Embed Size (px)
Citation preview

Spatiotemporal distribution of spectrin breakdownproducts induced by anoxia in adult rat optic nervein vitro
Nathalie Jette1, Elaine Coderre1, Maria A Nikolaeva1, Paula D Enright1, Akira Iwata2,Douglas H Smith2, Qiubo Jiang1 and Peter K Stys1
1Ottawa Health Research Institute, Division of Neuroscience, University of Ottawa, Ottawa, Ontario, Canada;2Department of Neurosurgery, University of Pennsylvania, Philadelphia, Pennsylvania, USA
Hypoxic/ischemic and traumatic injury to central nervous system myelinated axons is heavilydependent on accumulation of Ca ions in the axoplasm, itself promoted by Na influx from theextracellular space. Given the high density of nodal Na channels, we hypothesized that nodes ofRanvier might be particularly vulnerable to Ca overload and subsequent damage, as this is theexpected locus of maximal Na influx. Adult rat optic nerves were exposed to in vitro anoxia andanalyzed immunohistochemically for the presence of spectrin breakdown. Cleavage of spectrinbecame detectable between 15 and 30 mins of anoxia, and increased homogeneously along thelengths of fibers; localized breakdown was not observed at nodes of Ranvier at any time pointanalyzed. Spectrin breakdown was also found in glial processes surrounding axons. Confocalimaging of axoplasmic Ca also revealed a gradual and nonlocalized increase as anoxia progressed,without evidence of Ca ‘hot-spots’ anywhere along the axons at any time between 0 and 30 mins ofanoxic exposure in vitro. Calculations of Ca diffusion rates indicated that even if Ca entered or wasreleased focally in axons, this ion would diffuse rapidly into the internodes and likely producediffuse injury by activating Ca-dependent proteases. Western blot analysis for voltage-gated Nachannel protein revealed that key functional proteins such as these are also degraded by anoxia/ischemia. Thus, proteolysis of structural and functional proteins will conspire to irreversibly injurecentral axons and render them nonfunctional, eventually leading to transection, degradation, andWallerian degeneration.Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786. doi:10.1038/sj.jcbfm.9600226; published online14 September 2005
Keywords: anoxia; calcium; confocal microscopy; ichemia; axon; spectrin; glia
Introduction
Disorders of white matter may cause serious dis-ability if vital tracts are disrupted. Examples includeneurotrauma, with both traumatic brain and spinalcord injuries invariably damaging white mattertracts, stroke which almost always affects myeli-nated fibers in the brain, and inflammatory demye-lination such as multiple sclerosis. Much work hasbeen performed on the pathophysiology of ischemic
gray matter damage, and in the last decade sig-nificant insight has also been gained into themechanisms of white matter damage during is-chemic and traumatic injury. A complex interplayof voltage-gated and transmitter-activated ion chan-nels, transporters, mitochondrial failure, nitricoxide (NO) production, and free radical generationis set in motion, resulting in tissue destruction, withmechanisms sharing significant overlap betweenischemic and traumatic insults (for reviews, seeGoldberg and Ransom, 2003; Maxwell et al, 1997;Park et al, 2004; Stys, 2004). As with gray matterinjury, a final common step appears to be cellularoverload of Ca, an ion that is normally very tightlyregulated in all mammalian cells. Excessive cytoso-lic Ca will trigger a variety of deleterious Ca-dependent biochemical pathways, with importanteffectors being the Ca-activated proteases, thecalpains.
Received 8 July 2005; accepted 4 August 2005; published online14 September 2005
Correspondence: Dr PK Stys, Division of Neuroscience, OttawaHealth Research Institute, 725 Parkdale Avenue, Ottawa, ON,Canada K1Y 4K9. E-mail: [email protected]
This work was supported in part by the CIHR (operating and
equipment), National MS Society (operating), and the Heart and
Stroke Foundation of Ontario Career Investigator Award (salary)
grants to PKS, and NIH grant to DHS.
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786& 2006 ISCBFM All rights reserved 0271-678X/06 $30.00
www.jcbfm.com

The calpains are cysteine proteases that requireCa for activation, and function normally in growth,synaptic plasticity, and normal cellular remodeling(Croall and DeMartino, 1991; Melloni and Pontre-moli, 1989; Ray and Banik, 2003; Suzuki et al, 1995).Given the ubiquitous nature of this protease, and itsability to degrade a large variety of key cellularproteins, such as spectrin, neurofilament, ionchannels and receptors, myelin proteins, and manyothers (Buki et al, 1999; Deshpande et al, 1995; Jiangand Stys, 2000; McCracken et al, 2001; Schaecher etal, 2001; Schumacher et al, 2000; Shields et al,1999), it has also been implicated in destruction ofcentral nervous system (CNS) tissue during patho-logical conditions such as ischemia and trauma, inboth gray and white matter regions (Buki et al, 1999;McCracken et al, 1999; Roberts-Lewis et al, 1994;Saatman et al, 1996). Unlike neurons, myelinatedaxons are unique in that their surface is coveredalmost entirely by the myelin sheath, with onlyE1% of their surface area exposed to the extra-cellular space at nodes of Ranvier. Given that Cainflux appears to be a key step in axonal injury, andthat Ca-dependent calpain activation is an impor-tant consequence (Jiang and Stys, 2000), we hy-pothesized that nodes would be particularlyvulnerable to calpain-mediated injury during anox-ia. We performed a series of experiments in an invitro model of anoxic white matter to examine thespatiotemporal profile of calpain-mediated cytoske-letal damage to see if nodes are initially orpreferentially damaged. We also examined whethernodal Na channels, critical for supporting actionpotential conduction and present at very highdensities at nodes of Ranvier, are damaged duringanoxia.
Materials and methods
Tissue Preparation
Optic nerves were dissected from adult Long-Evans malerats (200 to 250 g; Charles River) as previously described(Stys et al, 1991). Nerves were equilibrated in 95% O2/5%CO2 for 60 mins in an interface chamber maintained at371C and were perfused at 2 mL/min with artificialcerebrospinal fluid (aCSF) containing the following (inmmol/L): NaCl 126, KCl 3.0, NaHCO3 26, MgSO4 2.0,NaH2PO4 1.25, CaCl2 2.0, and glucose 10 (pH 7.4). Anoxiawas induced by switching to 95% N2/5% CO2 for 15 to60 mins.
