Embed Size (px)
Citation preview

B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /b ra in res
Research Report
S-Allyl L-cysteine diminishes cerebral ischemia-inducedmitochondrial dysfunctions in hippocampus
Fahim Atif⁎,1, Seema Yousuf1, Sandeep Kumar AgrawalDepartment of Surgery, Section of Neurosurgery, 6009 Poynter Hall, University of Nebraska Medical Center, Omaha,Nebraska 68198-6250, USA
A R T I C L E I N F O
⁎ Corresponding author. Present address: DeGeorgia 30322 USA. Fax: +1 402 559 7779.
E-mail address: [email protected] (F. At1 Authors contributed equally to the paper
0006-8993/$ – see front matter © 2009 Elsevidoi:10.1016/j.brainres.2008.12.077
A B S T R A C T
Article history:Accepted 29 December 2008Available online 15 January 2009
Ischemic brain is highly vulnerable to free radicals mediated secondary neuronal damageespecially mitochondrial dysfunctions. Present study investigated the neuroprotectiveeffect of S-allyl L-cysteine (SAC), a water soluble compound from garlic, against cerebralischemia/reperfusion (I/R)-induced mitochondrial dysfunctions in hippocampus (HIP). Weused transient rat middle cerebral artery occlusion (MCAO) model of brain ischemia. SAC(300 mg/kg) was given twice intraperitoneally: 15 min pre-occlusion and 2 h post-occlusionat the time of reperfusion. SAC significantly restored ATP content and the activity ofmitochondrial respiratory complexes in SAC treated group which were severely altered inMCAO group. A marked decrease in calcium swelling was observed as a result of SACtreatment. Western blot analysis showed a marked decrease in cytochrome c release as aresult of SAC treatment. The status of mitochondrial glutathione (GSH) and glucose 6-phosphate dehydrogenase (G6-PD) was restored by SAC treatment with a significantdecrease in mitochondrial lipid peroxidation (LPO), protein carbonyl (PC) and H2O2 content.SAC significantly improved neurological deficits assessed by different scoring methods ascompared to MCAO group. Also, the brain edema was significantly reduced. The findings ofthis study suggest the ability of SAC in functional preservation of ischemic neurovascularunits and its therapeutic relevance in the treatment of ischemic stroke.
© 2009 Elsevier B.V. All rights reserved.
Keywords:SACCerebral ischemiaHippocampusMitochondriaFree radical
1. Introduction
Stroke and traumatic brain injuries are conditions thatcompromise mitochondrial function and contribute to neuro-nal death. Due to high energy demand, neurons are solelydependent on mitochondrial ATP production. Any damageeither by stroke or trauma to mitochondria causes alterationsin ATP production and an increase in reactive oxygen species(ROS) that are considered key events leading to both necrosisand apoptosis (Moro et al., 2005). The ischemic penumbra is
partment of Emergency
if)..
er B.V. All rights reserved
the major site for therapeutic interventions, though it iselectrically silent but has significant blood flow. Antioxidantshave a propensity to block or delay apoptosis. There are plentyof reports that suggest therapeutic effect of various naturalantioxidants against cerebral ischemic damage (van Leyenet al., 2006; Yousuf et al., 2007; Mukherjee et al., 2007).
S-allyl-L-cysteine (SAC), an active organosulfur compoundof garlic, has been reported to possess antioxidant activity.Several mechanisms of action of SAC have been proposed. Itwas suggested that SAC exhibits its potent antioxidant
Medicine, Brain Research Laboratory, Emory University, Atlanta,
.

Fig. 1 – Calcium uptake in brain non-synaptic mitochondriain various groups. Mitochondria (25 μg/reaction) werede-energized via incubation in isotonic buffer at roomtemperature for 3min.After incubation, calcium (100μM)wasadded to the reactionmixture and absorbancewasmeasuredat 540 nm for 5 min at every 1 min time interval. Results areshown as % change in absorbance. Values in parenthesisshow percent (%) decrease in absorbance corresponding tothe absorbance of de-energized mitochondria.
129B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
property by scavenging superoxide radical, hydroxyl radicalsand hydrogen peroxide (Kim et al., 2001; Ide and Lau, 2001).Anothermechanism suggests that SAC prevents oxidized low-density lipoprotein (LDL)-induced endothelial cell injury andLDL oxidation (Ide and Lau, 1997; Lau, 2001). Also, SAC hasbeen reported to regulate nitric oxide (NO) productionwhich isassociated with anti-inflammatory responses (Kim et al.,2001). Additionally, some researchers have reported theinhibitory effect of SAC against edema formation in theischemic rat brain due to its lipid peroxidation (LPO) loweringeffect (Numagami et al., 1996; Numagami and Ohnishi, 2001).There are several reports on neuroprotective effect of SACagainst various brain insults. SAC has been reported to exertits neuroprotective effect by scavenging peroxynitrite andinhibiting the ERK signaling pathway activated during initialhypoxic/ischemic insults (Kim et al., 2006). In vitro studies alsosuggested the neuroprotective effect of SAC against neuro-toxicity in cultured hippocampal neurons (Kosuge et al., 2003;Ito et al., 2003; Kosuge et al., 2006).
We report here another possible mechanism of SACmediated neuroprotection against cerebral ischemic damagein hippocampus. We hypothesize that SAC exerts its neuro-protective effect by modulating mitochondrial dysfunctionsand associated cell death during cerebral ischemia/reperfu-sion (I/R). In the present study, we used transient rat middlecerebral artery occlusion (MCAO) model of brain ischemia toinduce brain infarction and conducted experiments in rathippocampus as major biochemical alterations occur in thisregion. We have previously reported severe alterations inmitochondrial functions, mitochondrial antioxidant defense,activation of stress proteins (Hsp 70 and metallothionein) andcaspase-3 as well as histological changes in hippocampusafter transient focal cerebral ischemia (Yousuf et al., 2007,2009).
2. Results
2.1. Effect of SAC on mitochondrial LPO, PC andintracellular H2O2 level
A significant increase (P<0.001) in LPO level was observed inMCAO group as compared to sham values (Table 1). Also, thelevel of protein oxidation was significantly increased (P<0.01)in MCAO group as evidenced by level of PC formation (Table1). Intracellular H2O2 level was increased significantly
Table 1 – Status of various parameters measured in hippocamp
Parameter
Sham
LPO (nmol TBARS/mg protein) 29.21±1.19PC (nmol DNPH/mg protein) 1.35±0.13H2O2 (nmol H2O2/gm tissue) 271.37±6.74GSH (nmol GSH/gm tissue) 0.442±0.021G6-PD (nmol NADP reduced/mg protein) 71.87±1.31Edema (μg/gm dried weight) 80.90±0.905
Values are expressed as mean±SE. Significance was determined as ⁎Pcompared with MCAO group.
