Embed Size (px)
Citation preview

, , ,
*Department of Biochemistry and Molecular Biology, Monash University, Victoria, Australia
�Brain Injury & Repair Group, Howard Florey Institute, University of Melbourne, Parkville, Victoria, Australia
�Centre for Neurosciences University of Melbourne, Parkville, Victoria, Australia
Amyotrophic lateral sclerosis (ALS) represents 85% of allmotor neuron disease in humans and is characterized by thedegeneration and death of motor neurons (Cleveland andRothstein 2001; Talbot 2002; Boillee et al. 2006). In ALS,degenerating motor neurons characteristically contain pro-teinaceous cytoplasmic inclusions, leading to the view thatALS is a protein aggregation disorder. The inclusions infamilial ALS associated with mutant superoxide dismutase 1(mSOD1) are predominantly ubiquitinylated and contain p62(Mizuno et al. 2006), 14-3-3 (Kawamoto et al. 2004), andTDP-43 (Winton et al. 2008), and importantly, Cu, Zn-SOD1(Rosen et al. 1993). Abnormal intracellular protein inclu-sions are present both in spinal cords of familial ALS patientsand in the most widely accepted animal model of disease,transgenic mice over-expressing G93A mSOD1 proteins(Gurney et al. 1994; Kunst et al. 1997). Furthermore,
intracellular inclusions containing SOD1 are also found insporadic ALS cases, suggesting that the wildtype (WT)enzyme can aggregate, possibly under conditions of oxida-tive stress (Ezzi et al. 2007).
Received October 10, 2008; revised manuscript received October 28,2008; accepted October 31, 2008.Address correspondence and reprint requests to Professor Phillip
Nagley, Department of Biochemistry and Molecular Biology, MonashUniversity, PO Box 13D, Victoria 3800, Australia.E-mail: [email protected] used: ALS, amyotrophic lateral sclerosis; CHAPS, 3-(3-
cholamidopropyl)-dimethylammoniopropane sulfonate; cyt c, cyto-chrome c; DAPI, 4,6-diamidino-2-phenylindole; DIC, differentialinterference contrast; EGFP, enhanced green fluorescent protein; ER,endoplasmic reticulum; mSOD1, mutant SOD1; NPC, nuclear porecomplex; PBS, phosphate-buffered saline; SOD1, superoxide dismutase1; STS, staurosporine; WT, wildtype.
Abstract
Mutations in Cu, Zn-superoxide dismutase 1 (SOD1) are
associated with degeneration of motor neurons in the dis-
ease, familial amyotrophic lateral sclerosis. Intracellular pro-
tein inclusions containing mutant SOD1 (mSOD1) are
associated with disease but it is unclear whether they are
neuroprotective or cytotoxic. We report here that the forma-
tion of mSOD1 inclusions in a motor neuron-like cell line
(NSC-34) strongly correlates with apoptosis via the mito-
chondrial death pathway. Applying confocal microscopic
analyses, we observed changes in nuclear morphology and
activation of caspase 3 specifically in cells expressing
mSOD1 A4V or G85R inclusions. Furthermore, markers of
mitochondrial apoptosis (activation and recruitment of Bax,
and cytochrome c redistribution) were observed in 30% of
cells bearing mSOD1 inclusions but not in cells expressing
dispersed SOD1. In the presence of additional apoptotic
challenges (staurosporine, etoposide, and hydrogen perox-
ide), cells bearing mSOD1 inclusions were susceptible to
further apoptosis suggesting they were in a pro-apoptotic
state, thus confirming that inclusions are linked to toxicity.
Surprisingly, cells displaying dispersed SOD1 [both wildtype
(WT) and mutant] were protected against apoptosis upstream
of mitochondrial apoptotic signaling, induced by all agents
tested. This protection against apoptosis was unrelated to
SOD1 enzymatic activity because the G85R that lacks
enzymatic function protected cells similarly to both WT SOD1
and A4V that possesses WT-like activity. These findings
demonstrate new aspects of SOD1 in relation to cellular via-
bility; specifically, mSOD1 can be either neuroprotective or
cytotoxic depending on its aggregation state.
Keywords: apoptosis, mitochondria, motor neuron disease,
neuroprotection, superoxide dismutase 1.
J. Neurochem. (2009) 108, 578–590.
JOURNAL OF NEUROCHEMISTRY | 2009 | 108 | 578–590 doi: 10.1111/j.1471-4159.2008.05799.x
578 Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors

More than 130 SOD1mutations are known (Valentine et al.2005) and these mutants encompass various enzymaticactivities and aggregation propensities. One predominantview is that the disease-associatedmutations destabilize SOD1proteins, leading to the formation of non-native oligomers,eventually resulting in intracellular protein aggregation andthe formation of inclusions (Boillee et al. 2006; Pasinelli andBrown 2006; Shaw and Valentine 2007). However, it is stillcontroversial as to whether or not the inclusions are inthemselves cytotoxic. The appearance of mSOD1 aggregatesin transgenic mice with disease progression (Johnston et al.2000; Wang et al. 2002) suggest a link to toxicity (Shibataet al. 1996; Kato et al. 2000). Several theories have beenproposed for the possible toxicity of SOD1 aggregates (Shaw2005; Shaw and Valentine 2007), including impaired axonaltransport, decreased chaperone activity, mitochondrial andproteosomal dysfunction, and most recently the involvementof endoplasmic reticulum (ER) stress (Atkin et al. 2006; Ohet al. 2008). Furthermore, many essential components, such asubiquitin proteasome components or chaperone proteins arerecruited to the aggregates, compromising vital cellularfunctions (Chaudhuri and Paul 2006). However, it is alsounclear if aggregation is a cause or a consequence of otherupstream cellular event. Furthermore, the exact mechanism ofmotor neuron death caused by the toxicity of mSOD1 has notbeen precisely defined, although it is clear that the activation ofcaspases subsequently leads to motor neuron death (Satha-sivam et al. 2005; Shaw 2005).
Others, however, report that increased formation ofmSOD1 inclusions did not correlate with neuronal deathin vitro (Lee et al. 2002; Oh et al. 2008) raising thepossibility that the inclusions may even be neuroprotectiveby the sequestering of toxic misfolded proteins. It remainsunclear as to the significance of biologically relevantdifferences associated with specific features of SOD1 proteinaggregation, or technical differences, such as the specific cellmodel used, the level of protein expression, or detailedmethodologies of analysis of the cellular response.
In this study, by using single cell analyses, we analyzedthe engagement of cells in apoptotic signaling involving themitochondrial death pathway, in relation to the generation ofmSOD1 inclusions within cells. We used a cell modelappropriate to ALS, namely the motor neuron-like cell lineNSC-34, which is a hybrid of mouse motor neuron andneuroblastoma cells (Cashman et al. 1992). We characterizedthe apoptotic signaling in NSC-34 cells expressing either WTor mSOD1-enhanced green fluorescent proteins (EGFP)constructs. We examined the susceptibility of cells express-ing mSOD1 to further apoptotic challenge with staurosporine(STS) (a protein kinase inhibitor) (Bertrand et al. 1994),etoposide (a topoisomerase inhibitor) (Froelich-Ammon andOsheroff 1995) or hydrogen peroxide (H2O2) (producingoxidative stress). Our results show that the generation ofmSOD1 inclusions is linked tightly to cell death involving
the mitochondrial apoptotic signaling pathway, which isexacerbated by further apoptotic challenge. Unexpectedly,however, NSC-34 cells expressing SOD1 in the dispersedform (either WT or mutant), display significantly less celldeath when the cells are exposed to various apoptotic insults,suggesting that even mSOD1 is protective against apoptosiswhen dispersed.
