Embed Size (px)
Citation preview

Available online at www.sciencedirect.comEUROPEAN
European Polymer Journal 44 (2008) 1378–1389
www.elsevier.com/locate/europolj
POLYMERJOURNAL
Site-selective protein glycation and PEGylation
Stefano Salmaso, Alessandra Semenzato, Sara Bersani, Francesca Mastrotto,Anna Scomparin, Paolo Caliceti *
Department of Pharmaceutical Sciences, University of Padua, Via F. Marzolo, 5-35131 Padua, Italy
Received 19 December 2007; received in revised form 19 February 2008; accepted 20 February 2008Available online 29 February 2008
Abstract
A two step protocol has been set up to selectively conjugate PEG to buried amino acids of proteins. The processinvolves site-specific glycation followed by PEGylation of the oxidized glycosides. Aimed at glycating the cysteine groupsof proteins, two maleimide-glycosylic linkers have been synthesised: galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide and maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide. The linkers were extensivelycharacterized by 1H NMR, FT-IR, ESI–TOF mass spectrometry and colorimetric assays. Complete conjugation of theactivated linkers to Cys34 of human serum albumin was obtained in about 2 h. The selective oxidation of the galactosyland maltosyl moieties by periodate treatment yielded two and three available aldehyde groups, respectively. The PEG-hydrazide conjugation to the aldehyde groups was found to be 100% in about 40 h, whereas less than 30% protein mod-ification was obtained by direct conjugation of commercial PEG-maleimide to Cys34. The pH dependent PEG-glycosylhydrazone bond hydrolysis at various pH values was verified. PEG release was faster under mild acidic and basic condi-tions than at neutral pH. Furthermore, the maltosyl derivatives, by virtue of the higher number of coupled PEG chains,showed a slower protein release as compared to the galactosyl counterpart, indicating that the choice of the glycosylic lin-ker allows for control of protein release kinetics.� 2008 Elsevier Ltd. All rights reserved.
Keywords: PEG; Protein conjugation; Selective PEGylation; Protein glycation; Glycosylic linkers
1. Introduction
Over the past decades, various effective PEGyla-tion procedures have been set up to produce proteinderivatives with enhanced biopharmaceutical andphysicochemical properties [1–3]. However, the lackof conjugation selectivity has often been found to
0014-3057/$ - see front matter � 2008 Elsevier Ltd. All rights reserved
doi:10.1016/j.eurpolymj.2008.02.021
* Corresponding author. Tel.: +39 0498077646; fax: +390498275366.
E-mail address: [email protected] (P. Caliceti).
limit the development of active therapeutic biocon-jugates. Random polymer conjugation yields, infact, inhomogeneous derivatives with undefinedcomposition, significant loss in biological activityand unpredictable in vivo behaviour. In particular,signaling proteins, namely interleukins and hor-mones, have been found to undergo dramatic activ-ity decreases upon random PEGylation [4,5].
In order to overcome these problems, new andcreative approaches for site selective polymer conju-gation have been developed. Basically, these strate-gies rely on the exploitation of rare chemoselective
.

S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1379
anchors, which may be either naturally presentor artificially introduced into the protein backbone[6].
Amino acids seldom present in the protein struc-ture are natural sites for selective polymer coupling.Cysteine, for example, can be targeted for site-spe-cific conjugation with thiol reactive maleimide orvinylsulfone activated PEGs because the reducedform of this amino acid is infrequently available innatural proteins [1,3]. Furthermore, cysteine resi-dues can be properly introduced into the proteinsequence by engineering techniques without signifi-cant loss of biological activity, thus providing fortailor made polymer conjugation [7,8]. Cys–Cys res-idues can be PEGylated by reduction and bis-alkyl-ation to give a three-carbon bridge to which PEG iscovalently attached [9], while NH2-terminal serinesor threonines, can be PEGylated after oxidation[10]. PEGylation selectivity can also be achievedby discriminating the amino acid reactivity underdefined conjugation conditions. Typically, the differ-ences among the pKa values of the a-NH2 of the N-terminal amino acid (7.6–8.0), the e-NH2 of lysines(9.3–10.2) and the amino group of the histidine ring(5.5–7.5) have been exploited for production ofmono-PEGylated derivatives, such as PEG-filgas-trim or PEG-interferon [11,12]. Other site-specificPEGylation techniques involve the use of moresophisticated protein engineering procedures, tag-based technologies, or enzymes, namely transgluta-minase and carboxypeptidase, which can PEGylatedefined glutamines exposed on the protein surfaceor terminal carboxylic moieties [13–16]. Finally, gly-coproteins can be selectively oxidized at the level ofthe glycosylic functionality and then PEGylatedwith PEG-hydrazide (PEG-Hz) [17,18].
However, the inaccessibility of buried anchorsites as well as the large hydrodynamic size and highflexibility of PEG may hamper the selective polymerconjugation of targeted amino acids. Cysteine, forexample, when available in the reduced form, is pro-tected in cavities that prevent Cys–Cys mediatedprotein dimerization [19]. Terminal amino groupsof aromatic amino acids are often located in hydro-phobic pockets or involved in hydrophobic interac-tions, which prevent their direct PEGylation [20].Finally, enzyme mediated PEGylation may be lim-ited by the steric hindrance of the macromoleculesinvolved in the conjugation process. Nevertheless,techniques that enhance the coupling site exposure,namely by reversible unfolding, may, on one hand,reduce the site-directed specificity and, on the other,
induce protein inactivation by misfolding or aggre-gation [21,22].
Hetero-bifunctional or multifunctional lowmolecular weight linkers may offer a proper solutionto these problems as they can easily react with bur-ied protein functional groups and act as PEGylationsites [23].
Recently, Neose disclosed a fascinating PEGyla-tion procedure, GlycoPEGylation, which involvesthe selective glycosylation of serines and threoninesin proteins expressed without glycosylation in Esch-
erichia coli, followed by the conjugation of sialicacid derivatized PEG to the introduced N-acetylgalactosamine (GalNAc) residues by sialyltransfer-ase [24].
