Embed Size (px)
Citation preview

ELSEVIER
Life Sciences. Vol. 65, No. 12. pp. 1217-1235, 1999 Copyright 0 1999 Elsevicr Science Inc. Printed in the USA. All rights reserved
m24-3205/99/s-~ fmnt matter
PI1 SOO24-3205(99)00169-l
MIN1REwEw
SELECTIVE SEROTONIN REUPTAKE INHIBITORS AND
NEUROENDOCRINE FUNCTION
Dani K. Raap and Louis D. Van de Kar
Department of Pharmacology, Loyola University Chicago, Stritch
School of Medicine, Maywood, IL 60153, USA
(Received in final form May 12, 1999)
Selective serotonin (WIT) reuptake inhibitors (SSRIs) are effective drugs for the treatment of several neuropsychiatric disorders associated with reduced serotonergic function. Serotonergic neurons play an important role in the regulation of neuroendocrine function. This review will discuss the acute and chronic effects of SSRIs on neuroendocrine function. Acute administration of SSRIs increases the secretion of several hormones, but chronic treatment with SSRIs does not alter basal blood levels of hormones. However, adaptive changes are induced by long-term treatment with SSRIs in serotonergic, noradrenergic and peptidergic neural function. These adaptive changes, particularly in the function of specific post-synaptic receptor systems, can be examined from altered adrenocorticotrophic hormone (ACTH), cortisol, oxytocin, vasopressin, prolactin, growth hormone (GH) and renin responses to challenges with specific agonists. Neuroendocrine challenge tests both in experimental animals and in humans indicate that chronic SSRIs produce an increase in serotonergic terminal function, accompanied by desensitization of post-synaptic 5-HT,, receptor-mediated ACI’H, cortisol, GH and oxytocin responses, and by supersensitivity of post-synaptic 5-HT2, (and/or 5HT,,) receptor-mediated secretion of hormones. Chronic exposure to SSRIs does not alter the neuroendocrine stress-response and produces inconsistent changes in a, adrenoceptor-mediated GH secretion. Overall, the effects of SSRIs on neuroendocrine function are dependent on adaptive changes in specific neurotransmitter systems that regulate the secretion of specific hormones.
Key Words: antidepressants, SSRIs. serotonin, 5-HT. fluoxetine, paroxetine. fluvoxamine, sertraline, ACTH, cortisol, corticosterone, growth hormone, prolactin, oxytocin, vasopressin, renin, stress, hormone
Corresponding author: Louis D. Van de Kar. Ph.D., Department of Pharmacology. Loyola University Chicago, Snitch School of Medicine, 2160 S. First Ave. Maywood, IL 60153, phone: (708) 216-3263, fax: (708) 216-6596. e mail: [email protected]

1218 SSRIs and Neuroendocrine Function Vol. 65, No. 12, 1999
Drugs such as fluoxetine (ProzacB), paroxetine (Paxil@), fluvoxamine (Luvox@), sertraline (Z.oloft@), and citalopram (CelexaB) are selective serotonin (5HT) reuptake inhibitors (SSRIs). The use of SSRIs has revolutionized psychiatry not only because of their effectiveness in treating depression with relatively few side effects, but also because of their ability to alleviate several other disorders associated with serotonergic dysfunction. These disorders include anxiety, obesity, bulimia, aggression, obsessive compulsive disorder, premenstrual syndrome, premature ejaculation, and post- traumatic stress disorder (1-l 1). The SEW-induced inhibition of MIT reuptake increases 5-HT levels in the synaptic cleft, thereby prolonging the activation of post-synaptic 5-HT receptors (12-17). Since 5-HT plays a major role in neuroendocrine function, this review will examine the acute and chronic effects of SSRIs on basal and stimulus-induced changes in hormone levels.
Therapeutic relief in patients taking SSRIs is usually not attained until 2-3 weeks after the beginning of treatment (18,19). Hence, adaptive changes in neural function likely underlie the therapeutic effectiveness of SSRIs. This characteristic delay in the onset of clinical improvement is a major concern since this period is associated with a high risk of suicide (20-22). Neuroendocrine challenge tests (i.e. measuring plasma hormone levels after the acute administration of a challenge drug) can provide an indication of adaptive changes in receptor function in the brain; hence, it may be possible to use these tests to predict which patients will respond to SSRIs (4,23-3 1).
In the following sections, the changes in neuroendocrine function induced by acute and long-term exposure to SSRIs will be outlined. In addition, this review will discuss the use of neuroendocrine challenge tests, in experimental animals and in patients, as a diagnostic tool and as a monitor of neural function in the brain during and after treatment with SSRIs.
Neuroendocrine Pathways
Hypothalamic neurons regulate the secretion of most hormones from both the posterior (neural) and anterior lobes of the pituitary gland. For example, neurons in the hypothalamic paraventricular nucleus contain corticotropin releasing hormone (CRH), oxytocin and vasopressin. The CRH-containing neurons and vasopressin-containing neurons play major roles in the secretion of ACTH from the anterior lobe of the pituitary gland. Other neurons in the paraventricular nucleus are responsible for the secretion of oxytocin, renin and vasopressin (32-34). The secretion of prolactin is regulated by a prolactin releasing factor and by inhibitory dopaminergic neurons in the hypothalamus (35-38). The secretion of GH is regulated by both inhibitory (somatostatin) and stimulatory (GH-RH) hypothalamic peptides.
Hormone release is controlled by hypothalamic neurons, many of which are innervated by serotonergic nerve terminals originating in the midbrain raphe nuclei (Figure 1). Serotonergic neurons send collaterals to several brain areas, including the hypothalamus and limbic areas of the brain such as the amygdala (39-41). For example, one study used double labeled micro-spheres, combined with a retrogradely transported fluorescent tracer, to demonstrate that dorsal raphe serotonergic cells send collaterals to the hypothalamic paraventricular nucleus (PVN) and the central amygdaloid nucleus (40). Thus, changes in serotonergic function in limbic areas could also be detected in hypothalamic neurons that are innervated by collaterals of the same serotonergic neurons (42-45).
Substantial evidence indicates a role for central serotonergic neurons in the regulation of the secretion of ACTH (and the adrenal hormones corticosterone or cortisol), oxytocin, prolactin, GH, renin and vasopressin (46,47). Of the many 5-HT receptor subtypes, only a few have been characterized with respect to their role in neuroendocrine function. The best information available suggests the following
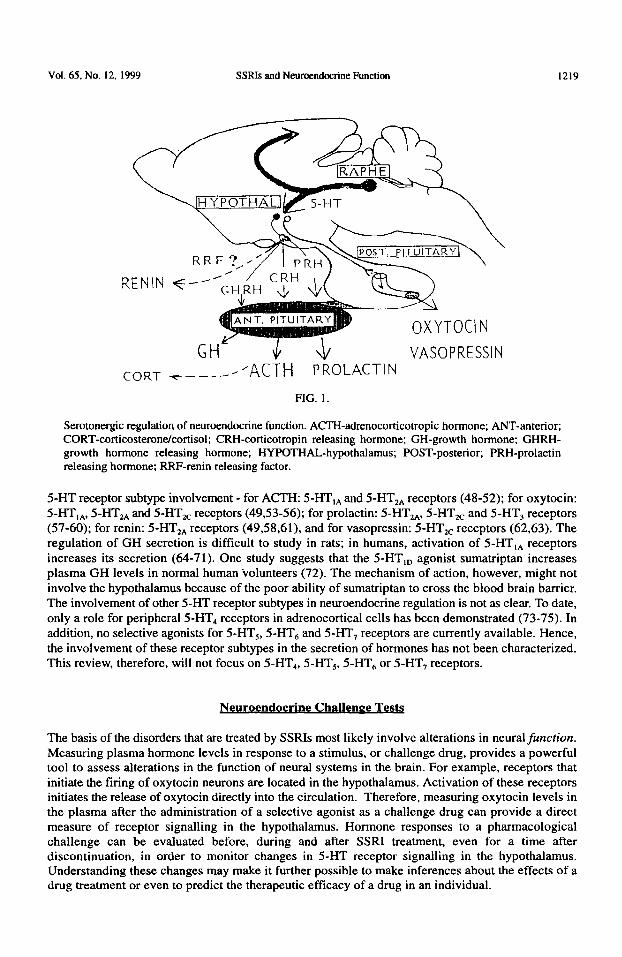
Vol. 65. No. 12, 1999 SSRIs and Neuroendocrine Function 1219
CORT -c-----’ +Cl-H PROLACTIN VASOPRESSIN
FlG. 1.
Serotonergic regulation of neuroendocrine function. ACEI-adrenocorticotropic hormone; ANT-anterior; CORT-corticosteronekortisol; CRH-corticotropin releasing hormone; GH-growth hormone; GHRH- growth hormone releasing hormone; HYPOTHAL-hypothalamus; POST-posterior; PRH-prolactin releasing hormone; RRF-renin releasing factor.
5-HT receptor subtype involvement - for ACTH: 5-HT,, and 5-HT,, receptors (48-52); for oxytocin: 5-I-IT,,, 5-HT,, and 5-HT,, receptors (49,53-56); for prolactin: 5-HT,,, 5-HT,, and 5-HT, receptors (57-60); for renin: 5-HT,, receptors (49,58,61), and for vasopressin: 5-HTzc receptors (62,63). The regulation of GH secretion is difficult to study in rats; in humans, activation of 5-HT,, receptors increases its secretion (64-71). One study suggests that the 5-HT,, agonist sumatriptan increases plasma GH levels in normal human volunteers (72). The mechanism of action, however, might not involve the hypothalamus because of the poor ability of sumatriptan to cross the blood brain barrier. The involvement of other 5-I-IT receptor subtypes in neuroendocrine regulation is not as clear. To date, only a role for peripheral 5-HT4 receptors in adrenocortical cells has been demonstrated (73-75). In addition, no selective agonists for 5-HT,, 5-HT, and 5-HT, receptors are currently available. Hence, the involvement of these receptor subtypes in the secretion of hormones has not been characterized. This review, therefore, will not focus on 5-HT,, 5-HT,, 5-HT, or 5-HT, receptors.
The basis of the disorders that are treated by SSRIs most likely involve alterations in neural function.
Measuring plasma hormone levels in response to a stimulus, or challenge drug, provides a powerful tool to assess alterations in the function of neural systems in the brain. For example, receptors that initiate the firing of oxytocin neurons are located in the hypothalamus. Activation of these receptors initiates the release of oxytocin directly into the circulation. Therefore, measuring oxytocin levels in the plasma after the administration of a selective agonist as a challenge drug can provide a direct measure of receptor signalling in the hypothalamus. Hormone responses to a pharmacological challenge can be evaluated before, during and after SSRI treatment, even for a time after discontinuation, in order to monitor changes in 5-HT receptor signalling in the hypothalamus. Understanding these changes may make it further possible to make inferences about the effects of a drug treatment or even to predict the therapeutic efficacy of a drug in an individual.