Materials and methods
Cell culture and transfectionWildtype SOD1 and mSOD1 (A4V and G85R) constructs encoding
EGFP tagged human SOD1 at the C-terminus were generated as
previously described (Turner et al. 2005). Motor neuron-like NSC-
34 cells were supplied by Dr Neil Cashman (University of Toronto,
Canada). Cell lines were maintained in Dulbecco’s modified Eagle’s
medium supplemented with 2 mM L-glutamine, 10 mM HEPES
buffer, and 10% (v/v) fetal calf serum. Cells were maintained in a
humidified 37�C incubator with 5% CO2. Cells were subcultured in
24-well plates at a density of 5 · 104 cells per well and were
transfected transiently with plasmids (1 lg DNA per well) using a
1 : 1 ratio of lipofection reagent (‘Transfast’; Promega, Sydney,
NSW, Australia) to DNA. For immunostaining experiments, cells
were seeded in 24-well plates containing 13 mm round coverslips.
MaterialsStaurosporine and etoposide were purchased from Sigma-Aldrich
(St Louis, MO, USA). H2O2 was purchased from Merck (Kilsyth,
Victoria, Australia).
Apoptotic inductionFor apoptotic induction, 5 · 104 of cells were seeded in 24-well
plates without coverslip the day before transfection. The next day,
transfection was performed. After 72 h post-transfection, the
medium was replaced new culture medium containing 25 nM
STS, 250 lM etoposide or 250 lM H2O2. Cells were treated with
STS, etoposide, or H2O2 for 24 h in 37�C.
Collection of treated cellsFollowing treatment of transfected NSC-34 cells with STS,
etoposide or H2O2 after 24 h, adherent cells (i.e. viable) were
trypsinized and combined with non-adherent cells (i.e. non-viable)
to collect the total cell population for each sample. Cells were
pelleted by centrifugation at 800 g for 5 min, then resuspended in
appropriate volume of culture media. The cellular content of each
sample was immobilized onto coverslips pre-coated with poly-L-
lysine (5 lg/mL) by centrifugation of the suspension at 112 g for
3 min in a Cytospin centrifuge (Shandon Scientific Ltd, Waltham,
MA, USA), so that both viable and non-viable cells were collected
on coverslips. The transfection efficiency was about 30% after the
Cytospin step, which was no different from the non-treated controls.
Immunocytochemistry and confocal imagingAfter 72 h post-transfection, cells were washed with phosphate-
buffered saline (PBS) once and then fixed with 3.5% paraformalde-
hyde for 10 min at 25�C. After that, cells were permeabilized with
0.1% Triton-X-100 for 10 min [except for tests of Bax activation,
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 579

where cells were permeabilized with CHAPS buffer [150 mM NaCl,
10 mM HEPES, 1.0% 3-(3-cholamidopropyl)-dimethylammoniopro-
pane sulfonate (CHAPS)] after fixation]. Cells were washed three
times with PBS after each step and then blocked with 3.5% bovine
serum albumin for at least 1 h at 25�C. Cells were then incubated withthe mouse monoclonal anti-Bax antibody (clone 6A7) (BD Bio-
sciences Pharmingen�, San Diego, CA, USA) at 4�C overnight. To
investigate cytochrome c (cyt c) redistribution, caspase 3 activation
and the presence of the nuclear pore complex (NPC), primary mouse
monoclonal anti-cyt c antibody (BD Biosciences Pharmingen�),
rabbit anti-cleaved caspase 3 (Neuromics, Edina, MN, USA) and
mouse monoclonal anti-NPC (clone MAb 414) (Covance, Berkeley,
CA, USA) antibodies were used at this stage. After overnight
incubation, cells were washed three times with PBS to remove the
primary antibody and were subsequently incubated with a suitable
secondary antibody conjugated to Alexa 568 (BD Biosciences
Pharmingen�) at 4�C overnight. Nuclear morphology was also
monitored by staining fixed cells with 0.5 lg/mL 4,6-diamidino-2-
phenylindole (DAPI) for 10 min at 25�C and then cells were washed
three times with PBS. Coverslips were mounted onto slides. Cells
were imaged with an Olympus Fluoview 500 inverted confocal
laser scanning microscope (Olympus, Mount Waverley, Victoria,
Australia) equipped with an Argon/HeNe laser light source. Images
were collected using a 60·/1.2 water immersion lens (UPlanApo60·/1.20w, Olympus). In dual-channel imaging, photomultiplier sensitiv-
ities and offsets were set to a level at which bleed through effects from
one channel to another were negligible. Differential interference
contrast (DIC) images were collected from all fields under Nomarski
optics. For cell counting, 200 cells of untransfected and dispersed
green fluorescent cells were counted, and > 60 cells of inclusion-
positive cells were counted randomly of each well under a confocal
microscopy. The data were obtained from three independent
experiments unless specified in the figure legends.
Statistical analysisAll data are expressed as the mean ± SD values. The data were
analyzed for statistical significance by an ANOVA followed by Turkey
post hoc test (GraphPad Prism, San Diego, CA, USA). The
differences were considered significant at p < 0.05.
Results
mSOD1 inclusions are associated with apoptosis in NSC-34cellsNSC-34 cells transfected with WT, A4V, or G85R SOD1-EGFP constructs were examined by confocal microscopy.WT SOD1 was observed in a dispersed form, such thatfluorescence was homogeneously by distributed throughoutthe cells in all cases (Fig. 1a). On the other hand, mSOD1transfectants (both A4V and G85R) formed cytoplasmicfluorescent inclusions in 20–30% of cells; in the remainingcells, fluorescence was seen in dispersed form. Such dualityof morphology may be due to the intrinsic heterogeneity oftransiently expressing cells or to differential levels of proteinexpression and cell-to-cell variation in protein homeostasis(Matsumoto et al. 2005). The proportion of cells bearing
inclusions seen here is consistent with other studies using thiscell line (Turner et al. 2005). Note that while the NSC-34cells show relatively short neurite projections under cultureconditions used here, evident in some panels of Fig. 1aand b, these cells have many other properties characteristicof authentic motor neurons (Cashman et al. 1992; Eggettet al. 2000).
Cells expressing dispersed SOD1 (both WT and mutant)show normal nuclear morphologies (Fig. 1a), similar to bothuntransfected control cells and non-transfected bystandercells (the latter are defined here as untransfected cells in apopulation of cells exposed to a vector under transfectionconditions). In virtually all cells expressing dispersedmSOD1 shows the fluorescence was excluded from thenuclear region, unlike those expressing WT SOD1-EGFP(Fig. 1a), consistent with a previous report (Sau et al. 2007).
Significantly, in some of the cells bearing mutant A4V orG85R inclusions, the DAPI-stained nuclei were fragmented,whilst others were without detectable DAPI stain (Fig. 1a),in both cases suggesting that apoptosis was occurring. Toaccount for loss of DAPI staining, we treated untransfectedcells with STS (Fig. S1), and found that a significantproportion of cells lost the ability to be stained with DAPIsuggesting that this is a feature of apoptosis in NSC-34 cells.Further evidence for this notion was obtained by immuno-staining with antibody specific for the NPC, confirming thepresence of nuclei (albeit with condensed morphology) insuch cells (Fig. S1). We infer that in many apoptotic NSC-34cells, some changes in nuclear organization occur thatrenders DAPI unable to bind to chromosomal DNA underthe conditions normally used for DAPI staining.
Quantification of apoptotic nuclei in the various cellpopulations showed that there is negligible cell death inuntransfected cells nor in cells expressing dispersed WT,A4V, or G85R SOD1 (Fig. 1c). However, 30% of cellsexpressing mSOD1 A4V or G85R, with clearly visibleinclusions, contained apoptotic nuclei (Fig. 1c).