Immunostaining for Spectrin Breakdown Products(SBP), Neurofilament NF-160, and Na Channels
We used qualitative confocal immunofluorescence toexamine which optic nerve elements were damaged byanoxia. Antiserum against SBPs (SBP 38-2 – a generousgift from Dr Robert Siman, University of Pennsylvania)was used to examine Ca-dependent calpain-mediated
degradation of the structural protein spectrin in opticnerves. We did not attempt to precisely delineate thesubcellular distributions of spectrin breakdown patterns,but instead used this marker to distinguish whether nodalregions were initially and preferentially damaged, whichwas our initial hypothesis. After surgical preparation,optic nerve dissection, incubation in aCSF, and exposureto normoxia or anoxia as described above, samples werefixed in 4% paraformaldehyde for 3 h, and then cryopro-tected in 20% sucrose phosphate buffer at 41C overnight.Tissues were then rinsed twice for 10 mins in 0.05 mol/LTris, rinsed for 20 mins in methanol, rinsed again threetimes for 10 mins in Tris 0.05 mol/L. The sample was thenblocked with 10% normal goat serum in 0.3% Tris-tritonX-100, and incubated overnight in primary antiserumdiluted in Tris-triton/normal goat serum at concentrationsof 1:1000 for NF-160 (Sigma, St Louis), 1:5000 for SBP 38-2, and 1:500 for Na channels (Sigma). After 3 quick rinsesin 0.05 mol/L Tris buffer, tissues were incubated for 2 h ingoat anti-mouse Alexa-488 (Molecular Probes, Eugene,OR) at a concentration of 1:100 (against NF), goat anti-rabbit Alexa-594 at 1:100 (against SBP), and anti-mouseAlexa-488 at 1:250 (against Na channels) in 0.05 mol/LTris/0.9% NaCl. Tissues were then quickly rinsed in0.05 mol/L Tris three times, and mounted on microscopeslides in Prolong Antifade Reagent (Molecular Probes).Images were collected on a Bio-Rad 1024 confocallaser-scanning microscope with an � 60 oil immersionobjective.
Na Channel Western Blots
Frozen optic nerves (n = 4, each group) were homogenizedin 200 mL of lysis buffer (M-PER, Pierce, Rockford, IL,USA) with protease inhibitor mixtures (pepstatin A, 1mg/mL;leupeptin, 1mg/mL; aprotinin, 1mg/mL; Pefabloc SC,0.2 mmol/L; benzemidine, 0.1 mg/mL; calpain inhibitors Iand II, 8mg/mL each; all from Sigma) on ice for 30 mins.The homogenates were centrifuged at 15,000g for 30 minsat 41C. The supernatants were frozen at �801C until use.The samples (15 mg/lane) were boiled for 5 mins in SDSsample buffer (glycerol 1 mL, b-mercaptoethanol 0.5 mL,10% SDS 3 mL, 1.0 mol/L Tris-HCl 1.25 mL, pH 6.7, andbromophenol blue 2 mg) and run on 7% Tris acetateNuPAGE gel (Invitrogen, Carlsbad, CA, USA) at 30 V for1.5 h. The polypeptides were electrotransferred to immu-nobilon membranes (Millipore) at 150 V for 1.5 h. Non-specific binding was blocked using 5% non-fat milk inPBST (9.1 mmol/L dibasic sodium phosphate, 1.7 mmol/Lmonobasic sodium phosphate, 150 mmol/L NaCl, and0.1% Tween 20) for 30 mins at room temperature (RT).The membranes were incubated with polyclonal rabbitanti-pan Na channel antibody (#06–811, Upstate, LakePlacid, NY; 1:80) specific for the intracellular III–IV loopof the Na channel a-subunit (amino acids (a.a.) 1491 to1508 of full length a.a. 1–2005; P04775) overnight at 41C,then incubated with HRP-conjugated secondary antibody(1:20,000, Vector Laboratories, Burlingame, CA, USA) for1 h each at RT. The membranes were rinsed three timeswith TTBS (100 mmol/L Tris, 0.9% NaCl, and 0.1% Tween
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
778
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786

20) for 5 mins at RT, incubated with WestDura Chemilu-minescent Substrate (Pierce) for 5 mins, and then exposedto X-ray film (Biomax MS-1, Kodak, Rochester, NY, USA).The film was scanned and analyzed using ImageTraksoftware (written by PKS).
Ca Imaging in Live Optic Nerve
Adult Long-Evans male rats were anesthetized with 80%CO2/20% O2 and decapitated. Optic nerves were dissectedout, immersed in Ca2 +-free aCSF at 41C. Nerves wereplaced in an interface perfusion chamber and loaded for1.5 h with fluorescent dyes through a suction pipetteapplied to the distal end. For Ca2 + imaging nerves wereplaced in a custom-built perfusion chamber, mounted onan upright Nikon C1 confocal laser-scanning microscope.Imaging was performed at 361C with an � 60 immersionobjective, itself maintained at the same temperature toavoid cooling of the sample. Images were analyzed usingImageTrak: axonal fluorescence changes were normalizedto average basal levels and reported as a ratio of signalcollected from Ca2 +-sensitive and -insensitive fluoro-phores plotted against time.
Oregon Green 488 BAPTA-1 Dextran (KdE450 nmol/L)was used as a Ca2 + indicator. For visualization of axonalprofiles, nerves were coloaded with red Alexa Fluor 594Dextran. The red Ca2 +-independent fluorescence was usedto outline regions of interest (ROIs) from which green Ca2 +
-dependent fluorescence was measured. The loadingbuffer contained low Na+ concentration (NaCl wasreplaced by 126 mmol/L of N-methyl-D-glucamine) andlow Ca2 + concentration (CaCl2 was omitted) to mimicintraaxonal concentrations. Dye concentrations inthe loading pipette were: 75 mmol/L for Oregon Green488 BAPTA-1 Dextran and 100mmol/L Alexa Fluor594 Dextran. Fluorescence dyes were purchased fromMolecular Probes.
