Embed Size (px)
Citation preview

European Journal of Pharmacology 607 (2009) 213–219
Contents lists available at ScienceDirect
European Journal of Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /e jphar
Pulmonary, Gastrointestinal and Urogenital Pharmacology
RBx 6198: A novel α1-adrenoceptor antagonist for the treatment of benignprostatic hyperplasia
Kamna Nanda a,⁎, Krishna S. Naruganahalli a, Suman Gupta a, Shivani Malhotra a, Atul Tiwari a,Laxminarayan G. Hegde a, Sanjay Jain b, Neelima Sinha b, Jung B. Gupta a, Anita Chugh a,Nitya Anand c, Abhijit Ray a
a Department of Pharmacology, Ranbaxy Research Laboratories, Plot -20, Sector-18, Gurgaon, 122015, Indiab Department of Medicinal Chemistry, Ranbaxy Research Laboratories, Plot -20, Sector-18, Gurgaon 122015, Indiac Central Drug Research Institute, Chattar Manzil Palace, Post Box No.173, Lucknow 226001, India
⁎ Corresponding author. Tel.: +91 124 2342001 10x52E-mail address: [email protected] (K. Nan
0014-2999/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.ejphar.2009.02.026
a b s t r a c t
a r t i c l e i n f oArticle history:
The present study, investi Received 5 May 2008Received in revised form 2 February 2009Accepted 9 February 2009Available online 23 February 2009Keywords:α1A-adrenoceptorα1B-adrenoceptorUroselectiveα1D-adrenoceptorBenign prostatic hyperplasia
gates the effect of RBx 6198, 2-{3-[4-(2-Isopropoxy-phenyl)-piperazin-1-yl]-propyl}-3a, 4, 7, 7a-tetrahydro-isoindole-1, 3,-dione, a novel α1-adrenoceptor antagonist, in both in vitro andin vivo test systems. RBx 6198 is a potent (nanomolar affinity) α1A-adrenoceptor antagonist withdemonstrable uroselectivity in anaesthesized dog model. In radioligand binding studies using humanrecombinant receptors, RBx 6198 exhibited high selectivity (~50 fold) for the α1A-adrenoceptor subtype ascompared to α1B-adrenoceptor subtype. In order to assess tissue selectivity, the antagonistic effect of RBx6198 on the phenylephrine induced contractile response of isolated rat prostate, spleen and aorta wascharacterized. RBx 6198 was 8 fold more potent in inhibiting phenylephrine-evoked contractions of isolatedtissues compared to tamsulosin. However, the compound was non-selective for α1A vs. α1D-adrenoceptorlike tamsulosin. In anaesthetized beagle dogs RBx 6198 suppressed the intraurethral pressure response tophenylephrine to a greater extent than the mean arterial pressure response thereby demonstratinguroselectivity consistent with in vitro binding and functional data. RBx 6198 was 6.4 fold more uroselective ascompared to tamsulosin after i.v. route dose administration. Taken together all results from preclinicalstudies, it is suggested that RBx 6198 is a novel α1-adrenoceptor antagonist that exhibited improvedpharmacological profile over tamsulosin in both in vitro and in vivo.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
Benign prostatic hyperplasia, is a condition common in elderlymale population (40 years and older) that manifests as bladder outletobstruction and consequent difficulty in micturition. Benign prostatichyperplasia is characterized by static and dynamic components,which contribute to lower urinary tract symptoms. The staticcomponent refers to the enlargement of the prostate gland, whichmay result in the compression of the urethra and obstruction of urineflow from the bladder (Tiwari et al., 2005). The dynamic componentreflects the smooth muscle tone of the bladder neck and prostatesmooth muscle, which is regulated by the sympathetic nerve systemthrough α1-adrenoceptors (Pulito et al., 2000). Therefore, treatmentwith α1-adrenoceptors antagonists in patients would be an effectivetherapy to decrease intraurethral pressure and to increase urinaryflow rate (Yasunori et al., 1999). In fact, α1-adrenoceptors blockers areconsidered as the first-line therapy for the treatment of lower urinary
67; fax: +91 124 2343544.da).
ll rights reserved.
tract symptoms associated with clinical benign prostatic hyperplasia,with a proven clinical efficacy (Oesterling, 1995). However, the majorproblem with commonly used alpha adrenoceptor antagonist likeprazosin, terazosin, doxazosin and alfuzosin, is their inability todistinguish between vascular and urinary tract alpha adrenoceptors.This is potentially associated with a range of vascular side effects inthe patients particularly, postural hypotension, dizziness, fatigue andasthenia (Tiwari et al., 2005; Andersson, 1998).
With the well characterization of three distinct subtypes of α1-adrenoceptors – α1A, α1B and α1D (Hieble et al., 1995; Michel et al.,1995) in humans, prostatic tissue has been reported to predominantlyexpress α1A-adrenoceptors subtype with major blood vessels expres-sing a heterogeneous mix of all three subtypes. However, it is α1B-adrenoceptors, which has been reported to play an important role inthe elderly in vasoregulation (Schwinn and Michelotti, 2000). With apredominant expression of α1B-adrenoceptors in the vascular bedsand their role in maintaining vascular tone it is postulated that α1A-adrenoceptors selective antagonist with lesser affinity for α1B-adrenoceptors might exhibit an efficacy in alleviating the lowerurinary tract symptoms associated with Benign prostatic hyperplasiawith an improved vascular tolerability.

214 K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
Amongst currently available α1-adrenoceptor antagonists for thetreatment of benign prostatic hyperplasia, tamsulosin is the onlyuroselective drug with a moderate selectivity overα1B-adrenoceptors.However, antagonists, which are highly selective for α1A-adrenocep-tors relax prostate smooth muscle and reduce benign prostaticobstruction, appear to be incapable of treating filling symptomsrelated to the bladder.With an abundance ofα1D-adrenoceptors in thehuman bladder and non-existence of α1B-adrenoceptors it washypothesized that targetingα1D-adrenoceptors could be an importanttarget for improving symptoms related to bladder filling. Thereforeantagonists that exhibit high affinity for α1A-adrenoceptors withminimal affinity for α1B-adrenoceptors may demonstrate minimalcardiovascular side effects but may not be fully efficacious fortreatment of irritative symptoms associated with benign prostatichyperplasia. As another school of thought blockade of α1A-adreno-ceptors, α1D-adrenoceptors would be required for achieving optimalclinical efficacy. With the current information available, we atRanbaxy Research Laboratories decided to design and discover novelα-1 adrenoceptor antagonists with a balanced subtype selectivityprofile: α1A=α1DNα1B for optimal therapeutic efficacy with minimalcardiovascular side effects. The present study highlights the char-acterization of lead candidate RBx 6198 (Fig. 1) over variouspreclinical aspects under in vitro and in vivo systems.