In order to confirm the apoptotic events in NSC-34 cellsbearing mSOD1 inclusions, we determined whether caspaseactivation is involved in cell death. Accordingly, transfectedcells were subjected to immunocytochemistry for cleavedcaspase 3 (Fig. 1b). Caspase 3 activation was observed in20–30% of cells bearing mSOD1 inclusions (A4Vor G85R),but in < 2% of untransfected or bystander cells. Cellsexpressing dispersed WT, A4V, or G85R SOD1 (Fig. 1d)showed similarly negligible levels of caspase 3 activation.Therefore, these data support the view that mSOD1 inclu-sions are associated with apoptosis in NSC-34 cells.
Cells bearing mSOD1 inclusions are more susceptible toapoptosis upon STS treatment but cells expressingdispersed SOD1 are protectedThe increased incidence of apoptotic nuclei and caspase 3activation suggests that inclusion-forming cells are in a state
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
580 | K. Y. Soo et al.

that favors apoptosis. This was tested by exposing WT, A4V,or G85R SOD1-transfected cells to an inducer of apoptosisfor 24 h (STS). Apoptotic nuclei were detected amongst both
untransfected cells and cells bearing mSOD1 inclusions, butrarely in cells expressing dispersed SOD1 (Fig. 2a). Quan-tification revealed that apoptosis was induced in 30% of
(a) (b)
(c) (d)
Fig. 1 Nuclear morphology changes and caspase 3 activation in cells
expressing WT or mSOD1 proteins. (a) NSC-34 cells expressing WT,
A4V, or G85R SOD1-EGFP constructs (first column) were fixed and
stained with DAPI (second column). Overlays of the confocal images
of GFP fluorescence and DAPI (third column), and the DIC images
(last column) are shown. White arrows indicate fragmented or con-
densed nuclei and yellow arrows indicate presumed location of nuclei
undetectable by DAPI staining under conditions used. Scale bars
10 lm, applies to all fields. (b) Cells as in panel (a) were additionally
immunostained with antibodies recognizing cleaved caspase 3,
indicative of activation of the enzyme (second column). Other indica-
tions as for panel (a). (c) Quantified data for cells with apoptotic nuclei.
Nuclei were defined as apoptotic if they showed fragmented or con-
densed morphology, or if they failed to stain with DAPI. Horizontal axis
labels indicate cell populations or subpopulations analyzed; ‘byst’
indicates bystander cells (i.e. non-transfected cells in a population
subjected to transfection); ‘disp’ indicates cells containing dispersed
SOD1-EGFP; ‘incl’ indicates cells containing unambiguously identifi-
able inclusions of mSOD1-EGFP. (d) Quantified data for cells immu-
nostained for cleaved (and activated) caspase 3. Indications otherwise
are as for panel (c). Results are expressed as mean ± SD, n = 3,
*p < 0.001.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 581

untransfected cells (Fig. 2c). Strikingly, in cells bearingmSOD1 A4V, or G85R inclusions, 60–90% displayedapoptotic nuclei (Fig. 2c). Unexpectedly, however, only 5–10% of cells displaying apoptotic nuclei were observed incells expressing dispersed WT, A4V, or G85R SOD1
compared with bystander cells, which showed similar extentsof apoptosis as untransfected cells exposed to STS (Fig. 2c).
In parallel, caspase 3 immunofluorescence was examinedin the cells treated with STS (Fig. 2b). Caspase 3 activationwas observed in 20% of both untransfected cells and
(a) (b)
(c) (d)
Fig. 2 Nuclear morphology changes and caspase 3 activation in cells
expressing WT or mSOD1 proteins following treatment of cells with
STS. (a) NSC-34 cells expressing WT, A4V, or G85R SOD1-EGFP
constructs were treated with STS (25 nM) for 24 h before being sub-
jected to fixation and imaging. Images are shown for EGFP-tagged
proteins and DAPI as in Fig. 1(a). (b) Cells as in panel (a) were
additionally immunostained with antibodies specific for cleaved (acti-
vated) caspase 3. Images are shown for EGFP-tagged proteins,
activated caspase 3 and DAPI as in Fig. 1(b). Other indication as in
Fig. 1(a). (c) Quantified data for cells displaying apoptotic nuclear
morphology. (d) Quantified data for cells displaying activated caspase
3. Other indications as for Fig. 1(c). Results are expressed as
mean ± SD, n = 3, *p < 0.001.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
582 | K. Y. Soo et al.

bystander cells, but in 70% of cells bearing mSOD1inclusions (Fig. 2b and d). However, in cells expressingdispersed SOD1 (either WT or mutants A4V or G85R) therewas a significantly reduced proportion with caspase 3activation (< 5%) (Fig. 2b and d).
We conclude that mSOD1 inclusions are associated withtoxicity and a greater susceptibility to apoptosis in NSC-34cells. Surprisingly, however, dispersed SOD1 protects cellsagainst apoptosis upon STS treatment. The observed cyto-toxicity and neuroprotection do not require SOD1 enzymaticactivity, because in each situation we examined, concordantproperties of cells were observed for both mSOD1 A4V andG85R.
To test whether the neuroprotection afforded by dispersedSOD1 also occurs with other apoptotic stimuli, cells weretreated with etoposide (to induce genotoxic damage byinhibition of topoisomerase) or H2O2 (to induce oxidativestress). Following treatment with either etoposide or H2O2,approximately 30% of untransfected cells contained apopto-tic nuclei in each case (Fig. S2). In contrast, approximately60% of cells bearing mSOD1 inclusions (A4V or G85R)underwent apoptosis in the presence of etoposide (Fig. S2a),and this proportion was even greater (80%) with H2O2
treatment (Fig. S2b). However, < 5% of cells expressingdispersed SOD1 (WT, A4V, or G85R) contained apoptoticnuclei after etoposide treatment (Fig. S2a). Likewise, uponH2O2 treatment, there is a significantly reduced frequency ofapoptotic nuclei amongst cells expressing dispersed SOD1(WT or mutant) compared to their bystander cells (Fig. S2b).Such protection was evidently greater for cells expressingWT SOD1 than those expressing mSOD1.
To eliminate the possibility that the EGFP-moiety is itselfneuroprotective, we examined NSC-34 cells transfected withvector encoding EGFP alone. After treatment with STS,etoposide or H2O2, EGFP-expressing cells contained asimilar proportion with apoptotic nuclei compared tobystander untransfected cells (20–30% of total, data notshown). This finding confirms that the protection because ofdispersed SOD1 is not a consequence of the EGFP tag.
Cytochrome c redistribution as an indicator ofmitochondrial signaling in apoptosis because of mSOD1inclusions in NSC-34 cellsWe examined the role of mitochondrial apoptotic signalingassociated with the presence of mSOD1 inclusions. Theredistribution of cyt c from mitochondria to the cytosol wasmonitored by immunofluorescence and confocal microscopyin NSC-34 cells transfectants (WT, mSOD1 A4V, andG85R). In untransfected cells and also in cells expressingdispersed WT, A4V, or G85R SOD1, cyt c was observed in apunctate distribution but with negligible staining in thenuclear region (Fig. 3a). In contrast, in many cells bearingmutant A4V or G85R SOD1 inclusions, cyt c was barelydetectable (Fig. 3a). As argued below, this suggests that
apoptosis was occurring, even though one would expect toobserve dispersed cyt c across the whole cell (i.e. redistrib-uted from mitochondria) under these conditions.
To account for such loss of immunostaining for cyt c,untransfected cells were treated with STS. In such popula-tions, 80% of cells possessed mitochondrial cyt c; however,10% of cells possessed dispersed cyt c and 10% of cells didnot immunostain detectably for cyt c (Fig. S3a and b).Moreover, half of those cells that were classified as apoptotic(indicated by nuclear morphology) failed to show cyt cstaining (Fig. S3c). This failure to detect cyt c immuno-chemically was thus mostly associated with the presence ofapoptotic nuclei. A specific example is seen in Fig. 3a,bottom row, where the nucleus of the cell bearing inclusionsof G85R SOD1 fails to stain with DAPI and also is almostdevoid of cyt c immunostaining. Hence, we infer that, ingeneral, cyt c is rapidly degraded in the cytosol of apoptoticcells.