Results
Lack of Spectrin Breakdown in Control Nerves
Calpain-mediated spectrin breakdown is a reliableindex of calpain activity (Siman et al, 1989). Opticnerves were dissected from freshly killed rats,placed in an in vitro perfusion chamber at 371C,and allowed to equilibrate under normoxic condi-tions for E60 mins. Immunohistochemistry wasthen performed for SBP to examine if any elementsof the optic nerve were damaged by excision andequilibration. Figures 1A to 1D shows representativeconfocal micrographs of rat optic nerve doublestained with antineurofilament antibody to outlineaxon cylinders and SBP. Normoxic conditions didnot induce significant axonal damage, showing noevidence of accumulation of spectrin cleavageproducts either immediately after dissection or after60 mins of normoxic incubation. Figures 1E and 1Fshows double staining with the same two antisera,but with images taken from the cut end of normoxic
nerve: this traumatized region showed abundantspectrin breakdown as expected, serving as apositive control for this antibody. Oligodendrocytes
Figure 1 Confocal microscopic images of fixed normoxic controloptic nerves stained immunohistochemically with standardmarkers (neurofilament and CNPase to identify axon cylindersand oligodendrocytes, respectively) and for spectrin breakdown.(A–D) Sections taken from the middle of the nerve (away fromthe cut ends) show regular neurofilament profiles and very littlespectrin degradation. (E–F) Sections near the traumatized cutends show considerable spectrin breakdown as expected. (G–H)CNPase outlines oligodendroglia and their processes. As withaxons sampled from the middle of the nerve, there was littlespectrin breakdown within these cells or their processes. Scalebars in (A–F) 10mm. Scale bars in (G, H) 20mm.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
779
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786

labeled with anti-CNPase antibody (Trapp et al,1988) and SBP exhibited very little cytoskeletaldamage under normoxic conditions (Figures 1Gand 1H).
Figure 2 shows control optic nerve as above, butdouble labeled for Na channels and SBP. Numerouspunctate regions reflect the high density of Nachannels at nodes of Ranvier. Once again, controloptic nerves did not exhibit spectrin breakdown,either diffusely or more specifically at the nodesof Ranvier.
Spectrin Breakdown in Anoxic Optic Nerves
Optic nerves exposed to 15, 30, or 60 mins of anoxiaat 371C in the in vitro perfusion chamber wereprocessed for immunohistochemistry using thesame markers as for control nerves above. Comparedwith normoxic controls, no significant spectrinbreakdown was observed after 15 mins of anoxia(Figures 3A and 3B). However, significant spectrincleavage was seen after 30 mins of anoxia (Figures3C and 3D). The pattern exhibited a roughly linearappearance which followed the direction of thefibers, but was otherwise diffuse, with no indicationof any focal onset of spectrin proteolysis. Given theexpected ionic disturbances that may begin at nodalregions, we hypothesized that spectrin breakdownmay start in this location; however, nodal labelingwith Na channel antibody did not suggest prefer-ential colocalization of spectrin breakdown at nodesof Ranvier (Figures 3E and 3F).
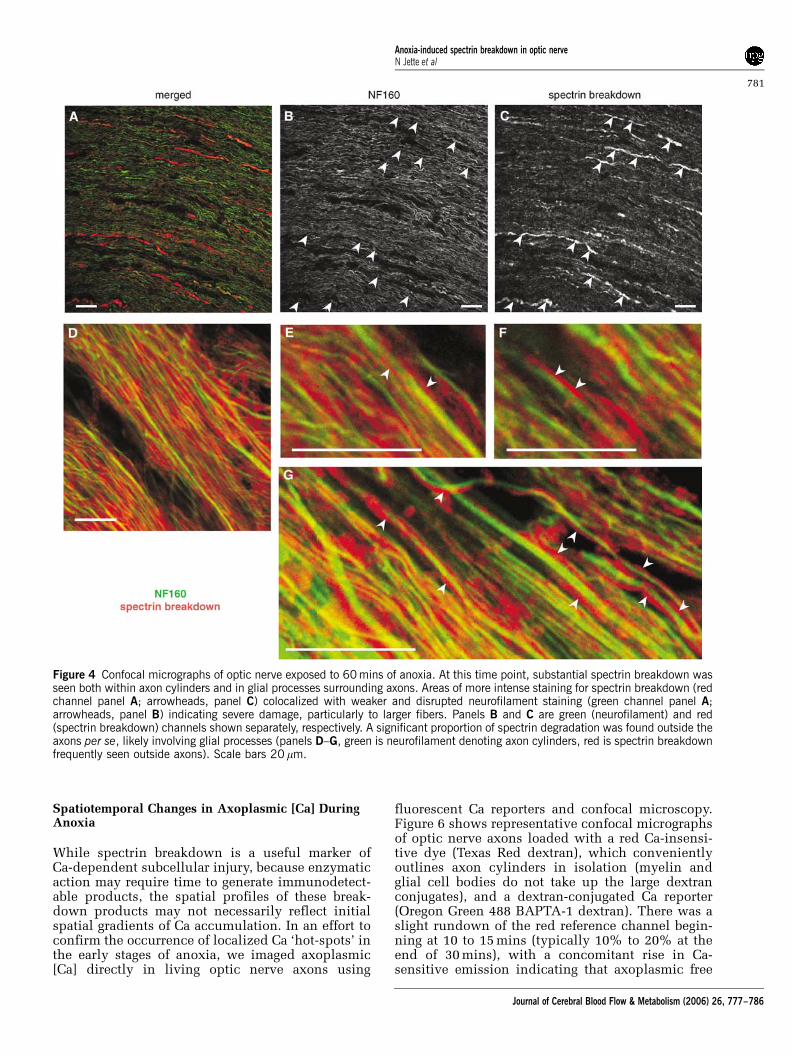
After 60 mins of anoxia, substantial spectrinbreakdown was seen as expected, but neurofilamentprofiles appeared more disrupted, indicating in-creasing damage to axon cylinders (Figure 4). Manyaxons (particularly larger diameter fibers) displayedstrong SBP signal colocalized with much weakerand disrupted neurofilament staining (arrowheadsin Figures 4B and 4C indicate identical regions of
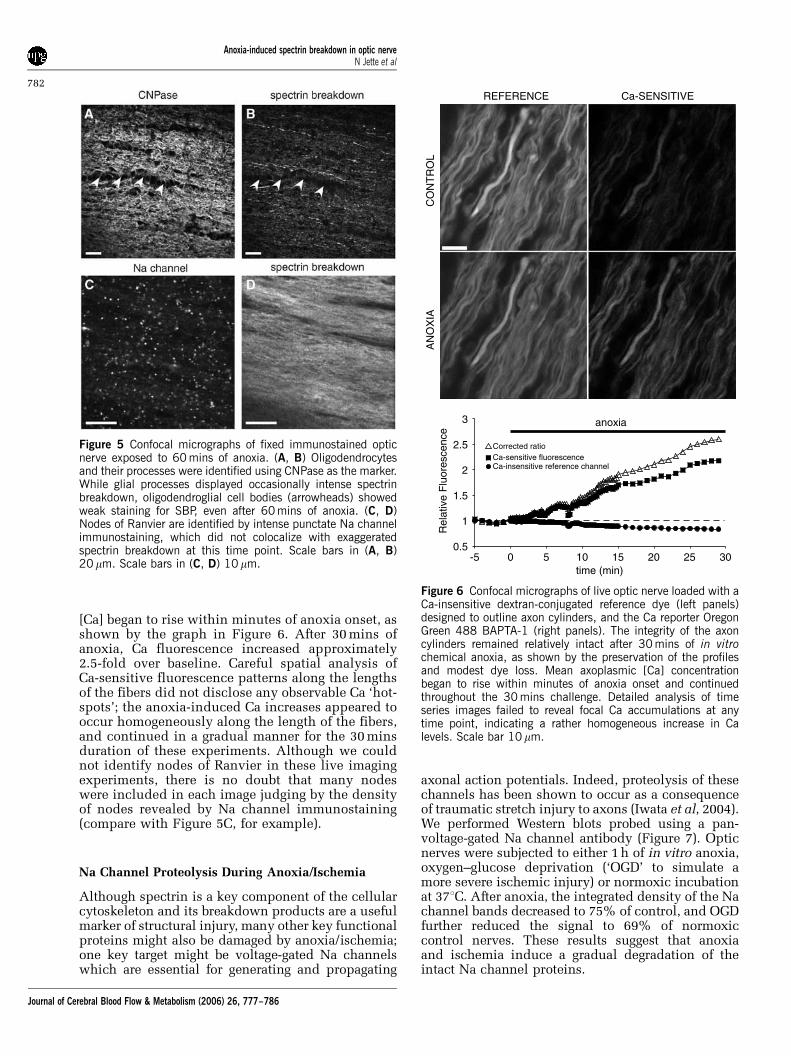
weak neurofilament colocalized with strong SBP).Interestingly, much of the SBP signal was found tolie outside axon cylinders, often immediately ad-jacent to these profiles (Figure 4E to 4G), even whileoligodendroglial cell bodies did not exhibit signifi-cant spectrin breakdown even after 60 mins ofanoxia (Figures 5A and 5B). Most of the SBP signalwas associated either with distal processes of thesecells and/or axon cylinders (Figures 4D to 4G and5B). Even after 60 mins of anoxia, double-labelingwith Na channel antibody and SBP did not indicatea preferential focus of spectrin cleavage at nodes ofRanvier (Figures 5C and 5D).