(P<0.001) in MCAO group as compared to sham values(Table 1). As a result of SAC supplementation, a significantdecrease (P<0.001) in LPO level, PC content and intracellularH2O2 level was observed in MCAO+SAC group as compared toMCAO group.
2.2. Effect of SAC on mitochondrial GSH content
As a result of cerebral I/R, a significant decrease (P<0.001) inGSH content was observed in MCAO group when comparedwith sham values (Table 1). Supplementation of SAC in MCAOgroup (MCAO+SAC) significantly attenuated (P<0.01) thedepleted GSH levels as compared to MCAO group.
2.3. Effect of SAC on the activity of G6-PD
A significant decrease (P<0.001) in the activity of G6-PD wasobserved in MCAO group when compared with sham values(Table 1). SAC supplementation showed a significant increase(P<0.001) the G6-PD activity in MCAO+SAC group over MCAOgroup.
us of different groups
Group
MCAO MCAO+SAC
40.48±0.93⁎ 32.6±0.65##
5.08±0.088⁎ 2.98±0.12##
546.85±6.51⁎ 327.32±5.81##
0.285±0.014⁎ 0.369±0.014#
40.57±0.912⁎ 52.12±1.23##
95.33±0.494⁎ 89.66±0.494##
<0.001 when compared with sham group; #P<0.01, ##P<0.001 when
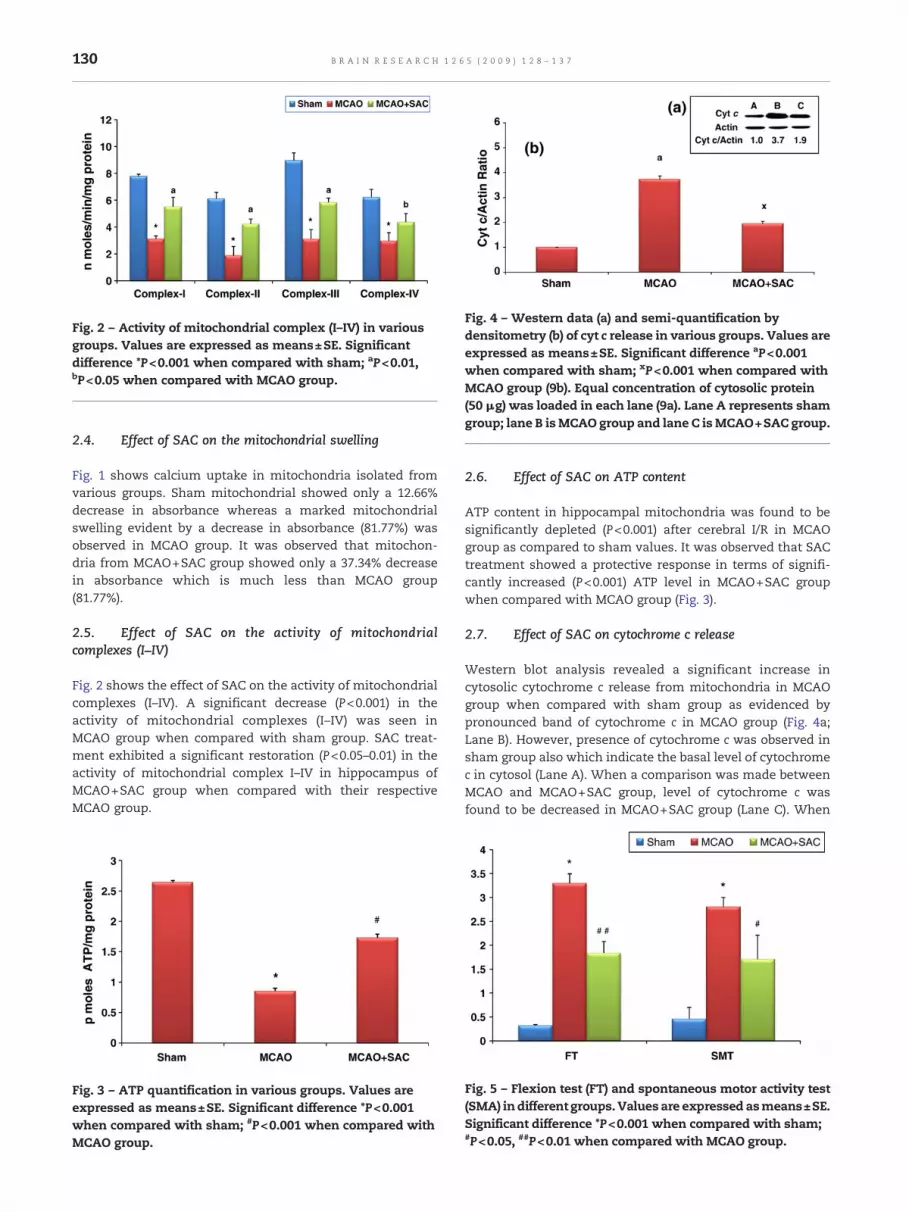
Fig. 2 – Activity of mitochondrial complex (I–IV) in variousgroups. Values are expressed as means±SE. Significantdifference *P<0.001 when compared with sham; aP<0.01,bP<0.05 when compared with MCAO group.
Fig. 4 – Western data (a) and semi-quantification bydensitometry (b) of cyt c release in various groups. Values areexpressed as means±SE. Significant difference aP<0.001when compared with sham; xP<0.001 when compared withMCAO group (9b). Equal concentration of cytosolic protein(50 μg) was loaded in each lane (9a). Lane A represents shamgroup; lane B isMCAO group and lane C isMCAO+SAC group.
130 B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
2.4. Effect of SAC on the mitochondrial swelling
Fig. 1 shows calcium uptake in mitochondria isolated fromvarious groups. Sham mitochondrial showed only a 12.66%decrease in absorbance whereas a marked mitochondrialswelling evident by a decrease in absorbance (81.77%) wasobserved in MCAO group. It was observed that mitochon-dria from MCAO+SAC group showed only a 37.34% decreasein absorbance which is much less than MCAO group(81.77%).
2.5. Effect of SAC on the activity of mitochondrialcomplexes (I–IV)
Fig. 2 shows the effect of SAC on the activity of mitochondrialcomplexes (I–IV). A significant decrease (P<0.001) in theactivity of mitochondrial complexes (I–IV) was seen inMCAO group when compared with sham group. SAC treat-ment exhibited a significant restoration (P<0.05–0.01) in theactivity of mitochondrial complex I–IV in hippocampus ofMCAO+SAC group when compared with their respectiveMCAO group.
Fig. 3 – ATP quantification in various groups. Values areexpressed as means±SE. Significant difference *P<0.001when compared with sham; #P<0.001 when compared withMCAO group.
