Human pharmaceuticals in the aquatic environ-ment: a review of recent toxicological studies and considerations for toxicity testing. Rev Environ Contam Toxicol 2012; 218: 1–99.
TABLE 7.6Monitoring Hazard Risk Assessment Based on Results of Chronic or Acute Studies
Risk Assessment Parameter Description
AHE Adverse health effect: change in body function or cell structure that might lead to disease or health problems
LOAEL Lowest observed adverse effect level: EPA’s lowest level of a stressor that causes statistically and biologically signifi cant differences in test samples as compared with other samples not subjected to stressor
MRL Minimal risk level: ATSDR estimate of daily human exposure to hazardous substance at or below a level that is unlikely to pose a measurable risk of harmful (adverse) noncancerous effects
NAPHH No apparent public health hazard: category used in ATSDR’s public health assessments for sites where human exposure to contaminated media may occur, may have occurred in past, or may occur in future, but exposure is not expected to cause any harmful health effects
NOAEL No observed adverse effect level: greatest concentration or amount of a substance, found by experiment or observation, that causes no detectable adverse alteration of morphology, functional capacity, growth, development, or life span of target organism under defi ned conditions of exposure
NOEL No observed effect level: EPA’s exposure level at which no statistically or biologically signifi cant differences in the frequency or severity of any effect in the exposed or control populations occur (Recently, NOEL has been replaced by NOAEL).
PHH Public health hazard: category used in ATSDR’s public health assessments for sites that pose public health hazards because of long-term exposures (more than 1 yr) to suffi ciently high levels of hazardous substances or radionuclides that could result in harmful health effects
RfD Reference dose: EPA estimate, with built-in uncertainty or safety factors, of daily lifetime dose of substance that is unlikely to cause harm in humans
UF Uncertainty factor: mathematical adjustments for reasons of safety when knowledge is incomplete; factors used in calculation of doses that are not harmful to a population; applied to LOAEL, NOAEL, MRL
UPHH Urgent public health hazard: category used in ATSDR’s public health assessments for sites where short-term exposures (less than 1 yr) to hazardous substances or conditions could result in harmful health effects requiring rapid intervention
ATSDR, Agency for Toxic Substances and Disease Registry; part of U.S. Department of Health and Human Services, http://www.atsdr.cdc.gov; EPA, U.S. Environmental Protection Agency http://www.epa.gov.

104 PRINCIPLES OF TOXICOLOGY TESTING
Bussiere JL. Species selection considerations for preclinical toxicology studies for biotherapeutics. Expert Opin Drug Metab Toxicol 2008; 4: 871.
Clarke J, Hurst C, Martin P, et al. Duration of chronic toxicity studies for biotechnology-derived pharmaceuticals: is 6 months still appropriate? Regul Toxicol Pharmacol 2008; 50: 2–22.
Clift MJ, Gehr P, Rothen-Rutishauser B. Nanotoxicology: a perspective and discussion of whether or not in vitro testing is a valid alternative. Arch Toxicol 2011; 85: 723.
Combes R, Gaunt I, Balls M. A scientifi c and animal welfare assessment of the OECD health effects test guidelines for the safety testing of chemicals under the European union REACH sys-tem. Altern Lab Anim 2006; 34: 77.
Creton S, Douglas M, Wheeler JR, Hutchinson TH. Challenging the requirement for chronic fi sh toxicity studies on formulated plant protection products. Toxicol Lett 2010; 199: 111.
Cooper RL, Lamb JC, Barlow SM, et al. A tiered approach to life stages testing for agricultural chemical safety assessment. Crit Rev Toxicol 2006; 36: 69.
Lai MW, Klein-Schwartz W, Rodgers GC, et al. Annual report of the American association of poison control centers’ national poisoning and exposure database. Clin Toxicol 2006; 44: 803.
Lin KK. Progress report on the guidance for industry for statistical aspects of the design, analysis, and interpretation of chronic rodent carcinogenicity studies of pharmaceuticals. J Bio-pharm Stat 2000; 10: 481.
Séralini GE, Cellier D, Sultan C, et al. How subchronic and chronic health effects can be neglected for GMOs, pesticides or chemicals. Int J Biol Sci 2009; 5: 438.
Scholz S, Fischer S, Gündel U, et al. The zebrafi sh embryo model in environmental risk assessment--applications beyond acute toxicity testing. Environ Sci Pollut Res Int 2008; 15: 394.
Trosko JE, Chang CC. Factors to consider in the use of stem cells for pharmaceutic drug development and for chemical safety assessment. Toxicology 2010; 270: 18.
U.S. Food and Drug Administration. International Conference on Harmonisation, Guidance on the duration of chronic toxicity testing in animals (rodent and nonrodent toxicity testing), availability. Fed Regist 1999; 64: 34259.
Van Cauteren H, Bentley P, Bode G, et al. The industry view on long-term toxicology testing in drug development of human pharmaceuticals. Pharmacol Toxicol 2000; 86: 1.
REVIEW ARTICLES
Cunha GC, van Ravenzwaay B. Evaluation of mechanisms inducing thyroid toxicity and the ability of the enhanced OECD Test Guideline 407 to detect these changes. Arch Toxicol 2005; 79: 390.
Doe JE, Lewis RW, Botham PA. Comments on a scientifi c and animal welfare assessment of the OECD Health Effects Test Guidelines for the safety testing of chemicals under the Euro-pean Union REACH system. Altern Lab Anim 2006; 34: 111.
Embry MR, Belanger SE, Braunbeck TA, et al. The fi sh embryo toxicity test as an animal alternative method in hazard and risk assessment and scientifi c research. Aquat Toxicol 2010; 97: 79.
Fako VE, Furgeson DY. Zebrafi sh as a correlative and predictive model for assessing biomaterial nanotoxicity. Adv Drug Deliv Rev 2009; 61: 478.
Gallardo E, Queiroz JA. The role of alternative specimens in toxicological analysis. Biomed Chro-matogr 2008; 22: 795.
Gelbke HP, Hofmann A, Owens JW, Freyberger A. The enhancement of the subacute repeat dose toxicity test OECD TG 407 for the detection of endocrine active chemicals: comparison with toxicity tests of longer duration. Arch Toxicol 2007; 81: 227.
Grindon C, Combes R, Cronin MT, Roberts DW, Garrod JF. An integrated decision-tree testing strat-egy for repeat dose toxicity with respect to the requirements of the EU REACH legislation. Altern Lab Anim 2008; 36: 93.
Gwiazda R, Lucchini R, Smith D. Adequacy and consistency of animal studies to evaluate the neu-rotoxicity of chronic low-level manganese exposure in humans. J Toxicol Environ Health A 2007; 70: 594.

SUBCHRONIC AND CHRONIC TOXICOLOGY TESTING 105
Hard GC, Khan KN. A contemporary overview of chronic progressive nephropathy in the laboratory rat, and its signifi cance for human risk assessment. Toxicol Pathol 2004; 32: 171.
Hoffmann S, Hartung T. Toward an evidence-based toxicology. Hum Exp Toxicol 2006; 25: 497.Hutchinson TH, Shillabeer N, Winter MJ, Pickford DB. Acute and chronic effects of carrier solvents
in aquatic organisms: a critical review. Aquat Toxicol 2006; 76: 69.Kroes R, Galli C, Munro I, et al. Threshold of toxicological concern for chemical substances present
in the diet: a practical tool for assessing the need for toxicity testing. Food Chem Toxicol 2000; 38: 255.
Lai DY. Toward toxicity testing of nanomaterials in the 21st century: A paradigm for moving forward. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2012; 4: 1.
McCarty LS, Borgert CJ. Review of the toxicity of chemical mixtures: theory, policy, and regulatory practice. Regul Toxicol Pharmacol 2006; 45: 119.
Tetko IV, Bruneau P, Mewes HW, Rohrer DC, Poda GI. Can we estimate the accuracy of ADME-Tox predictions? Drug Discov Today 2006; 11: 700.
Tomatis L. Role of experimental and epidemiological evidence of carcinogenicity in the primary prevention of cancer. Ann Ist Super Sanita 2006; 42: 113.
Waters M, Jackson M. Databases applicable to quantitative hazard/risk assessment–towards a predic-tive systems toxicology. Toxicol Appl Pharmacol 2008; 233: 34.

106
8 Acute dermal toxicity testing
INTRODUCTION
Regulatory agencies in the United States currently have testing guidelines for chemicals and materials with high potential for accidental, inadvertent, or clinical human exposure. These substances are intended for cutaneous (and ocular) administration as drugs and biologicals to humans and animals and for commercial chemical (industrial, environmental, occupa-tional, and domestic) applications.
The need for local and systemic in vivo cutaneous toxicity evaluations are mandated by U.S. and European Union regulations. Thus, the designs of studies to evaluate acute dermal toxicities require animal models to ensure safety of product formulations before arrival on the market for human consumption. The procedures incorporated into in vivo studies are unique to cutaneous toxicity evaluation and the methods used for in vivo toxicity testing of these products are discussed in the chapter.
ACUTE DERMAL TOXICITY TESTS
DESCRIPTION
All products for human and veterinary consumption are regulated for evaluation for toxicol-ogy testing. With dermal toxicity studies, rabbits serve as standards for evaluating local toxicity (irritation). Guinea pigs are generally listed in testing guidelines as an acceptable species, but their use is uncommon for dermal toxicology evaluations. They have been the species of choice for sensitization studies until recently. The local lymph node assay (LLNA) performed in CBA/Ca or CBA/J mice is a refi nement of the guinea pig assay. It is discussed in both the in vivo and in vitro toxicity testing sections of this book because it is an alterna-tive to the traditional guinea pig assay.
PRIMARY IRRITATION
Dermal acute studies are designed to provide information on local effects, particularly skin irritation and corrosion. In the aftermath of World War II and the development of chemical warfare research, the need to ensure the safety of consumer products generated signifi cant interest. Thus the Draize skin test was born in the mid-twentieth century, motivated by the hazards of unsafe cosmetics. Justifi ed by an urgency for public protection, the Draize test became a government-endorsed method for evaluating the safety of materials meant for local contact uses. The Draize eye test is discussed in Chapter 9.
Draize et al. (1944) published a quantitative assessment of skin irritation as a guideline for product safety. They defi ned a primary local irritant as a substance that produced an infl ammatory dermal reaction. The infl ammatory process as it pertained to dermal irritation

ACUTE DERMAL TOXICITY TESTING 107
was characterized by the presence of edema1 and erythema.2 Table 8.1 outlines the grading of primary reactions of rabbit skin based on Draize observations. Because of the potential for pain and skin damage resulting from cutaneously applied materials, careful monitoring of cutaneous irritation and toxicity studies is now mandated by animal welfare regulations.
The Animal Welfare Act (amended 1985, U.S. Department of Agriculture) regulates the care and use of most animals used for research and toxicity testing. Although rats and mice are not covered by the act, most institutions have adopted similar internal standards for these species. The act states that skin irritancy testing is an example of a procedure that can be expected to cause more than momentary or slight pain; although the dosing proce-dure is generally not painful, the reaction caused by a product may elicit pain.
The rabbit is the species of choice for dermal irritation because it has relatively sensi-tive skin as compared with human skin. This species is also easier to handle than larger species and rabbit skin has high permeability. Because of the rabbit’s enhanced sensitivity to dermal insult, it is generally considered to be overpredictive of human irritation, thus bringing into question its relevance to human risk assessment. Many current regulations prefer albino rats to rabbits although the latter still represent the standard for local toxicity (irritation) evaluations. The guinea pig is generally listed in testing guidelines as an accept-able species, but is rarely used in dermal irritation toxicity evaluations.
Study Design and Procedures
Dermal irritation studies are designed to mimic human exposure and are generally per-formed on one to three albino rabbits. A test area is prepared by shaving the fur (2–3 cm2) on the back or abdomen. Initially, a single animal is exposed sequentially (3 minutes and 1 and 4 hours) to the test material (0.5 mL or 0.5 g). Most drugs and biological materials are left uncovered. Applications may be covered if a material is applied under a bandage
1 Accumulation of fl uid in subcutaneous and interstitial spaces. 2 Redness of skin as a result of increased local blood fl ow.
TABLE 8.1Grading of Skin Reactions: Acute Dermal Irritation
Dermal Irritation Skin Reaction Score
Erythema and eschar formation No erythema 0Very slight erythema, barely perceptible 1Well defi ned erythema 2Moderate to severe erythema 3Severe erythema (beet redness) to eschar formation
(injuries in depth)4
(maximum)Edema formation No edema 0
Very slight edema (barely perceptible) 1Well defi ned edema 2Moderate edema (raised ~1 mm) 3Severe edema (extending beyond area of exposure and
raised >1 mm)4
Source: Courtesy of Draize, J.H. and Kelley, E.A., Toxicology, 1, 267, 1959.

108 PRINCIPLES OF TOXICOLOGY TESTING
(burn treatment) or covered with clothing (to mimic occupational exposure to chemicals). A semiocclusive covering generally consists of a porous dressing such as gauze held in place with non-irritating tape. If dermal corrosion is seen after exposure, the test is terminated and the material is classifi ed as corrosive. If no corrosion is seen, two additional animals are exposed to the material for up to 4 hours and irritation is scored according to the Draize irritation potential classifi cation, as detailed in Table 8.2. The sum of the mean erythema and edema scores is computed and listed as the primary dermal irritation index (PDII).
Other procedural details to be considered include the (1) choice of a solvent vehicle for administration, (2) amount of sample administered to the skin site, (3) need for animal restraining equipment, and (4) preparation of intact and abraded skin sites. These details are summarized in Table 8.3.
SKIN SENSITIZATION
Evaluation of the potential to produce sensitization is required for many chemicals. Upon application to and penetration of the skin, a chemical can elicit a subsequent reaction, usually
TABLE 8.2Classification of Dermal Irritation Potential
PDII Classifi cation Score
Non-irritant 0.0Negligible irritant >0.0–0.5Mild irritant >0.5–2.5Moderate irritant >2.5–5.0Severe irritant >5.0–8.0
Note: Score based on observation of area at 60 min, 24, 48, and 72 hrs from application. Observations for reversibility made for up to 14 days.
Source: Courtesy of Auletta, C.S., Basic Clin. Pharmacol. Toxicol., 95, 201, 2004.
TABLE 8.3Other Factors Involved in Dermal Irritation Study Design
Factor Description
Solvent vehicle Inert (CMC, saline); oil based only according to particular application; paste for solids
Sample volume Minimized and uniform coverage to areaRestraining devices Only in longer studiesPreparation of test site Intact or abradeda
Post-treatment observation Up to 4 hrs; scored as described above
aThick skin is gently scraped with a steel comb or punctated with a hypodermic needle to allow increased access to underlying subcutaneous structures.

ACUTE DERMAL TOXICITY TESTING 109
days or weeks after repeated exposures. This type of skin sensitization is known as allergic contact sensitivity or dermatitis. The response requires at least 24–72 hours for development and a reaction is characterized as a type IV (delayed-type hypersensitivity) cell-mediated immunity. Responses range from mild irritation resulting in erythema and induration, to the development of eczema, eruptions, and fl aring, to full systemic immune manifestations. The response to a chemical is typically dose-dependent and self-limiting (days to weeks) and of varying intensity. Furthermore, the process requires initial sensitization, adaptation, and sub-sequent rechallenge.
Immunologically, a reaction involves antigen-specifi c T cell activation and begins with an intradermal or mucosal challenge (sensitization stage). CD4+ T cells then recognize MHC-II (major histocompatibility class II) antigens on antigen-presenting cells (APCs; Langerhans cells, for example) and differentiate to T
H1 cells. This sensitization stage
requires prolonged (at least 2 weeks) local contact with an agent. A subsequent repeat chal-lenge stage (elicitation) induces differentiated T
H1 (memory) cells to release cytokines,
further stimulating attraction of phagocytic monocytes and granulocytes. The release of lysosomal enzymes from the phagocytes results in local tissue necrosis. Contact hypersen-sitivity resulting from prolonged exposure to plant resins and jewelry, for example, is caused by the lipophilicity of a chemical in oily skin secretions, thus acting as a hapten. Although most reactions are self-limiting, extensive exposure results in tissue necrosis.
Study Design and Procedures
Invariably, the test animals used for skin sensitization studies are guinea pigs, particularly because of their known susceptibility to a variety of chemical sensitizers. Consequently, the most common skin sensitization methods include the guinea pig maximization test (GPMT) of Magnusson and Kligman and the Buehler test. Studies utilize 10–20 animals in a treated group and 5–10 in a control group. For the GPMT, induction is started according to the adju-vant method by a set of intradermal injections (test article, Freund’s adjuvant, and test article in adjuvant), followed 1 week later by topical application, and topical challenge 2 weeks later.
In the closed-patch Buehler test, three topical applications 1 week apart constitute the induction phase, followed by topical challenge 2 weeks later. The appearance of edema or erythema after the challenge dose, greater than that resulting from the sensitizing dose, is indicative of sensitization. For both studies, periodic reliability checks are performed with a known mild to moderate skin sensitizer.
The murine LLNA is a scientifi cally validated alternative used to assess the potentials of substances to produce skin sensitization.3 The LLNA uses young adult female CBA mice instead of guinea pigs. The procedure is completed in a week and causes less pain and distress. It is based on the visual evaluation of the ability of a chemical to cause allergic reactions after repeated applications to the skins of about 20–30 animals. Three days after the fi nal application, animals receive intravenous injections of 3H-thymidine or 125I. After 5 hours, auricular lymph nodes are excised, drained, and radioactivity is standardized according to a stimulation index. A cutoff value greater than or equal to 3 is considered indicative of sensitization. Although the validity of the assay was endorsed by independent peer review evaluation (ICCVAM/NICEATM 2004; OECD Guideline 429, 2002), some
3 Because it is an in vivo assay, the OECD considers it of equal merit but not a required replacement for animal toxicity tests.

110 PRINCIPLES OF TOXICOLOGY TESTING
concerns involve the potential for false-positive results resulting from irritation and poor absorption of aqueous-based formulations.
The revised OECD Guideline 404 also suggests that data on skin irritation or corrosion can be obtained from other animal studies. For instance, it is possible to determine the irritation potential or lack of irritation potential by reviewing the results of a dermal toxic-ity study. In addition, if a material is highly toxic by the dermal route, it is not necessary to assess its irritation potential also. Although not specifi cally mentioned, it may also be pos-sible to gain information on the irritation potential of a material from results of studies assessing dermal sensitization or the studies commonly performed to confi rm skin safety in humans.
PHOTOALLERGIC AND PHOTOTOXIC REACTIONS
Dermal phototoxicity of a chemical is defi ned as a toxic response elicited after dermal or systemic exposure to a chemical following subsequent exposure to light, particularly ultraviolet (UV) radiation from sunlight. The biological effects of UV rays alone are of lower frequency and longer wavelength (about 10–8 to 10–6 m) than ionizing radiation (10–7 to 10–12 m for X-rays). Thus the effects of UV radiation are less penetrating and benign.
Unlike ionizing radiation, skin damage induced by UV rays is mediated principally by the generation of reactive oxygen species and the interruption of melanin production. Like ionizing radiation, however, cumulative or intense exposure to UV rays precipitates DNA mutations: base pair insertions, deletions, single-strand breaks, and DNA-protein cross-links. DNA repair mechanisms play an important role in correcting UV-induced DNA damage and in preventing further consequences of excessive UV exposure (as with sunburn).
Melanin production by melanocytes increases and the epidermis thickens in an attempt to prevent future damaging effects. The protective ability of antioxidant enzymes and DNA repair pathways diminishes with age, thus setting the conditions for development of skin neoplasms later in life. In addition, UV light can convert chemicals into reactive oxygen species that elicit local cellular toxicity.
The extent of solar injury depends on the type of UV radiation, the duration and inten-sity of exposure, clothing, season, altitude and latitude, and the amount of melanin pig-ment present in the skin. In fact, individuals differ greatly in their responses to sun exposure. Also, harmful effects of UV rays are fi ltered out by glass, smog, and smoke and enhanced when refl ected off snow and sand. The indiscriminate use of chlorofl uorocarbons in aerosol propellants depletes the UV-blocking properties of ozone in the stratosphere, thus allowing greater intensities of UV rays to penetrate through the protective upper atmospheric layers.
Table 8.4 summarizes the chronic effects of sunlight and photosensitivity reactions. Actinic keratoses, described as precancerous keratotic lesions, result from years of sun exposure, especially in fair-skinned individuals. They present as pink, poorly marginated, scaly, or crusted superfi cial growths on skin.
Photosensitive reactions involve the development of unusual responses to sunlight, sometimes exaggerated in the presence of a variety of chemicals or precipitating circum-stances. Erythema (redness) and dermatitis (infl ammation of the skin or mucous mem-branes) are acute responses to direct UV light-induced photoallergic reactions. They are mediated by dilation and congestion of superfi cial capillaries and a type IV hypersensitiv-ity cell-mediated reaction. In addition, macules, papules, nodules, and target (bull’s eye shaped) lesions are seen in multiform variations of erythematous reactions.

ACUTE DERMAL TOXICITY TESTING 111
Urticaria is a pruritic skin eruption characterized by transient wheals of varying shapes and sizes, with well defi ned erythematous margins and pale centers. Development of urti-caria is mediated by capillary dilation in the dermis in response to release of vasoactive amines (histamine and kinins) upon sun exposure, especially subsequent to chemical expo-sure. More severe photosensitivity reactions lead to the development of bullae—thin walled blisters on the skin or mucous membranes larger than 1 cm in diameter and containing clear serous fl uid. Dehydration, scaling, scarring, fi brosis, and necrosis develop as the exposed areas heal.
Among the exogenous factors that contribute to the development of photosensitivity reactions are ingestion or application of antibacterial and antifungal antibiotics (sulfon-amides, tetracyclines; griseofulvin, respectively) and thiazide diuretics. Contact with der-mal products containing coal tar, salicylic acid, plant derivatives, and ingredients of colognes, perfumes, cosmetics and soaps are additional sources of the problem.
In Vivo Study Design and Procedures for Photosensitivity and Phototoxicity
In vivo tests for photosensitivity and phototoxicity are conducted in guinea pigs or rabbits. Test articles are administered orally or parenterally for 10–14 days or applied topically. The challenge phase starts 2–3 weeks later with another dose plus exposure to a UV lamp.4 Fac-tors that affect photosensitivity test results include a chemical’s propensity to absorb light, length of UV exposure, and proximity of light source to shaved area. As with other dermal tests, control groups incorporate positive and negative photoallergic agents. Lesions are scored as described earlier.
Methods for Pyrogen Testing In Vivo
Rabbits have a similar pyrogen5 tolerance to humans; therefore, observing a change in body temperature in rabbits correlates with the presence of pyrogens in humans. The rabbit pyro-gen test is a method that can detect nonbacterial endotoxin pyrogens as well as bacterial endotoxins.
4 The shorter wavelengths of sunburn-producing UVB radiation (280–320 nm λ) are more damaging than those of UVA rays (320–400 nm λ). 5 A fever-producing substance
TABLE 8.4Chronic Effects of Sunlight and Photosensitivity Reactions
Reaction to Excessive Sunlight Clinical Effects
General dermatologic reactions Dermatoheliosis: aging of skin due to chronic exposure to sunlightElastosis: yellow discoloration of skin with accompanying small
nodules; wrinkling, hyperpigmentation, atrophy, dermatitisActinic keratoses Precancerous keratotic lesions after many years of exposure to UV raysSquamous/basal cell carcinomas Occur more commonly in light-skinned individuals exposed to extensive
UV rays (sun tanning) during adolescenceMalignant melanomas Cancerous dermal lesions associated with increased, intense, prolonged
exposure to UV lightPhotosensitive reactions Erythema and erythema multiform lesions; urticaria, dermatitis, bullae;
thickened, scaling patches

112 PRINCIPLES OF TOXICOLOGY TESTING
The U.S. Pharmacopeia describes the pyrogen test as an in vivo method “designed to limit to an acceptable level the risks of febrile reaction in the patient to the administration, by injection, of the product concerned.” The test involves measuring the rise in temperature of healthy, mature rabbits following the intravenous injection of a test solution. The prod-ucts are substances that can be tolerated by the experimental rabbit in a dose not to exceed 10 mL per kg injected intravenously within a period of not more than 10 minutes. Further-more, the U.S.Pharmacopeia describes the apparatus, type, and number of test animals, method for temperature recording, and the specifi c protocol procedure.
Briefl y, each of three rabbits, weighing individually not less than 1.5 kg, receives 10 mL per kg of the test solution. Prior to injection of rabbits for the fi rst time, the animals are conditioned in the housing quarters for not more than 7 days. A sham test is performed that includes all of the steps as directed below except injection of the test substance. Ani-mals are not used more frequently than once every 48 hours. During the acclimation period, rectal baseline temperatures are recorded as the control temperatures of each rabbit. This is the basis for the determination of any temperature increase resulting from the injection of a test solution. In anyone group of test rabbits, only rabbits whose control temperatures do not vary by more than 1°C from each other are included in the protocol, and the tempera-ture of any individual rabbit should not exceed 39.8°C.
The test substance is administered via the ear vein and should be completed within 10 minutes. The rectal temperature is recorded at 1, 2, and 3 hours post injection. Interpre-tation of the results (Decision Statements) specifi es a limit on the temperature rise allowed for any one rabbit as 0.5°C. If no rabbit shows an individual rise in temperature of 0.5°C or more above its respective control temperature, the product meets the requirements for the absence of pyrogens. A decrease in temperature is recorded as zero increase. If the tem-perature of a single rabbit rises 0.5°C or above, then the test is expanded to include fi ve more rabbits (total eight rabbits). If not more than three of the eight rabbits show individual rises in temperature of 0.5°C or more, and if the sum of the eight individual maximum temperature rises does not exceed 3.3°C, the material under examination also meets the requirements for the absence of pyrogens.
In Vivo Methods for Detecting Allergic Contact Dermatitis
Allergic contact dermatitis is a type IV (delayed type hypersensitivity) cell-mediated reac-tion of the immune system to either allergens or irritants on the skin (e.g., poison ivy, drugs, or metals).6 The hypersensitivity reaction usually occurs as a result of subcutaneous, low dose contact exposure to an allergen (sensitization phase), followed by T-helper cell elicita-tion (challenge phase). T-cell expansion results in erythema, cellular infi ltrates, and forma-tion of vesicles and abscesses further contributing to immune tissue destruction. Clinically, contact dermatitis results in edema, wheals, fl ares, pruritis (burning, itchy rashes), which resolves only after several days to weeks. Prevention and avoidance of the potential allergen can prevent recurrence. Persistent chronic sensitization is a hallmark of the condition.
The Guinea Pig Maximization Test (GPMT) is an example of an in vivo test to screen for substances with the potential for causing human skin sensitization). It was fi rst proposed by Magnusson ad Kligmann (1969). OECD guideline 406 (1981) describes the procedure in
6 This hypersensitivity reaction should not be confused with type I antibody-mediated reactions, characterized by an initial sensitization reaction followed by mast cell activation and increased vascular permeability. Recruitment of neutrophils and eosinophils completes the mechanism of immune tissue destruction in type I hypersensitivity (e.g., atopic dermatitis or eczema).

ACUTE DERMAL TOXICITY TESTING 113
detail. Briefl y, the test animals are exposed intradermally to the test substance, along with an adjuvant to enhance the immune reaction of the guinea pig (sensitization phase). After 1 week, the guinea pigs are exposed to a lower concentration of the test material, and their allergic reaction is measured (challenge, elicitation phase). The test is considered positive when 15% of guinea pigs display a reaction. To ensure against false negative results, a min-imum of 20 animals are typically used.
The GPMT has been largely superseded by the murine local nymph node assay (LLNA; Basketter and Scholes, 1992). The REACH Regulation (Annex VII, paragraph 8.3) states “The Murine Local Lymph Node Assay (LLNA) is the fi rst-choice method for in vivo test-ing. Only in exceptional circumstances should another test be used. Justifi cation for the use of another test shall be provided.”
OECD test guideline (TG) 429 for the testing of chemicals, Skin Sensitization: Local Lymph Node Assay (2002), describes the protocol for the LLNA in the mouse. As with the GPMT, the LLNA is a test for the induction phase of skin sensitization and provides quan-titative data suitable for dose–response assessment. The basic principle underlying the LLNA is that sensitizers induce proliferation of lymphocytes in the lymph nodes draining the site of chemical application. This proliferation is proportional to the dose and to the potency of the applied allergen. Assessment of results is calculated as the proliferation in test groups compared with that in vehicle-treated controls. The ratio of the proliferation in treated groups to that in the concurrent vehicle control group (Stimulation Index), should be 3 before a test substance is further evaluated as a potential skin sensitizer. Most of the methods described in the OECD TGs incorporate the use of in vivo radioactive labeling to measure an increased number of proliferating cells in the draining auricular lymph nodes. It is important to note that the method provides certain advantages with regard to animal welfare and provides data for confi rming that chemicals lack a signifi cant potential to cause skin sensitization. Although the LLNA will not eliminate the use of animals in the assess-ment of allergic contact sensitizing activity, it has the potential to reduce the number of animals required for this purpose.
The methodology is based upon consideration of immunological events stimulated by chemicals during the induction phase of sensitization. Unlike TG 406, the LLNA does not require that challenge-induced dermal hypersensitivity reactions be elicited, nor does it require the use of an adjuvant. Further developments of the murine LLNA, such as the LLNA-DA (2009) and the LLNA:BrdU-ELISA (2009) are in vitro modifi cations using non-radiolabeled components. These tests reduce the number of animals used and stress associated with the traditional guinea pig tests, and are described in Section III of this book, In Vitro Toxicology Tests.
CONCLUSIONS
In vivo skin toxicology testing methods have been used for decades to provide data for formulating risk assessment programs. The information generated using these tests is hampered by the scientifi c limitations and ethical objections posed, with the animal care issue at the forefront. The development of in vitro alternatives for this and other toxicity tests (discussed in Section III of this book) is necessarily based on comparisons to in vivo data. Thus validation of alternative methods depends on the reliability of in vivo data. It is logical to conclude that until the issues concerning animal skin testing methods are addressed, development of alternative in vitro tests will be more diffi cult than initially assumed.

114 PRINCIPLES OF TOXICOLOGY TESTING
SUGGESTED READINGS
Basketter DA, Lea LJ, Cooper K, et al. Threshold for classifi cation as a skin sensitizer in the local lymph node assay: a statistical evaluation. Food Chem Toxicol 1999; 37: 1.
Draize JH, Kelley EA. The urinary excretion of boric acid preparations following oral administration and topical applications to intact and damaged skin of rabbits. Toxicology 1959; 1: 267.
Draize JH, Woodward G, Calvery HO. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J Pharmacol Exp Ther 1944; 82: 377.
Friedenwald JS, Buschke W. Some factors concerned in the mitotic and wound-healing activities of the corneal epithelium. Trans Am Ophthalmol Soc 1944; 42: 371.
ICCVAM. The murine local lymph node assay, NIH Publication 99-4494, Research Triangle 1999.Kerr A, Ferguson J. Photoallergic contact dermatitis. Photodermatol Photoimmunol Photomed 2010;
26: 56.Neumann NJ, Blotz A, Wasinska-Kempka G, et al. Evaluation of phototoxic and photoallergic poten-
tials of 13 compounds by different in vitro and in vivo methods. J Photochem Photobiol B 2005; 79: 25.
Jester JV. Extent of corneal injury as a biomarker for hazard assessment and the development of alternative models to the Draize rabbit eye test. Cutan Ocul Toxicol 2006; 25: 41.
Magnusson B, Kligman AM. The identifi cation of contact allergens by animal assay: the guinea pig maximization test. J Invest Dermatol 1969; 52: 268.
Martinez V, Corsini E, Mitjans M, et al. Evaluation of eye and skin irritation of arginine-derivative surfactants using different in vitro endpoints as alternatives to in vivo assays. Toxicol Lett 2006; 164: 259.
REVIEW ARTICLES
Auletta CS. Current in vivo assays for cutaneous toxicity: local and systemic toxicity testing. Basic Clin Pharmacol Toxicol 2004; 95: 201.
Basketter DA, Scholes EW. Comparison of the local lymph node assay with the guinea-pig maxi-mization test for the detection of a range of contact allergens. Food Chem Toxicol 1992; 30: 65.
Basketter DA, Andersen KE, Liden C, et al. Evaluation of the skin sensitizing potency of chemicals by using the existing methods and considerations of relevance for elicitation. Contact Der-matitis 2005; 52: 39.
Bouvier d’Yvoire M, Bremer S, Casati S, et al. ECVAM and new technologies for toxicity testing. Adv Exp Med Biol 2012; 745: 154.
Buehler EV. Occlusive patch method for skin sensitization in guinea pigs: the Buehler method. Food Chem Toxicol 1994; 32: 97.
Chatterjee A, Babu RJ, Ahaghotu E, Singh M. The effect of occlusive and unocclusive exposure to xylene and benzene on skin irritation and molecular responses in hairless rats. Arch Toxicol 2005; 79: 294.
Elkeeb D, Elkeeb L, Maibach H. Photosensitivity: a current biological overview. Cutan Ocul Toxicol 2012; Epub ahead of print.
Frankild S, Volund A, Wahlberg JE, Andersen KE. Comparison of the sensitivities of the Buehler test and the guinea pig maximization test for predictive testing of contact allergy. Acta Derm Venereol 2000; 80: 256.
Glatz M, Hofbauer GF. Phototoxic and photoallergic cutaneous drug reactions. Chem Immunol Allergy 2012; 97: 167.
Kimber I, Basketter DA, Berthold K, et al. Skin sensitization testing in potency and risk assessment. Toxicol Sci 2001; 59: 198.
Maurer JK, Parker RD, Carr GJ. Ocular irritation: microscopic changes occurring over time in the rat with surfactants of known irritancy. Toxicol Pathol 1998; 26: 217.

ACUTE DERMAL TOXICITY TESTING 115
McDougal JN, Boeniger MF. Methods for assessing risks of dermal exposures in the workplace. Crit Rev Toxicol 2002; 32: 291.
OECD, Guideline 406: Guinea Pig Maximization Test (GPMT), 1981. [Available from: http://www.oecd.org]
OECD, Guideline 429: Murine Local Lymph Node Assay, 2002. [Available from: http://www.oecd.org]
Robinson MK, Nusair TL, Fletcher ER, Ritz HL. A review of the Buehler guinea pig skin sensitiza-tion test and its use in a risk assessment process for human skin sensitization. Toxicology 1990; 61: 91.
Robinson MK, Perkins MA. A strategy for skin irritation testing. Am J Contact Dermatitis 2002; 13: 21.
Svendsen O. The minipig in toxicology. Exp Toxicol Pathol 2006; 57: 335.

116
9 Acute ocular toxicity testing
INTRODUCTION
No issue in toxicology testing has provoked more controversy or protest than the Draize eye test. Published in the 1940s as a quantitative analysis of the effects of chemicals on rabbit eyes, it followed a similar manuscript by Friedenwald et al. (1944) that proposed a quantita-tive method for evaluating the severities of ocular testing methods. Today, advances in ocular toxicology are challenging the validity, precision, relevance, and need of the Draize eye test.
Preclinical product safety tests with rabbits and other mammals also raise ethical con-cerns about animal welfare. In the U.S., the Draize eye test, however, still remains the only offi cial, regulatory-approved procedure for predicting ocular irritants. With the develop-ment of alternative non-animal procedures to replace the Draize test, the data generated in the Draize procedure are also used as standards against which the performances of alterna-tive methods are measured (see chap. 14). Nevertheless, since the publishing of the last edition of this text, signifi cant progress has been achieved in the development of in vitro ocular toxicology tests for use in supplementing in vivo testing methods.
EYE IRRITATION AND CORROSION TESTING
The Draize test involves a standardized protocol for instilling agents onto the corneas and conjunctivae of laboratory animals, particularly rabbits. A sum of ordinal-scale items of the outer eye gives an index of ocular morbidity. The test uses a scoring system that describes reactions produced in the cornea, conjunctiva, and iris. Since its inception, the test has under-gone several modifi cations, including the addition of other descriptive parameters such as thickness of the eyelids, induction of erythema, edema, discharge, corneal opacity, capillary damage, and pannus (vascularization) of the cornea.
In an attempt to standardize ocular testing, Organization for Economic Cooperation and Development (OECD) Guideline 4051 recommends that all existing information on a test substance should be evaluated prior to undertaking in vivo eye irritation and corrosion stud-ies of that substance and that sequential testing strategies be considered for each new and revised test guideline. Movement toward a weight-of-the-evidence approach to hazard assessment, including multistep tiered testing strategies, is suggested prior to commencing animal tests. [Table 9.1 outlines the stepwise approach as suggested in the supplement to OECD TG 405]. Furthermore, since in vivo eye irritancy and corrosion tests are federally regulated, the revised guidelines recommend performing tests sequentially, one animal at a time. This allows reassessment of data as the results accumulate to avoid duplication of ani-mal tests. In addition, corrosive classifi cation of a test material precludes further testing with
1 “Acute Eye Irritation/Corrosion” was adopted in 1981 and revised in 1987 and 2002.

ACUTE OCULAR TOXICITY TESTING 117
additional animals and allows for immediate euthanasia of injured or suffering animals. Additional experimentation is necessary only if data are insuffi cient to support classifi cation.
OBJECTIONS TO AND LIMITATIONS OF EYE IRRITATION AND CORROSION TESTING
Several major sources of variability in animal eye irritation and corrosion testing have led to the conclusion that the rabbit eye is an unsatisfactory model for the human eye. The indi-vidual testing of animals creates small group sizes for multiple dose testing, thus necessarily disqualifying adequate statistical comparisons. Increasing the group size, however, will undermine the impetus toward reduction of animal testing and development of in vitro alter-natives to the Draize eye method. Also, grade defi nitions of ocular irritancy and corrosion are relatively oversimplifi ed. Thus the existing scoring systems do not refl ect the complexities of the total in vivo response. Also, the use of algorithms to simplify the in vivo data is not suffi ciently accurate for comparison with in vitro data. These and other considerations that add to the variability and objections to the use of the Draize eye test are defi ned in Table 9.2.
TABLE 9.1Weight-of-the-Evidence Approach to Hazard Assessment
Step Number Description Explanation
1 Evaluation of existing human and animal data
Clinical, occupational studies, case reports, and animal test data from ocular studies; human/animal studies of dermal corrosion/irritation; evaluation of corrosivity, irritancy, or lack of corrosivity from previously performed ocular studies
2 Analysis of SARs Negative data of structurally related chemicals do not constitute suffi cient evidence of non-corrosivity/non-irritancy; validated SAR approaches are used to identify the corrosion and irritation potential for both dermal and ocular effects
3 Physicochemical properties and chemical reactivity
Demonstration of pH extremes (e.g., pH < 2.0 or > 11.5) suggests strong local effects; evaluation of buffering capacity
4 Consideration of other existing information
Determination of acute systemic toxicity via dermal route
5, 6 Results from in vitro (5) or ex vivo (6) tests
Demonstration of corrosive or severe irritant properties in a validated in vitro test (step 5) or ex vivo test (step 6) for the specifi c assessment of eye or skin corrosivity/irritation
7 Assessment of in vivo dermal irritancy or corrosivity of the substance
Insuffi cient evidence for ocular toxicity may require eval-uation for in vivo skin irritation/corrosion potential fi rst (TG 404)
8, 9 In vivo test in rabbits Initial test using one animal: step 8, severe irritant effects to the eyes, does not require further testing; step 9, lack of corrosive or severe irritant effects requires a confi rmatory test with two additional animals
Abbreviation: SAR, structure activity relationship. Source: OECD supplement to TG 405, http://www.oecd-ilibrary.org/.

118 PRINCIPLES OF TOXICOLOGY TESTING
METHODS FOR IN VIVO OCULAR TOXICOLOGY TESTING
OECD test guideline 405 (TG 405; 2002) replaces the original TG 405, adopted in 1981, for Acute Eye Irritation/Corrosion testing of chemicals. This method provides informa-tion on health hazards likely to arise from exposure to test substances by application on the eye. An essential component of this TG includes the recommendation that, prior to undertaking the described in vivo test for acute eye irritation and corrosion, a weight-of-the-evidence analysis should be performed on the existing relevant data. This ensures that unnecessary suffering, stress, or pain is not infl icted on an inordinate number of animals.
TG 405 is intended preferably for use with albino rabbits. The test substance is applied as a single dose in the conjunctival sac of one eye of each animal. The other eye, which remains untreated, serves as a control. The initial test uses one animal; the dose level depends on the nature of the test substance. A negative result requires confi rmation by using up to two additional animals. The testing protocol should proceed in a sequen-tial manner with one animal at a time, rather than exposing the two additional animals simultaneously. The duration of the observation period should be suffi cient to evaluate the magnitude and reversibility of the effects observed; that is, the eyes are examined at 1, 24, 48, and 72 hours after test substance application. The ocular irritation scores are evaluated in conjunction with the nature and severity of lesions, and their reversibility or lack thereof. Individual scores do not represent an absolute standard for the irritant properties of a material, because other effects of the test material are also evaluated.2
2 The testing strategy includes the performance of validated and accepted in vitro tests.
TABLE 9.2Sources of Variability and Objections to Draize Eye Irritation and Corrosion Testing
Source Clinical Effects and Objections
Laboratory variability Intra- and inter-laboratory variations observed during the subjective scoring of test substances; standardization of time-to-heal criteria for assessment of severity of damage; grade defi nitions oversimplifi ed; existing scoring systems do not refl ect complexities of in vivo responses; lack of consistent data collection and interpretation of inadequate algorithms
Animal-related variability Variations in individual responses of animals exposed to same test substance; signifi cant differences in rabbit versus human eye anatomy: presence of nictitating membrane and lower mean corneal thickness in rabbit eye; differences in tearing mechanisms; increased sensitivity of rabbit (vs. human or primate) eye to chemical irritants; sensory response at time of instillation
Standardization of experimental variability
Concentration of test substance; use of anesthesia altering ocular responses; irrigation procedures to remove excess or particulates; adoption of standard dose volume (10 μL) to improve reproducibility; small numbers of animals used per group; duration of exposure; time to heal after exposure
Ethical considerations Minimizing acute and chronic pain and suffering; proper implementation of euthanasia

ACUTE OCULAR TOXICITY TESTING 119
PROCEDURE FOR ACUTE EYE IRRITATION AND/OR CORROSION
Details of the ocular test are outlined accordingly. The test substance3 is applied to the con-junctival sac of one eye of each animal after pulling the lower lid away from the eyeball. The lids are then gently held together for about 1 second in order to prevent loss of the material. The other eye, which remains untreated, serves as a control. Eyes of the test animals are not washed for at least 24 hours following instillation, except for solids or in the case of immedi-ate corrosive or irritating effects.
Experimental eyes are examined at 1, 24, 48, and 72 hours after test substance applica-tion. For the purposes of the evaluation, animals are maintained under experimental condi-tions no longer than necessary once defi nitive information has been obtained. Humane disposal of animals is warranted in the following circumstances or when animals display the following signs or symptoms: presence of continuing severe pain or distress; appearance of corneal perforation or ulceration; detection of blood in the anterior chamber of the eye; mea-surement of grade 4 corneal opacity4; appearance of ulceration or necrosis of the conjunctival membrane.
Animals that do not develop ocular lesions are terminated not earlier than 3 days post instillation. In general, rabbits with mild to moderate lesions are observed until the lesions
3 Application of 0.1 mL of a liquid; not greater than 100 mg of a solid. 4 Detection of opaque cornea, where the iris is not discernible through the opacity.
TABLE 9.3Grading of Ocular Lesions
Ocular Area Evaluation Score Description
Corneal Opacitya Opacity 0 No ulceration or opacity1 Scattered or diffuse areasb; iris clearly visible2 Discernible translucent areas; iris slightly obscured3 Size of pupil barely discernible; no details of
iris visible4 Opaque cornea; iris not discernible
Iris Observation of the appearance of the iris and its reaction to light
0 Normal1 Markedly deepened rugae, congestion, swelling, moderate
circumcorneal hyperaemia; iris reactive to light2 Hemorrhage, gross destruction, or no reaction to light
Conjunctivae Redness (excluding cornea and iris)
0 Normal1 Some hyperemic blood vessels2 Diffuse, crimson color; individual vessels not easily
discernible3 Diffuse deep red
Chemosis Swelling (refers to lids and/or nictitating membranes)
0 Normal1 Some swelling, above normal2 Obvious swelling with partial eversion of lids3 Swelling with lids about half closed4 Swelling with lids more than half closed
aOpaque areas.bDegree of density, Areas of opacity.Source: OECD supplement to TG 405, http://www.oecd-ilibrary.org/

120 PRINCIPLES OF TOXICOLOGY TESTING
clear or demonstrate reversibility. Observations continue at 7, 14, and 21 days, at which time the project is terminated.
Table 9.3 outlines the grading of ocular reactions to the conjunctiva, cornea and iris. Other adverse systemic effects are also noted at this time during the scoring. After record-ing the observations at 24 hours, further examination is enhanced with the instillation of fl uorescein.5
It is important to note that the grading of ocular responses is subjective. Training of personnel is an important component of performing, recording, and interpreting the obser-vations. Also, ocular irritation scores are evaluated in conjunction with the nature and severity of the lesions, and their ability for reversibility. In addition, the scores are not absolute and the total scores should not be interpreted as standards for any irritant proper-ties of a material. In fact, scores are generally regarded when compared with other known values, chemicals, and standards by which an appropriate evaluation and risk assessment can be presented. Consequently, a full description and evaluation of all observations during the testing period is essential for valid interpretation of the results.
CONCLUSIONS
In vivo ocular toxicology testing methods have been used for decades to provide data for formulating risk assessment programs. As with in vivo dermal testing, the information gener-ated during ocular tests is hampered by the scientifi c limitations and ethical objections posed, with the animal care issue at the forefront of ocular testing. The development of in vitro alternatives for this and other toxicity tests (discussed in Section III of this book) is necessar-ily based on comparisons to in vivo data. Thus validation of alternative methods depends on the reliability of in vivo data. Remarkable advancements have occurred in the development of in vitro ocular methods. It is conservative enough to state that only in vitro dermal toxicol-ogy methods have shown more utility in the replacement of animal toxicology tests as in vitro ocular models.
SUGGESTED READINGS
Jester JV. Extent of corneal injury as a biomarker for hazard assessment and the development of alternative models to the Draize rabbit eye test. Cutan Ocul Toxicol 2006; 25: 41.
Martinez V, Corsini E, Mitjans M, et al. Evaluation of eye and skin irritation of arginine-derivative surfactants using different in vitro endpoints as alternatives to in vivo assays. Toxicol Lett 2006; 164: 259.
Park NC. 1999.[Available from: http:/iccvam.niehs.nih.gov/methods/epiddocs/cwgfi nal/cwgfi nal.html] [last accessed December 2, 2012].
Secchi A, Deligianni V. Ocular toxicology: the Draize eye test. Curr Opin Allergy Clin Immunol 2006; 6: 367.
Wilhelmus KR. The Draize eye test. Surv Ophthalmol 2001; 45: 493.Yang TH, Liu SH, Wang SJ, Young YH. An animal model of ocular vestibular-evoked myogenic
potential in guinea pigs. Exp Brain Res 2010; 205: 145.
5 An orange dye instilled in the eye to detect damage to the cornea or outer surface. Briefl y, the eye is blotted or instilled with the dye, and spread on to the surface. Subsequently, a blue fl uorescent light directed at the eye illu-minates as a green fl uorescence and determines the location, size, and shape of the staining.

ACUTE OCULAR TOXICITY TESTING 121
REVIEW ARTICLES
Abraham MH, Hassanisadi M, Jalali-Heravi M, et al. Draize rabbit eye test compatibility with eye irritation thresholds in humans: a quantitative structure-activity relationship analysis. Toxi-col Sci 2003; 76: 384.
Barile FA. Validating and troubleshooting ocular in vitro toxicology tests. J Pharmacol Toxicol Methods 2010; 61: 136.
Kishore AS, Surekha P, Murthy PB. Assessment of the dermal and ocular irritation potential of multi-walled carbon nanotubes by using in vitro and in vivo methods. Toxicol Lett 2009; 191: 268.
Maurer JK, Parker RD, Carr GJ. Ocular irritation: microscopic changes occurring over time in the rat with surfactants of known irritancy. Toxicol Pathol 1998; 26: 217.
OECD, Test Guideline 405: (TG 405), Acute Eye Irritation/Corrosion, 2002. [Available from: http://www.oecd.org]
Pauly A, Brignole-Baudouin F, Labbé A, et al. New tools for the evaluation of toxic ocular surface changes in the rat. Invest Ophthalmol Vis Sci 2007; 48: 5473.
Prinsen MK. The Draize eye test and in vitro alternatives: a left-handed marriage? Toxicol in Vitro 2006; 20: 78.
Roberts JE. Screening for ocular phototoxicity. Int J Toxicol 2002; 21: 491.Svendsen O. The minipig in toxicology. Exp Toxicol Pathol 2006; 57: 335.York M, Steiling W. A critical review of the assessment of eye irritation potential using the Draize
rabbit eye test. J Appl Toxicol 1998; 18: 233.

122
10 Toxicity testing for fertility and reproduction
INTRODUCTION
Teratogens are chemicals or drugs that cause toxicity to an embryo by inducing structural mal-formations, physical dysfunction, behavioral alterations, or genetic abnormalities in the fetus. The expression of the teratogenic response—the embryotoxic effect of a compound on the growth and development of the fetus—is usually manifested at birth or in the immediate post-natal period. A chemical may also interfere with the reproduction by inducing toxicity early in the reproductive cycle (fi rst trimester) by impairing fertilization or by interfering with implanta-tion. Thus, the initial interaction of a fertilized ovum and exposure to a suspected teratogen may result in the inability to establish pregnancy or induction of its spontaneous termination.
Reproductive toxicology also involves the study of adverse effects of xenobiotic agents on events prior to fertilization of the ovum including spermatogenesis and oogenesis. That is, chemicals may alter the development of sperm, may interfere with the interaction of sperm and oocytes, or may preclude proper transport of oocytes within the reproductive tract. These responses create a need to defi ne, develop, and properly perform tests aimed at evaluating chemicals with the potentials to interfere with human reproduction.
Therapeutic drug use and exposure to environmental and occupational agents during pregnancy have increased dramatically in recent years. Epidemiological studies indicate that about 90% of women ingest one or more medications while pregnant (FDA, 1995). In addition, the rates of environmental and occupational exposure to hazardous chemicals before or during pregnancy have increased substantially. Excluding the commonly pre-scribed prenatal vitamins, iron supplements, and tocolytic1 drugs, women under 35 years of age take three prescriptions during the course of their pregnancies on an average; the num-ber increases to fi ve in women over 35.
Some of the environmental chemicals to which women are inadvertently exposed dur-ing pregnancy include insecticides, herbicides, fungicides, metals (such as mercury in sea-food), and growth hormones (in processed foods). For most of these chemicals and drugs, well-controlled studies are not conducted prior to marketing nor are the studies mandated for most of the therapeutic or nontherapeutic agents.
One complicating factor arises from the extensive abilities of therapeutic drugs, envi-ronmental chemicals, and herbal products to infi ltrate the maternal–fetal environment. Although several therapeutic drugs are screened for their embryotoxic or teratogenic poten-tials in animal reproductive studies, many chemicals are not. U.S.regulations do not man-date that pharmaceutical and chemical companies provide such information. Also, most drug development programs do not routinely incorporate screening or follow-up studies of drugs for potential embryotoxic effects in pregnant women during preclinical and clinical
1 Drugs that are used to suppress premature labor.

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 123
phases or in post-marketing periods. Finally, environmental chemicals have generally not been subjected to teratogenic testing. This means no human embryotoxic data exist for a multitude of chemicals used in the marketplace.
HISTORY AND DEVELOPMENT OF TERATOGENICITY TESTING
The thalidomide disaster of 1961 initiated a signifi cant effort by U.S. and European legislative bodies to establish programs for teratogenicity testing. Developed as a sedative–hypnotic agent with no particular advantage over drugs of the same class, thalidomide was initially shown to lack teratogenic effects in all species tested except rabbits. Soon after its introduction in the European market, it was linked to the development of a relatively rare birth defect known as “phocomelia.” The epidemic proportions of the teratogenic effect of thalidomide prompted the passage of the Harris–Kefauver Amendment in the United States in 1962, one of the many additions to the Pure Food and Drug Act. The amendment requires extensive preclinical phar-macological and toxicological research before a therapeutic compound is marketed.
BRIEF DESCRIPTION OF MATERNAL–FETAL PHYSIOLOGY
FETAL PRENATAL DEVELOPMENT
Prenatal growth and development involves the beginning, growth, and maturity of the anat-omy and physiology from fertilization (conception) to parturition (birth). The fi rst trimester (fi rst 10 weeks of pregnancy in humans) is characterized as the period for embryological development during which differentiation of precursor stem cells progresses, culminating in the appearance of the fetal membranes and embryonic disc. The remainder of the pregnancy (11–40 weeks) is dedicated to fetal development during which the established blueprints of organs and tissues undergo further growth and maturity. These processes constitute prenatal growth and development. Thus, in humans, the duration of the gestational period is normally 9 months (or 280 days from the fi rst day of the last menstrual period, assuming a regular 28-day cycle). Postnatal development begins at birth and progresses to adolescence.
Figure 10.1 illustrates the cycle of reproductive life. The major stages of reproduction (shaded areas) are composed of the events that occur during formation and development of the embryo within those stages (darker shaded area), along with the parallel time sequences (gray boxes). Consequently, the most sensitive periods of exposure to toxic substances cor-respond to the events occurring during the reproductive cycles (mating, cleavage, gestation, and parturition). In general, exposure during the earlier stages of fetal development is asso-ciated with more severe teratogenic consequences. In addition, dose and duration of expo-sure are critical in evaluating the mechanisms of reproductive toxicity.
FIRST TRIMESTER
The fi rst trimester is the period corresponding to embryological and early fetal development. Following are the main events occurring during the fi rst trimester:
Cleavage (days 1 through 6): A series of cell divisions following fertilization of the ovum by the sperm cell, resulting in the formation and transport of the morula (early cell division stage) and blastocyst (late cell division stage) through the fallopian tube.
Implantation (days 7 through 10): Adhesion, attachment, and penetration of the uter-ine endometrial lining by the blastocyst; trophoblast, amniotic cavity, and inner cell mass form and expand. Further development of the yolk sac, chorion, allantois,

124 PRINCIPLES OF TOXICOLOGY TESTING
amnion, and embryonic disc (from the inner cell mass) ensues during weeks 3 and 4 (beginning of embryogenesis).
Placentation (weeks 2 through 10): Starting as early as day 10, blood vessels form around the periphery of the blastocyst to establish the presence of the placenta; this multilayered membranous structure is composed of infi ltrating chorionic villi and highlighted by the appearance of fetal blood vessels.
Embryogenesis (weeks 5 through 10, concurrent with placentation): The differen-tiation and folding of the embryonic disc produce a physically and developmentally distinct and recognizable embryo. Because of the critical events occurring during this phase, as well as during fertilization and formation of germ layers (Fig. 10.1), the fi rst trimester ies the most sensitive period of prenatal growth and development to external stimuli, especially to drugs and chemicals.
SECOND AND THIRD TRIMESTERS
The second and third trimesters range from the 10th to 40th week (4th to 9th month) of ges-tation. The second trimester is characterized by the development of organ systems (organo-genesis) while the third trimester exhibits a rapid growth and maturation of established organs in preparation for parturition (birth).
The next section summarizes the criteria necessary to develop well-planned reproduc-tive toxicity tests according to the mammalian species under consideration. Extrapolation of the data derived from animal studies to human hazard assessment is also discussed. The current focus on the development of in vitro alternative toxicity tests for reproductive stud-ies is covered in chapter 17.
Sexualmaturation
Fertilization
Weaning Morula
b
Lactation Blastocyst
Parturition I Implantation
Fetaldevelopment Placentation
Embryogenesis
Fertility
Germ layersand transport
Gestation
Perinatal/postnatal
development
Mating
Cleavage
Prenatalgrowth and
development
Parturition
Wk 5–10
Yrs
Wk 10–40
Wk 2–10
Adolescence
Day 0
Day 1–4
Day 4–7
Day 7–10
FIGURE 10.1 Cycle of reproductive life and periods of potential embryo and reproductive toxicity.

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 125
MECHANISMS OF DEVELOPMENTAL TOXICITY
SUSCEPTIBILITY
As noted earlier, the fi rst 8–10 weeks of embryogenesis qualify as a sensitive period for the exposure of a fetus to drugs or chemicals. For instance, exposure to exogenous compounds within 21 days after fertilization presents the greatest potential for embryotoxicity resulting in the death of the fetus. Exposure to compounds between the 3rd and 8th weeks is potentially teratogenic, that is, the agents may interfere with organogenesis. The effects are distinguished by the development of gross anatomic, metabolic, or functional defects and precipitation of spontaneous abortion.
Because the schematic for embryonic development is already established by the begin-ning of the second trimester, exposure to toxic agents during the second or third trimesters is less likely to be teratogenic. However, exposure may still interfere with the growth and maturation of critical organ systems, resulting in underdevelopment or alterations of normal organ function.
DOSE RESPONSE AND THRESHOLD
Exchange of nutrients, gases, and waste materials between the maternal and fetal circula-tions commences from the 5th week after fertilization, with the initiation of the formation of the placenta. Maternal and fetal circulations do not mix. Instead, maternal blood enters inter-villous spaces (sinuses) of the placenta through ruptured maternal arteries, then drains into uterine veins for return to the maternal circulation.
Fetal blood enters the placenta via a pair of umbilical arteries and returns to the fetal circulation by the umbilical vein. The umbilical arteries branch into capillaries, are sur-rounded by the syncytial trophoblasts, and form a network of chorionic villi. Solutes, gases, and nutrients from the maternal circulation enter the sinuses, surround and bathe chorionic villi, and traverse the epithelium and connective tissues of the villi before penetrating the fetal capillary endothelial cells. The materials are then carried toward the embryo through the umbilical vein.
Chemicals enter the fetal circulation as they traverse any other membrane barrier. The rate of exchange between maternal and fetal circulation depends on the established equilib-rium across the placenta and the rate of penetration of the chemical through the endothelial and epithelial barriers. Lipid-soluble drugs and chemicals and dissolved gases readily infi l-trate placental membranes and establish high concentrations of the substances in venous cord blood. Thus, compounds may freely circulate around the embryo within 30–60 minutes of maternal administration. In addition, exogenous substances can alter the dynamic tone of placental blood vessels, altering nutrient, gas, and waste exchange. Depending on the toxi-cokinetics, some chemicals such as gases restrict oxygen transport to fetal tissues, change the biochemical dynamics of the two circulations, and interfere with the endocrine function of the placenta. The degree of chemical penetration of placental barriers relies less on the number and thickness of the layers and more on the dynamic physicochemical interactions of the chemical with the placenta.
It is important to consider that because embryonic tissues grow rapidly and have high DNA-turnover rates, they are vulnerable to many agents that may not promote toxic reac-tions in children or adults. As explained earlier, the most sensitive period for embryonic development is within the fi rst trimester of pregnancy, when the fetus is in the early stages of embryonic development. Some classes of agents require repeated, continuous, or high-dose exposure to elicit untoward reactions. Other more potent chemicals, such as suspected

126 PRINCIPLES OF TOXICOLOGY TESTING
carcinogens, target rapidly proliferating cells and tissues. Their effects are signifi cant and occur with higher frequencies.
TERATOGENICITY
TERATOGENIC AGENTS
According to the defi nition of a teratogen cited at the beginning of this chapter, a teratogenic response to a chemical involves an embryotoxic effect on the growth and development of a fetus. The effect is manifested at birth or in the immediate postnatal period. Approximately 25% of all teratogenic responses are due to genetic susceptibilities and chromosomal aber-rations, whereas less than 75% arise from unknown origins. The remaining, apparently small, percentage of teratogenic responses arises from known environmental and therapeutic exposures and sociobehavioral habits. Consequently, these recognized human teratogens are subject to intensive scrutiny as a result of their continuous presence and important uses in society. The American College of Occupational and Environmental Medicine recognizes several classes of known human teratogens, as listed in Table 10.1.
FACTORS ASSOCIATED WITH TERATOGENICITY
Any chemical agent should be suspected of causing a teratogenic response if the conditions of exposure are suffi cient for eliciting a toxic reaction. For instance, dose response and time of exposure to a particular agent often determine the severity of the damage and the type of defect that occurs. In general, the teratogenic effect of a chemical is directly proportional to the toxic response. Consequently, a relatively higher dose of a chemical is more likely to trigger a teratogenic effect than a lower dose. Time of exposure is another critical component because vulnerability to chemical exposure is greater early in embryonic development (fi rst trimester) than during the fetal development period (second and third trimesters).
The experience with thalidomide provides a classic example of critical exposure during organogenesis. The specifi city of the malformations was linked to the time of exposure to the
TABLE 10.1Classes of Teratogens Recognized by American College of Occupational and Environmental Medicine
Classes of Known Human Teratogens Examples
Ionizing radiation Atomic weapons, radioiodine, radiation therapy Maternal infections Cytomegalovirus, herpes viruses I and II, parvovirus B-19, rubella virus,
syphilis, toxoplasmosis, Venezuelan equine encephalitis virus Maternal disorders Alcoholism, endemic cretinism, diabetes, folic acid defi ciency, hyperthermia,
phenylketonuria, rheumatic disease and congenital heart block, virilizing tumors
Additive behavioral habits Chronic alcoholism, drug abuse, cigarette smokingEnvironmental chemicals Metals (including mercury and lead compounds), herbicides, industrial solvents,
polychlorobiphenyls Drugs Many therapeutic agents

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 127
sedative–hypnotic drug during pregnancy (after the last menstrual period): 35–37 days, ears; 39–41 days, arms; 41–43 days, uterus; 45–47 days, tibia; 47–49 days, triphalangeal thumbs.
The types and severity of abnormalities caused by a teratogenic agent are also more dependent on the genetic susceptibility of a pregnant woman than on the susceptibility of the fetus. For example, variation in maternal metabolism toward a particular drug deter-mines what metabolites eventually reach the fetus. Differences in placental membranes, rate of formation, placental transport, and biotransformation also affect fetal exposure.
Finally, as noted earlier, the principles of toxicokinetics and pharmacokinetics of dif-ferent classes of compounds infl uence the degree of penetration of the placental membrane. Generally, the greater the lipid solubility of an agent, the more likely is the potential for fetal exposure through the maternal–fetal circulation.
Thus, these factors as well as the characteristics of individual compounds determine the probability that a chemical will cause a genetic defect. The probabilities are categorized accordingly:
Absolute risk: The rate of occurrence of an abnormal phenotype among individuals exposed to an agent. Chronic maternal alcoholism increases the risk of an infant born with fetal alcohol syndrome up to 45%.
Relative risk: The ratio of the rate of the condition among those exposed to a chemical and the non-exposed general population. Cigarette smokers have twice the relative risk of the general population. Thus the risk of delivering a low birth weight infant is twice as likely in smokers than non-smokers. It is important to note, however, that a high relative risk may still indicate a low absolute risk in rare conditions such that a high relative risk of 10 for a rare condition with an incidence of 1 in 100,000 calcu-lates to a low absolute risk of 1 in 10,000.
Attributable risk: The rate of a condition attributed to exposure to an agent, especially to known teratogens (90% risk of development of phocomelia in patients exposed to thalidomide in utero).
The increasing awareness of teratogenic potential from exposure to hazardous chemi-cals and therapeutic drugs during pregnancy has facilitated dissemination of information among regulatory agencies, healthcare professionals, and the public. As a result, teratogen information services are easily accessed in most states through national databases and the efforts of applicable societies and organizations (see Suggested Readings and Review Articles).
ANIMAL TESTS FOR TERATOGENICITY
Developmental toxicity testing is primarily intended to provide hazard identifi cation con-cerning the potential effects of prenatal exposure on a developing human or animal organism. For human pharmaceuticals, this information can be used to limit patient population expo-sure. In addition to hazard identifi cation, in certain circumstances the no observed effect level (NOEL) determined by developmental toxicity studies for veterinary pharmaceutical compounds can be used as the basis for establishment of acceptable daily intake.
In order to mimic a suspected or known human teratogenic response, the test system involves the administration of a specifi c chemical encompassing a range of doses sus-pected of eliciting toxic effects on an animal species. Ideally, the species should be genet-ically susceptible to a variety of chemical classes and the animals should be at a sensitive stage of embryonic development. The degree of susceptibility, however, is less associated

128 PRINCIPLES OF TOXICOLOGY TESTING
with a genetically programmed species for chemical sensitivity and more a function of factors associated with the properties of biotransformation peculiar to an animal group. Consequently, maternal and/or fetal ability for biotransformation of compounds may alter the net effect of a teratogenic substance. Thus, depending on the parent chemical, active biotransformation reactions may predispose an embryo for teratogenic effects by forming reactive intermediates. Alternatively, although the absence of rapid biotransformation reactions may reduce the production of active metabolites, the lack of inactivating mecha-nisms may prolong exposure of the fetus to the active parent compound.
The most common approach to identifying teratogens is to perform developmental toxicity tests in two species, usually rodents and rabbits, for evaluation of human therapeu-tic drugs (International Conference on Harmonization, 1994) and for testing of pesticides and environmental chemicals [U.S. Environmental Protection Agency (EPA), 1998]. The basis for the two-species approach is that no single species is defi nitively predictive for human teratogenicity nor is any single species more sensitive to teratogens than others. Rodents and rabbits are particularly useful because
1. they appear to demonstrate low incidences of spontaneous congenital anomalies.2. care and costs are reasonable.3. they produce large litters.4. they have relatively short gestation times (several weeks).
For instance, rodent and rabbit gestation times are limited to 21 days and 32 days, respectively. Thus, in order to detect the potential teratogenicity of a chemical, the agent is administered within 7–10 days (for rodents and rabbits, respectively) after implantation. This period of cell cleavage and embryogenesis represents a narrow window for detecting teratogens and developmental and reproductive toxicity in these species.
The use of a tiered approach was suggested for developmental toxicity evaluation of vet-erinary drugs for food-producing animals that may also be useful for a variety of chemical and pharmacological classes (International Conference on Harmonization, 1994; EPA, 1998). The tiered method begins with evaluation and observation for developmental toxicity in a rodent species. If the testing proves positive, no further testing is required in a second species (unless the acceptable daily intake is determined based on the NOEL from this study). A negative or equivocal result for teratogenicity in a rodent prompts a developmental test in a second species, customarily the rabbit. Other signs of developmental toxicity in the rodent species in the absence of teratogenicity—visceral or skeletal malformations, fetal mortality, or intrauterine growth retardation—requires a developmental toxicity test in a second species. Thus, the tiered approach provides thorough hazard identifi cation based on the use of a second species for compounds negative for teratogenicity in a rodent. This scheme for stepwise testing ensures adequate screening of potential teratogens and attempts to limit unnecessary animal testing.
Standard Protocols
Table 10.2 summarizes the OECD Guidelines for the Testing of Chemicals, Section 4: Health Effects, for in vivo animal testing of developmental, embryotoxic, and reproductive toxicity. Some representative individual methodologies are described in detail in the individual sec-tions below.
Standard methodologies for screening for teratogenicity are performed to ensure suf-fi cient exposure at critical times post implantation. The protocol is outlined in Table 10.3. Essentially, administration of a chemical is proportioned about 6 days post implantation to

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 129
TAB
LE 1
0.2
OEC
D G
uide
lines
for
Tes
ting
of
Che
mic
als,
Sec
tion
4,
Hea
lth
Effe
cts:
In
Viv
o A
nim
al T
esti
ng f
or D
evel
opm
enta
l, Em
bryo
toxi
c,
and
Rep
rodu
ctiv
e To
xici
ty (
Acc
ordi
ng t
o th
e Ye
ar S
ubm
itte
d)
Test
Gui
delin
e N
umbe
rD
ate
Subm
itte
d (Y
ear)
Titl
eD
escr
ipti
on
415
1983
One
-Gen
erat
ion
Rep
rodu
ctio
n To
xici
ty S
tudy
Gen
eral
info
rmat
ion
conc
erni
ng th
e ef
fect
s of
a te
st s
ubst
ance
on
mal
e an
d fe
mal
e re
prod
uctiv
e pe
rfor
man
ce47
8 19
84G
enet
ic T
oxic
olog
y: R
oden
t Dom
inan
t Let
hal T
est
Indu
ctio
n of
a d
omin
ant l
etha
l eve
nt a
fter
exp
osur
e to
a te
st s
ubst
ance
, w
hich
cau
ses
embr
yoni
c or
fet
al d
eath
421
1995
Rep
rodu
ctio
n/D
evel
opm
enta
l Tox
icity
Scr
eeni
ng T
est
Test
sub
stan
ce is
adm
inis
tere
d in
gra
duat
ed d
oses
to s
ever
al g
roup
s of
mal
es
and
fem
ales
; scr
eeni
ng te
st42
219
96C
ombi
ned
Rep
eate
d D
ose
Toxi
city
Stu
dy w
ith th
e R
epro
duct
ion/
Dev
elop
men
tal T
oxic
ity S
cree
ning
Tes
tA
s w
ith T
G 4
21; m
ales
are
dos
ed f
or a
min
imum
of
4 w
eeks
; fem
ales
are
do
sed
thro
ugho
ut th
e st
udy
(~54
day
s)48
319
97M
amm
alia
n Sp
erm
atog
onia
l Chr
omos
ome
Abe
rrat
ion
Test
Test
mea
sure
s ch
rom
osom
e ev
ents
in s
perm
atog
onia
l ger
m c
ells
; pre
dict
ive
of in
duct
ion
of in
heri
tabl
e m
utat
ions
in g
erm
cel
ls41
420
01Pr
enat
al D
evel
opm
ent T
oxic
ity S
tudy
Dev
elop
men
tal t
oxic
ity te
st d
esig
ned
to p
rovi
de g
ener
al in
form
atio
n on
the
effe
cts
of p
rena
tal e
xpos
ure
on p
regn
ant t
est a
nim
als
and
on th
e de
velo
ping
or
gani
sm41
620
01Tw
o-G
ener
atio
n R
epro
duct
ion
Toxi
city
Two-
gene
ratio
n re
prod
uctio
n te
st d
esig
ned
to p
rovi
de g
ener
al in
form
atio
n on
the
effe
cts
of a
test
sub
stan
ce o
n th
e in
tegr
ity a
nd p
erfo
rman
ce o
f th
e m
ale
and
fem
ale
repr
oduc
tive
syst
ems
440
2007
Ute
rotr
ophi
c B
ioas
say
in R
oden
tsA
n in
viv
o sh
ort-
term
scr
eeni
ng te
st b
ased
on
the
incr
ease
in u
teri
ne w
eigh
t or
ute
rotr
ophi
c re
spon
se44
320
11E
xten
ded
One
-Gen
erat
ion
Rep
rodu
ctiv
e To
xici
ty
Stud
yE
valu
atio
n of
rep
rodu
ctiv
e an
d de
velo
pmen
tal e
ffec
ts th
at m
ay o
ccur
as
a re
sult
of p
re-
and
post
nata
l che
mic
al e
xpos
ure
as w
ell a
s an
eva
luat
ion
of
syst
emic
toxi
city
in p
regn
ant a
nd la
ctat
ing
fem
ale
rode
nts
488
2011
Tra
nsge
nic
Rod
ent S
omat
ic a
nd G
erm
-Cel
l Gen
e M
utat
ion
Ass
ays
Det
ects
che
mic
als
that
indu
ce g
ene
mut
atio
ns. U
ses
tran
sgen
ic r
oden
ts th
at
cont
ain
mul
tiple
cop
ies
of c
hrom
osom
ally
inte
grat
ed p
lasm
id o
r ph
age
shut
tle v
ecto
rs

130 PRINCIPLES OF TOXICOLOGY TESTING
determine teratogenic potential. Dose–response relationships are based on observable and measured defects according to treatment group.
A frequently used test for detecting, screening, and measuring developmental toxicity is OECD TG 414, Prenatal Development Toxicity study. Submitted for approval in 2001, this protocol for developmental toxicity testing is designed to provide general information concerning the effects of prenatal exposure on the pregnant test animal and on the develop-ing organism. The test substance is administered to established pregnant animals from implantation to one day prior to the day of scheduled sacrifi ce (generally as close as pos-sible to the normal day of delivery). Rats or rabbits are the preferred species. The study is designed so as to yield at least 20 female animals with implantation sites at necropsy. The agents are orally administered in three concentrations by gavage. A limit test may be per-formed if no effects are expected at a dose of 1000 mg/kg body weight. Clinical observa-tions are measured and recorded accordingly throughout the project. Before caesarean section, the females are killed, the uterine contents are examined, and the fetuses are evalu-ated for soft tissue and skeletal changes. A variety of histopathological analyses are avail-able that allow for procedures to dissolve skin and visceral organs, for preserving skeletal structure, and for differential staining of the bones and connective tissue. The specimen is then preserved in 10% neutral buffered formalin or any other suitable preservative. In any study that demonstrates an absence of toxic effects, further investigation should be consid-ered to establish absorption and bioavailability of the test substance.
EMBRYOTOXICITY
As with teratogenic studies, classical methods for testing embryotoxic substances involve rodents and rabbits, principally because of their relatively short gestational periods. The procedures involve exposure of the female either before or immediately after conception for the length of gestation. The beginning of the exposure sequence depends on the desired toxicity data. For instance, to determine the effects of a chemical on growth and develop-ment of the fetus, the exposure period begins several days after fertilization, at about implan-tation. The time sequence must be carefully determined so as not to interfere with mating.
Rodents and rabbits are convenient for embryotoxic experiments because of their large litter sizes, usually about 8–10 embryos per litter, thus allowing for chemical scoring according to the number of fetuses affected. For instance, a particular indicator is selected as a measure of toxicity and the end point is then monitored in each embryo. The minimal accepted value for this indicator is regarded as toxic and the experiment is completed on the rest of the litter. The greater the number of embryos that exhibit toxicity by this criteria, the
TABLE 10.3Standard Methodology for Teratogenicity Testing in Animals
Time Period Protocol
Day 0 Breeding of animals; determination of successful impregnation by the presence of vaginal plug
Days 6–15: rodent; days 6–18: rabbit
Chemical administration to females (3 dosage groups, 10 rats or 5 rabbits per group)
~Day 20: rodent; ~day 30 to 31: rabbit
Euthanize animals, necropsy dams and fetuses; identify gross anomalies; perform pathological, histological, physiological, and biochemical analyses of fetuses

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 131
higher the score and the more toxic the chemical. For a given number of chemicals that can be screened at different dosage levels for various indicators in each fetus, the number of experiments is planned to optimize resources and effort. The following section outlines the various protocols available for embryotoxicity testing.
FETOTOXICITY MEASUREMENTS AND END POINTS
One of the fi rst in vivo reproductive toxicology tests to be submitted for validation is OECD TG 415, one-generation reproduction toxicity study (Table 10.2). This TG for reproduction testing is designed to provide general information concerning the effects of a test substance on male and female reproductive performance. The test substance is administered orally in the drinking water in graduated doses to several groups of males and females. Males are dosed during growth and for at least one complete spermatogenic cycle; females are dosed for at least two complete estrous cycles. During the mating period, the test substance is administered to both sexes, followed by administration only to females during pregnancy and for the duration of the nursing period.
Rodents are the primary animal species for TG 415. Each test and control group should yield about 20 pregnant females, for at least three test groups. A limit test is performed if no effects are expected at a dose of 1000 mg/kg body weight. Detailed measurements are taken of weight, food consumption, gross necropsy and histopathology. Properly con-ducted, this reproduction test should provide a satisfactory estimation of a NOEL and an understanding of adverse effects on reproduction, parturition, lactation and postnatal growth. As the test chemical is administered before mating, the substance may also interefere with mating. Thus the utility of the test is that not only is it possible to determine the toxic effects of a chemical on genetic events, but also on mating and behavioral aspects of reproduction.
An extended version of TG 415 was submitted for review and validation in 2011. TG 443, the Extended One-Generation Reproductive Toxicity study, evaluates the reproductive and developmental effects that may occur as a result of pre- and postnatal chemical expo-sure, as well as an evaluation of systemic toxicity in pregnant and lactating female rodents.
An updated version of TG 415 was submitted in 2001 as TG 416, the Two-Generation Reproduction Toxicity. This TG is designed to provide general information concerning the effects of a test substance on the integrity and performance of the male and female repro-ductive systems for two generations, as well as on the growth and development of the off-spring. The test substance is administered daily in graduated doses to several groups of males and females. Males and females of the parent generation (5–9 weeks old) are exposed to the test agent dosed during growth, their mating, during the resulting pregnancies, and throughout the weaning of their fi rst-generation offspring. The administration of the sub-stance is continued to fi rst-generation offspring during their growth into adulthood, mating and production of a second generation (until weaning).
As with TG 415, the rat is the preferred species, yielding not less than 20 pregnant females at or near parturition. At least three dose levels and a concurrent control is admin-istered orally (by drinking water or gavage). Limit tests and results are interpreted as men-tioned earlier. The main difference from the one-generation study is that the fi ndings are evaluated at the NOEL and include information and interpretation of data to present con-clusions on the effects of chemicals on reproduction, parturition, lactation, postnatal devel-opment, as well as growth and sexual development in subsequent generations.
Another in vivo test that measures embryotoxic effects is TG 483, the Mammalian Spermatogonial Chromosome Aberration Test. Introduced in 1997, this test measures

132 PRINCIPLES OF TOXICOLOGY TESTING
chromosome events in spermatogonial germ cells and is, therefore, expected to be predic-tive of induction of inheritable mutations in germ cells. The preferred species, male Chi-nese hamsters and mice, are exposed to a single or repeated dose of the test substance by gavage or by intraperitoneal injection. Animals are treated with a metaphase-arresting agent (mitomycin-C), after which they are sacrifi ced. A karyotype analysis is performed and chromosome preparations are stained with Giemsa. Metaphase cells are analyzed for chromosome aberrations.
Positive results from the in vivo spermatogonial chromosome aberration test indicate that a substance induces chromosome aberrations in the germ cells of the species tested.
More recently, the Transgenic Rodent Somatic and Germ Cell Gene Mutation Assay (TG 488, 2011) was submitted to detect chemicals that induce gene mutations. The test uses transgenic rodents that contain multiple copies of chromosomally integrated plasmid or phage shuttle vectors. The transgenes contain reporter genes for the detection of various types of mutations induced by test substances. Groups of transgenic animals are treated for 28 consecutive days, followed by a 3-day off-period prior to sacrifi ce; during this time unrepaired DNA lesions are fi xed into stable mutations. Animals are then sacrifi ced, genomic DNA is isolated from the tissues of interest and purifi ed. Mutations that have arisen during treatment are scored by recovering the transgene and analyzing the pheno-type of the reporter gene in a bacterial host defi cient for the reporter gene. Mutant fre-quency, the reported parameter in these assays, is calculated by dividing the number of plaques per plasmids containing mutations in the transgene by the total number of plaques per plasmids recovered from the same DNA sample.
WHOLE-EMBRYO CULTURE
Preimplantation Techniques
As the fi rst description of the culture of preimplantation embryos was reported in 1913, the method has since shown its importance in experimental embryology and reproductive biol-ogy. The technique is described for the preimplantation cultures of embryos from mice, rabbits, and humans. In general, 3- to 6-day old embryos (3-day morulae to 6-day blasto-cysts) are removed from superovulated sexually mature animals and transplanted into cul-ture dishes containing media supplemented with fetal bovine serum or serum-free conditioned medium.
The cultures are exposed to the test substance during the incubation period and exam-ined microscopically or monitored for growth and proliferation using biochemical proce-dures. A variety of cell viability indicators are then used to evaluate the cytotoxicities of xenobiotics in cultured embryonic stem cells derived from the mouse blastocysts. Thus, preimplantation embryo cultures may provide interesting stem cell systems for testing embryonic toxicity after exposure to xenobiotics.
Postimplantation Embryo Culture
A classical method for embryotoxic potential that requires separation of whole embryos from females is known as postimplantation embryo culture. The technique involves the surgical removal of 1- or 2-week-old rodent embryos, after which they are allowed to acclimate in a medium under cell culture conditions for several hours. The viable fetuses are then exposed to different doses of chemicals and the effects on growth and development are determined.
The method is dramatic because structural changes occur quickly, within several days, and embryotoxicity is monitored in culture as growth and differentiation progress. This

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 133
method has the additional benefi t of allowing for further investigation of substances with teratogenic activities as well. In addition, culturing embryos in vitro provides the opportu-nity to study the direct actions of suspected toxins free of maternal hormonal and pharma-cokinetic infl uences. Dose-related growth retardation and frequency of malformations are monitored macroscopically and depend on the periods of gestation of the explanted embryos.
Postimplantation whole-embryo culture is suitable for screening embryotoxic sub-stances because it involves the period of fetal development that includes morphogenesis and early organogenesis. The procedure is adequate for screening chemicals that also have teratogenic potential as well as embryotoxicity because it monitors multiple cellular pro-cesses that refl ect the complex nature of embryonic development. The period for identify-ing toxicity, however, is limited up to week 2 because development of cultured rodent embryos requires placental development. Consequently, the technique refl ects activities of chemicals during several coordinated cellular events, especially proliferation, migration, association, differentiation, and cell death. Ideally, proper performance of the postimplan-tation embryo culture screening technique involves the following:
1. A suffi cient number of viable fetuses explanted for testing2. Unobstructed, progressive, and continuous embryonic development3. Knowledge of the responses and mechanisms of chemical toxicity4. Ease of manipulation and interpretation of the protocol and a low frequency of false
negatives5. Administration of approximate equimolar concentrations as those encountered in
therapeutic or environmental exposures, with systematic control of dose and time of exposure
6. Elimination of maternal factors that may interfere with the test system’s response to the agent
Occasionally, responsiveness of postimplantation rodent embryos to cytotoxic agents may require metabolic activation. Thus modifi cations of the protocols may require incuba-tion of embryos in a medium containing the test agent plus hepatic S9 fractions.
Although the procedure uses growth in culture as a component of the protocol, it should not be confused with in vitro cell culture methods (chap. 16). The embryo cultures still require extensive animal preparation and sacrifi ce and offer few advantages for large-scale prescreening studies.
Organ Cultures
Fetal organs are maintained in culture in the primary cell stage by establishing whole-organ cultures and partially or totally submerging the tissue in the growth culture medium. Briefl y, the fetal tissue explant is placed on micron fi lter inserts or in tissue culture plastic coated with extracellular matrix components. This approach has been useful in the culture of limb buds from several species of animals in a serum-containing or chemically defi ned medium. Morphological development and differentiation are monitored microscopically; biochemical indicators are also employed.
Several organs have been extensively investigated for embryotoxic phenomena includ-ing kidney development from suspected nephrotoxic agents and primary organotypic cul-tures of type II epithelial cells from fetal rat lungs. Thus, as with other organ-specifi c primary cultures, the advantage of organ culture lies in the ability of the in vivo–in vitro system to act as a model for detecting target organ toxicity.

