Embed Size (px)
Citation preview

CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
REVIEW
CURRENTOPINION Recent advances in the antiphospholipid antibody
syndrome
Copyright © Lippincott W
1065-6251 � 2014 Wolters Kluwer
a b,c
Shruti Chaturvedi and Keith R. McCraePurpose of review
The antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by recurrentthrombosis and/or obstetrical morbidity in the presence of persistently positive antiphospholipid antibodies.Recent insights into the pathogenesis of APS have begun to elucidate pathophysiology and led to theidentification of potential therapeutic interventions. The objective of this review is to examine the advancesin this field and highlight the areas of further investigation.
Recent findings
Several mechanisms of thrombosis and pregnancy loss in APS have been proposed. These includeactivation of endothelial cells, monocytes, and platelets, and/or inhibition of natural anticoagulant andfibrinolytic systems by antiphospholipid antibodies. However, in many cases the underlying molecularmechanisms and their relevance to the human disorder remain uncertain. New therapeutic agents such asstatins, hydroxychloroquine, rituximab, complement inhibitors, and interventions aimed at disruption ofintracellular signaling pathways have shown promise in preclinical and clinical studies.
Summary
Indefinite anticoagulation remains the mainstay of treatment for thrombotic APS. Despite advances indiagnostic techniques, it remains difficult to predict thrombotic risk in asymptomatic patients withantiphospholipid antibodies. Further mechanistic and clinical studies are needed to predict thrombotic riskand develop improved therapies for this devastating illness.
Keywords
b2-glycoprotein I, antiphospholipid, lupus anticoagulant, thrombosis
aDepartment of Internal Medicine, bTaussig Cancer Institute andcDepartment of Cellular and Molecular Medicine, Cleveland Clinic,Cleveland, Ohio, USA
Correspondence to Keith R. McCrae, MD, Taussig Cancer Institute, R4-018, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195, USA.Tel: +1 216 445 7809; fax: +1 216 445 7809; e-mail: [email protected]
Curr Opin Hematol 2014, 21:000–000
DOI:10.1097/MOH.0000000000000067
INTRODUCTION
The antiphospholipid syndrome (APS) is a systemicautoimmune disorder characterized by recurrentthrombosis and/or obstetrical morbidity in thepresence of antiphospholipid antibodies (aPL),including lupus anticoagulant, anti-b2-glycoproteinI (anti-b2GPI), and/or anticardiolipin (aCL) anti-bodies [1]. Thrombosis in APS can affect any vascularsite, with the deep veins of the lower extremities andthe cerebral arterial circulation most commonlyaffected [2]. A small number of patients (<1%)develop catastrophic antiphospholipid syndrome(CAPS) [2], defined as small-vessel thrombosis inthree or more organs in less than 1 week in thepresence of aPL, with histopathologic confirmationof small-vessel thrombosis in the absence of inflam-mation [3]. CAPS is associated with high (50%)mortality, mostly because of cerebral and cardiacthrombosis, infections, and multiorgan failure [4
&
].Obstetrical morbidity in APS includes the unex-plained death of one or more morphologically nor-mal fetuses at or beyond the 10th week of gestation,
illiams & Wilkins. Unau
Health | Lippincott Williams & Wilk
the premature birth of one or more morphologicallynormal neonates before the 34th week of gestationbecause of either eclampsia or severe preeclampsia,and/or three or more unexplained, consecutive spon-taneous abortions before the 10th week of gestation[1]. Other clinical associations of aPL include throm-bocytopenia, livedo reticularis, transient ischemicattacks and skin ulcers [4
&
].
DIAGNOSIS AND THROMBOTIC RISKASSESSMENT
The diagnosis of APS rests on the presence of at leastone clinical and laboratory criterion (Table 1).
thorized reproduction of this article is prohibited.
ins www.co-hematology.com

C
CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
KEY POINTS
� Antiphospholipid antibodies are associated with arterialand venous thrombosis as well as recurrent and latefetal loss.
� There have been numerous mechanisms proposed toaccount for the effects of these antibodies, although theprimary mechanistic pathways remain uncertain.
� Interactions of antiphospholipid antibodies withvascular cells, mediated through recognition of cell-bound b2GPI, may play a central role in pathogenesis.
� Guidelines recommend that patients withantiphospholipid antibody-associated thrombosis shouldbe treated with long-term anticoagulation.
Hemostasis and thrombosis
According to the revised classification guidelines(2006), the laboratory criteria for APS require thepersistence (for>12 weeks) of a lupus anticoagulant,detected according to the guidelines of the Inter-national Society of Thrombosis and Hemostasis [5]and/or high titers of IgG or IgM autoantibodiesagainst cardiolipin and/or b2GPI detected usingstandardized ELISAs [1]. Patients with other aPLssuch as antiprothrombin antibodies, IgA aCL oranti-b2GPI antibodies, or those with clinical mani-festations other than thrombosis or pregnancy mor-bidity cannot be formally diagnosed with APS usingthese criteria. It remains difficult to predict throm-botic risk in patients with aPL, and patients may beencountered who develop thrombi yet do not meetthe criteria for APS due to the presence of only low ormoderate levels of aCL or anti-b2GPI antibodies.
In most reports, lupus anticoagulants are morestrongly associated with thrombotic risk and preg-nancy complications than aCL or anti-b2GPI anti-
opyright © Lippincott Williams & Wilkins. Unautho
Table 1. Revised classification criteria for definite APS
Clinical criteria (one or more)
Vascular thrombosis
One or more objectively confirmed episodes of arterial, venous orsmall vessel thrombosis in any tissue or organ
Pregnancy morbidity
One or more unexplained deaths of a morphologically normal fetusat or beyond the 10th week of gestation; or one or morepremature births of a morphologically normal neonate before the34th week of gestation because of placental insufficiency,preeclampsia, or eclampsia; or three or more unexplainedconsecutive spontaneous abortions before the 10th week ofgestation
At least one clinical and one laboratory criteria must be present for the diagnosis ofaCL, anticardiolipin; APS, antiphospholipid syndrome; b2GPI, b2-glycoprotein I; ISTAdapted with permission [1].
2 www.co-hematology.com
bodies alone [6]. The risk of a first thrombotic eventin triple-positive patients, that is, those with posi-tivity for lupus anticoagulant, aCL, and anti–b2GPIantibodies, may be as high as 5.3% per year [7].Antibodies directed toward b2GPI domain 1 maycorrelate more strongly with thrombotic riskand pregnancy morbidity [8]. Antibodies againstprothrombin also are associated with thrombosis,but with lower odds ratios than those against b2GPI[9
&
]. A better understanding of the thrombotic riskprofile of patients with aPL is needed to help definethe need for, and the optimal type and duration of,antithrombotic therapy.
