Embed Size (px)
Citation preview

ARTHRITIS & RHEUMATISMVol. 54, No. 9, September 2006, pp 2872–2885DOI 10.1002/art.22077© 2006, American College of Rheumatology
Soluble Human p55 and p75 Tumor Necrosis Factor ReceptorsReverse Spontaneous Arthritis in Transgenic Mice Expressing
Transmembrane Tumor Necrosis Factor �
Carl K. Edwards, III,1 Alison M. Bendele,2 Leonid I. Reznikov,3 Giamila Fantuzzi,3
Elizabeth S. Chlipala,2 Li Li,4 Lyle L. Moldawer,5 John D. Mountz,6
Yi-Yang Yvonne Li,4 and Charles A. Dinarello3
Objective. The roles of the transmembrane andsecreted forms of tumor necrosis factor � (TNF�) inrheumatoid arthritis (RA) remain unclear. Agents usedto inhibit TNF� have shown varying efficacy in RApatients, suggesting that anti-TNF� agents possess dis-similar mechanisms of action, including the ability toneutralize transmembrane (tmTNF�) and secretedTNF�. In this study, TNF�-knockout (TNF�-KO) micethat were genetically altered to express elevated levels oftmTNF� were constructed to further understand theroles of the 17-kd secreted, trimeric, and 26-kd trans-membrane forms of TNF�.
Methods. A speed-congenic mating scheme was
used to generate 3 unique strains of mice: 1) transgenictmTgA86 mice overexpressing 26-kd tmTNF� and alsosecreting 17-kd trimeric TNF� (tmTNF�-transgenic),2) TNF��/� mice (TNF�-KO), and 3) transgenic miceoverexpressing tmTNF� backcrossed to TNF�-KO mice(tmTNF�-transgenic/TNF�-KO). Mice were treatedwith phosphate buffered saline (as vehicle control),dexamethasone (as positive control), or modified recom-binant human soluble TNF receptor (sTNFR) p55 orp75, and were assessed clinically and histopathologi-cally for signs of inflammation and development ofarthritis.
Results. The tmTNF�-transgenic/TNF�-KO micewere born with crinkled tails and spinal deformitiessimilar to those in ankylosing spondylitis. By 2–4 weeks,these mice developed symmetric inflammatory arthritis,characterized by tissue swelling, pannus formation, andbone deformities. The tmTNF�-transgenic mice alsodeveloped spontaneous-onset arthritis, but at a slowerrate (100% incidence by 10–12 weeks). Clinical andhistologic progression of arthritis in the tmTNF�-transgenic/TNF�-KO mice was reduced by treatmentwith dexamethasone or with the p55 or p75 sTNFR (69%and 63% reduction in total histologic score, respec-tively).
Conclusion. These data show that arthritis issufficiently initiated and maintained in tmTNF�-transgenic/TNF�-KO mice, and that it can be neutral-ized by recombinant human p55 or p75 sTNFR, result-ing in amelioration of the biologic and subsequenthistologic destructive effects of tmTNF�.
Numerous investigations have pointed to a keyrole of the proinflammatory pleiotropic cytokines tumornecrosis factor � (TNF�) and interleukin-1 (IL-1) in
Dr. Edwards’ work was supported by the National PsoriasisFoundation, the National Alopecia Areata Foundation, and the Uni-versity of Colorado at Denver and Health Sciences Center AcademicEnrichment Funds. Dr. Dinarello’s work was supported by NIH grantsAI-15614, HL-68743, and CA-04 6934.
1Carl K. Edwards III, PhD: Amgen, Inc., Thousand Oaks,California, and University of Colorado Health Sciences Center atDenver and Health Sciences Center, Aurora, Colorado; 2Alison M.Bendele, DVM, PhD, Elizabeth S. Chlipala, MS: Bolder Biopath, Inc.,and University of Colorado at Boulder; 3Leonid I. Reznikov, MD,Giamila Fantuzzi, PhD, Charles A. Dinarello, MD: University ofColorado Health Sciences Center at Denver; 4Li Li, MD, PhD,Yi-Yang Yvonne Li, MD, PhD: University of Colorado Health Sci-ences Center at Denver, and Health Sciences Center, Aurora, Colo-rado; 5Lyle L. Moldawer, PhD: University of Florida College ofMedicine, Gainesville; 6John D. Mountz, PhD, MD: University ofAlabama at Birmingham.
Drs. Edwards and Bendele conducted a portion of thisresearch while employees of Amgen Boulder, Boulder, Colorado. Dr.Dinarello has previously received consulting fees (less than $10,000)from Amgen.
Address correspondence and reprint requests to Carl K.Edwards, III, PhD, University of Colorado Health Sciences Center atDenver, Department of Dermatology, Mailstop 8127, 12801 East 17thAvenue, PO Box 6511, Aurora, CO 80045. E-mail: [email protected].
Submitted for publication August 16, 2005; accepted inrevised form June 13, 2006.
2872

host defense and inflammatory disease processes (1).Expression of TNF� and IL-1 has been observed indisease-associated tissues and in the circulation of pa-tients with acute or chronic inflammatory diseases (2).In the last 15 years, several approaches to inhibit theactivities of TNF� and IL-1 have been developed andtested (3). These include the use of neutralizing antibod-ies to TNF�, as well as soluble TNF receptors (sTN-FRs), which exhibit properties enabling them to bind thesecreted, 17-kd, soluble trimeric form of TNF� as well asthe 26-kd transmembrane (membrane-bound, an-chored) TNF� (tmTNF�) (4). Clinical trials have dem-onstrated significant effects with these agents, and re-duction of TNF� activities has become an importanttherapeutic option utilized by clinicians to treat inflam-matory diseases (5).
The role of tmTNF� in the initiation and pro-gression of chronic inflammatory diseases remains anunresolved issue (6–8). The diseases in which a possiblerole of tmTNF� has been implicated include rheumatoidarthritis (RA) (6,9), sepsis (10), brain inflammation (11),Crohn’s disease (8,12–14), colitis (15,16), hepatitis(17,18), psoriasis (19), and human immunodeficiencyvirus type 1 (20). The importance of tmTNF� in thesediseases is highly relevant, particularly in patients withRA, in whom reduced TNF� activity is associated withan increase in routine and opportunistic infections (21–23). Therefore, understanding how tmTNF� or secretedTNF� initiates disease may influence the developmentof new therapeutic agents with improved efficacy andsafety profiles (3,24).
In original studies led by Dr. George Kollias(Hellenic Pasteur Institute, Athens, Greece), investiga-tors generated a strain of transgenic mice expressing amutant transmembrane form of murine TNF� protein(muTNF�1312), which accumulated on the cell surfaceof monocytic cells and resulted in arthritogenic changesin the mouse joints, involving cooperative signalingthrough both the p55 and the p75 sTNFRs (25,26).These mixed-strain background (129/SvEv � C57BL/6)mice, which were heterozygous for the TNF transgene(25), were termed TgA86�/� and TgA86�/� � TNF�/�
mice (26,27), and they developed macroscopic swellingof the ankles and histopathologic evidence of polyarthri-tis by �4–6 weeks of age (25). However, recent studiesin our laboratories, which assessed herpes simplex virus1 (HSV-1) infection in different mouse backcrosses(mainly, C57BL/6 and 129/SvEv), revealed a differentialexpression of the genetic locus termed herpes resistancelocus, which is closely linked to the TNF p55 receptorgene (Tnfsf1a) on mouse chromosome 6 (28). Noncon-
genic BL/6 � 129/SvEv p55�/� null mice infected withHSV-1 were highly susceptible to infection, whereascongenic BL/6 p55�/� null mice were highly resistant toHSV-1 infection (28), supporting the concept that vari-ations in genetically stable, congenic mice as comparedwith unstable, noncongenic mice are important in dis-ease susceptibility and outcome (29).
In the present study, we utilized homozygous,transgenic mice overexpressing murine tmTNF�; thesemice were additionally backcrossed to homozygousTNF�-knockout (TNF�-KO) mice (generating a strainherein referred to as tmTNF�-transgenic/TNF�-KO) inorder to further understand the roles of the secreted17-kd and membrane 26-kd forms of TNF� in thepathogenesis of inflammation and autoimmunity. Thesegenetically altered tmTNF�-transgenic/TNF�-KO micedid not secrete the soluble 17-kd form of murine TNF�,but overexpressed the 26-kd form of murine tmTNF�.Importantly, because of our observations in the above-mentioned viral infection studies, we utilized transgenicmice that were bred congenically on a stable C57BL/6genetic background, effectively minimizing mixed-genetic phenotypes (30). Our results indicate that miceoverexpressing 26-kd tmTNF� and lacking the release of17-kd, soluble trimeric TNF� provide a suitable modelfor the initiation of aggressive inflammation leading tosevere arthritis. Moreover, the findings suggest thatmodified, recombinant forms of either the human p55 orthe human p75 sTNFR are able to neutralize thebiologic and destructive effects of tmTNF� and inhibitinflammatory disease in vivo.
MATERIALS AND METHODS
Role of the funding source. Amgen, Inc., supplied therecombinant human PEGylated sTNFR (p55-PEG) pegsuner-cept. All authors wrote the manuscript, agreed to submit themanuscript for publication, and approved the content of themanuscript prior to submission.
