Embed Size (px)
Citation preview

Regular Article
TRANSPLANTATION
Siglec-G–CD24 axis controls the severity of graft-versus-host diseasein miceTomomi Toubai,1 Guoqing Hou,1 Nathan Mathewson,1 Chen Liu,2 Ying Wang,1 Katherine Oravecz-Wilson,1
Emily Cummings,1 Corinne Rossi,1 Rebecca Evers,1 Yaping Sun,1 Julia Wu,1 Sung Won Choi,3 Dexing Fang,4 Pan Zheng,5
Yang Liu,5 and Pavan Reddy1
1Department of Internal Medicine, University of Michigan Comprehensive Cancer Center, Ann Arbor, MI; 2Department of Pathology, University of Florida
College of Medicine, Gainesville, FL; 3Department of Pediatrics and Communicable Diseases, University of Michigan Medical School, Ann Arbor, MI;4OncoImmune, Inc., Rockville, MD; and 5Center for Cancer and Immunology Research, Children’s National Medical Center, Washington, DC
Key Points
• Demonstrates a role fornegative regulator of innateimmunity, Siglec-G, incontrolling GVHD.
• Shows that enhancing theinteraction between hostSiglec-G and CD24 on donorT cells with a novel CD24fusion protein mitigatesGVHD.
Activation of sialic-acid–binding immunoglobulin-like lectin-G (Siglec-G) by non-
infectious damage-associated molecular patterns controls innate immune responses.
However, whether it also regulates T-cell–mediated adaptive immune responses is not
known. Graft-versus-host reaction is a robust adaptive immune response caused by
allogeneic hematopoietic cell transplantation that have been activated by antigen-
presenting cells (APCs) in the context of damaged host tissues following allogeneic
hematopoietic cell transplantation. The role of infectious and noninfectious pattern
recognition receptor–mediated activation in the induction and aggravation of graft-
versus-host disease (GVHD) is being increasingly appreciated. But the role of pathways
that control innate immune responses to noninfectious stimuli in modulating GVHD has
heretofore not been recognized. We report that Siglec-G expression on host APCs,
specifically on hematopoietic cells, negatively regulates GVHD in multiple clinically
relevant murine models. Mechanistic studies with various relevant Siglec-G and CD24
knockout mice and chimeric animals, along with rescue experiments with novel CD24
fusion protein demonstrate that enhancing the interaction between Siglec-G on host APCs with CD24 on donor T cells attenuates
GVHD. Taken together, our data demonstrate that Siglec-G–CD24 axis, controls the severity ofGVHDand suggest that enhancing this
interaction may represent a novel strategy for mitigating GVHD. (Blood. 2014;123(22):3512-3523)
Introduction
Innate immune response is initiated by the evolutionarily con-served Toll-like receptors (TLRs), nod-like receptors, and otherpattern recognition receptors that respond to damage-associatedmolecular patterns (DAMPs) and pathogen-associated molecularpatterns (PAMPs). Immune activation by these pathways initiatesand/or accentuates T-cell–mediated immunity.1 However, it is lessclear whether a dedicated recognition pathway serves as a negativeregulator for innate immune responses and may also attenuateT-cell–mediated responses.
Members of the family of sialic-acid–binding immunoglobulin-like lectins (Siglecs) have emerged as potential negative regulators ofinnate immunity.2 A number of homologous members of the Siglecfamily have been identified in humans andmice.2Most Siglec familymembers have immunoreceptor tyrosine-based inhibitory motifs(ITIMs) or ITIM-like regions in their intracellular domains.2,3 Recentdata demonstrated that Siglec-G deletion in mice (Siglec-G2/2)exacerbated the production of inflammatory cytokines and acuteorgan failure in response to DAMPs, such as high-mobility group
box 1 (HMGB-1) in acetaminophen-induced liver necrosis,4 andcecal ligation and puncture models.5 In contrast to their exacerbatedinflammatory response to DAMPs, Siglec-G2/2 mice showed sim-ilar inflammatory responses to PAMPs, such as lipopolysaccharide(LPS) or polyinosinic:polycytidylic acid, which are agonists ofTLR 4 and 3, respectively.4,5 Thus, recent data have identifiedSiglec-G expression on antigen-presenting cells (APCs), such asdendritic cells (DCs), as an important negative regulator of innateimmunity. However, whether Siglec-G expression on APCs has anyimpact on the modulation of T-cell–mediated disease processes hasheretofore not been appreciated.
Host tissue injuries caused by hematopoietic cell transplantation(HCT) conditioning regimens, including high-dose chemotherapyand/or total body irradiation (TBI), is considered to be the first step inthe development of acute graft-versus-host disease (GVHD),6 a life-threatening complication of allogeneic HCT (allo-HCT).7 Host tissueinjuries caused by the conditioning regimen leads to the release ofproinflammatory cytokines, such as TNF-a, IL-1b, and IL-6, as well
Submitted December 19, 2013; accepted March 23, 2014. Prepublished online
as Blood First Edition paper, April 2, 2014; DOI 10.1182/blood-2013-12-
545335.
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge
payment. Therefore, and solely to indicate this fact, this article is hereby
marked “advertisement” in accordance with 18 USC section 1734.
© 2014 by The American Society of Hematology
3512 BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom

as the release of DAMPs and PAMPs.6,8-11 BothDAMPs and PAMPscan activate APCs, such as DCs,6,8-11 which are critical for the devel-opment of acute GVHD.12-14 Recent experimental data have demon-strated that targeting certain DAMPs and the activation of APCsinduced by them, can lead to aggravation of acute GVHD.15-17
Alloreactive donor T lymphocytes, activated by both donor and hostAPCs, are absolutely critical for induction and perpetuation ofGVHD.7 Activation of the innate immune system, such as the APCs,plays a key role in enhancing the severity of donor T-cell–mediatedGVHD. However, the role of negative regulators of innate immunityin regulating the severity of GVHD has not been recognized. This isparticularly critical in light of the recent observations, which demon-strate that the absence of any one subset of professional host-derivedhematopoietic APCs, such as DCs or macrophages, contrary to theexpectations of reducing donor T-cell responses, are either irrelevant,or actually enhance donor T-cell responses and accentuate GVHDseverity.18-22 These newer observations suggest that pathways thatmitigate the hematopoietic APCs may be as critical for attenuatingGVHD.
Following conditioning for allo-HCT, several DAMPs are re-leased, including uric acid and adenosine triphosphate (ATP) thathave been demonstrated to contribute to activation of host APCs andenhanceGVHD.However, it is less clearwhether a dedicatedDAMPrecognition pathway may serve as a negative regulator for innateimmune responses, and control the responses of donor T cells andthe severity of GVHD. Herein, utilizing a multimodal approach inseveral well-defined, clinically relevant murine models of allo-HCT,we determined the role of a defined negative regulator of responses toDAMPs and Siglec-G, in modulating T-cell responses and GVHDseverity. We used Siglec-G–deficient animals and its ligand knock-outs, the CD24 deficient donor T cells, along with rescue experimentswith novel CD24 fusion protein inmultiple animals and chimeras, andfound that Siglec-G expression on hostAPCs controls GVHDseverityby regulating CD24-dependent donor T-cell responses. Our datatherefore, demonstrates a novel role for Sigleg-G–CD24 axis incontrolling GVHD and suggests that enhancing this interaction mayrepresent a novel pathway for mitigating GVHD.
Materials and methods
Mice
C57BL/6 C3H.sw BALB/c mice were purchased from the Jackson Lab-oratory (BarHarbor,ME) andCharlesRiver Laboratories (Wilmington,MA).B6 Ly5.2 mice were purchased from NCI-Frederick (Frederick, MD).B6-background Siglec-G2/2GFP1/1 and CD242/2 have been previouslydescribed.23,24 All animals were cared for according to regulations reviewedand approved by the University Committee on the Use and Care of Animalsper University Laboratory Animal Medicine guidelines of the Universityof Michigan. Bone marrow (BM) chimeras were generated as previouslydescribed and detailed in the supplemental Methods on the BloodWeb site.14
BM transplantation (BMT)
BMTs were performed as in the supplemental Methods and as previouslydescribed.14,25 Wild-type (WT) B6 and Siglec-G2/2 animals received either8 Gy or 13 Gy (137Cs source) on day21 and 2.53 106 CD901 T cells, alongwith 53 106 T-cell–depleted BM (TCD-BM) cells from either syngeneic B6or allogeneic BALB/c donors. For studies with BM chimeras, they received9GyTBI on day21 andwere injectedwith 53 106 BMand 2.53 106 CD901
T cells from syngeneic or allogeneic donors on day 0 as previouslydescribed.14,26 Survival was monitored daily, and the recipients’ body weight
and GVHD clinical scores were measured weekly as in the supplementalMethods and as previously described.27 Histopathologic analysis of the liverand gastrointestinal (GI) tract, the primary GVHD target organs, wasperformed as described.28 For experiments with CD24 fusion protein,comprising of the extracellular domain of CD24 and IgG Fc (5 mg/kg)(OncoImmune, Ann Arbor, MI), an equivalent dose of nonspecific IgGwere administered IP on day 21 of allo-HCT.29
In vitro cultures and fluorescence-activated cell sorter
(FACS) analysis
BMDCs were obtained from WT-B6, Siglec-G2/2, and CD242/2 B6animals as detailed in the supplemental Methods. FACS analyses,including carboxyfluorescein diacetate succinimidyl ester (CFSE) andapoptosis analysis with Annexin staining, were performed as previouslydescribed.21,26 For mixed leukocyte reaction (MLR) cultures, splenic T cellswere magnetically separated and cultured with BMDCs from WT-B6,Siglec-G2/2, or B6-background CD242/2 mice. The incorporation of3H-thymidine (1 mCi/well) by proliferating T cells during the final 16 hourswas measured (see the supplemental Methods).