2. Materials and methods
2.1. In-vitro receptor binding assays
2.1.1. Human recombinant clonesChinese hamster ovary (CHO) cells stably transfected with human
α1A, α1B, or α1D-adrenoceptors (Keffel et al., 2000) were cultured inan atmosphere of 5% CO2/95% O2 at 37 °C in F-12 HAM mediumsupplemented with 10% heat-inactivated fetal calf serum,1 mMglutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin. Selectionpressure was maintained by regular addition of geneticin (G 418;Roche Biochemicals, Mannheim, Germany) to the culture medium.Cells were pelleted by centrifugation at 500 ×g for 5 min washed andhomogenized in ice-cold buffer A (50 mM Tris, 1 mMMgCl2) followedby centrifugation of homogenate at 35,000 ×g for 20 min at 4 °C. Thecell homogenate was suspended in buffer A and frozen at −80 °C tilluse.
2.1.2. Rat tissuesRat sub maxillary gland and liver were employed as source of α1A
andα1B adrenoceptor subtypes respectively (Michel et al., 1989). MaleWistar rats weighing 200–250 g were sacrificed (CO2 asphyxiation) toisolate sub maxillary glands and liver. Sub maxillary glands wereisolated immediately after sacrifice whereas the liver was firstlyperfused with buffer (Tris HCl 50 mM, pH 7.4) before isolation. Thetissues were homogenized in 10 volumes of buffer (Tris HCl 50 mM,NaCl 100 mM, EDTA 1 mM, pH 7.4) with a polytron homogenizer(Kinematica). The homogenate was filtered through two layers of wetgauze and filtrate was centrifuged at 500 ×g for 10 min. Thesupernatant was subsequently centrifuged at 40,000 ×g for 20 min.The pellet thus obtained was resuspended in same volume of assay
Fig. 1. 2-{3-[4-(2-Isopropoxy-phenyl)-piperazin-1-yl]-propyl}-3a,4,7,7a-tetrahydro-isoindole-1,3-dione.
buffer (Tris HCl 50 mM, EDTA 1 mM, pH 7.4) and were stored at−70 °C until the time of assay. Protein was estimated using ModifiedLowry's Method.
Competition radioligand binding assay in recombinant cellmembranes was performed by using [3H] prazosin as the radioligand(Michel et al., 1996). Briefly, experiments were performed in bindingbuffer containing 50 mM Tris, 10 mM MgCl2 and 0.5 mM EDTA at pH7.5 in a total assay volume of 1000 µl. The protein content typicallywas 40–60 µg/assay. Themixtures were incubated at 25 °C for 45 min.The binding assays for native tissues were performed according toU'Prichard et al. (1978) with minor modifications. The membranehomogenates of native tissues (100 µg protein) were incubated in titreplates in 250 µl of assay buffer (Tris HCl 50mM, EDTA 1mM, pH 7.4) at25 °C for 1 h. Non-specific binding was determined in the presence of10 µM terazosin. The reaction was terminated by vacuum filtrationover 0.1% polyethylenimine pre-treated filtermat (Wallac) using a cellharvester (Skatron). The filters were then washed with ice-cold50 mM Tris HCl buffer (pH 7.4). The filtermats were dried andtransferred to 24 well plates (PET A. No cross talk). Radioactivityretained on filters was counted in 500 µl of supermix in microbetacounter (Wallac Microbeta Trilux).
The data was analysed by sigmoidal non-linear curve fitting underPrism program (Graph pad Software, San Diego, CA, USA). ResultingIC50 values from the binding studies were converted into affinityestimates (Ki values) based on the affinity (Kd values) of [3H] prazosinas determined from saturation binding studies. Results are expressedas mean±S.E.M for each group. Number of experiments for each groupis represented as n. Statistical significance for pKi(−log Ki) values wasassessed by analysis of variance followed by multiple comparisons byTukey Kramer test. P value b0.05 was considered as significant.
Subtype selectivity α1A vs. α1B and α1A vs. α1D was expressed as anantilogarithmof difference ofmeanpKi atα1A andα1B adrenoceptors andantilogarithm of difference of mean pKi at α1A and α1D adrenoceptorsrespectively.
2.2. Rat isolated tissue functional assays
Male Wistar rats (250–400 g) were euthanized with an overdoseof thiopentone sodium (~300 mg/kg, i.p.). Aortic rings, prostate andspleen strips were removed and kept in aerated Krebs Henseleit bufferof the following composition (mM): NaCl, 118; KCl 4.7; CaCl2, 2.5;MgSO4, 1.2; NaHCO3, 25; KH2PO4, 1.2, glucose 11.1. These tissues werethen mounted in the buffer maintained at 37 °C and aerated withcarbogen (95% oxygen and 5% carbon dioxide) during the entirelength of experiment. A resting tension of 2 g (rat aorta and spleen) or1 g (rat prostate) was applied. The tissues were equilibrated for90 min with frequent washes at 15-min interval. After equilibration,the tissues were depolarized by 60 mM KCl and the same process wasrepeated till two reproducible contractile responses were recorded ona grass polygraph Model 7 or letica chart recorder system andexpressed as tension in grams. Cumulative concentration responsecurves to norepinephrine in rat aorta (Bucker et al., 1996) wereobtained following, which the tissues were allowed to relax to thebaseline while they were given a wash every 15 min. Tissues wereincubated with different concentrations of test compound (RBx 6198),with tamsulosin as a reference standard and vehicle for 20 min, afterwhich the second norepinephrine cumulative concentration responsecurve was obtained in aorta. In prostatic and hemispleen strips onlyone phenylephrine cumulative concentration response curve wasobtained in the absence or presence of RBx 6198 or tamsulosinincubated for 20 min.
The data was analyzed using a nonlinear curve-fitting program tocalculate EC50 using the Prism program (Graph pad Software, SanDiego, CA, USA). Functional antagonism, in terms of pKB, wascalculated from EC50 data using the relationship: − log [antagonist(M)/(EC50 agonist in the presence of antagonist/EC50 agonist in the

215K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
absence of antagonist)−1]. In tissues where Emax attained was lessthan 50% pKB was calculated by Kenakin's double reciprocal plot. One-way ANOVA followed by Tukey's Studentized Range (HSD) test wasused for statistical analysis.
