Embed Size (px)
Citation preview

Experimental Neurology 167, 282–289 (2001)doi:10.1006/exnr.2000.7573, available online at http://www.idealibrary.com on
Retinal Ganglion Cell and Nonneuronal Cell Responsesto a Microcrush Lesion of Adult Rat Optic Nerve
Inmaculada Selles-Navarro,*,1 Benjamin Ellezam,†,1 Raul Fajardo,† Mathieu Latour,† and Lisa McKerracher†*Laboratorio de Oftalmologia Experimental, Facultad de Medicina, Universidad de Murcia, Murcia, Spain; and†Departement de pathologie et biologie cellulaire, Universite de Montreal, Montreal, Quebec H3C 3J7, Canada
Received July 7, 2000; accepted September 26, 2000; published online December 19, 2000
bt1
blaaadawfmewmpa
tsCeelctc
Injury of the optic nerve has served as an importantmodel for the study of cell death and axon regenera-tion in the CNS. Analysis of axon sprouting and regen-eration after injury by anatomical tracing are aided bylesion models that produce a well-defined injury site.We report here the characterization of a microcrushlesion of the optic nerve made with 10-0 sutures tocompletely transect RGC axons. Following microcrushlesion, 62% of RGCs remained alive 1 week later, and28% of RGCs, at 2 weeks. Optic nerve sections stainedby hematoxylin-based methods showed a thin line ofintensely stained cells that invaded the lesion site at24 h after microcrush lesion. The lesion site becameincreasingly disorganized by 2 weeks after injury, andboth macrophages and blood vessels invaded the le-sion site. The microcrush lesion was immunoreactivefor chondroitin sulfate proteoglycans (CSPG), and anadjacent GFAP-negative zone developed early afterthe lesion, disappearing by 1 week. Luxol fast bluestaining showed a myelin-free zone at the lesion site,and myelin remained distal to the lesion at 8 weeks. Tostudy the axonal response to microcrush lesion, an-terograde tracing was used. Within 6 h after injury allRGC axons retracted back from the site of lesion. By 1week after injury, axons regrew toward the lesion, butmost stopped abruptly at the injury scar. The few ax-ons that were able to cross the injury site did notextend further in the optic nerve white matter by 8weeks postlesion. Our observations suggest that boththe CSPG-positive scar and the myelin-derived growthinhibitory proteins contribute to the failure of RGCregeneration after injury. © 2001 Academic Press
Key Words: retinal ganglion cell; axotomy; myelin;chondroitin sulfate proteoglycans; cell death.
INTRODUCTION
Injury in the CNS causes permanent impairmentbecause injured neurons do not regenerate their
1 Co-first authors.
2820014-4886/01 $35.00Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
transected axons. Following injury in the CNS thereare a number of changes in nonneuronal cells, includ-ing the formation of a glial scar by astrocytes andinvasion of macrophages and microglia (11). Many dif-ferent growth inhibitory proteins present in the CNSblock axon growth. Both tenascin and chondroitin sul-fate proteoglycans (CSPGs) are expressed following in-jury to the CNS (14, 21, 24, 25). Myelin-derived growthinhibitory proteins that remain distal to the injury arealso important barriers to axon regeneration (7, 26, 28,30). The relative contributions of myelin and the glialscar to blocking regeneration is controversial (9, 10).Nonetheless, many in vivo studies clearly indicate thatoth myelin proteins and glial scar proteoglycans con-ribute to regeneration failure in adult mammals (9,7, 19, 32).The extent of necrosis at the site of lesion is affected
y both ischemia and the inflammatory response fol-owing trauma. The type of injury or lesion can greatlyffect the amount of secondary damage that followsxonal transection in the CNS. Anatomical studies ofxon regeneration are aided by lesion models that re-uce necrosis and cavitation so that the distances ofxonal growth can be accurately followed. Recently, itas shown that the glial scar is greatly reduced by the
ormation of a small focal lesion at the site of neuronalicrotransplantation (9, 10). Transplanted neurons
xtended axons across the lesion site into the distalhite matter when scar-associated proteoglycan im-unoreactivity was reduced. In animals with greater
roteoglycan expression at the scar, axons were notble to cross the lesion (9, 10).The optic nerve has served as an important model for
he study of the nonneuronal response to injury and totudy axon regeneration in the CNS (4, 12, 36, 38).omplete transection of the optic nerve results in isch-mia and necrosis, and a large region of cavitationxtends toward the optic nerve head (14, 15). Crushesions cause less damage, and cell survival is betterompared to transection injury (3). However, after op-ic nerve crush, an area of cavitation develops in theentral optic nerve that extends toward the optic nerve

(wl2
t2wbCtnmhwtF(aniafa
npt
A
g
283MICROCRUSH LESION OF OPTIC NERVE
head (3). The lack of a clear and defined injury site andthe variable extent of retrograde axon degenerationafter crush lesion obscure the ability to assess axonregeneration after injury. Here we report studies on amicrocrush lesion of the optic nerve, which creates awell-defined injury site. We show that within 24 h afterinjury all RGC axons retract back from the site oflesion. By 1 week, axons sprout back to stop abruptly atthe injury scar. We further document the morphologi-cal changes in the scar from 6 h to 8 weeks after lesionand show that most axons that are able to cross thescar do not extend for long distances in the whitematter of the optic nerve.