1220 SSRls and Neuroe.ndocrine Function Vol. 65. No. 12. 1999
The use of neuroendocrine challenge tests does have limitations. Hormone release is controlled by many factors and care must be taken with interpretation of the results. These confounding factors can include circadian rhythms of hormones, the stress of the individual at the time of the test, individual variations in basal hormone levels, possible undiagnosed endocrine disorders, and the specificity of the challenge drug. Ways of overcoming some of these variables include obtaining multiple time points for each individual, measuring several different hormones, and administering a placebo (70).
The choice of stimulus is a major factor in determining which neurotransmitter systems are altered and how they are altered both before and after sustained exposure to SSRIs. Challenge drugs can act presynaptically to induce the release of an endogenous neurotransmitter, which will activate post- synaptic receptors and lead to hormone release. Alternatively, the challenge drug can directly activate post-synaptic receptors to induce hormone release (70).
By virtue of their ability to inhibit the reuptake of 5-HT, SSRIs elevate the extracellular levels of M-IT in the synapse. Consequently, the activation of post-synaptic 5-HT receptors in the hypothalamus stimulates the secretion of several hormones. Indeed, several studies using rats have indicated that a single injection of fluoxetine increases plasma levels of ACTH or corticosterone (76-78). In addition, fluoxetine was reported to increase the secretion of growth hormone (GH) in young rat pups (79). Acute administration of fluoxetine and paroxetine also increases plasma cortisol levels in humans (80,81). Increases in prolactin release, both in humans and in rats have been noted after acute administration of citalopram and fluvoxarnine (82-84). Fluoxetine alone does not seem to consistently increase prolactin secretion (85-87). Little information is available regarding the acute effects of SSRIs on the secretion of oxytocin and renin, and some studies suggest that SSRIs increase the secretion of vasopressin (88-91). In addition, human studies on the acute effect of intravenously injected citalopram indicate an increase in plasma cortisol and prolactin (82,83).
SSRIs are substantially less efficacious in increasing plasma levels of hormones than 5-HT releasing drugs such as d-fenfluramine (92-96). One explanation for this weak neuroendocrine response to an
POST. PITUITARY
FIG. 2.
A proposed mechanism of action for selective serotonin reuptake inhibitors on somatodendritic and post- synaptic S-HT,, receptors.

Vol. 65, No. 12, 1999 SSRIs and Neuroendocrine Function 1221
acute administration of SSRIs is that, by blocking 5-HT reuptake in the cell body region in the raphe, they subsequently activate somatodendritic 5-HT,, autoreceptors. These 5-HT,, autoreceptors in the raphe nuclei provide negative feedback inhibition of serotonergic tiring (Figure 2). Therefore, the tendency of SSRIs to increase the levels of 5-HT in the synapse are negated by activation of the negative feedback, leading to reduced release of 5-HT in the forebrain. Consequently, less activation of post-synaptic 5-HT receptors occurs and a less robust neuroendocrine response to acute administration of SSRIs is observed.
While a single injection of SSRIs alone may have a weak stimulatory effect on the secretion of hormones, SSRIs can be combined with 5-HT precursors to potentiate their effect. Studies using rats provided early evidence that combined administration of 5-hydroxytryptophan (5-HTP) with fluoxetine increases the secretion of both corticosterone and prolactin (76,85,87). Because fenfluramine and its d-isomer have been recently banned from the market in the U.S., there is a need to find new pre-synaptic challenges in biological psychiatry. Combining SSRIs with low doses of 5- HTP may substitute for d-fenfluramine.
Chronic Effects of SSR&
Basal Hormone Levels
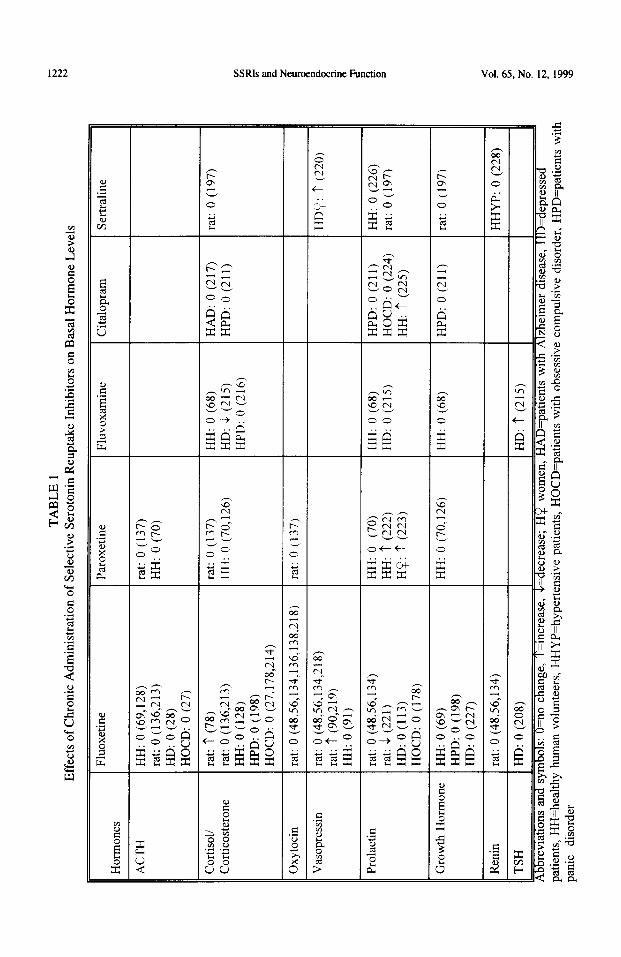
Several studies have measured basal hormone levels in rats, in healthy human volunteers, and in patients before and after repeated exposure to SSRIs (Table 1). In general, no consistent alterations in basal levels of any hormone were observed after long-term exposure to SSRIs, particularly in rats or in healthy human volunteers. Reports about basal levels in patients are inconsistent, partly because the superimposition of SSRI treatment on the treated disorder could result in great variations in basal levels. Such variations are particularly conspicuous in the hypothalamic-pituitary-adrenal axis which is altered in many depressed patients (97-99).
The homeostatic regulation of hormone levels involves complex interactions among several systems, including multiple neurotransmitters and several feedback loops (47,66,100-107). Changes in hormone levels due to dysfunction in one system associated with a particular disorder, or due to the repeated administration of a drug, are commonly compensated for by other brain or feedback mechanisms to maintain basal hormone levels within a narrow homeostatic range. Hence, measurement of basal hormone levels would not be expected to serve as a useful tool for examining altered function of a particular neural system before, during or after treatment with SSRIs.
Presynaptic Challenges
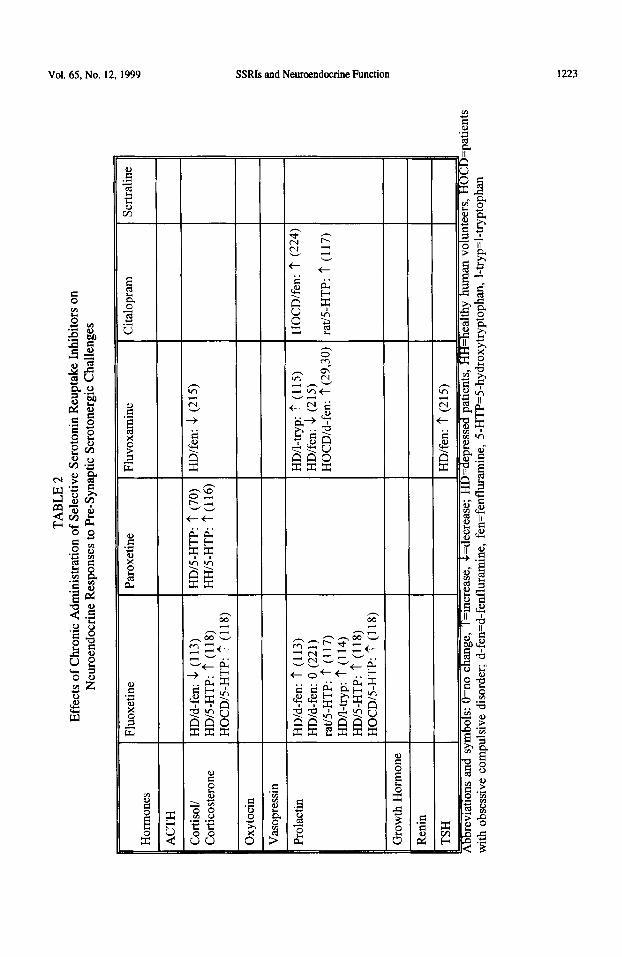
The function of serotonergic nerve terminals can be examined with the use of challenge drugs that act to increase levels of S-I-IT in the synaptic cleft, thereby inducing a subsequent release of hormones into the circulation (Table 2). The challenges that have been used in SSRI research to induce the release of hormones include (a) serotonin precursors, e.g. 5-hydroxytryptophan (5-HTP) or I-tryptophan, which increase the synthesis (and thereby the concentration) of 5-HT in the nerve terminals (70,108-l 11). (b) SSRIs themselves (such as intravenously injected citalopram (82)) which, by blocking the reuptake of 5-I-IT into the nerve terminal, induce an increase of 5-HT in the synaptic cleft, and (c) 5-I-IT releasing drugs like p-chloroamphetamine (used in animals only), fenfluramine and its active isomer d-fenfluramine, that are taken up through the serotonin transporter into the nerve terminal and induce the release of 5-HT ( 112,113).
Studies using 5-HTP or I-tryptophan challenges have indicated a potentiated cortisol and prolactin response in patients and in rats after long-term SSRI treatment (70,114-l 18). As SSRIs block 5-HT reuptake, it would follow that administration of a precursor would result in a build up of 5-HT in the
Hor
mon
es
TA
BL
E
1 E
ffec
ts
of C
hron
ic
Adm
inis
trat
ion
of S
elec
tive
Sero
toni
n R
eupt
ake
Inhi
bito
rs
on B
asal
H
orm
one
Lev
els
AC
TH
Cor
tisol
i C
ortic
oste
rone
Oxy
toci
n
Vas
opre
ssin
Prol
actin
Gro
wth
H
orm
one
Ren
in
TSH
bbre
vlat
ions
an
d S
J
Fluo
xetin
e Pa
roxe
tine
HH
: 0
(69,
128)
ra
t: 0
(136
,213
) H
D:
0 (2
8)
HO
CD
: 0
(27)
rat:
‘? (
78)
rat:
0 (1
36,2
13)
HH
: 0
(128
) H
PD:
0 (1
98)
HO
CD
: 0
(27,
178,
214)
rat:
0 (1
37)
HH
: 0
(70)
rat:
0 (1
37)
HH
: 0
(70,
126)
rat:
0 (4
8,56
,134
,136
,138
,218
) ra
t: 0
(137
)
rat:
0 (4
8,56
,134
.2
18)
rat:
? (9
0,21
9)
HH
: 0
(91)
rat:
0 (4
8,56
,134
) ra
t: -1
(221
) H
D:
0 (1
13)
HO
CD
: 0
(178
)
HH
: 0
(70)
H
H:
t (2
22)
HP:
t
(223
)
HH
: 0
(69)
H
PD:
0 (1
98)
HD
: 0
(227
)
HH
: 0
(70,
126)
rat:
0 (4
8,56
,134
)
HD
: 0
(208
)
nbol
s:
O=n
o ch
ange
, I =
mcr
ease
, ,
rdec
reas
e;
H!#
wom
en,
Fluv
oxam
ine
Cita
lopr
am
HH
: 0
(68)
H
D:
& (
215)
H
PD:
0 (2
16)
HA
D:
0 (2
17)
HPD
: 0
(211
)
HH
: 0
(68)
H
D:
0 (2
15)
HPD
: 0
(211
) H
OC
D:
0 (2
24)
HH
: t
(225
)
HH
: 0
(68)
HD
: t
(215
)
IAD
=pat
ient
s w
ith
Alz
heim
er
dise
ase,
I
Sert
ralin
e
rat:
0 (1
97)
HD
i;:
? (2
20)
HH
: 0
(226
) ra
t: 0
(197
)
rat:
0 (1
97)
HH
YP:
0
(228
)
)=de
pres
sed
patie
nts,
H
H=h
ealth
y hu
man
vo
lunt
eers
, H
HY
P=hy
pert
ensi
ve
patie
nts,
H
OC
D=p
atie
nts
with
ob
sess
ive
com
puls
ive
diso
rder
, H
PD=p
atie
nts
with
pa
nic
diso
rder
Vol. 65. No. 12. 1999 SSRIs and Neuroemdocrine Function 1223