2.3. Efficacy study in anaesthetized dogs
This study was conducted on male beagle dogs weighing between12 and 20 kg. Dogs were obtained from the experimental animalfacility of Ranbaxy Research Laboratories. They were housed instandard kennels individually and used in a crossover design. Eachanimal in a single study was given incremental doses of testcompound (ranging from 1 to 30 µg/kg etc.). Between each study, awashout period of one week was allowed. Allocation of treatment toeach animal was randomly determined before the start of the study. Asingle animal was given more than one dose of the test or standarddrug as mentioned above. The experimental procedures describedbelowwere reviewed and approved by the Institutional Animal EthicsCommittee of Ranbaxy Research Laboratories.
2.3.1. Implantation of telemetric transmittersDogs were instrumented with TL11M2D70PCT telemetric trans-
mitters for blood pressure measurement. Implantationwas performedunder anesthesia using pentobarbitone sodium 35 mg/kg, givenintravenously. The telemetric transmitter implanted subcutaneouslyin the flank region and the pressor sensor catheter of transmitter wasintroduced into the femoral artery. Telemetric transmitters wereimplanted at least 10 days before use. During recovery period,antibiotics and analgesics were given for 5 days.
2.3.2. Intraurethral pressure measurementOn the day of the study, a 7F Swan–Ganz pulmoball balloon
catheter, lubricated with a water-soluble jelly, was inserted into theurethral orifice. The balloon was placed at the prostatic level andinflated with 1.5 ml of room air. The balloon port was connected to aGrass Polygraph (Model 7) via Statham P32XL transducer forintraurethral pressure measurement. The Intraurethral pressure wasrecorded manually on a chart paper.
2.3.3. Study protocolThe day prior to the study, animals, which have been previously
implanted with telemetry implants, were put under fasting(wateronly) and anesthesia was induced by pentobarbitone sodium (35 mg/kg, iv). The animal was placed in dorsal horizontal position on athermostatically controlled operating table. The animal was allowedto breathe spontaneously and when required the trachea wasintubated with an endotracheal tube to facilitate easy normalbreathing. A scalp vein set needle was introduced into the femoralvein for the administration of the drug solutions and for theintravenous fluids.
All preparations were completed within 20–30 min after theinduction of anesthesia. At the end of this preparatory phase, aminimum period of 30 min was allowed to verify the stability of thecardiovascular parameters measured. At the end of the stabilizationperiod, phenylephrine response (4 µg/kg, iv.) on blood pressure andintraurethral pressure was taken. Subsequently, a cumulative doseresponse with phenylephrine (1, 2, 4, 8 and 16 µg/kg, iv.) was taken.Once the basal level of hemodynamic parameters and intraurethralpressure were obtained, the test compound or vehicle was adminis-tered as defined under the study design. Twenty minutes after theadministration of the test compound the cumulative response withphenylephrine was repeated. The dose of phenylephrine wasincreased till the maximal response of phenylephrine on intraurethralpressure is nearly equal to that of the response obtained at 16 µg/kgbefore the administration of the test compound. Similarly, the next
dose response was taken 20 min after the administration of thehighest dose of the test compound as described in the study design.
The absolute change from basal blood pressure and intraurethralpressure in response to different doses of phenylephrine (mm Hg andmm respectively) was computed before and after treatment with testor standard compound. Since it is not possible to increase the dose ofphenylephrine more than 16 µg/kg because of onset of cardiacarrhythmias, blood pressure and intraurethral pressure data wasexpressed as percent of pseudomaximal response as described byKenny et al. (1996). Pseudomaximal responsewas computed using thefollowing formula:
10k of response obtained at 16 μg= kg + actual response at 16 μg = kg
Dose of phenylephrine (µg/kg) was expressed as molar weight/kg.ED50 of phenylephrinewas calculated by plotting the data to nonlinearregression analysis in the Graph pad prism software. pA2 of theantagonist was calculated by plotting log(Dose ratio −1) againstantagonist dose (molar weight /kg) and subjecting the data to Schildregression where intercept of x-axis represents pA2 value. Dose ratiowas calculated by using the following formula:
ðED50 of phenylephrine in the presence of test compound= ED50 ofphenylephrine in controlÞ− 1: Uroselectivity was calculated as :antilog ½pA2ðintraurethral pressureÞ− pA2ðmean arterial pressureÞ�:
2.4. Drugs and chemicals
All drugs and chemicals used in the study were of AR grade.Phenylephrine and norepinephrine were procured from SigmaChemicals; U.S.A. RBx 6198 and tamsulosin were received fromChemical Research Department (Ranbaxy Research Laboratories,Gurgaon) with a certificate of analysis. Stock solutions of RBx 6198and tamsulosin were prepared freshly. Subsequent dilutions wereprepared from the stock.
3. Results
3.1. Radioligand binding assays
3.1.1. Binding studies with human recombinant α1-adrenoceptorsRBx 6198was found to be a potentα1A/1D-adrenoceptor antagonist
with sub nanomolar affinity represented by pKi values of 9.21±0.07,7.5±0.1 and 8.4±0.09 for human recombinant α1A, α1B and α1D-adrenoceptors, respectively as shown in Table 1. RBx 6198 exhibitedgood selectivity over α1B-adrenoceptors (51 fold). The affinity of RBx6198 for α1B and α1D was significantly different compared to affinityfor α1A-adrenoceptors. In comparison tamsulosin exhibited moderateselectivity (14 fold) for α1A over α1B and was non-selective over α1D
adrenoceptors (Table 1).
3.1.2. Binding studies with rat α1-adrenoceptorsIn rat native tissue based competitive binding assay, RBx 6198 was
a potent inhibitor of [3H] prazosin binding with pKi of 9.8±0.04 and7.62±0.08 for α1A and α1B-adrenoceptors subtypes, respectively. RBx6198 exhibited high selectivity for α1A over α1B-adrenoceptorssubtype, which was statistically significant (Pb0.05). Tamsulosinalso exhibited high affinity for rat α1-adrenoceptor with pKi values of10.2±0.08 and 9.28±0.07 for α1A and α1B-adrenoceptors subtypes,respectively. Tamsulosin exhibited moderate selectivity for α1A overα1B-adrenoceptors adrenoceptors subtype, which was statisticallysignificant (Pb0.05). The fold selectivity for α1A over α1B-adrenocep-tors was significantly different between RBx 6198 compared withtamsulosin in the same experiment (Table 2).