Cytokine enzyme-linked immunosorbent assay (ELISA)
Concentrations of IL-1b, IL-2, IL-6, IL-10, IL-12, TNF-a, IL-17A, IFN-g,and HMGB-1 were measured in the serum and culture supernatants byELISA, performed according to the manufacturer’s protocol (see thesupplemental Methods for details), and read at 450 nm using a microplatereader (Model 3550; Bio-Rad Laboratories, Hercules, CA). All samples andstandards were run in duplicate.
Statistical analysis
TheMann-WhitneyU test was used for the statistical analysis of in vitro data,and the Wilcoxon signed-rank test was used to analyze survival data.A P value,.05 was considered to be statistically significant.
Results
Expression of Siglec-G after radiation
Because radiation induces tissue damage and Siglec-G is critical fornegatively regulating inflammatory responses to DAMPs,4,5 we firstexploredwhether conditioningwith radiation affected the expressionof Siglec-G on APCs. B6 animals were irradiated with 8 and 13 Gy,and splenic DCswere analyzed 8 and 24 hours later for expression ofSiglec-G. We observed a significant reduction in the expression ofSiglec-G (Figure 1A) in a radiation dose-dependentmanner.We nextdetermined whether the reduction in Siglec-G is associated withincreased inflammation following high-dose irradiation. We ana-lyzed the secretion of proinflammatory cytokines, such as TNF-a,following high-intensity (13 Gy) and low-intensity (8 Gy) con-ditioning. Consistent with previous reports,30 radiation caused a dose-dependent increase in the release of proinflammatory cytokines TNF-a(supplemental Figure 1). Siglec-G negatively regulates the secretion ofproinflammatory cytokine release through its signaling via intracellularITIMdomains that are associatedwithSHP-1.4Therefore, todeterminewhether the reduction in the expression of Siglec-G had functionalconsequences, we analyzed the expression of intracellular markersof Siglec-G–dependent ITIM signaling. As shown in Figure 1B,irradiation caused a significant reduction in the expression of pSHP-1expression, demonstrating a significant reduction in theSiglec-G ITIMsignaling after radiation. These data show that irradiation reduced theexpression of negative regulator of DAMP responses, Siglec-G andits signaling in APCs.
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3513
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
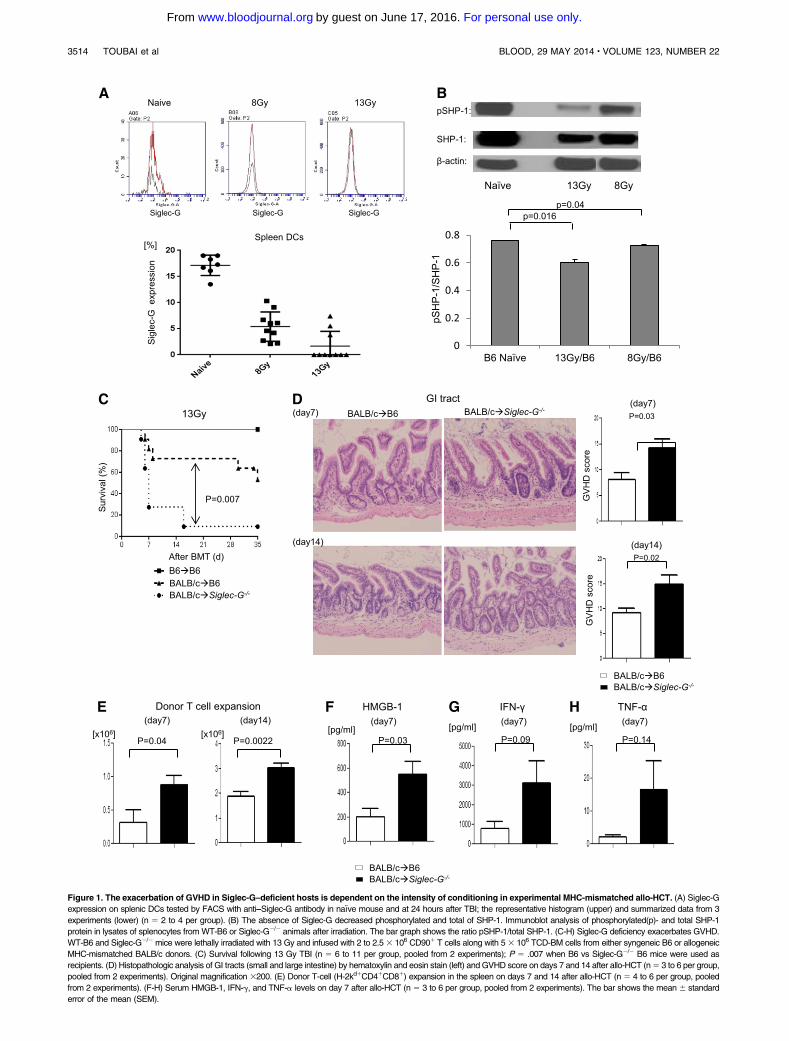
Figure 1. The exacerbation of GVHD in Siglec-G–deficient hosts is dependent on the intensity of conditioning in experimental MHC-mismatched allo-HCT. (A) Siglec-G
expression on splenic DCs tested by FACS with anti–Siglec-G antibody in naıve mouse and at 24 hours after TBI; the representative histogram (upper) and summarized data from 3
experiments (lower) (n 5 2 to 4 per group). (B) The absence of Siglec-G decreased phosphorylated and total of SHP-1. Immunoblot analysis of phosphorylated(p)- and total SHP-1
protein in lysates of splenocytes fromWT-B6 or Siglec-G2/2 animals after irradiation. The bar graph shows the ratio pSHP-1/total SHP-1. (C-H) Siglec-G deficiency exacerbates GVHD.
WT-B6 and Siglec-G2/2mice were lethally irradiated with 13 Gy and infused with 2 to 2.53 106 CD901 T cells along with 53 106 TCD-BM cells from either syngeneic B6 or allogeneic
MHC-mismatched BALB/c donors. (C) Survival following 13 Gy TBI (n 5 6 to 11 per group, pooled from 2 experiments); P 5 .007 when B6 vs Siglec-G2/2 B6 mice were used as
recipients. (D) Histopathologic analysis of GI tracts (small and large intestine) by hematoxylin and eosin stain (left) and GVHD score on days 7 and 14 after allo-HCT (n5 3 to 6 per group,
pooled from 2 experiments). Original magnification 3200. (E) Donor T-cell (H-2kd1CD41CD81) expansion in the spleen on days 7 and 14 after allo-HCT (n 5 4 to 6 per group, pooled
from 2 experiments). (F-H) Serum HMGB-1, IFN-g, and TNF-a levels on day 7 after allo-HCT (n 5 3 to 6 per group, pooled from 2 experiments). The bar shows the mean 6 standard
error of the mean (SEM).
3514 TOUBAI et al BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom

Siglec-G–deficient hosts show greater severity of GVHD
We hypothesized that the absence of Siglec-G in the irradiated re-cipients of allo-HCTwould lead toan increased inflammatory responseresulting in more severe GVHD. We used the well-established majorhistocompatibility complex (MHC)-mismatched (BALB/c→B6)model of allo-HCT, and conditioned the WT and Siglec-G2/2 B6recipients with 13 Gy radiation. They were then transplanted withTCD-BM and splenic T cells from syngeneic B6 or allogeneicBALB/c donors. Compared with WT allogeneic B6 animals, theallogeneic Siglec-G2/2 mice demonstrated significantly greatermortality from GVHD after allo-HCT (Figure 1C; P5 .007). Theenhanced mortality of the allogeneic Siglec-G2/2 mice was asso-ciated with a more severe histopathologic GVHD of the GI tract ondays17 and114 than theWT-B6 allogeneic mice (Figure 1D). Theincrease in GVHD severity and mortality was also associated withgreater donor T-cell expansion and serum levels of HMGB-1, andproinflammatory cytokines IFN-g and TNF-a in the allogeneicSiglec-G2/2 mice, compared with the allogeneic WT-B6 mice(Figure 1E-H).
To rule out strain- and model-dependent artifacts, we next eval-uated the role of Siglec-G in another well-established,MHC-matched,minor histocompatibility antigen (MiHA)-mismatched GVHDmodel(C3H.SW→B6),12 and found that Siglec-G deficiency in the hostsenhanced GVHD severity (supplemental Figure 2). Because the in-crease in DAMPs was dependent on the radiation dose, we reasonedthat reduced intensity of conditioning might not enhance GVHDdespite the absence of Siglec-G. Consistent with this notion, whenrecipient animals were conditioned with a reduced dose of irradiation(8 Gy), Siglec-G2/2 mice demonstrated a propensity toward highermortality and severity of GVHD that did not reach statisticalsignificance when compared with WT-B6 mice (supplementalFigure 3A-B).