PATHOGENESIS
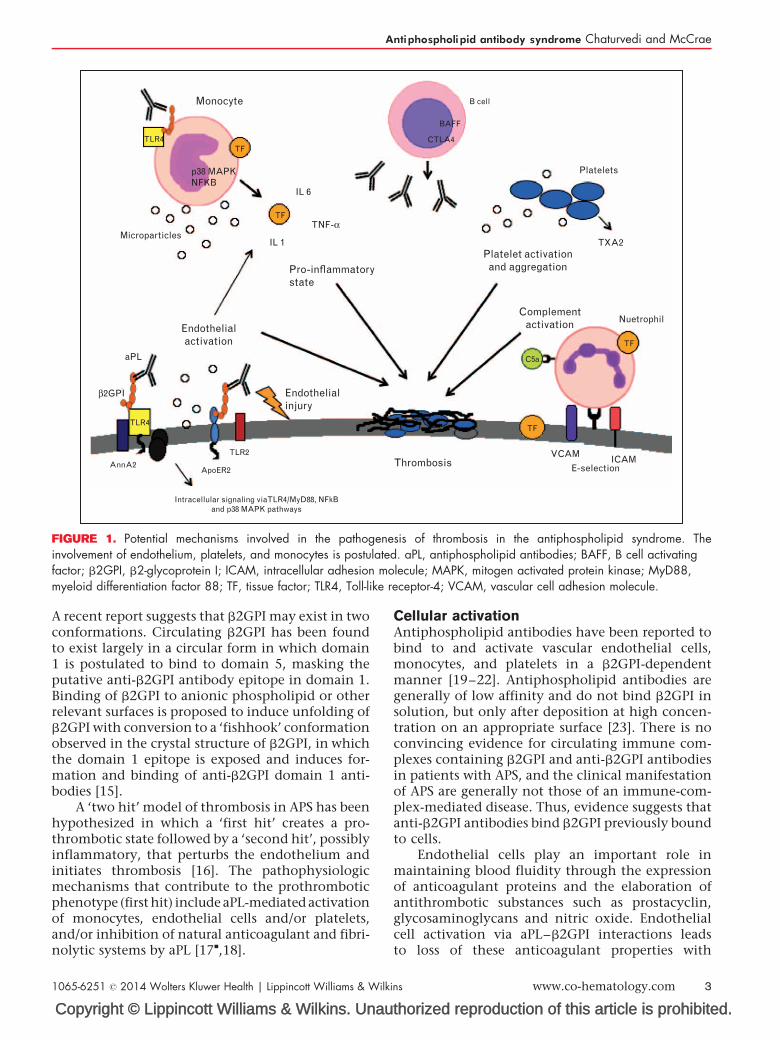
Antiphospholipid antibodies directed against phos-pholipid binding proteins bound to the surfaceof endothelial cells, monocytes and platelets arelikely central to the pathogenesis of APS. b2GPI isrecognized as the primary antigenic target of aPL[10] (Fig. 1). Affinity-purified human anti-b2GPIautoantibodies potentiate arterial and venousthrombus formation in a mouse model [11], andlupus anticoagulants whose effects are mediated viainteractions with b2GPI confer a higher risk ofthrombosis than those due to aCL or anti-prothrom-bin antibodies [6,7]. b2GPI is also expressed on thesurface of placental trophoblasts, where anti-b2GPIantibody binding to b2GPI results in inhibition ofgrowth and differentiation of trophoblasts, andinflammatory changes leading to fetal loss [12]. Dis-placement of annexin V from these cells may alsoallow exposure of anionic phospholipid, providing anidus for coagulation complex assembly [13].
b2GPI is a plasma glycoprotein containing five‘sushi’ domains, the fifth of which is atypical andmediates binding to anionic phospholipid [14].
rized reproduction of this article is prohibited.
Laboratory criteria (one or more should be present on two ormore occasions at least 12 weeks apart)
Lupus anticoagulant detected according to the ISTHguidelines
aCL antibody of IgG and IgM isotype, present in medium orhigh titer (>40 IgG or IgM phospholipid units or >99thpercentile) measured on standardized ELISA. Anti-b2GPIantibody of IgG and IgM isotype present in titer >99thpercentile measured on standardized ELISA
definite APS.H, International Society of Thrombosis and Hemostasis.
Volume 21 � Number 00 � Month 2014
CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Monocyte
Thrombosis
Platelet activationand aggregation
Complementactivation
Platelets
TXA2
Nuetrophil
VCAMICAM
E-selection
Microparticles
Endothelialactivation
Pro-inflammatorystate
Endothelialinjury
IL 6
IL 1
aPL
TNF-α
β2GPI
TLR4TF
B cell
BAFF
CTLA4
p38 MAPKNFKB
TF
TF
TF
C5a
TLR4
TLR2
AnnA2ApoER2
Intracellular signaling viaTLR4/MyD88, NFkBand p38 MAPK pathways
FIGURE 1. Potential mechanisms involved in the pathogenesis of thrombosis in the antiphospholipid syndrome. Theinvolvement of endothelium, platelets, and monocytes is postulated. aPL, antiphospholipid antibodies; BAFF, B cell activatingfactor; b2GPI, b2-glycoprotein I; ICAM, intracellular adhesion molecule; MAPK, mitogen activated protein kinase; MyD88,myeloid differentiation factor 88; TF, tissue factor; TLR4, Toll-like receptor-4; VCAM, vascular cell adhesion molecule.
Antiphospholipid antibody syndrome Chaturvedi and McCrae
A recent report suggests that b2GPI may exist in twoconformations. Circulating b2GPI has been foundto exist largely in a circular form in which domain1 is postulated to bind to domain 5, masking theputative anti-b2GPI antibody epitope in domain 1.Binding of b2GPI to anionic phospholipid or otherrelevant surfaces is proposed to induce unfolding ofb2GPI with conversion to a ‘fishhook’ conformationobserved in the crystal structure of b2GPI, in whichthe domain 1 epitope is exposed and induces for-mation and binding of anti-b2GPI domain 1 anti-bodies [15].