134 PRINCIPLES OF TOXICOLOGY TESTING
International Guidelines
In 1999, the Organization for Economic Cooperation and Development (OECD) proposed international recommendations as harmonization guidelines of chemical classifi cation sys-tems for reproductive toxicity (OECD Series On Testing and Assessment, Number 15; OECD Guidelines for Testing Chemicals, Section 4, Health Effects). The guidelines describe the systems currently in use by member organizations and countries including the European Union, United States, Canada, and Australia. They also provide overviews and comparisons of the systems in use, hazard classifi cation methods, data requirements, mechanisms for classifying substances, and regulatory consequences of classifi cation. An attachment lists substances classifi ed in the European Union as toxic to reproduction.
In the United States, test guidelines for developmental toxicity testing are designed to provide general information concerning the effects of exposure of pregnant test animals on developing organisms. The guidelines were published by the EPA and meet the labeling requirements of the Federal Hazardous Substances Act, the Federal Insecticide, Fungicide and Rodenticide Act and the Toxic Substances Control Act. Developmental neurotoxicity testing guidelines are also designed to address functional and behavioral effects of prenatal exposure, including teratogenicity and embryotoxicity information.
Reproduction and fertility testing is designed to incorporate gonadal function, the estrus cycle, mating behavior, conception, gestation, parturition, lactation and weaning, and growth and development of offspring. Proposed revisions to the developmental and reproductive toxicity testing guidelines should increase the sensitivity of the studies in identifying other effects such as cartilaginous development in fetuses, spermatogenesis, and sexual maturation.
In general, the regulatory systems are broadly similar, are based on classifi cations of chemicals according to their intrinsic properties, and involve the consideration of reproduc-tive effects seen in humans and those demonstrated in suitable animal tests. Some differ-ences among the systems include coverage of specifi c reproductive effects, a number of classifi cation categories for each type of reproductive effect, the possibility of classifying on the basis of limited evidence, and concentration cutoffs for classifying mixtures. Label-ing requirements may also pose formidable obstacles to harmonization of classifi cation.
MALE REPRODUCTIVE TOXICOLOGY
SPERMATOGENESIS
The testis is composed of a series of highly convoluted seminiferous tubules enclosed in the tunica albuginea, tunica vaginalis, and supporting connective tissue and vascula-ture. The tubules are composed of three cell types: spermatogenic (germ) cells, Sertoli cells, and the interstitial cells of Leydig. As the germ cells proliferate during spermato-genesis, they undergo meiotic division and migrate from the basement membrane of the tubule toward the lumen.
Development of germ cells arises with the appearance of spermatogonia, embryonic stem cells, near the basement membrane. As spermatogonia continue their progression toward the tubular lumen, they differentiate to primary spermatocytes, secondary sper-matocytes, and spermatids. Spermiogenesis follows for an additional 5 weeks in humans and involves the differentiation of spermatids into mature spermatozoa. Sperm cells undergo a metamorphosis from rounded cells that adhere onto the apical surfaces of semi-niferous tubules into the characteristically shaped mature spermatozoa.
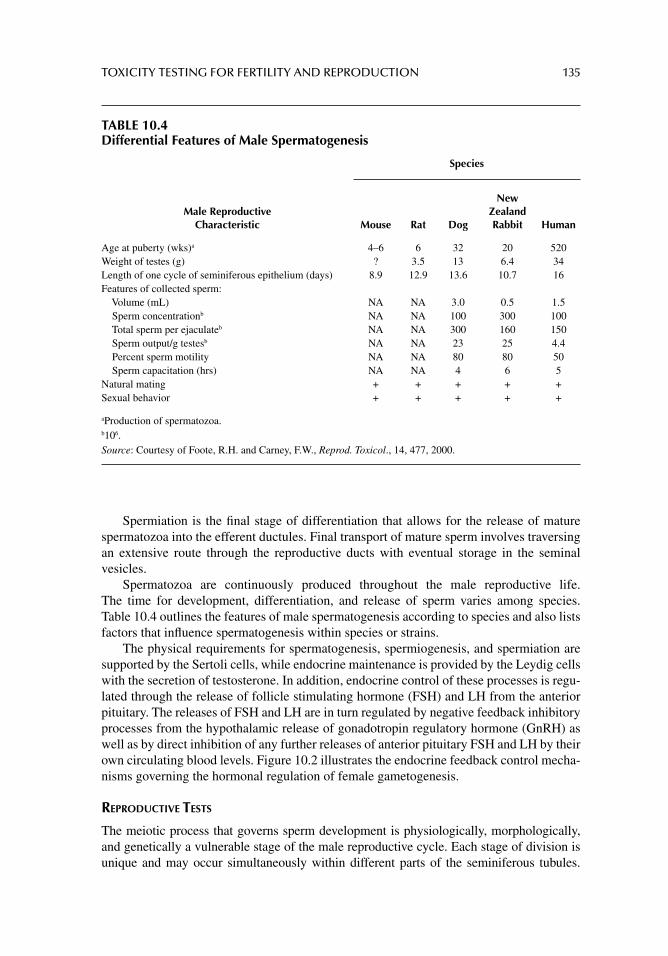
TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 135
Spermiation is the fi nal stage of differentiation that allows for the release of mature spermatozoa into the efferent ductules. Final transport of mature sperm involves traversing an extensive route through the reproductive ducts with eventual storage in the seminal vesicles.
Spermatozoa are continuously produced throughout the male reproductive life. The time for development, differentiation, and release of sperm varies among species. Table 10.4 outlines the features of male spermatogenesis according to species and also lists factors that infl uence spermatogenesis within species or strains.
The physical requirements for spermatogenesis, spermiogenesis, and spermiation are supported by the Sertoli cells, while endocrine maintenance is provided by the Leydig cells with the secretion of testosterone. In addition, endocrine control of these processes is regu-lated through the release of follicle stimulating hormone (FSH) and LH from the anterior pituitary. The releases of FSH and LH are in turn regulated by negative feedback inhibitory processes from the hypothalamic release of gonadotropin regulatory hormone (GnRH) as well as by direct inhibition of any further releases of anterior pituitary FSH and LH by their own circulating blood levels. Figure 10.2 illustrates the endocrine feedback control mecha-nisms governing the hormonal regulation of female gametogenesis.
REPRODUCTIVE TESTS
The meiotic process that governs sperm development is physiologically, morphologically, and genetically a vulnerable stage of the male reproductive cycle. Each stage of division is unique and may occur simultaneously within different parts of the seminiferous tubules.
TABLE 10.4Differential Features of Male Spermatogenesis
Species
Male Reproductive Characteristic Mouse Rat Dog
New Zealand Rabbit Human
Age at puberty (wks)a 4–6 6 32 20 520Weight of testes (g) ? 3.5 13 6.4 34Length of one cycle of seminiferous epithelium (days) 8.9 12.9 13.6 10.7 16Features of collected sperm: Volume (mL) NA NA 3.0 0.5 1.5 Sperm concentrationb NA NA 100 300 100 Total sperm per ejaculateb NA NA 300 160 150 Sperm output/g testesb NA NA 23 25 4.4 Percent sperm motility NA NA 80 80 50 Sperm capacitation (hrs) NA NA 4 6 5Natural mating + + + + +Sexual behavior + + + + +
aProduction of spermatozoa.b106.Source: Courtesy of Foote, R.H. and Carney, F.W., Reprod. Toxicol., 14, 477, 2000.
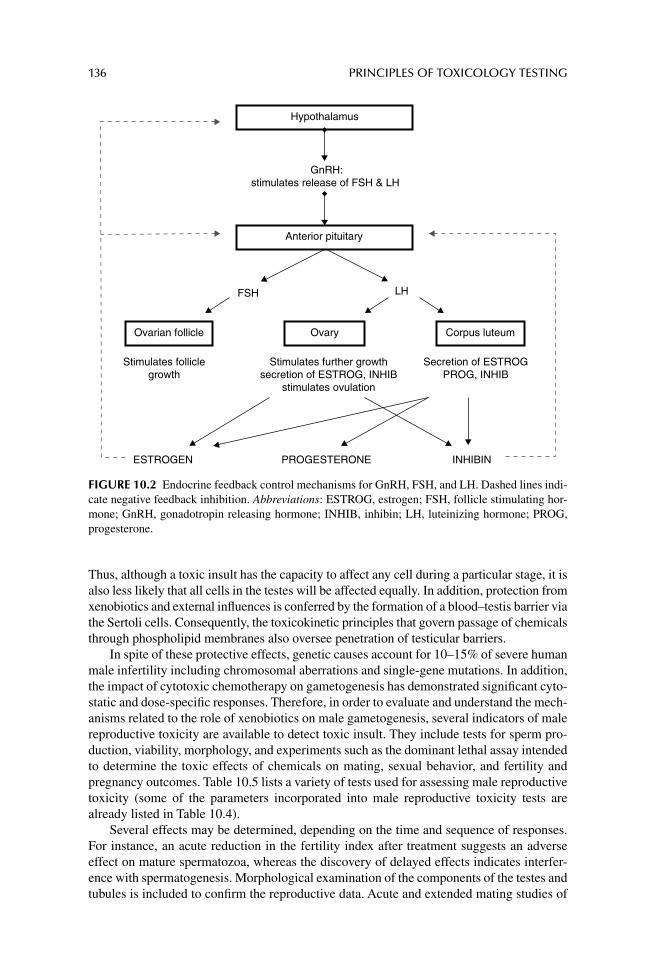
136 PRINCIPLES OF TOXICOLOGY TESTING
Thus, although a toxic insult has the capacity to affect any cell during a particular stage, it is also less likely that all cells in the testes will be affected equally. In addition, protection from xenobiotics and external infl uences is conferred by the formation of a blood–testis barrier via the Sertoli cells. Consequently, the toxicokinetic principles that govern passage of chemicals through phospholipid membranes also oversee penetration of testicular barriers.
In spite of these protective effects, genetic causes account for 10–15% of severe human male infertility including chromosomal aberrations and single-gene mutations. In addition, the impact of cytotoxic chemotherapy on gametogenesis has demonstrated signifi cant cyto-static and dose-specifi c responses. Therefore, in order to evaluate and understand the mech-anisms related to the role of xenobiotics on male gametogenesis, several indicators of male reproductive toxicity are available to detect toxic insult. They include tests for sperm pro-duction, viability, morphology, and experiments such as the dominant lethal assay intended to determine the toxic effects of chemicals on mating, sexual behavior, and fertility and pregnancy outcomes. Table 10.5 lists a variety of tests used for assessing male reproductive toxicity (some of the parameters incorporated into male reproductive toxicity tests are already listed in Table 10.4).
Several effects may be determined, depending on the time and sequence of responses. For instance, an acute reduction in the fertility index after treatment suggests an adverse effect on mature spermatozoa, whereas the discovery of delayed effects indicates interfer-ence with spermatogenesis. Morphological examination of the components of the testes and tubules is included to confi rm the reproductive data. Acute and extended mating studies of
GnRH:stimulates release of FSH & LH
FSH
Stimulates folliclegrowth
Secretion of ESTROGPROG, INHIB
Stimulates further growthsecretion of ESTROG, INHIB
stimulates ovulation
ESTROGEN PROGESTERONE INHIBIN
Hypothalamus
Anterior pituitary
LH
Ovarian follicle Ovary Corpus luteum
FIGURE 10.2 Endocrine feedback control mechanisms for GnRH, FSH, and LH. Dashed lines indi-cate negative feedback inhibition. Abbreviations: ESTROG, estrogen; FSH, follicle stimulating hor-mone; GnRH, gonadotropin releasing hormone; INHIB, inhibin; LH, luteinizing hormone; PROG, progesterone.

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 137
rodents and rabbits require one or several turnover cycles of the seminiferous epithelium, resulting in several weeks to months of treatment, in order to achieve a steady state between agent and target site.
Combinations of these assays are capable of screening both strong and weak toxicants based on dose–response parameters. Antifertility effects, infl uence on implantation, pre- and postspermatogenic effects, and fetal lethality are assessed with appropriately planned and executed timing of protocols. Consequently, based on the evaluation of the results, mecha-nistic information is achievable. Conclusions about toxic effects are derived from direct action on the meiotic process to the infl uence on the neuroendocrine system, including hypothalamic–hypophyseal feedback, effects on autonomic regulation of erection and ejac-ulation, and mating behavior.
While these protocols provide valuable information for the determination of potential reproductive toxicity, the methods for conducting the assessments have not been well developed in all laboratories and are continually evolving. The lack of standardization of methods does not allow for smooth transition of data among laboratories. To address the differences in methods, however, several recent working groups have convened to discuss optimization of methods for assessing sperm parameters in animal species. The sections suggested readings and review articles at the end of this chapter provide more information and list summaries and outlines of individual protocols.
FEMALE REPRODUCTIVE TOXICOLOGY
OOGENESIS
The ovaries are the paired female gonads that contain primordial follicles in which oogenesis occurs. Oogenesis is defi ned as the development and formation of mature secondary oocytes. The follicles arise from primordial germ cells and represent the corresponding embryonic stem cells in the female reproductive cycle. The meiotic process is more complex in female mammals, principally because of the interruptions in cycles.
Meiosis I begins during fetal life and stops at birth with the development of oogonia. A genetically determined number of oogonia develop and remain dormant until puberty
TABLE 10.5Recommended Male Reproductive Toxicity Tests
Test Parameter Features
Sperm production Measurements of ejaculatory volume, sperm concentration, total sperm count, motility, morphology, capacitation
Serial mating Males treated with single or repeated doses of agent and mated continuously with untreated females
Extended mating studies Subchronic administration of test agent over several spermatogenic cycles prior to serial mating with untreated female animals
Other specifi c parameters: Fertility index Percentage of matings resulting in successful pregnancy during the
time period Dominant lethal assay Single, repeated, acute or chronic exposure of males throughout a
complete spermatogenic cycle, then serially mated with untreated females; objective: monitoring for severe chromosomal damage or lethal germ-cell mutations
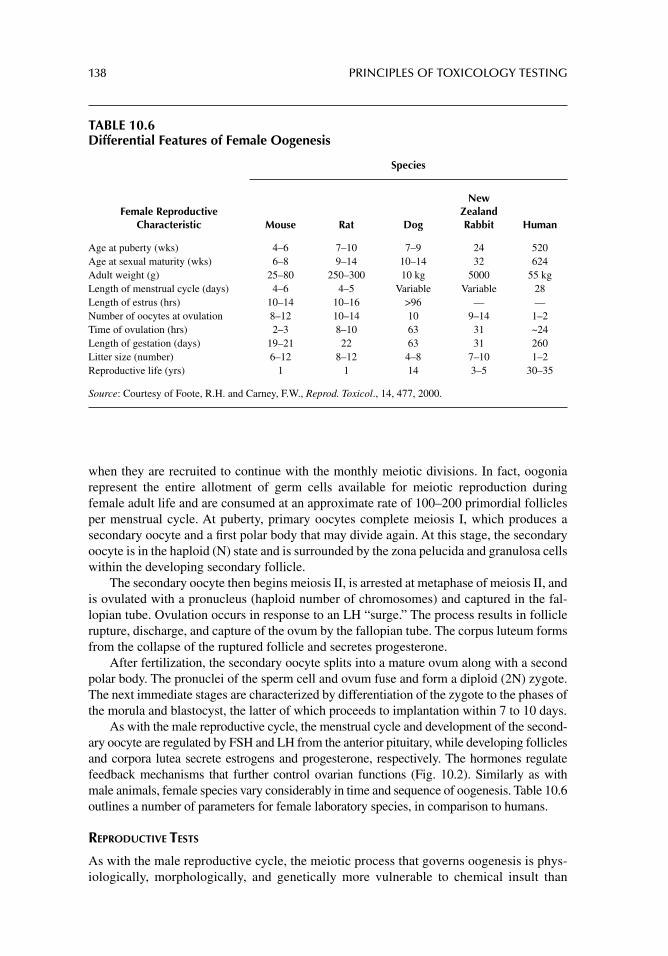
138 PRINCIPLES OF TOXICOLOGY TESTING
when they are recruited to continue with the monthly meiotic divisions. In fact, oogonia represent the entire allotment of germ cells available for meiotic reproduction during female adult life and are consumed at an approximate rate of 100–200 primordial follicles per menstrual cycle. At puberty, primary oocytes complete meiosis I, which produces a secondary oocyte and a fi rst polar body that may divide again. At this stage, the secondary oocyte is in the haploid (N) state and is surrounded by the zona pelucida and granulosa cells within the developing secondary follicle.
The secondary oocyte then begins meiosis II, is arrested at metaphase of meiosis II, and is ovulated with a pronucleus (haploid number of chromosomes) and captured in the fal-lopian tube. Ovulation occurs in response to an LH “surge.” The process results in follicle rupture, discharge, and capture of the ovum by the fallopian tube. The corpus luteum forms from the collapse of the ruptured follicle and secretes progesterone.
After fertilization, the secondary oocyte splits into a mature ovum along with a second polar body. The pronuclei of the sperm cell and ovum fuse and form a diploid (2N) zygote. The next immediate stages are characterized by differentiation of the zygote to the phases of the morula and blastocyst, the latter of which proceeds to implantation within 7 to 10 days.
As with the male reproductive cycle, the menstrual cycle and development of the second-ary oocyte are regulated by FSH and LH from the anterior pituitary, while developing follicles and corpora lutea secrete estrogens and progesterone, respectively. The hormones regulate feedback mechanisms that further control ovarian functions (Fig. 10.2). Similarly as with male animals, female species vary considerably in time and sequence of oogenesis. Table 10.6 outlines a number of parameters for female laboratory species, in comparison to humans.
REPRODUCTIVE TESTS
As with the male reproductive cycle, the meiotic process that governs oogenesis is phys-iologically, morphologically, and genetically more vulnerable to chemical insult than
TABLE 10.6Differential Features of Female Oogenesis
Species
Female Reproductive Characteristic Mouse Rat Dog
New Zealand Rabbit Human
Age at puberty (wks) 4–6 7–10 7–9 24 520Age at sexual maturity (wks) 6–8 9–14 10–14 32 624Adult weight (g) 25–80 250–300 10 kg 5000 55 kgLength of menstrual cycle (days) 4–6 4–5 Variable Variable 28Length of estrus (hrs) 10–14 10–16 >96 — —Number of oocytes at ovulation 8–12 10–14 10 9–14 1–2Time of ovulation (hrs) 2–3 8–10 63 31 ~24Length of gestation (days) 19–21 22 63 31 260Litter size (number) 6–12 8–12 4–8 7–10 1–2Reproductive life (yrs) 1 1 14 3–5 30–35
Source: Courtesy of Foote, R.H. and Carney, F.W., Reprod. Toxicol., 14, 477, 2000.
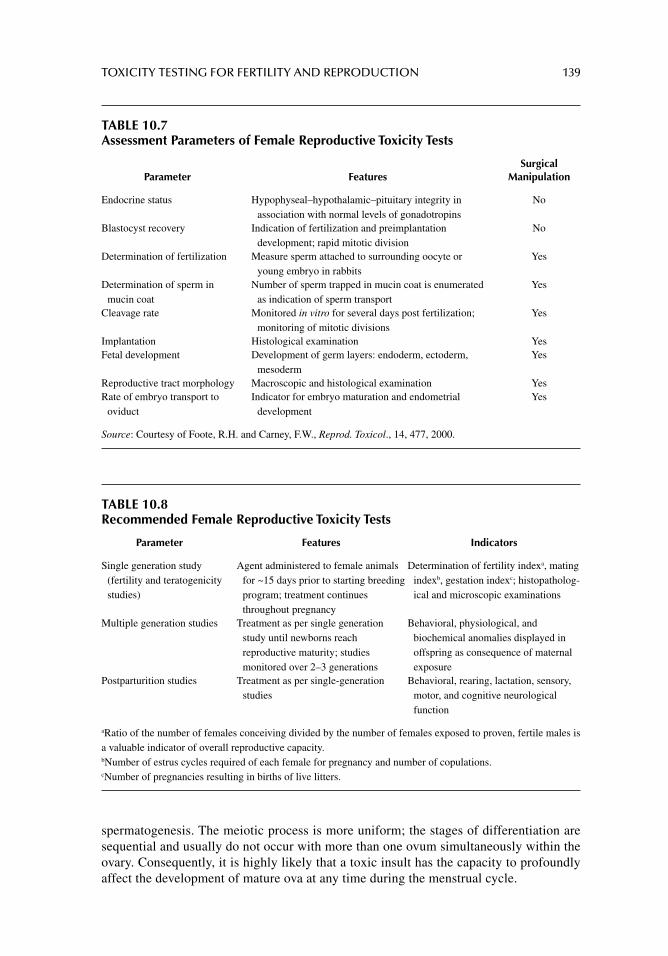
TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 139
TABLE 10.7Assessment Parameters of Female Reproductive Toxicity Tests
Parameter FeaturesSurgical
Manipulation
Endocrine status Hypophyseal–hypothalamic–pituitary integrity in association with normal levels of gonadotropins
No
Blastocyst recovery Indication of fertilization and preimplantation development; rapid mitotic division
No
Determination of fertilization Measure sperm attached to surrounding oocyte or young embryo in rabbits
Yes
Determination of sperm in mucin coat
Number of sperm trapped in mucin coat is enumerated as indication of sperm transport
Yes
Cleavage rate Monitored in vitro for several days post fertilization; monitoring of mitotic divisions
Yes
Implantation Histological examination YesFetal development Development of germ layers: endoderm, ectoderm,
mesodermYes
Reproductive tract morphology Macroscopic and histological examination YesRate of embryo transport to
oviductIndicator for embryo maturation and endometrial
developmentYes
Source: Courtesy of Foote, R.H. and Carney, F.W., Reprod. Toxicol., 14, 477, 2000.
spermatogenesis. The meiotic process is more uniform; the stages of differentiation are sequential and usually do not occur with more than one ovum simultaneously within the ovary. Consequently, it is highly likely that a toxic insult has the capacity to profoundly affect the development of mature ova at any time during the menstrual cycle.
TABLE 10.8Recommended Female Reproductive Toxicity Tests
Parameter Features Indicators
Single generation study (fertility and teratogenicity studies)
Agent administered to female animals for ~15 days prior to starting breeding program; treatment continues throughout pregnancy
Determination of fertility indexa, mating indexb, gestation indexc; histopatholog-ical and microscopic examinations
Multiple generation studies Treatment as per single generation study until newborns reach reproductive maturity; studies monitored over 2–3 generations
Behavioral, physiological, and biochemical anomalies displayed in offspring as consequence of maternal exposure
Postparturition studies Treatment as per single-generation studies
Behavioral, rearing, lactation, sensory, motor, and cognitive neurological function
aRatio of the number of females conceiving divided by the number of females exposed to proven, fertile males is a valuable indicator of overall reproductive capacity.bNumber of estrus cycles required of each female for pregnancy and number of copulations.cNumber of pregnancies resulting in births of live litters.

140 PRINCIPLES OF TOXICOLOGY TESTING
In order to assess the mechanisms related to the role of xenobiotics on female gameto-genesis, several indicators of female reproduction are outlined in Table 10.7. These features represent important parameters in the evaluation of normal reproductive status and can then be monitored after administration of a toxic substance.
Table 10.8 summarizes a variety of recommended female reproductive toxicity test combinations that are capable of screening both strong and weak toxic substances based on dose–response parameters. Antifertility effects, infl uence on female gametogenesis, implantation, effects on pre- and postorganogenesis, and fetal abnormalities are assessed with appropriately planned and executed timing of protocols. Conclusions on toxic effects are derived from analysis of the steps involved in female reproduction.
Finally, Table 10.9 outlines a description of segment testing and assessment of repro-ductive toxicity. This sequential conduct of studies enables a systematic evaluation of toxi-cology testing data while minimizing unnecessary duplication of laboratory and animal resources.
SUGGESTED READINGS
Anonymous. International Conference on Harmonization: guidelines on detection of toxicity to reproduction for medicinal products. Fed Reg 1994; 59: 48746.
Augustine-Rauch K. Alternative experimental approaches for interpreting skeletal fi ndings in safety studies. Birth Defects Res B Dev Reprod Toxicol 2007; 80: 497.
de Jong E, Doedée AM, Reis-Fernandes MA, Nau H, Piersma AH. Potency ranking of valproic acid analogues as to inhibition of cardiac differentiation of embryonic stem cells in comparison to their in vivo embryotoxicity. Reprod Toxicol 2011; 31: 375.
Eckardt K, Kaltenhäuser J, Kilb C, Seiler A, Stahlmann R. Relative potency of albendazole and its sulfoxide metabolite in two in vitro tests for developmental toxicity: the rat whole embryo culture and the mouse embryonic stem cell test. Reprod Toxicol 2012; 34: 378–84.
Flick B, Klug S. Whole embryo culture: an important tool in developmental toxicology today. Curr Pharm Des 2006; 12: 1467.
Fucic A, Stojkovic R, Miškov S, et al. Transplacental genotoxicity of antiepileptic drugs: animal model and pilot study on mother/newborn cohort. Reprod Toxicol 2010; 30: 613.
Hurtt ME, Cappon GD, Browning A. Proposal for a tiered approach to developmental toxicity testing for veterinary pharmaceutical products for food-producing animals. Food Chem Toxicol 2003; 41: 611.
Knudsen TB, Kavlock RJ, Daston GP, et al. Developmental toxicity testing for safety assessment: new approaches and technologies. Birth Defects Res B Dev Reprod Toxicol 2011; 92: 413.
Kultima K, Jergil M, Salter H, et al. Early transcriptional responses in mouse embryos as a basis for selection of molecular markers predictive of valproic acid teratogenicity. Reprod Toxicol 2010; 30: 457.
TABLE 10.9Segment Testing
Segment Protocols
I Separate treatment of males or females with test substance, but not both or together II Normal fertilization: Untreated females mated with untreated males; administration of agent
post fertilizationIII Assessment for teratogenic and embryotoxic effects during prenatal development; examination
of postnatal development including effects on parturition and fetal survival, lactation, weaning, and maturity

TOXICITY TESTING FOR FERTILITY AND REPRODUCTION 141
Organization of Teratogen Information Services. [Available from: http://www.otispregnancy.org/] [Last accessed December, 2012].
Presibella KM, Kita DH, Carneiro CB, Andrade AJ, Dalsenter PR. Reproductive evaluation of two pesticides combined (deltamethrin and endosulfan) in female rats. Reprod Toxicol 2005; 20: 95.
U.S. Environmental Protection Agency. Health effects test guidelines, OPPTS 870.3700, Prenatal Developmental Toxicity Study. EPA 1998; 712: 207.
REVIEW ARTICLES
Bignami G. Economical test methods for developmental neurobehavioral toxicity. Environ Health Perspect 1996; 104: 285.
Brachet A. Recherches sur le determinism hereditaire de l’ouef des manniferes: development in vitro de jeunes vesicules blastodermique du lapin. Arch Biol 1913; 28: 447.
Buschmann J. Critical aspects in reproductive and developmental toxicity testing of environmental chemicals. Reprod Toxicol 2006; 22: 157.
Cohen SM, Robinson D, MacDonald J. Alternative models for carcinogenicity testing. Toxicol Sci 2001; 64: 14.
Detailed Review Document on Classifi cation Systems for Reproductive Toxicity in OECD Member Countries, OECD Series on Testing and Assessment, Number 15, 1999.
Foote RH, Carney EW. The rabbit as a model for reproductive and developmental toxicity studies. Reprod Toxicol 2000; 14: 477.
Hurtt ME, Cappon GD, Browning A. Proposal for a tiered approach to developmental toxicity testing for veterinary pharmaceutical products for food-producing animals. Food Chem Toxicol 2003; 41: 611.
Jelinek R. The contribution of new fi ndings and ideas to the old principles of teratology. Reprod Toxicol 2005; 20: 295.
Organisation for Economic Cooperation and Development (OECD) Guidelines for the Testing of Chemicals. [Available from: http://www.oecd.org/document/40/0,3746,en_2649_34377_37051368_1_1_1_1,00.html] [Last accessed July 2012].
Riecke K, Stahlmann R. Test systems to identify reproductive toxicants. Andrologia 2000; 32: 209.Seed J, Chapin RE, Clegg ED, et al. Methods for assessing sperm motility, morphology, and counts
in the rat, rabbit, and dog: a consensus report. Reprod Toxicol 1996; 10: 237.Storgaard L, Bonde JP, Olsen J. Male reproductive disorders in humans and prenatal indicators of
estrogen exposure: a review of published epidemiological studies. Reprod Toxicol 2006; 21: 4.
Tam PP. Post-implantation mouse development: whole embryo culture and micro-manipulation. Int J Dev Biol 1998; 42: 895.
Tyl RW. In honor of the Teratology Society’s 50th anniversary: the role of Teratology Society mem-bers in the development and evolution of in vivo developmental toxicity test guidelines. Birth Defects Res C Embryo Today 2010; 90: 99.
Walker R. The signifi cance of excursions above the ADI: duration in relation to pivotal studies. Regul Toxicol Pharmacol 1999; 30: S114.
Webster WS, Brown-Woodman PD, Ritchie HE. A review of the contribution of whole embryo cul-ture to the determination of hazard and risk in teratogenicity testing. Int J Dev Biol 1997; 41: 329.

142
11 Carcinogenicity and mutagenicity testing in vivo
INTRODUCTION AND DEFINITIONS
Cell proliferation occurs in most tissues throughout a life span and is infl uenced by a variety of circumstances. During this normal physiological state, the delicate balance between cell proliferation and apoptosis (programmed cell death) is perpetuated to ensure the integrity and proper function of organs and tissues. Mutations in DNA that lead to cancer develop-ment interfere with this orderly process by disrupting its regulation. Consequently, the initiation of a cancer is due to an abnormal and uncontrolled progression of cell prolifera-tion characterized by unregulated cell division and metastasis (spreading) of foci of cells to distant tissues.
Carcinogenesis represents the unwarranted appearance or increased incidence of abnormal cell proliferation occurring in an age-matched low-risk group whose propensity for development of cancer is statistically lower. A carcinogen is any chemical or viral agent that increases the frequency or distribution of new tumors, results in their appearance within a low-risk or otherwise early age group, or results in the introduction of new patho-logical growths otherwise absent in experimental controls.
Most chemical carcinogens require metabolic activation before demonstrating carcino-genic potential. As with most toxic phenomena, a minimum dosage is necessary to elicit a carcinogenic event. Epigenetic or nongenotoxic carcinogens enhance the growth of tumors by mechanisms other than through alteration of DNA. These chemicals infl uence absorp-tion, biotransformation, or reduce elimination of the initiating agent. Cocarcinogens redirect hormonal action on cell proliferation and inhibit intercellular communication, thus allowing for greater proliferative capacity. Alternatively, they induce immunosuppression of protec-tive immunological pathways.
The carcinogenic process is on occasion a response to a mutation occurring within the genetic materials of normal cells, resulting in uncontrolled cell division and transformation to the immortal phenotype. The unrestrained and often rapid proliferation of cells is further characterized as benign or malignant tumors. Benign tumors do not metastasize, are usually confi ned to local target organ areas, are clinically susceptible to therapeutic intervention, and carry more favorable prognoses. Alternatively, malignant tumors metastasize to distant organ locations, are not necessarily amenable to therapeutic intervention, and possess less favorable prognoses.
Mutagenesis refers to the ability of a virus or chemical agent to induce changes in the genetic sequences of mammalian or bacterial cells, thus altering the phenotypic expression of cell characteristics. Genotoxicity refers to the ability of an agent to induce heritable

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 143
changes in genes that exercise homeostatic control in somatic cells while increasing the risk of infl uencing benign or malignant transformation. Genotoxic substances induce genotox-icity by either binding directly to DNA or by indirectly altering the DNA sequence, causing irreversible damage. It is also important to note, however, that genotoxic substances are not necessarily carcinogenic.
In addition, interaction of a chemical, physical, or viral agent with nucleic acids results in the disruption of the transfer of genetic information and the development of genotypic and phenotypic consequences. Lastly, mitogenesis is the induction of cell division (mitosis) within eukaryotic or prokaryotic cells, stimulating transit through the cell cycle. Prolonged and continuous exposure to growth factors is required to commit cells to the cell cycle.
MULTISTAGE CARCINOGENESIS
The process of carcinogenesis is divided into three experimentally defi ned stages: tumor initiation, tumor promotion, and tumor progression. This multistage development requires the malignant conversion of benign hyperplastic cells to a malignant state, involving inva-sion and metastasis as manifestations of further genetic and epigenetic changes.
TUMOR INITIATION
The early concept of tumor initiation indicated that the initial changes in chemical carcino-genesis involved irreversible genetic alterations. Recent data from molecular studies of pre-neoplastic human lung and colon tissues; however, implicate epigenetic changes as an early event in carcinogenesis. DNA methylation of promoter regions of genes can transcription-ally silence tumor suppressor genes. Thus, carcinogen–DNA adduct formation is central to the theory of chemical carcinogenesis and may be a necessary but not suffi cient prerequisite for tumor initiation.
DNA adduct formation that causes either the activation of a proto-oncogene or the inac-tivation of a tumor suppressor gene is categorized as a tumor-initiating event. One important characteristic of this stage is its irreversibility: the genotype or phenotype of the initiated cell is conferred during the process. Chemicals capable of initiating cells are referred to as initiating agents. Furthermore, initiation without the subsequent promotion and progression rarely yields malignant transformation.
TUMOR PROMOTION
In multistage carcinogenesis, tumor promotion comprises the selective clonal expansion of initiated cells through a mechanism of gene activation. Because the accumulation rate of mutations is proportional to the rate of cell division, clonal expansion of initiated cells pro-duces a larger population of cells that are at risk of further genetic changes and malignant conversion. Tumor promoters are generally nonmutagenic, are not solely carcinogenic, and often are able to mediate their biologic effects without metabolic activation. In addition, they do not directly interact with DNA.
Interestingly, tumor promotion is reversible, although the continued presence of the promoting agent maintains the state of the promoted cell population (preneoplastic lesion). Thus the stage appears to have a long duration, especially in humans, and is a preferred target for experimental manipulation. Examples of typical tumor promoters include tetra-decanoyl phorbol acetate, phenobarbital, and 2,3,7,8-tetrachlorodibenzodioxin.
During tumor promotion, malignant conversion may occur in which a preneoplastic cell is transformed into the malignant phenotype. The process requires further genetic changes.

144 PRINCIPLES OF TOXICOLOGY TESTING
Frequent repeated administration of the tumor promoter is more important than the total dose. In addition, if the tumor promoter is discontinued before malignant conversion has occurred, premalignant or benign lesions may regress. Tumor promotion contributes to the process of carcinogenesis by the expansion of a population of initiated cells that are then at risk for malig-nant conversion. Conversion of a fraction of these cells to malignancy is accelerated in propor-tion to the rate of cell division and the quantity of dividing cells in the benign tumor or preneoplastic lesion. In part, these genetic changes result from the infi delity of DNA synthesis.
TUMOR PROGRESSION
Tumor progression comprises the expression of the malignant phenotype and the tendency of malignant cells to acquire more aggressive characteristics over time. Also, metastasis may involve the ability of tumor cells to secrete proteases that allow invasion beyond the immedi-ate primary tumor location. A prominent characteristic of the malignant phenotype is the propensity for genomic instability and uncontrolled growth. Further genetic and epigenetic changes occur, including the activation of proto-oncogenes and the functional loss of tumor suppressor genes.
Loss of function of tumor suppressor genes usually occurs in a bimodal fashion, and most frequently involves point mutations in one allele and loss of the second allele by a deletion, recombinational event, or chromosomal nondisjunction. These phenomena confer growth advantage to the cells along with the capacity for regional invasion, and ultimately distant metastatic spread. Despite evidence for an apparent scheduling of certain muta-tional events, the accumulation of these mutations appears to be the determining factor and not the order or the stage of tumorigenesis in which they occur.
Figure 11.1 illustrates the events that occur during the three stages of chemical carci-nogenesis and the requirements necessary for the completion of the multistage process.
Normal cell Initiated cell
Preneoplastic cellsNeoplastic cells
Results from DNAmutations
Initiated cells remain“dormant’’, and rarelybecome neoplastic cellswithout undergoingpromotion and progression
Irreversible
Cell signaling and geneexpression are altered, leading to increased cell mitogensis
Induces formation ofpreneoplastic cells
Long duration and reversible,especially at an earlier stage
Results from additional genomicstructural alterations due tokaryotypic instability
Leads to the formation of benignor malignant neoplasms
Cells may acquire malignantphenotypes such as invasiveness, angiogenesis, and/or metastasis
Irreversible
Initiating agents Promoting agents Progressor agents
Initiation Promotion Progression
Mutations occur inoncogenes, proto-oncogenes,or tumor suppressor genes
Results from clonal expansion of initiated cells
FIGURE 11.1 The three stages of chemical carcinogenesis. Source: From Chemical Carcinogenesis and Mutagenesis, in: Clinical Toxicology: Principles and Mechanisms, 2nd edition, Barile, F.A., Informa HealthCare Publishers, 2010, Ch. 31.

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 145
CARCINOGENIC AND GENOTOXIC AGENTS
Both collective and individual exposures often defi ne environmental toxicology. Epidemio-logical research supported by experimental investigations led to the determination of profi les of various risk factors. These profi les are perpetually incomplete because humans inces-santly modify the environment, thus increasing the risks of interactions with agents against which there may be no protective mechanisms. The multiplicity of chemical and physical carcinogenic agents does not render them easy to study since the development of carcinoma is, in all probability, not due to contact with a single carcinogenic factor but rather to the sum of many possible causative agents.
Of the various environmental hazardous compounds, cigarette smoke enjoys the high-est causal relationship with cancer risk in humans. Tobacco smoking plays a major role in the etiology of lung, oral cavity, and esophageal cancers and a variety of other chronic degenerative diseases. Although cigarette smoke is a mixture of about 4000 chemicals including more than 60 known human carcinogens, 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (nicotine-derived nitrosamino ketone, NNK) is the most carcinogenic tobacco-specifi c nitrosamine. NNK induces lung tumors in mice, rats, and hamsters. The International Agency for Research on Cancer has designated NNK and N-nitrosonornicotine as known human carcinogens. NNK is metabolically activated by cytochrome P-450 enzymes in the lungs and generates O6-methylguanine in DNA. The reaction generates G:C to A:T muta-tions with the subsequent activation of the K-ras proto-oncogene and development of tumor initiation.
Radiation also plays a signifi cant role as one of the most causative physical factors inducing human cancers. Radiation promotes double-strand breaks in DNA that lead to chromosome aberrations and cell death and also generates a variety of oxidative DNA dam-age. Because of genotoxicity, radiation at high doses evidently results in the appearances of various tumors in humans. Even at low doses, residential exposure to radioactive radon and its decay products, for instance, may account for about 10–20% of all lung cancer deaths worldwide.
CARCINOGENICITY TESTING IN VIVO
PRINCIPLES OF IN VIVO CARCINOGENICITY TESTING
The toxicology community generally agrees that carcinogenicity testing and its application to human carcinogenic risk assessment need improvement. In addition, more information is necessary about the incorporation of mechanisms and modes of action into the risk assessment process. Advances in molecular biology have identifi ed a growing number of proto-oncogenes and tumor suppressor genes that are highly conserved across species and are associated with an extensive variety of mammalian cancers.
In vivo transgenic rodent models that incorporate these mechanisms are used to iden-tify pathways involved in tumor formation. Transgenic methods are considered exten-sions of genetic manipulation by selective breeding—a technique that has long been employed in science and agriculture. With carcinogenicity testing, the use of two rodent species is especially important for identifying trans-species carcinogens. The capacity of a substance to induce neoplastic events among species suggests that the carcinogenic mechanisms are conserved and therefore may have signifi cance for humans. Based on available information, there is suffi cient experience with some in vivo transgenic rodent carcinogenicity models to support their application as complementary second species studies in conjunction with a single 2-year rodent carcinogenicity study. Following are

146 PRINCIPLES OF TOXICOLOGY TESTING
the examples of assessment factors in the consideration of the adequacy of the test model and test compound:
1. Adequacy of doses included in the study and suitability of the route of administration.2. Bioavailability of the test compound in rodent models.3. Toxicokinetic profi le of the compound in rodent models and humans.4. Biotransformation profi les of the compound in rodent models and humans.5. Appropriate application of International Conference on Harmonization guidelines.
Risk-benefi t considerations are as follows:
1. Comparison of systemic exposure at an estimated maximum daily recommended dose to systemic exposure in rodent test models.
2. If systemic exposure information is lacking, the estimated exposure ratio should be based on surface area (mg/m2) estimation of dose in the test species and the maxi-mum daily recommended clinical dose.
3. Severity of malignancy.4. Size and composition of patient population and frequency and extent of exposure.
Even the smallest in vivo carcinogenicity studies, when performed properly, are expen-sive and time consuming. Consequently, only a relatively small fraction of new and existing chemicals has been tested accordingly. Therefore, the development and validation of alterna-tive approaches to offset the resources necessary for in vivo identifi cation and classifi cation of human carcinogens and mutagens are important parts of carcinogenicity testing. Current methods used for this evaluation, however, such as the Ames test and long-term in vivo stud-ies, suffer from several shortcomings:
1. The need to extrapolate the results of the Ames test from prokaryotes to humans.2. The need to extrapolate in vivo testing results from rodents to humans.3. The prolonged time required for the conduction of in vivo tests.
The current methods also suffer from limited validation comparing the agreement of various methods with each other and applying laboratory results to human populations.
In 1996, the National Institute of Environmental Health Sciences (NIEHS, Research Triangle Park, North Carolina, USA) initiated the predictive toxicology evaluation project to develop and validate improved methods to predict the carcinogenicities of compounds. The project conducts collaborative experiments that subject the perfor-mance of predictive toxicology methods to objective evaluation and incorporate ongoing testing conducted by the National Toxicology Program (NTP) to estimate the true errors of models applied to make prospective predictions on previously untested chemical substances.
The study fi rst describes the identifi cation of a group of standardized NTP chemical bioassays, then announces and advertises the evaluation experiment. The information about the bioassay is distributed to encourage researchers to publish their predictions in peer-reviewed journals, using unsolicited approaches and methods, after which the collection of data is published to compare and contrast predictive toxicology approaches and models to applied situations. Several chemicals’ carcinogenesis bioassays have been presented since

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 147
the fi rst supplement publication of the predictive toxicology evaluation, along with a table of prediction sets, challenges associated with estimating the true error of models, and a summary.
IN VIVO GENOTOXICITY AND CYTOGENETIC TESTS
Several validated in vivo models are currently accepted for assessment of genotoxicity. These include the bone marrow or peripheral blood cytogenetic assay. When a compound demonstrates negative results in vitro, performance of a single in vivo cytogenetics assay is usually suffi cient. A compound that induces a biologically relevant positive result in one or more in vitro tests requires further testing with an in vivo model in addition to the cytogenetic assay. The test usually incorporates a tissue other than the bone marrow or peripheral blood that can provide further clarifi cation of effects. Compounds that screen positively in any of the in vitro tests, particularly the bone marrow cytogenetic assay, require confi rmation with an in vivo method accordingly:
1. Determining any signifi cant change in the proportion of immature erythrocytes among total erythrocytes in the bone marrow at the doses and sampling times used in the micronucleus test or measuring a signifi cant reduction in mitotic index for the chromosomal aberration assay.
2. Providing evidence of bioavailability of substance-related material by measuring blood or plasma levels.
3. Directly measuring substance-related material in the bone marrow.4. Performing autoradiographic assessment of tissue exposure.
With international agreement and scientifi c collaboration, the Organization for Economic Cooperation and Development (OECD) has proposed several in vivo carcinogenicity and mutagenicity protocols for testing of chemicals. Table 11.1 summarizes the OECD Guide-lines for the Testing of Chemicals, Section 4: Health Effects, for in vivo animal testing carci-nogenicity and mutagenicity. Some representative individual methodologies are described in detail in the individual sections below.
To date, no validated, widely used in vivo system measures gene mutation. In vivo gene mutation assays using endogenous genes or transgenes in several tissues of rats and mice are at various stages of development; until such tests for mutation are accepted, results from other in vivo tests for genotoxicity in tissues other than the bone marrow can provide valuable additional data. Disagreements between in vivo and in vitro test results performed in parallel must be analyzed separately.
OECD In Vivo Carcinogenicity and Mutagenicity Test Guidelines for the Testing of Chemicals
The Draize test for bacterial mutagenesis presented a breakthrough in the testing of chemi-cals for mutagenicity and genotoxicity. As an in vitro test, this methodology is discussed in Section III, as OECD TG 471, Bacterial Reverse Mutation Test (chap. 17). One of the fi rst in vivo toxicology tests, however, for testing carcinogenicity and mutagenicity of chemicals is OECD TG 486, Unscheduled DNA Synthesis (UDS) Test with Mammalian Liver Cells In Vivo (Table 11.1).
The purpose of the UDS test with mammalian liver cells in vivo is to identify sub-stances that induce DNA repair after excision and removal of a stretch of DNA containing a region of damage. The damaged region is suspected to be induced by chemical substances

148 PRINCIPLES OF TOXICOLOGY TESTING
or physical agents in the liver. The test is based on the convenient incorporation of tritium-labeled thymidine (3H-TdR) for 3–8 hours, into the DNA of liver cells which have a low frequency of cells in the S-phase1 of the cell cycle. Uptake of the radioactive beta-emitter is determined by autoradiography or scintillation counting. Rats are the most common spe-cies; an adequate “N” number of animals should be at least three animals per group, at a minimum of two dose levels. As with previous validated in vivo tests (chaps. 6 and 7) a limit test may be performed if no effects are expected at a dose of 2000 mg/kg body weight. Test substances are administered as single treatments by gavage using a stomach tube or a suitable intubation cannula. Livers are extirpated from animals 12–16 hours after adminis-tration of the test substance, and liver cells are prepared for autoradiography. Normally 100 cells are scored from each animal from at least two slides. Simultaneously, cells are homog-enized and washed and radioactive incorporation is monitored by scintillation counting.
A positive result from the UDS test with mammalian liver cells in vivo indicates that a substance induces DNA damage which has the potential for repair by unscheduled DNA synthesis in vitro. A negative result occurs when a test substance which, compared with controls, does not stimulate unscheduled DNA synthesis, is thus classifi ed as not capable of inducing DNA damage.
More recently, animal carcinogenicity studies have advanced not only the methodol-ogy of screening for chemical carcinogens and mutagens, but have also allowed for the
1 DNA synthesis phase.
TABLE 11.1OECD Guidelines: In Vivo Guidelines for Animal Testing of Chemicals for Carcino-genicity and Mutagenicity (According to the Year Submitted)
Test Guideline Number
Date Submitted (Year) Title Description
471 1997 Bacterial Reverse Mutation Test
Uses amino acid requiring at least fi ve strains of Salmonella typhimurium and Escherichia coli to detect point mutations by base substitutions or frameshifts
486 1997 Unscheduled DNA Synthesis Test with Mammalian Liver Cells in vivo
Identifi cation of substances that induce DNA repair after excision and removal of DNA regions of damage induced by chemical substances
451 2009 Carcinogenicity Studies Intended primarily for use with rats and mice, both sexes, and for oral administration (see text)
453 2009 Combined Chronic Toxicity/Carcinogenicity Studies
Identify the majority of chronic and carcinogenic effects and determine dose–response relationships following prolonged and repeated exposure
488 2011 Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays
Describes an in vivo assay that detects chemicals that may induce gene mutations
Abbreviation: OECD, Organization for Economic Cooperation and Development.

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 149
understanding of the mechanisms involved with the pathology and cell biology of chem-ically mediated cancer development. For instance, OECD TG 451, Carcinogenicity Stud-ies, is a long-term carcinogenicity study (24 months)2 whose objective is to observe test animals for a major portion of their life span for the development of neoplastic lesions during or after exposure to various doses of a test substance administered via the oral, dermal, or inhalation route. The TG is intended for use with both sexes of rats and/or mice, and for oral administration. Each dose group and concurrent control group must contain at least 50 animals of each sex, at a minimum of three dose levels with a control group. Animals are dosed with the test substance daily and the mode of exposure is adjusted according to the toxicokinetic profi le of the test substance. Termination of the study should be considered when the number of survivors in the lower dose groups or the control group falls below 25%. Results and indicators are interpreted accordingly, throughout the length of the project, and at termination, and include measurements of basic parameters (weight, food and water consumption, behavior), and at least a daily, detailed histopathologic and hematologic analysis, examination of organ functions, blood and urine chemistries, and gross necropsy.
Similarly, OECD TG 453, the Combined Chronic Toxicity/Carcinogenicity Studies, was also submitted in 2009 as a modifi cation of TG 451. This group of protocols identi-fi es the majority of chronic and carcinogenic effects and determines dose-response rela-tionships following prolonged and repeated exposure of a suspected carcinogen/mutagen. The objective of this methodology is to identify carcinogenic effects as well as the majority of chronic effects of the test agent. In addition, a properly conducted study can be used to determine dose-response relationships following prolonged and repeated exposure.
The design is similar to TG 451; that is, the rat is the species of choice, containing 50 animals of each sex per three dosing groups with a concurrent control group (for the carcinogenicity phase). The chronic toxicity phase of the study contains at least 10 ani-mals of each sex. The agent is administered via three main routes, namely oral, dermal, and inhalation. The period of dosing and duration of the study are normally 12 months and 24 months for the chronic and carcinogenicity phases, respectively. Results are ana-lyzed and interpreted as noted earlier. Adequate studies allow for conclusions concerning the neoplastic/carcinogenic potential of the compound, as well as the general toxicity. Propensity of the compound for mutagenicity and genotoxicity may also be evaluated if the proper genetic analyses are performed throughout the length of the project.
Lastly, OECD TG 488, Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays, was introduced in 2011. The methodologies describe an in vivo assay that detects chemicals with the potential for inducing gene mutations. The study is designed to detect and monitor reporter genes, in transgenic rats or mice, that contain multiple copies of chro-mosomally integrated plasmid or phage shuttle vectors. The reporter genes allow for the identifi cation of various types of mutations induced by unknown test substances.
Specifi cally, positive and negative control groups, and at least three treatment groups of transgenic animals, are exposed to the test substance for 28 consecutive days. At the end of the exposure period and prior to sacrifi ce, a 3-day period is incorporated into the study dur-ing which the agent is not administered. Presumably it is during this period that unrepaired DNA lesions are fi xed into stable mutations. Genomic DNA is then isolated from the tis-sues of interest and purifi ed. Mutations that presumably have arisen during treatment are
2 The normal duration of 24 months for rodents may be adjusted to 18 months depending on the particular strain of mice.

150 PRINCIPLES OF TOXICOLOGY TESTING
scored by recovering the transgene and analyzing the phenotype of the reporter gene in a bacterial host defi cient for the reporter gene. Mutant frequency, the reported parameter in these assays, is calculated by dividing the number of plaques per plasmids containing mutations in the transgene by the total number of plaques/plasmids recovered from the same DNA sample.
CONCLUSIONS
The assessment of the genotoxic and carcinogenic potential of a compound should exam-ine all of the relevant fi ndings and acknowledge the intrinsic values and limitations of both in vitro and in vivo tests.
In vivo tests have an important role in genotoxicity and carcinogenicity testing strate-gies. The signifi cance of in vivo results in genotoxicity testing strategies is proportional to the demonstration of adequate exposure of the target tissue to the test compound. This is especially true for negative in vivo test results and when in vitro tests show convincing evidence of genotoxicity. In addition, a dose-limiting toxic response can occur in a tissue other than the genotoxic target organ. In such cases, toxicokinetic data are used to provide evidence of bioavailability. If adequate exposure is not achieved, for example with com-pounds demonstrating poor target tissue availability, extensive protein binding and other conventional in vivo genotoxicity tests may be of limited value. Details of available in vitro carcinogenicity and mutagenicity assays are discussed in chapter 15.
BIOASSAY PROTOCOLS FOR IN VIVO CARCINOGENICITY TESTING
STUDY DURATION
Based on the mechanisms of carcinogenesis described above and the unpredictable nature of dose–response relationships in the induction of tumors, in vivo carcinogenicity studies are necessarily performed for 18–24 months. European Union guidelines specify a minimum of 18 months for mice and 24 months for rats; U.S. and OECD regulations require studies for at least 18–24 months in mice and 24–30 months in rats or for the life span of a species in the event of a high survival rate.
These chronic assays ensure that the time interval of exposure to a potential carcinogen is suffi cient to induce a carcinogenic response within the major part of a rodent’s life span. The studies generally combine qualitative data with quantitative results for human risk assessments, the purpose of which is to demonstrate that a chemical lacks the ability to induce or promote cancer formation.
INDICATORS FOR DETECTION OF CARCINOGENS
In contrast to acute or chronic toxicity tests, animal carcinogenicity assays rely on what appears to be a single end point: the development of neoplasia. Consequently, more attention is focused on the morphological examinations of organs and tissues in an effort to evaluate the carcinogenic response. The criteria include (1) determining the onset of tumors, (2) evaluating for appearance and number of different types of tumors, and (3) determining the number of tumor-bearing animals.
Various physiological and toxicological indicators are also monitored throughout the length of a study when economically feasible, especially monitoring of growth and devel-opment, food and water consumption, mortality rate, morbidity, and intermittent hemato-logical and biochemical evaluation of blood and urine samples. Representative target and

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 151
nontarget organs are extirpated from animals periodically and at study termination and subjected to morphological and histological studies. Other specimens are also processed for clinical evaluation.
SELECTION OF ANIMAL SPECIES AND NUMBERS
Similar factors that affect the selection of animals in chronic toxicity studies (chap. 7) also infl uence the selection of animal models for carcinogenic projects. In general, regulatory guidelines require 50 animals of each sex per treatment group, preferably from both species of rodents. The mouse and the rat are the species of choice, principally because of the cumu-lative favorable factors that they afford to chronic studies: availability, relatively fewer resources needed during the length of the study, smaller size of animals and ease of handling, routine housing requirements, shorter life spans that more closely mimic chronic exposure, and an abundance of literature available on their carcinogens.
EXPOSURE LEVELS
The maximum tolerated dose (MTD) of chemical exposure without demonstration of adverse noncarcinogenic effects is selected as the upper limit dose. Two proportionately linear or logarithmic lower doses plus controls complete the treatment groups. In a 12-week preliminary investigation, the MTD should not cause losses of body weight greater than 10% in comparison with matched control groups. In addition, the MTD should not cause signifi cant mortality, clinical signs of toxicity, or pathologic lesions other than those related to carcinogenicity among the treatment groups. Animals in the control groups are also monitored for normal frequency of tumor incidence, especially toward the latter stages of a chronic study.
Although the MTD is associated with several controversial features that render the results diffi cult to interpret, it is still a feasible general guideline for establishing dosage levels. One of the major problems associated with the assignment of the MTD as the upper limit dose is that by attempting to correlate the interaction between a toxicological response and a carcinogenic observation in relation to assigned dose, the individual mechanisms may not have enough commonality to expect a carcinogenic phenomenon even when the dose is toxicologically tolerated.
Alternatively, in the absence of a noticeable toxic effect such as with a weak toxicant (e.g., food additive), the MTD selected may be exceedingly high so that the toxicological mechanism associated with the chemical at the high dose is responsible for the induction of a carcinogenic event. The situation is analogous to the induction of a carcinogenic effect by a mitogenic chemical.
Another important criterion in the selection of doses is to estimate the lowest dose at which carcinogenicity is not observed: the no-observable-adverse-effect level. The objec-tive of targeted exposure levels, therefore, is to estimate the incidence of tumor induction within a range of doses for a test chemical without producing signifi cant noncarcinogenic (toxic) events.
The chemical is then administered through an appropriate route, usually either in drink-ing water or formulated as a component of diet over the duration of the study. Administration via inhalation, gastric gavage, dermal, or parenteral routes is reserved for specialty studies.
SOURCES OF VARIABILITY
Because of the extended duration and complexity of carcinogenicity studies, several factors may infl uence tumor incidence in experiments and contribute to inconsistent

152 PRINCIPLES OF TOXICOLOGY TESTING
or unreliable interpretation of the data factors that may not be of concern in chronic toxicity studies. The condition of the animal-care quarters, genetic differences among strains, amounts of food consumption and weight gain, survival ages of the animals, identifi cation of gross lesions, pathology sampling procedures, preparation of the spec-imens for histological and morphological analysis are all factors that contribute to the variability of results during a project. Evaluation of data depends on objective and sub-jective observations and analyses and statistical applications throughout the duration of a study.
Another complicating factor necessarily involves the signifi cant background incidence of spontaneous tumor formations in rodents. Rodents demonstrate a marked predisposition toward the production of specifi c tumor types at particular sites, especially dermal, pulmo-nary, and hepatic neoplasms. This suggests that a rodent possesses an initiated genotype that biases the animal strain toward the formation of preneoplastic lesions.
Interestingly, new questions have surfaced about a particular feature of the design, conduct, and interpretation of the data from original carcinogenicity studies performed with animals fed ad libitum diets (food is available 24 hr/day without restriction through-out the course of a study). In the past 20 years, evidence indicates that animals on restricted calorie diets are leaner, have higher basal metabolic rates, increased survival times, decreased tumor incidence, and fewer pathological complications with age. The infl uence of calorie restriction on subchronic and chronic toxicology projects is dis-cussed in detail in chapter 7. These factors have similar consequences in the conduct of carcinogenicity programs.
CARCINOGENICITY BIOASSAYS
Chapter 17 outlines a series of in vitro tests as screening methodologies to aid in the pre-liminary planning for chronic in vivo carcinogenicity studies. In general, three systems are currently in practice that allow for progression to long-term animal projects. These include (1) short-term test batteries, (2) tier testing, and (3) decision point analysis.
Short-Term Test Batteries
This popular approach includes the incorporation of a battery of short-term tests with differ-ent carcinogenic indicators. The use of assays with various mechanistic targets is the basis for the ability of the short-term tests to detect a greater range of compounds with carcino-genic potentials. Although the principle of the system appears sound, concern about the technical aspects of the various tests and applications of the results to human risk assessment requires cautious interpretation of data.
Tier Testing
The principle of tier testing relies on a systematic sequence of ranked levels used in a testing strategy: a positive screen at the fi rst level qualifi es for confi rmation at subse-quent levels. The Ames test has frequently been employed as a screening test system in the fi rst tier, followed by performance of genotoxicity tests in the second and third tiers.
Decision Point Analysis
Table 11.2 offers a scheme for the decision point analysis for carcinogen testing. The approach is based on systematically delineating the mechanism of toxicity of a chemical.

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 153
The sequential levels commence with structure–activity analysis of the chemical as a preliminary screen, followed by a battery of in vitro short-term tests. These tests are then evaluated at decision point 1. If results are negative or inconclusive, tests for carcinogenic promoters are continued, after which decision point 2 is determined. Depending on the conclusions obtained, the process continues through decision points 3 and 4. If results are defi nitive about the carcinogenic potential of a compound at any level, no further testing is needed. Interpretation of the results from combinations of the bioassays may involve test-ing of the chemical in chronic studies. Nevertheless, conclusions concerning risk assess-ment about carcinogenic potential are accomplished with a minimal number of animals and reduced number of protocols.
Rodent Liver Altered Foci Induction
The altered foci induction test system measures the regenerative capacity of rat liver following partial hepatectomy in the presence of initiator or promoter chemicals. The indicator relies on the formation of foci from rapidly replicating, phenotypically altered cells. The foci are suggestive of preneoplastic lesions that produce hyperplastic nodules or transform to malignant growths. In addition, the higher levels of phase I enzyme activity in the altered hepatocyte foci enable the biotransformation of procar-cinogens. Histochemical, immunochemical, and histological assays are employed for the detection of chemical and enzyme biomarkers and pathology, respectively, that are associated with tumor induction. The technique is also modifi ed to detect tumor pro-moters. Thus the protocol takes advantage of a marked reduction in the time and an increase in the sensitivity required for the demonstration of carcinogenic initiation and promotion.
TABLE 11.2Decision Point Analysis for Detection of Chemicals with Carcinogenic Potentials
Level Procedure Description
A Structure–activity relationship Analysis of structural features of a chemical and molecular similarities that suggest similar mechanisms of toxicity of chemicals in structurally related classes
B Short-term in vitro assays Bacterial mutagenesis; mammalian mutagenesis; cell transformation; mammalian DNA repair assays
Decision point 1: Evaluation of results from tests at levels A and BC Test for promoters In vitro assays; in vivo assays
Decision point 2: Evaluation of results from tests at levels A, B, and CD Limited in vivo bioassays Rodent liver altered foci induction; mouse skin neoplasm
induction; mouse pulmonary neoplasm induction; female Sprague–Dawley rat breast cancer induction
Decision point 3: Evaluation of results from tests at levels A, B, C, and appropriate test from DE Chronic bioassay Chronic carcinogenic studies in rodents
Decision point 4: Final evaluation of results from tests at all levels for risk assessment analysis
Source: From Williams, GM, Weisburger, JH, Mutat Res, 1988, 205, 79.

154 PRINCIPLES OF TOXICOLOGY TESTING
MUTAGENICITY TESTING IN VIVO
Mutations are hereditary genotypic changes that occasionally express phenotypic altera-tions. Thus a defect in the nucleotide sequence leads to misreading of the genetic code. In the absence of adequate DNA repair mechanisms, the consequence of such a mutation may result in carcinogenic sequelae. The spectrum of genetic damage is dependent on those factors delineated earlier in the conduction of carcinogenic studies, including level and duration of exposure, extent of genotypic alteration, and response of the organism to the initiation event.
The bases of tests for mutagenic potential are discussed in detail in chapter 17. Briefl y, the bacterial tests incorporate the ability to detect reverse mutations from the nutrient-dependent strain to a “wild-type” capable of sustaining itself on minimal medium or chem-ically induced forward mutations resulting in measurable phenotypes. Forward mutations occur at several loci within one gene, represent a larger target for mutagenic chemicals, and consequently are relatively easier to detect. Reverse mutation assays rely on the abilities of organisms to induce mutations at a specifi c locus, thus providing a smaller selective target site for a chemical.
SUGGESTED READINGS
Ames BN. Identifying environmental chemicals causing mutations and cancer. Science 1979; 204: 587.
Ames BN, Gold LS, Willett WE. The causes and prevention of cancer. Proc Natl Acad Sci USA 1995; 92: 5258.
Assem FL, Holmes P, Levy LS. The mutagenicity and carcinogenicity of inorganic manganese com-pounds: a synthesis of the evidence. J Toxicol Environ Health B Crit Rev 2011; 14: 537.
Benigni R, Bossa C, Tcheremenskaia O, Giuliani A. Alternatives to the carcinogenicity bioassay: in silico methods, and the in vitro and in vivo mutagenicity assays. Expert Opin Drug Metab Toxicol 2010; 6: 809.
Billington R, Lewis RW, Mehta JM, Dewhurst I. The mouse carcinogenicity study is no longer a scientifi cally justifi able core data requirement for the safety assessment of pesticides. Crit Rev Toxicol 2010; 40: 35.
Custer LL, Sweder KS. The role of genetic toxicology in drug discovery and optimization. Curr Drug Metab 2008; 9: 978.
European Economic Community. Genotoxicity: Specifi c aspects of regulatory genotoxicity tests for phar-maceuticals. Legislative Basis Directive 75/318/EEC, Brussels, 1995. International Agency for Research on Cancer. [Available from: http://www.iarc.fr] [Last accessed July 2012].
Fowler P, Smith K, Young J, et al. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. I. Choice of cell type. Mutat Res 2012; 742: 11.
Hernández LG, Slob W, van Steeg H, van Benthem J. Can carcinogenic potency be predicted from in vivo genotoxicity data?: a meta-analysis of historical data. Environ Mol Mutagen 2011; 52: 518.
Madden JC, Hewitt M, Przybylak K, et al. Strategies for the optimisation of in vivo experiments in accordance with the 3Rs philosophy. Regul Toxicol Pharmacol 2012; 63: 140.
Spjuth O, Eklund M, Ahlberg Helgee E, Boyer S, Carlsson L. Integrated decision support for assess-ing chemical liabilities. J Chem Inf Model 2011; 51: 1840.
Tavtigian SV, Pierotti MA, Borresen-Dale AL. International Agency for Research on Cancer Workshop on expression array analyses in breast cancer taxonomy. Breast Cancer Res 2006; 8: 303.
Weisburger JH, Williams GM. The decision-point approach for systematic carcinogen testing. Food Cosmet Toxicol 1981; 19: 561.
Williams GM, Weisburger JH. Application of a cellular test battery in the decision point approach to carcinogen identifi cation. Mutat Res 1988; 205: 79.

CARCINOGENICITY AND MUTAGENICITY TESTING IN VIVO 155
REVIEW ARTICLES
Benigni R, Giuliani A. Putting the predictive toxicology challenge into perspective: refl ections on the results. Bioinformatics 2003; 19: 1194.
Bennett LM, Davis BJ. Identifi cation of mammary carcinogens in rodent bioassays. Environ Mol Mutagen 2002; 39: 150.
Bristol DW, Wachsman JT, Greenwell A. The NIEHS Predictive Toxicology Evaluation Project: Chemcarcinogenicity bioassays. Environ Health Perspect 1996; 104: 1001.
Carr CJ, Kolbye AC Jr. A critique of the use of the maximum tolerated dose in bioassays to assess cancer risks from chemicals. Regul Toxicol Pharmacol 1991; 14: 78.
Chico-Galdo V, Massart C, Jin L, et al. Acrylamide, an in vivo thyroid carcinogenic agent, induces DNA damage in rat thyroid cell lines and primary cultures. Mol Cell Endocrinol 2006; 257: 6.
Cimino MC. Comparative overview of current international strategies and guidelines for genetic toxicology testing for regulatory purposes. Environ Mol Mutagen 2006; 47: 362.
Cohen SM, Ellwein LB. Cell proliferation in carcinogenesis. Science 1990; 249: 1007.Contrera J, DeGeorge J. In vivo transgenic bioassays and assessment of the carcinogenic potential of
pharmaceuticals. Environ Health Perspect 1998; 106: 71.Doe JE, Boobis AR, Blacker A, et al. A tiered approach to systemic toxicity testing for agricultural
chemical safety assessment. Crit Rev Toxicol 2006; 36: 37.Duerksen-Hughes PJ, Yang J, Ozcan O. Induction as a genotoxic test for 25 chemicals undergoing in
vivo carcinogenicity testing. Environ Health Perspect 1999; 107: 805.Gregory AR. Species comparisons in evaluating carcinogenicity in humans. Regul Toxicol Pharma-
col 1988; 8: 160.Helma C, Kramer S. A survey of the predictive toxicology challenge 2000–2001. Bioinformatics
2003; 19: 1179.Ikeda M, Masumura K, Sakamoto Y, et al. Combined genotoxic effects of radiation and a tobacco-
specifi c nitrosamine in the lungs of gpt-delta transgenic mice. Mutat Res 2007; 626: 15–25.
Jacobson-Kram D, Sistare FD, Jacobs AC. Use of transgenic mice in carcinogenicity hazard assess-ment. Toxicol Pathol 2004; 32: 49.
Jones S, Kazlauskas A. Growth-factor-dependent mitogenesis requires two distinct phases of signal-ing. Nat Cell Biol 2001; 3: 165.
Kane AB. Animal models of malignant mesothelioma. Inhal Toxicol 2006; 18: 1001.Keenan KP, Laroque P, Dixit R. Need for dietary control by caloric restriction in rodent toxicology
and carcinogenicity studies. J Toxicol Environ Health B Crit Rev 1998; 1: 135.MacDonald J, French JE, Gerson RJ, et al. The utility of genetically modifi ed mouse assays for
identifying human carcinogens: a basic understanding and path forward. Toxicol Sci 2004; 77: 188.
Mebust M, Crawford-Brown D, Hofmann W, Schollnberger H. Testing extrapolation of a biologically based exposure-response model from in vitro to in vivo conditions. Regul Toxicol Pharma-col 2002; 35: 72.
Moolgavkar SH, Knudson AG. Mutation and cancer: a model for human carcinogen-esis. J Natl Cancer Inst 1981; 66: 1037.
Perera EP. Perspectives on the risk assessment of nongenotoxic carcinogens and tumor promotors. Environ Health Perspect 1991; 94: 231.
Rao GN, Huff J. Refi nement of long-term toxicity and carcinogenicity studies. Fundam Appl Toxicol 1990; 15: 33.
Rao PM, Laconi E, Vasudevan S, et al. Dietary and metabolic manipulations of the carcinogenic process: role of nucleotide pool imbalances in carcinogenesis. Toxicol Pathol 1987; 15: 190.
Roe FJ. Refi nement of long term toxicity and carcinogenesis studies. Fundam Appl Toxicol 1991; 16: 616.

156 PRINCIPLES OF TOXICOLOGY TESTING
Slamenova D. Contemporary trends in in vivo and in vitro testing of chemical carcinogens. Neo-plasma 2001; 48: 425.
Snyder RD. An update on the genotoxicity and carcinogenicity of marketed pharmaceuticals with reference to in silico predictivity. Environ Mol Mutagen 2009; 50: 435.
Tharappel JC, Lee EY, Robertson LW, Spear BT, Glauert HP. Regulation of cell proliferation, apop-tosis, and transcription factor activities during the promotion of liver carcinogenesis by polychlorinated biphenyls. Toxicol Appl Pharmacol 2002; 179: 172.
Waddell WJ, Fukushima S, Williams GM. Concordance of thresholds for carcinogenicity of N-nitros-odiethylamine. Arch Toxicol 2006; 80: 305.

157
12 Introduction to in vitro toxicology testing
HISTORY OF IN VITRO METHODS
It was not until the development of cell and tissue culture techniques in the 1950s that allowed the scientifi c community to understand the principles and mechanisms of the bio-medical sciences, particularly cell biology of cellular functions. By the 1960s, the develop-ment of cell culture methods allowed for the growth of animal and human cells and tissues on plastic. This was the fi rst example that demonstrated the survival of cells outside intact organisms. With further refi nement of the culture conditions, and insights into the require-ments for maintaining viability of organs, tissues, and cells in an artifi cial environment, a transformation in cell biology was underway.
Soon after, a variety of scientifi c disciplines began to appreciate the breakthroughs realized with these new techniques. In the 1970s, toxicologists adapted cell and tissue cul-ture methodologies as pathways for revealing mechanistic toxicology—the mechanisms of actions of toxicants. Cell culture broadened the scope of cell and cancer biology and other toxicologists came to realize that cells grown in vitro also retained the properties of their organs of origin. Perhaps these surrogate “tissues” grown in plastic could mimic animal tissues and organs in their responses to toxic insults.
Thus the term in vitro toxicology was born which generally referred to the handling of tissues outside of intact organ systems under conditions that support their growth, differen-tiation, and stability. Since the seminal experiments established the validity of cell culture techniques in toxicology, the methods have proven fundamental for understanding and developing critical procedures in cellular and molecular biology, as well as in pharmacol-ogy, genetics, reproductive biology, and oncology.1 Cell culture technology has improved dramatically as a result of interest in the methods and through its broad applications.
Although much of the mystery originally surrounding the techniques has waned, the doctrine underlying the establishment of the methodology remains. For example, improve-ments in our knowledge of serum requirements in media have fostered continued excite-ment in the roles of growth factors and cell differentiation. Without these techniques, the recent discoveries in the fi eld of stem cell biology may not have been realized. Conse-quently, the discipline of cell culture and its application to in vitro toxicology and biology represent a tool poised to answer questions in the biomedical sciences that can only be addressed by examining the proliferation of isolated cells without the infl uence of other organ systems. With this understanding, the use of cell culture does not purport to represent the whole human organism, but can signifi cantly contribute to our understanding of the workings of its components.
1 The terms cell culture and tissue culture are used interchangeably in this section.

158 PRINCIPLES OF TOXICOLOGY TESTING
HISTORY OF CELL CULTURE
The use of cell culture techniques in toxicological investigations is known as in vitro cyto-toxicology or in vitro toxicology. The latter term also includes noncellular test systems, such as isolated organelles or high-throughput microarrays (see Section III, chap. 17). The increasing interest and success of these techniques in toxicology are consequences of the considerable advancements realized since the advent of the technology:
1. Since the growth of the fi rst mammalian cells in capillary glass tubes was described, the technology has progressed and has been refi ned extensively.
2. The mechanisms of toxicity of chemicals in humans and animals have been eluci-dated through the use of in vitro toxicological methods.
3. The necessity for determining the toxic effects of industrial chemicals and pharma-ceuticals that are developed and marketed at rapid and unprecedented rates has made it compulsory to develop fast, simple, and effective alternative test systems.
4. Although the normal rate of progression of any scientifi c discipline is determined by progress within the scientifi c community, some areas have received more encouragement than others.
Specifi cally, the public objections and disapproval of animal testing have forced aca-demic institutions, industrial concerns, and regulatory agencies to direct research initiatives toward the development of alternative models for animal toxicology testing.
To understand the possibilities for and, more importantly, the limitations of cell culture methods in toxicology, it is necessary to become acquainted with the main features of the techniques for culturing cells and tissues. These techniques require specialized training, separate laboratory facilities, and steadfast attention to details. In addition, unlike other types of biomedical research, these techniques require continuous care of cells in culture for extended periods which necessitates planning and coordination.
TISSUE EXPLANTS
In the fl edgling years of cell culture, U.S. scientists removed tissue explants from animals and allowed them to adhere to glass cover slips or placed them in capillary tubes in clots formed from lymph or plasma. Figure 12.1 shows an early attempt to grow animal cells in glass tubes or plates following what appeared to be aseptic technique. The discovery was made early on that a tissue or organ that was once an intact biological specimen would exhibit a breakdown in the supporting matrix, followed by migration of individual cells from the specimen as a consequence.
With the addition of serum or whole blood, the resulting clot formed a hanging drop, making it possible to see these cells through an ordinary light microscope. With some refi nement, the explants demonstrated that cells migrated out of a dissected specimen. This discovery advanced the cultivation of individual cells bathed in plasma or embryonic serum extract in small glass tubes. Aseptic techniques were further refi ned and the refi ne-ments allowed cells to be maintained in culture for longer periods. As the cells prolifer-ated in higher-quality tissue culture fl asks, they could be transferred from one fl ask to another by scraping and loosening the growing monolayers with a rubber “policeman” (cell scraper).
Synthetic media (Earle, Parker, and Eagle) were developed quickly and used with vari-ous serum additives. The main problem encountered was bacterial and fungal contamination

INTRODUCTION TO IN VITRO TOXICOLOGY TESTING 159
which, because of the more rapid rate of mitosis of the microorganisms, usually outpaced the growth of the mammalian cells. This contamination generally resulted in overwhelming bacterial cell growth and disintegration of the cell cultures. This setback was largely over-come by the addition of liquid antibiotics to the media.
Later on, the development of better aseptic techniques such as the incorporation of sterile disposable glassware, autoclave units, and laminar air fl ow hoods made antibiotics superfl uous in most cases. With an increase in the understanding of the infl uences of pH, buffers, gases, and ambient environments and the incorporation of chemically inert plastics and microprocessor-controlled incubators came the realization of the full potential of in vitro technology.
RECENT DEVELOPMENTS
Today, cell biologists have further developed culture techniques to aid in their understanding of cellular and extracellular interactions such as mesenchymal–epithelial relationships, epithelium–cell matrix interactions, and stem cell biology. These in vitro studies have materialized largely through the development of additions to accepted protocols such as chemically defi ned cell culture media, the addition of cellular substrata to culture fl asks, the introduction of porous mem-branes and fi lter inserts that allow for the passage of low-molecular-weight soluble substances, the development of treated plastic surfaces, and incubation of cells with cocultures.
IN VITRO SYSTEMS
APPLICATIONS IN RISK ASSESSMENT AND PREDICTIVE TOXICOLOGY
Prediction of chemically induced toxicity in humans from in vitro data continues to be a signifi cant challenge for the biomedical community, and pharmaceutical and toxicological industries. Generally, conventional in vitro cell culture systems are limited by their inability to maintain phenotypic characteristics over time in culture, including stable expression of metabolic and bioactivation pathways. Even their complex adaptive responses to chemical exposure are sometimes diffi cult to interpret. In addition, these systems require signifi cant
1
2
3
(A) (B)
FIGURE 12.1 Scan of the original diagram showing examples of glass (A) and fabric (B) envelopes and photographs of a Leighton tube (1) and envelopes (2 and 3) for the preparation of primary explants or dispersed animal cells. Source: Courtesy Robertson, AL, Tissue Cult Assoc Man 1, 167, 1975.