METHODS
Surgical Procedures and RGC Survival Studies
Female Sprague–Dawley rats weighing 180–200 gwere anesthetized with intraperitoneal injections of0.6 ml/kg Hypnorm, 2.5 mg/kg diazepam, and 35 mg/kgketamine. The left optic nerve was exposed through asuperior temporal approach, and the dural sheath wasslit longitudinally, taking care to avoid the ophthalmicartery. Microcrush lesions were made with 10-0 su-tures used to completely constrict the optic nerve byholding a tight knot for 60 s and then releasing thesuture. The skin was closed with 4-0 silk sutures andthe fundis oculi was examined to verify the integrity ofthe retinal blood circulation. Animals whose retinasshowed ischemic damage were discarded. Animalswere studied by anterograde labeling 6 h (n 5 3), 24 h(n 5 4), 1 week (n 5 3), 2 weeks (n 5 6), and 8 weeksn 5 2) after microcrush lesion. Additional animalsithout anterograde labeling were examined by histo-
ogical and immunostaining methods at 1 week (n 5), 2 weeks (n 5 5), and 8 weeks (n 5 3).To study RGC survival after the microcrush lesion,
he RGCs were examined 1 week (n 5 5 animals) andweeks (n 5 4 animals) after lesion, and the right eyeas used for controls. RGCs were retrogradely labeledy applying 2% FluoroGold (Fluorochrome, Englewood,O) in 10% dimethyl sulfoxide, 0.9% NaCl bilaterally
o the superior colliculi and dorsal lateral geniculateuclei after removing the overlying pia (37, 39). Ani-als were fixed by perfusion with 4% paraformalde-yde, 0.1 M phosphate buffer; the retinas of both eyesere removed and prepared as flattened whole mounts
o examine by epifluorescence. The mean densities ofluoroGold-labeled RGCs were estimated as described
33, 39). Briefly, RGCs were counted on 12 standardreas (0.45 3 0.35 mm each) located beside the opticerve head and at 1.35 and 2.7 mm from the optic disk
n each of the retinal quadrants. In another group ofnimals (n 5 3) the RGCs were retrogradely labeledrom the superior colliculus with FluoroGold immedi-tely after the microcrush lesion to verify the complete-
ess of the lesion. In this case, the FluoroGold does notass the lesion site to label the retina when axons areransected.
nterograde Labeling
To identify RGC axons in the optic nerve by antero-rade labeling, 5 ml 1% cholera toxin b subunit (CTB)
(List Biological Labs, Campbell, CA) was injected intothe vitreous with a Hamilton syringe. Animals werefixed by perfusion with 4% paraformaldehyde, 0.1 Mphosphate buffer, 4 h after CTB injection in the eye.The eyes with attached optic nerves were removed,postfixed in 4% paraformaldehyde, 0.1 M phosphatebuffer, and cryoprotected in 30% sucrose overnight.Longitudinal cryostat sections of retina with attachedoptic nerve were mounted on gelatin-coated slides andstored at 4°C before use.
To detect CTB in RGC axons, slides were blockedwith 5% normal rabbit serum, 0.2% Triton X-100. Theywere incubated overnight with goat anti-cholera toxinantibody (1:5,000 List Biological Labs, Campbell, CA),washed, and incubated with biotinylated rabbit anti-goat antibody (1:200, Vector Lab, Burlingame, CA),followed by DTAF-conjugated streptavidin (1:500,Jackson Immunoresearch Labs, West Grove, PA). Insome experiments a horseradish peroxidase (HRP)-linked second antibody and enzymatic color reactionwere used to detect CTB (20).
Histological and Immunostaining Procedures
Fixed optic nerves were embedded in paraffin andcut in longitudinal sections at 4 to 6 mm. The lesion sitewas followed by staining sections with two differenthematoxylin-containing stains: hematoxylin/phloxineB/safran (HPS) stain (23) or Masson’s trichrome (35).Additional sections were stained using the Luxol fastblue method to reveal myelin (23). All slides weremounted using an automatic system and coverslippingfilm (TissueTek, Somagen Diagnostics, Edmonton, Al-berta, Canada).
Immunohistochemical localization of GFAP andCSPG was performed on freshly cut cryostat sectionswith an anti-GFAP polyclonal antibody (1:250, Sigma,St. Louis, MO) and the CS56 mouse IgM anti-CSPGantibody (1:100, Sigma), respectively. A FITC-conju-gated goat anti-rabbit secondary antibody (1:40, Pro-mega, Madison, WI) was used to reveal GFAP staining,and a biotinylated rabbit anti-mouse IgM antibody(1:100, Chemicon, Temecula, CA) followed by DTAF-conjugated streptavidin (1:500, Jackson Immunore-search Labs, West Grove, PA) was used for CSPGstaining. Immunostaining of laminin to localize bloodvessels was performed with a monoclonal antibody(1:200, Sigma) followed by an FITC-conjugated anti-mouse antibody (1:100, Promega).