Quantification of cyt c redistribution and nuclear mor-phology revealed that almost all untransfected cells displayednormal nuclei and retain cyt c in the mitochondria (Fig. 3c).Moreover, nearly 100% of cells expressing dispersed WT oreither mSOD1 have similar characteristics, consistent withthe normal, non-apoptotic state (Fig. 3c). However, in cellsbearing A4Vor G85R SOD1 inclusions, 30% contained bothredistributed cyt c (or disappearance of cyt c) and apoptoticnuclei (Fig. 3c). Further, 10% of cells with A4V inclusionsshowed cyt c redistribution (or disappearance) but displayednormal nuclei after DAPI staining (Fig. 3c). However, forcells with either A4V or G85R inclusions, very few showedapoptotic nuclei together containing cyt c in its typicalmitochondrial distribution (Fig. 3c). The data indicate thatmitochondrial apoptotic signaling is responsible for the deathof cells bearing mSOD1 inclusions.
The relationship between mitochondrial apoptotic signal-ing and the neuroprotective effect of dispersed SOD1 wasthen examined by STS treatment of cells (Fig. 3b). Ingeneral, in the STS-treated populations, including bothuntransfected and inclusion-bearing cells, some cells failingto stain for cyt c and also displaying apoptotic nuclei wereobserved (Fig. 3b). The proportion of cells with both cyt credistribution (or disappearance) and apoptotic nucleiincreased to 30% for both untransfected cultures andbystander cells (Fig. 3d). In untreated control cells, theseapoptotic indices were negligible. Approximately 60–80% ofSTS-treated cells bearing A4Vor G85R inclusions show bothcyt c redistribution (or disappearance) and apoptotic nuclei(Fig. 3d). In contrast, in 95% of cells expressing dispersedSOD1 (WT or either mutant), both the presence of cyt c inthe mitochondria and normal nuclear morphology indicatedthat apoptosis was not induced (Fig. 3d). This is consistentwith neuroprotective effects of dispersed SOD1. On the otherhand, a small proportion of cells bearing mSOD1 inclusions(about 15% for A4V and 5% for G85R) showed apoptotic
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 583
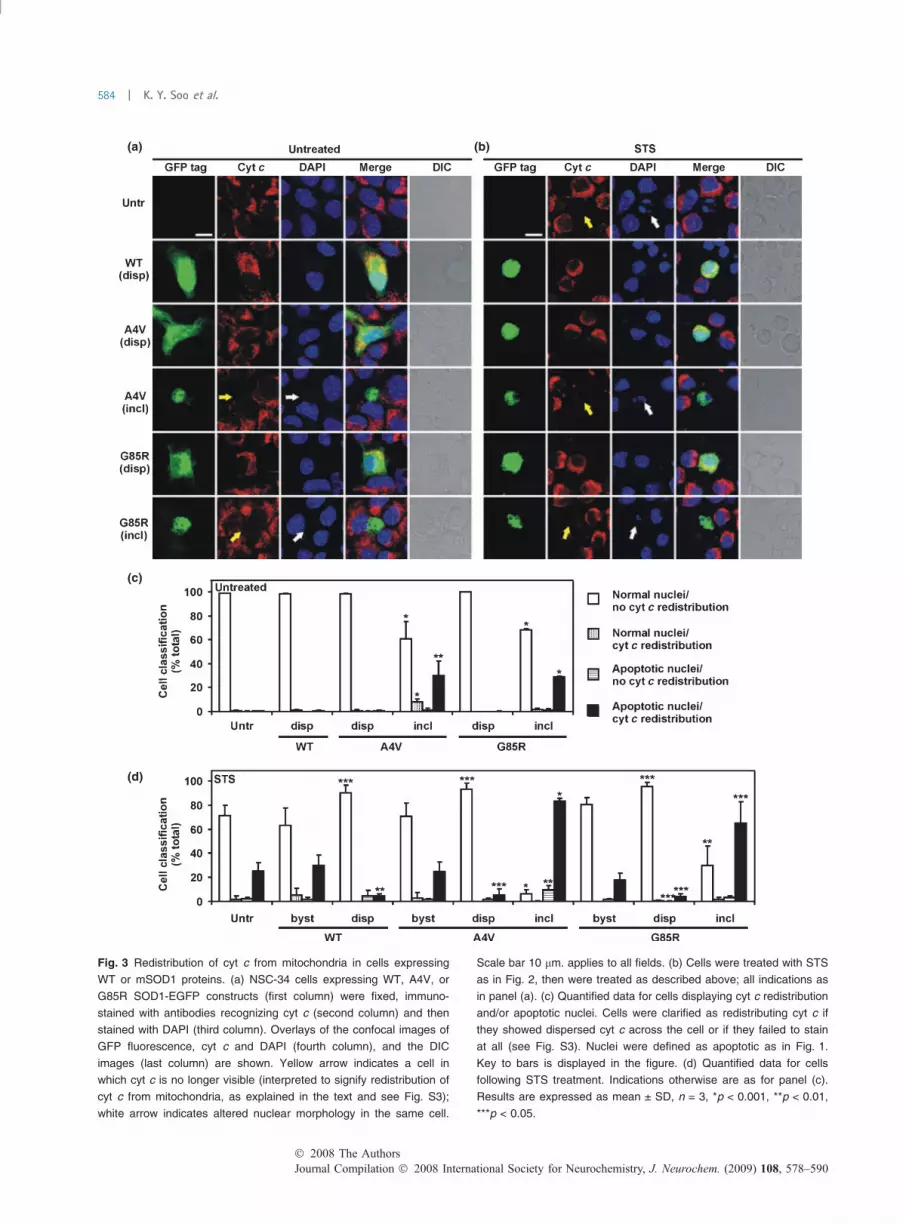
(a) (b)
(c)
(d)
Fig. 3 Redistribution of cyt c from mitochondria in cells expressing
WT or mSOD1 proteins. (a) NSC-34 cells expressing WT, A4V, or
G85R SOD1-EGFP constructs (first column) were fixed, immuno-
stained with antibodies recognizing cyt c (second column) and then
stained with DAPI (third column). Overlays of the confocal images of
GFP fluorescence, cyt c and DAPI (fourth column), and the DIC
images (last column) are shown. Yellow arrow indicates a cell in
which cyt c is no longer visible (interpreted to signify redistribution of
cyt c from mitochondria, as explained in the text and see Fig. S3);
white arrow indicates altered nuclear morphology in the same cell.
Scale bar 10 lm. applies to all fields. (b) Cells were treated with STS
as in Fig. 2, then were treated as described above; all indications as
in panel (a). (c) Quantified data for cells displaying cyt c redistribution
and/or apoptotic nuclei. Cells were clarified as redistributing cyt c if
they showed dispersed cyt c across the cell or if they failed to stain
at all (see Fig. S3). Nuclei were defined as apoptotic as in Fig. 1.
Key to bars is displayed in the figure. (d) Quantified data for cells
following STS treatment. Indications otherwise are as for panel (c).
Results are expressed as mean ± SD, n = 3, *p < 0.001, **p < 0.01,
***p < 0.05.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
584 | K. Y. Soo et al.

nuclei but cyt c was still localized in mitochondria (Fig. 3d).Thus, apoptotic signaling independent of mitochondria mightbe occurring in a minority of such cells after exposure toSTS.