In order to expand the potential advantages ofthis two step procedure, we set up a method forselective modification of buried amino acids, whichrelies on the chemical glycation of proteins followedby attachment of PEG chains to the glycosylic func-tional groups. Two glycosylic linkers were properlysynthesised and activated in order to glycate thethiol group of Cys34 of human serum albumin(HSA), which was used as protein model. TheHSA glycation and PEGylation of the oxidized link-ers was compared to the direct protein PEGylationperformed using commercial PEG-maleimide. Thecompositions of the PEGylated derivatives as wellas their stabilities were evaluated.
2. Experimental section
Lactose (O-p-D-galactopyranosyl-[1,4]-a-D-glu-copyranose), maltotriose (O-a-D-glucopyranosyl-[1,4]-O-a-D-glucopyranosyl-[1,4]-D-glucopyranose),1,12-diaminododecane, N-succinimidyl 3-maleimi-dopropionate, triethylamine (Et3N) and humanserum albumin (HSA) were purchased from FlukaBioChemika (Buchs SG, Switzerland). Potassiumhydroxide, iodine and sodium m-periodate werepurchased from Riedel-de-Haen (Seelze, Germany).Monomethoxy-PEG hydrazide (10 kDa PEG-Hz)and 10 kDa monomethoxy-PEG maleimide (10kDa PEG-maleimide) were purchased from NektarTherapeutics (Huntsville, AL, USA). 2,4-Dinitro-phenylhydrazine (DNPH), 5,50-dithiobis(2-nitro-benzoic acid) (DTNB), 2,4,6-trinitrobenzensulfonicacid (TNBS) and all other reagents were purchasedfrom Sigma–Aldrich (St. Louis, MO, USA). Thesolvents were furnished by Carlo Erba (Milan,Italy). Solvents and reagents were of analytical orHPLC grade.

1380 S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389
Fourier transform infrared (FT-IR) analyseswere carried out with a Perkin–Elmer FT-IR 1600series spectrophotometer (Norworlk, CT, USA)using KBr salt. 1H NMR spectra were recordedon a Bruker Spectrospin AMX 300 MHz (Fallan-den, Switzerland) using D2O as internal standard(4.79 ppm). NMR signals were indicated as singlet(s) broad singlet (bs), doublet (d), triplet (t),multiplet (m) and broad multiplet (bm); couplingconstants of peak multiplicities (J) have beenexpressed in Hz. ESI–TOF mass spectra (ESI-MS)of the linkers were recorded on an Applied Biosys-tems Mariner ESI–TOF instrument (Monza, MI,Italy) operating in the positive ion mode.
2.1. Synthesis and characterization of maleimide-
activated glycosyl-spacer arm linkers
2.1.1. Synthesis of galactosyl-glucono-CO–NH–(CH2)12–NH2 and maltosyl-glucono-CO–NH–
(CH2)12–NH2
1,12-Diaminedodecane (9.0 g, 45.8 mmol) andtriethylamine (Et3N) (638 ll, 4.6 mmol) were dis-solved in methanol (18 ml) and the reaction mixturewas heated at refluxing temperature (65 �C). Lac-tonolactone (520 mg, 1.5 mmol) or maltotrionolac-tone (769 mg, 1.5 mmol), prepared according tothe procedure described in the literature [25], weredissolved in methanol (7 ml) and then introduceddropwise into the reaction mixture under magneticstirring. After 2 h, the mixture was cooled at roomtemperature and filtered to remove triethylammo-nium chloride. The methanol solution was concen-trated under vacuum and the crude product wasrecovered by precipitation in ice-cold diethyl ethercontaining 0.1% Et3N. The precipitate was dis-solved in methanol and reprecipitated in diethylether to remove the unreacted 1,12-diaminedode-cane. The galactosyl-glucono-CO–NH–(CH2)12–NH2 and maltosyl-glucono-CO–NH–(CH2)12–NH2
compounds were collected as white powders by cen-trifugation at 5000 rpm, left under reduced pressureuntil complete residual solvent removal and storedunder anhydrous conditions in desiccators. Theproduct recovery yields were 87% molar for galacto-syl-glucono-CO–NH–(CH2)12–NH2 [galactosyl-glucono-CO–NH–(CH2)12–NH2/lactonolactonemolar ratio, %] and 65% molar for maltosyl-glucono-CO–NH–(CH2)12–NH2 [maltosyl-glucono-CO–NH–(CH2)12–NH2/maltotrionolactone molar ratio,%]. The products were analysed by FT-IR, 1H
NMR (D2O), ESI-MS and TNBS assay for theamino group determination [26].
2.1.2. Galactosyl-glucono-CO–NH–(CH2)12–NH2
FT-IR (KBr): the disappearance of the galacto-syl-gluconolactone cyclic ester peaks at m 1741cm�1 (stretching C@O) and 1240 cm�1 (stretchingC–O) was observed. 1H NMR (300 MHz, D2O):1.32 (bs, 16H), 1.59 (bm, 4H), 2.87 (m, 2H), 3.28(m, 2H), 3.5–4.2 (bm, 12H), 4.40 (d, 1H, J = 3.3)ppm. ESI-MS: m/e calculated for C24H48N2O11,540.33; found 541.31 (M+H)1+ and 271.16(M+2H)2+. TNBS assay: 92%.
2.1.3. Maltosyl-glucono-CO–NH–(CH2)12–NH2
FT-IR (KBr): the disappearance of the maltosyl-gluconolactone corresponding to the cyclic esterbonds at m 1739 cm�1 (stretching C@O) and 1231cm�1 (stretching C–O) was observed. 1H NMR(300 MHz, D2O): 1.33 (bs, 16 H), 1.59 (bm, 4H),2.89 (m, 2H), 3.28 (m, 2H), 3.5–4.8 (bm, 22H),5.20 (d, 1H, J = 3.9), 5.43 (d, 1H, J = 3.9) ppm.ESI-MS: m/e calculated for C30H58N2O16, 702.79;found 703.41 (M+H)1+ and 352.21 (M+2H)2+.TNBS assay: 95%.