1224 SSRls and Neuroendocrine Function Vol. 65. No. 12, 1999
synaptic cleft, consequently increasing the secretion of hormones. A severe deficiency in serotonergic nerve terminal function (e.g. reduced expression of synthesizing enzymes) might prevent I-tryptophan from elevating the levels of 5-HT in the nerve terminals and thus plasma hormone levels. Since differences exist in the pharmacokinetics of SSRIs, as well as variations from one individual to another, it would be difficult to interpret the resulting elevation in hormone levels. However, the use of I-tryptophan in humans was discontinued in the U.S. in 1990; 5-HTP is still available as a presynaptic challenge.
Another question associated with this indirect strategy, upon discovering altered neuroendocrine responses, is where in the neurotransmission and/or signalling process the alterations may occur. The elevation in synaptic levels of 5-HT can lead to activation of several 5-HT receptor subtypes, increasing the secretion of several hormones. It is not likely that one could determine with certainty which 5-HT receptor system(s) was (were) altered during exposure to SSRIs by examining the hormone responses to 5-HT precursors. Therefore, follow-up studies would be necessary in which challenges with specific 5-HT receptor agonists would determine whether the SSRI-induced alterations involve only nerve terminal function or changes in post-synaptic 5-HT receptor subtypes.
An acute challenge with a SSRI, such as the intravenous challenge with 20 mg citalopram, has advantages as a diagnostic tool before treatment with SSRIs (82,83). The specificity of such a drug is dependent on its high affinity for the 5-HT transporter. However, it is less likely that it will be of use as a diagnostic tool during treatment with SSRIs or for the duration that SSRIs, or their active metabolites, remain in the brain to inhibit 5-HT uptake. As the 5-HT transporters are already blocked by SSRIs, any additional challenge with a SSRI is not likely to produce a further inhibition of 5-HT reuptake.
The use of 5-HT releasing drugs (e.g. d-fenfluramine) requires an intact 5-HT uptake mechanism to exert their effect. These drugs can be used prior to the beginning of treatment with a SSRI. However, these drugs can not be used reliably during SSRI treatment because they can not enter the nerve terminals if the transporters are blocked. Furthermore, a challenge with 5-HT releasing drugs after discontinuation of treatment should be delayed until the SSRI and any active metabolite(s) have been cleared from the system. This issue is of particular concern with SSRIs that have a long half-life and active metabolites, such as fluoxetine. Finally, the only 5-HT releasers that were approved for use in humans, d-fenfluramine and d,l-fenfluramine, have recently been pulled from the shelves in the U.S. No substitutes are currently approved for use in humans in the U.S.
Post-Synaptic Challenges
A direct means of assessing receptor system function in the hypothalamus is by administering selective agonists that activate post-synaptic receptor signalling to directly induce the release of hormones. This strategy can be a powerful tool to investigate alterations in specific receptor systems induced by chronic SSRI exposure. Most studies, so far, have focused either on post-synaptic 5-HT,, or 5-HT,,,, receptor systems. Because these 5-HT receptors are linked through G proteins to second messenger enzymes, activation of each receptor leads to the secretion of multiple molecules of oxytocin or CRH. CRH receptors also are linked via G proteins to effector enzymes. Hence, activation of each CRH receptor on corticotrophs in the pituitary will lead to release of multiple molecules of ACTH, which can further stimulate the release of even more molecules of corticosterone (or cortisol in humans). This amplification of responses means that these hormones can be used as highly sensitive peripheral markers of the overall functional status of particular receptor systems in the hypothalamus. It should be noted that corticosterone has been shown to alter the levels of several G proteins in the brain (119,120). Such changes could thereby modify the response of the hypothalamic-pituitary-adrenal axis to pharmacological challenges. These complex interactions provide further support for the necessity of measuring several different hormones when using neuroendocrine challenge tests.