Figure 2 Confocal micrographs of control normoxic optic nervelabeled with anti-pan Na channel antibody (A) and spectrinbreakdown. (B) Numerous punctate regions reflect the highdensity of Na channels at nodes of Ranvier. Uninjured opticnerves did not exhibit spectrin breakdown, either diffusely ormore specifically at the nodes of Ranvier. Scale bars 10mm.
Figure 3 Confocal micrographs of optic nerves harvested after15 or 30 mins of in vitro anoxia. (A, B) Neurofilament stainoutlines healthy axon cylinders, with little evidence of spectrindegradation after 15 mins of anoxia. Nor was there evidence offocal accumulation of spectrin degradation at this time pointthat may reflect early localized cytoskeletal damage at nodes ofRanvier. (C, D) After 30 mins of anoxia, significant spectrinbreakdown was observed; the distribution appeared diffusealong the lengths of the fibers with no evidence of focalpredilection. (E, F) Na channel immunostaining identifies nodesof Ranvier. After 30 mins of anoxia, spectrin degradationappeared relatively homogeneous with no obvious predilectionfor nodal regions. Scale bars 10 mm.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
780
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786
Spatiotemporal Changes in Axoplasmic [Ca] DuringAnoxia
While spectrin breakdown is a useful marker ofCa-dependent subcellular injury, because enzymaticaction may require time to generate immunodetect-able products, the spatial profiles of these break-down products may not necessarily reflect initialspatial gradients of Ca accumulation. In an effort toconfirm the occurrence of localized Ca ‘hot-spots’ inthe early stages of anoxia, we imaged axoplasmic[Ca] directly in living optic nerve axons using
fluorescent Ca reporters and confocal microscopy.Figure 6 shows representative confocal micrographsof optic nerve axons loaded with a red Ca-insensi-tive dye (Texas Red dextran), which convenientlyoutlines axon cylinders in isolation (myelin andglial cell bodies do not take up the large dextranconjugates), and a dextran-conjugated Ca reporter(Oregon Green 488 BAPTA-1 dextran). There was aslight rundown of the red reference channel begin-ning at 10 to 15 mins (typically 10% to 20% at theend of 30 mins), with a concomitant rise in Ca-sensitive emission indicating that axoplasmic free
Figure 4 Confocal micrographs of optic nerve exposed to 60 mins of anoxia. At this time point, substantial spectrin breakdown wasseen both within axon cylinders and in glial processes surrounding axons. Areas of more intense staining for spectrin breakdown (redchannel panel A; arrowheads, panel C) colocalized with weaker and disrupted neurofilament staining (green channel panel A;arrowheads, panel B) indicating severe damage, particularly to larger fibers. Panels B and C are green (neurofilament) and red(spectrin breakdown) channels shown separately, respectively. A significant proportion of spectrin degradation was found outside theaxons per se, likely involving glial processes (panels D–G, green is neurofilament denoting axon cylinders, red is spectrin breakdownfrequently seen outside axons). Scale bars 20 mm.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
781
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786
[Ca] began to rise within minutes of anoxia onset, asshown by the graph in Figure 6. After 30 mins ofanoxia, Ca fluorescence increased approximately2.5-fold over baseline. Careful spatial analysis ofCa-sensitive fluorescence patterns along the lengthsof the fibers did not disclose any observable Ca ‘hot-spots’; the anoxia-induced Ca increases appeared tooccur homogeneously along the length of the fibers,and continued in a gradual manner for the 30 minsduration of these experiments. Although we couldnot identify nodes of Ranvier in these live imagingexperiments, there is no doubt that many nodeswere included in each image judging by the densityof nodes revealed by Na channel immunostaining(compare with Figure 5C, for example).