2.1.4. Synthesis of galactosyl-glucono-CO–NH–
(CH2)12–NH–CO–(CH2)2-maleimide and maltosyl-glucono-CO–NH–(CH2)12–NH–CO–
(CH2)2-maleimide
Galactosyl-glucono-CO–NH–(CH2)12–NH2 (21.8mg, 40.2 lmol) or maltosyl-glucono-CO–NH–(CH2)12–NH2 (28.2 mg, 40.2 lmol) and Et3N(5.6 ll, 40.2 lmoles) were dissolved in anhydrousdimethylsulfoxide (1.0 ml) and added to N-succin-imidyl 3-maleimidopropionate [NHS-CO-(CH2)2-maleimide] (15 mg, 56.3 lmol). After 15 min ofvigorous stirring, the solutions were precipitated inice-cold diethyl ether and the galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide ormaltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide were recovered by centrifugation at5000 rpm and washed twice in diethyl ether. Theproduct recovery yields were 50% molar for galacto-syl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide [galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide/galactosyl-glucono-CO–NH–(CH2)12–NH2 molar ratio, %] and 84%molar for the maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide [maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2–maleimide/malto-syl-glucono-CO–NH–(CH2)12–NH2 molar ratio, %].

S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1381
The products were analysed by 1H NMR (D2O),ESI-MS, TNBS test and DTNB assay for evalua-tion of the maleimide groups [27].
2.1.5. Galactosyl-glucono-CO–NH–(CH2)12–NH–
CO–(CH2)2–maleimide1H NMR (300 MHz, D2O): 1.30 (bs, 16H), 1.42
(bt, 2H), 1.54 (bt, 2H), 2.50 (t, 2H, J = 6.3), 3.11 (t,2H, J = 6.6), 3.5–4.2 (bm, 16H), 4.40 (d, 1H,J = 3.3), 6.88 (s, 2H) ppm. ESI-MS: m/e calculatedfor C31H53N3O14, 691.76; found 692.37 (M+H)1+
and 346.70 (M+2H)2+. TNBS assay: 4%. DTNBassay: 95%.
2.1.6. Maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide
1H NMR (300 MHz, D2O): 1.31 (bs, 16H), 1.44(bt, 2H), 1.56 (bt, 2H), 2.52 (t, 2H, J = 6.3), 3.13 (t,2H, J = 6.6), 3.28 (bm, 4H), d 3.5–4.8 (bm, 22H),5.20 (d, 1H, J = 3.9), 5.43 (d, 1H, J = 3.9), 6.92 (s,2H) ppm. ESI-MS: m/e calculated for C37H63-N3O19, 853.41; found 854.41 (M+H)1+ and 427.71(M+2H)2+. The TNBS assay: 1%. DTNB assay:92%.
2.2. High performance liquid chromatography
analysis
Size-exclusion analyses (SEC-HPLC) were runon a Jasco PU-1580 system with the UV-1575 detec-tor set at 280 nm. The system was equipped with aBio-Gel SEC 40-XL column (5 lm, 300 � 7.8 mm;BioRad, Milan, Italy) and isocratically eluted with63 mM phosphate buffer, 3% isopropanol, pH 7.2,at a flow rate of 0.6 ml/min.
2.3. PEGylation of HSA at Cys34 with PEG-
maleimide
Ten kiloDalton PEG-maleimide (15 mg, 1.5 lmol)was added to HSA solutions (10 mg/ml, 0.144 mM)in 50 mM phosphate, 10 mM EDTA (PBS/EDTA),pH 6.5, to obtain a 1:10 protein:polymer molar ratio.The reaction solution was maintained at room tem-perature with mild shaking and analysed at scheduledtimes by SEC-HPLC.
2.4. HSA glycation and PEG-Hz conjugation
2.4.1. HSA glycation
Ten microliters of a 144 mM galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide or
maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide solution in methanol were taken andprocessed under vacuum to eliminate the organicsolvent. Then, 1 ml of a 10 mg/ml solution ofHSA (144 lM) in PBS/EDTA, pH 6.5, was addedto the dried product to yield a 10:1 linker:HSAmolar ratio. The reaction solution was maintainedat room temperature with mild shaking for 2 hand then the excess of linker was removed by gel fil-tration chromatography performed with a SepahdexG-25 resin. The elution fractions containing the pro-tein were collected and concentrated by ultra-filtra-tion using a cut-off membrane of 10 kDa. Theprotein content in the concentrated solution wasdetermined by optical density at 280 nm and theresidual protein sulfhydryl groups were determinedby DTNB assay.
2.4.2. Periodate oxidation of glycosylated HSA
The glycosylated HSA was selectively oxidizedaccording to the adapted protocol reported in theliterature [19]. Sodium m-periodate (200 mM in mil-liQ water) was added to the protein solution to givea final concentration of 10 mM. The reaction solu-tion was maintained in the dark in an ice bath for90 min and then quenched by addition of 500 mMmannitol solution in milliQ water to yield a15 mM mannitol concentration. The solution wasultrafiltered with Centricon-10 tubes (3 kDa cut-off) at 2500 rpm and washed at least 6 times withPBS/EDTA, pH 6.5. The protein concentration inthe ultrafiltered solution was estimated by measur-ing the UV absorbance at 280 nm (e0.1% = 0.545mg/ml). The aldehyde groups were estimated byreaction with 2,4-dinitrophenylhydrazine (DNPH)according to the procedure described by Jain andco-workers [28]. The oxidized carbohydrate albuminwas analyzed by SEC-HPLC.
2.4.3. PEGylation of glycosylated HSA
PEG-Hz was added to 1 ml of a 10 mg/ml solu-tion of oxidized glycosyl-HSA in PBS/EDTA, pH6.5, in order to yield a 10:1 PEG:aldehyde molarratio. The reaction mixture was shaken gently atroom temperature and analysed at scheduled timesby SEC-HPLC. The degree of protein PEGylationwas determined from the peak area of the PEGylat-ed and unPEGylated protein peaks and expressed asfollows:
%PEGylation
¼ PEGylated HSA=total HSAð Þ � 100

1382 S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389
The reaction kinetic constants (k) were determinedaccording to a simplified first order equation (Eq.(1)).