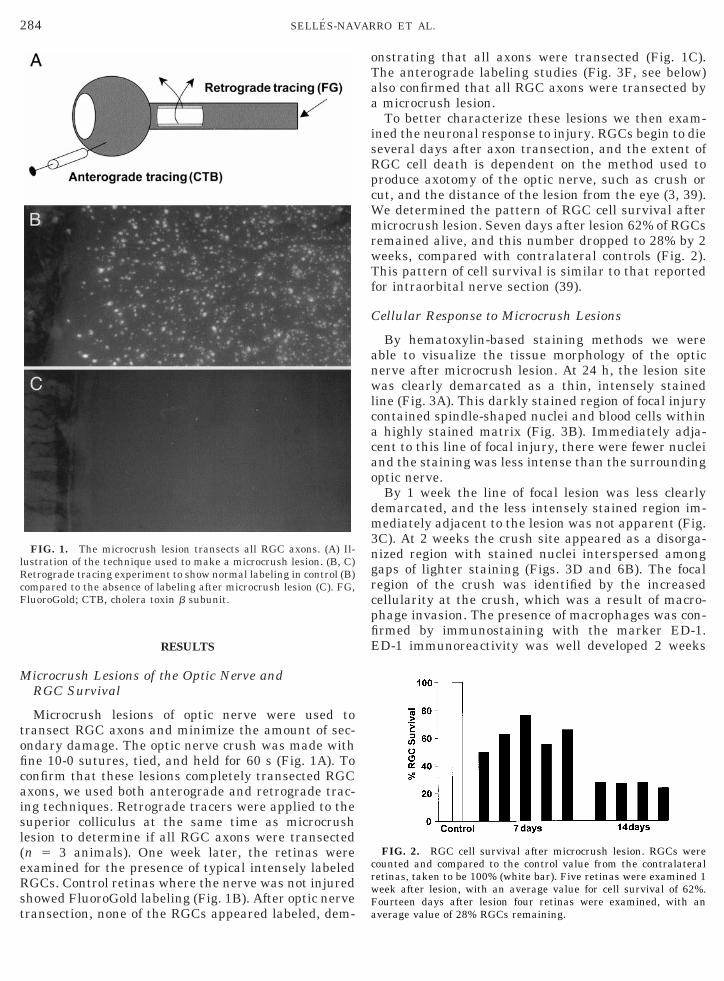
284 SELLES-NAVARRO ET AL.
RESULTS
Microcrush Lesions of the Optic Nerve andRGC Survival
Microcrush lesions of optic nerve were used totransect RGC axons and minimize the amount of sec-ondary damage. The optic nerve crush was made withfine 10-0 sutures, tied, and held for 60 s (Fig. 1A). Toconfirm that these lesions completely transected RGCaxons, we used both anterograde and retrograde trac-ing techniques. Retrograde tracers were applied to thesuperior colliculus at the same time as microcrushlesion to determine if all RGC axons were transected(n 5 3 animals). One week later, the retinas wereexamined for the presence of typical intensely labeledRGCs. Control retinas where the nerve was not injuredshowed FluoroGold labeling (Fig. 1B). After optic nervetransection, none of the RGCs appeared labeled, dem-
FIG. 1. The microcrush lesion transects all RGC axons. (A) Il-lustration of the technique used to make a microcrush lesion. (B, C)Retrograde tracing experiment to show normal labeling in control (B)compared to the absence of labeling after microcrush lesion (C). FG,FluoroGold; CTB, cholera toxin b subunit.
onstrating that all axons were transected (Fig. 1C).The anterograde labeling studies (Fig. 3F, see below)also confirmed that all RGC axons were transected bya microcrush lesion.
To better characterize these lesions we then exam-ined the neuronal response to injury. RGCs begin to dieseveral days after axon transection, and the extent ofRGC cell death is dependent on the method used toproduce axotomy of the optic nerve, such as crush orcut, and the distance of the lesion from the eye (3, 39).We determined the pattern of RGC cell survival aftermicrocrush lesion. Seven days after lesion 62% of RGCsremained alive, and this number dropped to 28% by 2weeks, compared with contralateral controls (Fig. 2).This pattern of cell survival is similar to that reportedfor intraorbital nerve section (39).
Cellular Response to Microcrush Lesions
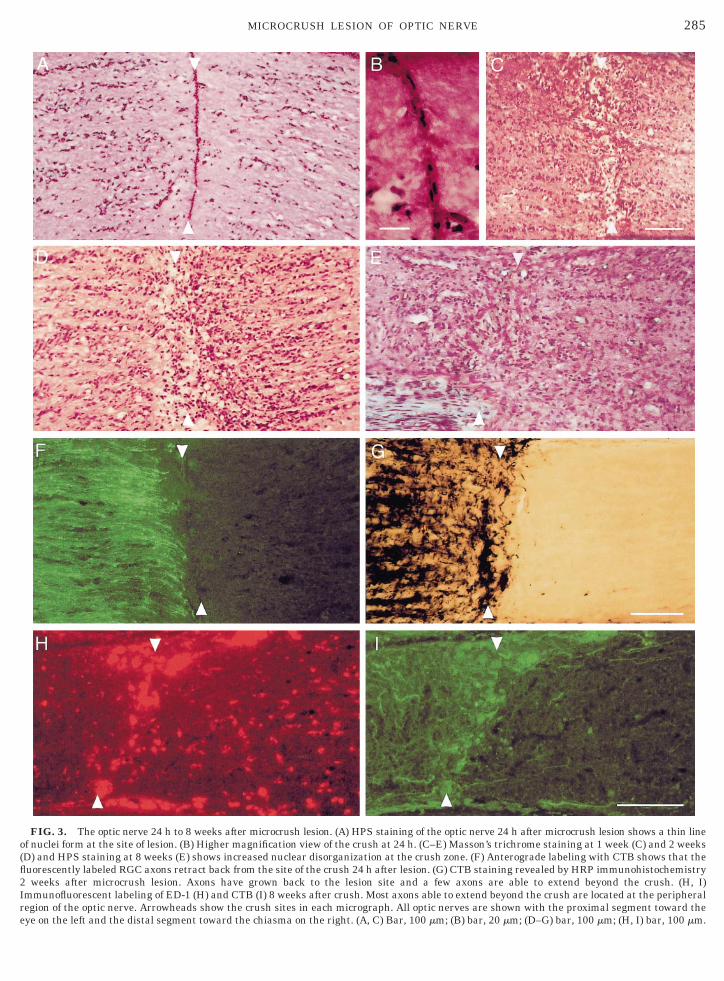
By hematoxylin-based staining methods we wereable to visualize the tissue morphology of the opticnerve after microcrush lesion. At 24 h, the lesion sitewas clearly demarcated as a thin, intensely stainedline (Fig. 3A). This darkly stained region of focal injurycontained spindle-shaped nuclei and blood cells withina highly stained matrix (Fig. 3B). Immediately adja-cent to this line of focal injury, there were fewer nucleiand the staining was less intense than the surroundingoptic nerve.
