Embed Size (px)
Citation preview

1
Institute of Biophysics of the CAS Department of Biophysical Chemistry and Molecular Oncology
Monika Hermanová
Electrochemical methods for detection of DNA-
protein interactions and for monitoring of DNA
enzymatic processing
Ph.D. Dissertation
Supervisor: Doc. RNDr. Miroslav Fojta, CSc. Brno 2018

2
Bibliographic entry
Author: Mgr. Monika Hermanová
Title of Dissertation: Electrochemical methods for detection of DNA-protein interactions
and for monitoring of DNA enzymatic processing
Degree Program: Biology
Field of Study: Molecular and cell biology
Supervisor: doc. RNDr. Miroslav Fojta, CSc.
Academic Year: 2017/2018
Number of Pages: 128
Key words: Electrochemistry, DNA-protein interactions, p53 protein, DNA labeling,
terminal deoxynucleotidyl transferase

3
Bibliografický záznam
Jméno a příjmení autora: Mgr. Monika Hermanová
Název disertační práce: Elektrochemické metody pro detekci interakcí DNA s proteiny a pro
sledování enzymatických přeměn DNA
Název disertační práce anglicky: Electrochemical methods for detection of DNA-protein
interactions and for monitoring of DNA enzymatic processing
Studijní program: Biologie
Studijní obor: Molekulární a buněčná biologie
Školitel: doc. RNDr. Miroslav Fojta, CSc.
Akademický rok: 2017/2018
Počet stran: 128
Klíčová slova v češtině: Elektrochemie, DNA-protein interakce, protein p53, značení DNA,
terminální deoxynukleotidyl transferáza
Klíčová slova v angličtině: Electrochemistry, DNA-protein interactions, p53 protein, DNA
labeling, terminal deoxynucleotidyl transferase

4
© Monika Hermanová, Masaryk University, 2018

5
I would like to thank my supervisor, doc. RNDr. Miroslav Fojta, CSc. for all the
support that I received from him during my Ph.D. studies, for a lot of useful advice and ideas
and for being such a scientist I would like to become one day.
My thanks also go to all current and former colleagues at the Department of
Biophysical Chemistry and Molecular Oncology, especially Hanka Pivoňková, Peťa Orság,
Pavlínka Havranová, Luděk Havran and Honza Špaček for being extremely helpful at any
occasion I needed something and for creating a wonderful working environment where it has
always been fun to work.
I would also like to thank my whole family because without them, this work would
never come into existence. My mom and dad for being very supportive during my childhood
and my studies, my husband Michal for helping me with anything that I did not manage to do
and all of them - my parents, Michal and my mother-in-law for all the babysitting they did.
And finally, my biggest thanks of all go to Toník, who had a great patience with his
mom when she was too busy to play with him and who was often the last kid in the
kindergarten in the afternoon because mom had to finish her experiments in the lab. Despite
all of this, he has always been in a good mood and has helped me handle everything with
ease.

6
Abstract
Three novel applications of DNA electrochemistry and redox DNA labeling are
presented. Electrochemical signals of DNA are based either on redox activity of DNA bases,
which can be observed as CA and G peak at mercury electrodes and G and A peak at carbon
electrodes or on adsorption/desorption and reorientation processes which can be observed as
tensammetric peaks at mercury electrodes. Another option for studying DNA using
electrochemical methods lies in its redox labeling, which can be achieved using covalently
bound labels, such as osmium tetroxide complexes, or by enzymatic incorporation of
modified nucleobases into DNA.
Redox labeling of DNA via enzymatic incorporation was used in development of a
novel approach for detection of DNA-protein interactions. This approach is based on dual
labeling of DNA probes using two different electroactive labels, which yield reduction peaks
at distinct potentials and therefore enable detection of protein binding in a competition
experiment. We show that using this approach, specific and non-specific binding of the p53
protein can be distinguished as a strong preference of the p53 protein was observed towards
DNA probes bearing a specific p53 binding site (p53CON).
Label-free detection using intrinsic DNA signals was used in studies of terminal
deoxynucleotidyl transferase (TdT) tailing reactions. We found out that electrochemical
detection can be very useful for monitoring the TdT tailing reactions, especially with
pyrolytic graphite electrode (PGE) being suitable for remarkably precise determination of the
tailing reaction products length. On the other hand, hanging mercury drop electrode (HMDE)
revealed formation of various DNA structures, such as DNA hairpins and G-quadruplexes,
which influence behavior of DNA molecules at the negatively charged surface of HMDE as
well as progress of the TdT tailing reaction.
Modification with osmium tetroxide complex was applied in development of a new
two-step technique of DNA modification, which comprised enzymatic construction of DNA
bearing butyl acrylate (BA) moieties followed by chemical modification of a reactive C=C
double bond in the acrylate residue. Such approach enabled modification of the BA
conjugates in both single- and double-stranded (ds) DNA under conditions precluding
modification of nucleobase residues in ds DNA.

7
Abstrakt
V této práci představujeme tři nové aplikace využívající elektrochemii DNA a
redoxní značení DNA. Elektrochemické signály, které poskytuje DNA, jsou založeny buď na
redoxní aktivitě bází DNA, v důsledku které můžeme pozorovat píky CA a G na rtuťových
elektrodách a píky G a A na uhlíkových elektrodách, nebo na adsorpčně-desorpčních jevech a
reorientaci DNA na povrchu elektrody, které můžeme sledovat prostřednictvím
tenzametrických píků na rtuťových elektrodách. Další možnost pro využití elektrochemických
metod ke studiu DNA představuje redoxní značení DNA, kterého může být dosaženo
například pomocí kovalentně se vázajících značek, jako jsou komplexy oxidu osmičelého,
nebo pomocí enzymatické inkorporace modifikovaných nukleobází do DNA.
Značení DNA prostřednictvím enzymatické inkorporace redoxních značek do DNA
bylo využito v nové metodě pro detekci interakcí proteinů s DNA. Tato metoda je založena na
duálním značení DNA sond s využitím dvou různých elektroaktivních značek, které poskytují
redukční píky při odlišných potenciálech a umožňují tak detekci vazby proteinu na značené
sondy v kompetičním upořádání. S použitím tohoto přístupu bylo možné rozlišit sekvenčně
specifickou a nespecifickou vazbu proteinu p53 na DNA, jelikož protein p53 vykazoval silnou
preferenci pro sondy obsahující specifickou vazebnou sekvenci pro protein p53 (p53CON).
Detekce DNA pomocí jejích vlastních signálů byla využita pro studium syntézy
jednořetězcových úseků DNA katalyzované terminální deoxynukleotidyl transferázou (TdT).
Zjistili jsme, že elektrochemická detekce může být úspěšně použita pro sledování této reakce.
Obzvláště elektroda z pyrolytického grafitu se ukázala jako velmi vhodná pro pozoruhodně
přesné určení délky produktů reakce. Na druhou stranu, visící rtuťová kapková elektroda
(HMDE) odhalila tvorbu různých struktur DNA, jako jsou vlásenky nebo G-kvadruplexy,
které ovlivňují chování molekul DNA na negativně nabitém povrchu HMDE a také průběh
syntézy DNA katalyzované TdT.
Modifikace s využitím komplexu oxidu osmičelého byla uplatněna v nové
dvoukrokové technice modifikace DNA, která spočívala v enzymatické konstrukci DNA
nesoucí butylakrylátové (BA) skupiny a následné modifikaci reaktivní dvojné vazby C=C
v akrylátovém zbytku. Tento přístup umožnil modifikaci BA jak v jednořetězcové, tak
dvouřetězcové DNA i za podmínek, za kterých modifikace bází v dvouřetězcové DNA
neprobíhá.

8
Contents
1. INTRODUCTION .............................................................................................................. 10
2. PROTEIN-NUCLEIC ACIDS INTERACTIONS ........................................................... 10
2.1 Proteins interacting with nucleic acids ........................................................................... 10
2.2 p53 protein ........................................................................................................................ 11
2.2.1 p53 function ................................................................................................................. 12
2.2.2 p53 structure ................................................................................................................ 13
2.2.3 p53-DNA interactions .................................................................................................. 15
2.3 Terminal Deoxynucleotidyl Transferase ........................................................................ 16
2.4 Methods for studying protein-nucleic acids interactions .............................................. 19
2.4.1 Electrophoretic mobility shift assay ............................................................................ 20
2.4.2 Immunoprecipitation techniques .................................................................................. 21
2.4.3 DNA footprinting ......................................................................................................... 21
2.4.4 Other methods .............................................................................................................. 21
2.4.5 Electrochemical techniques ......................................................................................... 21
3. ELECTROANALYTICAL CHEMISTRY ...................................................................... 22
3.1 Types of electrodes ........................................................................................................... 23
3.1.1 Mercury electrodes ...................................................................................................... 24
3.1.2 Solid electrodes ............................................................................................................ 25
3.2 Electrochemical methods ................................................................................................. 26
3.2.1 Cyclic Voltammetry (CV) ........................................................................................... 27
3.2.2 Linear Sweep Voltammetry (LSV) .............................................................................. 27
3.2.3 Differential-pulse voltammetry (DPV) ........................................................................ 28
3.2.4 Square-wave voltammetry (SWV) ............................................................................... 28
3.2.5 Alternating current voltammetry (ACV) ..................................................................... 29
3.3 Electrochemistry of nucleic acids .................................................................................... 29
3.3.1 Adsorptive transfer stripping in DNA analysis ............................................................ 30

9
3.3.2 Analysis of DNA structure at mercury electrodes ....................................................... 31
3.3.3 DNA signals at mercury electrodes ............................................................................. 31
3.3.4 DNA signals at carbon electrodes ................................................................................ 33
3.4 Electrochemical labeling of nucleic acids ....................................................................... 34
3.4.1 Noncovalently bound redox indicators ........................................................................ 35
3.4.2 Osmium tetroxide complexes ...................................................................................... 35
3.4.3 Redox labels and their enzymatic incorporation .......................................................... 37
3. AIMS OF THE DISSERTATION .................................................................................... 40
4. RESULTS AND DISCUSSION ......................................................................................... 41
4.2 Label-free voltammetric detection of products of terminal deoxynucleotidyl
transferase tailing reaction .................................................................................................... 41
4.3 Butylacrylate-nucleobase Conjugates as Targets for Two-step Redox Labeling of
DNA with an Osmium Tetroxide Complex .......................................................................... 47
5. CONCLUSIONS ................................................................................................................. 51
6. REFERENCES ................................................................................................................... 53
LIST OF ABBREVIATIONS ................................................................................................ 67
LIST OF PUBLICATIONS AND CONFERENCES .......................................................... 70
APPENDICES ......................................................................................................................... 72
APPENDIX 2 .......................................................................................................................... 73
APPENDIX 3 .......................................................................................................................... 91

10
1. Introduction
Electrochemistry of nucleic acids is a booming field of study, although its beginning
dates back to 1950s when the first electrochemical signals of DNA were discovered by Emil
Paleček at the Institute of Biophysics in Brno. Since then, many applications have been
introduced which take advantage of either intrinsic electrochemical activity of DNA bases at
various electrodes or chemical modifications of DNA. These applications involve
development of DNA hybridization sensors, approaches enabling studies of DNA structure
and damage or detection of DNA-protein interactions.
Proteins including enzymes interacting with DNA are of crucial importance as they
influence and regulate many key cellular processes therefore new methodologies for studying
such interactions are required. We can monitor either the interaction itself (like in the case of
p53 protein interactions with DNA presented in this work) or results of the enzymatic
processing of DNA (like in the case of terminal deoxynucleotidyl transferase tailing reaction
also presented in this work). These two approaches presented here are based on
electrochemistry of nucleic acids, with the first one utilizing redox DNA labeling and the
second one taking advantage of label-free detection using intrinsic signals provided by DNA.
A novel approach for labeling DNA based on modification with osmium tetroxide complexes
also presented in this work can be potentially applied for similar applications in the future.
2. Protein-nucleic acids interactions
2.1 Proteins interacting with nucleic acids
Interactions of proteins with nucleic acids (NA) are of particular significance since
they occur in the most important processes in the cell, such as replication, transcription or
DNA repair. Proteins can interact with either RNA or DNA during these processes,
nevertheless, in this work, only DNA is subject of the study. Protein–DNA interactions can
vary a lot in their nature: some proteins are able to bind non-specifically to any DNA
sequence while other proteins are only able to bind to precisely defined genomic regions.
Sequence-specific DNA binding is typical for transcription factors or restriction
nucleases – proteins which need to locate the precise sequence in the genome in order to
perform its function. Nevertheless, slight base pair alterations in the DNA can be often
overcome. During sequence-specific DNA binding, protein interactions with NA sequences is

11
primarily determined by recognition of hydrogen-bonding determinants situated in the major
and minor grooves of the DNA that interact with complementary recognition of the amino
acids of the protein itself (Seeman et al., 1976). Hence, interactions are promoted when DNA
bases are arranged in an optimal sequence. The hydrogen bond donator and acceptor patterns
in the DNA grooves are recognized by complementary hydrogen bond donator and acceptors
on the protein surface (Bowater et al., 2015).
The most frequent mode of DNA binding by proteins is the non-specific DNA
binding. The main component providing the necessary free energy to stabilize the interaction
is the electrostatic attraction between positively charged amino acids and the negatively
charged phosphodiester DNA backbone (Jen-Jacobson, 1997). These electrostatic, non-
specific binding affinities are based on the displacement of counter-ions from the DNA
(Hippel, 1994) and, thus, are not as tight as the previously described sequence-specific
interactions. Although the proteins binding DNA in a non-specific manner do not require a
specific sequence, they may prefer or demand specific DNA structures. Such DNA regions
can be essential, specialized sites that require a non-B structure, some arise accidently during
various cellular processes, and others can be damage induced (Bowater et al., 2015).
There is a wide range of proteins that can bind DNA, including histones, proteins
involved in replication or transcription such as helicases, single-stranded DNA binding
proteins or transcription factors, proteins involved in DNA repair and various enzymes acting
on DNA such as DNA polymerases, nucleases, ligases or topoisomerases. In this work, two
proteins belonging to different classes of proteins capable of DNA binding were used – one of
them, the p53 protein, is a transcription factor, the other one, terminal deoxynucleotidyl
transferase, is a special type of a DNA polymerase. Both of them are therefore described in
more detail.
2.2 p53 protein
The p53 protein, also called the “guardian of the genome” (Lane, 1992), is a tumor
suppressor which has been extensively studied over the past couple of decades. It was first
described in 1979 as a protein that binds to the simian virus (SV40) large T antigen (Lane and
Crawford, 1979). Its importance for maintaining genetic stability of a cell and preventing
cancer transformation is evident from the fact that more than 50 % of human cancers contain
mutations in this gene (Levine, 1997). p53 functions largely as a transcription factor, and can
trigger a variety of antiproliferative programs by activating or repressing key effector genes
(Zilfou and Lowe, 2009), which can act for example in cell cycle, DNA repair or apoptosis.

12
The biological activities of p53 are closely connected with its ability to interact with DNA
(Fojta et al., 2004).
There are two structural and functional homologs of the p53 protein, p63 and p73. As
a result of sharing similar domain architecture and sequence identity with p53, p73 and p63
isoforms can form oligomers, bind DNA and transactivate majority of the p53-responsive
genes (Dötsch et al., 2010; Khoury and Bourdon, 2010).
2.2.1 p53 function
P53 is a short-lived protein that is maintained at low, often undetectable, levels in
normal cells. Stabilization of the protein in response to a stress signal (e.g. DNA damage)
results in a rapid rise in p53 levels (Bates and Vousden, 1996; Kubbutat et al., 1997) and
subsequent activation of target genes. Changes in the rate of transcription of the p53 gene thus
play a minor role, if any, in induction of p53 (Oren, 1999). One of the key mechanisms by
which the p53 function is controlled, is through interaction with MDM2 protein (Kubbutat et
al., 1997). MDM2 protein acts as E3 ubiquitin ligase, which enables it to ubiquitinate the p53
protein and target it for degradation in proteasome (Michael and Oren, 2003). The p53 protein
positively regulates the MDM2 gene at the level of transcription and the MDM2 protein
negatively regulates the p53 protein at the level of its activity. This creates a feedback loop
that regulates both the activity of the p53 protein and the expression of the MDM2 gene (Wu
et al., 1993).
After p53 activation, several outcomes are possible, depending on type and extent of
the stress factors affecting the cell, all of which may under appropriate circumstances
contribute to its tumor suppressive properties. In the case of DNA damage, p53 can arrest cell
cycle in G1 or G2 phase in order to allow DNA damage repair occur prior to replication of
DNA or beginning of mitosis. p53 induces a G1 arrest primarily through the transactivation of
p21Waf1/Cip1
, a cyclin-dependent kinase inhibitor (Brugarolas et al., 1995; Deng et al., 1995),
which causes activation of Rb protein, resulting in negative regulation of the E2F family of
transcription factors and blockage of the transfer from G1 to S phase (Giono and Manfredi,
2006).
After the induction of cell cycle arrest, p53 can activate genes involved in DNA repair
or can directly interact with proteins taking part in the DNA repair process (Latonen and
Laiho, 2005). One of the most studied proteins, which take part in the DNA repair and are
transactivated by p53, is the GADD45 protein (Smith et al., 2000). If the DNA damage

13
cannot be repaired, p53 can induce p53-dependent apoptosis. There are three mechanisms of
apoptosis induced by p53: binding of a ligand to death receptors from the TNF-R (tumor
necrosis factor receptor) family, a mechanism involving translocation of p53 to mitochondrial
membrane and induction of apoptosis in a transcription-independent manner by p53 cytosolic
activities (Bourdon et al., 2002; Wang et al., 2005; Green and Kroemer, 2009). Several p53
target genes including Bax, PUMA, Noxa, p53-AIP, PIG3, Fas/APO1, and KILLER/DR5
have been implicated in p53-induced apoptosis (Owen-Schaub et al., 1995; Toshiyuki and
Reed, 1995; Polyak et al., 1997; Oda, 2000).
In the case that the p53 protein is mutated, the cell processes in which p53 is involved
can be altered by different mechanisms: a) loss of the wtp53 function, b) dominantly negative
inhibition of the wtp53 function and c) gain of function, which can lead to transcriptional
activation of genes involved in cell proliferation, cell survival and angiogenesis.
Consequently, cells expressing some forms of mutant p53 show enhanced tumorigenic
potential with increased resistance to chemotherapy and radiation (Cadwell and Zambetti,
2001).
2.2.2 p53 structure
Human p53 is a nuclear phosphoprotein of MW 53 kDa, encoded by a 20-Kb gene
containing 11 exons and 10 introns, which is located on the small arm of chromosome 17
(Lamb and Crawford, 1986). Wild-type p53 protein contains 393 amino acids and is
composed of several structural and functional domains (Fig. 1): a N-terminus containing an
amino-terminal domain (residues 1-42) and a proline-rich region with multiple copies of the
PXXP sequence (residues 61-94, where X is any amino acid), a central core domain (residues
102-292), and a C- terminal region (residues 301-393) containing an oligomerization domain
(residues 324-355), a strongly basic carboxyl-terminal regulatory domain (residues 363-393),
a nuclear localization signal sequence and a nuclear export signal sequence (Bai and Zhu,
2006).

14
Figure 1. Structure of the p53 protein. The protein has three main domains: N-terminal
activation domain, DNA binding domain and C-terminal tetramerization domain.The N-
terminal domain includes transactivation subdomain and a proline-rich fragment. The central
DNA binding domain is required for sequence-specific DNA binding. The C-terminal region
is considered to perform a regulatory function. Numbers indicate residue number. NLS,
nuclear localization signal sequence; NES, nuclear export signal sequence. (Bai and Zhu,
2006)
The N-terminal domain consists of two transactivation domains and a proline-rich
region. It is required for transactivation activity and interacts with various transcription factors
including acetyltransferases and MDM2 (Fields and Jang, 1990). The proline-rich region
plays a role in p53-dependent apoptosis and in p53 stability regulated by MDM2 (Sakamuro
et al., 1997).
Central domain is responsible for the sequence-specific DNA binding to the consensus
binding site. Amino acid residues within this domain are frequently mutated in human cancer
cells and tumor tissues. The Arg175, Gly245, Arg248, Arg249, Arg273, and Arg282 (see Fig.
X) are reported to be mutation hot spots in various human cancers (Bai and Zhu, 2006). The
central region of p53 is its most highly conserved region, not only when p53 is compared with
its homologues from Drosophila and Caenorhabditis elegans, but also as compared with its
mammalian family members, p63 and p73 (Kaelin, 1999).
C-terminus of the p53 protein contains an oligomerization domain, which is located
within the amino acids 324 and 355 and is responsible for tetramerization of the protein and
the C-terminal regulatory and DNA-binding domain. This C-terminal domain of p53 can act
as a negative regulator of the core DNA binding domain (Hupp, 1999). If the interaction
between the C-terminus and the core DNA binding domain is disrupted by posttranslational
modification (such as phosphorylation and acetylation), the DNA binding domain will
become active, thus induce an enhanced transcriptional activity (Bai and Zhu, 2006). The C-
terminal domain can bind DNA in a sequence-nonspecific manner.

15
2.2.3 p53-DNA interactions
As has been mentioned above, p53 acts as a transcription factor and therefore its
function is closely related to its ability to bind DNA. p53 can bind DNA either in a sequence-
specific manner, which is mediated by the central DNA binding domain, or in a sequence-
nonspecific (structure-selective) manner, mediated by the C-terminal DNA binding domain.
2.2.3.1 Sequence-specific DNA binding
For binding DNA in a sequence-specific manner, p53 protein requires a consensus
binding site consisting of two copies of the 10 base pair motif (p53CON) 5‘-
PuPuPuC(A/T)(T/A)GPyPyPy-3‘ separated by 0-13 base pairs (El-Deiry et al., 1992), which
is found in the vicinity of many of its target gene promoters (McKinney and Prives, 2002).
The internal symmetry of the motif and the fact that the consensus binding site contains two
copies of the motif imply that p53 binds DNA as a tetramer. One copy of the motif is
insufficient for the binding, and subtle alterations of the motif, even when present in multiple
copies, result in loss of affinity for p53 (El-Deiry et al., 1992). Sequences, that match the
consensus binding site, are often bound with different efficacy, on the other hand, some
sequences, which do not correspond to the consensus site, are bound with high affinity (Foord
et al., 1993; Halazonetis et al., 1993). Intensity of the p53 binding also depends on number of
nucleotides which are inserted between the two copies of the motif. While insertion of 5 or 15
nucleotides inhibits p53 binding to DNA, insertion of 10 nucleotides does not affect the
binding, which corresponds to the fact that after insertion of 10 nucleotides, the half-sites
remain on the same face of the double helix (Wang et al., 1995). DNA topology is also an
important parameter for regulating the specific interaction of p53 with its target binding sites
(Göhler et al., 2002).
Aside from the central DNA binding site, the C-terminal domain of the p53 protein is
also very important for the sequence-specific DNA binding as it regulates the sequence-
specific binding of the central DNA binding domain. If not modified, the C-terminal domain
inhibits the central DNA binding domain but after phosphorylation of serines or acetylation of
lysines in the C-terminal domain, the sequence-specific DNA binding is activated (Hupp and
Lane, 1994), which is caused by the diminution of the negative regulatory effect of the C-
terminal domain. Same effect can be achieved by antibodies, peptides or other molecules
interacting with the C-terminal domain (Selivanova et al., 1998) or by deletion of the last 30
amino acids (Hupp and Lane, 1994).

16
2.2.3.2 Sequence-nonspecific DNA binding
Besides the sequence-specific DNA binding, p53 is able to bind also DNA not
containing the consensus binding site. It was found that p53 binds selectively to supercoiled
DNA, which has been denominated as supercoil-selective (SCS) DNA binding (Paleček et al.,
1997; Palecek et al., 2001). Negatively supercoiled (sc) DNA, regardless of the presence or
absence of the p53CON, is bound preferentially by both wt (Paleček et al., 1997; Mazur et al.,
1999; Fojta et al., 2004) and mutant (Brázdová et al., 2009) p53. Basic segment of the C-
terminal DNA binding domain (amino acids 363–382) in an oligomeric state is responsible
for the SCS binding (Brázdová et al., 2002; Fojta et al., 2004).
The C-terminal DNA binding domain can recognize non-canonical DNA structures
stabilized by supercoiling, such as hairpins (Palecek et al., 2001; Fojta et al., 2004), cruciform
structures (Jagelská et al., 2010), bent DNA (McKinney and Prives, 2002), three- and four-
way junctions (Subramanian and Griffith, 2005), structurally flexible chromatin DNA (Kim
and Deppert, 2003), telomeric t-loops (Stansel et al., 2002) or triplexes (Brázdová et al.,
2016) and bind them with substantial preference.
The sequence-nonspecific DNA binding comprises p53 binding to DNA exhibiting
various types of damage. This can include single-stranded DNA ends (Bakalkin et al., 1995),
DNA with insertion-deletion mismatches, DNA containing single- or double-strand breaks
resulting from UV or ionizing radiation or DNA modified with anticancer drugs, such as
cisplatin (Fojta et al., 2003; Pivoňková et al., 2006).
2.3 Terminal Deoxynucleotidyl Transferase
Terminal Deoxynucleotidyl transferase (TdT) is a DNA polymerase, which is unique among
other polymerases as it possesses an unusual ability to incorporate nucleotides in a template-
independent manner using only single-stranded DNA as the nucleic acid substrate (Michelson
and Orkin, 1982). TdT was first isolated from a calf thymus gland and was described as
enzyme with polymerase activity (Bollum, 1960). Although the biochemical mechanism of
TdT reaction was described soon (Kato et al., 1967), physiological role of TdT remained
unclear for many years. However, later it was discovered that its physiological role lies in
random addition of nucleotides to single-stranded DNA during V(D)J recombination
(Baltimore, 1974). During the assembly of immunoglobulin and T cell receptor variable
region genes from variable (V), diversity (D), and joining (J) segments, the germline-encoded
repertoire is further diversified by processes that include template-independent addition of
nucleotides (N regions) by TdT at gene segment junctions. TdT deficient lymphocytes have

17
no N regions in their variable region genes, which shows that TdT is responsible for N region
addition (Komori et al., 1993). The ability of TdT to randomly incorporate nucleotides
increases antigen receptor diversity and aids in generating the ∼1014
different
immunoglobulins and ∼1018
unique T cell antigen receptors that are required for the
neutralization of potential antigens (Sadofsky, 2001; Motea and Berdis, 2010). TdT is thus
crucial for evolution and adaptation of the vertebrate immune system (Kunkel et al., 1986;
Komori et al., 1993).
For its catalytic activity, the enzyme requires a single-stranded initiator that is at least three
nucleotides long and a free 3'-OH end for extension (Kato et al., 1967; Bollum, 1974). The
order by which TdT binds DNA and dNTP is random; studies performed in the absence or
presence of product inhibitors suggest a rapid equilibrium random mechanism in which TdT
forms the catalytic competent ternary complex via binding of dNTP prior to DNA or vice
versa (Deibel and Coleman, 1980). This also implies that TdT catalyzes DNA synthesis in a
distributive mode. Single-stranded DNA is generally the preferred primer for the reaction but
under certain conditions, TdT can add tails to double-stranded DNA as well (Roychoudhury
et al., 1976; Deng and Wu, 1981). The enzyme also catalyzes a limited polymerization of
ribonucleotides at the 3'-end of oligodeoxynucleotides (Roychoudhury et al., 1976; Boulé et
al., 2001).
Since the DNA synthesis by TdT is template-independent, it could be expected that TdT
incorporates all natural nucleotides with equal efficiencies. Nevertheless, although in vitro the
TdT can incorporate all four nucleotides onto single-stranded DNA, in vivo, a bias was
observed for the incorporation of dGMP and dCMP versus dAMP and dTMP (Basu et al.,
1983; Mickelsen et al., 1999). This preference may offer explanation for high G/C content of
the Ig and T cell receptor N regions. For example, it has been demonstrated that Km for dGTP
and dATP can differ very significantly, with the Km for dGTP being more than 4-fold lower
than Km for dATP (Modak, 1978; Yang et al., 1994). At the molecular level, the preference in
nucleotide utilization could reflect favorable hydrogen-bonding interactions between the
incoming dNTP and active site amino acids that guide nucleotide binding (Motea and Berdis,
2010).
Another unique feature attributed to TdT, aside from the template-independent activity, is the
ability to perform de novo synthesis of short fragments of DNA ranging in size from 2- to 15-
mers when provided with dNTPs in the absence of a primer (Ramadan et al., 2004). These
fragments are hypothesized to act as signals for DNA repair or recombination machinery

18
(Ramadan et al., 2004). However, this hypothesis has yet to be tested, in order to prove if de
novo DNA synthesis indeed occurs in vivo and to show biological relevance of this
phenomenon.
TdT, like all DNA polymerases, requires divalent metal ions to catalyze the phosphoryl
transfer reaction associated with nucleotide incorporation. However, TdT is unique in its
ability to use a variety of divalent cations such as Co2+
, Mn2+
, Zn2+
and Mg2+
(Deibel and
Coleman, 1980). Each metal ion has different effects on the kinetics of nucleotide
incorporation. For example, Mg2+
facilitates the preferential utilization of dGTP and dATP
whereas Co2+
increases the catalytic polymerization efficiency of the pyrimidines, dCTP and
dTTP (Chang and Bollum, 1990). Zn2+
behaves as a unique positive effector for TdT since
reactions with Mg2+
are stimulated by addition of micromolar quantities of Zn2+
. However,
Zn2+
is not an intrinsic part of the enzyme (Chang and Bollum, 1990). X-ray structures and
modeling of the 3’-end during the pre-catalytic and post-catalytic state for aforementioned
divalent cations are depicted in Fig. 2.