PEGylation rate ¼ k½aldehyde� ð1Þ
The kinetic constants (k0) were extrapolated asthe slope of the plot of the natural logarithm ofthe concentration of native protein vs. time
ln c ¼ ln c0 � k0 � t ð2Þ
where c0 is the initial concentration of the glycosyl-ated protein expressed as percentage and c is theconcentration at time t.
PEGylated HSA was purified by fast proteinliquid chromatography (FPLC) using a Hiload 26/60 Superdex 200 semi-preparative column (Pharma-cia, Piscataway, NJ) eluted with 0.3 ml/min of10 mM phosphate buffer, pH 7.4. The eluted frac-tions were analysed by UV spectroscopy at280 nm and by iodine assay [29] for protein andPEG determination, respectively. Positive fractionsin both tests were collected and concentrated byultra-filtration using a 10-kDa cut-off membrane(Amicon, Beverly, MA, USA).
The purified products were analysed by SEC-HPLC, DTNB and iodine assays to estimate thedegree of modification (number of polymer chains/protein molecule).
2.5. Reversibility of hydrazone linkage of PEGylated
HSA
PEGylated HSA samples (100 ll, 5 mg/ml pro-tein) were added to a 500 mM carbohydrazide solu-tion in PBS, pH 7.4, to yield a 100:1carbohydrazide:conjugated PEG ratio. At sched-uled times, the samples were analysed by SEC-HPLC and the areas of the peaks correspondingto the PEGylated and the unPEGylated proteinwere determined.
2.6. Hydrolysis of PEGylated HSA in aqueoussolution at different pHs
PEGylated HSA samples were prepared by dilu-tion of 5 mg/ml protein solutions to 1 mg/ml pro-tein with A. 100 mM citrate buffer, pH 4.5; B.100 mM phosphate, pH 7.4 or C. 100 mM borate,pH 8.4. The samples were incubated in closed vialsat 25 �C with slight shaking and analysed at sched-uled times by SEC-HPLC. The areas of the peaks
corresponding to the PEGylated and the unPEGy-lated protein were estimated and expressed asfollows:
%hydrolysis ¼ ½glycated HSA=total HSA� � 100
3. Results and discussion
The site-specific PEGylation procedure reportedin the present study involves the selective attach-ment of glycosylic linkers to proteins followed byPEGylation of the oxidized glycosydes. These link-ers can be selectively conjugated (glycation) to hid-den protein sites and provide for multi-polymerattachment.
The two glycosylic linkers synthesized for site-specific protein conjugation, galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide andmaltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide, were designed to be conjugatedto the thiol groups of Cys, whose location in hydro-phobic clefts of proteins usually limit their accessi-bility to conjugation. Human serum albumin(HSA) was used as a protein model since it possessesa Cys residue in position 34 (Cys34), which is locatedin a shallow crevice around 9.5 A deep [30]. Thisposition makes the Cys available for conjugationto a number of small lipophylic molecules [31] butit set hurdles to the interaction with large hydro-philic molecules.
To promote docking of the linker into thehydrophobic cavity, a hydrophobic dodecylalkylspacer was introduced between the reactive malei-mide group and the glycosyl residues. Since thealkyl chain is unable to coordinate water mole-cules as compared to the hydrophilic PEG [3],the linker can penetrate the Cys34 cleft with lim-ited hindrance. The length and the volume ofthe –NH–(CH2)12–NHCO(CH2)2-maleimide groupwere, in fact, calculated to be 25 A and 370 A3,respectively, while the hydrodynamic volume ofa (OCH2CH2)n-maleimide group with comparablelength (n = 5–6) was calculated to be 690 A3
(MOE 2007.09, Chemical Computing Group,Inc., Montreal, Quebec, Canada). Therefore, theformer can better fit the cavity where the Cys34
is located than the latter.The two glycosyl moieties were selected because
their different length and complexity can result indifferent exposition on the protein surface that

S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1383
can yield a different number of polymer cou-plings.
3.1. Synthesis and characterization of maleimide-
activated glycosyl-alkyl linkers
Scheme 1 describes the synthesis of the two differ-ent glycosyl linkers used for selective glycation ofhuman serum albumin.
In order to conjugate 1,12-diaminododecane tothe glycosyl moieties, lactose and maltotriose wereactivated to lactonolactone and maltotrionolactoneby oxidation of the reducing ends, followed by ringclosure. This convenient procedure produces glyco-syl derivatives that are highly reactive towards pri-mary amines with high reaction yield [25].
The activated glycosyl residues were reacted witha large molar excess of 1,12-diaminododecane,which prevented crosslinking. The FT-IR spectrashowed the disappearance of the carbonyl bond ofthe lactone derivatives. The amine content in thepurified maltosyl and galactosyl products deter-mined by a colorimetric assay was 92% and 95%(determined NH2/glycosyl derivative moles/moles,%), respectively, which were consistent with thatof the expected monoamino derivatives. The 1HNMR and ESI-MS analyses confirmed the chemical
O
O
OH
OH
OHR
OH1. I2
M
2. H
MeOH, reflux NH
H2N-(CH2)12-NH2, Et3NOH
OOH
OH
OHO
NH
OH
OOH
OH
OHO
anhydrous DMSO, 30 min,r.t.
NHS-CO-(CH2)2-maleimide R
R
OH O
OH
OH
OH
Galactosyl-
R =
Scheme 1. Schematic description of the maleimide-glycosidic linker smaleimide and maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-m
structure of the derivatives. As an example Fig. 1reports the spectra obtained with the maltosyl deriv-ative. The 1H NMR spectra showed the disappear-ance of the signals relative to the reducing endproton of the sugar, d 4.706 and d 4.680 of lactoseand maltotriose, respectively. The mass/charge(m/z) signals obtained by ESI-MS analysis of thepurified galactosyl-glucono-CO–NH–(CH2)12–NH2
[541.30 (M+H)1+ and 271.16 (M+2H)2+] andmaltosyl-glucono-CO–NH–(CH2)12–NH2 [703.41(M+H)1+ and 352.21 (M+2H)2+] correspond tothe calculated masses of the two derivatives whichare 540.33 (M+H)1+ and 702.79 (M+H)1+, respec-tively. The ESI-MS spectra did not show the 201(M+H)1+ m/z signal of the unreacted 1,12-diami-nododecane, indicating that the product was exten-sively purified by precipitation in diethyl ether.