By 1 week the line of focal lesion was less clearlydemarcated, and the less intensely stained region im-mediately adjacent to the lesion was not apparent (Fig.3C). At 2 weeks the crush site appeared as a disorga-nized region with stained nuclei interspersed amonggaps of lighter staining (Figs. 3D and 6B). The focalregion of the crush was identified by the increasedcellularity at the crush, which was a result of macro-phage invasion. The presence of macrophages was con-firmed by immunostaining with the marker ED-1.ED-1 immunoreactivity was well developed 2 weeks
FIG. 2. RGC cell survival after microcrush lesion. RGCs werecounted and compared to the control value from the contralateralretinas, taken to be 100% (white bar). Five retinas were examined 1week after lesion, with an average value for cell survival of 62%.Fourteen days after lesion four retinas were examined, with anaverage value of 28% RGCs remaining.
o(fl2Ire
285MICROCRUSH LESION OF OPTIC NERVE
FIG. 3. The optic nerve 24 h to 8 weeks after microcrush lesion. (A) HPS staining of the optic nerve 24 h after microcrush lesion shows a thin linef nuclei form at the site of lesion. (B) Higher magnification view of the crush at 24 h. (C–E) Masson’s trichrome staining at 1 week (C) and 2 weeksD) and HPS staining at 8 weeks (E) shows increased nuclear disorganization at the crush zone. (F) Anterograde labeling with CTB shows that theuorescently labeled RGC axons retract back from the site of the crush 24 h after lesion. (G) CTB staining revealed by HRP immunohistochemistryweeks after microcrush lesion. Axons have grown back to the lesion site and a few axons are able to extend beyond the crush. (H, I)
mmunofluorescent labeling of ED-1 (H) and CTB (I) 8 weeks after crush. Most axons able to extend beyond the crush are located at the peripheralegion of the optic nerve. Arrowheads show the crush sites in each micrograph. All optic nerves are shown with the proximal segment toward theye on the left and the distal segment toward the chiasma on the right. (A, C) Bar, 100 mm; (B) bar, 20 mm; (D–G) bar, 100 mm; (H, I) bar, 100 mm.
286 SELLES-NAVARRO ET AL.
after injury (not shown), becoming less intense by 8weeks (Fig. 3H), with most macrophages concentratedjust at the lesion site.
At 8 weeks postinjury, in the hematoxylin-stainedsections the lesion site remained less well defined thanat 24 h (Figs. 3E and 6C). At this time point the lesionsite was identified by the presence of blood vessels thathad invaded to fill the focal region of microlesion (Fig.3E) and by the thin band of macrophages (Fig. 3H).
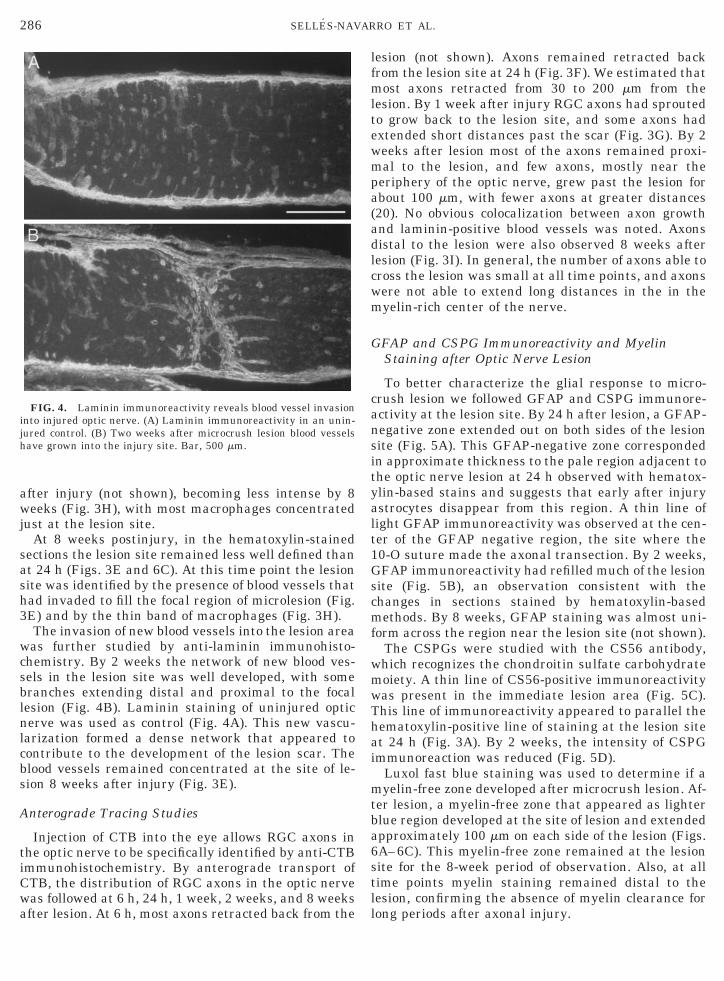
The invasion of new blood vessels into the lesion areawas further studied by anti-laminin immunohisto-chemistry. By 2 weeks the network of new blood ves-sels in the lesion site was well developed, with somebranches extending distal and proximal to the focallesion (Fig. 4B). Laminin staining of uninjured opticnerve was used as control (Fig. 4A). This new vascu-larization formed a dense network that appeared tocontribute to the development of the lesion scar. Theblood vessels remained concentrated at the site of le-sion 8 weeks after injury (Fig. 3E).