2.6. Effect of SAC on ATP content
ATP content in hippocampal mitochondria was found to besignificantly depleted (P<0.001) after cerebral I/R in MCAOgroup as compared to sham values. It was observed that SACtreatment showed a protective response in terms of signifi-cantly increased (P<0.001) ATP level in MCAO+SAC groupwhen compared with MCAO group (Fig. 3).
2.7. Effect of SAC on cytochrome c release
Western blot analysis revealed a significant increase incytosolic cytochrome c release from mitochondria in MCAOgroup when compared with sham group as evidenced bypronounced band of cytochrome c in MCAO group (Fig. 4a;Lane B). However, presence of cytochrome c was observed insham group also which indicate the basal level of cytochromec in cytosol (Lane A). When a comparison was made betweenMCAO and MCAO+SAC group, level of cytochrome c wasfound to be decreased in MCAO+SAC group (Lane C). When
Fig. 5 – Flexion test (FT) and spontaneous motor activity test(SMA) indifferent groups.Values are expressed asmeans±SE.Significant difference *P<0.001 when compared with sham;#P<0.05, ##P<0.01 when compared with MCAO group.

131B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
the data of cyt c were normalized with actin data it wasobserved that MCAO group showed highly significant(P<0.001) increase in cyt c level as compared to sham values.SAC treatment resulted in a significant decrease (P<0.001) incyt c release when compared with MCAO group (Fig. 4b).
2.8. Effect of SAC on brain edema
In the MCAO group, a significant increase (P<0.001; 95.33±0.494) in brain water content was observed compared to shamgroup (80.9±0.905). Whereas, a marked depletion in the extentof brain edemawas seen inMCAO+SAC group (89.66±0.494) ascompared to MCAO group (Table 1).
2.9. Effect of SAC on behavior testes flexion test (FT) andspontaneous motor activity (SMA) and open field tests
Evaluation by both the methods (A and B) showed noneurological deficits in sham group (Fig. 5). While in theMCAO group, the neurological deficits were severe. The MCAOrats showed very consistent forelimb flexion (P<0.00l). Asignificant reduction (P<0.01) of the severity of this behavioralabnormality was seen in MCAO+SAC group as compared toMCAO group. In MCAO group, rats consistently showed circlingmovements and postural abnormalities besides other neurolo-gical signs like less to no spontaneous movements and severepaw flexions. Conversely, MCAO+SAC group significantly(P<0.05) improved the neurological outcome as compared tothe MCAO group. Also, fewer disturbances in posture and nocircling movements were observed when compared to theMCAO group. In SMA test, the rats of MCAO group spend mostof the time in the center of the cage with posture curvedtowards the paretic side, while MCAO+SAC group rats com-paratively moved around in the cage and explored theirenvironment. Open field activities showed that MCAO grouprats quickly developed decreased locomotor and stereotypicevents (P<0.01–0.001) with simultaneous expected increase inresting time as compared to sham group (Fig. 6). In contrast, themagnitude and the duration of these effects were found to beimproved in MCAO+SAC group. This alteration in the explora-tory behavior of rats was ameliorated by SAC treatment withsignificant increase (P<0.05) in the period of immobility orresting time too when compared with MCAO group (Fig. 6).
Fig. 6 – Open field activity in different groups. Values areexpressed as means±SE. Significant difference aP<0.001,bP<0.01 when compared with sham; xP<0.05 whencompared with MCAO group.
Fig. 7 – Neuroprotective effect of SAC on brain infarctionfollowing transient focal cerebral ischemia. Panel (A):Representative photographs of brain sections stained with2% TTC; Panel (B): Infarct volume measurement; Panel (C):Neurological scoring in various groups.MCAOgroupdevelopeda significant lesion (*P<0.001) compared to shamgroup,whereasMCAO+SAC group showed a significant decrease in tissuedamage over MCAO group (#P<0.001).
2.10. Effect of SAC on infarct size
TTC staining in sham brain did not show any detectable lesionor non-viable tissue damage (Fig. 7A). Conversely, brainsections obtained from MCAO group showed detectable

132 B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
lesions as white patches in the areas that are supplied by theMCA. The lesionswere present in both the lateral striatumandthe overlying cortex. It was observed that SAC treatmentsignificantly decreased the lesions in striatum and overlyingcortex of MCAO+SAC group brain as compared to sham group.Measurement of infarct volume showed a significant (P<0.001)increase in infarct area as compared to sham group whereas;MCAO+SAC group showed a significant (P<0.001) reduction ininfarct area over MCAO group (Fig. 7B).
3. Discussion
Our data represent neurochemical and neurobehavioralevidences that suggest SAC as a potent neuroprotectiveagent against cerebral I/R-induced secondary neuronaldamage by modulating mitochondrial dysfunctions in hippo-campus. In ischemic brain, ROS generation is the hallmark ofsecondary neuronal damage that alters structural and func-tional properties of brain lipid and proteins (Slemmer et al.,2008). In the present study, a marked increase in mitochon-drial LPO and PC formation was observed in MCAO group thatcould be attributed to the ROS action generated duringcerebral I/R. Mitochondria have their own enzymatic as wellas non-enzymatic antioxidant defense systemwhich combatsROS generated in mitochondria during oxidative phosphor-ylation. Mitochondrial reduced GSH is considered a primaryline of defense against ROS as it is involved in the eliminationof H2O2 by glutathione peroxidase. G6-PD maintains intracel-lular reduced GSH content. We observed a significant increasein intracellular H2O2 level and depleted GSH content and G6-PD activity in MCAO group. GSH content and activity of G6-PDhas been reported to decrease in ischemic conditions (Sarkarand Das, 2006).
As a result of SAC treatment, a significant decrease inmitochondrial LPO, PC level and intracellular H2O2 level wasobserved. Moreover, GSH content and G6-PD activity inMCAO+SAC group was significantly increased. This response of SACcould be attributed to its potential property of scavenging offree radicals that are generally increased in post-ischemicbrain (Kim et al., 2001; Kim et al., 2006). Inhibitors of LPO andfree radicals scavengers have been reported to exhibitneuroprotective effect against I/R-mediated brain injury(Young et al., 1988; Floyd and Carney, 1992). SAC has beenreported as a strong scavenger of ROS and inhibitor of ROSmediated LPO in ischemic brain (Numagami and Ohnishi,2001). In a report on structure–activity relationship of neuro-protective and ROS scavenging effect, it was observed thatpresence of alanyl group and lack of oxo group with inbetween molecular properties of SAC makes it a potentscavenger of ROS and inhibitor of LPO and neuronal death(Kim et al., 2006). As we observed the restoration in mitochon-drial GSH content and G6-PD activity and intracellular H2O2
level in MCAO+SAC group, this could be attributed to theantioxidant potential of SAC.