The activation of the glycosyl linkers as malei-mide derivatives was carried out by the amino reac-tion of galactosyl-glucono-CO–NH–(CH2)12–NH2
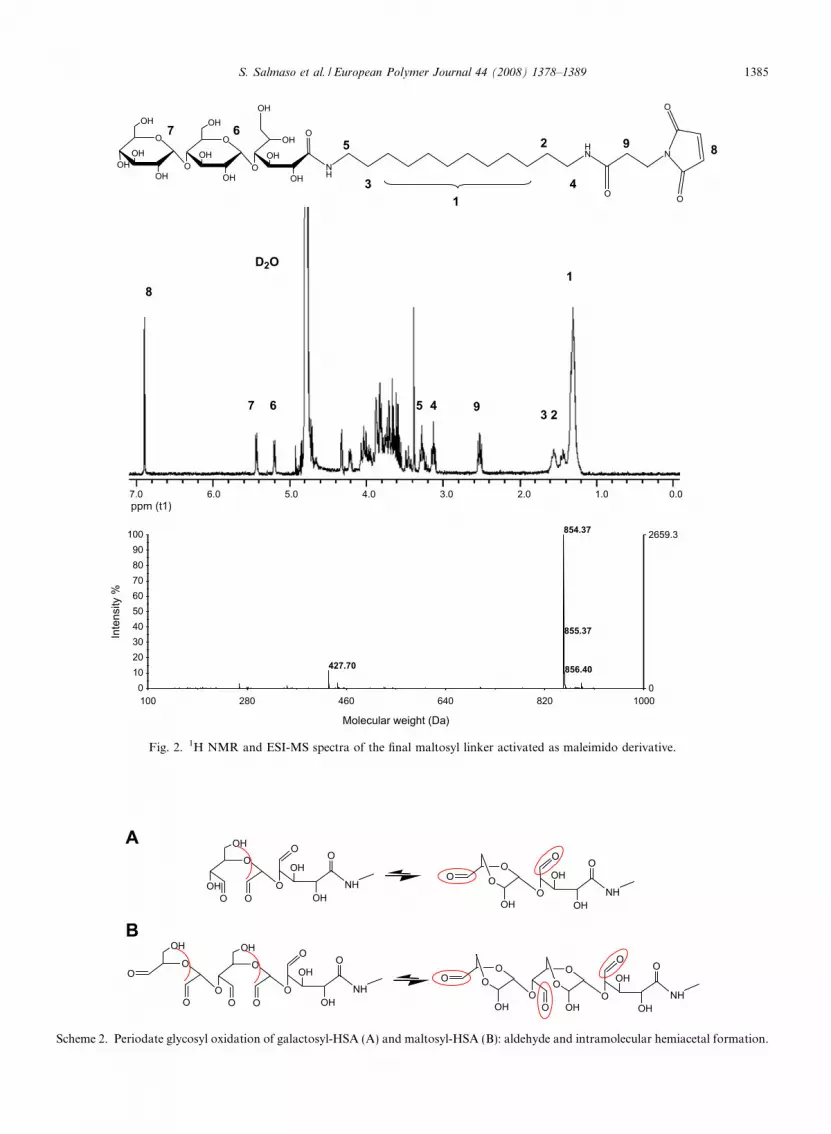
and maltosyl-glucono-CO–NH–(CH2)12–NH2 withN-succinimidyl 3-maleimidopropionate. Fig. 2reports, as an example, the 1H NMR and ESI-MSspectra obtained with the maltosyl derivative. TheESI-MS spectra showed that the m/z signalsobtained with galactosyl-glucono-CO–NH–(CH2)12
–NH–CO–(CH2)2–maleimide [692.37 (M+H)1+
, KOHeOH, reflux+ exchange
NH2
HN
O
N
O
O
O
OOH
OH
OH
OR
O
OH
OH
OH
OH O
O
OH
OH
OH
Maltosyl-
yntheses: galactosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-aleimide.

1D2O
7 54
2, 36
OH
OOH
OH
O
OH
OH
OHOH
OH O
O
OH
OH
OHO
NH
NH2
6
4
2
3 1
7 5
100 280 460 640 820 10000
1.7E+
0102030405060708090
100 703.41
704.41
352.21
705.42
Molecular weight (Da)
Inte
nsity
(%)
7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0ppm (t1)
Fig. 1. 1H NMR and ESI-MS spectra of the maltosyl-alkylamino intermediate.
1384 S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389
and 346.70 (M+2H)2+] and maltosyl-glucono-CO–NH–(CH2)12–NH–CO–(CH2)2-maleimide [854.40(M+H)1+ and 427.70 (M+2H)2+] correspond tothe calculated values: 691.76 (M+H)1+ and 853.37(M+H)1+ for the maleimide-activated galactosyl-glucono and maltosyl-glucono derivatives, respec-tively. The structure identity of the linkers wasconfirmed by 1H NMR spectroscopy and specificcolorimetric assays. The integration of the 1HNMR signals belonging to the alkyl chain (16H atd 1.3) and to the vinyl bond of maleimide (2H at d6.9) gave a degree of activation of 98–99%, whichwas in fair agreement with the data obtained bythe colorimetric –SH retro-titration performed withDTNB and cysteine which yielded 98% and 96%maleimide/glycosyl linker (moles/moles, %) for thegalactosyl and maltosyl derivative, respectively.
3.2. Preparation and characterization of PEGylated
HSA
The Cys glycation was performed at pH 6.5because the reactivity of the maleimide groupstowards thiol groups is about 1000-fold faster at thispH than that of primary amines [32]. The Ellmanassay showed that after 2 h both linkers yieldedcomplete functionalization of the available Cys.