Anterograde Tracing Studies
Injection of CTB into the eye allows RGC axons inthe optic nerve to be specifically identified by anti-CTBimmunohistochemistry. By anterograde transport ofCTB, the distribution of RGC axons in the optic nervewas followed at 6 h, 24 h, 1 week, 2 weeks, and 8 weeksafter lesion. At 6 h, most axons retracted back from the
FIG. 4. Laminin immunoreactivity reveals blood vessel invasioninto injured optic nerve. (A) Laminin immunoreactivity in an unin-jured control. (B) Two weeks after microcrush lesion blood vesselshave grown into the injury site. Bar, 500 mm.
lesion (not shown). Axons remained retracted backfrom the lesion site at 24 h (Fig. 3F). We estimated thatmost axons retracted from 30 to 200 mm from thelesion. By 1 week after injury RGC axons had sproutedto grow back to the lesion site, and some axons hadextended short distances past the scar (Fig. 3G). By 2weeks after lesion most of the axons remained proxi-mal to the lesion, and few axons, mostly near theperiphery of the optic nerve, grew past the lesion forabout 100 mm, with fewer axons at greater distances(20). No obvious colocalization between axon growthand laminin-positive blood vessels was noted. Axonsdistal to the lesion were also observed 8 weeks afterlesion (Fig. 3I). In general, the number of axons able tocross the lesion was small at all time points, and axonswere not able to extend long distances in the in themyelin-rich center of the nerve.
GFAP and CSPG Immunoreactivity and MyelinStaining after Optic Nerve Lesion
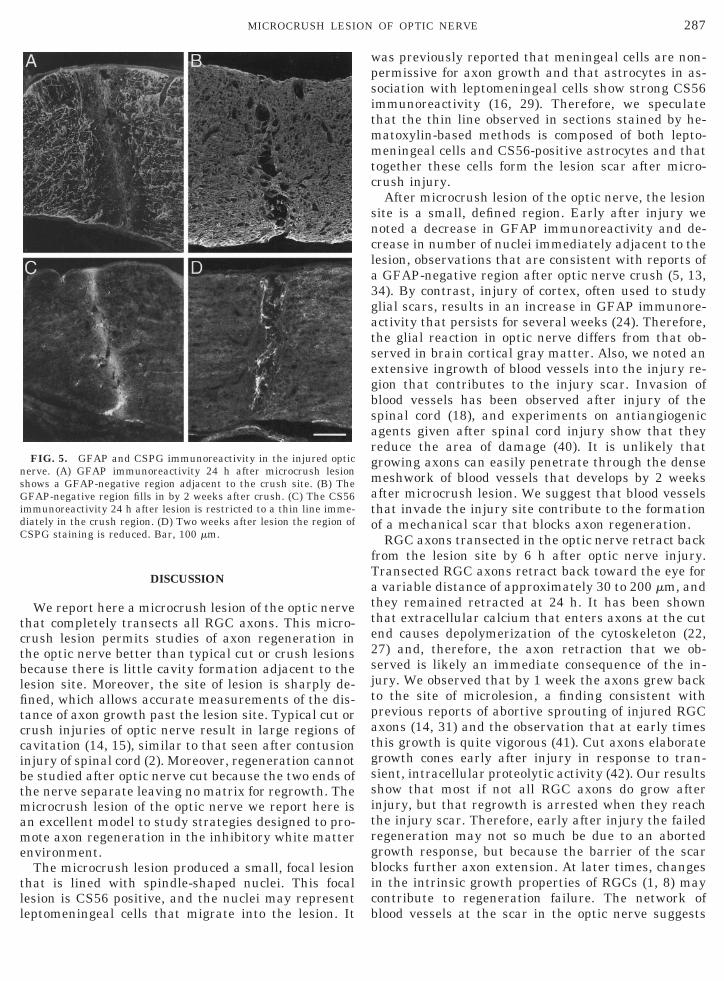
To better characterize the glial response to micro-crush lesion we followed GFAP and CSPG immunore-activity at the lesion site. By 24 h after lesion, a GFAP-negative zone extended out on both sides of the lesionsite (Fig. 5A). This GFAP-negative zone correspondedin approximate thickness to the pale region adjacent tothe optic nerve lesion at 24 h observed with hematox-ylin-based stains and suggests that early after injuryastrocytes disappear from this region. A thin line oflight GFAP immunoreactivity was observed at the cen-ter of the GFAP negative region, the site where the10-O suture made the axonal transection. By 2 weeks,GFAP immunoreactivity had refilled much of the lesionsite (Fig. 5B), an observation consistent with thechanges in sections stained by hematoxylin-basedmethods. By 8 weeks, GFAP staining was almost uni-form across the region near the lesion site (not shown).
The CSPGs were studied with the CS56 antibody,which recognizes the chondroitin sulfate carbohydratemoiety. A thin line of CS56-positive immunoreactivitywas present in the immediate lesion area (Fig. 5C).This line of immunoreactivity appeared to parallel thehematoxylin-positive line of staining at the lesion siteat 24 h (Fig. 3A). By 2 weeks, the intensity of CSPGimmunoreaction was reduced (Fig. 5D).