160 PRINCIPLES OF TOXICOLOGY TESTING
advancement and improvement before long-term toxicity studies can deliver understanding of key cellular and molecular events involved in human risk assessment. In spite of these drawbacks, cell culture models have been able to explain primary and secondary adaptation to chemical exposure, have been useful in identifying the important mediators of infl amma-tion, proliferation, and apoptosis. Considerable progress has also been achieved in imple-menting a more effective strategy for in vitro–in vivo extrapolation and human risk assessment, particularly with the signifi cant advances in tissue culture technology and the improvement in the level of biological complexity of the models. Several of the foregoing chapters describe the current and ongoing need for more relevant, in vitro surrogate systems of human and animal organs. Some chapters will emphasize the advances made in acute and chronic toxicity testing using cell culture techniques, particularly with the use of organotypic models, their ability to re-create the multicellular architecture, and how these strive to mimic and hemodynamic properties of the intact organism using novel culture platforms. As these systems become more widely used for chemical and drug toxicity testing, there will be a corresponding need to establish standardized testing conditions, end point analyses and acceptance criteria. In the future, a balanced approach between sample throughput and bio-logical relevance should provide better in vitro tools that are complementary with animal testing and assist in conducting more predictive human risk assessment.
APPLICATIONS WITH BIOMARKERS AND DRUG DEVELOPMENT
The goal of the National Academy of Sciences, National Research Council’s strategy, Toxicology Testing in the 21st Century (2007), is to overcome the preponderance of currently used animal tests for toxicity testing and replace, reduce, and refi ne (3Rs) these methods with techniques that can identify novel molecular pathways for prediction of human toxicity. In addition, in vitro systems, whether of human or animal origin, can provide the most relevant mechanistic information for human risk assessment. As a corollary, advanced and sensitive indicators of toxicity, in the form of appropriate biomarkers, are expected to interpret the very systems that animal toxicology has found elusive for decades. These biomarkers can also be used for novel types of toxicity-related high-throughput screening methods for thousands of compounds that require testing during the cumbersome phases of drug development.
The fi eld has not only already advanced logarithmically in this positive direction, but other disciplines have also been established as a consequence of these developments, namely stem cell biology; systems biology; toxicogenomic, transcriptomic, proteomic, and metabo-lomic technologies; and epigenetic toxicology. The challenges, of course, involve the han-dling of vast quantities of data generation, statistical treatment, bioinformatic integration, interpretation of the results, and discussion of the mechanisms and principles underlying the information. Currently, the search for biomarkers of toxicity may require sophisticated strat-egies for time-resolved quantitative differential approaches. Or the elusive biomarker may actually be derived from a simpler model that has been overlooked in the quest for accurate indicators (MTT2 assay as proof positive). Finally, statistical principles, normalization of primary data, and principal component and cluster analysis are well developed for analyzing most of these technologies, but may require adaptation to fi elds such as metabolomics.
CORRELATION WITH ANIMAL TESTING AND WITH HUMAN EXPOSURE
As noted above, in 2007 the U.S. National Academy of Sciences released a report that envisions a not-so-distant future in which virtually all routine toxicity testing would be
23-(4, 5-Dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide.

INTRODUCTION TO IN VITRO TOXICOLOGY TESTING 161
conducted with in vitro systems by evaluating cellular responses using a suite of toxicity path-way assays. Based on these results, human risk assessment would be predicted not from his-torically static models of animal testing that are mandatory for regulatory compliance, but would be based on dynamic interactions of dose–response technologies that reveal important information on pathway mechanisms, conduits for computational systems, and explanations of the biological principles underlying the circuitry of each toxic pathway. The model systems of the twenty fi rst century will be used for in vitro to in vivo extrapolations with correlations from other analogous systems such as pharmacokinetic models and epigenetic toxicology.
The scientifi c tools needed to effect the changes in regulatory toxicity testing are cur-rently available or can be manipulated to reach this advanced stage. Therefore, the scien-tifi c discussion of this vision for the future of toxicity testing should be focused not on whether the technology is available for implementation, but on what is the force required to move the fi eld from the traditional high-dose animal-based tests. As with many advances in the biomedical sciences, developments are not gradual. They appear in the form of scal-able peaks until realization occurs that the challenges in implementing the vision have been identifi ed and the obstacles have been surmounted.
CONCLUSION
The following chapters highlight the principles, mechanisms, and current developments in the fi eld of in vitro toxicology testing. In particular, the in vitro techniques described here corre-late in principle with the in vivo toxicology testing techniques described in Section II and emphasize the importance of the former as alternative technologies for animal toxicity testing.
SUGGESTED READINGS
Barile FA. Introduction to In Vitro Cytotoxicology: Mechanisms and Methods. Boca Raton, FL: CRC Press, 1994.
Blaauboer BJ. The contribution of in vitro toxicity data in hazard and risk assessment: current limita-tions and future perspectives. Toxicol Lett 2008; 180: 81.
Eagle H. Media for animal cell culture. Tissue Cult Assn Man 1977; 3: 517.Freshney RI. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications,
6th edn. New York: Wiley-Blackwell, 2007.Freshney RI, Stacey GN, Auerbach JM. Culture of Human Stem Cells. New Jersey: John Wiley &
Sons, Inc, 2007.McGarrity GJ. Serum quality control. Tissue Cult Assn Man 1975; 1: 167.National Research Council (NRC). Toxicity Testing in the 21st Century: A Vision and A Strategy.
Washington, DC: National Academy Press, 2007. National Toxicology Program (NTP).NTP. A National Toxicology Program for the 21st Century: A Roadmap for the Future. Research
Triangle Park, NC: NTP, NIEHS, 2004. [Available from: http://ntp.niehs.nih.gov/?objectid=EE4AED80-FIF6-975E-7317D7CB17625A15]
Robertson AL. Envelope technique for selective isolation of cells from multilayer organ cultures for metabolic studies. Tissue Cult Assn Man 1975; 1: 139.
Weiss L. Cell contact phenomena. In: Waymouth C, ed. Advances in Tissue Culture. Baltimore: Williams & Wilkins, 1970: 48.
REVIEW ARTICLES
Abir R, Nitke S, Ben-Haroush A, Fisch B. In vitro maturation of human primordial ovarian follicles: clinical signifi cance, progress in mammals, and methods for growth evaluation. Histol Histopathol 2006; 21: 887.

162 PRINCIPLES OF TOXICOLOGY TESTING
Aschner M, Fitsanakis VA, dos Santos AP, et al. Blood–brain barrier and cell-cell interactions: methods for establishing in vitro models of the blood–brain barrier and transport mea-surements. Methods Mol Biol 2006; 341: 1.
Lecluyse EL, Witek RP, Andersen ME, Powers MJ. Organotypic liver culture models: Meeting cur-rent challenges in toxicity testing. Crit Rev Toxicol 2012; 42: 501.
Lin H, Bu Q, Cen X, Zhao YL. Current methods and research progress in nanomaterials risk assess-ment. Curr Drug Metab 2012; 3: 354.
Rippon HJ, Bishop AE. Embryonic stem cells. Cell Prolif 2004; 37: 23.Seth G, Hossler P, Yee JC, Hu WS. Engineering cells for cell culture bioprocessing: physiological
fundamentals. Adv Biochem Eng Biotechnol 2006; 101: 119.Turksen K, Troy TC. Human embryonic stem cells: isolation, maintenance, and differentiation.
Methods Mol Biol 2006; 331: 1.

163
13 Cell culture methodology
CELL CULTURE LABORATORY
ENVIRONMENT
The cell culture laboratory is a sophisticated, meticulous, detail-oriented environment that relies on well-practiced features of cell biology while incorporating the latest instrumenta-tion of biotechnology. In general, good laboratory practice is critical for maintaining an environment for optimum cell growth and proliferation. In spite of the sophisticated tech-niques and instrumentation required to establish and maintain a cell culture laboratory, the work, at least in part, is repetitive and requires considerable amounts of time. Consequently, most of the techniques focus on the procedural steps in the protocol. It is also important that the physical laboratory is dedicated to cell culture exclusively. Using the laboratory for other than cell culture techniques and incubation increases the risk of microbiological and cross-contamination from other parts of the environment.
In general, cultured cells require a sterile environment and a supply of nutrients for growth. The immediate culture environment, namely the incubator, must be able to main-tain stable pH, temperature, and humidity. Over the last few decades, media have been developed to accommodate the requirements needed by cultured cells and to harmonize the incubator with the nutrients. For example, various defi ned basal media, defi ned serum-free media, and a variety of balanced salt solutions are now available commercially. The media have benefi tted from advancements in cell biology, and have since been modifi ed to incor-porate essential nutrients, such as amino acids, vitamins, fatty acids, and lipids. Conse-quently, media suitable for supporting the growth of a wide range of cell types are now available.
Maintaining proper technique is paramount, and it is important for researchers to audit their processes as they work, disbursing special attention to procedure and details. For instance, the area of the biological safety cabinet (i.e., the sterile hood), must be wiped down completely with 70% alcohol, both before and after any procedure. Hoods should not be used for storage; investigators often conveniently house canisters, bottles, and pipettors under the hood. Also, when not in use, the bench top of the safety cabinet should be exposed to ultraviolet light to ensure a sterile environment during off hours. All nonsterile equip-ment, instruments, and containers should be disinfected before transporting them into the confi nes of the sterile atmosphere. In addition, it is important to establish an uninterrupted, fi ltered airfl ow within the room. This is particularly important when manipulating liquids, removing caps from bottles, or lifting lids from tissue culture plates. Any interruption of airfl ow from pedestrians crossing the room can overcome the fi ltered air fl ow from the hood. In addition, all attention is focused on the procedure; the laboratory personnel should not be distracted by alarms, phones, opening of doors, or other individuals in the immediate confi nes.
Lab coats are generally left in the lab, hand washing is essential, and nonsterile instru-ments are kept in clean order. Sterile gloves are optional, since the gloves necessarily

164 PRINCIPLES OF TOXICOLOGY TESTING
touch nonsterile items in the hood. When moving cells from the cabinet to the incubator, and vice-versa, the tissue culture labware must be properly covered and transport time is minimized.
Incubators provide a synthetic environment that mimics the in vivo milieu and encour-ages cell growth. Interestingly, these favorable conditions also allow for microorganisms to thrive. Thus, incubators must be kept clean—the inside as well as the outside of the equip-ment should be thoroughly wiped, or preferably disinfected. Isopropyl alcohol (70%) is safe to use, in limited amounts, and only on work surfaces.1
Shared incubators present their own challenges, particularly that of cross-contamination. It is recommended that all personnel authorized to use a common cell culture space, be instructed as to the proper procedures in the laboratory, so as to prevent forward contamina-tion. For instance, incubator doors should not be left open for inordinate amounts of time. This habit allows for escape of gases, and invites environmental pathogens to enter. Loca-tions within the chamber are assigned to those sharing the unit. Moving plates and fl asks around and relocating fl asks and plates of other personnel while looking for one’s own cul-ture will increase the risk of contamination.
Emphasis is also placed on preventative maintenance. For example, HEPA fi lters within biological safety cabinets need to be changed routinely as agreed to by the authorized work-force. The HEPA fi lters require time to change according to the instructions manual. CO
2 sensors are recalibrated as recommended by the manufacturer. And, 100% humidity is
mandatory for sustaining proper isotonic and pH criteria. Ultradry, heated winter air draws all of the humidity out of an incubator when the door is opened. Incubators are kept in a location away from direct sunlight, especially in warmer climates. Note the location of heating and air conditioning ducts; the incubator and hood should be positioned away from these units.
Attention to detail, rigorous adherence to standards and processes, and investment in quality disposable supplies and chemicals, work spaces and instruments, offer long-term benefi ts. The following discussion focuses on the equipment used in the cell culture labora-tory and its importance in proper cell culture technique.
EQUIPMENT
The equipment, supplies, and analytical instruments in the cell culture laboratory are impor-tant aspects of the cell culture laboratory and are relied on for sensitive detection of cellular events. As the technology has advanced, so has the laboratory environment. Consequently, several important features of the cell culture laboratory are essential, including the following and other aspects of cell and tissue culture discussed in subsequent sections:
1. Sterile air fl ow work station.2. Temperature-controlled incubator.3. Autoclave.4. Cell counter.5. Custom gas tanks.6. Source of ultrapure water.
Biological safety cabinets (Class II) reduce microbial contamination and also protect opera-tors from exposure (Fig. 13.1). These units are used for handling mammalian cells and also potentially infectious microorganisms, thus removing microbes within the sterile atmosphere
1 Bleach is not recommended because, even in small amounts, it may corrode stainless steel.

CELL CULTURE METHODOLOGY 165
while simultaneously protecting the operator from exposure. Alternatively, laminar fl ow hoods are unidirectional air fl ow vents that isolate the operating environment by blowing sterile air over the surface of the working platform or table top out toward the operator, ren-dering the operator vulnerable to exposure. This apparatus is generally reserved for prepara-tion of sterile cell culture media, pharmaceutical injectables, and solutions.
Automatic glassware washing facilities, an incubator with digital humidity and tem-perature controls, and a dependable inverted microscope facilitate experiments and allow for less troublesome, routine operations in a cell culture laboratory (Fig. 13.2). Thus, ade-quate tissue culture facilities fi ll a critical and dedicated role in toxicological investigations.
(A) (B)
FIGURE 13.1 Biological safety cabinet: 5-ft purifi er digital series class II, type A2. (A) Full view show-ing blower housing, above, and working space, below; (B) Close-up view showing 5-ft work space. Cabinet can be mounted on a stand (as shown), or on a bench top. Source: Photo from personal fi les.
FIGURE 13.2 Air-jacketed automatic CO2 incubator. Source: Photo from personal fi les.

166 PRINCIPLES OF TOXICOLOGY TESTING
In general, in order to maintain sterility, viability, identifi cation, and integrity of cultured cells, the room containing the tissue culture facilities is dedicated to this function. How-ever, complete separation of all functions is not always possible or necessary. For instance, an analytical balance, pH meter, hot plate, and magnetic stirrer can be shared with other laboratory areas, whereas media preparation facilities, cell culture equipment, and glass-ware are generally reserved for the cell culture laboratory.
Most supplies and plastic ware used in handling cell cultures routinely are sterile and disposable. These include borosilicate glass or plastic pipettes and bottles, polyethylene tis-sue culture fl asks, petri dishes, polypropylene and polyethylene centrifuge tubes, and fi lter-ing supplies. The disposable supplies reduce microbiological contamination and integration of other cultured cells present in the laboratory. In addition, sterile, ready-formulated media may be purchased and save the inconvenience of preparation time.
CULTURED CELLS
PRIMARY CULTURES
In general, establishing cells in culture from intact, viable tissue explants2 is easily facilitated from younger donors; that is, the younger the age of the mammalian or human donor, the more replications and shorter replication time are expected from the resultant cells. In addi-tion, cells from embryonic tissue are quickly established in culture, usually progressing through more population doublings in vitro than cells from adult donors. Some cells derived from neoplastic tissue are similar to embryonic cells in this respect.
Ideally, cell cultivation begins with the aseptic removal of an explant (1/2 × 1 mm) from a tissue or organ specimen. The cells of the explant are mechanically and/or enzy-matically separated from the matrix and are allowed to grow in a medium in contact with the bottom surface of the culture vessel. While the central core of the explant often atro-phies due to less favorable diffusion of nutrients through the tissue, peripheral cells migrate outward and proliferate. The cells migrating from the tissue establish a primary culture. Two important and parallel processes occur during this transformation:
1. The differentiated cells of the original explant may divide, depending on the organ of origin; also, as the culture progresses, the established primary cells suc-cessively lose some of their specialized functions—a process referred to as dedifferentiation.
2. Less specialized cells such as mesenchymal cells (e.g., fi broblasts) divide rapidly and eventually outpace the specialized cells (Fig. 13.3).
FINITE CONTINUOUS CELL LINES
As the cells from the primary culture proliferate, they eventually occupy the entire vessel surface exposed to the medium. Thus, the culture reaches a state of confl uency and micro-scopically appears as a uniform, closely attached carpet of cells known as a monolayer. The cells are ready for transfer to new culture vessels containing fresh medium. With this manip-ulation, the culture ceases to be in a primary state and is designated a continuous cell line. Subcultivation, or passaging, is achieved by detaching the cells from the glass or plastic
2 Explant cultures are viable anatomical specimens removed from a mammalian donor and prepared for cell cul-ture; they usually measure 1–3 mm in diameter. They have the capability of generating primary cultures.

CELL CULTURE METHODOLOGY 167
using a calcium chelator solution such as EDTA or by exposing the monolayer surface to enzymatic methods.3
The cells are dispersed, collected, centrifuged, resuspended, counted, and inoculated in several new vessels. Because of the selective survival of viable cells, the cell line is more homo-geneous and dedifferentiates with time. A culture that enjoys adequate proliferation is har-vested as above every 3–7 days, depending on the cells, and subdivided to a new environment.
Cell lines established from non-neoplastic tissue have fi nite aging processes in vitro and ultimately stop dividing. They exhibit a specifi c number of cell divisions, usually deter-mined experimentally.4 Such a fi nite cell line has a constant (diploid) number of chromo-somes and exhibits an orderly oriented growth pattern including inhibition of growth of individual cells by contacts with their neighboring cells (contact inhibition).
It is possible to select a single cell from a confl uent primary culture and transfer it to a culture fl ask. The proliferation of cells from a single primary cell is designated a clonal cell line. Cell culture cloning, according to the traditional defi nition, allows for the selection of one type of cell (e.g., a cell with a specifi c functional marker) and the establishment of a subculture in which the desired characteristic has been passed on to the progeny.
IMMORTAL CONTINUOUS CELL LINES
Cell lines that do not age in culture with continuous passage are transformed to immortal continuous cell lines, principally because their growth patterns do not exhibit typical signs of aging in culture. Transformation occurs either spontaneously or is induced as a result of incubation with viruses or carcinogenic or mutagenic chemicals.
The exact mechanism of the transformation involves the initiation and promotion of stages of carcinogenesis. Thus a continuous cell line acquires a set of characteristics such as a varied chromosome number (heteroploidy) and loss of contact inhibition. Moreover, immortal continuous cell lines are able to form colonies in soft agar media, that is, without the help of glass or plastic contact, and induce tumors if implanted in immunologically
3 Because the enzyme of choice is trypsin, the process of subcultivation of monolayers is often referred to as trypsinization . 4 Finite replications are also referred to as a population doubling level (PDL) and may require months in culture, depending on growth rate.
(A) (B)
FIGURE 13.3 Human lung fi broblasts derived from normal human lung parenchyma. (A) Sub-confl uent cells proliferating in logarithmic phase; (B) Confl uent cells in stationary phase (400×). Source: Photos from personal fi les.

168 PRINCIPLES OF TOXICOLOGY TESTING
nude animals. In spite of this, some highly differentiated cell functions that mimic those of specialized cells persist in immortal cell lines.
These differentiated functional markers are used to characterize the cells and are relied upon to monitor the progress of the monolayers in culture, particularly because the low-power morphological appearances of fi nite and immortal cells in vitro are not easily distin-guishable. The criteria are also used as parameters for assessing in vitro toxicity. Table 13.1 presents some frequently used indicators of toxicity as general markers of cell viability and function.
STEM CELL LINES
Embryonic stem (ES) cells are derived from pluripotent cells of the early mammalian embryo and are capable of unlimited, undifferentiated proliferation in vitro. In chimeras with intact embryos, ES cells contribute to a wide range of adult tissues including germ cells, providing a powerful approach for introducing specifi c genetic changes into a cell line. Plu-ripotency allows cells to differentiate into many cell types and tissues, including all three embryonic germ layers. Furthermore, experimental evidence has shown that organogenesis of embryonic and adult stem cells to neural, muscle, dermal, and bone marrow lineages is possible. Figure 13.4 illustrates a general outline of the steps involved in the derivation of differentiated ES cells in culture.
The concept of deriving ES cells from mice and humans is relatively recent. The protocols developed to maintain ES cells in continuous culture continue to emerge. Some progress, however, has demonstrated the transformation of ES cells to different organ and tissue structures including in vitro organogenesis and differentiation of human, pri-mate, and mouse ES cells into functional gut-like organs, smooth muscle cells, cardio-myocytes, neurons, and hematopoietic entities. In addition, soluble factors infl uence direct differentiation of mouse ES cells. For example, IL-3 directs cells toward macro-phage, mast cell, neutrophil, or erythroid lineages. Retinoic acid induces neuron forma-tion, and transforming growth factor induces myogenesis. Differentiation is also encouraged by culturing ES cells on different feeder layers and extracellular matrix (ECM) components that exhibit morphological and physiological properties characteris-tic of the target organ.
TABLE 13.1General Viability Toxicity Criteria for Cell Lines
Indicator Methods for Determining Degree of Toxicity
Cell morphology Histological, ultrastructural, immunohistochemical analysis Cell proliferation Cell count, mitotic frequency, DNA synthesis, karyotypic analysis Cell division Plating effi ciency, clonal formation Cell metabolism Uptake of fl uorescent or isotope-labeled precursors, fl uorescent luminescence
assays for enzymatic activity Cell membrane Leakage of enzymes (LDH), uptake of trypan blue Cell staining Immunohistochemical or cytochemical stains for cell markers Cell differentiation PCR analysis for gene markers Mitochondria Mitochondrial reduction (MTT assay)Lysosomes Vital staining, NRU assay
Abbreviations: LDH, lactate dehydrogenase; NRU, neutral red uptake.

CELL CULTURE METHODOLOGY 169
The increasing awareness of the infl uence of growth factors and basement membrane substrata has improved our understanding of the role of cell regeneration in wound repair and in preneoplastic tumor growth. In their ability to form tight junction strands, differen-tiating epidermal and epithelial cells act to prevent free interchange of most solutes between luminal and interstitial fl uids along the paracellular route. Breakdowns in the barrier func-tions of these membranes may contribute to cellular neoplasia. Thus, continuous progress in stem cell biology and biotechnology is based on the understanding that in vivo epithelial, epidermal, and mesenchymal cells undergo continuous renewal by multipotent stem cells that remain anchored to the organ basement membrane. Thus a single stem cell migrates and commits to all of the embryonic lineages.
Infl uence of Culture Environment on ES Cells
When ES cells are allowed to differentiate in suspension culture or in the absence of feeder layers, they form spherical multicellular aggregates or embryoid bodies (EBs) that contain a variety of cell types. By manipulating combinations of growth factors (GFs) in the presence of ECM components, epithelial or epidermal-specifi c gene expression and differentiation are induced.
Fertilized ovum
Day 6
Endoderm Ectoderm Mesoderm Ovum/Spermatozoaliver skin bloodGIT brain musclepancreas hair bonelung cartilage
Blastocyst
ES cell (pluripotent)
In vitro expansion
ES cell (EB)
Differentiation
Germ cell lineagesSomatic cell lineages
Inner cell mass(pluripotent)
FIGURE 13.4 Embryonic stem (ES) cells isolated from the inner cell mass of a blastocyst. Under the infl uence of different growth factors, ES cells have the potential to differentiate to cells of different lineages. Abbreviations: EB, embryoid body; GIT, gastrointestinal tract.

170 PRINCIPLES OF TOXICOLOGY TESTING
The mechanism by which GFs and ECM infl uence cell differentiation or expression of differentiation markers is the subject of much interest. In the presence of GFs in culture, mouse ES cells acquire different expression profi les and display evidence for tight junc-tions. It is understood that without GFs, leukemic inhibitory factor (LIF), or feeder layers, mouse embryonic stem cells spontaneously differentiate into a variety of uncontrolled cell colonies, and properties for cellular differentiation are delayed.
Thus, GFs and ECM are capable of directing linear specifi c differentiation. For instance, epidermal growth factor (EGF) is a mitogenic polypeptide that allows for differ-entiation into ectoderm (including skin) and mesoderm, while keratinocyte growth factor (KGF) is an epithelial cell-specifi c mitogen responsible for normal proliferation and dif-ferentiation of epithelial cells. Consequently, differentiation of ES cells in culture to spe-cifi c lineages may be mediated by enrichment of the environment with GFs that infl uence either cell expansion, improved survival, or differentiation.
Culture of Human or Mouse ES Cells
Currently, most experimental stem cells are derived from blastocysts of humans or mice. The cells spontaneously differentiate into embryonic structures in the absence of a feeder layer or conditioned medium. They are maintained in the undifferentiated state by frequent sub-culture (every 2 days) on confl uent mitomycin-C treated or irradiated mouse embryonic feeder (MEF) layers or in complete culture medium to which is added LIF, or both.5
In Figure 13.5, undifferentiated mouse ES cultures are distinguished from differentiated populations and from the underlying MEFs by the formation of an EB. These aggregates are 4–10 cell layers thick over the feeder layer, are oval or round, and have defi ned borders. As undifferentiated mouse ES cell colonies propagate, they maintain their aggregate morphology
5 Recombinant human LIF is a lymphoid factor that promotes long-term maintenance of mouse ES cells by sup-pressing spontaneous differentiation; it is not effective in human ES cells.
FIGURE 13.5 Phase contrast image of embryoid bodies from mouse ES-D3 cells after removal of leukemic inhibitory factor. Aggregates are round and have well-defi ned borders. They maintain their aggregate morphology and are distinguished from the underlying feeder layer (400×).
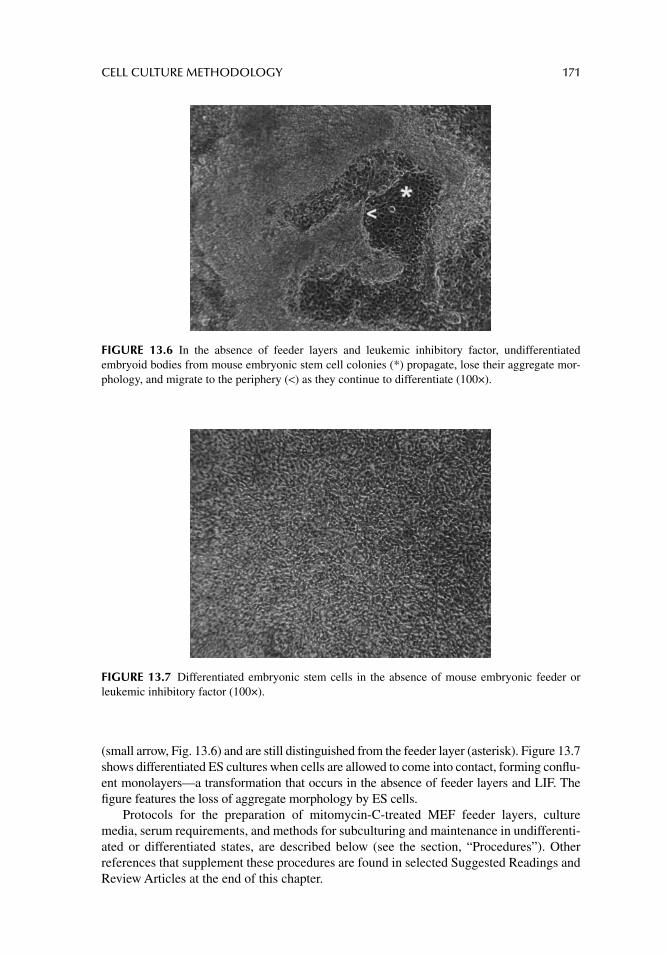
CELL CULTURE METHODOLOGY 171
(small arrow, Fig. 13.6) and are still distinguished from the feeder layer (asterisk). Figure 13.7 shows differentiated ES cultures when cells are allowed to come into contact, forming confl u-ent monolayers—a transformation that occurs in the absence of feeder layers and LIF. The fi gure features the loss of aggregate morphology by ES cells.
Protocols for the preparation of mitomycin-C-treated MEF feeder layers, culture media, serum requirements, and methods for subculturing and maintenance in undifferenti-ated or differentiated states, are described below (see the section, “Procedures”). Other references that supplement these procedures are found in selected Suggested Readings and Review Articles at the end of this chapter.
FIGURE 13.6 In the absence of feeder layers and leukemic inhibitory factor, undifferentiated embryoid bodies from mouse embryonic stem cell colonies (*) propagate, lose their aggregate mor-phology, and migrate to the periphery (<) as they continue to differentiate (100×).
FIGURE 13.7 Differentiated embryonic stem cells in the absence of mouse embryonic feeder or leukemic inhibitory factor (100×).

172 PRINCIPLES OF TOXICOLOGY TESTING
CLONAL GROWTH AND MAINTENANCE CULTURES
Highly differentiated fi nite cell lines from explanted or enzymatically treated tissue are maintained in primary culture by manipulating the liquid medium environment. Such tech-niques include the use of serum-free medium, arginine-free medium, and coating of the plastic culture surfaces with ECM (laminin or fi bronectin) that promotes growth and adhe-sion of either epithelial or mesenchymal cells, respectively. In addition, cell types are mechanically separated by employing clonal growth or differential adhesion manipula-tions—in whole tissue, epithelial cells are separated from fi broblasts based on the ability of the latter cells to settle and adhere to the surface more quickly. One to three hours after an initial inoculation of the culture with the cell suspension, as determined empirically, the medium is removed and the epithelial cells that have not adhered are plated in a separate vessel. The primary culture now serves as a maintenance culture of specialized cells.
Proliferation and growth of mortal maintenance cultures depends on several factors including organ of origin, age of donor, and cell type. For instance, normal primary cultures of specialized cells, including those derived from adult rodent liver, may be kept for weeks, while cells derived from embryonic liver, neuronal, or lung epithelium are viable for months. An established culture always has a tendency to lose specifi c functions with time, but this process may be counteracted by culturing the specialized cells on MEF feeder lay-ers or by using serum-free medium. Isolation of primary cultures, however, requires repeated use of animals for extraction and establishment of explants, the costs of which are described in chapters 6 and 7 in Section II.
CRITERIA FOR IDENTIFICATION AND MONITORING OF CULTURED CELLS
Various criteria, cell markers, and indicators are used for the identifi cation and classifi cation of cells in culture. These methods are employed when a cell line is established and periodi-cally thereafter to monitor the genetic purity of the cells. The criteria are described below (section, “Procedures”).
Karyotypic Analysis
When a designated cell line is derived from normal tissue, the chromosome complement should be identical to the parent cell or the species of origin. In this way, the cell line is classifi ed as diploid (2n chromosomes). Any other designation would classify the cells as aneuploid or heteroploid. Continuous cell lines derived from tumors or transformed cells fall into the latter categories.
Aging in Culture
The life spans of cells in culture are measured according to the population doubling level (pdl). This calculation monitors the frequency of mitotic events characteristic of proliferating cells after their original establishment in primary culture. The general formula for assessing pdl is:
pdlf = 3.32 (log F − log I) + pdl
i
where pdlf represents the fi nal pdl at the time of trypsinization or at the end of a given subcul-
ture; F is the fi nal cell count; I is the initial cell number used to initiate the individual culture vessel at the beginning of the subculture; and pdl
i equals the doubling level of the cells used
to initiate the subculture.Cells with fi nite life spans generally show signs of aging such as loss of cell morphol-
ogy, decrease in rate of proliferation, increase in cytoplasmic lipid content, and other

CELL CULTURE METHODOLOGY 173
evidence of apoptosis. The amount of pdl exhibited by any particular fi nite cell line or primary culture depends largely on the age of the organ of origin. Thus the pdl is ascer-tained from the literature or determined empirically. Immortal continuous cell lines are capable of indefi nite multiplication in vitro, provided they are maintained under optimum conditions. Monitoring of the pdl is not always necessary with immortal cell lines.
Anchorage-Dependent Cultures
Some cells are capable of growing in suspension culture, while others require attachment to plastic. This plastic support may take the form of polyethylene or polypropylene treated so that its electrostatic properties allow isolated cells to attach and migrate. Other attach-ment matrices include components of the ECM such as laminin and fi bronectin. These components mimic the in vivo attachment substrata normally present in the epithelial lining and the interstitium, respectively. Thus, they may be selective for cells of epithelial or mes-enchymal origin. Still other support matrices use microporous fi lter membranes that both allow for cellular attachment and also permit passage of macromolecules through the mem-branes to the basolateral surface of the cell layer.
Contact Inhibition
As mortal continuous cells proliferate in a sparse culture environment, they migrate and occupy the available surface that is inundated with medium. As the surface becomes congested, the cells necessarily come into cell membrane-to-membrane contact. Although the monolayer is viable, proliferation eventually ceases and the cells exhibit contact inhi-bition characterized by arrest of the cell cycle in the G
0 phase. Figure 13.8 illustrates the
cell cycle exhibited by proliferating cells in vitro or in vivo.
G0
G2phase
G1phase
S phase
M phase
FIGURE 13.8 Four successive phases of cell cycle for proliferating cells in vitro or in vivo. Mortal proliferating somatic cells spend most of their existence in interphase. As a cell prepares to divide, it enters the G
1 phase lasting from several hours to days and characterized by synthesis of cell orga-
nelles and centriole replication (the G0 phase represents a resting stage or subphase of G
1). The
S phase begins when DNA synthesis starts and the chromosomes have replicated. The G2 phase starts
when DNA replication is complete and the content of the nucleus has doubled. The extended part of interphase then continues in the M phase with the stages of cell division: prophase, metaphase, ana-phase, and telophase. The M phase begins with mitosis (nuclear division) and ends with cytokinesis (cytoplasmic division). Cell division terminates with the completion of cytokinesis.

174 PRINCIPLES OF TOXICOLOGY TESTING
Cells that do not exhibit contact inhibition grow in multilayers, successively loosen-ing attached cells from the support as space for growth and proliferation becomes limited and competitive. Consequently, although confl uent multilayer cultures do not exhibit contact inhibition, cells are often discovered fl oating in the surrounding medium. Most continuous immortal cell lines do not succumb to growth restrictions demonstrated by mortal cells.
Other methods of cell classifi cation are not customarily performed but are incorpo-rated into mechanistic experiments. These include analysis of tissue-specifi c differenti-ated properties such as secretion of macromolecules or the presence of intracellular enzymes; ultrastructural identifi cation of structure–function activity; fl uorescent label-ing for identifi able structural proteins; reverse transcriptase polymerase chain reaction (RT-PCR) analysis for expression of gene-specifi c primers; and, the ability of the cells to form an invasive malignant tumor when injected into immunologically defi cient (nude) mice.
CULTURE REQUIREMENTS
MEDIA COMPONENTS
Cells are cultured in a liquid medium whose components are precisely controlled and moni-tored. The medium is composed of a buffered solution of physiological ion concentration containing soluble amino acids, carbohydrates, vitamins, minerals, fatty acids, and other cofactors. Optional ingredients include a pH indicator, separate buffering systems such as HEPES, and some non-essential amino acids that are incorporated when required by a par-ticular cell type. The formulas for the media are readily available (see websites listed in Suggested Readings) in soluble powdered or liquid formulas.
Some of the more commonly used media are modifi ed Eagle’s medium, basal medium Eagle, Dulbecco’s modifi ed Eagle’s medium (DMEM), and Ham’s F12 medium. These solutions are generally designed for use with serum or serum proteins with the addition of fetal or newborn mammalian serum as needed.
Serum is a complex mixture of poorly defi ned biological components consisting of soluble cell growth factors, transport proteins, hormones, essential trace metals, lipids, and ECM adhesion factors. Without the addition of a minimum percentage of serum (5–20%) as part of the formulation, cellular proliferation does not proceed favorably.
In cell culture, balanced salt solutions function as washing, irrigating, transporting, and diluting fl uids while maintaining intracellular and extracellular osmotic balance. In addition, the solutions provide cells with water and inorganic ions essential for normal cell metabolism and are formulated to provide a buffering system to maintain the liquid within the physiological pH range (7.2–7.6). The solutions are also combined with carbo-hydrates such as glucose, thus supplying an energy source for cell metabolism during extensive wash procedures. The most commonly used prepared salt solutions include Dul-becco’s phosphate buffered saline (PBS), Earle’s balanced salts, and Hanks’ balanced salt solution.
In addition to satisfying nutrient requirements, the cells must be placed in an environ-ment that mimics the in vivo situation. Table 13.2 summarizes the minimum conditions that support survival, maintenance, and proliferation of cells in culture. The parameters include temperature, pH, carbon dioxide tension, buffering, osmolality, humidifi cation, and the absence of microbial contamination (sterile technique). They vary only slightly, depending on the cell type.

CELL CULTURE METHODOLOGY 175
TEMPERATURE
For most mammalian cells, the optimum temperature for cell proliferation and differentia-tion is 37ºC. Cells withstand falls in temperature more easily than rises. In fact, temperatures as low as 4ºC may reduce metabolic activity but will not irreversibly inhibit biological func-tions. Temperatures as high as 39ºC may stimulate the formation of heat shock proteins, resulting in irreversible functional and ultrastructural damage and apoptotic events.
PH
The pH range that supports optimum cellular multiplication is 7.2–7.4, with some cell growth occurring at the extremes of 7.0 and 7.6. Some cells are more tolerant of slightly acidic conditions than corresponding basic conditions, and transformed cells are often more resistant to changes in pH than diploid cells. In addition, rapidly proliferating cells with high metabolic requirements eliminate excessive acidic waste material more quickly, result-ing in rapid falls in pH even in the presence of strong buffers. These types of cells often require more frequent media washes. In general, pH is readily monitored in an incubator as well as during routine manipulation with the addition of pH indicators (e.g., phenol red) in the growth media and salt solutions. The indicators do not interfere with culture stability.
CARBON DIOXIDE TENSION
Most culture media contain bicarbonate as part of the buffering system to prevent large and rapid changes in pH. At room temperature and standard incubator pressures, bicarbonate and carbonic acid are in equilibrium in solution at physiological pH. At 37ºC, however, soluble CO
2 is driven out of solution, thus disrupting the equilibrium established among bicarbonate,
carbonic acid, and CO2. This condition disturbs the critical balance in the concentrations of
carbonic acid and bicarbonate in the culture medium.Thus, balanced CO
2 tension and maintenance of pH are satisfi ed by maintaining an
increased partial pressure of CO2 in the gas phase above the liquid. Modern water-jacketed
CO2 incubators are designed to adequately maintain the gaseous CO
2 phase in a chamber at
TABLE 13.2General Minimum Requirements for Maintenance and Proliferation of Cultured Cells
Requirement Parameters
Culture conditions pH, buffered growth media, wash solutions, temperature, humidity, osmolality, ultrapure water
Technical Sterile technique, patienceEquipment Biological safety cabinet or laminar fl ow hood, CO
2 incubator, autoclave, water
purifi cation systemIn vitro medium
supplementationFBS, FCS, NCS, NHS, ECM components, MEFs, culture inserts, defi ned media
additives, pretreated culture vesselsSupplies Plastic culture wells, plates, fl asks, and other disposable suppliesOptional components pH indicators, HEPES buffer, NEAA, antibiotics, cocultures
Abbreviations: ECM, extracellular matrix; FBS, fetal bovine serum; FCS, fetal calf serum; MEFs, mouse embry-onic feeder cells; NCS, newborn calf serum; NEAA, non-essential amino acids; NHS, newborn horse serum.

176 PRINCIPLES OF TOXICOLOGY TESTING
controlled levels. Therefore, most culture media are formulated for use with 2–15% CO2 so
that the appropriate pH is maintained throughout the life cycles of the cells.
BUFFERING
When petri dishes, fl asks, or multiwell plates are removed from an incubator chamber, CO2
necessarily escapes from the chamber and from the warm liquid medium. This inevitably results in the inability of pH to be adequately maintained in the absence of the gas phase. This occurrence is not usually a problem when cell cultures are removed during washing steps with physiological salt solutions or during replacement of expended medium. When in vitro experiments necessitate long periods outside an incubator, however, a buffering system is required. Many laboratories routinely incorporate organic buffers such as HEPES in media formulations to prevent the rapid shift of pH when cultures are removed from a CO
2 incuba-
tor. Additionally, suffi cient sodium hydroxide or sodium bicarbonate is added to reach the working pH.
When a gas or vapor environment must be maintained without interruption, as in exper-iments using gaseous toxins, cells are grown in screw-capped tissue culture fl asks. The fl asks are then perfused with specially formulated custom gas mixtures separate from the incubator environment and sealed with screw caps, thus preventing escape of the gaseous phase. Although tedious, the method is useful when analyzing the effects of volatile organic liquids or gases in toxicology testing protocols.
OSMOLALITY
The optimum range of osmolality in a culture medium depends on the cell type, but it is generally between 250 and 325 milliosmoles per kilogram (mOsm/kg). The osmolality of a solution is standardized during the preparation of the powdered medium. Concerns arise during the time of incubation when the relative humidity in the chamber is not kept at or near saturation. When an incubator door is opened, dry air enters and cools the chamber, thus releasing humidifi ed air by evaporation. An incubator that does not provide for adequate heat transfer will remain cool. If the liquid medium in the cultures is at a higher temperature, the water in the medium evaporates, leaving a solution with a higher osmolality.
To prevent evaporation and to maintain the proper osmolality of a culture medium, rapid equilibration of heat throughout the chamber is necessary. Modern water-jacketed incubators are equipped with internal humidity control devices and fans that distribute heat. These chambers are precisely insulated, heated, and humidifi ed to prevent the formation of cool spots that may prevent the humidifi cation of the chamber. Alternatively, the chamber humidity is maintained by leaving an open trough of water (preferably with a large surface area) at the bottom of the instrument. In older generation incubators, adequate humidifi ca-tion is determined visually by the presence of condensation on the glass door of the incuba-tor, but no condensation should be formed on the culture dishes. Some incubators are equipped with heated inner doors that prevent condensation.
Humidifi cation is also monitored by measuring the osmolality of control media incu-bated in parallel to cell cultures. With evaporation, the osmolality increases, as compared with fresh medium at the start of a cell cycle, indicating that the humidity is inadequate.
WATER REQUIREMENT
A fundamental requirement of cell culture systems, and one that is often overlooked in the initial steps of establishing a cell culture laboratory, is the quality of the water used to prepare media and salt solutions. Ordinarily, distilled water or a single passage of water

CELL CULTURE METHODOLOGY 177
through a deionizing column is not suffi cient to remove all the impurities that remain from feed water. Contaminants in the water may take the form of trace metals, organics, variable amounts of divalent cations such as magnesium or calcium, and metabolic prod-ucts of microorganisms. Such contaminants interfere with cell growth and functional processes.
The impurities are essentially removed with systems that recirculate water through a series of deionizing and organic exchange columns. Most of these systems are equipped with digital sensing monitors that display the purity of the collected water in megaohms (mega- ). As ions and impurities are removed in the fi ltration columns, water resistance increases. A resistance measuring at least 10 mega- is suitable for culturing cells; the limit of purifi cation with many ion exchange columns is about 18 mega- . Some water purifying systems also yield water which is sterilized by gamma irradiation at the col-lecting end.
GLASSWARE REQUIREMENT
Another aspect of maintaining a cell culture laboratory at optimum effi ciency is the manner in which the glassware is handled, washed, and stored. All glassware designated for use in a cell culture area should not be used for storage of other chemicals or for experiments unre-lated to the growth of cells. Glassware is clearly marked for cell or tissue culture only and is washed in a detergent compatible with the objectives of the laboratory. Essentially, the deter-gent should leave no residue adhering to the pores of the glass. It should rinse easily with deionized water. The glassware is also stored separately from other equipment and materials kept in the laboratory.
SERUM-FREE MEDIUM
In investigations dedicated to understanding the infl uences of nanomaterials on growth and proliferation, mechanisms of toxicity, and actions of growth factors or hormones on cultured cells, the use of batch fetal or newborn serum is not acceptable. Serum is a complex and poorly characterized mixture, the composition of which varies according to the commercial lot. In addition, serum contains naturally occurring substances or microbiological contami-nants (e.g., mycoplasma, viruses, endotoxins, and prions) that may prove toxic or induce genetic or structural transformations for most cells.
Thus, the use of serum-free medium replaces serum in standard growth solutions by incorporating a mixture of synthetic or otherwise defi ned factors that substitute for essential culture requirements. The factors include hormones (insulin, transferrin), non-essential amino acids, trace metals (selenium, manganese, and zinc), growth factors (epi-dermal growth factor, fi broblast growth factor, and keratinocyte growth factor), and ECM components (iaminin and fi bronectin). Selective serum-free media facilitate adaptation of cells to the culture environment and allow for better standardization of experimental conditions.
A serum-free medium also facilitates the isolation of the desired cell type during pro-cedures for the establishment of primary cell cultures. Incorporation of serum-free medium as a culture requirement essentially eliminates the overgrowth of fi broblasts that usually proliferate rapidly in serum-supplemented media. Serum-free medium is individually pre-pared and adapted to each cell type. The advantages of integrating serum-free (or even low serum) media are well documented and have been described for use in maintenance cul-tures of many primary and specialized cells and for fi nite and continuous cell lines (see Suggested Readings and Review Articles at the end of this chapter).

178 PRINCIPLES OF TOXICOLOGY TESTING
PROCEDURES
STATIC AND PERFUSION CELL SYSTEMS
In general, culture conditions during an experiment or assay differ from those used during routine growth and maintenance of stock cultures. All stock cultures and most experiments are static cell systems that entertain a change of cell medium before or during subcultivation. Dur-ing normal incubation, depletion of nutrients, production of metabolic acids, and accumulation of excretion products occur in the medium, resulting in less favorable cell culture conditions.
Cell types that are anchorage dependent are cultured as monolayers on the bottom sur-face of a fl ask and covered with medium in addition to an overlying gas phase consisting of 2–15% CO
2 in air. Cells may also be anchored on fi lter inserts or polystyrene particles.
Rotating fl asks or tubes are used to increase surface area by providing contact of the sides and tops of the vessel with media, which also permits better oxygenation of cells. Some cell types (continuous cell lines) need not adhere to a substratum and may be cultured in mag-netically stirred suspension cultures or roller bottles.
Perfusion cultures are more advanced compared with static cell cultures because they allow for the uniform fl ow of fresh medium by continuous irrigation of the fl ask. These systems also use positive pressure pumps and micron fi lters to provide the cultures with oxygenated, waste-free medium. Although maintenance of these systems is fastidious, they may present some distinct advantages in metabolic toxicity testing studies. Note that most cell culture test systems are much less oxygenated than animal tissues and thus predomi-nantly permit anaerobic cell metabolism.
DISPERSION OF TISSUES
Many methods exist for the establishment of cell suspensions. The choice of protocol largely depends on the cell type desired and the experience of the investigator. Most of the com-monly used procedures for isolating primary cultures or continuous cell lines employ the same principles underlying tissue dispersion. Briefl y, the tissue is surgically extirpated and the cells are dispersed mechanically, chemically, and/or enzymatically.
Ordinarily, a typical protocol will use all these manipulations. A tissue is mechanically dissected, minced, or sheared and passed through nylon mesh or sterile gauze. Initial disper-sion of tissue is further completed with repetitive pipetting, magnetic stirring, or gentle vor-texing. Chemical dispersion involves the removal of calcium from the dispersant solution by the addition of chelators such as ethylenediamine tetraacetic acid (EDTA). Enzymatic dis-persion requires the use of trypsin, chymotrypsin, collagenase, deoxyribonuclease (DNAse), or pronase. Figure 13.9 illustrates a typical protocol for isolating cells from animals and outlines preparation of the animal for anesthesia and surgical manipulation, general procure-ment of the organ, preparation and mechanical treatment of the tissue, dispersal in an enzy-matic solution, and fi nal recovery and plating of the suspended cells in growth medium.
In general, this procedure is typical for isolating cells from any intact visceral organ or when obtaining biopsy specimens and should be performed in a biological safety cabinet. Explants are also established by placing individual dissected tissues in a drop of medium and allowing the specimens to adhere. Maintenance of aseptic conditions is essential for the establishment of successful cell cultures.
SUBCULTURING
Subculturing or passaging of cells involves the mechanical or enzymatic disruption of the cell monolayer, and dividing and transferring the cells to new culture vessels. The

CELL CULTURE METHODOLOGY 179
procedure is required when a cell population has occupied most or all of the available surface area in a vessel. At this density level, as explained earlier, confl uent normal dip-loid cells exhibit contact inhibition—that is, cell proliferation and migration slow or cease as cells come in contact with each other. Common methods used for the dispersion of cells include mechanical and enzymatic disruption.
Mechanical disruption involves the use of sterile silicone or plastic cell scrapers to dislodge the monolayer in aggregates. The method is not satisfactory for maintaining con-tinuous cultures whose cell membranes are sensitive to physical manipulation because the process often compresses or lyses cells. The technique, however, is often used when termi-nating experiments for preparatory analyses.
Dissect into 1mm3 pieces; stirwith trypsin, elastase, DNAse in
PBS × 20 min @ 37°C
Neutralize enzyme solution withDME-10; centrifuge cell
suspension; transfer to flasks;allow to settle using differential
adhesion
Anesthetize animal according tostandard IACUC protocol
Disinfect surface with 70%alcohol
Surgically remove organ intact(perfuse organ in situ withbuffer/enzyme solution if
necessary)
Lavage organ with cold buffersolution
Filter undigested tissue throughgauze; re-incubate with enzymes;
repeat twice
Incubate @ 37°C in 2-15% CO2and 100% humidity
FIGURE 13.9 Typical procedure for mechanical and enzymatic isolation of cells from intact animals. All procedures are performed at 4ºC using aseptic techniques. Abbreviations: IACUC, Institutional Animal Care and Use Committee; PBS, Dulbecco’s phosphate buffered saline; DME-10, Dulbecco’s modifi ed Eagle’s medium containing 10% fetal bovine serum (see section “Clonal Growth and Main-tenance Cultures”).

180 PRINCIPLES OF TOXICOLOGY TESTING
Enzymatic disruption uses proteolytic enzymes such as trypsin, collagenase, and/or DNAses that yield satisfactory cell numbers and afford less cell destruction. The enzyme solu-tion is formulated as 0.5–2.0% in Ca+2 and Mg+2-free phosphate-buffered balanced salt solution (PBS) and supplemented with a cation chelator (EDTA). The cells are not left in contact with the enzyme for more than the prescribed time (about 10 minutes) because enzymes are capable of damaging cell membranes. Termination of enzyme activity is accomplished by neutralizing the activity of the solution by the addition of a fresh medium containing serum. Figure 13.10 highlights the protocol generally used for enzymatic dispersion of cell monolayers.
After counting, the cell suspension is diluted and the cells are inoculated (seeded) into appropriate culture vessels. In most cases, cells are seeded at 104 cells/cm2 of surface area, a seeding density corresponding to about one-fourth to one-third the confl uent density, thus allowing for optimum cell proliferation. The newly seeded culture plates, wells, or fl asks are incubated undisturbed for several hours to facilitate cell attachment.
MEASUREMENT OF GROWTH AND VIABILITY
Some important considerations when determining the integrity of a cell line or newly estab-lished primary cultures rest upon the knowledge and experience with cell culture in general and the cell type in particular. Overall, several criteria are assessed to determine the potential utility of the cells for future toxicology investigations including the following:
1. The viability of the cultures and appearance of the cells under a phase contrast (PC) microscope, especially as a fi nite cell line approaches the anticipated maximum number of doublings.
Confluent monolayer rinsed twicewith PBS
Rinse with 1:10 trypsin-EDTA*solution × 1min
Aspirate, discard; incubate withtrypsin-EDTA solution × 10 min at
37°C
Examine cells
Neutralize with equal amount ofDME-10
Pellet, resuspend, count cells, adjust volume of suspension to seed new culture vessels at 104
cells/cm2
FIGURE 13.10 Outline of general protocol used for enzymatic dispersion of cell monolayers. *1:10 dilution of 10× trypsin–EDTA concentrate in PBS. Abbreviations: DME-10, Dulbecco’s modifi ed Eagle’s medium containing 10% fetal bovine serum; PBS, phosphate buffered saline.

CELL CULTURE METHODOLOGY 181
2. The ability of the cells to retain as many of the morphological and biochemical features as the original parent cell or primary culture. Most of these features are discussed in subsequent chapters since toxicological evaluations depend on the retention of these features.
More common criteria used to monitor cell proliferation and viability routinely include cell counting (as described earlier); the exclusion of the vital trypan blue dye; the ability to sur-vive or proliferate under normal culture conditions; and the percentage of cells inoculated into the culture vessel that will attach to the culture surface. Each procedure is performed as part of the routine monitoring of culture status. Most of these features are determined under the PC microscope, assuming that the individual responsible for the cultures has a familiarity with the cells in question. Figure 13.11 outlines the method of vital dye exclusion.
CRYOPRESERVATION
Most cell lines are frozen indefi nitely and later thawed for continued cultivation. Frozen stock cultures are necessary prerequisites for standardized in vitro toxicology testing where frozen stocks of the same passage of a cell line are maintained independent of time and place. Depending on the extent of shared use and availability to other investigators, the pro-curement of an ultralow temperature freezer is useful. These units maintain temperatures just below the critical point of ice formation (–130ºC) and are equipped with alarms and backup power units in the event of electrical power failure. For independent laboratories, a liquid nitrogen (LN
2) refrigerator is convenient but requires a standing order for the liquid due to
constant evaporation.Cell lines and primary cultures are cryopreserved when the optimal conditions for the
culture are attained. The procedure for subculturing of monolayers is launched as described and outlined in Figure 13.12. Monolayers are dispersed by immersing in a buffered trypsin
Select representative vesselsfrom cultures
Rinse and preserve in 70%ethanol or isopropanol × 10 min
Add 1% trypan blue/70% alcoholsolution × 10 min
Examine cells
Rinse with PBS × 3
Count an aliquot and calculatepercentage of cells stained
FIGURE 13.11 Outline of protocol using trypan blue vital dye exclusion method. Abbreviation: PBS, phosphate buffered saline.

182 PRINCIPLES OF TOXICOLOGY TESTING
solution, neutralized with a growth medium containing serum (complete growth medium), and centrifuged (400× g for 10 minutes). The supernatant is discarded and the pellet is resuspended in complete growth medium containing an antifreeze such as dimethylsulfox-ide (DMSO) or glycerin. Cells are resuspended in DME-10 plus 7.5% DMSO at a concen-tration of 1 to 3 × 106 cells/mL in sterile 2-mL screw-capped cryoampules. The ampules are labeled and slowly frozen at a controlled rate of –1ºC/min in an automated freezing cham-ber. These chambers are usually available as accessory units accompanying liquid nitrogen refrigerators. Alternatively, the ampules are frozen in LN
2 vapors by placing them in a
styrofoam container and positioning it in the neck of the LN2 refrigerator overnight. It is
important to ensure that the level of the nitrogen is not so high as it will freeze the cells too rapidly. The next day, the ampules are snapped into cryocanes and immersed in the LN
2.
Adequate cell stocks of each frozen passage are maintained and a representative vial of cells is thawed and cultured soon after freezing to evaluate the freezing process. This ensures against the loss of a line should contamination or another unanticipated event occurs.
Rinse with 1:10 trypsin-EDTA*solution × 1 min
Confluent monolayers rinsedtwice with PBS
Aspirate, discard; incubate withtrypsin-EDTA solution × 10 min
at 37°C
Examine cells
Neutralize with equal amount ofDME-10
Pellet, resuspend, adjust volumeof suspension to count cells
Centrifuge again and resuspendin DME-10 plus 7.5% DMSO;
adjust to a cell concentration of1 to 3 × 106 cells/ml-vial
Freeze in sterile, 2ml, screw-capped cryoampules at a
controlled rate of –1°C/min
FIGURE 13.12 Outline of protocol for freezing cell lines. DME-10 = Dulbecco’s modifi ed Eagle’s medium containing 10% fetal bovine serum. *1:10 dilution of 10× trypsin–EDTA concentrate in PBS. Abbreviations: DMSO, dimethyl sulfoxide; PBS, phosphate buffered saline.

CELL CULTURE METHODOLOGY 183
An ampule is thawed rapidly by agitating in water at 37°C for not more than 3–4 minutes and then swabbed with 70% alcohol, allowed to dry, and the contents are dispensed into warm growth medium using a sterile plugged Pasteur pipette. For most cell types, the cul-ture may be left undisturbed in the incubator in the appropriate atmosphere overnight, after which the medium is replaced.
CELL IDENTIFICATION
In a laboratory that is accustomed to handling several cell lines of many individuals, it is necessary to verify a cell line once initiated, especially when in the presence of risk of cross contamination of cell lines. Some methods used for identifi cation of characteristic cell mark-ers include labeling with fl uorescent antibodies, cytogenetic karyotypic studies, isoenzyme profi les, and expression of gene primers using RT-PCR. It is also occasionally necessary to confi rm the absence of mycoplasma infections of cultures. This microbial contamination is described as bacterium that lacks a cell wall, tenaciously adheres to cell membranes, and is not eliminated by routine addition of antibiotics to the media.
In fact, the indiscriminate addition of antibiotics to growth and saline-buffered wash media is responsible for routine mycoplasma contamination. The presence of mycoplasma is easily confi rmed with the availability of commercial RT-PCR kits containing primers for gene expression of mycoplasma-specifi c sequences (see web sites listed in Suggested Readings at the end of this chapter).
CULTURE OF MOUSE EMBRYONIC STEM CELLS AND MOUSE EMBRYONIC FEEDER LAYERS IN TISSUE CULTURE
Preparation of Mitomycin-C Treated Mouse Embryonic Fibroblasts (3T3) Feeder Layers
Mouse embryonic fibroblasts (MEFs, 3T3 Swiss mouse fi broblasts, CCL-92, ATCC) are seeded at 104 cells/cm2 in T-75 fl asks and grown in Dulbecco’s modifi ed Eagle’s medium supplemented with 10% calf bovine serum (DMEM-10), 4.5 g/L glucose, 1.5% g/L sodium bicarbonate, 4 mM glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin in an atmo-sphere of 5% CO
2 in air, according to general cell culture procedures described previously
and the product information sheet available for these cells (CCL-92; ATCC). When mono-layers are 70% confl uent, cells are treated with 10 μg/mL mitomycin-C in DMEM-10 for 2 hours at 37°C. The plate is washed and passaged with trypsin/EDTA. Pellets are replated for immediate use or allowed to attach overnight at 1:3 dilution in T-75 fl asks. Alternatively, several T-75 fl asks containing 70% confl uent monolayers are treated with 10 μg/mL mito-mycin as above, and prepared for immediate use.
Culturing and Subculturing Mouse Embryonic Stem Cells
ES-D3 (CRL-1934, ATCC) are derived from blastocysts of a 129S2/SvPas mouse. The cells spontaneously differentiate into embryonic structures in the absence of a feeder layer or conditioned medium. They are maintained in the undifferentiated state by frequent subcul-ture (every 2–3 days) on confl uent irradiated (12,000 rads) or mitomycin-C treated feeder layers, as described. ES-D3 cells are propagated in DMEM containing 2 mM L-alanine-L-glutamine, 1000 U/mL LIF (recombinant human LIF), a lymphoid factor that promotes long-term maintenance of mouse embryonic stem (mES) cells by suppressing spontaneous differentiation). The medium is adjusted to contain 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, and supplemented with 0.1 mM 2-mercaptoethanol, 0.1% non-essential amino

184 PRINCIPLES OF TOXICOLOGY TESTING
acids, and 15% fetal bovine serum (ES-DMEM). Mitomycin C-treated feeder layers are seeded as a monolayer at approximately 1.5–2.0 x 106 cells/T-75 fl ask at least one day before plating the cells. ES-D3 cells are transferred to a 15 mL centrifuge tube containing 5 mL complete ES-DMEM and added to the fl ask containing feeder cells. The fl ask is incubated at 37°C in a humidifi ed 5% CO
2/95% air incubator. Medium change is performed every day,
and cells are passaged every 2 days.For subculturing, the volumes used in this protocol are for T-75 fl asks. A split ratio of
1:4 to 1:7 every 1–2 days is recommended. Plating densities range from 3 to 4 × 106 cells/T-75. If the colonies are close to or touching each other, then the culture is overgrown, resulting in differentiation. Enough fl asks with MEFs are prepared as noted above. The medium is aspirated from the fl ask with the cells, washed with PBS (Ca+2/Mg+2-free), and trypsinized. After about 1 minute, ES colonies preferentially dissociate and detach from the fl ask. Cells are dislodged, centrifuged, and resuspended in 50 mL fresh ES-DMEM, depending on the split ratio. Medium from 4–7 feeder layer fl asks are aspirated and replaced with 15 mL/fl ask of mES cell suspension. Cultures are incubated at 37°C in a humidifi ed 5% CO
2/95% air incubator. Medium is changed every day. Stock cultures for plating are
maintained in the undifferentiated state by propagating them on MEFs and in the presence of LIF.
Induction of Differentiation in mES Cells
When mES cells are allowed to differentiate in suspension culture, they form spherical mul-ticellular aggregates, or EBs, that contain a variety of cell types. Limited evidence exists for the differentiation of mES cells into particular populations, such as hematopoietic cells, cardiomyocytes, smooth muscle cells, and neurons, in an EB environment. Morphological observation of mES cells is used primarily to determine differentiation in cultures as follows:
Using PC microscopy6, undifferentiated cultures are distinguished from differentiated populations, as well as from underlying MEFs. ES-D3 cells form aggregates with 4–10 cell layer thickness over the feeder layer. The aggregates are not round and are poorly defi ned. Electron microscopy (EM) can also be used to confi rm PC observations. EM examination generally reveals undifferentiated D3 cells with a large nucleus-to-cytoplasm ratio and numerous ribosomes. In general, undifferentiated mES cell colonies have clearly polarized structures with numerous villi, many apoptotic bodies, and autophagosomes facing the feeding medium. Tight junctions and gap junctions are present in mES cell lines, especially close to the surface. Highly organized intermediate fi lamentous structures, such as neuro-fi laments, and small aggregates of microtubular bundles on the perimeter of the nuclei are suggestive of differentiation.
Detection of Expression Markers of Differentiation in mES Cells by RT-PCR
Several markers are used that can distinguish undifferentiated mES cells from their differen-tial progeny. These include markers for the presence of SSEA antigens, OCT3/4, SOX-2 REX-1, and TERF. In addition, trophectoderm and germ layer markers are negative indica-tors that confi rm absence of differentiation.
Total RNA is isolated from cell pellets using any protocol for total RNA (RNAeasy Mini® from Qiagen, Hilden, Germany is suitable), and cDNA is synthesized using 100 ng–1 μg of total RNA in a 20-μL reaction. Superscript II (from Invitrogen, Inc., Carlsbad,
6 Phase contrast (PC) microscopic inspection is routinely performed with an inverted PC microscope equipped with at least a 5.0 megapixel microscope camera and processed with advanced photographic software.

CELL CULTURE METHODOLOGY 185
California, USA), a modifi ed Maloney murine leukemia virus RT, and Oligo(dT) 12–18 primers, are used according to the manufacturer’s instructions. Aliquots of cDNA, equivalent to 1/20 of the above reaction, are used in a 20-μL reaction volume. PCR ampli-fi cation of different genes is performed using DNA polymerase. PCR reactions are run for 35 cycles (94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute) and incubated at 72°C for 10 minutes at the end to ensure complete extension. PCR products are subjected to electrophoresis in 4–6% TBE (Tris buffer) polyacrylamide gels containing 0.5 μg/mL ethidium bromide. One hundred base pairs (bp) DNA ladder is used to estimate size of the amplifi ed bands. Forward and reverse primers are used for RT-PCR for selected gene expression. Several other markers exist which can be tested as negative control markers (see Ginis et al. 2004, Review Articles).
LARGE SCALE BIOPROCESSES
Large scale engineering efforts in mammalian cell culture have made it possible to produce therapeutic biologics including vaccines, ECM components, skin and tissue grafts for surgi-cal manipulations and wound healing, antibodies and hybridomas, diagnostics, and other products for research and clinical applications. These industrial cell culture methods have improved productivity of metabolically engineered cells, increasing the properties favorable for large-scale bioprocessing.
The fundamental physiological basis for cell engineering involves a set of characteris-tics that select for favorable growth and proliferation and for properties of cellular mecha-nisms including metabolism, protein processing, and pathways of apoptosis. Some cell culture engineering developments incorporate enhanced process effi ciency procedures for large scale cultures of animal cells such as bioreactor processing, feedback culture, and dynamic nutrient feeding. These principles and methods are further described in articles and books listed in the Suggested Readings and Review Articles sections at the end of this chapter and subsequent chapters.
SOURCES OF CELL LINES
Premier sources of cell lines, cell strains, and other organisms for cell biology and genetics studies are certifi ed cell banks such as the American Type Culture Collection (ATCC, Manas-sas, Virginia, USA), and the European Collection of Cell Cultures (ECACC, Porton Down, Wiltshire, UK). These facilities operate as nonprofi t institutions and stock thousands of cell types. The ATCC classifi es cultures as certifi ed cell lines (CCLs) or cell repository lines. More information is available about CCLs: donor, karyotypic analysis, expected population doublings, cell structural features, and enzyme and gene markers. In addition, both cell banks house repositories for viruses, fungi, bacteria, genetic probes, and vectors. Other cell repositories and federal registries are noted below.
SUGGESTED READINGS
American Type Culture Collection (ATCC), Manassas, VA. [Available from: http://www.atcc.org] [Last accessed August 2012].
Andrei G. Three-dimensional culture models for human viral diseases and antiviral drug develop-ment. Antiviral Res 2006; 71: 96.
Drab M, Haller H, Bychkov R, et al. From totipotent embryonic stem cells to spontaneously contract-ing smooth muscle cells: a retinoic acid and db-cAMP in vitro differentiation model. FASEB J 1997; 11: 905.
El-Ali J, Sorger PK, Jensen KF. Cells on chips. Nature 2006; 442: 403.

186 PRINCIPLES OF TOXICOLOGY TESTING
European Collection of Cell Cultures (ECACC), Porton Down, Wiltshire, U.K. [Available from: http://www.ecacc.org.uk] [Last accessed September 2012].
Hay RJ. Human cells and cell cultures: availability, authentication and future prospects. Hum Cell 1996; 9: 143.
Klug MG, Soonpaa MH, Koh GY, Field LJ. Genetically selected cardiomyocytes from differentiating embryonic stem cells form stable intracardiac grafts. J Clin Invest 1996; 98: 216.
McDonald JW, Liu XZ, Qu Y. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med 1999; 12: 1410.
National Disease Research Interchange (NDRI), Philadelphia. [Available from: http://www.ndriresource.org]
National Institutes of Health, Human Embryonic Stem Cell Registry, Bethesda, MD. [Available from: http://stemcells.nih.gov/research/registry]
Potocnik AJ, Kohler H, Eichmann K. Hemato-lymphoid in vivo reconstitution potential of subpopu-lations derived from in vitro differentiated embryonic stem cells. Proc Natl Acad Sci USA 1997; 94: 10295.
Seth G, Hossler P, Yee JC, Hu WS. Engineering cells for cell culture bioprocessing: physiological fundamentals. Adv Biochem Eng Biotechnol 2006; 101: 119.
Williams DJ, Sebastine IM. Tissue engineering and regenerative medicine: manufacturing chal-lenges. IEE Proc Nanobiotechnol 2005; 152: 207.
Wlaschin KF, Hu WS. Feedbatch culture and dynamic nutrient feeding. Adv Biochem Eng Biotech-nol 2006; 101: 43.
Yamada T, Yoshikawa M, Takaki M, et al. In vitro functional gut-like organ formation from mouse embryonic stem cells. Stem Cells 2002; 20: 41.
REVIEW ARTICLES
Alonso L, Fuchs E. Stem cells of the skin epithelium. Proc Natl Acad Sci USA 2003; 100: 11830.Bradley A, Evans M, Kaufman M, Robertson E. Formation of germ-line chimaeras from embryo-
derived teratocarcinoma cell lines. Nature 1984; 309: 255.Butler M. Animal cell cultures: recent achievements and perspectives in the production of biophar-
maceuticals. Appl Microbiol Biotechnol 2005; 68: 283.Evans M, Kaufman M. Establishment in culture of pluripotential cells from mouse embryos. Nature
1981; 92: 154.Even MS, Sandusky CB, Barnard ND. Serum-free hybridoma culture: ethical, scientifi c and safety
considerations. Trends Biotechnol 2006; 24: 105.Findikli N, Candan NZ, Kahraman S. Human embryonic stem cell culture: current limitations and
novel strategies. Reprod Biomed Online 2006; 13: 581.Garner JP. Tissue engineering in surgery. Surgeon 2004; 2: 70.Ginis I, Luo Y, Miura T, et al. Differences between human and mouse embryonic stem cells. Dev Biol
2004; 269: 360.Kim JB. Three-dimensional tissue culture models in cancer biology. Semin Cancer Biol 2005; 15: 365.Kuwahara M, Ogaeri T, Matsuura R, et al. In vitro organogenesis of gut-like structures from mouse
embryonic stem cells. Neurogastroenterol Motil 2004; 16: 14.Mackenzie IC. Stem cell properties and epithelial malignancies. Eur J Cancer 2006; 42: 1204.Rosolowsky M, McKee R, Nichols W, Garfi nkle B. Chromosomal characterization of MRC-5 cell
banks utilizing G-banding technique. Dev Biol Stand 1998; 93: 109.Thorgeirsson SS, Grisham JW. Overview of recent experimental studies on liver stem cells. Semin
Liver Dis 2003; 23: 303.Turksen K, Troy TC. Human embryonic stem cells: isolation, maintenance, and differentiation.
Methods Mol Biol 2006; 331: 1.Verlinsky Y, Strelchenko N, Kukharenko V, et al. Repository of human embryonic stem cell lines and
development of individual specifi c lines using stembrid technology. Reprod Biomed Online 2006; 13: 547.

187
14 Cell culture methods for acute toxicology testing
INTRODUCTION
Data obtained from animal experiments yield information pertaining to the dose for lethal or sublethal toxicity that corresponds to many different general toxic mechanisms and effects. Similarly, in vitro cell systems also detect a wide spectrum of unspecifi ed mechanisms and effects. In contrast to animal experiments, however, all cell tests of acute toxicity in current use measure the concentration of a substance that interferes with or alters components, struc-tures, or biochemical pathways within cells.
This range of injury is further specifi ed by the length of exposure to the chemical (incu-bation time), thus allowing a test to be predictive for risks associated with toxic effects in vivo for those doses of the tested substance. This assumes that similar concentrations are measured in corresponding human tissue for comparable exposure periods. When undif-ferentiated (or dedifferentiated) cell lines are used for acute testing, the results are limited to determining the risk for the basal cytotoxicology1 of the measured concentrations. Con-versely, the use of primary cultures of differentiated cells (such as those of liver, lung, heart, or kidney) yields information capable of predicting cellular toxicity for each organ.
TESTING PRINCIPLES
Cell tests of acute or chronic2 toxicity used to mimic local or systemic effects are different from cell tests of mutagenicity and carcinogenicity. The latter have the ability to suggest a known underlying mechanism such as mutation, transformation, initiation, promotion, or progression based on bacterial cell responses to a chemical. They are designated, therefore, as short-term tests and are applied to the prediction of mutagenicity or carcinogenicity of chemicals in humans or animals.
In comparison, assessing the potential carcinogenicity or mutagenicity of a chemical retrospectively in humans normally takes much longer and is plagued with clinical and ethical considerations. Acute cytotoxicology tests instead simulate injuries from tested sub-stances caused by a number of incomplete mechanisms during periods of exposure that are realistic for acute toxicity. This allows for direct extrapolation of the data from the quantita-tive test to the analogous in vivo situation. Thus, although cytotoxicology tests may be acute, they are not short-term tests. In addition, they are less expensive and time consuming in comparison with animal tests.
1 Effects of a chemical on fundamental processes that involve structures and functions common to all mammalian cells including membranes, mitochondria, and chromosomes, among other organelles. 2 Terminology used in describing in vitro cell culture models, such as acute or chronic, has replaced the traditional terms of general toxicity.

188 PRINCIPLES OF TOXICOLOGY TESTING
There are similarities between short-term mutagenicity tests and cytotoxicology tests in cell culture. The latter tests are performed using a few standardized techniques whose relevance to human toxicity is pertinent. Also, the concentrations ultimately correspond to human toxic or lethal blood concentrations, whether acute, chronic, systemic, or local. For mutagenicity tests, the relevance applies to the mechanisms that are appropriate for humans.
Historically, cell cultures are employed in mechanistic studies identifying the underly-ing mechanisms of toxicity or action in response to exposure of cultured cells to low con-centrations of chemicals or drugs. Because the development of alternative models for cytotoxicology testing is newer, the protocols are often compared with those of mechanistic studies. As a result, some differences between studies of toxic mechanisms in cell cultures and cytotoxicology testing have prompted interesting discussions. In mechanistic studies for example, the parameters and systems vary, depending on the desired information, such that empirical and statistical relevance of the system is secondary to understanding the mechanism of toxicity. With cell methods for acute testing, however, rigorous interlabora-tory evaluation prior to standardization proposals is essential and the procedures have well-defi ned criteria for toxic assessment such as standards for cell viability and cell function. In addition, the protocols are exacting so as to ensure minimal variability.
CRITERIA
Many methods used for assessing systemic and local toxicity are available, some of which are derived from traditional mechanistic models. Their validity was usually established as a result of frequent use in one laboratory, followed by repetition and confi rmation by others. This method of evaluation thus proceeds more as a matter of convenience for a laboratory, particularly if the assay is easily accommodated to routine testing facilities. The methods are necessarily similar; cells are exposed to different concentrations of a test substance for a specifi c time period, after which the degree of inhibition of viability or functional parameter is measured. These criteria for standards of measurement represent the toxic end point. The most common criteria are listed in Table 14.1.
Some indicators of toxicity detect the net effects of different chemicals and have the advantage of demonstrating a toxic end point that is common to many types of mecha-nisms. Such indicators include damage to cell membranes or loss of viability—generally
TABLE 14.1Basal Cytotoxic Criteria for Assessment of Acute Systemic or Local Toxicity in Cultured Cells
CriteriaMethods Used to Determine Acute Toxicity
in Cultured Cells
Cell culture characteristics Plating effi ciency, PDL, loss of phenotypic appearanceMorphology Phase contrast microscopic analysisProliferation Cell counting, mitotic frequency, DNA synthesisMetabolism Glycolysis, protein and lipid synthesis, enzyme activityCytoplasmic membrane Leakage of markers, retention of dyesMitochondria Mitochondrial integrity, cell viability (MTT assay)Lysosomes Uptake of dyes (NRU assay)Chromosomal evaluation Karyotypic analysis, SCE
Abbreviations: MTT, mitochondrial reduction assay; NRU, neutral red uptake; PDL, population doubling level; SCE, sister chromatid exchange. See text for description of assays.
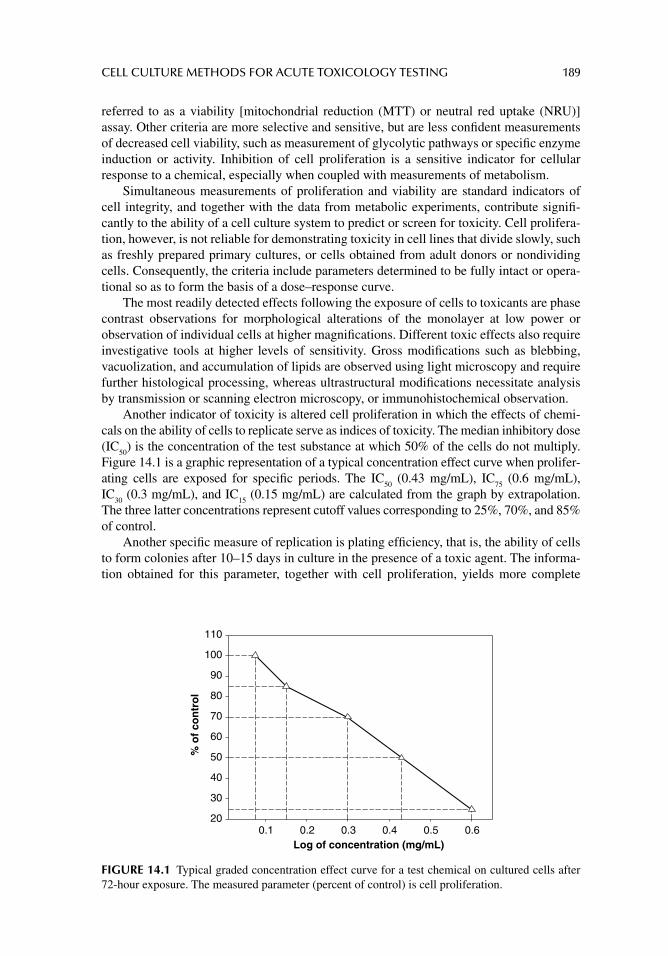
CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 189
referred to as a viability [mitochondrial reduction (MTT) or neutral red uptake (NRU)] assay. Other criteria are more selective and sensitive, but are less confi dent measurements of decreased cell viability, such as measurement of glycolytic pathways or specifi c enzyme induction or activity. Inhibition of cell proliferation is a sensitive indicator for cellular response to a chemical, especially when coupled with measurements of metabolism.
Simultaneous measurements of proliferation and viability are standard indicators of cell integrity, and together with the data from metabolic experiments, contribute signifi -cantly to the ability of a cell culture system to predict or screen for toxicity. Cell prolifera-tion, however, is not reliable for demonstrating toxicity in cell lines that divide slowly, such as freshly prepared primary cultures, or cells obtained from adult donors or nondividing cells. Consequently, the criteria include parameters determined to be fully intact or opera-tional so as to form the basis of a dose–response curve.
The most readily detected effects following the exposure of cells to toxicants are phase contrast observations for morphological alterations of the monolayer at low power or observation of individual cells at higher magnifi cations. Different toxic effects also require investigative tools at higher levels of sensitivity. Gross modifi cations such as blebbing, vacuolization, and accumulation of lipids are observed using light microscopy and require further histological processing, whereas ultrastructural modifi cations necessitate analysis by transmission or scanning electron microscopy, or immunohistochemical observation.
Another indicator of toxicity is altered cell proliferation in which the effects of chemi-cals on the ability of cells to replicate serve as indices of toxicity. The median inhibitory dose (IC
50) is the concentration of the test substance at which 50% of the cells do not multiply.
Figure 14.1 is a graphic representation of a typical concentration effect curve when prolifer-ating cells are exposed for specifi c periods. The IC
50 (0.43 mg/mL), IC
75 (0.6 mg/mL),
IC30
(0.3 mg/mL), and IC15
(0.15 mg/mL) are calculated from the graph by extrapolation. The three latter concentrations represent cutoff values corresponding to 25%, 70%, and 85% of control.
Another specifi c measure of replication is plating effi ciency, that is, the ability of cells to form colonies after 10–15 days in culture in the presence of a toxic agent. The informa-tion obtained for this parameter, together with cell proliferation, yields more complete
Log of concentration (mg/mL)0.1 0.2 0.3 0.4 0.5 0.6
% o
f co
ntr
ol
20
30
40
50
60
70
80
90
100
110
FIGURE 14.1 Typical graded concentration effect curve for a test chemical on cultured cells after 72-hour exposure. The measured parameter (percent of control) is cell proliferation.