The glycosyl oxidation was performed withsodium m-periodate, which can selectively cleavethe carbon–carbon bonds containing vicinal diolsto generate pairs of aldehyde groups. Scheme 2shows that the formation of intramolecular hemi-acetals between the hydroxyl group on C6 and thealdehydic group on C4 of the glycosilic residuereduces the number of chemically reactive aldehydes
100 280 460 640 820 10000
2659.3
0102030405060708090
100 854.37
855.37
427.70 856.40
1D2O
OH
OOH
OH
O
OH
OH
OHOH
OH O
O
OH
OH
OHO
NH
HN N
O
O
O
8
896
7
4
2
31
5
75
9 3 26 4
Inte
nsity
%
Molecular weight (Da)
7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0ppm (t1)
Fig. 2. 1H NMR and ESI-MS spectra of the final maltosyl linker activated as maleimido derivative.
O
OOO
OO
O
OHOH
O
OOHO
OO
OH
OOO
OH
OH
OO
NHO
OH
OH
OO
NH
O
OHOO
OH
O
OH
O OOOH
OH
OO
NHO
OH
OH
OO
NH
A
B
Scheme 2. Periodate glycosyl oxidation of galactosyl-HSA (A) and maltosyl-HSA (B): aldehyde and intramolecular hemiacetal formation.
S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1385

1386 S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389
in the oxidized protein [33]. Accordingly, the quan-titative analysis of aldehydes, carried out withDNPH, showed that under the optimized oxidativeconditions two and three free aldehydic groups wereobtained in the case of galactosyl-HSA and malto-syl-HSA, respectively.
Scheme 3A shows the PEGylation of oxidizedHSA carried out with PEG-Hz, which can reactwith the aldehyde moieties to form reversiblehydrazone bonds [34]. The SEC-HPLC analysisperformed to monitor the protein PEGylation(Scheme 3) showed the appearance of high-molec-ular weight derivatives (lower retention time),which was concomitant to the disappearance ofthe native HSA (higher retention time). Fig. 3shows the PEGylation profiles obtained by proteinglycation and, for comparison, the PEGylationprofile obtained with PEG-maleimide. After 50 hof the reaction, the degree of conversion to PEGy-lated species was 93% for both of the glycated pro-teins, while only 27% PEGylation was obtainedwith the PEG-maleimide. The extrapolated kineticconstants for the direct PEGylation of native
Retention Time0 5 10
Rel
ativ
e Ab
sorb
ance
at 2
80 n
m
B
A
PEGylated HSA
Scheme 3. Chemical structure representation of PEGylated maltosyl-HPEGylated maltosyl-HSA (B).
HSA with PEG-maleimide was 7.4 � 10�3 h�1,while 63.9 � 10�3 h�1 and 86.0 � 10�3 h�1 wereobtained with galactosyl-HSA and maltosyl-HSA,respectively. According to the limited accessibilityof Cys34 to hydrophilic molecules with largehydrodynamic sizes, the Cys34 conjugation withPEG-maleimide was slow and incomplete. On thecontrary, the glycated protein derivatives under-went faster and almost complete protein conjuga-tion, which was attributed to the exposition ofthe glycosylic anchors on the protein surface. Thedifferent conjugation profiles of the two glycosylderivatives are attributable to the length of the gly-cosyl function. The polymer approach and cou-pling to the aldehyde groups is, in fact, expectedto be favoured when the longer maltosyl linker isused, as it is more exposed on the protein surfaceand presents a higher number of coupling pointsthan the shorter galactosyl linker.
The gel filtration analysis showed that the appar-ent molecular mass of the PEGylated galactosyl-HSA and maltosyl-HSA corresponded to 140 kDaand 170 kDa globular protein sizes, respectively.
(min)15 20 25
Native HSA
SA (A) and gel permeation chromatography profile of native and

0
20
40
60
80
100
0 20 40 60 80 100 120Time (hours)
% P
EG-H
SA
0
1
2
3
4
5
0 20 40 60 80 100 120Time (hours)
ln (%
HSA
)
A
B
Fig. 3. HSA PEGylation profiles: (A) PEGylation of native HSAwith commercial 10 kDa PEG-maleimide (j); PEGylation ofperiodate-oxidized galactosyl-HSA with 10 kDa PEG-Hz (e);PEGylation of periodate-oxidized maltosyl-HSA (d) with10 kDa PEG-Hz. Time point mean values and standard devia-tions (±SD) were calculated from five experiments. (B) Disap-pearance of native HSA by reaction with 10 kDa PEG-maleimide(j) and disappearance of oxidized galactosyl-HSA (e) andoxidized maltosyl-HSA (d) by reaction with 10 kDa PEG-Hz.
80
100
pro
tein
S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1387
These results are in reasonable agreement with thetheoretical molecular masses calculated on the basisof the molecular weight of the native protein(69 kDa) and the apparent size of 10 kDa PEG. Itis noteworthy that due to its ability to coordinatethree water molecules per oxyethylene monomer,the 10 kDa PEG possesses a hydrodynamic volumecorresponding to a globular protein of about30 kDa [35].
0
20
40
60
0 100 200 300 400Time (hours)
% P
EGyl
ated
Fig. 4. Displacement of 10 kDa PEG-Hz from PEGylatedgalactosyl-HSA (j) and PEGylated maltosyl-HSA (h) bycarbohydrazide. Time point mean values and standard deviations(±SD) were calculated from four experiments.
3.3. Kinetic profiles of hydrolysis of PEGylated HSA
The PEG-Hz conjugation to oxidized glycosidesyields reversible hydrazone bonds that undergopH dependent hydrolysis [36]. The combination ofsite-specific PEGylation with releasable polymerconjugation may have numerous applications inthe perspective of therapeutic use of bioconjugates,since it exploits the beneficial attributes of protein
hyper-PEGylation with the preservation of biologi-cal activity [37]. Indeed, the activity of the proteinresulting from PEG detachment is not hamperedby the bulky polymer on the protein surface.Furthermore, the detached PEG can be rapidlyeliminated from the body via glomerular ultra-filtra-tion, which prevents polymer accumulation in thebody. These advantages are of particular relevancein the case of multivalent protein PEGylation,which may yield a significant decrease in bioactivityand reduced PEG elimination.
