Embed Size (px)
Citation preview

Subcellular localization of ERGIC-53 under endoplasmicreticulum stress condition
Sheng-Ying Qin, Norihito Kawasaki, Dan Hu,Hideto Tozawa, Naoki Matsumoto, and Kazuo Yamamoto1
Department of Integrated Biosciences, Graduate School of Frontier Sciences,University of Tokyo, Kashiwa 277-8562 Chiba, Japan
Received on May 17, 2012; revised on July 4, 2012; accepted on July 16,2012
Newly synthesized glycoproteins destined for secretion aretransported from the endoplasmic reticulum (ER),through the Golgi and toward the cell surface. In this se-cretion pathway, several intracellular ER- or Golgi-resi-dent transmembrane proteins serve as cargo receptors.ER–Golgi intermediate compartment (ERGIC)-53, VIP36and VIPL, which have an L-type lectin domain within theluminal portion, participate in the vectorial transport ofglycoproteins via sugar–protein interactions. To under-stand the nature of these receptors, monoclonal antibodieswere generated against human ERGIC-53, VIP36 andVIPL using 293T cells expressing these receptors on cellsurfaces. These cells were used to immunize rats and forscreening antibody-producing clones. Flow cytometricanalysis and immunoprecipitation studies showed that theobtained monoclonal antibodies bound specifically to thecorresponding cargo receptors. Immunostaining of HeLacells using the monoclonal antibodies showed that the lo-calization of ERGIC-53 changed from relatively broad dis-tribution in both the ER and the Golgi under normalconditions to a compact distribution in the Golgi underER stress conditions. This redistribution was also observedby the overexpression of ERGIC-53 and abrogated by co-expression with VIPL but not VIP36. Real-time polymer-ase chain reaction revealed that ERGIC-53 along withseveral chaperone proteins was up-regulated after tunica-mycin treatment; however, the expression of VIPL was un-changed. Furthermore, ERGIC-53 co-precipitated withVIPL but not VIP36, indicating that ERGIC-53 may inter-act with VIPL in the ER, which may regulate the localiza-tion of ERGIC-53 inside cells. Taken together, theseobservations provide new insights into the regulation ofthese cargo receptors and the quality control of glycopro-teins within cells.
Keywords: cargo receptor / ERGIC-53 / ER stress /glycoprotein quality control / VIPL
Introduction
In many cases, glycosylation directly affects the properties ofproteins and cells, sometimes with important biological conse-quences. The biological functions of glycosylation are mostlymediated outside the cell (Ashwell and Morell 1974; Crockerand Varki 2001; Ley 2003). In contrast, oligosaccharides, es-pecially N-glycans, function as tags for the quality control ofglycoproteins within cells. Recently, the biological mechan-isms regulating folding, transport and endoplasmic reticulum(ER)-associated degradation of glycoproteins have becomebetter understood (Helenius and Aebi 2004; Moremen andMolinari 2006; Ruddock and Molinari 2006; Yamamoto2009). The extension of protein folding, or the onset of dis-posal, is regulated by several ER- and Golgi-resident sugar-binding proteins (called lectins), which recognize specificN-glycan structures attached to proteins. We recently deter-mined the sugar-binding specificity of the intracellular cargoreceptors, ER–Golgi intermediate compartment (ERGIC)-53,VIP36 and VIPL, using a flow cytometry-based method in-corporating soluble lectin tetramers (Kawasaki et al. 2007,2008; Yamaguchi et al. 2007; Yamamoto and Kawasaki 2010)and frontal affinity chromatography (Kamiya et al. 2005,2008). We found that progressive trimming of terminal sugarresidues is required to recruit cargo receptors. However, theintracellular localization of these receptors is necessary if weare to understand their biological roles, particularly withrespect to the transport of cargo proteins. The cargo receptors,ERGIC-53, VIP36 and VIPL, are homologous at the aminoacid level; however, monoclonal antibodies that distinguishbetween these cargo receptors have not yet been reported.VIP36 localization to either the pre-Golgi secretory pathwayor the post-Golgi pathway was shown using endogenouslyexpressed Myc-tagged or FLAG-tagged receptors and anti-tagantibodies (Fiedler et al. 1994; Fullekrug et al. 1999), and arecent report identified VIP36 mainly on the cell surface(Shirakabe et al. 2011). ERGIC-53, which was first identified asa marker of the ERGIC, is distributed between the ER and theGolgi (Vollenweider et al. 1998; Appenzeller et al. 1999),whereas VIPL localized predominantly in the ER and may be anon-cycling ER-resident protein (Nufer, Mitrovic, et al. 2003);however, there may be the possibility that exogenouslyexpressed tagged protein does not co-localized with endogenous
1To whom correspondence should be addressed: Tel: +81-4-7136-3614; Fax:+81-4-7136-3619; e-mail: [email protected]
Glycobiology vol. 22 no. 12 pp. 1709–1720, 2012doi:10.1093/glycob/cws114Advance Access publication on July 20, 2012
© The Author 2012. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: [email protected] 1709
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

one or that overexpression causes altered intracellular localiza-tion, since the intracellular localization of proteins is regulatedby cytoplasmically associated coat proteins. Furthermore, up- ordown-regulation accompanied by cellular activation (or othercell responses) cannot be monitored using exogenouslyexpressed tagged proteins, since regulated transcription dependson both cis-regulatory elements on the gene and trans-elementssuch as miRNAs. Therefore, to rule out these artifacts and toidentify the endogenous receptors involved in the transport ofglycoproteins within the cells, it is important to track endogen-ous ERGIC-53, VIP36 and VIPL, using specific monoclonalantibodies that recognize these proteins in their nativeconformation.Here, we describe an easy and simple method of generating
monoclonal antibodies against the intracellular cargo recep-tors, ERGIC-53, VIP36 and VIPL, using a cell surfacedisplay technique (Lemaire et al. 2011). Using these monoclo-nal antibodies, we elucidated the intracellular distribution ofendogenous ERGIC-53 changed under conditions of ERstress induced by tunicamycin or dithiothreitol (DTT). Thesame distribution of ERGIC-53 to the Golgi was also causedby overexpression of ERGIC-53, which was abrogated byco-expression with VIPL, but not by co-expression withVIP36. Furthermore, ERGIC-53 co-precipitated with VIPLbut not VIP36, indicating that ERGIC-53 can interact withVIPL in the ER, which may regulate the localization ofERGIC-53 inside cells. These observations provide newinsights into the regulation of these cargo receptors during thequality control of glycoproteins within cells, particularlyunder conditions of ER stress.