For both etoposide and H2O2 treatments, as for STS(Fig. 3d), untransfected and bystander cells showed apopto-tic indices (cyt c redistribution and apoptotic nuclei) in about20–30% of the population (Fig. S4a). Hence, these additionalpharmacological agents do elicit mitochondrial participationin cell death signaling. However, 95% of cells expressingdispersed SOD1 (WT or either mutant) are unaffected interms of these apoptotic indices (Fig. S4). In contrast, therewere substantially increased frequencies of cells bearingmSOD1 inclusions with cyt c redistribution and apoptoticnuclei after cellular treatment with etoposide (50%) or H2O2
(80%) (Fig. S4b and c). Together, these data suggest thatredistribution of mitochondrial cyt c accounts for theactivation of caspase 3 (Fig. 2) in inclusion-bearing cellsundergoing apoptosis by a range of insults. Moreover, theneuroprotective effect of dispersed SOD1 is evidentlyupstream of the redistribution of mitochondrial cyt c.
Bax activation is involved in both the cytotoxicity of SOD1inclusions and neuroprotection of dispersed SOD1in NSC-34 cellsIn order to characterize the mitochondrial participation inapoptosis related to SOD1 inclusions, we examined whetherBax activation is involved in the cytotoxicity or neuropro-tection of either form of SOD1. Bax is a pro-apoptoticmember of the Bcl-2 family, which is involved in permea-bilization of the outer mitochondrial membrane. We used aconformation-specific Bax antibody which recognizes onlyactivated Bax, using conditions where cells are permeabi-lized with CHAPS buffer. Neither untransfected cells northose expressing dispersed WT or mSOD1 demonstratedsignificant frequencies of Bax activation and apoptotic nuclei(Fig. 4a and c). By contrast, 20–30% of cells bearing eitherA4V or G85R inclusions contained activated Bax andapoptotic nuclei (Fig. 4a and c). Similar data were obtainedin studies of recruitment of Bax to mitochondria (Fig. S5aand c). In some cells bearing SOD1 inclusions, apoptoticnuclei were seen but no Bax activation was detected(Fig. 4c); these cells were apparently more prevalent whenbearing inclusions of mSOD1 A4V rather than G85R.However, this cell type may contribute to an under-estima-tion of Bax activation as it was sometimes difficult toconclusively determine if a cell was stained positively or notwhen there is extensive cell death (Fig. 4a). Nevertheless, theresults suggest that the formation of mSOD1 inclusions isassociated with Bax activation in NSC-34 cells.
After STS treatment, approximately 30% of untransfectedcontrol cells and relevant bystander cells have apoptoticnuclei but only 10% of these cells have Bax activation(Fig. 4b and d). In contrast, in STS-treated cells expressing
dispersed WT, A4V, or G85R SOD1, Bax activation andapoptotic nuclei were rarely observed (Fig. 4b and d).Suppression of Bax recruitment to mitochondria was alsoevident (Fig. S5b and d). On the other hand, 30% of cellsbearing mSOD1 inclusions (A4V or G85R) have both Baxactivation and apoptotic nuclei (Fig. 4b and d). However,60% or 30% of inclusion-bearing cells with A4V or G85R,respectively, had apoptotic nuclei but no Bax activation(Fig. 4d). This suggests a possible reduced involvement ofmitochondria in apoptosis or may reflect difficulty inimmunostaining apoptotic cells for activated Bax (Fig. 4d)perhaps akin to the loss of cyt c immunostaining, asmentioned above.
Discussion
We have clearly demonstrated that the formation of intracel-lular mSOD1 inclusions is closely linked to cytotoxicity viamitochondrial apoptosis in a motor neuronal cell line.Surprisingly, expression of dispersed SOD1, either WT ormutant, protects cells against cell death induced by a range ofapoptogenic agents.
mSOD1 inclusions correlate with cytotoxicity in NSC-34cellsPrevious studies have implied that there was a correlationbetween toxicity and the formation of mSOD1 inclusions(Matsumoto et al. 2005; Kabuta et al. 2006). However,others have reported that mSOD1-mediated cell death wasindependent of aggregate formation (Lee et al. 2002;Takeuchi et al. 2002). Our single cell analyses showconclusively that there is a strong relationship betweenmSOD1 inclusions and toxicity. Through the examination ofspecific mitochondrial apoptotic markers in relation tomSOD1 inclusions, this study demonstrates that the toxicityof inclusions is mediated via mitochondrial signaling path-ways and hence offers a new perspective on this relationship.
Mitochondrial apoptotic signaling occurs selectively inNSC-34 cells bearing mSOD1 inclusionsMitochondrial involvement in the pathogenesis of bothsporadic and familial ALS, as well as in mouse models ofdisease, has been suggested by many authors. Mitochondrialswelling and vacuolization are early signs of motor neurondeath in SOD1G93A transgenic mice (Bendotti et al. 2001;Okato-Matsumoto and Fridovich 2002; Higgins et al. 2003).Decreased activity of cyt c oxidase was observed in thespinal cord of ALS patients (Fujita et al. 1996). Impairedmitochondrial respiration was also seen in the brain andspinal cord of SOD1G93A transgenic mice (Kirkinezos et al.2005). SOD1 is normally considered to be cytosolic;however, it was also found mislocalized in the mitochondriaof NSC-34 cells and tissues of SOD1G93A mice (Kirkinezoset al. 2005; Raimondi et al. 2006; Son et al. 2007).
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 585

(a)
(c)
(d)
(b)
Fig. 4 Bax activation in cells expressing WT and mSOD1 proteins. (a)
NSC-34 cells expressing WT, A4V, or G85R SOD1-EGFP constructs
(first column) were fixed, permeabilized with CHAPS buffer and sub-
jected to immunostaining under conditions whereby only activated Bax
is detected by the conformation-specific Bax antibody (second col-
umn). Nuclear morphology was indicated by staining cells with DAPI
(third column). Overlays of the confocal images of GFP fluorescence,
activated Bax and DAPI (fourth column), and the DIC images (last
column) are shown. Yellow arrow indicates a cell in which Bax is acti-
vated (interpreted to signify strong fluorescence around the nucleus);
white arrow indicates altered nuclear morphology in the same cell.
Scale bar 10 lm, applies to all fields. (b) Cells were treated with STS as
in Fig. 2, then were treated as described above; all indications as in
panel (a). (c) Quantified data for cells displaying activated Bax and/or
apoptotic nuclei. Nuclei were defined as apoptotic as in Fig. 1. Key to
bars is displayed in the figure. (d) Quantified data for cells following
STS treatment. Indications otherwise are as for panel (a). Results are
expressed as mean ± SD, n = 3, *p < 0.001, **p < 0.01, ***p < 0.05.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
586 | K. Y. Soo et al.

Aggregation of mSOD1 within mitochondria was reported tooccur in the cytotoxicity of motor neurons in ALS (Takeuchiet al. 2002; Liu et al. 2004).
Nonetheless, a distinction in relation to cytotoxicityshould be made between notions of mitochondrial dysfunc-tion as a consequence of abnormal SOD1 biology andmitochondrially mediated apoptosis. Our findings of relativeprotection against H2O2 (oxidative stressor) provided by WTor mutant dispersed SOD1 argues against the former notion.The latter possibility has been previously considered, withother studies showing that cyt c redistribution andsubsequent caspase activation occur with concomitantlymSOD1 expression (Cozzolono et al. 2005; Sathasivamet al. 2005). Further, application of the broad-spectrumcaspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(O-Me)fluoromethyl ketone (zVAD-fmk) inhibited cell death in vitroand also in SOD1G93A mice (Sathasivam et al. 2005; Wootzet al. 2006; Tokuda et al. 2007). However, interpretation ofthese observations was complicated by the use of heteroge-neous cell populations and the consequent inability todetermine the exact relationships between expression ofmSOD1 and the cell death indicators examined. In thepresent study, by single cell analyses, we are able todemonstrate the direct relationship between mSOD1 inclu-sions and the activation of mitochondrially mediated apop-tosis. This involves Bax recruitment and activation, upstreamof cyt c redistribution to cytosol, which requires theactivation of Bax on mitochondria (Smith et al. 2008).Furthermore, by using additional apoptogenic challenges, weshow that cells bearing mSOD1 inclusions are much morepre-disposed to apoptosis than those cells that do not forminclusions. In our study, similar findings were obtained incomparisons between cells transfected with mSOD1 G85Rand A4V, substantiating the notion that toxicity is unrelatedto enzymatic function.