Cerebral I/R-induced ROS mediated oxidative damagecontributes in the exacerbation of intracellular calcium levelsleading to mitochondrial swelling that subsequently leads tothe opening of the mitochondrial permeability transitionpores (MPTP) and depolarization of mitochondrial membrane
potential (Okabe et al., 2000; Xiong et al., 2007). All thesephenomena eventually affect oxidative phosphorylation andresulting ATP production (Halestrap, 2005). Mitochondrialrespiratory chain has strongly been suggested susceptible tocerebral I/R. Thus, during cerebral I/R mitochondrial com-plexes (I–IV) are damaged (Allen et al., 1995; Almeida et al.,1995). Since mitochondria are the prime target of ROS/RNSduring cerebral I/R, mitochondrial dysfunctions result in therelease of cytochrome c which induces further cascade of celldeath (Korde et al., 2007; Cheng et al., 2007). In the presentstudy, MCAO group showed a significant increase in mito-chondrial swelling, altered activity of mitochondrial com-plexes (I–IV) and decreased ATP content. Conversely, SACtreatment reduced mitochondrial swelling and restored thestatus of mitochondrial complexes (I–IV) and ATP level inMCAO+SAC group. Also, a significant increase in cytosolicconcentration of cytochrome c in MCAO group was observed.This increase in cytochrome c level was markedly subsided inSAC treated group (MCAO+SAC). This could be hypothesizedthat SAC effectively scavenged ROS/RNS generation inischemic hippocampus and hence maintained mitochondrialintegrity which subsequently inhibited mitochondrial dys-functions. During focal cerebral ischemia the duration ofischemia and the reperfusion play a vital role in the develop-ment of brain edema which further leads to ischemic cascade(Li et al., 2000). In the present study, a significant increase inbrain water content was observed in MCAO group which wasfound to be markedly decreased in MCAO+SAC group. Thereare reports suggesting the role of SAC treatment in thereduction in brain edema during cerebral ischemia by inhibit-ing ROS-mediated LPO (Numagami et al., 1996; Numagami andOhnishi, 2001). Our findings are in agreement with theprevious reports.
Cerebral I/R causes necrosis mainly in the frontal sensor-imotor cortices and caudate-putamen which comprise a widerange of motor and sensorimotor deficits including partialparalysis, poor locomotor activity and lack of coordination(Hunter et al., 1998). Uncontrolled ROS/RNS generation isthought to play a critical role in behavioral deficits (Fukui etal., 2002). Oxidative stress and aging are the factors that alsoaccount for impaired memory functions (Fukui et al., 2001). Ithas been well established fact that balanced antioxidantshelps in controlling the cognitive and motor functions of thecerebral cortex and the hippocampus (Stadtman, 1992;Yousuf et al., 2007). In the present study, MCAO groupshowed significant neurobehavioral deficits. We observedthat the spontaneous motor activity and the flexion testswere severely altered in the MCAO group. This may bebecause cerebral I/R results in increased free radicals genera-tion leading to oxidant stress in hippocampus that havecommand over learning and motor activities. On the otherhand, SAC treatment reduced neurobehavioral deficits inMCAO+SAC group which were significantly higher in MCAOgroup. This could be attributed to the antioxidant potential ofSAC that protected hippocampus against I/R mediatedoxidative damage. There are previous reports that suggestthe use of antioxidants helps in recovery of neurobehavioraldeficits (Yousuf et al., 2007; Shuaib et al., 2007). In conclusion,the data suggest SAC as an excellent antioxidant whichexhibits its neuroprotective effect against cerebral ischemic

133B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
damage in hippocampus by modulating mitochondrialdysfunctions.
4. Experimental procedures
4.1. Drugs and chemicals
S-allyl L-cysteine was procured from Fisher Scientific, USA.Sucrose, percoll, HEPES, phenylmethanesulfonylfluoride(PMSF), 2,4-dinitrophenylhydrazine (DNPH), dithiothreitol(DTT), NADH, NADP, lecithin, rotenone, ubiquinone, succinicacid, 2,6-dichlorophenolindophenol, Tween-20, bovine serumalbumin (BSA) were obtained from Sigma, USA. Nitrocellulosemembrane, acrylamide, bis-acrylamide, TEMEDwere obtainedfrom BioRad. Anti cytochrome c mouse monoclonal IgG2b (sc-13560) and Goat anti mouse IgG HRP (sc-2005) were purchasedfrom Santa Cruz Biotech. USA.
4.2. Experimental design
4.2.1. Experiment # 1Male Wistar rats were divided into three groups (n=10 eachgroup): “Sham” (without MCAO and vehicle treated only);“MCAO” (2 h occlusion followed by 22 h reperfusion andvehicle treated); “MCAO+SAC” (with MCAO and 300 mg/kgSAC treated). The dose of SAC was determined according tothe previous report of Numagami et al. (1996). SAC (300mg/kg)in saline solution or equal volumes of vehicle were adminis-tered intraperitoneally (i.p.) in animals twice: 15 min pre-occlusion and 2 h post-occlusion at the time of reperfusion.After the completion of the reperfusion period, the animalswere assessed for neurobehavioral activity and then sacrificedfor the isolation of non-synaptic mitochondria from hippo-campus. ATP content; activity of mitochondrial complexes (I–IV); lipid peroxidation (LPO); reduced glutathione (GSH) andprotein carbonyl (PC) content were estimated in isolatedmitochondria in various groups.
4.2.2. Experiment # 2Animals (n=12 each group) were grouped and treated in thesame manner as in experiment 1. After reperfusion hippo-campi were processed to obtain cytosolic fraction for theestimation of glucose 6-phosphate dehydrogenase (G6-PD)and cytochrome c (Cyt c) release. H2O2 content was estimatedin post-mitochondrial supernatant.
4.2.3. Experiment # 3A separate experiment was conducted for the evaluation ofbrain edema. Animals (n=4) were grouped and treated in thesame manner as in experiment 1. After ischemia andreperfusion, brains were harvested for the estimation ofedema in various groups.
4.3. Induction of cerebral ischemia/reperfusion (I/R)
The right middle cerebral artery occlusion (MCAO) wasproduced using an intraluminal filament model by themethod of Longa et al. (1989). In brief, the rats wereanesthetized with pentobarbital (40 mg/kg, i.p.), a 4-0 nylonmonofilament with blunt end coated with poly-L-lysine wasintroduced into the external carotid artery (ECA) and
advanced into the middle cerebral artery via the internalcarotid artery (ICA) (17–20 mm) until a slight resistance wasfelt. Such resistance indicated that the filament had passedbeyond the proximal segment of the anterior cerebral artery(ACA). At this point, the intraluminal suture blocks the originof middle cerebral artery (MCA) and occluded all sources ofblood flow from ICA, anterior cerebral artery and the posteriorcerebral artery. Two hours after the induction of ischemia,the filament was slowly withdrawn and the animals werethen returned to their cages for a period of 22 h ofreperfusion. Throughout the procedure, a thermocouple wasinserted into the rectum and the body temperature wasmaintained at 37±0.5 °C with a thermostatiscally controlledwater blanket. In sham-operated rats, the external carotidartery was surgically prepared for insertion of the filamentbut the filament was not inserted.