Na Channel Proteolysis During Anoxia/Ischemia
Although spectrin is a key component of the cellularcytoskeleton and its breakdown products are a usefulmarker of structural injury, many other key functionalproteins might also be damaged by anoxia/ischemia;one key target might be voltage-gated Na channelswhich are essential for generating and propagating
axonal action potentials. Indeed, proteolysis of thesechannels has been shown to occur as a consequenceof traumatic stretch injury to axons (Iwata et al, 2004).We performed Western blots probed using a pan-voltage-gated Na channel antibody (Figure 7). Opticnerves were subjected to either 1 h of in vitro anoxia,oxygen–glucose deprivation (‘OGD’ to simulate amore severe ischemic injury) or normoxic incubationat 371C. After anoxia, the integrated density of the Nachannel bands decreased to 75% of control, and OGDfurther reduced the signal to 69% of normoxiccontrol nerves. These results suggest that anoxiaand ischemia induce a gradual degradation of theintact Na channel proteins.
Figure 5 Confocal micrographs of fixed immunostained opticnerve exposed to 60 mins of anoxia. (A, B) Oligodendrocytesand their processes were identified using CNPase as the marker.While glial processes displayed occasionally intense spectrinbreakdown, oligodendroglial cell bodies (arrowheads) showedweak staining for SBP, even after 60 mins of anoxia. (C, D)Nodes of Ranvier are identified by intense punctate Na channelimmunostaining, which did not colocalize with exaggeratedspectrin breakdown at this time point. Scale bars in (A, B)20mm. Scale bars in (C, D) 10mm.
REFERENCE Ca-SENSITIVE
CO
NT
RO
LA
NO
XIA
0.5
1
1.5
2
2.5
3
-5 0 5 10 15 20 25 30time (min)
Rel
ativ
e F
luor
esce
nce
anoxia
Ca-insensitive reference channelCa-sensitive fluorescenceCorrected ratio
Figure 6 Confocal micrographs of live optic nerve loaded with aCa-insensitive dextran-conjugated reference dye (left panels)designed to outline axon cylinders, and the Ca reporter OregonGreen 488 BAPTA-1 (right panels). The integrity of the axoncylinders remained relatively intact after 30 mins of in vitrochemical anoxia, as shown by the preservation of the profilesand modest dye loss. Mean axoplasmic [Ca] concentrationbegan to rise within minutes of anoxia onset and continuedthroughout the 30 mins challenge. Detailed analysis of timeseries images failed to reveal focal Ca accumulations at anytime point, indicating a rather homogeneous increase in Calevels. Scale bar 10mm.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
782
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786

Discussion
Central axons suffer damage in a number ofdisorders, such as traumatic injury to the brainand spinal cord; acute stroke, neonatal periventri-cular leukomalacia and more chronic subcorticalischemia; inflammatory demyelinating diseasessuch as multiple sclerosis; and the encephalopathyassociated with HIV infection (Back, 2001; Maxwellet al, 1997; Raja et al, 1997; Schwartz and Fehlings,2002; Trapp et al, 1998). As distinct as the initialinciting events might be, these conditions result inoverlapping white matter neuropathology, whichincludes demyelination, axonal damage, and finallyirreversible loss of brain and spinal fibers. Indeed, itis thought that loss of central axons is a keypredictor of clinical outcome (Medana and Esiri,2003). Neuropathological examination reveals axo-nal damage that is surprisingly focal, at least early inthe course before Wallerian degeneration results incomplete disappearance of the fiber: axonal spher-oids and swellings are characteristic findings inbrains of patients with multiple sclerosis, neuro-trauma victims, and those with ischemic injury(Povlishock and Christman, 1995; Trapp et al, 1998;Yam et al, 1998). Such swellings display strongstaining for beta-amyloid precursor protein (suggest-ing focal failure of axonal transport) (Stone et al,2004; Yam et al, 1998) and spectrin breakdown(Buki et al, 1999), indicating a Ca-mediated disrup-tion of the axonal cytoskeleton. Thus, there might beselective regions of the axon that are vulnerable tospheroid formation and ultimately transection com-pared with other areas. The unanswered question iswhether the initial locus of injury is random orwhether it is governed by intrinsic structural/functional inhomogeneities of myelinated axons.
The most striking feature of myelinated axons isthe highly specialized node of Ranvier (for reviews,see Poliak and Peles, 2003; Scherer and Arroyo,2002). This interruption of the ensheathing myelin
occupies only E1% of the axon’s surface area, yetcontains sufficient channel densities to rapidlyconduct inward action currents to support saltatoryconduction. Nodes contain high densities of Nachannels, and thus would be expected to experiencesignificant axoplasmic fluctuations of [Na] duringnormal action potential propagation, and alsoduring pathological conditions such as anoxia/ischemia. Because much of the pathological accu-mulation of axonal Ca is driven both by depolariza-tion and a rise in axoplasmic [Na] (for a recentreview, see Stys 2004), we hypothesized that Caderegulation and accumulation would begin atnodes of Ranvier, leading to preferential damage tothe cytoskeleton and possibly to Na channelsthemselves, in this region.
Examining the spatio-temporal distributions of Caions as a function of injury in small-diameter centralaxons is very difficult. We adopted two comple-mentary strategies: the first was to immunohisto-chemically assay the distribution of calpain-cleavedbreakdown products of spectrin, a ubiquitouscytoskeletal structural protein. The calpains are afamily of cysteine proteases whose enzymaticactivity is stimulated by ambient free Ca ions (Croalland DeMartino, 1991; Melloni and Pontremoli,1989); therefore, locally elevated Ca levels shouldactivate calpain, and in turn result in preferentialaccumulation of breakdown products in regions ofhigh [Ca]. Rat optic nerves exposed to 0, 15, 30, and60 mins of in vitro anoxia failed to display prefer-ential ‘hot-spots’ of spectrin cleavage: the pattern ofspectrin breakdown evolved from virtually unde-tectable signal in normoxic control specimens andthose exposed to 15 mins of anoxia (Figures 1 and3B), to a fairly homogeneous increase in SBPs thatbegan between 15 and 30 mins of anoxia (Figures 3Dand 3F). Careful examination of sections double-labeled with Na channel antibodies which clearlyidentified nodes of Ranvier (Figures 2A, 3E and 5C)failed to reveal any preferential accumulation ofSBPs in this area. The cut (and therefore trauma-tized) ends of control nerves, and nerves exposed tolonger periods of anoxia (30 and 60 mins), exhibitedclear and strong spectrin breakdown staining,indicating that the antibody was reliable and couldeasily detect these products in damaged regions.