Mice. Original breeding pairs of C57BL/6 and 129/SvEv (B6 � 129) inbred mice were obtained from The JacksonLaboratory (Bar Harbor, ME). Breeding pairs of 129S6/SvEv(previously known as 129SvEvTac), (B6 � 129)F1, and (B6 �129)F2 mice were obtained from Taconic Farms (German-town, NY). The following lines of mice, maintained on agenetically mixed B6 � 129 background, were originally ob-tained from Dr. George Kollias and have been previouslydescribed elsewhere (25,31,32): homozygous TNF��/�
(knockout) mice (line 549), also known as (129/SvEv �C57BL/6)-[KO]TNF� or TNF��/�KO; heterozygous murinetmTNF�–transgenic mice (line 551), also known as (129/SvEv � C57BL/6)-[TG]A86 or tmTgA86�/�TNF�; and het-erozygous murine tmTNF�–transgenic tmTgA86�/� � ho-mozygous TNF��/� (knockout) mice (line 555), also known as
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2873

(129/SvEv � C57BL/6)-[TG]A86�/� � (129/SvEv � C57BL/6)-[KO]TNF��/� or tmTgA86�/�TNF��/�KO. The heterozy-gous B6 � 129 tmTgA86�/� mice carry �50 copies of a 3.2-kbhybrid/globin transgene construct containing the promoter ofthe murine TNF� gene, the coding sequence of the murineTNF�1312 gene (25), and the 3�-untranslated region andpolyadenylation site of the human �-globin gene (25).
Because of the possibility of generating conflictingresults with mixed-strain inbred mice, we developed a speed-congenic mating scheme using mouse lines 549, 551, and 555.This resulted in 3 new congenic strains: 1) homozygousTNF��/� (knockout) mice (line 549-N12; herein referred to asTNF�-KO), also known as (C57BL/6N12)-[KO]TNF� orTNF�KON12; 2) homozygous murine tmTNF�–transgenic micewhich also secrete soluble TNF� at levels comparable withthose in wild-type mice (line 551-N10; herein referred to astmTNF�-transgenic), also known as C57BL/6N10-[Tg]A86 ortmTgA8TNF�N10; and 3) homozygous murine tmTNF�–transgenic � homozygous TNF��/� (knockout) mice whichonly express the biologically active 26-kd tmTNF� (line 555-N8; herein referred to as tmTNF�–transgenic/TNF�-KO), alsoknown as C57BL/6N10-[Tg]A86 � C57BL/6 N12-[KO]TNF�or tmTgA86TNF��/�KON8 (Table 1). Speed-congenic matingresulted in single- and double-homozygous mice that weresubsequently identified by routine polymerase chain reactionanalysis of genomic tail DNA (28), and in later generations(N5–N12), by clinical evaluation of male and female pups withobviously deformed, crinkled tails and hunched-back pheno-types at birth.
The presence of tmTNF� on thioglycollate-elicitedperitoneal macrophages was confirmed using either immuno-fluorescence (33) or fluorescence-activated cell sorter analysis(34). Peritoneal macrophages were harvested with phosphatebuffered saline (PBS), washed, and incubated with a rat
anti-mouse TNF� monoclonal antibody (Dako, Glostrup,Denmark). A goat anti-rat fluorescein isothiocyanate–conjugated IgG was used for staining and analysis on aFACScan flow cytometer (Becton Dickinson, Mountain View,CA) (34). Immunoreactive, secreted, trimeric TNF� was mea-sured by enzyme-linked immunosorbent assay (ELISA) (R&DSystems, Minneapolis, MN) using supernatants obtained fromthioglycollate-elicited peritoneal macrophages (3 individualmice from each strain), which were stimulated with 250 ng/mllipopolysaccharide for 12 hours (35). Confirmation of tmTNF�bioactivity was carried out using the WEHI-164 clone 13cytotoxicity assay as previously described (36,37). All mouselines described were housed in sterile micro-isolator housingunits (Lab Products, Maywood, NJ) under specific-pathogen–free conditions at Taconic Farms.
Treatment of mice. Mice were treated subcutaneouslywith either vehicle (PBS), dexamethasone (as a positive con-trol) (Sigma, St. Louis, MO), p55-PEG (pegsunercept [38–40],a kind gift from Dr. Alan Solinger at Amgen), or p75-Fc(etanercept [40–42], purchased from the pharmacy at theUniversity of Colorado Health Sciences Center at Denver).After administration of the sTNFRs, groups of 3 mice each pertime point were killed at 2, 8, 24, 48, 72, and 96 hourspostinjection. Blood was then collected in tubes containingEDTA for processing. Specific sandwich ELISAs measuringserum levels of the sTNFRs and containing antibodies to eachagent were conducted as previously described (43). The limit ofquantification of serum sTNFR levels with both assays was 0.78ng/ml. Results are expressed as the mean � SEM �g/ml pertime point. Serum levels of p55-PEG or p75-Fc after a singleintraperitoneal or subcutaneous injection in the tmTNF�-transgenic/TNF�-KO mice were measured as described previ-ously (39,44).
Table 1. Strains of congenic mice*
tmTNF� Secreted TNF� Bioactivity Disease onset
Background congenic strainC57BL/6 � 22.1 � 6.0 10.1 � 2.5 Not observed129/SvEv (B6 � 129) � 20.1 � 3.0 11.4 � 3.0 Not observed(B6 � 129)F1 � 23.6 � 2.0 12.3 � 3.0 Not observed(B6 � 129)F2 � 21.1 � 3.3 12.1 � 2.2 Not observed129S6/SvEv � 22.3 � 2.5 13.2 � 2.0 Not observed
Speed-congenic strainHomozygous TNF�-KO (N12) � 0.0 � 0.0 0.0 � 0.0 Not observedHomozygous tmTNF�-transgenic (N10) �� 17.6 � 4.2† 53.6 � 9.3 �4–6 weeks after birthDouble-homozygous tmTNF�-transgenic/TNF�-KO (N8) ��� 11.2 � 2.4‡ 69.6 � 7.3 �2–4 weeks after birth
* The presence or absence of transmembrane tumor necrosis factor � (tmTNF�) on thioglycollate-elicited peritoneal macrophages was assessedusing a semiquantitative scoring system of no immunofluorescence (�), low immunofluorescence (�), or elevated immunofluorescence (�� or���) (36) and confirmed using FACScan analysis (35). Immunoreactive, secreted, trimeric TNF� was assessed by enzyme-linked immunosorbentassay using supernatants obtained from thioglycollate-elicited peritoneal macrophages (3 individual mice from each strain; triplicate wells ofmacrophages [1 � 106 ml] assessed after stimulation with 250 ng/ml lipopolysaccharide [LPS] for 12 hours [35]). Values are the mean � SEM �g/ml.In vitro bioactivity was assessed using thioglycollate-elicited peritoneal macrophages (1 � 106/ml) from 3 individual mice each, after stimulation with250 ng/ml LPS. After 12 hours, macrophages were fixed with cold 1% paraformaldehyde and tested for tmTNF� bioactivity using the WEHI-164clone 13 cytotoxicity assay. Values are the mean � SEM percentage of cytotoxic cells (36,37). Disease onset was assessed at birth through 9 monthsof age (days 0, 5, 15, 20, 25, 30, 40, 50, 60, 70, 80, 100, 120, 140, 160, 200, 225, 250, and 275). A total of 3–5 animals per mouse strain per month(n � 60–100 mice total) were killed over the course of 9 months for histopathologic assessment to observe disease onset and severity. KO �knockout; N � number of speed congenic crosses.† P not significant (but nearly different) versus mean bioactivity, by Duncan’s new multiple range two-way analysis of variance.‡ P 0.05 versus mean bioactivity, by Duncan’s new multiple range two-way analysis of variance.
2874 EDWARDS ET AL

Scoring and clinical assessment of inflammatory ar-thritis. Volume measurements for assessment of inflammationof both hind paws (paw scores assessed on a scale of 0–4 [45]using water plethysmography) were performed prior to onsetof arthritis and every other day thereafter until the studieswere completed. At termination, the tibiotarsal joint wastransected at the level of the medial and lateral malleolus, forthe determination of paw weight as another measure ofinflammation. Paws were then collected into formalin forhistopathologic evaluation. Body weights were also deter-mined, as previously described (39,46,47).
Histopathologic assessment. Ankle joints were kept in10% neutral buffered formalin for at least 24 hours prior toplacement in Decalcifier I (Surgipath Medical Industries,Richmond, IL) for �1 week. When decalcification was com-plete, the digits were trimmed and the ankle joint wastransected in the longitudinal plane to yield approximatelyequal halves. These samples were processed for paraffinembedding, sectioned, and stained with hematoxylin and eosin.Multiple sections were prepared to ensure that the distal tibiawith both cortices was present, and that abundant distal tibialmedullary space was available for evaluation.