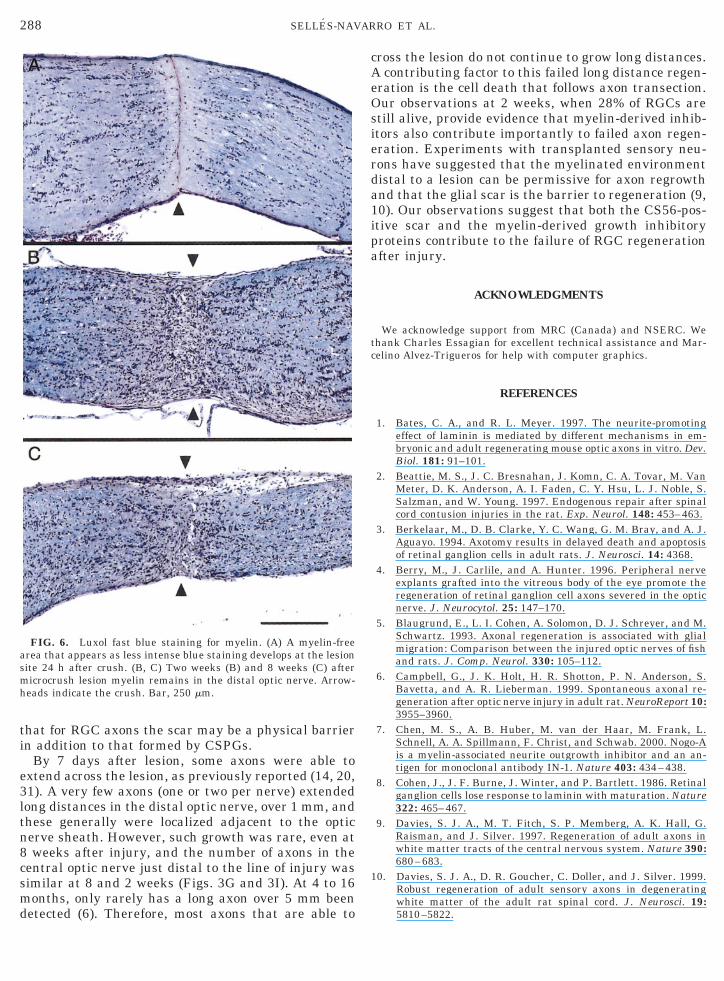
Luxol fast blue staining was used to determine if amyelin-free zone developed after microcrush lesion. Af-ter lesion, a myelin-free zone that appeared as lighterblue region developed at the site of lesion and extendedapproximately 100 mm on each side of the lesion (Figs.6A–6C). This myelin-free zone remained at the lesionsite for the 8-week period of observation. Also, at alltime points myelin staining remained distal to thelesion, confirming the absence of myelin clearance forlong periods after axonal injury.
287MICROCRUSH LESION OF OPTIC NERVE
DISCUSSION
We report here a microcrush lesion of the optic nervethat completely transects all RGC axons. This micro-crush lesion permits studies of axon regeneration inthe optic nerve better than typical cut or crush lesionsbecause there is little cavity formation adjacent to thelesion site. Moreover, the site of lesion is sharply de-fined, which allows accurate measurements of the dis-tance of axon growth past the lesion site. Typical cut orcrush injuries of optic nerve result in large regions ofcavitation (14, 15), similar to that seen after contusioninjury of spinal cord (2). Moreover, regeneration cannotbe studied after optic nerve cut because the two ends ofthe nerve separate leaving no matrix for regrowth. Themicrocrush lesion of the optic nerve we report here isan excellent model to study strategies designed to pro-mote axon regeneration in the inhibitory white matterenvironment.
The microcrush lesion produced a small, focal lesionthat is lined with spindle-shaped nuclei. This focallesion is CS56 positive, and the nuclei may representleptomeningeal cells that migrate into the lesion. It
FIG. 5. GFAP and CSPG immunoreactivity in the injured opticnerve. (A) GFAP immunoreactivity 24 h after microcrush lesionshows a GFAP-negative region adjacent to the crush site. (B) TheGFAP-negative region fills in by 2 weeks after crush. (C) The CS56immunoreactivity 24 h after lesion is restricted to a thin line imme-diately in the crush region. (D) Two weeks after lesion the region ofCSPG staining is reduced. Bar, 100 mm.
was previously reported that meningeal cells are non-permissive for axon growth and that astrocytes in as-sociation with leptomeningeal cells show strong CS56immunoreactivity (16, 29). Therefore, we speculatethat the thin line observed in sections stained by he-matoxylin-based methods is composed of both lepto-meningeal cells and CS56-positive astrocytes and thattogether these cells form the lesion scar after micro-crush injury.
After microcrush lesion of the optic nerve, the lesionsite is a small, defined region. Early after injury wenoted a decrease in GFAP immunoreactivity and de-crease in number of nuclei immediately adjacent to thelesion, observations that are consistent with reports ofa GFAP-negative region after optic nerve crush (5, 13,34). By contrast, injury of cortex, often used to studyglial scars, results in an increase in GFAP immunore-activity that persists for several weeks (24). Therefore,the glial reaction in optic nerve differs from that ob-served in brain cortical gray matter. Also, we noted anextensive ingrowth of blood vessels into the injury re-gion that contributes to the injury scar. Invasion ofblood vessels has been observed after injury of thespinal cord (18), and experiments on antiangiogenicagents given after spinal cord injury show that theyreduce the area of damage (40). It is unlikely thatgrowing axons can easily penetrate through the densemeshwork of blood vessels that develops by 2 weeksafter microcrush lesion. We suggest that blood vesselsthat invade the injury site contribute to the formationof a mechanical scar that blocks axon regeneration.
RGC axons transected in the optic nerve retract backfrom the lesion site by 6 h after optic nerve injury.Transected RGC axons retract back toward the eye fora variable distance of approximately 30 to 200 mm, andthey remained retracted at 24 h. It has been shownthat extracellular calcium that enters axons at the cutend causes depolymerization of the cytoskeleton (22,27) and, therefore, the axon retraction that we ob-served is likely an immediate consequence of the in-jury. We observed that by 1 week the axons grew backto the site of microlesion, a finding consistent withprevious reports of abortive sprouting of injured RGCaxons (14, 31) and the observation that at early timesthis growth is quite vigorous (41). Cut axons elaborategrowth cones early after injury in response to tran-sient, intracellular proteolytic activity (42). Our resultsshow that most if not all RGC axons do grow afterinjury, but that regrowth is arrested when they reachthe injury scar. Therefore, early after injury the failedregeneration may not so much be due to an abortedgrowth response, but because the barrier of the scarblocks further axon extension. At later times, changesin the intrinsic growth properties of RGCs (1, 8) maycontribute to regeneration failure. The network ofblood vessels at the scar in the optic nerve suggests
1
288 SELLES-NAVARRO ET AL.
that for RGC axons the scar may be a physical barrierin addition to that formed by CSPGs.