19
Figure 2. Pre- and post-catalytic active sites with different metal ions (Gouge et al., 2013).
TdT is able to tolerate various bulky modifications of the nucleotides. This feature has been
utilized in in vivo and in vitro labeling of double-strand DNA breaks in a technique called
TUNEL (TdT-mediated dUTP-biotin nick end-labeling) (Motea and Berdis, 2010). The
nucleotide analogs, which can be incorporated by TdT, include 2′,3′-dideoxynucleotides
(Ono, 1990), p-nitrophenylethyl triphosphate or p-nitrophenyl triphosphate (Arzumanov et
al., 2000) and recently in our lab it was shown that TdT can incorporate 3-nitrophenyl-7-
deazaG (Horáková et al., 2011), butyl acrylate modified dATP or dUTP (Havranová-
Vidláková et al., 2017) or vinyl (unpublished results).
2.4 Methods for studying protein-nucleic acids interactions
As already mentioned, protein-nucleic acids interactions are of crucial importance in
many key cellular processes, which leads to the need for extensive studies of these

20
interactions. Many traditional methods are used for this purpose along with new methods,
which are being developed.
Each technique can provide a wealth of knowledge of such interactions, but each has
limitations that usually restrict it from elucidating a full description of the mode of interaction
between the protein and NA. Hence, alternative, complementary techniques are usually
applied to the same system to provide a more complete description of these interactions
(Bowater et al., 2015).
2.4.1 Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) is based on electrophoretic
separation of protein–NA mixtures. The speed, at which different molecules move through the
gel, is determined by their size, charge and shape. After the protein binding to the NA, the
complex of NA bound to protein becomes less mobile and is shifted up the gel compared to
the NA alone. The fraction of free and bound NA molecules can thus be determined from the
ratio of bound and unbound NA.
Competition assay is a type of an EMSA experiment, which enables to determine the
most favorable DNA sequence for the binding protein. Different oligonucleotides of defined
sequence are used as competitors. Choice of appropriate competitors allows identification of
the precise binding site of the protein. Protein binding to different DNAs can be also
indirectly evaluated using indicator oligonucleotide or longer DNA substrate as competitor
(Brázda et al., 2006).
Another type of the EMSA experiment comprises use of antibody that recognizes the
protein. When the antibody is added to the mixture, larger complex containing the NA,
protein and antibody is created and is even more shifted. This method is referred to as a
supershift assay and is used to unambiguously identify a protein present in the protein–NA
complex (Bowater et al., 2015).
EMSA analysis is quite limited by the fact that experimental environments are
restricted by the conditions required for electrophoresis, which constitutes quite a
disadvantage of this method. The ionic strength of a solution has a substantial impact on the
modes of protein–NA interactions, meaning non-specific interactions that generally rely
heavily on charge are reduced in high-salt environments, but stronger specific interactions that
are dependent on other factors, such as base sequence, can remain (Jen-Jacobson, 1997). This

21
can result in misinterpretation of the observed data when DNA binding is studied using
EMSA analysis.
2.4.2 Immunoprecipitation techniques
In immunoprecipitation techniques, antibodies are used to pull down their antigen out
of solution; the antigen can be either the protein binding to NA or a specific sequence or
structure in the NA. To allow recovery and analysis of the sample after the precipitation, the
agent that recognizes the complex is often immobilized to a substrate or surface. An important
advantage of precipitation approaches is their significant degree of flexibility in terms of how
the experiment can be set up, allowing for diverse reaction conditions to be studied.
Chromatin immunoprecipitation (ChIP) is a widespread method for identification of
sequences to which proteins bind in genomic DNA within cells (Furey, 2012; Christova,
2013).
2.4.3 DNA footprinting
Footprinting assays are based on the principle of protection of protein-bound DNA
from degradation. The procedure employs chemical (using e.g. hydroxyl radicals) or
enzymatic (using e.g. DNase I) digestion of naked- and protein bound-DNA oligomers. Both
the reactions are then compared using gel electrophoresis. Footprinting has been a valuable
tool for elucidating sequence specificity and dissociation constants of a variety of ligands
binding to DNA (Dey et al., 2012). Its advantage lies in the fact that the protein-DNA binding
can occur under defined reaction conditions.
2.4.4 Other methods
Many other methods have been employed in studies of protein-nucleic acids
interactions. These include high-resolution techniques such as X-ray crystallography or
nuclear magnetic resonance (Bowater et al., 2015). Further examples of techniques used for
studying protein-NA interactions are isothermal titration calorimetry (Bowater et al., 2015),
yeast one-hybrid assay, phage display for DNA-binding proteins, proximity ligation assay,
fluorescence resonance energy transfer (FRET), circular dichroism, atomic force microscopy
(AFM), surface plasmon resonance (SPR) or various in silico tools for prediction and
identification of DNA–protein interactions (Dey et al., 2012).
2.4.5 Electrochemical techniques
Among the widely used biophysical methods based on optical detection, new methods
utilizing electrochemical detection have been presented. They can take advantage of the

22
intrinsic electrochemical or electrocatalytic activity of either proteins or DNA. Approaches
utilizing signals specific for proteins enable detection of their DNA binding, for example
lysozyme binding to DNA aptamers (Kawde et al., 2005; Ostatná et al., 2017) or MutS
protein recognizing mispaired and unpaired bases in duplex DNA (Paleček et al., 2004;
Masařík et al., 2007). Other approaches are based on detection of DNA, both natural and
labeled. DNA can be immobilized on the electrode surface, which has been used in the study
of T3-RNA polymerase (Meunier-Prest et al., 2010), protein MutH (Ban et al., 2004), anti-
DNA antibodies (Evtugyn et al., 2008), -thrombin (Evtugyn et al., 2008; Ding et al., 2010)
or lysozyme (Huang et al., 2009) binding to DNA. Another approach utilizes separation of
protein-DNA complexes at magnetic beads (MB) (Paleček and Fojta, 2007); subsequent
electrochemical detection can be label-free, where p53 protein binding to supercoiled and
linearized plasmid DNA can be distinguished (Němcová et al., 2010) or can use DNA
labeling by various methods such as terminal deoxynucleotidyl transferase tail-labeling
(Horáková et al., 2011) or labeling by oxoosmium complexes (Němcová et al., 2014).
Magnetic beads-based assay was also used for detection of recognition of a specific aptamer
by thrombin (Cheng et al., 2010; Zheng et al., 2010). Another possibility for detection of
protein-DNA binding lies in use of redox-labelled click-transformable DNA probe, which has
been applied in a study using click reaction of azidophenyl for detection of p53-DNA binding
(Balintová et al., 2015). Electrochemical detection can be also employed in studying damaged
DNA and enzymes involved in its processing (Fojta et al., 2016) such as T4 endonuclease V
and E.Coli exonuclease III (Fojta, 2005; Havran et al., 2008), 8-oxoguanine DNA glycosylase
(Liu et al., 2015), photolyase (DeRosa et al., 2005) or ligase (Zauner et al., 2005; Vacek et
al., 2008).
3. Electroanalytical chemistry
Electroanalytical chemistry is the branch of chemistry concerned with the interrelation
of electrical and chemical effects. A large part of this field deals with the study of chemical
changes caused by the passage of an electric current and the production of electrical energy by
chemical reactions. In electrochemical systems, we are concerned with the processes and
factors that affect the transport of charge across the interface between chemical phases, which
are an electronic conductor (an electrode) and an ionic conductor (an electrolyte). Since one
interface is not enough to perform the measurement, electrochemical experiments take place
in collections of interfaces called electrochemical cells. These systems comprise at least two
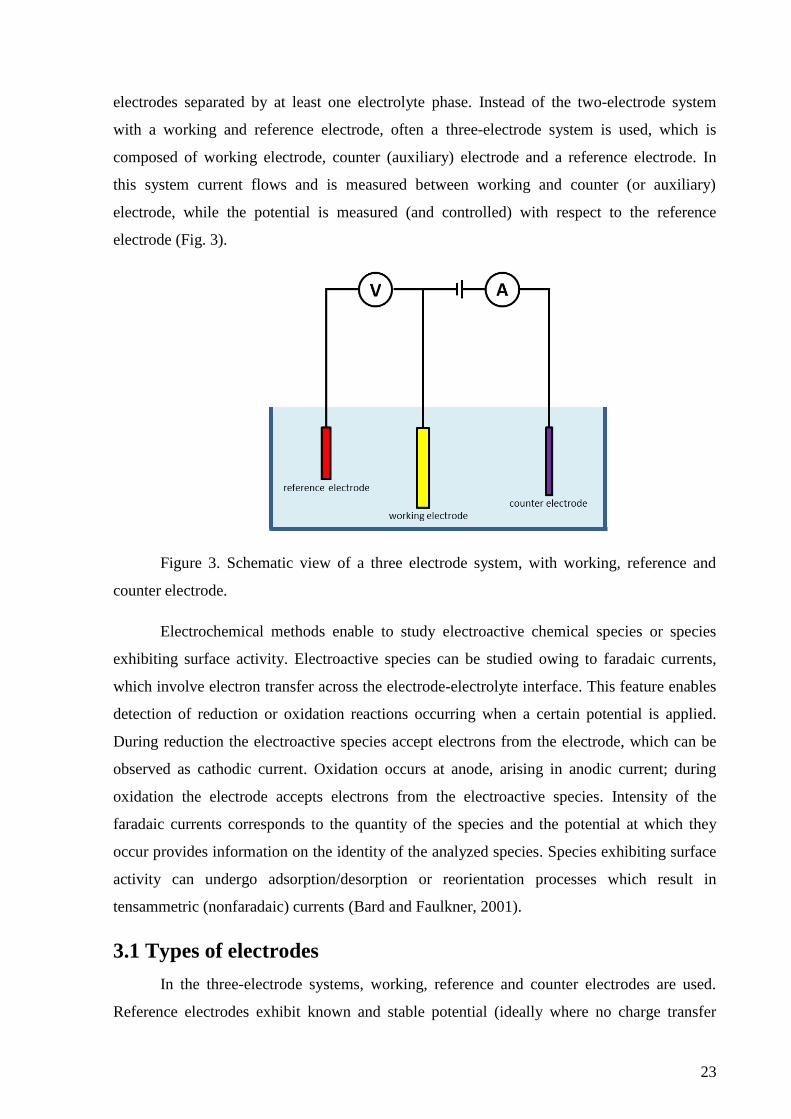
23
electrodes separated by at least one electrolyte phase. Instead of the two-electrode system
with a working and reference electrode, often a three-electrode system is used, which is
composed of working electrode, counter (auxiliary) electrode and a reference electrode. In
this system current flows and is measured between working and counter (or auxiliary)
electrode, while the potential is measured (and controlled) with respect to the reference
electrode (Fig. 3).
Figure 3. Schematic view of a three electrode system, with working, reference and
counter electrode.
Electrochemical methods enable to study electroactive chemical species or species
exhibiting surface activity. Electroactive species can be studied owing to faradaic currents,
which involve electron transfer across the electrode-electrolyte interface. This feature enables
detection of reduction or oxidation reactions occurring when a certain potential is applied.
During reduction the electroactive species accept electrons from the electrode, which can be
observed as cathodic current. Oxidation occurs at anode, arising in anodic current; during
oxidation the electrode accepts electrons from the electroactive species. Intensity of the
faradaic currents corresponds to the quantity of the species and the potential at which they
occur provides information on the identity of the analyzed species. Species exhibiting surface
activity can undergo adsorption/desorption or reorientation processes which result in
tensammetric (nonfaradaic) currents (Bard and Faulkner, 2001).
3.1 Types of electrodes
In the three-electrode systems, working, reference and counter electrodes are used.
Reference electrodes exhibit known and stable potential (ideally where no charge transfer

24
over a wide potential range occurs) and are made of metal covered with a layer of its salt in a
solution containing an anion of this salt. Typical examples are the silver chloride electrode,
mercury-mercurous sulfate electrode or calomel electrode. Counter electrodes are made of
electrochemically inert materials such as platinum or carbon. Its surface area is often much
larger than that of the working electrode to ensure that the half-reaction occurring there can
proceed fast enough and does not limit the process at the working electrode.
Working electrodes are made of plenty of materials involving mercury, carbon, or
noble metals such as gold or platinum. Material of the working electrode strongly influences
its use and suitability for various electrochemical techniques. Working electrode should
provide high signal-to-noise characteristics, as well as a reproducible response. Thus, its
selection depends primarily on two factors: the redox behavior of the target analyte and the
background current over the potential region required for the measurement. Other
considerations include the potential window, electrical conductivity, surface reproducibility,
mechanical properties, cost and toxicity (Wang, 2001a). Potential window is usually limited
by electrolysis of water; anodic potential range is limited by the oxygen evolution, cathodic
potential range by the hydrogen evolution. Both these processes are pH-dependent.
3.1.1 Mercury electrodes
Mercury is a traditional material used in electroanalytical chemistry. Using mercury
electrodes is highly advantageous as their surface is atomically smooth, can be easily renewed
and is highly reproducible. Another advantage lies in the fact that mercury electrodes exhibit
high values of hydrogen overvoltage and therefore enable measurements at very negative
potentials. However, at mildly positive potentials, mercury is oxidized and the anodic range is
thus limited. Therefore, mercury electrodes are convenient for analysis of reduction processes.
Mercury electrodes are liquid and as such are not suitable for use in biosensors, where solid
materials are more favorable.
Various types of mercury electrodes are being used, the most common of which are
dropping mercury electrode (DME), hanging mercury drop electrode (HMDE) or mercury
film electrode (MFE). Besides these types of liquid mercury electrodes, solid amalgam
electrodes (SAE) have been used, which are alloys of mercury with another metal, such as
silver or copper. They constitute a non-toxic alternative of mercury electrodes, suitable for use
in sensors and exhibiting similar electrochemical characteristics as mercury electrodes such as
highly negative potentials enabling to observe reduction and catalytic hydrogen evolution.

25
SAE have been used in electrochemical analysis of many species, including DNA
(Yosypchuk et al., 2002; Fadrná et al., 2004; Hasoň and Vetterl, 2006).
3.1.2 Solid electrodes
Most frequently used solid working electrodes are made of various types of carbon or
a metal, such as gold or platinum. Each material has its own specific features but a common
characteristic of the solid electrodes (with some exceptions) is an anodic potential window
enabling analysis of oxidizable compounds. Compared to mercury electrodes, solid electrodes
exhibit good mechanical stability and therefore are suitable for use in portable devices. Solid
electrodes can be easily miniaturized and occur in many variants, such as rotating disk
electrodes or screen-printed electrodes. They offer inexpensive mass production and their
surface can be modified with a broad spectrum of films and layers.
An important factor in using solid electrodes is the dependence of the response on the
surface state of the electrode. Accordingly, the use of such electrodes requires precise
electrode pretreatment and polishing to obtain reproducible results. The nature of these
pretreatment steps depends on the materials involved. Mechanical polishing and potential
cycling are commonly used for metal electrodes, while various chemical or electrochemical
procedures are added for activating carbon-based electrodes. Unlike mercury electrodes, solid
electrodes present a heterogeneous surface with respect to electrochemical activity (Wang,
2001b).
Solid electrodes based on carbon are currently in widespread use in electroanalysis,
primarily because of their broad potential window, low background current, rich surface
chemistry, low cost, chemical inertness, and suitability for various sensing and detection
applications. Renewal of their surface is easier than in the case of metal electrodes. In
contrast, electron transfer rates observed at carbon surfaces are often slower than those
observed at metal electrodes. Microstructure of the electrode surface (edge- or basal-plane
orientation of the graphite sheets) has a profound effect on the electrochemical reactivity at
carbon electrodes as well as other factors, such as cleanliness of the surface and presence of
surface functional groups. The most commonly used carbon electrodes are:
- Pyrolytic graphite electrode (PGE) is made by heat treatment of pyrolytic carbon
or by chemical vapor deposition. PGE can occur in edge- or basal-plane
orientation. The basal-plane electrode consists of graphite layers which lie parallel

26
to the surface. In comparison, edge-plane electrodes are fabricated in a way that
the layers of graphite lie perpendicular to the surface (Banks and Compton, 2005).
- Glassy carbon electrode exhibits wide potential window, chemical inertness
(solvent resistance) and relatively reproducible performance. The structure of
glassy carbon involves thin, tangled ribbons of cross-linked graphite-like sheets.
The electrode material has a high density and small pore size and its surface can be
polished to achieve a shiny “mirror-like” appearance.
- Carbon paste electrode uses graphite powder mixed with various water-
immiscible nonconducting organic binders (pasting liquids such as mineral oil) and
offer an easily renewable and modified surface and very low background current
contributions. Disadvantage of carbon pastes is the tendency of the organic binder
to dissolve in solutions containing organic solvent.
- Diamond electrodes are fabricated by chemical vapor deposition methods. Since
diamond itself is an insulator, it has to be doped with boron to become conductive.
Diamond electrodes have wide potential window (approaching 4 V), low and
stable background currents and relatively low adsorption of organic molecules.
Platinum and gold electrodes are the most widely used metallic electrodes. Such
electrodes offer very favorable electron transfer kinetics and a large anodic potential range. In
contrast, the low hydrogen overvoltage at these electrodes limits the cathodic potential
window. Another limiting factor lies in the high background currents associated with the
formation of surface oxide or adsorbed hydrogen layers. Compared to platinum electrodes,
gold ones are more inert, and hence are less prone to the formation of stable oxide films or
surface contamination. Therefore the gold electrodes are often preferred in electroanalytical
chemistry. Gold electrodes are widely used as substrates for self-assembled organosulfur
monolayers, including thiol-derivatized DNA oligonucleotides (Herne and Tarlov, 1997).
3.2 Electrochemical methods
Since the discovery of polarography by Jaroslav Heyrovský in 1922, many
electrochemical methods have been presented, taking advantage of various approaches –
chronopotentiometry, chronoamperometry, voltammetry, or impedance spectroscopy. The
most frequently used method for DNA analysis is voltammetry. Voltammetry is based on the
same principle as polarography, the difference is in the fact that in polarography, the working
electrode is dropping mercury electrode (DME). During voltammetry (or polarography),

27
potential is applied to the working electrode and the actual current value is measured as the
dependent variable. The most common voltammetric methods are listed below.
3.2.1 Cyclic Voltammetry (CV)
Cyclic voltammetry is the most widely used voltammetric technique. The power of
cyclic voltammetry results from its ability to rapidly provide considerable information on the
thermodynamics of redox processes and the kinetics of heterogeneous electron transfer
reactions and on coupled chemical reactions or adsorption processes. Cyclic voltammetry is
often the first experiment performed in an electroanalytical study (Wang, 2001b).
Cyclic voltammetry consists of scanning linearly the potential of working electrode,
using a triangular potential waveform (Fig. 4). Depending on the information sought, single or
multiple cycles can be used. Scan rate (the rate of a potential change) is a very important
factor in CV. By adjusting a scan rate, nature of the electrode process and its reversibility can
be explored and it is possible to discriminate between redox, tensammetric or catalytic
processes.
Figure 4. Potential-time waveform used in cyclic voltammetry (three successive
potential cycles).
3.2.2 Linear Sweep Voltammetry (LSV)
Linear sweep voltammetry is very similar to cyclic voltammetry, the difference being
that in LSV a scan in only one direction is performed. The potential waveform thus resembles
that of CV but it lacks the reverse scan (Fig. 5). During the potential sweep, the potentiostat
measures the current resulting from the applied potential.
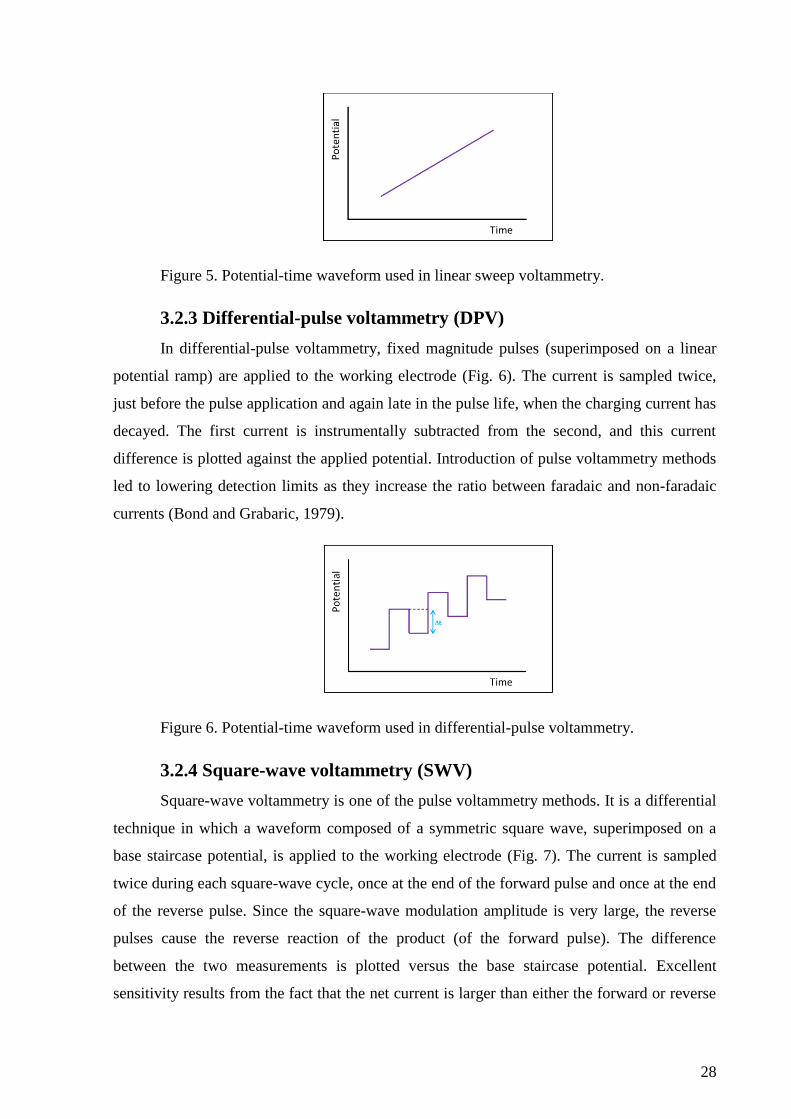
28
Figure 5. Potential-time waveform used in linear sweep voltammetry.
3.2.3 Differential-pulse voltammetry (DPV)
In differential-pulse voltammetry, fixed magnitude pulses (superimposed on a linear
potential ramp) are applied to the working electrode (Fig. 6). The current is sampled twice,
just before the pulse application and again late in the pulse life, when the charging current has
decayed. The first current is instrumentally subtracted from the second, and this current
difference is plotted against the applied potential. Introduction of pulse voltammetry methods
led to lowering detection limits as they increase the ratio between faradaic and non-faradaic
currents (Bond and Grabaric, 1979).
Figure 6. Potential-time waveform used in differential-pulse voltammetry.
3.2.4 Square-wave voltammetry (SWV)
Square-wave voltammetry is one of the pulse voltammetry methods. It is a differential
technique in which a waveform composed of a symmetric square wave, superimposed on a
base staircase potential, is applied to the working electrode (Fig. 7). The current is sampled
twice during each square-wave cycle, once at the end of the forward pulse and once at the end
of the reverse pulse. Since the square-wave modulation amplitude is very large, the reverse
pulses cause the reverse reaction of the product (of the forward pulse). The difference
between the two measurements is plotted versus the base staircase potential. Excellent
sensitivity results from the fact that the net current is larger than either the forward or reverse
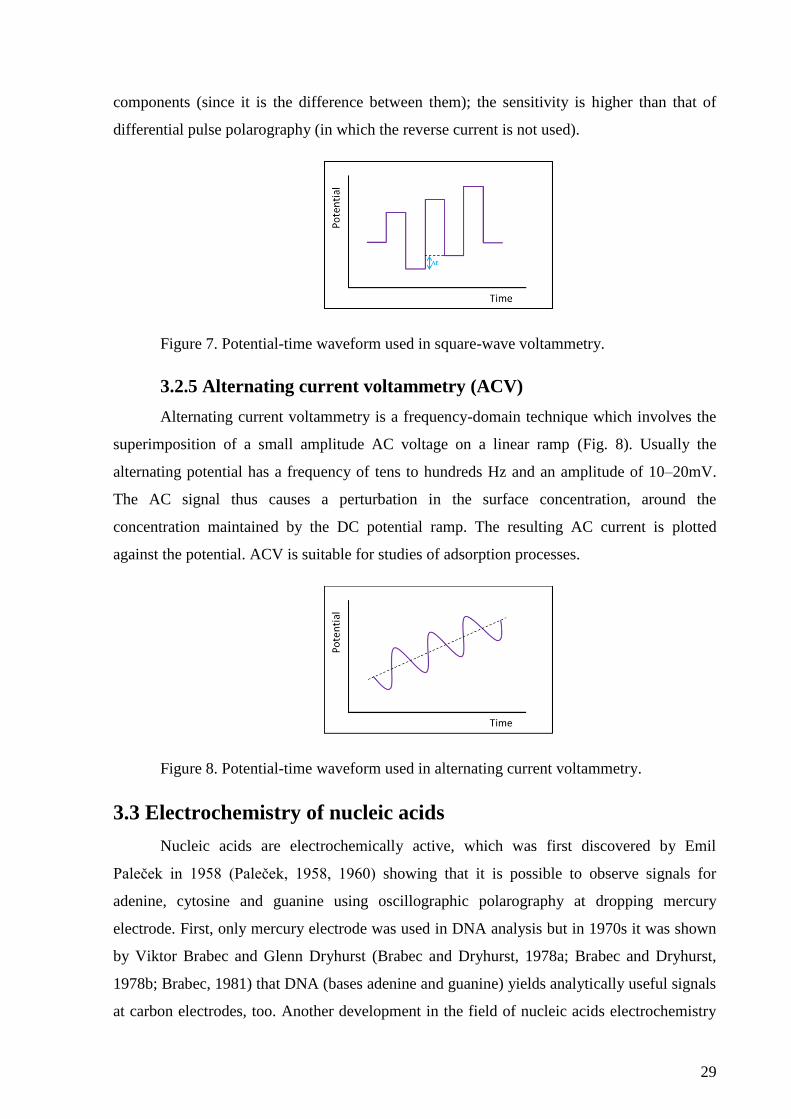
29
components (since it is the difference between them); the sensitivity is higher than that of
differential pulse polarography (in which the reverse current is not used).
Figure 7. Potential-time waveform used in square-wave voltammetry.
3.2.5 Alternating current voltammetry (ACV)
Alternating current voltammetry is a frequency-domain technique which involves the
superimposition of a small amplitude AC voltage on a linear ramp (Fig. 8). Usually the
alternating potential has a frequency of tens to hundreds Hz and an amplitude of 10–20mV.
The AC signal thus causes a perturbation in the surface concentration, around the
concentration maintained by the DC potential ramp. The resulting AC current is plotted
against the potential. ACV is suitable for studies of adsorption processes.
Figure 8. Potential-time waveform used in alternating current voltammetry.
3.3 Electrochemistry of nucleic acids
Nucleic acids are electrochemically active, which was first discovered by Emil
Paleček in 1958 (Paleček, 1958, 1960) showing that it is possible to observe signals for
adenine, cytosine and guanine using oscillographic polarography at dropping mercury
electrode. First, only mercury electrode was used in DNA analysis but in 1970s it was shown
by Viktor Brabec and Glenn Dryhurst (Brabec and Dryhurst, 1978a; Brabec and Dryhurst,
1978b; Brabec, 1981) that DNA (bases adenine and guanine) yields analytically useful signals
at carbon electrodes, too. Another development in the field of nucleic acids electrochemistry