Ankles obtained from arthritic mice were given scoresof 0–5 for bone resorption and inflammation according topreviously published criteria (46,48–52). For bone resorption,scores were as follows: 0 � normal, 1 � minimal (small areasof resorption in the distal tibial trabecular or cortical bone, notreadily apparent on low magnification, and rare osteoclasts),2 � mild (increasing areas of resorption in the distal tibialtrabecular or cortical bone, not readily apparent on lowmagnification, with osteoclasts more numerous), 3 � moderate(obvious resorption of the medullary trabecular and corticalbone, without full-thickness defects in the cortex, loss of somemedullary trabeculae, lesion apparent on low magnification,and osteoclasts more numerous), 4 � marked (full-thicknessdefects in the cortical bone, often with distortion of the profileof the remaining cortical surface, marked loss of medullarybone of the distal tibia, numerous osteoclasts, and no resorp-tion in smaller tarsal bones), and 5 � severe (full-thicknessdefects in the cortical bone, often with distortion of the profileof the remaining cortical surface, marked loss of medullarybone of the distal tibia, numerous osteoclasts, and resorptiondefinitely present in smaller tarsal bones) (52). For inflamma-tion, scores were as follows: 0 � normal, 1 � minimalinfiltration of inflammatory cells in the periarticular tissue, 2 �mild infiltration with mild edema, 3 � moderate infiltrationwith moderate edema, 4 � marked infiltration with markededema, and 5 � severe infiltration with severe edema (52).
Histopathologic scoring was expressed as the compos-ite total score of the histologic parameters of inflammation,pannus formation, cartilage damage, and bone resorption (46);the total score was used because of the destructive pathologicprocesses observed in the tmTNF�-transgenic/TNF�-KO ani-mals. Cartilage damage was scored according to the followingcriteria (0–5 scale): 0 � normal, 1 � minimal-to-mild loss oftoluidine blue staining, with no obvious chondrocyte loss orcollagen disruption, 2 � mild loss of toluidine blue staining,with mild focal (superficial) chondrocyte loss and/or collagendisruption, 3 � moderate loss of toluidine blue staining, withmultifocal moderate (to middle-zone depth) chondrocyte lossand/or collagen disruption, 4 � marked loss of toluidine blue
staining, and 5 � severe, diffuse loss of toluidine blue staining,with severe multifocal (no tidemark depth) chondrocyte lossand/or collagen disruption. A small number of animals (n � 2)were killed at the time of initiation of treatment to histologi-cally confirm the presence or absence of arthritis at studyinitiation and also to evaluate the morphologic characteristicsof the spleen, to differentiate between TNF�-KO animals andtmTNFA�-transgenic/TNF�-KO animals throughout thetreatment and observation period.
Statistical analysis. Clinical data on the knee, paw, andankle-joint diameter and volume were analyzed by determiningthe area under the curve (AUC), with subsequent comparisonsby analysis of variance (ANOVA) (52). For calculation of theAUC, the daily volume of the ankle joints (determined using awater displacement system) for each mouse was entered andplotted with SAS software (SAS Institute, Cary, NC), and theperiod of time spanning the treatment days after the onset ofdisease to the termination day was computed in relation toankle volume. Mean values for each group were determinedand the percentage inhibition of arthritis in relation to thevalues in arthritis controls was calculated by comparing themean values between the treated and normal control animals.
ANOVA was performed to test for differences amongthe treatment groups. If the ANOVA showed significantdifferences (P 0.05), Student’s pairwise t-tests were per-formed to test for differences between the treatment groupsand the arthritis control group, and between each treatmentgroup. A similar nonparametric analysis was performed forscores of bone resorption and inflammation between groups,using Kruskal-Wallis and Wilcoxon’s pairwise tests. Pawweights (expressed as the mean � SEM paw volume on waterdisplacement) for each group were analyzed for differencesusing Student’s t-test. The percentage inhibition of paw volumeand the AUC were calculated using the formula ([A �B)]/A) � 100, where A indicates the difference in mean valuesbetween the disease controls and normal animals, and Bindicates the difference in mean values between the treatedanimals and normal animals. Duncan’s new multiple rangetwo-way ANOVA was used to assess significant (P 0.05)differences among the mean values (43).
RESULTS
Generation of transgenic mice overexpressingtmTNF�. Because of the possibility of conflicting resultswith mixed-strain inbred mice, we developed a speed-congenic mating scheme and applied it to the originalmixed-strain tmTgA86�/� and TNF��/� mice, whichresulted in double-homozygous tmTNF�-transgenic/TNF�-KO mice (Figures 1A–C). This new strain of mice(tmTNF�-transgenic/TNF�-KO) developed spontaneous-onset arthritis by 2–4 weeks of age, with full severity ofarthritis by 4–6 weeks of age. In comparison, thetmTNF�-transgenic mice (which also secreted solubleTNF� levels comparable with those in wild-type mice[32]) also developed spontaneous-onset arthritis, but at aslower rate (i.e., by �4–6 weeks of age, 100% incidence
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2875
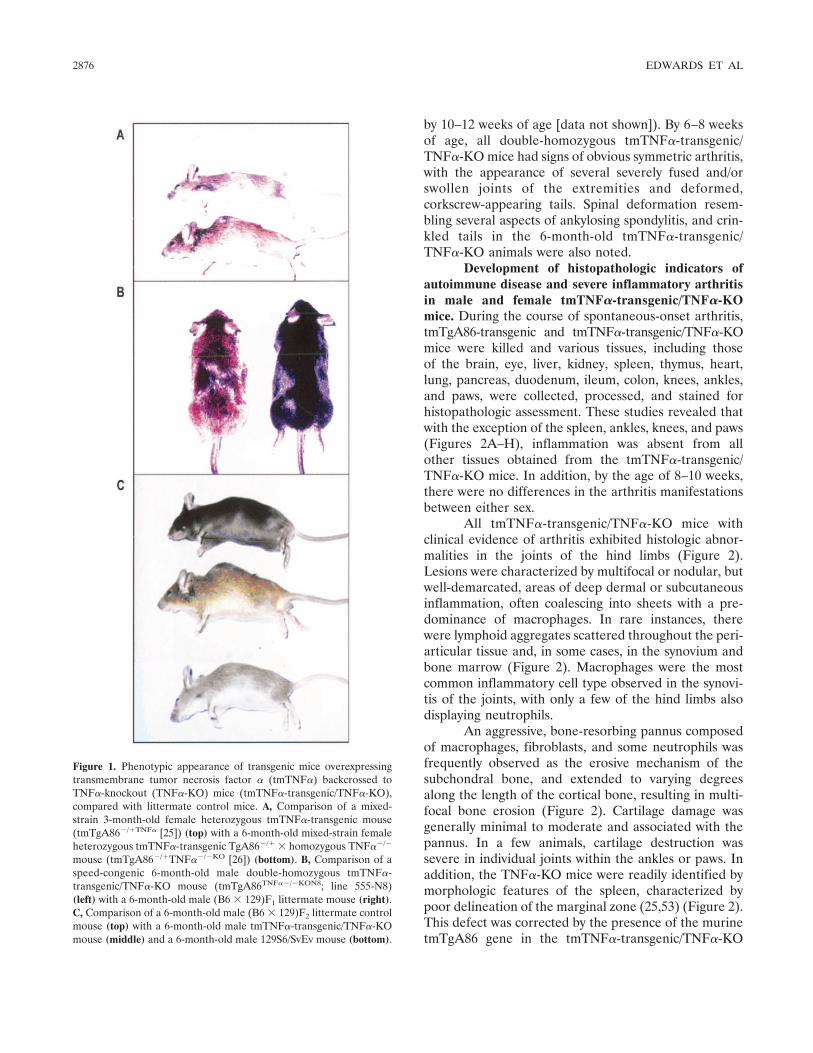
by 10–12 weeks of age [data not shown]). By 6–8 weeksof age, all double-homozygous tmTNF�-transgenic/TNF�-KO mice had signs of obvious symmetric arthritis,with the appearance of several severely fused and/orswollen joints of the extremities and deformed,corkscrew-appearing tails. Spinal deformation resem-bling several aspects of ankylosing spondylitis, and crin-kled tails in the 6-month-old tmTNF�-transgenic/TNF�-KO animals were also noted.
Development of histopathologic indicators ofautoimmune disease and severe inflammatory arthritisin male and female tmTNF�-transgenic/TNF�-KOmice. During the course of spontaneous-onset arthritis,tmTgA86-transgenic and tmTNF�-transgenic/TNF�-KOmice were killed and various tissues, including thoseof the brain, eye, liver, kidney, spleen, thymus, heart,lung, pancreas, duodenum, ileum, colon, knees, ankles,and paws, were collected, processed, and stained forhistopathologic assessment. These studies revealed thatwith the exception of the spleen, ankles, knees, and paws(Figures 2A–H), inflammation was absent from allother tissues obtained from the tmTNF�-transgenic/TNF�-KO mice. In addition, by the age of 8–10 weeks,there were no differences in the arthritis manifestationsbetween either sex.
All tmTNF�-transgenic/TNF�-KO mice withclinical evidence of arthritis exhibited histologic abnor-malities in the joints of the hind limbs (Figure 2).Lesions were characterized by multifocal or nodular, butwell-demarcated, areas of deep dermal or subcutaneousinflammation, often coalescing into sheets with a pre-dominance of macrophages. In rare instances, therewere lymphoid aggregates scattered throughout the peri-articular tissue and, in some cases, in the synovium andbone marrow (Figure 2). Macrophages were the mostcommon inflammatory cell type observed in the synovi-tis of the joints, with only a few of the hind limbs alsodisplaying neutrophils.