Unexpectedly, we found that intracellularly dispersedSOD1 (both WT and mutant) protects cells against deathinduced by a range of different challenges. Previousobservations indicated that WT SOD1 proteins have anti-apoptotic properties, both in yeast and neural cells understress conditions (Rabizadeh et al. 1995; Zhai et al. 2005).What is surprising here, however, is the finding that thedispersed mSOD1 had the same protective capacity as that ofWT SOD1 against apoptotic induction. Furthermore, aftercellular treatment with apoptotic stimuli, we also found thatcyt c redistribution and Bax activation, as well as Baxrecruitment to mitochondria, were reduced in cells express-ing dispersed SOD1 compared to untransfected cells. There-fore, we suggest that the protective effect elicited bydispersed SOD1 is upstream of Bax recruitment to mito-chondria. It should be noted that these conclusions couldonly have been reached by studies such as ours, whereanalysis of single cells allowed a population to be stratifiedfor those expressing the different forms of SOD1, or no
SOD1 at all. In analyzing cell populations as a whole (as inmany previous studies), cells expressing the neuroprotectivedispersed SOD1 would not have been identified.
Possible mechanisms involved in the cytotoxicity associatedwith mSOD1 inclusions and the neuroprotection bydispersed SOD1What are the processes upstream of mitochondria that lead toapoptosis in cells bearing inclusions? Furthermore, whatproperties of dispersed SOD1 protect cells against stressesleading to apoptosis?
In relation to the first question, BH3-only proteins, such asBid, Bim, Bik, or Puma/Noxa, act as activators of pro-apoptotic proteins Bax and Bak (Ward et al. 2004; Hackerand Weber 2007). The latter proteins are those that permea-bilize the outer mitochondrial membrane, allowing redistri-bution to cytosol of apoptogenic proteins in theintermembrane space (Smith et al. 2008). A recent studyhas demonstrated that deletion of Bim promotes cell survivalin NSC-34 cells expressing mSOD1 and that mSOD1transgenic mice, in which Bim was knocked out, had adelayed onset of disease and extended lifespan (Hetz et al.2007). ER stress is known to be a trigger for Bimtranslocation to mitochondria and the activation of apoptosis(Puthalakath et al. 2007). Significantly, ER stress has alsorecently been implicated in the pathogenesis of ALS (Wootzet al. 2004; Atkin et al. 2006; Kikuchi et al. 2006; Nagataet al. 2007; Oh et al. 2008). Up-regulation of the fullspectrum of proteins of the unfolded protein responseinduced by ER stress, including apoptotic protein CHOPand caspase 12, has been observed in NSC-34 cellsexpressing mSOD1, SOD1G93A transgenic mice (Atkin et al.2006) and human patients with sporadic ALS (Atkin et al.2008). Interestingly, inhibitors of ER stress delayed theformation of mSOD1 inclusions and blocked cell death in amouse neuroblastoma cell model (Oh et al. 2008). One couldconsider that the aggregation of mSOD1 could activate ERstress responses that lead to apoptosis in NSC-34 cells.
On the other hand, Steckley et al. (2007) showed thatPuma, induced by oxidative stress, can induce conforma-tional changes in Bax leading to its activation and subsequentpermeabilization of the outer mitochondrial membrane.Moreover, deletion of the BH3-only protein Puma protectsmotor neurons from ER stress-induced apoptosis and delaysmotor neuron loss in SOD1G93A transgenic mice (Keiranet al. 2007). Therefore, it is possible that Bim and Puma actin concert to promote mitochondrial death signaling whencells expressing mSOD1 are liable to generate inclusions.
In consideration of the second question concerning theneuroprotective mechanism afforded by dispersed SOD1proteins, one possibility is that over-expression of WT ormSOD1 in NSC-34 cells leads to a mild degree of cellularstress, but with sub-lethal consequences. The potentialactivation of stress responses, such as those observed by
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 587

Kirby et al. (2002), could generate a temporary anti-apop-totic signaling regime in the cells, as they endeavor to dealwith over-expressed SOD1 (whether WT or mutant). Thismay involve engagement of SOD1 with the unfolded proteinresponse. ER stress in its initial phases promotes cell survivalby increasing the cellular capacity for protein folding anddegradation by up-regulation of chaperones, and by reducinggeneral protein synthesis. It is only when ER stress isprolonged or unresolved that the switch to the apoptotic stateoccurs (Ron and Walter 2007). Cellular stress responses maysuppress apoptotic signaling, particularly at the levels ofactivation or mobilization of BH3-only proteins whose rolesare to recruit mitochondria into apoptotic signaling. Perhapsthe cells that show manifest inclusions of mSOD1 have lostthis battle, and instead of behaving in an anti-apoptoticmode, they are now switched strongly to pro-apoptoticsignaling through the mitochondrial pathway.
Whilst we cannot state with confidence that the inclusionsdirectly cause apoptosis, the tight linkage we observedsuggests that formation of inclusions and the final deathsignals are closely entwined, even if the causal events may beupstream. We are as yet unable to define, in precisemolecular terms, the critical cellular decision point when acell chooses not to die or allows itself to die. A relevanttrigger may be the propensity of mSOD1 to form solubleoligomers, which may be the toxic entities, perhaps latergoing on to form inclusions (Sau et al. 2007). We note that inthe cellular system studied here, the dispersed form of WTSOD1 was found in both the cytoplasmic and the nucleus ofthe cell. In contrast, dispersed mSOD1 (either A4V orG85R), was frequently distributed exclusively in the cyto-plasmic area. One possible explanation for this observation isthat mSOD1-EGFP could form oligomers which by their sizeare excluded from entry into the nucleus. The NPC has a sizelimit of approximately 50 kDa for the free passage ofproteins. Hence the 43 kDa SOD1-EGFP fusion proteinsused in this study should be able to access the nucleus. Notethat it is the SOD1 moiety of the SOD1-EGFP fusion that isresponsible for protection, not EGFP as such. We note alsothat it is formally possible that the absence of dispersedSOD1 could generate the evident toxicity associated withinclusions rather than the presence of potentially aggregatingforms of SOD1. While SOD1 knockout mice show a range ofchronic degenerative conditions (Turner and Talbot 2008),not all symptoms are typical of ALS. Moreover, culturedcells depleted of SOD1 by siRNA prematurely age reachingaccelerated senescence (Blander et al. 2003), this process isslower than the apoptosis characterized here.
The question that remains, however, is how nuclearexclusion relates to the cytotoxicity or neuroprotectionmediated by SOD1 in NSC-34 cells? The situation may becomplex in light of recent observations where the presence ofSOD1 in the nucleus has a protective effect on the suscepti-bility of nuclear DNA to damage (Sau et al. 2007). The
relevance of DNA damage to the apoptotic markers used in thepresent work remains to be determined. A more centralquestion in relation to the demonstrated neuroprotectiveeffects of SOD1 (WT or mutant) is to relate this finding tothe cellular and molecular events in the pathogenesis of ALS.Collectively, these approaches based on cellular studies willprovide new aspects of SOD1 biology in relation to cytotox-icity and neuroprotection, and the consequent link to ALS.