Vol. 65, No. 12, 1999 SSRIs and Neurcendocrine Function 1225
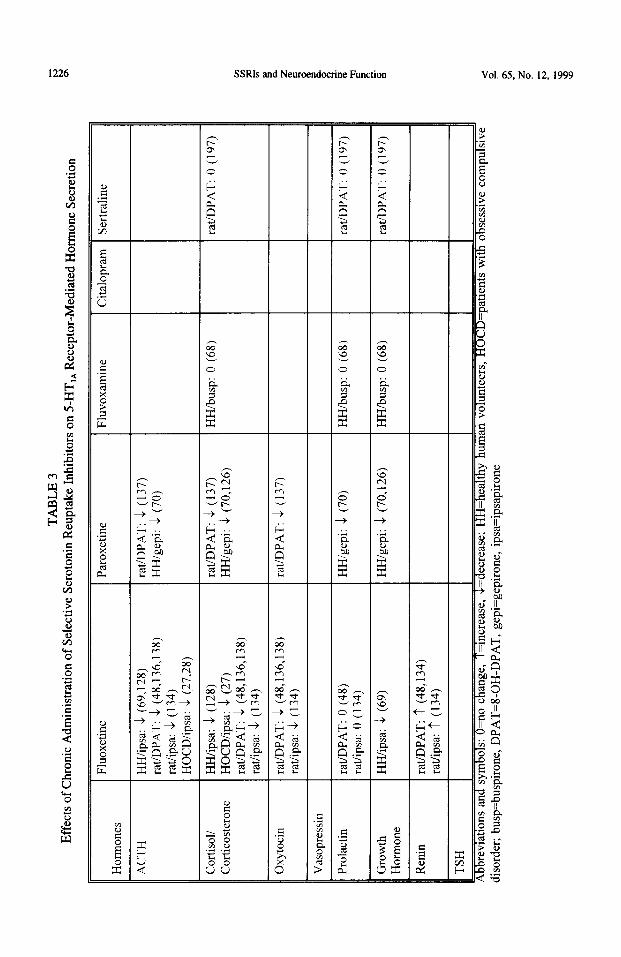
S-HT,, receptors. Several 5-HT,, receptor agonists that are available for use in humans (flesinoxan, buspirone, ipsapirone, gepirone and tandospirone) and in animals (8-OH-DPAT and alnespirone) induce the release of ACTH, cortisol, corticosterone, oxytocin, GH and prolactin (27,28,48,51,53,64,65,69,70,121-131). The ACTH, corticosterone, cortisol and oxytocin responses to 5-HT,, agonists can be prevented by 5-I-IT,, antagonists such as WAY-100635, pindolol, spiperone and NAN-190 (48,53,71,121,129-138). Activation of 5-HT,* receptors also increases the secretion of GH in humans, while this phenomenon is not seen in rats with consistency (28,65-70,126).
The prolactin response to 5-HT,, agonists is not likely mediated by activation of 5-HT,, receptors. In humans, buspirone and gepirone increase plasma prolactin levels, but ipsapirone and tandospirone do not increase plasma prolactin levels (28,64,121,122,125). In rats, gepirone and alnespirone do not increase plasma prolactin levels while 8-OH-DPAT and ipsapirone produce a very short-lived increase which, unlike ACTH and oxytocin, disappears within 30 minutes (67,139-145). The antagonists WAY- 100635 and pindolol do not consistently inhibit the prolactin responses to several agonists, including 8-OH-DPAT, alnespirone, flesinoxan and buspirone (71,125,129,130,146). Thus, the most reliable neurocndocrine markers of central 5-I-IT,, receptor signalling in humans are ACIH, cortisol, GH and, likely, oxytocin.
In healthy humans, in patients with mood disorders and in rats, repeated exposure to SSRIs reduces the ACTH, cortisol/corticosterone, oxytocin (in rats) and GH (in humans) responses to several 5-HT,, agonists. These agonists include buspirone, ipsapirone, gepirone and (in rats) 8-OH-DPAT (Table 3). These observations suggest a desensitization of post-synaptic 5-HT,, receptors in the hypothalamus. This desensitization develops gradually in rats over 2 weeks of repeated exposure to SSRIs (136.137), a time course which is similar to the development of therapeutic relief in patients. The time-course studies using rats indicate that SSRIs produce a partial 5-HT,, receptor-mediated desensitization of ACTH and oxytocin responses to 8-OH-DPAT after 3 daily injections. The maximum desensitization occurs after 7-14 daily injections (136,137).
In rats, the effects of fluoxetine are dose-dependent (5-10 mgikg/day), but the dose producing maximal desensitization of hypothalamic 5-HT,, receptor signalling is 10 mg/kg/day (138). In humans, much lower fluoxetine doses (about 0.3-l mg/kg/day) are required to produce 5-HT,, receptor desensitization (69). The difference in doses is likely due to differences in the pharmacokinetics of fluoxetine between rats and humans. In rats, the half-life after a 10 mg/kg dose of fluoxetine is 7.7 hours for the parent drug and 15.8 hours for its pharmacologically active metabolite, norfluoxetine (147). In contrast, the half-life of fluoxetine in humans is 4 to 6 days, and of norfluoxetine is 4 to 16 days (148). Since humans take fluoxetine (20-60 mg) daily, the accumulation of fluoxetine and norfluoxetine in their brain could be as high as it would be with higher doses in rats.
Termination of fluoxetine administration to rats is followed by a very slow recovery of function of hypothalamic 5-I-IT,, receptor systems. Even 60 days after discontinuation of fluoxetine, the oxytocin response to the S-OH-DPAT was still significantly reduced by 26% from controls (149). In contrast, the suppressed ACTH response to 8-OH-DPAT (a less direct indicator of desensitization) gradually returned to control levels by day 14 of withdrawal from fluoxetine (149).
The exact site in the 5-HT,, receptor system that is altered during exposure to SSRIs remains to be examined. No change was observed in the density of 5-HT,, receptors, but a decrease in the levels of G, protein occurs during exposure to SSRIs (137,138,150-152). G, proteins mediate the ACTH and oxytocin responses to 5-HT,, agonists (153,154). Thus, these observations suggest that chronic exposure to SSRIs reduces the coupling of 5-I-D”,,, receptors to their effector mechanisms. Supporting this conclusion is the observation that 5-I-IT,* receptor desensitization in rats is manifested by a shift to the right in the dose-response curve of 8-OH-DPAT, with no decrease in the maximal hormone response to the highest doses (48). These observations also suggest that studies employing agonist
TA
BL
E
3 E
ffec
ts
of C
hron
ic
Adm
inis
trat
ion
of S
elec
tive
Sero
toni
n R
eupt
ake
Inhi
bito
rs
on 5
HT
,, R
ecep
tor-
Med
iate
d H
orm
one
Secr
etio
n
- Fl
uoxe
tine
Hor
mon
es
AC
TH
Cor
tisol
l C
ortic
oste
rone
Oxy
toci
n
Paro
xetin
e
HH
/ipsa
: -1
(69
.128
) ra
t/DPA
T:
k (4
8,13
6,13
8)
rat/i
psa:
&
(13
4)
HO
CD
/ipsa
: k
(27,
28)
HH
/ipsa
: &
(12
8)
HO
CD
iipsa
: k
(27)
ra
t/DPA
T:
& (
48,1
36,1
38)
rat/i
psa:
k
(134
)
rat/D
PAT
: -1
(48
,136
,138
) ra
t/ips
a:
k (1
34)
rat/D
PAT
: $
(137
) H
H/g
epi:
k (7
0)
rat/D
PAT
: &
(13
7)
HH
/gep
i: 4
(70,
126)
rat/D
PAT
: 4
(137
)
rat/D
PAT
: 0
(48)
ra
t/ips
a:
0 (1
34)
HH
/ipsa
: -1
(69
)
rat.D
PAT
: ‘?
(48
,134
) ra
t/ips
a:
t (1
34)
HH
igep
i: 1
(70)
HH
/gep
i: -1
(70
,126
)
I’ I
Abb
revi
atio
ns
and
sym
bols
: O
=no
chan
ge,
T=m
crea
se,
&=
decr
ease
; H
H=h
ealth
y hu
n di
sord
er;
busp
=bus
piro
ne,
DPA
T=I
-OH
-DPA
T,
gepi
=gep
iron
e,
ipsa
=ips
apir
one
Fluv
oxam
ine
/ C
italo
pram
1
Sert
ralin
e
n vo
lunt
eers
, H
OC
D=p
atie
nts
with
ob
sess
we
com
puls
iv

Vol. 65. No. 12. 1999 SSRls and Neumendocrine Function 1221
doses substantially above the ED, dose might provide misleading information on changes in 5-HT,, receptor function. Taken together, these studies suggest that adaptive changes resulting in desensitization of post-synaptic S-HT,, receptors may be involved in the mechanisms that underlie the therapeutic activity of SSRIs for several neuropsychiatric disorders.
5-HTmc receptors. Activation of 5-HT,, and/or 5-HT,, receptors increases the secretion of ACTH, corticosterone, cortisol, prolactin, oxytocin and renin (53,54,59,61,135,155-159). In addition, 5-HT,, receptor activation also increases the secretion of vasopressin in rats, while the effects in humans are not clear (62,63,160-165). The agonists most commonly used in humans are m-CPP and MK-212, while quipazine has been tested in a few studies (70,124,166- 179). These drugs have been considered to have a relatively low affinity for 5-HT,, receptors, although recent studies in NIH-3T3 cells suggest that m-CPP has a moderate affinity for agonist-labeled 5-HT,, receptors (180). Because MK-212 and m-CPP have been considered for a long time as 5-HT,, agonists, many investigators interpreted their hormone responses to either drug as peripheral markers of activated 5-HTzc receptors. However, recent data in rats indicate that the selective 5-HT,, antagonist MDL 100,907 dose-dependently blocks the effects of MK-212 and quipazine on plasma corticosterone levels, suggesting an exclusive role of 5-HTzA receptors in mediating the corticosterone responses to these two agonists (52). MDL 100,907 at very low doses (lo-100 pg/kg, SC) and the less specific 5-HT,, antagonist spiperone (100 &kg SC) block the effect of DO1 on the secretion of ACTH, corticosterone, oxytocin, prolactin and renin in rats (54,61,157, Javed, et al., Sot. Neurosci. Abstracts, 1999). This effect of the 5-HTlNvzc agonist DO1 on the hypothalamic-pituitary-adrenal axis and on oxytocin, prolactin and renin also is mediated by 5- HT,, receptors. To our knowledge, no comparable studies with MDL 100,907 have been undertaken with m-CPP. Consequently, it is still possible that the hormone responses to m-CPP are mediated by 5-HT,, receptors.
Few studies have described the influence of chronic exposure to SSRIs on the neuroendocrine response to 5-HT,,,, agonists, particularly when compared with the amount of data using 5-HT,, agonist challenges. The composite data suggest that long-term exposure of rats to fluoxetine increases the sensitivity or maximal responsiveness of ACTH, oxytocin, and, to a lesser extent, prolactin release after challenges with DO1 and MK-212 (56). The vasopressin response to MK-212 also was potentiated by fluoxetine, while DO1 did not increase plasma vasopressin levels either in saline or fluoxetine treated rats (56). Consistent with these observations, treatment of patients suffering from obsessive compulsive disorder with fluoxetine potentiated the prolactin and cortisol responses to m- CPP (178). On the other hand, treatment of healthy human volunteers with paroxetine (30 mg/day, 3 weeks) reduced the prolactin response to m-CPP (70,18 1). The differences between the two studies in humans could be ascribed to the different drugs used (paroxetine vs fluoxetine) or to the different condition of the humans tested (healthy vs depressed). With very few exceptions, most other molecular/biochemical markers of 5-HT,, and/or 5-HT, receptors support increased sensitivity and/or density of 5-HT?,, receptors after SSRI treatments (mostly fluoxetine) (182-l 89).
Other 5-HT receptors. Sumatriptan is a 5-HTlI, agonist with poor penetration of the blood-brain barrier, and is clinically useful in the treatment of migraine headaches. Administration of sumatriptan as a challenge has had mixed results. Some, but not all studies suggest that sumatriptan reduces plasma levels of prolactin, increases plasma levels of GH and does not alter the hypothalamic-pituitary adrenal axis (72,190- 193). Furthermore, treatment of healthy human volunteers with paroxetine (20 mg/day) for 16 days did not alter the effect of sumatriptan in reducing the secretion of prolactin (191). The interpretation of these data needs to await further characterization of the exact mechanism and site of action of sumatriptan on the secretion of hormones.
a, Adrenoceptors. Inconsistent changes in hormone responses to a2 adrenoceptor agonists are observed after SSRI treatment. Chronic treatment of rats with fluoxetine reduced the GH response to the a2 adrenoceptor agonist clonidine (194). Another study suggested that treatment of fawn hooded rats with