30
was achieved by introduction of adsorptive transfer stripping technique which led to reduced
volume of the sample required for analysis and increased sensitivity (Paleček and
Postbieglová, 1986).
3.3.1 Adsorptive transfer stripping in DNA analysis
Generally, stripping techniques are used for analyses of traces of metals. The
procedure is based on preconcentration of the analyte at the electrode surface and its
subsequent stripping. It is an extremely sensitive technique and it can lower the detection
limits by orders of magnitude compared to solution-phase voltammetric measurements
(Wang, 2001b). Besides metals, many inorganic and organic molecules that exhibit surface-
active properties can be analyzed using stripping techniques, including nucleic acids and
proteins. Depending on their redox activity, the adsorbed organic compounds can be observed
owing to their oxidation or reduction. Nonelectroactive macromolecules may also be
determined following their interfacial accumulation from tensammetric peaks.
In DNA analysis, adsorptive transfer stripping voltammetry (AdTSV) is often used,
which involves adsorption of DNA at the electrode surface from a small droplet of sample
(usually a few microliters) during open circuit, transfer of the electrode with the adsorbed
layer into a new medium containing blank electrolyte and subsequent voltammetric analysis
(Fojta et al., 2008). This approach takes advantage of the adsorption (strong enough to resist
exchange of the media) of DNA bases and sugar-phosphate backbone on surfaces of both
mercury and carbon electrodes (Brabec and Paleček, 1972; Brabec and Dryhurst, 1978a). This
technique has many advantages compared to conventional electrochemical analysis with the
analyte diluted in the background electrolyte. Probably the most important feature is that it
substantially reduces amount of the sample required for the analysis, which is often difficult
to obtain. Another advantage lies in the fact that DNA can be adsorbed from a solution which
differs from the electrolyte and is not suitable for electrochemical measurements and therefore
it is possible to separately optimize conditions for the adsorption and for the electrochemical
measurement. During the washing step possible interfering low molecular weight substances
can be removed. AdTSV also enables to exploit the differences in adsorbability of substances
on the electrode surface and to separate them accordingly. Moreover, it makes it possible to
study the interaction of biomacromolecules immobilized on the surface of the electrode with
substances contained in the solution without the results of the voltammetric measurement
being affected by the interactions in the bulk of the solution and to study the effect of
electrode potential on the properties and interactions of the adsorbed macromolecules

31
(Paleček and Postbieglová, 1986). Electrodes with adsorbed DNA layer can be used as simple
electrochemical biosensors (Paleček and Bartošík, 2012).
3.3.2 Analysis of DNA structure at mercury electrodes
When applying negative potentials at the mercury electrode, DNA adsorbed at the
electrode surface undergoes adsorption/desorption and reorientation processes which can be
observed as tensammetric peaks. Such peaks can provide valuable information on properties
of the studied DNA, especially its structure. For this purpose, alternating current voltammetry
(ACV) in weakly alkaline media is used.
At negative potentials, repulsion between negatively charged electrode surface and
negatively charged sugar-phosphate backbone of the DNA occurs, which can result in partial
desorption of the DNA molecules from the electrode surface. This leads in changes in
capacity of the electrode double layer, which can be observed as tensammetric signals. The
DNA structure influences which part of the DNA molecule participates in the
adsorption/desorption processes, which results in various tensammetric peaks (Fojta, 2004;
Palecek and Fojta, 2005).
At the AC voltammogram, three distinct tensammetric peaks can be observed. Peak 1
occurs at a potential around -1.2 V and is caused by desorption of the sugar-phosphate
backbone. At a potential around -1.4 V peak 3, which is caused by desorption of DNA bases,
can be observed. This peak occurs only if the DNA molecule is single stranded or contains
single stranded regions. Another tensammetric peak, peak 2, occurs at -1.3 V and corresponds
to changes in double stranded DNA during which distorted regions in the dsDNA molecule
are adsorbed via bases (Fojta et al., 1998; Fojta, 2004; Paleček and Bartošík, 2012).
The tensammetric signals can be applied as a sensitive tool for detection of DNA
damage. Covalently closed circular DNA such as supercoiled plasmid DNA (scDNA) does
not provide peak 3 since the bases are not accessible to the electrode surface. After
introduction of a strand break into DNA the DNA strands begin to unwind, which allows
adsorption of bases onto the electrode surface and peak 3 can thus be observed. This approach
enables to study various genotoxic agents and factors such as -radiation or hydroxyl radicals
and their influence on formation of single stand breaks (Fojta and Paleček, 1997).
3.3.3 DNA signals at mercury electrodes
Since their potential window is very wide at the negative side, mercury electrodes are
suitable for studying reduction of nucleic acids and their components. Adenine, cytosine, 5-

32
methylcytosine and guanine can be reduced at mercury electrodes. Adenine and cytosine are
reduced at very similar potential around -1.5 V (against Ag|AgCl|3M KCl reference electrode,
as all other potentials stated here), depending on pH of the electrolyte, and provide a common
cathodic peak CA (Paleček and Bartošík, 2012). Under certain conditions, especially when C
and A form homooligonucleotide blocks, it is possible to resolve their overlapping reduction
peaks using elimination voltammetry with linear scan (Trnková et al., 2003, 2006; Mikelová
et al., 2007). 5-methylcytosine is reduced at the same potential as cytosine, therefore it is not
possible to distinguish them when naturally occurring in DNA. Nevertheless, when cytosine
(unlike 5-methylcytosine) is converted to uracil using bisulfate treatment, decrease in C
reduction peak can be detected, which enables to observe levels of DNA methylation
(Bartošík et al., 2012).
Guanine can be reduced at highly negative potentials but its reduction peak cannot be
observed as it is overlapped by hydrogen background discharge. Guanine is reduced to 7,8-
dihydroguanine which can be oxidized during anodic scan in cyclic voltammetry providing
peak G at about -0.25 V (Trnková et al., 1980; Studničková et al., 1989). Recently it has been
proposed that electrochemically or electrocatalytically generated hydrogen radicals are
involved in chemical reduction of guanine to 7,8-dihydroguanine (Daňhel et al., 2016).
Thymine and uracil can be reduced at highly negative potentials at mercury electrodes
only in nonaqueous solution – it was shown that reduction of thymine (Cummings and Elving,
1979) and uracil (Cummings and Elving, 1978) can be observed in dimethylsulfoxide, which
is not convenient for the common DNA analysis.
Reduction signals of DNA bases at mercury electrodes are strongly dependent on the
DNA structure. While in single-stranded DNA (ssDNA) cytosine and adenine are accessible
to the electrode surface, in double-stranded DNA (dsDNA) their reduction sites are hidden
inside the double helix as they participate in the Watson-Crick base pairing (Fig. 9). This
results in substantially lower CA peak in dsDNA compared to ssDNA. The same applies to
guanines when forming G-quadruplexes. Hoogsteen-paired guanine residues in the
quadruplexes have only limited accessibility for the reduction process at the negatively
charged surface of the mercury electrode, which can be observed as decrease in the G peak
intensity when G-quadruplexes are formed (Vidláková et al., 2015).

33
Figure 9. Location of primary reduction sites of G, C and A at mercury electrodes (magenta
rectangles) and primary oxidation sites of G and A at carbon electrodes (blue circles).
3.3.4 DNA signals at carbon electrodes
Carbon electrodes are (unlike mercury electrodes) suitable for studying oxidation
processes when applying positive potentials. Adenine and guanine are oxidized at carbon
electrodes and they provide oxidation signals at potentials about 1.1 V for guanine and 1.3-1.4
V for adenine, which was first discovered in the 1970s (Dryhurst and Pace, 1970; Brabec and
Dryhurst, 1978a, 1978b; Brabec, 1981). These signals are the most frequently analytically
used signals provided by DNA at carbon electrodes (Paleček and Bartošík, 2012). Pyrimidine
bases can be also oxidized but their oxidation occurs at more positive potentials and the
signals thus interfere with the background currents caused by anodic oxygen evolution, which
hinder the detection of the oxidation peaks (Brotons et al., 2016). However, several
approaches have been developed that enable simultaneous detection of oxidation of all four
natural nucleotides, involving use of differential pulse voltammetry at glassy carbon electrode
(Oliveira-Brett et al., 2004) or use of chemically reduced graphene oxide modified glassy
carbon electrode, which enables to study DNA bases in both single-stranded DNA and
double-stranded DNA without a prehydrolysis step (Zhou et al., 2009).
Recently it has been shown in our lab for the first time that carbon electrodes, namely
pyrolytic graphite electrode in basal plane orientation, are suitable for studying not only
anodic oxidation processes but also cathodic reduction of the nucleobases up to potential of -
2.0 V. Moreover, products of irreversible oxidation/reduction of the parent bases were shown

34
to yield analytically useful, base-specific cathodic/anodic signals, making it possible to
distinguish between the canonical bases (adenine, cytosine, guanine and thymine), uracil and
5-methylcytosine in DNA without need of DNA hydrolysis or electrode modification, which
makes it an excellent tool for simultaneous label-free analysis of bases in DNA (Špaček et al.,
2017).
Although they are not natural DNA bases, it is worth mentioning that 7-deazapurines
(analogs of natural purine bases in which N7 atom is replaced by CH group) provide
oxidation signals at carbon electrodes, too. Both 7-deazaguanine (G*) and 7-deazaadenine
(A*) exhibit significantly lower potentials of their oxidation, compared to the respective
natural nucleobases. G* is oxidized at a potential around 0.8 V, which is about 300 mV less
positive than oxidation signal of G, A* at a potential around 1.1 V, which is also about 200-
300 mV less positive than potential at which A is oxidized and moreover, it overlaps with the
oxidation peak of G (Pivoňková et al., 2010). At mercury electrodes, A* can be reduced
giving rise to a similar irreversible cathodic peak as the natural A and G* does not yield any
peak analogous to the peak G due to guanine, in agreement with a loss of corresponding redox
site in G* (Dudová et al., 2016).
Generally, oxidation peaks gained at carbon electrodes are not as sensitive to changes
in DNA structure as reduction and tensammetric peaks obtained at mercury electrodes
(Paleček and Bartošík, 2012; Brotons et al., 2016).
3.4 Electrochemical labeling of nucleic acids
Although DNA possesses intrinsic electroactivity and their redox and tensammetric
signals can be used for various analytical applications, sometimes it is advantageous to use
DNA labeling by various chemical moieties which can be electrochemically detected. Using
electroactive DNA labeling has the following advantages compared to analysis of natural
DNA:
- Using DNA labels increases both sensitivity and selectivity of the analysis (Fojta
et al., 2007; Hocek and Fojta, 2011), enabling detection of the labeled DNA
species in the sample even if the unlabelled DNA is overabundant.
- DNA labels usually provide their signals at potentials that are not as extreme as in
the case of DNA bases, which are reduced or oxidized at very high negative or
positive potentials (Paleček and Bartošík, 2012).

35
- Unlike unlabeled DNA, which has been analyzed practically only at mercury- and
carbon-based electrodes, the DNA labels offer a wider range of possibilities as to
which electrode material can be used for the analysis. For example gold electrodes,
not very suitable for measuring DNA as only guanine signal can be studied
(Ferapontova and Domínguez, 2003), can be used for construction of DNA
hybridization sensors using probe oligonucleotides immobilized at the electrode
surface using thiol linkers (Flechsig and Reske, 2007; Surkus and Flechsig, 2009;
Jacobsen and Flechsig, 2013).
DNA can be labeled in various ways: DNA labels can either noncovalently interact
with DNA through groove binding or intercalation, or can covalently bind accessible reactive
groups in DNA (or RNA) such as in the case of osmium tetroxide complexes (Flechsig and
Reske, 2007; Fojta et al., 2007, 2011; Trefulka et al., 2007; Havran et al., 2008). Another
possibility lies in enzymatic incorporation of electrochemically modified nucleotides (Hocek
and Fojta, 2011). Many applications have been presented that utilize DNA labeling, with the
most common ones being used in construction of DNA hybridization sensors (Jelen et al.,
2002; Fojta et al., 2003, 2004; Fojta et al., 2007; Horáková et al., 2011), sensors of DNA
structure (Palecek and Hung, 1983; Paleček, 1992a) or DNA damage (Fojta, 2002; Fojta et
al., 2016) and in detection of DNA-proteins interactions (for more details see section 2.4).
3.4.1 Noncovalently bound redox indicators
Generally, noncovalently bound indicators are structure-specific and therefore suitable
for discrimination of ssDNA and dsDNA immobilized at the electrode surface. They can bind
DNA in various modes: indicators with cationic nature that interact electrostatically with the
polyanionic DNA chain; indicators binding to DNA groove, such as Hoechst 33258; planar
aromatic molecules capable of intercalation between adjacent bases in the DNA doublehelix,
such as echinomycin (Jelen et al., 2002), actinomycin D and proflavine (Gebala et al., 2009)
or anthraquinone (Wong and Gooding, 2003); bisintercalators and threading intercalators with
increased selectivity for dsDNA compared to regular intercalators.
3.4.2 Osmium tetroxide complexes
Complexes of osmium tetroxide with nitrogenous ligands (Os,L) were the first used
covalently binding electroactive DNA labels. Osmium tetroxide reacts with C=C double
bonds. The process involves [3+2] addition of osmium tetroxide across the C=C double bond
giving rise to an osmic acid diester (glycolate), that is subsequently hydrolyzed to the glycol
moiety and osmate. Analogous reactions are given by various compounds possessing the C=C

36
double bonds, including pyrimidine nucleobases (at C5=C6) and indole moiety featuring side
group of amino acid tryptophan (W, at C2=C3). It has been established that tertiary amines
stabilize the osmium(VI) glycolates upon coordination of the central osmium atom by the
nitrogenous ligands. Thus, products of modification of the pyrimidine or W residues with
osmium tetroxide complexes bearing the nitrogenous ligands are stable adducts retaining the
osmium moiety and the given ligand (Paleček, 1992b; Deubel, 2003; Fojta et al., 2011).
Pyridine (py) was the first nitrogenous ligand used in complexes with osmium tetroxide, later
more ligands have been presented such as 2,2’-bipyridine (bpy), 1,10- phenanthroline (phen)
derivatives or N,N,N’,N’-tetramethyl ethylenediamine (TEMED). These ligands influence the
potentials at which the Os,L complexes provide voltammetric signals, which was used in
“multicolor” DNA labeling enabling parallel analysis of multiple DNA targets (Fojta et al.,
2007).
In DNA, osmium tetroxide complexes react preferentially with pyrimidine
nucleobases, with thymine being most reactive, followed by uracil and cytosine (Reske et al.,
2009). The reaction proceeds only when the C5=C6 double bond is accessible to the Os,L
complex, which happens only in the case of single-stranded DNA. This makes Os,L
complexes excellent tool for probing DNA structure as they can very precisely discriminate
between ssDNA and dsDNA (Jelen et al., 1991; Paleček, 1992b). Six-valent osmium in
complex with nitrogenous ligands has also been used for labeling nucleic acids but instead of
reacting with pyrimidine nucleobases, it condensates with cis-diols of sugar residues, which
narrows it down (in the sense of NA labeling) to labeling of RNA. This was utilized for
labeling ribose at the 3’ end of RNA oligonucleotides (Trefulka et al., 2010).
Adducts of Os,L with DNA can be detected using different types of electrodes due to
the electrochemical activity of the central osmium atom, which can undergo several redox
processes. At mercury-based electrodes, Os,bpy modified DNA provides three reversible
faradaic peaks between 0 and -1 V and a catalytic peak, which appears close to the hydrogen
background discharge and is caused by catalytic hydrogen evolution by Os,L (Fig. 10). This
peak allows determination of labeled DNA in very low concentrations (Palecek and Hung,
1983; Yosypchuk et al., 2006; Paleček et al., 2009; Fojta et al., 2011). At carbon electrodes,
osmium tetroxide complexes also yield faradaic signals, which are analytically useful (Fojta et
al., 2003; Fojta et al., 2007). There is a sufficient difference between the potentials of free
Os,bpy and Os,bpy-DNA adducts, which enables to determine labeled DNA even in the
presence of unreacted Os,bpy when using an adsorptive transfer stripping voltammetric

37
procedure involving extraction of free Os,bpy from the electrode by chloroform (Fojta et al.,
2002).
Figure 10. Schematic representation of redox potentials of natural DNA bases, 7-
deazapurines and examples of electroactive moieties coupled to the nucleobases (Hocek and
Fojta, 2011).
3.4.3 Redox labels and their enzymatic incorporation
Another option for introduction of covalently bound label lies in enzymatic
incorporation of base-modified nucleotides into DNA. It has been shown already in 1981 that
DNA and RNA polymerases are able to incorporate modified nucleotides into the growing
chain (Langer et al., 1981). There are many polymerases, which have been tested for
incorporation of diverse modified nucleoside triphosphates (dNXTPs). In general, B-family
thermo-stable polymerases (Vent (exo-), Pwo, KOD) are the best enzymes tolerating the

38
presence of most modifications, A-family polymerases are much less suitable (Hocek and
Fojta, 2011). Apart from that, other enzymes have been found applicable for incorporation of
modified dNTPs, such as terminal deoxynucleotidyl transferase (TdT, for more details see
section 2.3) or reverse transcriptase (Adelfinskaya and Herdewijn, 2007).
Several methods can be used for incorporation of dNXTPs into DNA, such as primer
extension (PEX) during which the DNA polymerase incorporates nucleotides at the 3’-OH
end of the growing primer in the presence of a longer template. When one (or more) of the
natural dNTPs is replaced by a modified one (dNXTP), the polymerase incorporates the
modification at the position of the particular nucleobase. Using this method, one or several
modifications can be introduced at the end of the primer strand (Riedl et al., 2009; Hocek and
Fojta, 2011; Balintová et al., 2013; Simonova et al., 2014). Other methods suitable for
incorporation of dNXTPs into DNA comprise polymerase chain reaction (PCR) (Jäger et al.,
2005; Čapek et al., 2007), Nicking Enzyme Amplification Reaction (NEAR) (Menova et al.,
2013) or TdT tailing reaction (Arzumanov et al., 2000; Anne et al., 2007; Horáková et al.,
2011; Havranová-Vidláková et al., 2018).
Synthesis of the dNXTPs is done either in a classical way consisting of the synthesis of
a modified nucleoside followed by chemical triphosphorylation or by modification of
halogenated dNTPs by aqueous-phase cross-coupling reactions, which is often more efficient.
Modifications are usually linked to C5 of pyrimidine nucleotides or to C7 of 7-deazapurine
nucleotides because modifications in these positions affect DNA structure and function less
than in other positions. For example 8-substituted purine dNTPs are poor substrates for
polymerases. Modifications are either attached directly or via acetylene or phenylene linkers
(Hocek and Fojta, 2008, 2011).
The redox labels used for modification of DNA are chosen with respect to their
electroactivity, which means that their signals should not interfere with reduction or oxidation
signals of natural nucleobases or with the background electrolyte discharge. The redox
processes often include multiple electrons and can be reversible. Examples of suitable redox
labels (for some of them, see Fig. 10) used for enzymatic incorporation into DNA in our lab
in cooperation with Michal Hocek’s lab at IOCB Prague, are: ferrocene (Brázdilová et al.,
2007), Os(bpy)3 and Ru(bpy)3 complexes (Vrábel et al., 2009), aminophenyl and nitrophenyl
(Cahová et al., 2008), tetrathiafulvalene (Riedl et al., 2009), anthraquinone (Balintová et al.,
2011), benzofurazane (Balintová et al., 2013), methoxyphenol and dihydrobenzofuran

39
(Simonova et al., 2014), azidophenyl (Daňhel et al., 2016) and phenothiazine (Simonova et
al., 2017).

40
3. Aims of the dissertation
1) Development of a new method for detection of protein-DNA interactions based on dual
redox labeling of DNA.
2) Electrochemical study of products of terminal deoxynucleotidyl transferase tailing
reaction.
3) Modification of butylacrylate conjugates in single- and double-stranded DNA with
osmium tetroxide complex.

41
4. Results and discussion
4.2 Label-free voltammetric detection of products of terminal
deoxynucleotidyl transferase tailing reaction
Monika Hermanová, Pavlína Havranová-Vidláková, Anna Ondráčková, Swathi Senthil
Kumar, Richard Bowater, Miroslav Fojta
Appendix 2
In this work, terminal deoxynucleotidyl transferase (TdT) tailing reaction has been studied
using electrochemical methods. For the TdT tailing reaction, three types of
homooligonucleotides – A30, C30 and T30 – were used as primers to which various dNTPs
(dATP, dCTP, dGTP, dTTP, dA*TP and dG*TP or their combinations) were added to form
the tail. The tailing reaction was performed in five different primer:dNTP molar ratios: 1:1,
1:5, 1:10, 1:20 and 1:50; no enzyme control was performed in order to compare signals
gained for the tailing reaction products to those of the sole primer.
Guanine (G) and 7-deazaguanine (G*) were used for the tailing reaction with all three primers
- A30, C30 and T30 (Figure 16). A30+G combination revealed a significant difference between
the results gained at HMDE and PGE for the same reaction products (Fig. 16A). At PGE,
height of the GOX
peak increased with the increasing primer:dNTP ratio. In contrast, signals
gained at the HMDE showed a decreasing tendency from 1:5 to 1:50 ratio after the initial
growth between 1:1 and 1:5 ratio. This behavior at HMDE in contrast to what was observed at
PGE has been previously observed and was ascribed to formation of G-quadruplexes (G4)
(Vidláková et al., 2015). The denaturing PAGE autoradiogram also indicated formation of
G4. The formation of G4 structures has two aspects. Firstly, when the primer:nucleotide ratio
is sufficient to produce G-tails capable of G4 formation, the tailing reaction is stopped when
the G4 is formed because of the inaccessibility of the 3’-OH end to TdT. Secondly, the G4
formation changes behavior of the oligonucleotides at the mercury electrode surface since
mercury-containing electrodes are sensitive to changes in DNA structure. On the other hand,
oxidation of guanine at PGE depends merely on the length of the oligonucleotide molecules
and does not reflect any structural changes occurring in the molecules. Electrochemical
analysis at PGE thus provided comparable results as PAGE analysis. In the case of T30+G
(Figure 16C), the PGE and denaturing PAGE revealed the same pattern of the length of the