4.4. Preparation of post mitochondrial supernatant (PMS)and cytosolic fraction
A 10% homogenate of hippocampus in ice cold 0.1 Mphosphate buffer (pH 7.4) containing KCl (1.17%) was firstcentrifuged at 3150 ×g for 10 min to remove nuclei andunbroken cells. The supernatant was then centrifuged at10,500 ×g for 20 min. The resulting supernatant was postmitochondrial supernatant (PMS) was further centrifuged at100,000 ×g to obtain cytosolic fraction as supernatant. PMS andcytosolic fractionwere used for the estimation of H2O2 contentand estimation of G6-PD activity respectively.
4.5. Isolation of non-synaptic mitochondria fromhippocampus
The mitochondria were prepared according to the method ofNagy et al. (1984). A 10% homogenate (w/v) was prepared inisolation buffer I (0.32 M sucrose; 5 mM N-2-hydroxyethylpi-perazine-N-2-ethane sulfonic acid and 0.1 mM EDTA-K+; pH7.5) with a Teflon-glass homogenizer at approximately1000 rpm with 5 strokes up and 5 strokes down. Thehomogenate was centrifuged at 1000 ×g for 10 min at 4 °Cand pellet (P1) was discarded. Supernatant (S1) was furthercentrifuged at 4 °C for 20 min at 12,000×g. Pellet (P2) were re-suspended in isolation buffer (3 ml/gm of original wet tissue).The pure mitochondria and synaptosomes were isolated onthe percoll gradients of 8.5, 10 and 20% (2.0, 4.0 and 4.0 ml) inbuffer II (0.25 M sucrose, 5 mMHEPES, and 0.1mM EDTA-K+ pH7.2). Samples were centrifuged at 15,000 ×g for 20 min at 4 °C.The pure mitochondria were obtained at the bottom layer ofpercoll (20%), which was resuspended in 10 ml of isolationbuffer and further centrifuged at 15,000 ×g for 10 min at 4 °C.The mitochondrial pellet was resuspended in appropriateamount of buffer and used for the various mitochondrialestimations.
4.6. Mitochondrial lipid peroxidation (LPO)
The level of LPO in isolated non-synaptic mitochondria wasmeasured as thiobarbituric acid-reactive substances (TBARS)by the method of Buege and Aust (1978). Briefly, the reactionmixture contained purified non-synaptic mitochondria (4 mg/ml) with an equal volume of Buege and Aust reagent

134 B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
(trichloroacetic acid, 15% w/v in 0.25 M HCl and thiobarbituricacid, 0.37% w/v in 0.25 M HCl) and kept in boiling water for15 min. The samples were then centrifuged at 1000 ×g for10 min after cooling. The absorbance of the supernatant wasmeasured at 532 nm. The concentration of TBARS wasdetermined using an extinction coefficient of 1.56×105 andresults were expressed as nmol of TBARS/mg ofmitochondrialprotein.
4.7. Mitochondrial protein oxidation (protein carbonyl; PC)
Protein carbonyls content was measured in mitochondriausing the method of Sohal et al. (2006). The mitochondrialpreparation was divided into two portions each containing 2–4 mg of protein. To one portion, 4 ml of 2 N HCl was addedand incubated at room temperature shaking intermittentlyfor 1 h. The other portion was treated with 4 ml of 10 mMDNPH in 2 N HCl and incubated by shaking intermittently for1 h at room temperature. After incubation the mixture wasprecipitated with 10% trichloroacetic acid and centrifuged.The precipitate was washed thrice with 4 ml of ethanol:ethylacetate (1:1). The final protein precipitate was dissolved in6 M guanidine hydrochloride and the absorption of solutionwas measured at 370 nm. The values were expressed as nmolcarbonyls/mg mitochondrial (molar extinction coefficient22,000).
4.8. Mitochondrial reduced glutathione (GSH)
Mitochondrial GSH was measured using the method of HissinandHilf (1976). Mitochondria (1mg protein)were suspended in25 μl of 25% HPO3 and 90 μl of sodium phosphate buffer (0.1 M,pH 8.0, with 5 mM EDTA). The samples were centrifuged at15,000 ×g for 10 min at 4 °C and the supernatant was collectedfor measurement of GSH. Supernatant (100 μl) was incubatedwith 100 μl of o-phthalaldehyde (0.1% in methanol) and 1.8 mlof 0.1 M phosphate buffer (pH 8.0) for 15 min at roomtemperature in dark. Fluorescence was measured with afluorescence spectrophotometer at an excitation excitationwavelength of 350 nm and an emission wavelength of 420 nm.
4.9. Assay of glucose-6-phosphate dehydrogenase (G6-PD)
The activity of G6-PD was assayed by the method of Zaheer etal. (1965). The reaction mixture in a total volume of 3.0 mlconsisted of 0.05 M Tris–HCl buffer (pH 7.6), 0.1 mM NADH(0.1 mM), 0.1 ml glucose 6-phosphate (0.8 mM), 0.1 ml MgCl2(8 mM) 0.1 ml cytosol and 2.3 ml distilled water. The changesin absorbance was recorded for 3 min at 340 nm and theenzyme activity was calculated as nmol NADP reduced/mgprotein using molar extinction coefficient of 6.22×103 M1 cm1.
4.10. Hydrogen peroxide (H2O2) generation
Hydrogen peroxide was estimated by themethod described byPick and Keisari (1981). Briefly, a 0.5 ml solution of phenol red(0.1 mg/ml phosphate buffer; 0.1 M pH 7.4) was mixed with0.5 ml 10% PMS and incubated at 37 °C for 10 min. The 1 mlsodium hydroxide (1 M) was added and the absorbance wasread at 610 nm. The quantity of H2O2 produced was expressedas nmol of H2O2 generated/mg protein.
4.11. Measurement of mitochondrial swelling
Ca2+ induced mitochondrial swelling of the de-energizedmitochondria was done by the method of Halestrap andDavidson (1990). Mitochondria (25 μg protein) were added1.1 ml of isotonic buffer containing 150 mM KSCN, 5 mM Tris,0.5 μl rotenone/ml, and 0.5 μg antimycin/ml (pH 7.2) at 30 °C.Swelling was initiated by addition of Ca2+ (100 μM) to thecuvette and the absorbance was monitored for 5 min at540 nm. Change in absorbance was monitored as percentchange compared with the control values.