190 PRINCIPLES OF TOXICOLOGY TESTING
information on cell survival, replication, and recovery from toxic insult. Cell proliferation is measured using several methods including cell counting, DNA content, protein content, and enzyme activity. The assays for DNA content using biochemical methods and monitor-ing the incorporation of radio-labeled precursors into DNA represent two different methods supplying similar information.
Another general index of toxicity is cell viability. This end point is measured by using vital stains such as trypan blue, which enters only compromised membranes of dead cells, and neutral red uptake, which is actively absorbed by living cells. The latter is commonly used for biomaterial testing in the agar overlay method and in cell tests for acute systemic toxicity. Counts of dead and vital cells are compared with controls and provide indices of lethality of the test compound.
Cells derived from different organs or tissues that retain some specialized functions in vitro or maintain specialized structures are also widely used in in vitro toxicology. For cells that retain differentiated features, effects on more specialized functions and structures are monitored in addition to effects on fundamental processes, including the measurements for specifi c end products, metabolic pathways, and membrane integrity. Table 14.2 lists some general applications of specifi c end points used for primary and continuous cells that have retained some features from the organ of origin.
Because some cell systems do not possess effi cient metabolic activities or have dedif-ferentiated3 with continuous passage in vitro, testing in culture may require some form of metabolic activation usually performed in one of three ways:
1. The addition of S9 fractions from rat liver. Mixed function oxidases are induced in cultured cells by treating animals with phenobarbital or β-naphthofl avone before isolation of primary cultures.
2. Preincubation of the test substance with a primary hepatocyte culture and addition of preincubated medium to the test culture.
3. Co-culture of target cells with hepatocytes in the presence of the test substance.
In addition to the differences in toxic criteria, cell culture methods incorporate different incubation and exposure times and require varying media volumes, cell densities, serum concentrations, and gas phases. Most experiments are performed in 6-, 12-, 24-, 96-, and 384-well microtiter plates (384-well plate has a capacity of 0.1 mL/well). This allows the use of microliter quantities of the test substances and permits easier automation of procedures. The test solution is diluted in serial or logarithmic steps using a microdiluter and many cul-tures are analyzed simultaneously with plate scanners with spectrophotometric, fl uorescent, or luminescent capabilities (Fig. 14.2).
One critical variable during the course of an experiment is exposure time. As with in vivo studies, longer exposure times require lower doses of a chemical than the concentra-tions needed to cause acute toxicity. Toxicity for many substances therefore may increase 10- to 100-fold if the toxic end point is measured after 96 hours rather than after 24 hours.
Other factors that alter toxic concentrations include serum components in the medium and whether the cultures are proliferating or static. For instance, high serum concentrations generally decrease toxicity of substances that are protein bound. Confl uent monolayers with established intercellular contacts show more resistance to otherwise comparable toxic con-centrations than cells that are in the logarithmic phase of proliferation or in primary cultures.
3 Mortal or immortal cells maintained in continuous culture through several passages lose some of their original genotypic and phenotypic characteristics while retaining organ-specifi c functions.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 191
The physicochemical properties of test compounds infl uence exposure conditions and the concentrations of toxic agents in the culture medium. The medium is usually an aque-ous salt solution containing various soluble additives. Only hydrophilic test compounds are completely solubilized. For volatile toxic agents, special incubation steps are necessary to ensure a constant exposure with time, provided the partition coeffi cients and solubilities are known. Similar problems apply to hydrophobic test compounds and mineral particulates. Lipophilic substances are generally insoluble in water solutions but are solubilized with ethanol, acetone, or dimethylsulfoxide (DMSO) before addition to the medium. Under these conditions, an additional control group receiving the carrier solvent alone is usually included in a study. These practical considerations are further discussed in chapter 18.
EXPERIMENTAL ASSAYS
VIABILITY (CYTOTOXICITY) ASSAYS
In order to increase comparability of results and optimize testing procedures, standardization and adherence to the experimental protocols are desirable. The use of well-characterized cell
TABLE 14.2Toxicology Indicators or Biomarkers for Cells Retaining Some Differentiated Features in Culture
Descriptive Method Examples of Indicators Monitored
Synthesis, release, or incorporation of specifi c molecules
Protein, carbohydrate, and lipid synthesis, metabolic end-products
Synthesis, release, or activity of specifi c enzymes Enzyme release or activityInteractions of chemicals with cells Intracellular accumulation, phagocytosis, mitogenic
responseAlterations of metabolic pathways Effect on carbohydrate and lipid energy storage or
utilizationCell surface activities Receptor binding, membrane polarization, chemotactic
response
Incubate withmedium + test
chemical
Add 10 μL MTTstock solution
(5 mg/mL in PBS)
+24–96 hr Last 1–2 hr ofincubation
Aspirate, rinsewith PBS, add
DMSO
Shake × 5 min
Read absorbanceat 550 nm
FIGURE 14.2 Outline of MTT assay procedure. Cells are incubated with increasing concentrations of test chemical (gray fi lled circles) plus blanks (clear, fi lled circles, no cells). Abbreviations: DMSO, dimethylsulfoxide; MTT, methylthiazol tetrazolium salt; PBS, phosphate buffered saline. Photo shows BioTek FLx800 Multidetection Microplate Reader®. Source: Courtesy of www.biotek.com.

192 PRINCIPLES OF TOXICOLOGY TESTING
lines, possibly of human origin, is important. Specifi c toxicity end points and the most suit-able assay methods are uniformly incorporated into cytotoxicology screens. Cell cultures are examined periodically for possible contamination with microorganisms, for cross contami-nation with other mammalian cell types, and for genetic integrity.
Adequate documentation provides details of exposure conditions and information on the purity and source of a test compound. Moreover, measurement of the concentration of the compound at the beginning and at the end of the experiment, particularly for insoluble or volatile substances, facilitates interlaboratory comparisons of results. Two frequently used microtiter methods, the NRU and MTT assays, are described below. Both are suitable for rapid screening of proliferation and viability in primary and continuous cell lines.
Trypan Blue Exclusion Test of Cell Viability
The trypan blue exclusion test is used to determine the number of viable cells present in a cell population. The test presumes that live cells possess intact cell membranes that exclude certain high-molecular-weight dyes, such as trypan blue, eosin, or propidium. Dead or nonviable cells do not exclude the dyes. In this protocol, the dye is applied to a monolayer of cells in culture or mixed with a cell suspension. The specimen is then visu-ally examined under phase contrast or light microscopy for the presence or absence of the dye. In general, viable cells do not endocytose the dye and have actually excluded the chemical. Therefore, these cells display a clear cytoplasm. Nonviable cells that have lost cell membrane integrity are not capable of excluding the dye. Thus the chemical perme-ates the cytoplasm and the dye has not been excluded; thus the cells appear with a blue cytoplasm.
Figure 14.3 outlines the steps involved in the protocol for trypan blue exclusion, as detailed below. The procedure can be performed on cell culture monolayers or with a cell suspension. The latter is handled as if the cell monolayer has been trypsinized4 from the culture plate. The monolayer is fi rst rinsed with enough phosphate buffered saline (PBS) to cover the monolayer and performed three times. The PBS is aspirated after each wash.5 The monolayer is then covered with 0.4% trypan blue solution in PBS, and either 70% ethanol, or 70% isopropanol. The culture is allowed to incubate for 3 minutes at room temperature. Cells are trypsinized (as described in the section, “Subculturing” of chapter 13) and counted within 3–5 minutes of mixing with trypan blue—longer incubation periods result in cell death and reduce viability. A drop of the trypan blue/cell suspension is placed on a hema-cytometer and the cells are counted accordingly. Percent (%) viable cells are calculated according to the total number of viable cells per ml of aliquot; that is, the percent of viable cells of the total number of cells in the hemacytometer fi eld, where:
= ×total number of viable cells per ml of aliquot% viable cells 100
total number of cells per ml of aliquot
Dye exclusion is a simple and rapid technique measuring cell viability. However, as with the cell viability assays described below (MTT, NRU), measurement of cell membrane integrity, through the process of dye permeability, is indirectly used to monitor viability. Thus, in instances where membrane integrity is maintained, cell viability may still be compromised,
4 Refers to cells enzymatically treated and removed from the plastic culture plate surface. 5 Serum proteins from the growth media stain with trypan blue and can interfere with interpretation of observations.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 193
thus leading to a false-positive conclusion. Another potential problem is the subjective nature of observing for partial dye uptake, which contributes to inaccurate readings. An alternative method is one requiring the ability of the cell to scatter light, using propidium iodide uptake. This technique, however, requires quantitative fl uorescence measurements with a fl uorom-eter or with a fl ow cytometer, the latter of which is described in subsequent chapters.
NRU Assay
The NRU cytotoxicology assay is currently in validation studies to determine its suitability for use as an alternative for the prediction of acute systemic toxicity in vivo (see ICCVAM in the Review Articles section at the end of this chapter). The assay uses neutral red (3-amino-7-dimethylamino-2-methyl phenazine hydrochloride), a weak cationic dye, that is preferen-tially absorbed into lysosomes. The results rely on the premise that a cytotoxic chemical, regardless of site or mechanism of action, interferes with normal lysosomal uptake, which refl ects the number of viable cells. Since only viable cells are capable of maintaining the lysosomal process intact, the degree of inhibition of viability in proportion to the concentra-tion of the test compound provides an indication of toxicity.
3–10 min @ RT
Cell monolayer rinsed three timeswith PBS
Submerge monolayer with 0.4% TB
Rinse 3 times with 1:10 trypsin-EDTA solution × 1 min
Aspirate, discard; incubate withtrypsin-EDTA solution × 10 min at
37°C
Neutralize with equal amount ofDME-10
Collect, resuspend cells
Count total cellsCount cells stained with TB
FIGURE 14.3 Outline of trypan blue (TB) exclusion assay for cell viability. See text for details of the protocol. Abbreviations: EDTA, ethylene diamine tetraacetate; DME-10, Dulbecco’s Modifi ed Eagle’s Medium supplemented with 10% fetal bovine serum.

194 PRINCIPLES OF TOXICOLOGY TESTING
Any chemical that demonstrates a selective effect on lysosomes produces a false- positive response. Alternatively, this dynamic renders the system useful for detecting chemicals that selectively affect lysosomes and thus more readily suggests a specifi c mechanism of action. For example, chloroquine phosphate specifi cally alters lysosomal pH and thus has a greater effect on NRU than comparably toxic chemicals.
Cells are seeded in microtiter plates and allowed to settle and adhere for 24 hours (Fig. 14.2). The growth medium is replaced with fresh incubating medium containing the test chemical at increasing concentrations. Three hours before the end of the exposure period, the medium is aspirated from the wells and replaced with neutral red solution (to a fi nal concentration of 50 μg/mL in the incubating medium). The cultures are incubated for an additional 3 hours at 37ºC, after which the medium is removed and the cells are rinsed with PBS, fi xed, and de-stained with acetic acid/50% ethanol. The plates are shaken for 10 minutes and the absorbance is read at 540 nm with a microplate reader against a reference control and blank wells. Absorbance data correlate linearly with cell viability over a specifi c optical density range of 0.2–1.0.
One major drawback of the assay is the precipitation of the dye into visible, fi ne, needle-like crystals, yielding inaccurate readings. The precipitation is induced by some chemicals and thus a visual inspection stage during the procedure is important.
MTT Assay
In this assay, the tetrazolium salt, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bro-mide (MTT), is actively absorbed into cells and reduced in a mitochondrial-dependent reac-tion to yield a formazan product. The product accumulates within cells because it cannot pass through intact cell membranes. Upon addition of DMSO, isopropanol, or other suitable solvent, the sterile product is solubilized, released from intracellular stores, and is readily quantifi ed colorimetrically. Since only viable cells are capable of reducing MTT, the amount of reduced MTT is proportional to the intensity of blue-violet color formation and greatest cell viability. Thus cell viability correlates with cytotoxicology. This assay is suitable for a variety of culture systems displaying relatively high levels of mitochondrial activity. It should be noted that some compounds selectively affect mitochondria, resulting in an over-estimation of toxicity.
The assay begins the same way as the NRU procedure (Fig. 14.4). During the fi nal hour of incubation with the test chemical, 10 μL of 0.5% MTT solution is added to the test medium and incubated for 1–2 hours at 37°. The medium plus MTT is then replaced with DMSO and agitated for 5 minutes. The plates are read at 550 nm with a microplate reader against reference blanks (wells containing no cells). Absorbance correlates linearly with cell number (viability) over a specifi c optical density range of 0.2–1.0. Recently, XTT has been substituted for MTT because XTT is already soluble in aqueous media and obviates the need for DMSO.
SEARCH FOR BIOMARKERS IN TOXICOLOGY
One of the most important goals of acute toxicology testing is to predict the adverse effects of drugs, chemicals, and other xenobiotics on mammalian systems. Traditional toxicology tests generally provide descriptive information regarding the potential toxicity of chemicals, drugs, and physical agents. The tests provide mechanistic information for the determination of the pathway of toxic events. Historically, in vivo tests are the predominant models for predicting toxicity. However, they are limited in their ability to assess risk to humans because of the inherent incongruities with the rodent models. The tests necessarily involve imperfect

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 195
correlations between humans and the animal models. The advent and development of in vitro tests has supported and improved the predictive ability of the animal models. And the in vitro tests have contributed to a more ethical approach to toxicology testing. The tre-mendous complexity of the results and data obtained from these models, however, often leads to confusion of the interpretation of the results. Furthermore, risk analysis and conclu-sions may not be reliable based on the methodology of the tests. In order to ensure that data obtained from in vitro tests are reliable, and correlate with the abundance of animal data, the quest for accurate and predictive biomarkers has opened the fi eld to extraordinary technical developments.
Several fi elds which have heretofore been nonexistent 10 years ago, include the realms of systems biology, in silico systems, kinetic modeling, gene expression, and physiologi-cally based pharmacokinetic modeling. These newly emerging fi elds integrate data con-cerning chemical molecules and their interactions, and position them into a network behavior. Consequently, these models, together or systematically, should be able to assist toxicology in its predictive role.
In addition, multiple in vivo parameters have been used for many years to measure target organ functionality to assess pathological events. It is also important to note that, besides histopathology assessment, serum-based protein biomarkers have been the most common and traditional parameters. Urine has also been incorporated into the toxicology profi le to obtain additional information regarding the status of different organs. Thus, serum protein indicators, urine electrolytes, and constituents, and gene expression profi ling with microRNA identifi cation, together have led to the development of toxicogenomics—a relatively new discipline that combines the fi elds mentioned above with emerging tech-nologies of genomics, proteomics, and bioinformatics, the purpose of which is to identify and characterize mechanisms of toxic action of known and suspected toxicants. To date, the most popular submissions for these tests have been applied to chemically induced liver injury, developmental toxicity studies, nephrotoxicity, endocrine disruptors, toxicokinetics, and extrapolation of the effects of in vitro concentrations to in vivo dose levels. Other areas that require further investigation involve indicators for oxidative stress, epigenetic markers, and DNA adduct markers for hazard identifi cation. With further technical advances and assay improvements, a signifi cant number of applicable biomarkers will be developed that
Incubate withmedium + test
chemical
Replace with NRstock solution
(5 mg/mL in PBS)
+24–96 hr Last 1–3 hr ofincubation
Aspirate, rinsewith PBS, addNR destainer
Shake × 10 min
Read absorbanceat 540 nm
FIGURE 14.4 Outline of NRU assay procedure. Cells are incubated with increasing concentrations of test chemical (gray fi lled circles) plus blanks (clear, fi lled circles, no cells). Abbreviations: NR, neutral red; NR destainer, 1% acetic acid/50% ethanol; PBS, phosphate buffered saline. Photo dis-plays BioTek FLx800 Multidetection Microplate Reader®. Source: Courtesy of www.biotek.com.

196 PRINCIPLES OF TOXICOLOGY TESTING
can be used to estimate, predict, and forecast toxicity events that, until now, have had to rely on provisional risk analysis methodologies.
CONCLUSIONS
Several multilaboratory validation studies and programs sponsored by regulatory agencies have demonstrated that cytotoxicology tests with cell lines yield similar results when the inhibitory concentrations are compared from experiments using similar incubation times and assays. This indicates that different cell types and toxicity criteria measure fundamental processes that allow comparisons of otherwise dissimilar methods.
The relatively small differences among cell lines probably depend on variations encountered with cellular metabolic activity, protocols, and technical variability, leading to differences in the sensitivities of the experimental methods. Thus acute or chronic toxicity detected with these test methods is often referred to as basal cytotoxicology (see Ekwall, B., Suggested Readings), suggesting that any reasonable method that incorporates an established cell line will accurately measure the fundamental processes inherent in all cells that are affected by toxic insult.
ACUTE TARGET ORGAN TOXICOLOGY TESTING
USE OF SPECIALIZED PRIMARY CULTURES
Isolation of homogeneous cell populations from the structural framework of an intact organ is a formidable endeavor, yet presents a unique opportunity to study the metabolism of a single cell type. Numerous systems for primary cultures of specialized cell types—cells isolated from an organ as a primary culture that still retain suffi cient phenotypic and geno-typic features of the parent cells—have been described. Even after several passages, the cultured cells are capable of responding to the toxic effects of compounds as does the in vivo target organ of origin. In most cases, the target organs of interest to toxicologists that can explain the mechanistic roles of toxicity include the lungs, liver, skin, kidneys, hematopoi-etic system, nerve tissue, cells of the immune system, and skeletal and cardiac muscles.
With the exception of techniques that use keratinocytes or ocular models of toxicity, stan-dardized or validated test methods have not been developed to date.6 The reason is that pri-mary cultures have several disadvantages for the assessment of organ specifi c cytotoxicology:
1. Repeated and renewable sources of animals are often needed to establish primary cultures.
2. Isolation of organs and establishing primary cultures are laborious procedures.3. When used for detecting and quantifying systemic toxicity in target organs, primary
cultures are limited to only a few of the many possible organ injuries that could be determined using animals. This is a fundamental limitation of the technology. In conventional whole animal experiments, any or all of the affected organs that show gross pathology are analyzed at the time of sacrifi ce. The organs are then frozen or preserved indefi nitely with fi xatives according to the analytical protocol.
4. To standardize testing protocols and generate comparable data, primary cultures are derived from identical animal species and strains.
6 Only select tests for local toxicity are currently in validation programs or have been validated.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 197
Development of primary cultures from human donors is also fraught with theoretical, technical, and ethical obstacles. Even as efforts are made to match the ages and sexes of donors, a primary culture established from one individual is not necessarily identical to another. This impediment is overcome by performing the necessary basic tests to charac-terize a primary culture, including karyotypic analysis and identifi cation of functional markers. This battery of procedures in the analytical method ensures progress toward development of standardized methodologies and facilitates interlaboratory comparisons.
Despite the disadvantages of establishing initial cultures for in vitro testing, many types of specialized cells are used to detect target organ toxicities of chemicals. Often, a prototype chemical and its analogues are screened so that their toxicity is compared in the same sys-tem. This permits established toxicity data for some of the substances to be used as a refer-ence for previously unknown toxicity of unrelated materials. Hepatocytes fulfi ll this testing role principally because of the importance that the liver demonstrates in xenobiotic metabo-lism; that is, the metabolism of exogenous substances. Other cell types that have historical or current utility in target organ toxicology testing include lung macrophages and visceral epithelial cells, blood, cardiac, renal, neuronal, and muscle cells. Procedures describing the isolation of cells from liver, kidneys, and lungs are briefl y described next.
ESTABLISHMENT OF PRIMARY CULTURES OF HEPATOCYTES
In vivo, the liver plays an important role in drug and xenobiotic metabolism. The liver per-forms preliminary screening of most compounds following administration of a drug or chemical. According to the principles of the fi rst pass phenomenon, a chemical is susceptible to biochemical metabolic processes immediately after oral absorption because the circula-tion of the upper intestinal tract is intimately associated with that of the biliary tract. These toxicokinetic processes7 alter the chemical structure of the parent compound, rendering it, for the most part, inactive, water soluble, and ready for renal elimination. Thus, the study of toxicokinetics can yield useful information about the disposition of a toxic compound fol-lowing exposure. The information can be used to predict or monitor the metabolic products of the parent compound, whether toxic or inactive, the duration of the effect, sequestration into body compartments, and the route of elimination.
The process of metabolizing or detoxifying a chemical in vivo usually follows a two-step mechanism which, in sequence, results in the formation of a water-soluble, inactive metabolite. The fi rst step in the process, phase I biotransformation, involves a group of cellular enzymes including mono-oxygenases, esterases, and hydrolases, collectively known as the cytochrome P450 enzymes. Through a series of reduction/oxidation reactions, the active sites of chemicals are converted to expose electrophilic polar substituents. With the completion of these reactions, phase II biotransformation reactions follow to couple circulating plasma and interstitial constituents to the electrophilic sites of the phase I metab-olite. These constituent groups consist of acetyl, glucuronyl, and glutathione moieties that convert the phase I metabolites to water-soluble compounds, thus preparing the nontoxic products for elimination. It is important to note that although the majority of substances are detoxifi ed and inactivated through this series of reactions, some substances are activated from nontoxic or inactive parent compounds to active toxic metabolites.8
The fact that the liver has the greatest capacity of all organs to alter chemical sub-stances does not preclude the ability of other organs to contribute to the toxicokinetics of a
7 The qualitative and quantitative study of the time course of absorption, distribution, biotransformation, and elimination of an agent in an intact organism. 8 In fact, the mechanisms of many therapeutic drugs, especially cancer chemotherapeutic agents, rely on the conversions of inactive parent compounds to active metabolites.

198 PRINCIPLES OF TOXICOLOGY TESTING
compound. The rationale for using hepatocyte cultures, therefore, is based on the abundant metabolic potential of the liver for biotransformation in vivo. In addition, the implication is that a drug that exerts an effect on an intact organ will also similarly target the isolated cells. Based on this premise, many of the initial studies using hepatocyte cultures presented conclusions for selected target organ toxicity. Admittedly, target organ toxicity can be cor-related with the ability of the liver from a particular species to biotransform an agent and effectively detoxify it. The extrapolation from in vitro models to the in vivo situation, how-ever, does not depend exclusively on the fi nal products of metabolism nor on the selective role of the liver to alter the chemical, but on the expression of basal cytotoxicology that occurs as a result of accumulation of the compound in the liver. Consequently, hepatocyte cultures are suitable for use as screening methods for systemic toxicity because they can provide information on basal and organ-specifi c cytotoxicology.
Because of the extensive metabolic capacity of the liver to screen most circulating xenobiotics, hepatocytes have the propensity to screen toxicants that are selectively altered or sequestered by the liver. A signifi cant disadvantage to the use of mortal diploid hepato-cytes, however, is that they cannot be routinely maintained as differentiated continuous cell lines and must be established as primary cultures for each experiment. Some of the various methods used for the isolation and maintenance of primary cultures of hepatocytes include in situ liver perfusion and digestion, primary hepatocyte enrichment, and hepatocyte cell culture (suspension and attachment cultures).
In Situ Liver Perfusion
In situ liver perfusion relies on the infusion of fl uids through a freshly isolated intact rat liver and collecting the cells in the effl uent at the end of the last wash step. Initially, a wash perfu-sion medium formulated in HBSS and containing heparin as the active ingredient is infused through the inferior vena cava at 25–35 mL/minute through one arm of a Y tube connector. The solution removes blood from the portal circulation. After 10 minutes, the digestion per-fusion medium, also formulated with HBSS containing collagenase, is infused through the other arm of the connector for 10 minutes. The liver is then transferred to a petri dish and 20 mL of a fresh cell wash medium is injected into the organ.
Further release of hepatocytes is stimulated by mechanically agitating the dish, surgi-cally manipulating small incisions in the organ, and scraping the lobes of the liver. The procedure is repeated and the solutions are collected and centrifuged. Primary cultures are initiated by inoculating culture fl asks with the cell suspensions. All solutions are prepared, and manipulations are performed under aseptic conditions.
Perfusion of explanted liver specimens from a human donor requires performance of aseptic technique in a manner not unlike that described for in situ perfusion. Briefl y, fresh biopsy material is perfused through ostensibly exposed blood vessels on the outer surfaces of the organ. Perfusion, digestion, and wash-out media are similar in composition as above, and fl ow rates are maintained constant and recirculated. Further release of primary hepato-cytes is accomplished by gentle combing and shredding of the intact organ after transfer-ring to sterile petri dishes.
Primary Hepatocyte Enrichment
Suspensions of isolated liver cells are manipulated so as to remove nonhepatocyte and nonviable cells from the desired cultures. A low-speed, isodensity Percoll™ (Sigma-Aldrich Co. LLC, St. Louis, MO, USA) centrifugation procedure used to enrich the population of primary hepatocytes from liver cell isolates has been described (Gomez-Lechon, M.J. et al.).

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 199
Using aseptic technique, a suspension of freshly isolated and washed cells is mixed with and then layered on a stock solution of iso-osmotic Percoll and centrifuged at 50× g for 10 min-utes at 4ºC. The parenchymal hepatocytes are separated and removed from the bottoms of the tubes while contaminating and nonviable cells fl oat on top. The primary cells are then resuspended in appropriate medium in preparation for primary cultures.
Hepatocyte Cell Culture
As with most cell cultures, freshly isolated hepatocytes are maintained using routine culture procedures. The criteria for selecting any system are based on the degree of differentiation that the primary cultures undergo once exposed to the artifi cial culture conditions and the consequent loss of differentiated functions. Primary cultures of parenchymal cell suspension cultures are not desirable because these cells result in a rapid loss of differentiated functions and cytochrome P450 levels. Thus cytotoxicology tests must be performed generally within 12 hours.
Attachment cultures, using fl asks or wells to which basement membrane substrata have been layered, are more suitable. Establishing primary cultures on plastic cell culture plates coated on cellular attachment components maintains differentiated features, improves via-bility and membrane integrity, especially after the traumatic isolation procedures, and also preserves cytochrome P450 activity for several weeks. The substrata include collagen types I and IV, laminin, fi bronectin, or combinations of these, and are applied as soluble solutions to the culture plate surfaces.
Functional Markers of Primary Hepatocyte Cultures
Table 14.3 summarizes some of the important criteria used for assessing functional and dif-ferentiated features of freshly isolated liver parenchymal cells. These criteria are also incor-porated in cytotoxicology testing of chemicals that exhibit organ-specifi c toxicities. In fact, one or more test methods are used to screen chemicals for potential liver toxicity. In addition, the ability of a chemical to undergo biotransformation is effi ciently evaluated using primary cultures of hepatocytes.
ESTABLISHMENT OF PRIMARY CULTURES FROM OTHER ORGANS
Principles of isolation of cells and for establishing primary cultures are described in the coming sections. Detailed methods for isolation procedures are described in several review articles listed at the end of this chapter.
TABLE 14.3Functional Markers of Primary Hepatocyte Cultures
Functional Marker Descriptive Indicators
Cell membrane integrity Trypan blue exclusion, lactate dehydrogenase leakage, aspartate aminotransferase leakage
Phase I metabolic reactions 7-ethoxycoumarin-O-deethylation activityPhase II conjugation reactions Glutathione determination, glutathione-S-transferase activityPeroxisomal-oxidation capacity Cyanide-insensitive acyl-CoA oxidase activity, carnitine
acyltransferase activity

200 PRINCIPLES OF TOXICOLOGY TESTING
Isolation of Renal Cortical Cells
Several methods effectively yield suffi cient quantities and purity of cortical renal cells from a variety of animal species. The cortical cells stem from the renal cortex and constitute the most metabolically active cells of the kidney. Of these, proximal tubule and distal tubule cells are more frequently used for the assessment of in vitro renal toxicity. The methods of isolating and obtaining enriched populations of tubular cells rely on the ability of the procedures to selec-tively exclude other contaminating cell types such as cells associated with the glomeruli and cortical collecting duct. The most popular protocols include enzymatic methods, mechanical methods and historically, microdissection techniques. Any of these may be coupled with den-sity gradient centrifugation on Percoll to enrich the fractions for proximal or distal tubule cells.
Although developed as one of the original methods for isolating cortical cells, micro-dissection has limited use because of poor yields of tubule cells and the prolonged times required. Essentially, the method requires precise manipulative skills using microdissect-ing capability in order to focus on nephron tubules. The method is protracted and, for the purposes of screening nephrotoxic chemicals, does not afford enough cells for a suitable cytotoxicology testing procedure.
Mild enzymatic digestion with purifi ed collagenase in a balanced salt medium is the method of choice for releasing cortical cells from the basement membrane. In general, the procedure is similar to that described above for enzymatic digestion for hepatocyte isolation.
Mechanical separation of proximal cells relies on the in situ perfusion of the kidney with a suspension of iron oxide particles. These micronized pellets lodge in the glomeruli and allow for the separation of proximal tubule cells from the fi ltering apparatus. Ideally, after perfusion, portions of the cortex are hand homogenized, coarsely fi ltered, and the suspension is exposed to a mechanical stirring bar to which the iron-laden glomeruli are attracted and separated.
Thus, selection of a particular isolation procedure must primarily yield a suffi cient number of cells adequate for toxicity testing and, in addition, the cells must be of suffi cient purity so that reliable data are generated. Underlying this premise is the need to assess the functional characteristics of the cell types.
Use of Continuous Renal Cell Lines
Over the past 30 years, improved methods for maintaining continuous cultures of cells have allowed for the realization of general mechanistic cytotoxicology studies. Renal cell culture technology has benefi ted primarily from the improved methodology in much the same way as in other fi elds. The problems associated with other continuous cell lines, however, have also plagued the maintenance of continuous cultures of renal cells (and include those dis-cussed in chapter 13, the most important of which include the loss of in vivo characteristics following isolation and subsequent passage over several generations in vitro. This dedifferen-tiation may be followed by a decrease in the levels of functional markers necessary for iden-tifying the cell type and for assessing cytotoxicology. Nevertheless, propagation of continuous cell lines has allowed for important contributions to understanding renal cell biology and physiology. It may be necessary to approach renal cytotoxicology, however, more cautiously when using cell lines. As with the use of continuous cell lines from any organ, the apparent toxicity from a chemical may be a refl ection of basal cytotoxicology of the cell rather than a specifi c renal toxic effect.
Among the most frequently used permanent cell lines used in renal studies are the transformed cells including Madin–Darby canine kidney and LLC-PK renal epithelial cell lines.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 201
Madin–Darby canine kidney cells maintain morphological features and functional characteristics consistent with cells of distal tubule/collective duct origin. In addition, they demonstrate biochemical evidence of transporting epithelia, although several strains and clones are available that differ considerably from the parent cell line.
LLC-PK cells are derived from pig proximal tubule cells and share morphological features characteristic of their parent cells, including the ability to form domes, the pres-ence of apical membrane microvilli, and several junctional complexes. Physiologically they express high activities of brush border membrane marker enzymes, retain the ability to transport small molecules, and express high transepithelial electrical resistance when cultured on microporous membrane fi lter culture inserts.
Assessment of functional features for identifi cation of primary and continuous cultures of differentiated renal cortical cells include general cell culture markers for membrane integrity and cell proliferation and morphological examination for phenotypic appearance. Assessing the abilities of cells for toxicokinetic studies requires intact and suffi ciently high levels of enzymatic activity including the presence of cytochrome P450 isozymes and other oxidative enzymes present in proximal tubular brush borders (phosphorylases, dehydroge-nases, and ATPases). Collectively, these features provide evidence for the origins of the cells and lend support for data generated during metabolic investigations.
Establishment of Primary Cultures of Lung Cells
Mechanistic studies of organ-specifi c toxicity in mammalian lungs have been hampered by the heterogeneity of lung composition. Over 40 different cell types are identifi ed in this organ, not all of which have been extensively characterized. This complex cellular organiza-tion, together with cell interactions with the extensive vascular, lymphatic, and neuronal networks, presents formidable challenges to the performance and interpretation of mecha-nistic studies of lung-specifi c toxicity. Table 14.4 lists some of the major cell types currently used in pulmonary studies, along with their unique biochemical features. These cells have also found utility in in vitro cytotoxicology studies.
The isolation and maintenance of relatively pure populations of alveolar and bronchio-lar epithelial and endothelial cell lines from mammalian lungs utilize enzymatic digestion of normal rodent lung followed by centrifugal elutriation, laser fl ow cytometry, or differen-tial adhesion procedures. Type II pneumocytes have been maintained in continuous culture following isolation from rats, rabbits, cats, hamsters, and mice. They retain high levels of enzymatic activity, display cytoplasmic lamellar inclusions, and synthesize and secrete sur-factant. These features are maintained in culture for many weeks and, as a result, are used in mechanistic cytotoxicology studies for pulmonary alveolar specifi c toxins. Derivation of continuous cell lines of alveolar epithelial cells from primary cultures, however, has gener-ally resulted in the disappearance of characteristic features such as the loss of cytochrome P450 enzyme activity and reduction in proliferation within 2 weeks of initial isolation.
The organotypic culture represents a model system suitable for the investigation of in vitro cytotoxic responses. Isolated cells are layered on Gelfoam squares, reaggregate, and form alveolar-like structures, thus resembling their in vivo appearance. Generally, the cultures are established from fetal, newborn, and postnatal rats. The cultures retain differ-entiated characteristics for longer periods of time in culture, and maintain their capacities to produce and secrete pulmonary surfactant. Organotypic models of type II pneumocytes are useful in identifying agents with potential capacity for oxidant lung injury.
Clara cells are examples of nonciliated bronchiolar epithelial cells that possess a secre-tory role. Ultrastructurally, as with pulmonary endothelial cells, the presence of extensive
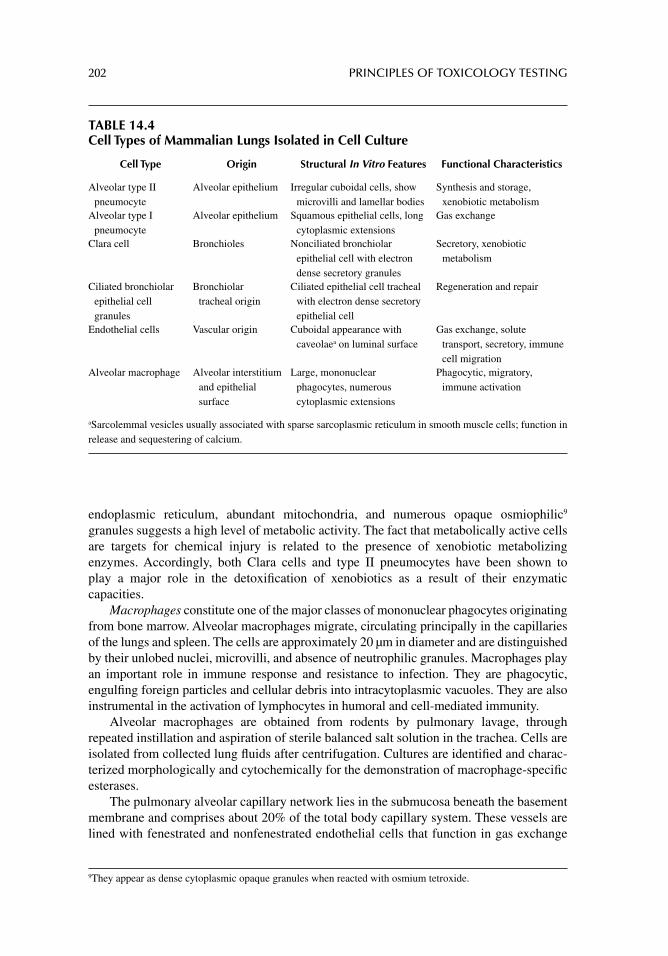
202 PRINCIPLES OF TOXICOLOGY TESTING
endoplasmic reticulum, abundant mitochondria, and numerous opaque osmiophilic9 granules suggests a high level of metabolic activity. The fact that metabolically active cells are targets for chemical injury is related to the presence of xenobiotic metabolizing enzymes. Accordingly, both Clara cells and type II pneumocytes have been shown to play a major role in the detoxifi cation of xenobiotics as a result of their enzymatic capacities.
Macrophages constitute one of the major classes of mononuclear phagocytes originating from bone marrow. Alveolar macrophages migrate, circulating principally in the capillaries of the lungs and spleen. The cells are approximately 20 μm in diameter and are distinguished by their unlobed nuclei, microvilli, and absence of neutrophilic granules. Macrophages play an important role in immune response and resistance to infection. They are phagocytic, engulfi ng foreign particles and cellular debris into intracytoplasmic vacuoles. They are also instrumental in the activation of lymphocytes in humoral and cell-mediated immunity.
Alveolar macrophages are obtained from rodents by pulmonary lavage, through repeated instillation and aspiration of sterile balanced salt solution in the trachea. Cells are isolated from collected lung fl uids after centrifugation. Cultures are identifi ed and charac-terized morphologically and cytochemically for the demonstration of macrophage-specifi c esterases.
The pulmonary alveolar capillary network lies in the submucosa beneath the basement membrane and comprises about 20% of the total body capillary system. These vessels are lined with fenestrated and nonfenestrated endothelial cells that function in gas exchange
9 They appear as dense cytoplasmic opaque granules when reacted with osmium tetroxide.
TABLE 14.4Cell Types of Mammalian Lungs Isolated in Cell Culture
Cell Type Origin Structural In Vitro Features Functional Characteristics
Alveolar type II pneumocyte
Alveolar epithelium Irregular cuboidal cells, show microvilli and lamellar bodies
Synthesis and storage, xenobiotic metabolism
Alveolar type I pneumocyte
Alveolar epithelium Squamous epithelial cells, long cytoplasmic extensions
Gas exchange
Clara cell Bronchioles Nonciliated bronchiolar epithelial cell with electron dense secretory granules
Secretory, xenobiotic metabolism
Ciliated bronchiolar epithelial cell granules
Bronchiolar tracheal origin
Ciliated epithelial cell tracheal with electron dense secretory epithelial cell
Regeneration and repair
Endothelial cells Vascular origin Cuboidal appearance with caveolaea on luminal surface
Gas exchange, solute transport, secretory, immune cell migration
Alveolar macrophage Alveolar interstitium and epithelial surface
Large, mononuclear phagocytes, numerous cytoplasmic extensions
Phagocytic, migratory, immune activation
aSarcolemmal vesicles usually associated with sparse sarcoplasmic reticulum in smooth muscle cells; function in release and sequestering of calcium.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 203
and movement of solutes. The cells regulate the transport into the circulation of important chemical mediators such as biogenic amines, kinins, angiotensins, and prostaglandins. In addition, they monitor the migration of monocytes, granulocytes, neutrophils, and lympho-cytes from the lymphatic to systemic circulations.
Endothelial cells are ideally suited to conduct a variety of specifi c metabolic activities important in the pulmonary processing of vasoactive substances. Enzymes, chemically mediated inhibitors, cytoplasmic receptors, and transport systems of endothelial cells determine the level of hormones entering the circulation. Through the regulation of these mechanisms the pulmonary endothelium plays an important role in the response of the lung to xenobiotics.
Endothelial cells from large and small vessels of the lung possess specialized proper-ties and characteristics. The presence of fenestrations, organ-specifi c antigens on surface membranes, and cell membrane-specifi c glycoproteins are examples of differences among cells of various origins. The cells are generally isolated by enzymatic digestion of the greater vessels from rodent or rabbit lungs. Cells grow to form confl uent monolayers using routine cell culture techniques.
In culture, endothelial cells demonstrate typical cuboidal arrangements when confl uent and they possess caveolae on their luminal surfaces. Functionally, detectable activity of angiotensin converting enzyme on the plasma membrane and intracytoplasmic xenobiotic metabolizing enzymes are hallmarks of the cells in culture.
DIFFERENTIAL TOXICOLOGY TESTING
It is important to note that differentiated cells derived from passages of primary cultures are inclined to refl ect both organ-specifi c and indirect effects of basal cytotoxicology when chal-lenged with a toxic insult. It is possible, however, that by studying specialized functions of organ-specifi c cells, fi ne adjustments in the dosages used during exposure are required to distinguish basal cytotoxicology from target organ effects. Alternatively, toxicity is demon-strated by selecting a narrower range of dosages or incubation for shorter periods of expo-sure. This requires analysis at intervals during the exposure period rather than at the end of the incubation.
SUMMARY
The development of in vitro methods for assessing acute systemic toxicity has progressed considerably in the past 20 years as a result of similarities of test methods to the traditional in vivo protocols. Also, many biomarkers have now been identifi ed in vitro as a result of the development of cell culture models, in order to predict toxicity of a variety of unknown sub-stances. However, caution must be exercised with certain aspects of cell culture methodol-ogy when screening for acute systemic toxicity. For example, false-positive and false-negative results are usually errors of estimation and are closely related to the physicochemical proper-ties of a test substance. Diffi culty with solubility of a test substance in a solvent may under-estimate true toxic concentrations. And the contribution of the solvent to the overall effect on the cell cultures must be taken into consideration.
While no single in vitro method accounts for all of these parameters, the combination of individual tests addresses these end points. The anticipation for success relies on diligent collective cooperation among the scientifi c, regulatory, industrial, commercial, and public representatives, using the concept of a battery of validated assays to screen chemicals with potential for human toxicity.

204 PRINCIPLES OF TOXICOLOGY TESTING
SUGGESTED READINGS
Barile FA. Continuous cell lines as a model for drug toxicity assessment. In: Castell JV, Gómez-Lechón MJ, eds. In Vitro Methods in Pharmaceutical Research. London: Academic Press, 1997.
Bartsch H, Arab K, Nair J. Biomarkers for hazard identifi cation in humans. Environ Health 2011; 10: 1.Ekwall B. Basal cytotoxicity data (BC data) in human risk assessment. In Proceedings of Workshop
on Risk Assessment and Risk Management of Toxic Chemicals, National Institute for Envi-ronmental Studies. Ibaraki, Japan, 1992, 137.
Ekwall B. The basal cytotoxicity concept. In: Goldberg A, van Zupten LFM, eds. Alternative Meth-ods in Toxicology and the Life Sciences. New York: Mary Ann Liebert, 1995; 11: 721.
Ekwall B, Barile FA. Standardization and validation. In: Barile FA, ed. Introduction to In Vitro Cyto-toxicology: Mechanisms and Methods. chap. 11 Boca Raton, FL: CRC Press, 1994.
Koturbash I, Beland FA, Pogribny IP. Role of microRNAs in the regulation of drug metabolizing and transporting genes and the response to environmental toxicants. Expert Opin Drug Metab Toxicol 2012; 8: 597.
Koturbash I, Beland FA, Pogribny IP. Role of epigenetic events in chemical carcinogenesis—A justifi cation for incorporating epigenetic evaluations in cancer risk assessment. Toxicol Mech Methods 2011; 21: 289.
Martyniuk CJ, Alvarez S, Denslow ND. DIGE and iTRAQ as biomarker discovery tools in aquatic toxicology. Ecotoxicol Environ Saf 2012; 76: 3.
Pogribny IP. Epigenetic events in tumorigenesis: putting the pieces together. Exp Oncol 2010; 32: 132.
Tajbakhsh J. DNA methylation topology: Potential of a chromatin landmark for epigenetic drug toxicology. Epigenomics 2011; 3: 761.
REVIEW ARTICLES
Afshari CA, Hamadeh HK, Bushel PR. The evolution of bioinformatics in toxicology: Advancing toxicogenomics. Toxicol Sci 2011; 120: S225.
Aufderheide M, Knebel JW, Ritter D. Novel approaches for studying pulmonary toxicity in vitro. Toxicol Lett 2003; 140: 205.
Barile FA, Cardona M. Acute cytotoxicity testing with cultured human lung and dermal cells. In Vitro Cell Dev Biol Anim 1998; 34: 631.
Barile FA, Arjun S, Hopkinson D. In vitro cytotoxicity testing: biological and statistical signifi cance. Toxicol In Vitro 1993; 7: 111.
Bishop AE, Rippon HJ. Stem cells: potential for repairing damaged lungs and growing human lungs for transplant. Expert Opin Biol Ther 2006; 6: 751.
Borenfreund E, Puerner JA. Cytotoxicity of metals, metal–metal and metal–chelator combinations assayed in vitro. Toxicology 1986; 39: 121.
Braun A, Hämmerle S, Suda K, et al. Cell cultures as tools in biopharmacy. Eur J Pharm Sci 2000; 11: S51.
Bruner LH, Carr GJ, Curren RD, Chamberlain M. Validation of alternative methods for toxicity test-ing. Environ Health Perspect 1998; 106: 477.
Casciano DA. The use of genomics in model in vitro systems. Adv Exp Med Biol 2012; 745: 210.Chapin RE, Creasy DM. Assessment of circulating hormones in regulatory toxicity studies ii. Male
reproductive hormones. Toxicol Pathol 2012; 40: 1063–78.Cockshott A, Evansl P, Ryan CA, et al. The local lymph node assay in practice: a current regulatory
perspective. Hum Exp Toxicol 2006; 25: 387.Dolbeare F, Vanderlaan M. Techniques for measuring cell proliferation. In: Tyson CA, Frazier JM,
eds. In Vitro Toxicity Indicators, Methods in Enzymology. San Diego: Academic Press, 1994; 1B: 178.
Elaut G, Henkens T, Papeleu P, et al. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr Drug Metab 2006; 7: 629.
Forbes B, Ehrhardt C. Human respiratory epithelial cell culture for drug delivery applications. Eur J Pharm Biopharm 2005; 60: 193.

CELL CULTURE METHODS FOR ACUTE TOXICOLOGY TESTING 205
Freed LE, Vunjak-Novakovic G. Space fl ight bioreactor studies of cells and tissues. Adv Space Biol Med 2002; 8: 177.
Fuchs TC, Hewitt P. Biomarkers for drug-induced renal damage and nephrotoxicity—An overview for applied toxicology. AAPS J 2011; 13: 615.
Fuchs TC, Hewitt P. Preclinical perspective of urinary biomarkers for the detection of nephrotoxicity: what we know and what we need to know. Biomark Med 2011; 5: 763.
Geenen S. Systems biology tools for toxicology. Arch Toxicol 2012; 86: 1251.Ginis I, Luo Y, Miura T, et al. Differences between human and mouse embryonic stem cells. Dev Biol
2004; 269: 360.Gomez-Lechon MJ, Donato MT, Castell JV, Jover R. Human hepatocytes in primary culture: the
choice to investigate drug metabolism in man. Curr Drug Metab 2004; 5: 443.Guha C, Lee SW, Chowdhury NR, Chowdhury JR. Cell culture models and animal models of viral
hepatitis, Part II: hepatitis C. Lab Anim 2005; 34: 39.Huggins J. Alternatives to animal testing: research, trends, validation, regulatory acceptance. ALTEX
2003; 20: 3.Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), Report of
International Workshop on In Vitro Methods for Assessing Acute Systemic Toxicity, NIH Publication 01-4499. National Institute of Environmental Health Sciences, Research Tri-angle Park, NC, 2001. [Available from: http://iccvam.niehs.nih.gov/docs/guide-lines/subguide.htm]
Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), Guid-ance Document on Using in Vitro Data to Estimate in Vivo Starting Doses for Acute Toxic-ity, NIH Publication 01-4500. National Institute of Environmental Health Sciences, Research Triangle Park, NC, 2001. [Available from: http://iccvam.niehs.nih.gov/docs/guide lines/subguide.htm]
Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), Biennial Progress Report of National Toxicology Program, NIH Publication 04-4509, National Institute of Environmental Health Sciences, Research Triangle Park, NC, 2003. [Available from: http://iccvam.niehs.nih.gov/about/annrpt/bienrpt044509.pdf]
Jones B, Stacey G. Safety considerations for in vitro toxicology testing. Cell Biol Toxicol 2001; 17: 247.
Kia R. Stem cell-derived hepatocytes as a predictive model for drug-induced liver injury: Are we there yet? Br J Clin Pharmacol 2012; Epub ahead of print.
Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. MicroRNAs in cancer management. Lancet Oncol 2012; 13: e249–58.
Louekari K, Sihvonen K, Kuittinen M, Somnes V. In vitro tests within the REACH information strat-egies. Altern Lab Anim 2006; 34: 377.
Louisse J, Verwei M, Woutersen RA, Blaauboer BJ, Rietjens IM. Toward in vitro biomarkers for developmental toxicity and their extrapolation to the in vivo situation. Expert Opin Drug Metab Toxicol 2012; 8: 11.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55.
Rasoulpour RJ, LeBaron MJ, Ellis-Hutchings RG, Klapacz J, Gollapudi BB. Epigenetic screening in product safety assessment: are we there yet? Toxicol Mech Methods 2011; 21: 298.
Roth RA, Ganey PE. Animal models of idiosyncratic drug-induced liver injury—Current status. Crit Rev Toxicol 2011; 41: 723.
Van Summeren A, Renes J, van Delft JH, Kleinjans JC, Mariman EC. Proteomics in the search for mechanisms and biomarkers of drug-induced hepatotoxicity. Toxicol In Vitro 2012; 26: 373.
Vermeir M, Annaert P, Mamidi RN, et al. Cell-based models to study hepatic drug metabolism and enzyme induction in humans. Expert Opin Drug Metab Toxicol 2005; 1: 75.
Waring WS, Moonie A. Earlier recognition of nephrotoxicity using novel biomarkers of acute kidney injury. Clin Toxicol (Phila) 2011; 49: 720.
Yokoi T, Nakajima M. Toxicological implications of modulation of gene expression by microRNAs. Toxicol Sci 2011; 123: 1.

206
15 Acute local toxicology testing in vitro
INTRODUCTION
No other fi eld in in vitro toxicology testing has driven academic, industrial, and government resources to develop cell modeling systems as much as the need for alternatives to local toxicity testing. This is principally due to the ubiquitous presence of cosmetics, toiletries, and dermal and ocular pharmaceuticals marketed for worldwide use. Furthermore, the results obtained with the original models for dermal and ocular toxicity were not successful in predicting effects encountered in vivo, primarily because of the inconsistencies between data generated from animal models and human risk exposure. However, technical advances and improved knowledge of cell culture technology have allowed improvements in the development of cellular systems, with some tests undergoing full validation evaluation in the United States and European Union (EU).
The development of a cellular model of local toxicity involves the establishment of primary cultures from specialized tissues, especially dermal and ocular epithelium. These primary cultures possess specifi c functional markers that can distinguish selective toxic effects. In particular, they assess the effects of a chemical on a specifi c functional target that is unique for ocular epithelia and distinguish the chemical’s effect on ocular cells from effects on other cell types. Other isolated in vitro models used to screen for local toxicity incorporate tissue types that are not limited to organ of origins and include supportive structures such as connective tissue cells. For instance, corneal epithelial cell culture mod-els have been introduced to test for chemicals with potential for detecting ocular effects.
Most in vitro tests in development involve cell and tissue cultures of human or animal origin. The systems are used to evaluate the effects of substances in isolated environments devoid of hormonal, immune, or neural infl uence. These advantages have also been noted as drawbacks—the elimination of other biological factors in the system does not allow the method to mimic interactions occurring in the whole organism. Nevertheless, many in vitro methods have been developed since the early 1990s; widespread regulatory acceptance, however, especially in the United States, is limited. The problem centers on the time and cost necessary to develop the tests and the arduous validation process.
OCULAR TOXICOLOGY TESTING
The current in vivo Draize rabbit eye test method identifi es both irreversible (e.g., corrosion) and reversible ocular effects. It also provides scoring that allows relative categorization of severity for reversible effects such as those caused by mild, moderate, or severe irritants. The Draize test, however, has been criticized for its lack of reproducibility, its subjective nature of assessment, the variable interpretation of results, the high dose of test material used, the overprediction of human response, and the infl iction of pain to animals. The normal end

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 207
points of toxicity for the in vivo rabbit eye test include corneal opacity, infl ammation, and cytotoxicology. The procedure is described in detail in “Section II.”
TIERED TESTING APPROACH
Tiered testing, standardization and validation of methods, and risk assessment are concepts which have been discussed and reviewed extensively (see Review Articles in References), particularly since they must be continuously updated to accommodate the large infl ux of chemicals, methods, and data into the regulatory arena. The tiered testing approach relies on a systematic sequence of ranked levels used in a testing strategy. A positive result from a test indicator at the fi rst or primary level qualifi es the substance for confi rmation of that positive result at the subsequent level, usually in order of complexity of effects detected with the indicator. For instance, analysis of a test agent for structure–activity relationship and physi-cochemical properties is frequently employed as a screening test in the fi rst tier. The analysis of structural features of a chemical may suggest similar mechanisms of toxicity of chemicals in structurally related classes. This is followed by a second set (second tier) of tests incorpo-rating short-term bacterial or mammalian assays, after which a decision point is determined, based on the evaluation of results from data accumulated at levels 1 and 2. The process continues through a third level in assigned categories of irritation toward the evaluation of human and animal risk assessment.
Specifi cally, current alternative approaches to the assessment of eye irritation make routine use of tiered testing strategies based initially on the weight of evidence (WoE) approach (McNamme et al., 2009). The principle is that all available information is con-sidered in the evaluation of eye irritation, which may include physicochemical properties of the test chemical, historical in vivo animal data, in vitro and human data, and exposure. A tiered testing strategy using a decision tree approach follows whether the data from the WoE approach are deemed insuffi cient. The three-step decision tree evaluates ingredients incorporating all available data (step 1), an in vitro severe eye irritancy test (step 2), and an in vitro mild to moderate irritancy test (step 3). A more complex alternative decision tree evaluates non-irritants, irritants, and severe eye irritants using validated in vitro assays as well as scientifi cally applicable in vitro methods with acceptable industrial experience. Current Environmental Protection Agency (EPA) ocular testing guidelines and the United Nations (UN) Globally Harmonized System (GHS) of Classifi cation and Labeling of Chemicals (2003) indicate that if serious ocular damage is anticipated,1 then a test on a single animal may be considered. When anticipated damage is observed, no further animal testing is necessary (EPA, 1998); if it is not observed, additional test ani-mals may be evaluated sequentially until concordant irritant or non-irritant responses are observed.
DEVELOPMENT OF IN VITRO OCULAR TOXICOLOGY TESTING METHODS
One of the original models developed in the early 1990s as a biochemical procedure to evaluate ocular irritation was the EYTEX™ system (Courtellemont et al., 1999). The test was based on the concept that the normal transparent state of the cornea depends on the relative degree of hydration and organization of corneal proteins. Corneal opacifi cation, therefore, is a result of a decrease in protein hydration associated with changes in protein conformation and aggregation. The EYTEX test simulated corneal opacifi cation by using alterations in the
1 Serious ocular damage to the rabbit eye is described as an irreversible adverse effect still occurring on day 21 after exposure.

208 PRINCIPLES OF TOXICOLOGY TESTING
hydration and conformation of an ordered macromolecular matrix to predict in vivo ocular irritancy. Although the model did not prove reliable as an in vitro prescreening system for determining eye irritation potential (IP) of cosmetic formulations, it set the foundation for further development of ocular toxicology testing models.
Gradual improvement to the methodologies involved the development of cell models of local toxicity using the establishment of primary cultures from specialized tissues, espe-cially cells of ocular epithelial origin. These primary cultures possess specifi c functional markers that can distinguish selective toxic effects. In particular, the effects of a chemical on a specifi c functional target, which is unique for ocular epithelia, is assessed and distin-guished from the chemical’s effect on ocular cells or any other cell type. Other isolated in vitro models used to screen for local toxicity incorporate tissue types that are not limited to the organ of origin but include supportive structures, such as keratinocyte epithelial cells, connective tissue cells, or in vitro compartmental models (Barile, 1997; Calabro et al., 2008; Konsoula and Barile, 2007; Barile, 2010). These cells retain suffi cient physiological properties similar to the mammalian eye that allow for a response to a test substance. For instance, corneal epithelial cell culture models have been introduced to test for chemicals with potential for detecting ocular effects.
More recently, organotypic models have warranted suffi cient interest as methods that maintain short-term normal physiological and biochemical function of the mammalian cor-nea in an isolated system. In these test methods, damage by the test substance is assessed by quantitative measurements of changes in corneal opacity and permeability using opaci-tometry and spectrophotometry, respectively. Both measurements are used quantitatively for irritancy classifi cation for prediction of in vivo ocular IP of a test substance. Examples of organotypic models that incorporate these criteria include the bovine corneal opacity and permeability (BCOP) assay; the isolated chicken eye (ICE) test method (or the chicken enucleated eye test method); and, the isolated rabbit eye (IRE) assay. A fourth method, the hen’s egg test-chorioallantoic membrane (HET-CAM) assay, differs in the evaluation crite-ria but is also normally included among this class of in vitro protocols.
Each of these protocols is discussed in the chapter in detail as representative candidate in vitro organotypic methods for assessing ocular irritation and corrosion. The methodolo-gies, protocol details, applications, and their validation status are outlined. A brief histori-cal perspective of the development of original in vitro ocular testing models is also mentioned. More importantly, improving upon and troubleshooting the current techniques, in order to present the models as stand-alone in vitro tools for ocular toxicity assessment, is emphasized. By 2003, the EPA nominated four in vitro ocular toxicology testing meth-ods for evaluation by the Interagency Coordinating Committee on the Validation of Alter-native Methods (ICCVAM, NIEHS):
1. Hen’s egg test–chorioallantoic membrane assay.2. Bovine corneal opacity and permeability assay [Section 4, OECD Test Guideline
(TG) 437].3. Isolated chicken eye test method or chicken enucleated eye test method (Section 4,
OECD TG 438).4. Isolated rabbit eye assay.
ICCVAM endorsed the pre-evaluation of these methods by the National Toxicology Pro-gram Interagency Center for the Evaluation of Alternative Toxicological Methods and rec-ommended background review documents for each method. The procedures are used as part

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 209
of a preclinical battery of tests by many EU countries for identifi cation of ocular irritants. The principles of the procedures and their validation statuses are listed in Table 15.1 and reviewed below.
HET-CAM TEST
Background
The HET-CAM test provides information on the effects that may occur in the conjunctiva following exposure to a test substance. The test method was an extension of traditional chicken embryo models used by embryo toxicologists and virologists (Luepke and Kemper 1986; Parish 1985) and was based on the observation that the CAM of an embryonated hen’s egg is similar to the vascularized mucosal tissues of the human or rabbit eye. Initial results in the 1990s, using modifi cations of the current HET-CAM test, showed high rank order cor-relation between data from testing chemicals and cosmetic formulations with Draize in vivo results. Later studies included classifi cation of several classes of irritants, ranging from non-irritant to severe, with high correlations between the systems. This development was encour-aging since it suggested that in vitro models could represent valid alternative constructs to predict in vivo ocular toxicity. Thus, it was reasonable to conclude that adverse effects on the CAM could correlate to irritation or corrosion in vivo. Consequently, the HET-CAM was validated for its ability to predict ocular corrosives and severe or irreversible effects as defi ned by the U.S. EPA (1996), the EU (2001), and the UN GHS of Classifi cation and Labeling of Chemicals (UN, 2003).
Overview of the Protocol
The major components of the HET-CAM test method involve exposing and treating the CAM of chicken eggs with a test substance. The CAM is a vascular fetal membrane com-posed of the fused chorion and allantois. The development of end points is observed and scored, and the data are evaluated in relation to a prediction model. The end points monitor the changes that occur in blood vessels in the CAM.
The allantois functions in maintaining chick embryo viability by serving as an embry-onic respiratory, excretory, and storage organ. During chicken embryo development, the allantois increases in size, wraps around the embryo, and fuses with the chorion to form the CAM. The fusion of the two membranes allows for a free exchange of gases between the embryo and the environment, after which there is a rapid increase in the surface area for
TABLE 15.1Current Organotypic Models of Alternative Methods for Ocular Toxicology Testing
Name Validation Status Test Method Indicator Test Objective
HET-CAM Validated (US) Hemorrhage, coagulation, cytolysis Ocular sensitivity and corrosionBCOP Validated (US) Increase in corneal opacity and thickness Ocular sensitivity and corrosionICE Validated (US) Increase in corneal opacity and thickness Ocular sensitivity and corrosionIRE Validated (US) Increase in corneal opacity and thickness Ocular sensitivity and corrosionEpiOcular Not validated MTT cell viability Ocular sensitivity and corrosion
Abbreviations: BCOP, bovine corneal opacity and permeability; HET-CAM, hen’s egg test–chorioallantoic mem-brane; ICE, isolated chicken eye; IRE, isolated rabbit eye; US, United States.

210 PRINCIPLES OF TOXICOLOGY TESTING
the fi rst 10 days of incubation. Thus the procedure monitors blood vessel changes in the CAMs of chicken eggs.
Briefl y, fertilized hen’s eggs are incubated under optimized conditions for 9 days. On the 10th day, the eggs are opened and the CAM is exposed. The test substance is applied to the surface of the CAM for 20 seconds, rinsed with water, and evaluated for development of irritant end points (hyperemia, hemorrhage, and coagulation) at 0.5, 2, and 5 minutes after rinsing. Irritant effects in the CAM blood vessels and albumen are subjectively assessed and a score is assigned based on the time required for development of each end point. The scores are totaled to yield a total irritation score (IS; maximum of 21) for the tested substance.
Details of the Typical Protocol
Fertilized White Leghorn hen’s eggs are incubated at 37ºC ± 1ºC, at an average relative humidity of 62.5%, for 9 or 10 days. The eggs usually weigh between 50 and 80 g and are rotated several times daily until the procedure is performed. In general, based on the observation of the development of the CAM, the earliest incubation day for test applica-tion is day 7. On day 10, the eggs are surgically cut with a rotary saw or tool with a cut-ting disk, a portion of the eggshell is removed, and the inner membrane and CAM are exposed.
A 200–300 μL of test substance is applied to the surface of the CAM for 20 seconds and rinsed with water. Appropriate test substance controls are incorporated, including pos-itive, negative, solvent, and benchmark controls. At 0.5, 2, and 5 minutes after rinsing, visual inspection is used for evaluating development of irritant end points, including hyper-emia, hemorrhage, and coagulation. The development of hemorrhage and coagulation is evaluated in combination with other end points including injection (mild hemorrhage), vasoconstriction, dilation, and lysis.
Scoring is assigned based on the time required for the development of each end point and evaluated according to a variety of user-defi ned scoring scales. In general, the scores range from 0 (no effect) to 3 (severe irritant effect) (Balls et al. 1995; Steiling et al. 1999). For instance, the IS represents the IP of the test substance based on the time (seconds) of the fi rst appearance of blood hemorrhage (hemorrhage time), vessel lysis (lysis time), or protein coagulation (coagulation time).
Decision Criteria
Several computations of relative scores are interpreted in the decision analysis. The Q-score represents a comparison of the IP of the test substance with that of a reference substance. This score is calculated using the IP of both the test substance and a reference substance. The irritation threshold concentration (ITC) is defi ned as the lowest concentration required to produce a slight or weak response after application of the test substance. The S-score (sever-ity score) is the highest total score for any end point evaluated for a test substance. Based on a range of 0–3, severity scores assigned for each end point are totaled for all of the replicate eggs per test substance, and calculated to produce an end point total score. The severity irri-tation score (SIS) is based on the potency of the antiangiogenic effect of the test substance. The scores are summarized to yield an IS for the test substance (maximum score of 21), for which they are subsequently assigned to an irritancy category according to Table 15.2. Alter-natively, the Federal Hazardous Substance Act classifi cation system (Gettings et al. 1996) assigns irritation classifi cation based on Q-scores, mean coagulation detection time, combi-nation of IS and ITC, and S-scores.

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 211
Based on the IS classifi cation, the HET-CAM is proposed to provide information on the effects that may occur in the human or animal conjunctiva following exposure to a test substance as a response to local injury, rather than monitoring direct cytotoxicology. The test also assumes that acute effects induced by a test substance on capillaries and proteins in the membrane are similar to effects induced by the same test substance in the eye of a treated rabbit and, moreover, correlates to irritation and/or corrosion in human eyes.
Validation
The HET-CAM test method is currently used by a variety of toxicology testing consortia for the identifi cation of ocular corrosives and severe irritants in a tiered testing strategy. In this approach, positive in vitro test results are considered in a WoE decision in order to classify the substance as an ocular corrosive or a severe irritant. The ability of the test to correctly identify ocular corrosives and severe irritants, as defi ned by the EPA (1996), the EU (2001), and the GHS (UN 2003), has been investigated in controlled validation studies (ICCVAM, NIH Publication No. 06-4511, 2006). Based on the results of these studies, the HET-CAM was not recommended for screening and identifying ocular corrosives and severe irritants, as a replacement for the Draize rabbit eye test.
The HET-CAM test method, however, has shown potential to refi ne and reduce animal use in eye irritation testing, when used in the GHS tiered testing scheme. In addition, for assessing severe eye damage, this method reduces the number of animals subjected to test-ing and reduces the pain and suffering of rabbits by their exclusion from the testing of corrosives and severe irritants.
BOVINE CORNEAL OPACITY AND PERMEABILITY ASSAY
Background
The BCOP assay is a modifi ed organotypic model (i.e., isolated whole organ) in vitro eye irritation test method that uses isolated eyes from slaughtered cattle. The assay provides for short-term maintenance of normal physiological and biochemical function of the cornea in an isolated system. The basis of the test relies on the role of the cornea as an indicator of visual impairment resulting from damage occurring during accidental eye exposure to chem-icals. In addition, results obtained with the BCOP assay are comparable to those obtained with the in vivo Draize eye test, since the corneal effects with the latter are of considerable signifi cance in the scoring system for ocular irritancy.
TABLE 15.2Current Models of Alternative Methods for Dermal Toxicology Testing
Name Validation Status Test Method Indicator Test Objective
LLNA ICCVAM/US Allergic skin reaction Skin sensitizationCorrositex ICCVAM/US Visual color changes Skin corrosionEpiDerm EU/US MTT Skin corrosionEpiSkin ECVAM/EU MTT Skin corrosionTER ECVAM/EU Electrical resistance Skin corrosion3T3 phototoxicity ECVAM/EU NRU Phototoxicity
Abbreviations: BCOP, bovine corneal opacity and permeability; EU, European Union; HET-CAM, hen’s egg test–chorioallantoic membrane; ICE, isolated chicken eye; IRE, isolated rabbit eye; US, United States.

212 PRINCIPLES OF TOXICOLOGY TESTING
Overview of the Protocol
As an important transparent tissue in visual functioning, the cornea is the major refracting element in the optical path for light passage through the lens and toward the retina. The BCOP assay measures two important components of ocular irritation affecting the cornea: opacity and permeability. Opacity, which is experimentally determined by the amount of light transmission through the cornea, was initially investigated as the only corneal end point graded in many in vivo ocular irritancy assays. Irritant-induced opacity in the cornea is an indicator of protein denaturation, swelling, vacuolization, or damage in the epithelial or stromal layers. Permeability is measured by the amount of sodium fl uorescein dye that passes through all corneal cell layers. However, since ocular irritation can also involve the iris and conjunctiva, this method originally underrepresented the complex response of the eye to irritants. To overcome the criticisms, several additional end points were added to the BCOP assay, including corneal swelling and histological evaluation of morphological alterations. In addition, the use of opacity and permeability together was purported to better predict ocular irritancy.
Details of the Typical Protocol
The basic procedure has undergone some modifi cations since fi rst described by Gautheron et al. (1992). The BCOP assay uses isolated corneae from the eyes of freshly slaughtered cattle obtained from a local slaughterhouse. The abattoir should be in close proximity to allow for transport of the eyes to the laboratory within 2–4 hours after the ani-mals are killed. Immersion in Hanks’ Balanced salt solution (HBSS) is adequate for storage during transport at either ambient or cold temperatures.
Initial experimental preparatory steps require dissection of the cornea from the eye structure and mounting on a plastic holder fi tted into a two-compartment cell capable of measuring light transmission. Corneae are surgically dissected to avoid epithelial and endothelial damage. A 2–3 mm rim of sclera should remain to assist in subsequent han-dling. Isolated corneae are mounted in corneal holders consisting of anterior and posterior compartments (5 mL volume) which interface with the epithelial and endothelial sides of the cornea, respectively. Both chambers are fi lled with a medium and then the cornea is mounted over an O-ring that is positioned around the opening of the posterior chamber. The chambers are clamped together and the dosing holes located on the top of each chamber allow the epithelial and endothelial sides of the cornea to be treated independently. The assembly is equilibrated at 32°C ± 1°C for 1 hour, after which the medium is changed and baseline opacity measurements are established. After the equilibration period, corneae are examined for defects and are discarded if baseline readings register above 10. With the epithelial side of the cornea in the anterior position, the test substance is introduced in the upper chamber, after which the transmission is measured spectrophotometrically.
Changes in light passage through the cornea are commonly assessed with a white light, dual-beam opacitometer that provides a center-weighted reading of light transmission. The two compartments, each with its own light source and photocell, are individually used for treated corneae and for calibration and standardization.
Permeability measurements are based on the amount of sodium fl uorescein (NaFL) that permeates through the cornea into the posterior chamber. Immediately after completing the fi nal opacity measurements, 1 mL of NaFL (0.4% for liquids and surfactants, 0.5% for solids) is added to the anterior compartment of the corneal holder. The apparatus is incu-bated horizontally for 90 minutes and the amount of dye that penetrates the cornea is deter-mined by measuring the optical density of the medium in the posterior chamber (compared

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 213
with controls) using a microplate reader or UV/VIS spectrophotometer set at 490 nm. Mea-surement of diffusion of dye to the lower chamber is correlated with the opacity index (see the next section, “Decision Criteria”) and with in vivo Draize test scores.
Treatment protocols are separately employed depending on test substance characteris-tics (e.g., for liquids, surfactants, or solids). Application of 750 μL dilutions of the solutions are inserted through the dosing holes and corneae are incubated horizontally for 10 ± 1 minutes at 32°C ± 1°C. The chemical is removed from the anterior compartment and the epithelial surface is washed at least three times. After refi lling both chambers with fresh medium, a second opacity measurement is similarly recorded, after which the corneae are incubated for an additional 2 hours prior to fi nal opacity measurements. Solids are tested as solutions or suspensions for 4 hours at 32°C ± 1°C as 20% dilutions in saline or deionized water. After examination for visible insoluble or suspended particles, test materials are subsequently manipulated as with liquids.
Although the BCOP assay can accommodate a wide range of physicochemical charac-teristics, low density water insoluble substances do not adequately contact the cornea dur-ing treatment. Also, opaque or colored test substances interfere with opacity and permeability measurements. Appropriate controls must also be included that monitor for positive, negative, solvent, and benchmark activity.
Decision Criteria
Mean corrected opacity values and mean corrected permeability values (OD units ± SD) are calculated for each treatment group. As with the HET-CAM assay, an in vitro irritancy score is computed combining both opacity and permeability values. The substance is assigned to categories based on a threshold in vitro irritancy score of 55.1 and above, or a permeability value greater than 0.600, for the identifi cation of severely irritating chemicals. Substances are assigned to categories according to the classifi cation system outlined in Table 15.3.
Alternatively, opacity and permeability values are evaluated independently and com-pared with benchmark materials. Histological observations, if necessary, are used to determine the type and depth of corneal injury, as well as the reversibility of tissue damage. After determination of scores, the cornea is fi xed in 10% neutral buffered formalin (NBF-10) at room temperature for at least 24 hours before processing as per paraffi n sec-tions. Corneal sections are stained in hematoxylin and eosin and examined for lesions in the epithelium, stroma, and endothelium. Thus, histopathological analysis may be of importance for ocular irritants with questionable observable irritation, for clarifi cation of reversibility, to determine occult changes or to reduce false-negative results.
Validation
EU regulatory agencies accept positive outcomes from the BCOP ocular irritation test method for classifying and labeling severe eye irritants. When a negative result is obtained, an in vivo test is subsequently required, as BCOP has not been shown to adequately dis-criminate between eye irritants and non-irritants (EU 2004). The assay is currently used by some U.S. and EU companies as an in-house method to assess the ocular IP of a wide range of substances. In addition, the BCOP test method could potentially be used to identify irreversible, corrosive, and severe IP of drugs and chemicals, in a tiered testing strategy (e.g., GHS; UN 2003). EPA, GHS, and EU have also evaluated and recommended its pos-sible future use for the identifi cation of mild and moderate ocular irritants based on histo-pathological evaluation of corneal tissue; use of a corneal holder that maintains normal

214 PRINCIPLES OF TOXICOLOGY TESTING
TAB
LE 1
5.3
OEC
D T
esti
ng G
uide
lines
(TG
): In
Vit
ro G
uide
lines
for A
cute
, Ocu
lar,
and
Der
mal
Tes
ting
of C
hem
ical
s (A
ccor
ding
to th
e Ye
ar S
ubm
itte
d,
Not
Inc
ludi
ng T
ests
for
In V
itro
Mut
agen
icit
y)
TG N
o an
d th
e Ye
ar S
ubm
itte
dTi
tle
Obj
ecti
veD
escr
ipti
on
429
(201
0)Sk
in S
ensi
tizat
ion,
LL
NA
Indu
ctio
n of
pro
lifer
atio
n of
lym
phoc
ytes
in
mou
se a
uric
ular
lym
ph n
odes
dra
inin
g th
e si
te o
f ch
emic
al a
pplic
atio
n
Prol
ifer
atio
n is
pro
port
iona
l to
the
dose
app
lied;
pro
vide
s a
mea
sure
men
t of
sen
sitiz
atio
n; u
ses
radi
oact
ive
labe
ling
to m
easu
re c
ell p
rolif
erat
ion.
A
red
uced
LL
NA
app
roac
h us
es 4
0% f
ewer
ani
mal
s.43
9 (2
010)
In V
itro
Ski
n Ir
rita
tion
For
haza
rd id
entifi
cat
ion
of ir
rita
nt c
hem
ical
s in
acc
orda
nce
with
the
UN
GH
S C
lass
ifi ca
tion
and
Lab
elin
g (C
ateg
ory
2)
Bas
ed o
n re
cons
truc
ted
hum
an e
pide
rmis
(R
hE);
cel
l via
bilit
y is
mea
sure
d us
ing
the
MT
T a
ssay
. Irr
itant
test
sub
stan
ces
are
iden
tifi e
d by
thei
r ab
ility
to d
ecre
ase
cell
viab
ility
bel
ow d
efi n
ed th
resh
old
leve
ls.
442A
(20
10)
Skin
Sen
sitiz
atio
n L
ocal
Lym
ph
Nod
e A
ssay
: DA
(L
LN
A: D
A)
LL
NA
: DA
is a
non
radi
oact
ive
mod
ifi ca
tion
to th
e L
LN
A m
etho
d fo
r id
entif
ying
pot
entia
l sk
in s
ensi
tizin
g te
st s
ubst
ance
s.
Mea
sure
s th
e pr
olif
erat
ion
of ly
mph
ocyt
es in
duce
d by
test
sub
stan
ces
in
the
auri
cula
r ly
mph
nod
es o
f C
BA
/J m
ouse
str
ain;
bas
ed o
n m
easu
re-
men
t of A
TP
cont
ent b
y bi
olum
ines
cenc
e as
an
indi
cato
r of
pro
lifer
atio
n.44
2B (
2010
)Sk
in S
ensi
tizat
ion
Loc
al L
ymph
N
ode
Ass
ay: B
rdU
-EL
ISA
LL
NA
: Brd
U-E
LIS
A is
a n
onra
dioa
ctiv
e m
odifi
catio
n to
the
LL
NA
met
hod
for
iden
tifyi
ng p
oten
tial s
kin
sens
itizi
ng te
st
subs
tanc
es.
Mea
sure
s th
e pr
olif
erat
ion
of ly
mph
ocyt
es in
duce
d by
test
sub
stan
ces
in
the
auri
cula
r ly
mph
nod
es o
f C
BA
/JN
mou
se s
trai
n; b
ased
on
mea
sure
-m
ent o
f B
rdU
con
tent
, an
anal
ogue
of
thym
idin
e, a
s an
indi
cato
r of
pr
olif
erat
ion.
437
(200
9)B
ovin
e C
orne
al O
paci
ty a
nd
Perm
eabi
lity
Test
M
etho
d fo
r id
entif
ying
ocu
lar
corr
osiv
es a
nd
seve
re ir
rita
nts
Use
d to
cla
ssif
y su
bsta
nces
as
ocul
ar c
orro
sive
s an
d se
vere
irri
tant
s; to
xic
effe
cts
to th
e co
rnea
are
mea
sure
d as
opa
city
and
per
mea
bilit
y (s
ee te
xt).
438
(200
9)Is
olat
ed C
hick
en E
ye (
ICE
) Te
st
Met
hod
for
iden
tifyi
ng o
cula
r co
rros
ives
and
se
vere
irri
tant
sTo
xic
effe
cts
to th
e co
rnea
are
mea
sure
d by
a o
paci
ty, a
sses
smen
t of
dam
age
to e
pith
eliu
m, m
easu
rem
ent o
f in
crea
sed
thic
knes
s, a
nd
eval
uatio
n of
mac
rosc
opic
mor
phol
ogic
al d
amag
e to
the
surf
ace
(see
text
).

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 215
435
(200
6)In
Vit
ro M
embr
ane
Bar
rier
Tes
t M
etho
d fo
r id
entif
ying
ski
n co
rros
ives
Use
s an
art
ifi ci
al m
embr
ane
desi
gned
to r
espo
nd to
cor
rosi
ve s
ubst
ance
s,
base
d on
the
subs
tanc
e pe
netr
atio
n tim
e th
roug
h th
e m
embr
ane
barr
ier.
Com
pose
d of
two
com
pone
nts,
a s
ynth
etic
mac
rom
olec
ular
bio
barr
ier
and
a ch
emic
al d
etec
tion
syst
em. M
easu
res
elap
sed
time
betw
een
appl
icat
ion
of th
e te
st s
ubst
ance
to th
e m
embr
ane
barr
ier
and
barr
ier
pene
trat
ion.
428
(200
4)Sk
in A
bsor
ptio
n: I
n Vi
tro
Met
hod
Prov
ides
info
rmat
ion
on a
bsor
ptio
n of
a
radi
olab
eled
test
sub
stan
ceV
iabl
e hu
man
or
anim
al s
kin
used
to m
easu
re th
e re
cept
or fl
uid
and
the
dist
ribu
tion
and
abso
rptio
n pr
ofi le
of
the
test
sub
stan
ce in
a tw
o-ch
ambe
r di
ffus
ion
test
sys
tem
, with
tim
e.43
0 (2
004)
In V
itro
Ski
n C
orro
sion
: T
ER
Tes
tM
etho
d fo
r id
entif
ying
ski
n co
rros
ives
Te
st m
ater
ial i
s ap
plie
d to
epi
derm
al s
urfa
ces
of s
kin
disk
s se
para
ting
com
part
men
ts. L
oss
of n
orm
al s
trat
um c
orne
um in
tegr
ity a
nd b
arri
er
func
tion
are
the
indi
cato
rs, a
ided
by
redu
ctio
n in
TE
R b
elow
a th
resh
old
leve
l (5
k f
or r
at).
431(
2004
)In
Vit
ro S
kin
Cor
rosi
on: H
uman
Sk
in M
odel
Tes
t Id
entifi
cat
ion
of c
orro
sive
and
non
corr
osiv
e ch
emic
al s
ubst
ance
s w
hen
supp
orte
d by
a
wei
ght-
of-e
vide
nce
dete
rmin
atio
n
Test
mat
eria
l is
appl
ied
to a
3-D
hum
an s
kin
mod
el, c
onsi
stin
g of
a
reco
nstr
ucte
d ep
ider
mis
with
a f
unct
iona
l str
atum
cor
neum
. Ide
ntifi
ed b
y a
decr
ease
in c
ell v
iabi
lity,
and
bas
ed o
n ab
ility
to p
enet
rate
the
stra
tum
co
rneu
m b
y di
ffus
ion/
eros
ion.
432
(200
4)In
Vit
ro 3
T3
NR
U
Phot
otox
icity
Tes
tE
valu
ates
pho
to-c
ytot
oxic
ity b
y th
e re
lativ
e re
duct
ion
in v
iabi
lity
of c
ells
exp
osed
to th
e ch
emic
al in
the
pres
ence
ver
sus
abse
nce
of li
ght
Use
s B
alb/
c 3T
3 fi b
robl
ast m
onol
ayer
s. C
ytot
oxic
ity b
ased
on
conc
entr
atio
n-de
pend
ent r
educ
tion
of ly
soso
mal
upt
ake
of th
e vi
tal d
ye
NR
; pos
sibl
e to
dis
tingu
ish
viab
le, d
amag
ed, o
r de
ad c
ells
fro
m e
ach
othe
r.
Abb
revi
atio
ns: A
TP,
ade
nosi
ne tr
ipho
spha
te; B
rdU
, 5-b
rom
o-2-
deox
yuri
dine
; LL
NA
, loc
al ly
mph
nod
e as
say;
NR
, neu
tral
red
; TE
R, t
rans
cuta
neou
s el
ectr
ical
res
ista
nce.