1228 SSRIs and Neuroendocrine Function Vol. 65, No. 12, 1999
fluoxetine (2.5 mg/kg/day) for I6 days potentiated the GH response to a clonidine challenge (195). However, this dose of lluoxeline is too low to produce a sustained inhibition of .5-HT uptake (138,196). Treatment of rats with sertraline (7.5 or 10 mg/kg/day) for 21 days had little effect on the GH response to clonidine (50 &kg) (197). In humans suffering from panic disorder, the GH response to clonidine is blunted when compared with control human subjects. Treatment with fluoxetine for 12 weeks did not restore the blunted GH response to clonidine in spite of clinical improvement (198). Finally, depressed patients treated with fluoxetine and challenged with the norepinephrine uptake inhibitor desipramine exhibited a blunted GH response. This latter effect, although it could be mediated by post-synaptic ~1~ receptors, also reflects released norepinephrine which could be altered by chronic treatment with fluoxetine. Patients suffering from panic disorder showed a greater effectiveness of clonidine to suppress plasma cortisol levels than a control group. Treatment of these patients with fluoxetine, although producing moderate improvement, did not restore the cortisol response to clonidine to levels observed in the control group (199). Taken together, the data so far do not support a major role of 01~ adrenoceptors in the adaptive changes induced by SSRIs.
Other challenges. Thyrotropin releasing hormone (TRH) can be used as a stimulus to increase the secretion of prolactin (200-206). In depressed patients suffering from anger attacks, treatment with fluoxetine increased the prolactin response to the TRH challenge (207). In another study, fluoxetine treatment did not alter the thyroid stimulating hormone (TSH) response to a TRH challenge (208). There is a lack of insight into the exact mechanism through which TRH regulates the secretion of prolactin. Consequently, it is not clear how these observations can be used to diagnose adaptive changes in brain neural function occurring during SSRI treatment.
Stress-Induced Hormone Release
Most of the hormones discussed in this review are considered “stress hormones.” These hormones play an important role in survival mechanisms during exposure to adverse conditions and are therefore secreted during such conditions (209). While SSRIs are prescribed for anxiety and panic disorders, little information is available regarding their effects on stress-induced release of hormones. Daily injections of rats with fluoxetine (5 mg/kg/day) for 21 days did not alter the corticosterone response to forced swim stress (210). Recently, our laboratory has demonstrated that daily injections of fluoxetine (10 mg/kg/day) to rats for 14 days did not alter the effect of conditioned fear stress on the secretion of ACTH, corticosterone, oxytocin, prolactin or renin (Zhang et al., manuscript in preparation). Patients who suffer from panic disorder often respond adversely (with increased panic episodes) to a challenge with cholecystokinin (CCK4). Treatment of patients who suffer from panic disorder with citalopram for 8 weeks showed improvement within 2 weeks. Yet, the ability of CCK4 to stimulate the release of several hormones (GH, prolactin, cortisol) and increase cardiovascular responses was not altered by citalopram treatment (211). Similarly, treatment of patients with fluvoxamine, for 8 weeks, did not alter the cortisol response to anxiety induced by the CQ adrenoceptor antagonist yohimbine (iv) (212). Combined, these data suggest that the neuroendocrine stress responses are not altered by chronic SSRI treatment. The importance of hormones such as corticosterone/cortisol and renin for survival probably requires multiple neurotransmitter mechanisms to mediate the effects of stress on the secretion of these hormones. It is unlikely that all these neural mechanisms would be altered in a similar manner by SSRIs.
Conclusions
Acute administration of selective serotonin reuptake inhibitors may produce only minor alterations in neuroendocrine regulation. After chronic administration, however, SSRIs induce adaptive changes in 5HT and other receptor systems. While there are negligible effects on basal levels of hormones, long-

Vol. 65, No. 12, 1999 SSRIs and Neuroendocrine Function 1229
term exposure to SSRIs initiates a cascade of events that leads to changes in receptor signalling. These alterations are easily detected by measuring stimulus-induced hormone release. The most consistent changes observed are desensitization of the 5HT,, receptor-mediated hormone responses and a potentiation of the 5HT,, receptor-mediated hormone responses.
Using neuroendocrine challenge tests in psychiatric patients may provide a tool for understanding the mechanisms underlying the disorders. These tests may also be a means for investigating the mechanisms involved in the adaptive changes produced by long-term SSRI exposure. More importantly, neuroendocrine challenge tests may allow physicians to predict whether a patient will respond to SSRIs without having to wait several weeks after the initiation of treatment.
1.
2. 3. 4.
5.
6.
7. 8.
9. 10.
11.
12. 13.
14. 1.5. 16. 17. 18. 19.
20.
21.
22. 23.
24. 25. 26. 27.
SM. HAENSEL, T.M.A.L. KLEM, W.C.J. HOP and A.K. SLOB, J. Clin. Psychopharmacol. 18 72-77 (1998). S.C. KIM and K.K. SEO, J. Urol. 159 425-427 (1998). E.F. COCCARO, R.J. KAVOUSSI and R.L. HAUGER, Biol. Psychiatry 42 546-552 (1997). J.D. COPLAN, L.A. PAPP, D. PINE, J. MARTINEZ, T. COOPER, L.A. ROSENBLUM, D.F. KLEIN and J.M. GORMAN, Arch. Gen. Psychiatry 54 643-648 (1997). T. PEARLSTEIN, K. ROSEN and A.B. STONE, Endocrinol. Metabol. Clin. North Am. 26 279-294 (1997). V. PEREZ, I. GILABERTE, D. FARIES, E. ALVAREZ and F. ARTIGAS, Lancet 349 1594-I 597 (1997). K.A. YONKERS, J. Clin. Psychiatry 58 (suppl. 14) 4-l 3 (1997). N.H. DODMAN, R. DONNELLY, L. SHUSTER, P. MERTENS, W. RAND and K. MICZEK, J. Am. Vet. Med. Assoc. 209 1585- 1587 (1996). C.G. GREEN0 and R.R. WING, American Journal of Clinical Nutrition 64 267-273 (1996). D.J. GOLDSTEIN, M.G. WILSON, V.L. THOMPSON, J.H. POTVIN, A.H. RAMPEY, JR. and FLUOXETINE BULIMIA NERVOSA RES GRP, Br. J. Psychiatry 166 660-666 (1995). L.C. BARR, W.K. GOODMAN, C.J. MCDOUGLE, P.L. DELGADO, G.R. HENINGER, D.S. CHARNEY and L.H. PRICE, Arch. Gen. Psychiatry 51309-3 17 (1994). T. SHARP, V. UMBERS and S.E. GARTSIDE, Br. J. Pharmacol. 121941-946 (1997). A.M. GARDIER, I. MALAGIE, AC. TRILLAT, C. JACQUOT and F. ARTIGAS, Fundam. Clin. Pharmacol. 10 16-27 (1996). S. HJORTH, H.J. BENGTSSON and S. MILANO, Eur. J. Pharmacol. 316 43-47 (1996). C. MORET and M. BRILEY, Eur. J. Pharmacol. 295 189-197 (1996). S.B. AUERBACH and S. HJORTH, Naunyn Schmiedebergs Arch. Pharmacol. 352 597-606 (1995). D.S. KREISS and I. LUCKI, J. Pharrnacol. Exp. Ther. 274 866-876 (1995). F. ARTIGAS, L. ROMERO, C. DE MONTIGNY and P. BLIER, Trends Neurosci. 19 378-383 (1996). F.M. QUITKIN, P.J. MCGRATH, J.W. STEWART, B.P. TAYLOR and D.F. KLEIN, Neuropsychopharmacology 15 390-394 (1996). R. GROSS-ISSEROFF. A. BIEGON, H. VOET and A. WEIZMAN, Neurosci. Biobehav. Rev. 22 653-661 (1998). R.J. VERKES, R.C. VAN DER MAST, M.W. HENGEVELD, J.P. TUYL, A.H. ZWINDERMAN and G.M.J. VAN KEMPEN, Am. J. Psychiatry 155 543-547 (1998). C.A. STOCKMEIER, Ann. NY Acad. Sci. 836 220-232 (1997). A.J. CLEARE, R.M. MURRAY, R.A. SHERWOOD and V. O’KEANE, Psychol. Med. 28 295-300 (1998). A.J. CLEARE and A.J. BOND, Psychiatry Res. 69 89-95 (1997). A.J. CLEARE, R.M. MURRAY and V. O’KEANE, Psychopharmacology (Berl) 134 406-410 (1997). K. O’FLYNN, V. O’KEANE, J.V. LUCEY and T.G. DINAN, Biol. Psychiatry 30 377-382 (1991). K.P. LESCH, H.M. SCHULTE, M. OSTERHEIDER and T. MULLER, Psychopharmacology 105 415-420 (1991).
28. K.P. LESCH, Prog. Neuropsychopharmacol. Biol. Psychiat. 15 723-733 (1991).
Reference8