42
tailing reaction products as A30+G, indicating formation of G4, however these results could
not be complemented with analysis at HMDE because of prevailing behavior of T30
oligonucleotide at the mercury electrode surface. Unlike A30+G and T30+G, the C30+G tailing
reaction products (Figure 16B) did not form G4, and formed another DNA structure – DNA
hairpin since bases C and G are complementary. For comparison with behavior of the natural
guanine, 7-deazaguanine (G*) was used, which is an analogue of natural guanine incapable of
forming multi-stranded DNA structures such as triplexes or quadruplexes. Both voltammetric
analysis at PGE and PAGE analysis (Figure 16D) showed that, unlike in the case of A30+G
tailing products, length of the tailing products grew gradually with the rising ratio, which was
caused by the fact that G* cannot form G4. Practically the same results as for the A30+G*
were obtained for the T30+G* tailing products (Figure 16F) with similar pattern of the tailing
products lengths visible at PAGE autoradiogram and intensities of the G*OX
signal gained at
PGE. On the other hand, the pattern observed for C30+G* (Figure 16E) resembled more that
of the C30+G tailing products than those of A30+G* and T30+G*, which is in accordance with
the fact that the ability of G* to pair with C is not violated, which leads to hairpin formation
after a certain length of the tail is reached.
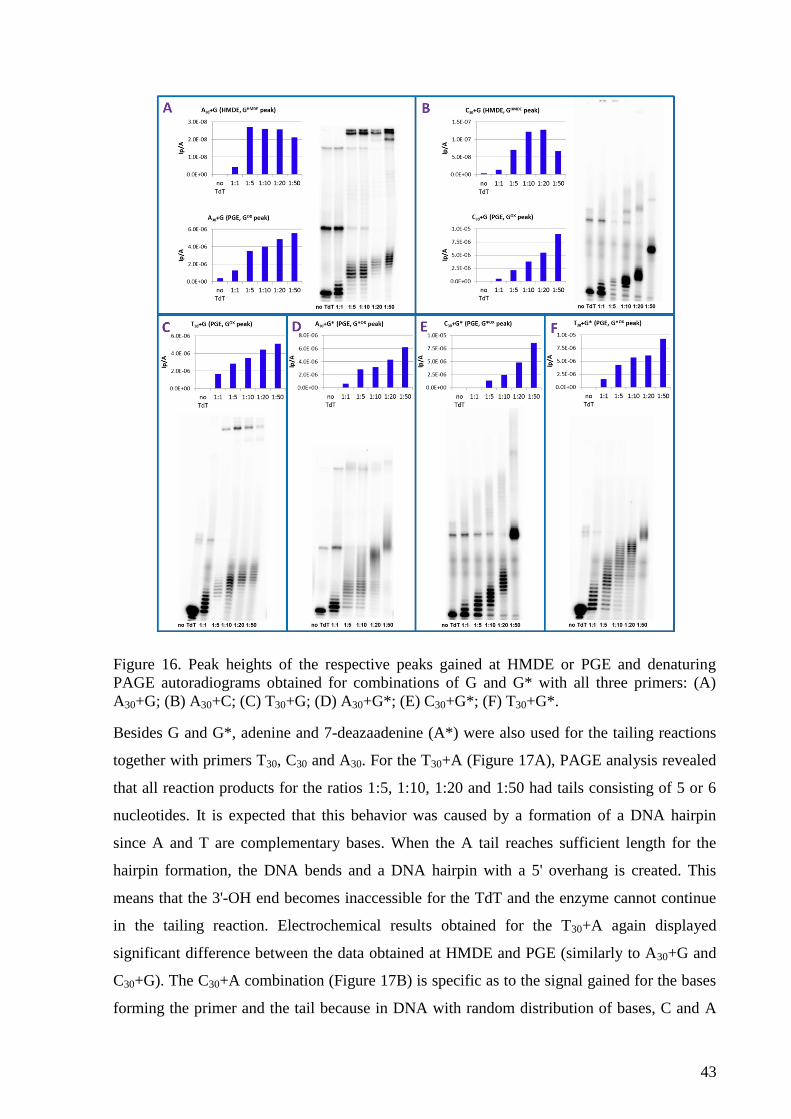
43
Figure 16. Peak heights of the respective peaks gained at HMDE or PGE and denaturing
PAGE autoradiograms obtained for combinations of G and G* with all three primers: (A)
A30+G; (B) A30+C; (C) T30+G; (D) A30+G*; (E) C30+G*; (F) T30+G*.
Besides G and G*, adenine and 7-deazaadenine (A*) were also used for the tailing reactions
together with primers T30, C30 and A30. For the T30+A (Figure 17A), PAGE analysis revealed
that all reaction products for the ratios 1:5, 1:10, 1:20 and 1:50 had tails consisting of 5 or 6
nucleotides. It is expected that this behavior was caused by a formation of a DNA hairpin
since A and T are complementary bases. When the A tail reaches sufficient length for the
hairpin formation, the DNA bends and a DNA hairpin with a 5' overhang is created. This
means that the 3'-OH end becomes inaccessible for the TdT and the enzyme cannot continue
in the tailing reaction. Electrochemical results obtained for the T30+A again displayed
significant difference between the data obtained at HMDE and PGE (similarly to A30+G and
C30+G). The C30+A combination (Figure 17B) is specific as to the signal gained for the bases
forming the primer and the tail because in DNA with random distribution of bases, C and A
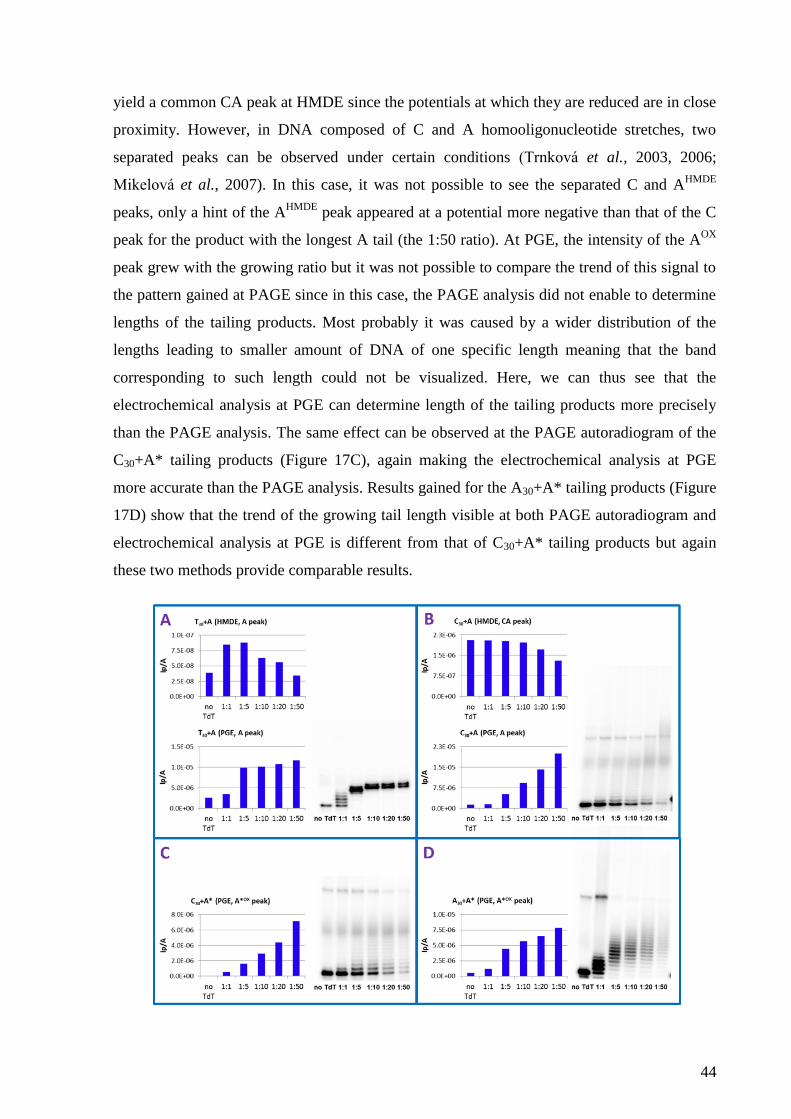
44
yield a common CA peak at HMDE since the potentials at which they are reduced are in close
proximity. However, in DNA composed of C and A homooligonucleotide stretches, two
separated peaks can be observed under certain conditions (Trnková et al., 2003, 2006;
Mikelová et al., 2007). In this case, it was not possible to see the separated C and AHMDE
peaks, only a hint of the AHMDE
peak appeared at a potential more negative than that of the C
peak for the product with the longest A tail (the 1:50 ratio). At PGE, the intensity of the AOX
peak grew with the growing ratio but it was not possible to compare the trend of this signal to
the pattern gained at PAGE since in this case, the PAGE analysis did not enable to determine
lengths of the tailing products. Most probably it was caused by a wider distribution of the
lengths leading to smaller amount of DNA of one specific length meaning that the band
corresponding to such length could not be visualized. Here, we can thus see that the
electrochemical analysis at PGE can determine length of the tailing products more precisely
than the PAGE analysis. The same effect can be observed at the PAGE autoradiogram of the
C30+A* tailing products (Figure 17C), again making the electrochemical analysis at PGE
more accurate than the PAGE analysis. Results gained for the A30+A* tailing products (Figure
17D) show that the trend of the growing tail length visible at both PAGE autoradiogram and
electrochemical analysis at PGE is different from that of C30+A* tailing products but again
these two methods provide comparable results.

45
Figure 17. Peak heights of the respective peaks gained at HMDE or PGE and denaturing
PAGE autoradiograms obtained for combinations of A and A* with all three primers: (A)
T30+A; (B) C30+A; (C) C30+A*; (D) A30+A*.
PAGE analysis of tailing products having tails consisting of cytosines (T30+C – Fig. 18A and
A30+C – 18B) showed that the length of the products grows with the growing ratio but even at
the highest ratios, the products are not as long as for other bases. Cyclic voltammetry at
HMDE was used for their analysis. For T30+C, a tensammetric peak caused by the T30 primer
(TTENS
) was observed but although it appeared at a potential close to that of the C peak, it was
possible to distinguish these two peaks as the potential at which it occurred was about 400
mV more negative than that of the C peak. In the case of A30+C, similar situation occurred as
for the C30+A, with C and A yielding a common reduction CA peak at HMDE. However, here
it was possible to determine the C peak for the higher ratios. With a growing number of
cytosines forming the tail, it was observed that the C peak appeared at a less negative
potential than the AHMDE
peak. For the 1:50 ratio, the peak was already well developed. The
peak heights gained for the CA peak showed no obvious trend and remained similar for the
primer and all the ratios. Nevertheless, an evident tendency to grow with the increasing ratio
was seen when the C peak was analyzed separately. Primers A30 and C30 were also used for
tailing reaction with thymines. The PAGE analysis of the A30+T tailing products (Fig. 18C)
showed that unlike for T30+A, the tailing reaction was not stopped after a few added
nucleotides and the tailing products gradually grew up to the 1:50 ratio. When analyzed at
HMDE, height of the A peak displayed decreasing tendency, which reflects decreasing
amount of the A30 parts of the tailing products that were adsorbed at the electrode surface due
to growing portion of the T-tails. The PAGE analysis of the C30+T (Fig. 18D) revealed the
same problem as for the C30+A and C30+A* combinations – no visible tailing products at the
autoradiogram. The voltammetric analysis of C30+T at HMDE confirmed that the cytosines
adsorb very strongly at the mercury surface (Hasoň et al., 2008), which is evident from the
fact that the C peak height remains unchanged for all the ratios.
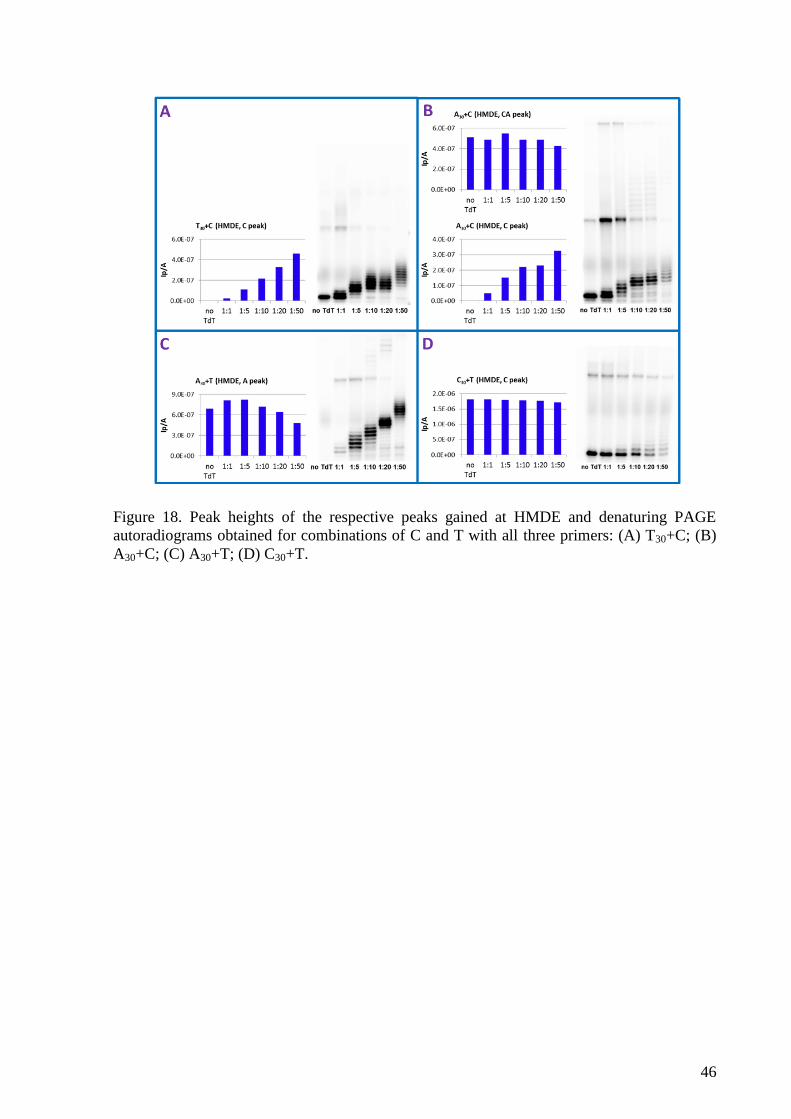
46
Figure 18. Peak heights of the respective peaks gained at HMDE and denaturing PAGE
autoradiograms obtained for combinations of C and T with all three primers: (A) T30+C; (B)
A30+C; (C) A30+T; (D) C30+T.

47
4.3 Butylacrylate-nucleobase Conjugates as Targets for Two-step
Redox Labeling of DNA with an Osmium Tetroxide Complex
Pavlína Havranová-Vidláková, Jan Špaček, Lada Vítová, Monika Hermanová, Jitka
Dadová, Veronika Raindlová, Michal Hocek, Miroslav Fojta and Luděk Havran
Appendix 3
In this work, butylacrylate- (BA) nucleobase (7-deaza adenine or uracil) conjugates
incorporated into DNA oligonucleotides have been tested for use in modification with
osmium tetroxide complex (Os,bpy). Single-stranded oligonucleotides bearing the dNBA
conjugates were prepared using terminal deoxynucleotidyl transferase (TdT) tailing reaction
(Figure 19, path a). In contrast to primer extension (PEX) technique with a template
dependent DNA polymerase (Figure 19, path b), which was successfully applied to construct
the dNBA
-modified DNA in our previous work (Dadová et al., 2013), the TdT tailing
technique has been tested here for the first time.
Fig. 19. Scheme of introduction of a single dNBA
nucleotide at the 3’-terminus of a
single-stranded initiator (path a) or templated PEX synthesis of ds ON bearing four dNBA
nucleotides (path b); and osmylation of the acrylate moieties in ss and ds DNA.

48
Denaturing polyacrylamide gel electrophoresis analysis showed that TdT tailing
reaction was successful in adding both unlabeled and BA-labeled dNTPs to the dA20 primer
(Figure 20). Ex situ AdTS SWV at PGE was used to analyze electrochemically the TdT
tailing products after treating them with 2 mM Os,bpy (Figure 20b,c). The dA20 initiator, in
agreement with the absence of any Os,bpy-reactive nucleobases in it, did not give any adduct-
specific peak and the same applies to the TdT tailing product with dATP (Fig, 20b). In
contrast, all the other TdT products (with dT, dU, dA*, dABA
and dUBA
) yielded well
developed signals in the region of peak , which differed in peak potentials as well as in
intensities. Peak heights obtained after Os,bpy modification of individual TdT tailing
products exhibited significant differences, with the most intense response recorded for dABA
and only negligible peak a observed for dU (Figure 20b,c). For both dABA
and dUBA
conjugates, significant increase of the signal was detected when compared to the respective
parent nucleotides dA* or dU, suggesting a considerable reactivity for the acrylate moiety
and/or modification of both C7=C8 and C2’’=C3’’ double bonds in the dABA
conjugate.
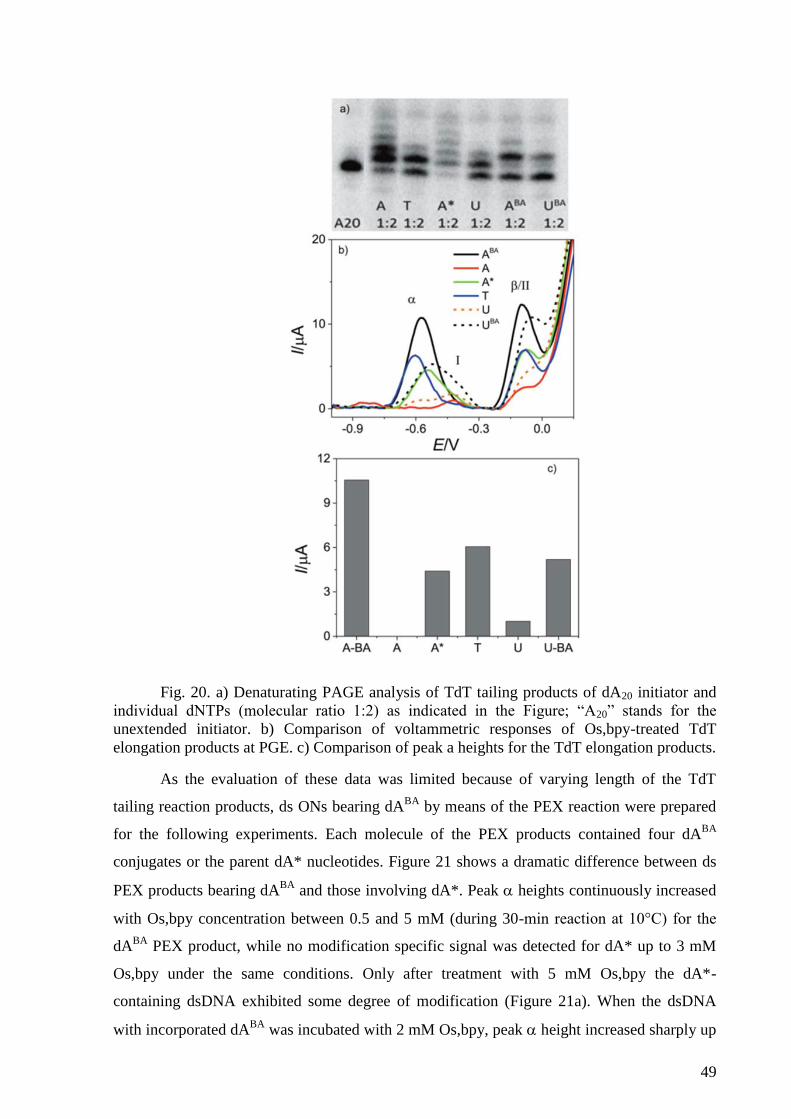
49
Fig. 20. a) Denaturating PAGE analysis of TdT tailing products of dA20 initiator and
individual dNTPs (molecular ratio 1:2) as indicated in the Figure; “A20” stands for the
unextended initiator. b) Comparison of voltammetric responses of Os,bpy-treated TdT
elongation products at PGE. c) Comparison of peak a heights for the TdT elongation products.
As the evaluation of these data was limited because of varying length of the TdT
tailing reaction products, ds ONs bearing dABA
by means of the PEX reaction were prepared
for the following experiments. Each molecule of the PEX products contained four dABA
conjugates or the parent dA* nucleotides. Figure 21 shows a dramatic difference between ds
PEX products bearing dABA
and those involving dA*. Peak heights continuously increased
with Os,bpy concentration between 0.5 and 5 mM (during 30-min reaction at 10°C) for the
dABA
PEX product, while no modification specific signal was detected for dA* up to 3 mM
Os,bpy under the same conditions. Only after treatment with 5 mM Os,bpy the dA*-
containing dsDNA exhibited some degree of modification (Figure 21a). When the dsDNA
with incorporated dABA
was incubated with 2 mM Os,bpy, peak height increased sharply up
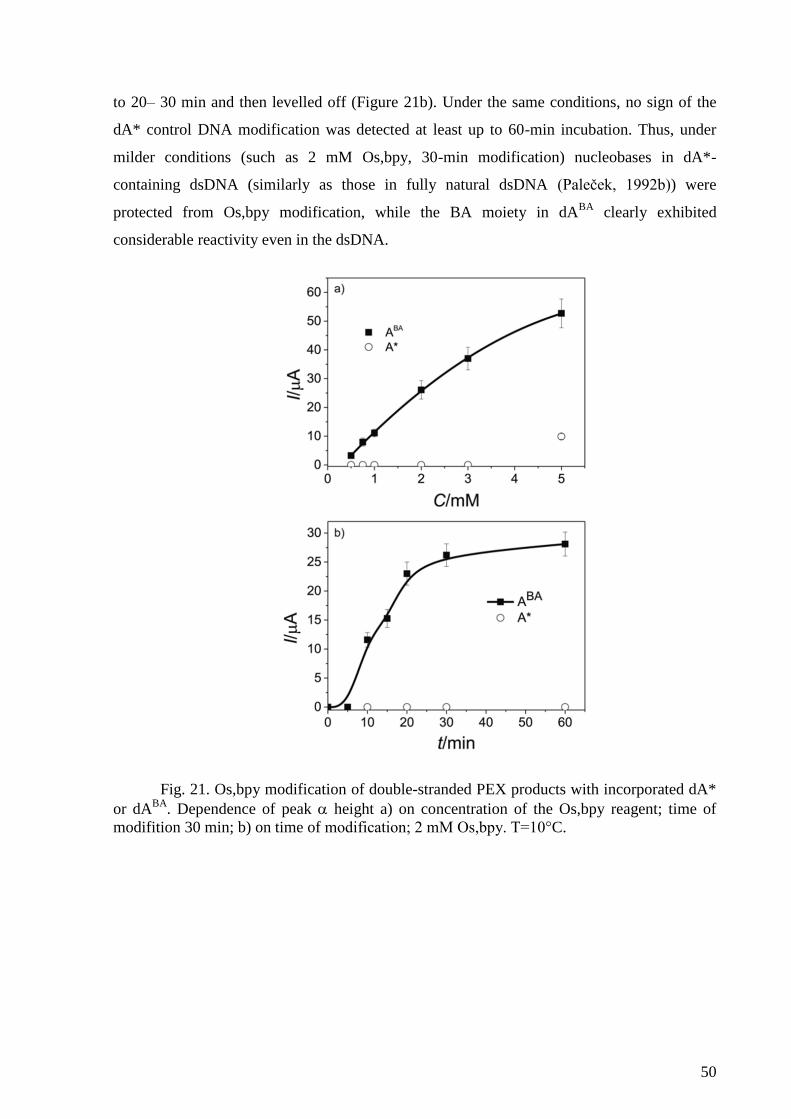
50
to 20– 30 min and then levelled off (Figure 21b). Under the same conditions, no sign of the
dA* control DNA modification was detected at least up to 60-min incubation. Thus, under
milder conditions (such as 2 mM Os,bpy, 30-min modification) nucleobases in dA*-
containing dsDNA (similarly as those in fully natural dsDNA (Paleček, 1992b)) were
protected from Os,bpy modification, while the BA moiety in dABA
clearly exhibited
considerable reactivity even in the dsDNA.
Fig. 21. Os,bpy modification of double-stranded PEX products with incorporated dA*
or dABA
. Dependence of peak height a) on concentration of the Os,bpy reagent; time of
modifition 30 min; b) on time of modification; 2 mM Os,bpy. T=10°C.

51
5. Conclusions
We have presented three novel applications based on electrochemistry of DNA, taking
advantages of either intrinsic signals provided by DNA at mercury and carbon electrodes or
labeling of DNA with various electroactive markers.
First application can detect interactions of p53 protein with DNA. The p53 protein was
used as model protein as it can bind DNA in various modes therefore its use is appropriate for
development of a new methodology for studying DNA-protein interactions. Moreover its
DNA binding modes are very well described and especially in our laboratory, its DNA
binding properties have been intensively studied. The developed approach is based on dual
labeling of DNA probes and magnetic beads-based immunoprecipitation assay with
subsequent electrochemical detection. The DNA probes were labeled with two redox markers
– one label was used for a DNA probe bearing a specific binding site for the p53 protein
(p53CON), second label for a DNA probe lacking such site. We showed that using the
competition arrangement with these two labeled DNA probes, the specific and non-specific
binding of the p53 protein could be easily distinguished. The competition binding experiment
can thus provide information on relative affinities of the protein to the different DNA probes.
Furthermore, the p53 binding to DNA can be modulated by specific monoclonal antibodies.
This approach can be, after selection of appropriate DNA probes and suitable monoclonal
antibodies, potentially applied to study DNA-binding properties of other proteins.
Second approach comprised monitoring of terminal deoxynucleotidyl transferase
tailing reactions using intrinsic electroactivity of nucleobases. Various combinations of
homooligonucleotide primers and dNTPs were used for the tailing reactions and signals of the
respective bases forming the tail were studied using hanging mercury drop electrode (HMDE)
and pyrolytic graphite electrode (PGE). PGE showed to be very suitable for precise
determination of the tailing reaction products length as it corresponded to results obtained
using denaturing polyacrylamide gel electrophoresis (PAGE) and in some cases it even
outperformed the PAGE analysis. However, this approach enabled not only to study the
tailing reaction as such but also to observe formation of DNA structures, which was possible
due to use of HMDE for the analysis since it is very sensitive to DNA structure. Signals
obtained for some of the tailing products at HMDE showed decreasing trend, which reflected
structural changes occurring in the tailing reaction products and formation of various DNA
structures, such as DNA hairpins and G-quadruplexes. Thus, this approach proves to be an

52
excellent tool for studying the TdT tailing reactions but also for exploring electrochemical
behavior of the DNA oligonucleotides and influence of their structure at electrode surfaces.
In the third application, a new two-step technique of DNA modification with the
electroactive osmium tetroxide complex (Os, bpy) has been developed. The technique consists
of polymerase construction of DNA bearing butylacrylate (BA) moieties attached to uracil or
to 7-deaza adenine, followed by chemical modification of a reactive C=C double bond in the
acrylate residue. The BA-functionalized oligonucleotides were prepared either by TdT tailing
reaction or primer extension reaction. This approach enabled modification of the BA
conjugates in both single- and double-stranded (ds) oligonucleotides under conditions when
modification within the nucleobase rings in ds DNA was not possible. Moreover, Os,bpy
adducts formed with particular nucleobases and the BA conjugates can be electrochemically
distinguished on the basis of different redox potentials. These results are promising for the
application of the general two-step procedure in redox labeling of double-stranded DNAs and
in studies of DNA and interactions changing the accessibility of reactive groups located
within the DNA major groove.

53
6. References
Adelfinskaya, O. and Herdewijn, P. (2007) ‘Amino acid phosphoramidate nucleotides as
alternative substrates for HIV-1 reverse transcriptase’, Angewandte Chemie - International
Edition, 46(23), pp. 4356–4358. doi: 10.1002/anie.200605016.
Anne, A. et al. (2007) ‘Enzymatic redox 3’-end-labeling of DNA oligonucleotide monolayers
on gold surfaces using terminal deoxynucleotidyl transferase (TdT)-mediated single base
extension’, J Am Chem Soc, 129(10), pp. 2734–2735. doi: 10.1021/ja067954v.
Arzumanov, A. A. et al. (2000) ‘Terminal deoxynucleotidyl transferase catalyzes the reaction
of DNA phosphorylation.’, Nucleic acids research, 28(5), pp. 1276–81. Available at:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=102600&tool=pmcentrez&rendert
ype=abstract%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/10666473%5Cnhttp://www.pubmed
central.nih.gov/articlerender.fcgi?artid=PMC102600.
Bai, L. and Zhu, W. (2006) ‘p53 : Structure , Function and Therapeutic Applications’, Journal
of Cancer Molecules, 2(4), pp. 141–153. doi: 10.1007/978-94-017-9211-0_16.
Bakalkin, G. et al. (1995) ‘p53 binds single-stranded DNA ends through the C-terminal
domain and internal DNA segments via the middle domain’, Nucleic Acids Research, 23(3),
pp. 362–369. doi: 10.1093/nar/23.3.362.
Balintová, J. et al. (2011) ‘Anthraquinone as a redox label for DNA: Synthesis, enzymatic
incorporation, and electrochemistry of anthraquinone-modified nucleosides, nucleotides, and
DNA’, Chemistry - A European Journal, 17(50), pp. 14063–14073. doi:
10.1002/chem.201101883.
Balintová, J. et al. (2013) ‘Benzofurazane as a new redox label for electrochemical detection
of DNA: Towards multipotential redox coding of DNA bases’, Chemistry - A European
Journal, 19(38), pp. 12720–12731. doi: 10.1002/chem.201301868.
Balintová, J. et al. (2015) ‘Azidophenyl as a click-transformable redox label of DNA suitable
for electrochemical detection of DNA–protein interactions’, Chem. Sci., 6(1), pp. 575–587.
doi: 10.1039/c4sc01906g.
Baltimore, D. (1974) ‘Is terminal deoxynucleotidyl transferase a somatic mutagen in
lymphocytes?’, Nature, 248, pp. 409–411.
Ban, C. et al. (2004) ‘Detection of protein-DNA interaction with a DNA probe: distinction
between single-strand and double-strand DNA-protein interaction.’, Nucleic acids research,
32(13), p. e110. doi: 10.1093/nar/gnh109.
Banks, C. E. and Compton, R. G. (2005) ‘Edge Plane Pyrolytic Graphite Electrodes in
Electroanalysis: An Overview’, Analytical Sciences, 21(11), pp. 1263–1268. doi:
10.2116/analsci.21.1263.
Bard, A. J. and Faulkner, L. R. (2001) Electrochemical methods: fundamentals and
applications, JohnWilley & Sons.
Bartošík, M., Fojta, M. and Paleček, E. (2012) ‘Electrochemical detection of 5-

54
methylcytosine in bisulfite-treated DNA’, Electrochimica Acta, 78, pp. 75–81. doi:
10.1016/j.electacta.2012.05.115.
Basu, M., Hegde, M. V. and Modak, M. J. (1983) ‘Synthesis of compositionally unique DNA
by terminal deoxynucleotidyl transferase’, Biochemical and Biophysical Research
Communications, 111(3), pp. 1105–1112. doi: 10.1016/0006-291X(83)91413-4.
Bates, S. and Vousden, K. H. (1996) ‘P53 in Signaling Checkpoint Arrest or Apoptosis’,
Current Opinion in Genetics and Development, 6(1), pp. 12–18. doi: 10.1016/S0959-
437X(96)90004-0.
Bollum, F. J. (1960) ‘Calf thymus polymerase.’, The Journal of biological chemistry, 235(8),
pp. 2399–2403.
Bollum, F. J. (1974) ‘Terminal Deoxynucleotidyl Transferase’, Enzymes, 10, pp. 145–171.
doi: 10.1016/S1874-6047(08)60137-7.
Bond, A. M. and Grabaric, B. S. (1979) ‘Fast Sweep Differential Pulse Voltammetry at a
Dropping Mercury Electrode with Computerized Instrumentation’, Analytical Chemistry,
51(1), pp. 126–128.
Boulé, J. B., Rougeon, F. and Papanicolaou, C. (2001) ‘Terminal Deoxynucleotidyl
Transferase Indiscriminately Incorporates Ribonucleotides and Deoxyribonucleotides’,
Journal of Biological Chemistry, 276(33), pp. 31388–31393. doi: 10.1074/jbc.M105272200.
Bourdon, J. C. et al. (2002) ‘Scotin, a novel p53-inducible proapoptotic protein located in the
ER and the nuclear membrane’, Journal of Cell Biology, 158(2), pp. 235–246. doi:
10.1083/jcb.200203006.
Bowater, R. P. et al. (2015) ‘Biophysical and electrochemical studies of protein–nucleic acid
interactions’, Monatshefte für Chemie - Chemical Monthly, 146(5), pp. 723–739. doi:
10.1007/s00706-014-1405-4.
Brabec, V. (1981) ‘Nucleic Acid Analysis by Voltammetry at Carbon Electrodes’,
Bioelectrochemistry and Bioenergetics, 128(8), pp. 437–449. doi: 10.1016/0302-
4598(81)80005-0.
Brabec, V. and Dryhurst, G. (1978a) ‘Electrochemical Behavior of Natural and Biosynthetic
Polynucleotides at Pyrolytic-Graphite Electrode a New Probe for Studies of Polynucleotide
Structure and Reactions’, Journal of Electroanalytical Chemistry, 89(1), pp. 161–173. doi:
Doi 10.1016/S0022-0728(78)80041-2.
Brabec, V. and Dryhurst, G. (1978) ‘Electrochemical behavior of natural and biosynthetic
polynucleotides at the pyrolytic graphite electrode. A new probe for studies of polynucleotide
structure and reactions’, Journal of Electroanalytical Chemistry, 89, pp. 161–173.
Brabec, V. and Dryhurst, G. (1978b) ‘Electrochemical Oxidation of Polyadenylic-Acid at
Graphite Electrodes’, Journal of Electroanalytical Chemistry, 91(2), pp. 219–229. doi: Doi
10.1016/S0022-0728(78)80102-8.
Brabec, V. and Paleček, E. (1972) ‘Adsorption of single‐ stranded and double‐ helical
polynucleotides on the mercury electrode’, Biopolymers, 11(12), pp. 2577–2589. doi:
10.1002/bip.1972.360111215.