A ‘two hit’ model of thrombosis in APS has beenhypothesized in which a ‘first hit’ creates a pro-thrombotic state followed by a ‘second hit’, possiblyinflammatory, that perturbs the endothelium andinitiates thrombosis [16]. The pathophysiologicmechanisms that contribute to the prothromboticphenotype (first hit) include aPL-mediated activationof monocytes, endothelial cells and/or platelets,and/or inhibition of natural anticoagulant and fibri-nolytic systems by aPL [17
&
,18].
Copyright © Lippincott Williams & Wilkins. Unau
1065-6251 � 2014 Wolters Kluwer Health | Lippincott Williams & Wilk
Cellular activationAntiphospholipid antibodies have been reported tobind to and activate vascular endothelial cells,monocytes, and platelets in a b2GPI-dependentmanner [19–22]. Antiphospholipid antibodies aregenerally of low affinity and do not bind b2GPI insolution, but only after deposition at high concen-tration on an appropriate surface [23]. There is noconvincing evidence for circulating immune com-plexes containing b2GPI and anti-b2GPI antibodiesin patients with APS, and the clinical manifestationof APS are generally not those of an immune-com-plex-mediated disease. Thus, evidence suggests thatanti-b2GPI antibodies bind b2GPI previously boundto cells.
Endothelial cells play an important role inmaintaining blood fluidity through the expressionof anticoagulant proteins and the elaboration ofantithrombotic substances such as prostacyclin,glycosaminoglycans and nitric oxide. Endothelialcell activation via aPL–b2GPI interactions leadsto loss of these anticoagulant properties with
thorized reproduction of this article is prohibited.
ins www.co-hematology.com 3

C
CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Hemostasis and thrombosis
transformation to a pro-adhesive, procoagulantphenotype characterized by increased expressionof adhesion molecules (E-selectin, intracellularadhesion molecule-1, and vascular cell adhesionmolecule-1) and tissue factor, enhanced secretionof pro-inflammatory cytokines and chemokines,and release of procoagulant and proinflammatorymicroparticles [24]. Annexin A2, a cell-surface recep-tor for plasminogen and plasminogen activator,mediates the binding of b2GPI to cells [20,25].However, annexin A2 does not have a transmem-brane domain; therefore, other co-receptors must beinvolved in cellular activation. A recent studysuggested that anti-b2GPI antibodies induce signal-ing through a multiprotein complex that includesannexin A2, calreticulin, nucleolin, and Toll-likereceptor 4 (TLR4) [25], with TLR4 inducing acti-vation of a TLR4/myeloid differentiation factor 88(MyD88)-dependent pathway culminating in NF-kBactivation [26,27
&&
]. The involvement of p38mitogen activated protein kinase in endothelialactivation has also been demonstrated [28]. In addi-tion to annexin A2, other potential receptors such asapoER2 [29] and TLR2 may also play a role in endo-thelial activation [30].
b2GPI co-localizes with annexin 2 and TLR4 onthe lipid rafts of monocytes and anti-b2GPI anti-bodies stimulate monocyte tissue factor expression[31]. Antiphospholipid antibody-induced tissue fac-tor expression in monocytes occurs through phos-phorylation of MEK-1/ERK proteins, and the p38mitogen activated protein kinase-dependent nucleartranslocation and activation of NF-kB/Rel proteins[32]. Tissue factor is the main initiator of the extrinsiccoagulation pathway and is likely a key mediator ofAPS-related thrombosis.
Recombinant dimerized b2GPI and preformed,immobilized complexes of b2GPI and anti-b2GPImonoclonal antibodies induce platelet adhesionto collagen-coated surfaces in a glycoprotein IBand apoER2-dependent manner [33,34]. Yet, despiteevidence for platelet activation in patients with APS,specific binding of monomeric b2GPI to unacti-vated platelets has not been convincingly demon-strated.
Anticoagulant and fibrinolytic systems
Antiphospholipid antibodies promote thrombosisvia interference with the anticoagulant activity ofprotein C, protein S [35], annexin V [36], and antith-rombin [37]. Antiphospholipid antibodies inhibitthe activation and activity of protein C, both inthe fluid phase and on cell surfaces, and in doingso may enhance thrombin generation [38]. Anti-prothrombin antibodies from APS patients have
opyright © Lippincott Williams & Wilkins. Unautho
4 www.co-hematology.com
lupus anticoagulant activity, inhibit the inactivationof thrombin by antithrombin, induce tissue factorexpression, and display prothrombotic propertiesin vivo [39]. Antiphospholipid antibodies may alsoinhibit the interactions of antithrombin with anti-coagulant glycosaminoglycans on cell surfaces [40].
Elevated levels of coagulation factor XI havebeen identified as a risk factor for thrombosis inthe general population. APS patients have higherlevels of the active free thiol form of factor XI thanage and sex-matched controls [41]. The free thiolform of b2GPI is formed by the action of thiore-doxin-I and protein disulfide isomerase. WhetheraPL and antib2GPI antibodies have direct effects onprotein disulfide isomerase is unknown.
Annexin A5 binds to phosphatidylserine on cellsurfaces and forms a shield that inhibits the for-mation of coagulation complexes. Anti-b2GPI anti-bodies complexed with b2GPI can disrupt thisshield, exposing procoagulant phosphatidylserineand promoting thrombosis [13]. Hydroxychloro-quine has been reported to protect the annexin Vshield and has shown efficacy against aPL-mediatedthrombosis in a murine model [42,43].
b2GPI may have intrinsic fibrinolytic activity,and several studies have suggested that aPL mayinhibit fibrinolysis through interactions with tissueplasminogen activator and/or plasminogen [18,44].
Complement activation
Activation of the complement system has beenimplicated in the development of thrombosis andfetal loss in APS [45]. Activated complement frag-ments bind to and activate cells through the C5b-9membrane attack complex or C5a-receptor-medi-ated effects. Complement activation by aPL maygenerate the potent inflammatory mediator C5a,which recruits neutrophils and monocytes and leadsto exposure of tissue factor by endothelial cells andneutrophils [46]. Mice deficient in C3, C4, C5, orC5a receptor are protected from fetal loss induced byaPL IgG [47]. Complement deficiency is also protec-tive against thrombosis in some murine models [48].However, although elevated levels of complementsplit products have been identified in patients withAPS, these have not been demonstrated to correlatewith thrombosis [49]. Several recent case reportsdocument the successful use of eculizumab (human-ized anti-C5a monoclonal antibody) in patientswith CAPS and APS complicating renal transplan-tation [50
&&
].