ResultsProduction of monoclonal antibodies against ERGIC-53,VIP36 and VIPLThe cDNAs coding the luminal portion of human ERGIC-53,VIP36 and VIPL were cloned into pFLAG-CMV-CD8TM toexpress the proteins on mammalian cell surfaces asFLAG-tagged CD8α-fusion proteins. To confirm the expres-sion of the FLAG-tagged proteins on cell surfaces, 293T cellstransfected with each plasmid were stained with ananti-FLAG antibody and analyzed by flow cytometry. Morethan 97% of the transfected cells displayed FLAG-taggedERGIC-53, VIP36 or VIPL on the cell surface (Figure 1).Next, we immunized a rat with a mixture of transfected cellsexpressing ERGIC-53, VIP36 and VIPL and used harvestedspleen cells to produce monoclonal antibodies against allthree proteins. Screening of the culture supernatant from eachcloned hybridoma was performed by staining each of theFLAG-tagged ERGIC-53, VIP36 and VIPL-expressing celllines with each of the supernatants and analyzing them byflow cytometry. Of the 384 cloned cells screened, severalclones were identified as producing antibodies specific forFLAG-tagged ERGIC-53, VIP36 or VIPL. Three monoclonalantibodies, named ER1, ER2 and ER3, bound toFLAG-tagged ERGIC-53-expressing 293T cells, but not tocells expressing FLAG-tagged VIP36 or VIPL. The VIP1monoclonal antibody was specific for FLAG-taggedVIP36-expressing cells, and two monoclonal antibodies, VL1
and VL2, were specific for VIPL-expressing cells(Figure 2A). To further confirm the specificity of the anti-bodies, ER1, VIP1 and VL1, we performed immunoprecipita-tion of ERGIC-53, VIP36 and VIPL from cell lysatesexpressing myc-tagged ERGIC-53, VIP36 or VIPL, respect-ively. Though VIP36 dimmer was partially detected, myc-tagged ERGIC-53, VIP36 and VIPL were precipitated withER1, VIP1 and VL1, respectively (Figure 2B). Monoclonalantibodies, ER1, ER2, ER3, VIP1, VL1 and VL2 were puri-fied from the hybridoma culture supernatants and all wereidentified as rat IgG2a with κ light chains (data not shown).
Distinct intracellular distribution of ERGIC-53 aftertunicamycin or DTT treatmentLabeled ER1, VIP1 and VL1 monoclonal antibodies wereused to study the intracellular localization and distribution ofendogenous ERGIC-53, VIP36 and VIPL in HeLa cells byimmunocytochemistry. HeLa cells were fixed with paraformal-dehyde, permeabilized with TX-100 and then stained with theanti-ERGIC-53, anti-VIP36 and anti-VIPL monoclonal anti-bodies. The microscopic observation of endogenous VIPLshowed that the protein localized in the ER and co-localizedwith the ER marker protein, calnexin (CNX; data not shown),which is consistent with a previous report showing thatHA-tagged VIPL localized to the ER (Nufer, Mitrovic, et al.2003). Endogenous ERGIC-53 (Figure 3, control) and VIP36(data not shown) were distributed around the ER and theGolgi, and partially co-localized with both the ER markerprotein, CNX and the Golgi marker protein, Golgi 58K.These data are also in good agreement with previous studies(Vollenweider et al. 1998; Fullekrug et al. 1999; Shirakabeet al. 2011). Next, we observed the distribution of ERGIC-53in HeLa cells after the induction of ER stress. Tunicamycincauses ER stress by causing the accumulation of non-glycosylated proteins in the ER lumen (Morris et al. 1997).After treatment with 5 µg/mL of tunicamycin for 24 h, theERGIC-53, which distributed in both the ER and the Golgiunder normal conditions, became mainly localized to theGolgi apparatus with a morphological change to form acompact shape [Figure 3, tunicamycin (TM)]. The same distri-bution of ERGIC-53 was observed when the cell was treatedwith 2 mM DTT for 24 h, which also causes ER stress to thecell (Oliveira et al. 2009; Figure 3, DTT). Overexpression ofERGIC-53 also caused a similar distribution of the protein tothe Golgi within the cell, whereas the localization ofERGIC-53 to the ER was enhanced by co-expression withVIPL (Figure 4A) but not by co-expression with VIP36(Figure 4B), which was also reported by Nufer, Mitrovic,et al. (2003). These results suggest that the distribution ofERGIC-53 is strictly regulated under ER stress conditions andVIPL may be involved in regulating the localization ofERGIC-53 inside the cells.
Expression of ERGIC-53 and VIP36, but not VIPL, isup-regulated by tunicamycinAn immunofluorescence study revealed a curious distributionof ERGIC-53 induced by tunicamycin treatment. Under ERstress conditions, several chaperones and intracellular lectins,which are associated with ER-associated protein degradation
S-Y Qin et al.
1710
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

(ERAD), are up-regulated via the unfolded protein responsein the ER (Schroder and Kaufman 2005). To determinewhether ERGIC-53, VIP36 and VIPL were similarlyup-regulated in HeLa cells under the same conditions, thetunicamycin-mediated induction of the mRNAs encodingthese proteins was examined by real-time polymerase chainreaction (PCR). Immunoglobulin-binding protein (a majorprotein of the Hsp70 family), CNX and calreticulin, which areboth calcium-dependent lectins, were up-regulated more than10-fold after 24 h of tunicamycin treatment (Figure 5), whichis in agreement with previous reports (Zhang and Kaufman
2004; Hebert and Molinari 2007; Coe et al. 2008). ER deg-radation enhancing α-mannosidase-like protein 1 and the ubi-quitin ligase subunit FBG3, which were both involved inERAD, are also induced �4-fold 14 h after tunicamycin treat-ment (Hosokawa et al. 2001; Ilyin et al. 2002). VIP36,ERGIC-53 and MCFD2 (which is associated with ERGIC-53)mRNAs were also increased 2–6-fold after 14 h and by>6-fold after 42 h (Figure 5). In contrast, VIPL mRNA levelsdid not change, even after 24 h and increased <2-fold after42 h (Figure 5). The same results were obtained whenHEK293 cells and human testicular carcinoma (DU145) cells
Fig. 1. Expression of intracellular cargo receptors on the cell surface of 293T cells. (A) Topology of the cargo receptor–CD8α fusion protein. The luminal part ofthe cargo receptor is fused with the membrane proximal, transmembrane and intracellular domains of CD8α. (B) 293T cells expressing each FLAG-tagged cargoreceptor–CD8α fusion protein were stained with or without (none) an anti-FLAG monoclonal antibody (anti-FLAG Ab) or an isotype-matched controlmonoclonal antibody (isotype Ab), followed by PE-labeled goat anti-mouse IgG F(ab′)2.