55
Brázda, V. et al. (2006) ‘Searching for target sequences by p53 protein is influenced by DNA
length’, Biochemical and Biophysical Research Communications, 341(2), pp. 470–477. doi:
10.1016/j.bbrc.2005.12.202.
Brázdilová, P. et al. (2007) ‘Ferrocenylethynyl Derivatives of Nucleoside Triphosphates:
Synthesis, Incorporation, Electrochemistry, and Bioanalytical Applications’, Chemistry - A
European Journal. WILEY‐ VCH Verlag, 13(34), pp. 9527–9533. doi:
10.1002/chem.200701249.
Brázdová, M. et al. (2002) ‘Role of tumor suppressor p53 domains in selective binding to
supercoiled DNA.’, Nucleic acids research, 30(22), pp. 4966–74. Available at:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=137164&tool=pmcentrez&rendert
ype=abstract.
Brázdová, M. et al. (2009) ‘Modulation of gene expression in U251 glioblastoma cells by
binding of mutant p53 R273H to intronic and intergenic sequences’, Nucleic Acids Research,
37(5), pp. 1486–1500. doi: 10.1093/nar/gkn1085.
Brázdová, M. et al. (2016) ‘P53 specifically binds triplex DNA in vitro and in cells’, PLoS
ONE, 11(12), pp. 1–25. doi: 10.1371/journal.pone.0167439.
Brotons, A. et al. (2016) ‘Carbon materials for the electrooxidation of nucleobases,
nucleosides and nucleotides toward cytosine methylation detection: a review’, Analytical
Methods, 8(4), pp. 702–715. doi: 10.1039/c5ay02616d.
Brugarolas, J. et al. (1995) ‘Radiation-induced cell cycle arrest compromised by p21
deficiency’, Nature, 377, pp. 552–557.
Cadwell, C. and Zambetti, G. P. (2001) ‘The effects of wild-type p53 tumor suppressor
activity and mutant p53 gain-of-function on cell growth.’, Gene, 277(1–2), pp. 15–30. doi:
Doi 10.1016/S0378-1119(01)00696-5.
Cahová, H. et al. (2008) ‘Aminophenyl- and nitrophenyl-labeled nucleoside triphosphates:
Synthesis, enzymatic incorporation, and electrochemical detection’, Angewandte Chemie -
International Edition, 47(11), pp. 2059–2062. doi: 10.1002/anie.200705088.
Čapek, P. et al. (2007) ‘An Efficient Method for the Construction of Functionalized DNA
Bearing Amino Acid Groups through Cross-Coupling Reactions of Nucleoside Triphosphates
Followed by Primer Extension or PCR’, Chemistry - A European Journal. Wiley-Blackwell,
13(21), pp. 6196–6203. doi: 10.1002/chem.200700220.
Chang, M. S. and Bollum, F. J. (1990) ‘Multiple Roles of Divalent
Deoxynucleotidyltransferase Cation in the Terminal Reaction *’, The Journal of Biological
Chemistry, 265(29), pp. 17436–17440.
Cheng, G. et al. (2010) ‘A new electrochemically active-inactive switching aptamer
molecular beacon to detect thrombin directly in solution’, Biosensors and Bioelectronics.
Elsevier B.V., 25(10), pp. 2265–2269. doi: 10.1016/j.bios.2010.03.008.
Christova, R. (2013) Detecting DNA-protein interactions in living cells - ChIP approach. 1st
edn, Advances in Protein Chemistry and Structural Biology. 1st edn. Elsevier Inc. doi:
10.1016/B978-0-12-411637-5.00004-4.

56
Cummings, T. E. and Elving, P. J. (1978) ‘Electrochemical Reduction of Uracil in Dimethyl-
Sulfoxide - Autoprotonation of Anion Radical’, Journal of Electroanalytical Chemistry,
94(2), pp. 123–145. doi: Doi 10.1016/S0022-0728(78)80329-5.
Cummings, T. E. and Elving, P. J. (1979) ‘Electrochemical Reduction of Thymine in
Dimethyl-Sulfoxide Auto-Protonation of the Radical-Anion and Reduced Free-Radical’,
Journal of Electroanalytical Chemistry, 102(2), pp. 237–248.
Dadová, J. et al. (2013) ‘Aqueous heck cross-coupling preparation of acrylate-modified
nucleotides and nucleoside triphosphates for polymerase synthesis of acrylate-labeled DNA’,
Journal of Organic Chemistry, 78(19). doi: 10.1021/jo4011574.
Daňhel, A., Havran, L., et al. (2016) ‘Hydrogen Evolution Facilitates Reduction of DNA
Guanine Residues at the Hanging Mercury Drop Electrode: Evidence for a Chemical
Mechanism’, Electroanalysis, 28(11), pp. 2785–2790. doi: 10.1002/elan.201600242.
Daňhel, A., Trošanová, Z., et al. (2016) ‘Voltammetric analysis of 5-(4-Azidophenyl)-2’-
deoxycytidine nucleoside and azidophenyl-labelled single- and double-stranded DNAs’,
Electrochimica Acta, 215(August), pp. 72–83. doi: 10.1016/j.electacta.2016.08.096.
Deibel, M. R. and Coleman, M. S. (1980) ‘Biochemical properties of purified human terminal
deoxynucleotidyltransferase.’, The Journal of biological chemistry, 255(9), pp. 4206–12.
Available at: http://www.ncbi.nlm.nih.gov/pubmed/7372675.
Deng, C. et al. (1995) ‘Mice lacking p21CIP1/WAF1 undergo normal development, but are
defective in G1 checkpoint control.’, Cell, 82(4), pp. 675–684. doi: 10.1016/0092-
8674(95)90039-X.
Deng, G. and Wu, R. (1981) ‘An improved procedure for utilizing terminal transferase to add
homopolymers to the 3’ termini of DNA’, Nucleic Acids Research, 9(16), pp. 4173–4188.
DeRosa, M. C., Sancar, A. and Barton, J. K. (2005) ‘Electrically monitoring DNA repair by
photolyase.’, Proceedings of the National Academy of Sciences of the United States of
America, 102(31), pp. 10788–92. doi: 10.1073/pnas.0503527102.
Deubel, D. V. (2003) ‘Reactivity of osmium tetraoxide towards nitrogen heterocycles:
Implications for the molecular recognition of DNA mismatch’, Angewandte Chemie -
International Edition, 42(17), pp. 1974–1977. doi: 10.1002/anie.200250462.
Dey, B. et al. (2012) ‘DNA-protein interactions: Methods for detection and analysis’,
Molecular and Cellular Biochemistry, 365(1–2), pp. 279–299. doi: 10.1007/s11010-012-
1269-z.
Ding, C., Ge, Y. and Lin, J. M. (2010) ‘Aptamer based electrochemical assay for the
determination of thrombin by using the amplification of the nanoparticles’, Biosensors and
Bioelectronics, 25(6), pp. 1290–1294. doi: 10.1016/j.bios.2009.10.017.
Dötsch, V. et al. (2010) ‘P63 and P73, the Ancestors of P53.’, Cold Spring Harbor
perspectives in biology, 2(9). doi: 10.1101/cshperspect.a004887.
Dryhurst, G. and Pace, G. F. (1970) ‘Electrochemical Oxidation of Guanine at the Pyrolytic
Graphite Electrode’, Journal of The Electrochemical Society, 117(10), p. 1259. doi:
10.1149/1.2407283.

57
Dudová, Z. et al. (2016) ‘Electrochemical behavior of 7-deazaguanine- and 7-deazaadenine-
modified DNA at the hanging mercury drop electrode’, Monatshefte fur Chemie, 147(1), pp.
3–11. doi: 10.1007/s00706-015-1584-7.
El-Deiry, W. S. et al. (1992) ‘Definition of a consensus binding site for p53’, Nature
Genetics. Nature Publishing Group, 1(1), pp. 45–49. doi: 10.1038/ng0492-45.
Evtugyn, G. A. et al. (2008) ‘Potentiometric DNA sensor based on electropolymerized
phenothiazines for protein detection’, Electroanalysis, 20(12), pp. 1300–1308. doi:
10.1002/elan.200704186.
Fadrná, R. et al. (2004) ‘Voltammetric Determination of Adenine, Guanine, and DNA Using
Liquid Mercury Free Polished Silver Solid Amalgam Electrode’, Analytical Letters, 37(3), pp.
399–413. doi: 10.1081/AL-120028615.
Ferapontova, E. E. and Domínguez, E. (2003) ‘Direct electrochemical oxidation of DNA on
polycrystalline gold electrodes’, Electroanalysis, 15(7), pp. 629–634. doi:
10.1002/elan.200390079.
Fields, S. and Jang, S. K. (1990) ‘Presence of a Potent Transcription Activating Sequence in
the p53 Protein’, Science, 249(4972), pp. 1046–1049. doi: 10.1126/science.2144363.
Flechsig, G.-U. and Reske, T. (2007) ‘Electrochemical detection of DNA hybridization by
means of osmium tetroxide complexes and protective oligonucleotides’, Analytical
Chemistry. American Chemical Society, 79(5), pp. 2125–2130. doi: 10.1021/ac062075c.
Fojta, M. et al. (1998) ‘Two superhelix density-dependent DNA transitions detected by
changes in DNA adsorption/desorption behavior’, Biochemistry, 37(14), pp. 4853–4862. doi:
10.1021/bi9729559.
Fojta, M. (2002) ‘Electrochemical Sensors for DNA Interactions and Damage’,
Electroanalysis, 14(21), pp. 1449–1463.
Fojta, M. et al. (2002) ‘Voltammetric microanalysis of DNA adducts with osmium
tetroxide,2,2’-bipyridine using a pyrolytic graphite electrode’, Talanta, 56(5), pp. 867–874.
Fojta, M., Pivonkova, H., et al. (2003) ‘Recognition of DNA modified by antitumor cisplatin
by “latent” and “active” protein p53’, Biochemical Pharmacology, 65(8), pp. 1305–1316. doi:
10.1016/S0006-2952(03)00078-9.
Fojta, M., Havran, L., et al. (2003) ‘Two-surface strategy in electrochemical DNA
hybridization assays: Detection of osmium-labeled target DNA at carbon electrodes’,
Electroanalysis, 15(5–6), pp. 431–440. doi: 10.1002/elan.200390050.
Fojta, M., Pivonkova, H., et al. (2004) ‘Investigations of the supercoil-selective DNA binding
of wild type p53 suggest a novel mechanism for controlling p53 function’, European Journal
of Biochemistry, 271(19), pp. 3865–3876. doi: 10.1111/j.1432-1033.2004.04323.x.
Fojta, M. (2004) ‘Mercury Electrodes in Nucleic Acid Electrochemistry: Sensitive Analytical
Tools and Probes of DNA Structure. A Review’, Collection of Czechoslovak Chemical
Communications, 69(4), pp. 715–747. doi: 10.1135/cccc20040715.
Fojta, M., Havran, L., et al. (2004) ‘Multiply osmium-labeled reporter probes for

58
electrochemical DNA hybridization assays: Detection of trinucleotide repeats’, Biosensors
and Bioelectronics, 20(5), pp. 985–994. doi: 10.1016/j.bios.2004.06.015.
Fojta, M. (2005) ‘Use of DNA Repair Enzymes in Electrochemical Detection of Damage to
DNA Bases in Vitro and in’, Bioelectrochemistry, 77(9), pp. 2920–2927.
Fojta, M. et al. (2007) ‘“Multicolor” electrochemical labeling of DNA hybridization probes
with osmium tetroxide complexes’, Analytical Chemistry, 79(3), pp. 1022–1029. doi:
10.1021/ac0616299.
Fojta, M. et al. (2008) ‘Electrochemical stripping techniques in analysis of nucleic acids and
their constituents’, Current Analytical Chemistry, 4(3), pp. 250–262. doi:
10.2174/157341108784911415.
Fojta, M. et al. (2011) ‘Osmium tetroxide complexes as versatile tools for structure probing
and electrochemical analysis of biopolymers’, Current Analytical Chemistry, 7(1), pp. 35–50.
doi: 10.2174/157341111793797653.
Fojta, M. et al. (2016) ‘Recent progress in electrochemical sensors and assays for DNA
damage and repair’, TrAC - Trends in Analytical Chemistry, 79, pp. 160–167. doi:
10.1016/j.trac.2015.11.018.
Fojta, M. and Paleček, E. (1997) ‘Supercoiled DNA-modified mercury electrode: A highly
sensitive tool for the detection of DNA damage’, Analytica Chimica Acta, 342(1), pp. 1–12.
doi: 10.1016/s0003-2670(96)00551-x.
Foord, O., Navot, N. and Rotter, V. (1993) ‘Isolation and characterization of DNA sequences
that are specificially bound by wild-type p53 protein’, Molecular and Celluar Biology, 13, pp.
1378–1384.
Furey, T. S. (2012) ‘ChIP-seq and Beyond: new and improved methodologies to detect and
characterize protein-DNA interactions’, Nature Reviews Genetics, 13(12), pp. 840–852. doi:
10.1038/nrg3306.ChIP-seq.
Gebala, M. et al. (2009) ‘Label-Free Detection of DNA Hybridization in Presence of
Intercalators Using Electrochemical Impedance Spectroscopy’, Electroanalysis. Wiley-
Blackwell, 21(3–5), pp. 325–331. doi: 10.1002/elan.200804388.
Giono, L. E. and Manfredi, J. J. (2006) ‘The p53 Tumor Suppressor Participates in Multiple
Cell Cycle Checkpoints’, Journal of cellular physiology, 209(1), pp. 13–20. doi: 10.1002/JCP.
Göhler, T. et al. (2002) ‘Specific interaction of p53 with target binding sites is determined by
DNA conformation and is regulated by the C-terminal domain’, Journal of Biological
Chemistry, 277(43), pp. 41192–41203. doi: 10.1074/jbc.M202344200.
Gouge, J. et al. (2013) ‘Structures of intermediates along the catalytic cycle of terminal
deoxynucleotidyltransferase: Dynamical aspects of the two-metal ion mechanism’, Journal of
Molecular Biology. Elsevier Ltd, 425(22), pp. 4334–4352. doi: 10.1016/j.jmb.2013.07.009.
Green, D. R. and Kroemer, G. (2009) ‘Cytoplasmic functions of the tumor suppressor p53’,
Nature, 458(7242), p. 1127. doi: 10.1038/nature07986.Cytoplasmic.
Halazonetis, T. D., Davis, L. J. and Kandil, A. N. (1993) ‘Wild-type p53 adopts a ’mutant’-

59
like conformation when bound to DNA.’, The EMBO journal, 12(3), pp. 1021–8. doi:
10.1002/j.1460-2075.1993.tb05743.x.
Hasoň, S. and Vetterl, V. (2006) ‘Microanalysis of oligodeoxynucleotides by cathodic
stripping voltammetry at amalgam-alloy surfaces in the presence of copper ions’, Talanta,
69(3), pp. 572–580. doi: 10.1016/j.talanta.2005.10.029.
Hasoň, S., Vetterl, V. and Fojta, M. (2008) ‘Two-dimensional condensation of pyrimidine
oligonucleotides during their self-assemblies at mercury based surfaces’, Electrochimica Acta,
53(6), pp. 2818–2824. doi: 10.1016/j.electacta.2007.10.065.
Havran, L. et al. (2008) ‘Sensitive voltammetric detection of DNA damage at carbon
electrodes using DNA repair enzymes and an electroactive osmium marker’, Analytical and
Bioanalytical Chemistry, 391(5), pp. 1751–1758. doi: 10.1007/s00216-008-1850-1.
Havranová-Vidláková, P. et al. (2017) ‘Butylacrylate-nucleobase Conjugates as Targets for
Two-step Redox Labeling of DNA with an Osmium Tetroxide Complex’, Electroanalysis, pp.
371–377. doi: 10.1002/elan.201700702.
Havranová-Vidláková, P. et al. (2018) ‘Butylacrylate-nucleobase Conjugates as Targets for
Two-step Redox Labeling of DNA with an Osmium Tetroxide Complex’, Electroanalysis,
30(2). doi: 10.1002/elan.201700702.
Herne, T. M. and Tarlov, M. J. (1997) ‘Characterization of DNA Probes Immobilized on Gold
Surfaces’, J. Am. Chem. Soc., 119(13), pp. 8916–8920. doi: 10.1021/ja9719586.
Hippel, P. H. von (1994) ‘Protein-DNA Recognition: New Perspectives and Underlying
Themes’, Science, 263, pp. 769–770.
Hocek, M. and Fojta, M. (2008) ‘Cross-coupling reactions of nucleoside triphosphates
followed by polymerase incorporation. Construction and applications of base-functionalized
nucleic acids’, Org Biomol Chem, 6(13), pp. 2233–2241. doi: 10.1039/b803664k.
Hocek, M. and Fojta, M. (2011) ‘Nucleobase modification as redox DNA labelling for
electrochemical detection’, Chemical Society Reviews, 40(12), p. 5802. doi:
10.1039/c1cs15049a.
Horakova, P. et al. (2011) ‘Tail-labelling of DNA probes using modified deoxynucleotide
triphosphates and terminal deoxynucleotidyl transferase. Application in electrochemical DNA
hybridization and protein-DNA binding assays’, Org Biomol Chem. 2011/01/05, 9(5), pp.
1366–1371. doi: 10.1039/c0ob00856g.
Horáková, P. et al. (2011) ‘Tail-labelling of DNA probes using modified deoxynucleotide
triphosphates and terminal deoxynucleotidyl transferase. Application in electrochemical DNA
hybridization and protein-DNA binding assays.’, Organic & biomolecular chemistry, 9(5),
pp. 1366–71. doi: 10.1039/c0ob00856g.
Huang, H. et al. (2009) ‘DNA aptamer-based detection of lysozyme by an
electrochemiluminescence assay coupled to quantum dots’, Electrochemistry
Communications. Elsevier B.V., 11(4), pp. 816–818. doi: 10.1016/j.elecom.2009.01.009.
Hupp, T. R. (1999) ‘Regulation of p53 protein function through alterations in protein-folding
pathways’, Cellular and Molecular Life Sciences, 55(1), pp. 88–95. doi:

60
10.1007/s000180050272.
Hupp, T. R. and Lane, D. P. (1994) ‘Regulation of the cryptic sequence-specific DNA-
binding function of p53 by protein kinases’, Cold Spring Harbor Symposia on Quantitative
Biology, 59, pp. 195–206. doi: 10.1101/SQB.1994.059.01.024.
Jacobsen, M. and Flechsig, G.-U. (2013) ‘Hybridization Detection of Osmium Tetroxide
Bipyridine-Labeled DNA and RNA on Heated Gold Wire Electrodes’, Electroanalysis.
Wiley-Blackwell, 25(2), pp. 373–379. doi: 10.1002/elan.201200460.
Jagelská, E. B. et al. (2010) ‘The potential of the cruciform structure formation as an
important factor influencing p53 sequence-specific binding to natural DNA targets’,
Biochemical and Biophysical Research Communications, 391(3), pp. 1409–1414. doi:
10.1016/j.bbrc.2009.12.076.
Jäger, S. et al. (2005) ‘A versatile toolbox for variable DNA functionalization at high
density’, Journal of the American Chemical Society, 127(43), pp. 15071–15082. doi:
10.1021/ja051725b.
Jelen, F. et al. (1991) ‘Osmium tetroxide reactivity of DNA bases in nucleotide sequencing
and probing of DNA structure’, General Physiology and Biophysics, 10(5), pp. 461–473.
Jelen, F., Erdem, A. and Paleček, E. (2002) ‘Cyclic voltammetry of echinomycin and its
interaction with double-stranded and single-stranded DNA adsorbed at the electrode’,
Bioelectrochemistry, 55(1–2), pp. 165–167. doi: 10.1016/S1567-5394(01)00143-8.
Jen-Jacobson, L. (1997) ‘Protein-DNA recognition complexes: Conservation of structure and
binding energy in the transition state’, Biopolymers - Nucleic Acid Sciences Section, 44(2), pp.
153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U.
Kaelin, W. G. (1999) ‘The p53 gene family.’, Oncogene, 18(53), pp. 7701–7705. doi:
10.1038/sj.onc.1202955.
Kato, K.-I. et al. (1967) ‘Deoxynucleotide-polymerizing Thymus Gland Enzymes of Calf’,
The Journal of Biological Chemistry, 242(11), pp. 2780–2789.
Kawde, A. N. et al. (2005) ‘Label-free bioelectronic detection of aptamer-protein
interactions’, Electrochemistry Communications, 7(5), pp. 537–540. doi:
10.1016/j.elecom.2005.03.008.
Khoury, M. P. and Bourdon, J. (2010) ‘The Isoforms of the p53 Protein’, Cold Spring Harbor
perspectives in biology, pp. 1–11. doi: 10.1101/cshperspect.a000927.
Kim, E. and Deppert, W. (2003) ‘The complex interactions of p53 with target DNA : we learn
as we go’, Cell, 150, pp. 141–150. doi: 10.1139/O03-046.
Komori, T. et al. (1993) ‘Lack of N regions in antigen receptor variable region genes of TdT-
deficient lymphocytes’, Science, 261(5125), pp. 1171–1175. doi: 10.1126/science.8356451.
Kubbutat, M. H. G., Jones, S. N. and Vousden, K. H. (1997) ‘Regulation of p53 stability by
Mdm2’, Nature, 387, pp. 299–303.
Kunkel, T. a et al. (1986) ‘Rearrangements of DNA mediated by terminal transferase.’,
Proceedings of the National Academy of Sciences of the United States of America, 83(6), pp.

61
1867–71. doi: 10.1073/pnas.83.6.1867.
Lamb, P. and Crawford, L. (1986) ‘Characterization of the Human p53 Gene’, Molecular and
Cellular Biology, 6(5), pp. 1379–1385.
Lane, D. P. (1992) ‘Cancer. p53, guardian of the genome.’, Nature, pp. 15–16. doi:
10.1038/358015a0.
Lane, D. P. and Crawford, L. V (1979) ‘T antigen is bound to a host protein in SV40-
transformed cells.’, Nature, 278, pp. 261–263. doi: 10.1038/278261a0.
Langer, P. R., Waldrop, A. A. and Ward, D. C. (1981) ‘Enzymatic synthesis of biotin-labeled
polynucleotides: novel nucleic acid affinity probes.’, Proceedings of the National Academy of
Sciences, 78(11), pp. 6633–6637. doi: 10.1073/pnas.78.11.6633.
Latonen, L. and Laiho, M. (2005) ‘Cellular UV damage responses - Functions of tumor
suppressor p53’, Biochimica et Biophysica Acta - Reviews on Cancer, 1755(2), pp. 71–89.
doi: 10.1016/j.bbcan.2005.04.003.
Levine, A. J. (1997) ‘P53, the Cellular Gatekeeper for Growth and Division’, Cell, 88(3), pp.
323–331. doi: 10.1016/S0092-8674(00)81871-1.
Liu, F. et al. (2015) ‘An electrochemical method to assay human 8-oxoguanine DNA
glycosylase 1’, Electrochemistry Communications. Elsevier B.V., 50, pp. 51–54. doi:
10.1016/j.elecom.2014.11.011.
Masařík, M. et al. (2007) ‘Label-free voltammetric detection of single-nucleotide mismatches
recognized by the protein MutS’, Analytical and Bioanalytical Chemistry, 388(1), pp. 259–
270. doi: 10.1007/s00216-007-1181-7.
Mazur, S. J. et al. (1999) ‘Preferential binding of tumor suppressor p53 to positively or
negatively supercoiled DNA involves the C-terminal domain.’, Journal of molecular biology,
292(2), pp. 241–249. doi: 10.1006/jmbi.1999.3064.
McKinney, K. and Prives, C. (2002) ‘Efficient specific DNA binding by p53 requires both its
central and C-terminal domains as revealed by studies with high-mobility group 1 protein.’,
Molecular and cellular biology, 22(19), pp. 6797–808. doi: 10.1128/MCB.22.19.6797.
Menova, P., Raindlova, V. and Hocek, M. (2013) ‘Scope and Limitations of the Nicking
Enzyme Amplification Reaction for the Synthesis of Base-Modified Oligonucleotides and
Primers for PCR’, Bioconjugate Chemistry, 24(6), pp. 1081–1093. doi: 10.1021/bc400149q.
Meunier-Prest, R. et al. (2010) ‘Electrochemical probe for the monitoring of DNA-protein
interactions’, Biosensors and Bioelectronics. Elsevier B.V., 25(12), pp. 2598–2602. doi:
10.1016/j.bios.2010.04.023.
Michael, D. and Oren, M. (2003) ‘The p53-Mdm2 module and the ubiquitin system’,
Seminars in Cancer Biology. Elsevier, 13(1), pp. 49–58. doi: 10.1016/S1044-579X(02)00099-
8.
Michelson, A. M. and Orkin, S. H. (1982) ‘Characterization of the homopolymer tailing
reaction catalyzed by terminal deoxynucleotidyl transferase. Implications for the cloning of
cDNA.’, Journal of Biological Chemistry, 257(24), pp. 14773–14782.