4.12. ATP level
The isolated mitochondria (1 mg/ml) were re-suspended in areaction mixture containing 0.25 M sucrose, 1 mM MgCl2,10 mM HEPES and 1 mM EDTA. The suspension was brieflysonicated and centrifuged at 5000 ×g for 5 min. The super-natant was (100 μl) incubated with luciferin-luciferase (5 mg/ml) and the bioluminescence was measured in a Lumines-cence spectrometer microplate reader (Perkin-Elmer, S 50 B)using ATP Bioluminescent Assay Kit (Sigma, USA). The resultswere expressed as pmol of ATP mg−1 protein.
4.13. Mitochondrial respiratory chain complexes (I–IV)
Mitochondrial complexes (I–IV) were measured spectro-photometrically according to the method of Ragan et al.(1987) as modified by Desai et al. (1996). MitochondrialComplex-I (NADH-ubiquinone oxidoreductase, EC 1.6.99.3)activity was monitored by the oxidation of NADH to NAD byubiquinone-1 at 30 °C which was reduced to ubiquinol-1. Thereaction mixture contained potassium phosphate buffer(KPB, 10 mM pH 8.0), NADH (5 mM), lecithin (2 mM) andmitochondria (1 mg/ml). The reaction was started by theaddition of ubiquinone-1 (10 mM) to the reaction mixture(3 ml) and the decrease in absorbance was measured at340 nm for 3 min. Mitochondrial Complex-II (succinate-ubiquinone oxidoreductase, EC 1.3.5.1) activity was mea-sured as the rate of reduction of ubiquinone-2 to ubiquinol-2by succinic acid at 30 °C followed by secondary reduction of2, 6-dichlorophenolindophenol (DPIP) by the ubiquinolformed. The reaction mixture contained potassium phos-phate buffer 10 mM, pH 7.4), succinic acid (1 M, pH 7.4), EDTA(10 mM, pH 7.4) and distilled water. The decrease inabsorbance was measured for 3 min at 600 nm after theaddition of a mixture of DPIP (4.65 mM), ubiquinone-2(2.5 mM) and mitochondria (1 mg/ml) to the above reactionmixture. Mitochondrial Complex-III (ubiquinol-cytochromec reductase, EC 1.10.2.2) activity was measured by thefollowing rate of reduction of cytochrome c by ubiquinol-2at 30 °C. The reaction was started by the addition ofubiquinol-2 (1 mM) to the reaction mixture containingpotassium phosphate buffer (10 mM, pH 7.4), EDTA(100 mM), cytochrome c (1 mM) and the mitochondria(1 mg/ml). The reduced cytochrome c was measured bymonitoring the increase in absorbance at 550 nm. Mitochon-drial Complex-IV (cytochrome oxidase, EC 1.9.3.1) activitywas measured as the rate of oxidation of reduced cyto-chrome c bymitochondria (1 mg/ml) at 30 °C. The decrease inreduced cytochrome c was monitored at 550 nm.

135B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
4.14. Sample preparation and Western blot analysis forcytochrome c
Brains were harvested to remove hippocampi. Tissues wereweighed and homogenized (10%) in ice cold homogenizationmedium (5 mMHEPES with 0.32 M sucrose, 1 mMMgCl2, 2 mMEGTA, and 0.1 mM PMSF). The homogenates were centrifugedat 700 ×g for 10 min at 4 °C. The pellet (P1) was resuspended inthe same volume of homogenization buffer and the super-natant was centrifuged at 10,000 ×g for 10 min at 4 °C. Theresulting pellet (P2), which consisted of mitochondrial andmembrane, was resuspended in the same volume of homo-genization buffer. The supernatant was removed and ultra-centrifuged at 75,000 ×g for 1 h at 4 °C. The sub-cellular fractionS3, containing the cytosol, was used for the detection ofcytochrome c. Western blot analysis was carried out on 8%sodium dodecyl sulphate-polyacrylamide gel electrophoresis(SDS-PAGE) according to the method of Ouyang et al. (1999)with some modifications. Protein (50 μg) was transferred to anitrocellulose membrane in transfer buffer (15.6 mM Tris,120 mM glycine; pH 8.2) at 30 V for overnight at 4 °C. Themembrane was then blocked with 5% non-fat dried milk for1 h. After three washing with TBS-T (200 mM tris-base, 1.5 MNaCl, 0.1% Tween-20, pH 7.5), the membrane was hybridizedwith primary antibody against cytochrome c (1:1000, mousemonoclonal IgG2b) for 3 h at room temperature. Subsequently,the membrane was then incubated with horseradish perox-idase conjugated secondary antibodies (1:2000; goat antimouse IgG HRP) for 1 h. Blots were developed on X-ray filmusing ECL method (enhanced chemiluminisence reagents,BioRad).
4.15. Evaluation of brain edema
The water content in whole brain was determined by wetweight to dry weight ratios. The whole brain was weighed,dried for 24 h at 105 °C, and re-weighed. The percentage ofwater was calculated according to the method of Demediuk etal. (1990). Results were expressed as μg/gm dried weight usingthe following formula: [%H2O=(wet weight−dry weight)/wetweight×100].
4.16. Protein estimation
Mitochondrial protein concentrations were determined spec-trophotometrically using a BSA as standard by the method ofLowry et al. (1951).
4.17. Neurological severity score
After 22 h of reperfusion, the neurological status of theanimals was evaluated using two different methods: methodA was used as previously described by Bederson et al. (1986).Accordingly, four categories of neurological findings werescored: 0=no observed neurological deficit; 1=contralateralforelimb flexion with wrist flexion and shoulder adduction;2=reduced resistance to lateral push; 3=circling movementstowards the paretic side. In method B, spontaneous motoractivity (SMA) was evaluated for 5 min by placing theanimals in their normal environment (cage). Neurologicalscoring was given as: 0=rats moved around in the cage andexplored the environment; 1=rats moved in the cage but did
not approach to all the sides and hesitated to move; 2=ratsbarely moved in the cage and showed postural abnormal-ities (curved towards the paretic side); 3=rats unable tomove at all with their posture curved towards the pareticside.
4.18. Open field tests (locomotor activity)
Open field tests were done according to the method ofYamamoto et al. (1988) with slight modification at ambienttemperature ranging between 26 and 32 °C. Mean of sessiontotals of vehicle and treatment groups were compared forlocomotion (s), rest (s), distance travelled (cm), average speed(cm/s). Videopath analyser (Coulbourn Instrument, Allen-town, PA, USA) consists of an open field chamber(50×50×35 cm), and a video camera was fixed over thechamber by an adjacent rod, an activity monitor, a program-mer and a printer. Sessions total for all parameters weretaken. Observations were recorded for 10 min. Open field testwas operated and scored by trained and experienced obser-vers who were blind to the condition of the animals. Eachanimal was tested individually and only once.