An aggressive, bone-resorbing pannus composedof macrophages, fibroblasts, and some neutrophils wasfrequently observed as the erosive mechanism of thesubchondral bone, and extended to varying degreesalong the length of the cortical bone, resulting in multi-focal bone erosion (Figure 2). Cartilage damage wasgenerally minimal to moderate and associated with thepannus. In a few animals, cartilage destruction wassevere in individual joints within the ankles or paws. Inaddition, the TNF�-KO mice were readily identified bymorphologic features of the spleen, characterized bypoor delineation of the marginal zone (25,53) (Figure 2).This defect was corrected by the presence of the murinetmTgA86 gene in the tmTNF�-transgenic/TNF�-KO
Figure 1. Phenotypic appearance of transgenic mice overexpressingtransmembrane tumor necrosis factor � (tmTNF�) backcrossed toTNF�-knockout (TNF�-KO) mice (tmTNF�-transgenic/TNF�-KO),compared with littermate control mice. A, Comparison of a mixed-strain 3-month-old female heterozygous tmTNF�-transgenic mouse(tmTgA86�/�TNF� [25]) (top) with a 6-month-old mixed-strain femaleheterozygous tmTNF�-transgenic TgA86�/� � homozygous TNF��/�
mouse (tmTgA86�/�TNF��/�KO [26]) (bottom). B, Comparison of aspeed-congenic 6-month-old male double-homozygous tmTNF�-transgenic/TNF�-KO mouse (tmTgA86TNF��/�KON8; line 555-N8)(left) with a 6-month-old male (B6 � 129)F1 littermate mouse (right).C, Comparison of a 6-month-old male (B6 � 129)F2 littermate controlmouse (top) with a 6-month-old male tmTNF�-transgenic/TNF�-KOmouse (middle) and a 6-month-old male 129S6/SvEv mouse (bottom).
2876 EDWARDS ET AL

Figure 2. Histopathologic features and clinical progression of arthritis in tmTNF�-transgenic/TNF�-KO mice. Groups of representative mice (n �5 per group) were killed at various time points, and tissues were harvested and processed for hematoxylin and eosin (H&E) and/or toluidine blue(TB) staining. A and B, H&E staining of the spleen of an 8-week-old TNF�-KO (A) and a tmTNF�-transgenic/TNF�-KO (B) female mouse revealspoor delineation of the marginal zone (arrows in A) compared with normal morphologic features with a well-defined marginal zone (arrows in B)in the periarteriolar lymphoid areas. C and D, TB staining of ankle joint sections from an 8-week-old TNF�-KO (C) and a tmTNF�-transgenic/TNF�-KO (D) male mouse reveals normal morphologic features in the distal tibial (T) bone and marrow, normal articular cartilage (arrowheads),and normal periarticular tissue/synovium (S) in C compared with severe arthritis with pannus-mediated cartilage destruction (arrowheads), severeinfiltration of synovium with inflammatory cells, and periosteal bone resorption (arrows) in D. E and F, TB staining of tibiotarsal ankle joints froman 8-week-old TNF�-KO (E) and a tmTNF�-transgenic/TNF�-KO (F) female mouse shows normal morphologic features with normal cartilage(arrowheads), synovium, and cartilage (arrows) in E compared with severe pannus and cartilage loss (arrowheads) with synovitis and bone erosions(arrows) in F. G and H, TB-stained sections of the ankle digit joint from an 8-week-old TNF�-KO (G) and a tmTNF�-transgenic/TNF�-KO (H)female mouse display normal morphologic features with normal synovium and medullary bone in G compared with synovitis and bone erosions(arrows) with marked medullary new bone formation in H. I, Clinical progression of the developing inflammatory arthritis in male and femaledouble-homozygous tmTNF�-transgenic/TNF�-KO mice, assessed as the mean � SEM clinical paw score (determined by water plethysmography[52]) (n � 20 per group). The tmTNF�-transgenic/TNF�-KO mice with minimal clinical evidence of arthritis at 4 weeks of age were kept underbarrier-free specific-pathogen–free conditions throughout the course of the study (46,52). Results are representative of 1 of 2 separately conductedstudies using a total of 40 tmTNF�-transgenic/TNF�-KO mice, with similar experimental results. (Original magnification � 25 in C and D; � 50 inA, B, and E–H.) See Figure 1 for other definitions.

mice, which resulted in a return to normal morphologicappearance (Figure 2).
Faster development of spontaneous-onset in-flammatory arthritis in male compared with femaletmTNF�-transgenic/TNF�-KO mice. During the devel-opment of arthritis in male and female tmTNF�-transgenic/TNF�-KO mice, we observed that by 36 dayspostinjection, all of the male mice had individual clinicalinflammation scores of �2.0 in the paws (Figure 2I). Incontrast, female mice showed a slower progression ofdisease, and individual clinical inflammation scores of�2.0 in the paws were observed only after 8 weeks of age(Figure 2I). The B6, B6 � 129, (B6 � 129)F1, (B6 �129)F2, 129S6/SvEv, and TNF�-KO mice did not de-velop spontaneous arthritis through 9 months of age,thus supporting the original observations of Kollias andcolleagues (31,54) (data not shown). These results dem-onstrate that when murine tmTNF� is overexpressed invivo in a genetically stable mouse background, this formof TNF� is sufficient to induce early-onset, spontaneousarthritis even in the absence of endogenously secretedTNF�. Furthermore, the presence of secreted TNF�does not add significantly to the overall severity ofdisease over time in these genetically manipulated mice,which suggests that tmTNF� is the major disease-meditating cytokine in this model.
Reduction in clinical progression of inflamma-tory arthritis in tmTNF�-transgenic/TNF�-KO mice bytreatment with dexamethasone or p55-PEG. We nextassessed the effects of dexamethasone or p55-PEGtreatment in this animal model (40,43). Mixed-sex, 3–4-week-old tmTNF�-transgenic/TNF�-KO mice with onlyminimal clinical evidence of arthritis were randomlyseparated into 1 of 3 treatment groups (Figure 3). By day2 of vehicle (PBS) control treatment (i.e., 30 days ofage), tmTNF�-transgenic/TNF�-KO animals exhibitedaccelerated clinical inflammation scores of the paws,which consistently worsened throughout the course ofthe experiment (to day 50) (Figure 3). In comparison,both the dexamethasone-treated and p55-PEG–treatedtmTNF�-transgenic/TNF�-KO mice exhibited limitedclinical progression of arthritis in comparison with thevehicle-treated mice, and the arrest of progression lastedthroughout the course of the study (Figure 3). Thesedata reveal that dexamethasone is effective in this modelof tmTNF�-driven experimental arthritis. In addition,p55-PEG, shown earlier to be nonimmunogenic in ro-dents (39,43), limits the progression of the disease tothe same extent as that of dexamethasone in thesemice. Thus, the results suggest that a modified, re-combinant form of the human p55 sTNFR is capable
of neutralizing the biologic effects of overexpressedtmTNF� in vivo.
Reduction in clinical and histologic severity ofarthritis in tmTNF�-transgenic/TNF�-KO mice bytreatment with p55-PEG. We next addressed the effectsof treatment on the histopathologic parameters of thespontaneous-onset arthritis that developed in tmTNF�-transgenic/TNF�-KO mice, using previously describedcriteria (39,46,47,52). Mixed-sex, 8-week-old tmTNF�-transgenic/TNF�-KO mice with established clinical ar-thritis were randomly separated into 1 of 3 treatmentgroups. Mice received PBS vehicle, dexamethasone, orp55-PEG (38,40,43). After 4 weeks of treatment, ani-mals were killed and the paws and ankles (right and left)were examined.
Treatment with p55-PEG significantly (P 0.05)reduced the individual histologic scores, including scoresof inflammation, pannus formation, cartilage damage,and bone damage, in comparison with these parameterscores for the vehicle-treated mice (Figure 4A andFigure 5G). Dexamethasone significantly reduced theinflammation and pannus formation in this group ofmice compared with the vehicle control mice (Figure 4Aand Figure 5E). Similarly, both dexamethasone andp55-PEG significantly reduced the mean histologic
Figure 3. Clinical progression of inflammatory arthritis (mean �SEM paw score) in mixed-sex double-homozygous tmTNF�-transgenic/TNF�-KO mice treated with phosphate buffered salinevehicle control (50 �l/mouse intraperitoneally [IP] each day; n � 20),dexamethasone (0.5 mg/kg IP each day; n � 20), or recombinanthuman PEGylated soluble TNF receptor (p55-PEG) (10 mg/kg IPeach day; n � 20]). The tmTNF�-transgenic/TNF�-KO mice had onlyminimal, if any, clinical evidence of arthritis on day 29. Each treatmentwas administered over a 3-week period. Results are representative of1 of 2 separately conducted studies using a total of 125 tmTNF�-transgenic/TNF�-KO mice, with similar experimental results. SeeFigure 1 for other definitions.