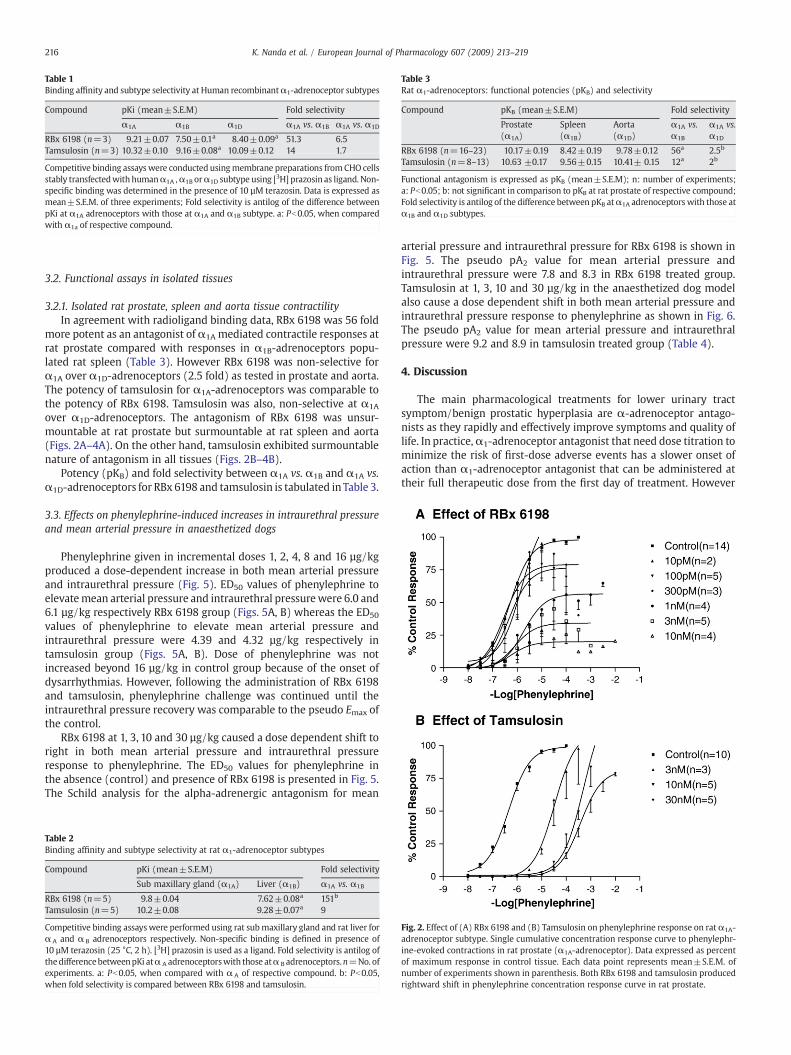
Table 1Binding affinity and subtype selectivity at Human recombinantα1-adrenoceptor subtypes
Compound pKi (mean±S.E.M) Fold selectivity
α1A α1B α1D α1A vs. α1B α1A vs. α1D
RBx 6198 (n=3) 9.21±0.07 7.50±0.1a 8.40±0.09a 51.3 6.5Tamsulosin (n=3) 10.32±0.10 9.16±0.08a 10.09±0.12 14 1.7
Competitive binding assayswere conducted usingmembrane preparations fromCHO cellsstably transfectedwith humanα1A ,α1B orα1D subtype using [3H] prazosin as ligand. Non-specific binding was determined in the presence of 10 µM terazosin. Data is expressed asmean±S.E.M. of three experiments; Fold selectivity is antilog of the difference betweenpKi at α1A adrenoceptors with those at α1A and α1B subtype. a: Pb0.05, when comparedwith α1a of respective compound.
Table 3Rat α1-adrenoceptors: functional potencies (pKB) and selectivity
Compound pKB (mean±S.E.M) Fold selectivity
Prostate(α1A)
Spleen(α1B)
Aorta(α1D)
α1A vs.α1B
α1A vs.α1D
RBx 6198 (n=16–23) 10.17±0.19 8.42±0.19 9.78±0.12 56a 2.5b
Tamsulosin (n=8–13) 10.63 ±0.17 9.56±0.15 10.41± 0.15 12a 2b
Functional antagonism is expressed as pKB (mean±S.E.M); n: number of experiments;a: Pb0.05; b: not significant in comparison to pKB at rat prostate of respective compound;Fold selectivity is antilog of the difference between pKB atα1A adrenoceptors with those atα1B and α1D subtypes.
216 K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
3.2. Functional assays in isolated tissues
3.2.1. Isolated rat prostate, spleen and aorta tissue contractilityIn agreement with radioligand binding data, RBx 6198 was 56 fold
more potent as an antagonist of α1A mediated contractile responses atrat prostate compared with responses in α1B-adrenoceptors popu-lated rat spleen (Table 3). However RBx 6198 was non-selective forα1A over α1D-adrenoceptors (2.5 fold) as tested in prostate and aorta.The potency of tamsulosin for α1A-adrenoceptors was comparable tothe potency of RBx 6198. Tamsulosin was also, non-selective at α1A
over α1D-adrenoceptors. The antagonism of RBx 6198 was unsur-mountable at rat prostate but surmountable at rat spleen and aorta(Figs. 2A–4A). On the other hand, tamsulosin exhibited surmountablenature of antagonism in all tissues (Figs. 2B–4B).
Potency (pKB) and fold selectivity between α1A vs. α1B and α1A vs.α1D-adrenoceptors for RBx 6198 and tamsulosin is tabulated inTable 3.
3.3. Effects on phenylephrine-induced increases in intraurethral pressureand mean arterial pressure in anaesthetized dogs
Phenylephrine given in incremental doses 1, 2, 4, 8 and 16 µg/kgproduced a dose-dependent increase in both mean arterial pressureand intraurethral pressure (Fig. 5). ED50 values of phenylephrine toelevatemean arterial pressure and intraurethral pressurewere 6.0 and6.1 µg/kg respectively RBx 6198 group (Figs. 5A, B) whereas the ED50
values of phenylephrine to elevate mean arterial pressure andintraurethral pressure were 4.39 and 4.32 µg/kg respectively intamsulosin group (Figs. 5A, B). Dose of phenylephrine was notincreased beyond 16 µg/kg in control group because of the onset ofdysarrhythmias. However, following the administration of RBx 6198and tamsulosin, phenylephrine challenge was continued until theintraurethral pressure recovery was comparable to the pseudo Emax ofthe control.