Origin of antiphospholipid
Loss of immune tolerance is thought to be respon-sible for the origin of pathogenic aPL, which appears
rized reproduction of this article is prohibited.
Volume 21 � Number 00 � Month 2014

CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Antiphospholipid antibody syndrome Chaturvedi and McCrae
to be predominantly antigen driven. Bacterial andviral infections have been implicated in the develop-ment of aPL and shown to induce pathogenic anti-bodies against b2GPI. Antiphospholipid antibodiesdevelop in mice immunized with a cytomegalovi-rus-derived peptide [51], and a recent study demon-strated that protein H of Streptococcus pyogenes canbind b2GPI and expose neoepitopes that induceproduction of anti-b2GPI antibodies [52]. Oxidationof b2GPI by reactive oxygen and nitrosative speciesalso increase in its immunogenicity [53].
Rauch et al. [54] demonstrated that TLR4 isinvolved in inducing a break in immune toleranceand production of aPL. Dysregulation of other TLRsincluding TLR7, TLR8, and TLR9 may also contrib-ute to the development of aPL [55]. Hydroxychlor-oquine inhibits TLR7 and is associated with reducedpersistence of aPL in patients with systemic lupuserythematosus (SLE) [56
&&
].
THERAPEUTIC ADVANCES
Anticoagulation with heparin followed by long-termanticoagulation with a vitamin K antagonist is themainstay of therapy for thrombotic APS. However, asignificant proportion of patients have recurrentthrombosis despite antithrombotic therapy [57].Vitamin K antagonists are also problematic becauseof food and drug interactions, bleeding compli-cations, and need for frequent monitoring. Fur-thermore, aPL interact differently with differentthromboplastin reagents affecting monitoring ofthe prothrombin time and international normalizedratio [58].
The oral direct thrombin inhibitors (dabigatran)and direct factor Xa inhibitors (rivaroxaban andapixaban) overcome some of these disadvantages– they are fixed dose, do not need routine monitor-ing, and have few drug or food interactions. How-ever, they are irreversible and there is limitedexperience in patients with APS. The Rivaroxabanin AntiPhospholipid Syndrome (RAPS) trial is anongoing, open-label, prospective, noninferiorityrandomized trial evaluating the efficacy of rivarox-aban in patients with thrombotic APS. Oral directinhibitors should be considered in APS patients witha first or recurrent venous thromboembolis (VTE)only when there is vitamin K antagonist intoleranceor poor anticoagulant control. There are no data torecommend their use in APS patients with recurrentVTE occurring on therapeutic anticoagulation or inAPS-related arterial thrombosis [59
&
].Advances in the understanding of patho-
genic mechanisms involved in APS have led tothe identification of new therapeutic approaches.These include inhibition of cellular activation and
Copyright © Lippincott Williams & Wilkins. Unau
1065-6251 � 2014 Wolters Kluwer Health | Lippincott Williams & Wilk
intracellular signaling pathways, antiplatelet agents,and immunomodulatory therapies (Table 2).
Statins
Statins display anti-inflammatory properties andinhibit cellular activation by aPL. Fluvastatin pre-vents the expression of adhesion molecules andtissue factor by aPL-treated endothelial cells in vitro[60]. Simvastatin and pravastatin also reduced fetalloss in a mouse model [61]. In a recent prospectivestudy, Erkan et al. [62
&
] demonstrated that fluvasta-tin treatment reduced the levels of biomarkers ofinflammation and thrombosis in patients with aPL.Modulation of aPL effects on vascular cells by statinscould be a valuable approach in the management ofAPS. At this time, however, statins are not recom-mended in patients with APS in the absence ofhyperlipidemia [59
&
]. Clinical studies are neededto evaluate their role in adjuvant therapy andfor primary thromboprophylaxis in aPL-positivepatients.
Hydroxychloroquine
Hydroxychloroquine is an established treatment forSLE due to its anti-inflammatory and immuno-modulatory effects. Hydroxychloroquine has beenreported to protect against arterial and venousthrombosis in SLE patients with and without aPL[63]. This is likely mediated by a decrease in lupusactivity and cardiovascular risk factors as well asmodulation of aPL effects. Hydroxychloroquineinhibits platelet aggregation and arachidonic acidrelease from stimulated platelets. It also inhibitsbinding of anti-b2GPI antibodies to purified phos-pholipid membranes. Recent studies by Rand andcolleagues have shown that hydroxychloroquineprotects the annexin A5 shield on endotheliumand placental syncytiotrophoblast from disruptionby aPL and preserves anticoagulant activity [64].Observational studies have shown that hydroxy-chloroquine decreased aPL titers and lowered theodds of having persistently positive aPL [56
&&
].Hydroxychloroquine is currently recommended
for all aPL-positive patients with SLE [59&
]. There areno strong data to support the use of hydroxychlor-oquine in patients with aPL without systemic auto-immune diseases. A recently published prospectivetrial comparing oral anticoagulation (fluindione)with or without hydroxychloroquine in primaryAPS patients reported that there were six (30%)VTE events in the monotherapy group versus zeroevents in the hydroxychloroquine group at 6 and36 months [65
&
]. A multicenter, randomized controltrial (NCT 01784523) to determine the efficacy of
thorized reproduction of this article is prohibited.
ins www.co-hematology.com 5

Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Tab
le2
.N
ovel
targ
eted
trea
tmen
tsin
antip
hosp
holip
idsy
ndro
me
Dru
gTa
rget
sIn
-vit
ro/i
n-vi
vost
ud
ies
Clin
ica
lst
ud
ies
Stat
ins
Fluv
asta
tinRe
duce
sce
llula
rac
tivat
ion,
TF,
IL-6
,IL-1
b,
adhe
sion
mol
ecul
eex
pres
sion
.U
preg
ulat
eseN
OS
Dec
reas
edex
pres
sion
ofTF
and
adhe
sion
mol
ecul
eson
endo
thel
ialc
ells
invi
tro,
redu
ced
feta
llos
sin
the
mou
sem
odel
ofA
PS
Dec
reas
edin
flam
mat
ory
mar
kers
inA
PSpa
tient
s,on
goi
ngcl
inic
altri
alN
CT0
0674297
Hyd
roxy
chlo
roqu
ine
Ann
exin
A5,
TLR7
Prot
ects
Ann
A5
shie
ldon
endo
thel
ium
and
sync
ytio
troph
obla
stD
ecre
ased
thro
mbo
sis
rate
inSL
Epa
tient
sw
ith/w
ithou
taPL
.In
prim
ary
aPL,
decr
ease
daP
Ltit
ers
and
pers
iste
nce.