4.19. Infarct assessment
The animals were euthanized after reperfusion, under chloralhydrate anesthesia followed by decapitation. The brains wererapidly dissected out and the forebrains were cut into sixcoronal sections, 1.5 mm thick, using a rat brain matrix(Activational Systems, MI, USA). The sections were stained byincubating them in a solution of 2% of 2,3,5-triphenyltetrazo-lium chloride (TTC) at 37 °C for 15 min. For imaging, thesections were scanned by a high-resolution scanner (HewlettPackard Scanjet automatic document feeder). The total meaninfarct area of each section was observed by the change incoloration.
4.20. Statistical analysis of data
The statistical analysis of data was done using analysis ofvariance (ANOVA) with post-hoc analysis. The Tukey–Kramerpost-hoc test was applied to serve as significant amonggroups. The significance of results was ascertained atp<0.05. All the data are presented as means±standard error(S.E.) of the means.
R E F E R E N C E S
Allen, K.L., Almeida, A., Bates, T.E., Clark, J.B., 1995. Changes ofrespiratory chain activity in mitochondrial and synaptosomalfractions isolated from the gerbil brain after graded ischemia.J. Neurochem. 64, 2222–2229.
Almeida, A., Allen, K.L., Bates, T.E., Clark, J.B., 1995. Effect ofreperfusion following cerebral ischemia on the activity of themitochondrial respiratory chain in the gerbil brain.J. Neurochem. 65, 1698–1703.
Bederson, J.B., Pitts, L.H., Tsuji, M., Nishimura, M.C., Davis, R.L.,Bartkowski, H., 1986. Rat middle cerebral artery occlusion:evaluation of the model and development of a neurologicexamination. Stroke 17, 472–476.
Buege, J.A., and Aust, S.D., eds 1978. Methods in Enzymology, vol.52: Microsomal Lipid Peroxidation.

136 B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
Cheng, A., Arumugam, T.V., Liu, D., Khatri, R.G., Mustafa, K.,Kwak, S., Ling, H.P., Gonzales, C., Xin, O., Jo, D.G., Guo, Z.,Mark, R.J., Mattson, M.P., 2007. Pancortin-2 interacts withWAVE1 and Bcl-xL in a mitochondria-associated proteincomplex that mediates ischemic neuronal death. J. Neurosci.27, 1519–1528.
Demediuk, P., Lemke, M., Faden, I., 1990. Spinal cord edema andchanges in tissue content of Na, K, and Mg after impact traumain rats. In: Long, D., et al. (Ed.), Advanced. Neurology, Vol. 52.Raven Press, New York, pp. 225–232.
Desai, V.G., Feuers, R.J., Hart, R.W., Ali, S.F., 1996. MPP+ inducedneurotoxicity in mouse is age-dependent: evidenced by theselective inhibition of complexes of electron transport. BrainRes. 715, 1–8.
Floyd, R.A., Carney, J.M., 1992. Free radical damage to proteinand DNA: mechanisms involved and relevant observationson brain undergoing oxidative stress. Ann. Neurol. 32,S22–S27.
Fukui, K., Onodera, K., Shinkai, T., Suzuki, S., Urano, S., 2001.Impairment of learning and memory in rats caused byoxidative stress and aging, and changes in antioxidativedefense systems. Ann. N. Y. Acad. Sci. 928, 168–175.
Fukui, K., Omoi, N.O., Hayasaka, T., Shinnkai, T., Suzuki, S., Abe, K.,Urano, S., 2002. Cognitive impairment of rats caused byoxidative stress and aging, and its prevention by vitamin E.Ann. N. Y. Acad. Sci. 959, 275–284.
Halestrap, A., 2005. Biochemistry: a pore way to die. Nature 434,578–579.
Halestrap, A.P., Davidson, A.M., 1990. Inhibition of Ca2+-inducedlarge-amplitude swelling of liver and heart mitochondria bycyclosporine is probably caused by the inhibitor binding tomitochondrial-matrix peptidyl-prolyl cis-trans isomerase andpreventing it interacting with the adenine nucleotidetranslocase. Biochem. J. 268, 153–160.
Hissin, P.J., Hilf, R., 1976. A fluorometric method for determinationof oxidized and reduced glutathione in tissues. Anal Biochem.74, 214–226.
Hunter, A.J., Mackay, K.B., Rogers, D.C., 1998. To what extent havefunctional studies of ischemia in animals been useful in theassessment of potential neuroprotective agents. TrendsPharmacol. Sci. 19, 59–66.
Ide, N., Lau, B.H., 1997. Garlic compounds protect vascularendothelial cells fromoxidized low density lipoprotein inducedinjury. J. Pharm. Pharmacol. 49, 908–911.
Ide, N., Lau, B.H., 2001. Garlic compounds minimize intracellularoxidative stress and inhibit nuclear factor-kB activation.J. Nutr. 131, 1020S–1026S.
Ito, Y., Ito, M., Takagi, N., Saito, H., Ishige, K., 2003. Neurotoxicityinduced by amyloid beta-peptide and ibotenic acid inorganotypic hippocampal cultures: protection byS-allyl-L-cysteine, a garlic compound. Brain Res. 985, 98–107.
Kim, K.M., Chun, S.B., Koo, M.S., Chio, W.J., Kim, T.W., Kwon, Y.T.,Chung, H.T., Billiar, T.R., Kim, Y.M., 2001. Differential regula-tion of NO availability from macrophages and endothelial cellsby the garlic component S-allyl cysteine. Free Radic. Biol. Med.30, 747–756.
Kim, J.M., Lee, J.C., Chang, N., Chun, H.S., Kim, W.K., 2006.S-Allyl-L-cysteine attenuates cerebral ischemic injury byscavenging peroxynitrite and inhibiting the activity ofextracellular signal-regulated kinase. Free Radic. Res. 40,827–835.
Korde, A.S., Pettigrew, L.C., Craddock, S.D., Pocernich, C.B.,Waldmeier, P.C., Maragos, W.F., 2007. Protective effects ofNIM811 in transient focal cerebral ischemia suggestinvolvement of the mitochondrial permeability transition.J. Neurotrauma 24, 895–908.
Kosuge, Y., Koen, Y., Ishige, K., Minami, K., Urasawa, H., Saito, H.,Ito, Y., 2003. S-allyl-L-cysteine selectively protects cultured rathippocampal neurons from amyloid beta-protein- and
tunicamycin-induced neuronal death. Neuroscience 122,885–895.
Kosuge, Y., Sakikubo, T., Ishige, K., Ito, Y., 2006. Comparative studyof endoplasmic reticulum stress-induced neuronal death in ratcultured hippocampal and cerebellar granule neurons.Neurochem. Int. 49, 285–293.