2878 EDWARDS ET AL

scores for medullary bone retention when comparedwith those in the vehicle-treated mice (Figure 4B andFigures 5E–H). Interestingly, neither the steroid-treatednor the p55-PEG–treated animals manifested signifi-cantly reduced bone resorption scores, although therewas a clear trend toward improvement in histologicdisease (Figure 4B and Figures 5E–H). Moreover, dexa-methasone and p55-PEG were shown to significantlyreduce the mean total histologic scores in the tmTNF�-transgenic/TNF�-KO mice (Figure 4C). Thus, these
results suggest that p55-PEG is as effective as dexameth-asone in reducing the histologic parameters of estab-lished arthritis in tmTNF�-transgenic/TNF�-KO mice.
Reduction in clinical progression of developingarthritis in tmTNF�-transgenic/TNF�-KO mice bytreatment with either p55-PEG or p75-Fc sTNFR. Sincewe previously showed that the p55-PEG and p75-Fc
Figure 5. Histologic changes in the joints of tmTNF�-transgenic/TNF�-KO mice treated with dexamethasone or p55-PEG. Histopatho-logic assessments were conducted in arthritic ankle joints with severityscores for inflammation and bone resorption. Tissues were harvestedat 84 days (i.e., at the completion of the treatment period). A and B,Normal ankle from a TNF�-KO male mouse. C and D, Ankle from atmTNF�-transgenic/TNF�-KO male mouse treated with vehicle,showing moderate periarticular inflammation with severe retention ofmedullary bone in the area of the physis and, in D, calcified cartilagebone in the area of the physis. E and F, Ankle from a tmTNF�-transgenic/TNF�-KO male mouse treated with dexamethasone, show-ing mild periarticular inflammation with moderate retention of themedullary bone in the area of the physis and, in F, calcified cartilagein the area of the physis. G and H, Ankle from a tmTNF�-transgenic/TNF�-KO male mouse treated with p55-PEG, showing mild periar-ticular inflammation with minimal retention of the medullary bone inthe area of the physis and, in H, calcified cartilage bone in the area ofthe physis. (Stained with hematoxylin and eosin in A, C, E, and G, andwith toluidine blue in B, D, F, and H; original magnification � 25.) SeeFigure 4 for other definitions.
Figure 4. Effects of phosphate buffered saline vehicle (50 �l/mouseintraperitoneally [IP] each day; n � 12), dexamethasone (Dex) (0.5mg/kg IP each day; n � 12), or recombinant human PEGylated solubletumor necrosis factor receptor (p55-PEG) (10 mg/kg IP each day; n �12) on established inflammatory arthritis in transgenic mice overex-pressing transmembrane tumor necrosis factor � (tmTNF�) back-crossed to TNF�-knockout (TNF�-KO) mice (tmTNF�-transgenic/TNF�-KO). Mixed-sex, 8-week-old double-homozygous tmTNF�-transgenic/TNF�-KO mice with clinical evidence of arthritis wererandomly assigned to receive treatment administered over a 4-weekperiod, after which animals were killed and the paws and ankles (rightand left) were histologically assessed. A, Effects of treatment onindividual histologic score. B, Effects of treatment on individualhistologic bone scores, including bone resorption and medullary boneretention inflammation. C, Effects of treatment on total histologicscores representing 6 individual joints with possible scores of 0–5 foreach joint. Results are representative of 1 of 2 experiments using atotal of 125 tmTNF�-transgenic/TNF�-KO mice, with similar experi-mental results. Q2D � every other day.
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2879

sTNFRs have different mechanisms for reducing TNF�-driven IL-8 release in whole blood (40), we assessed theability of p55-PEG and p75-Fc to reduce disease severityin tmTNF�-transgenic/TNF�-KO mice with developingarthritis. To ensure that both molecules were adminis-tered at the appropriate, most effective doses over time,pharmacokinetic assays were performed to measure theblood levels of each sTNFR over time. The p55-PEGand p75-Fc sTNFRs were administered to separategroups of tmTNF�-transgenic/TNF�-KO male and fe-male mice (n � 18 each) and to groups of 3 mice each atthe 2-, 8-, 24-, 72-, and 96-hour time points post–subcutaneous injection. We have previously reportedthat the ELISAs used to measure both of these agents inrodents are sensitive and specific (40). Thepharmacokinetic/pharmacodynamic data indicated thatat the 1 mg/kg dose, serum levels of p75-Fc weresubstantially higher than those of p55-PEG at the samedose through 48 hours (mean � SEM serum levels 90 �5 �g/ml for p75-Fc versus 42 � 6 �g/ml for p55-PEG).The differential mean levels of these sTNFRs in micewere even more pronounced at the 10 mg/kg dose (datanot shown).
Mixed-sex, 3–4-week-old tmTNF�-transgenic/TNF�-KO mice with only minimal, if any, clinical evi-
dence of arthritis were randomly separated into 1 of the3 treatment groups. Mice treated with p55-PEG orp75-Fc had markedly reduced severity of clinical diseaseas compared with control mice treated with vehicle(Figures 6 and 7). There was no effect of treatment withp55-PEG or p75-Fc on body weight gain (data not
Figure 6. Comparison of histopathologic changes in ankle joints aftertreatment of developing arthritis with soluble tumor necrosis factorreceptors in tmTNF�-transgenic/TNF�-KO mice. Groups of represen-tative male mice (n � 7 per group) were killed at various time pointsand tissues were harvested and processed using toluidine blue staining.A and B, Metacarpophalangeal joint from a normal C57BL/6 wild-typemouse (A) and a vehicle-treated tmTNF�-transgenic/TNF�-KOmouse (B) on day 28 of the treatment period. C and D, Metacarpo-phalangeal joint from a p55-PEG–treated tmTNF�-transgenic/TNF�-KO mouse (C) and a p75-Fc–treated tmTNF�-transgenic/TNF�-KO mouse (D) on day 28 of the treatment period. Treatmentschedule was similar to that shown in Figure 3. (Original magnifica-tion � 100.) See Figure 4 for other definitions.
Figure 7. Comparison of histopathologic scores in ankle joints aftertreatment of developing arthritis with soluble tumor necrosis factorreceptors in tmTNF�-transgenic/TNF�-KO mice. A, Clinical pawscore. B, Histologic scores, including scores of inflammation, pannusformation, cartilage damage, and bone damage. C, Histologic bonescores, including bone resorption and medullary bone retention in-flammation. D, Total histologic scores representing 6 individual jointswith possible scores of 0–5 for each joint. Results are representative of1 of 2 experiments using a total of 75 tmTNF�-transgenic/TNF�-KOmale mice, with similar experimental results. See Figure 4 for defini-tions.
2880 EDWARDS ET AL

shown). The marked beneficial effects of treatment withp55-PEG or p75-Fc were evident in the scores of eachhistologic parameter (Figure 7).
Metacarpophalangeal joints from the normal B6wild-type mice showed growth plates as closed andmedullary cavities containing normal adipose tissue andtrabecular bone (Figure 6A). In comparison, the med-ullary cavity of the tmTNF�-transgenic/TNF�-KO micehad abundant new bone (with retained cartilage) origi-nating from the growth plates (both physeal andarticular-epiphyseal), as well as periarticular and syno-vial inflammation (Figure 6B); in addition, markedresorption of cortical bone was present and the marrowcavity contained proliferating mesenchymal tissue ratherthan adipose bone marrow (Figure 6B).
Inhibition of both bone resorption and medullarybone retention was evident in mice treated with p55-PEG or p75-Fc (Figures 6C and D). Examination of themetacarpophalangeal joints from p55-PEG–treatedtmTNF�-transgenic/TNF�-KO mice on day 28 of thetreatment period showed that the medullary cavity con-tained small amounts of new bone (with retained carti-lage) originating from the growth plate, with minimalperiarticular and synovial inflammation and mild boneresorption (Figure 6C). Similarly, analysis of the meta-carpophalangeal joints from p75-Fc–treated tmTNF�-transgenic/TNF�-KO mice showed that the medullarycavity contained abundant new bone (with retainedcartilage) originating from the growth plates; however,there was minimal periarticular and synovial inflamma-tion with minimal bone resorption (Figure 6D). Thesefindings suggest that the tmTNF�-transgenic/TNF�-KOmouse model is sufficient for initiating and maintaininginflammation and arthritis, and that both p55-PEG andp75-Fc are able to neutralize the biologic and subse-quent histologic destructive effects specifically caused bytmTNF� in vivo.
DISCUSSION
There are several new observations in this studythat extend earlier seminal observations made by Dr.Kollias and colleagues (25,53,54). First, the present datashow that genetically stable inbred mice produced byspeed-congenic breeding (that is, mice that are nearly100% homozygous for the B6 genotype [28]) that arealso deficient in secreted, soluble murine TNF� and aremade transgenic to overexpress tmTNF� rapidly de-velop symmetric inflammatory arthritis by 2–4 weeks ofage. In earlier published studies, heterozygoustmTgA86�/� mice developed arthritis at a slower rate
(by 8 weeks of age) and did not have an obviousankylosing spondylitis hunched-back phenotype (25).