Siglec-G expression on host hematopoietic cells is crucial for
regulating GVHD
We next evaluated the role of Siglec-G expression on host hema-topoietic- and nonhematopoietic-derived APCs in enhancing theseverity of GVHD. WT-B6 Ly5.2 or the Siglec-G2/2 B6 animalswere lethally irradiated with 11 Gy and infused with 5 3 106 BMcells and 5 3 106 splenocytes from syngeneic Ly5.1 WT-B6 orSiglec-G2/2 donors, such that Siglec-G deficiency was confined toonly the hematopoietic or the nonhematopoietic cell compartments,respectively. Chimerism analyses of the donor hematopoietic cellswere complete donor types (.95%), 3 months after first trans-plantation. The [B6→B6Ly5.2], [B6 Ly5.2→Siglec-G2/2], and the[Siglec-G2/2→B6Ly5.2] animals were then used as recipients in anallo-HCT about 3 to 4months later. The chimericmice received 9Gyandwere injected IVwith 2.53106CD901T cells alongwith 53106
BM cells from either syngeneic B6 or MHC-mismatched allogeneicBALB/c donors. All chimeras that received syngeneic T cells andBM cells survived the duration of the observation period with nosigns ofGVHD(Figure 2A).Bycontrast, the allogeneic [B6→B6Ly5.2]and the [B6Ly5.2→Siglec-G2/2] chimeras showed similar GVHD,while the [Siglec-G2/2→B6Ly5.2] chimeras showed more severeGVHD (P5 .03).
To preclude strain-specific responses, we also used the[B6→B6Ly5.2], [B6Ly5.2→Siglec-G2/2], and [Siglec-G2/2
→B6Ly5.2] chimeras as recipients in an MHC-matched, multi-ple MiHA-mismatched C3H.sw→B6 model of allo-HCT. The[B6→B6Ly5.2], [B6Ly5.2→Siglec-G2/2], and [Siglec-G2/2
→B6Ly5.2] chimeras were irradiated and transplanted with
TCD-BM and splenic T cells from B6 or allogeneic C3H.swdonors. As expected, all of the syngeneic recipients survived. Onceagain, the allogeneic [Siglec-G2/2→B6Ly5.2] animals that lackedSiglec-G expression on the host hematopoietic-derived APCs,showed greater mortality and more severe GVHD than theallogeneic [B6→B6Ly5.2] or the [B6Ly5.2→Siglec-G2/2] recipientsafter HCT (Figure 2B; P5 .02). The enhanced mortality and severityof GVHD in the allogeneic [Siglec-G2/2→B6Ly5.2] animals wasassociated with greater donor T-cell expansion on days 7 and 14in both MHC-mismatched and MHC-matched minor disparatesystems (Figure 2C-D), and was also associated with an increase inserum levels of IFN-g on day 14 after allo-HCT (supplementalFigure 4).
The enhanced mortality in these multiple models was apparentearly, after BMT. To gain further insight into the enhancement ofearly mortality, we next determined whether the presence of Siglec-Gon hostAPCs led to the regulation of only inflammation, early afterBMT, and/or if it also caused a retention or reduced the expansionof donor T cells in the secondary lymphoid organs (eg, spleenand regional lymph nodes [LN] such as mesenteric LN [MLN]),preventing T-cell egress into target tissues. Even very early afterBMT, on day11 (Figure 2E-F), the absence of Siglec-G on host cellsled to increased recovery of donor T cells in secondary LN such asMLN, and also increased the levels of proinflammatory cytokineTNFa. To further determine the role and kinetics of the early increasein donor T cells and inflammatory cytokines, we also assessed thedonor T-cell expansion and recovery in the secondary LN, andadditionally in the GVHD target tissues and GI tract on day13 afterBMT. In the absence of Siglec-G in the host APCs, allogeneic donorT cells showed greater expansion and recovery in both the secondaryLNs (Figure 2G) and the GVHD target tissues, as well as the GI tract(Figure 2H); and also showed greater levels of T-cell proinflamma-tory cytokines such as INF-g (Figure 2I). These data suggest thathost Siglec-G attenuates early mortality after allo-HCT, both byregulating enhanced donor T-cell expansion and proinflammatorycytokine milieu early after HCT. There were no detectable levels ofserum LPS early after BMT on day 1 and no increase on day 3 (datanot shown). This, when taken in light of the absence of earlymortalityin syngeneic animals despite increased conditioning, suggests thatSiglec-G–mediated regulation in early GVHD mortality is not solelydue to condition-related inflammation.
Siglec-G deficiency enhances DAMP-induced inflammatory
responses from hematopoietic APCs
To evaluate the mechanisms of enhanced GVHD in the absence ofSiglec-G on hematopoietic-derived cells, we focused on professionalhematopoietic APC subsets, namely DCs25,26 and macrophages. Wefirst determined the effect of Siglec-G deficiency on the numberand phenotype of DCs. No significant differences were observedbetween the WT or Siglec-G2/2 animals in either the percent or theabsolute numbers of CD11c1 DCs, or the absolute numbers andpercent expression of co-stimulatory molecules such as CD80, CD86,CD40, and PD-L1 (supplemental Figure 5A-F). The expression ofMHCclass II and the number of plasmacytoidDCswere also similar inWT-B6 and Siglec-G2/2mice (supplemental Figure 5G-H). Becausegranulocyte macrophage–colony stimulating factor-cultured BMDCsare considered to be a model of in vivo inflammatory monocyte-derived DCs that differentiate from circulatingmonocytes in responseto inflammation,31 we next examined the impact of Siglec-G on thegeneration of DCs from BM. The phenotype (CD80, CD86, andI-Ab) of granulocyte macrophage–colony stimulating factor-induced
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3515
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
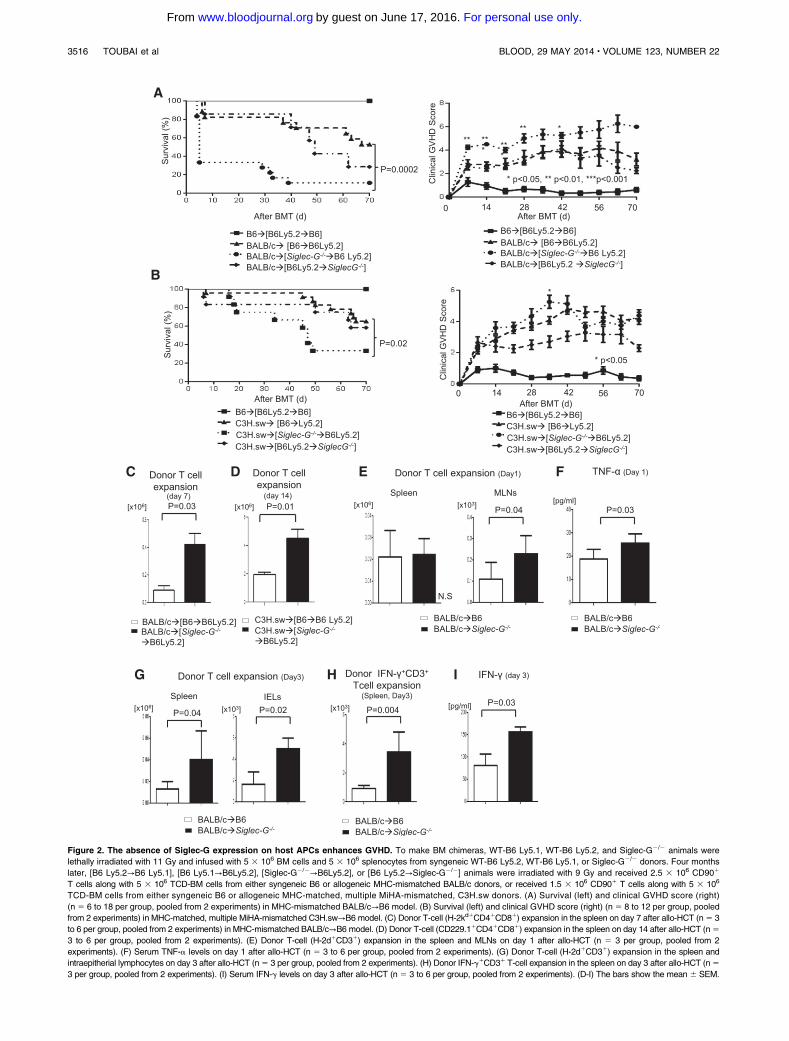
Figure 2. The absence of Siglec-G expression on host APCs enhances GVHD. To make BM chimeras, WT-B6 Ly5.1, WT-B6 Ly5.2, and Siglec-G2/2 animals were
lethally irradiated with 11 Gy and infused with 5 3 106 BM cells and 5 3 106 splenocytes from syngeneic WT-B6 Ly5.2, WT-B6 Ly5.1, or Siglec-G2/2 donors. Four months
later, [B6 Ly5.2→B6 Ly5.1], [B6 Ly5.1→B6Ly5.2], [Siglec-G2/2→B6Ly5.2], or [B6 Ly5.2→Siglec-G2/2] animals were irradiated with 9 Gy and received 2.5 3 106 CD901
T cells along with 5 3 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c donors, or received 1.5 3 106 CD901 T cells along with 5 3 106
TCD-BM cells from either syngeneic B6 or allogeneic MHC-matched, multiple MiHA-mismatched, C3H.sw donors. (A) Survival (left) and clinical GVHD score (right)
(n 5 6 to 18 per group, pooled from 2 experiments) in MHC-mismatched BALB/c→B6 model. (B) Survival (left) and clinical GVHD score (right) (n 5 8 to 12 per group, pooled
from 2 experiments) in MHC-matched, multiple MiHA-mismatched C3H.sw→B6 model. (C) Donor T-cell (H-2kd1CD41CD81) expansion in the spleen on day 7 after allo-HCT (n5 3
to 6 per group, pooled from 2 experiments) in MHC-mismatched BALB/c→B6 model. (D) Donor T-cell (CD229.11CD41CD81) expansion in the spleen on day 14 after allo-HCT (n5
3 to 6 per group, pooled from 2 experiments). (E) Donor T-cell (H-2d1CD31) expansion in the spleen and MLNs on day 1 after allo-HCT (n 5 3 per group, pooled from 2
experiments). (F) Serum TNF-a levels on day 1 after allo-HCT (n 5 3 to 6 per group, pooled from 2 experiments). (G) Donor T-cell (H-2d1CD31) expansion in the spleen and
intraepitherial lymphocytes on day 3 after allo-HCT (n5 3 per group, pooled from 2 experiments). (H) Donor IFN-g1CD31 T-cell expansion in the spleen on day 3 after allo-HCT (n5
3 per group, pooled from 2 experiments). (I) Serum IFN-g levels on day 3 after allo-HCT (n 5 3 to 6 per group, pooled from 2 experiments). (D-I) The bars show the mean 6 SEM.