RBx 6198 at 1, 3, 10 and 30 µg/kg caused a dose dependent shift toright in both mean arterial pressure and intraurethral pressureresponse to phenylephrine. The ED50 values for phenylephrine inthe absence (control) and presence of RBx 6198 is presented in Fig. 5.The Schild analysis for the alpha-adrenergic antagonism for mean
Table 2Binding affinity and subtype selectivity at rat α1-adrenoceptor subtypes
Compound pKi (mean±S.E.M) Fold selectivity
Sub maxillary gland (α1A) Liver (α1B) α1A vs. α1B
RBx 6198 (n=5) 9.8±0.04 7.62±0.08a 151b
Tamsulosin (n=5) 10.2±0.08 9.28±0.07a 9
Competitive binding assayswere performed using rat submaxillary gland and rat liver forα A and α B adrenoceptors respectively. Non-specific binding is defined in presence of10 µM terazosin (25 °C, 2 h). [3H] prazosin is used as a ligand. Fold selectivity is antilog ofthedifferencebetweenpKi atα A adrenoceptorswith those atα B adrenoceptors.n=No. ofexperiments. a: Pb0.05, when compared with α A of respective compound. b: Pb0.05,when fold selectivity is compared between RBx 6198 and tamsulosin.
arterial pressure and intraurethral pressure for RBx 6198 is shown inFig. 5. The pseudo pA2 value for mean arterial pressure andintraurethral pressure were 7.8 and 8.3 in RBx 6198 treated group.Tamsulosin at 1, 3, 10 and 30 µg/kg in the anaesthetized dog modelalso cause a dose dependent shift in both mean arterial pressure andintraurethral pressure response to phenylephrine as shown in Fig. 6.The pseudo pA2 value for mean arterial pressure and intraurethralpressure were 9.2 and 8.9 in tamsulosin treated group (Table 4).
4. Discussion
The main pharmacological treatments for lower urinary tractsymptom/benign prostatic hyperplasia are α-adrenoceptor antago-nists as they rapidly and effectively improve symptoms and quality oflife. In practice,α1-adrenoceptor antagonist that need dose titration tominimize the risk of first-dose adverse events has a slower onset ofaction than α1-adrenoceptor antagonist that can be administered attheir full therapeutic dose from the first day of treatment. However
Fig. 2. Effect of (A) RBx 6198 and (B) Tamsulosin on phenylephrine response on ratα1A-adrenoceptor subtype. Single cumulative concentration response curve to phenylephr-ine-evoked contractions in rat prostate (α1A-adrenoceptor). Data expressed as percentof maximum response in control tissue. Each data point represents mean±S.E.M. ofnumber of experiments shown in parenthesis. Both RBx 6198 and tamsulosin producedrightward shift in phenylephrine concentration response curve in rat prostate.
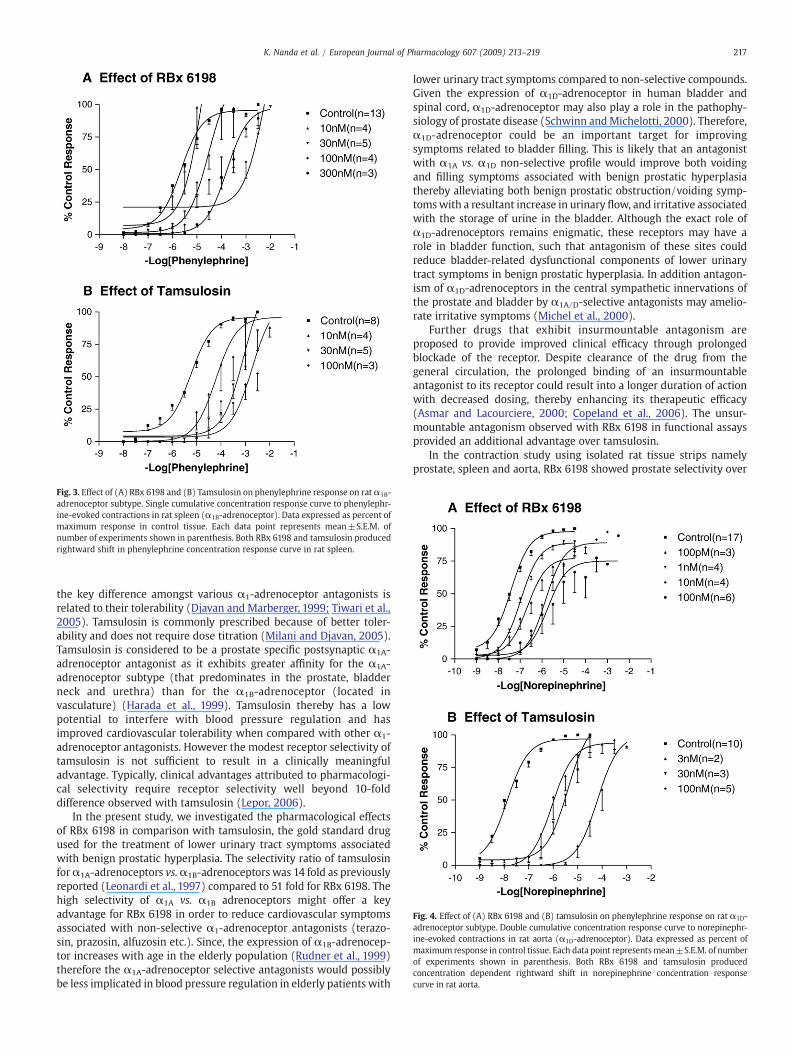
Fig. 3. Effect of (A) RBx 6198 and (B) Tamsulosin on phenylephrine response on ratα1B-adrenoceptor subtype. Single cumulative concentration response curve to phenylephr-ine-evoked contractions in rat spleen (α1B-adrenoceptor). Data expressed as percent ofmaximum response in control tissue. Each data point represents mean±S.E.M. ofnumber of experiments shown in parenthesis. Both RBx 6198 and tamsulosin producedrightward shift in phenylephrine concentration response curve in rat spleen.
Fig. 4. Effect of (A) RBx 6198 and (B) tamsulosin on phenylephrine response on rat α1D-adrenoceptor subtype. Double cumulative concentration response curve to norepinephr-ine-evoked contractions in rat aorta (α1D-adrenoceptor). Data expressed as percent ofmaximum response in control tissue. Each data point representsmean±S.E.M. of numberof experiments shown in parenthesis. Both RBx 6198 and tamsulosin producedconcentration dependent rightward shift in norepinephrine concentration responsecurve in rat aorta.