ERGIC-53 localization under ER stress condition
1711
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

Fig. 2. Specificity of the generated monoclonal antibodies. (A) 293T cells expressing cargo receptor-CD8α fusion proteins were stained with each monoclonalantibody (ER1, ER2, ER3, VL1, VL2 and VIP1) followed by staining with PE-labeled anti-mouse IgG. Lines indicate staining with a secondary antibody alone.Filled histograms show staining with the indicated monoclonal antibodies. (B) Cell lysates from 293T cells expressing myc-tagged cargo receptors andimmunoprecipitates of the cell lysates with a VIP1, VL1 or ER1 monoclonal antibody were electrophoresed under reduced conditions, blotted onto a membraneand stained with an anti-myc antibody. The white arrow head indicates myc-tagged ERGIC-53, and the black arrowhead indicates myc-tagged VIPL and VIP36.
S-Y Qin et al.
1712
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

were treated with tunicamycin under the same conditions(data not shown). This suggests that different expressionlevels of ERGIC-53 and VIPL (i.e. a different ratio ofERGIC-53 to VIPL) could be induced during ER stress,which may be a reason for the redistribution of ERGIC-53 tothe Golgi.
ERGIC-53 co-precipitated with VIPL but not VIP36The relationship of VIPL with the distribution of ERGIC-53made us speculate that ERGIC-53 may interact with VIPL,either directly or indirectly, in the ER, which then regulates
the localization of ERGIC-53 inside the cells. Next, we per-formed immunoprecipitation experiment using cell lysatesexpressing myc-tagged ERGIC-53 and/or HA-tagged VIPLand anti-myc and anti-HA antibodies. When the lysate ofcells expressing myc-ERGIC-53 plus HA-VIPL was precipi-tated with an anti-HA antibody, myc-ERGIC-53 wasco-precipitated (Figure 6A, lane 12). However, the signal cor-responding to myc-ERGIC-53 was detected neither from thecells expressing HA-VIPL only (Figure 6A, lane 10) nor fromthe cells expressing myc-ERGIC-53 only (Figure 6A, lane11), though HA-VIPL was actually precipitated with ananti-HA antibody (Figure 6B, lane 10). Similarly, when lysate
Fig. 3. Intracellular distribution of endogenous ERGIC-53 in HeLa cells after tunicamycin or DTT treatment. After HeLa cells were untreated (upper two rows,control) or treated with 5 µg/mL of tunicamycin (middle two rows, TM) or 2 mM DTT (lower two rows) for 24 h, they were fixed and co-stained with ananti-CNX antibody and an anti-ERGIC-53 antibody ER1 (ERGIC-53) for the visualization of the distribution of ERGIC-53 in the ER or co-stained with ananti-Golgi 58K protein antibody (Golgi 58K) and an anti-ERGIC-53 antibody ER1 (ERGIC-53) for the visualization of the distribution of ERGIC-53 in theGolgi. The nucleus was stained with DAPI and a merged image is shown in the right panel. White scale bars: 10 µm.
ERGIC-53 localization under ER stress condition
1713
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

Fig. 4. Intracellular distribution of overexpressed ERGIC-53 in HeLa cells with or without co-expression with VIPL or VIP36. After HeLa cells were transfectedwith plasmid(s) for 24 h, cells were fixed and co-stained with an anti-CNX antibody and an anti-ERGIC-53 antibody ER1 (ERGIC-53) for the visualization ofthe distribution of ERGIC-53 in the ER or co-stained with an anti-Golgi 58K protein antibody (Golgi 58K) and an anti-ERGIC-53 antibody ER1 (ERGIC-53)for the visualization of the distribution of ERGIC-53 in the Golgi. The nucleus was stained with DAPI and a merged image is shown in the right panel. Whitescale bars: 10 µm. (A) HeLa cells were transfected with pRcCMV1-ERGIC-53 (upper two rows, ERGIC-53) or co-transfected with both pRcCMV1-ERGIC-53and pRcCMV1-VIPL (lower two rows, ERGIC-53 and VIPL). (B) HeLa cells were transfected with pRcCMV1-ERGIC-53 (upper two rows, ERGIC-53) orco-transfected with both pRcCMV1-ERGIC-53 and pRcCMV1-VIP36 (lower two rows, ERGIC-53 and VIP36).
S-Y Qin et al.
1714
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

of cells expressing myc-ERGIC-53 plus HA-VIPL was preci-pitated with an anti-myc antibody, HA-VIPL wasco-precipitated (Figure 6B, lane 8). However, the signal corre-sponding to HA-VIPL was not detected from the cells expres-sing myc-ERGIC-53 only (Figure 6B, lane 7) and from thecells expressing HA-VIPL only (Figure 6B, lane 6), eventhough myc-ERGIC-53 was equally precipitated with ananti-myc antibody (Figure 6A, lane 7). This indicates that theprecipitation of myc-ERGIC-53 depends on the presence ofHA-VIPL and vise versa. The same data were obtained whenwe precipitated endogenous VIPL from cell lysates expressingmyc-ERGIC-53 using an anti-VIPL antibody, VL1(Figure 2B, lane 12). These data together indicated thatERGIC-53 interacts with VIPL, either directly or indirectly.