3516 TOUBAI et al BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
BMDCs generated from Siglec-G2/2mice was similar to the pheno-type of WT BMDCs (supplemental Figure 6A).
We next evaluated the functional responses of conventional DCsfrom WT and Siglec-G2/2 mice. We tested the effect of Siglec-Gdeficiency on the innate immune responses of DCs that are triggeredthrough TLRs. BMDCs from either WT or Siglec-G2/2 mice werestimulated with LPS, a potent stimulator of the innate immuneresponse via TLR4. Siglec-G–deficient DCs andWTDCs respondedin a comparable manner, as determined by the secretion of IL-6,TNF-a, IL-10, IL-1b, and IL-12, as shown in a previous report4
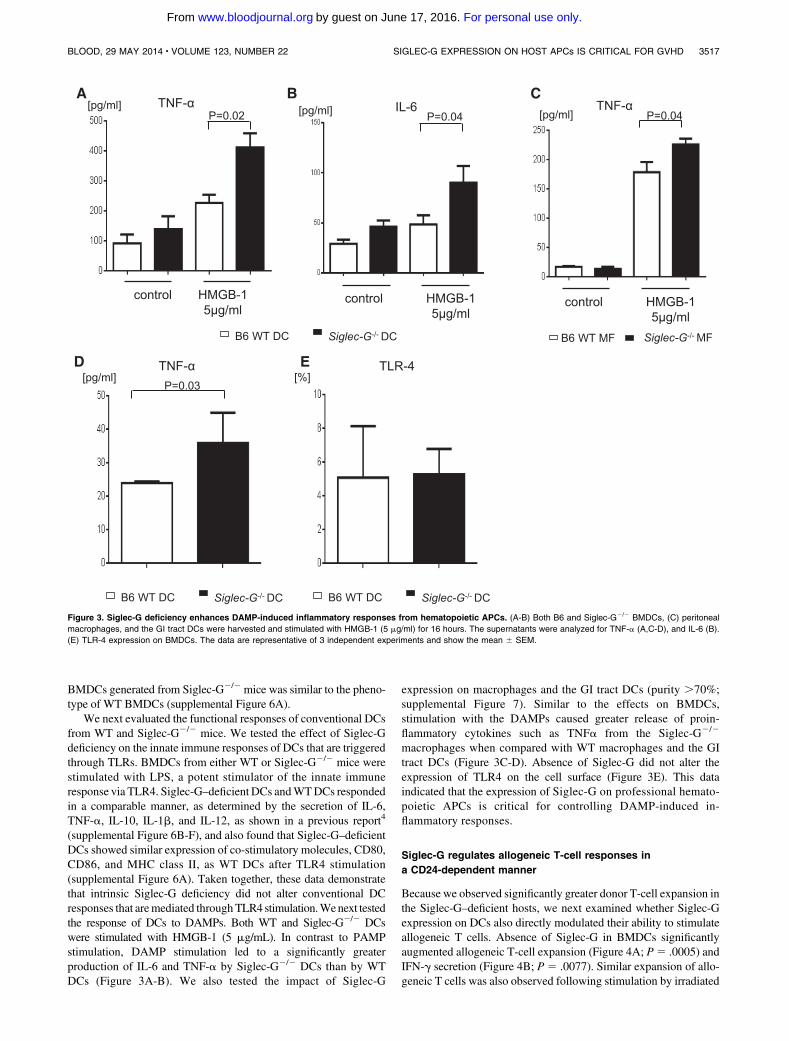
(supplemental Figure 6B-F), and also found that Siglec-G–deficientDCs showed similar expression of co-stimulatory molecules, CD80,CD86, and MHC class II, as WT DCs after TLR4 stimulation(supplemental Figure 6A). Taken together, these data demonstratethat intrinsic Siglec-G deficiency did not alter conventional DCresponses that aremediated throughTLR4 stimulation.Wenext testedthe response of DCs to DAMPs. Both WT and Siglec-G2/2 DCswere stimulated with HMGB-1 (5 mg/mL). In contrast to PAMPstimulation, DAMP stimulation led to a significantly greaterproduction of IL-6 and TNF-a by Siglec-G2/2 DCs than by WTDCs (Figure 3A-B). We also tested the impact of Siglec-G
expression on macrophages and the GI tract DCs (purity .70%;supplemental Figure 7). Similar to the effects on BMDCs,stimulation with the DAMPs caused greater release of proin-flammatory cytokines such as TNFa from the Siglec-G2/2
macrophages when compared with WT macrophages and the GItract DCs (Figure 3C-D). Absence of Siglec-G did not alter theexpression of TLR4 on the cell surface (Figure 3E). This dataindicated that the expression of Siglec-G on professional hemato-poietic APCs is critical for controlling DAMP-induced in-flammatory responses.
Siglec-G regulates allogeneic T-cell responses in
a CD24-dependent manner
Because we observed significantly greater donor T-cell expansion inthe Siglec-G–deficient hosts, we next examined whether Siglec-Gexpression on DCs also directly modulated their ability to stimulateallogeneic T cells. Absence of Siglec-G in BMDCs significantlyaugmented allogeneic T-cell expansion (Figure 4A; P5 .0005) andIFN-g secretion (Figure 4B; P5 .0077). Similar expansion of allo-geneic T cells was also observed following stimulation by irradiated
Figure 3. Siglec-G deficiency enhances DAMP-induced inflammatory responses from hematopoietic APCs. (A-B) Both B6 and Siglec-G2/2 BMDCs, (C) peritoneal
macrophages, and the GI tract DCs were harvested and stimulated with HMGB-1 (5 mg/ml) for 16 hours. The supernatants were analyzed for TNF-a (A,C-D), and IL-6 (B).
(E) TLR-4 expression on BMDCs. The data are representative of 3 independent experiments and show the mean 6 SEM.