217K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
the key difference amongst various α1-adrenoceptor antagonists isrelated to their tolerability (Djavan and Marberger, 1999; Tiwari et al.,2005). Tamsulosin is commonly prescribed because of better toler-ability and does not require dose titration (Milani and Djavan, 2005).Tamsulosin is considered to be a prostate specific postsynaptic α1A-adrenoceptor antagonist as it exhibits greater affinity for the α1A-adrenoceptor subtype (that predominates in the prostate, bladderneck and urethra) than for the α1B-adrenoceptor (located invasculature) (Harada et al., 1999). Tamsulosin thereby has a lowpotential to interfere with blood pressure regulation and hasimproved cardiovascular tolerability when compared with other α1-adrenoceptor antagonists. However the modest receptor selectivity oftamsulosin is not sufficient to result in a clinically meaningfuladvantage. Typically, clinical advantages attributed to pharmacologi-cal selectivity require receptor selectivity well beyond 10-folddifference observed with tamsulosin (Lepor, 2006).
In the present study, we investigated the pharmacological effectsof RBx 6198 in comparison with tamsulosin, the gold standard drugused for the treatment of lower urinary tract symptoms associatedwith benign prostatic hyperplasia. The selectivity ratio of tamsulosinforα1A-adrenoceptors vs.α1B-adrenoceptors was 14 fold as previouslyreported (Leonardi et al., 1997) compared to 51 fold for RBx 6198. Thehigh selectivity of α1A vs. α1B adrenoceptors might offer a keyadvantage for RBx 6198 in order to reduce cardiovascular symptomsassociated with non-selective α1-adrenoceptor antagonists (terazo-sin, prazosin, alfuzosin etc.). Since, the expression of α1B-adrenocep-tor increases with age in the elderly population (Rudner et al., 1999)therefore the α1A-adrenoceptor selective antagonists would possiblybe less implicated in blood pressure regulation in elderly patients with
lower urinary tract symptoms compared to non-selective compounds.Given the expression of α1D-adrenoceptor in human bladder andspinal cord, α1D-adrenoceptor may also play a role in the pathophy-siology of prostate disease (Schwinn andMichelotti, 2000). Therefore,α1D-adrenoceptor could be an important target for improvingsymptoms related to bladder filling. This is likely that an antagonistwith α1A vs. α1D non-selective profile would improve both voidingand filling symptoms associated with benign prostatic hyperplasiathereby alleviating both benign prostatic obstruction/voiding symp-tomswith a resultant increase in urinary flow, and irritative associatedwith the storage of urine in the bladder. Although the exact role ofα1D-adrenoceptors remains enigmatic, these receptors may have arole in bladder function, such that antagonism of these sites couldreduce bladder-related dysfunctional components of lower urinarytract symptoms in benign prostatic hyperplasia. In addition antagon-ism of α1D-adrenoceptors in the central sympathetic innervations ofthe prostate and bladder by α1A/D-selective antagonists may amelio-rate irritative symptoms (Michel et al., 2000).
Further drugs that exhibit insurmountable antagonism areproposed to provide improved clinical efficacy through prolongedblockade of the receptor. Despite clearance of the drug from thegeneral circulation, the prolonged binding of an insurmountableantagonist to its receptor could result into a longer duration of actionwith decreased dosing, thereby enhancing its therapeutic efficacy(Asmar and Lacourciere, 2000; Copeland et al., 2006). The unsur-mountable antagonism observed with RBx 6198 in functional assaysprovided an additional advantage over tamsulosin.
In the contraction study using isolated rat tissue strips namelyprostate, spleen and aorta, RBx 6198 showed prostate selectivity over
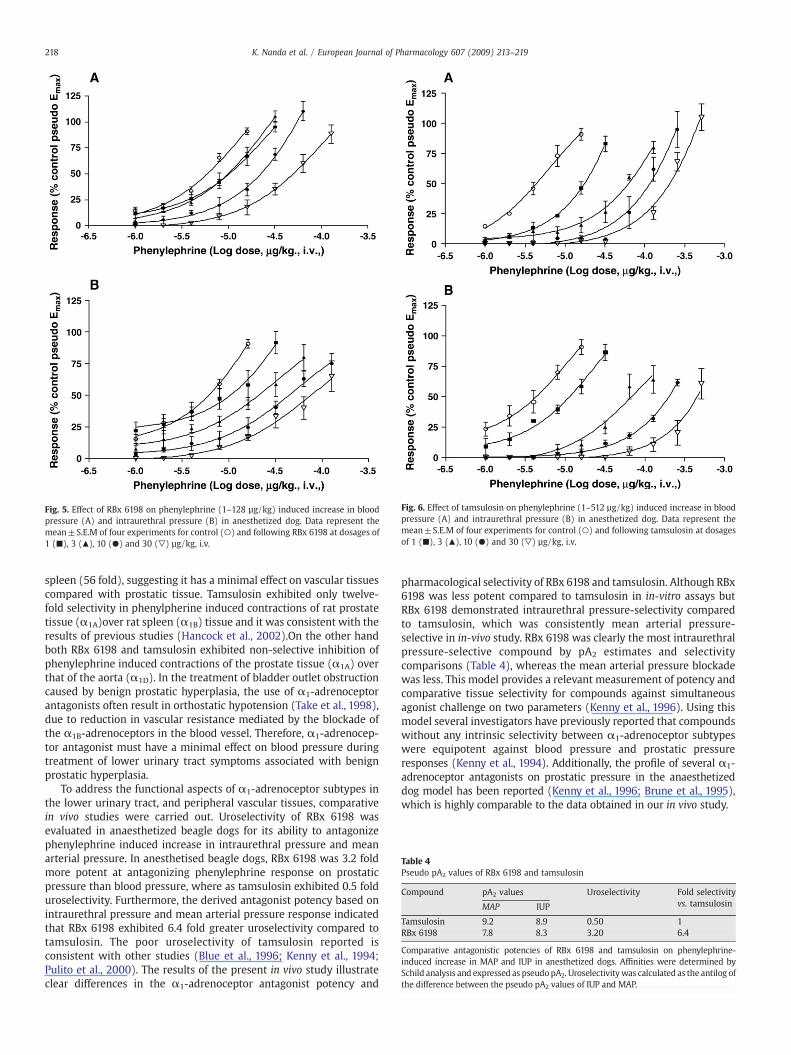
Fig. 5. Effect of RBx 6198 on phenylephrine (1–128 µg/kg) induced increase in bloodpressure (A) and intraurethral pressure (B) in anesthetized dog. Data represent themean±S.E.M of four experiments for control (○) and following RBx 6198 at dosages of1 (■), 3 (▲), 10 (●) and 30 (▽) µg/kg, i.v.