By 7 days after lesion, some axons were able toextend across the lesion, as previously reported (14, 20,31). A very few axons (one or two per nerve) extendedlong distances in the distal optic nerve, over 1 mm, andthese generally were localized adjacent to the opticnerve sheath. However, such growth was rare, even at8 weeks after injury, and the number of axons in thecentral optic nerve just distal to the line of injury wassimilar at 8 and 2 weeks (Figs. 3G and 3I). At 4 to 16months, only rarely has a long axon over 5 mm beendetected (6). Therefore, most axons that are able to
FIG. 6. Luxol fast blue staining for myelin. (A) A myelin-freearea that appears as less intense blue staining develops at the lesionsite 24 h after crush. (B, C) Two weeks (B) and 8 weeks (C) aftermicrocrush lesion myelin remains in the distal optic nerve. Arrow-heads indicate the crush. Bar, 250 mm.
cross the lesion do not continue to grow long distances.A contributing factor to this failed long distance regen-eration is the cell death that follows axon transection.Our observations at 2 weeks, when 28% of RGCs arestill alive, provide evidence that myelin-derived inhib-itors also contribute importantly to failed axon regen-eration. Experiments with transplanted sensory neu-rons have suggested that the myelinated environmentdistal to a lesion can be permissive for axon regrowthand that the glial scar is the barrier to regeneration (9,10). Our observations suggest that both the CS56-pos-itive scar and the myelin-derived growth inhibitoryproteins contribute to the failure of RGC regenerationafter injury.
ACKNOWLEDGMENTS
We acknowledge support from MRC (Canada) and NSERC. Wethank Charles Essagian for excellent technical assistance and Mar-celino Alvez-Trigueros for help with computer graphics.
REFERENCES
1. Bates, C. A., and R. L. Meyer. 1997. The neurite-promotingeffect of laminin is mediated by different mechanisms in em-bryonic and adult regenerating mouse optic axons in vitro. Dev.Biol. 181: 91–101.
2. Beattie, M. S., J. C. Bresnahan, J. Komn, C. A. Tovar, M. VanMeter, D. K. Anderson, A. I. Faden, C. Y. Hsu, L. J. Noble, S.Salzman, and W. Young. 1997. Endogenous repair after spinalcord contusion injuries in the rat. Exp. Neurol. 148: 453–463.
3. Berkelaar, M., D. B. Clarke, Y. C. Wang, G. M. Bray, and A. J.Aguayo. 1994. Axotomy results in delayed death and apoptosisof retinal ganglion cells in adult rats. J. Neurosci. 14: 4368.
4. Berry, M., J. Carlile, and A. Hunter. 1996. Peripheral nerveexplants grafted into the vitreous body of the eye promote theregeneration of retinal ganglion cell axons severed in the opticnerve. J. Neurocytol. 25: 147–170.
5. Blaugrund, E., L. I. Cohen, A. Solomon, D. J. Schreyer, and M.Schwartz. 1993. Axonal regeneration is associated with glialmigration: Comparison between the injured optic nerves of fishand rats. J. Comp. Neurol. 330: 105–112.
6. Campbell, G., J. K. Holt, H. R. Shotton, P. N. Anderson, S.Bavetta, and A. R. Lieberman. 1999. Spontaneous axonal re-generation after optic nerve injury in adult rat. NeuroReport 10:3955–3960.
7. Chen, M. S., A. B. Huber, M. van der Haar, M. Frank, L.Schnell, A. A. Spillmann, F. Christ, and Schwab. 2000. Nogo-Ais a myelin-associated neurite outgrowth inhibitor and an an-tigen for monoclonal antibody IN-1. Nature 403: 434–438.
8. Cohen, J., J. F. Burne, J. Winter, and P. Bartlett. 1986. Retinalganglion cells lose response to laminin with maturation. Nature322: 465–467.
9. Davies, S. J. A., M. T. Fitch, S. P. Memberg, A. K. Hall, G.Raisman, and J. Silver. 1997. Regeneration of adult axons inwhite matter tracts of the central nervous system. Nature 390:680–683.
0. Davies, S. J. A., D. R. Goucher, C. Doller, and J. Silver. 1999.Robust regeneration of adult sensory axons in degeneratingwhite matter of the adult rat spinal cord. J. Neurosci. 19:5810–5822.

1
1
1
2
2
2
2
2
289MICROCRUSH LESION OF OPTIC NERVE
11. Fawcett, J. W., and R. A. Asher. 1999. The glial scar and centralnervous system repair. Brain Res. Bull. 49: 377–391.
12. Fournier, A. E., and L. McKerracher. 1997. Expression of spe-cific tubulin isotypes increases during regeneration of injuredCNS neurons, but not after the application of brain-derivedneurotophic factor (BDNF). J. Neurosci. 17: 4623–4632.
13. Frank, M., and H. Wolburg. 1998. Cellular reactions at thelesion site after crushing of the rat optic nerve. Glia 16: 227–240.
4. Giftochristos, N., and S. David. 1988. Laminin and heparinsulfate proteoglycan in the lesioned adult mammalian centralnervous system and their possible relationship to axonalsprouting. J. Neurocytol. 17: 385–397.
15. Graftstein, B., and N. A. Ingoglia. 1982. Intracranial transec-tion of the optic nerve in adult mice: Preliminary observations.Exp. Neurol. 76: 318–330.
16. Hirsch, S., and M. Bahr. 1999. Immunocytochemical character-ization of reactive optic nerve astrocytes and meningeal cells.Glia 26: 36–46.
7. Huang, D. W., L. McKerracher, P. Braun, and L. McKerracher.1999. A therapeurtic vaccine approach to stimulate axon regen-eration in the adult mammalian spinal cord. Submitted forpublication.
18. Imperato-Kalmar, E., A. McKinney, L. Schnell, B. R. Rubin,and M. E. Schwab. 1997. Local changes in vascular architecturefollowing partial spinal cord lesion in the rat. Exp. Neurol. 140:322–328.