The arthritis observed in the present study wascharacterized by moderate soft tissue swelling in thepaws, knees, and ankles of tmTNF�-transgenic/TNF�-KO animals; they also developed extensive syno-vial inflammation, pannus formation, and bone deformi-ties with nearly 100% incidence by 4–6 weeks of age.Extensive histologic characterization of bone lesions inthe tmTNF�-transgenic/TNF�-KO mice indicated acombination of bone resorption (associated with thepresence of osteoclasts) and medullary bone retention(associated with the physeal retention of calcifiedcartilage/bone) originating from the growth plates. ThetmTNF�-transgenic/TNF�-KO mice in our study did notshow consistent cartilage damage by histology, support-ing the notion that IL-1 is the dominant cartilage-destructive cytokine (52,55,56). The precise role(s) oftmTNF�-induced IL-1� (i.e., cell-bound) and IL-1�(i.e., soluble) production and subsequent cartilage de-struction have not been well characterized in animalmodels of arthritis, but findings in a recent reportsuggest that TNF� plays an important role in theactivation of autoreactive T cells through the inductionof OX40 (57).
We also demonstrated that the homozygoustmTgA86 mice, which also secreted soluble TNF� levelscomparable with those in B6 wild-type mice, developedinflammatory arthritis, but the development of thisdisease occurred at a later age (4–6 weeks) and tooklonger (�10–12 weeks) to fully manifest itself. Thesedata suggest that 1) transgenic mice expressing solely the26-kd tmTNF� are a sufficient model for the initiationand maintenance of inflammation and arthritis indepen-dent of soluble, trimeric, 17-kd TNF�, and 2) mice thatare produced and bred on a mixed-strain background(such as B6 � 129) may potentially mask authentictransgenic and/or knockout mouse phenotypes. Thefindings in the original studies by Dr. Kollias andcolleagues, in which mixed-strain tmTgA86�/� micewere used, also supported these concepts; however, itwas not clear from those previous studies (25,54) whythe presence of tmTNF� or its secreted form wasrequired to cause inflammation. In the present study,this issue has been resolved.
It has previously been shown that geneticallymixed mice made deficient in TNF� do not developarthritis (31,58,59). The speed-congenically producedTNF�-KO mice in our study did not develop arthritis,but clearly had a splenic architecture with abnormalgerminal centers that could be readily differentiated
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2881

from that in either the tmTNF�–expressing and solubleTNF�–secreting tmTNF�-transgenic mice or thetmTNF�-transgenic/TNF�-KO mice used in our studies.To our knowledge, these findings are the first to dem-onstrate these phenotypic distinctions.
In addition, it is likely that this study is the first todemonstrate that tmTNF�-induced arthritis could bearrested by treatment with either p55-PEG (38,40,43) orp75-Fc (41,42,60,61) sTNFRs when administered at themost appropriate doses and time periods. Treatmentwith both of these sTNFRs resulted in limitation of thedevelopment and clinical severity of spontaneous-onsetinflammatory arthritis, as well as reduction in the his-topathologic parameters of inflammation, including thescores of inflammation, pannus formation, cartilagedamage, and bone destruction.
In adjuvant-induced (39,47,50), collagen-induced(43), and streptococcal cell wall–induced (43,62) arthri-tis, p55-PEG reduced clinical disease as well as histo-logic evidence of inflammation, pannus formation, andbone damage, similar to our observations in tmTNF�-transgenic/TNF�-KO mice. Both aspects of the bonelesions in the diseased mice were ameliorated by eitherp55-PEG or p75-Fc treatment. We observed that p55-PEG treatment resulted in slightly greater inhibition ofeach histologic parameter examined; this may be due tothe ability of this molecule to enter the synovial spacemore effectively than the p75-Fc molecule (63), al-though the blood levels of p75-Fc were higher through48 hours post–subcutaneous injection (43,63). Neverthe-less, the present studies demonstrate the marked abilityof both p55-PEG and p75-Fc to inhibit the joint patho-logic changes caused solely by overexpressed tmTNF�.
The phenomenon of cooperative receptor signal-ing was originally observed by Kollias and colleagues(25). In those studies, the p55 and p75 receptors coop-erated positively to mediate signaling pathways anddrive the joint inflammation and joint damage of arthri-tis observed in noncongenic p75�/�, p55�/�, p55�/�/p75�/�, and tmTgA86�/� mice (25). The presence of thep55 TNFR was required for the tmTNF�-mediatedarthritis to occur. However, in the absence of the p75TNFR, the disease was significantly delayed, suggestingthat the 2 TNFRs cooperate in mediating the diseaseprocess (25). The use of tmTNF�-transgenic/TNF�-KOmice allowed us to confirm that this cooperative signal-ing occurs, since there are no other genetic loci in thesemice that could theoretically influence the function ofthese receptors with the cell-bound membrane ligand(28).
In a model of concanavalin A–induced, CD4� T
cell–dependent experimental hepatitis, both TNFRswere required for the development of disease (26). Micedeficient in either of the TNFRs did not develop hepa-titis. It was proposed that the principal ligand for p55TNFR is secreted TNF�, whereas tmTNF� is the pri-mary signaling ligand for p75 TNFR. Although theconcept is controversial, it is now well accepted that thep55 TNFR mediates apoptosis through the death do-main signaling pathway, and that the p75 receptormediates inflammation (11,64). Because of the closejuxtaposition of the tmTNF� to the p75 TNFR thatoccurs during direct cell–cell contact, tmTNF�/p75 com-plexes may form with increased stability and signalingpotential. Steric hindrance by transmembrane TNF�would prevent ligand passing from the p75 receptor andpermit signal transduction to occur. Therefore, tmTNF�is proposed to be the primary activator of the p75receptor, implying that this receptor contributes to thejuxtacrine TNF� response in tissues, as shown in murineexperimental models of RA and hepatitis (26,59).
PEG-p55 and p75-Fc are effective in similarsubpopulations of RA patients (65), and our studydemonstrated that either sTNFR is effective in blockingthe activity of tmTNF� in tmTNF�-transgenic/TNF�-KO mice. It is conceivable that when systemicTNF� concentrations are low, the p75 TNFR and/or thesTNFR serve as a “ligand passer” to the p55 TNFR andsTNFR, and that the p55 sTNFR is entirely essential forreducing soluble and tmTNF� activity due to its near-irreversible properties of binding affinity to TNF�(32,40,66,67). Although this has not been demonstratedto date, it is also conceivable that the p55 sTNFR bindsto the unoccupied TNF� site in p75-Fc–treated RApatients and helps facilitate its clearance (40). Finally,this suggests that p55-PEG may prove useful for thetreatment of RA patients who no longer respond top75-Fc. It is clear that the tmTNF�-transgenic/TNF�-KO mouse model provides an extremely usefultool to dissect the signaling pathways mediated bytmTNF� binding to either p55 or p75 TNFR–inducedinflammatory responses.
As discussed above, the “off” rate of TNF�binding to p75 is rapid when compared with the bindingto p55 (34,66). In many ways, the same type of rapid“off” rate is observed when one considers tmTNF� andp75-Fc binding. The p75-Fc as the Fc bivalent construct(etanercept) binds to membrane TNF�, but this inter-action is reversible, in that the inhibition of biologicactivity is less than that produced by infliximab and thebinding is of a lower affinity (68–70). In contrast,infliximab forms more stable complexes with membrane
2882 EDWARDS ET AL

TNF� relative to the complexes formed with etanercept(4). Moreover, a greater number of infliximab moleculesare bound to tmTNF� with higher avidity comparedwith p75-Fc. This was supported by the observation thatinfliximab exhibited a greater reduction in the biologicactivity of membrane TNF�–bearing cells on endothelialtarget cells compared with the effects of p75-Fc (68).Those studies are consistent with the concept that theaction of p75-Fc on tmTNF� as well as soluble TNF� isreversible, whereas the binding of anti-TNF� monoclo-nal antibodies is more effective for soluble TNF� in theextracellular space as well as for tmTNF� (4,68–70).
In contrast to anti-TNF� monoclonal antibodiesof the IgG1 class, p75-Fc does not appear to lyse cells oractivate caspase 3 in cells expressing tmTNF�, althoughthe results of a recent report suggest otherwise (71).Although p75-Fc is constructed with the complementreceptor domains of human IgG1, the hinge region ofthe fusion of the Fc chain to the p75-Fc is missing one ofthe CH2 groups, and hence the structure is predicted tobe more rigid than the Fc of natural antibodies. Recom-binant human soluble p55 monomeric TNFR has not, todate, been shown to lyse cells (3), and this is also the casefor other monomeric non–Fc-containing p55 sTNFRs(PEG-p55) (65) or CHO-derived p55 (72). Thus, onemust consider that in any clinical event indicative ofdecreased host defense against infection associated withtherapy using any method of TNF� neutralization (in-fliximab, adalimumab, anti-TNF� monoclonal antibod-ies of the IgG4 class, p75-Fc, p55-Fc, or PEG-p55),fixation of complement also results in loss of cells (suchas T cells and macrophages) bearing tmTNF� (4).