Ong
oing
clin
ical
trial
NC
T01784523
Com
plem
enti
nhib
itors
Ecul
izum
abC
5a
Cas
ere
ports
inC
APS
,on
goi
ngcl
inic
altri
alN
CT0
1029587
Def
ibro
tide
Ade
nosi
nere
cept
or,
neut
roph
ilTF
expr
essi
onC
ase
repo
rtsin
refrac
tory
CA
PS
B-ce
ll-di
rect
edth
erap
yC
ase
repo
rts,
clin
ical
trial
(RIT
APS
)
Ritu
xim
abC
D20
Cas
ere
ports
ofaP
Lsu
ppre
ssio
n
Co-
stim
ulat
ory
bloc
kade
CTL
A4
CTL
A4-Ig
prev
ente
daP
Lpr
oduc
tion
ina
mur
ine
mod
el
BAFF
anta
goni
sts
BAFF
Prev
ente
dA
PSon
seta
ndim
prov
essu
rviv
alin
am
ouse
mod
el
NF-
kB
inhi
bito
r(M
G132)
NFK
BD
ownr
egul
ated
proi
nfla
mm
ator
yan
dpr
othr
ombo
ticef
fect
sof
antib
odie
sfrom
APS
patie
nts
ina
mou
sem
odel
TLR4
inhi
bitio
n(T
AK242)
TLR4
Redu
ced
B2G
PI/a
nti-B
2G
PIin
duce
dTF
,p3
8M
APK
,M
yD88
and
TRIF
expr
essi
onin
THP-
1ce
lls
p38
MA
PKin
hibi
tion
p38
MA
PKBl
ocke
dN
F-kB
signa
ling
and
TFex
pres
sion
inaP
Lst
imul
ated
THP-
1ce
lls
Fact
or-X
I-direc
ted
ther
apy
Fact
orXI
/PD
IBl
ocke
dth
rom
bus
form
atio
nin
am
ouse
mod
elw
ithno
incr
ease
inbl
eedi
ngrisk
aPL,
antip
hosp
holip
id;
APS
,an
tipho
spho
lipid
synd
rom
e;BA
FF,
Bce
llac
tivat
ing
fact
or;
b2G
PI,
b2-g
lyco
prot
ein
I;C
APS
,ca
tast
roph
ican
tipho
spho
lipid
synd
rom
e;eN
OS,
endo
thel
ialn
itric
oxid
esy
ntha
seM
APK
,m
itogen
activ
ated
prot
ein
kina
se;
PDI,
prot
ein
disu
lfide
isom
eras
e;RI
TAPS
,Ri
tuxi
mab
inA
ntiP
hosp
holip
idSy
ndro
me;
SLE,
syst
emic
lupu
ser
ythe
mat
osus
;TF
,tis
sue
fact
or;
TLR,
Toll-
like
rece
ptor
;TR
IF,
TIR
dom
ain
cont
aini
ngad
apto
rin
duci
ngIL-1
b.
Hemostasis and thrombosis
6 www.co-hematology.com Volume 21 � Number 00 � Month 2014

CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Antiphospholipid antibody syndrome Chaturvedi and McCrae
hydroxychloroquine for primary thrombosis pre-vention in patients with aPL without systemic auto-immune diseases is currently underway.
Complement inhibition
Complement is implicated in the pathogenesis ofthrombosis and fetal loss in APS via generationof C5a. Anti-C5a monoclonal antibodies and C5a-receptor antagonist peptides have demonstratedefficacy in attenuating thrombosis and preventingfetal loss in murine models of APS. Clinical studies ofcomplement inhibition in APS are limited to casereports describing the successful use of eculizumabin patients with refractory CAPS and APS-relatedpostrenal transplant thrombotic microangiopathy[50
&&
]. An ongoing phase II study (NCT01029587)is evaluating the efficacy of eculizumab to preventrecurrent CAPS after kidney transplantation inpatients with a prior history of CAPS. Eculizumabcarries a risk of infection with encapsulated organ-isms and patients should be immunized againstmeningococcus before starting treatment [59
&
].
Defibrotide
Defibrotide is an adenosine receptor antagonist withantithrombotic, profibrinolytic, and anti-inflamma-tory effects on vascular endothelial cells that alsoblocks neutrophil tissue factor expression. It is beensuccessfully used in hepatic veno-occlusive diseaseand multiorgan failure after stem cell transplant.Defibrotide could potentially decrease endothelialactivation in APS and has been used for the treat-ment of refractory CAPS [66].
B-cell-directed therapy
B cells contribute to APS pathogenesis through theproduction of pathogenic aPL. Rituximab (anti-CD20 chimeric monoclonal antibody) inducesB-cell depletion and has been used with success insome patients with refractory CAPS [67]. The Ritux-imab in AntiPhospholipid Syndrome (RITAPS) trial,a prospective, open-label, phase II trial of rituximabin primary APS patients, reported that rituximab iseffective in controlling some noncriteria manifes-tations of APS such as thrombocytopenia, hemolyticanemia, skin ulcers, and nephropathy [68
&&
].Two studies have shown a benefit of B-cell mod-
ulatory therapies in murine models of APS. The firstfocused on co-stimulatory blockage with cytotoxic Tlymphocyte antigen 4 immunoglobulin (CTLA4-Ig)and demonstrated that CTLA4-Ig could not treatAPS but was able to prevent B-cell activationand aPL production if administered prior to aPL
Copyright © Lippincott Williams & Wilkins. Unau
1065-6251 � 2014 Wolters Kluwer Health | Lippincott Williams & Wilk
antibody development [69]. The second approachtargeted B cell activating factor (BAFF), a tumornecrosis factor (TNF)-like cytokine that supportsB-cell survival and differentiation. In contrast toco-stimulatory blockade, BAFF antagonists pre-vented APS onset and prolonged survival. Basedon these studies, B-cell modulation shows somepromise as a therapeutic approach in APS [70]. Aba-tacept, a CTLA4 blocker, and belimumab, a BAFFantagonist, are approved for use in rheumatoidarthritis and SLE, respectively, but have not beenused in patients with APS.