In order to evaluate the reversibility of the hydra-zone bond, a competition study has been carried outby incubating the HSA-PEG conjugates with carbo-hydrazide. Fig. 4 shows that PEG can be slowly dis-placed from the two glycated bioconjugates byaddition of the competing agent, indicating thatthe reversible character of the hydrazone bondscan be exploited to control in vivo protein and poly-mer release.
The hydrolysis kinetics of the bioconjugates atpH 4.5, 7.4 and 8.4 are reported in Fig. 5 and thehydrolysis rate constants. The hydrazone hydrolysisdisplayed first order kinetics. According to the datareported in the literature, the hydrolysis of thehydrazone linkage was faster in mild acidic or basicenvironments than at physiological pH. However,the hydrazone cleavage was slow also at acidicpH, though the literature reports that at this pHprompt hydrolysis takes place [36]. At pH 4.5 and7.4, the maltosyl derivative showed slower proteinrelease with respect to the galactosyl counterpart,probably because the former is composed of threePEG chains while the latter is composed of twopolymer chains. Therefore, the protein exposure inthe body can be fairly controlled by the degree of

Table 1PEG release kinetic constants from the PEGylated galactosyl-HSA and maltosyl-HSA bioconjugates at pH 4.5, 7.4 and 8.4
pH 4.5 (h�1) pH 7.4 (h�1) pH 8.4 (h�1)
Galactosyl-HSA 1.4 � 10�3 (R2 = 0.975) 0.8 � 10�3 (R2 = 0.968) 1.3 � 10�3 (R2 = 0.963)Maltosyl-HSA 0.9 � 10�3 (R2 = 0.947) 0.5 � 10�3 (R2 = 0.940) 1.3 � 10�3 (R2 = 0.962)
A
0
20
40
60
80
100
0 100 200 300 400 500 600 700
Time (hours)
% P
EGyl
ated
pro
tein A1
3
3.5
4
4.5
5
0 100 200 300 400 500 600 700
Time (hours)
ln %
PEG
ylat
ed p
rote
in
B
0
20
40
60
80
100
0 200 400 600 800 1000
Time (hours)
% P
EGyl
ated
pro
tein B1
3
3.5
4
4.5
5
0 100 200 300 400 500 600
Time (hours)
ln %
PEG
ylat
ed p
rote
in
Fig. 5. PEG release profiles from the PEGylated galactosyl-HSA (A and A1) and PEGylated maltosyl-HSA (B and B1) at pH 4.5 (h), pH7.4 (d) and pH 8.4 (N). Time point mean values and standard deviations (±SD) were calculated from four experiments.
1388 S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389
PEGylation that can be, in turn, controlled by theglycosyl linker.
4. Conclusions
Glycation represents a versatile approach forpreparation of tailor made PEGylated proteins, asit combines the benefits of site selective conjugationwith multiple polymer coupling. This procedureexpands the advantages of selective glycosyl PEGy-lation to human recombinant proteins, which areusually manufactured by recombinant expressionin E. coli as non-glycosylated proteins.
As compared to enzyme mediated glycosylation,the protein glycation may be properly set up formodification of protein sites that are not suitablesubstrates for glycosyl transferases. Therefore, thischemical approach may provide for new opportuni-ties in site-specific protein derivatization. A variety
of glycosylic linkers can, in fact, be properlydesigned to selectively glycate amino acids that arenot accessible to direct PEGylation, since they areburied in the protein structure.
The glycosylic linkers are multi-point anchors forPEG coupling, but the polymer is confined at thepending glycosylic sites of the proteins, thus limitingthe hurdles due to the polymer on the proteinsurface.
Finally, the releasable PEGylation of glycatedproteins represents a further advantage of this tech-nique, as it can provide for slow active proteinrelease by polymer detachment according to kineticsthat depend on the structural properties of the lin-ker (see Table 1).
Acknowledgment
This work has been financially supported by theItalian Ministry of Research (2006030353).

S. Salmaso et al. / European Polymer Journal 44 (2008) 1378–1389 1389
References
[1] Roberts MJ, Bentley MD, Harris JM. Chemistry for peptideand protein PEGylation. Adv Drug Deliver Rev2002;54:459–76.
[2] Fee CJ, Van Alstine JM. PEG-proteins: reaction engineeringand separation issues. Chem Eng Sci 2006;6:924–39.
[3] Caliceti P, Veronese FM. Pharmacokinetic and biodistribu-tion properties of poly(ethylene glycol)-protein conjugates.Adv Drug Deliver Rev 2003;55:1261–77.
[4] Pasut G, Veronese FM. Polymer-drug conjugation, recentachievements and general strategies. Prog Polym Sci2007;32:933–61.
[5] Bowen S, Tare N, Inoue T, Yamasaki M, Okabe M, Horii I,et al. Relationship between molecular mass and duration ofactivity of polyethylene glycol conjugated granulocyte col-ony-stimulating factor mutein. Exp Hematol1999;27:425–32.
[6] Kochendoerfer GG. Site-specific polymer modification oftherapeutic proteins. Curr Opin Chem Biol 2005;9:555–60.
[7] Zappe H, Snell ME, Bossard MJ. PEGylation of cyanovirin-N, an entry inhibitor of HIV. Adv Drug Deliver Rev2008;60:79–87.
[8] Long DL, Doherty DH, Eisenberg SP, Smith DJ, RosendahlMS, Christensen KR, et al. Design of homogeneous,monopegylated erythropoietin analogs with preservedin vitro bioactivity. Exp Hematol 2006;34:697–704.
[9] Balan S, Cho J, Godwin A, Teo I, Laborde CM, Heidel-berger S, et al. Site-specific PEGylation of protein disulphidebonds using a tri-carbon bridge. Bioconjugate Chem2007;18:61–76.