It has recently been shown that tmTNF�, in theabsence of soluble TNF�, is sufficient to control acute,but not chronic, Mycobacterium tuberculosis infection, inpart through its expression on T cells (73). This isimportant, since numerous recent studies have shownthat facultative intracellular microorganisms, includingM tuberculosis, can be reactivated upon treatment withhigh-affinity anti-TNF� antibodies, which presumablybind to tmTNF� (4,6,18). Finally, clinical observationsare especially relevant, since Vedanta and colleagueshave recently shown that reverse signaling of tmTNF�differentially modulates CD4� and CD8� T cell activity(74). Thus, our results support the suitability of a mousemodel overexpressing 26-kd tmTNF� and lacking therelease of 17-kd, soluble trimeric TNF� for the initiationof aggressive inflammation leading to severe arthritis,which can be ameliorated with modified, recombinantforms of either the human p55 or the human p75sTNFR.
ACKNOWLEDGMENTS
We thank Danette Barron for her expert assistance onthe illustrations, David A. Norris, MD, for his constructivereview of the manuscript, and Nancy Gunzner for her assis-tance with the typing and preparation of the manuscript.
REFERENCES
1. Dinarello CA. Inflammatory cytokines: interleukin-1 and tumornecrosis factor as effector molecules in autoimmune diseases. CurrOpin Immunol 1991;3:941–8.
2. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood1996;87:2095–147.
3. Edwards CK III. Drug discovery and development for inflamma-tory diseases. Exp Opin Ther Targets 2004;8:1–13.
4. Dinarello CA. Differences between anti-tumor necrosis factor-�monoclonal antibodies and soluble TNF receptors in host defenseimpairment. J Rheumatol 2005;32:40–7.
5. Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001;358:903–11.
6. Wagner U, Pierer M, Wahle M, Moritz F, Kaltenhauser S,Hantzschel H. Ex vivo homeostatic proliferation of CD4� T cellsin rheumatoid arthritis is dyregulated and driven by membrane-anchored TNF�. J Immunol 2004;173:2825–33.
7. Zou J, Rudwaleit M, Brandt J, Theiling A, Braun J, Sieper J.Down-regulation of the nonspecific and antigen-specific T cellcytokine response in ankylosing spondylitis during treatment withinflixamab. Arthritis Rheum 2004;48:780–90.
8. Van Deventer AJ. Transmembrane TNF� induction of apoptosis,and the efficacy of the TNF-targeting therapies in Crohn’s disease.Gastroenterology 2004;121:1242–6.
9. Weinblatt M, Keystone E, Furst DE, Moreland LW, Weisman M,Birbara CA, et al. Adalimumab, a fully human anti–tumor necrosisfactor � monoclonal antibody, for the treatment of rheumatoidarthritis in patients taking concomitant methotrexate. ArthritisRheum 2005;48:35–45.
10. Dekkers PE, Lauw FN, Ten-Hove T, Velde AA, Lumley P,Becherer D, et al. The effect of a metalloproteinase inhibitor(GI5402) on tumor necrosis factor-� (TNF-�) and TNF-� recep-tors during human endotoxemia. Blood 1999;94:2252–8.
11. Akassoglou K, Douni E, Bauer J, Lassmann H, Kollias G, ProbertL. Exclusive tumor necrosis factor (TNF) signaling by the p75TNFreceptor triggers inflammatory ischemia in the CNS of transgenicmice. Proc Natl Acad Sci U S A 2003;100:709–14.
12. Agnholt J, Dahlerup JF, Kaltoft K. The effect of etanercept andinfliximab on the production of tumour necrosis factor-�, interfer-on-� and GM-CSF in in vivo activated intestinal T lymphocytecultures. Cytokine 2003;23:76–85.
13. Sandborn WJ, Hanauer SB, Katz SD, Safdi M, Wolf DG, BaergRD, et al. Etanercept for active Crohn’s disease: a randomized,double-blind, placebo-controlled trial. Gastroenterology 2001;121:1088–94.
14. Van den Brande J, Braat H, van den Brink GR, Versteeg HH,Bauer CA, Hoedemaker I, et al. Inflixamab, but not etanercept,induces apoptosis in lamina propria T-lymphocytes from patientswith Crohn’s disease. Gastroenterology 2003;124:1774–85.
15. Corazza N, Brunner T, Buri C, Rihs S, Imboden MA, Seibold I, etal. Transmembrane tumor necrosis factor is a potent inducer ofcolitis even in the absence of its secreted form. Gastroenterology2004;127:816–25.
16. Marzo-Ortega H, McGonagle D, O’Connor P, Emery P. Efficacyof etanercept for treatment of Crohn’s related spondyloarthritisbut not colitis. Ann Rheum Dis 2004;62:74–6.
17. Willuweit A, Sass G, Schoneberg A, Eisel UL, Tiegs G, Clauss M.Chronic inflammation and protection from acute hepatitis in
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2883

transgenic mice expressing TNF in endothelial cells. J Immunol2001;167:3944–52.
18. Murray DA, Crispe IN. TNF� controls intrahepatic T cell apopto-sis and peripheral T cell numbers. J Immunol 2004;173:2402–9.
19. Kawaguchi M, Mitsuhashi Y, Kondo S. Overexpression of tumornecrosis factor-� converting enzyme in psoriasis. Br J Dermatol2005;152:915–9.
20. Lazdins JK, Grell M, Walker MR, Woods-Cook K, Scheurich P,Pfizenmaier K. Membrane tumor necrosis factor (TNF) inducedcooperative signaling of TNFR60 and TNFR80 favors induction ofcell death rather than virus production in HIV-infected T cells. JExp Med 1997;185:81–90.
21. Neven N, Vis M, Voskuyl AE, Nurmohamed MT, Dijkmans BA,Lems WF. Adverse events in patients with rheumatoid arthritistreated with inflixamab in daily clinical practice. Ann Rheum Dis2005;64:645–6.
22. Dimakou K, Papaioannides D, Latsi P, Katsimboula S, Korantzo-poulos P, Orphananidou D. Disseminated tuberculosis complicat-ing anti-TNF treatment. Int J Clin Pract 2004;58:1052–5.
23. Wallis RS, Broder M, Wong J, Lee A, Hoq L. Reactivation oflatent granulomatous infections by inflixamab. Clin Infect Dis2005;41:S194–8.
24. Dinarello CA. Anti-cytokine therapeutics and infections. Vaccine2003;S2:24–34.
25. Alexopoulou L, Pasparakis M, Kollias G. A murine transmem-brane tumor necrosis factor transgene induces arthritis by coop-erative p55/p75 tumor necrosis factor receptor signaling. EurJ Immunol 1997;27:2588–92.
26. Kusters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E,Kunstle G, et al. In vivo evidence for a functional role of bothtumor necrosis factor (TNF) receptors and transmembrane TNFin experimental hepatitis. Eur J Immunol 1997;27:2870–5.
27. Alexopoulou L, Pasparakis M, Kollias G. Mutant mice expressinguncleavable transmembrane TNF are resistant to low dose listeriainfection but develop lethal meningeal inflammation with highincidence. Cytokine 1997;9:869–71.
28. Lundberg P, Welander H, Openshaw C, Nalbandion A, EdwardsCK III, Moldawer LL, et al. A locus on mouse chromosome 6 thatdetermines resistance to herpes simplex viruses also influencesreactivation, while an unlinked locus augments resistance offemale mice. J Virol 2004;77:11661–73.
29. Wakeland EK, Morel L, Achey K, Yui M, Longmate J. Speedcongenics: a classic technique in the fast lane (relatively speaking).Immunol Today 1997;18:472–7.
30. Wong HL. Speed congenics: applications for transgenic andknock-out mouse strains. Immunol Today 2002;18:472–7.
31. Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immuneand inflammatory responses in TNF�-deficient mice: a criticalrequirement for TNF � in the formation of primary B cell follicles,follicular dendritic cell networks and germinal centers, and in thematuration of the humoral immune response. J Exp Med 1996;184:1397–411.
32. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B,et al. The transmembrane form of tumor necrosis factor is theprime activating ligand of the 80kDa tumor necrosis factor recep-tor. Cell 1995;83:793–802.
33. Shurety W, Pagan JK, Prins JB, Stow JL. Endocytosis of uncleavedtumor necrosis factor-� in macrophages. Lab Invest 2001;81:107–17.
34. Gerspach J, Gotz A, Zimmermann G, Kolle C, Bottinger H, Grell M.Detection of membrane-bound tumor necrosis factor (TNF): ananalysis of TNF-specific reagents. Micros Res Tech 2000;50:243–50.
35. Josephs MD, Bahjat FR, Fukuzuka K, Ksontini R, Solorzano CC,Edwards CK III, et al. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-� and not by Fasligand. Am J Physiol Regul Integr Comp Physiol 2000;278:R1196–201.
36. Solorzano CC, Ksontini R, Pruitt JH, Hess PJ, Edwards PD,Kaibara A, et al. Involvement of 26 kD cell associated TNF� inexperimental hepatitis and exacerbation of liver injury with amatrix metalloproteinase inhibitor. J Immunol 1997;158:414–9.
37. Solorzano CC, Kaibara A, Hess PJ, Edwards PD, Ksontini R,Abouhamze A, et al. Pharmacokinetics, immunogenicity andefficacy of dimeric TNFR binding proteins in healthy and bacte-remic baboon. J Appl Physiol 1998;84:1119–30.