Inhibition of intracellular signaling pathways
As described above, several intracellular signalingpathways are involved in aPL-mediated pathogeniceffects. Several of these pathways pose potentialtherapeutic targets. For example, NF-kB inhibitioninhibits aPL-induced cellular activation and throm-bosis [71], and TLR4 inhibition decreases tissuefactor, MyD88, and TNF expression in in-vitro andin-vivo studies [72
&
].
CONCLUSION
Lifelong anticoagulation remains the mainstay oftherapy for thrombotic APS. The managementof asymptomatic patients with aPL is still contro-versial, as the pathogenesis of this disorder is stillnot well understood or supported by a unifyinghypothesis. Innovative therapeutic approaches suchas immune modulation, complement inhibition,and inhibiting intracellular signaling pathwayshave shown promising results in preclinical studies.Further mechanistic and clinical studies are neededto develop and implement improved therapies forthis devastating illness.
Acknowledgements
This work was supported by a Bridge Grant from theAmerican Society of Hematology (to K.R.M.) and NIHGrant P50 HL081011 (K.R.M. project leader).
Conflicts of interest
The authors have no conflicts of interest to disclose.
REFERENCES AND RECOMMENDEDREADINGPapers of particular interest, published within the annual period of review, havebeen highlighted as:
& of special interest&& of outstanding interest1. Miyakis S, Lockshin MD, Atsumi D, et al. International consensus statement onan update of the preliminary classification criteria or antiphospholipid syn-drome (APS). J Thromb Haemost 2006; 4:295–306.
thorized reproduction of this article is prohibited.
ins www.co-hematology.com 7

C
CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Hemostasis and thrombosis
2. Cervera R, Piette J, Font J, et al. Antiphospholipid syndrome: clinical andimmunologic manifestations and patterns of disease expression in a cohort of1000 patients. Arthritis Rheum 2002; 46:1019–1027.
3. Asherson RA, Cervera R, Piette J, et al. Catastrophic antiphospholipidsyndrome: international consensus statement on classification criteria andtreatment guidelines. Lupus 2003; 12:530–534.
4.&
Cervera R, Serrano R, Pons-Estel GJ, et al. Morbidity and mortality in theantiphospholipid syndrome during a 10-year period: a multicentre prospectivestudy of 1000 patients. Ann Rheum Dis 2014. [Epub ahead of print]
This 10-year multicenter observational cohort study involving 1000 patientsinvestigated the major causes of morbidity and mortality in patients with APS.5. Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus
anticoagulants: an update. On behalf of the Subcommittee on Lupus Anti-coagulant/Antiphospholipid Antibody of the Scientific and StandardisationCommittee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
6. Galli M, Borrelli G, Jacobsen EM, et al. Clinical significance of differentantiphospholipid antibodies in the WAPS (warfarin in the antiphospholipidsyndrome) study. Blood 2007; 110:1178–1183.
7. Devreese K, Peerlinck K, Hoylaerts MF. Thrombotic risk assessment in theantiphospholipid syndrome requires more than the quantification of lupusanticoagulants. Blood 2010; 115:870–878.
8. Banzato A, Pozzi N, Frasson R, et al. Antibodies to domain I of beta(2)gly-coprotein I are in close relation to patients risk categories in antiphospholipidsyndrome (APS). Thromb Res 2011; 128:583–586.
9.&
Sciascia S, Sanna G, Murru V, et al. Antiprothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT)antibodies and the risk of thrombo-sis in the antiphospholipid syndrome. Thromb Haemost 2014; 111:354–364.
This study examined the role of aPS/PT antibodies in establishing thrombotic risk inAPS patients.10. McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Antiphospholipid anti-
bodies are directed against a complex antigen that includes a lipid-bindinginhibitor of coagulation: b 2-glycoprotein I (apolipoprotein H). Proc Natl AcadSci USA 1990; 87:4120–4124.
11. Arad A, Proulle V, Furie RA, et al. b(2)-Glycoprotein-1 autoantibodies frompatients with antiphospholipid syndrome are sufficient to potentiate arterialthrombus formation in a mouse model. Blood 2011; 117:3453–3459.
12. Redecha P, Franzke CW, Ruf W, et al. Neutrophil activation by the tissuefactor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model ofantiphospholipid syndrome. J Clin Invest 2008; 118:3453–3461.
13. Rand JH, Wu XX. Antibody-mediated disruption of the annexin-V antithrom-botic shield: a new mechanism for thrombosis in the antiphospholipid syn-drome. Thromb Haemost 1999; 82:649–655.
14. Hunt JE, Simpson RJ, Chesterman CN, et al. Identification of a region of beta-2-glycoprotein I critical for lipid binding and anticardiolipin antibody cofactoractivity. Proc Natl Acad Sci USA 1993; 90:2141–2145.
15. Agar C, van Os GM, Morgelin M, et al. Beta2-glycoprotein I can exist in 2conformations: implications for our understanding of the antiphospholipidsyndrome. Blood 2010; 116:1336–1343.
16. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antipho-spholipid syndrome: understanding the antibodies. Nat Rev Rheumatol2011; 7:330–339.
17.&
Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipidsyndrome. N Engl J Med 2013; 11:1033–1044.
This recent review covers the recent advances in the pathogenesis of APS,including mechanisms of cellular activation, intracellular signaling, and develop-ment of pathogenic aPL.18. Krone KA, Allen KL, McCrae KR. Impaired fibrinolysis in the antiphospholipid
syndrome. Curr Rheumatol Rep 2010; 12:53–57.19. Simantov R, LaSala JM, Lo SK, et al. Activation of cultured vascular endothelial
cells by antiphospholipid antibodies. J Clin Invest 1995; 96:2211–2219.20. Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by
antiphospholipid/antibeta2 glycoprotein I antibodies. Blood 2005; 105:1964–1969.
21. Nojima J, Masuda Y, Iwatani Y, et al. Tissue factor expression on monocytesinduced by antiphospholipid antibodies as a strong risk factor for thromboem-bolic complications in SLE patients. Biochem Biophys Res Commun 2008;365:195–200.
22. Martinuzzo ME, Maclouf J, Carreras LO, Levy-Toledano S. Antiphospholipidantibodies enhance thrombin-induced platelet activation and thromboxaneformation. Thromb Haemost 1993; 70:667–671.
23. Roubey RA, Eisenberg RA, Harper MF, Winfield JB. ‘Anticardiolipin’ auto-antibodies recognize beta 2-glycoprotein I in the absence of phospholipid.Importance of Ag density and bivalent binding. J Immunol 1995; 154:954–960.