After 60 mins of anoxia, many axons displayedfragmentation of their cylinders, as shown by a moremottled appearance of neurofilament staining,which often overlapped with stronger spectrinbreakdown signal (Figures 4A to 4C). This wasparticularly evident in large-diameter fibers. Neuro-filament breakdown is expected, as this structuralprotein is also a substrate for calpain (Banik et al,1997; Schumacher et al, 2000) (as are many otherssuch as tau, tubulin, ankyrin (Rami et al, 1997)), andcleavage of these proteins can be blocked by calpaininhibitors (Stys and Jiang, 2002). Interestingly, littlespectrin breakdown was observed in the cell bodiesof oligodendroglia, even after 60 mins of anoxia.
Figure 7 Immunoblot probed for the alpha subunit of thevoltage-gated Na channel using a pan-specific antibody.Samples were obtained from optic nerves exposed to either60 mins of OGD or anoxia in vitro. Anoxia reduced theintegrated density of the Na channel bands to 75% of control,and OGD to 69% of normoxic control nerves, indicatingproteolysis of Na channel subunits during anoxic/ischemicinjury.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
783
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786
Although these cells are known to be vulnerable toanoxic/ischemic injury (Dewar et al, 2003), ourrelatively limited 60 mins exposure to anoxia (com-pared with ischemia) might have produced only aminor degree of structural injury to these cellswhich can be detected by our antibody. In contrast,glial processes running parallel to axons (probablyoligodendrocytic, e.g. Figures 4D to 4G) displayedsignificant spectrin degradation products, indicat-ing that these distal glial extensions appear morevulnerable to anoxia than the parent cell bodies,possibly due to preferential localization of m-calpain to these regions (Ray et al, 2002).
Analyzing Ca-dependent enzymatic activity is areliable, albeit indirect, method of inferring fluctua-tions in cytosolic Ca levels. It was also possible thatCa does accumulate focally at nodes, but because itmay take time for the enzyme to be activated and todegrade spectrin, the resulting injury (as evidencedby SBP staining) might have been spatially‘smeared’, and focal Ca rises missed. For this reason,we complemented the immunohistochemical stu-dies with direct measurements of axoplasmic Causing a high-affinity Ca indicator and confocalmicroscopy. Images were initially acquired every15 secs after the start of anoxia in case any focal Caelevations were brief, and could be missed withlonger sampling intervals. Axoplasmic [Ca] began torise within minutes of anoxia onset, and continuedfor the duration of the experiment (30 mins). Nofocal Ca elevations were observed in any axons atany time point: Ca levels rose gradually andhomogeneously. Although we did not identify nodesof Ranvier in the live axon-imaging experiments, thehigh density of nodes found in fixed tissue impliesthat we had many nodes in each imaging field, sothe absence of potential nodal Ca ‘hot-spots’ was notbecause we might have missed nodal regions in oursampling of random areas. As our Ca measurementscould not be calibrated (due to the overlying myelinsheath which would prevent clamping axoplasmic[Ca] with ionophores), we cannot provide absolute[Ca]’s. However, based on previous experimentsfrom our lab (Nikolaeva and Stys, 2002) and the Calevels required to activate m-calpain (Croall andDeMartino, 1991; Melloni and Pontremoli, 1989), weestimate that axoplasmic [Ca] rose into the micro-molar range.
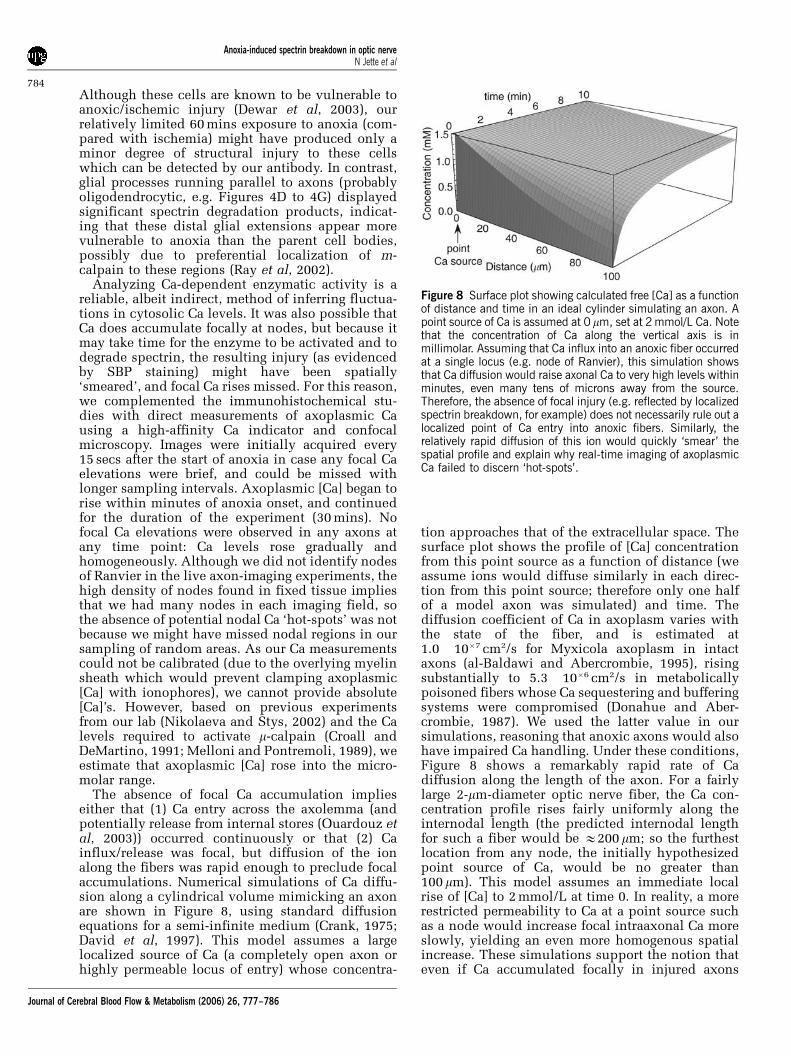
The absence of focal Ca accumulation implieseither that (1) Ca entry across the axolemma (andpotentially release from internal stores (Ouardouz etal, 2003)) occurred continuously or that (2) Cainflux/release was focal, but diffusion of the ionalong the fibers was rapid enough to preclude focalaccumulations. Numerical simulations of Ca diffu-sion along a cylindrical volume mimicking an axonare shown in Figure 8, using standard diffusionequations for a semi-infinite medium (Crank, 1975;David et al, 1997). This model assumes a largelocalized source of Ca (a completely open axon orhighly permeable locus of entry) whose concentra-