To further confirm the specificity of the interaction betweenERGIC-53 and VIPL, we analyzed the interaction betweenERGIC-53 and VIP36 by co-precipitation from myc-ERGIC-53 and FLAG-tagged VIP36 (FLAG-VIP36)-expressing 293Tcells. We did not observe any co-precipitation with myc-ERGIC-53 and FLAG-VIP36 (Figure 6C and D). These datasuggested that ERGIC-53 specifically interacted with VIPLbut not VIP36.To address the physiological relevance of ERGIC-53/VIPL
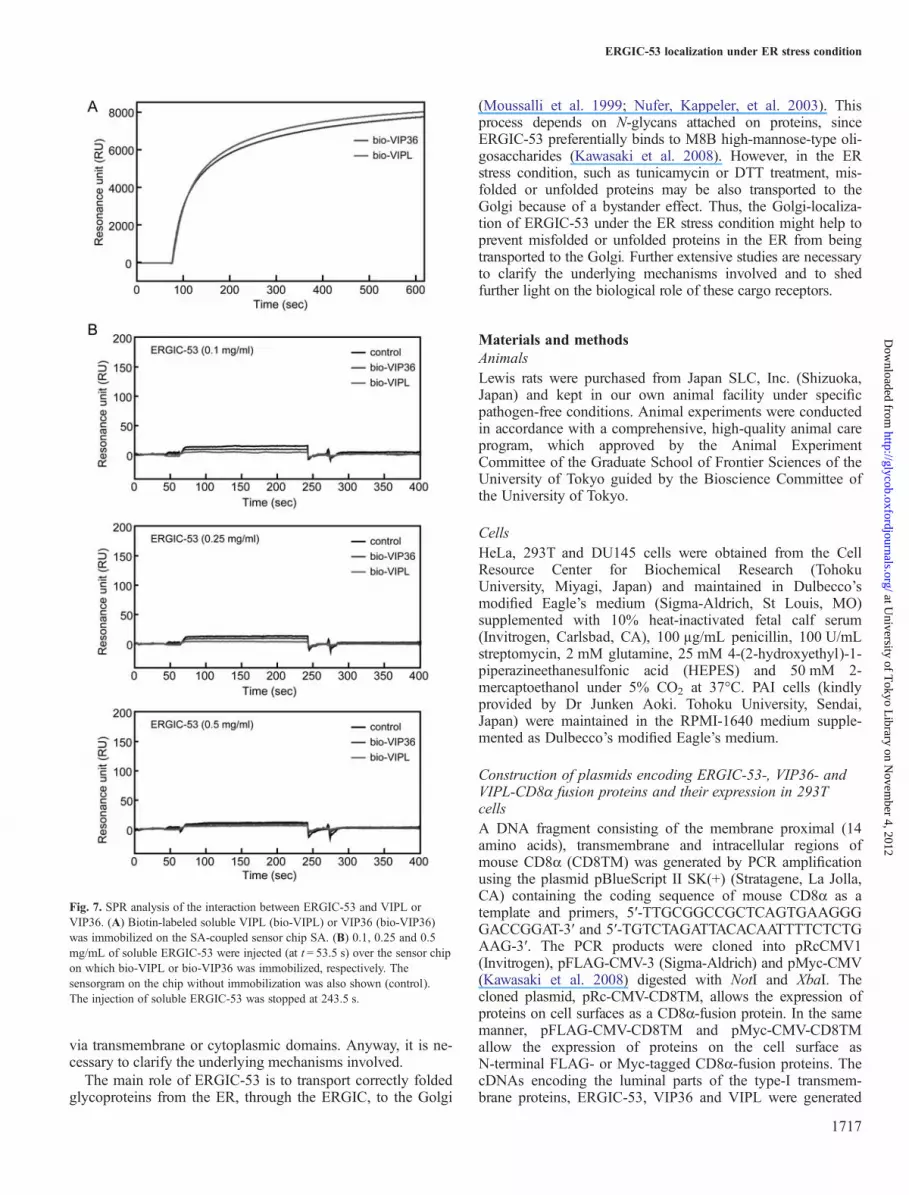
association, we investigated the interaction of ERGIC-53 withVIPL or VIP36 by the surface plasmon resonance (SPR)method using their luminal portions. However, no significantbinding of ERGIC-53 to either VIPL or VIP36 was observed(Figure 7). This finding may provide two possible explana-tions. First, the interaction between ERGIC-53 and VIPL maybe mediated by the transmembrane or cytoplasmic domains ofthese cargo receptors. Second, ERGIC-53 may interact withVIPL indirectly via another molecule.
Discussion
In the present study, we successfully generated monoclonalantibodies specific for ERGIC-53, VIP36 and VIPL, whichare cargo receptors distributed on the membranes of the ERand the Golgi. For this purpose, we expressed the ER- andthe Golgi-resident receptors on the cell surface of 293T cellsand these cells were used for both immunization and screen-ing of monoclonal antibodies. As shown in the present study,this procedure has many advantages when preparing monoclo-nal antibodies without the need for the purification of theimmunized protein. All of the established monoclonal anti-bodies (ER1, ER2, ER3, VIP1, VL1 and VL2) could be usedfor immunoprecipitation and immunostaining. Western blot-ting and immunostaining with each of the antibodies werefurther performed to test the reactivity of each monoclonalantibody with FLAG-tagged proteins transferred on polyviny-lidene fluoride (PVDF) membrane. Although FLAG-taggedproteins were clearly stained by an anti-FLAG antibody, notall monoclonal antibodies reacted with denaturedFLAG-tagged ERGIC-53, VIP36 or VIPL; even when theantibody concentration was increased to 50 µg/mL (data notshown), indicating these antibodies reacted with target pro-teins with native conformation. Thus, this method appears togenerate monoclonal antibodies that recognize proteins in thenative conformation.In this study, we identified the intracellular localization of
endogenous cargo receptors using established monoclonalantibodies. VIP36 and VIPL have the same cytoplasmic ERexit motif (KRFY) on their C termini, but VIPL has an add-itional arginine at the fifth position from the C terminus, creat-ing a diarginine ER localization motif (RKR). As a result,VIPL is a resident of the ER (Neve et al. 2003; Nufer,Mitrovic, et al. 2003), unlike ERGIC-53 and VIP36, whichcycle in the secretory pathway. In the case of ERGIC-53, theoverexpression of ERGIC-53 in the cell interfered with theintracellular distribution of the protein itself (Figure 4). Thisappeared to occur physiologically when ER stress was trig-gered in the cell. When cells were treated with tunicamycin orDTT, ERGIC-53, which distributed in both the ER and the
Fig. 5. Induced expression of chaperones, intracellular lectins and cargoreceptors after tunicamycin treatment in HeLa cells. Quantitative real-timePCR was performed as described in Materials and methods. Glyceraldehydephosphate dehydrogenase (GAPDH) mRNAwas used as an endogenouscontrol. The relative intensity of expression was normalized against GAPDHbefore tunicamycin treatment. Closed bars indicate the cells treated with 5 µg/mL of tunicamycin in 0.05% DMSO for the indicated times and open barswere the cells treated with 0.05% DMSO alone under the same condition.Results shown represent the means of triplicate experiments and error barsindicate SD. The P-values were calculated using a paired Student’s t-test andstatistical significance was determined when a P-value was <0.05 (*P < 0.05).