24. Williams FM, Parmar K, Hughes GR, Hunt BJ. Systemic endothelial cellmarkers in primary antiphospholipid syndrome. Thromb Haemost 2000;84:742–746.
25. Allen KL, Fonseca FV, Betapudi V, et al. A novel pathway for humanendothelial cell activation by antiphospholipid/antib2 glycoprotein I antibo-dies. Blood 2012; 119:884–893.
26. Raschi E, Testoni C, Bosisio D, et al. Role of the MyD88 transductionsignaling pathway in endothelial activation by antiphospholipid antibodies.Blood 2003; 101:3495–3500.
opyright © Lippincott Williams & Wilkins. Unautho
8 www.co-hematology.com
27.&&
Zhou H, Sheng L, Wang H, et al. Antib2GPI/b2GPI stimulates activation ofTHP-1 cells through TLR4/MD-2/MyD88 and NF-kB signaling pathways.Thromb Res 2013; 132:742–749.
This study showed that antib2GPI/b2GPI-complex-induced IL-6, IL-8, and TNF-aexpression in THP-1 cells is mediated through TLR4/MD-2/MyD88 and NF-kBsignaling pathways. This suggests new therapeutic targets such as TLR4 and NF-kB.28. Vega-Ostertag ME, Ferrara DE, Romay-Penabad Z, et al. Role of p38 mitogen-
activated protein kinase in antiphospholipid antibody-mediated thrombosisand endothelial cell activation. J Thromb Haemost 2007; 5:1828–1834.
29. Ramesh S, Morrell CN, Tarango C, et al. Antiphospholipid antibodies promoteleukocyte-endothelial cell adhesion and thrombosis in mice by antagonizingeNOS via b2GPI and apoER2. J Clin Invest 2011; 121:120–131.
30. Alard JE, Gaillard F, Daridon C, et al. TLR2 is one of the endothelial receptorsfor beta 2-glycoprotein I. J Immunol 2010; 185:1550–1557.
31. Sorice M, Longo A, Capozzi A, et al. Antibeta2-glycoprotein I antibodiesinduce monocyte release of tumor necrosis factor alpha and tissue factor bysignal transduction pathways involving lipid rafts. Arthritis Rheum 2007;56:2687–2697.
32. Lopez-Pedrera C, Buendıa P, Cuadrado MJ, et al. Antiphospholipid antibodiesfrom patients with the antiphospholipid syndrome induce monocyte tissuefactor expression through the simultaneous activation of NF-kB/Rel proteinsvia the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERKpathway. Arthritis Rheum 2006; 54:301–311.
33. Urbanus RT, Pennings MT, Derksen RH, de Groot PG. Platelet activation bydimeric beta2-glycoprotein I requires signaling via both glycoprotein Ib alphaand apolipoprotein E receptor 20 . J Thromb Haemost 2008; 6:1405–1412.
34. Shi T, Giannakopoulos B, Yan X, et al. Antibeta2-glycoprotein I antibodies incomplex with beta2-glycoprotein I can activate platelets in a dysregulatedmanner via glycoprotein Ib–IX–V. Arthritis Rheum 2006; 54:2558–2567.
35. Malia RG, Kitchen S, Greaves M, Preston FE. Inhibition of activated protein Cand its cofactor protein S by antiphospholipid antibodies. Br J Haematol1990; 76:101–107.
36. Rand JH, Wu XX, Lapinski R, et al. Detection of antibody-mediated reductionof annexin A5 anticoagulant activity in plasmas of patients with the antipho-spholipid syndrome. Blood 2004; 104:2783–2790.
37. Chamley LW, McKay EJ, Pattison NS. Inhibition of heparin/antithrombin IIIcofactor activity by anticardiolipin antibodies: a mechanism for thrombosis.Thromb Res 1993; 71:103–111.
38. Marciniak E, Romond EH. Impaired catalytic function of activated protein C: anew in vitro manifestation of lupus anticoagulant. Blood 1989; 74:2426–2432.
39. Vega-Ostertag M, Liu X, Kwan-Ki H, et al. A human monoclonal antipro-thrombin antibody is thrombogenic in vivo and upregulates expression oftissue factor and E-selectin on endothelial cells. Br J Haematol 2006;135:214–219.
40. Hwang KK, Grossman JM, Visvanathan S, et al. Identification of antithrombinantibodies in the antiphospholipid syndrome that interfere with the inactivationof thrombin by antithrombin. J Immunol 2001; 167:7192–7198.
41. Giannakopoulos B, Gao L, Qi M, et al. Factor XI is a substrate for oxidor-eductases: enhanced activation of reduced FXI and its role in antiphospho-lipid syndrome thrombosis. J Autoimmun 2012; 39:121–129.
42. Rand JH, Wu XX, Quinn AS, et al. Hydroxychloroquine protects the annexinA5 anticoagulant shield from disruption by antiphospholipid antibodies:evidence for a novel effect for an old antimalarial drug. Blood 2010;115:2292–2299.
43. Edwards MH, Pierangeli S, Liu X, et al. Hydroxychloroquine reverses throm-bogenic properties of antiphospholipid antibodies in mice. Circulation 1997;96:4380–4384.
44. Chen PP, Yang CD, Ede K, et al. Some antiphospholipid antibodies bind tohemostasis and fibrinolysis proteases and promote thrombosis. Lupus 2008;17:916–921.
45. Fischetti F, Durigutto P, Pellis V, et al. Thrombus formation induced byantibodies to beta 2-glycoprotein I is complement dependent and requiresa priming factor. Blood 2005; 106:2340–2346.
46. Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor-tissue factorcrosstalk in neutrophils links innate immunity to coagulation pathways. JImmunol 2006; 177:4794–4802.
47. Salmon JE, Girardi G, Holers VM. Activation of complement mediates antipho-spholipid antibody-induced pregnancy loss. Lupus 2003; 12:535–538.
48. Carrera-Marın A, Romay-Penabad Z, Papalardo E, et al. C6 knock-out miceare protected from thrombophilia mediated by antiphospholipid antibodies.Lupus 2012; 21:1497–1505.
49. Breen KA, Seed P, Parmar K, et al. Complement activation in patients withisolated antiphospholipid antibodies or primary antiphospholipid syndrome.Thromb Haemost 2012; 107:423–429.
50.&&
Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrentantiphospholipid antibody syndrome and enables successful renal transplan-tation. Am J Transplant 2014; 14:459–465.