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3517
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
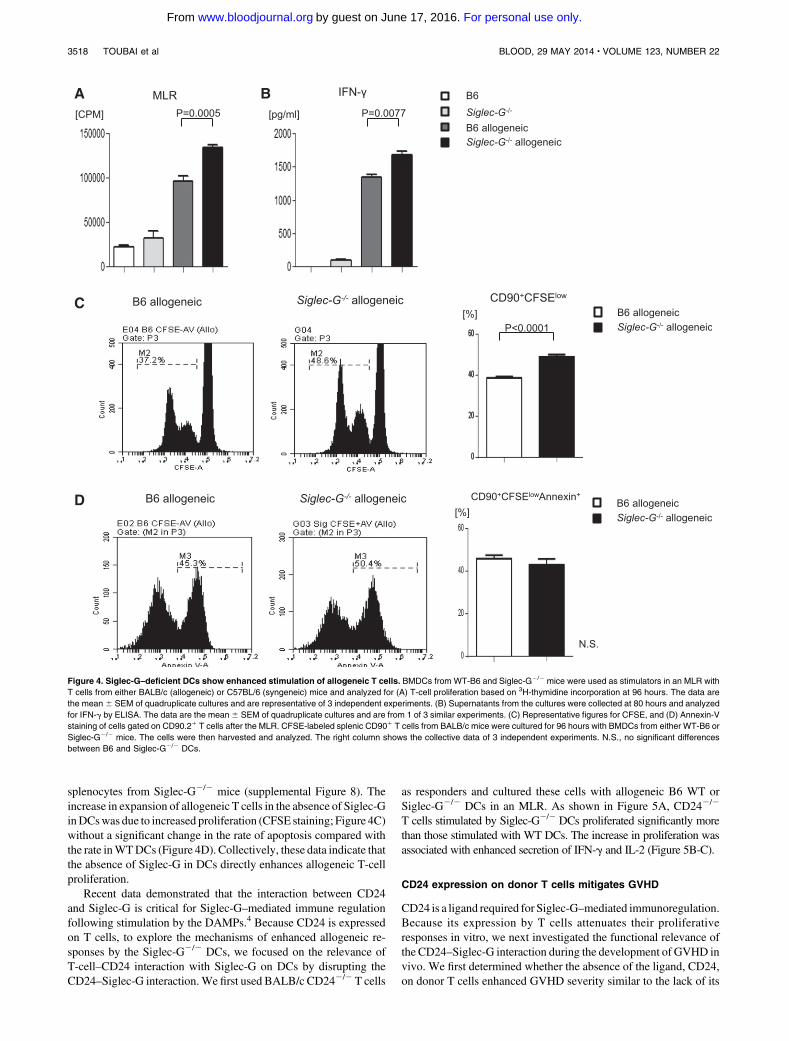
splenocytes from Siglec-G2/2 mice (supplemental Figure 8). Theincrease in expansion of allogeneic T cells in the absence of Siglec-GinDCswas due to increased proliferation (CFSE staining; Figure 4C)without a significant change in the rate of apoptosis compared withthe rate inWTDCs (Figure 4D). Collectively, these data indicate thatthe absence of Siglec-G in DCs directly enhances allogeneic T-cellproliferation.
Recent data demonstrated that the interaction between CD24and Siglec-G is critical for Siglec-G–mediated immune regulationfollowing stimulation by the DAMPs.4 Because CD24 is expressedon T cells, to explore the mechanisms of enhanced allogeneic re-sponses by the Siglec-G2/2 DCs, we focused on the relevance ofT-cell–CD24 interaction with Siglec-G on DCs by disrupting theCD24–Siglec-G interaction.We first used BALB/c CD242/2 T cells
as responders and cultured these cells with allogeneic B6 WT orSiglec-G2/2 DCs in an MLR. As shown in Figure 5A, CD242/2
T cells stimulated by Siglec-G2/2 DCs proliferated significantly morethan those stimulated with WT DCs. The increase in proliferation wasassociated with enhanced secretion of IFN-g and IL-2 (Figure 5B-C).
CD24 expression on donor T cells mitigates GVHD
CD24 is a ligand required for Siglec-G–mediated immunoregulation.Because its expression by T cells attenuates their proliferativeresponses in vitro, we next investigated the functional relevance ofthe CD24–Siglec-G interaction during the development of GVHD invivo. We first determined whether the absence of the ligand, CD24,on donor T cells enhanced GVHD severity similar to the lack of its
Figure 4. Siglec-G–deficient DCs show enhanced stimulation of allogeneic T cells. BMDCs from WT-B6 and Siglec-G2/2 mice were used as stimulators in an MLR with
T cells from either BALB/c (allogeneic) or C57BL/6 (syngeneic) mice and analyzed for (A) T-cell proliferation based on 3H-thymidine incorporation at 96 hours. The data are
the mean 6 SEM of quadruplicate cultures and are representative of 3 independent experiments. (B) Supernatants from the cultures were collected at 80 hours and analyzed
for IFN-g by ELISA. The data are the mean6 SEM of quadruplicate cultures and are from 1 of 3 similar experiments. (C) Representative figures for CFSE, and (D) Annexin-V
staining of cells gated on CD90.21 T cells after the MLR. CFSE-labeled splenic CD901 T cells from BALB/c mice were cultured for 96 hours with BMDCs from either WT-B6 or
Siglec-G2/2 mice. The cells were then harvested and analyzed. The right column shows the collective data of 3 independent experiments. N.S., no significant differences
between B6 and Siglec-G2/2 DCs.
3518 TOUBAI et al BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
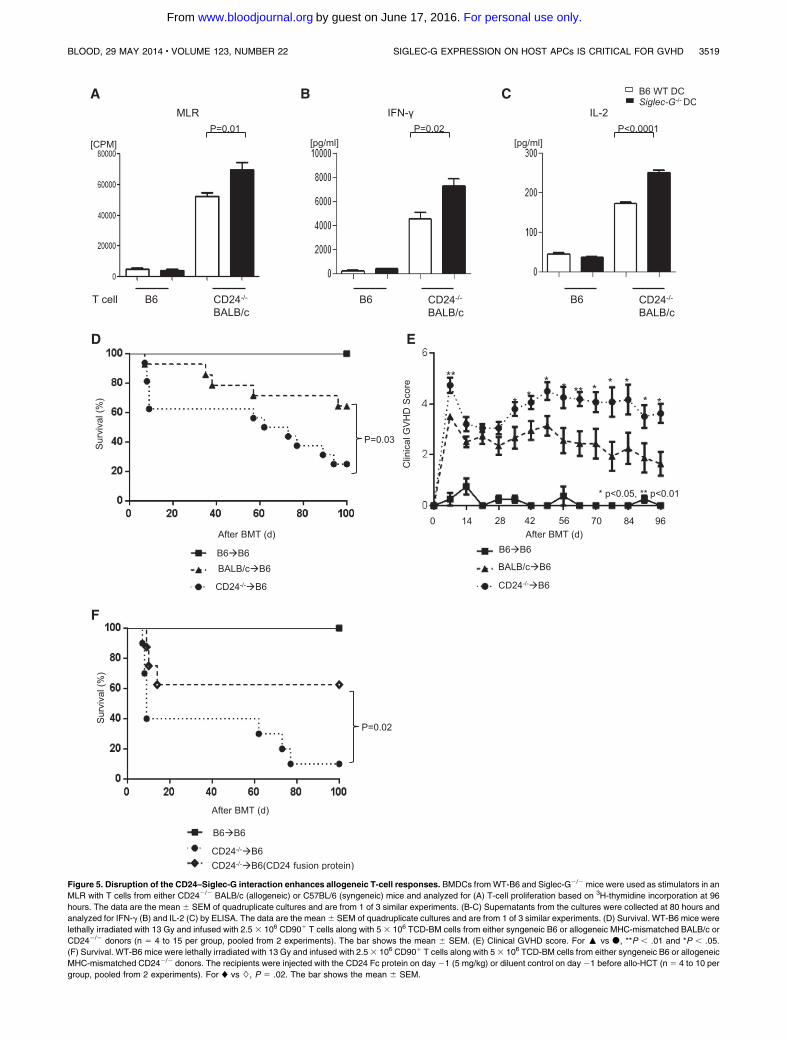
Figure 5. Disruption of the CD24–Siglec-G interaction enhances allogeneic T-cell responses. BMDCs fromWT-B6 and Siglec-G2/2 mice were used as stimulators in an
MLR with T cells from either CD242/2 BALB/c (allogeneic) or C57BL/6 (syngeneic) mice and analyzed for (A) T-cell proliferation based on 3H-thymidine incorporation at 96
hours. The data are the mean 6 SEM of quadruplicate cultures and are from 1 of 3 similar experiments. (B-C) Supernatants from the cultures were collected at 80 hours and
analyzed for IFN-g (B) and IL-2 (C) by ELISA. The data are the mean6 SEM of quadruplicate cultures and are from 1 of 3 similar experiments. (D) Survival. WT-B6 mice were
lethally irradiated with 13 Gy and infused with 2.5 3 106 CD901 T cells along with 5 3 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-mismatched BALB/c or
CD242/2 donors (n 5 4 to 15 per group, pooled from 2 experiments). The bar shows the mean 6 SEM. (E) Clinical GVHD score. For : vs d, **P , .01 and *P , .05.
(F) Survival. WT-B6 mice were lethally irradiated with 13 Gy and infused with 2.53 106 CD901 T cells along with 53 106 TCD-BM cells from either syngeneic B6 or allogeneic
MHC-mismatched CD242/2 donors. The recipients were injected with the CD24 Fc protein on day21 (5 mg/kg) or diluent control on day 21 before allo-HCT (n 5 4 to 10 per
group, pooled from 2 experiments). For ♦ vs ◊, P 5 .02. The bar shows the mean 6 SEM.
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3519
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
receptor, Siglec-G, on host APCs. The B6 recipients were lethallyirradiated and transplanted with TCD- BM and splenic T cells fromthe syngeneic B6 animals, or TCD-BM from allogeneicWTBALB/c,along with splenic T cells from either WT or CD242/2 BALB/cdonors. As shown in Figure 5D-E, all of the syngeneic animalssurvivedwith no signs ofGVHD. The allogeneic recipients that weretransplanted with WT T cells demonstrated GVHD, which wassignificantly less severe than in those animals that were transplantedwith CD242/2 T cells (P5 .03).