Lau, B.H.S., 2001. Suppression of LDL oxidation by garlic. J. Nutr.131, 985S–988S.
Li, H.F., Chen, S.A., Wu, S.N., 2000. Evidence of the stimulatoryeffect of resveratrol on Ca-activated K+ current in vascularendothelial cells. Cardiovasc. Res. 45, 1035–1045.
Longa, E.Z., Weinstein, P.R., Carlson, S., Cummins, R., 1989.Reversible middle cerebral artery occlusion withoutcraniectomy in rats. Stroke 20, 84–91.
Lowry, O.H., Rosenbrough, N.J., Farr, A.L., Randall, R.J., 1951.Protein measurement with folin phenol reagent. J. Biol. Chem.193, 265–275.
Moro, M.A., Almeida, A., Bolaños, J.P., Lizasoain, I., 2005.Mitochondrial respiratory chain and free radical generation instroke. Free Radic. Biol. Med. 39, 1291–1304.
Mukherjee, P.K., Ahamed, K.F., Kumar, V., Mukherjee, K.,Houghton, P.J., 2007. Protective effect of biflavones fromAraucaria bidwillii Hook in rat cerebral ischemia/reperfusioninduced oxidative stress. Behav. Brain Res. 178, 221–228.
Nagy, A., Antonio, V., Delgado-Escueta, 1984. Rapid preparation ofsynaptosomes from mammalion brain using nontoxicisoosmotic gradient material (percol) J. Neurochem. 43,1114–1123.
Numagami, Y., Ohnishi, S.T., 2001. S-Allylcysteine inhibits freeradical production, lipid peroxidation and neuronal damage inrat brain ischemia. J. Nutr. 131, 1100S–1105S.
Numagami, Y., Sato, S., Ohnishi, S.T., 1996. Attenuation of ratischemic brain damage by aged garlic extracts: a possibleprotecting mechanism as antioxidants. Neurochem. Int. 29,135–143.
Okabe, E., Tsujimoto, Y., Kobayashi, Y., 2000. Calmodulinand cyclic ADP-ribose interaction in Ca2+ signaling related tocardiac sarcoplasmic reticulum: superoxide anionradical-triggered Ca2+ release. Antioxid. Redox. Signal. 2,47–54.
Ouyang, Y.B., Tan, Y., Comb, M., Liu, C.L., Martone, M.E., Siesjo, B.R., Hu, B.R., 1999. Survival and death-promoting events aftertransient cerebral ischemia: phosphorylation of Akt, release ofcytochrome c, and activation of caspase like proteases. J. Cereb.Blood. Flow Metab. 19, 1126–1135.
Pick, A., Kesari, Y., 1981. Superoxide anion and H2O2 production bychemically elicited peritonealmacrophages-induction bymultiplenon-phagocytic stimulus. Cellular Immunol. 59, 301–308.
Ragan, C.L., Wilson, M.T., Darley-Usmar, M., Lowe, P.N., 1987.Subtractionation of mitochondria, and isolation of the proteinof oxidative phosphorylation. In: Darley-Usmar, V.M.,Rickwood, D., Wilson, M.T. (Eds.), Mitochondria a PracticalApproach. RL Press, London, pp. 79–112.
Sarkar, S., Das, N., 2006. Mannosylated liposomal flavonoid incombating age-related ischemia-reperfusion inducedoxidative damage in rat brain. Mech. Ageing Dev. 127, 391–397.
Shuaib, A., Lees, K.R., Lyden, P., Grotta, J., Davalos, A., Davis, S.M.,Diener, H.C., Ashwood, T., Wasiewski, W.W., Emeribe, U., SaintII, T.I., 2007. NXY-059 for the treatment of acute ischemicstroke. N. Engl. J. Med. 357, 562–571.
Slemmer, J.E., Shacka, J.J., Sweeney, M.I., Weber, J.T., 2008.Antioxidants and free radical scavengers for the treatment ofstroke, traumatic brain injury and aging. Curr. Med. Chem. 15,404–414.
Sohal, R.S., Kamzalov, S., Sumien, N., Ferguson, M., Rebrin, I.,Heinrich, K.R., Forster, M.J., 2006. Effect of coenzyme Q10 intakeon endogenous coenzyme Q content, mitochondrial electrontransport chain, antioxidative defenses, and life span of mice.Free Radic. Biol. Med. 40, 480–487.

137B R A I N R E S E A R C H 1 2 6 5 ( 2 0 0 9 ) 1 2 8 – 1 3 7
Stadtman, E., 1992. Free radicals in the genesis of Alzheimer'sdisease. Ann. N.Y. Acad. Sci. 695, 73–76.
van Leyen, K., Kim, H.Y., Lee, S.R., Jin, G., Arai, K., Lo, E.H., 2006.Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke37, 3014–3018.
Xiong, Z.G., Chu, X.P., Simon, R.P., 2007. Acid sensing ionchannels—novel therapeutic targets for ischemic brain injury.Front. Biosci. 12, 1376–1386.
Yamamoto, M., Tamura, A., Kirino, T., Shimizu, M., Sano, K., 1988.Behavioral changes after focal cerebral ischemia by left middlecerebral artery occlusion in rats. Brain Res 452, 323–328.
Young, W., Wojak, J.C., DeCrescito, V., 1988. 21-Aminosteroidreduces ion shifts and edema in the rat middle cerebral
artery occlusion model of regional ischemia. Stroke 19,1013–1019.
Yousuf, S., Atif, F., Ahmad, M., Hoda, M.N., Khan, M.B., Ishrat, T.,Islam, F., 2007. Selenium plays a modulatory role againstcerebral ischemia-induced neuronal damage in rathippocampus. Brain Res. 1147, 218–225.
Yousuf, S., Atif, F., Ahmad, M., Hoda, M.N., Ishrat, T., Khan, M.B.,Islam, F., 2009. Resveratrol exerts its neuroprotective effect bymodulating mitochondrial dysfunctions and associated celldeath during cerebral ischemia. Brain Res. 1250, 242–253.
Zaheer, N., Tewari, K.K., Krishnan, P.S., 1965. Exposer andsolubilization of hepatic mitochondrial shunt dehydrogenases.Arch. Biochem. Biophys. 109, 646–648.









![Nucleophilic attack at the central allyl carbon atom in [(.eta.3-allyl)ML2]+ complexes (M = palladium, platinum). Experimental facts and new theoretical insights](https://img.pdfslide.net/doc/110x75/6334e53fcd4bf2402c0ad2d8/nucleophilic-attack-at-the-central-allyl-carbon-atom-in-eta3-allylml2-complexes.jpg)



