This study reports eculizumab use in patients with CAPS and renal failure inpreventing recurrent thrombosis, and allowing successful renal transplantation.51. Uthman IW, Gharavi AE. Viral infection and antiphospholipid antibodies.
Semin Arthritis Rheum 2002; 31:256–263.52. Van Os GM, Meijers JC, Agar C, et al. Induction of anti-b2 glycoprotein I
autoantibodies in mice by protein H of Streptococcus pyogenes. J ThrombHaemost 2011; 9:2447–2456.
rized reproduction of this article is prohibited.
Volume 21 � Number 00 � Month 2014

CE: Alpana; MOH/210504; Total nos of Pages: 9;
MOH 210504
Antiphospholipid antibody syndrome Chaturvedi and McCrae
53. Passam FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular patho-physiology of the antiphospholipid syndrome: the role of oxidative posttran-slational modification of beta 2 glycoprotein I. J Thromb Haemost 2011;9:275–282.
54. Rauch J, Dieude M, Subang R, Levine JS. The dual role of innate immunity inthe antiphospholipid syndrome. Lupus 2010; 19:347–353.
55. Aguilar-Valenzuela R, Nickerson K, Romay-Penabad Z, et al. Involvement ofTLR7 and TLR9 in the production of antiphospholipid antibodies. ArthritisRheum 2011; 63:s281.
56.&&
Broder A, Putterman C. Hydroxychloroquine use is associated with lowerodds of persistently positive antiphospholipid antibodies and/or lupus anti-coagulant in systemic lupus erythematosus. J Rheumatol 2013; 40:30–33.
This is the first study to show that hydroxychloroquine use is associated with lowerodds of having persistently positive lupus anticoagulant and aPL.57. Cervera R, Khamastha MA, Shoenfeld Y, et al. Morbidity and mortality in the
antiphospholipid syndrome during a 5 year period: a multicenter prospectivestudy of 1000 patients. Ann Rheum Dis 2009; 68:1428–1432.
58. Tripodi A, Chantarangkul V, Clerici M, et al. Laboratory control of oral anti-coagulant treatment by the INR system in patients with the antiphospholipidsyndrome and lupus anticoagulant. Br J Haematol 2001; 115:672–678.
59.&
Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress onAntiphospholipid Antibodies: Task Force report on antiphospholipid syn-drome treatment trends. Autoimmun Rev 2014; 13:685–696.
The report of the APS Treatment Trends Task Force, created as part of the 14thInternational Congress on aPL, systematically reviewed the potential future treat-ment strategies for aPL-positive patients.60. Ferrara DE, Swerlick R, Casper K, et al. Fluvastatin inhibits up-regulation of
tissue factor expression by antiphospholipid antibodies on endothelial cells.J Thromb Haemost 2004; 2:1558–1563.
61. Redecha P, Franzke CW, Ruf W, et al. Neutrophil activation by the tissuefactor/factor VIIa/PAR2 axis mediates fetal death in a mouse model ofantiphospholipid syndrome. J Clin Invest 2008; 118:3453–3461.
62.&
Erkan D, Willis R, Murthy VL, et al. A prospective open-label pilot study offluvastatin on proinflammatory and prothrombotic biomarkers in antiphospho-lipid antibody positive patients. Ann Rheum Dis 2013; 73(6):1176–1180.
This prospective clinical study demonstrated that proinflammatory and prothrom-botic biomarkers, which are upregulated in persistently aPL-positive patients, canbe reversibly reduced by fluvastatin.
Copyright © Lippincott Williams & Wilkins. Unau
1065-6251 � 2014 Wolters Kluwer Health | Lippincott Williams & Wilk
63. Petri M. Use of hydroxychloroquine to prevent thrombosis in systemic lupuserythematosus and in antiphospholipid antibody-positive patients. Curr Rheu-matol Rep 2011; 13:77–80.
64. Wu XX, Guller S, Rand JH. Hydroxychloroquine reduces binding of antipho-spholipid antibodies to syncytiotrophoblasts and restores annexin A5 expres-sion. Am J Obstet Gynecol 2011; 205:e7–e14.
65.&
Schmidt-Tanguy A, Voswinkel J, Henrion D, et al. Antithrombotic effects ofhydroxychloroquine in primary antiphospholipid syndrome patients. J ThrombHaemost 2013; 11:1927–1929.
This is the first prospective study investigating the use of hydroxychloroquine forsecondary venous thrombosis prophylaxis in primary APS.66. Espinosa G, Berman H, Cervera R. Management of refractory cases of
catastrophic antiphospholipid syndrome. Autoimmun Rev 2011; 10:664–668.
67. Berman H, Rodriguez-Pinto I, Cervera R, et al. Rituximab use in the cata-strophic antiphospholipid syndrome: descriptive analysis of the CAPS reg-istry patients receiving rituximab. Autoimmun Rev 2012; 12:1085–1090.
68.&&
Erkan D, Vega J, Ramon G, et al. A pilot open-label phase II trial of rituximab fornoncriteria manifestations of antiphospholipid syndrome. Arthritis Rheum2013; 65:464–471.
This phase II pilot study showed that rituximab is well tolerated and effective intreating some noncriteria manifestations of APS.69. Akkerman A, Huang W, Wang X, et al. CTLA4Ig prevents initiation but not
evolution of antiphospholipid syndrome in NZW/BXSB mice. Autoimmunity2004; 37:445–451.
70. Kahn P, Ramanujman M, Bethunaickan R, et al. Prevention of murine antipho-spholipid syndrome by BAFF blockade. Arthritis Rheum 2008; 58:2824–2834.
71. Montiel-Manzano G, Romay-Penabad Z, Papalardo de Martinez E, et al.In vivo effects of an inhibitor of nuclear factor kappa B on thrombogenicproperties of antiphospholipid antibodies. Ann N Y Acad Sci 2007; 1108:540–553.
72.&
Xie H, Zhou H, Wang H, et al. Antib(2)GPI/b(2)GPI induced TF and TNF-aexpression in monocytes involving both TLR4/MyD88 and TLR4/TRIF signal-ing pathways. Mol Immunol 2013; 53:246–254.
This study showed that antib(2)GPI/b(2)GPI complex induced tissue factor andTNF-a expression involves TLR4/MyD88 and TLR4/TRIF signaling pathways, andthis can be blocked by inhibiting TLR4-mediated signal transduction.
thorized reproduction of this article is prohibited.
ins www.co-hematology.com 9

