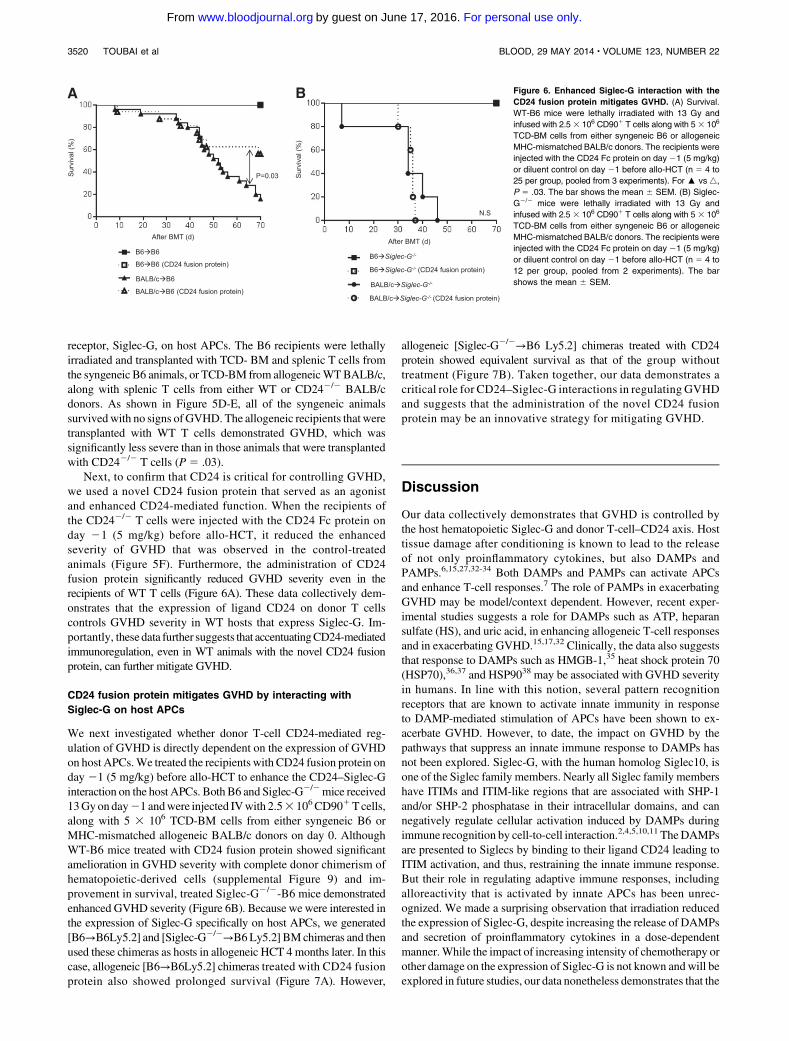
Next, to confirm that CD24 is critical for controlling GVHD,we used a novel CD24 fusion protein that served as an agonistand enhanced CD24-mediated function. When the recipients ofthe CD242/2 T cells were injected with the CD24 Fc protein onday 21 (5 mg/kg) before allo-HCT, it reduced the enhancedseverity of GVHD that was observed in the control-treatedanimals (Figure 5F). Furthermore, the administration of CD24fusion protein significantly reduced GVHD severity even in therecipients of WT T cells (Figure 6A). These data collectively dem-onstrates that the expression of ligand CD24 on donor T cellscontrols GVHD severity in WT hosts that express Siglec-G. Im-portantly, these data further suggests that accentuatingCD24-mediatedimmunoregulation, even in WT animals with the novel CD24 fusionprotein, can further mitigate GVHD.
CD24 fusion protein mitigates GVHD by interacting with
Siglec-G on host APCs
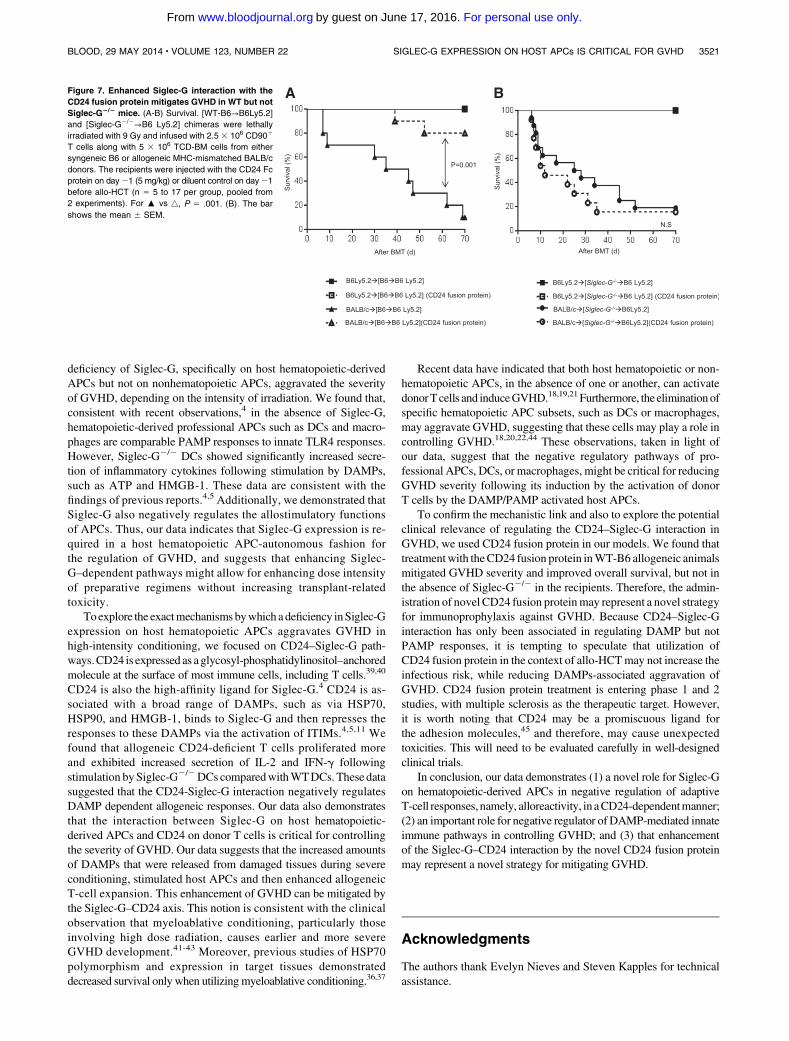
We next investigated whether donor T-cell CD24-mediated reg-ulation of GVHD is directly dependent on the expression of GVHDon host APCs.We treated the recipients with CD24 fusion protein onday 21 (5 mg/kg) before allo-HCT to enhance the CD24–Siglec-Ginteraction on the host APCs. BothB6 and Siglec-G2/2mice received13Gyonday21 andwere injected IVwith 2.53 106CD901Tcells,along with 5 3 106 TCD-BM cells from either syngeneic B6 orMHC-mismatched allogeneic BALB/c donors on day 0. AlthoughWT-B6 mice treated with CD24 fusion protein showed significantamelioration in GVHD severity with complete donor chimerism ofhematopoietic-derived cells (supplemental Figure 9) and im-provement in survival, treated Siglec-G2/2-B6 mice demonstratedenhanced GVHD severity (Figure 6B). Because we were interested inthe expression of Siglec-G specifically on host APCs, we generated[B6→B6Ly5.2] and [Siglec-G2/2→B6Ly5.2] BMchimeras and thenused these chimeras as hosts in allogeneic HCT 4 months later. In thiscase, allogeneic [B6→B6Ly5.2] chimeras treated with CD24 fusionprotein also showed prolonged survival (Figure 7A). However,
allogeneic [Siglec-G2/2→B6 Ly5.2] chimeras treated with CD24protein showed equivalent survival as that of the group withouttreatment (Figure 7B). Taken together, our data demonstrates acritical role for CD24–Siglec-G interactions in regulating GVHDand suggests that the administration of the novel CD24 fusionprotein may be an innovative strategy for mitigating GVHD.
Discussion
Our data collectively demonstrates that GVHD is controlled bythe host hematopoietic Siglec-G and donor T-cell–CD24 axis. Hosttissue damage after conditioning is known to lead to the releaseof not only proinflammatory cytokines, but also DAMPs andPAMPs.6,15,27,32-34 Both DAMPs and PAMPs can activate APCsand enhance T-cell responses.7 The role of PAMPs in exacerbatingGVHD may be model/context dependent. However, recent exper-imental studies suggests a role for DAMPs such as ATP, heparansulfate (HS), and uric acid, in enhancing allogeneic T-cell responsesand in exacerbating GVHD.15,17,32 Clinically, the data also suggeststhat response to DAMPs such as HMGB-1,35 heat shock protein 70(HSP70),36,37 and HSP9038 may be associated with GVHD severityin humans. In line with this notion, several pattern recognitionreceptors that are known to activate innate immunity in responseto DAMP-mediated stimulation of APCs have been shown to ex-acerbate GVHD. However, to date, the impact on GVHD by thepathways that suppress an innate immune response to DAMPs hasnot been explored. Siglec-G, with the human homolog Siglec10, isone of the Siglec family members. Nearly all Siglec family membershave ITIMs and ITIM-like regions that are associated with SHP-1and/or SHP-2 phosphatase in their intracellular domains, and cannegatively regulate cellular activation induced by DAMPs duringimmune recognition by cell-to-cell interaction.2,4,5,10,11 TheDAMPsare presented to Siglecs by binding to their ligand CD24 leading toITIM activation, and thus, restraining the innate immune response.But their role in regulating adaptive immune responses, includingalloreactivity that is activated by innate APCs has been unrec-ognized. We made a surprising observation that irradiation reducedthe expression of Siglec-G, despite increasing the release of DAMPsand secretion of proinflammatory cytokines in a dose-dependentmanner.While the impact of increasing intensity of chemotherapy orother damage on the expression of Siglec-G is not known andwill beexplored in future studies, our data nonetheless demonstrates that the
Figure 6. Enhanced Siglec-G interaction with the
CD24 fusion protein mitigates GVHD. (A) Survival.