1230 SSRIs and Neuroendocrine Function Vol. 65, No. 12, 1999
29.
30.
31.
32. 33.
34. 35.
36.
37. 38. 39. 40. 41. 42. 43.
44. 45.
46. 47.
48.
49. 50. 51.
52. 53. 54.
55. 56.
57.
58.
59.
60.
61.
62. 63.
64.
P. MONTELEONE, F. CATAPANO, F. BORTOLOTTI and M. MAJ, Biol. Psychiatry 42 175-180 (1997). P. MONTELEONE, F. CATAPANO, S. DI MARTINO, C. FERRARO and M. MAJ, British Journal of Psychiatry 170 554-557 (1997). P. MONTELEONE, F. CATAPANO, A. TORTORELLA and M. MAJ, Neuropsychobiology 36 8-12 (I 997). J.P. HERMAN and W.E. CULLINAN, Trends Neurosci. 20 78-84 (1997). B. BOHUS, J.M. KOOLHAAS, SM. KORTE, B. ROOZENDAAL and A. WIERSMA, Clin. Exp. Pharmacol. Physiol. 23 177-182 (1996). L.D. VAN DE KAR, Clin. Exp. Pharmacol. Physiol. 23 166-170 (1996). S. HINUMA, Y. HABATA, R. FUJI, Y. KAWAMATA, M. HOSOYA, S. FUKUSUMI, C. KITADA, Y. MASUO, T. ASANO, H. MATSUMOTO, M. SEKIGUCHI, T. KUROKAWA, 0. NISHIMURA, H. ONDA and M. FUJINO, Nature 393 272-276 (1998). R.A. DURHAM, M.J. EATON, K.E. MOORE and K.J. LOOKINGLAND, Eur. J. Pharmacol. 335 37-42 (1997). J.Y. LIN and J.T. PAN, Brain Res. 727 182-186 (1996). G. BAGDY and G.B. MAKARA, Eur. J. Pharmacol. 275 301-305 (1995). E.J. VAN BOCKSTAELE, A. BISWAS and V.M. PICKEL, Brain Res. 624 188-198 (1993). T. PETROV, T.L. KRUKOFF and J.H. JHAMANDAS, Cell Tissue Res. 277 289-295 (1994). T. PETROV, T.L. KRUKOFF and J.H. JHAMANDAS, J. Comp. Neurol. 318 18-26 (1992). Z. LIPOSITS, C. PHELIX and W.K. PAULL, Histochemistry 86 541-549 (1987). P.E. SAWCHENKO, L.W. SWANSON, H.W.M. STEINBUSCH and A.A.J. VERHOFSTAD, Brain Res. 277 355-360 (1983). L.D. VAN DE KAR and S.A. LORENS, Brain Res. 162 45-54 (1979). L.D. VAN DE KAR, S.A. LORENS, A. VODRASKA, G. ALLERS, M. GREEN and L.S. VAN ORDEN, Neuroendocrinology 31309-3 15 (1980). F. CHAOULOFF, Brain Res. Rev. 18 l-32 (1993). L.D. VAN DE KAR, Handbook of experimental pharmacology. Serotoninergic neurons and 5-HT receptors, H.G. Baumgarten and M. Gothert (eds), 537-562, Springer, Berlin (1997). Q. LI, A.D. LEVY, T.M. CABRERA, MS. BROWNFIELD, G. BA’ITAGLIA and L.D. VAN DE KAR, Brain Res. 630 148-156 (1993). L.D. VAN DE KAR and MS. BROWNFIELD, NIPS 8 202-207 (1993). L. PAN and F. GILBERT, Neuroendocrinology 56 797-802 (1992). K.P. LESCH, A. HOH, J. DISSELKAMP-TIETZE, M. WIESMANN, M. OSTERHEIDER and H.M. SCHULTE, Arch. Gen. Psychiatry 48 540-547 (1991). S.K. HEMRICK-LUECKE and R.W. FULLER, Eur. J. Pharmacol. 311207-211 (1996). G. BAGDY and K.T. KALOGERAS, Brain Res. 611330-332 (1993). J.A. SAYDOFF, P.A. RITTENHOUSE, L.D. VAN DE KAR and M.S. BROWNFIELD, J. Pharmacol. Exp. Ther. 257 95-99 (1991). G. BAGDY, K.T. KALGGERAS and K. SZEMEREDI, Eur. J. Pharmacol. 229 9-14 (1992). Q. LI, M.S. BROWNFIELD, G. BA’ITAGLIA, T.M. CABRERA, A.D. LEVY, P.A. RI’ITENHOUSE and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 266 836-844 (1993). A.D. LEVY, Q. LI, P.A. RITTENHOUSE and L.D. VAN DE KAR, Neuroendocrinology 58 65-70 (1993). Q. LI, P.A. RITI-ENHOUSE, A.D. LEVY, M.C. ALVAREZ SANZ and L.D. VAN DE KAR, Neuropharmacology 31983-989 (1992). P.A. RITTENHOUSE, A.D. LEVY, Q. LI, CL. BETHEA and L.D. VAN DE KAR, Endocrinology 133 661-667 (1993). L.D. VAN DE KAR, S.A. LORENS, J.H. URBAN and C.L. BETHEA, Neuropharmacology 28 299-305 (1989). P.A. RIT’TENHOUSE, E.A. BAKKUM and L.D. VAN DE KAR, J. Ph armacol. Exp. Ther. 259 58-65 (1991). I.K. ANDERSON, G.R. MARTIN and A.G. RAMAGE, Br. J. Pharmacol. 107 1020-1028 (1992). M.S. BROWNFIELD, J. GREATHOUSE, S.A. LGRENS, J. ARMSTRONG, J.H. URBAN and L.D. VAN DE KAR, Neuroendocrinology 47 277-283 (1988). K.P. LESCH, R. RUPPRECHT, B. POTEN, U. MULLER, K. SOHNLE and J. FRI’IZE, Biol. Psychiatry 26 203-205 (1989).

Vol. 65. No. 12, 1999 SSRls and Neurcendocrine Function 1231
65.
66.
67.
68.
69.
70.
71.
72.
73. 74.
75. 76. 77. 78.
79.
80.
81.
82.
83.
84.
85. 86. 87. 88. 89. 90.
91. 92.
93. 94. 95. 96.
97. 98.
P.J. COWEN, A.C. POWER, C.J. WARE and I.M. ANDERSON, Br. J. Psychiatry 164 372-379 ( 1994). A.D. LEVY, M.H. BAUMANN and L.D. VAN DE KAR, Frontiers in Neuroendocrinology 15 l-72 (1994). E. MELLER and K. BOHMAKBR, J. Phatmacol. Exp. Ther. 271 1246-1252 (1994). I.M. ANDERSON, J.F.W. DEAKIN and H.E.J. MILLER, Psychopharmacology (Bed) 128 74-82 (1996). B. LERER, Y. GELFIN and M. GORFINE, Psychoneuroendocrinology 22 S 166 ( 1997). P.J. COWEN, Merhods in Neuroendocrinology, L.D. Van de Kar (ed), 205223, CRC Press, Boca Raton (1998). B. SELE’ITI, C. BENKELFAT, P. BLIER, L. ANNABLE, F. GILBERT and C. DE MONTIGNY, Neuropsychopharmacology 13 93-104 (1995). J.R.E. HERDMAN, N.J. DELVA, R.E. HOCKNEY, G.M. CAMPLING and P.J. COWEN, Psychopharmacology (Bet-l) 113 561-564 (1994). S. IDRES, C. DELARUE, H. LEFEBVRE and H. VAUDRY, Mol. Brain Res. 10 251-258 (1991). H. LEFEBVRE, V. CONTESSE, C. DELARUE, C. SOUBRANE, A. LEGRAND, J.-M. KUHN, L.-M. WOLF and H. VAUDRY, J. Clin. Endocrinol. Metab. 77 1662-1666 (1993). C.A. RIZZI, Eur. J. Clin. Phartnacol. 47 377-378 (1994). R.W. FULLER, H.D. SNODDY and B.B. MOLLOY, Life Sci. 19 337-346 (1976). M. BIANCHI, P. SACERDOTE and A.E. PANERAI, Eur. J. Pharmacol. 263 81-84 (1994). R.W. FULLER, K.W. PERRY, S.K. HEMRICK-LUECKE and E. ENGLEMAN, J. Pharrn. Pharmacol. 48 68-70 (1996). L.M. KATZ, L. NATHAN, C.M. KUHN and SM. SCHANBERG, Psychoneuroendocrinology 21 219-235 (1996). U. VON BARDELEBEN, A. STEIGER, A. GERKEN and F. HOLSBOER, International Clinical Psychopharmacology 4 SuppI 1 l-5 ( 1989). C. REIST, D. HELMESTE, L. ALBERS, H. CHHAY and S.W. TANG, Psychiatry Research 60 177-184 (1996). E. SEIFRITZ, P. BAUMANN, M.J. MULLER, 0. ANNEN, M. AMEY, U. HEMMETER, M. HATZINGER, F. CHARDON and E. HOLSBOER-TRACHSLER, Neuropsychopharrnacology 14 253-263 (1996). E. SEIFRI’IZ, M.J. MULLER, 0. ANNEN, R. NIL, M. HATZINGER, U. HEMMETER, P. MOORE and E. HOLSBOER-TRACHSLER, Journal of Psychiatric Research 31 543-554 (1997). S. CELLA, A. PENALVA, V. LOCATELLI, A. NOVELLI, D. COCCHI and E.E. MULLER, Br. J. Clin. Pharmacol. 15 SuppI 3 357S-364s (1983). L. KRULICH, Life Sci. 17 1141-l 144 (1975). J.A. CLEMENS, B.D. SAWYER and B. CERIMELE, Endocrinology 100 692-698 (1977). J.A. CLEMENS, M.E. ROUSH and R.W. FULLER, Life Sci. 22 2209-2214 (1978). P.F. ARAVICH, T.S. RIEG, I. AHMED and T.J. LAUTERIO, Brain Res. 612 180-189 (1993). D.M. GIBBS and W. VALE, Brain Res. 280 176-179 (1983). C.M. FAULL, J.A. CHARLTON, E. PHILLIPS, S. THORNTON, T. BUTLER and P.H. BAYLIS, Ann. NY Acad. Sci. 689 484-488 (1993). C.M. FAULL, P. ROOKE and P.H. BAYLIS, Clin. Endocrinol. 35 423-430 (1991). E.F. COCCARO, R.J. KAVOUSSI, T.B. COOPER and R.L. HAUGER, Neuropsychopharmacology 15 595-607 (1996). S.B.G. PARK, D.J. WILLIAMSON and P.J. COWEN, Psychol. Med. 26 1191-l 196 (1996). J. BANCROFI and A. COOK, J. Affect. Disord. 36 57-64 (1995). A.J. CLEARE, A. MCGREGOR and V. O’KEANE, Clin. Endocrinol. (Oxf. ) 43 713-719 (1995). J.V. LUCEY, V. O’KEANE, G. BUTCHER, A.W. CLARE and T.G. DINAN, Br. J. Psychiatry 161 517-521 (1992). H.D. ERLING, E.T. MELLERUP and O.J. RAFAELSEN, Psychiatry Res. 17 309-316 (1985). K.L. BLOOMINGDALE, R.G. VASILE, J.E. GUDEMAN, B. GERSON and J.J. SCHILDKRAUT, Biol. Psychiatry 21390-393 (1986).
99. W.C. DUNCAN, JR., Pharmacol. Ther. 71253-312 (1996). 100. J.J. EVANS, Eur. J. Endocrinol. 137 559-571 (1997). 101. F. HOLSBOER and N. BARDEN, Endocr. Rev. 17 187-205 (1996). 102. P.J. MULROW and R. FRANCO-SAENZ, J. Hypertens. 14 173-176 (1996).