ERGIC-53 localization under ER stress condition
1715
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

Golgi under normal conditions, became mainly localized tothe Golgi apparatus (Figure 3). Further, we also demonstratedthat co-expression with VIPL caused increased localization ofERGIC-53 to the ER (Figure 4A). Real-time reverse tran-scriptase–PCR indicated that the levels of ERGIC-53 mRNAincreased greatly upon ER stress, whereas VIPL mRNA levelsdid not change, even after 24 h (Figure 5). These observationsmade us speculate that ERGIC-53 may interact with VIPL inthe ER and such interaction causes distinct intracellular distri-bution of ERGIC-53 under the ER stress condition.Interestingly, orthologs for human ERGIC-53 and VIPL arebroadly distributed in many eukaryotes from human to fissionyeast, whereas VIP36 is restricted to higher organisms (Ilyinet al. 2002), which suggested that ERGIC-53 and VIPL mightbe functionally associated each other. Based on this hypoth-esis, we immunoprecipitated myc-tagged ERGIC-53 andHA-tagged VIPL from cell lysates using anti-myc andanti-HA antibodies. As shown in Figure 6A and B, myc-
tagged ERGIC-53 co-precipitated with VIPL and vise versa.This supports the hypothesis that ERGIC-53 interacts withVIPL, either directly or indirectly. To further confirm theinteraction between ERGIC-53 and VIPL, we also investi-gated the interaction of ERGIC-53 with VIPL by the SPRmethod using their soluble forms. However, no significantbinding of ERGIC-53 to VIPL was observed (Figure 7). Theresult indicates that the interaction between ERGIC-53 andVIPL is not mediated via the luminal portions or thatERGIC-53 interacts with VIPL indirectly via another mol-ecule. One of the latter possibilities is that glycoproteins con-taining both M8B and M9 high-mannose-type glycansmediate the interaction between ERGIC-53 and VIPL, sinceERGIC-53 and VIPL bind strongly to M8B and M9 glycans,respectively (Yamaguchi et al. 2007; Kawasaki et al. 2008).However, ERGIC-53 could interact with VIPL but not VIP36,which also binds to high-mannose-type glycans, indicating thatthe interaction between ERGIC-53 and VIPL may be mediated
Fig. 6. ERGIC-53 co-precipitated with VIPL, but not with VIP36. Cell lysates expressing myc-tagged ERGIC-53 and/or HA-tagged VIPL wereimmunoprecipitated with an anti-myc antibody (lanes 5–8) or an anti-HA antibody (lanes 9–12) and the precipitates were stained with (A) an anti-myc antibodyor (B) an anti-HA antibody, respectively. Expression of myc-ERGIC-53 and/or HA-VIPL in 293T cells were confirmed by western blotting of the cell lysatesusing anti-myc and anti-HA antibodies, respectively (lanes 1–4). The black arrow heads in (A) and (B) indicate myc-tagged ERGIC-53 and HA-tagged VIPL,respectively. Cell lysates expressing myc-tagged ERGIC-53 and/or FLAG-tagged VIP36 were immunoprecipitated with an anti-myc antibody (lanes 5–8) or ananti-FLAG antibody (lanes 9–12) and the precipitates were stained with (C) an anti-myc antibody or (D) an anti-FLAG antibody, respectively. Expression ofmyc-ERGIC-53 and/or FLAG-VIP36 in 293T cells were confirmed by western blotting of the cell lysates using anti-myc and anti-FLAG antibodies, respectively(lanes 1–4). The black arrow heads in (C) and (D) indicate myc-tagged ERGIC-53 and FLAG-tagged VIP36, respectively.
S-Y Qin et al.
1716
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from
via transmembrane or cytoplasmic domains. Anyway, it is ne-cessary to clarify the underlying mechanisms involved.The main role of ERGIC-53 is to transport correctly folded
glycoproteins from the ER, through the ERGIC, to the Golgi
(Moussalli et al. 1999; Nufer, Kappeler, et al. 2003). Thisprocess depends on N-glycans attached on proteins, sinceERGIC-53 preferentially binds to M8B high-mannose-type oli-gosaccharides (Kawasaki et al. 2008). However, in the ERstress condition, such as tunicamycin or DTT treatment, mis-folded or unfolded proteins may be also transported to theGolgi because of a bystander effect. Thus, the Golgi-localiza-tion of ERGIC-53 under the ER stress condition might help toprevent misfolded or unfolded proteins in the ER from beingtransported to the Golgi. Further extensive studies are necessaryto clarify the underlying mechanisms involved and to shedfurther light on the biological role of these cargo receptors.
Materials and methodsAnimalsLewis rats were purchased from Japan SLC, Inc. (Shizuoka,Japan) and kept in our own animal facility under specificpathogen-free conditions. Animal experiments were conductedin accordance with a comprehensive, high-quality animal careprogram, which approved by the Animal ExperimentCommittee of the Graduate School of Frontier Sciences of theUniversity of Tokyo guided by the Bioscience Committee ofthe University of Tokyo.
CellsHeLa, 293T and DU145 cells were obtained from the CellResource Center for Biochemical Research (TohokuUniversity, Miyagi, Japan) and maintained in Dulbecco’smodified Eagle’s medium (Sigma-Aldrich, St Louis, MO)supplemented with 10% heat-inactivated fetal calf serum(Invitrogen, Carlsbad, CA), 100 µg/mL penicillin, 100 U/mLstreptomycin, 2 mM glutamine, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 50 mM 2-mercaptoethanol under 5% CO2 at 37°C. PAI cells (kindlyprovided by Dr Junken Aoki. Tohoku University, Sendai,Japan) were maintained in the RPMI-1640 medium supple-mented as Dulbecco’s modified Eagle’s medium.
Construction of plasmids encoding ERGIC-53-, VIP36- andVIPL-CD8α fusion proteins and their expression in 293TcellsA DNA fragment consisting of the membrane proximal (14amino acids), transmembrane and intracellular regions ofmouse CD8α (CD8TM) was generated by PCR amplificationusing the plasmid pBlueScript II SK(+) (Stratagene, La Jolla,CA) containing the coding sequence of mouse CD8α as atemplate and primers, 5′-TTGCGGCCGCTCAGTGAAGGGGACCGGAT-3′ and 5′-TGTCTAGATTACACAATTTTCTCTGAAG-3′. The PCR products were cloned into pRcCMV1(Invitrogen), pFLAG-CMV-3 (Sigma-Aldrich) and pMyc-CMV(Kawasaki et al. 2008) digested with NotI and XbaI. Thecloned plasmid, pRc-CMV-CD8TM, allows the expression ofproteins on cell surfaces as a CD8α-fusion protein. In the samemanner, pFLAG-CMV-CD8TM and pMyc-CMV-CD8TMallow the expression of proteins on the cell surface asN-terminal FLAG- or Myc-tagged CD8α-fusion proteins. ThecDNAs encoding the luminal parts of the type-I transmem-brane proteins, ERGIC-53, VIP36 and VIPL were generated
Fig. 7. SPR analysis of the interaction between ERGIC-53 and VIPL orVIP36. (A) Biotin-labeled soluble VIPL (bio-VIPL) or VIP36 (bio-VIP36)was immobilized on the SA-coupled sensor chip SA. (B) 0.1, 0.25 and 0.5mg/mL of soluble ERGIC-53 were injected (at t = 53.5 s) over the sensor chipon which bio-VIPL or bio-VIP36 was immobilized, respectively. Thesensorgram on the chip without immobilization was also shown (control).The injection of soluble ERGIC-53 was stopped at 243.5 s.