62
Mickelsen, S. et al. (1999) ‘Modulation of terminal deoxynucleotidyltransferase activity by
the DNA-dependent protein kinase.’, Journal of immunology, 163(2), pp. 834–843.
Mikelová, R. et al. (2007) ‘Resolution of overlapped reduction signals in short hetero-
oligonucleotides by elimination voltammetry’, Electroanalysis, 19(2–3), pp. 348–355. doi:
10.1002/elan.200603739.
Modak, M. J. (1978) ‘Biochemistry of Terminal Deoxynucleotidyltransferase: Mechanism of
Inhibition by Adenosine 5ʹ-Triphosphate’, Biochemistry, 17(15), pp. 3116–3120. doi:
10.1021/bi00608a027.
Motea, E. A. and Berdis, A. J. (2010) ‘Terminal deoxynucleotidyl transferase: The story of a
misguided DNA polymerase’, Biochimica et Biophysica Acta - Proteins and Proteomics.
Elsevier B.V., 1804(5), pp. 1151–1166. doi: 10.1016/j.bbapap.2009.06.030.
Němcová, K. et al. (2010) ‘A label-free electrochemical test for DNA-binding activities of
tumor suppressor protein p53 using immunoprecipitation at magnetic beads’, Analytica
Chimica Acta, 668(2), pp. 166–170. doi: 10.1016/j.aca.2010.04.018.
Němcová, K. et al. (2014) ‘Electrochemical detection of DNA binding by tumor suppressor
p53 protein using osmium-labeled oligonucleotide probes and catalytic hydrogen evolution at
the mercury electrode’, Analytical and Bioanalytical Chemistry, 406(24), pp. 5843–5852. doi:
10.1007/s00216-014-7996-0.
Oda, E. (2000) ‘Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of
p53-Induced Apoptosis’, Science, 288(5468), pp. 1053–1058. doi:
10.1126/science.288.5468.1053.
Oliveira-Brett, A. M. et al. (2004) ‘Voltammetric determination of all DNA nucleotides’,
Anal Biochem, 332(2), pp. 321–329. doi: 10.1016/j.ab.2004.06.021.
Ono, K. (1990) ‘Inhibitory effects of various 2′, 3′-dideoxynucleoside 5′-triphosphates on the
utilization of 2′-deoxynucleoside 5′-triphosphates by terminal deoxynucleotidyltransferase
from calf thymus’, BBA - Gene Structure and Expression, 1049(1), pp. 15–20. doi:
10.1016/0167-4781(90)90078-G.
Oren, M. (1999) ‘Regulation of the p53 Tumor Suppressor Protein’, Journal of Biological
Chemistry, 274(51), pp. 36031–36034. doi: 10.1074/jbc.274.51.36031.
Ostatná, V. et al. (2017) ‘Chronopotentiometric sensing of specific interactions between
lysozyme and the DNA aptamer’, Bioelectrochemistry, 114, pp. 42–47. doi:
10.1016/j.bioelechem.2016.12.003.
Owen-Schaub, L. B. et al. (1995) ‘Wild-type human p53 and a temperature-sensitive mutant
induce Fas/APO-1 expression.’, Molecular and Cellular Biology, 15(6), pp. 3032–3040. doi:
10.1128/MCB.15.6.3032.
Palecek, E. et al. (2001) ‘Binding of p53 and its core domain to supercoiled DNA’, European
Journal of Biochemistry, 268(3), pp. 573–581. doi: 10.1046/j.1432-1327.2001.01898.x.
Paleček, E. (1958) ‘Oszillographische Polarographie der Nucleeinsäuren und ihrer
Bestandteile’, Naturwissenchaften, 45(8), pp. 186–187.

63
Paleček, E. (1960) ‘Oscillographic polarography of highly polymerized Deoxyribonucleic
acid’, Nature, 188, pp. 656–657. doi: 10.1038/188656a0.
Paleček, E. (1992a) ‘Probing DNA structure with osmium tetroxide complexes in vitro’,
Methods in Enzymology. Academic Press, 212(C), pp. 139–155. doi: 10.1016/0076-
6879(92)12010-N.
Paleček, E. (1992b) ‘Probing DNA structure with osmium tetroxide complexes in vitro’,
Methods in Enzymology, 212, pp. 139–155. doi: 10.1016/0076-6879(92)12010-N.
Paleček, E. et al. (1997) ‘Tumor suppressor protein p53 binds preferentially to supercoiled
DNA’, Oncogene, 15(18), pp. 2201–2209. doi: 10.1038/sj.onc.1201398.
Paleček, E. et al. (2004) ‘Sensitive electrochemical determination of unlabeled MutS protein
and detection of point mutations in DNA.’, Analytical chemistry, 76(19), pp. 5930–5936. doi:
10.1021/ac049474x.
Paleček, E. and Bartošík, M. (2012) ‘Electrochemistry of nucleic acids’, Chemical Reviews,
112(6), pp. 3427–3481. doi: 10.1021/cr200303p.
Palecek, E. and Fojta, M. (2005) ‘Electrochemical DNA Sensors’, in Bioelectronics.
Weinheim, FRG: Wiley-VCH Verlag GmbH & Co. KGaA, pp. 127–192. doi:
10.1002/352760376X.ch5.
Paleček, E. and Fojta, M. (2007) ‘Magnetic beads as versatile tools for electrochemical DNA
and protein biosensing’, Talanta, 74(3), pp. 276–290. doi: 10.1016/j.talanta.2007.08.020.
Palecek, E. and Hung, M. A. (1983) ‘Determination of nanogram quantities of osmium-
labeled nucleic acids by stripping (inverse) voltammetry.’, Analytical biochemistry, 132(2),
pp. 236–42. Available at: http://www.ncbi.nlm.nih.gov/pubmed/6353997 (Accessed: 5 April
2017).
Paleček, E. and Postbieglová, I. (1986) ‘Adsorptive stripping voltammetry of
biomacromolecules with transfer of the adsorbed layer’, Journal of Electroanalytical
Chemistry, 214(1–2), pp. 359–371. doi: 10.1016/0022-0728(86)80108-5.
Paleček, E., Trefulka, M. and Fojta, M. (2009) ‘End-labeling of peptide nucleic acid with
osmium complex. Voltammetry at carbon and mercury electrodes’, Electrochemistry
Communications, 11(2), pp. 359–362. doi: 10.1016/j.elecom.2008.11.049.
Pivoňková, H. et al. (2006) ‘Recognition of cisplatin-damaged DNA by p53 protein: Critical
role of the p53 C-terminal domain’, Biochemical and Biophysical Research Communications,
339(2), pp. 477–484. doi: 10.1016/j.bbrc.2005.11.038.
Pivoňková, H. et al. (2010) ‘Direct voltammetric analysis of DNA modified with
enzymatically incorporated 7-deazapurines’, Analytical Chemistry, 82(16), pp. 6807–6813.
doi: 10.1021/ac100757v.
Polyak, K. et al. (1997) ‘A model for p53-induced apoptosis’, Nature, 389(6648), pp. 300–
305. doi: 10.1038/38525.
Ramadan, K. et al. (2004) ‘De Novo DNA synthesis by human DNA polymerase λ, DNA
polymerase μ and terminal deoxyribonucleotidyl transferase’, Journal of Molecular Biology,

64
339(2), pp. 395–404. doi: 10.1016/j.jmb.2004.03.056.
Reske, T. et al. (2009) ‘Kinetics of the labeling reactions of thymine, cytosine and uracil with
osmium tetroxide bipyridine’, Microchimica Acta. Springer Vienna, 166(3–4), pp. 197–201.
doi: 10.1007/s00604-009-0195-6.
Riedl, J. et al. (2009) ‘Tetrathiafulvalene-Labelled Nucleosides and Nucleoside
Triphosphates: Synthesis, Electrochemistry and the Scope of Their Polymerase Incorporation
into DNA’, European Journal of Organic Chemistry, 21, pp. 3519–3525. doi:
10.1002/ejoc.200900392.
Roychoudhury, R., Jay, E. and Wu, R. (1976) ‘Terminal labeling and addition of
homopolymer tracts to duplex DNA fragments by terminal deoxynucleotidyl transferase.’,
Nucleic acids research, 3(4), pp. 863–77. doi: 10.1093/nar/3.1.101.
Sadofsky, M. J. (2001) ‘The RAG proteins in V(D)J recombination: more than just a
nuclease’, Nucleic Acids Research, 29(7), pp. 1399–1409. doi: 10.1093/nar/29.7.1399.
Sakamuro, D. et al. (1997) ‘The polyproline region of p53 is required to activate apoptosis but
not growth arrest.’, Oncogene, 15(8), pp. 887–898. doi: 10.1038/sj.onc.1201263.
Seeman, N. C., Rosenberg, J. M. and Rich, A. (1976) ‘Sequence-specific recognition of
double helical nucleic acids by proteins.’, Proceedings of the National Academy of Sciences,
73(3), pp. 804–808. doi: 10.1073/pnas.73.3.804.
Selivanova, G. et al. (1998) ‘Reactivation of mutant p53: a new strategy for cancer therapy.’,
Seminars in cancer biology, 8(5), pp. 369–378. doi: 10.1006/scbi.1998.0099.
Simonova, A. et al. (2014) ‘Methoxyphenol and Dihydrobenzofuran as Oxidizable Labels for
Electrochemical Detection of DNA’, ChemPlusChem. WILEY‐ VCH Verlag, 79(12), p. n/a-
n/a. doi: 10.1002/cplu.201402194.
Simonova, A. et al. (2017) ‘Phenothiazine-linked Nucleosides and Nucleotides for Redox
Labelling of DNA’, Org. Biomol. Chem., (July). doi: 10.1039/C7OB01439B.
Smith, M. L. et al. (2000) ‘p53-mediated DNA repair responses to UV radiation: studies of
mouse cells lacking p53, p21, and/or gadd45 genes.’, Molecular and cellular biology, 20(10),
pp. 3705–3714. doi: 10.1128/MCB.20.10.3705-3714.2000.
Špaček, J. et al. (2017) ‘Label-free detection of canonical DNA bases, uracil and 5-
methylcytosine in DNA oligonucleotides using linear sweep voltammetry at a pyrolytic
graphite electrode’, Electrochemistry Communications, 82(June), pp. 34–38. doi:
10.1016/j.elecom.2017.07.013.
Stansel, R. M., Subramanian, D. and Griffith, J. D. (2002) ‘P53 Binds Telomeric Single
Strand Overhangs and T-Loop Junctions in Vitro’, Journal of Biological Chemistry, 277(14),
pp. 11625–11628. doi: 10.1074/jbc.C100764200.
Studničková, M. et al. (1989) ‘Reduction of guanosine at a mercury electrode’,
Bioelectrochemistry and Bioenergetics, 275(21), pp. 83–86.
Subramanian, D. and Griffith, J. D. (2005) ‘Modulation of p53 binding to Holliday junctions
and 3-cytosine bulges by phosphorylation events’, Biochemistry, 44(7), pp. 2536–2544. doi:

65
10.1021/bi048700u.
Surkus, A. E. and Flechsig, G.-U. (2009) ‘Electrochemical detection of DNA melting curves
by means of heated biosensors’, Electroanalysis, 21(10), pp. 1119–1123. doi:
10.1002/elan.200804539.
Toshiyuki, M. and Reed, J. C. (1995) ‘Tumor suppressor p53 is a direct transcriptional
activator of the human bax gene’, Cell, 80(2), pp. 293–299. doi: 10.1016/0092-
8674(95)90412-3.
Trefulka, M. et al. (2007) ‘Voltammetry of osmium end-labeled oligodeoxynucleotides at
carbon, mercury, and gold electrodes’, Electroanalysis. Wiley-Blackwell, 19(12), pp. 1334–
1338. doi: 10.1002/elan.200703849.
Trefulka, M., Bartošík, M. and Paleček, E. (2010) ‘Facile end-labeling of RNA with
electroactive Os(VI) complexes’, Electrochemistry Communications. Elsevier, 12(12), pp.
1760–1763. doi: 10.1016/J.ELECOM.2010.10.016.
Trnková, L., Jelen, F. and Postbieglová, I. (2003) ‘Application of Elimination Voltammetry to
the Resolution of Adenine and Cytosine Signals in Oligonucleotides. I. Homo-
oligodeoxynucleotides dA9 and dC9’, Electroanalysis, 15(19), pp. 1529–1535. doi:
10.1002/elan.200302719.
Trnková, L., Jelen, F. and Postbieglová, I. (2006) ‘Application of elimination voltammetry to
the resolution of adenine and cytosine signals in oligonucleotides II. Hetero-
oligodeoxynucleotides with different sequences of adenine and cytosine nucleotides’,
Electroanalysis, 18(7), pp. 662–669. doi: 10.1002/elan.200503447.
Trnková, L., Studničková, M. and Paleček, E. (1980) ‘Electrochemical behaviour of guanine
and its derivatives. I. Fast cyclic voltammetry of guanosine and calf thymus DNA’, Journal of
Electroanalytical Chemistry, 116(7), pp. 643–658. doi: 10.1016/S0022-0728(80)80294-4.
Vacek, J. et al. (2008) ‘Label-Free Electrochemical Monitoring of DNA Ligase Activity’,
Analytical Chemistry, 80(19), pp. 7609–7613.
Vidláková, P. et al. (2015) ‘G-quadruplex-based structural transitions in 15-mer DNA
oligonucleotides varying in lengths of internal oligo(dG) stretches detected by voltammetric
techniques’, Analytical and bioanalytical chemistry, 407(19), pp. 5817–5826. doi:
10.1007/s00216-015-8768-1.
Vojtĕsek, B. et al. (1992) ‘An immunochemical analysis of the human nuclear
phosphoprotein p53. New monoclonal antibodies and epitope mapping using recombinant
p53.’, Journal of immunological methods, 151(1–2), pp. 237–44. doi: 10.1016/0022-
1759(92)90122-A.
Vrábel, M. et al. (2009) ‘Base-modified DNA labeled by [Ru(bpy)3]2+ and [Os(bpy)3]2+
complexes: Construction by polymerase incorporation of modified nucleoside triphosphates,
electrochemical and luminescent properties, and applications’, Chemistry - A European
Journal, 15(5), pp. 1144–1154. doi: 10.1002/chem.200801538.
Wang, J. (2001a) Analytical Electrochemistry, Analytical Electrochemistry. doi:
10.1002/0471790303.ch3.

66
Wang, J. (2001b) Analytical Electrochemistry, Analytical Electrochemistry. doi:
10.1002/0471790303.ch3.
Wang, W. et al. (2005) ‘Acridine derivatives activate p53 and induce tumor cell death through
bax’, Cancer Biology and Therapy, 4(8), pp. 893–898. doi: 10.4161/cbt.4.8.2134.
Wang, Y. et al. (1995) ‘Interaction of p53 with its consensus DNA-binding site.’, Molecular
and Cellular Biology, 15(4), pp. 2157–2165. doi: 10.1128/MCB.15.4.2157.
Wong, E. L. S. and Gooding, J. J. (2003) ‘Electronic detection of target nucleic acids by a 2,6-
disulfonic acid anthraquinone intercalator’, Analytical Chemistry, 75(15), pp. 3845–3852. doi:
10.1021/ac034129d.
Wu, X. et al. (1993) ‘The p53-mdm-2 autoregulatory feedback loop’, Genes & Development,
53(7), pp. 1126–1132. doi: 10.1101/gad.7.7a.1126.
Yang, B., Gathy, K. N. and Coleman, M. S. (1994) ‘Mutational analysis of residues in the
nucleotide binding domain of human terminal deoxynucleotidyl transferase’, Journal of
Biological Chemistry, 269(16), pp. 11859–11868.
Yosypchuk, B. et al. (2002) ‘Use of solid amalgam electrodes in nucleic acid analysis’,
Electroanalysis, 14(21), pp. 1488–1493. doi: 10.1002/1521-4109(200211)14:21<1488::AID-
ELAN1488>3.0.CO;2-X.
Yosypchuk, B. et al. (2006) ‘Voltammetric behavior of osmium-labeled DNA at mercury
meniscus-modified solid amalgam electrodes. Detecting DNA hybridization’, Electroanalysis,
18(2), pp. 186–194. doi: 10.1002/elan.200503392.
Zauner, G. et al. (2005) ‘Tethered DNA hairpins facilitate electrochemical detection of DNA
ligation’, The Analyst. The Royal Society of Chemistry, 130(3), p. 345. doi:
10.1039/b413556c.
Zheng, J. et al. (2010) ‘An aptamer-based assay for thrombin via structure switch based on
gold nanoparticles and magnetic nanoparticles’, Talanta. Elsevier B.V., 80(5), pp. 1868–
1872. doi: 10.1016/j.talanta.2009.10.036.
Zhou, M., Zhai, Y. and Dong, S. (2009) ‘Electrochemical sensing and biosensing platform
based on chemically reduced graphene oxide’, Analytical Chemistry, 81(14), pp. 5603–5613.
doi: 10.1021/ac900136z.
Zilfou, J. T. and Lowe, S. W. (2009) ‘Tumor suppressive functions of p53.’, Cold Spring
Harbor perspectives in biology, 1(5). doi: 10.1101/cshperspect.a001883.

67
List of abbreviations
scDNA supercoiled DNA
ACV alternating current voltammetry
AdTSV adsorptive transfer stripping voltammetry
AFM atomic force microscopy
BA butylacrylate
BF benzofurazane
bpy 2,2’-bipyridine
ChIP chromatin immunoprecipitation
CON DNA probe containing p53CON
CTDBS C-terminal DNA binding site of the p53 protein
CV cyclic voltammetry
DME dropping mercury electrode
DPV differential-pulse voltammetry
dsDNA double-stranded DNA
EMSA electrophoretic mobility shift assay
FRET fluorescence resonance energy transfer
G4 G-quadruplexes
HMDE hanging mercury drop electrode
Km Michaelis constant
LSV linear sweep voltammetry
MB magnetic beads

68
MBIP magnetic beads-based immunoprecipitation assay
MFE mercury film electrode
NA nucleic acids
NEAR nicking enzyme amplification reaction
NO2 nitrobenzene
noCON DNA probe not containing p53CON
Os,bpy complex of osmium tetroxide with 2,2’-bipyridine
Os,L complex of osmium tetroxide with nitrogenous ligand
p53CON consensus binding site for the p53 protein
PAGE polyacrylamide gel electrophoresis
PCR polymerase chain reaction
PEX primer extension reaction
PGE pyrolytic graphite electrode
phen 1,10- phenanthroline
Pu purine
Py pyrimidine
SAE solid amalgam electrodes
SCS supercoil-selective DNA binding
SPR surface plasmon resonance
ssDNA single-stranded DNA
SWV square-wave voltammetry
TdT terminal deoxynucleotidyl transferase
TEMED N,N,N’,N’-tetramethyl ethylenediamine
69
TUNEL TdT-mediated dUTP-biotin nick end-labeling
V(D)J variable, diversity, and joining segments
wtp53 wild type p53

70
List of publications and conferences
1) Dual Redox Labeling as a Tool for Electrochemical Detection of p53 Protein-DNA
Interactions
Monika Hermanová, Petr Orság, Jana Balintová, Michal Hocek, Miroslav Fojta
2) Label-free voltammetric detection of products of terminal deoxynucleotidyl
transferase tailing reaction
Monika Hermanová, Pavlína Havranová, Anna Ondráčková, Swathi Senthil Kumar, Richard
Bowater and Miroslav Fojta
Electroanalysis (2018)
3) Butylacrylate-nucleobase Conjugates as Targets for Two-step Redox Labeling of
DNA with an Osmium Tetroxide Complex
Pavlína Havranová-Vidláková, Jan Špaček, Lada Vítová, Monika Hermanová, Jitka Dadová,
Veronika Raindlová, Michal Hocek, Miroslav Fojta and Luděk Havran
Electroanalysis (2018) 30, 371–377
Conferences:
Monika Hermanová, Jan Špaček, Petr Orság, Miroslav Fojta. Tail-labeled
oligonucleotide probes for a dual electrochemical magnetic immunoprecipitation assay of
DNA-protein binding. 11th
International Interdisciplinary Meeting on Bioanalysis, Brno,
Czech Republic, October 20-22 2014.
Monika Hermanová, Jan Špaček, Petr Orság, Miroslav Fojta. A new electrochemical
DNA binding competition assay using oligonucleotide substrates tail-labeled with two
different redox tags. CEITEC Annual Conference “Frontiers in Material and Life
Sciences”, Brno, Czech Republic, October 21-24 2014. Abstract Book, Page 131.
Monika Hermanová, Jan Špaček, Hana Pivoňková and Miroslav Fojta. Studies of
protein-DNA interactions using immunoprecipitation with DNA probes labelled with

71
electroactive groups. XXXV. Modern Electrochemical Methods, Jetřichovice, Czech
Republic, May 18-22 2015. Abstract Book, Pages 57-59.
Monika Hermanová, Jan Špaček, Miroslav Fojta. Electrochemical detection of DNA-
protein binding using dual redox tail-labeling of oligonucleotide probes. XXIII International
Symposium on Bioelectrochemistry and Bioenergetics of the Bioelectrochemical Society,
Malmö, Sweden, June 14-18 2015. Abstract Book, Page 215.
Monika Hermanová, Petr Orság, Miroslav Fojta. Elektrochemická detekce DNA-protein
interakcí využívající oligonukleotidy značené pomocí PEX reakce. XVI. Mezioborové setkání
mladých biologů, biochemiků a chemiků, Milovy, Czech Republic, May 10-13 2016.
Abstract Book, Page 65.
Monika Hermanová, Petr Orság, Miroslav Fojta. Electrochemical methods for detection of
protein-DNA interactions. XXV. Biochemický sjezd, Praha, Czech Republic, September 13-
16 2016. Abstract Book, Page 179.
Monika Hermanová, Petr Orság and Miroslav Fojta. Electrochemical methods for
detection of protein-DNA interactions. XXXVII. Modern Electrochemical Methods,
Jetřichovice, Czech Republic, May 15-19 2017. Abstract Book, Pages 58-61.
Monika Hermanová and Miroslav Fojta. Electrochemical analysis of products of terminal
deoxynucleotidyl transferase tailing reaction using intrinsic electroactivity of nucleobases. 69.
Zjazd chemikov, Vysoké tatry, Horný Smokovec, Slovakia, September 11-15 2017. Abstract
Book, Page 75.
Monika Hermanová, Pavlína Havranová, Anna Ondráčková and Miroslav Fojta.
Analysis of products of nontemplate enzymatic synthesis of DNA oligonucleotides using
voltammetric methods. XXXVIII. Modern Electrochemical Methods, Jetřichovice, Czech
Republic, May 21-25 2018. Abstract Book, Pages 76-79.
Monika Hermanová, Pavlína Havranová, Anna Ondráčková and Miroslav Fojta. Label-
free detection of products of terminal deoxynucleotidyl transferase tailing reaction. 17th
International Conference on Electroanalysis, Rhodes, Greece, June 3-7 2018. Abstract
Book, Page 156.