1232 SSRIs and Neuroendocrine Function Vol. 65, No. 12, 1999
103. 104. 105. 106. 107.
108. 109. 110.
111.
112.
113. 114.
115.
116.
117.
118.
119. 120.
121.
122.
123.
i24. 12.5. 126.
127.
128.
129.
130.
131.
132.
133.
134.
M. ALTEMUS, Ann. NY Acad. Sci. 771 697-707 (1995). S. FELDMAN and J. WEIDENFELD, Prog. Neurobiol. 45 129-141 (1995). S. FELDMAN, N. CONFORTI and J. WEIDENFELD, Neurosci. Biobehav. Rev. 19 235-240 (1995). S. AL-DAMLUJI, Baillieres Clinical Endocrinology & Metabolism 7 355-392 (1993). E.O. JOHNSON, T.C. KAMILARIS, G.P. CHROUSOS and P.W. GOLD, Neurosci. Biobehav. Rev. 16 115-130 (1992). H.Y. MELTZER and M. MAES, Psychopharmacology (Berl) 114 635-643 (1994). M.A. LEE, J.F. NASH, M. BARNES and H.Y. MELTZER, Psychopharmacology 103 258-264 (1991). D.S. CHARNEY, G.R. HENINGER, J.F. REINHARD, D.E. STERNBERG and K.M. HAFSTEAD, Psychopharmacology 78 38-43 (1982). A. WINOKUR, N.D. LINDBERG, I. LUCKI, J. PHILLIPS and J.D. AMSTERDAM, Psychopharmacology 88 2 13-219 (1986). S.B. PARK, D.J. WILLIAMSON and P.J. COWEN, International Clinical Psychopharmacology 10 215-220 (1995). V. O’KEANE. D. MCLOUGHLIN and T.G. DINAN, J. Affect. Disord. 26 143-I 50 (1992). L.H. PRICE, D.S. CHARNEY, P.L. DELGADO, W.K. GOODMAN, J.H. KRY STAL, SW. WOODS and G.R. HENINGER, Progress in Neuro-Psychopharmacology & Biological Psychiatry 14 459-472 (1990). L.H. PRICE. D.S. CHARNEY, P.L. DELGADO, G.M. ANDERSON and G.R. HENINGER, Arch. Gen. Psychiatry 46 625-631 (1989). P.A. SARGENT, D.J. WILLIAMSON and P.J. COWEN, British Journal of Psychiatry 172 49-52 (1998). H.Y. MELTZER, M. SIMONOVIC, R.D. STURGEON and VS. FANG, Acta Psychiatrica Scandinavica, Supplementum 290 1 OO- 12 1 ( 198 1). H. MELTZER, B. BASTANI, K. JAYATI-IILAKE and M. MAES, Neuropsychopharmacology 17 l-1 1 (1997). D.Y. OKUHARA, S.G. BECK and N.A. MUMA, Brain Res. 745 144-151 (1997). N. SAITO, X. GUITART, M. HAYWARD, J.F. TALLMAN, R.S. DUMAN and E.J. NESTLER, Proc. Natl. Acad. Sci. 86 3906-3910 (1989). P.J. COWEN, I.M. ANDERSON and D.G. GRAHAME-SMITH, J. Clin. Psychopharmacol. 10 (Suppl.) 2 1 S-25s ( 1990). T.G. DINAN, S. BARRY, L.N. YATHAM, M. MOBAYED and M. O’HANLON, lnt. Clin. Psychopharmacol. 5 119-123 (1990). K.P. LESCH, M. MAYER, J. DISSELKAMP-TIE’IZE, A. HOH, M. WIESMANN, M. OSTERHEIDER and H.M. SCHULTE, Biol. Psychiatry 28 620-628 (1990). D.L.MURPHY,K.P.LESCH,C.S.AULAKHandT.A.PIGO~,Pharmacol..Rev.43527-552(1991). I.M. ANDERSON and P.J. COWEN, Psychopharmacology 106 428-432 (1992). P. SARGENT, D.J. WILLIAMSON, G. PEARSON, J. ODONTIADIS and P.J. COWEN, Psychopharmacology (Berl) 132 296-302 (1997). I.S. SHIAH, L.N. YATHAM, R.W. LAM and A.P. ZIS, Neuropsychopharmacology 17 382-390 (1997). 1. BERLIN, D. WAROT, V. LEGOUT, S. GUILLEMANT, G. SCHGLLNHAMMER and A.J. PUECH, Clin. Pharmacol. Ther. 63 428-436 (1998). A. VICENTIC, Q. LI, G. BATTAGLIA and L.D. VAN DE KAR, Eur. J. Pharmacol. 346 261-266 (lY98). A.D. LEVY, Q. LI, M. GUSTAFSON and L.D. VAN DE KAR, Eur. J. Pharmacol. 274 141-149 (1995). L.D. VAN DE KAR, A.D. LEVY, Q. LI and M.S. BROWNFIELD, Pharmacol. Biochem. Behav. 61 677-683 (1998). F. GILBERT, C. BRAZELL, M.D. TRICKLEBANK and SM. STAHL, Eur. J. Pharmacol. 147 431-439 (1988). K.P. LESCH, K. SOHNLE, B. POTEN, G. SCHOELLNHAMMER, R. RUPPRECHT and H.M. SCHULTE, J. Clin. Endocrinol. Metab. 70 670-674 (1990). Q. Ll, M.S. BROWNFIELD, A.D. LEVY, G. BATTAGLIA, T.M. CABRERA and L.D. VAN DE KAR, Biol. Psychiatry 36 300-308 (1994).
135. G. BAGDY, Behav. Brain Res. 73 277-280 ( 1996). 136. Q. LI, N.A. MUMA and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 279 1035-1042 (1996).

Vol. 65, No. 12, 1999 SSRIs aad Neuroendocrine Function 1233
137.
138.
139. 140. 141.
142. 143. 144.
145.
146.
147.
148. 149.
150. 151. 152.
153.
154. 155.
156. 157.
158.
159. 160.
161.
162.
163.
164. 165.
166.
167.
168. 169. 170. 171. 172.
Q. LI, N.A. MUMA, G. BATTAGLIA and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 282 1581-1590 (1997). D.K. RAAP, S. EVANS, F. GARCIA, Q. LI, N.A. MUMA, W.A. WOLF, G. BATTAGLIA and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 288 98-106 (1999). M. SIMONOVIC, G.A. GUDELSKY and H.Y. MEL’IZER, J. Neural Transm. 59 143-149 (1984). J.F. NASH and H.Y. MELTZER, J. Pharmacol. Exp. Ther. 249 236-241 (1989). K.J. KELLAR, B.A. HULIHAN-GIBLIN, S.E. MULRONEY, M.D. LUMPKIN and C.M. FLORES, Neuropharmacology 31643-647 (1992). G. BAGDY and G.B. MAKARA, Endocrinology 134 1127-l 13 1 (1994). J.T. PAN and I.C.H. YANG, Life Sci. 58 1189-l 194 (1996). L. GROENINK, J. VAN DER GUGTEN. J.C. COMPAAN, R.A.A. MAES and B. OLIVIER, Psychopharmacology (Berl) 13193-100 (1997). A. DI SCIULLO, M.T. BLUEI PAJOT, F. MOUNIER, C. OLIVER, B. SCHMIDT and C. KORDON, Endocrinology 127 567-572 (1990). C.S. AULAKH, K.M. WOZNIAK, M. HAAS, J.L. HILL, J. ZOHAR and D.L. MURPHY, Eur. J. Pharmacol. 146 253-259 (1988). S. CACCIA, M. CAPPI, C. FRACASSO and S. GARA’ITINI, Psychopharmacology 100 509-514 (1990). C.L. DEVANE, Am. J. Med. 97 SuppI. 6A 13S-23s (1994). D.K. RAAP, F. GARCIA, N.A. MUMA, W.A. WOLF, G. BA’ITAGLIA and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 288 561-567 (1999). J.G. HENSLER, G.B. KOVACHICH and A. FRAZER, Neuropsychopharmacology 4 13 1 - 144 (199 1). Q. LI, G. BATTAGLIA and L.D. VAN DE KAR, Brain Res. 769 141-151 (1997). E. LE POUL, N. LAARIS, E. DOUCET, A.-M. LAPORTE, M. HAMON and L. LANFUMEY, Naunyn Schmiedebergs Arch. Pharmacol. 352 141-148 (1995). F. SERRES, D.K. RAAP, N.A. MUMA, F. GARCIA, G. BATTAGLIA, Y. ZHANG, Q. MA and L.D. VAN DE KAR, Sot. Neuroscience Abst. 24 539.18 (1998).(Abstract) L.D. VAN DE KAR, Q. LI and G. BATTAGLIA, Sot. Neuroscience Abst. 24 539.17 (1998).(Abstract) M.J. OWENS, D.L. KNIGHT, J.C. RITCHIE and C.B. NEMEROFF, J. Pharmacol. Exp. Ther. 256 795-800 (1991). S.E. GARTSIDE, P.M. ELLIS, T. SHARP and P.J. COWEN, Eur. J. Pharmacol. 221 27-33 (1992). P.A. RITTENHOUSE, E.A. BAKKUM, A.D. LEVY, Q. LI, M. CARNES and L.D. VAN DE KAR, J. Pharmacol. Exp. Ther. 271 1647-1655 (1994). P. MAZZOLA-POMIE’ITO, C.S. AULAKH, S.J. HUANG and D.L. MURPHY, J. Pharmacol. Exp. Ther. 279 782-789 (1996). R.H. ALPER and J.M. SNIDER, J. Pharmacol. Exp. Ther. 243 829-833 (1987). G. BAGDY, A.F. SVED, D.L. MURPHY and K. SZEMEREDI, Eur. J. Pharmacol. 210 285-289 (1992). R.S. KAHN, M.A. KLING, S. WETZLER, G.M. ASNIS and H. VAN PRAAG, Psychopharmacology (Berl) 108 225-228 (1992). J.A. SAYDOFF, F. LEANZA, M. CARNES and M.S. BROWNFIELD, Sot. Neurosci. Abstr. 18 823 (#346.5) (1992). L.D. VAN DE KAR, P.A. RIT-TENHOUSE, P. O’CONNOR, T. PALIONIS, M.S. BROWNFIELD, S.J. LENT, M. CARNES and CL. BETHEA, Biol. Psychiatry 32 258-269 (1992). P.E. PERGOLA, A.F. SVED, J.L. VOOGT and R.H. ALPER, Neuroendocrinology 57 550-558 (1993). J.A. SAYDOFF, P.A. RITI’ENHOUSE, M. CARNES, J. ARMSTRONG, L.D. VAN DE KAR and M.S. BROWNFIELD, Am. J. Physiol. Endocrinol. Metab. 270 E513-E521 (1996). E.A. PARATI, P. ZANDARI, D. COCCHI, T. CARACENI and E.E. MUELLER, J. Neural Transm. 47 273-297 (1980). H.Y. MELTZER and M.T. LOWY, Psychophumtacology: The Third Generation of Progress, H.Y. Meltzer (ed), 513-526, Raven Press, New York (1987). M.T. LOWY and H.Y. MELTZER, Biol. Psychiatry 23 818-828 (1988). M.A. LEE and H.Y. MELTZER, Drug Alcohol Depend. 35 217-222 (1994). H.Y. MELTZER and M. MAES, Biol. Psychiatry 38 310-318 (1995). B. BASTANI, J.F. NASH and H.Y. MELTZER, Arch. Gen. Psychiatry 47 833-839 (1990). A. BROOCKS, T.A. PIGOTT, J.L. HILL, S. CANTER, T.A. GRADY, F. L’HEUREUX and D.L. MURPHY, Psychiatry Res. 79 1 l-20 (1998).