[10] Geoghegan KF, Stroth JG. Site-directed conjugation ofnanopeptide groups to peptides and proteins via periodateoxidation of a 2-amino alcohol. Application, modification atN-terminal serine. Bioconjugate Chem 1992;3:138–46.
[11] Kinstler O, Molineux G, Treuheit M, Ladd D, Gegg C.Mono-N-terminal poly(ethyleneglycol)-protein conjugates.Adv Drug Deliver Rev 2002;54:477–85.
[12] Wang YS, Youngster S, Grace M, Bausch J, Bordens R,Wyss DF. Structural and biological characterization ofpegylated recombinant interferon alpha-2b and its thera-peutic implications. Adv Drug Deliver Rev 2002;54:547–70.
[13] Tirat A, Freuler F, Stettler T, Mayr LM, Leder LE.Evaluation of two novel tag-based oabelling technologiesfor site-specific modification of proteins. Int J Biol Macro-mol 2006;39:66–76.
[14] Deiters A, Cropp TA, Summerer D, Mukherji M, SchultzPG. Site-specific PEGylation of proteins containing unnat-ural amino acids. Bioorg Med Chem Lett 2004;14:5743–5.
[15] Sato H. Enzymatic procedure for site-specific PEGylation ofproteins. Adv Drug Deliver Rev 2002;54:487–504.
[16] Peschke B, Zundel M, Bak S, Clausen TR, Blume N,Pedersen A, et al. C-terminally PEGylated hGH derivatives.Bioorg Med Chem 2007;15:4382–95.
[17] Youn YS, Na DH, Yoo SD, Song SC, Lee KC. Carbohy-drate-specifically polyethylene glycol-modified ricin A-chainwith improved therapeutic potential. Int J Biochem Cell Biol2005;37:1525–33.
[18] Hansen CB, Kao GY, Moase EH, Zalipsky S, Allen TM.Attachment of antibodies to sterically stabilized liposomes:evaluation, comparison and optimization of coupling pro-cedures. Biochim Biophys Acta 1995;1239:133–44.
[19] Pettersen MTN, Jhonson PH, Pettersen SB. Detailed anal-ysis of the amino acid neighbours and conformation ofcysteines in proteins. Protein Eng 1999;12:535–48.
[20] Esposito P, Barbero L, Caccia P, Caliceti P, D’Antonio M,Piquet G, et al. PEGylation of growth hormone-releasinghormone (GRF) analogues. Adv Drug Deliver Rev2003;55:1279–91.
[21] Wetzel R, Becker M, Behlke J, Billwitz H, Bohlm S, Ebert B,et al. Temperature behaviour of human serum albumin. EurJ Biochem 1980;104:469–78.
[22] Wang W. Instability, stabilization and formulation of liquidprotein pharmaceuticals. Int J Pharm 1999;185:129–88.
[23] Natarjan A, Xiong CY, Albrecht H, DeNardo GL,DeNardo SJ. Characterization of site-specific ScFv PEGy-lation for tumor-targeting pharmaceuticals. BioconjugateChem 2005;16:113–21.
[24] DeFrees S, Wang ZG, Xing R, Scott AE, Wang J, Zopf D,et al. GlycoPEGylation of recombinant therapeutic proteinsproduced in Escherichia coli. Glycobiology 2006;16:833–43.
[25] Kobayashi K, Sumitomo H, Ina Y. Synthesis and functionof polyetyrene derivatives having pendant oligosaccharides.Polym J 1985;17:567–75.
[26] Snyder SL, Sobocinsky PZ. An improved 2,4,6-trinitroben-zensulfonic acid method for the determination of amines.Anal Biochem 1975;64:284–8.
[27] Riddles PW, Blakeley RL, Zerner B. Ellman’s reagent: 5,50-dithiobis(2-nitrobenzoic acid) – a re-examination. AnalBiochem 1979;94:75–81.
[28] Jain S, Hreczuk-Hirst DH, McCormack B, Mital M,Epenetos A, Laing P, et al. Polysialylated insulin: synthesis,characterization and biological activity in vivo. BBA2003;1622:42–9.
[29] Sims GE, Snape TJ. A method for the estimation ofpolyethylene glycol in plasma protein fractions. Anal Bio-chem 1980;107:60–3.
[30] Stewart AJ, Blindauer CA, Berezenko S, Sleep D, Tooth D,Sadler PJ. Role of Tyr84 in controlling the reactivity ofCys34 of human albumin. FEBS J 2005;272:353–62.
[31] Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K.Crystal structure of human serum albumin at 2.5 A resolu-tion. Protein Eng 1999;12:439–46.
[32] Buechler KF, Banaszczyk MG, Noar JB. US Patent6967107; 2005.
[33] Bruneel D, Schatch E. Chemical modification of pullulan: 1.periodate oxidation. Polymer 1993;34:2628–32.
[34] King TP, Zhao SW, Lam T. Preparation of proteinconjugates via intermolecular hydrazone linkage. Biochem-istry 1986;25:5774–9.
[35] Sherman MR, Williams LD, Saifer MGP, French JA, KwakLW, Oppenheim JJ. Conjugation of high-molecular weightpoly(ethylene glycol) to cytokines: granulocyte-macrophagecolony – stimulating factors as model substrates. In: HarrisMJ, Zalipsky S, editors. ACS Symposium Series 680, ACS,Washington, DC; 1997. p. 155–69.
[36] Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E,Levchenko TS, et al. ‘‘SMART” drug delivery systems:double-targeted pH-responsive pharmaceutical nanocarriers.Bioconjugate Chem 2006;17:943–9.
[37] Li S, Yang Z, Sun X, Tan Y, Yagi S, Hoffman RM. Proteincarboxyl amidation increases the potential extent of proteinpolyethylene glycol conjugation. Anal Biochem 2004;330:264–71.


















![[Aging: role and control of glycation]](https://img.pdfslide.net/doc/110x75/635f0913ac6942764f03d5ce/aging-role-and-control-of-glycation.jpg)