9. Keirstead, H. S., S. J. Hasan, G. D. Muir, and J. D. Steeves.1992. Suppression of the onset of myelination extends the per-missive period for the functional repair of embryonic spinalcord. Proc. Natl. Acad. Sci. USA 89: 11664–11668.
0. Lehmann, M., A. E. Fournier, I. Selles-Navarro, P. Dergham, N.Leclerc, G. Tigyi, and L. McKerracher. 1999. Inactivation of thesmall GTP-binding protein Rho promotes CNS axon regenera-tion. J. Neurosci. 19: 7537–7547.
1. Levine, J. 1998. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J. Neurosci. 14: 4716–4730.
2. LoPachin, R. M., and E. J. Lehning. 1997. Mechanism of cal-cium entry during axon injury and degeneration. Toxicol. Appl.Pharmacol. 143: 233–244.
23. Luna, L. G. 1992. Histopathologic Methods and Color Atlas ofSpecial Stains and Tissue Artifacts. Johnson Printers, DownersGrove, IL.
4. McKeon, R. J., R. C. Schreiber, J. S. Rudge, and J. Silver. 1991.Reduction of neurite outgrowth in a model of glial scarringfollowing CNS injury is correlated with the expression of inhib-itory molecules on reactive astrocytes. J. Neurosci. 11: 3398–3411.
5. McKeon, R. J., and J. Silver. 1995. Injury-induced proteogly-cans inhibit the potential for laminin-mediated axon growth onastrocytic scars. Exp. Neurol. 136: 32–43.
26. McKerracher, L., S. David, J. L. Jackson, V. Kottis, R. Dunn,and P. E. Braun. 1994. Identification of myelin-associated gly-coprotein as a major myelin-derived inhibitor of neurite out-growth. Neuron 13: 805–811.
27. Meller, K. 1987. Early structural changes in the axoplasmiccytoskeleton after axotomy studies by cryofixation. Cell TissueRes. 250: 663–672.
28. Mukhopadhyay, G., P. Doherty, F. S. Walsh, P. R. Crocker, andM. T. Filbin. 1994. A novel role for myelin-associated glycopro-tein as an inhibitor of axonal regeneration. Neuron 13: 805–811.
29. Ness, R., and S. David. 1997. Leptomenigeal cells modulate theneurite growth promoting properties of astocytes in vitro. Glial19: 47–57.
30. Prinjha, R., S. E. Moore, M. Vinson, S. Blake, R. Morrow, G.Christie, D. Michalovich, D. L. Simmons, and F. Walsh. 2000.Inhibitor of neurite outgrowth in humans. Nature 403: 383–384.
31. Richardson, P. M., V. M. K. Issa, and S. Shemie. 1982. Regen-eration and retrograde degeneration of axons in the rat opticnerve. J. Neurocytol. 11: 949–966.
32. Schwab, M. E., J. P. Kapfhammer, and C. E. Bandtlow. 1993.Inhibitors of neurite outgrowth. Annu. Rev. Neurosci. 16: 565–595.
33. Selles-Navarro, I., M. P. Villegas-Perez, M. Salvador-Silva,J. M. Ruiz-Gomez, and M. Vidal-Sanz. 1996. Retinal ganglioncell death after different transient periods of pressure-inducedischemia and survival intervals. Invest. Opthal. Vis. Sci. 37:2002–2014.
34. Solomon, A. S., V. Lavie, U. Hauben, A. Monsonego, E. Yoles,and M. Schwartz. 1996. Complete transection of rat optic nervewhile sparing the meninges and the vasculature: an experimen-tal model for optic nerve neuropathy and trauma. J. Neurosci.Methods 70: 21–25.
35. Thompson, S. W., and R. D. Hunt. 1966. Selected Histochemicaland Histopathological Methods. Thomas, Springfield, IL.
36. Vidal-Sanz, M., G. M. Bray, M. P. Villegas-Perez, S. Thanos,and A. J. Aguayo. 1987. Axonal regeneration and synapse for-mation in the superior colliculus by retinal ganglion cells in theadult retina. J. Neurosci. 7: 2894–2909.
37. Vidal-Sanz, M., M. P. Villegas-Perez, G. M. Bray, and A. J.Aguayo. 1988. Persistent retrograde labelling of adult rat reti-nal ganglion cells with the carbocyanine dye DiI. Exp. Neurol.102: 92–101.
38. Villegas-Perez, M. P., M. Vidal-Sanz, G. M. Bray, and A. J.Aguayo. 1988. Influences of peripheral nerve grafts on the sur-vival and regrowth of axotomized retinal ganglion cells in adultrats. J. Neurosci. 8: 265.
39. Villegas-Perez, M. P., M. Vidal-Sanz, M. Rasminsky, G. M.Bray, and A. J. Aguayo. 1993. Rapid and protracted phases ofretinal ganglion cell loss follow axotomy in the optic nerve ofadult rats. J. Neurobiol. 24: 23–36.
40. Wamil, A. W., B. D. Wamil, and C. G. Hellerqvist. 1998. CM101-mediated recovery of walking abiity in adult mice paralyzed byspinal cord injury. Proc. Natl. Acad. Sci. USA 95: 13188–13193.
41. Zeng, B.-Y., P. N. Anderson, G. Campbell, and A. R. Lieberman.1994. Regenerative and other responses to injury in the retinalstump of the optic nerve in adult albino rats: Transection of theintraorbital optic nerve. J. Anat. 185: 643–661.
42. Ziv, N. E., and M. E. Spira. 1998. Induction of growth coneformation by transient and localized increases of intracellularproteolytic activity. J. Cell Biol. 140: 223–232.





