72
Appendices

73
Appendix 2
LABEL-FREE VOLTAMMETRIC DETECTION OF PRODUCTS OF TERMINAL
DEOXYNUCLEOTIDYL TRANSFERASE TAILING REACTION
Monika Hermanová1, Pavlína Havranová-Vidláková
1, Anna Ondráčková
1, Swathi Senthil
Kumar2, Richard Bowater
2, Miroslav Fojta
1,3,*
1 Institute of Biophysics, Czech Academy of Sciences, Kralovopolska 135, 612 65 Brno, Czech
Republic
2 School of Biological Sciences, University of East Anglia, Norwich, Norwich Research Park,
NR4 7TJ, United Kingdom
3 Central European Institute of Technology, Masaryk University, Kamenice 753/5, CZ-625 00
Brno, Czech Republic
*Corresponding author: [email protected]

74
Abstract
A label-free approach that takes advantage of intrinsic electrochemical activity of nucleobases
has been applied to study the products of terminal deoxynucleotidyl transferase (TdT) tailing
reaction. DNA homooligonucleotides A30, C30 and T30 were used as primers for the tailing
reaction to which a dNTP - or a mixture of dNTPs - and TdT were added to form the tails.
Electrochemical detection enabled study of the tailing reaction products created by various
combinations of primers and dNTPs, with pyrolytic graphite electrode (PGE) being suitable
for remarkably precise analysis of the length of tailing reaction products. Furthermore, the
hanging mercury drop electrode (HMDE) was able to reveal formation of various DNA
structures, such as DNA hairpins and G-quadruplexes, which influence the behavior of DNA
molecules at the negatively charged surface of HMDE. Thus, the described approach proves
to be an excellent tool for studying the TdT tailing reactions and for exploring how various
DNA structures affect both the tailing reactions and electrochemical behavior of DNA
oligonucleotides at electrode surfaces.
Key words: terminal deoxynucleotidyl transferase; oligonucleotide tailing; DNA
electrochemistry; label free; nucleobase; reduction; oxidation

75
1. Introduction
Terminal Deoxynucleotidyl Transferase (TdT) is a unique DNA polymerase as it catalyzes
random polymerization of deoxynucleotides at the 3'-OH ends of DNA in a template
independent manner [1,2]. The physiological role of TdT lies in random addition of
nucleotides to single-stranded DNA during V(D)J recombination. The ability of TdT to
randomly incorporate nucleotides increases antigen receptor diversity and, thus, TdT plays a
crucial role in the evolution and adaptation of the vertebrate immune system [3]. For its
catalytic activity, the enzyme requires a free 3'-OH group and a minimum of three nucleotide
residues to constitute the primer [4]. In vitro, TdT can incorporate all four natural
deoxyribonucleotides into single-stranded DNA. However, in vivo, a bias was observed
towards the incorporation of pyrimidine versus purine nucleotides [2], with the incorporation
efficiency of G being more than 4-fold higher than that of A [5,6]. TdT, like all DNA
polymerases, requires divalent metal ions for the catalysis. However, TdT is unique in its
ability to use a variety of divalent cations, such as Co2+
, Mn2+
, Zn2+
and Mg2+
[7]. Each metal
ion has different effects on the kinetics and mechanism of dNTP (deoxynucleoside
triphosphates; N stands for A, adenine, C, cytosine, G, guanine or T, thymine) utilization. For
example, Mg2+
facilitates the preferential utilization of dGTP and dATP whereas Co2+
increases the catalytic polymerization efficiency of the pyrimidines, dCTP and dTTP [8].
Nucleic acids are electrochemically active, which was first shown in the second half of the
1950s [9], and yield analytically useful signals at various electrodes [10,11]. Mercury-based
electrodes (such as hanging mercury drop electrode, HMDE) are traditionally used to measure
DNA signals related to reduction of its natural components (although it should be noted that
reduction of nucleobases at a graphite electrode has recently been reported on [12]). Here, we
use the HMDE as a well-established tool for DNA structure-sensitive measurements. In
mixed nucleotide sequences, adenine (A) and cytosine (C) yield the common cathodic CA
peak resulting from their reduction; guanine (G) gives the anodic GHMDE
peak, which results
from re-oxidation of a reduction product of guanine, 7,8-dihydroguanine, which is formed at
very negative potentials and, therefore, cannot be observed under usually applied conditions
due to an overlap of its signal with the cathodic background discharge at the mercury
electrode [13,14]. At carbon electrodes, A and G can be oxidized at positive potentials, giving
rise to AOX
and GOX
peaks, respectively. Analogues of natural purines, 7-deazaadenine (A*)
and 7-deazaguanine (G*), can also be studied using carbon electrodes as they produce
oxidation signals at positive potentials [15]. Moreover, adsorption/desorption processes or

76
reorientation of parts of the DNA chains occur at the negatively charged surface of mercury
electrodes, which can be observed as tensammetric (capacitive) signals [10]. Generally,
signals gained at mercury-containing electrodes are remarkably sensitive to changes in DNA
structure, which enables detection of different types of changes, such as formation of strand
breaks [16], DNA superhelicity-induced structural transitions [17], binding of DNA
intercalators [18] or formation of non-canonical DNA structures such as triplexes [19] and
quadruplexes [20,21].
Methods for studying the TdT tailing reaction have been based on polyacrylamide gel
electrophoresis (PAGE), usually along with radioactive labeling of DNA [22–27]. Here, for
the first time we apply an electrochemistry-based approach to study the TdT tailing reactions,
taking advantage of the intrinsic electroactivity of nucleobases. A set of DNA
homooligonucleotides were used as primers for the tailing reactions, to which a dNTP (or
a mixture of dNTPs) and TdT were added to form the tails. Taking account of the primer and
dNTP used in the particular reaction, an appropriate electrode was chosen for the tailing
reaction products analysis - HMDE or pyrolytic graphite electrode (PGE). We show that
electrochemical analysis enables a very precise study of the TdT tailing reaction products and,
furthermore, it can detect formation of DNA structures occurring in the TdT products.
2. Experimental section
2.1 Terminal Deoxynucleotidyl Transferase (TdT) reaction
As primers for the TdT reaction, single stranded oligonucleotides composed of one nucleotide
type, A30, C30 and T30 (Generi Biotech, Czech Rep.) were used. The reaction mixture
contained 5 M DNA primer (A30, C30 or T30), 5 M, 25 M, 50 M, 100 M or 250 M
dNTP (dGTP, dG*TP, dATP, dA*TP, dCTP or dTTP, Sigma-Aldrich, USA), 10 U Terminal
Deoxynucleotidyl Transferase (calf thymus, recombinant expressed in E. coli; New England
Biolabs, USA) and CoCl2 in a total volume of 20 l. Reaction mixtures were
incubated at 37 °C for 60 min. Products of the TdT reaction were purified using the
Nucleotide Removal Kit (Qiagen, the Netherlands) and dissolved in 30 l deionized water.
2.2 Denaturing PAGE
TdT reaction products were labeled by 32
P, dried out and dissolved in 5 l loading buffer
(80% formamide, 10 mM EDTA, 1 mg.ml-1
xylene cyanol FF, 1 mg.ml-1
bromphenol blue).
Aliquots (2.5 l) of the reaction mixtures were loaded into a 15% denaturing polyacrylamide

77
gel containing 1x TBE pH 8 and 7 M urea, which had been preheated at 25 W for 30 min, and
then electrophoresis continued at 25 W for 90 min. After drying, the gel was
autoradiographed and visualized using a Typhoon FLA 9000.
2.3 Electrochemical analysis
All electrochemical measurements were performed at room temperature in a three-electrode
setup (with the hanging mercury drop electrode, HMDE, or basal-plane pyrolytic graphite
electrode, PGE, as the working electrode, Ag/AgCl/3 M KCl as the reference electrode and
platinum wire as the auxiliary electrode) using an Autolab analyzer (Ecochemie, the
Netherlands) in connection with VA-Stand 663 (Metrohm, Switzerland). The measurements
were done in an adsorptive transfer stripping (AdTS) mode. DNA (10 g.ml-1
) was
accumulated at the electrode surface from 4 l aliquots (containing 0.2 M NaCl) for 60 s, then
the electrode was rinsed by deionized water and placed into the electrochemical cell
containing blank electrolyte. Cyclic voltammetry (CV) measurements at HMDE were
performed in 0.3 M ammonium formate with 0.05 M sodium phosphate, pH 6.9 as electrolyte;
CV settings were: initial potential 0.0 V, switching potential -1.85 V, final potential 0.0 V,
scan rate 1 V.s-1
, step potential 5 mV. Before each CV measurement the solution of the
background electrolyte was purged with argon. Square wave voltammetry (SWV)
measurements at PGE were performed in 0.2 M acetate buffer, pH 5.0; SWV settings were:
initial potential -1 V, final potential 1.6 V, frequency 200 Hz, amplitude 50 mV, scan rate 1
V.s-1
. Before each SWV measurement the PGE was pretreated by applying a potential of 1.8
V for 30 s in the background electrolyte, and its surface was renewed by peeling off the
graphite top layer using sticky tape.
3. Results and discussion
For the tailing reaction, three types of homooligonucleotides – A30, C30 and T30 – were used as
primers to which various dNTPs (dATP, dCTP, dGTP, dTTP, 7-deazaadenine - dA*TP and 7-
deazaguanine - dG*TP or their combinations) were added to form the tail. G30 was not used as
a primer since DNA oligonucleotides containing longer stretches of guanines tend to form G-
quadruplexes (G4) [21,28] and such structure would not be a suitable substrate for TdT
because of its inability to access the 3'-OH end folded within the G4 structure. Figure 1 shows
cyclic voltammograms at HMDE and square wave voltammograms at PGE gained for the A30,
C30 and T30 primers compared to blank electrolyte. For the A30 primer, the A reduction peak at
HMDE (AHMDE
) and A oxidation peak at PGE (AOX
) can be observed. C30 yields only a
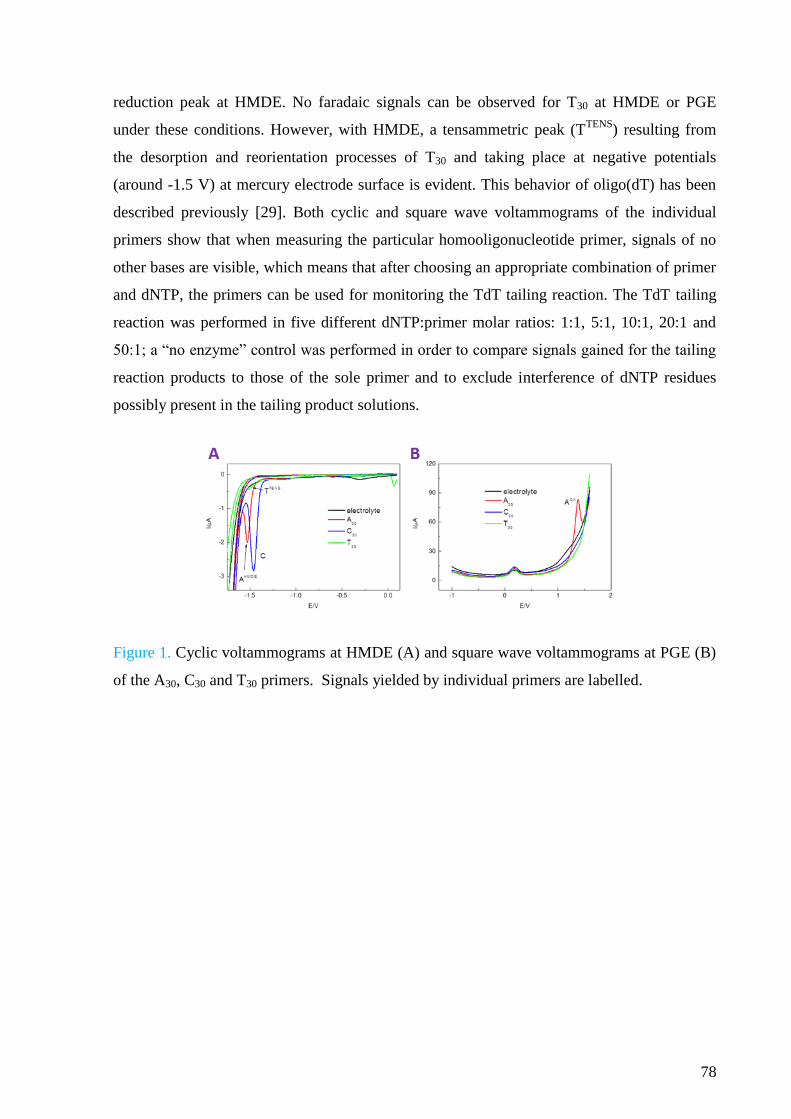
78
reduction peak at HMDE. No faradaic signals can be observed for T30 at HMDE or PGE
under these conditions. However, with HMDE, a tensammetric peak (TTENS
) resulting from
the desorption and reorientation processes of T30 and taking place at negative potentials
(around -1.5 V) at mercury electrode surface is evident. This behavior of oligo(dT) has been
described previously [29]. Both cyclic and square wave voltammograms of the individual
primers show that when measuring the particular homooligonucleotide primer, signals of no
other bases are visible, which means that after choosing an appropriate combination of primer
and dNTP, the primers can be used for monitoring the TdT tailing reaction. The TdT tailing
reaction was performed in five different dNTP:primer molar ratios: 1:1, 5:1, 10:1, 20:1 and
50:1; a “no enzyme” control was performed in order to compare signals gained for the tailing
reaction products to those of the sole primer and to exclude interference of dNTP residues
possibly present in the tailing product solutions.
Figure 1. Cyclic voltammograms at HMDE (A) and square wave voltammograms at PGE (B)
of the A30, C30 and T30 primers. Signals yielded by individual primers are labelled.
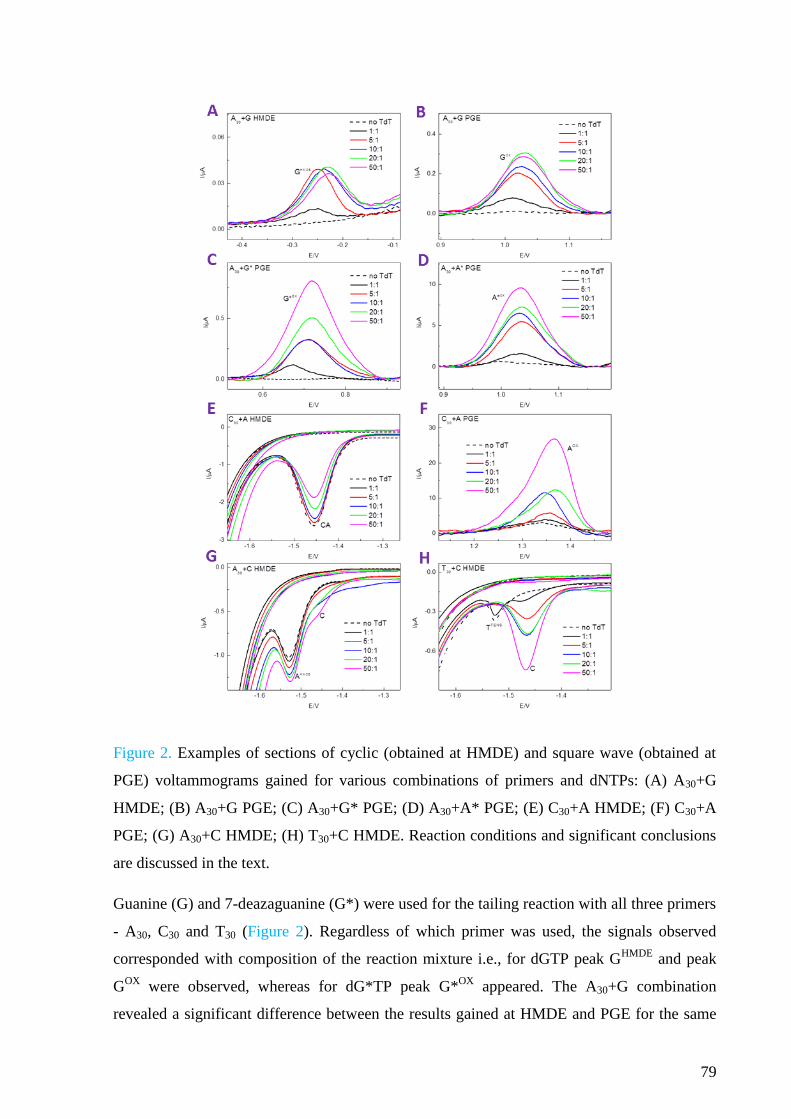
79
Figure 2. Examples of sections of cyclic (obtained at HMDE) and square wave (obtained at
PGE) voltammograms gained for various combinations of primers and dNTPs: (A) A30+G
HMDE; (B) A30+G PGE; (C) A30+G* PGE; (D) A30+A* PGE; (E) C30+A HMDE; (F) C30+A
PGE; (G) A30+C HMDE; (H) T30+C HMDE. Reaction conditions and significant conclusions
are discussed in the text.
Guanine (G) and 7-deazaguanine (G*) were used for the tailing reaction with all three primers
- A30, C30 and T30 (Figure 2). Regardless of which primer was used, the signals observed
corresponded with composition of the reaction mixture i.e., for dGTP peak GHMDE
and peak
GOX
were observed, whereas for dG*TP peak G*OX
appeared. The A30+G combination
revealed a significant difference between the results gained at HMDE and PGE for the same

80
reaction products (Figures 2A, B and 3A). With PGE the height of the GOX
peak continuously
increased with the increasing dNTP:primer ratio. In contrast, peak GHMDE
showed a
decreasing tendency from a 5:1 to 50:1 ratio after the initial growth between 1:1 and 5:1 ratio.
This behavior at HMDE is in contrast to what was observed at PGE. An analogous effect,
decrease of peak GHMDE
with increasing length of Gn stretches, has been previously observed
and ascribed to the formation of intermolecular parallel G-quadruplexes (G4) [21]. Since the
G-rich sequences are generally known to adopt the structure of G4 and this ability is more
pronounced in longer Gn stretches, it can be assumed that the G-tails formed G4 at
dNTP:primer molar ratios of 5:1 and higher. This assumption is further supported by the fact
that K+ ions, which stabilize formation of G4, are present in the TdT reaction buffer, in which
the tailing reaction takes place.
The denaturing PAGE autoradiogram showed that the length of the tailing reaction products
did not grow significantly at dGTP:primer ratios higher than 5:1. Moreover, for the ratios 5:1
and higher, heavier species were formed (detected at starts of the gel, see Figure 3A), whose
mass was too big to enter the gel; these species can be expected to be formed by the
intermolecular G4. This suggested that DNA obtained from the G-tailing reactions occurred in
two states: G4, which were resistant to the denaturing conditions during the PAGE, and
unstructured oligonucleotides that create a ladder typical for TdT tailing reaction products. In
principle, formation of G4 structures influences the intensity of the measured peak GHMDE
in
two ways. Firstly, when the dNTP:primer ratio is sufficient to produce G-tails that are capable
of stable G4 formation, the tailing reaction is stopped when the G4 is formed because of the
inaccessibility of the 3'-OH end to TdT. Secondly, the G4 formation changes behavior of the
oligonucleotides at the mercury electrode surface, which is related to the fact that DNA
structures existing in solution can be preserved after adsorption at the electrode surface in
adsorptive transfer techniques [10]. Moreover, mercury-containing electrodes are sensitive to
changes in DNA structure. It has been well established that the accessibility of nucleobase
residues for electrode reactions is strongly dependent on the DNA structure, particularly at
mercury electrodes. It can be assumed that this feature is responsible for the decrease of the
signals at the HMDE, together with the fact that electrostatic repulsion of the rigid quadruplex
structures from the negatively charged surface of the HMDE may hinder reduction of guanine
residues involved [21]. On the other hand, the oxidation of guanine at the PGE takes place at
the positively charged surface of the electrode to which the oligonucleotide molecules are
attracted. This facilitates the oxidation process even in the quadruplex structure. Along with
bigger accessibility of the primary oxidation site of guanine to the electrode surface, the
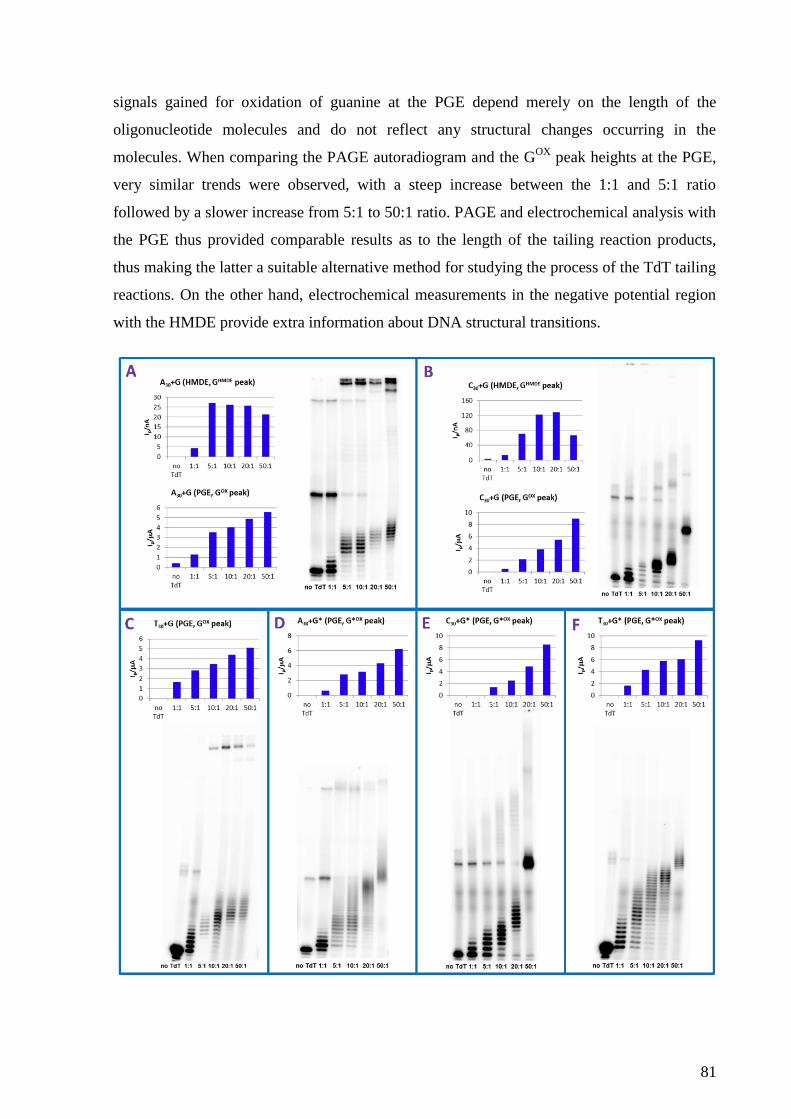
81
signals gained for oxidation of guanine at the PGE depend merely on the length of the
oligonucleotide molecules and do not reflect any structural changes occurring in the
molecules. When comparing the PAGE autoradiogram and the GOX
peak heights at the PGE,
very similar trends were observed, with a steep increase between the 1:1 and 5:1 ratio
followed by a slower increase from 5:1 to 50:1 ratio. PAGE and electrochemical analysis with
the PGE thus provided comparable results as to the length of the tailing reaction products,
thus making the latter a suitable alternative method for studying the process of the TdT tailing
reactions. On the other hand, electrochemical measurements in the negative potential region
with the HMDE provide extra information about DNA structural transitions.

82
Figure 3. Heights of the respective peaks gained at HMDE or PGE and denaturing PAGE
autoradiograms obtained for combinations of G and G* with all three primers: (A) A30+G; (B)
A30+C; (C) T30+G; (D) A30+G*; (E) C30+G*; (F) T30+G*. Reaction conditions and significant
conclusions are discussed in the text.
In the case of T30+G (Figure 3C), the denaturing PAGE revealed the same pattern of the
length of the tailing reaction products as A30+G, indicating formation of G4 at ratios higher
than 5:1, and a very similar trend as for A30+G was also observed using voltammetric analysis
at PGE. Nevertheless, voltammetric responses of the T30+G tailing products at HMDE could
not be compared to those of A30+G because behavior of the former at HMDE is
predominantly affected by the T30 oligonucleotide due to strong interactions of
homopyrimidine stretches with the negatively charged HMDE surface [29], which can further
affect adsorption and redox processes of the G-tails, especially if G4 are formed. These
phenomena, whose detailed description is out of scope of this report, are intensively studied in
our laboratory (S. Hasoň, H. Pivoňková et al., manuscripts in preparation).
Unlike A30+G and T30+G, the C30+G tailing reaction products (Figure 3B) did not form G4,
which is evident from the PAGE autoradiogram showing that the tail length does not stop
growing after a particular ratio and the longest products are obtained for the highest ratio
(50:1). Voltammetric analysis at PGE again reveals a good correlation with the PAGE
analysis. However, at HMDE, the signal grows only until the 20:1 ratio, then for the 50:1 ratio
the signal drops significantly. This can be explained by formation of another DNA structure,
such as a DNA hairpin (or, alternatively, duplex dimer of self-complementary GnCm
oligonucleotides). Bases C and G are complementary and tend to form duplex DNA when
possible, therefore when the G-tail reaches sufficient length for the hairpin formation (in this
case around 30 added nucleotides) it can bend over and form a hairpin with the
complementary C30 primer. The formation of such alternative structures can make signals at
HMDE decrease. It can also explain why only one band corresponding to two or three lengths
of the tailing products occurs in the PAGE autoradiogram, suggesting that the tailing reaction
stops when the DNA hairpin is formed.
For comparison with the behavior of the natural guanine, 7-deazaguanine (G*) was used,
which is an analogue of natural guanine in which the N7 atom is replaced by CH group. This
feature implicates that while the Watson-Crick base pairing (with C) of the 7-deazaguanine

83
remains unaffected, the Hoogsteen base pairs cannot be formed due to the involvement of the
N7 atom in the formation of this type of base pair. Thus, DNA molecules containing G* are
incapable of forming multi-stranded DNA structures, such as triplexes or quadruplexes. This
fact is utilized, for example, in PCR amplification of G-rich sequences where the quadruplex
formation could affect the amplification process. For analysis of the A30+G* tailing reaction
products (Figure 2C and 3D), only oxidation at PGE was used since, with HMDE, G* does
not yield any peak analogous to the GHMDE
peak, in agreement with absence of the
corresponding redox site in G* [30]. Both voltammetric and PAGE analysis showed that,
unlike in the case of A30+G tailing products, the length of the tailing products grew gradually
with the rising dG*TP:primer ratio, with the average tail length much higher than in the case
of dGTP, especially for the 20:1 and 50:1 ratios and the G*OX
signal for the 50:1 ratio being
~10-fold higher than that for the 1:1 ratio. This was in agreement with the fact that, unlike
natural guanine, G* is not capable of forming G4 and, therefore, the TdT tailing reaction was
not inhibited in contrast to what was observed in experiments with G (see above). G* is thus
suitable for studying how TdT incorporates an analogue of G into DNA as it does not exhibit
limitations related to formation of G4 structures described above for G.
Practically the same results as for the A30+G* were obtained for the T30+G* tailing products
(Figure 3F) with a similar pattern of the tailing products lengths visible on the PAGE
autoradiogram and intensities of the G*OX
signal gained at PGE. On the other hand, the
pattern observed for C30+G* (Figure 3E) resembled more that of the C30+G tailing products
than those of A30+G* and T30+G*, which is in accordance with the fact that the ability of G*
to form Watson-Crick pairs with C is not violated, which allows hairpin formation after a
certain length of the tail is reached.
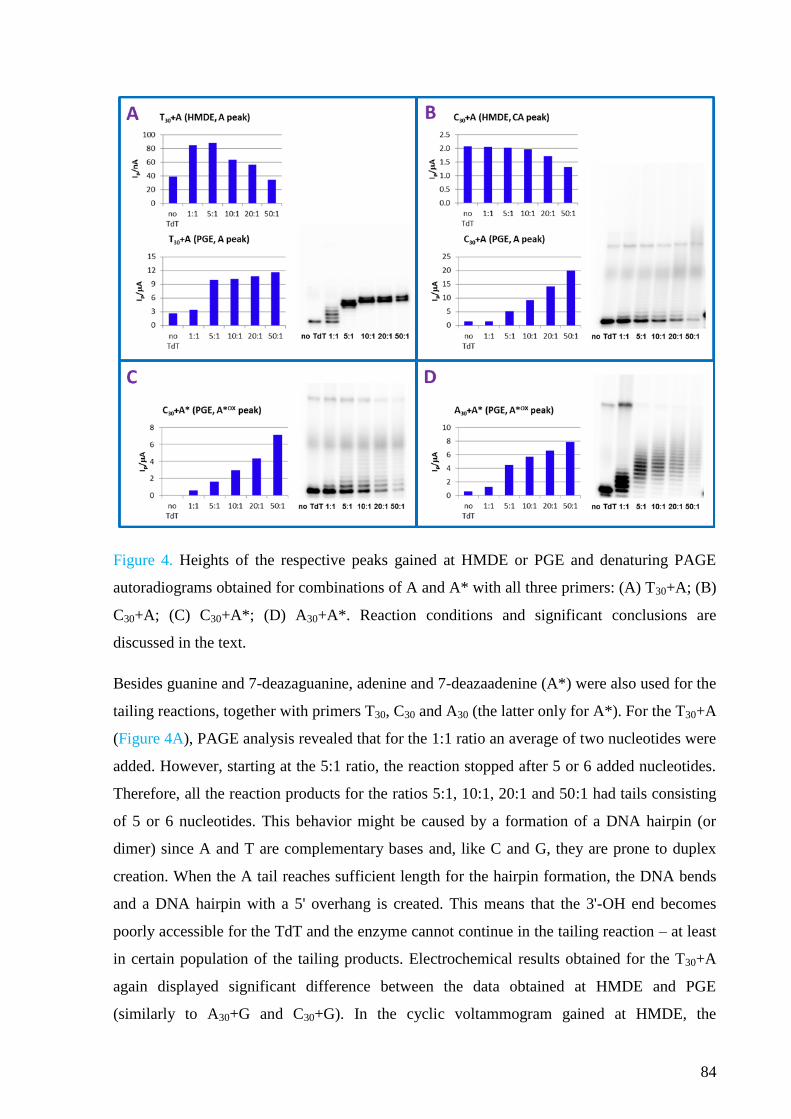
84
Figure 4. Heights of the respective peaks gained at HMDE or PGE and denaturing PAGE
autoradiograms obtained for combinations of A and A* with all three primers: (A) T30+A; (B)
C30+A; (C) C30+A*; (D) A30+A*. Reaction conditions and significant conclusions are
discussed in the text.
Besides guanine and 7-deazaguanine, adenine and 7-deazaadenine (A*) were also used for the
tailing reactions, together with primers T30, C30 and A30 (the latter only for A*). For the T30+A
(Figure 4A), PAGE analysis revealed that for the 1:1 ratio an average of two nucleotides were
added. However, starting at the 5:1 ratio, the reaction stopped after 5 or 6 added nucleotides.
Therefore, all the reaction products for the ratios 5:1, 10:1, 20:1 and 50:1 had tails consisting
of 5 or 6 nucleotides. This behavior might be caused by a formation of a DNA hairpin (or
dimer) since A and T are complementary bases and, like C and G, they are prone to duplex
creation. When the A tail reaches sufficient length for the hairpin formation, the DNA bends
and a DNA hairpin with a 5' overhang is created. This means that the 3'-OH end becomes
poorly accessible for the TdT and the enzyme cannot continue in the tailing reaction – at least
in certain population of the tailing products. Electrochemical results obtained for the T30+A
again displayed significant difference between the data obtained at HMDE and PGE
(similarly to A30+G and C30+G). In the cyclic voltammogram gained at HMDE, the