tion approaches that of the extracellular space. Thesurface plot shows the profile of [Ca] concentrationfrom this point source as a function of distance (weassume ions would diffuse similarly in each direc-tion from this point source; therefore only one halfof a model axon was simulated) and time. Thediffusion coefficient of Ca in axoplasm varies withthe state of the fiber, and is estimated at1.0� 10�7 cm2/s for Myxicola axoplasm in intactaxons (al-Baldawi and Abercrombie, 1995), risingsubstantially to 5.3� 10�6 cm2/s in metabolicallypoisoned fibers whose Ca sequestering and bufferingsystems were compromised (Donahue and Aber-crombie, 1987). We used the latter value in oursimulations, reasoning that anoxic axons would alsohave impaired Ca handling. Under these conditions,Figure 8 shows a remarkably rapid rate of Cadiffusion along the length of the axon. For a fairlylarge 2-mm-diameter optic nerve fiber, the Ca con-centration profile rises fairly uniformly along theinternodal length (the predicted internodal lengthfor such a fiber would be E200 mm; so the furthestlocation from any node, the initially hypothesizedpoint source of Ca, would be no greater than100 mm). This model assumes an immediate localrise of [Ca] to 2 mmol/L at time 0. In reality, a morerestricted permeability to Ca at a point source suchas a node would increase focal intraaxonal Ca moreslowly, yielding an even more homogenous spatialincrease. These simulations support the notion thateven if Ca accumulated focally in injured axons
Figure 8 Surface plot showing calculated free [Ca] as a functionof distance and time in an ideal cylinder simulating an axon. Apoint source of Ca is assumed at 0 mm, set at 2 mmol/L Ca. Notethat the concentration of Ca along the vertical axis is inmillimolar. Assuming that Ca influx into an anoxic fiber occurredat a single locus (e.g. node of Ranvier), this simulation showsthat Ca diffusion would raise axonal Ca to very high levels withinminutes, even many tens of microns away from the source.Therefore, the absence of focal injury (e.g. reflected by localizedspectrin breakdown, for example) does not necessarily rule out alocalized point of Ca entry into anoxic fibers. Similarly, therelatively rapid diffusion of this ion would quickly ‘smear’ thespatial profile and explain why real-time imaging of axoplasmicCa failed to discern ‘hot-spots’.
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
784
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786

(be it from the influx at the nodes or focal releasefrom cisternae of axoplasmic reticulum (Ouardouzet al, 2003)), this ion would rapidly diffuse into theinternodes and not result in focal accumulations. Inthe absence of focal Ca accumulations in anoxicaxons, it is interesting to speculate why focalstructural damage (in the form of axonal spheroids)is often observed in anoxic/ischemic or traumatizedaxons. One possibility might be that the source of Cais not focal, but that the vulnerability of axons to adamaging increase in axoplasmic [Ca] is inhomoge-neous, with certain areas being intrinsically moresusceptible than others.
Although our results and those from previousstudies (Buki et al, 1999; Saatman et al, 2003;Schumacher et al, 2000; Stys and Jiang, 2002) clearlyindicate extensive injury to structural proteins ininjured axons, we also wanted to examine whetherkey functional proteins were damaged by anoxia/ischemia as well. Voltage-gated Na channels aredamaged by proteases in a Ca-dependent mannerafter mechanical stretch injury to axons, with thesuggestion raised of an upregulation of the persis-tent component of Na currents resulting from thisproteolytic cleavage of the alpha subunit (Iwata etal, 2004). Our results from anoxic and ischemicoptic axons parallel these observations; while ourresults are only qualitative, we found a consistentreduction in the amount of immunodetectable Nachannel protein after exposure to in vitro anoxia orsimulated ischemia. Degradation of this key proteinlikely contributes to a significant degree to theoverall functional injury caused by anoxia/ischemiain optic nerve.
In summary, the present results indicate thatanoxia produces a sustained increase in axoplasmic[Ca], and degradation of the spectrin cytoskeleton inaxons and distal glial processes beginning between15 and 30 mins of anoxia. Contrary to our initialhypothesis, structural injury does not begin at nor isit preferentially localized to nodes of Ranvier, whereexaggerated ionic dysregulation might be expectedto occur. While we cannot exclude a focal source ofCa (either influx at nodal regions and/or focalrelease from internal stores), it appears that diffu-sion of this ion along the axoplasm in energy-depleted fibers is rapid enough to raise [Ca] fairlyhomogeneously and induce injury continuouslyalong the length of fibers in our in vitro model ofanoxia. Equally important is our finding that keyfunctional proteins such as voltage-gated Na chan-nels are also targets of proteolytic cleavage, whichwill conspire to render an axon nonfunctional,eventually leading to its degeneration.