216 PRINCIPLES OF TOXICOLOGY TESTING
corneal curvature; or, examining the effect of modifying various test method protocol com-ponents, such as duration of exposure, on the accuracy and reliability of the test method.
Based on the results of current validation studies, there is suffi cient amount of data to support the use of the BCOP test method, in appropriate circumstances and with certain limitations, as a screening test to identify substances as ocular corrosives and severe irri-tants (i.e., EPA Category I, UN GHS Category 1, EU Category R41) in a tiered-testing strategy (ICCVAM, NIH Publication No. 06-4511, 2006). However, ICCVAM proposed that the BCOP test method is not recommended to identify substances from all hazard cat-egories as defi ned by the GHS, EPA, and EU classifi cation systems (EPA 1996; EU 2001; UN 2003), particularly because of the signifi cant lesions associated with 50% of the EPA Category III substances that were false negative in BCOP. In addition, the BCOP test method can be used as a screening test to identify substances not labeled as irritants (i.e., EU Not Labeled, GHS Not Classifi ed), from all other hazard categories (GHS Category 1, 2A, or 2B) (ICCVAM 2009).
ISOLATED CHICKEN EYE TEST
Background
The ICE protocol was fi rst developed based on the IRE method (see the section, “Isolated Rabbit Eye Test”) in order to obviate the need for laboratory animals as the source for test eyes. For this reason it has remained essentially the same as the IRE assay. The ICE test method is an organotypic in vitro bioassay where the test substance is applied to the cornea of eyes isolated from chickens that have been processed for human consumption. The model provides short-term maintenance of normal physiological and biochemical function of the chicken eye in an isolated system.
Overview of the Protocol
Enucleated chicken eyes are immediately dissected and irrigated with isotonic saline. Test substances are applied as single doses for 10 seconds, followed by rinsing with isotonic saline. Corneal reactions are measured at regular intervals up to 4 hours post-treatment. As with the BCOP assay, damage by the test substance is assessed by determination of corneal indicators: (1) corneal swelling; (2) opacity; and (3) fl uorescein retention (FR).
1. Corneal swelling is determined by calculating the increase in corneal thickness, a quantitative and reliable end point of corneal injury, from a baseline measurement.
2. The degree of corneal damage, as determined by measuring corneal opacity, pro-vides an indicator that is directly correlated to the in vivo rabbit eye test.
3. FR provides an assessment of corneal permeability, indicative of damage to the corneal surface. For each substance, a quantitative score is calculated to determine an overall Irritation Index or the chemical is qualitatively assigned to an in vitro irritancy classifi cation.
Details of the Typical Protocol
Enucleated chicken eyes are isolated from spring chickens of either sex, approximately 7 weeks old and weighing 2.5–3.0 kg. The laboratory should be located close to the breeder or slaughterhouse so that transportation to the laboratory and processing are accomplished within 2 hours. The temperature range during transport is not considered critical. Eyes are

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 217
immediately dissected and placed in a superfusion apparatus supplied with isotonic saline (32°C ± 1.5°C) through a steel tube attached to a peristaltic pump. Each eye is mounted in a custom-built stainless steel clamp with the cornea positioned vertically and then transferred to a chamber in the superfusion apparatus. Eyes are examined with a slit-lamp microscope for these minimum viability requirements and baseline measurements: corneal thickness should not deviate by more than 10% from the mean value, should not have an FR score of greater than 0.5 (indicating corneal permeability), and should not show corneal opacity or any other signs of damage.
After an equilibration period, the eye is removed from the superfusion apparatus and placed horizontally in the holder. Thirty microliters of a test substance is applied as a single dose for 10 seconds, followed by rinsing with isotonic saline (20 mL). Following exposure to the chemical substance, the extent of damage to the cornea is determined by measuring the corneal indicators. Corneal reactions are measured at regular intervals up to 4 hours post treatment, and mean values for each parameter (corneal swelling and opacity) are determined. FR is evaluated at 30 minutes post treatment. Test eyes and appropriate con-trols, including positive, negative, solvent, and benchmark controls, as well as control eyes, are examined prior to treatment and at 30, 75, 120, 180, and 240 minutes after treatment.
Quantitative measurements, expressed as percent of control, are determined from anal-ysis of corneal swelling only, thus supporting improved precision and reduced interlabora-tory variability. Corneal thickness measurements, as indicators of corneal swelling, are performed with an optical pachymeter on a slit-lamp microscope, measured at the corneal apex. Corneal opacity is also determined with slit-lamp examination, by scoring the area of the cornea that is most densely opacifi ed. Based on the highest mean score for corneal opacity observed at any time point, an overall category score is assigned for each test sub-stance. Mean FR values for all test eyes are calculated at 30-minute time points post treat-ment only. Substances that adhere to the cornea must be adequately removed before FR is determined. After the fi nal examination at 4 hours, eyes are typically fi xed in 4% NBF for histopathological examination.
Decision Criteria
For the ICE assay method, the IP of the test substance is based on the maximum mean values of the measurements and is defi ned within categories ranging from non-irritant to severely irritant. The four irritancy categories, therefore, are interpreted from corneal thickness, cor-neal opacity, and fl uorescein retention measurements, monitored alone or in combination, according to the classifi cations listed in Table 15.4.
TABLE 15.4ICE Irritancy Classification System
Parameter Measurement Score Category*
Corneal thickness Mean corneal swelling Percentage I to IVCorneal Opacity Mean maximum score 0 to 4.0 I to IVFR Mean FR score 0 to 3.0 I to IV
*Category assignments for test substances include: I, non-irritant; II, slight irritant; III, moderate irritant; or, IV, severe irritant.Abbreviations: ICE = isolated chicken eye test method; FR = fl uorescein retention.

218 PRINCIPLES OF TOXICOLOGY TESTING
Validation
In validation studies conducted by ICCVAM, the accuracy of the ICE test method for iden-tifying ocular corrosives and severe irritants, as defi ned by the EPA (1996), EU (2001), and GHS (UN 2003), was evaluated and assessed in separate in vitro–in vivo comparative studies and found to be comparable among the three hazard classifi cation systems (83–87%). The validation studies concluded that the method evaluates only corneal effects. It does not take into account the infl uence on the iris and the conjunctiva and does not measure reversibility of corneal effects. Systemic effects following ocular instillation are not observed and the assay will not identify slow-acting irritants. However, there are suffi cient data to support the use of the ICE test method as a screening test to identify substances as ocular corrosives and severe irritants (i.e., EPA Category I, UN GHS Category 1, EU R41) in a tiered-testing strat-egy, as part of a WoE approach. Furthermore, the study recommended that histopathological evaluation of the corneal tissue, using a standardized scoring scheme, should be included to characterize and improve the usefulness of the method (ICCVAM, NIH Publication No. 06-4511, 2006).
ISOLATED RABBIT EYE TEST
Background
The IRE test was developed as an in vitro alternative to the in vivo Draize rabbit eye test method for the assessment of eye irritation (rabbit enucleated eye test; Burton et al. 1981). As with the ICE, the IRE is an organotypic test method that eliminates the use of live animals for ocular irritancy testing and thus obviates the pain and suffering potentially associated with the in vivo Draize rabbit eye test. The method is also purported to incorporate rabbit eyes isolated from euthanized animals used for other purposes, such as a food source.
Overview of the Protocol and Validation Summary
In the IRE test method, candidate substances are applied over the corneae of enucleated rab-bit eyes in a manner similar to the ICE test. As with the BCOP, the effects of the test sub-stance on the cornea of the isolated eye are measured quantitatively as an increase in thickness (swelling), opacity, and fl uorescein penetration, as well as for morphological changes to the corneal epithelium. The lack of a widely accepted standardized protocol for detecting ocular corrosives and severe irritants, however, has hampered the acceptance of this test method as a replacement for the Draize rabbit eye test. In addition, based on the accuracy, false-negative and false-positive rates (35–40%), ICCVAM did not recommend the method for screening and identifying ocular corrosives, severe irritants, or for distin-guishing non-irritants from mild irritants (ICCVAM 06-4511, 2006; ICCVAM 2009). Also, there are insuffi cient data using all four recommended IRE end points (corneal opacity, corneal swelling, fl uorescein penetration, and histological observations of signifi cant effect on corneal epithelium). Thus, any further information about the model and the protocols are found in other reviews (Guerriero et al. 2004).
EPIOCULAR™ MODEL
The EpiOcular (OCL-200, MatTek Corporation, Ashland, Massachusetts, USA) is an in vitro human corneal model designed to replace the traditional animal Draize eye test. The model consists of a three-dimensional in vitro human corneal epithelium (HCE) composed of nor-mal human-derived epidermal keratinocytes cultured on a permeable polycarbonate mem-brane that forms a stratifi ed squamous multilayered epithelium similar to that of the cornea.

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 219
The tissue construct has an air–liquid interface and exhibits morphological and growth char-acteristics that mimic in vivo conditions.
Cellular viability is measured with the MTT colorimetric assay using an IC50
end point for comparison among different potential toxicants. The model provides data measuring cellular cytotoxicology as a refl ection of ocular irritation. The test is purported to yield eye irritation data comparable to responses to the in vivo rabbit eye test. Although not validated, the test can distinguish among the IP of mildly toxic products.
HCE™ METHOD
Similar to the EpiOcular™ model, the HCE™ (SkinEthic Laboratories, Nice, France) model consists of immortalized human corneal epithelial cells cultured at the air–liquid interface on a polycarbonate substrate membrane. The air–epithelial tissue lacks a stratum corneum and thus morphologically resembles the corneal mucosa of the human eye. Cellular viability is measured with the MTT colorimetric assay, as well as other in vitro indicators, particularly LDH release, histology, quantifi cation of cytokine release, and gene expression. ECVAM sponsored in vivo–in vitro comparisons in formal validation studies, the results of which currently indicate the usefulness of the model as a prescreen for eye irritation of test ingre-dients and raw materials. Although the method has not satisfi ed a complete formal validation study, the HCE model is suitable to detect corneal repair and recovery in vitro.
DERMAL TOXICOLOGY TESTING
TIERED TESTING APPROACH
Screening techniques to detect local toxicity are used to investigate the effects of various irritating substances on the skin and in the lungs. Substances studied in lung cell cultures include tobacco smoke, diesel exhausts, nitrogen dioxide, aldehydes, phorbol esters, and paraquat. The isolation of alveolar epithelial cells and nonciliated bronchiolar epithelial (Clara) cells and their establishment in primary culture greatly accelerated the underlying mechanisms of the local pulmonary responses to xenobiotics. The culturing conditions, however, are not without problems and are often labor intensive (see previous sections on establishing primary cell cultures). In addition, continuous cultures of these cells are diffi cult to maintain in their differentiated state. Despite this, the need for isolating dermal and lung cells is especially warranted, particularly because they are known to be instrumental in the infl ammatory response.
DEVELOPMENT OF IN VITRO DERMAL TOXICOLOGY TESTING METHODS
Earlier attempts to assess skin irritation of chemicals in vitro have met with considerable setbacks. The defi ciencies associated with dermal fi broblasts as models for skin toxicity studies center around the continuing controversy of the dedifferentiation of the cells once they are maintained in serial culture. They lose their phenotypic appearance and characteris-tic features of their original parent cells. Recent progress in dermal toxicological studies has concentrated on the development of organotypic models that mimic mammalian skin. Table 15.2 lists current models for in vitro dermal toxicology testing.
As with in vivo testing, the OECD TG for in vitro testing are a collection of relevant test methods used by government, industry, and academic laboratories to determine the safety of chemicals and chemical preparations. The methods have been designed by international cooperation and include tests for physicochemical properties of chemicals, human health

220 PRINCIPLES OF TOXICOLOGY TESTING
effects, environmental effects, and degradation and accumulation in the environment. Table 15.3 lists and summarizes OECD in vitro TG for dermal in vitro toxicity testing (Section 4, OECD Guidelines for the Testing of Chemicals), not including in vitro testing for mutagenicity (Chapter 16). Although most of these tests are classifi ed as in vitro alter-natives to animal testing protocols, some of them, such as OECD TG 442A, are modifi ed versions of original animal methods (local lymph node assay, LLNA) that reduce and refi ne the use of animals for those applications.
Murine Local Lymph Node Assay
LLNA (OECD TG 429; Table 15.3) is a scientifi cally accepted protocol used to assess the potential of substances to produce skin sensitization. LLNA uses mice as replacements for guinea pigs in the conventional methodology and is based on the ability of a chemical to cause allergic reactions after repeated application to the skins of 20–40 animals. Although not a true in vitro method, the validity of the assay was endorsed by independent peer review evaluation based on its ability to cause less pain and distress to animals (ICCVAM, 2005). A reduced LLNA approach is described in OECD TG 429 which uses up to 40% fewer ani-mals. It incorporates radiolabeling with [3H]-methyl thymidine as the indicator (Stimulation Index).
Corrositex™ Assay
The Corrositex assay (InVitro International, Irvine, California, USA) is a standardized and quantitative in vitro test for assessing skin corrosion. It is based on the time required for a test chemical to penetrate a biobarrier membrane. The membrane is composed of a reconstructed collagen matrix, developed to mimic the physicochemical properties of rat skin. The time needed for visual color change is inversely proportional to the corrosivity level of the test chemical. Corrositex was granted regulatory approval by the U.S. Department of Transporta-tion, EPA, Food and Drug Administration, Occupational Safety and Health Administration, and the Consumer Product Safety Commission. The method is a replacement for the in vivo Draize rabbit skin test and its use is limited to assessing corrosivity of specifi c classes of chemicals including organic bases and inorganic acids.
EpiDerm™, Episkin™, TER, and NRU
The European Commission on the Validation of Alternative Methods (ECVAM) has vali-dated three alternative in vitro test models for detecting skin corrosivity: EpiDerm (EPI-200, MatTek Corporation, Ashland, Massachusetts, USA), Episkin (Imedex, Inc., Alpharetta, Georgia, USA), and the rat skin transcutaneous electrical resistance (TER) method.
The EPI-200 skin corrosion test is a validated alternative method to the Draize rabbit skin corrosion test in both the United States and EU. Episkin is validated for identifying corrosive properties of chemicals. It is an accepted test in the EU and for two of the three UN packing group classifi cations (groups I and II/III, ICCVAM, 2002).2 Both EpiDerm and Episkin use a three-dimensional system consisting of a reconstructed human epidermis and functional stratum corneum. Analysis of the stratum corneum using transmission elec-tron microscopy reveals intercellular lamellae characteristic of normal human epidermis. Unlike the previous models, EpiDerm appears to have hemidesmosomes arranged in a well-organized basement membrane. The tests are validated in Canada and the EU as total
2 Group assignment is based on a chemical’s ability to cause skin corrosion after 3 minutes, 1 hour, and 4 hours, respectively.

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 221
replacements for animal-based skin corrosion studies. In addition, EpiDerm is the only commercially available organotypic in vitro model for assessing dermal corrosivity approved by both EU and U.S. regulatory agencies.
The TER method monitors changes in transcutaneous electrical resistance (TER; OECD TG 430) in a rat skin disk as a result of chemical exposure over time. Substances with resistance values greater than 5 k /cm are considered noncorrosive. The TER method is validated and is part of a battery of tests for skin corrosion in the EU.
ECVAM, in collaboration with the EU and European Cosmetics, Toiletry and Perfum-ery Association (COLIPA), has completed validation studies on other tests including the 3T3 neutral red uptake (NRU; OECD TG 432) phototoxicity test and its application to ultraviolet fi lters (ECVAM, 1998–2001). The in vitro NRU test, a standard cell viability assay, uses BALB/c 3T3 mouse fi broblasts to determine the cytotoxic effect of a chemical in the presence or absence of non-cytotoxic ultraviolet radiation. The NRU test is validated in the EU as a replacement for acute animal phototoxicity studies.
SUMMARY
The development of in vitro methods for assessing acute local toxicity has progressed con-siderably in the past 20 years as a result of similarities of the mechanisms of the test methods to the traditional in vivo protocols. In fact, the cell culture models for local toxicology test-ing, in particular, incorporate several features consistent with, and at times superior to, the animal testing methods for dermal and ocular toxicology, specifi cally:
1. Incubation times in vitro are comparable to those used in vivo.2. The test substance comes in direct contact with the ocular or dermal epithelium.3. With the exception of the uveal layer of the cornea, the lack of blood vessels allows
for attainment of steady-state chemical concentrations in contact with tissue; thus results obtained from test protocols are not skewed by interruptions in absorption or local pharmacokinetic phenomena.
Alternatively, caution must be exercised with certain aspects of cell culture methodology when screening for ocular irritancy. For example, false-positive and false-negative results are usually errors of estimation and are closely related with the physicochemical proper-ties of a test substance. Severely irritating properties of a substance in vivo, for instance, may be accounted for by extreme pH, which may not be detected in vitro. In addition, diffi culty with solubility of a test substance in a solvent may underestimate true toxic concentrations.
While no single in vitro method accounts for all of these parameters, the combination of individual tests addresses these end points. The anticipation for success relies on diligent collective cooperation among the scientifi c, regulatory, industrial, commercial, and public representatives, using the concept of a battery of validated assays to screen chemicals with potential for human toxicity.
SUGGESTED READINGS
Barile FA. Cell culture methods for acute toxicology testing. Chapter 13 In: Principles of Toxicology Testing. New York: Informa HealthCare, 2007: 175–202.
Barile FA. Continuous cell lines as a model for drug toxicity assessment. In: Castell JV, Gómez-Lechón MJ, eds. In Vitro Methods in Pharmaceutical Research. London: Academic Press, 1997.

222 PRINCIPLES OF TOXICOLOGY TESTING
Calabro AR, Konsoula R, Barile FA. Evaluation of in vitro cytotoxicity and paracellular permeability of intact monolayers with mouse embryonic stem cells. Toxicol In Vitro 2008; 22: 1273.
Cooper KJ, Earl LK, Harbell J, Raabe H. Prediction of the ocular irritancy of prototype shampoo formulations by the isolated rabbit eye (IRE) test and bovine corneal opacity and permea-bility assay (BCOP) assay. Toxicol In Vitro 2001; 15: 95.
Ekwall B. Basal cytotoxicity data (BC data) in human risk assessment. In Proceedings of Workshop on Risk Assessment and Risk Management of Toxic Chemicals. National Institute for Envi-ronmental Studies, Ibaraki, Japan, 1992: 137.
Gautheron P, Dukic M, Alix D, Sina JF. Bovine corneal opacity and permeability test: an in vitro assay of ocular irritancy. Fundam Appl Toxicol 1992; 18: 442.
McNamme P, Hibatallah J, Costabel-Farkas M, et al. A tiered approach to the use of alternatives to animal testing for the safety assessment of cosmetics: eye irritation. Regul Toxicol Pharma-col 2009; 54: 197.
REVIEW ARTICLES
Atkinson KA, Fentem JH, Clothier RH, Balls M. Alternatives to ocular irritation testing in animals. Lens Eye Toxicol Res 1992; 9: 247.
Barile FA, Cardona M. Acute cytotoxicity testing with cultured human lung and dermal cells. In Vitro Cell Dev Biol 1998; 34: 631.
Basketter DA, Chamberlain M, Griffi ths HA, et al. The classifi cation of skin irritants by human patch test. Food Chem Toxicol 1997; 35: 845.
Burton AB, York M, Lawrence RS. The in vitro assessment of severe eye irritants. Food Cosmet Toxicol 1981; 19: 471.
Cockshott A, Evansl P, Ryan CA, et al. The local lymph node assay in practice: a current regulatory perspective. Hum Exp Toxicol 2006; 25: 387.
Courtellemont P, Pannetier M, Biesse JP, et al. Evaluation of the Eytex(TM) system in the COLIPA eye irritation program. Toxicology in Vitro 1999; 13: 295.
Dearman RJ, Basketter DA, Kimber I. Local lymph node assay: use in hazard and risk assessment. J Appl Toxicol 1999; 19: 299.
Fentem JH, Botham PA. ECVAM’s activities in validating alternative tests for skin corrosion and irritation. Altern Lab Anim 2002; 30: 61.
Hoffman J, Heisler E, Karpinski S, et al. Epidermal Skin Test 1000 (EST-1000): a new reconstructed epidermis for in vitro skin corrosivity testing. Toxicol In Vitro 2005; 19: 925.
Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), Biennial Progress Report of National Toxicology Program, NIH Publication 04-4509, National Institute of Environmental Health Sciences, Research Triangle Park, NC, 2003. [Available from: http://iccvam.niehs.nih.gov/about/annrpt/bienrpt044509.pdf]
ICCVAM Test method evaluation report: In vitro ocular toxicity test methods for identifying severe irritants and corrosives. NIH Publication No.: 06-4511, National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2006.
ICCVAM, Background review document: Current status of in vitro test methods for identifying ocu-lar corrosives and severe irritants: bovine corneal opacity and permeability test method. (NIH Publication No.: 06-4512, National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2006.
ICCVAM, Background review document: Current status of in vitro test methods for identifying ocu-lar corrosives and severe irritants: isolated chicken eye test method. NIH Publication No.: 06-4513, National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2006.
ICCVAM, Background review document: Current status of in vitro test methods for identifying ocular corrosives and severe irritants: isolated rabbit eye test method. NIH Publication No.: 06-4514,

ACUTE LOCAL TOXICOLOGY TESTING IN VITRO 223
National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2006.
ICCVAM, Background review document: Current status of in vitro test methods for identifying ocu-lar corrosives and severe irritants: hen’s egg test - chorioallantoic membrane test method. NIH Publication No.: 06-4515, National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2006.
ICCVAM. Draft proposed ICCVAM test method recommendations: Evaluation of the validation status of alternative ocular safety testing methods and approaches. National Toxicology Program (NTP), NICEATM, NIEHS, NIH, U.S. Public Health Service, Department of Health and Human Services, 2009.
Jones PA, Budynsky E, Cooper KJ, et al. Comparative evaluation of fi ve in vitro tests for assessing the eye irritation potential of hair care products. Altern Lab Anim 2001; 29: 669.
Kimber I, Dearman RJ, Betts CJ, et al. The local lymph node assay and skin sensitization: a cut-down screen to reduce animal requirements? Contact Dermatitis 2006; 54: 181.
Robinson MK, Perkins MA. A strategy for skin irritation testing. Am J Contact Dermat 2002; 13: 21.Robinson MK, Cohen C, de Fraissinette Ade B, et al. Non-animal testing strategies for assessment of
the skin corrosion and skin irritation potential of ingredients and fi nished products. Food Chem Toxicol 2002; 40: 573.
Stobbe JL, Drake KD, Maier KJ. Comparison of in vivo (Draize method) and in vitro (Corrositex assay) dermal corrosion values for selected industrial chemicals. Int J Toxicol 2003; 22: 99.
Ubels JL, Ditlev JA, Clousing DP, Casterton PL. Corneal permeability in a redesigned corneal holder for the bovine cornea opacity and permeability assay. Toxicol In Vitro 2004; 18: 853.
Ubels JL, Pruis RM, Sybesma JT, Casterton PL. Corneal opacity, hydration and endothelial morphol-ogy in the bovine cornea opacity and permeability assay using reduced treatment times. Toxicol In Vitro 2000; 14: 379.
U.S. Environmental Protection Agency, Health Effects Test Guidelines, OPPTS 870.2400, Acute Eye Irritation, 1998.

224
16 Toxicokinetic studies in vitro
INTRODUCTION
Toxicokinetics refers to the events surrounding the metabolic fate of a compound adminis-tered in vivo or in vitro and how this metabolism affects the concentration of the compound in the target organ. Organs, tissues, and cells, therefore, are the objective of chemical distribu-tion. Toxicokinetics therefore refers to numerous events that affect the absorption of a toxic dose of a chemical in the gastrointestinal tract, through the skin, via the lungs, or across the mucous membranes. To whatever extent absorption occurs, the chemical is modifi ed to a related compound, albeit more or less toxic. This metabolism occurs primarily in the liver, but also takes place in other metabolically active organs such as the lungs or kidneys.
Metabolism notwithstanding, and depending on its physicochemical properties, the chemical either circulates or is then distributed to and sequestered in various organs. Finally, the substance is eliminated either actively or passively through the extensive fi ltra-tion processes of the kidney. Alternatively, the chemical is selectively eliminated through the lungs, skin, or intestinal tract.
Toxicokinetics contributes to the risk assessment estimation for the chemical based on calculated, potential exposure to humans or animals. In vitro methods participate in toxico-kinetic modeling and risk assessment by supplementing data derived from in vivo studies. Not surprisingly, most toxicokinetic effects are measured in cell culture systems as rela-tively simple interactions between chemical substances and cell functions.
The following discussion introduces the concepts of kinetic distribution and metabo-lism of test compounds in in vitro systems and coordinates the information currently avail-able with future studies. Chapter 3, entitled “Toxicokinetics”, describes the general principles of absorption, distribution, metabolism, and elimination.
METABOLIC STUDIES IN CELL CULTURE
The liver plays a key role in the detoxifi cation of toxic substances but it can also transform a chemical into a metabolite more toxic than the parent compound. It is possible for the toxic metabolite to induce damage to the liver and other target organs. The underlying mechanism for liver injury from many chemicals whose targets are not specifi c for that organ may be explained through a toxicokinetic distribution.
In primary cultures of hepatocytes, it is possible to study both metabolite-induced liver toxicity and the pharmacological aspect of liver metabolism: the detoxifi cation or activa-tion of chemicals. Cells from rodents and humans are used and maintained in cultures either as organ cultures, monolayers, or isolated cells in suspension. The cells from mice and rats are obtained from organ perfusion of the liver, using collagenase to separate the cells rapidly and gently. Cytochrome P450 enzymes are functional and are maintained in

TOXICOKINETIC STUDIES IN VITRO 225
these systems from a few hours to several days. Cultures of human hepatocytes are also realized with co-culture of feeder layers of other continuous cell lines. Recently, functional human hepatocytes have been maintained in a viable state after freezing, very important because liver metabolism is species specifi c. Another development is the use of cell lines with preserved P450 activity instead of primary cultures of liver cells. Finally, several sys-tems co-culturing hepatocytes and other target cells are used for toxicokinetic modeling studies.
Among the different cell types of cell cultures described, metabolic and hepatocellular transporting properties are studied with a large number of substances. Examples of specifi c research areas include (1) comparison of metabolizing capacities of liver cells and S9 mix-tures, (2) cytochrome P450 activity preservation in hepatocytes, (3) species-specifi c metab-olism, (4) the protective effects of reduced glutathione (GSH) in the presence of liver damage, and (5) the comparison of the metabolizing capacity of the liver and those of other organs such as kidneys, lungs, and intestine.
BIOTRANSFORMATION IN CELL CULTURE
Enzymatic Metabolism
Most cells in vivo have the capacity to enzymatically metabolize chemicals to less toxic, more water-soluble compounds, that are subsequently excreted through the urine. This capacity is clearly most pronounced in liver cells, but kidney, lung, intestine, and skin have extensive capacities for xenobiotic metabolism. Most other organs, such as blood cells, endothelial cells, muscles, and connective tissues, have relatively low capacities for biotrans-formation. The main aspect of the biotransformation of chemicals by the most active cells is the ability to add functional groups (phase I reactions) that later are bound to endogenous substances such as glucuronic acid or GSH, resulting in increased water solubility of the metabolite (phase II biotransformation).
The most important phase I enzyme is cytochrome P450 oxygenase, inducible by such chemicals as phenobarbital. Examples of other more commonly found enzymes are epox-ide hydrolase (phase I) and the transferases (phase II). It is important to note that the described biotransformation may result in more reactive (toxic) metabolites. Details of these reactions are discussed in chapter 3.
Maintenance cultures of specifi c cells with high metabolic activity display biotransfor-mation capacity, including signifi cant P450 activity for several hours or days. Whether this short period is suffi cient to mimic clinically relevant metabolism of all types of chemicals is questionable. Early passages of fi nite lines, especially of fetal cells, may display certain metabolic activities. Most other cultures of both fi nite and continuous cell lines, however, demonstrate low or immeasurable P450 levels that are sometimes coupled with hydrolase or glucuronyl transferase activity. The P450 activity found in some cell lines is increased by induction in vitro. Thus, few cell lines have some biotransformation capacity compared with the least competent cell types in vivo.
Primary cultures of rat hepatocytes are used as metabolic activation systems in a variety of toxicity assays. As mentioned earlier, freshly isolated cells are used to ensure metabolic integrity since continuous cultures lose metabolic capacity with time. Furthermore, some dif-ferences exist in the abilities of animal and human tissues to metabolize chemicals, although variations among individual donors may circumvent the intrinsic variations encountered between species. Consequently, primary cultures are suitable models for assessing biotrans-formation in vitro.

226 PRINCIPLES OF TOXICOLOGY TESTING
Use of S9 Mixture
Two methods are presently available to increase the capacity of biotransformation of cell cultures: (1) target cells are cultured on a feeder layer of more metabolically competent cells, for example, BHK21 cells irradiated to inhibit mitotic division, and (2) whole micro-somes are added to the culture medium or preferably an ultracentrifuged fraction of homog-enized liver from rats or mice pretreated with Arochlor 1254, thus containing P450 and other important microsomal enzymes. This fraction plus the NADPH cofactor is called the S9 mixture.
The fi rst attempts to overcome the obstacle of limited metabolic capacity in short-term test systems were based on the observation that the metabolism required to activate many carcinogens to their active form was localized primarily with mixed function oxidases in the endoplasmic reticula of liver cells. The shortcomings of the Ames test conclusively demonstrated that the predictivity of the bacterial test for carcinogens substantially increased with the addition of a crude fraction of rat liver homogenate. These fi ndings established the hepatic S9 fraction as the activation system of choice in most short-term testing protocols.
S9-reinforced cell line cultures offer moderate advantages over primary cultures of hepa-tocytes: (1) the system is fl exible, easy to prepare and can be used with a variety of cell cul-ture models, (2) drug-metabolizing enzymes are relatively stable in storage and are characterized biochemically prior to incorporation into the experiment, and (3) the system enhances phase I reactions such as the formation of reactive metabolites, but induces relative decreases of the biosynthetic phase II reactions since energy-rich phase II cofactors are not included in the mix.
Signifi cant disadvantages often discourage the use of the mix: (1) S9-supplemented cultures require the addition of exogenous cofactors such as NADPH, (2) supplementation is cytotoxic to many systems and test results and enzyme activities per milligram of protein vary considerably, (3) the mixtures often exhibit technical variations in the enzymes, cofac-tors, exposure conditions, concentrations of the S9 in the mix, and enzyme inducers, and (4) the enzyme activity may not yield results that refl ect the species, sex, and specifi city of the organ of origin.
Preparation of S9 Fractions
The S9 activation system is derived from the supernatant of the 9000× g spin from Aroclor 1254-induced rat liver homogenates. Upon injection in rats, Aroclor 12541 induces a broad spectrum of drug metabolizing enzymes. The liver is then homoge-nized, and the microsomes, containing the phase I mixed function oxidase enzymes, are isolated from the continuous endoplasmic reticulum fragments using differential centrifugation.
The supernatant S9 fraction is used as the crude homogenate or is further enriched for microsomal activity by additional high speed centrifugation. This separates a highly enriched microsomal pellet from the supernatant cystolic fraction. The former contains phase I biotransformation enzymes while the supernatant is rich in many of the enzymes of phase II (conjugative) metabolism. An S9 buffer mixture is then formulated with cofactors as illustrated in Figure 16.1.
1 A mixture of polychlorinated biphenyls or a combination of phenobarbital and β-naphthafl avone is also used.

TOXICOKINETIC STUDIES IN VITRO 227
IN VITRO STUDIES OF ABSORPTION, DISTRIBUTION, METABOLISM, AND ELIMINATION
The absorption, distribution, and elimination (ADME) of a chemical in vivo are infl uenced by the interaction of biotransformation processes with the substance. Listed below are some of the factors that affect these processes:
1. The ability and time that a substance requires to cross cellular barriers such as the intestinal mucous membranes, blood–brain barrier, and skin.
2. The ability of the substance to enter and bind to the target site. 3. The extent of binding to blood proteins and accumulation and sequestration in lipid
compartments. 4. The passage through the capillary endothelium.5. The water–lipid solubility ratio of the substance and its distribution through the
intestine, blood, extracellular, and intracellular fl uids.
The capacity to predict the mechanisms of toxicity and biotransformation of a chemical from its structural formula is feasible from structure–activity relationship models. It is also possible to predict absorption, distribution, and elimination from the structure of a sub-stance and its reactivity with specifi c cellular transport proteins.
A chemical’s physicochemical properties such as water and lipid solubility, ionization constant in acid or basic pH, and protein-binding capacity are also instrumental in predict-ing its toxicokinetic characteristics. For example, large and highly ionized molecules have diffi culty entering or traversing through cell membranes and are therefore poorly absorbed. Alternatively, small and moderately acidic or basic molecules are absorbed and distributed to the extra- and intracellular spaces. Moreover, highly lipophilic agents are readily absorbed and rapidly distributed to all physiological compartments. In addition, they are
Rodent treated with Aroclor 1254
Remove and homogenize liver inKCl solution
Centrifuge at 9000 × g for 10 min@4°C
Remove supernatant (S9)fraction, mix with cofactor
solution* (3:7 ratio)
Store at 4°C up to 6 hrs; add tocultured cells, maximum of 2 hrs
FIGURE 16.1 Protocol for preparation of S9 mixture. Cofactor solution consists of MgClz, glucose-
6-phosphate, NADPH in phosphate buffer, 0.2 mol/L, pH 7.4. Source: From Flint OP, Orton TC. Toxicol Appl Pharmacol, 383, 76, 1984.

228 PRINCIPLES OF TOXICOLOGY TESTING
often bound to tissues in high concentrations, and this may also account for the high degree of intracellular protein binding.
Most mutagenic and carcinogenic substances affect cells at relatively low concentra-tions. This is accounted for by biotransformation of the substance, and therefore is the reason most mutagens are classifi ed as indirect acting. Substances that have general toxic effects, however, especially after acute toxic doses, demonstrate toxicity at higher concen-trations that saturate the biotransforming capacity of the liver. This concentration can be estimated if the dose and physicochemical properties of the substance are known. In sum-mary, in vitro tests to determine distribution coeffi cient, protein binding, dissociation, and cellular transport are readily performed, and the physicochemical data generated from these studies supplement other types of in vivo toxicity data. Thus in vitro models for ADME are receiving increasing attention because they provide information for screening large numbers of lead compounds, especially when performed with high-throughput auto-mation and analysis. These models are discussed in the literature cited in the next sections.
SUGGESTED READINGS
During A, Harrison EH. Intestinal absorption and metabolism of carotenoids: insights from cell cul-ture. Arch Biochem Biophys 2004; 430: 77.
Flint OP, Orton TC. An in vitro assay for teratogens with cultures of rat embryo midbrain and limb bud cells. Toxicol Appl Pharmacol 1984; 76: 383.
U.S. Food and Drug Administration. Guidance for Industry-Drug Metabolism/Drug Interaction Stud-ies in the Drug Development Process: Studies In Vitro. Rockville, MD, 1997.
REVIEW ARTICLES
Andersen ME. Toxicokinetic modeling and its applications in chemical risk assessment. Toxicol Lett 2003; 138: 9.
Bakand S, Winder C, Khalil C, Hayes A. Toxicity assessment of industrial chemicals and airborne contaminants: transition from in vivo to in vitro test methods: a review. Inhal Toxicol 2005; 17: 775.
Blaauboer BJ. Biokinetic and toxicodynamic modelling and its role in toxicological research and risk assessment. Altern Lab Anim 2003; 31: 277.
Ding S, Yao D, Burchell B, Wolf CR, Friedberg T. High levels of recombinant CYP3A4 expression in Chinese hamster ovary cells are modulated by co-expression of human reductase and hemin supplementation. Arch Biochem Biophys 1997; 348: 403.
Ekins S, Waller CL, Swaan PW, et al. Progress in predicting human ADME parameters in silico. J Pharmacol Toxicol Methods 2000; 44: 251.
Gonzales FJ, Korzekwa KR. Cytochrome P450 expression systems. Annu Rev Pharmacol Toxicol 1995; 35: 369.
Groneberg DA, Grosse-Siestrup C, Fischer A. In vitro models to study hepatotoxicity. Toxicol Pathol 2002; 30: 394.
Guillouzo A. Liver cells models in in vitro toxicology. Environ Health Perspect 1998; 106: 511.Hayashi Y. Designing in vitro assay systems for hazard characterization: basic strategies and related
technical issues. Exp Toxicol Pathol 2005; 57: 227.Kedderis GL, Lipscomb JC. Application of in vitro biotransformation data and pharmacokinetic
modeling to risk assessment. Toxicol Ind Health 2001; 17: 315.Lin JH. Tissue distribution and pharmacodynamics: a complicated relationship. Curr Drug Metab
2006; 7: 39.McCarley KD, Bunge AL. Pharmacokinetic models of dermal absorption. J Pharm Sci 2001; 90: 1699.Zucco F, De Angelis I, Testai E, Stammati A. Toxicology investigations with cell culture systems: 20 years
after. Toxicol In Vitro 2004; 18: 153.

229
17 Mutagenicity and carcinogenicity testing in vitro
INTRODUCTION
The last several decades have seen signifi cant progress in the development of short-term tests for the detection of chemical mutagens and carcinogens. This advancement relies on understanding the complex processes leading to the potential carcinogenic actions of these chemicals and how short-term tests screen for human hazards. The in vitro tests encompass a wide variety of methods that are quicker, more economical, and more convenient than whole-animal tests for the detection of mutations, chromosome breakage, and other genetic effects.
Mutations are changes in deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) resulting in major consequences on normal cell proliferation, reproduction, physiology, and cell biology. Mutations exhibit immediate or delayed consequences but may also rep-resent stable inherited changes in gene sequences resulting in phenotypic alterations. The type and extent of consequences of mutations depend on the dose, frequency, duration of exposure, and subsequent secondary effects caused by the response of the organism to the initial perturbation. Several terms that are used to defi ne the effect of chemicals on the chromosome apparatus are listed here:
Mitogenesis: An immediate or delayed cellular response resulting in the induction of increased cell proliferation.
Genotoxicity: The effect of a chemical or its metabolites that induces changes to the genome and infl uences the production of malignant transformation.
Chemical carcinogenicity: The effect of a chemical or its metabolites capable of inducing irreversible changes to normal cellular biology resulting in reversible or irreversible cellular transformation and uncontrolled cell proliferation. These changes result from genetic mutations, interfere with transfers of genetic information, and are usually manifested as lethal effects.
Considerable effort has been devoted to the development of short-term tests as methods for detecting, screening, and understanding the bases of chemical carcinogens. Short-term test-ing methods, therefore, are based on the accepted knowledge that mutations are the results in part of exposures to mutagenic and carcinogenic chemicals. Therefore, in vitro test sys-tems use a variety of indicators that refl ect mutational events in the presence of carcinogens and mutagens including microorganisms, plants, insects, and cultured mammalian cells to screen for potentially mutagenic, teratogenic, and carcinogenic agents. The advantages of

230 PRINCIPLES OF TOXICOLOGY TESTING
in vitro tests depend on the rapid assessment of large numbers of chemicals, the suggestion of a mechanism for carcinogenicity or mutagenicity, the reduction, replacement, and refi ne-ment of animal testing, and a contribution to human and animal risk assessment that corre-lates as well as the predictive ability of animal toxicology testing.
The fi rst few sections of this chapter discuss the mechanisms of mutagenicity and car-cinogenicity, as well as the principles of multistage carcinogenesis, which will establish the foundation for understanding the rapid development of the in vitro tests.
CARCINOGENIC PROCESS
Cancer is a group of diseases in which there is an uncontrolled proliferation of cells that express varying degrees of fi delity to their precursor cell of origin. This cell proliferation occurs in almost all tissues throughout a life span and is infl uenced by a variety of circum-stances. During the normal physiological state, the delicate balance between cell prolifera-tion and apoptosis (programmed cell death) is perpetuated to ensure the integrity and proper function of organs and tissues. Mutations in DNA that lead to cancer development interfere with this orderly process by disrupting its regulation. Consequently, the initiation of a cancer is due to an abnormal and uncontrolled progression of cell proliferation char-acterized by unregulated cell division and metastasis (spreading) of foci of cells to distant tissues.
Carcinogenesis represents the unwarranted appearance or increased incidence of abnormal cell proliferation occurring in an age-matched low-risk group whose propensity for development of cancer is statistically lower. A carcinogen is any chemical or viral agent that increases the frequency or distribution of new tumors, results in their appearance within a low-risk or otherwise early age group, or results in the introduction of new patho-logical growths otherwise absent in experimental controls.
Most chemical carcinogens require metabolic activation before demonstrating carcino-genic potential. As with most toxic phenomena, a minimum dosage is necessary to elicit a carcinogenic event. Epigenetic or nongenotoxic carcinogens enhance the growth of tumors by mechanisms other than through alteration of DNA. These chemicals infl uence absorp-tion and biotransformation, or reduce elimination of the initiating agent. Co-carcinogens redirect hormonal action on cell proliferation and inhibit intercellular communication, thus allowing for greater proliferative capacity. Alternatively they induce immunosuppression of protective immunological pathways.
The carcinogenic process is, therefore, a response to a mutation occurring within the genetic material of normal cells, resulting in uncontrolled cell division and transformation to the immortal phenotype. The unrestrained and often rapid proliferation of cells is further characterized as benign or malignant. Benign tumors do not metastasize, are usually con-fi ned to local target organ areas, are clinically susceptible to therapeutic intervention, and carry a more favorable prognosis. Alternatively, malignant tumors metastasize to distant organ locations, are not necessarily amenable to therapeutic intervention, and possess a less favorable prognosis. Mutagenesis refers to the ability of a virus or chemical agent to induce changes in the genetic sequence of mammalian or bacterial cells, thus altering the pheno-typic expression of cell characteristics. Genotoxicity refers to the ability of an agent to induce heritable changes in genes that exercise homeostatic control in somatic cells, while increasing the risk of infl uencing benign or malignant transformation. Genotoxic sub-stances induce genotoxicity by either binding directly to DNA or by indirectly altering the DNA sequence resulting in irreversible damage. It is also important to note, however, that genotoxic substances are not necessarily carcinogenic.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 231
In addition, interaction of a chemical, physical, or viral agent with nucleic acids results in the disruption of the transfer of genetic information and the development of genotypic and phenotypic consequences. Lastly, mitogenesis is the induction of cell division (mitosis) within eukaryotic or prokaryotic cells by stimulating transit through the cell cycle. Pro-longed and continuous exposure to growth factors is required to commit cells to the cell cycle.
By defi nition, cancer must have at least six criteria: (1) self-suffi ciency in growth sig-nals, (2) insensitivity to antigrowth signals, (3) evasion of apoptosis, (4) tissue invasion and metastasis, (5) sustained angiogenesis, and (6) limitless replicative potential. The induction of a neoplasm is a multistage process that occurs over a long period of time, the stages of which include initiation, promotion, and progression (see the section, “Multistage Carcinogenesis”). The causes of most human cancers remain unidentifi ed; however, con-siderable evidence suggests that environmental and life-style factors, and chemical agents, are important contributors. For example, tobacco smoking appears to be responsible for approximately 30% of all cancer deaths in developed countries. The infl uence of environ-mental factors on the incidence of cancer in a population is supported by epidemiological observations:
1. While overall cancer incidence is reasonably constant between countries, inci-dences of specifi c tumor types vary considerably.
2. Large differences in cancer incidence exist within populations of a single country.3. Migrant populations assume the cancer incidence of their new environment within
one to two generations.4. Cancer incidence within a population changes rapidly.
Thus, understanding the molecular and cellular process underlying chemical carcinogenesis is of critical importance for carcinogenic risk assessment as well as development of mecha-nistically based chemopreventive and therapeutic strategies for the management of human cancers.
MECHANISMS OF CHEMICAL CARCINOGENESIS
METABOLISM
Carcinogenic agents initiate cancer progression by utilizing one of two pathways:
1. Parent chemicals can cause cancers directly, such as with heavy metals.2. Some chemicals require metabolic activation to reactive intermediates in order to
affect the carcinogenic process; for example organic chemicals, such as benzo(a)pyrene is fi rst converted to an electrophilic intermediate which then covalently bind to cellular macromolecules. Accordingly, chemical carcinogens that require meta-bolic activation to exert their carcinogenic effects are termed procarcinogens, whereas their highly reactive metabolites are designated ultimate carcinogens. Met-abolic intermediates between procarcinogens and ultimate carcinogens are called proximate carcinogens.
Cytochrome P450 phase 1 and phase 2 enzymes are involved in the metabolism of carcino-gens and usually result in the formation of reactive metabolites. Phase 2 enzyme-catalyzed

232 PRINCIPLES OF TOXICOLOGY TESTING
reactions often lead to detoxifi cation and elimination, by which it effectively protects against chemical carcinogenesis. Over time, the constant assault of cells with reactive metabolites that linger at the cellular level probably leads to the interaction with cellular macromolecules and the carcinogenic process.
CHEMICAL STRUCTURE AND CARCINOGENIC ACTIVITY
It is recognized that there is a relationship between the chemical structure of a suspected carcinogen and the carcinogenic activity. Molecules that contain aromatic moieties, such as aromatic nitro groups, N-oxides, mono- and dialkylamino groups, and aliphatic and aro-matic epoxides, are the chemicals most associated with carcinogenic activity. These struc-tural “alerts” are apparently useful for carcinogenic risk assessment.
FREE RADICALS AND REACTIVE OXYGEN SPECIES
As with the chemical activation of procarcinogens to proximate and ultimate carcinogens, bioactivation of chemicals also results in the formation of free radicals and reactive oxygen species (ROS), that is chemical species that cause oxidative damage to biomolecules includ-ing nucleic acids (DNA), proteins, and lipids. These reactive species are generated from ion-izing radiation, ultraviolet light, and a variety of exogenous and endogenous cellular sources (Fig. 17.1).
Endogenous SourcesMitochondria
NAD(P)H oxidaseXanthine oxidaseTransition metals
Exogenous sourcesUV light & Ionizing radiation
Organic chemicalsHeavy metals
Airborne nanoparticles
Genomic DNANucleotide base modifications
Strand breakageInhibition of DNA repair
enzymes
Cell signal transductionOxidation of:
transcriptional activators protein kinase cascades
DNA mutations Dysregulation of cell growth,differentiation, and death
Multistage carcinogenesis
Reactive oxygen species
FIGURE 17.1 Involvement of free radicals and reactive oxygen species in multistage carcinogenesis.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 233
ROS interaction with DNA induces formation of adducts, which may lead to the activa-tion of proto-oncogenes and/or the inactivation of tumor suppressor genes. Free radicals and ROS are also able to target cell signaling molecules, including transcription factors and protein kinase cascades, resulting in altered cell signal transduction and gene expression. The ability of free radicals and ROS to elicit DNA mutations and cause dysregulated cell signaling contributes to the multistage carcinogenesis pathway (Fig. 17.1).
MUTAGENESIS
As mentioned previously, mutagenesis occurs when the mammalian or bacterial cell genetic sequence is altered such that transcription and translation are transformed. This induces a modifi cation of the phenotypic expression of cell characteristics, which more often results in permanent pathological changes rather than benefi cial adjustments. In general, carcinogen-ics are separated into genotoxic and nongenotoxic classifi cations. Thus a genotoxic carcino-gen is able to interact with DNA directly, leading to mutations. The production of mutations is due primarily to chemical or physical alterations in the structure of DNA that result in inaccurate replication of that gene. The process includes two major steps:
1. Structural DNA alteration and cell proliferation lead to permanent DNA alteration. Interaction of reactive intermediates derived from bioactivation of carcinogens with DNA results in formation of DNA adducts, DNA strand breaks, and DNA-protein cross-links. Carcinogens that result in the formation of bulky DNA adducts often specifi cally react with the sites in the purine ring, particularly the N7 position of guanine. This target is the most nucleophilic site in DNA where many ultimate carcinogens form covalent adducts. Direct alkylation of DNA bases by several alkylating carcinogenic agents has also been demonstrated.
2. DNA modifi cation resulting from the hydroxylation of DNA nucleotide bases is often a manifestation of ROS reactivity. While changes occur in all four bases, the most understood step is the formation of 8-hydroxy-2′-deoxyguanosine.
Interaction of carcinogens with DNA produces adducts not only on DNA bases, but also on the sugar and phosphate backbone. The covalent modifi cations of DNA by carcinogenic agents may ultimately lead to the development of cancers. Although the modifi cations of DNA by carcinogens are critically involved in the induction of DNA mutations, the muta-genesis process is also greatly affected by various DNA repair mechanisms.
DNA REPAIR
As the interaction of ultimate carcinogens with DNA results in the formation of various DNA adducts, the continued presence of the adduct prompts the cellular machinery to repair the structural alteration. It is estimated that more than a hundred genes are dedicated to DNA repair, the purpose of which is to correct the structural damage induced by carcinogens. In this way, mutagenesis is prevented. Alternatively, the persistence of DNA–carcinogen adducts is indicative of the insuffi ciency of DNA repair.
EPIGENETIC CARCINOGENESIS
As discussed earlier, genotoxic carcinogens directly interact with DNA through the pres-ence of reactive electrophilic metabolites in the chemical, leading to mutations. In contrast,

234 PRINCIPLES OF TOXICOLOGY TESTING
nongenotoxic (or epigenetic) carcinogens do not directly affect DNA in the induction of cancer.1 In fact, nongenotoxic carcinogens:
1. are nonmutagenic;2. show no evidence of direct chemical reactivity with DNA;3. do not possess common chemical structural features within the class;4. exhibit a well-established dose-threshold effect;5. exhibit a lower carcinogenic potential than genotoxic carcinogens.
While DNA damage is described as a general mechanism of the carcinogenic effect of geno-toxic carcinogens, nongenotoxic mechanisms are more heterogeneous. Proposed mecha-nisms of nongenotoxic carcinogenesis are summarized in Table 17.1.
MULTISTAGE CARCINOGENESIS
The process of carcinogenesis is divided into three experimentally defi ned stages: tumor initiation, tumor promotion, and tumor progression. This multistage development of carci-nogenesis requires the conversion of benign hyperplastic cells to the malignant state, involv-ing invasion and metastasis as manifestations of further genetic and epigenetic changes. Figure 17.2 correlates with the discussion below and illustrates the events that occur during the three stages of chemical carcinogenesis. The diagram also outlines the requirements necessary for the completion of the multistage process.
TUMOR INITIATION
The early concept of tumor initiation indicated that the initial changes in chemical carcino-genesis involved irreversible genetic alterations. Recent data from molecular studies of pre-neoplastic human lung and colon tissues, however, implicate epigenetic changes as an early event in carcinogenesis. DNA methylation of promoter regions of genes can transcription-ally silence tumor-suppressor genes. Thus, carcinogen–DNA adduct formation is central to the theory of chemical carcinogenesis, and may be a necessary, but not a suffi cient, prereq-uisite for tumor initiation. The formation of DNA adducts, causing either the activation of a proto-oncogene or the inactivation of a tumor-suppressor gene, is the tumor-initiating event. One important characteristic of this stage is its irreversibility, such that the genotype/pheno-type of the initiated cell is conferred during the process. Chemicals capable of initiating cells are referred to as initiating agents. Furthermore, initiation without the subsequent promotion and progression rarely yields malignant transformation.
TUMOR PROMOTION
In multistage carcinogenesis, tumor promotion comprises the selective clonal expansion of initiated cells through a mechanism of gene activation. Because the accumulation rate of mutations is proportional to the rate of cell division, clonal expansion of initiated cells pro-duces a larger population of cells that are at risk of further genetic changes and malignant conversion. Tumor promoters are generally nonmutagenic, are not solely carcinogenic, and often are able to mediate their biological effects without metabolic activation. In addition,
1 The National Toxicology Program (NTP) Report on Chemical Carcinogenicity estimates that 40% of chemicals classifi ed as carcinogens are nongenotoxic (epigenetic) carcinogens, with the liver as the most common target organ (NTP, 2009).

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 235
they do not directly interact with DNA. Interestingly, tumor promotion is reversible, although the continued presence of the promoting agent maintains the state of the promoted cell pop-ulation (preneoplastic lesion). Thus the stage appears to have a long duration and is a pre-ferred target for experimental manipulation. Examples of typical tumor promoters include tetradecanoyl phorbol acetate, phenobarbital, and 2,3,7,8-tetrachlorodibenzo-ρ-dioxin.
During tumor promotion, malignant conversion may occur in which a preneoplastic cell is transformed into the malignant phenotype, but requires further genetic changes. Frequent repeated administration of the tumor promoter is more important than the total dose. In addition,
TABLE 17.1OECD Testing Guidelines (TG): In Vitro Guidelines for Genetic Toxicology Testing of Chemicals (By the Year Submitted, Not Including Tests Involving In Vivo Genetic Testinga)
TG No. TitleYear
Submitted Objective
471 Bacterial reverse mutation test 1983b Detects mutations which revert mutations present in the test strains and restores the functional capability of the bacteria to synthesize an essential amino acid.
473 In vitro mammalian chromosome aberration test
1983b Identifi es agents that cause structural chromosome or chromatid aberrations in cultured mammalian somatic cells.
476 In vitro mammalian cell gene mutation test
1984b Detects common gene mutations at TK, HPRT, and XPRT induced by chemical substances in cell lines
477 Genetic toxicology: Sex-Linked Recessive Lethal Test in Drosophila melanogaster
1984 Detects mutations in the X-chromosome of D.melanogaster are phenotypically expressed in males carrying the mutant gene.
479 Genetic toxicology: In vitro sister chromatid exchange assay in mammalian cells
1986 Detects reciprocal exchanges of DNA between two sister chromatids of a duplicating chromosome.
480 Genetic toxicology: Gene mutation assay in Saccharomyces cerevisiae
1986 Measures forward or reverse gene mutations in yeast strains of S.cerevisiae.
481 Genetic toxicology: mitotic recombination in S.cerevisiae
1986 Measures mitotic recombination (gene conversion or crossing over) in S.cerevisiae.
482 Genetic toxicology: DNA damage and repair, unscheduled DNA synthesis in mammalian cells in vitro
1986 Detects DNA repair synthesis after excision and removal of a stretch of DNA containing the region of damage induced by an agent.
487 In Vitro Mammalian Cell Micronucleus Test
2010 Detection of micronuclei in the cytoplasm of interphase cells that have undergone cell division during or after exposure to the test substance.
aOECD TG 474, 475, 478, 483, 484, 485, 486, and 488 are in vivo genetic tests requiring animal involvement prior to analysis. bRecent update, 1997. Abbreviations: HPRT, hypoxanthine–guanine phosphoribosyl transferase; TK, thymidine kinase; XPRT, transgene of xanthineguanine phosphoribosyl transferase.

236 PRINCIPLES OF TOXICOLOGY TESTING
if the tumor promoter is discontinued before malignant conversion has occurred, premalignant or benign lesions may regress. Tumor promotion contributes to the process of carcinogenesis by the expansion of a population of initiated cells that are then at risk for malignant conversion. Conversion of a fraction of these cells to malignancy is accelerated in proportion to the rate of cell division and the quantity of dividing cells in the benign tumor or preneoplastic lesion. In part, these genetic changes result from the infi delity of DNA synthesis.
TUMOR PROGRESSION
Tumor progression comprises the expression of the malignant phenotype and the tendency of malignant cells to acquire more aggressive characteristics over time. Also, metastasis may involve the ability of tumor cells to secrete proteases that allow invasion of secondary organs beyond the immediate primary tumor location. A prominent characteristic of the malignant phenotype is the propensity for genomic instability and uncontrolled growth. Further genetic and epigenetic changes occur, including the activation of proto-oncogenes and the functional loss of tumor suppressor genes. Loss of function of tumor-suppressor genes usually occurs in a bimodal fashion, and most frequently involves point mutations in one allele and loss of the second allele by a deletion, recombinational event, or chromosomal disjunction. These phenomena confer growth advantage to the cells as well as the capacity for regional inva-sion, and ultimately, distant metastatic spread. Despite evidence for an apparent scheduling of certain mutational events, it is the accumulation of these mutations, and not the order or the stage of tumorigenesis in which they occur, that appears to be the determining factor.
CARCINOGENIC CHARACTERISTICS
Experimentally, a carcinogen is an agent that produces a statistically signifi cant increased incidence of malignant neoplasms, compared with untreated control animals, when admin-istered to previously untreated animals. Carcinogens are chemicals, physical forces (such as ultraviolet light and gamma radiation), and biological catalysts (such as viruses). Chemical carcinogens are the most commonly investigated toxicological mediators.
Normal cell Initiated cell Preneoplastic cells
Neoplastic cells
Initiation
DNA mutations may, occur in oncogenes, proto-oncogenes, or tumor suppressor genes
Initiated cells remain “dormant”; rarely become neoplastic cells without undergoing promotion and progression
Irreversible
Promotion
Clonal expansion of initiated cells
Cell signaling and gene expression are altered, leading to increased cell mitogenesis and reduced apoptosis
Leads to the formation of pre-neoplastic cells
Long duration and reversible, especially at an earlier stage
Progression
Additional genomic structural alterations due to karyotypicinstability
Leads to the formation of benign or malignant neoplasms
Cells acquire malignant phenotype such as invasiveness, angiogenesis, and/or metastasis
Irreversible
Initiating agents Promoting agents Progressor agents
FIGURE 17.2 A schematic illustration of the three stages of chemical carcinogenesis.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 237
CLASSIFICATION
Chemical carcinogens are classifi ed according to their chemical characteristics, such as:
1. organic chemicals: benzo(a)pyrene, afl atoxin B1, and benzene;2. inorganic chemicals: arsenic, cadmium, chromium and nickel;3. hormonal substances: estrogens, anabolic, and androgenic steroids.
Furthermore, chemical carcinogens are classifi ed as genotoxic or nongenotoxic (epigene-tic) carcinogens, based on reactivity with DNA, or as animal or human carcinogens, depend-ing on the evidence of carcinogenicity in animals and humans. The International Association for Research on Cancer evaluates and categorizes human carcinogens accordingly:
Group 1: These agents have suffi cient epidemiological evidence of carcinogenicity in humans; as such they are labeled as known human carcinogens; for example, arse-nic, afl atoxin B1, benzene, estrogens, vinyl chloride, nickel, and chromium;
Group 2A: These substances possess suffi cient evidence of carcinogenicity in animals, but have limited evidence of carcinogenicity in humans; therefore, they are labeled as probably carcinogenic to humans; for example, benz(a)anthracene, polychlorinated biphenols, and styrene oxide;
Group 2B: They possess either limited evidence of carcinogenicity in humans or have suffi cient evidence of carcinogenicity in animals and inadequate evidence of carcino-genicity in humans; labeled as possibly carcinogenic to humans; for example sty-rene, N-methyl-N-nitroso urethane;
Group 3: It is not possible to classify this group as per carcinogenicity; for example, benzo[e]pyrene, and dibenz[a,c]anthracene;
Group 4: These chemicals possess inadequate evidence of carcinogenicity in both ani-mals and humans, and thus are probably not carcinogenic to humans; for example, caprolactam.
IDENTIFICATION
Identifi cation of carcinogenic agents is a critical step in carcinogenic risk assessment. While epidemiological studies provide the most defi nitive means of establishing carcinogenicity to humans from exposure to an agent, they are not protective of the human health, since conclu-sions on carcinogenicity are typically derived after occurrence. In addition, epidemiological studies have limited sensitivity for identifi cation of carcinogens. Thus, potential carcinogens are identifi ed based on the execution of in vitro and animal experiments, and are classifi ed according to the following scheme:
1. Short-term bioassays: Include mutagenesis assays and neoplastic transformation in cell culture. Duration of these assays is from several weeks to 3 months.
2. Medium-term bioassays: Target qualitative and quantitative analysis of preneoplas-tic evidence. These assays are performed in animals for 2–8 months.
3. Long-term animal bioassays: Time frame is 18–24 months.
U.S. FDA regulations require chronic 2-year bioassay rodent studies for identifying potential carcinogens. Further information on in vivo testing requirements is described in chapter 11, Carcinogenicity and Mutagenicity Testing In Vivo.

238 PRINCIPLES OF TOXICOLOGY TESTING
CARCINOGENIC POTENTIAL
Not all carcinogenic chemicals are equally successful in inducing neoplasia, that is, they exhibit different carcinogenic potencies. The carcinogenic potential of a chemical is defi ned as the slope of the dose–response curve for induction of neoplasms. This defi nition, however, is not suffi cient for estimating the carcinogenic potential based on data from chronic carci-nogenesis bioassays. Historically, several methods have been developed to measure the car-cinogenic potential, particularly the tumorigenesis dose rate 50% (TD
50). This value has
been popular for the estimation of the carcinogenic potential for a number of chemicals. The TD
50 is calculated as the dose rate (mg/kg body weight/day) of carcinogen, which when
administered chronically for a standard period, induces neoplasms in half of the test animals. The value is adjusted for spontaneous neoplasms and is an important component of carcino-genic risk assessment.
CARCINOGENIC RISK ASSESSMENT
The process of carcinogenic risk assessment closely parallels general toxicologic risk assess-ment as outlined in chapter 4. Specifi cally, the steps include the following:
1. Identifi cation and evaluation of the carcinogen and its potential.2. Mechanism elucidation.3. Exposure assessment.4. Dose–response assessment.5. Qualitative and quantitative estimation of risk in humans.
As previously noted, information and conclusions generated from epidemiological studies are tempered by the use of various laboratory experimental methods due to the nonprotec-tive feature and limited sensitivity of the former. Several limitations, however, are encoun-tered in the scientifi c and practical application of the information derived from laboratory bioassays to estimate human risk. For instance, approximating carcinogenic risk based upon uncomplicated laboratory data requires major assumptions in order to extrapolate carcinogenic information from bioassays to humans, examples of which are outlined in chapter 2. Another signifi cant problem is the dose–response relationship. High doses, such as the maximum tolerated dose of a test agent, are used in the 2-year animal bioassay for identifi cation of carcinogenicity. The actual levels of human exposure to a particular potential carcinogenic chemical are generally much lower than those used in laboratory animal experiments. Therefore, applying the carcinogenic data obtained in animals treated with high doses of a test substance to the carcinogenic potential in humans whose expo-sure to that agent is considerably lower poses a signifi cant hurdle in extrapolation statis-tics. In addition, extrapolation from high dose to low dose in carcinogenic risk assessment is further complicated by the assumption of the existence of a threshold for genotoxic carcinogens.
Qualitative and quantitative risk estimation presents another consideration in determi-nation of carcinogenic risk in humans. Qualitative risk estimation is easier to develop based on the qualitative analysis of the information obtained from various bioassays. In contrast, quantitative risk estimation is more cumbersome and subject to greater variations. In order to validate quantitative risk analysis, the process should integrate biological data, toxicoki-netic information, and toxicodynamic parameters of specifi c carcinogenic chemicals or mixtures. To satisfy this objective, various mathematical models and algorithms have been

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 239
developed and utilized for the quantitative estimation of carcinogenic risk in humans. Among them are the one-hit (linear), multihit (k-hit), multistage, extreme value, log-probit, and the MKV biomathematical models, as well as biologically based cancer modeling, some of which have been projected by the U.S. Environmental Protection Agency (EPA) for quantitative analysis of carcinogenic risk.
CARCINOGENIC AND GENOTOXIC AGENTS
Both collective and individual exposures often defi ne environmental toxicology. Epidemio-logical research, supported by experimental investigations, has led to the determination of a “profi le” of various risk factors. This profi le is perpetually incomplete since humans inces-santly modify the environment, thus increasing the risk of interaction with agents against which there may be no protective mechanisms. The multiplicity of chemical or physical carcinogens does not render them amenable for study as the development of carcinoma is, in all probability, not due to contact with a single carcinogenic factor but rather to the sum of many possible causative forces.
Of the various environmental hazardous compounds, cigarette smoke enjoys the high-est causal relationship with cancer risk in humans. Tobacco smoking plays a major role in the etiology of lung, oral cavity and esophageal cancers, as well as with a variety of chronic degenerative diseases. Although cigarette smoke is a mixture of about 4000 chemicals, it includes more than 60 known human carcinogens, 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (nicotine-derived nitrosamino ketone, NNK) is the most carcinogenic tobacco-specifi c nitrosamine. NNK induces lung tumors in mice, rats, and hamsters, and the International Agency for Research on Cancer (IARC) has designated NNK and N-nitro-sonornicotine (NNN) as known human carcinogens. NNK is metabolically activated by CYP P450 enzymes in the lung and generates O6-methylguanine in DNA. The reaction generates G:C to A:T mutations with the subsequent activation of K-Ras proto-oncogene and development of tumor initiation.
Radiation also contributes to the causative physical factors that induce human cancers. Radiation promotes double-strand breaks (DSBs) in DNA that leads to chromosome aber-rations and cell death, and generates a variety of oxidative DNA damage. Because of the genotoxicity potential, radiation at high doses evidently results in the appearance of various tumors in humans. Even at low doses, residential exposure to radioactive radon and its decay products, for instance, may account for about 10–20% of all lung cancer deaths worldwide.
Several important international programs are directed at the harmonization of genetic toxicology testing strategies and testing standards. The Organization for Economic Coop-eration and Development (OECD) has a major role in developing recommendations for internationally harmonized testing protocols. The protocols developed by OECD are updated periodically (http://www.oecd.org/). Table 17.1 lists OECD guidelines on genetic toxicology testing and guidance on the selection and application of in vitro assays. Many of the tests in this table are described in detail in the next sections. In vivo mutagenicity and genotoxicity tests are presented in Section II.
BACTERIAL CELL SYSTEMS
Short-term tests use both prokaryotic and eukaryotic cells to detect the vast majority of potential carcinogens and mutagens. The prokaryotes include bacteria and blue-green algae (cyanobacteria). The bacterial test systems use auxotrophic organisms whose growth and

240 PRINCIPLES OF TOXICOLOGY TESTING
proliferation depend on the presence of an essential rate-limiting nutrient component in the growth medium, usually an amino acid. The auxotrophic organism thus represents a mutant bacterium with a highly specifi c defect in a gene locus. In contrast, the normal or wild-type prototrophic organism lacks the mutation; its growth is not dependent on the addition of the essential rate-limiting nutrient. Prototrophic organisms are capable of maintaining normal cell proliferation from the minimal essential growth medium components. Figure 17.3 out-lines typical growth properties of auxotrophic and prototrophic bacteria as preliminary experiments in preparation for the Ames test.
The foundation of the bacterial tests is based on the induction of (1) a reverse mutation from the component-dependent strain to a wild-type capable of sustaining growth on mini-mal essential medium and (2) a forward mutation in which additional agent-induced phe-notypic genetic changes are identifi ed.
Detection of forward mutations results in larger genetic targets for a chemical, present-ing several loci within specifi c genomes for identifi cation of phenotypic changes. Reverse mutation assays provide specifi c and selective target sites for chemical action, relying on a second agent-induced mutation to modify the effect of a pre-existing mutation.
AMES TEST
The bacterial mutagenicity assay for a direct-acting mutagen, the reverse-mutation plate test, plate-incorporation test, or agar overlay test, uses bacterial strains mixed with known
Auxotrophic cells plated–Require HIS
Incubated
Auxotrophic mutant colonies, missing frommedium minus HIS
Prototrophic“wild-type” bacterialcolonies
Growth medium plus HIS Growth medium minus HIS
FIGURE 17.3 Growth properties of auxotrophic and prototrophic bacteria. Abbreviation: HIS, histidine.
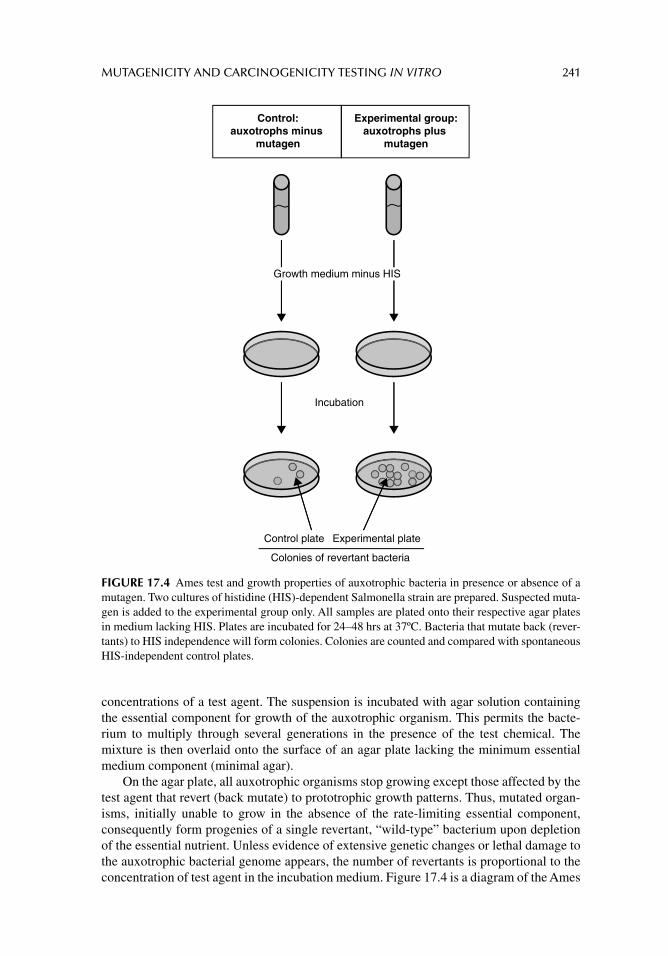
MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 241
concentrations of a test agent. The suspension is incubated with agar solution containing the essential component for growth of the auxotrophic organism. This permits the bacte-rium to multiply through several generations in the presence of the test chemical. The mixture is then overlaid onto the surface of an agar plate lacking the minimum essential medium component (minimal agar).
On the agar plate, all auxotrophic organisms stop growing except those affected by the test agent that revert (back mutate) to prototrophic growth patterns. Thus, mutated organ-isms, initially unable to grow in the absence of the rate-limiting essential component, consequently form progenies of a single revertant, “wild-type” bacterium upon depletion of the essential nutrient. Unless evidence of extensive genetic changes or lethal damage to the auxotrophic bacterial genome appears, the number of revertants is proportional to the concentration of test agent in the incubation medium. Figure 17.4 is a diagram of the Ames
Incubation
Control plate Experimental plate
Colonies of revertant bacteria
Control:auxotrophs minus
mutagen
Experimental group:auxotrophs plus
mutagen
Growth medium minus HIS
FIGURE 17.4 Ames test and growth properties of auxotrophic bacteria in presence or absence of a mutagen. Two cultures of histidine (HIS)-dependent Salmonella strain are prepared. Suspected muta-gen is added to the experimental group only. All samples are plated onto their respective agar plates in medium lacking HIS. Plates are incubated for 24–48 hrs at 37ºC. Bacteria that mutate back (rever-tants) to HIS independence will form colonies. Colonies are counted and compared with spontaneous HIS-independent control plates.