Acknowledgements
This work was supported by the National Health and Medical
Research Council of Australia, the Bethlehem Griffiths Research
Foundation, a Henry H. Roth Charitable Foundation grant for MND
Research, and a Fellowship (to JDA) from the Motor Neuron
Disease Research Institute of Australia.
Supporting information
Additional Supporting Information may be found in the online
version of this article:
Fig. S1 Staining of nuclei in apoptotic NSC-34 cells with nuclear
pore complex (NPC) antibodies reveals presence of nuclei unable to
be stained with DAPI.
Fig. S2 Nuclear morphology changes in cells expressing WT or
mSOD1 proteins after treatment with etoposide and H2O2.
Fig. S3 Loss of immunostaining for cyt c in apoptotic NSC-34
cells.
Fig. S4 Redistribution of cyt c from mitochondria in cells
expressing WT or mSOD1 proteins after exposure to various
apoptotic stimuli.
Fig. S5 Mobilization of Bax to mitochondria in cells expressing
WT or mSOD1 proteins.
Please note: Wiley-Blackwell are not responsible for the content
or functionality of any supporting materials supplied by the authors.
Any queries (other than missing material) should be directed to the
corresponding author for the article.
References
Atkin J. D., Farg M. A., Turner B. J. et al. (2006) Induction of theunfolded protein response in familial amyotrophic lateral sclerosisand association of protein-disulfide isomerase with superoxidedismutase 1. J. Biol. Chem. 281, 30152–30165.
Atkin J. D., Farg M. A., Walker A. K., McLean C., Tomas D. and HorneM. K. (2008) Endoplasmic reticulum stress and induction of theunfolded protein response in human sporadic amyotrophic lateralsclerosis. Neurobiol. Dis. 30, 400–407.
Bendotti C., Calvaresi N., Chiveri L., Prelle A., Moggio M., Braga M.,Silani V. and Biasi S. D. (2001) Early vacuolization and mito-chondrial damage in motor neurons of FALS mice are not asso-ciated with apoptosis or with changes in cyt oxidase histochemicalreactivity. J. Neurol. Sci. 191, 25–33.
Bertrand R., Solary E., OConnor P., Kohn K. W. and Pommier Y. (1994)Induction of a common pathway of apoptosis by staurosporine.Exp. Cell Res. 211, 314–321.
Blander G., de Oliveira R. M., Conboy C. M., Haigis M. and GuarenteL. (2003) Superoxide dismutase 1 knock-down induces senescencein human fibroblasts. J. Biol. Chem. 278, 38966–38969.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
588 | K. Y. Soo et al.

Boillee S., VeldeC.V. andClevelandD.W. (2006)ALS: a disease ofmotorneurons and their nonneuronal neighbours. Neuron 52, 39–59.
Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., ShawI. T., Dahrouge S. and Antel J. P. (1992) Neuroblastoma · spinalcord (NSC) hybrid cell lines resemble developing motor neurons.Dev. Dyn. 194, 209–221.
Chaudhuri T. K. and Paul S. (2006) Protein-misfolding diseases andchaperone-based therapeutic approaches. FEBS J. 273, 1331–1349.
Cleveland D. W. and Rothstein J. D. (2001) From Charcot to LouGehrig: deciphering selective motor neuron death in ALS. Nature2, 806–819.
Cozzolono M., Ferri A., Ferraro E., Rotilio G., Cecconi F. and CarriM. T. (2005) Apaf1 mediates apoptosis and mitochondrial damageinduced by mutant human SOD1s typical of familial amyotrophiclateral sclerosis. Neurobiol. Dis. 21, 69–79.
Eggett C. J., Crosier S., Manning P., Cookson M. R., Menzies F. M.,McNeil C. J. and Shaw P. J. (2000) Development and characteri-sation of a glutamate-sensitive motor neurone cell line. J. Neuro-chem. 74, 1895–1902.
Ezzi S. A., Urushitani M. and Julien J. P. (2007) Wild-type superoxidedismutase acquires binding and toxic properties of ALS-linkedmutant forms through oxidation. J. Neurochem. 102, 170–178.
Froelich-Ammon S. J. and Osheroff N. (1995) Topoisomerase poisons:harnessing the dark side of enzyme mechanism. J. Biol. Chem.270, 21429–21432.
Fujita K., Yamauchi M., Shibayama K., Ando M., Honda M. and NagataY. (1996) Decreased cytochrome c oxidase activity but unchangedsuperoxide dismutase and glutathione peroxidase activities in thespinal cord of patients with amyotrophic lateral sclerosis. J. Neu-rosci. Res. 45, 276–281.
Gurney M. E., Pu H., Chiu A. Y. et al. (1994) Motor neuron degener-ation in mice that express human Cu, Zn superoxide dismutasemutation. Science 264, 1772–1775.
Hacker G. and Weber A. (2007) BH3-only proteins trigger cytochrome crelease, but how? Arch. Biochem. Biophys. 462, 150–155.
Hetz C., Thielen P., Fisher J., Pasinelli P., Brown R. H., Korsmeyer S.and Glimcher L. (2007) The proapoptotic Bcl-2 family memberBim mediates motoneuron loss in a model of amyotrophic lateralsclerosis. Cell Death Differ. 14, 1386–1389.
Higgins C. M. J., Jung C. and Xu Z. (2003) ALS-associated mutantSOD1G93A causes mitochondrial vacuolation by expansion of theintermembrane space and by involvement of SOD1 aggregationand peroxisomes. BioMed. Central Neurosci. 4, 16–29.
Johnston J. A., Dalton M. J., Gurney M. E. and Kopito R. R. (2000)Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophiclateral sclerosis. Proc. Natl Acad. Sci. USA 97, 12571–12576.
Kabuta T., Suzuki Y. and Wada K. (2006) Degradation of amyotrophiclateral sclerosis-linked mutant Cu, Zn-superoxide dismutase pro-teins by macroautophagy and the proteosome. J. Biol. Chem. 281,30524–30533.
Kato S., Horiuchi S., Liu J. et al. (2000) Advanced glycation endprod-uct-modified superoxide dismutase-1 (SOD1)-positive inclusionsare common to familial amyotrophic lateral sclerosis patients withSOD1 gene mutations and transgenic mice expressing humanSOD1 with a G85R mutation. Acta Neuropathol. (Berl.) 1000,490–505.
Kawamoto Y., Akiguchi I., Nakamura S. and Budka H. (2004) 14-3-3proteins in Lewy body-like hyaline inclusions in patients withsporadic amyotrophic lateral sclerosis. Acta Neuropathol. (Berl.)108, 531–537.
Keiran D., Woods I., Villunger A., Strasser A. and Prehn J. H. (2007)Deletion of the BH3-only protein puma protects motor neurons
from ER stress-induced apoptosis and delays motor neuron loss inALS mice. Proc. Natl Acad. Sci. USA 104, 20606–20611.
Kikuchi H., Almer G., Yamashita S., Guegan C., Nagai M., Xu Z.,Sosunov A. A., McKhann G. M. and Przedborski S. (2006) Spinalcord endoplasmic reticulum stress associated with a microsomalaccumulation of mutant superoxide dismutase-1 in an ALS model.Proc. Natl Acad. Sci. USA 103, 6025–6030.
Kirby J., Menzies F. M., Cookson M. R., Bushby K. and Shaw P. J.(2002) Differential gene expression in a cell culture model ofSOD1-related familial motor neurone disease. Hum. Mol. Genet.11, 2061–2075.
Kirkinezos I. G., Bacman S. R., Hernandez D., Oca-Cossio J., Arias L.J., Perez-Pinzon M. A., Bradley W. G. and Moraes C. T. (2005)Cytochrome c association with the inner mitochondrial membraneis impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 25, 164–172.