ERGIC-53 localization under ER stress condition
1717
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

by PCR using plasmid pBlueScript II SK (+)-ERGIC-53,-VIP36 and -VIPL, respectively, and the primers listed below:
FLAG-ERGIC-53-forward:5′-ACAAGCTTGACGGCGTGGGA-3′;
ERGIC-53-forward:5′-GAAAGCTTATGGCGGGATCCAGG-3′;
ERGIC-53-reverse:5′-AGCGGCCGCCAGACAAACATGATGGA-3′;
FLAG-VIP36-forward:5′-TAAGCTTGATATAACTGACGGCAACAG-3′;
VIP36-forward:5′-GAAAGCTTATGGCGGCGGAAGGCT-3′;
VIP36-reverse:5′-AGCGGCCGCCCCGCCACCCCGTCAG-3′.
FLAG-VIPL-forward:5′-ACAAGCTTGGTCAAACGTTCGA-3′;
VIPL-forward: 5′-GAAAGCTTATGGCGGCGACTCT-3′; andVIPL-reverse: 5′-AGCGGCCGCCGGCCAGGCCACT-3′.
After digestion with HindIII and NotI, the PCR productswere cloned into pRc-CMV-CD8TM, pFLAG-CMV-CD8TMor pMyc-CMV-CD8TM digested with the same restrictionenzymes. 293T cells were transfected withpFLAG-CMV-CD8TM-ERGIC-53, -VIP36 or -VIPL usingLipofectamine 2000 (Invitrogen) according to the manufac-turer’s protocol. Transfected cells were used then for immun-ization and screening.
Flow cytometryFlow cytometry was performed to confirm the expression ofFLAG-tagged CD8α fusion proteins on the surface of 293Tcells. For staining with an anti-FLAG antibody(Sigma-Aldrich), 2 × 105 cells were incubated with 10 μg/mLof a primary antibody for 30 min on ice, then the cells werewashed twice with Hank’s balanced salt solution (NissuiPharmaceuticals, Tokyo, Japan) containing 0.1% BSA (Wako,Osaka, Japan) and 0.1% sodium azide (Wako) [fluorescence-activated cell sorting (FACS) buffer]. Washed cells werestained with 10 μg/mL of fluorescein isothiocyanate-labeledgoat anti-mouse IgG F(ab′)2 (Beckman Coulter, Fullerton,CA) or R-phycoerythrin (PE)-labeled goat anti-mouse IgG F(ab′)2 (Beckman Coulter), for 30 min on ice. The cells werewashed twice with FACS buffer and suspended in FACSbuffer containing 1 μg/mL of propidium iodide. The fluores-cence intensity of stained cells was measured using aFACSCalibur flow cytometer and the data were analyzedusing CellQuest software (BD Biosystems, San Jose, CA).
Generation of hybridomas and screeningBefore immunization, 293T cells (5 × 107) expressingFLAG-tagged ERGIC-53-, VIP36- or VIPL-CD8α on the cellsurface were emulsified with complete Freund’s adjuvant(Difco Laboratories, Detroit, MI; 38:62, v/v; total volume =1.3 mL). Two female Lewis rats were immunized in thefootpad with the emulsified immunogen mixture (100 µL/footpad). For the booster immunizations, the same amount ofimmunogen mixture emulsified with Freund’s incomplete ad-juvant (Difco Laboratories) was injected three times at 10-dayintervals. Animals were sacrificed 3 days after the last
injection and common iliac lymph node cells were collectedand fused at a 2:1 ratio with the non-Ig producing mousemyeloma cell line PAI using polyethylene glycol 1500 (50%w/v in 75 mM HEPES, pH 7.2; Roche, Mannheim, Germany).After fusion, hybridomas were selected in the hypoxanthine–aminopterin–thymidine medium containing 50–100 U/mLmouse IL-6 (culture supernatant from mouse IL-6-transfectedX63 cells, a generous gift from Dr H. Karasuyama, TokyoMedical and Dental University, Tokyo, Japan). For the firstscreening, hybridoma culture supernatants were assayed forbinding to 293T cells co-transfected with pFLAG-CMV-CD8TM-ERGIC-53, -VIP36 and -VIPL by flow cytometry.For the second screening, culture supernatants were assayedfor binding to 293T cells expressing N-terminal FLAG-taggedERGIC-53-, VIP36- or VIPL-CD8α. For the final screening,293T cells expressing ERGIC-53-, VIP36- or VIPL-CD8αwere used.
Purification of monoclonal antibodiesThe isotype of the antibodies was determined using a ratMonoAb ID/SP kit (Zymed Laboratories, San Francisco, CA).All antibodies generated in this study were rat IgG2aκ. Theculture supernatants from hybridoma producing monoclonalantibodies, ER1, ER2, ER3, VIP1, VL1 and VL2, were con-centrated with ammonium sulfate, dialyzed against 10 mMsodium phosphate, pH 7.4, containing 137 mM NaCl and2.68 mM KCl (PBS(−)) and purified by affinity chromatog-raphy using Hi-Trap Protein G HP (GE Healthcare,Buckinghamshire, UK). Approximately 20–30 mg of a puri-fied antibody was purified from 600 mL of the hybridomaculture supernatant.
ImmunoprecipitationTwenty-four hours after transfection ofpMyc-CMV-ERGIC-53, -VIP36 or -VIPL with or withoutpHA-CMV-VIPL or pFLAG-CMV-VIP36, 293T cells (2.0 ×106 cells) were washed with PBS(−) and suspended in lysisbuffer [50 mM Tris–HCl, pH 8.0, containing 150 mM NaCl,0.5% Triton X-100, 1 mM ethylenediaminetetraacetic acid(EDTA), 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1mg/mL leupeptin] at 2.0 × 107 cells/mL. After 1 h incubationon ice, the lysate was cleared by centrifugation at 12,000 × gfor 20 min. The supernatant was then mixed with proteinG-Sepharose beads (GE Healthcare) to which antibodies hadbeen bound and incubated with rotation at 4°C for 18 h. Thebeads were washed three times with wash buffer (50 mMTris–HCl, pH 7.5, containing 150 mM NaCl, 0.1% TritonX-100, 1 mM PMSF and 5 mM iodoacetamide). In the caseof co-immunoprecipitation experiment, anti-HA antibodyHA-7 (Sigma-Aldrich), anti-FLAG antibody (Sigma-Aldrich)and anti-Myc antibody which was purified from the culturesupernatant of hybridoma clone 9E10 purchased from theAmerican type culture collection (Manassas, VA) were used.
Polyacrylamide gel electrophoresis and western blottingPrecipitated proteins were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis according to themethod of Laemmli (1970) and transferred to PVDF
S-Y Qin et al.
1718
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

membrane (Millipore, Bedford, MA) at 15 V for 45 min usingsemi-dry blotting system (Bio-Rad Laboratories, Hercules,CA). The membrane was incubated with 20 mM Tris–HCl,pH 7.5, containing 150 mM NaCl, 0.005% Tween-20 [tris-buffered saline (TBS)-T] and 3% BSA for 1 h and then incu-bated with biotinylated anti-Myc, anti-FLAG or an anti-HAantibody for 1 h at 25°C followed by washing three times foreach 15 min with TBS-T. Next, the membrane was incubatedwith horseradish peroxidase-conjugated streptavidin (SA) for1 h at 25°C, followed by washing three times for each 15 minwith TBS-T. Finally, the membrane was processed usingenhanced chemiluminescence (ECL; GE Healthcare) andexposed to ECL films (GE Healthcare).