242 PRINCIPLES OF TOXICOLOGY TESTING
test and the growth properties of auxotrophic bacteria in the absence or presence of a mutagen.
Bacterial mutagenesis assays are the most widely used short-term tests for screening of potential mutagens and carcinogens. They are highly sensitive for genotoxic agents, are technically easy, fast, and economical. The most extensively used bacterial assay system uses mutant tester strains of Salmonella typhimurium developed by Ames (1972). The orig-inal method used strains that were genetically defective in their ability to synthesize histi-dine (HIS). Assay sensitivity was later improved by incorporating additional mutations in tester strains (Table 17.2) specifi cally designed to detect frame shift or base pair substitu-tion mutations induced by different classes of mutagens.
All strains contain some type of mutation in the HIS operon, while additional muta-tions increase their sensitivity to mutagens. For instance, the rfa mutation causes partial loss of the lipopolysaccharide surface coatings of bacteria and thus increases their perme-ability to large molecules that do not normally penetrate cell walls. Another mutation (uvrB) greatly increases the sensitivity of the bacteria to mutagens by deleting the gene coding for DNA excision repair.
Many of the standard tester strains also contain the R-factor plasmid, pKM101. These strains are sensitive to a number of mutagens that are inactive in non-R-factor parent strains. Thus, sensitivity for identifying weak mutagenic agents as well as specifi city for detecting different classes of carcinogens was a signifi cant advancement in the development of the test (Table 17.3). Today, the Ames test is the only short-term assay that is validated, popular, and sensitive for detecting carcinogens. Its fl exibility allows its adaptation to testing large numbers of chemicals with varying characteristics. Nevertheless, awareness of its limita-tions is important in the evaluation and interpretation of data. Probably the most important consideration is adherence to standardized protocols to limit inter- and intralaboratory vari-ability. Two variations of the Ames, the spot test and the standard text, are described next.
Spot Test
The spot test is the simplest technique for rapid screening of large numbers of chemicals. TA98, TA100, or TA1535 S. typhimurium tester strains are incubated in minimal medium. The organism is applied with an agar overlay containing a rate-limiting quantity of HIS to initiate growth (plate incorporation test). A fi lter disk soaked with known concentrations of the test agent is placed and incubated on the agar. The toxicity of the diffusible solution containing the dissolved chemical is proportional to the diameter of the zone of growth inhibition versus the growth of revertant bacteria around the application site. Although water-soluble agents do not easily penetrate the agar and dilution of the chemical concentration increases with outward
TABLE 17.2Incorporation of Mutations of Original Salmonella Strains Incurring Increased Sensitivity of Ames Test to Mutagens
Mutation Feature
uvrB Eliminates DNA excision repair systemrfa Interferes with synthesis of LPS components of cell walls resulting in increased
permeability of bacterial cell walls to mutagens
Abbreviation: LPS, lipopolysaccharide.
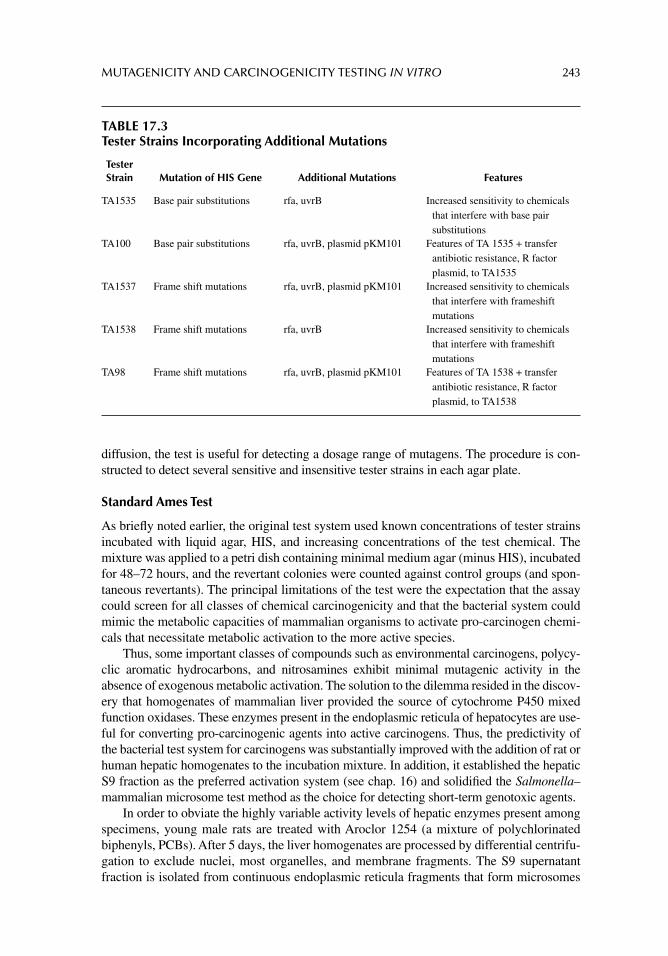
MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 243
TABLE 17.3Tester Strains Incorporating Additional Mutations
Tester Strain Mutation of HIS Gene Additional Mutations Features
TA1535 Base pair substitutions rfa, uvrB Increased sensitivity to chemicals that interfere with base pair substitutions
TA100 Base pair substitutions rfa, uvrB, plasmid pKM101 Features of TA 1535 + transfer antibiotic resistance, R factor plasmid, to TA1535
TA1537 Frame shift mutations rfa, uvrB, plasmid pKM101 Increased sensitivity to chemicals that interfere with frameshift mutations
TA1538 Frame shift mutations rfa, uvrB Increased sensitivity to chemicals that interfere with frameshift mutations
TA98 Frame shift mutations rfa, uvrB, plasmid pKM101 Features of TA 1538 + transfer antibiotic resistance, R factor plasmid, to TA1538
diffusion, the test is useful for detecting a dosage range of mutagens. The procedure is con-structed to detect several sensitive and insensitive tester strains in each agar plate.
Standard Ames Test
As briefl y noted earlier, the original test system used known concentrations of tester strains incubated with liquid agar, HIS, and increasing concentrations of the test chemical. The mixture was applied to a petri dish containing minimal medium agar (minus HIS), incubated for 48–72 hours, and the revertant colonies were counted against control groups (and spon-taneous revertants). The principal limitations of the test were the expectation that the assay could screen for all classes of chemical carcinogenicity and that the bacterial system could mimic the metabolic capacities of mammalian organisms to activate pro-carcinogen chemi-cals that necessitate metabolic activation to the more active species.
Thus, some important classes of compounds such as environmental carcinogens, polycy-clic aromatic hydrocarbons, and nitrosamines exhibit minimal mutagenic activity in the absence of exogenous metabolic activation. The solution to the dilemma resided in the discov-ery that homogenates of mammalian liver provided the source of cytochrome P450 mixed function oxidases. These enzymes present in the endoplasmic reticula of hepatocytes are use-ful for converting pro-carcinogenic agents into active carcinogens. Thus, the predictivity of the bacterial test system for carcinogens was substantially improved with the addition of rat or human hepatic homogenates to the incubation mixture. In addition, it established the hepatic S9 fraction as the preferred activation system (see chap. 16) and solidifi ed the Salmonella–mammalian microsome test method as the choice for detecting short-term genotoxic agents.
In order to obviate the highly variable activity levels of hepatic enzymes present among specimens, young male rats are treated with Aroclor 1254 (a mixture of polychlorinated biphenyls, PCBs). After 5 days, the liver homogenates are processed by differential centrifu-gation to exclude nuclei, most organelles, and membrane fragments. The S9 supernatant fraction is isolated from continuous endoplasmic reticula fragments that form microsomes

244 PRINCIPLES OF TOXICOLOGY TESTING
containing high concentrations of phase I mixed function oxidases. The fraction is used as a crude homogenate, is further enriched by additional high speed centrifugation for phase II enzymes, or is frozen.
To complete the short-term test, an aliquot of the S9 fraction is added to the Salmonella–test chemical–agar buffered suspension, overlaid onto a minimal medium agar plate, and incu-bated for 48–72 hours at 37°C. The revertant colonies are counted against controls (Fig. 17.4).
MAMMALIAN CELL SYSTEMS
BASIS OF MUTAGENICITY TESTING WITH MAMMALIAN SYSTEMS
Determining mutagenic profi les of chemicals such as pesticides and herbicides requires tests of high sensitivity and specifi city. An effective strategy uses tests that produce reproducible and biologically relevant data based upon three stages. Stage 1 uses bacterial gene mutation assays as described earlier; stage 2 assays are cytogenetic tests that monitor clastogenicity and aneugenicity (described later in this chapter); and, stage 3 assays record the induction of gene mutations in cultured mammalian cells.
Mutagenesis testing in mammalian cells is conducted as part of stage 3 to confi rm that a chemical is presumed mutagenic for higher animals. Testing in mammals supposes that higher degrees of DNA repair and xenobiotic metabolism are markedly different from those of bacterial systems. Consequently, several assays incorporate various continuous cell lines and primary cultures of animal and human origin. While primary cultures main-tain some biotransformation capability, continuous cell lines have limited potential. Cell culture systems thus permit a closer examination of the mechanisms of action responsible for gene mutation and chromosome aberrations.
Short-term mammalian tests detect mutations at the hypoxanthine–guanine phosphori-bosyl transferase (HGPRT) locus using Chinese hamster lung (V79) or Chinese hamster ovary (CHO) cells; mutations at the thymidine kinase (TK) locus, in the mouse lymphoma L5178Y cell line; or the TK6 human lymphoblastoid line. Chemical treatment of the cells induces a forward mutation at a specifi c locus, after which the cultures are incubated to allow phenotypic expression of the induced mutants. The mutants form colonies in a medium containing a selective agent that allows only mutants to proliferate while inhibit-ing wild-type cells. Mutagenic potential is proportional to mutant cell colony formation.
The V79, CHO, L5178Y, and TK6 cells lack the metabolic capacity to activate pro-mutagens and are therefore supplemented with an exogenous activation system including hepatic S9 fractions or intact cellular activation systems. The addition of exogenous factors and systems, however, introduces a major source of variability within the testing system.
CELL LINES
CHO and V79 Cells
These cells have stable karyotypes (2N = 20, 21, respectively), with a doubling time of 12–16 hours. The continuous cell lines are used with a variety of genetic markers including resistance to 8-azaadenine, cycloheximide, and methotrexate. The mutagenesis system involves a mutation at the HGPRT locus, the gene of which is present on the X chromosome. The locus encodes for the HGPRT enzyme responsible for the conversion of the purines, hypoxanthine, and guanine into nucleotides. Chemical agents such as 6-thioguanine (6-TG) and 8-azaguanine are cytotoxic to cells possessing HGPRT (HGPRT+), whereas a mutation at the HGPRT locus (HGPRT–) induces resistance.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 245
The assay relies on the metabolism of 6-TG and 8-azaguanine by HGPRT to toxic ribophosphorylated derivatives that cause cell death when incorporated into DNA. CHO or V79 cells (HGPRT+) are incubated with the test agent in the presence or absence of S9 supplementation. The cultures are incubated and replated in a medium containing 6-TG. Mutations at the HGPRT locus (HGPRT–) inactivate the enzyme and inhibit the catalytic formation of toxic metabolites from 6-TG. The mutant cells are able to survive using de novo purine biosynthesis and form colonies at concentrations of 6-TG normally cyto-toxic to wild-type cells.2
Mouse Lymphoma L5178Y Cells
The mouse lymphoma L5178Y mutagenesis system is based on quantifying forward muta-tions occurring at the heterozygous TK locus (L5178Y/TK±). The gene expresses the TK enzyme responsible for the incorporation of exogenous thymidine monophosphate. When trifl uorothymidine (TFT) is substituted for thymidine as the substrate, the resultant phos-phorylated TFT irreversibly inhibits thymidylate synthetase, resulting in cell death. Muta-tions at the locus result in the loss of TK activity and the formation of a homozygous strain (TK–/–). Incorporation of 5-bromodeoxyuridine (BrdUrd) results in cytotoxicity to the TK± cells, while the enzyme-defi cient cells (TK–/–) continue proliferation as they are unable to convert BrdUrd to toxic metabolites.
When lymphoma cells are exposed to a putative mutagen in the L5178Y mutagenesis system, forward mutations occur at the TK locus and inactivate the TK enzyme. As with the CHO and V79 cells, the inclusion of TFT in the culture medium selects for TK mutants that proliferate by synthesizing purines de novo, whereas wild-type cells do not survive.3
CYTOGENETIC TESTING
The second stage of tier testing for the detection of mutagenicity and carcinogenicity includes the cytogenetic tests. Three different types of in vivo and in vitro cytogenetic tests monitor clastogenicity and aneugenicity. These light microscopic tests are used to determine chemi-cally induced changes in a structure or number of chromosomes and include tests to determine (1) chromosome aberrations, (2) exchanges of chromosome materials among sister chroma-tids, and (3) chromosome fragments or micronuclei arising from chromosome damage.
Originally developed for in vivo testing, advances in cell and tissue culture technology have created advantages for in vitro protocols. In particular, the cell culture adaptations are more convenient, economical, and are capable of screening many compounds. In fact, some procedures incorporate sequential use of both in vivo and in vitro methods, thus allowing more applicability for human exposure to genotoxic agents.
Chromosome Aberrations
Congenital abnormalities and neoplastic growth have long been considered to result in part from chromosome aberrations. Most known carcinogens are also identifi ed as clastogens—agents that induce chromosome breakage. As described in chapter 6 “Acute Toxicology Testing,” in vivo methods may incorporate microscopic examinations of different cell types from animals previously treated with a test agent. In contrast, in vitro methods are more
2 The CHO systems are also used extensively to examine chromosomal aberrations and sister chromatid exchanges, thus allowing comparison of data from tests having different end points. 3 Interestingly, the TK mutagenesis assay is also useful in detecting the clastogenic potential of a test compound, that is, the induction of chromosome breakage.

246 PRINCIPLES OF TOXICOLOGY TESTING
sensitive but, as with bacterial mutagenic systems, usually require exogenous supplementa-tion for enzymatic activation. Nevertheless, CHO and V79 cell lines and human lympho-cytes are commonly used for short-term cytogenetic studies.
Protocols are similar for most cell types and are similar to the protocol described for karyotypic analysis (chapter 13). The procedure commences with the induction of prolif-eration of cells, when needed, to stimulate entry into the cell cycle. This is accomplished by the addition of a mitogen such as phytohemagglutinin. Initial dosage range determina-tion of the test chemical includes measurement of cell viability or mitotic index as a base-line control parameter for the cell line. Subconfl uent cultures in the logarithmic phase of growth are prepared in LabTek™ chambers and treated for an empirically determined period to encompassing all stages of the cell cycle.4 The cultures are washed; colchicine is added to arrest the cells in metaphase and the cultures are processed 2–3 hours later for analysis of chromosome aberrations. Figure 17.5A illustrates the principles of typical models of chromosome aberration, exchange, and reunion.
A metaphase spread relies on proper swelling of the cells by the addition of hypotonic potassium chloride solution. Cells are then fi xed, the chamber cover is removed to expose the fi xed cells on the plastic or glass slide, and the slide is stained with methylene blue or Giemsa counterstain. Chromosomes are examined microscopically (1000×, oil immer-sion), and numbers and types of aberrations are scored.
Sister Chromatid Exchange
Sister chromatid exchange (SCE) involves the crossover of replicating DNA between chro-matids at homologous loci. Unlike chromosome aberration, the interchange does not involve morphological alteration and is detected only by differential labeling of the sister chroma-tids (Fig. 17.5B and C). Traditional methods using [3H]-thymidine ([3H]-TdR) incorporation into newly transcribed DNA have been replaced by methods using BrdUrd labels combined with different stains.
The incorporation of BrdUrd into cells offers a more accurate estimation of the frac-tions of cells in S phase than does DNA measurement alone using the [3H]-TdR uptake. The optimal method is the simultaneous measurement of both DNA content and incorpo-rated BrdUrd into cells. Continuous exposure to BrdUrd label in a method analogous to those developed for [3H]-TdR provides an estimate of the fraction of noncycling cells, and pulse-chase experiments provide quantitative measures of cell cycle progression. For instance, pulse-chase exposure to BrdUrd (0.5–2 hours) labels that fraction of cells in S phase at the time of the pulse, whereas continuous exposure (more than 2 hours) tags all cells that pass through S phase during the exposure period.
The protocol is similar to karyotypic analysis and detection of chromosome aberra-tions, with the exception that the BrdUrd label is added to the culture medium for at least two population doubling levels. Cells are grown in LabTek chambers; pulse-chase and continuous exposures to the halopyrimidine are used after incubation with the test chemi-cals. After appropriate wash steps, sister chromatids are distinguished with a fl uorescent label (anti-BrdUrd IgG-FITC conjugated antibody) plus Giemsa stain (FPG method) that produces permanently and differentially stained chromosomes.5 Blocking controls and biological controls are performed in parallel sets to account for background nonspecifi c
4 In the presence of S9 supplementation, chemical exposures are reduced to 2 to 3 hours due to cytotoxicity of the additive. 5 Fluorescence has the advantage of determining labeling index defi ned as BrdUrd labeled cells per total cells observed in the fi eld.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 247
fl uorescence and autofl uorescence, especially that observed with G1 cells that tend to glow
with minimal green fl uorescence from antibodies to BrdUrd.It is commonly accepted that a strong correlation exists between the induction of SCE
and carcinogenicity. In addition, SCE testing is considerably less troublesome than tests for chromosome aberrations, thus validating SCE detection as a dominant and useful short-term test for carcinogens and genotoxic agents.
Detection of Micronuclei
Tests designed for the measurement of micronuclei are comparable in sensitivity and speed to detection of chromosome aberrations, although the former tests detect only chromosome
Aberration
Localhomology
Non-homologousend joining
Non-homologousend joining
DSB
Simple aberration
DSB
Complex aberration
Homologousrepair
DSB
Two chromosomes
Two chromosomes
One chromosome
(A)
(B)
(C)
FIGURE 17.5 Standard models of chromosome aberrations. (A) Simple aberration, two double-strand break (DSB) misjoin. (B) Rejoining of more than two DSBs to form complex aberration. (C) Recombinational misrepair where a single-strand break leads to enzymatically mediated homolo-gous misrepair.

248 PRINCIPLES OF TOXICOLOGY TESTING
breakage. Micronuclei arise from chromosome fragments that are not successfully passed to daughter nuclei during mitotic cell division, particularly due to the lack of a centromere.
The assays are also used with a variety of plant and mammalian cells in culture; prolif-erating cells are particularly useful. Slight variations in procedure from those used for chromosome aberrations are noted in the protocols for the detection of micronuclei. Briefl y, cultured cells are treated with a test agent for a selected period, then incubated in the pres-ence of cytochalasin B to prevent cytokinesis6; nuclear division is not blocked (colchicine is omitted from the procedure). The rest of the protocol proceeds as for chromosome aber-rations. Cells that contain two nuclei are scored for the presence of micronuclei that are not incorporated into daughter nuclei.
Limitations of Short-Term Cytogenetic Testing
As mentioned earlier, cultured cells lack the metabolic potential of biotransformation, thus limiting measurement of genotoxicity to nonmetabolized parent chemicals. Microsomal enzyme preparations, feeder layers, and co-cultures, however, may supplement for the lack of metabolizing enzymes. While the test for chromosome aberrations is more sensitive and dependable, it is more laborious and more expensive than the micronucleus test or the SCE test.
UNSCHEDULED DNA SYNTHESIS
Principles of Testing
As mammalian cells both in vivo and in vitro are continuously insulted with natural and experimental exposures to chemicals, they have by evolutionary selection developed defense systems that facilitate repair of the infl icted damage. The repair mechanism of the process of unscheduled DNA synthesis is well characterized. The principles of this toxicology test rely on the incorporation of [3H]-TdR into cultured cells during or after treatment with the test chemical. The cell preparations are fi xed and dried on microscope slides and exposed to a phosphorescent digital imaging system.7 Other methods involve quantitative measurement of uptake of [3H]-TdR into newly synthesized or repaired DNA from control and chemically treated groups.
Unscheduled DNA synthesis estimation is an indirect measurement of DNA damage, since the sequential enzymatic repair process is initiated by a cell’s recognition of the pres-ence of DNA adducts formed as a result of chemical insult. The primary DNA structure is restored through excision of the adducts, DNA strand polymerization, and subsequent liga-tion. The intranuclear incorporation of the radioactive nucleotide base during the repair process acts as a quantitative tracer for the repaired lesion during any period of anticipated or unanticipated (unscheduled) DNA synthesis. The assay generates semiquantitative or quantitative data by autoradiography or liquid scintillation counting, respectively.
CELL TRANSFORMATION
Principles of Testing
Short-term tests for cell transformation represent sensitive and mechanistically valid in vitro models for chemically induced cancers in vivo. Malignant transformation is
6 Cytoplasmic division. 7 Traditional autoradiographic methods involved exposure of slides to a photographic emulsion that usually required several days to visualize opaque radioactive spots in nuclei.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 249
accompanied by phenotypic alterations that are retrospectively related to carcinogenic induction. The tests use a variety of end points that rely on the apparent alterations to detect the transforming abilities and carcinogenic potentials of test compounds, includ-ing (1) loss of anchorage dependence and (2) alterations cellular morphology or growth characteristics.
The latter criterion is measurable using typical cell culture parameters such as loss of contact inhibition, altered growth in agar, appearance of immortal characteristics, proliferation in multilayer cultures, and viral dependence. In addition, transformed cells acquire the ability to induce cancer when transplanted into syngeneic immuno-logically defi cient animals. The tests however suffer from lack of reproducibility, tre-mendous variability, and subjective interpretation encountered with the scoring of some results.
Focus Transformation Assay
Immortal cell lines, C3H/1OT1/2 and BALB/c 3T3 fi broblasts, are used in focus transforma-tion assays. The cells display characteristics of immortal mammalian cultures, particularly their continuous proliferation and tumorigenic propensity in vivo. Unlike other human or rodent immortal cells such as A549 cells derived from human lung carcinomas and Caco-2 cells from human colon carcinomas, C3H/1OT1/2 and BALB/C 3T3 organize into a spindle shape morphology and exhibit density-dependent and contact inhibition of growth. In addi-tion, they form foci in monolayer cultures.
The assay consists of exposing cultures to test chemicals for 24–72 hours upon initial plating, after which an additional 4–6 weeks of passage and exposure to chemicals expresses the transformed phenotype (Fig. 17.6). Monolayers or multilayers are fi xed and stained to determine the presence of foci of transformed cells. Variability of results arises from the
Cells seeded at 1 × 104 cells/cm2
in LabTek® chambers
Incubate and allow to settle for 24 hrs
Expose to chemical:1–3 d without enzyme activation2–4 hrs with enzyme activation
Wash, change medium
Incubate for additional 4–6 wks,change medium as needed
Preserve in fixative and stain
FIGURE 17.6 Outline of focus transformation assay using C3H/1OT1/2 and BALB/c 3T3 fi broblasts.

250 PRINCIPLES OF TOXICOLOGY TESTING
subjective infl uence of scoring. Thus, attempts to standardize the assay are based on the recognition of foci according to the type observed:
• Type I foci are not scored as malignantly transformed; they are tightly packed foci and monolayer cultures are not tumorigenic after inoculation into host animals.
• Type II foci exhibit dense, overlapping, multilayer networks of cells.• Type III foci are densely stained overlapping groups of multilayers of cells; the
edges of the growth patterns are irregular and are tumorigenic upon injection into host animals.
The assay has particular value for understanding and screening mechanistic initiation and promotion of multistage carcinogens that occur in vivo.
Syrian Hamster Embryo Cell Transformation Assay
This assay incorporates the mortal cell line into both focus and clonal assays. The focus assay begins with target Syrian hamster embryo (SHE) cells seeded at 1 × 105 cells/cm2 in the absence of feeder layers. This represents a higher seeding density since normal cell inoculation densities for most mammalian cells requires about 10-fold fewer cells. The cells are incubated for 3 days followed by a chemical exposure for 3 days, and transformed foci are evaluated after an additional 20–25 days.
The clonal assay features initial plating of mitotically inactivated feeder layers up to the confl uent density and co-cultured with few hundred target SHE cells. After 24 hours, the cultures are exposed to the test chemical for 7–10 days and processed for histological transformation. SHE cells transform into condensed, separate colonies with clearly demar-cated boundaries in contrast to the appearance of traditional cell transformation assays. Figure 17.7 outlines the procedure.
Viral and Chemical Transformation Methods
Historically applied in mechanistic carcinogenic toxicology studies, viral and chemical transformation assays have also found some utility as short-term screening tests. Two well-categorized transformation methods include SHE/SA7 cell lines and Fisher rat embryo (FRE) cells (FRE/RLV transformation assay). Both cell lines are more susceptible to chem-ical transformation since the cells were previously exposed to infection with simian adeno-virus (SA7) and with Rauscher leukemia virus (RLV), respectively.
DEVELOPMENT OF SHORT-TERM TEST STRATEGIES
Important objectives in the development of in vitro short-term tests for the evaluation of muta-genic potential of chemicals include the identifi cation of potential germ cell and somatic cell mutagens as potential carcinogens. With the current understanding and developmental status of in vitro methods, it is unreasonable to assume that even a battery of in vitro tests alone can meet these objectives, especially for human risk assessment. Consequently, evaluation of mutagenic potential of chemicals, especially chemicals with direct human exposure, neces-sarily should incorporate multiple assays, the objective of which is to identify genotoxic and

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 251
cytogenetic carcinogens at an early stage. Thus, the ability of any testing scheme to detect potential carcinogens involves a variety of methods:
1. A preliminary screen using structure–activity relationship studies of the chemical.2. In vitro tests genetic tests with bacterial cells and chromosome tests with mamma-
lian cells.3. In vivo assays select carcinogenicity studies with animal models based on the pre-
liminary screens.
The performance of screening tests for the detection of mutagenic potential of chemicals is guided by the quantitative ability of the tests for sensitivity, specifi city, and concordance. Sen-sitivity is the percentage of carcinogens that screen positive in a given test; specifi city is the percentage of noncarcinogens that are negative; concordance refers to the percentage of results from both carcinogens and noncarcinogens from short-term tests that agree with in vivo data.
The European Scientifi c Committee on Cosmetics and Non-Food Products guidelines for testing of hair dyes for genotoxic, mutagenic, and carcinogenic potential include a bat-tery of six in vitro tests. The guidelines suggest that potential genotoxic activity of chemi-cals used in hair dyes may effectively be determined by the application of a limited number of well-validated test systems capable of detecting induced gene mutations and structural and numerical chromosome changes.
The recently updated guidelines for food additives by the European Community’s Scientifi c Committee on Food recommends a battery of three in vitro tests for the induc-tion of gene mutations in bacteria and in mammalian cells (which usually include the mouse lymphoma TK assay and assay for chromosome aberrations). Also, additional data
Incubate and allow to settle for24-hr
After 24-hr, expose to chemical:7–10 d without enzyme activation,12–24 hr, + 6–8 d, with enzyme
activation
Preserve in fixative and stain
Co-culture with 200–300 targetcells
Feeder cells mitoticallyinactivated and seeded in
LabTek® chambers at 1 × 105
cells/cm2
FIGURE 17.7 Outline of Syrian hamster embryo (SHE) cell transformation assay using SHE fi bro-blasts. Cells are mitotically inactivated using either ultraviolet light or mitomycin-C.

252 PRINCIPLES OF TOXICOLOGY TESTING
generated from physicochemical, structural, and metabolic properties may supplement the standard battery.
The EU also describes a number of tests similar to the OECD guidelines mentioned earlier (Table 17.1; http://europa.eu.int/) that include the morphological cell transformation assay. Among the national guidelines of the EU, those of the U.K. Department of Health are particularly noteworthy. The “Guidelines for the Testing of Chemicals for Mutagenicity” (http://www.doh.gov.uk/com.htm) are based on the recommendations of the Committee on Mutagenicity of Chemicals in Food, Consumer Products, and the Environment and provide background information and justifi cation for testing and assessment. The proposal extends to recommend the inclusion of an in vitro test for the potential to induce aneuploidy (micro-nucleus test or metaphase analysis).
The International Conference on Harmonization (ICH) of the Toxicological Require-ments for Registration of Pharmaceuticals for Human Use recommends a core testing bat-tery for evaluation of pharmaceuticals. The working group suggested that a battery include a test for gene mutation in bacteria for chromosome aberrations in mammalian cells in vitro or the L5178Y mouse lymphoma mammalian cell mutagenesis test and an in vivo test for chromosome damage in rodent hematopoietic cells. The ICH guidance documents address test procedures as well as strategy and test interpretation.
Reviews of genotoxicity testing programs and publications are documented as part of the GENETOX Program of the U.S. EPA (http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?GENETOX). The recently completed phases of its ongoing efforts to review the existing literature about chemically induced genetic toxicology were derived from peer-reviewed mutagenicity test data. The agency also gives direction to the diversity of the tests used to detect chemically induced genetic damage.
EPA’s Environmental Carcinogenesis Division is located within the National Health and Environmental Effects Research Laboratory. Its research focuses on the improved understanding of environmentally induced mutagenesis and carcinogenesis for incorpora-tion into human cancer risk assessment models. The program uses cellular, animal, and computer models for assessing responses to a broad range of environmental chemicals and mixtures. The objective of the research is to understand the chemical induction of somatic and germ cell mutations as a basis for improving risk assessments for cancer and heritable mutations, respectively. Goals and approaches of the research are directed at:
1. identifying hazards;2. assessing dose and tumor responses using applicable biomarkers;3. developing dose–response curves for tumor outcomes in humans using rodent
tumor data and cellular indicators of tumorigenicity;4. defi ning mechanisms of metabolic activation and detoxifi cation for mutagenic
agents;5. enabling comparison of human and laboratory animal and cellular test system
results;6. improving the performance of biologically based dose–response models used in
risk assessment;7. developing and validating molecular techniques, short-term bioassays, and whole-
animal bioassays for evaluating carcinogenic potential.
Thus, Environmental Carcinogenesis Division has the capability to infl uence the develop-ment of the cancer risk assessment process both nationally and globally.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 253
SUMMARY
A variety of factors contribute to the heterogeneous results obtained from in vitro mutagenic-ity tests and following are the most important of them:
1. Absence of cellular biotransformation systems in prokaryotic cells.2. Inconsistency in DNA repair mechanisms in prokaryotic cells.3. Sporadic presence of detoxifying enzymes in mammalian cell culture systems.4. Variable levels of sensitivity of in vitro methods to particular classes of mutagenic
agents.5. High sensitivity of in vitro tests whose results may not refl ect in vivo exposures.
Other confounding issues of human biology temper the validity of in vitro tests for detecting carcinogenic potential. Consider the infl uence of environmental factors including exposure circumstances, background rates of spontaneous mutations, and the relationship of gene struc-ture and biological function under normal circumstances of existence. These dynamics should enhance the development of regulatory testing schemes to prevent adverse effects associated with exposure to chemicals rather than confuse and inhibit progress toward this goal.
It is interesting that although general principles of mutagenicity risk assessment and guidelines are outlined and published, quantitative risk assessments are rarely made even within regulatory agencies. The volumes of genotoxicity data are currently used to make regulatory decisions based on genotoxic hazard classifi cation or to provide mechanistic information to support quantitative carcinogenicity risk assessments. Signifi cant mile-stones and progress were achieved with the development of in vitro mutagenicity testing technology and with their concordance with in vivo models. The future for using these models in human risk assessment portends that speculation is directed in favor of conserva-tive judgments. Consequently, the only guarantee that the controversies, limitations, and agreements among regulatory agencies, industrial concerns, and research institutions will be resolved is the achievement of perfect harmonization. Finally, developing acceptable in vitro tests systems to reduce unnecessary animal use is a noble goal, especially if the in vitro data for quantitative predictions of cell-specifi c effects are reliable. However, the goal is valid as long as human risk assessment is not compromised.
SUGGESTED READINGS
Ames BN, Gurney EG, Miller JA, Bartsch H. Carcinogens as frameshift mutagens: metabolites and derivatives of 2-acetylaminofl uorene and other aromatic amine carcinogens. Proc Natl Acad Sci USA 1972; 69: 3128.
Ames BN, Sims P, Grover PL. Epoxides of carcinogenic polycyclic hydrocarbons are frameshift mutagens. Science 1972; 176: 47.
Ames BN. Identifying environmental chemicals causing mutations and cancer. Science 1979; 204: 587.
Ames BN, Gold LS, Willett WE. The causes and prevention of cancer. Proc Natl Acad Sci USA 1995; 92: 5258.
Cohen SM, Ellwein LB. Cell proliferation in carcinogenesis. Science 1990; 249: 1007l.Jacobson-Kram D, Contrera JF. Genetic toxicity assessment: employing the best science for human
safety evaluation. Part I: early screening for potential human mutagens. Toxicol Sci 2007; 96: 16.
Jakóbisiak M, Lasek W, Gołab J. Natural mechanisms protecting against cancer. Immunol Lett 2003; 90: 103; Erratum in: Immunol Lett 2004; 91: 255.

254 PRINCIPLES OF TOXICOLOGY TESTING
Kirkland DJ, Aardema M, Banduhn N, et al. In vitro approaches to develop weight of evidence (WoE) and mode of action (MoA) discussions with positive in vitro genotoxicity results. Mutagen-esis 2007; 22: 161.
Klaassen CD. Casarett & Doull’s Toxicology: The Basic Science of Poisons, 7th edn. chap. 8 New York: McGraw-Hill Companies, Inc, 2007.
Klaunig JE. Acrylamide carcinogenicity. J Agric Food Chem 2008; 56: 5984.Little MP, Heidenreich WF, Moolgavkar SH, Schöllnberger H, Thomas DC. Systems biological and
mechanistic modelling of radiation-induced cancer. Radiat Environ Biophys 2008; 47: 39.Lorge E, Gervais V, Becourt-Lhote N, et al. Genetic toxicity assessment: employing the best science
for human safety evaluation part IV: a strategy in genotoxicity testing in drug development: some examples. Toxicol Sci 2007; 98: 39.
Marks F, Fürstenberger G, Müller-Decker K. Tumor promotion as a target of cancer prevention. Recent Results Cancer Res 2007; 174: 37.
National Toxicology Program (NTP). Department of Health and Human Services, Report on Car-cinogens (RoC). [Available from: http://ntp.niehs.nih.gov] [Last viewed Feb., 2009].
Nomura T. Transgenerational effects from exposure to environmental toxic substances. Mutat Res 2008; 659: 185.
Valavanidis A, Fiotakis K, Vlachogianni T. Airborne particulate matter and human health: toxico-logical assessment and importance of size and composition of particles for oxidative dam-age and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2008; 26: 339.
Weinstein IB. Cancer prevention: recent progress, and future opportunities. Cancer Res 1991; 51: 5080s.
REVIEW ARTICLES
Aardema MJ, Snyder RD, Spicer C, et al. SFTG international collaborative study on in vitro micro-nucleus test III. Using CHO cells. Mutat Res 2006; 607: 61.
Bolt HM, Foth H, Hengstler JG, Degen GH. Carcinogenicity categorization of chemicals: new aspects to be considered in a European perspective. Toxicol Lett 2004; 151: 29.
Brusick D. Evolution of testing strategies for genetic toxicity. Mutat Res 1988; 205: 69.Carere A, Stammati A, Zucco F. In vitro toxicology methods: impact on regulation from technical and
scientifi c advancements. Toxicol Lett 2002; 127: 153.Clare MG, Lorenzon G, Akhurst LC, et al. SFTG international collaborative study on in vitro micro-
nucleus test II. Using human lymphocytes. Mutat Res 2006; 607: 37.Decordier I, Kirsch-Volders M. The in vitro micronucleus test: from past to future. Mutat Res 2006;
607: 2.Farmer PB. Committee on Mutagenicity (COM) of Chemicals in Food, Consumer Products and the
Environment ILSI/HESI research programme on alternative cancer models: results of Syr-ian hamster embryo cell transformation assay. Toxicol Pathol 2002; 30: 536.
Hartley-Asp B, Wilkinson R, Venitt S, Harrap KR. Studies on the mechanism of action of LS 1727, a nitrosocarbamate of 19-nortestosterone. Acta Pharmacol Toxicol 1981; 48: 129.
Kirkland DJ, Henderson L, Marzin D, et al. Testing strategies in mutagenicity and genetic toxicol-ogy: an appraisal of the guidelines of the European Scientifi c Committee for Cosmetics and Non-Food Products for the evaluation of hair dyes. Mutat Res 2005; 588: 88.
Kowalski LA. In vitro carcinogenicity testing: present and future perspectives in pharmaceutical development. Curr Opin Drug Discov Devel 2001; 4: 29.
Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol 2008; 18: 322.
Mohan CG, Gandhi T, Garg D, Shinde R. Computer-assisted methods in chemical toxicity prediction. Mini Rev Med Chem 2007; 7: 499.
Müller L, Kikuchi Y, Probst G, et al. ICH-harmonized guidances on genotoxicity testing of pharma-ceuticals: evolution, reasoning and impact. Mutat Res 1999; 436: 195.

MUTAGENICITY AND CARCINOGENICITY TESTING IN VITRO 255
MacGregor JT, Casciano D, Muller L. Strategies and testing methods for identifying mutagenic risks. Mutat Res 2000; 455: 3.
Nesnow S. Complex mixtures of chemical carcinogens. In: Warshawsky D, Landolph J, eds. Molecu-lar Carcinogenesis and the Molecular Biology of Human Cancer. chap. 14 Boca Raton, FL: CRC Press, 2006.
Oliver J, Meunier JR, Awogi T, et al. SFTG international collaborative study on in vitro micronucleus test V. Using L5178Y cells. Mutat Res 2006; 607: 125.
Parry JM, Parry EM. The use of the in vitro micronucleus assay to detect and assess the aneugenic activity of chemicals. Mutat Res 2006; 607: 5.
Preston RJ. Genetic toxicology. In: Hodgson R, Smart R, eds. Introduction to Biochemical Toxicol-ogy, 3rd edn. chap. 16. New York: John Wiley & Sons, 2001.
Richardson SD, Plewa MJ, Wagner ED, Schoeny R, Demarini DM. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: a review and roadmap for research. Mutat Res 2007; 636: 178.
Rothfuss A, Steger-Hartmann T, Heinrich N, Wichard J. Computational prediction of the chromosome-damaging potential of chemicals. Chem Res Toxicol 2006; 19: 1313.
Schramke H, Meisgen TJ, Tewes FJ, Gomm W, Roemer E. The mouse lymphoma thymidine kinase assay for the assessment and comparison of the mutagenic activity of cigarette mainstream smoke particulate phase. Toxicology 2006; 227: 193.
Wexler P. The U.S. National Library of Medicine’s Toxicology and Environmental Health Informa-tion Program. Toxicology 2004; 198: 161.
Williams GM. Detection of chemical carcinogens by unscheduled DNA synthesis in rat liver primary cell cultures. Cancer Res 1977; 37: 1845.

256
18 Reproductive and teratogenicity studies in vitro
INTRODUCTION
Standard in vivo methods for screening substances that are potentially toxic to a fetus (see chap. 10) are laborious and expensive, and require extensive preliminary planning of exper-iments to determine the correct end point in the reproductive scheme.
Many chemicals cause a variety of pre- and postnatal developmental abnormalities and also interfere with mating and conception. Chemicals that induce structural malformations, physiological dysfunctions, behavioral alterations, and defi ciencies in the offspring, at birth or in the immediate postnatal period, are referred to as teratogens. Most chemicals are not extensively screened for their teratogenic potential, principally because of the many toxicity tests that are already required. Thus, most pharmaceutical and chemical companies do not routinely screen therapeutic drugs or industrial chemicals, respectively, for potential embryotoxicity during preclinical phases. Chemicals not destined to be used clinically are generally not subjected to teratogenic testing.
Consequently, there is dearth of embryotoxic data for the majority of substances used in the clinical marketplace. However, there is signifi cant pressure on regulatory agencies to accept models, such as the whole-embryo culture (WEC) described later, as screening sys-tems for safety evaluations. A sustained effort in the development of these and other in vitro methods into standardized, scientifi cally validated tools for developmental toxicology is important and crucial. The methodological development of these culture techniques is described in the following sections.
Classical methods for testing teratogenic substances utilize rodents and rabbits, princi-pally because of their relatively short gestational periods (21–22 and 32 days, respectively). Depending on the experimental objective, the procedures involve exposure of the female to a potential teratogen either before mating, after conception, during gestation, or in the immediate postnatal period. Alternatively, males are exposed to chemicals when the spe-cifi c objective is to determine the infl uence of a chemical on male reproductive capacity. Thus, the start of the exposure sequence depends on the toxicological questions of interest. Also, the trimester during which the chemical has its greatest toxic effects is determined by exposing gravid animals during a specifi c time of the gestational period. Using this proto-col provides answers about the mechanisms of action.
Rodents and rabbits are also convenient because of the large sizes of their litters, usu-ally about 8–10 embryos in a gravid animal. This allows for scoring of the chemicals according to the number of fetuses affected. For instance, a particular indicator is assessed in each embryo, the minimal accepted value of which is regarded as toxic. The greater the number of embryos that exhibit toxicity by this criteria, the higher the score and the more

REPRODUCTIVE AND TERATOGENICITY STUDIES IN VITRO 257
toxic the chemical. Given the potential number of chemicals that can be screened at the different dosage levels for various indicators in each fetus, the number of experiments can be technically and fi nancially exhausting.
Chapter 10, “Toxicity Testing for Fertility and Reproduction,” describes the mechanisms of developmental toxicity, susceptibility to teratogenic agents, and the factors associated with teratogenicity. This chapter also presents an introduction to maternal-fetal physiology. Thus, with the exception of the whole-embryo culture test for embryotoxicity described below, the current chapter directs the reader’s attention specifi cally so as to emphasize tests that have been developed for in vitro reproductive and teratogenicity studies.
ALTERNATIVE METHODS FOR EMBRYOTOXICITY TESTING
Alternative methods for embryotoxicity testing and developmental toxicology are divided into three types of systems: (1) WECs, (2) cell cultures, and (3) organ cultures. WEC and organ culture methods enjoy the advantages of representing higher levels of developmental mechanisms and end points that adequately refl ect organogenesis and morphogenesis. The methods, however, are laborious and rely on the use of animals. In contrast, while primary and continuous cell cultures are relatively easy to perform and depend on minimal or no animal use, they are mechanistically simpler. Thus, lower levels of sensitivity may not be suffi cient for detecting many potential teratogens that are more likely to be revealed with WEC methods. Table 18.1 summarizes the advantages and disadvantages of the three sys-tems that are further discussed in the coming text.
WHOLE-EMBRYO CULTURE
Postimplantation embryo culture is a classical method for the detection of potential embryo-toxic agents. The technique involves the removal of 1- or 2-week-old rodent embryos after which they are acclimated under cell culture conditions for several hours. The viable fetuses are then exposed to different concentrations of chemicals suspected of altering developmen-tal status. The method is dramatic: structural changes occur quickly and teratogenicity is grossly monitored in vitro as growth, development, and differentiation progress.
TABLE 18.1Advantages and Disadvantages of Alternative Methods for Embryotoxicity Testing
Method Advantages Disadvantages
Whole-embryo culture (WEC)
Mechanistically more complex; more sensitive to detecting potential teratogens; mimic different gestational periods
Requires signifi cant numbers of animals; laborious, expensive; serum concentration infl uences results; limited period of embryogenesis in culture
Organ culture Mechanistically not as complex as WEC; sensitive for detection of potential teratogens; mimics different gestational periods; detects organ-specifi c toxicity
Requires fewer animals than WEC methods; expensive and laborious
Primary and continuous cell culture
Technically easier; requires fewest animals; contributes to understand-ing mechanistic toxicity
Simpler biological system; less sensitive to embryotoxic effects; devoid of biotransformation capacity

258 PRINCIPLES OF TOXICOLOGY TESTING
One potential benefi t of the method is that it allows for further investigation of substances with teratogenic activities in vivo. In addition, culturing embryos in vitro provides the oppor-tunity to study the direct actions of teratogens free of maternal neuroendocrine, immunologi-cal, and metabolic infl uences, the advantage of which is viewed as a distinct disadvantage outside the realm of cell culture technology. In addition, dose-related growth retardation and frequency of malformations using WECs also depend on the periods of gestation of the explanted embryos.
Although the procedure uses growth in culture as a component of the protocol and the methods for growing whole-mouse embryos in vitro have largely improved in the past two decades, the technique should not be confused with in vitro cell culture methods. Embryo cul-tures still require extensive animal preparation and sacrifi ce and offer few advantages for large-scale prescreening studies. The technology, however, enables embryos to develop remarkably close to what is achieved in vivo from gastrulation to early organogenesis stages, while permit-ting direct chemical and micromanipulation of embryos. Lastly, procedures involving preim-plantation and early postimplantation of embryos during organogenesis (described later) may not be adequate for screening chemicals that have teratogenic potentials because they monitor single-cell processes that do not refl ect the complex nature of embryonic development.
CELL CULTURE METHODS
Because of the problems associated with traditional embryotoxicity testing, efforts have been made to develop in vitro cell culture teratogenicity tests. A prerequisite for the use of embry-onic tissue in culture is that the isolated organs or cells are maintained in culture for at least 48–72 hours. Several methods incorporating isolated cells or organs, for example the “micro-mass” method, are used. The technique is described for the culture of dissociated rodent arm and leg rudiment cells that are capable of synthesizing a variety of extracellular matrix pro-teins. Protein production is inhibited by most known teratogenic chemicals and measured by classical spectrophotometric analysis or by incorporation of radio-labeled amino acids.
Preimplantation Techniques
Since the fi rst description of the culture of preimplantation embryos by Brachet, the method has shown its importance in experimental embryology and reproductive biology. The tech-nique is described for the preimplantation culture of embryos from mice, rabbits, and humans. In general, 3- to 6-day-old embryos (4-day morulae and 6-day blastocysts) are removed from superovulated sexually mature animals and transplanted into culture dishes containing media supplemented with bovine serum albumin, serum, or serum-free condi-tioned medium. The cultures are exposed to the test substance, examined histologically, and monitored for growth and proliferation using biochemical procedures. With the advent of embryonic stem (ES) cell technology, however, the procedure may soon lose its popularity.
Continuous Cell Cultures
Finite or immortal continuous cell lines are used for monitoring teratogenicity in vitro. Sev-eral studies now attest to the importance of the potential for these cell lines for screening a variety of chemicals for teratogenic potential. The cell lines include differentiated early human embryonic cells (HFL1, MRC-5, and WI38 fetal human lung fi broblasts) derived from fi rst trimester fetuses and represent the period of gestation when the embryo is most susceptible to developmental anomalies. The fi broblasts are easily maintained in continuous culture, have fi nite life spans, and are characterized in aging studies. It is important to note,

REPRODUCTIVE AND TERATOGENICITY STUDIES IN VITRO 259
however, that these continuous cell lines may not be suitable cell culture models for screening chemicals with potential toxicity to fetal development because the lines are beyond the pri-mary culture stage. Consequently, their responses may refl ect basal cytotoxicity rather than a specifi c effect on growing fetuses.
Embryonic Stem Cells
ES cells are derived from pluripotent cells of early mammalian embryos and are capable of unlimited, undifferentiated proliferation in vitro. In chimeras with intact embryos, ES cells contribute to a wide range of adult tissues including germ cells, providing a powerful approach for introducing specifi c genetic changes into a mouse cell line. Pluripotency allows for the ability of the cells to differentiate to many types and tissues including all three embry-onic germ layers. Furthermore, experimental evidence is available for organogenesis of embryonic and adult stem cells to neural, muscle, dermal, epithelial, and bone marrow lin-eages. Progress on ES cells is prompted, in part, by the understanding that in vivo epithe-lium, epidermal, and mesodermal cells undergo continuous renewal by multipotent stem cells that remain anchored to basement membranes. Thus a single stem cell migrates, dif-ferentiates, and commits to all cell line lineages.
The study of ES cells began modestly in the 1960s with manipulation of early rabbit and mouse morulae and blastocysts that adhered readily to plastic tissue culture vessels. They were observed to form sheets of cells that were overgrown by stem cells from the inner cell masses. Whole-blastocyst cultures on collagen-coated surfaces produced aggre-gates of cells that eventually migrated and differentiated into neural, blood, and phagocytic cell types. Not until the inner cell mass was freed and cultured intact did the research com-munity realize that ES cells were capable of possessing good rates of cleavage and could demonstrate morphological and chromosomal stability. In mice, formations of cell aggre-gates with features of intermediate differentiation are known as embryoid bodies. Markers for differentiation of pluripotency reveal how neural, cardiac, hematological, and other cell lineages are derived in vitro.
The derivation of human ES cells is a phenomenon that came into existence in the late 1990s and these cells have clinical utility because of their rapid colonization and poten-tial for targeting tissues via fetal pathways. Thus, human ES cells offer broad therapeutic potential for clinical applications, the realization of which is a matter of time.
The fi rst description of a stem cell test as an in vitro assay for teratogenic potential outlined a method for maintaining mouse ES cells in culture in an undifferentiated state. Subsequently, manipulation of the culture conditions allowed establishment of a popula-tion of differentiated cells onto which chemicals could be tested, without the need for continuously establishing primary cultures from mouse embryos. Later, the embryonic stem cell test (EST) was used to detect teratogenic potentials of several compounds in undifferentiated cells. In 2004, ZEBET, in cooperation with the European Centre for Vali-dation of Alternative Methods (ECVAM), coordinated prevalidation and validation studies with three embryotoxicity tests including the EST. The studies concluded that correlation between in vitro EST data and in vivo data was good and that the EST was applicable for testing a diverse group of chemicals of different embryotoxic potentials.
ORGAN CULTURES
A variety of organ culture models have been developed as reproductive and developmen-tal toxicology test systems. The principles of these models are focused on the improve-ment of in vitro alternative methods that more closely represent the complex process of

260 PRINCIPLES OF TOXICOLOGY TESTING
embryogenesis in vivo. The methods require primary animal tissue, however, and have specifi c technical features that render them more laborious and more diffi cult to standard-ize among laboratories. Tests in this group include organotypic models for limb buds, digits, palatal shelves, lungs, intestines, and male and female reproductive organs.
Fetal organs have been maintained in culture in the primary cell stage by explanting them and partially or totally submerging their tissues in growth medium. A tissue explant is placed on a micron fi lter or in tissue culture plastic vessels coated with extracellular matrix components. This approach is useful in the culture of limb buds from several species of animals in a serum-supplemented or chemically defi ned medium. Morphological devel-opment, differentiation, and biochemical parameters are monitored. The variety of primary organotypic cultures isolated successfully include the following:
1. Cultures of mouse kidneys for studies of renal development and suspected nephro-toxic agents.
2. Limb buds for detecting chemical developmental anomalies.3. Gastrointestinal structures for potential teratogens for stomach and intestinal devel-
opment.4. Cultures of testes and ovaries for reproductive studies.5. Isolation of type II epithelial cells from fetal rat lungs.
Rat lung epithelial cells are specialized cells capable of synthesizing and storing a surfactant composed of phospholipids; its secretion is destined for the pulmonary alveolar space. In vivo, the surfactant coats the alveolar walls and decreases the surface tension present at the air–cell interface and thus allows oxygen to penetrate the thin alveolar structures. Con-sequently, the cells are used in toxicity testing to monitor embryotoxic effects in the lungs, particularly in relation to surfactant production and secretion. Thus, as with other organ-specifi c primary cultures, their advantage lies in their ability to act as models for detecting target organ toxicities. Interlaboratory validation studies involving alternative mammalian organotypic models provide the toxicological community with a variety of methods for inclusion in a battery of tests for testing chemicals with embryotoxic potential.
VALIDATION OF ALTERNATIVE METHODS FOR REPRODUCTIVE AND TERATOGENICITY STUDIES
The validation of alternative test systems relies on a comparison of sensitivity, specifi city, and concordance with documented in vivo studies. Since only those chemicals for which suffi cient in vivo data are available are used for comparisons of developmental toxicity, attempts at in vitro validation are limited by the number of test chemicals available. In addi-tion, the most relevant compounds for hazard assessment are those whose developmental toxicities are within marginal toxic ranges and clearly not at the extreme toxic or nontoxic ends. Thus, the ability of in vitro screening methods for predicting potential teratogens is hampered not by the development of the testing method, but by the lack of adequate com-parative in vivo information.
In order to standardize methodologies and defi ne end points for in vitro analysis, ECVAM defi ned a prediction model approach that attempts to extrapolate in vitro data to in vivo studies. The prediction model contains quantitative end points that are expected to predict in vivo responses. The project outcomes evaluate the performance of a test and are

REPRODUCTIVE AND TERATOGENICITY STUDIES IN VITRO 261
modifi ed when the database of chemicals tested in the in vitro system is suffi cient, espe-cially when performed with unknown classes of chemicals.
Currently, the most elaborate validation study of embryotoxicity assays performed was fi nanced by ECVAM and includes the EST, the limb bud micromass, and the WEC tech-nique. Each test involved 20 chemical compounds tested in four independent laboratories in a double-blind design. Preliminary data indicate that the WEC method displayed the best concordance of in vivo classifi cation and in vitro test results with 80% correct classifi ca-tions versus 78 and 71% for EST and micromass, respectively. Moreover, strong embryo-toxic compounds showed a predictivity of 100% in each of the test systems.
SUMMARY
The wealth of potential alternative tests available for developmental toxicology testing is in part a refl ection of the variety of biological mechanisms involved in embryonic and fetal development. Since it is widely accepted that embryogenesis is a remarkably complex phys-iological process, it is reasonable to conclude that, with our current biotechnology, some time is required before a single alternative test can replace whole-animal testing. Hence, it is more likely, at least in the immediate future, that combinations or batteries of tests will be able to address the complete range of mechanisms of embryonic and reproductive development.
Because of the unique features of this process, alternative testing in developmental toxicology differs signifi cantly from the principles of in vitro dermal and ocular toxicology testing areas that have already enjoyed substantial success in delivering alternative tests for regulatory acceptance. Scientifi c progress, however, is not ostensibly gradual but is more compelling and abrupt, so that realization of advanced alternative models for reproductive and embryo toxicology may appear on an unforeseeable, yet closer, horizon.
SUGGESTED READINGS
Bremer S, Hartung T. The use of embryonic stem cells for regulatory developmental toxicity testing in vitro: the current status of test development. Curr Pharm Des 2004; 10: 273.
Brown NA. Selection of test chemicals for the ECVAM international validation study on in vitro embryotoxicity tests. Altern Lab Anim 2002; 30: 177.
Calabro AR, Konsoula R, Barile FA. Evaluation of in vitro cytotoxicity and paracellular permeability of intact monolayers with mouse embryonic stem cells. Toxicol In Vitro 2008; 22: 1273.
Flint OP. In vitro tests for teratogens: desirable endpoints, test batteries and current status of the micromass teratogen test. Reprod Toxicol 1993; 7: 103.
Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modifi cation path-ways concurrently in mouse embryonic stem cells. Toxicol Lett 2012; 212: 169.
Genbacev O, White TE, Gavin CE, Miller RK. Human trophoblast cultures: models for implantation and peri-implantation toxicology. Reprod Toxicol 1993; 7: 75.
Genschow E, Spielmann H, Scholz G, et al. The ECVAM international validation study on in vitro embryotoxicity tests: results of the defi nitive phase and evaluation of prediction models. Altern Lab Anim 2002; 30: 151.
Genschow E, Spielmann H, Scholz G, et al. Validation of the embryonic stem cell test in the interna-tional ECVAM validation study on three in vitro embryotoxicity tests. Altern Lab Anim 2004; 32: 209.
Kirkland DJ, Henderson L, Marzin D, et al. Testing strategies in mutagenicity and genetic toxicol-ogy: an appraisal of the guidelines of the European Scientifi c Committee for Cosmetics and Non-Food Products for the evaluation of hair dyes. Mutat Res 2005; 588: 88.
Palmer AK. Introduction to (pre)screening methods. Reprod Toxicol 1993; 7: 95.

262 PRINCIPLES OF TOXICOLOGY TESTING
REVIEW ARTICLES
Augustine-Rauch K, Zhang CX, Panzica-Kelly JM. In vitro developmental toxicology assays: A review of the state of the science of rodent and zebrafi sh whole embryo culture and embryonic stem cell assays. Birth Defects Res C Embryo Today 2010; 90: 87.
Barile FA, Ripley-Rouzier C, Siddiqi ZE, Bienkowski RS. Effects of prostaglandin E1 on collagen production and degradation in human fetal lung fi broblasts. Arch Biochem Biophys 1988; 265: 441.
Barile FA, Siddiqi ZE, Ripley-Rouzier C, Bienkowski RS. Effects of puromycin and hydroxynorva-line on net production and intracellular degradation of collagen in human fetal lung fi bro-blasts. Arch Biochem Biophys 1989; 270: 294.
Barile FA, Arjun S, Hopkinson D. In vitro cytotoxicity testing: biological and statistical signifi cance. Toxicol In Vitro 1993; 7: 111.
Bechter R. The validation and use of in vitro teratogenicity tests. Arch Toxicol Suppl 1995; 17: 170.Brachet A. Recherches sur le determinism hereditaire de l’oeuf des mammiferes: development in
vitro de jeunes vesicules blastodermique du lapin. Arch Biol 1913; 28: 447.Brown NA. Teratogenicity testing in vitro: status of validation studies. Arch Toxicol 1987; 11: 105.Buesen R, Visan A, Genschow E, et al. Trends in improving the embryonic stem cell test (EST): an
overview. ALTEX 2004; 21: 15.Edwards RG. Stem cells today: Origin and potential of embryo stem cells. Reprod Biomed Online
2004; 8: 275.Evans M, Kaufman M. Establishment in culture of pluripotential cells from mouse embryos. Nature
1981; 92: 154.Fein A, Carp H, Torchinsky A, et al. Peri-implantation mouse embryos: an in vitro assay for assessing
serum-associated embryotoxicity in women with reproductive disorders. Reprod Toxicol 1998; 12: 155.
Flick B, Klug S. Whole embryo culture: an important tool in developmental toxicology today. Curr Pharm Des 2006; 12: 1467.
Flint O, Orton TC. An in vitro assay for teratogens with cultures of rat embryo midbrain and limb bud cells. Toxicol Appl Pharmacol 1984; 76: 383.
Kadereit S, Zimmer B, van Thriel C, Hengstler JG, Leist M. Compound selection for in vitro model-ing of developmental neurotoxicity. Front Biosci 2012; 17: 2442.
Lee HY, Inselman AL, Kanungo J, Hansen DK. Alternative models in developmental toxicology. Syst Biol Reprod Med 2012; 58: 10.
Marx-Stoelting P, Adriaens E, Ahr HJ, et al. A review of the implementation of the embryonic stem cell test (EST). The report and recommendations of an ECVAM/ReProTect Workshop. Altern Lab Anim 2009; 37: 313.
Newall DR, Beedles K. The stem cell test: a novel in vitro assay for teratogenic potential. Toxicol In Vitro 1994; 8: 697.
Pellizzer C, Bremer S, Hartung T. Developmental toxicity testing from animal toward embryonic stem cells. ALTEX 2005; 22: 47.
Piersma AH. Validation of alternative methods for developmental toxicity testing. Toxicol Lett 2004; 149: 147.
Pistollato F, Bremer-Hoffmann S, Healy L, Young L, Stacey G. Standardization of pluripotent stem cell cultures for toxicity testing. Expert Opin Drug Metab Toxicol 2012; 8: 239.
Riebeling C, Hayess K, Peters AK, et al. Assaying embryotoxicity in the test tube: current limitations of the embryonic stem cell test (EST) challenging its applicability domain. Crit Rev Toxicol 2012; 42: 443.
Riecke K, Stahlmann R. Test systems to identify reproductive toxicants. Andrologia 2000; 32: 209.Rohwedel J, Guan K, Hegert C, Wobus AM. Embryonic stem cells as an in vitro model for mutagen-
icity, cytotoxicity and embryotoxicity studies: present state and future prospects. Toxicol in Vitro 2001; 15: 741.
Spielmann H, Liebsch M. Validation successes: chemicals. Altern Lab Anim 2002; 30: 33.

REPRODUCTIVE AND TERATOGENICITY STUDIES IN VITRO 263
Tam PP. Post-implantation mouse development: whole embryo culture and micro-manipulation. Int J Dev Biol 1998; 42: 895.
Theunissen PT, Piersma AH. Innovative approaches in the embryonic stem cell test (EST). Front Biosci 2012; 17: 1965.
van der Laan JW, Chapin RE, Haenen B, Jacobs AC, Piersma A. Testing strategies for embryo-fetal toxicity of human pharmaceuticals. Animal models vs. in vitro approaches: a workshop report. Regul Toxicol Pharmacol 2012; 63: 115.
Webster WS, Brown-Woodman PD, Ritchie HE. A review of the contribution of whole embryo cul-ture to the determination of hazard and risk in teratogenicity testing. Int J Dev Biol 1997; 41: 329.
Wobus AM, Löser P. Present state and future perspectives of using pluripotent stem cells in toxicol-ogy research. Arch Toxicol 2011; 85: 79.

264
19 High-throughput screening and microarray analysis
HIGH-THROUGHPUT SCREENING IN PRECLINICAL DRUG TESTING
INTRODUCTION
High-throughput screening (HTS) incorporates a combination of modern robotics, data processing and control software, liquid handling devices, and sensitive detectors that allow effi cient and smooth processing of millions of biochemical, genetic, or pharmaco-logical tests in a short time. It provides a rapid, cost-effective method to screen potential sources for novel molecules often used in drug development. Unlike conventional or man-ual screening procedures, HTS allows for the handling of a wide variety of input samples; each sample is stamped with its own set of data and tracking issues and produces new experimental results. Thus, HTS makes it possible to collect large amounts of experimen-tal data related to observations about biological reactions to exposure to various chemical compounds.
Assay miniaturization is a key feature in effective HTS techniques. The smaller the area in which a reaction takes place, the more reactions are observed at a lesser cost. Screening techniques now facilitate analysis of up to 500,000 compounds per week. The invention which has served as the cornerstone for successful HTS-based technology in recent years is the gene microarray (MA), a procedure wherein DNA fragments are immo-bilized onto specifi c locations on glass or silicon surfaces. Stressors are often added to cells and gene expression is determined under various conditions.
The most commonly used type of gene MA is the cDNA MA. In this assay, the mes-senger RNA (mRNA) of interest is prepared and fl uorescently labeled with a variety of dyes. Red dyes, such as Cy5, are commonly used to determine overexpressed or active genes while green dyes, such as Cy3, are often used to determine underexpressed genes or controls. The labeled mRNAs are then pipetted into the wells of the MA and allowed to hybridize to any complementary cDNA that is immobilized to the surface of individual wells via covalently bonded cross-links. When evaluated by an MA scanner, the intensity of fl uorescence is proportional to the numbers of mRNA produced, so gene expression in the presence or absence of a xenobiotic is precisely and accurately quantifi ed. Large samples often contain many artifacts and outliers, and display great heterogeneity. Conse-quently, computational systems such as R-program are employed to normalize the data. The information is further analyzed for signifi cance for over- and under-expression of genes associated with different pathways by additional computational models, includ-ing Gene Ontology Term Analysis and Reactome (Berardini, 2010).

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 265
IMPACT OF HTS ASSAYS ON ANIMAL TESTING
The use of animal models has long been the standard for screening of therapeutic com-pounds for safety and effi cacy, and for studying the effects of chemicals on physiological systems. While animal models have the advantage of measuring effects of chemicals on a systems level, the drawbacks necessitate the expansion of in vitro screening methods during preclinical trials. Better preclinical screening methods equate to better lead compounds that are more likely to be successful in subsequent trials. Early detection of toxicity leads to better results from animal models and more successful agents overall. Thus, effective and accurate development of preclinical screening methods can lessen the economic impact associated with drug development.
In addition to economic interests, there are several other reasons for developing bet-ter HTS assays for toxicological testing. In the fi eld of environmental toxicity research, the National Research Council has proactively called for cell-based screening meth-ods to reduce animal testing (see Suggested Readings). For instance, as of 2009, ani-mal models for toxicity testing of cosmetics have been severely restricted in the European Union.
Other disadvantages to animal models include the typically lengthy period of time for acquiring data in in vivo studies; the signifi cant differences between human and animal models in metabolism, cancer, immunity, infection, and tissue-specifi c transcriptional reg-ulation do not always allow for good correlation of results between the systems; and, the high costs in animal procurement, staff training, veterinary fees, daily maintenance, and procedures for waste elimination and specimen disposal are prohibitive. High-throughput methods are automated, cost effi cient, help reduce the number of animal deaths, and refi ne the pain and suffering associated with animal testing. The latter methods thus aid in priori-tizing potentially promising agents for further testing while eliminating substances that would otherwise not have their toxicity manifested until late in the developmental process.
PROCEDURES
In preparation for an assay, microwell plates are fi lled with a biological target such as a pro-tein, intact cells, or an animal embryo. The target absorbs, binds to, or reacts with the com-pounds in the wells after a predetermined incubation time. Measurements are recorded manually or automatically across all the wells of the plate.1 Automated analysis is conducted on several simultaneous samples using indicators such as refl ectivity or transmission of polarized light through the target. The data output is recorded as a grid of numeric values, with each number mapping to the value obtained from a single well. High-capacity analysis equipment measures dozens of plates in minutes, thus generating thousands of experimental data points quickly.
Because physiological conditions have been so diffi cult to enable Cyp P450
metabolism using the cells and tissue that contain them, efforts have been attempted to provide an HTS assay containing all the enzymes necessary to metabolize compounds followed by stamping with a cell culture. This array, the MetaChip/DataChip® system mimics metabolic reactions of the liver to screen a large number of compounds with greater speed and effi ciency than in vivo traditional approaches. The MetaChip® set (Metabolizing Enzyme Toxicology Assay Chip, Solidus Biosciences, CA, USA) of the system induces high-throughput Cyp P
450 catal-
ysis integrated with human cell screening of metabolites formed on an MA. The DataChip®
1 Manual measurements are often necessary when microscopic examination is used to detect changes or defects in embryonic development caused by the test compounds.

266 PRINCIPLES OF TOXICOLOGY TESTING
(Data Analysis Toxicity Assay Chip, Solidus Biosciences, CA, USA) is an assay miniaturiza-tion of 1080 3-D human cell cultures on platforms encapsulated in alginate or collagen that emulate the native microenvironment of cells in a 2000-fold scale-down system.
The Cyp P450
isoenzymes on the MetaChip® are recombinant enzymes encapsulated in a sol-gel resulting in immobilization glass slides that react with compounds added to the wells. Sol-gels are advantageous in that their porosity is easily and precisely manipulated. They are optically transparent, are stable in most environments, and are highly compatible with a diverse array of moieties. Encapsulation is achieved by directly placing isolated Cyp P
450 isoenzymes into a small amount of MTMOS gel and spotting it onto 0.5-μm thick
MTMOS-coated glass slides. The chemical of interest is then spotted onto the sol-gel array and stamped with either a cell culture monolayer on a chamber slide or the cultures on the DataChip®. The sandwiched arrays are incubated for 6 hours at 37°C to allow unstable metabolites to diffuse into the monolayer. The MetaChip® is then discarded and the cell monolayer stained, washed with growth medium to remove excess unbound chemicals, incubated for 3 days, and then scanned by an MA scanner or fl uorescence microscope. Viability results are then compared with different drugs or drug concentrations, different types or concentrations of Cyp P
450 isoforms, or controls. This enables personalized screen-
ing based on toxicogenomic principles allowing for variations in Cyp P450
isoenzyme num-ber and activity. Depending on genetic characteristics, the ratios of enzymes in the chip are altered accordingly.
Applications of the DataChip® are particularly benefi cial in 1080 3-D human cell cul-tures. The cultures are then arranged comparatively with the physiological organs resulting in a fairly accurate representation of human anatomy and physiology for toxicological and pharmacological evaluation.2
EQUIPMENT
The HTS work station consists of liquid handling, plate replication, and plate sealing devices. A variety of microwell plates are used; 96- to 384-plate formats are the most com-mon. Each well is tagged with experimental targets, chemical compounds, or blanks. In addition, automated liquid handling devices, detection, and other robotic systems are fea-tured. Absorbance plate readers and detectors are used for kinetic measurements in enzyme or cellular assays. Laser scanning fl uorescence microplate cytometers offer high-content screening at throughputs compatible with primary and secondary screening protocols for fl uorescence-based assays.
The robotic features perform autonomous or preprogrammed tasks and are capable of screening combinatorial libraries against screening targets using cell-based assays. Daily average capacities of 480 microplates can generate over 180,000 data points.
APPLICATIONS
Biological and chemical applications for drug discovery and toxicological analysis are appli-cable for identifying toxicologically or pharmacologically active compounds, antibodies, and genes that modulate particular biomolecular pathways. These results provide starting points for drug designs and for understanding the mechanisms or interactions of target mol-ecules in biochemical processes. In addition, the technology is useful in screening novel
2 Traditional monolayer cultures are two dimensional. Cells are grown in 96- or 384-well microplates; the cultures are hindered by ineffi cient reagent removal, prolonged consumption of reagents, and diffi culty in washing.

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 267
drug candidates against libraries of chemicals for their abilities to modify targets (e.g., the ability of a chemical to modify a protein kinase target).
As a method for drug design, biological and physical properties of the targets are screened and a prediction model is constructed to categorize chemicals that interact with the active site (or receptor) of interest. Once a lead molecule series is established with suf-fi cient target potency, selectivity, and favorable drug-like properties, one or two compounds designated as “lead” compounds are proposed for drug development; the other compounds are considered “backups.” It is not unusual for research and development sectors to screen 100,000–300,000 compounds per screen to generate 100–300 “hits.” On average, one or two hits are assigned to the lead compound series. Larger screens of up to 2–3 million compounds (ultra-HTS) are required to generate to 7–10 leads. Improvements in lead gen-eration also come from optimizing library diversity.
FUTURE DIRECTIONS IN HTS
Designing drug candidates for therapeutic interventions and identifying toxic agents have increased the pressure to produce clinical candidates for new drugs and construct models for human risk analysis, respectively. The demand is creating a pressure to identify lead com-pounds at much faster rates than currently available. This imperative is forcing a techno-logical shift toward higher-density plate formats (1536- and 9600-well plates), nanoliter pipetting, and simultaneous imaging.
Assay miniaturization may be capable of facilitating screens of up to 500,000 com-pounds per week while minimizing reagent costs. Combinatorial chemistry is offering the twin opportunities of developing large compound libraries for screening with faster analogs of lead compounds. Finally, HTS is expanding toward target identifi cation, validation, and conversion of assay hits to qualifi ed leads via information generated either within screens or through downstream, high-throughput ADME (absorption, distribution, metabolism, and excretion) and toxicity testing.
MICROARRAY SYSTEMS
INTRODUCTION
MAs are miniature arrays of gene fragments attached to glass slides. The presentation of thousands of gene fragments in a single array allows for the detection of changes in gene expression within a signifi cant fraction of the total genome. The linear arrays of molecules are immobilized at discrete locations on an inert surface that permits simultaneous analysis.
MA technology is popular because it is conceptually simple, easy to implement, and is timed well to the growing interest in high-throughput parallel studies. In the past decade, MAs have evolved from membrane-based low-density arrays to silicon chip high-density oligonucleotide arrays.
TYPES OF ARRAYS
DNA MAs have DNA molecules immobilized at specifi c locations on glass or silicon sur-faces. They are developed as platforms for studying gene expression and mutation detection and are the subject of discussion in this chapter. Similarly, protein MAs that incorporate antibodies at immobilized positions have not yet been widely employed. Finally, siRNA platforms are relatively new systems which have great potential for HTS methodology. Both of the latter schema are discussed in this chapter.

268 PRINCIPLES OF TOXICOLOGY TESTING
DNA MICROARRAY DESIGN
DNA MAs are manufactured into two formats, depending on the type of molecule immobi-lized. Arrays containing PCR products of 200–2000 kilobase pairs are immobilized along the lengths of molecules by conveniently cross-linking to the surface of the array (often referred to as a complementary DNA or cDNA array). Alternately, oligonucleotide probes may be synthesized in situ on the array or presynthesized and then fi xed by covalent linkages to the termini. Figure 19.1 outlines a procedure for the production of and protocol associated with a cDNA MA plate.
The protocol in the cDNA MA technology begins with fl uorescent labeling of mRNA molecules present in the mammalian cell of interest. The labeled mRNA is then pipetted into the wells of the MA slide and hybridizes to the complementary sequences of the cDNA on the MA. The intensity of the fl uorescence is proportional to the number of mRNA mol-ecules produced by the degree of activity of the gene of interest, indicating hybridization to the corresponding cDNA.
FABRICATION
MAs are fabricated using a variety of technologies including photolithography, robotic spot-ting, and printing (pin-based and piezoelectric pin methods). Photolithography is the most widely used method for in situ DNA synthesis on arrays to form oligonucleotide MAs. The technique enables the synthesis of large numbers of high-density arrays. Robotic spotting uses a robotic arm to deposit presynthesized oligonucleotides and PCR-amplifi ed cDNA fragments on the surface of the array.
The device consists of a stage for holding the printed slides and a robotic arm that uses specialized pins to pick up solutions from a source, usually a 384-well plate. In the pin-based printing method, an example of two-system contact printing, steel pins are used
Prepare cDNA Probe
“Normal” Tumor
RT/PCR
Combineequal
amounts
Label withfluorescent dyes
SCAN
Hybridizeprobe to
microarray
Microarray technology
Prepare Microarray
FIGURE 19.1 Production and steps in cDNA microarray technology. Source: Courtesy of National Human Genome Research Institute, U.S. National Institutes of Health, public access at http://www.genome.gov/10000533.

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 269
to transfer fi xed volumes of solution as spots onto the array surface. The fl at tips of the pins form spots of a diameter corresponding to the diameter of the tip. The amount of solution absorbed by the pin tip is proportional to the surface tension of the liquid. Subsequently, when the pin is tapped against the array surface, fi xed volumes of the solution are dis-pensed. The volume of the dispensed liquid depends on many factors, most notably on the surface chemistry of the slide and the contact time between the two surfaces. Piezoelectric pin printing is an example of a noncontact method that consists of a reservoir for the print-ing solution and a capillary dispenser. Piezoelectric crystals are apposed closely with the capillary. When a voltage is applied to the crystals, they deform, squeezing together the walls of the capillary and forcing liquid from the orifi ce.
DETECTION TECHNOLOGY
Most of the current systems are based on fl uorescence detection. The formats are sensitive for biologically active transcripts, safer to handle than radionuclide based methods, and allow multiple sample detection. The two main types of detection systems available for MAs are scanners and imagers.
Scanners use moving optics or substrates to collect signals from small areas on a slide to compile an entire image. Alternatively, imagers capture the output from the entire array simultaneously. As with other fl uorescent detectors used frequently in cytotoxicology assays, the instrumentation is programmed to account for controls, blanks, normalization of data (equalization of data in comparative experiments), and autofl uorescence. The gen-erated data are usually analyzed as scatter plots that represent the comparison of gene expression profi les from different experimental conditions. Typically, fl uorescent intensi-ties from different dyes corresponding to different wavelengths are plotted against each other; red color is used to depict overexpression and green is used for underexpression. Figure 19.2 shows a typical cDNA MA fl uorescent glass slide with detection of fl uorescent data output.
Detectors for high-throughput cell and tissue culture arrays are ideally high throughput and multivariate to optimize the results obtained. Some detection techniques that are employed in HTS methods include multicolor fl ow cytometry, tissue cytometry, and sto-chastic sampling.
Multicolor fl ow cytometry uses measurements from multiple fl uorescent dyes and dual scattered light parameters to scan cells. In this method, lasers activate the dyes to higher-energy levels, resulting in scattered light emitted from biomolecules. The latter are coupled to antibody molecules. Computer-generated histograms quantify the intensity of fl uores-cence and the data from light scattering. This technique is advantageous in that it is both high throughput and multivariate. However, it may perturb the sample by inducing stress-related signaling. Accordingly, its use is restricted to cells outside their native 3-D environ-ment; that is, once cells are removed from host tissue and placed in a 2-D culture, cell morphology is altered, unanticipated cell–cell interactions occur, inputs from the extracel-lular matrix cease, and the new matrix leads to the emergence of different cell functions. Consequently, a relatively new 3-D technique, tissue cytometry, overcomes the 2-D limita-tions by allowing 3-D images of the tissue microenvironment with either a confocal micro-scope or two-photon microscope. The tissue cytometer achieves this by robotically removing the uppermost layer, scans deeper into tissues or organs, and systematically con-structs an accurate 3-D image of tissue and constituents.
Stochastic sampling couples laser-capture microdissection (LCM) with customized quantitative PCR. LCM consists of a laser coupled with a microscope, which is then

270 PRINCIPLES OF TOXICOLOGY TESTING
focused onto the tissue on a microscope slide. When moved by optics, the focus follows a specifi c trajectory (the element). The laser cuts a specifi c portion from the sample along the element so tissue extraction occurs for quantitative analysis with PCR.
APPLICATIONS OF CDNA MA TECHNOLOGY
Gene Expression and Genomic Toxicology
cDNA MAs are most widely used for studying gene expression. The mRNA is isolated from cells of interest in the absence or presence of a toxic agent or drug. The cDNA is prepared from both samples labeled using different fl uorescent dyes (most commonly fl uorescein green and rhodamine red). As described earlier, after hybridization of the cDNA molecules to the probe on the array, the ratio of red-to-green fl uorescence is proportional to the relative levels of the gene in the test and reference samples. Gene expression in traditional cDNA protocols has many research and clinical applications, some of which are described next.
siRNA Screens
Small interfering RNAs (siRNA) have been used to study genome-wide loss of function (LOF) in vitro from readily controllable gene knockdown. Three kinds of siRNA molecules are available for screening techniques.
1. Synthetic 21-nucleotide double-strand RNAs with two base overhangs on the 3′-end have been traditionally employed, since they resemble the hairpin structure of
FIGURE 19.2 Example of a microarray printed on the standard 1″ × 3″ glass slide format; the slides are accessible to standard 1″ × 3″ MA scanners. Source: Courtesy of Agilent Technologies, www.agilent.com.

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 271
endogenous single strand microRNA (miRNA) in appearance. They are pooled together in groups of 3–6 in HTS assays for optimization of gene silencing. Pooling of different siRNA molecules that silence the same gene results in increased throughput and reduced costs compared with individual siRNAs. However, a larger number of false-negative results are problematic. The pooling feature is enhanced by endonuclease-prepared siRNA (esiRNA), in which nucleotides are digested from a 200–500 base pair template to generate smaller siRNA molecules.
2. Vector expressed short hairpin RNA (shRNA) is a more novel technological devel-opment based on endogenous miRNA structure, in which single-strand RNA bends back upon itself to form a hairpin loop that resembles double-strand RNA. This miRNA backbone then allows for better recognition and processing by endogenous siRNA machinery, increasing effi ciency and exceeding the typical 5–7-day dura-tion of activity of synthetic siRNA.
3. The modifi er siRNA screen is exploited to identify drug-targeted genes and path-ways that affect specifi c phenotypes. Alterations are fi rst observed by screening a drug on an array without siRNA knockdown of genes, quickly followed by siRNA library screening without the drug, and fi nally by screening with both the library and drug. This three-tiered screening approach gathers information about affected molecular pathways, and provides valuable insights into the mechanistic activity of drugs. Additional information is also provided on biomarkers that leads to a dimin-ished response to a certain drug if certain silencing pathways show inhibition of the activity of the compound.
MAs based on siRNA technology are typically produced by dispensing the reagents roboti-cally into microplates, adding the cells through a liquid dispenser along with the culture medium, and any additional reagents required for the particular assay. Data are collected by plate readings or fl uorescent microscopy. Time-resolved live cell imaging is also performed in which siRNA transfection mixtures are drawn up by cells seeded on the top. This allows for the detection of a larger number of phenotypes.
Protein Production and Proteomic Toxicology
The 3-D matrices for protein binding onto microarrays are diffi cult to engineer. Unlike gene microarrays in which DNA is attached to wells, attachment of proteins to a plastic surface is more diffi cult. This is particularly due to the more complex 3-D structures of proteins com-pared with the linear helices of DNA molecules. If the protein structure is altered to any extent, it then develops the potential to interact with other unanticipated molecules. Therefore, it is diffi cult to integrate a protein with the surface of a chip without altering its 3-D conformation and thus affecting its activity. This accounts for the incorporation of the hydrogel layer in the DataChip® assay. It also explains why 3-D matrices use gels to hold enzymes onto the microchip surface. Modern hydrogel coatings contain high amounts of water as well as some important organic molecules, so as to resemble the normal physiolog-ical microenvironment. The coatings are only 100–500 nm thick, enabling many proteins to be bound to the surfaces of array slides. For instance, the DataChip® uses slides that are pretreated with 3-aminopropyltrimethoxysilane plus polystyrene-co-maleic anhydride. The latter chemical is coated with barium to promote gelation. It also contains poly-L-lysine, a positively charged amino acid that binds to negatively charged polysaccharides in alginate. These chemicals aid in supporting cell attachment to the matrix and confer a hydrophobic environment to the surface. Alginate is then evenly distributed and cells are incubated for

272 PRINCIPLES OF TOXICOLOGY TESTING
5 days to get linear growth and population doubling level3 rates comparable with those found in traditional tissue culture wells or plates.
One major limitation to the MetaChip/DataChip® assay is the possibility of interfer-ence of the hydrogel environment with typical cell proliferation and growth for the cell line in question. Gene expression and signal transduction pathways are also altered in modes that fail to resemble in vivo phenotypes. As a result, the microenvironment may not truly resemble the in vivo situation, despite the major refi nements in quantifi cation of metabolic activity.
Genotype/SNP Detection
MAs are also used for developing chips for detection and screening of single nucleotide polymorphisms (SNPs). An SNP is a DNA sequence variation occurring when a single nucleotide in a genome differs among members of a species or among paired chromosomes in an individual. The technique is applicable for the detection of a single nucleotide differ-ence in sequenced DNA fragments in two alleles from different individuals (e.g., AAGCCTA to AAGCTTA). The differences in hybridization due to single base pair changes are detected using oligonucleotide arrays, provided conditions of hybridization are rigidly controlled. Alternatively, primer extension-based methods are also employed. This method exploits the inability of many DNA polymerases to extend a strand where the 3’ terminal nucleotide is not perfectly matched.
Computational Assessment of Data
With the increased sophistication of in vitro cell culture models, it is vital that interpretation of such systems is scaled up to optimize their value. In order to achieve a successful high-throughput MA, data gathering techniques must also be high throughput. A systems biology approach is often incorporated where mathematical tools of bioinformatics and statistical analysis of data obtained from cell and tissue culture arrays are interpreted based on detect-able patterns. For example, systems pharmacology and multianalyte models are typically used for computational assessment. Systems pharmacology involves the study of the effects of drugs on biological systems, including the whole organism and its intracellular networks, tissues, organs, compartments, and individual cells, through a multianalyte or multisource model for the integration of data. Accordingly multianalyte models consist of gauges, such as gene transcripts, protein markers, and metabolic indicators, while multisource models are concerned with comparing samples from different body compartments. These models allow for integration by computational systems that measure changes in state through high-throughput analysis of thousands of measurements within a system to detect specifi c pat-terns of activity.
Because of the complexity of biological samples, data integration remains a challeng-ing endeavor. Advancements in bioinformatics, however, have helped approach the prob-lem systematically. Pharmaceutical bioinformatics is the measurement of multiple analytes per sample with the use of computer-generated images and statistical analysis to reveal drug response across physiological networks. Therefore, networks must be characterized in models that are dynamic, multivariate, and able to give quantitative data, despite a vast number of components that are involved and interconnected. With the enormous amount of information obtainable from network interactions, computational bioinformatic models are
3 Population doubling level is a calculation used to monitor the proliferation rate of cells in culture. It refers to the number of doublings a cell line has achieved in culture.

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 273
indeed critical in assessing the relevance of any particular perturbation. These models are reliant on the use of biomarkers, which are measurable characteristics that refl ect normal physiology, pathophysiology, or chemical response.
The use of correlation networks has substantially simplifi ed the arduous task of deduc-ing macroscale changes from measuring thousands of different biomarkers. Correlation networks assess statistical correlations between different measurements of biomarkers rep-resenting different samples. They fi lter random, unrelated correlations to include only those that are reversible in various states, including normal and disease states and before and after drug administration. For example, a set of analytes may be detected to only move together following a change induced by a specifi c chemical; this pattern of movement is then fl agged and a correlation is established. Additionally, multisource models are incorporated to track changes between compartments. Multiple measurements across many compartments are quantifi ed to contextualize patterns that are encountered on a molecular level that typically correlate with macroscale phenotypic effects taking place throughout the system. With this approach, correlative aspects of mechanisms are deduced, and quantitative predictions of alterations in the biological systems are arrived at with reasonable confi dence.
Some commonly used mathematical models that have allowed manipulations in culture to be measured in a high-throughput manner include partial least squares regression (PLSR) and Bayesian networks. PLSR is based on a signal-response mechanism. The model assumes that behavioral phenotypes are reliant on a combination of activities or states of key signaling regulators. A linear solution to a signal–response relationship is provided by the equation Y = ƒ(X), in which the signaling variable is the independent X-value and the response variable is represented by the dependent Y-value. PLSR has been operated to provide useful information regarding important combinations of signaling mechanisms controlling quantifi able behavioral phenotypes, such as growth factor activation of prolif-eration in tissues. Similarly, Bayesian network infers downstream effects of pathways fol-lowing an intervention at one level. This computational system operates through the use of infl uence diagrams, which correlate direct physical interactions between molecules with indirect infl uences less easily measured, based on unobserved intermediates acting in a system. The result is that pertubation at specifi c levels lead to probabilistic calculations representing signaling across multiple pathways to achieve an accurate representation of how interactions at one level can stimulate a cascade of events along interconnected path-ways, leading to the fi nal response.
SUMMARY
In the traditional sense, HTS not only allows for discovery and invention, but has created more necessities. This in turn leads to more inventions, thus completing the discovery cycle. But the cycle has not arrived “at full circle.” In the competitive toxicology and pharmaceuti-cal industries, overcoming the problems of time and cost of drug development and preclini-cal testing will lead to further advances in HTS. Not only will the technique help solve these hindrances, but will create entirely new avenues of discovery in the basic sciences as well as its applications. In addition, conventional industrial research approaches do not fulfi ll the demands of current drug development schedules. Thus, the applications of HTS have inte-grated the use of laboratory automation to collect vast amounts of toxicological and pharma-cological data and reliably profi le a range of biological activities. The information eventually is built in to large chemical libraries. The availability of these libraries and robotic systems for bioassays allows synthesis and testing of thousands of compounds daily, the net effect of which enhances our modern and healthy standard of living.