WT-B6 mice were lethally irradiated with 13 Gy and
infused with 2.53 106 CD901 T cells along with 53 106
TCD-BM cells from either syngeneic B6 or allogeneic
MHC-mismatched BALB/c donors. The recipients were
injected with the CD24 Fc protein on day 21 (5 mg/kg)
or diluent control on day 21 before allo-HCT (n 5 4 to
25 per group, pooled from 3 experiments). For : vs 4,
P 5 .03. The bar shows the mean 6 SEM. (B) Siglec-
G2/2 mice were lethally irradiated with 13 Gy and
infused with 2.5 3 106 CD901 T cells along with 5 3 106
TCD-BM cells from either syngeneic B6 or allogeneic
MHC-mismatched BALB/c donors. The recipients were
injected with the CD24 Fc protein on day 21 (5 mg/kg)
or diluent control on day 21 before allo-HCT (n 5 4 to
12 per group, pooled from 2 experiments). The bar
shows the mean 6 SEM.
3520 TOUBAI et al BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
deficiency of Siglec-G, specifically on host hematopoietic-derivedAPCs but not on nonhematopoietic APCs, aggravated the severityof GVHD, depending on the intensity of irradiation. We found that,consistent with recent observations,4 in the absence of Siglec-G,hematopoietic-derived professional APCs such as DCs and macro-phages are comparable PAMP responses to innate TLR4 responses.However, Siglec-G2/2 DCs showed significantly increased secre-tion of inflammatory cytokines following stimulation by DAMPs,such as ATP and HMGB-1. These data are consistent with thefindings of previous reports.4,5 Additionally, we demonstrated thatSiglec-G also negatively regulates the allostimulatory functionsof APCs. Thus, our data indicates that Siglec-G expression is re-quired in a host hematopoietic APC-autonomous fashion forthe regulation of GVHD, and suggests that enhancing Siglec-G–dependent pathways might allow for enhancing dose intensityof preparative regimens without increasing transplant-relatedtoxicity.
Toexplore the exactmechanismsbywhich a deficiency inSiglec-Gexpression on host hematopoietic APCs aggravates GVHD inhigh-intensity conditioning, we focused on CD24–Siglec-G path-ways.CD24 is expressed as a glycosyl-phosphatidylinositol–anchoredmolecule at the surface of most immune cells, including T cells.39,40
CD24 is also the high-affinity ligand for Siglec-G.4 CD24 is as-sociated with a broad range of DAMPs, such as via HSP70,HSP90, and HMGB-1, binds to Siglec-G and then represses theresponses to these DAMPs via the activation of ITIMs.4,5,11 Wefound that allogeneic CD24-deficient T cells proliferated moreand exhibited increased secretion of IL-2 and IFN-g followingstimulation bySiglec-G2/2DCs comparedwithWTDCs.These datasuggested that the CD24-Siglec-G interaction negatively regulatesDAMP dependent allogeneic responses. Our data also demonstratesthat the interaction between Siglec-G on host hematopoietic-derived APCs and CD24 on donor T cells is critical for controllingthe severity of GVHD. Our data suggests that the increased amountsof DAMPs that were released from damaged tissues during severeconditioning, stimulated host APCs and then enhanced allogeneicT-cell expansion. This enhancement of GVHD can be mitigated bythe Siglec-G–CD24 axis. This notion is consistent with the clinicalobservation that myeloablative conditioning, particularly thoseinvolving high dose radiation, causes earlier and more severeGVHD development.41-43 Moreover, previous studies of HSP70polymorphism and expression in target tissues demonstrateddecreased survival only when utilizingmyeloablative conditioning.36,37
Recent data have indicated that both host hematopoietic or non-hematopoietic APCs, in the absence of one or another, can activatedonorTcells and induceGVHD.18,19,21 Furthermore, the eliminationofspecific hematopoietic APC subsets, such as DCs or macrophages,may aggravate GVHD, suggesting that these cells may play a role incontrolling GVHD.18,20,22,44 These observations, taken in light ofour data, suggest that the negative regulatory pathways of pro-fessional APCs, DCs, or macrophages, might be critical for reducingGVHD severity following its induction by the activation of donorT cells by the DAMP/PAMP activated host APCs.
To confirm the mechanistic link and also to explore the potentialclinical relevance of regulating the CD24–Siglec-G interaction inGVHD, we used CD24 fusion protein in our models. We found thattreatmentwith theCD24 fusion protein inWT-B6 allogeneic animalsmitigated GVHD severity and improved overall survival, but not inthe absence of Siglec-G2/2 in the recipients. Therefore, the admin-istration of novel CD24 fusion proteinmay represent a novel strategyfor immunoprophylaxis against GVHD. Because CD24–Siglec-Ginteraction has only been associated in regulating DAMP but notPAMP responses, it is tempting to speculate that utilization ofCD24 fusion protein in the context of allo-HCTmay not increase theinfectious risk, while reducing DAMPs-associated aggravation ofGVHD. CD24 fusion protein treatment is entering phase 1 and 2studies, with multiple sclerosis as the therapeutic target. However,it is worth noting that CD24 may be a promiscuous ligand forthe adhesion molecules,45 and therefore, may cause unexpectedtoxicities. This will need to be evaluated carefully in well-designedclinical trials.
In conclusion, our data demonstrates (1) a novel role for Siglec-Gon hematopoietic-derived APCs in negative regulation of adaptiveT-cell responses,namely, alloreactivity, in aCD24-dependentmanner;(2) an important role for negative regulator of DAMP-mediated innateimmune pathways in controlling GVHD; and (3) that enhancementof the Siglec-G–CD24 interaction by the novel CD24 fusion proteinmay represent a novel strategy for mitigating GVHD.
Acknowledgments
The authors thank Evelyn Nieves and Steven Kapples for technicalassistance.
Figure 7. Enhanced Siglec-G interaction with the
CD24 fusion protein mitigates GVHD in WT but not
Siglec-G2/2 mice. (A-B) Survival. [WT-B6→B6Ly5.2]
and [Siglec-G2/2→B6 Ly5.2] chimeras were lethally
irradiated with 9 Gy and infused with 2.5 3 106 CD901
T cells along with 5 3 106 TCD-BM cells from either
syngeneic B6 or allogeneic MHC-mismatched BALB/c
donors. The recipients were injected with the CD24 Fc
protein on day21 (5 mg/kg) or diluent control on day21
before allo-HCT (n 5 5 to 17 per group, pooled from
2 experiments). For : vs 4, P 5 .001. (B). The bar
shows the mean 6 SEM.
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3521
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom

This work was supported by grants from the National In-stitutes of Health (National Cancer Institute, CA-0173878,CA143379, and National Heart, Lung and Blood Institute, HL-090775) (P.R.).
Authorship
Contribution: T.T. performed experiments, analyzed the data, andwrote the paper; G.H. and N.M. performed experiments; C.L.performed experiments and histopathologic analysis; Y.W.,K.O.W.,
E.C., C.R., R.E., Y.S., and J.W. performed experiments; S.W.C. andD.F. contributed reagents; P.Z. contributed reagents and helped towrite the paper; Y.L. analyzed the data, contributed reagents, andhelped to write the paper; and P.R. designed experiments, analyzedthe data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competingfinancial interests.
Correspondence: Pavan Reddy, Department of Internal Medi-cine, Division of Hematology and Oncology, Blood and MarrowTransplantation Program, University of Michigan ComprehensiveCancer Center, 3312 CCGC, 1500 East Medical Center Dr, AnnArbor, MI 48105-1942; e-mail: [email protected].
References
1. Iwasaki A, Medzhitov R. Regulation of adaptiveimmunity by the innate immune system. Science.2010;327(5963):291-295.
2. Crocker PR, Paulson JC, Varki A. Siglecs andtheir roles in the immune system. Nat RevImmunol. 2007;7(4):255-266.
3. Ravetch JV, Lanier LL. Immune inhibitoryreceptors. Science. 2000;290(5489):84-89.
4. Chen GY, Tang J, Zheng P, Liu Y. CD24 andSiglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323(5922):1722-1725.
5. Chen GY, Chen X, King S, et al. Amelioration ofsepsis by inhibiting sialidase-mediated disruptionof the CD24-SiglecG interaction. Nat Biotechnol.2011;29(5):428-435.
6. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561.
7. Blazar BR, Murphy WJ, Abedi M. Advances ingraft-versus-host disease biology and therapy.Nat Rev Immunol. 2012;12(6):443-458.