ImmunocytochemistryHeLa cells were cultured in 8-well culture slides (BDBiosciences, San Jose, CA) coated with 20 µg/mL of fibro-nectin and then treated with 5 µg/mL of tunicamycin in0.05% dimethyl sulfoxide (DMSO) for 24 h. As a control,DMSO was added at a final concentration of 0.05% to thecells instead of tunicamycin for the indicated times. The cellswere also treated with 2 mM DTT for 24 h. After treatment,the cells were fixed with 4% paraformaldehyde in PBS for 15min, washed three times with PBS and permeabilized with0.1% Triton X-100/PBS for 60 min. After blocking with 5%goat serum (Sigma-Aldrich) for 60 min, the cells were incu-bated with 5 µg/mL of an anti-CNX antibody (Abcam,Cambridge, UK) for the staining of the ER and 5 µg/mL ofan anti-Golgi 58K protein antibody (Sigma-Aldrich) for thestaining of the Golgi apparatus, respectively, and followed byincubation with 5 µg/mL of an Alexa Fluor 488-labeled anti-mouse antibody. After that, cells were stained with 8 µg/mLof a biotin-labeled anti-ERGIC-53 antibody (ER1) followedby incubation with 5 µg/mL of Alexa Fluor 568-labeled SA.Finally, the nucleus were stained with4′,6-diamidino-2-phenylindole (DAPI). The coverslips weremounted in Immersion oil (Leica Microsystems, Wetzler,Germany) and the cells were observed under a confocal laserscanning microscope LSM510 (Carl Zeiss, Gottingen,Germany) with LSM image browser software (Carl Zeiss).
Quantitative real-time PCRHeLa cells were treated with 5 µg/mL of tunicamycin for 14,24 and 42 h and then harvested with PBS/EDTA and washedtwice with PBS. Poly-A+ RNA was then extracted using a
µMACS mRNA isolation kit (Miltenyi Biotec, Germany).Each cDNA was generated using a SuperScript III first-strandsynthesis system (Invitrogen) according to the manufacturer’sinstructions using 0.2 mg of poly-A+ RNA. Quantitative real-time PCR was performed in a Smart cycler real-time PCRsystem (Takara Bio, Kyoto, Japan) using SYBR premix ExTaq (Takara Bio). The standard curve for each gene was con-structed using serial dilutions of control cDNA. The amplifi-cation conditions were: 35 cycles of denaturation at 95°C for15 s followed by annealing at 63°C for 15 s and extension at72°C for 30 s. The relative amounts of RNA for the targetgene transcripts were normalized against an endogenous gene,β-actin. Primer details are listed in Table I. The P-values werecalculated using a paired Student’s t-test and statistical signifi-cance was determined when a P-value was <0.05.
SPR analysis of the interaction of ERGIC-53 with VIPL orVIP36SPR experiments were performed with the Biacore 3000system (Biacore AB, Uppsala, Sweden). Biotinylated recom-binant soluble VIPL (Yamaguchi et al. 2007) or VIP36(Kawasaki et al. 2007) were immobilized to SA-coupledsensor chip SA (Biacore) until the resonance units reached ap-proximately saturation. The binding of soluble ERGIC-53(Kawasaki et al. 2008) to VIPL or VIP36 was measured in10 mM 2-(morpholino)ethanesulfonic acid (MES)–NaOH, pH7.0, containing 150 mM NaCl, 0.005% Tween-20 and 1.0mM CaCl2 at 25°C with a flow rate of 20 μL/min. ERGIC-53at 0.1, 0.25 and 0.5 mg/mL was applied to the sensor chip.The regeneration of the chip surface was carried out with 10mM MES–NaOH, pH 7.0, containing 150 mM NaCl, 0.005%Tween-20 and 10 mM EDTA.
Funding
This study was supported by a grant from Grants-in-Aid forScientific Research for K.Y. (21390173, 24390015) from theMinistry of Education, Culture, Sports, Science andTechnology, Japan. The funders had no role in study design,data collection and analysis, decision to publish or preparationof the manuscript.
Table I. Primers used for quantitative reverse transcriptase–PCR
Forward primer Reverse primer
CNX 5′-tccaaagccaagaaagacgatac-3′ 5′-gcccgagacatcaacacaag-3′Calreticulin 5′-agttccggcaagttctacgg-3′ 5′-tggccgacagagcataaaag-3′Immunoglobulin-binding protein 5′-aaccgctgaggcttatttgg-3′ 5′-tttggttgcttggcgttg-3′ER degradation enhancing α-mannosidase-like protein 1 5′-ttgactcttgttgatgcattgg-3′ 5′-cttggacggtggaatctttg-3′FBG3 5′-tcatattacacctgcctcaagtcc-3′ 5′-ccgtgtggtatccatcagctc-3′MCFD2 5′-agatgtcgccacaagaattgc-3′ 5′-ggtgcctgttcactcccttc-3′ERGIC-53 5′-ctctgctggaggcgtctatg-3′ 5′-ttcggcttttcatttgatgg-3′VIP36 5′-tgcccaccggctactacttc-3′ 5′-ttggtccagtcgatgctctc-3′VIPL 5′-tgtggggctgggagtatttg-3′ 5′-catagctgagggagccgttg-3′β-Actin 5′-gaggcactcttccagccttc-3′ 5′-cgtacaggtctttgcggatg-3′
ERGIC-53 localization under ER stress condition
1719
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from

Conflict of interest
None declared.
Abbreviations
CNX, calnexin; DAPI, 4′,6-diamidino-2-phenylindole;DMSO, dimethyl sulfoxide; DTT, dithiothreitol; ECL,enhanced chemiluminescence; EDTA, ethylenediaminetetraa-cetic acid; ER, endoplasmic reticulum; ERAD, ER-associateddegradation; ERGIC, ER and Golgi intermediate compart-ment; FACS, fluorescence-activated cell sorting; GAPDH, gly-ceraldehyde phosphate dehydrogenase; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; MES, 2-(mor-pholino)ethanesulfonic acid; PCR, polymerase chain reaction;PE, R-phycoerythrin; PMSF, phenylmethylsulfonyl fluoride;PVDP, polyvinylidene fluoride; SA, streptavidin; SPR, surfaceplasmon resonance; TBS, tris-buffered saline; TM,tunicamycin.
References
Appenzeller C, Andersson H, Kappeler F, Hauri HP. 1999. The lectinERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol.1:330–334.