274 PRINCIPLES OF TOXICOLOGY TESTING
SUGGESTED READINGS
Chan VS, Theilade MD. The use of toxicogenomic data in risk assessment: a regulatory perspective. Clin Toxicol 2005; 43: 121.
Chen T. DNA microarrays: an armory for combating infectious diseases in the new century. Infect Disord Drug Targets 2006; 6: 263.
Cosgrove BD, Griffi th LG, Lauffenburger DA. Fusing tissue engineering and systems biology toward fulfi lling their promise. Cell Mol Bioeng 2008; 1: 33.
Cummins JM, Velculescu VE. Implications of micro-RNA profi ling for cancer diagnosis. Oncogene 2006; 25: 6220.
Echeverri CJ, Perrimon N. High-throughput RNAi screening in cultured cells: A user’s guide. Nat Rev Genet 2006; 7: 373.
Griffi th LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol 2006; 7: 211.
Jessen BA, Mullins JS, de Peyster A, Stevens GJ. Assessment of hepatocytes and liver slices as in vitro test systems to predict in vivo expression. Toxicol Sci 2003; 75: 208.
Khaitovich P, Enard W, Lachmann M, Paabo S. Evolution of primate gene expression. Nat Rev Genet 2006; 7: 693.
Koczan D, Thiesen HJ. Survey of microarray technologies suitable to elucidate transcriptional networks as exemplifi ed by studying KRAB zinc fi nger gene families. Proteomics 2006; 6: 4704.
Krewski D, Andersen ME, Mantus E, Zeise L. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal 2009; 29: 474.
Leighton JK, Brown P, Ellis A, et al. Workgroup report: review of genomics data based on experience with mock submissions: View of the CDER Pharmacology Toxicology Nonclinical Phar-macogenomics Subcommittee. Environ Health Perspect 2006; 114: 573.
Lettieri T. Recent applications of DNA microarray technology to toxicology and ecotoxicology. Environ Health Perspect 2006; 114: 4.
Modlich O, Prisack HB, Bojar H. Breast cancer expression profi ling: the impact of microarray testing on clinical decision making. Expert Opin Pharmacother 2006; 7: 2069.
Neumann B, Held M, Liebel U, et al. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat Methods 2006; 3: 385.
Powers MJ, Domansky K, Kaazempur-Mofrad MR, et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol Bioeng 2002; 78: 257.
REVIEW ARTICLES
Atterwill CK, Wing MG. In vitro preclinical lead optimisation technologies (PLOTs) in pharmaceuti-cal development. Toxicol Lett 2002; 127: 143.
Berardini TZ. The gene ontology in 2010: Extensions and refi nements. Nucleic Acids Research, 2010; 38: D331.
Breitling R. Biological microarray interpretation: the rules of engagement. Biochim Biophys Acta 2006; 1759: 319.
Bugelski PJ. Gene expression profi ling for pharmaceutical toxicology screening. Curr Opin Drug Discov Devel 2002; 5: 79.
Coppola G, Geschwind DH. Microarrays and the microscope: Balancing throughput with resolution. J Physiol 2006; 575: 353.
Ekins S. Systems: ADME/Tox resources and network approaches. J Pharmacol Toxicol Methods 2006; 53: 38.
Garaizar J, Rementeria A, Porwollik S. DNA microarray technology: a new tool for the epidemio-logical typing of bacterial pathogens? FEMS Immunol Med Microbiol 2006; 47: 178.
Gibson NJ. Application of oligonucleotide arrays to high-content genetic analysis. Expert Rev Mol Diagn 2006; 6: 451.

HIGH-THROUGHPUT SCREENING AND MICROARRAY ANALYSIS 275
Guillouzo A. Applications of biotechnology to pharmacology and toxicology. Cell Mol Biol 2001; 47: 1301.
Irwin RD, Boorman GA, Cunningham ML, et al. Application of toxicogenomics to toxicology: basic concepts in the analysis of microarray data. Toxicol Pathol 2004; 32: 72.
Jafari P, Azuaje F. An assessment of recently published gene expression data analyses: Reporting experimental design and statistical factors. BMC Med Inform Decis Mak 2006; 6: 27.
Jubb AM, Pham TQ, Frantz GD, Peale FV Jr, Hillan KJ. Quantitative in situ hybridization of tissue microarrays. Methods Mol Biol 2006; 326: 255.
Lee M-Y, Park CB, Dordick JS, Clark DS. Metabolizing enzyme toxicology assay chip (MetaChip) for high-throughput microscale toxicology analyses. Proc Natl Acad Sci USA 2005; 102: 983.
Lee MY, Kumar RA, Sukumaran SM, et al. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc Natl Acad Sci USA 2008; 105: 59.
Li N, Tourovskaia A, Folch A. Biology on a chip: Microfabrication for studying the behavior of cul-tured cells. Crit Rev Biomed Eng 2003; 31: 423.
Low YL, Wedren S, Liu J. High-throughput genomic technology in research and clinical manage-ment of breast cancer: evolving landscape of genetic epidemiological studies. Breast Can-cer Res 2006; 8: 209.
Mans JJ, Lamont RJ, Handfi eld M. Microarray analysis of human epithelial cell responses to bacte-rial interaction. Infect Disord Drug Targets 2006; 6: 299.
Maurer HH. Screening procedures for simultaneous detection of several drug classes used for high throughput toxicological analyses and doping control: a review. Comb Chem High Throughput Screen 2000; 3: 467.
Meador V, Jordan W, Zimmermann J. Increasing throughput in lead optimization in in vivo toxicity screens. Curr Opin Drug Discov Devel 2002; 5: 72.
Merrick BA, Bruno ME. Genomic and proteomic profi ling for biomarkers and signature profi les of toxicity. Curr Opin Mol Ther 2004; 6: 600.
Nielsen TO. Microarray analysis of sarcomas. Adv Anat Pathol 2006; 13: 166.Petrik J. Diagnostic applications of microarrays. Transfus Med 2006; 16: 233.Seo D, Ginsburg GS, Goldschmidt-Clermont PJ. Gene expression analysis of cardiovascular diseases:
novel insights into biology and clinical applications. J Am Coll Cardiol 2006; 48: 227.Shioda T. Application of DNA microarray to toxicological research. J Environ Pathol Toxicol Oncol
2004; 23: 13.Shockcor JP, Holmes E. Metabonomic applications in toxicity screening and disease diagnosis. Curr
Top Med Chem 2002; 2: 35.Sievertzon M, Nilsson P, Lundeberg J. Improving reliability and performance of DNA microarrays.
Expert Rev Mol Diagn 2006; 6: 481.Sobek J, Bartscherer K, Jacob A, Hoheisel JD, Angenendt P. Microarray technology as a universal
tool for high-throughput analysis of biological systems. Comb Chem High Throughput Screen 2006; 9: 365.
Walmsley RM. Genotoxicity screening: the slow march to the future. Expert Opin Drug Metab Toxi-col 2005; 1: 261.

276
20 Toxicogenomics and epigenetic testing in vitro
INTRODUCTION
Toxicogenomics refers to the response of the entire genome to toxicants or environmental stressors. Toxicogenomics encompasses three major objectives: (1) elucidation of the con-nection between environmental stress and disease susceptibility; (2) assignment of the proper biomarkers of disease and their association to exposure to toxic substances; and (3) clarifi cation of the molecular mechanisms of toxicity. The fi eld has begun to incorporate traditional toxicological and histopathological end point evaluations with the data derived from transcriptomics, proteomics, metabolomics, and metabonomics. These terms are further described in the chapter and in Table 20.1.
TRANSCRIPTOMICS
Transcriptomics is the study of all RNA molecules in an organism or that of a particular cell type. Transcripts differ from genomics largely as a result of the ease with which alterations and mutations occur. Transcripts are transient molecules and are highly unstable when com-pared with DNA. Transcriptomics relies on complementary DNA (cDNA) microarray (MA) hybridization and analysis to study whole genomes (see chap. 19 for the discussion of MA technology). RNA transcripts from cDNA are labeled with dyes and spotted on to glass slides which hold analogous sequences.
PROTEOMICS
Proteomics is the systematic analysis of proteins in tissues of functional characterization in an intact organism. This requires identifi cation of proteins under toxic stresses that alter or disable the functions of some proteins. Furthermore, in the presence of toxins, it is important to note the location of post-translational modifi cations, protein complexes, binding sites, and structural complexes (Hamadeh, 2002). All of these aspects are likely to contribute to the dynamics of cell signaling pathways.
METABOLOMICS
Metabolomics is a quantitative analytical method that identifi es metabolites in pathways. Nuclear magnetic resonance has traditionally been used in metabolic fi ngerprinting where peaks in the spectra contain information regarding the structure of the metabolites. They can further be investigated through fragmentation and its patterns (Waters, 2004).

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 277
AN OVERVIEW OF COMPARATIVE AND FUNCTIONAL TOXICOGENOMICS
Two models have been useful in the progression of toxicogenomic studies.
1. Comparative toxicogenomics was developed as a model to deal with different aspects in collecting toxicity data for this burgeoning fi eld. Comparative toxicogenomics measures gene expression, protein levels, and metabolites in normal cells and toxicant-exposed cells. These comparisons are made upon initial exposure to the toxic agent. Data profi les are developed for each toxicant which can be compared to known chemicals in databases and their pathological effects. These data are subdivided into the class of the compound or the histopathology and chemistry that were observed from the animals that were tested with the substance. The objective of acquiring this database is to link it to phenotypic manifestations which can be used as early bio-markers to predict toxicant hazard or injury.
The strength of these comparisons rely on the variables of the test; that is, the consistency of the results of a test (reproducibility) and the ability of the test to represent a population (relevance). To achieve this objective, the databases at the National Center for Toxicogenomics, have helped to streamline and note differ-ences in variables including dosage, time, type of biological system, body weight of animal, and cell cycle data. These parameters help in the accuracy of predictive modeling and interpretation with the mechanism of action.
The fi rst stage of the comparative/predictive model is data collection. Data are obtained through in vitro and in vivo studies with hypotheses produced with regards to chemicals and their effects. Data collected are in the form of animal weight fl uc-tuations, serum markers, pathological differences, and mortality rates. The next stage deals with the development of a model; the model accumulates raw data and statistically analyzes correlations among gene, protein, and metabolite alterations, and toxicant-exposed and control experiments. The result is the establishment of correlative associations and identifi cation of variables in the study.
TABLE 20.1Terminology
Transcriptomics Techniques that assess the full complement of activated genes, mRNAs or transcripts; involve the use of cDNA or oligonucleotide microarrays
Proteomics A group of techniques used to assess the structural and functional attributes of proteins through the use of 2-D gel electrophoresis or LCMS
Metabonomics Techniques that ascertain alterations in the concentration of low-molecular-weight metabolites present in a cell or organism at a particular time (the metabonome) by using NMR or mass spectrometry coupled with LCMS or GCMS
Metabolomics The use of quantitative analytical methods for analyzing the whole metabolic composition of a cell or organism at a given time.
Biomarker A pharmacological or physiological assessment that is used to project a toxic event in an animal.
Abbreviations: GCMS, gas chromatography mass spectrometry; LCMS, liquid chromatography mass spectrome try; NMR, nuclear magnetic resonance.

278 PRINCIPLES OF TOXICOLOGY TESTING
Lastly, the utility stage analyzes the usefulness of these models by testing differ-ent models in silico. Data from different toxicants are subjected to further compari-sons with one another from the same or different classes of compounds. This information is used in the prediction of biological activity and toxicity and lessens the need for extensive experimentation in vivo if similarities are identifi ed with other known toxicants. For example, during drug development, hepatotoxicity is a major issue. Thus it has become increasingly useful for the inclusion of toxicogenomics in drug development studies. Zidek et al. (2007) have established a predictive screening system for acute hepatotoxicity by analyzing differential gene expression profi les of hepatotoxins and nontoxic compounds. Using MA analysis, the authors acquired 550 liver-specifi c gene expression profi les and analyzed them for histopathological differences and gene expression differences. In addition, the authors have identifi ed 64 potential biomarkers that are predictive of the chemical class. They speculated that the biomarkers involved are consequences of the deregulation of genes and path-ways due to the hepatotoxicants. These biomarkers can potentially be indicative of early hepatotoxicity. The study concluded that the novel, focused gene MA was able to classify compounds and predict them through gene expression profi les.
2. Functional toxicogenomics is the second modeling component that is concerned with the study of the functions of genes and proteins with respect to the stresses imposed by toxicants. The aim of functional toxicogenomics is to determine the mechanism of action of a compound and classifying the patterns along variables of time and dose. Gene and protein functionality are highly dependent on toxicant exposure and lead to the progression of variances of functionality within the same gene or protein. Cells are also known to manifest phenotypic differences at differ-ent points of exposure, thus creating diffi culties during the analytical method. Fur-thermore, proteins generally exhibit more than one phenotypic alteration. Consequently, analysis of patterns of change allows for the identifi cation of better biomarkers by limiting the responses to the toxicant.
BIOMARKERS
There is a need for new biomarkers in drug development studies. Yet many end points are not associated with conventional biomarkers, which elevates risk in clinical trials. For example, during a clinical trial for the development of therapeutic modalities for Alzheimer’s disease, an enzyme inhibitor was discovered to affect differentiation in intestinal epithelial cells. The compound inhibited cleavage of the Hes1 gene product through Notch1. Gene expression profi les and protein analysis led to the discovery of the biomarker adipsin, which was ulti-mately discovered to be associated with this toxicity (Searfoss et al., 2003).
INTEGRATION OF DATA
Integration of data is also important in determining the toxicological response of cells to chem-icals. Different analytical methods must also be refi ned in order to adequately compare results. For instance, the International Life Sciences Institute, Health and Environmental Institute sys-tematically began to report its data on the toxicity of hepatotoxicants and nephrotoxicants (2004). Similarly the Toxicogenomics Research Consortium standardized the results of toxi-cogenomics studies by categorizing known hepatotoxicants and nephrotoxicants. Together the

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 279
Institutes, in an attempt to understand and categorize these substances for clinical and animal studies, compiled information which has led to the classifi cation of thousands of toxic agents.
For example, acetaminophen (APAP) overdose has increasingly become a problem and a main cause of hospitalization in the United States. The integration of data of six different toxicogenomic studies regarding APAP has led to the identifi cation of 228 genes involved in the toxicological effects of the drug. Using mouse liver as the target organ, Fountoulakis et al. (2000) compared the proteomics of APAP and the nontoxic isomer AMAP. From a database of 256 proteins, the authors implicated 35 proteins with expression level differ-ences with concomitant histopathology, after an 8-hour exposure. Interestingly, all of the proteins had similar mechanisms of action. Heinloth et al. (2004) used an MA to analyze expression changes of 6000 genes in rats at various doses of APAP at which there is no toxicity, peak toxicity, and recovery. The reference time values were adopted from previous histopathological studies. The authors were able to focus on low doses that exhibited phe-notypic effects of energy loss through mitochondrial damage. In a related study, Reilly et al. (2001) examined livers from mice after exposure to APAP using high-density DNA arrays. They discovered 100 genes that exhibited altered expression levels. Similarly, Ruepp et al. (2002) exposed mice to APAP for 15 minutes to 4 hours and analyzed an MA of 450 genes, along with 2-D PAGE1 of liver proteins. The study concluded that there were signifi cant changes in glutathione S-transferase and infl ammatory signaling proteins. Coen et al. (2003) studied the metabolomics of liver and plasma of mice exposed to APAP and compared these results with MA analysis of the same proteins and pathways, while de Longueville et al. (2003) also performed a low-density MA and identifi ed changes in expression in rat hepatocytes for several toxicants, including APAP. Both studies concluded that there is a correlation between relative numbers of gene expression and cellular func-tionality, despite the differences in techniques and the applicable target organisms.
HUMAN GENOMIC VARIATION
Analysis of the human genome disclosed a high degree of DNA sequence homology (simi-larity) among individuals from around the planet; that is in parallel comparisons, any two humans are about 99.9% homologous in their DNA sequences. The differences in the genome are labeled as genetic variations and are triggered by different types of mechanisms includ-ing: (1) nucleotide substitutions; (2) insertion or deletion of nucleotides; (3) alterations in the number of tandem-repeat sequences; (4) variations in the copy number of genomic segments; and (5) combinations of these changes. A subtype of the genetic variations is known as genetic polymorphism, which is recognized at a frequency of about 1% in various popula-tions. Furthermore, DNA polymorphisms are divided into fi ve major classes:
1. Restriction fragment length polymorphism (RFLP).2. Variable number of tandem repeat or minisatellite (VNTR).3. Short tandem repeat (STR) or microsatellite.4. Single-nucleotide polymorphism (SNP).5. Copy-number variation (CNV).
1. RFLP is formed by variations in the size of the DNA fragment digested by a restriction endonuclease due to the base substitutions at the site identifi ed by the endonuclease.
1Two-dimensional polyacrylamide gel electrophoresis.

280 PRINCIPLES OF TOXICOLOGY TESTING
RFLP patterns allow differentiation between the parental alleles (whether the allele is of maternal or of paternal origin) of the specifi c loci in the genome.
2. VNTR is a point in a genome where a short nucleotide sequence is aligned as a tan-dem repeat (two or more nucleotides are duplicated and the repetitions are next to each other). VNTRs are signifi cantly polymorphic, with high heterozygosity in given populations because of a wide variety of the copy number of tandem-repeat DNA sequences.
3. Unlike VNTR loci, which constitute a few thousands in the human genome, STRs or microsatellite loci (where 10–60 nucleotides are repeated) are accessible at greater than 100,000 regions, comprising most of the genomic landscape. Thus, microsatellite markers have been successfully employed for linkage analysis or population genetics; one major advantage to performing the technique is that the analysis requires a relatively small quantity of DNA for identifi cation.
4. SNPs are the genetic variations that are ordinarily present in the human genome. On average, they are conferred in one of every 500–1000 base pairs (bp). SNPs are accepted as valuable tools for population genetics, particularly for analysis of genes prone to various diseases.
5. CNV, a mode of genomic structural variation, is a segment of DNA that is 1 kb or larger and exists as a variable copy number in comparison with a reference genome. Categories of CNVs include insertions, deletions, and duplications. The implication of CNVs in human phenotypes was fi rst introduced as genomic disorders. CNVs are frequently activated by de novo structural changes and account for the reporting of dozens of genomic disorders.
GENOMIC VARIATION IN TARGET MOLECULES
The objective of target identifi cation is to distinguish new targets, normally proteins, whose modulation might suppress or prevent disease progression. The advent of technologies that venture to associate changes in gene expression (genomics) and protein production (pro-teomics) with human disease, transformed many aspects of biomedicine. Toxicogenetics and toxicogenomics2 are acknowledged as the new “disciplines” of the biomedical sciences. The terminology associated with these emerging fi elds is presented in Table 20.2.
Toxicogenetics seeks to explain how genetic attributes result in disparity in individuals’ responses to drugs. A reaction to a specifi c medicine may be altered by factors that vary in agreement with the individual’s version of a gene or genes (alleles). These determinants incorporate the amount and rate of drug absorption, the rate of drug metabolism and elimi-nation, and the variability in the toxicological and pharmacological site of drug toxicity and action, respectively (Table 20.3).
Toxicogenomics is associated with the recognition of the genome and its products (RNA or proteins) as they are linked to drug toxicity. Toxicogenomics assesses how chemi-cal compounds alter the complete expression motif in defi ned tissues of interest. For instance, gene expression profi ling is used to establish an association of an explicit subset of genes with particular disease states or healthful tissues.
2The corresponding terms of pharmacogenetics and pharmacogenomics are used interchangeably with their counterpart terms.

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 281
TABLE 20.2A Comparison of Toxicogenetics and Toxicogenomics
Toxicogenetics Toxicogenomics
Differential effects of a drug in vivo, in different patients, that are dependent on the presence of inherited gene variants
Differential effects of compounds—in vivo or in vitro—on gene expression, among all expressed genes
Genetic approaches, as well as expression profi les and biochemical measures
Expression profi ling
Provides more patient- and disease-specifi c monitoring of biomedical issues
A tool for compound selection and drug discovery
One drug, many genomes Many drugs, one genomePatient variability Compound variability
Although both of these fi elds involve the assessment of drug effects using nucleic acid technology, the approaches are unambiguously different: toxicogenetics benefi ts from the analysis of differences between several individuals with attention to clinical response to a particular drug, whereas toxicogenomics outlines the study of differences between several compounds with respect to gene expression response in a single genome (such as a cell line). Clinically, pharmacogenetics can be used to expeditiously select the best medicine for an individual, whereas pharmacogenomics facilitates pharmaceutical research and development to select the best candidate therapy from a library of compounds undergoing evaluation.
GENOMIC VARIATION IN DRUG METABOLISM
The activity of cytochrome P450s determines the duration of action and the amount of a drug remaining in an organism. In humans, different forms of cytochrome P450 (i.e., CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) are accountable for eliminating drugs. Individual polymorphisms account for the majority of variations among CYP2C9, CYP2C19, and CYP2D6 individuals. Some individuals have polymorphisms (e.g., deletions) that cause enzyme defi ciencies; these individuals are recognized as “poor” or “slow” metab-olizers and are at a risk of concentration-associated toxicity. Others have polymorphisms (e.g., gene amplifi cations) that prompt enzyme activity; they are identifi ed as “extensive” or “ultrarapid” metabolizers and have the propensity to be unresponsive to therapy. Table 20.4 illustrates examples of polymorphisms documented to mediate drug activities.
TABLE 20.3Classification of Toxicogenetic Responses
Toxicokinetics Toxicodynamics
AbsorptionMetabolism Distribution Elimination
Causative drug action: related to molecular pathologyPalliative drug action: related to molecular physiology

282 PRINCIPLES OF TOXICOLOGY TESTING
A BRIEF REVIEW OF GENE STRUCTURE AND FUNCTION
Genes are the fundamental units of heredity, are composed of DNA, and provide directions for the creation of proteins. In humans, genes are arranged in sizes ranging from a few hun-dred DNA bases to more than 2 million bases. Thus, the genome consists of DNA, and encompasses all the information required to biologically assemble and physiologically sup-port an organism. In the cell nucleus, DNA molecules are wrapped into chromosomes; that is, thread-like structures comprised of DNA compactly folded around proteins (i.e., histones) that maintain its structure.
A nucleic acid is made up of a chain of nucleotides connected together through alter-nating series of sugar and phosphate residues from which nitrogenous bases project. The sugar residues in DNA and RNA are 2-deoxyribose and ribose, respectively. DNA is com-prised of four types of bases, including two purines [adenine (A) and guanine (G)], and two pyrimidines [cytosine (C) and thymine (T)]. The same bases exist in RNA with the excep-tion that uracil substitutes for thymine (Table 20.4).
The terminal positions of a protein-encoding genes are assigned as the locations at which transcription initiates and terminates. The coding domain is the nonterminal seg-ment of the gene, which consists of the nucleotide sequence that is translated into the chain of amino acids in the protein. The coding domain starts with the initiation codon3 (ATG) and concludes with one of the termination codons (TAA, TAG, or TGA). DNA sequences are located on both sides of the coding domain and are transcribed (not translated). Both the coding domain and the nontranslated domains are interrupted by introns, gene sequences that are removed from the primary transcript by splicing. Subsequently, exons are the com-ponents of the gene that are transcribed and are present in the mature transcript [messenger RNA (mRNA)].
EPIGENETIC TOXICOLOGY
Epigenetics is the study of heritable alterations in gene expression that are not coded in the DNA sequence. These changes infl uence the expression of a gene or the properties of its prod-uct. Fundamental to regulation of gene expression is the chromatin structure. The essential repeating unit of chromatin is the nucleosome, consisting of 147 bp of DNA wrapped around
3 A group of three nucleotide bases attached to and representing a specifi c amino acid.
TABLE 20.4Examples of Polymorphisms Reported to Mediate Drug Action
Gene product Drug Drug Action Linked to Minor Allele
CYP2D6 codeine Decreased analgesiaCYP2D6 propranolol Augmented -blockageCYP3A5 many Function not yet determinedCYP2C9 warfarin Reduced anticoagulant effectCYP2C9 phenytoin Increased toxicityP-glycoprotein digoxin Altered blood levelN-acetyltransferase isoniazid Increased risk of hepatoxicity
Abbreviation: CYP, cytochrome P450.

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 283
a core of eight histone proteins, the latter of which are designated H2A, H2B, H3, and H4. The histone modifi cations are interpreted by proteins committed to chromatin reconstruction, tran-scriptional stimulation or suppression. Two types of chromatin have been classifi ed: hetero-chromatin is conventionally compact and transcriptionally disengaged, whereas euchromatin retains an “open” confi guration and is inclined to gene transcription. The basic amino terminal tails of histones protrude out of the nucleosome and encounter numerous post-translational modifi cations, including (1) acetylation, (2) methylation, (3) phosphorylation, (4) ubiquitina-tion, and (5) sumoylation, which are described in the coming sections. In addition, DNA methylation has long been attributed to an epigenetic silencing mechanism of cardinal sig-nifi cance in transcription, chromatin structure, genomic imprinting, and chromosome instabil-ity. Modifi cation by DNA methylation arises by the covalent attachment of a methyl group to position 5 of the cytosine ring, forming 5’-methylcytosine. As mentioned earlier, DNA meth-ylation inhibits transcription, whereby the methyl group that extends out from the cytosine, attracts transcription factors that are attached to the DNA. These methyl-binding domains (MBDs) are, in turn, implicated in gene suppression and chromatin condensation.
THE HISTONE CODE
The “histone code” hypothesis suggests that specifi c histone modifi cations or combinations of modifi cations, confer unique biological functions to the region of the genome associated with them (Strahl and Allis, 2000). The majority of biological functions act to effect tran-scriptional activation, transcriptional silencing, chromatin rearrangement, as well as DNA damage repair, apoptosis, and mitosis. The MBDs act as “readers” to facilitate these func-tions. Thus the histone modifi cations interact with the MBDs yielding downstream protein recruitment, potentially activating enzyme and substrate reactions, resulting in the formation of unique chromatin domains. The histone modifi cations, and their associated functions include, but are not limited to the following.
Histone Acetylation/Deacetylation
Histone acetylation is the process where an acetyl group is presented to lysine residues at the N-terminus of the histone protein. The reverse is histone deacetylation, that is, the detach-ment of an acetyl group. Histone acetylation is triggered by histone acetyltransferases (HATs), while histone deacetylation is stimulated by histone deacetylases (HDACs). HATs and HDACs are mobilized to DNA by transcription factors that identify particular DNA sequences.
Transcriptionally active chromatin is associated with acetylated histones while inactive chromatin is linked to deacetylated histones. Acetylation is also implicated in functional replication, nucleosome adjustment, chromatin packing, and communications of non-histone proteins with nucleosomes.
Histone Methylation/Demethylation
As mentioned earlier, histone methylation is involved in the remodeling of amino acids in histone proteins by the joining of one, two, or three methyl groups. Histone methylation occurs on lysine or arginine residues and causes modifi cations in chromatin architecture by condensing or loosening its structure. Methylation of histones is modulated by histone methyltransferases and the donor of the methyl group is S-adenysolmethionine. Histone methylation plays a signifi cant role in determining between active chromatin (euchromatin) and silent states of chromatin (heterochromatin).

284 PRINCIPLES OF TOXICOLOGY TESTING
Histone Phosphorylation/Dephosphorylation
The addition and removal of phosphate groups also contribute to histone modifi cation path-ways. The attachment of negatively charged phosphate groups to histone tails abrogates their basic charge and depresses the histone inclination toward DNA. Phosphorylation is activated by histone kinases whereas dephosphorylation is stimulated by histone phosphatases.
Histone Ubiquitination/Deubiquitination
Histone ubiquitination is a modifi cation correlated with the attachment of ubiquitin, a 76-amino acid protein. Ubiquitin has been implicated in protein degradation, cell-cycle reg-ulation, endocytosis, and transcription.
Histone Sumoylation
The covalent attachment of a small ubiquitin modifi er (SUMO) to lysine residues also leads to histone modifi cations. Histone sumoylation is mediated via a peptide bond between the carboxyl terminus of SUMO and an epsilon (ε) amino group of a lysine residue of the target protein.
DNA ALTERATIONS AND GENOTOXIC EFFECTS
To a limited extent, all cells are capable of withstanding DNA damage from endogenous or exogenous sources.
Endogenous damage involves (1) wrong integration of nucleotides during DNA replication; (2) automatic or intended deamination of cytosine, adenine, guanine, or 5-methylcytosine modifying these bases to the miscoding uracil, hypoxanthine, xan-thine, and thymine, respectively; (3) automatic hydrolysis of the N-glycosylic bond, leading to depurination or depyrimidination; (4) various chemical conversions in nucle-otide bases, stimulated by reactive oxygen species (i.e., superoxide anions, hydroxyl radicals, and hydrogen peroxide).
Environmental (exogenous) damage includes ionizing radiation and ultraviolet radia-tion. Various genotoxic chemicals cause modifi cations in DNA structure, which may result in mutations that promote cancer risk.
Nucleotide-based damage triggers a variety of cellular responses in eukaryotic cells. These responses involve the (1) induction of cell-cycle-checkpoint pathways that prevent cell-cycle progression allows time for DNA repair; (2) transcriptional activation of an ele-vated number of genes; (3) activation of programmed cell death (apoptosis); (4) stimulation of biochemical pathways for circumventing nucleotide base damage; and (5) induction of explicit DNA repair pathways.
DNA REPAIR MECHANISMS
Eukaryotic cells are able to restore many types of DNA damage. The major DNA-repair pathways are (1) base excision repair; (2) nucleotide excision repair (NER); (3) mismatch repair; (4) homologous recombination (HR); and (5) nonhomologous end joining (NHEJ) repair. The fi rst three pathways lead to the excision of the damaged or mispaired bases, whereas HR and NHEJ enable the repair of double-strand breaks (DSBs). The latter are considered the most lethal of DNA lesions, and can be induced endogenously by cellular processes, including fracture during immunoglobulin gene rearrangement (V(D)J recombi-nation) and meiotic recombination. DSBs are also formed by exposure to ionizing radiation as well as radiomimetic drugs (e.g., bleomycin). Unrepaired DSBs will initiate cell cycle

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 285
checkpoint arrest that may eventually lead to cell death. Table 20.5 summarizes diseases associated with different DNA repair mechanisms.
Base Excision Repair
Base excision repair is induced by DNA repair-specifi c enzymes (DNA glycosylases) which distinguish single or subsets of chemically altered or false bases. For instance, uracil DNA glycosylase precisely selects uracil as an inappropriate base in DNA and hydrolyzes the N-glycosyl bond that conjugates the uracil base to the deoxyribose–phosphate backbone of DNA. Uracil is then removed as a free base, allowing for correction or rectifi cation in the DNA. This site is further repaired by an array of biochemical events.
Nucleotide Excision Repair
NER is associated with the removal of damaged bases from the genome. NER in mammalian cells is a complex process that implicates many proteins. The procedure includes the extrac-tion of bases as part of an oligonucleotide fragment. Humans with an inherited defi ciency in NER suffer from xeroderma pigmentosum and have an elevated propensity to skin cancer induced by sunlight exposure.
Mismatch Repair
Mismatch repair is linked to the excision of nucleotides that are inaccurately paired with the accurate nucleotide on the opposing DNA strand. Mispairing commonly occurs during DNA replication as a result of the restricted adhesion of the DNA replication process. The incor-rect base appears in the newly incorporated DNA strand.
Homologous Recombination
HR is an error-free DSB process that employs a sister chromatid as the template DNA to achieve exact repair. HR initially engages an Mre11– Rad50–Nbs1 complex succeeded by Rad51 and strand invasion. The Rad51 operates cooperatively with an array of other factors (e.g., Rad52 and Rad54 proteins) to foster strand invasion and sequential recombination. Strand invasion and migration are affi liated with the establishment of a structure known as Holliday Junction. Repair synthesis by DNA polymerase completes the repair process.
TABLE 20.5Human Syndromes with Defective Genome Maintenance
SyndromeAffected
Mechanism Type of Genome Instability Cancer Predisposition
BRCA1/BRCA2 HR Chromosome aberrations Breast/ovarian cancerHNPCC MMR Point mutations Colorectal cancerXeroderma pigmentosum NER Point mutation Ultraviolet radiation-
induced skin cancerAtaxia telangiectasia DSB response Chromosome aberrations LymphomaNijmegen breakage syndrome DSB response Chromosome aberrations Lymphoma
Abbreviations: DSB, double strand break; HNPCC, hereditary non-polyposis colorectal cancer; HR, homologous repair; MMR, mismatch repair; NER, nucleotide excision repair.

286 PRINCIPLES OF TOXICOLOGY TESTING
Non-homologous End-Joining
Unlike HR, NHEJ is an error-prone DSB repair mechanism that involves the DNA-dependent protein kinase (DNA-PK) and the DNA ligase IV complex, which together expedite the interconnection of broken inappropriate DNA ends. The DNA-PK complex incorporates the Ku70 and Ku80 proteins, which are responsible for locating the breach and negotiating the use of the DNA-PK catalytic subunit. DNA-PK is a nuclear serine/threonine protein kinase that consists of a catalytic subunit (DNA-PKcs), with the Ku subunits operating as the regulatory element.
EXPERIMENTAL MONITORING FOR EPIGENETIC TOXICITY
The profi ciency to delve into thousands of genes (or possibly all of the genes in a cell type) conveys new insights into the responses of chemicals or drugs on biological systems. MA technology will play an imperative role in validating toxic substances, appraising whether toxic effects appear at low doses, and predict responses from a variety of species.
The cDNA MA functions to examine gene expression changes as indications of chem-ical reactions. Following treatment with chemicals such as polycyclic aromatic hydrocar-bons, peroxisome proliferators, or endocrine disruptors, a gene expression “trademark” on a cDNA MA is presented, which corresponds with the cellular or tissue chain reactions to these chemicals. The response to different chemicals precipitates changes in the expression levels of many genes which denote a general toxic response. However, expression of a subpopulation of genes may be restricted to a distinct class of compounds, specifi cally at low doses. This application identifi es compounds as potential carcinogens/toxicants and reveals their mode of action by elucidating the signal transduction pathways. Table 20.6 summarizes the applications of MA in toxicology.
CLINICAL MONITORING FOR EPIGENETIC TOXICITY
By incorporating pharmacogenetics or toxicogenetics early into clinical evaluation of drug development studies, response characteristics for the determination of effi cacy could be understood early during large phase II clinical trials. For instance, in advance of the enroll-ment stage, the procedure could be used to identify patients with the SNP profi le related to effi cacy. This type of approach reduces subject variation and diminishes unwarranted expo-sure of “unresponsive” patients to a new drug. Thus, pharmacogenetics is useful in develop-ing medicines as well as what is currently used in advanced drug surveillance strategies.
TABLE 20.6Uses of Microarrays in Toxicology
Uses Description
Drug, chemical analysis Analyze toxicants on the basis of tissue-specifi c patterns of gene expression by establishing molecular signatures for chemical exposures
Mechanism of toxicity Assess mechanisms of effect of environmental agents through the analysis of gene expression networks
Biomarkers Use toxicant-induced gene expression as a biomarker to assess human exposureAdmixtures Examine interaction of mixtures of chemicalsHormesis effect Study the effects of low-dose exposures versus high-dose exposuresInterspecies variation Infer the effects of toxicants from one species to another

TOXICOGENOMICS AND EPIGENETIC TESTING IN VITRO 287
SUGGESTED READINGS
Bulera SJ, Eddy SM, Ferguson E, et al. RNA expression in the early characterization of hepatotoxi-cants in Wistar rats by high-density DNA microarrays. Hepatology 2001; 33: 1239.
Clayton TA, Lindon JC, Cloarec O, et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006; 440: 1073.
Coen M, Lenz EM, Nicholson JK, et al. An integrated metabonomic investigation of acetaminophen toxicity in the mouse using NMR spectroscopy. Chem Res Toxicol 2003; 16: 295.
Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene 2005; 24: 949.
Cutler P, Bell DJ, Birrell HC, et al. An integrated proteomic approach to studying glomerular neph-rotoxicity. Electrophoresis 1999; 20: 3647.
de Longueville F, Atienzar FA, Marcq L, et al. Use of a low-density microarray for studying gene expression patterns induced by hepatotoxicants on primary cultures of rat hepatocytes. Toxicol Sci 2003; 75: 378.
Fountoulakis M, Berndt P, Boelsterli UA, et al. Two-dimensional database of mouse liver proteins: changes in hepatic protein levels following treatment with acetaminophen or its nontoxic regioisomer 3-acetamidophenol. Electrophoresis 2000; 21: 2148.
Hamadeh HK, Amin RP, Paules RS, Afshari CA. An overview of toxicogenomics. Curr Issues Mol Biol 2002; 4: 45.
Heinloth AN, Irwin RD, Boorman GA, et al. Gene expression profi ling of rat livers reveals indicators of potential adverse effects. Toxicol Sci 2004; 80: 193.
Engle LJ, Simpson CL, Landers JE. Using high-throughput SNP technologies to study cancer. Onco-gene 2006; 25: 1594.
Friedberg EC. A brief history of the DNA repair fi eld. Cell Res 2008; 18: 3.Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modifi cation path-
ways concurrently in mouse embryonic stem cells. Toxicol Lett 2012; 212: 169.Health and Environmental Sciences Institute (HESI) of the ILSI, Washington, D.C., USA. [Available
from: http://www.hesiglobal.org/i4a/pages/index.cfm?pageid = 1] [Last accessed Septem-ber 2012].
Hellebrekers DMEI, Griffi oen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta 2007; 1775: 76.
International Life Sciences Institute (ILSI), Washington, D.C., USA. [Available from: http://www.ilsi.org/Pages/ HomePage.aspx] [Last accessed September 2012].
Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8: 253.
Luo W, Fan W, Xie H, et al. Phenotypic anchoring of global gene expression profi les induced by N-hydroxy-4-acetylaminobiphenyl and benzypyrene diolepoxide reveals correlations between expression profi les and mechanism of toxicity. Chem Res Toxicol 2005; 18: 619.
Paules RS. Phenotypic anchoring: linking cause and effect. Environ Health Perpsect 2003; 111: 6.Phillips ER, McKinnon PJ. DNA double-strand break repair and development. Oncogene 2007; 26: 7799.Reilly TP, Bourdi M, Brady JN, et al. Expression profi ling of acetaminophen liver toxicity in mice
using microarray technology. Biochem Biophys Res Commun 2001; 282: 321.Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetamino-
phen toxicity in mouse liver. Toxicol Sci 2002; 65: 135.Searfoss GH, Jordan WH, Calligaro DO, et al. Adipsin, a biomarker of gastrointestinal toxicity medi-
ated by a functional γ-secretase inhibitor. J Biol Chem 2003; 278: 46107.Sinha A, Singh C, Parmar D, Singh MP. Proteomics in clinical interventions: Achievements and
limitations in biomarker development. Life Sci 2007; 80: 1345.Strahl BD, Allis CD. The language of covalent histone modifi cations. Nature 2000; 403: 41.Toxicogenomics Research Consortium, National Institute of Environmental Health Sciences (NIEHS),
National Institutes of Health (NIH), U.S. Department of Health and Human Services (DHHS), U.S. [Available from: http://www.questia.com/read/1G1-84303305/niehs-initiates-national-toxicogenomics-research-consortium] [Last accessed January 2013].

288 PRINCIPLES OF TOXICOLOGY TESTING
Waring JF, Ciurlionis R, Jolly RA, Heindel M, Ulrich RG. Microarray analysis of hepatotoxins in vitro reveals a correlation between gene expression profi les and mechanisms of toxicity. Toxicol Lett 2001; 120: 359.
Waring JF, Jolly RA, Ciurlionis R, et al. Clustering of hepatotoxins based on mechanism of toxicity using gene expression profi les. Toxicol Appl Pharmacol 2001; 175: 28.
Waters M, Fostel J. Toxicogenomics and systems toxicology: aims and prospects. Nat Rev Genet 2004; 5: 936.
Zidek N, Hellmann J, Kramer PJ, Hewitt PG. Acute hepatotoxicity: a predictive model based on focused illumina microarrays. Toxicol Sci 2007; 99: 289.
REVIEW ARTICLES
Bártová E, Krejcí J, Harnicarová A, Galiová G, Kozubek S. Histone modifi cations and nuclear archi-tecture: A review. J Histochem Cytochem 2008; 56: 711.
Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9: 297.
Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet 2006; 7: 85.
Glasspool RM, Teodoridis JM, Brown R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J Cancer 2006; 94: 1087.
Huang TT, D’Andrea AD. Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol 2006; 7: 323.
Konsoula Z, Barile FA. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 2012; 66: 215.
Leon J. Pharmacogenomics: the promise of personalized medicine for CNS disorders. Neuropsycho-pharmacol Rev 2009; 34: 159.
Szyf M. The dynamic epigenome and its implications in toxicology. Toxicol Sci 2007; 100: 7.Waters MD, Foster JM. Toxicogenomics and systems toxicology: aims and prospects. Nat Rev Genet
2005; 5: 936.

289
21 Experimental design and statistics
ESTABLISHING AN EXPERIMENTAL PLAN
Several important considerations are fundamental in planning an in vitro experiment. Some of these steps are unique to the development of alternative testing models, while others are simply parts of an adequate plan for conducting any toxicology study (Table 21.1).
MATERIALS AND SUPPLIES
The choices of the materials and supplies are important in the preliminary preparations for a study. In fact, a particular manufacturer or distributor of the materials and chemicals is prob-ably not as important as obtaining them from the same supplier. For instance, the manufac-turer of cell culture supplies should be reputable, easily accessible, and referenced. In addition, the company should provide the researcher with details of the methods used to prepare, package, store, and ship supplies. The materials should be accompanied by material safety data sheets that list important packaging and handling precautions for shipping, receiving, and storage. It is also important that a research laboratory maintain records of dates and storage of chemicals. It is also benefi cial for consistency of a study protocol that one company provides the majority of the chemicals tested. Among the materials obtained and stocked routinely in suffi cient quantities are cell culture supplies, stock cell cultures, chemicals, isotopes, and materials for scintillation counting. Additionally, animal farms can provide an investigator with information about the breeding and housing of animals as well as physical examinations of the animals and a declaration of whether they are certifi ed as pathogen free.
METHODS
During manipulation of cells in culture, certain details are established and followed prudently and uniformly. The number of cells inoculated into culture vessels depends on the rate of inoculation and the size of the fl asks or wells. For instance, T-10, -25, -75, and -150 fl asks, corresponding to 10-, 25-, 75-, and 150-cm2 surface area, are suitable for individual experi-ments or for maintaining cells in continuous passage. Also 6-, 12-, 24-, 48-, 96-, and 384-well plates, corresponding to decreasing surface area per well, are routinely employed in screen-ing assays.
In general, cells are usually seeded at 104 cells per cm2. Contact-inhibited cultures are grown to stationary phase (confl uence), unless cell proliferation experiments are per-formed, and subcultured in appropriate complete medium supplemented with serum and antibiotics if needed. Alternatively, serum-free medium is used in a consistent manner. Depending on the doubling rate of the cell line, the time required for confl uent monolayers to grow can be estimated with considerable accuracy. For example, when seeded at one-third

290 PRINCIPLES OF TOXICOLOGY TESTING
the confl uent density or according to the formula above, most continuous cell lines with appreciable doubling rates reach the stationary phase within 4–8 days.
Monolayers of cells are then incubated with increasing concentrations of each chemi-cal in at least four doses (three to six wells per dose) for a predetermined period at 37ºC in a gaseous atmosphere defi ned by the requirements of the medium. The atmosphere may consist of 5–20% CO
2 in air unless otherwise stated. A stock solution of the test chemical
is prepared in the incubating medium and serial dilutions are made from the stock solution. This method of formulating a stock solution decreases the chances of introducing a repeti-tive error when adding chemical to each experimental group.
For experiments requiring longer incubation times, the media may be initially steril-ized so that aseptic conditions are easily maintained. This is especially important during incubation times of 72 hours or longer. A soluble solution of the chemical is sterilized using micron fi lters, while insoluble solids are fi rst exposed to ultraviolet light for several min-utes in an open test tube or on weighing paper before the addition of buffer.
In time–response experiments, the concentrations for all groups remain the same and the groups are terminated at preset incremental time periods. Before the experiment is ter-minated, indicators, dyes, fi xatives, and reactive substances are added as needed and allowed to incubate with the cultures. The cells are then processed and the reaction product is quantifi ed according to the protocol.
For isotope or fl uorescent labeling studies, a radioactive or fl uorescent tagged (labeled) precursor substrate is added to the cultures, usually 1 hour after the change from a growth medium to a labeling medium. The precursor may be an amino acid for protein synthesis, a monosaccharide for carbohydrate metabolism, a precursor for lipid biosynthesis, an enzyme substrate, or any molecule designed to trace an intracellular biosynthetic pathway. The labeling medium is a simpler version of the growth medium. Care is taken to avoid the presence of the precursor or parent molecule in the formula. In addition, the serum, gener-ally used during the growth stage is dialyzed to remove trace components that interfere with uptake of the precursor. Other supplements to the incubation medium include buffers, antibiotics, and cofactors necessary for the proper synthesis of the indicator molecules. Cell viability is usually measured in separate cultures and is expressed as a percentage of the control group results. Additional wells without cells are processed with the correspond-ing medium in parallel as reference blanks.
Several automated or manual cell-based assays are available as prepackaged kits for the spectrophotometric, fl uorescent, or luminescent detection of macromolecules represen-tative of cellular biochemical processes. The use of cell-based assays in toxicology and in drug discovery research is important for revealing the mechanisms of toxicological action, development of alternative in vitro assays for toxicology testing, and disease mechanisms.
TABLE 21.1Steps in Planning and Preparing In Vitro Experiments
Objective Considerations
Procuring materials Standard maintenance of readily available cell culture suppliesOrdering chemicals Anticipation of rate-limiting chemicals that should be always ready for usePlanning methods Anticipation of technical, scientifi c, and administrative scheduling critical for smooth
conduct of studyTime constraints Consideration of time required for initiating and maintaining cultures

EXPERIMENTAL DESIGN AND STATISTICS 291
Cell-based assays also are important in secondary screening studies of promising drug compounds in the course of drug development. The kits offer plate formats and membranes that allow a wide range of functional cell-based assay protocols to be performed, including the following:
1. Black multiscreen fl uorescent or luminescent plates with protocols ideal for cell viability and proliferation assays using fl uorescent detection or luminescent techniques.
2. Migration, invasion, and chemotaxis assay plates suitable for high-throughput mul-tiwell fi lter formats.
Some applications of cell-based assay kits are incorporated for studies involving cell viabil-ity, proliferation, cell migration, invasion, chemotaxis, whole-cell incubation, and whole-organism assays. The systems use conventional fl uorescent or time-resolved fl uorescent detection systems, as well as absorption-based protocols.
More recent fl uorescent and luminescent enzyme assays monitor intracellular enzyme activities. These systems are designed to determine cytotoxicity using a variety of fl uo-rescent or luminescent probes including assays for the detection of intracellular enzymes such as calcein-AM, rhodamine-based fl uorescent dyes (Molecular Probes, Inc., Eugene, Oregon, USA), CellTiter-Glo™, Caspase-3/7, and -8/9 assays (Promega Corp., Madison, Wisconsin, USA). Table 21.2 describes the functional principles of cell-based assays and their value in toxicology testing.
IC50
levels and concentration effect curves for each cell line are determined with the probes available in the kits. Results are coordinated by monitoring several indicators that
TABLE 21.2Cell-Based Fluorescent and Luminescent Assays and Their Utility in Toxicology Testing
Assay Description
Calcein-AM (Molecular Probes, Inc.) Soluble dye, freely enters intact cell membranes and fl uoresces upon cleavage with intracellular esterases; reliable viability indicator
SYTOX™ (Molecular Probes, Inc.) High-affi nity nucleic acid fl uorescent stain; penetrates compromised cell membranes but will not cross membranes of live cells; valuable dead-cell green fl uorescent indicator when used simultaneously with other measures of cytotoxicity
Rhodamine-110 series of dyes (Molecular Probes, Inc.)
Indicators for intracellular proteinases; upon cleavage with trypsin, plasmin/cathepsin, or other intracellular enzymes, nonfl uorescent bisamide derivatives of dyes are converted to fl uorescent molecules, with corresponding increases in fl uorescence emission
Caspase-3/7 and Caspase–Glo 8/9 (Promega Corp.)
Capable of detecting and differentiating cells undergoing apoptosis or necrosis in the presence of toxic compounds; assay based on the cleavage of profl uorescent peptide–rhodamine 110 substrate by caspase-3/7, resulting in green fl uorescent signal and cleavage of proluminogenic caspase 8/9, resulting in luminescence
CellTiter-Glo™ (Promega Corp., luminescent cell viability)
Homogeneous bioluminescent protocol designed for measurement of ATP; involves addition of luciferin substrate and stable luciferase directly to cells; bioluminescent glow is directly proportional to the amount of ATP released from viable cells

292 PRINCIPLES OF TOXICOLOGY TESTING
also contribute to an understanding of the mechanisms of toxicity. This information in combination with interlaboratory comparisons allows for the comparison of the reliability of cell-based assays as indicators for traditional in vitro cytotoxicity testing methods. Thus the convenient and rapid performance of a test is critical for incorporation into a battery of tests for screening acute human chemical toxicity.
EXPERIMENTAL DESIGN
MEDIUM AND CHEMICAL COMPATIBILITY
Most technical problems related to experimental design and execution of toxicology testing experiments involve the solubility or miscibility of the chemicals with the incubation medium. Solid chemicals that are insoluble in water, such as paracetamol (acetaminophen) and acetylsalicylic acid (aspirin) present with dissolution problems, especially at higher con-centration levels. In order to improve the solubility of these chemicals without the addition of a solvent that may potentially interfere with the interpretation of the fi nal results, a variety of techniques are employed, as described in Table 21.3.
Micronization
This procedure increases the surface area of a solid and can improve the solubility of a pow-der at low doses. A solution of the chemical is prepared in ethanol or dimethyl sulfoxide (DMSO) for each group and the solution is evaporated to dryness under a stream of nitrogen gas. The remaining lyophilized powder is then dissolved more readily in a medium, which is constantly stirred at 37°C for at least 1 hour prior to incubation.
Solvents
Occasionally it may be necessary to dissolve a test chemical in a solvent (such as ethanol, isopropanol, and DMSO) that is miscible with the incubation medium. A stock solution of the substance is prepared in the solvent and appropriate aliquots are then distributed to the
TABLE 21.3Technical Manipulations to Improve Solubility and Miscibility of Media and Chemicals
Manipulations Chemical Formulation Description
Micronization Solid dissolved in alcohol or DMSO
Evaporation of stock solution of chemical in alcohol, using constant stirring at 37ºC for 1 hr or under N
2 gas stream until powder is lyophilized
Stock solution in ethanol, IpOH, or DMSO
Solid dissolved in solvent
Concentrated stock solution of test chemical is formulated and aliquots are added to media of test groups as required; solvents are equally distributed among all groups including controls
Sonication Organic liquid Quantity of immiscible organic test liquid is added to medium and sonicated using an ultrasonic probe
Mineral oil overlay Organic liquid Quantity of volatile or organic test liquid is added to medium and mineral oil is layered over surface; fl asks are gassed and sealed individually
Abbreviations: DMSO, dimethyl sulfoxide; IpOH, isopropanol.

EXPERIMENTAL DESIGN AND STATISTICS 293
corresponding experimental groups. It is important, however, that the relative effect of the sol-vent alone is determined in control groups, even at its lowest concentration as part of the incu-bation medium. Also, all experimental groups are equilibrated with the same amounts of solvent including the controls. This will negate any minor effects of the solvent alone on the cells.
Sonication
Certain organic liquids such as xylene and carbon tetrachloride used as test chemicals are immiscible with water and culture media. Miscibility is improved by subjecting a stock solu-tion of the chemical to sonication. This manipulation requires the use of an ultrasonic proces-sor equipped with a longer probe for culture tubes that is inserted into the immiscible liquids for as few as 10 seconds at 10–50 W of power. The ultrasound energy completely homogenizes the mixture and allows enough time for adequately dispersing an aliquot of the “dissolved” test substance solution into the incubation medium. On occasion, the liquids separate out during the incubation time, in which case the mixture is resuspended and added again to the cultures.
Paraffi n (Mineral) Oil Overlay
More volatile chemicals such as alcohols, xylene, and chloroform are inconvenient because they permeate the incubator atmosphere and interfere with adjoining wells. The evaporation and thus the concentration of the chemical in the medium can be controlled by incubating the chemicals with cells previously grown in a 25-cm2 (T-25) fl ask that is then overlaid with a thin layer of paraffi n oil (light mineral oil). The oil suppresses evaporation of volatile mol-ecules at the surface by increasing the surface tension. The fl asks, however, must be indi-vidually permeated with a stream of CO
2 gas corresponding to the gaseous atmosphere of the
incubator and sealed with screw caps. This requires some forethought: it is necessary to anticipate which chemicals are tested and which vessels are used to grow the cells. In addi-tion, a separate gas tank formulated for the incubation medium is required for these studies.
The oil overlay produces enough surface tension to prevent evaporation of volatile chemicals incompletely dissolved in the medium. The partial pressure of the chemical in a liquid, however, determines to what extent the test substance partitions between the air and gas phases. Ultimately this partitioning affects the amount of substance dissolved in the incubating medium and in contact with the cell layer. Essentially, it is necessary to verify the actual concentration of the chemical in the liquid and the air space above the cell layer in a series of control fl asks. The method of choice includes head-space gas chromatography. Accordingly an accurate estimation of the kinetics of air–liquid distribution is computed.
CALCULATION OF CONCENTRATION RANGE
It is not always necessary to test wide ranges of logarithmic concentrations of chemicals, especially when resources are limited. For instance, the LD
50 value of a known substance is
used to estimate the human equivalent toxic concentrations (HETCs) in plasma—the con-centrations of a chemical used to initiate in vitro toxicology testing experiments are derived from rodent LD
50 values according to the following formula:
HETC = [(LD50
]/Vd] × 10−3
where HETC is the estimated human equivalent toxic concentration in plasma (mg/mL); LD
50 is the 50% lethal dose in rodents (mg/kg, intraperitoneal or oral); V
d is the volume of
distribution (L/kg); and 10–3 represents the constant for conversion into mL (L/mL).
294 PRINCIPLES OF TOXICOLOGY TESTING
Thus the calculated HETC value is an estimate of the toxic human blood concentration equivalent to the LD
50 dose administered to a rodent based on body weight. The HETC is
then used as a guideline for establishing the concentration range for each group of an experiment. HETC values are also used as a method for converting animal LD
50 data into
equivalent human toxicity information, which is valuable as a procedure for comparing in vitro IC
50 values against human lethal concentrations derived from clinical case studies
and poison control centers. Essentially, the formula offers a mechanism for converting in vivo rodent data to equivalent human toxicity concentrations that can be employed as a guideline to compare with in vitro data in their ability to predict human toxicity.
DETERMINATION OF INHIBITORY CONCENTRATIONS
Figure 21.1 shows a typical concentration effect curve generated for a representative chemi-cal using an indicator such as cell viability for basal cell functions. The concentration neces-sary to inhibit 50% (IC
50) of the newly synthesized macromolecules is then calculated based
on the slope and linearity of the regression line. The symmetry of the line is estimated from the regression analysis of the plot: percent loss of cell viability versus concentration.
The values for percent loss of cell viability are derived by converting the absolute value of the measured response to a fraction of the control value, that is, the measured value of the group at the lowest or highest chemical concentration. Using the control values as the 0% or 100% level, respectively, the responses from all subsequent groups are compared with these controls. Thus, absolute values are transformed into relative percentage values. This presents an added advantage such that different experiments are comparable even when the absolute values of the control groups are not identical. When the response is plot-ted against the concentration used for that group, the regression line is obtained. Similarly, the IC
50 is estimated by extrapolating from the 50% response.
The plots for some representative substances grouped according to chemical classifi ca-tion in Figure 21.1 show that the regression line is signifi cantly accurate so that the IC
50 may
Concentration of chemical X (10–3 or 10–6 M)
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
% L
oss
of
cell
viab
ility
0
10
20
30
40
50
60
70
80
90
100
FIGURE 21.1 Concentration effect curve for a representative chemical X in an in vitro cytotoxicol-ogy experiment using loss of cell viability as indicator. The inhibitory concentration at the 50% level (IC
50) is determined from this plot.

EXPERIMENTAL DESIGN AND STATISTICS 295
be computed with statistical confi dence.1 In a similar manner, the 25 and 75% inhibitory concentrations (IC
25 and IC
75, respectively, not shown) are also calculated to demonstrate
the lower and upper cutoff limits. In addition, the cutoff points aid in the determination of the range of toxicity. The greater the difference between the upper and lower inhibitory concentrations, the wider is the range of toxicity for a particular chemical. A chemical pos-sessing a smaller difference between the IC
25 and IC
75 has a narrower range of toxicity. This
implies a narrower margin between nontoxic and toxic doses or between toxicity and lethal-ity. When the IC
50 is not bracketed in the concentration range used for the chemical, the
experiment is repeated with adjusted sets of concentrations.
HYPOTHESIS TESTING AND STATISTICAL APPLICATIONS
Many statistical tests are available for analyzing a set of experimental data. The choice of a combination of statistical methods used, however, depends on the objectives of the study and the direction of the composition of the data set. Ostensibly, some forethought is required during the initial experimental planning so that the appropriate statistical approach is employed. The calculations are then incorporated into the study either alone or collec-tively with other statistical parameters, thus reinforcing the validity of the experimental conclusions.
In addition, the type and number of statistical tests engaged will reduce the chances that an insignifi cant result is declared signifi cant, especially when individual experiments are repeated. It is an innate consequence, however, that the more often the same statistical calculation is repeated for each experiment, the greater the chances of rejecting the true null hypothesis. Table 21.4 defi nes some of the fundamental terms used to describe and interpret statistical parameters in toxicology testing.
DEVELOPMENT OF HYPOTHESIS TEST
The purpose of hypothesis testing in cytotoxicity studies is to enable an investigator to reach a decision about a population by examining a sample of that population. The hypothesis, referred to as the null hypothesis (H
0) or hypothesis of no difference, is con-
structed as a statement about one or more populations. In the process of the statistical test, the null hypothesis is either rejected or accepted. If the testing procedure leads to rejec-tion, the data are construed as not compatible with the null hypothesis, but are supportive of some other as-yet-undetermined hypothesis. Any experimental treatment protocol in toxicology testing acts as an example of a hypothesis, the easiest of which are the toxico-logical changes incurred by a cell or organism as a result of the infl uence of a chemical. In general, the null hypothesis is established for the explicit purpose of being discredited or supported.
The hypothesis is also concerned with the parameters of the selected population. The form of the parameters established as quantifi able data information determines the choice of tests used. In hypothesis testing of univariate parametric tests, the populations are nor-mally distributed and the data are continuous. Each data point has a discrete number that implies a measurable relationship to other numbers among the populations. In hypothesis testing of nonparametric tests, the data are ranked or categorical. These types of data are not continuous, are presented in the form of contingency tables, and are collected and arranged so that they may be classifi ed according to one or more response categories.
1 The calculated correlation coeffi cient ( r value) for this theoretical plot is +1.00.

296 PRINCIPLES OF TOXICOLOGY TESTING
PARAMETRIC CALCULATIONS
The selection of parametric and nonparametric tests in toxicology testing studies is based on the initial determination of the availability of continuous or discrete data. Thus, parametric testing focuses on estimating or testing a hypothesis about one or more population parame-ters. A valid assumption of these tests is that the sampled populations should be approxi-mately normally distributed as in the “entire” population. Another requirement is the knowledge of the form of the population providing the samples for the basis of the infer-ences. Some of the more common parametric tests engaged in statistical analysis are discussed next.
Regression Analysis
Regression analysis or linear regression is one of two statistical tests, along with correla-tion analysis, that yields information about the relationship between two variables. The variables are usually normally distributed sample populations that take the form of experi-mental groups. For example, in toxicology testing, the experimental groups include a
TABLE 21.4Fundamental Statistical Terms for Interpreting In Vitro Data
Statistical Term Defi nition
Independent variable Characteristic or measurable parameter assigned to different values within test system; also known as random variable when values are obtained as a result of chance factors; e.g., designated concentration of a chemical
Dependent variable Value resulting from test measurement protocol; relies on the value of an independent variable; also known as observation or measurement, e.g., measurement of cell viability after exposure to a chemical
Continuous data Data plotted as discrete points on a line, including any measurable parameter; can assume any of an infi nite number of values between two fi xed points, e.g., body weight, protein concentration
Discontinuous data Measurable parameters as with continuous data; have only fi xed values, with no possible intermediate values, e.g., number of cells surviving treatment exposure
Probability Frequency with which a particular value is assigned specifi ed by an appropriately sized sample
Frequency distribution Table or graph in which values of variable are distributed among specifi ed class intervals; set of non-overlapping observations
Hypothesis testing Determination of whether one or more populations is compatible with available data; hypothesis is concerned with parameters of populations about which statement is made; testing aids investigator in reaching decision about population by examining sample from that population
Null hypothesis (hypothesis of no difference)
Statement about one or more populations supporting contention no signifi cant differences exist between or among groups of sample populations subjected to testing parameters
Signifi cance level All possible values a test statistic can assume are graphed as points on horizontal axis of distribution and divided into acceptance and rejection regions; experimental values (observations) are assigned to regions according to selected level of signifi cance; level specifi es area under curve of distribution of test statistic that is above values on horizontal axis constituting rejection region; the most common signifi cance levels are often set at 0.001, 0.01 or 0.05 corre-sponding to 99.9, 99.0, and 95% confi dence intervals, respectively

EXPERIMENTAL DESIGN AND STATISTICS 297
control group to which no chemical is added and each of the treatment groups is capable of generating data points. The variables in question consist of measurable responses of cells to a chemical (the Y or dependent variable) as a function of the concentration (the X or independent variable).
Thus regression analysis facilitates the identifi cation of the probable form of the rela-tionship between these variables, with the ultimate objective of predicting or estimating the value of the dependent variable in response to a known value of the independent variable. As in the case of in vitro cytotoxicology experiments, the IC
50 is estimated based on the regres-
sion analysis of the concentration effect curves. Thus, an estimate of the concentration nec-essary to inhibit 50% of the measured response is calculated within the confi dence intervals.
Often, the raw data of concentration effect relationships are not linear, necessitating transformation of the data into logarithmic value scales. Usually, the logs of the concentra-tions are plotted on the X axis against the probability (probit) scale, such as percentage response of the control value (Y axis). Such manipulations render the information more amenable to linear interpolation and allow for calculations of estimated IC
50 values with
greater statistical confi dence.
Correlation Analysis
Correlation analysis and the calculation of the correlation coeffi cient (r value) are computed to determine the strength or signifi cance of a regression analysis. The r value dictates the relative degree of correlation between the variables at a predetermined level of signifi cance. The largest absolute value that r can assume is ±1.0, resulting when all the variation in the dependent variable is explained by the regression analysis.
In this case, all the experimental observations are reduced to the regression line, within acceptable error limits. The lower limit of r is 0, which is obtained when the regression line is horizontal. The closer the r value approaches ±1.0 (–1.0 represents an inverse correla-tion), the greater the probability that the variation of the Y values is explained by the regres-sion. This indicates that the regression analysis accounts for a large proportion of the total variability in the observed values for Y. Conversely, a small r value (close to zero in the positive or negative direction) does not support the regression and is viewed with less con-fi dence. Additional objective statistical tests for signifi cance, however, are the hypothesis test for β = 0, the analysis of variance, and the Student’s t-test.
Hypothesis Test for b = 0
In addition to calculating the correlation coeffi cient (r value) for each concentration effect curve, the hypothesis test for β = 0 is applied as an alternative test of the null hypothesis of zero slope between the two variables, X (log of concentration) and Y (percentage response). The calculation is based on the slope of the sample regression equation. The test statistic, which is normally distributed as with the Student’s t, is calculated and compared with the minimum values of t at n minus two degrees of freedom for the 95 and 99% confi dence intervals or greater. Figure 21.2 illustrates concentration effect curves generated for repre-sentative chemicals using a cell viability indicator as a measure for cytotoxicity.
The correlation coeffi cient, the test statistic (t value), and inhibitory concentrations are calculated from the transform regression analysis of each plot (percent of control versus log of concentration). Different sets of concentration levels are repeated for each chemical until the test statistic for the plot is signifi cant at the 95% or 99% confi dence interval at this prob-ability. The test statistic is then compared with minimum values for t for the number of pairs of data involved minus two (or n – two degrees of freedom).
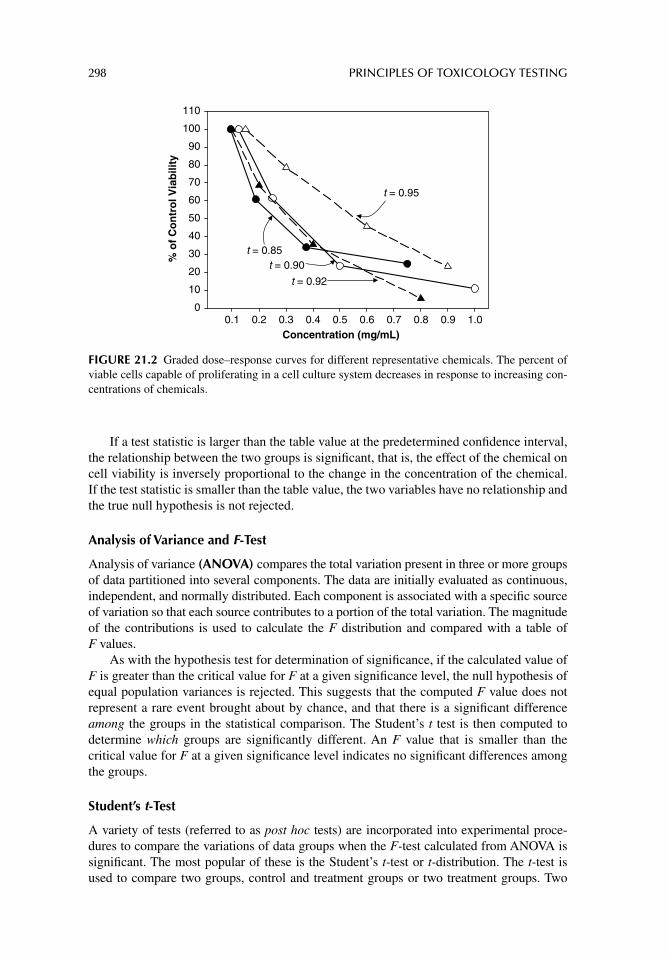
298 PRINCIPLES OF TOXICOLOGY TESTING
If a test statistic is larger than the table value at the predetermined confi dence interval, the relationship between the two groups is signifi cant, that is, the effect of the chemical on cell viability is inversely proportional to the change in the concentration of the chemical. If the test statistic is smaller than the table value, the two variables have no relationship and the true null hypothesis is not rejected.
Analysis of Variance and F-Test
Analysis of variance (ANOVA) compares the total variation present in three or more groups of data partitioned into several components. The data are initially evaluated as continuous, independent, and normally distributed. Each component is associated with a specifi c source of variation so that each source contributes to a portion of the total variation. The magnitude of the contributions is used to calculate the F distribution and compared with a table of F values.
As with the hypothesis test for determination of signifi cance, if the calculated value of F is greater than the critical value for F at a given signifi cance level, the null hypothesis of equal population variances is rejected. This suggests that the computed F value does not represent a rare event brought about by chance, and that there is a signifi cant difference among the groups in the statistical comparison. The Student’s t test is then computed to determine which groups are signifi cantly different. An F value that is smaller than the critical value for F at a given signifi cance level indicates no signifi cant differences among the groups.
Student’s t-Test
A variety of tests (referred to as post hoc tests) are incorporated into experimental proce-dures to compare the variations of data groups when the F-test calculated from ANOVA is signifi cant. The most popular of these is the Student’s t-test or t-distribution. The t-test is used to compare two groups, control and treatment groups or two treatment groups. Two
Concentration (mg/mL)0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
% o
f C
on
tro
l Via
bili
ty
0
10
20
30
40
50
60
70
80
90
100
110
t = 0.95
t = 0.85 t = 0.90
t = 0.92
FIGURE 21.2 Graded dose–response curves for different representative chemicals. The percent of viable cells capable of proliferating in a cell culture system decreases in response to increasing con-centrations of chemicals.

EXPERIMENTAL DESIGN AND STATISTICS 299
types of Student’s t-test are used, depending on the nature of the data points within the groups. If each observation generates corresponding numbers of continuous data and the number of observations is equal, the paired test is employed and the degree of freedom is represented as N – 1, where N is the number of observations made within groups. When the observations are not equal and the data are continuous but randomly distributed, the unpaired t-test is used. The degrees of freedom are represented as N
1 + N
2 – 2, where the number of
observations of data between groups is not equal or paired.As with the calculation of the F value, the value of t is compared with the values in the
t-distribution table at the appropriate signifi cance level (α) and degree of freedom. If the calculated t-value is greater than the critical value for t at a given signifi cance level, then the null hypothesis of equal sample variances is rejected, signifying a signifi cant difference between the two groups and the changes in the dependent variable are directly or indirectly proportional to changes of the independent variable. A t-value that is smaller than the criti-cal value for t at a given signifi cance level indicates no differences between the two groups.
NONPARAMETRIC OR GOODNESS-OF-FIT CALCULATIONS
Nonparametric statistical analysis is exactly analogous to parametric statistical methods but involves the analysis of ranked data. Traditionally known as goodness-of-fi t tests, these anal-yses are based on observed distribution of frequencies in treatment groups and their compat-ibility with a predetermined or hypothesized distribution. The procedures used for reaching a decision to reject or accept the null hypothesis consist of placing the observations into mutually exclusive categories and noting the frequency of observed values in each interval. The frequencies are then compared with available knowledge of normal frequency distribu-tions that would be expected if the samples were obtained from a normal distribution. The discrepancies between what was observed and what was expected are used to calculate the test statistic that is then compared with the minimum values in a standardized table of values for that particular test at a preassigned signifi cance level.
Chi-Square Test
The chi-square (χ2) test for independence of classifi cation is a widely used goodness-of-fi t test based on a cross tabulation of observed frequencies of two (2 × 2) or more (R × C) variables. This cross tabulation is also called a contingency table and arranges the discontinuous frequency data in rows (F) of control and treatment groups and in columns (C) of observed and expected frequencies. The degrees of freedom are calculated as (R – 1) × (C – 1). Chi-square is applied to normal, binomial, and Poisson distributions, depending on the number of categories. When χ2 is greater than the tabulated value of χ2 at a predetermined level of signifi cance (α), the null hypothesis is rejected at that level and the groups are considered signifi cantly different with respect to their treatment protocols. When the null hypothesis is true, the discrepancy between the observed and expected frequencies is distributed approximately as the calculated χ2.
Some underlying assumptions for the χ2 distribution include that the data are univariate and categorical and that the groups are approximately the same size with at least 50 total observations. As with other statistical analyses, the data should be collected by random independent sampling methods.
Wilcoxon Rank Sum
As with the χ2, the Wilcoxon rank sum test is used for comparison of groups of data not normally distributed when the data fall within the established groups with set ranges of

300 PRINCIPLES OF TOXICOLOGY TESTING
observations. Data for the experimental comparison groups are initially ranked in increasing order and assigned rank values. The sum-of-rank values are calculated and compared with the two limit table values to determine whether the groups are signifi cantly different at a given signifi cance level (α).
Kruskal–Wallis Nonparametric ANOVA
As with the parametric analysis of variance, the Kruskal–Wallis nonparametric ANOVA is used to reject or accept the null hypothesis that maintains that several group means are equal. The calculation is initially performed on discontinuous categorical groups of data to determine whether signifi cant differences exist among the groups. The test proceeds by arranging the observations of three or more groups in a single series in order of magnitude from smallest to largest. The observations are replaced by ranks starting from the fi rst. The ranks are then assigned to observations in each group and added separately to yield a sum of the ranks.
The test statistic (H) is computed and compared with a table of H values. When the calculated H value is greater than the table value for the number of observations in the study, the null hypothesis is rejected, suggesting a signifi cant difference between the groups. The test is usually followed by another nonparametric test such as a 2 × 2 χ2 or distribution-free multiple comparison test , depending on the number of groups, to determine which groups exhibit signifi cant differences. The Kruskal–Wallis ANOVA is used for both small and large samples. The test is weakened when many tied ranks occur.
SUGGESTED READINGS
Barile FA. Introduction to In Vitro Cytotoxicology: Mechanisms and Methods. Boca Raton, FL: CRC Press, 1994.
Barile FA. Clinical Toxicology: Principles and Mechanisms, 2nd edn. New York, London: Informa-Healthcare Publishers, 2010.
Baselt RC. Disposition of Toxic Drugs and Chemicals in Man, 7th edn. Foster City, CA: Biomedical Publications, 2004.
Chow SC, Shao J. Statistics in Drug Research: Methodologies and Recent Developments. Boca Raton, FL: CRC Press, 2002.
Daniel WW. Biostatistics: A Foundation for Analysis in the Health Sciences, 8th edn. New York: John Wiley & Sons, 2006.
Gad SC. Statistics and Experimental Design for Toxicologists and Pharmacologists, 4th edn. Boca Raton, FL: Taylor & Francis, 2005.
Lewis RJ Jr. Hazardous Chemicals Desk Reference. New York: John Wiley & Sons, 2002.Skillings JH, Weber D. A First Course in the Design of Experiments: A Linear Models Approach.
Boca Raton, FL: CRC Press, 1999.O’Neil MJ, Smith A, Heckelman PE, et al. eds. The Merck Index, 14th edn. White-house Station, NJ:
Merck Research Laboratories, 2006.
REVIEW ARTICLES
Bernauer U, Oberemm A, Madle S, Gundert-Remy U. The use of in vitro data in risk assessment. Basic Clin Pharmacol Toxicol 2005; 96: 176.
Elliott MR, Joffe MM, Chen Z. A potential outcomes approach to developmental toxicity analyses. Biometrics 2006; 62: 352.
Hayashi Y. Designing in vitro assay systems for hazard characterization: basic strategies and related technical issues. Exp Toxicol Pathol 2005; 57: 227.

EXPERIMENTAL DESIGN AND STATISTICS 301
Linkov I, Satterstrom FK. Weight of evidence: what is the state of the science? Risk Anal 2006; 26: 573.
Rietjens IM, Alink GM. Future of toxicology: low-dose toxicology and risk–benefi t analysis. Chem Res Toxicol 2006; 19: 977.
Suter W. Predictive value of in vitro safety studies. Curr Opin Chem Biol 2006; 10: 362.van der Hoeven N. Current issues in statistics and models for ecotoxicological risk assessment.
Acta Biother 2004; 52: 201.