Fig. 6. Effect of tamsulosin on phenylephrine (1–512 µg/kg) induced increase in bloodpressure (A) and intraurethral pressure (B) in anesthetized dog. Data represent themean±S.E.M of four experiments for control (○) and following tamsulosin at dosagesof 1 (■), 3 (▲), 10 (●) and 30 (▽) µg/kg, i.v.
Table 4Pseudo pA2 values of RBx 6198 and tamsulosin
Compound pA2 values Uroselectivity Fold selectivityvs. tamsulosinMAP IUP
Tamsulosin 9.2 8.9 0.50 1RBx 6198 7.8 8.3 3.20 6.4
Comparative antagonistic potencies of RBx 6198 and tamsulosin on phenylephrine-induced increase in MAP and IUP in anesthetized dogs. Affinities were determined bySchild analysis and expressed as pseudopA2. Uroselectivitywas calculated as the antilog ofthe difference between the pseudo pA2 values of IUP and MAP.
218 K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
spleen (56 fold), suggesting it has a minimal effect on vascular tissuescompared with prostatic tissue. Tamsulosin exhibited only twelve-fold selectivity in phenylpherine induced contractions of rat prostatetissue (α1A)over rat spleen (α1B) tissue and it was consistent with theresults of previous studies (Hancock et al., 2002).On the other handboth RBx 6198 and tamsulosin exhibited non-selective inhibition ofphenylephrine induced contractions of the prostate tissue (α1A) overthat of the aorta (α1D). In the treatment of bladder outlet obstructioncaused by benign prostatic hyperplasia, the use of α1-adrenoceptorantagonists often result in orthostatic hypotension (Take et al., 1998),due to reduction in vascular resistance mediated by the blockade ofthe α1B-adrenoceptors in the blood vessel. Therefore, α1-adrenocep-tor antagonist must have a minimal effect on blood pressure duringtreatment of lower urinary tract symptoms associated with benignprostatic hyperplasia.
To address the functional aspects of α1-adrenoceptor subtypes inthe lower urinary tract, and peripheral vascular tissues, comparativein vivo studies were carried out. Uroselectivity of RBx 6198 wasevaluated in anaesthetized beagle dogs for its ability to antagonizephenylephrine induced increase in intraurethral pressure and meanarterial pressure. In anesthetised beagle dogs, RBx 6198 was 3.2 foldmore potent at antagonizing phenylephrine response on prostaticpressure than blood pressure, where as tamsulosin exhibited 0.5 folduroselectivity. Furthermore, the derived antagonist potency based onintraurethral pressure and mean arterial pressure response indicatedthat RBx 6198 exhibited 6.4 fold greater uroselectivity compared totamsulosin. The poor uroselectivity of tamsulosin reported isconsistent with other studies (Blue et al., 1996; Kenny et al., 1994;Pulito et al., 2000). The results of the present in vivo study illustrateclear differences in the α1-adrenoceptor antagonist potency and
pharmacological selectivity of RBx 6198 and tamsulosin. Although RBx6198 was less potent compared to tamsulosin in in-vitro assays butRBx 6198 demonstrated intraurethral pressure-selectivity comparedto tamsulosin, which was consistently mean arterial pressure-selective in in-vivo study. RBx 6198 was clearly the most intraurethralpressure-selective compound by pA2 estimates and selectivitycomparisons (Table 4), whereas the mean arterial pressure blockadewas less. This model provides a relevant measurement of potency andcomparative tissue selectivity for compounds against simultaneousagonist challenge on two parameters (Kenny et al., 1996). Using thismodel several investigators have previously reported that compoundswithout any intrinsic selectivity between α1-adrenoceptor subtypeswere equipotent against blood pressure and prostatic pressureresponses (Kenny et al., 1994). Additionally, the profile of several α1-adrenoceptor antagonists on prostatic pressure in the anaesthetizeddog model has been reported (Kenny et al., 1996; Brune et al., 1995),which is highly comparable to the data obtained in our in vivo study.

219K. Nanda et al. / European Journal of Pharmacology 607 (2009) 213–219
Taken together all information, RBx 6198 may have a comparableor better efficacy with an improved cardiovascular safety profile overtamsulosin. A balanced antagonist profile exhibited by RBx 6198across α1-adrenoceptor subtypes implicated in the control of urinarybladder function and outlet resistance (α1A and α1D) withoutcardiovascular effects at effective doses may result into a superiortherapeutic modality over existing class of drugs. The improveduroselectivity of RBx 6198 demonstrated herein, particularly effects onintraurethral pressure versus mean arterial pressure, suggests thatRBx 6198 may have a high therapeutic potential in the treatment oflower urinary tract symptoms suggestive of benign prostatichyperplasia.
In conclusion, RBx 6198 is more selective than tamsulosin for thehuman recombinant α1A-adrenoceptor subtype over α1B-adrenocep-tor subtype and hence might offer a better cardiovascular tolerabilityover tamsulosin. The selectivity over α1D-adrenoceptor subtype washowever comparable to tamsulosin as demonstrated by in vitroreceptor binding data and functional data.We have also demonstratedthe high in vivo uroselectivity for RBx 6198 compared to tamsulosin. Inconclusion, RBx 6198 has the distinct potential and advantage overexisting drugs to be an effective agent in the treatment of benignprostatic hyperplasia with an improved tolerability/side effect profile.
References
Andersson, K.E., 1998. The concept of uroselectivity. Eur. Urol. 33, 7–11.Asmar, R., Lacourciere, Y., 2000. A new approach to assessing antihypertensive therapy:
effect of treatment on pulse pressure. Candesartan cilexetil in HypertensionAmbulatory Measurement of Blood Pressure (CHAMP) Study Investigators.J. Hypertens. 18, 1683–1690.
Blue, D., Zhu, O.-M., Isom, P., Young, S., Larson, M., Clarke, D., 1996. Effect of α1-adrenoceptor (α1-AR) antagonists in dog prostate/blood pressure models. FASEB J.10, A2454.
Brune, M.E., Buckner, S.A., Polakowski, J., Kerwin, J.F., Hancock, A., 1995. Pharmacolo-gical antagonism of an adrenergic agonist induced increases in canine intraurethralpressure in vivo. Drug Dev. Res. 34, 267–275.
Bucker, S.A., Oheim, K.W., Morse, P.A., Knepper, S.M., Hancock, A.A., 1996. Alpha1-adrenoceptor-induced contractility in rat aorta is mediated by the alpha 1Dsubtype. Eur. J. Pharmacol. 297, 241–248.