85
tensammetric peak mentioned above caused by the T30 primer can be observed. The potential
of the TTENS
peak coincides with that of AHMDE
peak. Thus, these two peaks cannot be
distinguished and there is a non-zero value for the primer (no TdT control). Then, for the 1:1
and 5:1 ratios the height of the collective peak at -1.52 V increased in comparison to the
signal gained for the sole primer, which agreed with the growing length of the A tails
observed at the PAGE autoradiogram. For the higher ratios, where the DNA hairpin is
expected to be formed, the signal sharply decreased, which was also observed for C30+G and
is in agreement with the fact that HMDE is sensitive to structural changes occurring in DNA.
On the other hand, the signals gained at PGE generally exhibit a poor sensitivity to variations
in DNA structure; therefore, the height of the AOX
peak displays a trend very similar to that
visible at the PAGE autoradiogram.
The C30+A combination generates a specific signal for the bases forming the primer and the
tail (Figure 2E,F and 4B). This occurs because in DNAs with a random distribution of bases,
C and A yield a common CA peak at HMDE since their reduction potentials are in close
proximity. However, in DNA composed of C and A homooligonucleotide stretches, two
separated peaks can be observed under certain conditions [31–33]. In this case, it was not
possible to see the separate C and AHMDE
peaks, only a hint of the AHMDE
peak appeared at a
potential more negative than that of the C peak for the product with the longest A tail (i.e. for
the 50:1 ratio). The height of the C peak decreased with the increasing dATP:primer ratio,
reflecting the decreasing proportion of cytosines in the tailing products. At PGE, the intensity
of the AOX
peak increased with higher ratios of dATP:primer, but it was not possible to
compare the trend of this signal to the pattern gained at PAGE since, in this case, the PAGE
analysis did not show distinct bands of the tailing products. Most probably this was caused by
a wider distribution of the lengths of tails, leading to smaller amounts of DNA of each
specific length, meaning that the bands corresponding to such lengths could not be visualized.
Nevertheless, the intensity of the band of the unextended C30 primer decreased with the higher
ratios of dATP:primer, which indicates that the primer has been consumed for the tailing
reaction. The same effect can be observed at the PAGE autoradiogram of the C30+A* tailing
products (Figure 4C), again making the electrochemical analysis at PGE more accurate than
the PAGE analysis. In both cases, the general trends of these signals are practically the same
(unlike in the case of A30+G), indicating that no unusual structure is formed in the A tail.
Thus, we can see that in some instances electrochemical analysis at PGE responds to the
increase of mean length of the tailing products more clearly than the PAGE analysis.
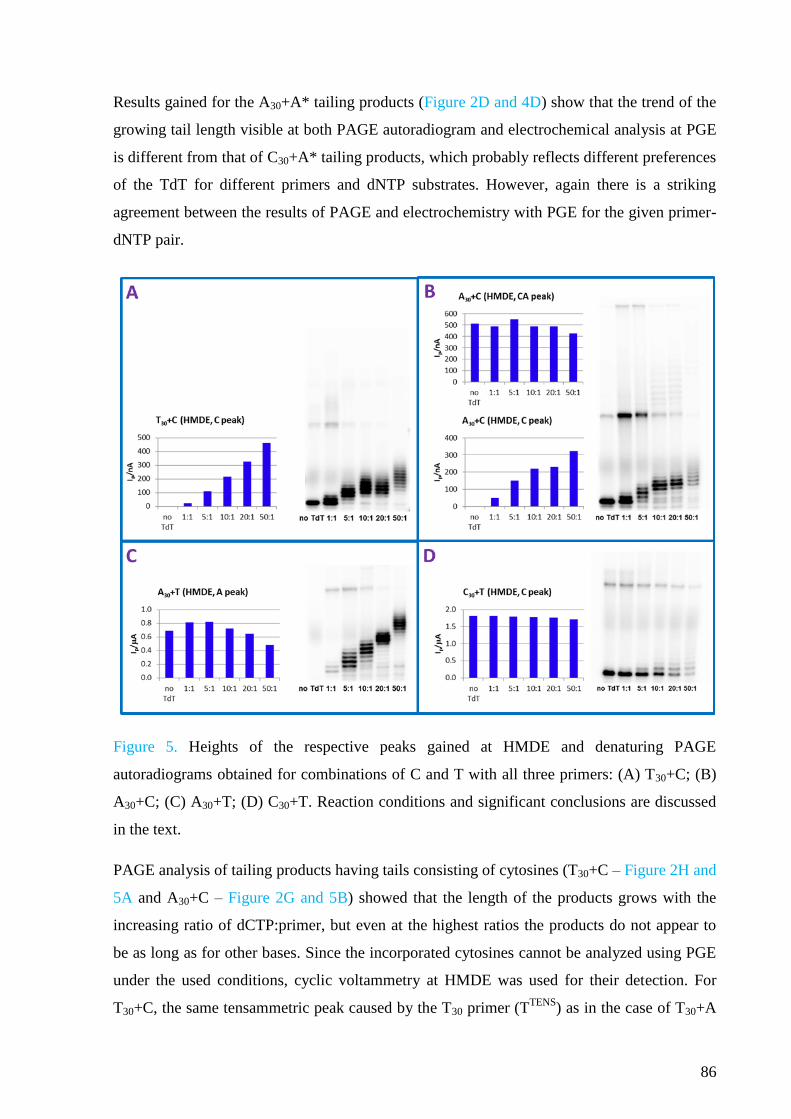
86
Results gained for the A30+A* tailing products (Figure 2D and 4D) show that the trend of the
growing tail length visible at both PAGE autoradiogram and electrochemical analysis at PGE
is different from that of C30+A* tailing products, which probably reflects different preferences
of the TdT for different primers and dNTP substrates. However, again there is a striking
agreement between the results of PAGE and electrochemistry with PGE for the given primer-
dNTP pair.
Figure 5. Heights of the respective peaks gained at HMDE and denaturing PAGE
autoradiograms obtained for combinations of C and T with all three primers: (A) T30+C; (B)
A30+C; (C) A30+T; (D) C30+T. Reaction conditions and significant conclusions are discussed
in the text.
PAGE analysis of tailing products having tails consisting of cytosines (T30+C – Figure 2H and
5A and A30+C – Figure 2G and 5B) showed that the length of the products grows with the
increasing ratio of dCTP:primer, but even at the highest ratios the products do not appear to
be as long as for other bases. Since the incorporated cytosines cannot be analyzed using PGE
under the used conditions, cyclic voltammetry at HMDE was used for their detection. For
T30+C, the same tensammetric peak caused by the T30 primer (TTENS
) as in the case of T30+A

87
was observed. Although TTENS
appeared at a potential close to that of the C peak, it was
possible to distinguish these two peaks as the potential at which it occurred was about 400
mV more negative than that of the C peak. The T30 peak was clearly discernible only in the
“no enzyme” control and the 1:1 ratio, with a slight hint at the 5:1 ratio; for the higher ratios
the peak diminishes, indicating that with the increasing ratio of dCTP:primer, the behavior of
the C-tail at the electrode surface prevails. The height of the C peak then grows gradually with
the increasing ratio. In the case of A30+C, similar situation occurred as for the C30+A, with C
and A being reduced at HMDE at close potentials. However, here it was possible to observe a
separate C peak for the higher ratios of dCTP:primer. With an increasing number of cytosines
forming the tail, it was observed that the C peak appeared at a less negative potential than the
AHMDE
peak. For the 50:1 ratio, the peak was already well developed. The peak heights gained
for the CA peak showed no obvious trend and remained similar for the primer and all ratios of
dCTP:primer. Nevertheless, an evident tendency to be higher with the increasing ratio was
seen when the C peak was analyzed separately. Moreover, this tendency matched the results
gained from the PAGE analysis, with a gradual increase from the 1:1 to 10:1 ratio and further
increase between 20:1 and 50:1 ratio.
Primers A30 and C30 were also used for tailing reaction with dTTP. The PAGE analysis of the
A30+T tailing products (Figure 5C) showed, that unlike for T30+A, the tailing reaction was not
stopped after a few added nucleotides and the tailing products gradually increased in length
up to the 50:1 ratio. It remains unclear why the behavior of TdT during the A30+T tailing
reaction differs from that of the T30+A combination. When analyzed at HMDE, the height of
the A peak displayed a decreasing tendency, which reflects a decreasing amount of the A30
parts of the tailing products that were adsorbed at the electrode surface due to a growing
portion of the T-tails. The PAGE analysis of the C30+T (Figure 5D) revealed the same
problem as for the C30+A and C30+A* combinations – no distinct tailing products at the
autoradiogram. However, the diminishing intensity of the C30 band indicates that the tailing
products were created but their length cannot be assessed. The voltammetric analysis of
C30+T at HMDE confirmed that the cytosines adsorb very strongly at the mercury surface
[29], which is evident from the fact that the C peak height remains unchanged for all ratios of
dTTP:primer.
Figure 6 shows the relative intensities of the peaks specific for the particular bases obtained
using square wave voltammetry at PGE or cyclic voltammetry at HMDE. Values of the peak
heights were normalized to the 1:1 dNTP:primer ratio, which enabled comparison of the
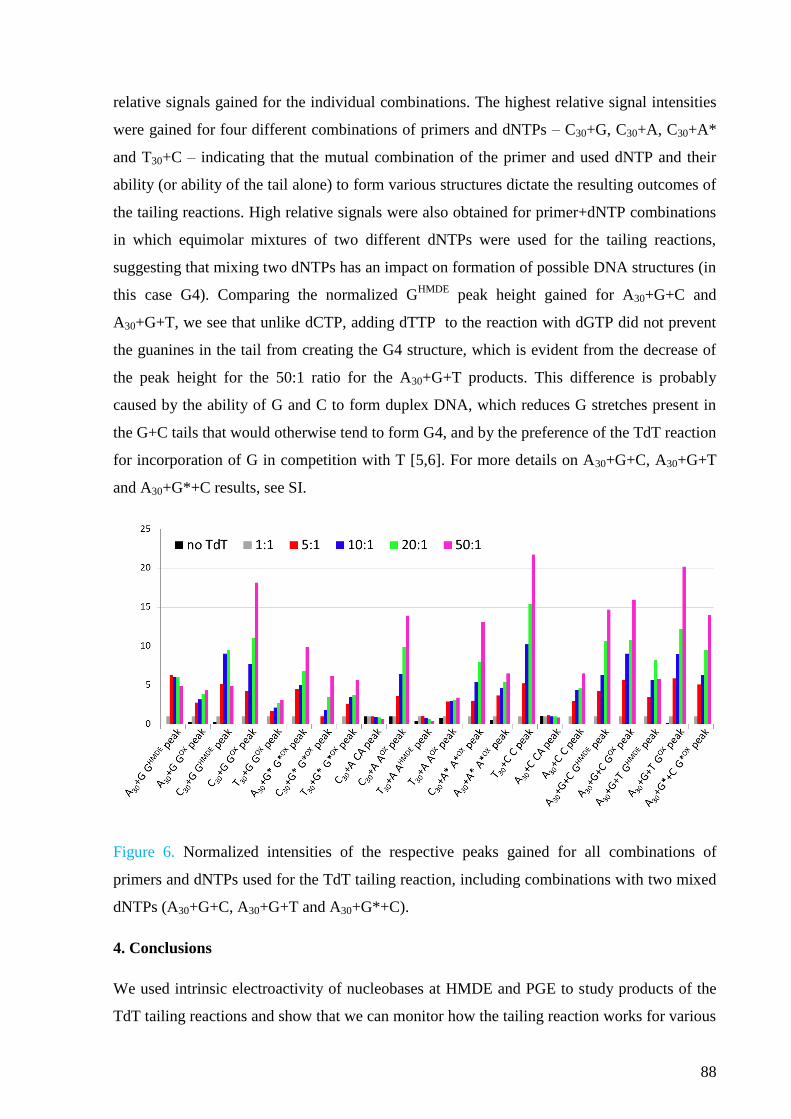
88
relative signals gained for the individual combinations. The highest relative signal intensities
were gained for four different combinations of primers and dNTPs – C30+G, C30+A, C30+A*
and T30+C – indicating that the mutual combination of the primer and used dNTP and their
ability (or ability of the tail alone) to form various structures dictate the resulting outcomes of
the tailing reactions. High relative signals were also obtained for primer+dNTP combinations
in which equimolar mixtures of two different dNTPs were used for the tailing reactions,
suggesting that mixing two dNTPs has an impact on formation of possible DNA structures (in
this case G4). Comparing the normalized GHMDE
peak height gained for A30+G+C and
A30+G+T, we see that unlike dCTP, adding dTTP to the reaction with dGTP did not prevent
the guanines in the tail from creating the G4 structure, which is evident from the decrease of
the peak height for the 50:1 ratio for the A30+G+T products. This difference is probably
caused by the ability of G and C to form duplex DNA, which reduces G stretches present in
the G+C tails that would otherwise tend to form G4, and by the preference of the TdT reaction
for incorporation of G in competition with T [5,6]. For more details on A30+G+C, A30+G+T
and A30+G*+C results, see SI.
Figure 6. Normalized intensities of the respective peaks gained for all combinations of
primers and dNTPs used for the TdT tailing reaction, including combinations with two mixed
dNTPs (A30+G+C, A30+G+T and A30+G*+C).
4. Conclusions
We used intrinsic electroactivity of nucleobases at HMDE and PGE to study products of the
TdT tailing reactions and show that we can monitor how the tailing reaction works for various

89
combinations of primers and dNTPs. This approach enabled study of the tailing reaction and
also allowed observation of the formation of DNA structures that may affect the tailing
reactions and electrochemical behavior of the respective oligonucleotides. This was
particularly evident in the case of HMDE since, for all tailing products capable of forming
unusual DNA structures, we observed a decrease in the corresponding signal, which reflected
the structural changes and their impact on the behavior of these molecules at the negatively
charged surface of HMDE. On the other hand, results obtained at PGE corresponded very
precisely to those obtained using denaturing PAGE, which implies that PGE can be used for
monitoring the lengths of the TdT tailing reaction products without dramatic interfering
effects of secondary DNA structure. Thus, combination of measurements at both types of
electrodes can provide complete (and complementary) information. In some cases, the
electrochemical analysis at PGE was even more accurate than the denaturing PAGE. Thus,
this approach proves to be an excellent tool for studying the TdT tailing reactions and also for
exploring electrochemical behavior of the DNA oligonucleotides at electrode surfaces.
5. Acknowledgements
This work was supported by the Czech Science Foundation (grant P206/12/G151), by the
ASCR (RVO 68081707), by the Ministry of Education, Youth and Sports of the Czech
Republic under project CEITEC 2020 (LQ1601), by the SYMBIT project reg. no.
CZ.02.1.01/0.0/0.0/15_003/0000477 financed from the ERDF, and from the European
Union’s Horizon 2020 research and innovation programme (project No 692068 BISON).
6. References
[1] K.-I. Kato, J. M. Goncalves, G. E. Houts, F. J. Bollum, J. Biol. Chem. 1967, 242,
2780–2789.
[2] E. A. Motea, A. J. Berdis, Biochim. Biophys. Acta - Proteins Proteomics 2010, 1804,
1151–1166.
[3] D. Baltimore, Nature 1974, 248, 409–411.
[4] F. J. Bollum, Enzymes 1974, 10, 145–171.
[5] B. Yang, K. N. Gathy, M. S. Coleman, J. Biol. Chem. 1994, 269, 11859–11868.
[6] M. J. Modak, Biochemistry 1978, 17, 3116–3120.
[7] M. R. Deibel, M. S. Coleman, J. Biol. Chem. 1980, 255, 4206–12.
[8] M. S. Chang, F. J. Bollum, J. Biol. Chem. 1990, 265, 17436–17440.

90
[9] E. Paleček, Naturwissenchaften 1958, 45, 186–187.
[10] E. Paleček, M. Bartošík, Chem. Rev. 2012, 112, 3427–3481.
[11] M. Fojta, F. Jelen, L. Havran, E. Paleček, Curr. Anal. Chem. 2008, 4, 250–262.
[12] J. Špaček, A. Daňhel, S. Hasoň, M. Fojta, Electrochem. Commun. 2017, 82, 34–38.
[13] L. Trnková, M. Studničková, E. Paleček, J. Electroanal. Chem. 1980, 116, 643–658.
[14] M. Studničková, L. Trnková, J. Zetěk, Z. Glatz, Bioelectrochem. Bioenerg. 1989, 275,
83–86.
[15] H. Pivoňková, P. Horáková, M. Fojtová, M. Fojta, Anal. Chem. 2010, 82, 6807–6813.
[16] M. Fojta, E. Palecek, Anal. Chim. Acta 1997, 342, 1–12.
[17] M. Fojta, R. P. Bowater, V. Staňková, L. Havran, D. M. J. Lilley, E. Paleček,
Biochemistry 1998, 37, 4853–4862.
[18] M. Fojta, L. Havran, J. Fulnečková, T. Kubičárová, Electroanalysis 2000, 12, 926–934.
[19] X. Cai, G. Rivas, H. Shirashi, P. Farias, J. Wang, M. Tomschik, F. Jelen, E. Paleček,
Anal. Chim. Acta 1997, 344, 65–76.
[20] A. M. Chiorcea-Paquim, A. M. Oliveira-Brett, Electrochim. Acta 2014, 126, 162–170.
[21] P. Vidláková, H. Pivoňková, I. Kejnovská, L. Trnková, M. Vorlíčková, M. Fojta, L.
Havran, Anal. Bioanal. Chem. 2015, 407, 5817–5826.
[22] R. Roychoudhury, E. Jay, R. Wu, Nucleic Acids Res. 1976, 3, 863–77.
[23] G. Deng, R. Wu, Nucleic Acids Res. 1981, 9, 4173–4188.
[24] A. M. Michelson, S. H. Orkin, J. Biol. Chem. 1982, 257, 14773–14782.
[25] J. B. Boulé, F. Rougeon, C. Papanicolaou, J. Biol. Chem. 2001, 276, 31388–31393.
[26] J. Gouge, S. Rosario, F. Romain, P. Beguin, M. Delarue, J. Mol. Biol. 2013, 425,
4334–4352.
[27] J. Loc’h, S. Rosario, M. Delarue, Structure 2016, 24, 1452–1463.
[28] E. P. Wright, J. L. Huppert, Z. A. E. Waller, Nucleic Acids Res. 2017, 45, 2951–2959.
[29] S. Hasoň, V. Vetterl, M. Fojta, Electrochim. Acta 2008, 53, 2818–2824.
[30] Z. Dudová, J. Špaček, M. Tomaško, L. Havran, H. Pivoňková, M. Fojta, Monatsh.
Chem. 2016, 147, 3–11.
[31] L. Trnková, F. Jelen, I. Postbieglová, Electroanalysis 2003, 15, 1529–1535.
[32] L. Trnková, F. Jelen, I. Postbieglová, Electroanalysis 2006, 18, 662–669.
[33] R. Mikelová, L. Trnková, F. Jelen, V. Adam, R. Kizek, Electroanalysis 2007, 19, 348–
355.

91
Appendix 3

92
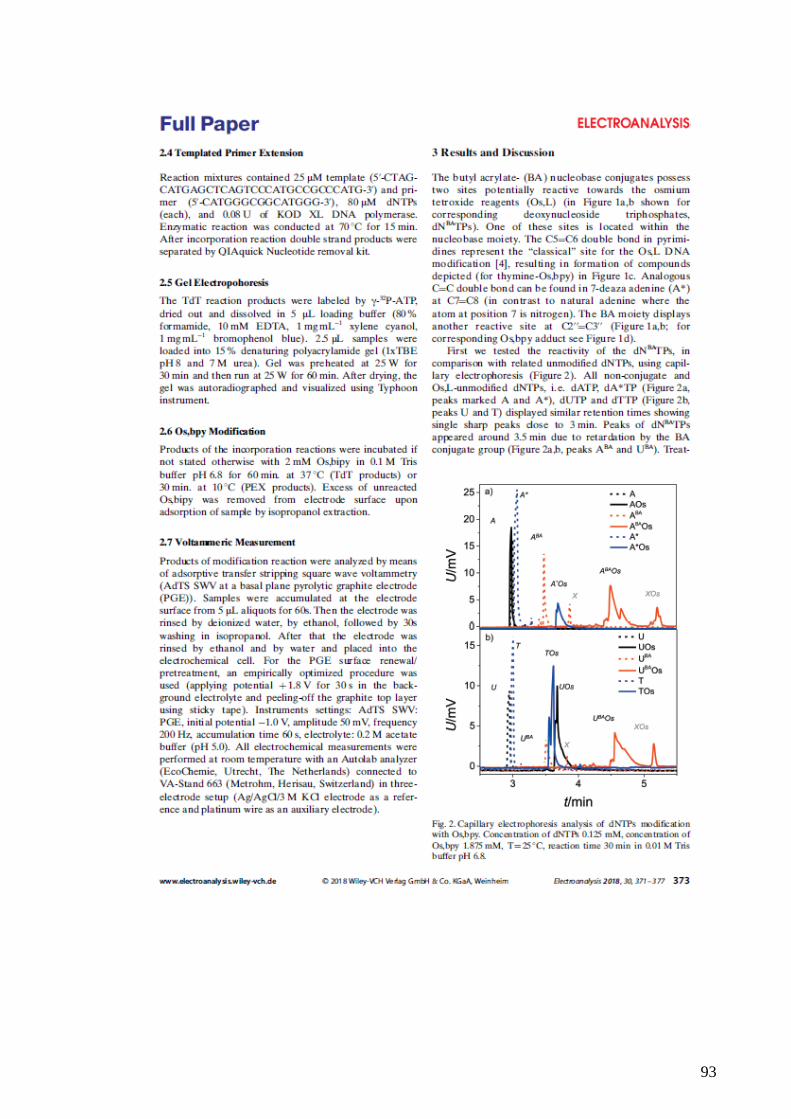
93

94
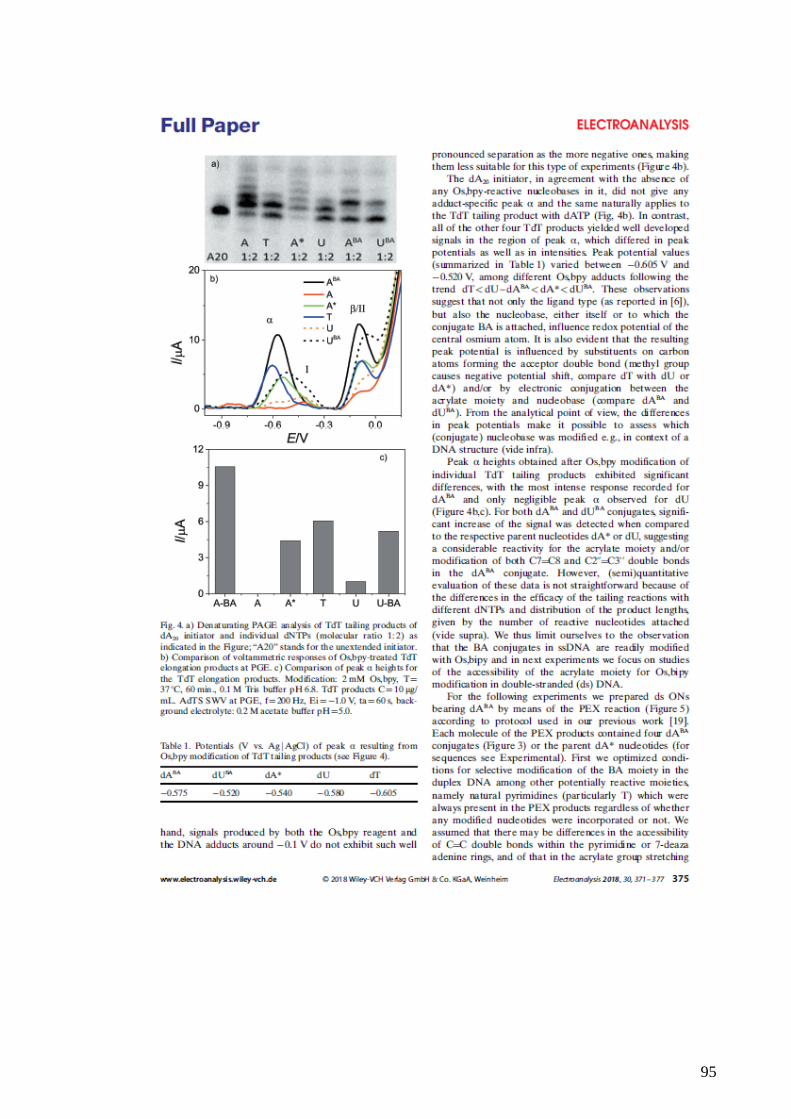
95
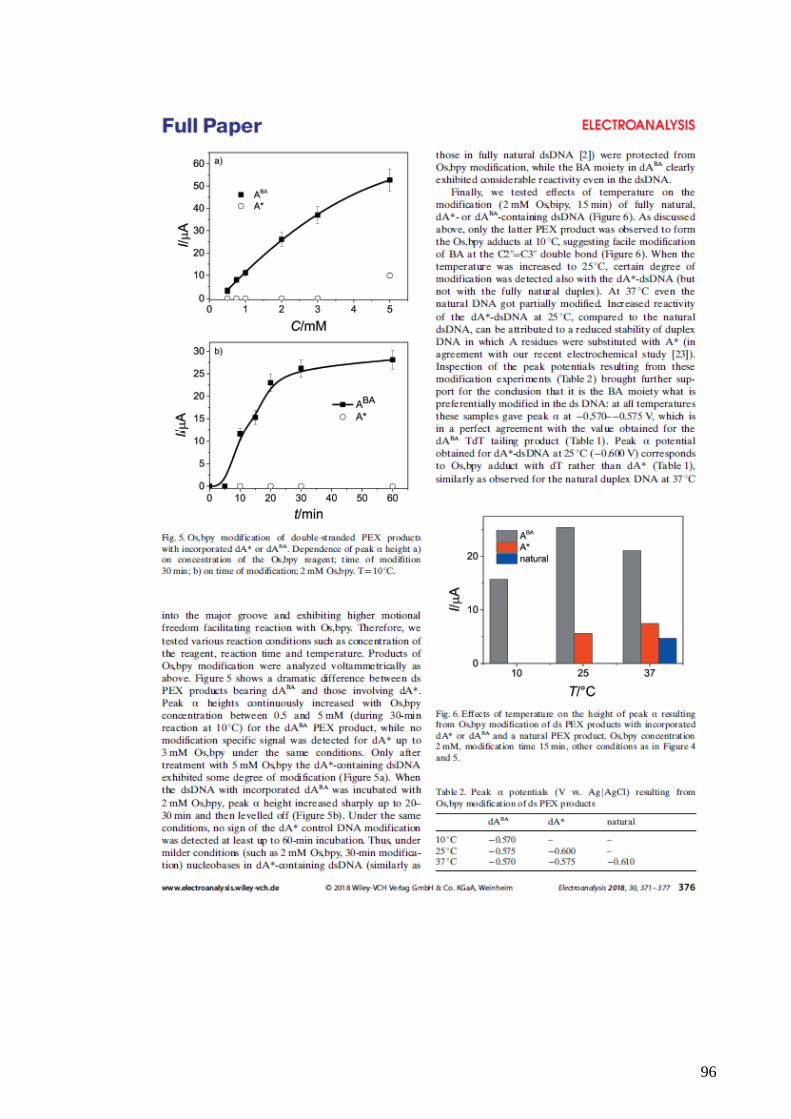
96

97





