302
22 Standardization and validation of alternative methods
INTRODUCTION
DEVELOPMENT OF ALTERNATIVE TEST METHODS
In the past twenty years, the toxicological community has witnessed an unprecedented bur-geoning of the development of various types of non-genetic, systemic and local in vitro methods as alternatives to animal testing. Although traditionally founded as mechanistically based cell culture procedures, many of them were adapted as empirical measures of many types of toxicity, based on the assumption that their targets are similar to those encountered in vivo. Some of these protocols are now well-established, such as the use of cell culture models to screen or predict for dermal and ocular local toxicity. Other proposed alternative in vitro tests are currently undergoing standardization of the procedures between single laboratories and institutions, such as the testing for acute systemic and target organ toxicity (including nervous system, liver, heart, and kidney), as well as various toxicokinetic tests. About 10 to 20 test methods have been developed for each system, now totaling about 100 to 200 different quantitatively reliable procedures.
Some of these techniques were rapidly adopted by the pharmaceutical and cosmetic industries as preliminary toxicology screening tools and used in parallel with conventional animal testing protocols. Based on the successful comparisons of the data with traditional in vivo procedures, the newly developed alternative tests are accepted, or acknowledged, by EU and US regulatory agencies for formal validation proposals. It is important to remem-ber, however, that many of the same methods have been in practical use for prediction of acute toxicity, eye irritancy, phototoxicity, and teratogenicity for several years, based on their ability to screen for potentially toxic substances. In fact, the US Pharmacopoeia rec-ommends cellular tests for local and systemic toxicity of plastic devices as a test of fi rst choice. Nevertheless these systems represent only a small fraction of the majority of proce-dures recently available and referenced in the literature and have not been subjected to eval-uation by regulatory, industrial, or academic concerns. Unlike the procedures that have been evaluated and have withstood scientifi c scrutiny, none of the latter methods have gained wide acceptance by the same scientifi c and governing communities.
STANDARDIZATION OF TEST PROTOCOLS
In general, individual proprietary and academic laboratories develop and propose in vitro pro-cedures aimed at specifi c toxic endpoints. The principle is to analyze a set of chemicals and their effects on cellular components. These evaluations involve testing for injury to basal cell or organ-specifi c functions, alteration to non-cellular structures, the infl ammatory reactions to

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 303
the injury, and the toxicokinetics of the chemical in the target tissue. A cell culture system is then constructed to simulate a composite of these factors, or perhaps only one factor, and thus becomes a partial test for the toxicity in question. In vitro eye irritancy procedures were devel-oped in this manner as composite protocols, such as the HET-CAM test. Other systems were based on the detection of toxicity on focused targets in the eye, such as tests with corneal epithelium, protein matrix simulating corneal structure, and tests of infl ammatory responses.
It is lengthy and expensive for individual laboratories to maintain this effort, especially when standardization of the test methods is essential for proposal as a toxicology screening procedure. In the case of eye irritancy, the laboratory must submit for peer review that the method will screen or predict eye toxicity in humans as well as the existing and accepted Draize procedure. Essentially, a fl edgling protocol requires validation—that is, evaluation of the technical reliability of the method as well as the relevance to its stated purpose. A composite method, for instance, requires testing of hundreds of carefully selected chem-icals, and groups of chemicals with similar structural classifi cation or application, with potential for human eye irritancy. The results are then compared to those obtained with the Draize method and to existing data for human toxicity. If the test has introduced only one component of the target injury, the other factors of the in vivo data are also incorporated into the validation scheme. For most laboratories, this demanding endeavor is not a realistic goal, since the availability of resources is often limited. Most laboratories, therefore, have performed preliminary and limited evaluation of their methods as feasibly possible. For instance, 20–50 chemicals are tested with the proposed protocol and compared with avail-able animal toxicity data for the same substances, thus presenting a method that has under-gone a cursory evaluation. The objective, therefore, is not to propose a scheme for regulatory acceptance, but rather to introduce a blueprint to the scientifi c and industrial community with the hope that it will be further evaluated for practical and general use as part of a toxicity testing protocol.
The bottleneck to implementation of alternative in vitro models into the general and local toxicity testing design is a lack of well-defi ned validation proposals, caused primarily by a paucity of resources of individual laboratories devoted to this purpose. Also, human toxicity data for most types of systemic and local toxicity are scarce, which makes it more diffi cult to accept screening tests for predicting acute or chronic toxicity. Finally, it is important to note that poorly defi ned validation processes are an impediment to future acceptance of effective in vitro testing.
Accordingly, it is not diffi cult to understand why so many alternative models for in vitro testing are not accepted into the general toxicity testing establishment. The explanation lies in a complex set of scientifi c and regulatory criteria where newer in vitro methods are rigorously and justifi ably compared to the theoretical basis that has formed the fundamentals of all tradi-tional toxicological inquiry, while the inscrutable organization of the regulatory agencies inev-itably leads to delay. The following discussion presents an explanation of the criteria and components of an alternative in vitro validation system, the status of current multi-laboratory validation programs, and a proposal for future validation schemes and legislative acceptance.
THE VALIDATION PROCESS
DEFINITIONS
The term validation is derived from the original application used for in vitro genotoxicity and mutagenicity tests and has been introduced into the non-genotoxic arena to denote relevance and reliability of a test for a specifi c purpose. Reliability refers to the reproducibility of a test

304 PRINCIPLES OF TOXICOLOGY TESTING
within the same laboratory and between laboratories—that is, intra- and inter- laboratory variation. Evaluation refers to a process whose aim is acceptance of the test by the scientifi c community, although it does not necessarily imply achievement of this goal. Although valida-tion suggests that this same goal of acceptance by the scientifi c community is an absolute, as defi ned by the regulatory agencies, the term is used most often by investigators as a process of appraising in vitro methods without the demand that the process be complete in order to reach this goal. Recently, some references recommend that the term should refer to a process used to evaluate the reliability and relevance of in vitro tests. This course of action includes the necessary investigatory steps aimed at acceptance of the method in the general scientifi c and industrial communities, rather than an absolute attainable objective. For clarifi cation, Table 22.1 defi nes some important terms used to understand the validation process and func-tion in the development of alternative test methods.
VITAL COMPONENTS OF A VALIDATION PROGRAM
Relevance and Reliability
The importance of the relationship between relevance and reliability is necessarily linked to the concept of validation. For instance, a test that demonstrates favorable results for a large number of chemicals, or chemical groups, implies considerable reliability. In contrast, a very reliable method may not be useful if it has not demonstrated some relevance to the in vivo situation. Many recently developed cytotoxicity testing protocols with objective end-point determinations from established laboratories are reproducible, and their results do not vary signifi cantly between laboratories. Closer inspection of the protocol, however, reveals that some of these methods lack relevance. Therefore, while relevance and reliability are equally important in the validation process, the concept of relevance is the “rate limiting” factor, that allows little tolerance for deviation from the goal of validation. Because of this criterion, most laboratory validation studies arising from individual laboratories have only evaluated relevance, and presented for scientifi c review with good reasons to believe that the reliability of the method is suffi cient as is.
TABLE 22.1Definitions of Some Fundamental Terms as Applied to the Validation Process
Term Defi nition
Validation A process for the determination of the relevance and reliability of a test for a specifi c purposeReliability Determination of the reproducibility of a test within the same laboratory and between
laboratories (intra- and inter-laboratory variation, respectively)Relevance Demonstration of the predictivity, applicability and comparability of a test method for a
corresponding in vivo situation; the “rate-limiting” factor that allows less tolerance for deviation from the goal of validation
Standardization Analysis of a test procedure and its components for reliability in comparison to internal and external controls
Validity Establishment of standards for routine intra- and inter-laboratory use of a toxicology test for the purpose of attributing credibility, effectiveness, or quality
Evaluation The process of acceptance of a test by the scientifi c community, although it does not necessarily imply achievement of this goal; appraisal of an in vitro method without the demand that the process be complete
Assessment In toxicology, the appraisal of a test with respect to its worth or value

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 305
Many of the pioneering projects that initiated the development of in vitro methods did not fulfi ll all of the criteria discussed above for optimal validation (see References). With the exception of acute systemic toxicity studies, most laboratories used many different compounds without regard to grouping chemicals in particular classes. In some studies, however, the numbers represent mixtures of products and materials that are impossible to request for retesting, while other reports did not present all the raw data. In many studies the in vitro test results were directly compared to dose-defi ned, gross animal toxicity data. For instance, comparisons of Draize test scores for validation against in vitro eye irritancy data only permitted rank correlations, thus negating a necessary in vitro/in vivo method of comparison (relevance). Some of the in vitro tests were also scored in a similar manner as rank correlations, which makes the linear correlation approach diffi cult.
With the exception of tests for local irritancy, validation by individual laboratories has not gained general acceptance for any other method. Nor have they replaced any currently accepted analogous animal toxicology test. As mentioned above, the explanation may reside in the lack of resources available to the smaller validation programs.
Table 22.2 presents some of the early approaches to the validation process and com-pares them to current methods and conditions necessary for refi ning validation programs. Three important considerations, which may determine the results of laboratory validation studies, are also discussed below.
Selection of Chemicals
The selection of chemicals used in a study is critical to the analysis of the results of any vali-dation program. A large number of chemicals are chosen to represent as many relevant chem-ical and therapeutic classes. The substances are not selected based only on their physical
TABLE 22.2Early and Current Approaches Aimed at Refining and Improving Validation Studies
Criteria Early approaches Recent, advanced approaches
Chemicals Few chemicals or from various classes Large number of chemicals from relevant classes
Water soluble and volatile chemicals; easy compatibility
Randomly selected chemicals
Proprietary formulations Defi ned chemicals from referenced databases
Data In vivo data with known toxicities from various protocols
In vivo data with known toxicities from similar or same protocols
Blind in vivo data Coded substancesComparisons to either animal or
human toxicitiesComparisons to both animal
and human toxicitiesStatistical
analysisSemi-quantitative in vitro comparisons Parallel linear comparisonsCorrelation, ad hoc analysis Correlation + toxicokinetic information
Laboratory evaluations
One or multi-laboratory evaluation of reliability
Multi-laboratory evaluation of reliability and relevance
Multicenter validation for single reliable tests (multivariate analysis)
Multicenter validation for selection of a battery of reliable tests (multivariate analysis)

306 PRINCIPLES OF TOXICOLOGY TESTING
properties in solution or their presumed cytotoxic action in vivo, for the results obtained may risk a technical bias toward the in vitro method. The dependable way to secure an unbiased selection is to devise a scheme for random selection from a chemical registry (e.g., the Reg-istry of Cytotoxicity, ICCVAM, or the NIOSH Registry). A procedure that is validated for a limited number of classes of chemicals is probably not of much practical value. Another prerequisite for a convincing validation study is to use defi ned chemicals (that is, by their Chemical Abstracts Service [CAS] formulas or their generic names) rather than proprietary mixtures or coded industrial products. Studies performed with defi ned chemicals allow for inter-laboratory testing and validation, thus permitting scientifi c legitimacy. In accordance with the selection of the chemicals and their labeling schema, presentation of the data is also important. Results of evaluation that are published without presentation of a tabulated data-base, for instance, risks irrelevant application and inconsistency with the target objectives.
Comparison to In Vivo Data
A second important factor in the validation program is the selection and comparison to analogous in vivo data. It is desirable to compare in vitro toxicity test results that have accompanying toxicokinetic parameters with in vivo toxicity data. Historically, earlier studies compared cell toxicity with animal LD50s and found relatively good correlations, indicating comparability of cell toxicity with rodent lethality.
Few studies are concerned with the prediction of acute human toxicity by animal lethality tests, or the prediction of human ocular and skin irritancy by the Draize technique. It is gener-ally accepted that these correlations are low, and that the poor comparisons are due to species differences. Ideally, in order to introduce an alternative in vitro test method for practical use as a procedure for screening for human toxicity, the validation step must use human toxicity data as the primary reference. The problem with human toxicity data, however, is its availability, coupled with the uneven and inconsistent quality of toxicity reference points that are available. Without a standardized database of these values, the in vitro methods are less reliable than the corresponding animal protocols used for comparison. Interestingly, the introduction of the EDIT program in 2000, that followed the successful completion of the MEIC program, pro-poses the compilation of a human toxicity database for comparison with alternative in vitro methods. The program, however, was replaced in 2008 by ACuteTox, as an integrative approach for prediction of acute human toxicity (see Suggested Readings).
It is now accepted that correlations between in vitro and in vivo toxicology testing for eye irritancy for different classes of chemicals depends on toxicokinetic information avail-able for that category. Thus, the addition of such information to the test batteries, such as the inclusion of absorption and elimination rates of chemicals, will improve future attempts at correlation. If animal LD50s or TD50s are used as reference points, the question of the precision of the animal method, however, and the variability introduced with the species gap, compounds the reliability of the cytotoxicity test. Proper use of animal data relies on comparisons to the human data for the same chemicals used in the study, thus establishing a baseline for determining if the in vitro method is comparable to the animal results. Ironi-cally, this may be construed as validation of animal tests, paradoxically included in the statistical analysis.
The bulk of available human data is currently obtained from selected references and poi-son control centers and is compiled from clinical case studies, hospital admissions, and emer-gency department visits. Although this information is not acquired systematically, it represents the most reliable source of human toxicity data available for commonly encountered chemi-cals. Thus, the clinical information is used as a basis for comparison against in vitro values.

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 307
Another source of reliable human toxicology data is generated through the testing on volun-teers for some types of toxicity, such as skin and eye irritancy. Human volunteers for skin irritation testing produces concentration–effect curves for fi xed end points, while in the case of eye irritancy, testing is limited to a minimal response (redness, itchiness).
Comparison to Other In Vitro Protocols
The third important factor for consideration in a validation program is the comparison to other in vitro data. Historically, the validation of the mechanistic mutagenicity tests was based on a qualitative in vitro/in vivo comparison, primarily because of the diffi culty associated with establishing a quantitative correlation. All in vitro tests of acute systemic toxicity, how-ever, are quantitative and, above all, are thought to simulate quantitative interactions between chemicals and cellular components in vivo. The tests therefore are validated quantitatively by linear regression analysis or similar statistical methods. This analysis considers the different mechanisms of toxicity and cellular targets associated with individual chemicals and includes categorization of outlines in the design of the validation program. For example, linear regres-sion analysis technically requires exact toxicity data without cutoff results—that is, an IC
50
value stated to be greater than a baseline value is not meaningful for the analysis.To date, most individual validation studies compare the results of in vitro toxicity tests
directly with some form of animal toxicity, such as acute systemic toxicity and skin or eye irritancy. Similarly, most tests are developed to account for only one of many interactions involved in the systemic toxic effect in animals, including toxicity to targets not measured by the test, and toxicokinetics (absorption, distribution, metabolism, and elimination). When the results from single tests are compared with general systemic toxicity, the com-parison is limited to a minor part of the general toxic phenomenon measured by the test, even if this determination is not obscured by toxicokinetic phenomenon. This type of vali-dation selectively discards procedures that measure relatively rare effects and, furthermore, cannot discriminate between important supplementary tests and others with similar infor-mation. In addition, it is erroneous to compare in vitro test results to doses administered in whole animals, whether local or systemic. Instead, comparisons to tissue concentrations can be realized if toxicokinetic data are incorporated in the analysis, or if the animal doses are converted to equivalent human toxicity data (see Chapter 21, Experimental Design and Statistics, Calculation of Dosage Range, HETC).
CURRENT VALIDATION PROGRAMS
HISTORY
The practical and economic advantages of centrally organized multi-laboratory validation programs have diminished the impetus to perform individual laboratory studies. Although individual laboratory validation studies maintain a competitive edge, especially with the comparatively higher fl exibility afforded to smaller operations, it appears that corresponding multi-laboratory approaches are more scientifi cally and economically feasible to satisfy regulatory requirements.
Early on, organized collaborative validation efforts enjoyed several advantages in com-parison to individual laboratory studies. Such multicenter validation was believed to enhance the ideas launched by the multi-laboratory evaluations of the reliability of isolated methods, performed in the 1960s. In its present form, multicenter programs consist of centrally directed testing of the same reference chemicals in many test systems and/or laboratories,

308 PRINCIPLES OF TOXICOLOGY TESTING
with the objective of evaluating both relevance and reliability of methods. Consequently, the development of multicenter policies and procedures is analogous to other important aspects of individual laboratory validation guidelines, such as improvement in the selection of chemicals, reference to in vivo data, and the methods of correlation.
To date, many proposed alternative in vitro toxicology tests in the USA have accumu-lated in the scientifi c literature and regulatory platforms without the benefi t of arriving at general acceptance. The situation is due in part to the diffi cult tasks and characteristics associated with introduction of a system from individual laboratories. The organization of multi-laboratory validation programs is prompted by in vitro toxicologists as a possi-ble remedy for the problem. In addition, public interest groups, academic, industrial and government agencies in the EU and USA, are actively engaged in the promotion of the development of in vitro toxicology tests by organized, well planned programs. Some organizations, such as FRAME (Fund for Replacement of Animals in Medical Experi-ments, UK), CAAT (Center for Alternatives to Animal Testing, Baltimore), and ERGATT (European Research Group for Alternatives to Toxicity Testing), were among the original organizations to coordinate efforts into proposals for future multicenter validation. The fi rst proposed multicenter programs were outlined by FRAME, which was soon followed by special conferences on validation arranged by the CAAT group. Later, the CAAT and ERGATT centers, in collaboration with FRAME, arranged a consensus conference on validation, the result of which was the development of a set of guidelines for validation of toxicity tests. The CAAT/ERGATT conference was followed by the ERGATT/CEC (Commission of the European Communities) conference on procedures for acceptance of validated tests. Thus, these efforts prompted the recognition of the importance of orga-nized and focused multi-laboratory validation programs.
ADVANTAGES OF ORGANIZED MULTI-LABORATORY VALIDATION PROGRAMS
The advantages of conducting multicenter validation programs to facilitate regulatory accep-tance of an alternative testing strategy include:
1. the evaluation of reliability of methods is performed in parallel with the evaluation of relevance;
2. the acceptance of results improves, compared with introduction of results from individual laboratories, if an independent group not involved in assay development has selected the chemicals;
3. multi-laboratory validation is considerably more economical than individual stud-ies because efforts and resources used to select chemicals and generate in vivo data, including the planning and execution of comparisons, is shared among laboratories and coordinated by a central organizing headquarter;
4. accessible databases for validation of relevance are established, thus fostering the simultaneous evaluation of many protocols, and limiting the possibility of omitting a valuable approach;
5. organized multi-laboratory studies validate the predictive ability of combined methods, facilitated by statistical multivariate analysis;
6. organized multi-laboratory programs effectively use models of various types of ani-mal and human toxicity, based on knowledge of the most critical targets for toxicity, mechanisms of action, and toxicokinetic data, and accordingly determine if addi-tional data is needed to contribute to parts of these models;

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 309
7. these programs model various in vitro methods and data as they are received, according to their application to different types of toxicity, such as acute systemic, ocular and skin irritancy, resulting in the discovery of distinct in vitro models for screening toxicity; and
8. comparison of results of different laboratories, using the same set of reference chemicals in different in vitro toxicity tests, with different targets and criteria, dis-tinguish differential application of contributed cytotoxicity studies.
CURRENT ORGANIZATIONS OR PROGRAMS DEDICATED TO THE DEVELOPMENT OF ALTERNATIVE MODELS
Several US, EU, and other international regulatory and scientifi c organizations are dedicated to the development of alternative models to animal toxicology testing. A summary of these organizations are listed in Table 22.3, and further described below.
The Belgium Platform for Alternative Methods (BPAM) was established in 1997 in Brussels. The main focus of BPAM is the development and validation of alternative methods.
TABLE 22.3Current Regulatory and Scientific Organizations Dedicated to the Development of Alternative Models to Animal Toxicology Testinga
OrganizationRepresentative
Country Description
BPAM Belgium Platform for Alternative Methods Belgium Regulatory, administrativeBUAV British Union for the Abolition of Vivisection UK Regulatory, administrativeCAAT Johns Hopkins Center for Alternatives to Animal
TestingUS Academic
ECVAM European Center for the Validation of Alternative Methods
EU Regulatory
EDIT Evaluation-Guided Development of In Vitro Tests Sweden Multi-laboratory programERGATT European Research Group for Alternatives to
Toxicity TestingEU Regulatory
FRAME Fund for the Replacement of Animals in Medical Experiments
UK Private foundation
ICCVAM Interagency Coordinating Committee on the Validation of Alternative Methods
US Regulatory, multi-laboratory program
IIVS Institute for In Vitro Sciences US Private, non-profi tIVAM In Vitro & Alternative Methods Specialty Section
of the US Society of ToxicologyUS US national toxicology
societyJaCVAM Japanese Center for the Validation of Alternative
MethodsJapan Regulatory, multi-laboratory
programMEIC Multicenter Evaluation of In Vitro Cytotoxicity Scandinavian Multi-laboratory programNCA Netherlands Center for Alternatives to Animal Use Netherlands Regulatory, administrativeNICA Nordic Information Center for Alternative
MethodsSweden Coordination of the EDIT
projectZEBET Center for Documentation and Evaluation of
Alternative Methods to Animal ExperimentsEU Regulatory
ZET Center for Alternative and Complementary Methods in Animal Experiments
Austria Administrative
aWebsites are listed in Review Articles.

310 PRINCIPLES OF TOXICOLOGY TESTING
The British Union for the Abolition of Vivisection (BUAV) has focused on the potential of in vitro methods to replace in vivo experiments. Its report, The Way Forward-A Non-Animal Testing Strategy for Chemicals, contains a review of the current status of in vitro methods and suggests that the EU provides suffi cient resources for the vast validation of in vitro methods in order to replace in vivo experiments.
The Johns Hopkins Center for Alternatives to Animal Testing (CAAT) was founded in the USA in 1981, with a grant from the Cosmetic, Toiletry and Fragrance Association (CTFA). Its main interest is in the administrative and educational promotion and support of techniques that replace the use of animals, reduce the need for animals for particular tests, or refi ne a technique in order to relieve or eliminate the amount of pain endured by an animal.
The European Center for the Validation of Alternative Methods (ECVAM) was estab-lished in 1986 by the EU. ECVAM coordinates its efforts or collaborates with several EU and US regulatory agencies for the development and regulatory acceptance of in vitro tests that refi ne, reduce, or replace the use of laboratory animals.
The European Research Group for Alternatives in Toxicity Testing (ERGATT) was estab-lished in 1985 in the Netherlands. One of the earliest founding organizations for the development and evaluation of in vitro methods, the group is represented on the ECVAM advisory committee and is involved with ECVAM and ICCVAM on the validation of non-animal tests in regulatory toxicity testing. In addition, ERGATT is involved in establishing risk assessment methods.
The Fund for the Replacement of Animals in Medical Experiments (FRAME) was established in the UK in 1969 to promote the replacement of in vivo experiments. In 1980, INVITTOX, a web database collection of protocols for in vitro methods, was established by FRAME and is now part of ECVAM’s Scientifi c Information Service. FRAME partici-pated in the validation study of the 3T3 NRU phototoxicity test in 1997 that was accepted by the EU in 2000, and which is undergoing validation by ICCVAM/NICEATM (see below). It also sponsors and publishes a leading toxicology journal for the development of alternative models, Alternatives to Laboratory Animals (ATLA).
The Interagency Coordinating Committee on the Validation of Alternative Methods ( ICCVAM) was established in 1997 in the USA as a standing committee by the Director of the National Institute of Environmental Health Sciences (NIEHS). By 2000, it received permanent status by US Congressional authorization. As a regulatory agency, its function is the organization, development and validation of alternative methods as well as the establish-ment of criteria and processes for the regulatory acceptance of in vitro methods. In 1998, the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) was established. In addition, ICCVAM and NICEATM work collaboratively, along with scientifi c and public advisory committees, such as the Sci-entifi c Advisory Committee for Alternative Toxicological Methods (SACATM–ICCVAM), for the establishment of methods that are predictive for adverse human toxicological effects.
The Institute for In Vitro Sciences (IIVS) was founded in 1997 in the USA. The goals of the non-profi t organization focus on evaluation of testing for skin and ocular toxicity, bioassay development, and the validation of in vitro methods.
The In Vitro and Alternative Methods (IVAM) Specialty Section within the US Society of Toxicology (US SOT) was founded in 1994. This special internal group is interested in the application of in vitro techniques to problems of cellular toxicity, understanding basic cellular processes involved in the induction of adverse outcomes of specifi c organs and the whole animal, and the development of cellular and sub-cellular systems to predict toxicity in vivo for risk assessment purposes. The group also supports in vitro test validation and all aspects of test development and acceptance for scientifi c or regulatory purposes.
The Multicenter Evaluation of In Vitro Cytotoxicity (MEIC) was initiated by Björn Ekwall in 1989 and was organized by a committee of the Scandinavian Society for Cell

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 311
Toxicology (SSCT). From 1989 to 1996, international laboratories voluntarily participated to test 50 representative chemicals with known and varied human and animal toxicity. The continuing and fi nal results of the MEIC studies were published in a series of reports indi-cating that IC50 values from 68 independent cytotoxicity assays, in combination with tox-icokinetic data, predicted human lethal dosage as effi cient as rodent LD50 values. These studies also suggested that primary cultures and human cell lines are more sensitive to chemical toxicity than continuous animal cell lines.
By 1998, the MEIC project was extended into the Evaluation-Guided Development of In Vitro Tests (EDIT) in order to organize global collaboration of laboratories for the estab-lishment of new in vitro tests. The EDIT project incorporated toxicokinetics studies in order to obtain a battery of tests for tier testing in animal toxicology. The Nordic Informa-tion Center for Alternative Methods (NICA) coordinated the EDIT project and was estab-lished in Sweden to promote the use of in vitro tests for regulatory toxicology. More recently, the ACuteTox program was developed as an integrated EU project under the Sixth Framework Program. The objective of the Program is to optimize, and prepare for valida-tion, an in vitro test strategy for predicting human acute toxicity. The aim is to demonstrate that animal tests for acute systemic toxicity, currently used for regulatory purposes, could be replaced by a combination of alternative assays (see Suggested Readings).
The Netherlands Center for Alternatives to Animal Use (NCA) was established in 1994. The main focus of this group is the development, acceptance, and promotion of alter-native methods in the Netherlands.
The Center for Documentation and Evaluation of Alternative Methods to Animal Experiments (ZEBET) was established in Germany in 1989 in order to reduce and replace animal experimentation with alternative models. ZEBET provides a database of 347 chem-icals for in vitro cytotoxicity data as well as their corresponding in vivo acute toxicity data. Known as the Registry of Cytotoxicity (RC) database, the information is used to develop regression models to determine starting doses for in vivo lethality. Moreover, ZEBET undertakes validation studies of in vitro methods in cooperation with ECVAM and the European Cosmetic, Toiletry and Perfumery Industry (COLIPA).1
The Center for Alternative and Complementary Methods in Animal Experiments (ZET) was established in 1996 in Austria. The main aim of the center is the development, improve-ment and validation of alternative methods using the “3 R’s” approach. ZET has released a series of publications, based on its sponsored international congresses, on alternative meth-ods to animal testing.
ORGANIZED PROGRAMS
Current or completed multi-laboratory validation studies of alternative in vitro ocular and dermal toxicology testing are discussed in chapter 15. These and other second generation validation studies are facilitated through collaborative international cooperation, particularly through the efforts of ICCVAM, NICEATM, ECVAM, and ZEBET. These partnerships pro-vide the foundation for validating alternative methods, as well as promotion and encourage-ment for the harmonization of scientifi c approaches to validation and review. In addition, both ICCVAM and ECVAM have outlined key objectives—to stimulate development of test meth-ods and testing strategies in prioritized areas by individual laboratories, and to facilitate the nomination of promising test methods for appropriate regulatory translation. Tables 22.4–22.8 summarize current in vitro validation programs, proposals, test method evaluations, and test-ing strategies currently sponsored by ICCVAM, from 1998 to date.
1COLIPA has since changed its name to Cosmetics Europe—The Personal Care Association.

312 PRINCIPLES OF TOXICOLOGY TESTING
TABLE 22.5Summary of Validation Programs and Evaluation of Testing Strategies and Test Methods Considered by ICCVAM-NICEATM: 2007–2009a
DescriptionNIH Publication
Number Year
The Reduced Murine Local Lymph Node Assay 09-6439 2009Murine Local Lymph Node Assay 09-7357 2009Biennial Progress Report: Interagency Coordinating Committee on the Validation
of Alternative Methods: 2006–200708-6529 2008
Validation Status of Five In Vitro Test Methods Proposed for Assessing Potential Pyrogenicity of Pharmaceuticals and Other Products
08-639108-6392
2008
Report on the ICCVAM-NICEATM/ECVAM Scientifi c Workshop on Alternative Methods to Refi ne, Reduce or Replace the Mouse LD50 Assay for Botulinum Toxin Testing
08-6416 2008
The NICEATM-ICCVAM Five-Year Plan (2008–2012) 08-6410 2008ICCVAM Test Method Evaluation Report: In Vitro Cytotoxicity Test for Estimating
Starting Doses for Acute Oral Systemic Toxicity Tests07-4519 2007
In Vitro Cytotoxicity Test Methods for Estimating Acute Oral Systemic Toxicity 07-4518 2007ICCVAM Test Method Evaluation Report – In Vitro Ocular Toxicity Test Methods for
Identifying Severe Irritants and Corrosives07-4517 2007
aSee http://iccvam.niehs.nih.gov/docs. Arranged according to NIH Publication number and year accepted.Abbreviation: BRD, background review document.
TABLE 22.4Summary of Validation Programs and Evaluation of Testing Strategies and Test Methods Considered by ICCVAM-NICEATM: 2010–2012a
DescriptionNIH Publication
Number Year
ICCVAM Biennial Progress Report 2010–2011 12-7873 2012Usefulness and Limitations of the Murine Local Lymph Node Assay for Potency
Categorization of Chemicals Causing Allergic Contact Dermatitis in Humans11-7709 2011
Recommendation to Discontinue Use of the Low Volume Eye Test for Ocular Safety Testing
10-7515 2010
Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products
10-7553 2010
Recommendations for Routine Use of Topical Anesthetics, Systemic Analgesics, and Humane Endpoints to Avoid or Minimize Pain and Distress in Ocular Safety Testing
10-7514 2010
Proposed In Vitro Testing Strategy for US Environmental Protection Agency: Ocular Hazard Classifi cation and Labeling of Antimicrobial Cleaning Products
10-7513 2010
Using the Murine Local Lymph Node Assay for Testing Pesticide Formulations, Metals, Substances in Aqueous Solutions, and Other Products
10-7512 2010
The Murine Local Lymph Node Assay: DA: A Nonradioactive Alternative Test Method to Assess the Allergic Contact Dermatitis Potential of Chemicals and Products
10-7551 2010
The Murine Local Lymph Node Assay: BrdU-ELISA 10-7552 2010ICCVAM 2008–2009 Biennial Report 10-7612 2010
asee http://iccvam.niehs.nih.gov/docs. Arranged according to NIH Publication number and year accepted.Abbreviation: BRD, background review document.

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 313
TABLE 22.6Summary of Validation Programs and Evaluation of Testing Strategies and Test Methods Considered by ICCVAM-NICEATM: 2004–2006a
DescriptionNIH Publication
Number Year
Biennial Progress Report 2004–2005: Interagency Coordinating Committee on the Validation of Alternative Methods
06-4516 2006
Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Isolated Rabbit Eye (IRE) Test Method
06-4514 2006
Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Isolated Chicken Eye (ICE) Test Method
06-4513 2006
Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Hen’s Egg Test - Chorioallantoic Membrane (Het-CAM) Test Method
06-4515 2006
Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Bovine Corneal Opacity and Permeability (BCOP) Test Method: Volumes 1& 2
06-4512 2006
ICCVAM Guidelines for the Nomination and Submission of New, Revised and Alternative Test Methods
03-4508 2004
Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion 04-4510 2004
aSee http://iccvam.niehs.nih.gov/docs. Arranged according to NIH Publication number and year accepted.Abbreviation: BRD, background review document.
TABLE 22.7Summary of Validation Programs and Evaluation of Testing Strategies and Test Methods Considered by ICCVAM-NICEATM: 2001–2003a
DescriptionNIH Publication
Number Year
ICCVAM Biennial Progress Report 2002–2003 04-4509 2003Evaluation of In Vitro Test Methods For Detecting Potential Endocrine Disruptors: Estrogen
Receptor and Androgen Receptor Binding and Transcriptional Activation Assays03-4503 2003
Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays
03-4504 2003
Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Transcriptional Activation Assays
03-4505 2003
Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Androgen Receptor Binding Assays
03-4506 2003
Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Androgen Receptor Transcriptional Activation Assays
03-4507 2003
ICCVAM Evaluation of EPISKIN™, EpiDerm™ (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) Assay – In Vitro Test Methods for Assessing the Dermal Corrosivity Potential of Chemicals
02-4502 2002
The Revised Up-and-Down Procedure: A Test Method for Determining the Acute Oral Toxicity of Chemicals; Volumes 1 & 2
02-4501 2001
EPISKINTM, EpiDermTM(EPI-200), and Rat Skin Transcutaneous Electrical Resistance (TER) – In Vitro Test Methods for Assessing the Dermal Corrosivity Potential of Chemicals
BRD 2001
ICCVAM Recommendations on In Vitro Methods for Assessing Acute Systemic Toxicity 01-4499 2001
aSee http://iccvam.niehs.nih.gov/docs. Arranged according to NIH Publication number and year accepted.Abbreviation: BRD, background review document.

314 PRINCIPLES OF TOXICOLOGY TESTING
SUMMARY
Current and future validation should not be hampered by theoretical considerations of the best way to achieve regulatory acceptance. The refi nement of the validation programs should be approached as a set of generally applicable rules. The most effective future validation will probably require studies directed at all levels, including the efforts of individual laboratories, reliability oriented multi-laboratory programs, and multiple-method multi-laboratory per-spectives focused on cell based toxicology. Combinations of different objectives will be evaluated in different studies. For instance, several methods could be analyzed for reliability in some programs, while simultaneously evaluated for specifi c or general relevance in other programs. It is conceivable that it may not be possible to combine some objectives in a single study, such as selection of chemicals, choice of in vivo data, and demands on correlative methods. Human data on acute systemic toxicity may not be compatible with a particular selection of some chemicals. There may necessarily be some confl ict of goals in the selec-tion of optimum objectives in the scheme of validation proposals.
It is important to understand that the progress of in vitro cytotoxicology testing relies on the continuous development of better strategies to evaluate new tests, rather than as a set of infl exible guidelines for the inclusion or exclusion of protocols. Thus, efforts to evaluate the scientifi c integrity of methods for validation purposes should be administered by inde-pendent investigative groups, rather than government sponsored organizations. The groups must be free from vested interests, with their services directed toward the development of scientifi cally valid programs.
SUGGESTED READINGS
Arias-Mendoza F, Zakian K, Schwartz A, et al. Methodological standardization for a multi-institutional in vivo trial of localized 31P MR spectroscopy in human cancer research. In vitro and normal volunteer studies. NMR Biomed 2004; 17: 382.
Balls M, Combes R. The need for a formal invalidation process for animal and non-animal tests. Altern Lab Anim 2005; 33: 299.
TABLE 22.8Summary of Validation Programs and Evaluation of Testing Strategies and Test Methods Considered by ICCVAM-NICEATM: 1997–2000a
DescriptionNIH Publication
Number Year
FETAX - Frog Embryo Teratogenesis Assay - Xenopus BRD 2000Evaluation of the Validation Status of Toxicological Methods 99-4496 1999The Murine Local Lymph Node Assay: A test Method for Assessing the Allergic
Contact Dermatitis Potential of Chemicals/Compounds.99-4494 1999
Corrositex®: An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals
99-4495 1999
Validation and Regulatory Acceptance of Toxicological Test Methods: A Report of the ad hoc Interagency Coordinating Committee on the Validation of Alternative Methods)
97-3981 1997
aSee http://iccvam.niehs.nih.gov/docs. Arranged according to NIH Publication number and year accepted.Abbreviation: BRD, background review document.

STANDARDIZATION AND VALIDATION OF ALTERNATIVE METHODS 315
Calabro AR, Gazarian DI, Barile FA. Effect of metals on β-actin and total protein synthesis in cul-tured human intestinal epithelial cells. J Pharmacol Toxicol Methods 2011; 63: 47.
Combes R, Barratt M, Balls M. An overall strategy for the testing of chemicals for human hazard and risk assessment under the EU REACH system. Altern Lab Anim 2006; 34: 15.
Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modifi cation path-ways concurrently in mouse embryonic stem cells. Toxicol Lett 2012; 212: 169.
Gartlon J, Kinsner A, Bal-Price A, et al. Evaluation of a proposed in vitro test strategy using neu-ronal and non-neuronal cell systems for detecting neurotoxicity. Toxicol In Vitro 2006; 20: 1569.
Hoffmann S, Hartung T. Designing validation studies more effi ciently according to the modular approach: retrospective analysis of the EPISKIN test for skin corrosion. Altern Lab Anim 2006; 34: 177.
Prieto P, Baird AW, Blaauboer BJ, et al. The assessment of repeated dose toxicity in vitro: a proposed approach. The report and recommendations of ECVAM workshop 56. Altern Lab Anim 2006; 34: 315.
Spielmann H, Seiler A, Bremer S, et al. The practical application of three validated in vitro embryo-toxicity tests. The report and recommendations of an ECVAM/ZEBET workshop (ECVAM workshop 57). Altern Lab Anim 2006; 34: 527.
REVIEW ARTICLES
Balls M, Clothier R. FRAME and the validation process. Altern Lab Anim 2009; 37: 631.Barile FA. Continuous cell lines as a model for drug toxicity assessment. In: Castell JV, Gómez-Lechón
M-J, eds. In Vitro Methods in Pharmaceutical Research. UK: Academic Press, 1997.Barile FA. Unifying the Effort Behind In Vitro Alternative Method Development. The Way Forward
2008: [Available from: AltTox.org, sponsored by Proctor & Gamble Company (P&G) and The Humane Society of the United States (HSUS)].
Barile FA. Validating and troubleshooting ocular in vitro toxicology tests. J Pharmacol Toxicol Meth-ods 2010; 61: 136.
Clemedson C, McFarlane-Abdulla E, Andersson M, et al. MEIC evaluation of acute systemic toxic-ity. Part I. Methodology of 68 in vitro toxicity assays used to test the fi rst 30 reference chemicals. Altern Lab Anim 1996; 24: 249.
Clemedson C, McFarlane-Abdulla E, Andersson M, et al. MEIC evaluation of acute systemic toxic-ity. Part II. In vitro results from 68 toxicity assays used to test the fi rst 30 reference chemi-cals and a comparative cytotoxicity analysis. Altern Lab Anim 1996; 24: 273.
Clemedson C, Barile FA, Ekwall B, et al. MEIC evaluation of acute systemic toxicity. Part III. In vitro results from 16 additional methods used to test the fi rst 30 reference chemicals and a com-parative cytotoxicity analysis. Altern Lab Anim 1998; 26: 93.
Clemedson C, Andersson M, Aoki Y, et al. MEIC evaluation of acute systemic toxicity. Part IV. In vitro results from 67 toxicity assays used to test reference chemicals 31–50 and a compara-tive cytotoxicity analysis. Altern Lab Anim 1998; 26: 131.
Clemedson C, Barile FA, Chesné C, et al. MEIC evaluation of acute systemic toxicity. Part VII. Pre-diction of human toxicity by results from testing of the fi rst 30 reference chemicals with 27 further in vitro assays. Altern Lab Anim 2000; 28: 161.
Clemedson C. The European ACuteTox project: a modern integrative in vitro approach to better pre-diction of acute toxicity. Clin Pharmacol Ther 2008; 84: 200.
Clothier RH. Phototoxicity and acute toxicity studies conducted by the FRAME Alternatives Labora-tory: a brief review. Altern Lab Anim 2007; 35: 515.
Curren R, Bruner L, Goldberg A, Walum E. 13th meeting of the Scientifi c Group on Methodologies for the safety evaluation of chemicals (SGOMSEC): validation and acute toxicity testing. Environ Health Perspect 1998; 106: 419.
Edler L, Ittrich C. Biostatistical methods for the validation of alternative methods for in vitro toxicity testing. Altern Lab Anim 2003; 31: 5.

316 PRINCIPLES OF TOXICOLOGY TESTING
Ekwall B. Overview of the fi nal MEIC results: II. The in vitro/in vivo evaluation, including the selec-tion of a practical battery of cell tests for prediction of acute lethal blood concentrations in humans. Toxicol In Vitro 1999; 13: 665.
Ekwall B, Barile FA. Standardization and validation. In: Introduction to In Vitro Cytotoxicology: Mechanisms and Methods, Chapter 11. Boca Raton, FL: CRC Press, 1994: 189.
Ekwall B, Clemedson C, Crafoord B, et al. MEIC evaluation of acute systemic toxicity. Part V. Rodent and human toxicity data for the 50 reference chemicals. Altern Lab Anim 1998; 26: 569.
Halle W, Spielmann H, Liebsch M. Prediction of human lethal concentrations by cytotoxicity data from 50 MEIC chemicals. ALTEX 2000; 17: 75.
Hellberg S, Eriksson L, Jonsson J, et al. Analogy models for prediction of human toxicity. Altern Lab Anim 1990; 18: 103.
Hill A, Mesens N, Steemans M, et al. Comparisons between in vitro whole cell imaging and in vivo zebrafi sh-based approaches for identifying potential human hepatotoxicants earlier in phar-maceutical development. Drug Metab Rev 2012; 44: 127.
Hollert H, Giesy J. The OECD validation program of the H295R steroidogenesis assay for the iden-tifi cation of in vitro inhibitors and inducers of testosterone and estradiol production. Phase 2: Inter-laboratory pre-validation studies. Environ Sci Pollut Res Int 2007; 14: 23.
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). Biennial Progress Report of the National Toxicology Program. NIH Publication No. 04-4509. Research Triangle Park, NC: National Institute of Environmental Health Sciences, 2003.[Available from: http://iccvam.niehs.nih.gov/about/annrpt/bienrpt044509.pdf]
Kinsner-Ovaskainen A, Maxwell G, Kreysa J, et al. Report of the EPAA-ECVAM workshop on the validation of Integrated Testing Strategies (ITS). Altern Lab Anim 2012; 40: 175.
Konsoula Z, Barile FA. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 2012; 66: 215.
Louekari K. Status and prospects of in vitro tests in risk assessment. Altern Lab Anim 2004; 32: 431.Paris M, Strickland J, Stokes W, et al. Phase I and II results of a validation study to evaluate in vitro
cytotoxicity assays for estimating rodent and human acute systemic toxicity. 43rd Annual Meeting of the Society of Toxicology, Toxicol. Sci, Abstract No. 240. 2004.
Rispin A, Stitzel K, Harbell J, Klausner M. Ensuring quality of in vitro alternative test methods: cur-rent practice. Regul Toxicol Pharmacol 2006; 45: 97.
Spielmann H, Genschow E, Leibsch M, Halle W. Determination of the starting dose for acute oral toxicity (ld50) testing in the up-and-down procedure (UDP) from cytotoxicity data. Altern Lab Anim 1999; 27: 957.
Spielmann H, Grune B, Liebsch M, et al. Successful validation of in vitro methods in toxicology by ZEBET, the National Centre for Alternatives in Germany at the BfR (Federal Institute for Risk Assessment). Exp Toxicol Pathol 2008; 60: 225.
Ubels JL, Clousing DP. In vitro alternatives to the use of animals in ocular toxicology testing. Ocul Surf 2005; 3: 126.
Ukelis U, Kramer PJ, Olejniczak K, Mueller SO. Replacement of in vivo acute oral toxicity studies by in vitro cytotoxicity methods: opportunities, limits and regulatory status. Regul Toxicol Pharmacol 2008; 51: 108.
Vanparys P, Corvi R, Aardema MJ, et al. Application of in vitro cell transformation assays in regula-tory toxicology for pharmaceuticals, chemicals, food products and cosmetics. Mutat Res 2012; 744: 111.

317
23 Applications of alternative models for toxicology testing
INTRODUCTION
In the past, the only systematic attempts to develop alternative models for animal toxicology testing were commandeered by academic endeavors aimed at conducting basic research, supplemented by their efforts at soliciting industrial support for launching in vitro assays. In the main, regulatory agencies displayed a cavalier attitude toward the fi eld, thus discounting the importance of changing perceptions within the pharmaceutical, cosmetic, and toxicology testing industries.
As increasing pressure has mounted from the scientifi c and public interest groups, the fi eld of alternative in vitro methods has witnessed favorable transformations and develop-ments including applications of the technology and its introduction into the regulatory arena. Although it is arguable that progress is unhurried and not extensive, consider the historical status of in vitro methods 20 years ago and the formidable obstacles that have been overcome. Several developments in the regulatory arena are presented as evidence of signifi cant advances in this direction including (1) the promulgation of the Organization for Economic Cooperation and Development (OECD) regulations in the European Union (EU), (2) the establishment of the Interagency Coordinating Committee on the Validation of Alter-native Methods (ICCVAM), National Toxicology Program (NTP) Inter-agency Center for the Evaluation of Alternative Toxicological Methods (NICEATM), and European Centre for the Validation of Alternative Methods (ECVAM) agencies and the Scientifi c Advisory Committee on Alternative Toxicological Methods advisory committee, (3) the founding of international societies for fostering the development of alternative methods, and (4) advances arising from workshops and validation programs.
PROMULGATION OF OECD AND FRAME REGULATIONS BY THE EUROPEAN UNION
The forerunner of the OECD was the Organisation for European Economic Cooperation that was formed to administer American and Canadian aid under the Marshall Plan for recon-struction of Europe after World War II. Since 1961, OECD’s goals have been the building of strong economies in its member countries and promoting regulatory and economic reforms in industrialized and developing nations. It also fosters policy recommendations, country reviews, thematic discussions, and cooperation with non-member countries.
OECD introduces and promotes guideline lists and reports based on physical chem-istry, ecotoxicology, degradation and accumulation, and short- and long-term toxicology testing by expert groups. It also publishes a series of documents on good laboratory

318 PRINCIPLES OF TOXICOLOGY TESTING
practices1 to ensure that in vitro tests used for regulatory purposes are reproducible, cred-ible, and acceptable.
Other efforts guided by the experiences from validation studies for alternative methods in toxicology and OECD initiatives include concepts of good cell culture practice that aim to defi ne minimum quality standards for in vitro techniques.
The Fund for the Replacement of Animals in Medical Experiments (FRAME) is a reg-istered U.K. charity whose objective is the promotion of alternatives to animal use in research, testing, and education. While it considers that the current scale of animal experi-mentation is unacceptable, it recognizes the need for adequately testing new consumer products, medicines, and industrial and agricultural chemicals to identify potential hazards to human and animal health. Consequently it advocates the “3R” approach (replacement, refi nement,2 and reduction) to animal experimentation and seeks to promote a moderate approach by encouraging realistic considerations of the ethical and scientifi c issues involved in animal toxicology testing.
FRAME recently introduced a number of suggestions for improving the initiatives of the E. U. REACH (registration, evaluation, and authorization of chemicals) system for safety testing and risk assessment, fi rst proposed as a White Paper in 2001. These initiatives considered the scientifi c and animal welfare issues raised by the REACH proposals and resulted in the adoption of legislative actions through the European Council and European Parliament.
ESTABLISHMENT OF ICCVAM, NICEATM, AND ECVAM REGULATORY AGENCIES
Over the past decade, based on provocation and encouragement from scientifi c and public interest groups, regulatory authorities in the United States and EU launched initiatives to validate new and improved toxicological test methods. In the United States, the ICCVAM and its supporting agency NICEATM were established by the federal government to work with test developers and federal agencies to facilitate the validation, review, and adoption of new scientifi cally sound test methods including alternatives that refi ne, reduce, and replace animal use.
ICCVAM was established in 1997 according to the ICCVAM Authorization Act of 20003 by the director of the National Institute of Environmental Health Sciences (NIEHS) to implement NIEHS directives.4 It is a permanent committee composed of representatives from 15 federal regulatory and research agencies and advisory groups. The committee coordinates cross-agency issues relating to development, validation, acceptance, and national and international harmonization of toxicology test methods.
NICEATM was established in 1998 to provide operational support for ICCVAM and carry out committee-related activities such as peer reviews and workshops for test methods of interest to federal agencies. NICEATM and ICCVAM coordinate scientifi c reviews of the validation statuses of proposed methods and provide recommendations regarding their use-fulness to appropriate agencies. Together, the agencies promote the validation and regulatory acceptance of toxicology test methods that are more predictive of adverse human and eco-logical effects than currently available methods and refi ne, reduce, and replace animal use.
1 Advisory document of Working Group on Good Laboratory Practice, The Application of the Principles of GLP to In Vitro Studies, OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring, 2004. 2Revised defi nition includes lessening or eliminating pain. 3 Public Law (P.L.) 106-545. 4 P.L. 103-43 directed NIEHS to develop and validate new test methods and establish criteria and processes for the validation and regulatory acceptance of toxicological testing methods.

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 319
ECVAM was created in the EU in 1992 by a communication from the Commission to the Council and the Parliament in October 19915 that requires the commission and mem-ber states to actively support the development, validation, and acceptance of methods that can reduce, refi ne, or replace the use of laboratory animals. ECVAM’s duties include the following:
1. Coordinating the validation of alternative test methods at the EU level.2. Promoting exchanges of information on the development of alternative test methods.3. Generating, maintaining, and managing a database of alternative procedures.4. Encouraging dialogs among legislators, industries, biomedical scientists, consumer
organizations, and animal welfare groups, with a view to the development, valida-tion, and international recognition of alternative test methods.
Both ICCVAM and ECVAM have adopted similar validation and regulatory accep-tance criteria for the adoption and use of alternative test methods. They have fostered and encouraged international collaborations to facilitate the adoption of new test methods by involving sharing of expertise and data for test method workshops and independent scien-tifi c peer reviews. Among their recent activities, NICEATM and ECVAM initiated a joint international validation study on in vitro methods for assessing acute systemic toxicity. These collaborations are expected to contribute to accelerated international adoption of harmonized new test methods that support improved public health and provide for reduced and more humane use of laboratory animals.
The feasibility and potential benefi ts of such a partnership between two large regula-tory organizations continue to evolve as the development of alternative methodologies pro-gresses. The fear, however, is that this extraordinary increase in the introduction of test methods from the scientifi c and industrial communities and the accompanying pressure to deliberate on and implement the procedures may strain the capacities of such institutions. Consequently, coordination of functions, maintenance of scientifi c standards, and commit-ments to the principles of validation support opportunities for effi cient execution of animal welfare objectives and the implementation of alternative tests on the regulatory agenda.
ESTABLISHMENT OF INTERNATIONAL EFFORTS FOR DEVELOPMENT OF ALTERNATIVE METHODS
Several other supportive agencies, societies, and congresses have assumed more active roles, particularly with fi nancial, political, and scientifi c encouragement to support the humane treatment of animals. Some of these organizations are listed in Table 23.1.
Among these, the In Vitro and Alternative Methods (IVAM) specialty section of the Society of Toxicology (SOT) in the United States is composed of members with expertise in the application of in vitro techniques to problems of cellular toxicity, with a special emphasis on safety evaluation. The interests of the specialty section include studies of the basic cellular processes involved in the induction of adverse outcomes of specifi c organs and the whole animal and the development of simple to complex cellular and subcellular systems to predict toxicity in vivo and to assess risk. IVAM holds regular meetings at the annual SOT congress to discuss topics of interest.
5The communication is a requirement in Directive 86/609/EEC on the protection of animals used for experimen-tal and other scientifi c purposes.

320 PRINCIPLES OF TOXICOLOGY TESTING
WORKSHOPS AND VALIDATION PROGRAMS
The ECVAM validation concept defi ned at two validation workshops held in Amden, Switzerland in 1990 and 1994 takes into account the essential elements of prevalidation and prediction models based on biostatistics criteria. The program has been accepted offi cially by EU member states, OECD, and the regulatory agencies of the United States. The ECVAM validation concept was introduced into the ongoing ECVAM/COLIPA6 validation study (see chap.19) of in vitro phototoxicity tests that ended successfully in 1998.
The 3T3 neutral red uptake in vitro phototoxicity test was the fi rst experimentally vali-dated in vitro toxicity test recommended for regulatory purposes by the ECVAM Scientifi c Advisory Committee and was accepted into EU legislation for testing of new chemicals in 2000. From 1996 to 1998, two in vitro skin corrosivity tests were successfully validated by ECVAM and were also offi cially accepted into EU regulations in 2010. From 1997 to 2000, an ECVAM validation study on three in vitro embryotoxicity tests was successfully com-pleted. The three tests (whole-embryo culture test, the micromass test on limb bud cells, and the embryonic stem cell test ) were encouraged for routine use in laboratories of the European pharmaceutical and chemical industries. Meanwhile, in 2010, the OECD Test Guidelines Program was considering the worldwide acceptance of the validated in vitro phototoxicity and corrosivity tests.
While trying to comply with the regulations established by the U.S. Animal Welfare Act and its amendments (1985), some research laboratories in the United States have been forced to terminate their efforts because of the high cost of maintaining and provid-ing for animals as required by the act. In response, the U.S. Public Health Service estab-lished the Offi ce of Animal Research and the Offi ce of Science Education to promote science education, coordinate policies, and distribute information on the use of animals in research. Historically, the use of animals in biomedical research has provided and will continue to provide valuable information about chemicals, drugs, and products at the vanguard of public health. Consequently, the burden of developing suitable protocols to
6Originally known as The European Cosmetics Association, as of January 2012, COLIPA is now known as Cosmetics Europe-The Personal Care Association
TABLE 23.1Additional Organizations Sponsoring Activities in Support of Humane Treatment of Animals
Organization Origin
ARDF Alternatives Research and Development Foundation Jenkintown, Pennsylvania, USA
EUROTOX Federation of European Toxicologists and European Societies of Toxicology
EU
FRAME Fund for Replacement of Animals in Medical Experiments UKHSUS Humane Society of the United States Washington, DC, USAIFER International Foundation for Ethical Research Chicago, Illinois, USAIVAM In Vitro and Alternative Methods Specialty Section
of the U.S. Society of ToxicologyReston, Virginia, USA
SSCT Scandinavian Society for Cell Toxicology SwedenWCAAUFS World Congress on Alternatives and Animal Use
in the Life SciencesEU

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 321
test the safety and effi cacy of thousands of drugs and chemicals used in medicine or introduced by the chemical industry is incumbent upon the scientifi c community.
SIGNIFICANCE OF ALTERNATIVE MODELS FOR ANIMAL TOXICOLOGY TESTING
IN VITRO TESTS AND THEIR SIGNIFICANCE
No single method is expected to cover the complexity of general toxicity in humans or ani-mals. The response of the mammalian organism to a chemical is known to involve various physiological targets and a variety of complex toxicokinetic factors. Chemicals induce injury through an assortment of toxic mechanisms. In addition, a number of questions have emerged as a result of previous and current validation studies of the prediction systemic and local toxicity by in vitro tests:
1. Are alternative methods capable of fulfi lling the need to model different types of quantitative general toxicity such as acute systemic toxicity or local irritancy? This goal may require several relevant systems such as human hepatocytes, heart, kid-ney, lung, nerve cells, and other cell lines of applicable and relevant importance.
2. Are the results generated from these methods predictive of basal cytotoxicity7 in general and local toxicity in particular?
3. Are quick, simple, and economic in vitro cell systems capable of sustaining the pres-sure and standards of safety and regulation demanded by the needs of public health?
FUNCTIONAL CLASSIFICATION OF CYTOTOXICITY BASED ON IN VITRO METHODOLOGIES
Other than the ability of a chemical to possess genotoxic or carcinogenic potential, cellular toxicity is classifi ed in several ways. One example is the distinction between local and sys-temic toxicity. Other classifi cations include, but are not limited to (1) acute versus chronic toxicity (see below), (2) immediate versus delayed toxicity, (3) diffuse versus targeted toxic-ity, and (4) high-dosage versus low-dosage toxicity. Figure 23.1 depicts the functional and cytotoxic classifi cations of primary toxic mechanisms based on different levels of physio-logic organization affected by chemicals. The ability of a toxic substance to target functional areas correlates this level with the corresponding cytotoxic classifi cation.
Basal Cell Functions
These fundamental processes involve structures and functions common to all cells in the human body and include the processes necessary to maintain cell membrane integrity, mito-chondrial oxidation, translation and transcription, and lysosomal enzyme activity. In cultured cells, these functions are generally delegated to undifferentiated or dedifferentiated cell lines that are maintained in continuous culture over several passages. Some cell lines do not require several passages but may dedifferentiate after several hours in culture. Hepatocytes, for instance, lose their high initial levels of enzyme activities after several hours in culture. These cell lines, however, are not necessarily without specifi c functional characteristics. Thus, most cell lines of mesenchymal origin (fi broblasts), for instance, maintain their protein secretory activities, although many of their original morphological and biochemical features are lost.
7 Toxicity of a chemical that affects basic cellular functions and structures common to all human and animal specialized cells regardless of organ of origin.

322 PRINCIPLES OF TOXICOLOGY TESTING
It must be remembered that these types of continuous cells have been traditionally used for mechanistic toxicological and pharmacological studies. Thus, regardless of their organs of origin, fi nite or immortal dedifferentiated continuous cell lines are grouped into different toxicological and pharmacological categories because of their varied mechanistic responses to chemicals. Cell lines with specialized persistent metabolic functions such as MDCK kidney cells or neuroblastoma 1300 cells demonstrate reactions to toxic agents based on specialized functions that persist to some extent. In cytotoxicity testing, however, most chemicals precipitate the same toxic alterations of basal cell functions in cell types of dif-ferent origin as well as in cells that are similar to those lacking most of the cell-specifi c functions.
Origin-Specifi c Cell Functions
These structures and functions distinguish cells of varied origin and are classifi ed as special-ized cells with specifi c cell functions. These include structures that are unique to the type of cell, help identify the cell type, and may require an energy source in addition to their basal functions. Examples of such structures and functions include the presence of cilia (tracheal epithelial cells), contractility (myocardial cells), hormone production (cells of glandular or intestinal origin), and cells with high enzymatic activity capable of demonstrating chemical detoxifi cation (hepatocytes or lung cells of epithelial origin).
Extracellular Functions
Certain cell pathways produce soluble mediators destined for extracellular activities. Their role in cytotoxic classifi cation, therefore, is limited primarily because interfer-ence with their activity at the destination does not refl ect their cell origins. Secretory processes such as the release of neurotransmitters by neurons and secretion of collagen by lung fi broblasts or cytokines by cells of immunity are diffi cult to trace to a target organ or cell within a cell culture system. The synthesis of extracellular matrix compo-nents is another example in which interference by a toxic agent at the sight of deposi-tion does not refl ect the effect of that chemical on epithelial cells. Of greater value as an indicator of toxicity is interference with these extracellular components by chemical substances either by nonspecifi c binding to the component or blocking receptors on the target cell. Nevertheless, the measurement of these functions in vitro overlaps with origin-specifi c functions, and thus may be an indication of organ-specifi c cytotoxicity (see the next section).
Functional classification Cytotoxic classification
Basal cell functions
Origin-specific cell functions
Basal cytotoxicity
Extracellular functions
Organ-specific cytotoxicity
Organizational cytotoxicity
FIGURE 23.1 Classifi cation of chemicals based on functional and cytotoxic levels of physiological organization.

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 323
CYTOTOXIC CLASSIFICATION BASED ON IN VITRO METHODOLOGIES
Analogous to functional classifi cation, there are three types of mechanisms for toxicity and thus a parallel system for cytotoxic classifi cation is described.
Basal Cytotoxicity
Basal cytotoxicity is an assault on fundamental processes, which involves structures and functions common to all cells in the body including cell membranes, mitochondria, ribo-somes, chromosomes, and lysosomes. For instance, a chemical that inhibits cell prolifera-tion at low or moderate concentrations is considered to have basal cytotoxic effects. Alternatively, a mutagen directly alters DNA sequences or interferes with chromosomal arrangement and thus also has the potential for basal cytotoxicity.
This concept should not be confused with the traditional ideas of systemic toxicity that excludes the contributions of genotoxicity to general systemic activity. In actuality, it is conceivable that a mutagen may act in a basal cytotoxic fashion because of chemical inter-ference with a fundamental structure common to all cells such as a chromosome.
Basal cell functions generally support organ-specifi c cell functions. Chemicals capable of affecting basal cell activities are also likely to interfere with specialized functions. Conse-quently, studies aimed at understanding basal cytotoxic phenomena are designed with either primary cultures or continuous cell lines. Continuous cultures offer the advantages of work-ing with more homogeneous populations, are well characterized, and are more easily manip-ulated in comparison to primary cultures. Among continuous maintenance cultures, embryonic or adult immortal cell lines are easily maneuvered and afford longer in vitro life spans. Although many continuous cell lines retain highly specialized biological activities in vitro that are characteristic of their original tissues, signifi cant loss of differentiation occurs with continued passages.
Basal cytotoxicity from a chemical, therefore, is demonstrated as a result of chemical insult upon cell membrane integrity, mitochondrial activity, or protein or DNA synthesis, since these are examples of fundamental metabolic functions common to all cells. In fact, the metabolic activities of cultured cells refl ect their adaptation to in vitro conditions rather than their primary sites of origin, which may account for different cell types dis-playing similar responses to toxins. The level of activity in these cells, however, varies from one cell type to another, depending on the cell’s metabolic rate and contribution to homeostatic mechanisms, but the processes do not differ qualitatively among organs. If chemical injury in an animal or human is a representation of the failure of the entire sys-tem to support the target organs, then all cells will display similar dysfunctions in their ability to maintain homeostasis. In many cases, target organ toxicity in humans is caused by basal cytotoxicity of chemicals distributed to the corresponding organ. If this hypoth-esis is true, tests using primary cultures and continuous differentiated cell lines could be developed to cover a large percentage of toxic effects and reduce the need to introduce many laborious systems with organ-specifi c cells that are structurally and mechanistically unrelated.
In order to show that a response to a toxic agent in a cell line is an expression of a basal cytotoxic phenomenon, several criteria must be met:
1. A substantial number of chemicals of various categories should be tested and shown to induce a degree of toxicity at moderate concentrations, especially those that are known to be toxic in vivo.

324 PRINCIPLES OF TOXICOLOGY TESTING
2. Indeed, metabolic functions of various cell lines removed from parent cells show similar responses to toxic agents, evidence of which is seen in several multicenter studies (see reference list at the end of this chapter).
3. Cytotoxic agents with similar mechanisms that show the same cytotoxic response in vivo and in vitro support the contention that this reaction is due to basal cytotoxicity.
Such experimental designs have also recently been incorporated into multicenter stud-ies and form the basis of current validation programs. The use of cultured cells for toxicity testing and screening has not progressed without objections. Among these, the most impor-tant are based on current technical limitations that have not been fully investigated.
Some types of cells require hormonal, neuronal, and immunological adjustments that are not readily present in routine monolayer cultures. Models designed to mimic extracel-lular functions and organizational toxicity, as with the incorporation of cocultures and cul-ture inserts, may improve the ability to distinguish among the three levels of cytotoxicity.
Many continuous cell lines are not capable of metabolizing xenobiotics. As a result, the effects of chemicals that require biotransformation for their toxicity will not be observed. The use of organ-specifi c primary cultures that retain high levels of enzymatic activity may overcome this drawback.
The signifi cance of exposure time has not been adequately addressed. At present, validated tests and tests undergoing evaluation measure cytotoxicity according to three types of exposure: (1) short-term tests involving exposure for periods up to 72 hours, (2) long-term tests, for periods of 4–7 days, and (3) special tests involving unstable, volatile, or insoluble materials. The criticisms imply that the exposure periods represent short incu-bation and observation times and are not predictive for acute in vivo effects. On the con-trary, cell culture systems are modeled to mimic chronic exposure in vivo through manipulation of subcultivation parameters (see below, measurements of acute and chronic toxicity).
Organ-Specifi c Cytotoxicity
Chemicals that exhibit selective toxicity, especially when in vitro concentrations are lower in comparison with in vivo concentrations, qualify for organ-specifi c toxicity—the effect of a chemical on a specialized function characteristic of a particular cell type, that is, origin-specifi c cell function. Measurement of these cell functions and extrapolation to organ- specifi c toxicity requires the use of primary cultures that have morphological and biochemical features similar to those of the original tissue. Therefore, they offer the possibility for comparative studies of specialized tissues obtained from a variety of mammalian species.
Primary cultures are generally more sensitive to the effects of toxic chemicals than cell lines because of their simultaneous role of adapting to the cell culture environment while infl uenced in the presence of a toxic substance. The main limitations of primary cultures include the diffi culty of technically establishing the culture, the low cell yields, the lack of homogeneity, and the rapid loss of organ- and cell-specifi c markers that determine organ-specifi c cytotoxicity.
Depending on the cell type, the use of primary cultures assures maintenance of special-ized functional markers present in the parent cell. Thus, by comparing similar classes of chemicals against primary and continuous cultures from the same organ, the differentiation of basal from organ-specifi c cytotoxicity is realized. In addition, primary and continuous cultures from the same organ incubated with structurally similar chemicals are used to dis-tinguish a substance with a predilection for a particular organ. For example, in order to

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 325
screen hepatotoxic agents, it is necessary to use a combination of primary hepatocytes and parallel cultures of dedifferentiated cells. If a considerably lower concentration of a sub-stance is needed to inhibit 50% of proteins synthesized (IC
50) in the primary hepatocyte
culture than in an analogous parenchymal hepatoma cell line, this suggests that the chemi-cal is mechanistically hepatotoxic. Similar moderate concentrations of a particular chemi-cal that cause a toxic response in both types of cultures could be explained as a basal cytotoxic phenomenon.
Organizational Cytotoxicity
Chemicals that obstruct metabolic or secretory pathways interfere with both organ-specifi c functions and organizational functions. Some examples of chemicals or drugs that cause organizational toxicity include receptor antagonists (muscarinic blockers such as atropine), immune modulators (immunosuppressants or inhibitors of cytokine release), or inhibitors of neurotransmitter metabolism (selective serotonin reuptake inhibitors). Therefore, in vitro tests to screen for these types of organizational toxicity incorporate systems capable of detecting the passage or movement of molecules throughout the media or across membranes. Models such as cocultures in fi lter inserts are used to predict the toxicities of chemical metabolites or secretory products of cells and their effects on the apposed monolayer. Mod-els for intestinal absorption employ this system. In addition, agents capable of preventing the binding of macromolecules with specifi c cell receptors qualify as interfering with organiza-tional toxicity.
These classifi cations are important for understanding the possibilities of cellular meth-ods in toxicology testing. Basal cytotoxic mechanisms are studied in systems with undif-ferentiated fi nite or continuous cell lines, while organ-specifi c cytotoxicity is analyzed in primary cultures with well-differentiated cells from different organs. Organizational toxic-ity is observed indirectly in cell cultures by examining the substrates or products of cell metabolism. It is important to note, however, that basal functions in cells are prerequisites for specifi c functions; basal cytotoxicity imitates the effects of organ-specifi c cellular tox-icity or even organizational toxicity by indirectly affecting metabolic functions. Also, the mode of toxicity of a chemical is specifi c within each type of toxicity. For example, basal cytotoxicity precipitated by a lipophilic chemical can act either by a specifi c action on the sodium ion pump or as a result of a nonspecifi c dissolution of the chemical to the nonpolar regions of the cell membrane. The net result of both mechanisms is loss of membrane integrity.
MEASUREMENTS OF ACUTE AND CHRONIC TOXICITY
A major criticism of in vitro cytotoxicity assays is that the procedures measure only acute toxicity, primarily because the exposure is of short duration and occurs through one cellular passage level. This is because occasionally it is diffi cult to determine the difference between acute and chronic exposures in culture. Since one culture cycle (one passage level) can extend from 3 to 7 days, depending on the cell line, it is conceivable to extend the exposure time, within the time of the cycle, to determine the response to a chemical. In this way, expo-sure to the chemical through at least one cell cycle differentiates between acute and subacute or subacute and chronic. This sequence for exposure also reveals any tolerance developed against a chemical.
The effects of chemicals in vivo at the cellular level also result from repeated exposures and follow familiar patterns, including (1) the slow accumulation of the chemical in target tissues until acute toxic concentrations are reached, (2) the slow accumulation of the chemical

326 PRINCIPLES OF TOXICOLOGY TESTING
in blood which subsequently distributes to target tissues, and (3) the chemical does not accumulate but causes toxicity through repeated insults until threshold injury results. To address these mechanisms, repeated and chronic exposure assays are performed, but are carefully outlined so as to mimic the in vivo situation. Examples of such experiments are outlined in Table 23.2.
RESULTS AND MECHANISTIC IMPLICATIONS
Basal cytotoxicity from a chemical, therefore, echoes the status of cell membrane integrity, mitochondrial activity, or protein synthesis, since these are examples of basic metabolic functions common to all cells. The level of activity, however, may vary from one organ to another, depending on its metabolic rate and contribution to homeostatic mechanisms, but the processes do not differ qualitatively among organs. In many cases, target organ toxicity in mammals refl ects basal cytotoxicity of chemicals distributed to the corresponding organ. If this hypothesis is true, tests using continuous cell lines (e.g., 3T3 mouse embryonic fi bro-blasts) cover a large percentage of toxic effects and reduce the need to introduce many labo-rious systems with specifi c cells.
Organ-specifi c primary and continuous cultures are incorporated into tests for systemic toxicity. These cultures are capable of demonstrating basal cytotoxicity, similar to the effects shown by undifferentiated cells, without the need to measure specifi c functional characteristics. Organ-specifi c cells also express genuine site-specifi c cellular toxicity. A chemical capable of altering a particular function that is unique to an organ may chal-lenge the basal or organ-specifi c origin of the mechanism of action. Comparison of the concentrations of chemicals in culture necessary to produce cytotoxic effects in cell lines of different origin may elucidate the underlying mechanisms of the toxic response.
TABLE 23.2Design of In Vitro Experiments to Mimic Corresponding Effects of Chemicals In Vivo at Cellular Level
Mechanism of In Vivo Effect Design of Corresponding Repeated Chronic Exposure Studies In Vitro Objective
A. Slow accumulation of chemical in target tissues until acute toxic concentrations are reached
Cultures exposed to low but increasing doses of chemical for at least two to three passage levels
To determine whether a chemical causes toxicity at concentrations below threshold or accumulation is necessary for toxic response
B. Slow increase in blood concentrations that subsequently distribute to target tissues
Prolonged repeated intermittent exposure experiments; proliferating cells are exposed to continuous low doses of chemical for three or more passages
To compare corresponding toxic responses between accumulation at low concentrations and steady state concentrations
C. Chemical does not accumulate but causes toxicity through repeated insults until threshold injury results
Repeated intermittent dosage assays: short repeated exposure to test chemical (up to 24 hrs) followed each time by substitution with fresh medium; incubations repeated in each of three successive passages
To determine the extent of insult occurring at the cellular level in the absence of an agent, after repeated insults by allowing suffi cient time for uncoupling of noncovalently bound substance

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 327
The use of organ-specifi c cells to test for organizational toxicity is also suitable as a screening system, since these cells provide information on basal and specifi c cytotoxic-ity. For example, primary cultures of hepatocytes are used for the determination of basal cytotoxicity and organ-specifi c toxicity, based on the extensive metabolic capacities of liver cells to screen most circulating xenobiotics. Of concern is that with the use of hepa-tocytes in particular and many organ-specifi c primary cultures in general, they are not routinely maintained in continuous culture and must be established for each experiment. In addition, primary cultures are more expensive than continuous cell lines and raise legitimate ethical questions when animals are needed as the sources of cells for repeated experiments. Thus, a long-term objective of effi cient cytotoxicity testing models is the determination of the role of continuous cultures originally derived from specifi c organs and their relationship to similar cells in primary culture. Further exploration of this objective will elucidate how continuous cell lines refl ect the in vivo organ’s response to a chemical insult.
EXTRAPOLATION TO HUMAN TOXICITY
Twenty years ago, the fl edgling perception that a standardized battery of in vitro tests could be developed for tier testing of acute local and systemic toxicity was speculative and ambi-tious. Progress in sensitivity, reliability, and technical feasibility of testing protocols, sup-ported by current validation studies, is a testament to the realization of those original objectives.
STANDARDIZED TEST BATTERIES
Certain methods assigned to the battery are used for systematic mapping of the different effects attributed to basal cytotoxicity. The mechanisms underlying basal cytotoxic phe-nomena explain how these effects are infl uenced by the physicochemical properties of the molecules and how the knowledge is useful for the interpretation of results in cellular testing. Alternative cell-based models are applied on actions of chemicals known to be cytotoxic to animals or humans by comparing the in vitro and in vivo concentrations of the same chemicals.
Figure 23.2 summarizes the organization and applications of in vitro cytotoxicity test-ing and assigns positions for basal cytotoxicity, organ-specifi c cytotoxicity, and organiza-tional toxicity within the general scheme. According to the diagram, carcinogenicity and mutagenicity data contribute to the cytotoxicology evaluation but this information can be circumvented. Overall, the objective of this information is to arrive at human toxic blood concentrations to assist in the prediction of human toxicity and evaluation of risk assess-ment. Note also that in vitro toxicokinetic studies contribute to the establishment of the model. Together with in vitro concentrations derived from the tests, a model for cytotoxic-ity is formulated.
A battery of selected protocols functions as a primary screening tool before the imple-mentation of routine animal testing. Subsequent animal tests are then performed to confi rm the results from tier testing, as well as to detect outliers that elude the screening protocols. Upon completion of specifi c animal tests to confi rm the in vitro data, a summary report of all available information is used as a basis for the screening and/or prediction of risk to humans.
With the refi nement of general in vitro–in vivo testing and the progress affording such studies with time, certain tests will be employed with greater confi dence as it becomes

328 PRINCIPLES OF TOXICOLOGY TESTING
apparent that the battery refl ects the acute toxicity of certain groups of substances, resulting in the inevitable elimination of particular aspects of animal testing.
HUMAN RISK ASSESSMENT
Together with genotoxic and mutagenic tests, in vitro cellular test batteries are applied as bio-logical markers of chemically induced risks, whether synthetic or naturally occurring. Such risks include biomonitoring of food and food additives and water and air analysis for environ-mental toxicants. If similar batteries of protocols are validated for their abilities to screen for or predict human toxic effects, these measurements will establish a signifi cant frame of refer-ence for monitoring of environmental, occupational, and commercial toxic threats.
Physicochemical data
In Vivo human and animal toxicology database
Tier testing, in vitro screening methods for:
1. basal cell functions 2. origin specific cell functions
3. extracellular functions
In Vivo human and animal toxicokinetic database
Tier testing, in vitro screening methods for:
biotransformation acute & chronic toxicity
cumulative toxicity
Human and animalgenotoxicity and
carcinogenicity database
Generation of in vitro IC50s Toxicokinetic modeling
Determination of:1. basal cytotoxicity
2. organ specific cytotoxicity3. organizational toxicity
Prediction of human:
Toxic blood concentrations Toxic exposure levels
Toxicokinetics
Prediction of animal:Starting doses for toxic studies
Toxic exposure levels Toxicokinetics
Human risk assessment
FIGURE 23.2 Organization and applications of in vitro cytotoxicology testing. Source: From Barile, Frank A., Clinical Toxicology: Principles and Mechanisms, Informa HealthCare Publishers, 2010.

APPLICATIONS OF ALTERNATIVE MODELS FOR TOXICOLOGY TESTING 329
USE OF ALTERNATIVE METHODS FOR HUMAN AND ANIMAL TOXICOLOGY
It is possible to apply the 3R principles to animal toxicity testing of existing chemicals in the environment with risk assessment based on the results of in vitro tier testing. This goal is achievable with a combination of data from reliable chemical and analytical measurements of human blood and tissue concentrations of the same chemicals, such as with volunteer testing of dermal and ocular irritancy or through the tabulation of databases with clinically relevant human toxicity information.
Based on observable trends and the practical experiences of many laboratories and regulatory agencies with in vitro cytotoxicity tests, it is conceivable and realistic that toxic-ity testing in the foreseeable future will be regularly performed with cell culture models. Furthermore, such testing will be more predictive of human toxicity than the current animal tests.
With the realization of this objective comes the benefi t that many new types of in vitro toxicology and toxicokinetic tests will be available and many of the current methods will be validated in refi ned programs. These programs are conducted as refi ned multifactorial model systems of large in vitro databases to account for human toxicity, including com-puterized physiologically based kinetic modeling. Simultaneously, the gradual accep-tance of new in vitro methods should not only depend on formal validation programs, but may be a consequence of the parallel experiences of various academic and industrial laboratories using both in vitro and in vivo methods. Finally, these achievements are not based only on political or societal attempts to reduce, refi ne, or replace the use of animals in research, but they complement a very effi cient and reliable system for protecting the public interest.
SUGGESTED READINGS
Barile FA. Mechanisms of Cytotoxicology. In: Introduction to In Vitro Cytotoxicology: Mechanisms and Methods. Boca Raton, FL: CRC Press, 1994; 27.
Basketter DA, Dearman RJ, Kimber I, Gerberick GF, Ryan CA. The impact of LLNA group size on the identifi cation and potency classifi cation of skin sensitizers: a review of published data. Cutan Ocul Toxicol 2009; 28: 19.
Ekwall B. Basal cytotoxicity data (BC data) in human risk assessment. In: Proceedings of Workshop on Risk Assessment and Risk Management of Toxic Chemicals. Ibaraki, Japan, National Institute for Environmental Studies, 1992; 137.
Ekwall B. The basal cytotoxicity concept. In: Goldberg A, van Zupten LFM, eds. Alternative Meth-ods in Toxicology and the Life Sciences. Vol. 11 Mary New York: Ann Liebert, 1995: 721.
Garthoff B. Alternatives to animal experimentation: the regulatory background. Toxicol Appl Phar-macol 2005; 207: 388.
Grindon C, Combes R. Introduction to the EU REACH legislation. Altern Lab Anim 2006; 34: 5.Gupta K, Rispin A, Stitzel K, Coecke S, Harbell J. Ensuring quality of in vitro alternative test meth-
ods: issues and answers. Regul Toxicol Pharmacol 2005; 43: 219.Koeter HB. Dialogue and collaboration: a personal view on laboratory animal welfare developments
in general, and on ECVAM’s fi rst decade in particular. Altern Lab Anim 2002; 30: 207.Schechtman LM, Stokes WS. ECVAM-ICCVAM: prospects for future collaboration. Altern Lab
Anim 2002; 30: 227.Schrage A, Hempel K, Schulz M, et al. Refi nement and reduction of acute oral toxicity testing: a
critical review of the use of cytotoxicity data. Altern Lab Anim 2011; 39: 273.Straughan D. Progress in applying the three Rs to the potency testing of botulinum toxin type
A. Altern Lab Anim 2006; 34: 305.

330 PRINCIPLES OF TOXICOLOGY TESTING
REVIEW ARTICLES
Basketter DA, Clewell H, Kimber I, et al. A roadmap for the development of alternative (non-animal) methods for systemic toxicity testing - t4 report. ALTEX 2012; 29: 3.
Bouvier d’Yvoire M, Bremer S, Casati S, et al. ECVAM and new technologies for toxicity testing. Adv Exp Med Biol 2012; 745: 154.
Combes R, Gaunt I, Balls MA. Scientifi c and animal welfare assessment of the OECD Health Effects Test Guidelines for the safety testing of chemicals under the European Union REACH system. Altern Lab Anim 2006; 34: 77.
Edler L, Ittrich C. Biostatistical methods for the validation of alternative methods for in vitro toxicity testing. Altern Lab Anim 2003; 31: 5.
Gruber FP, Hartung T. Alternatives to animal experimentation in basic research. ALTEX 2004; 21: 3.Kinsner-Ovaskainen A, Maxwell G, Kreysa J, et al. Report of the EPAA-ECVAM workshop on the
validation of Integrated Testing Strategies (ITS). Altern Lab Anim 2012; 40: 175.Kirkland DJ. Testing strategies in mutagenicity and genetic toxicology: an appraisal of the guidelines
of the european scientifi c committee for cosmetics and non-food products for the evalua-tion of hair dyes. Mutat Res 2005; 588: 88.
Louekari K. Status and prospects of in vitro tests in risk assessment. Altern Lab Anim 2004; 32: 431.Meyer O. Testing and assessment strategies, including alternative and new approaches. Toxicol Lett
2003; 140: 21.Riebeling C, Hayess K, Peters AK, et al. Assaying embryotoxicity in the test tube: current limitations
of the Embryonic Stem Cell Test (EST) challenging its applicability domain. Crit Rev Toxicol 2012; 42: 443.
Rispin A, Stitzel K, Harbell J, Klausner M. Ensuring quality of in vitro alternative test methods: cur-rent practice. Regul Toxicol Pharmacol 2006; 45: 97.
Scialli AR, Guikema AJ. REACH and reproductive and developmental toxicology: still questions. Syst Biol Reprod Med 2012; 58: 63.
Stokes WS, Schechtman LM, Hill RN. The Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM): a review of the ICCVAM test method evaluation process and current international collaborations with the European Centre for the Validation of Alternative Methods (ECVAM). Altern Lab Anim 2002; 30: 23.
Stokes W, McFarland R, Kulpa-Eddy J, et al. Report on the international workshop on alternative methods for human and veterinary rabies vaccine testing: State of the science and planning the way forward. Biologicals 2012; 40: 369.
van der Laan JW, Chapin RE, Haenen B, Jacobs AC, Piersma A. Testing strategies for embryo-fetal toxicity of human pharmaceuticals. Animal models vs. in vitro approaches: a workshop report. Regul Toxicol Pharmacol 2012; 63: 115.
Vanparys P, Corvi R, Aardema MJ, et al. Application of in vitro cell transformation assays in regula-tory toxicology for pharmaceuticals, chemicals, food products and cosmetics. Mutat Res 2012; 744: 111.

Completely revised and updated, this second edition of Principles of Toxicology continues to chart evolution of the field of toxicology from an applied and sup-
portive science, to its own refined and technical discipline. It begins with basic
toxicological principles, including absorption, distribution, metabolism, and elim-
ination of toxins, including chemicals and drugs. The book then discusses animal
(in vivo) and in vitro toxicology testing methods associated with toxicological
analysis and preclinical drug development.
See What’s New in the Second Edition:
Two new chapters on Chemical and Drug Receptor Interactions
and, Toxicogenomics.
corresponding in vitro systems that complement traditional animal
toxicology testing methods
principles of the disciplines rather than on the particular steps of the techniques.
The chapters discuss the universal application of the field as a scientific disci-
pline as opposed to amplification of laboratory techniques. In addition, the book
highlights contemporary issues in toxicology testing including the various means
of possible exposure to chemicals, high-throughput screening of chemicals for
preclinical drug development, and an overview of applications. It provides the
information necessary to interpret the significance of toxicology testing results
and construct a logical approach for the ultimate purpose of testing.
PHARMACOLOGY AND TOXICOLOGY
ISBN: 978-1-84214-528-9
9 781842 145289
9 0 0 0 0
H100468
P R I N C I P L E S O F
Toxicology Testing
S E C O N D E D I T I O N





