Copeland, R.A., Pompliano, D.L., Meek, T.D., 2006. Drug-target residence time and itsimplications for lead optimization. Nat. Rev. Drug Discov. 5, 730–739.
Djavan, B., Marberger, M., 1999. A meta-analysis on the efficacy and tolerability ofalpha1-adrenoceptor antagonists in patients with lower urinary tract symptomssuggestive of benign prostatic obstruction. Eur. Urol. 36, 1–13.
Hancock, A.A., Buckner, S.A., Brune, M.E., Esbenshade, T.A., Ireland, L.M., Katwala, S.,Milicic, I., Meyer, M.D., Kerwin, J.F., Williams, M., 2002. Preclinical pharmacology offiduxosin, a novel α1-adrenoceptor antagonist with uroselective properties.J. Pharmacol. Exp. Ther. 300, 478–548.
Harada, K., Ohmori, M., Kitoh, Y., Sugimoto, K., Fujimura, A., 1999. A comparison of theantagonistic activities of tamsulosin and terazosin against human vascular alpha1-adrenoceptors. Jpn. J. Pharmacol. 80, 209–215.
Hieble, J.P., Bylund, D.B., Clarke, D.E., Eikenburg, D.C., Langer, S.Z., Lefkowitz, R.J.,Minneman, K.P., Ruffolo Jr., R.R., 1995. International union of pharmacology.X. Recommendation for nomenclature of alpha 1-adrenoceptors: consensus update.Pharmacol. Rev. 47, 267–270.
Keffel, S., Alexandrov, A., Goepel, M., Michel, M.C., 2000. Alpha (1)-adrenoceptorsubtypes differentially couple to growth promotion and inhibition in Chinesehamster ovary cells. Biochem. Biophys. Res. Commun. 272, 906–911.
Kenny, B.A., Naylor, A.M., Carter, A.J., Read, A.M., Greengrass, P.M., Wyllie, M.G., 1994.Effect of alpha1 adrenoceptor antagonists on prostatic pressure and blood pressurein the anesthetized dog. Urology 44, 52–57.
Kenny, B.A., Miller, A.M., Williamson, I.J., O'Connell, J., Chalmers, D.H., Naylor, A.M., 1996.Evaluation of the pharmacological selectivity profile of alpha 1-adrenoceptorantagonists at prostatic alpha 1 adrenoceptors: binding, functional and in vivostudies. Br. J. Pharmacol. 118, 871–878.
Leonardi, A., Hieble, J.P., Guarneri, L., Naselsky, D.P., Poggesi, E., Sironi, G., Sulpizio, A.C.,Testa, R., 1997. Pharmacological characterization of the uroselective alpha-1antagonist Rec 15/2739(SB 216469): role of the alpha-1L adrenoceptor in tissueselectivity, part I. J. Pharmacol. Exp. Ther. 281, 1272–1283.
Lepor, H., 2006. The evolution of alpha-blockers for the treatment of benign prostatichyperplasia. Rev. Urol. 8, S3–S9.
Michel, A.D., Loury, D.H., Whiting, R.L., 1989. Identification of a single alpha1-adrenoceptor corresponding to the alpha-1A-subtype in rat sub maxillary gland.Br. J. Pharmacol. 98, 883–889.
Michel, M.C., Kenny, B., Schwinn, D.A., 1995. Classification of alpha1-adrenoceptorsubtypes. Naunyn-Schmiedebergs Arch. Pharmacol. 352, 1–10.
Michel, M.C., Grubbel, B., Taquchi, K., Verfurth, F., Otto, T., Kropff, D., 1996. Drugs for thetreatment of benign prostatic hyperplasia: affinity comparison at cloned alpha1-adrenoceptor subtypes and human prostate. J. Auton. Pharm. 16, 21–28.
Michel, M.C., Schafers, R.F., Goepel, M., 2000. Alpha-blockers and lower urinary tractfunction: more than smooth muscle relaxation? BJU Int. 86, 23–30.
Milani, S., Djavan, B., 2005. Lower urinary tract symptoms suggestive of benign prostatichyperplasia: latest update on alpha -adrenoceptor antagonists. Br. J. Urol. 95, 29–36.
Oesterling, J.E., 1995. Benign prostatic hyperplasia. Medical and minimally invasivetreatment options. N. Engl. J. Med. 332, 99–109.
Pulito, V.L., Li, Z., Varga, S.S., Mulcahy, L.S., Clark, K.S., Halbert, S.A., Reitz, A.B.,Murray,W.V.,Jolliffe, L.K., 2000. An investigation of the uroselective properties of four novel α1-adrenergic receptor subtype-selective antagonists. J. Pharmacol. Exp. Ther. 294,224–229.
Rudner, X.L., Berkowitz, D.E., Booth, J.V., Funk, B.L., Cozart, K.L., D'Amico, E.B., El-Moalem, H., Page, S.O., Richardson, C.D., Winters, B., Marucci, L., Schwinn, D.A.,1999. Subtype specific regulation of human vascular alpha (1)-adrenergic receptorsby vessel bed and age. Circulation 100, 2336–2343.
Schwinn, D.A., Michelotti, G.A., 2000.α1-adrenergic receptors in the lower urinary tractand vascular bed: potential role for the alpha1d subtype in filling symptoms andeffects of ageing on vascular expression. Br. J. Urol. 85, 6–11.
Take, H., Shibata, K., Awaji, T., Hirasawa, A., Ikegaki, I., Asano, T., Takada, T., Tsujimoto, G.,1998. Vascular alpha1-adrenoceptor subtype selectivity and alpha1-blocker-induced orthostatic hypotension. Jpn. J. Pharmacol. 77, 61–70.
Tiwari, A., Krishna, N.S., Nanda, K., Chugh, A., 2005. Benign prostatic hyperplasia: aninsight into current investigational medical therapies. Expert Opin. Investig. Drugs14, 1359–1372.
U'Prichard, D.C., Charness, M.E., Robertson, D., Snyder, S.H., 1978. Prazosin: differentialaffinities for two populations of alpha-noradrenergic receptor binding sites. Eur.J. Pharmacol. 50, 87–89.
Yasunori, S., Atsunori, K., Yuki, O., Kazua, A., Ikunoba, M., 1999. Effect of JTH-601, a novelα-adrenoceptor antagonist, on the function of lower urinary tract and bloodpressure. Eur. J. Pharmacol. 374, 495–502.





