Ashwell G, Morell AG. 1974. The role of surface carbohydrates in thehepatic recognition and transport of circulating glycoproteins. Adv EnzymolRelat Areas Mol Biol. 41:99–128.
Coe H, Bedard K, Groenendyk J, Jung J, Michalak M. 2008. Endoplasmic reticu-lum stress in the absence of calnexin. Cell Stress Chaperones. 13:497–507.
Crocker PR, Varki A. 2001. Siglecs, sialic acids and innate immunity. TrendsImmunol. 22:337–342.
Fiedler K, Parton RG, Kellner R, Etzold T, Simons K. 1994. VIP36, a novelcomponent of glycolipid rafts and exocytic carrier vesicles in epithelialcells. EMBO J. 13:1729–1740.
Fullekrug J, Scheiffele P, Simons K. 1999. VIP36 localisation to the early se-cretory pathway. J Cell Sci. 112(Pt 17):2813–2821.
Hebert DN, Molinari M. 2007. In and out of the ER: Protein folding, qualitycontrol, degradation, and related human diseases. Physiol Rev.87:1377–1408.
Helenius A, Aebi M. 2004. Roles of N-linked glycans in the endoplasmic re-ticulum. Annu Rev Biochem. 73:1019–1049.
Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A,Nagata K. 2001. A novel ER alpha-mannosidase-like protein acceleratesER-associated degradation. EMBO Rep. 2:415–422.
Ilyin GP, Serandour AL, Pigeon C, Rialland M, Glaise D, Guguen-GuillouzoC. 2002. A new subfamily of structurally related human F-box proteins.Gene. 296:11–20.
Kamiya Y, Kamiya D, Yamamoto K, Nyfeler B, Hauri HP, Kato K. 2008.Molecular basis of sugar recognition by the human L-type lectinsERGIC-53, VIPL, and VIP36. J Biol Chem. 283:1857–1861.
Kamiya Y, Yamaguchi Y, Takahashi N, Arata Y, Kasai K, Ihara Y, Matsuo I, Ito Y,Yamamoto K, Kato K. 2005. Sugar-binding properties of VIP36, an intracellu-lar animal lectin operating as a cargo receptor. J Biol Chem. 280:37178–37182.
Kawasaki N, Ichikawa Y, Matsuo I, Totani K, Matsumoto N, Ito Y,Yamamoto K. 2008. The sugar-binding ability of ERGIC-53 is enhancedby its interaction with MCFD2. Blood. 111:1972–1979.
Kawasaki N, Matsuo I, Totani K, Nawa D, Suzuki N, Yamaguchi D,Matsumoto N, Ito Y, Yamamoto K. 2007. Detection of weak sugar bindingactivity of VIP36 using VIP36-streptavidin complex and membrane-basedsugar chains. J Biochem. 141:221–229.
Laemmli UK. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature. 227:680–685.
Lemaire MM, Vanhaudenarde A, Nizet Y, Dumoutier L, Renauld JC. 2011.Induction of autoantibodies against mouse soluble proteins after immuniza-tion with living cells presenting the autoantigen at the cell surface infusion with a human type 2 transmembrane protein. J Immunol Methods.367:56–62.
Ley K. 2003. The role of selectins in inflammation and disease. Trends MolMed. 9:263–268.
Moremen KW, Molinari M. 2006. N-linked glycan recognition and process-ing: The molecular basis of endoplasmic reticulum quality control. CurrOpin Struct Biol. 16:592–599.
Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. 1997.Immunoglobulin binding protein (BiP) function is required to protect cellsfrom endoplasmic reticulum stress but is not required for the secretion ofselective proteins. J Biol Chem. 272:4327–4334.
Moussalli M, Pipe SW, Hauri HP, Nichols WC, Ginsburg D, Kaufman RJ.1999. Mannose-dependent endoplasmic reticulum (ER)-Golgi intermediatecompartment-53-mediated ER to Golgi trafficking of coagulation factors Vand VIII. J Biol Chem. 274:32539–32542.
Neve EP, Svensson K, Fuxe J, Pettersson RF. 2003. VIPL, a VIP36-like mem-brane protein with a putative function in the export of glycoproteins fromthe endoplasmic reticulum. Exp Cell Res. 288:70–83.
Nufer O, Kappeler F, Guldbrandsen S, Hauri HP. 2003. ER export ofERGIC-53 is controlled by cooperation of targeting determinants in allthree of its domains. J Cell Sci. 116:4429–4440.
Nufer O, Mitrovic S, Hauri HP. 2003. Profile-based data base scanning foranimal L-type lectins and characterization of VIPL, a novel VIP36-likeendoplasmic reticulum protein. J Biol Chem. 278:15886–15896.
Oliveira SJ, Pinto JP, Picarote G, Costa VM, Carvalho F, Rangel M, de SousaM, de Almeida SF. 2009. ER stress-inducible factor CHOP affects the ex-pression of hepcidin by modulating C/EBPalpha activity. PLoS One. 4:e6618.
Ruddock LW, Molinari M. 2006. N-glycan processing in ER quality control. JCell Sci. 119:4373–4380.
Schroder M, Kaufman RJ. 2005. The mammalian unfolded protein response.Annu Rev Biochem. 74:739–789.
Shirakabe K, Hattori S, Seiki M, Koyasu S, Okada Y. 2011. VIP36 protein isa target of ectodomain shedding and regulates phagocytosis in macrophageRaw 264.7 cells. J Biol Chem. 286:43154–43163.
Vollenweider F, Kappeler F, Itin C, Hauri HP. 1998. Mistargeting of the lectinERGIC-53 to the endoplasmic reticulum of HeLa cells impairs the secre-tion of a lysosomal enzyme. J Cell Biol. 142:377–389.
Yamaguchi D, Kawasaki N, Matsuo I, Totani K, Tozawa H, Matsumoto N, ItoY, Yamamoto K. 2007. VIPL has sugar-binding activity specific forhigh-mannose-type N-glycans, and glucosylation of the α1,2 mannotriosylbranch blocks its binding. Glycobiology. 17:1061–1069.
Yamamoto K. 2009. Intracellular lectins involved in folding and transport inthe endoplasmic reticulum. Biol Pharm Bull. 32:767–773.
Yamamoto K, Kawasaki N. 2010. Detection of weak-binding sugar activityusing membrane-based carbohydrates. Methods Enzymol. 478:233–240.
Zhang K, Kaufman RJ. 2004. Signaling the unfolded protein response fromthe endoplasmic reticulum. J Biol Chem. 279:25935–25938.
S-Y Qin et al.
1720
at University of T
okyo Library on N
ovember 4, 2012
http://glycob.oxfordjournals.org/D
ownloaded from





