8. Akira S, Uematsu S, Takeuchi O. Pathogenrecognition and innate immunity. Cell. 2006;124(4):783-801.
9. Kono H, Rock KL. How dying cells alert theimmune system to danger. Nat Rev Immunol.2008;8(4):279-289.
10. Liu Y, Chen GY, Zheng P. Sialoside-basedpattern recognitions discriminating infections fromtissue injuries. Curr Opin Immunol. 2011;23(1):41-45.
11. Liu Y, Chen GY, Zheng P. CD24-Siglec G/10discriminates danger- from pathogen-associatedmolecular patterns. Trends Immunol. 2009;30(12):557-561.
12. Shlomchik WD, Couzens MS, Tang CB, et al.Prevention of graft versus host disease byinactivation of host antigen-presenting cells.Science. 1999;285(5426):412-415.
13. Teshima T, Ordemann R, Reddy P, et al. Acutegraft-versus-host disease does not requirealloantigen expression on host epithelium. NatMed. 2002;8(6):575-581.
14. Reddy P, Maeda Y, Liu C, Krijanovski OI,Korngold R, Ferrara JL. A crucial role forantigen-presenting cells and alloantigenexpression in graft-versus-leukemia responses.Nat Med. 2005;11(11):1244-1249.
15. Wilhelm K, Ganesan J, Muller T, et al. Graft-versus-host disease is enhanced by extracellularATP activating P2X7R. Nat Med. 2010;16(12):1434-1438.
16. Jankovic D, Ganesan J, Bscheider M, et al. TheNlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med. 2013;210(10):1899-1910.
17. Brennan TV, Lin L, Huang X, et al. Heparansulfate, an endogenous TLR4 agonist, promotes
acute GVHD after allogeneic stem celltransplantation. Blood. 2012;120(14):2899-2908.
18. Koyama M, Kuns RD, Olver SD, et al. Recipientnonhematopoietic antigen-presenting cells aresufficient to induce lethal acute graft-versus-hostdisease. Nat Med. 2011;18(1):135-142.
19. Li H, Demetris AJ, McNiff J, et al. Profounddepletion of host conventional dendritic cells,plasmacytoid dendritic cells, and B cells does notprevent graft-versus-host disease induction.J Immunol. 2012;188(8):3804-3811.
20. Markey KA, Banovic T, Kuns RD, et al.Conventional dendritic cells are the critical donorAPC presenting alloantigen after experimentalbone marrow transplantation. Blood. 2009;113(22):5644-5649.
21. Toubai T, Tawara I, Sun Y, et al. Induction ofacute GVHD by sex-mismatched H-Y antigensin the absence of functional radiosensitive hosthematopoietic-derived antigen-presenting cells.Blood. 2012;119(16):3844-3853.
22. Hashimoto D, Chow A, Greter M, et al.Pretransplant CSF-1 therapy expands recipientmacrophages and ameliorates GVHD afterallogeneic hematopoietic cell transplantation.J Exp Med. 2011;208(5):1069-1082.
23. Ding C, Liu Y, Wang Y, et al. Siglecg limits thesize of B1a B cell lineage by down-regulatingNFkappaB activation. PLoS ONE. 2007;2(10):e997.
24. Nielsen PJ, Lorenz B, Muller AM, et al. Alterederythrocytes and a leaky block in B-celldevelopment in CD24/HSA-deficient mice. Blood.1997;89(3):1058-1067.
25. Reddy P, Sun Y, Toubai T, et al. Histonedeacetylase inhibition modulates indoleamine2,3-dioxygenase-dependent DC functions andregulates experimental graft-versus-host diseasein mice. J Clin Invest. 2008;118(7):2562-2573.
26. Toubai T, Sun Y, Tawara I, et al. Ikaros-Notchaxis in host hematopoietic cells regulatesexperimental graft-versus-host disease. Blood.2011;118(1):192-204.
27. Cooke KR, Hill GR, Crawford JM, et al.Tumor necrosis factor- alpha production tolipopolysaccharide stimulation by donor cellspredicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102(10):1882-1891.
28. Hill GR, Cooke KR, Teshima T, et al. Interleukin-11 promotes T cell polarization and preventsacute graft-versus-host disease after allogeneicbone marrow transplantation. J Clin Invest. 1998;102(1):115-123.
29. Zheng X, Wu W, Liu Y, Zheng P. Methods of useof soluble CD24 for therapy of rheumatoidarthritis. WIPO Patent 2011139820.
30. Hill GR, Crawford JM, Cooke KR, Brinson YS,Pan L, Ferrara JL. Total body irradiation andacute graft-versus-host disease: the role of
gastrointestinal damage and inflammatorycytokines. Blood. 1997;90(8):3204-3213.
31. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH.Differential development of murine dendritic cellsby GM-CSF versus Flt3 ligand has implications forinflammation and trafficking. J Immunol. 2007;179(11):7577-7584.
32. Tsukamoto H, Chernogorova P, Ayata K, et al.Deficiency of CD73/ecto-59-nucleotidase in miceenhances acute graft-versus-host disease. Blood.2012;119(19):4554-4564.
33. Nestel FP, Price KS, Seemayer TA, Lapp WS.Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alphaduring graft-versus-host disease. J Exp Med.1992;175(2):405-413.
34. Stenger EO, Turnquist HR, Mapara MY, ThomsonAW. Dendritic cells and regulation of graft-versus-host disease and graft-versus-leukemia activity.Blood. 2012;119(22):5088-5103.
35. Kornblit B, Masmas T, Petersen SL, et al.Association of HMGB1 polymorphisms withoutcome after allogeneic hematopoietic celltransplantation. Biol Blood Marrow Transplant.2010;16(2):239-252.
36. Jarvis M, Marzolini M, Wang XN, Jackson G,Sviland L, Dickinson AM. Heat shock protein 70:correlation of expression with degree of graft-versus-host response and clinical graft-versus-host disease. Transplantation. 2003;76(5):849-853.
37. Bogunia-Kubik K, Lange A. HSP70-hom genepolymorphism in allogeneic hematopoietic stem-cell transplant recipients correlates with thedevelopment of acute graft-versus-host disease.Transplantation. 2005;79(7):815-820.
38. Stuehler C, Mielke S, Chatterjee M, et al.Selective depletion of alloreactive T cells bytargeted therapy of heat shock protein 90: a novelstrategy for control of graft-versus-host disease.Blood. 2009;114(13):2829-2836.
39. Liu Y, Zheng P. CD24: a genetic checkpointin T cell homeostasis and autoimmunediseases. Trends Immunol. 2007;28(7):315-320.
40. Li O, Zheng P, Liu Y. CD24 expression on T cellsis required for optimal T cell proliferation inlymphopenic host. J Exp Med. 2004;200(8):1083-1089.
41. Lioure B, Bene MC, Pigneux A, et al; GOELAMS.Early matched sibling hematopoietic celltransplantation for adult AML in first remissionusing an age-adapted strategy: long-term resultsof a prospective GOELAMS study. Blood. 2012;119(12):2943-2948.
42. Jagasia M, Arora M, Flowers ME, et al. Riskfactors for acute GVHD and survival afterhematopoietic cell transplantation. Blood. 2012;119(1):296-307.
3522 TOUBAI et al BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom

43. Aoudjhane M, Labopin M, Gorin NC, et al; AcuteLeukemia Working Party (ALWP) of the Europeangroup for Blood and Marrow Transplantation(EBMT). Comparative outcome of reducedintensity and myeloablative conditioningregimen in HLA identical sibling allogeneichaematopoietic stem cell transplantation forpatients older than 50 years of age with acute
myeloblastic leukaemia: a retrospective survey
from the Acute Leukemia Working Party
(ALWP) of the European group for Blood and
Marrow Transplantation (EBMT). Leukemia.
2005;19(12):2304-2312.
44. Diamond MS, Kinder M, Matsushita H, et al. Type
I interferon is selectively required by dendritic cells
for immune rejection of tumors. J Exp Med. 2011;
208(10):1989-2003.
45. Friederichs J, Zeller Y, Hafezi-Moghadam A,
Grone HJ, Ley K, Altevogt P. The CD24/P-
selectin binding pathway initiates lung arrest of
human A125 adenocarcinoma cells. Cancer Res.
2000;60(23):6714-6722.
BLOOD, 29 MAY 2014 x VOLUME 123, NUMBER 22 SIGLEC-G EXPRESSION ON HOST APCs IS CRITICAL FOR GVHD 3523
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom
online April 2, 2014 originally publisheddoi:10.1182/blood-2013-12-545335
2014 123: 3512-3523
Choi, Dexing Fang, Pan Zheng, Yang Liu and Pavan ReddyWonOravecz-Wilson, Emily Cummings, Corinne Rossi, Rebecca Evers, Yaping Sun, Julia Wu, Sung
Tomomi Toubai, Guoqing Hou, Nathan Mathewson, Chen Liu, Ying Wang, Katherine mice
CD24 axis controls the severity of graft-versus-host disease in−Siglec-G
http://www.bloodjournal.org/content/123/22/3512.full.htmlUpdated information and services can be found at:
(2147 articles)Transplantation Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on June 17, 2016. by guest www.bloodjournal.orgFrom





