Kunst C. B., Mezey E., Brownstein M. J. and Patterson D. (1997)Mutations in SOD1 associated with amyotrophic lateral sclerosiscause novel protein interactions. Nat. Genet. 15, 91–94.
Lee J. P., Gerin C., Bindokas V. P., Miller R., Ghadge G. and Roos R. P.(2002) No correlation between aggregates of Cu/Zn superoxidedismutase and cell death in familial amyotrophic lateral sclerosis.J. Neurochem. 82, 1229–1238.
Liu J., Lillo C., Jonsson A. et al. (2004) Toxicity of familial ALS-linkedSOD1 mutants from selective recruitment to spinal mitochondria.Neuron 43, 5–17.
Matsumoto G., Stojanovic A., Holmberg C. I., Kim S. and MorimotoR. I. (2005) Structural properties and neuronal toxicity of amyo-trophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1aggregates. J. Cell Biol. 171, 75–85.
Mizuno Y., Amari M., Takatama M., Aizawa H., Mihara B. andOkamoto K. (2006) Immunoreactivities of p62, an ubiquitin-binding protein, in the spinal anterior horn cells of patients withamyotrophic lateral sclerosis. J. Neurosci. 249, 13–18.
Nagata T., Ilieva H., Murakami T., Shiote M., Narai H., Ohta Y., HayashiT., Shoji M. and Abe K. (2007) Increased ER stress during motorneuron degeneration in a transgenic mouse model of amyotrophiclateral sclerosis. Neurol. Res. 29, 767–771.
Oh Y. K., Shin K. S., Yuan J. and Kang S. J. (2008) Superoxidedismutase 1 mutants related to amyotrophic lateral sclerosisinduce endoplasmic stress in neuro2a cells. J. Neurochem. 104,993–1005.
Okato-Matsumoto A. and Fridovich I. (2002) Amyotrophic lateralsclerosis: a proposed mechanism. Proc. Natl Acad. Sci. USA 99,9010–9014.
Pasinelli P. and Brown R. H. (2006) Molecular biology of amyotrophiclateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723.
Puthalakath H., O’Reilly L. A., Gunn P. et al. (2007) ER stress triggersapoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349.
Rabizadeh S., Gralla E. B., Borchelt D. R. et al. (1995) Mutationsassociated with amyotrophic lateral sclerosis convert superoxidedismutase from an antiapoptotic gene to a proapoptotic gene:studies in yeast and neural cells. Proc. Natl Acad. Sci. USA 92,3024–3028.
Raimondi A., Mangolini A., Rizzardini M. et al. (2006) Cell culturemodels to investigate the selective vulnerability of motoneuronalmitochondria to familial ALS-linked G93ASOD1. Eur. J. Neuro-sci. 24, 387–399.
Ron D. and Walter P. (2007) Signal integration in the endoplasmicreticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8,519–529.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590
SOD1 and mitochondrial apoptotic signaling | 589

Rosen D. R., Siddique T., Patterson D. et al. (1993) Mutations in Cu/Znsuperoxide dismutase gene are associated with familial amyo-trophic lateral sclerosis. Nature 362, 59–62.
Sathasivam S., GriersonA. J. and ShawP. J. (2005) Characterization of thecaspase cascade in a cell culture model of SOD1-related familialamyotrophic lateral sclerosis: expression, activation and therapeuticeffects of inhibition. Neuropathol. Appl. Neurobiol. 31, 467–485.
Sau D., De Biasi S., Vitellaro-Zuccarello L. et al. (2007) Mutation ofSOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 16,1604–1618.
Shaw P. J. (2005) Molecular and cellular pathways of neurodegenerationin motor neuron disease. J. Neurol. Neurosurg. Psychiatry 76,1046–1057.
Shaw B. F. and Valentine J. S. (2007) How do ALS-associated mutationsin superoxide dismutase 1 promote aggregation of the protein.Trends Biochem. Sci. 32, 78–85.
Shibata N., Asayama K., Hirano A. and Kobayashi Y. (1996) Immu-nohistochemical study on superoxide dismutases in spinal cordsfrom autopsied patients with amyotrophic lateral sclerosis. Dev.Neurosci. 18, 492–498.
Smith D. J., Ng H., Kluck R. M. and Nagley P. (2008) The mitochondrialgateway to cell death. IUBMB Life 60, 383–389.
SonM., Puttaparthi K., Kawamata H., Rajendran B., Boyer P. J., ManfrediG. and Elliott J. L. (2007) Overexpression of CCS in G93A-SOD1mice leads to accelerated neurological deficits with severe mito-chondrial pathology. Proc. Natl Acad. Sci. USA 104, 6072–6077.
Steckley D., Karajgikar M., Dale L. B. et al. (2007) Puma is a dominantregulator of oxidative stress induced Bax activation and neuronalapoptosis. J. Neurosci. 27, 12989–12999.
Takeuchi H., Kobayashi Y., Ishigaki S., Doyu M. and Sobue G. (2002)Mitochondrial localization of mutant superoxide dismutase 1 trig-gers caspase-dependent cell death in a cellular model of familialamyotrophic lateral sclerosis. J. Biol. Chem. 277, 50966–50972.
Talbot K. (2002) Motor neuron disease. Postgrad. Med. J. 78, 513–517.Tokuda E., Ono S., Ishige K., Watanabe S., Okawa E., Ito Y. and Suzuki
T. (2007) Dysequilibrium between caspases and their inhibitors in a
mouse model for amyotrophic lateral sclerosis. Brain Res. 1148,234–242.
Turner B. J. and Talbot K. (2008) Transgenics, toxicity and therapeuticsin rodent models of mutant SOD1-mediated familial ALS. Pro-gress Neurobiol. 85, 94–134.
Turner B. J., Atkin J. D., Farg M. A., Zang D. W., Rembach A., LopesE. C., Patch J. D., Hill A. F. and Cheema S. S. (2005) Impairedextracellular secretion of mutant superoxide dismutase 1 associateswith neurotoxicity in familial amyotrophic lateral sclerosis.J. Neurosci. 25, 108–117.
Valentine J. S., Doucette P. A. and Potter S. Z. (2005) Copper-zincsuperoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev.Biochem. 74, 563–593.
Wang J., Xu G., Gonzales V., Coonfield M., Fromholt D., CopelandN. G., Jenkins N. A. and Borchelt D. R. (2002) Fibrillar inclusionsand motor neuron degeneration in transgenic mice expressingsuperoxide dismutase 1 with a disrupted copper-binding site.Neurobiol. Dis. 10, 128–138.
Ward M. W., Kogel D. and Prehn J. H. M. (2004) Neuronal apoptosis:BH3-only proteins the real killers? J. Bioenerg. Biomembr. 36,295–298.
Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q.and Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TARDNA-binding protein (TDP-43) induces disease-like redistribution,sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309.
Wootz H., Hansson I., Korhonen L., Napankangas U. and Lindholm D.(2004) Caspase-12 cleavage and increased oxidative stress duringmotorneuron degeneration in transgenic mouse model of ALS.Biochem. Biophys. Res. Commun. 322, 281–286.
Wootz H., Hansson I., Korhonen L. and Lindholm D. (2006) XIAPdecreases caspase-12 cleavage and calpain activity in spinal cord ofALS transgenic mice. Exp. Cell Res. 312, 1890–1898.
Zhai J., Lin H., Canete-Soler R. and Schlaepfer W. W. (2005) HoxB2binds mutant SOD1 and is altered in transgenic model of ALS.Hum. Mol. Genet. 14, 2629–2640.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2009) 108, 578–590� 2008 The Authors
590 | K. Y. Soo et al.





