References
al-Baldawi NF, Abercrombie RF (1995) Calcium diffusioncoefficient in Myxicola axoplasm. Cell Calcium 17:422–30
Back SA (2001) Recent advances in human perinatal whitematter injury. Prog Brain Res 132:131–47
Banik NL, Matzelle DC, Gantt WG, Osborne A, Hogan EL(1997) Increased calpain content and progressivedegradation of neurofilament protein in spinal cordinjury. Brain Res 752:301–6
Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999)The role of calpain-mediated spectrin proteolysis intraumatically induced axonal injury. J Neuropathol ExpNeurol 58:365–75
Crank J (1975) The mathematics of diffusion. New York:Oxford University Press
Croall DE, DeMartino GN (1991) Calcium-activated neutralprotease (calpain) system: structure, function, andregulation. Physiol Rev 71:813–47
David G, Barrett JN, Barrett EF (1997) Spatiotemporalgradients of intra-axonal [Na+] after transectionand resealing in lizard peripheral myelinated axons.J Physiol (Lond) 498:295–307
Deshpande RV, Goust JM, Hogan EL, Banik NL (1995)Calpain secreted by activated human lymphoid cellsdegrades myelin. J Neurosci Res 42:259–65
Dewar D, Underhill SM, Goldberg MP (2003) Oligoden-drocytes and ischemic brain injury. J Cereb Blood FlowMetab 23:263–74
Donahue BS, Abercrombie RF (1987) Free diffusioncoefficient of ionic calcium in cytoplasm. Cell Calcium8:437–48
Goldberg MP, Ransom BR (2003) New light on whitematter. Stroke 34:330–2
Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, MeaneyDF et al. (2004) Traumatic axonal injury inducesproteolytic cleavage of the voltage-gated sodium chan-nels modulated by tetrodotoxin and protease inhibi-tors. J Neurosci 24:4605–13
Jiang Q, Stys PK (2000) Calpain inhibitors confer bio-chemical, but not electrophysiological, protectionagainst anoxia in rat optic nerves. J Neurochem 74:2101–7
Maxwell WL, Povlishock JT, Graham DL (1997) Amechanistic analysis of nondisruptive axonal injury: areview. J Neurotrauma 14:419–40
McCracken E, Dewar D, Hunter AJ (2001) White matterdamage following systemic injection of the mitochon-drial inhibitor 3-nitropropionic acid in rat. Brain Res892:329–35
McCracken E, Hunter AJ, Patel S, Graham DI, Dewar D(1999) Calpain activation and cytoskeletal proteinbreakdown in the corpus callosum of head-injuredpatients. J Neurotrauma 16:749–61
Medana IM, Esiri MM (2003) Axonal damage: a keypredictor of outcome in human CNS diseases. Brain126:515–30
Melloni E, Pontremoli S (1989) The calpains. TrendsNeurosci 12:438–44
Nikolaeva MA, Stys PK (2002) Calcium changes inischemic rat optic nerve axons studied by confocalmicroscopy. Soc Neurosci Abstr: 299:296
Ouardouz M, Nikolaeva M, Coderre E, Zamponi GW,McRory JE, Trapp BD et al. (2003) Depolarization-induced Ca2+ release in ischemic spinal cord whitematter involves L-type Ca2+ channel activation ofryanodine receptors. Neuron 40:53–63
Park E, Velumian AA, Fehlings MG (2004) The role ofexcitotoxicity in secondary mechanisms of spinal cordinjury: a review with an emphasis on the implicationsfor white matter degeneration. J Neurotrauma 21:754–74
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
785
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786

Poliak S, Peles E (2003) The local differentiationof myelinated axons at nodes of Ranvier. Nat RevNeurosci 4:968–80
Povlishock JT, Christman CW (1995) Diffuse axonal injury.In: The axon: structure, function and pathophysiology(Waxman SG, Kocsis JD, Stys PK, eds), New York:Oxford University Press, 504–29
Raja F, Sherriff FE, Morris CS, Bridges LR, Esiri MM (1997)Cerebral white matter damage in HIV infection demon-strated using beta-amyloid precursor protein immunor-eactivity. Acta Neuropathol (Berl) 93:184–9
Rami A, Ferger D, Krieglstein J (1997) Blockade of calpainproteolytic activity rescues neurons from glutamateexcitotoxicity. Neurosci Res 27:93–7
Ray SK, Banik NL (2003) Calpain and its involvement inthe pathophysiology of CNS injuries and diseases:therapeutic potential of calpain inhibitors for preven-tion of neurodegeneration. Curr Drug Targets CNSNeurol Disord 2:173–89
Ray SK, Neuberger TJ, Deadwyler G, Wilford G, DeVriesGH, Banik NL (2002) Calpain and calpastatin expres-sion in primary oligodendrocyte culture: preferentiallocalization of membrane calpain in cell processes.J Neurosci Res 70:561–9
Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR,Siman R (1994) Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neuronsin the ischemic gerbil brain. J Neurosci 14:3934–44
Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH,Meaney DF (2003) Traumatic axonal injury results inbiphasic calpain activation and retrograde transportimpairment in mice. J Cereb Blood Flow Metab 23:34–42
Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R,McIntosh TK (1996) Prolonged calpain-mediated spec-trin breakdown occurs regionally following experimen-tal brain injury in the rat. J Neuropathol Exp Neurol 55:850–60
Schaecher KE, Shields DC, Banik NL (2001) Mechanism ofmyelin breakdown in experimental demyelination: aputative role for calpain. Neurochem Res 26:731–7
Scherer SS, Arroyo EJ (2002) Recent progress on themolecular organization of myelinated axons. J PeripherNerv Syst 7:1–12
Schumacher PA, Siman RG, Fehlings MG (2000) Pretreat-ment with calpain inhibitor CEP-4143 inhibits calpain Iactivation and cytoskeletal degradation, improves neuro-logical function, and enhances axonal survival aftertraumatic spinal cord injury. J Neurochem 74:1646–55
Schwartz G, Fehlings MG (2002) Secondary injurymechanisms of spinal cord trauma: a novel therapeuticapproach for the management of secondary patho-physiology with the sodium channel blocker riluzole.Prog Brain Res 137:177–90
Shields DC, Schaecher KE, Saido TC, Banik NL (1999) Aputative mechanism of demyelination in multiplesclerosis by a proteolytic enzyme, calpain. Proc NatlAcad Sci USA 96:11486–91
Siman R, Noszek JC, Kegerise C (1989) Calpain I activationis specifically related to excitatory amino acid induc-tion of hippocampal damage. J Neurosci 9:1579–90
Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK,Povlishock JT et al. (2004) Impaired axonal transportand altered axolemmal permeability occur in distinctpopulations of damaged axons followingtraumaticbrain injury. Exp Neurol 190:59–69
Stys PK (2004) White matter injury mechanisms. Curr MolMed 4:109–26
Stys PK, Jiang Q (2002) Ca2+-mediated calpain-dependentneurofilament breakdown in anoxic and ischemiccentral white matter axons. Neurosci Lett 328:150–4
Stys PK, Ransom BR, Waxman SG (1991) Compoundaction potential of nerve recorded by suction electrode:a theoretical and experimental analysis. Brain Res 546:18–32
Suzuki K, Sorimachi H, Yoshizawa T, Kinbara K, Ishiura S(1995) Calpain: novel family members, activation, andphysiologic function. Biol Chem 376:523–9
Trapp BD, Bernier L, Andrews SB, Colman DR (1988)Cellular and subcellular distribution of 2’,3’-cyclicnucleotide 3’-phosphodiesterase and its mRNA in therat central nervous system. J Neurochem 51:859–68
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, BoL (1998) Axonal transection in the lesions of multiplesclerosis. N Engl J Med 338:278–85
Yam PS, Dewar D, McCulloch J (1998) Axonal injurycaused by focal cerebral ischemia in the rat.J Neurotrauma 15:441–50
Anoxia-induced spectrin breakdown in optic nerveN Jette et al
786
Journal of Cerebral Blood Flow & Metabolism (2006) 26, 777–786





