38. Edwards CK III. PEGylated recombinant human soluble tumournecrosis factor receptor type I: a novel high affinity TNF receptordesigned for chronic inflammatory diseases. Ann Rheum Dis1999;58(Suppl 1):173–81.
39. Bendele AM, McComb J, Gould T, Frazier J, Chlipala ES, SeelyJ, et al. Combination benefit of PEGylated soluble tumor necrosisfactor receptor type I and dexamethasone or indomethacin inadjuvant arthritic rats. Inflamm Res 1999;48:453–60.
40. Frishman JI, Edwards CK III, Sonnenberg MG, Kohno T, CohenAM, Dinarello CA. Tumor necrosis factor (TNF)-�-induced in-terleukin-8 in human blood cultures discriminates neutralizationby the p55 and p75 TNF soluble receptors. J Infect Dis 2000;182:1722–30.
41. Moreland LW, Margolies G, Heck LW Jr, Saway A, Blosch C,Hanna R. et al. Recombinant soluble tumor necrosis factorreceptor (p80) fusion protein: toxicity and dose finding trial inrefractory rheumatoid arthritis. J Rheumatol 1996;23:1849–55.
42. Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleisch-mann RM, Bulpitt KJ, et al. Etanercept therapy in rheumatoidarthritis: a randomized, controlled trial. Ann Intern Med 1999;130:478–86.
43. Edwards CK III, Martin SW, Seely AJE, Kinstler O, Buckel SR,Bendele AM, et al. Design of PEGylated soluble tumor necrosisfactor receptor type I for chronic inflammatory diseases: sitespecific PEGylation of polypeptides. Adv Drug Del Rev 2004;55:1315–36.
44. Bendele AM, McComb J, Gould T, Chilipala L, Frazier J,Colagiovanni DB, et al. Comparative efficacy of sTNF-RI, a novelmonomeric, recombinant soluble TNF type I receptor, to dimeric’ssTNF-RI and sTNF-RII IgG1 Fc fusion proteins in animal modelsof rheumatoid arthritis [abstract]. Eur Cytokine Netw 1998;18:A89.
45. Campagnuolo J, Bolon B, Feige U. Kinetics of bone protection byrecombinant osteoprotegerin therapy in Lewis rats with adjuvantarthritis. Arthritis Rheum 2002;46:1926–36.
46. Bendele AM, McComb J, Gould T, McAbee T, Sennello G,Chlipala ES, et al. Animal models of arthritis: relevance to humandisease. Toxicologic Path 1999;27:134–42.
47. Bendele AM, McComb J, Gould T, Frazier J, Chlipala ES, SeelyJ, et al. Effects of PEGylated soluble tumor necrosis factorreceptor type I alone and in combination with methotrexate inadjuvant arthritic rats. Clin Exp Rheumatol 1999;17:553–60.
48. Bendele AM. Animal models of rheumatoid arthritis. J Musculo-skel Neuron Interact 2001;1:377–85.
49. Bendele AM. Animal models of osteoarthritis. J MusculoskelNeuron Interact 2001;1:363–76.
50. McComb J, Gould T, Chlipala ES, Sennelo G, Frazier J, Kieft G,et al. Antiarthritic activity of soluble tumor necrosis factor recep-tor type I forms in adjuvant arthritis: correlation of plasma levelswith efficacy. J Rheumatol 1999;26:1347–51.
51. Bendele AM, McAbee T, Sennello G, Frazier J, Chlipala ES,McCabe D. Efficacy of sustained blood levels of interleukin-1receptor antagonist in animal models of arthritis. Arthritis Rheum1999;42:498–506.
52. Bendele AM, Chlipala ES, Scherrer J, Frazier J, Sennello G, RichWJ, et al. Combination benefit of treatment with the cytokineinhibitors interleukin-1 receptor antagonist and PEGylated solu-ble tumor necrosis factor receptor type I in animal models ofrheumatoid arthritis. Arthritis Rheum 2000;43:2648–59.
2884 EDWARDS ET AL

53. Alexopoulou L, Pasparakis M, Kollias G. Complementation oflymphotoxin � knockout mice with tumor necrosis factor-express-ing transgenes rectifies defective splenic structure and function. JExp Med 1998;188:745–54.
54. Kassiotis G, Kollias G. TNF and receptors in organ-specificautoimmune disease: multi-layered functioning mirrored in animalmodels. J Clin Invest 2001;107:1507–8.
55. Bresnihan B, Alvaro-Gracia JM, Cobby M, Doherty M, DomljanZ, Emery P, et al. Treatment of rheumatoid arthritis with recom-binant human interleukin-1 receptor antagonist. Arthritis Rheum1998;41:2196–204.
56. Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW,et al. Treatment of rheumatoid arthritis with anakinra, a recom-binant human interleukin-1 receptor antagonist, in combinationwith methotrexate. Arthritis Rheum 2002;46:614–24.
57. Horai R, Nakajima A, Habiro K, Kotani M, Nakae S, Matsuki T,et al. TNF� is crucial for the development of autoimmune arthritisin IL-1 receptor antagonist-deficient mice. J Clin Invest 2004;114:1603–11.
58. Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al.Mice deficient in tumor necrosis factor-� are resistant to skincarcinogenesis. Nat Med 1999;5:828–31.
59. Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role oftumor necrosis factor and receptors in models of multiorganfailure, rheumatoid arthritis, multiple sclerosis and inflammatorybowel disease. Immunol Rev 1999;169:175–94.
60. Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleisch-mann RM, Weaver AL, et al. Treatment of rheumatoid arthritiswith a recombinant human tumor necrosis factor receptor(p75)-Fc fusion protein. N Engl J Med 1997;337:141–7.
61. Wooley PH, Dutcher J, Widmer MB, Gillis S. Influence of arecombinant human soluble tumor necrosis factor receptor FCfusion protein on type II collagen-induced arthritis in mice.J Immunol 1993;151:6602–7.
62. Kuiper S, Joosten LA, Bendele AM, Edwards CK III, Arntz OJ,Helsen MM, et al. Different roles of tumour necrosis factor � andinterleukin 1 in murine streptococcal cell wall arthritis. Cytokine1998;10:690–702.
63. Zinn KR, Chaudhuri TR, Edwards CK III, Liu HG, Mountz JD,Mountz JM. A novel Tc-99m–labeled soluble receptor for in vivoimaging of TNF-� expression in arthritis animal models [abstract].Arthritis Rheum 1998;41Suppl 9:S213.
64. Nagata S. DNA degradation in development and programmed celldeath. Annu Rev Immunol 2005;23:853–75.
65. Furst D, Fleischman R, Kopp E, Schiff M, Edwards CK III,Solinger A, et al. A phase 2 dose-finding study of PEGylatedrecombinant methionyl human soluble tumor necrosis factor typeI in patients with rheumatoid arthritis. J Rheumatol 2005;32:2303–10.
66. Grell M, Wajant H, Zimmerman G, Scheurich P. The type Ireceptor (CD120a) is the high-affinity receptor for soluble tumornecrosis factor. Proc Natl Acad Sci U S A 1998;95:570–5.
67. Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kdtumor necrosis factor (TNF) receptor recruits TNF for signalingby the 55 Kd TNF receptor. J Biol Chem 1993;268:18542–8.
68. Scallon B, Cai A, Solowski N, Rosenberg A, Song X-Y, Shealy D,et al. Binding and functional comparisons of two types of tumornecrosis factor antagonists. J Pharm Exp Ther 2002;301:418–26.
69. Scallon BJ, Moore MA, Trinh H, Knight DM, Ghrayeb J. Chi-meric anti-TNF-� monoclonal antibody cA2 binds recombinanttransmembrane TNF-� and activates immune effector functions.Cytokine 1995;7:251–9.
70. Scallon BJ, Trinh H, Nedelman M, Brennan FM, Feldmann M,Ghrayeb J. Functional comparisons of different tumour necrosisfactor receptor/IgG fusion proteins. Cytokine 1995;7:759–70.
71. Catrina AI, Trollmo C, Klint E, Engstrom M, Lampa J, Hermans-son Y, et al. Evidence that anti–tumor necrosis factor therapy withboth etanercept and inflixamab induces apoptosis in macrophages,but not lymphocytes, in rheumatoid arthritis joints. ArthritisRheum 2005;52:61–72.
72. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF� therapies: the next generation. Nat Drug Discovery 2003;5:736–46.
73. Saunders BM, Tran S, Ruuls S, Sedgwick JD, Briscoe H, BrittonWJ. Transmembrane TNF is sufficient to initiate cell migrationand granulomatous formation and provide acute, but not long-term, control of Mycobacterium tuberculosis infection. J Immunol2005;174:4852–9.
74. Vedanta NK, Holler E, Ewing P, Schulz U, Heffner S, Burger V,et al. Reverse signaling of membrane-integrated tumor necrosisfactor differentially regulates alloresponses of CD4� and CD8� Tcells against human microvascular endothelial cells. Immunology2005;115:536–43.
EFFICACY OF sTNFRs IN tmTNF�-TRANSGENIC MICE 2885





