1234 SSRls and Neuroendocrine Function Vol. 65, No. 12, 1999
173.
174. 175.
176.
177. 178.
179.
180.
181. 182.
183. 184.
18.5.
186.
187.
188.
189. 190. 191.
192.
193.
194. 195.
196.
197. 198.
199.
200. 201. 202. 203. 204.
205.
206.
A. BROOCKS, NC. BRIGGS, T.A. PIGOTT, J.L. HILL, SK. CANTER, T.J. TOLLIVER, D. BALDEMORE and D.L. MURPHY, Psychopharmacology (Berl) 130 91-103 (1997). S. WE’IZLER, G.M. ASNIS, J.M. DELECUONA and 0. KALUS, Psychiatry Res. 64 77-82 (1996). D. GARCIA-BORREGUERO, F.M. JACOBSEN, D.L. MURPHY, J.R. JOSEPH-VANDERPOOL, A. CHIARA and N.E. ROSENTHAL, Biol. Psychiatry 37 740-749 (1995). U. HALBREICH, N. ROJANSKY, S. PALTER, H. TWOREK, P. HISSIN and K. WANG, Biol. Psychiatry 37 434441 (1995). L.N. YATHAM and M. STEINER, Life Sci. 53 447-463 (1993). E. HOLLANDER, C. DECARIA, R. GULLY, A. NITESCU, R.F. SUCKOW, J.M. GORMAN, D.F. KLEIN and M.R. LIEBOWITZ, Psychiatry Res. 36 I- 17 (199 1). E.A. MUELLER, D.L. MURPHY and T. SUNDERLAND, J. Clin. Endocrinol. Metab. 61 1179- 1184 (1985). A.J. SLEIGHT, N.J. STAM, V. MUTEL and P.M.L. VANDERHEYDEN, Biochem. Pharmacol. 51 71-76 (1996). D.J. QUESTED, P.A. SARGENT and P.J. COWEN, Psychopharmacology (Berl) 133 305-308 (1997). J.M. MASSOU, C. TRICHARD, D. ATTAR-LEVY, A. FELINE, E. CORRUBLE. B. BEAUFILS and J.L. MARTINOT, Psychopharmacology (Berl) 133 99-101 (1997). Q. LI, N.A. MUMA, G. BATTAGLIA and L.D. VAN DER KAR, Brain Res. 775 225-228 (1997). A. LAAKSO, E.P. PALVIMAKI, M. KUOPPAMAKI, E. SYVALAHTI and J. HIETALA, Neuropsychopharmacology 15 143- 15 1 ( 1996). N. TILAKARATNE, Z.L. YANG and E. FRIEDMAN, Eur. J. Pharmacol. Mol. Pharmacol. 290 263-266 (1995). G.A. KENNE’IT, S. LIGHTOWLER, V. DE BIASI, N.C. STEVENS, M.D. WOOD, 1.F TULLOCH and T.P. BLACKBURN, Neuropharmacology 33 158 I- 1588 (1994). V. KLIMEK, J. ZAK-KNAPIK and M. MACKOWIAK, Journal of Psychiatry & Neuroscience 19 63-67 (1994). A.K. CADOGAN, C.A. MARSDEN, I. TULLOCH and D.A. KENDALL, Neuropharmacology 32 249-256 (1993). P.D. HRDINA and T.B. VU, Synapse 14 324-33 1 (1993). R. WHALE and P.J. COWEN, Psychopharmacology (Berl) 137 203-204 (1998). Y.K. WING, E.M. CLIFFORD, B.D. SHEEHAN, G.M. CAMPLING, R.A. HOCKNEY and P.J. COWEN, Psychopharmacology 124 377-379 (1996). M. MARKIANOS, L. LYKOURAS, J. HATZIMANOLIS and C. STEFANIS, Neuroendocrinol. Lett. 17 37-40 (I 995). R. FRANCESCHINI, A. CATALDI, A. GARIBALDI, P. CIANCIOSI, A. SCORDAMAGLIA, T. BARRECA and E. ROLANDI, Neuropharmacology 33 235-239 (1994). C.S. AULAKH, J.L. HILL and D.L. MURPHY, Neuroendocrinology 59 35-41 (1994). C.S. AULAKH, P. MAZZOLA-POMIETTO and D.L. MURPHY, Pharmacol. Biochem. Behav. 55 265-268 (1996). R.W. FULLER, H.D. SNODDY, K.W. PERRY, F.P. BYMASTER and D.T. WONG, Biochem. Pharmacol. 27 193-198 (1978). J.M. O’DONNELL and M. GREALY, Br. J. Pharmacol. 105 863-868 (1992). J.D. COPLAN, L.A. PAPP, J. MARTINEZ, D. PINE, L.A. ROSENBLUM, T. COOPER, M.R. LIEBOWITZ and J.M. GORMAN, American Journal of Psychiatry 152 619-622 (1995). J.D. COPLAN, D. PINE, L. PAPP, J. MARTINEZ, T. COOPER, L.A. ROSENBLUM and J.M. GORMAN, Neuropsychopharmacology 13 65-73 (I 995). R.R. GALA, J.A. PETERS, D.R. PIEPER and M.G. SUBRAMANIAN, Life Sci. 22 439-444 (1977). C.E. GROSVENOR and F. MENA, Endocrinology 107 863-868 (1980). N. BEN-JONATHAN, L.A. ARBOGAST and J.F. HYDE, Prog. Neurobiol. 33 399-447 (1989). B. COUZINET, F. DOREY and G. SCHAISON, Acta Endocrinol. 121 235-240 (1989). L. LYKOURAS, M. MARKIANOS, J. HATZIMANOLIS and C. STEFANIS, J. Affect. Disord. 23 191-197 (1991). I.M. ANDERSON, C.J. WARE, J.M. DA ROZA DAVIS and P.J. COWEN, Br. J. Psychiatry 160 372-378 (1992). J.C. GARBUTT, J.P. MAYO, K.Y. LITTLE, G.M. GILLETTE, G.A. MASON, B. DEW and A.J. PRANGE, JR., Alcohol. Clin. Exp. Res. 20 717-722 (1996).

Vol. 65, No. 12, 1999 SSRIs and Neuroendocrine Function 1235
207.
208.
209. 210.
211.
212.
213.
214.
21.5. 216. 217. 218. 219. 220. 221. 222. 223.
224. 225.
226. 227. 228.
J.F. ROSENBAUM, M. FAVA, J.A. PAVA, M.K. MCCARTHY, R.J. STEINGARD and E. BOUFFIDES, Am. J. Psychiatry 150 1164-l 168 (1993). J.D. AMSTERDAM, M. FAVA, G. MAISLIN, J. ROSENBAUM and M. HORNIG-ROHAN, J. Affect. Disord. 38 165-172 (1996). L.D. VAN DE KAR and M.L. BLAIR, Frontiers in Nemoendocrinology 20 l-48 (1999). G.E. DUNCAN, D.J. KNAPP, S.W. CARSON and G.R. BREESE, J. Pharmacol. Exp. Ther. 285 579-587 (1998). J. SHLIK, A. ALUOJA, V. VASAR, E. VASAR, T. PODAR and J. BRADWEJN, Journal of Psychiatry & Neuroscience 22 332-340 (1997). A.W. GODDARD, S.W. WOODS, D.E. SHOLOMSKAS, W.K. GOODMAN, D.S. CHARNEY and G.R. HENINGER, Psychiatry Res. 48 119-133 (1993). L.S. BRADY, P.W. GOLD, M. HERKENHAM, A.B. LYNN and H.J. WHITFIELD, JR., Brain Res. 572 117-125 (1992). P. MONTELEONE, F. CATAPANO, A. TORTORELLA, S. DI MARTIN0 and M. MAJ, Psychoneuroendocrinology 20 763-770 (1995). S. KASPER, A. VIEIRA, R. SCHMIDT and P. RICHTER, Pharmacopsychiatry 23 76-84 (1990). J.A. DEN BOER and H.G.M. WESTENBERG, Psychopharmacology 102 85-94 (1990). C.G. GOTTFRIES and A.L. NYTH, Ann. N. Y. Acad. Sci. 640 276-279 (1991). I.E. MARAR and J.A. AMICO, Endocrine 8 13-18 (1998). C.M. FAULL, J.A. CHARLTON, T.J. BUTLER and P.H. BAYLIS, J. Endocrinol. 139 77-87 (1993). M.E. BRADLEY, E.F. FOOTE, E.N. LEE and L. MERKLE, Pharmacotherapy 16 680-683 (1996). D.K. SOMMERS, M. VAN WYK and J.R. SNYMAN, Eur. J. Clin. Pharmacol. 46 441-444 (1994). P.J. COWEN and P.A. SARGENT, J. Psychopharmacol. 11 345-348 (1997). J.D. AMSTERDAM, F. GARCIA-ESPANA, D. GOODMAN, M. HOOPER and M. HORNIG-ROHAN, J. Affect. Disord. 46 151-156 (1997). V. PIDRMAN and I. TUMA, Acta Medica (Hradec Kralove) 40 99-102 (1997). K. LAINE, M. ANTI’ILA, E. HEINONEN, A. HELMINEN, R. HUUPPONEN, 0. MAKI-IKOLA, K. REINIKAINEN and M. SCHEININ, Clinical Neuropharmacology 20 419-433 (1997). C. GORDON, R. WHALE and P.J. COWEN, Psychopharmacology (Berl) 137 201-202 (1998). J.H. THAKORE and T.G. DINAN, American Journal of Psychiatry 152 616-618 (1995). J. JORDAN, F.H. MESSERLI, C.J. LAVIE, F.C. AEPFELBACHER and F. SORIA, Am. J. Cardiol. 75 743-744 (1995).





























