Embed Size (px)
Citation preview

Send Orders of Reprints at [email protected]
Current Organic Synthesis, 2013, 10, 411-424 411
Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic and � ,�-Dehydro-�-aminophosphinic Acid Derivatives
Anna Ku�nik*, Roman Mazurkiewicz and Nikodem Ku�nik
Department of Organic and Bioorganic Chemistry and Biotechnology, Silesian University of Technology, B. Krzywoustego 4, PL 44-100 Gliwice, Poland
Abstract: The synthesis of �,�-dehydro-�-aminophosphonic and �,�-dehydro-�-aminophosphinic acid derivatives is discussed. The most important synthetic pathways to the above-mentioned compounds have been divided into four groups depending on the type of bond formed as the last one. The configuration at the C=C double bond can be established based on the specific coupling constants 3JHP and 3JCP for the appropriate Z- and E-stereoisomers. The application of �,�-dehydro-�-aminophosphonates and �,�-dehydro-�-aminophosphinates, particularly in the enantioselective catalytic hydrogenation, is also discussed.
Keywords: �-aminophosphinic acids, �-aminophosphonic acids, asymmetric catalytic hydrogenation, �,�-dehydro-�-aminophosphinates, �,�-dehydro-�-aminophosphonates, Michaelis-Arbuzov reaction, Wadsworth-Emmons reaction.
1. INTRODUCTION �,�-Dehydro-�-aminophosphonic and �,�-dehydro-�-amino-
phosphinic acids can be considered as the phosphonic and phosphinic analogues of the well-known �,�-dehydro-�-amino acids (Fig. 1). The asymmetric catalytic hydrogenation of �,�-dehydro-�-amino acids is one of the most general methods for the asymmetric synthesis of �-amino acids [1, 2]. Similarly, �,�-dehydro-�-aminophosphonic and �,�-dehydro-�-aminophosphinic acids and their derivatives are widely used for the asymmetric synthesis of �-aminophosphonic and �-aminophosphinic acids by stereoselective catalytic hydrogenation [3-5]. The latter compounds,
as structural analogues and mimetics of �-amino acids, display a broad spectrum of biological activity and, therefore, have currently attracted the significant interest of organic chemists and biochem-ists. �,�-Dehydro-�-amino acids, as an important class of com-pounds, have been the subject of many comprehensive monographs [6-10]. In contrast, there are no similar reviews devoted to �,�-dehydro-�-aminophosphonic and �,�-dehydro-�-aminophos- phinic acids. In this paper we gathered data from the literature on
*Address correspondence to these authors at the Department of Organic and Bioor-ganic Chemistry and Biotechnology, Silesian University of Technology, B. Krzy-woustego 4, PL 44-100 Gliwice, Poland; Tel: +48 32 237 16 13; Fax: +48 32 237 20 94; E-mail: [email protected]
the synthesis, structure and applications of �,�-dehydro-�-aminophosphonic and �,�-dehydro-�-aminophosphinic acid deriva-tives.
2. SYNTHESIS OF �,�-DEHYDRO-�-AMINOPHOSPHONIC AND PHOSPHINIC ACID DERIVATIVES
The methods for the synthesis of �,�-dehydro-�-aminophosphonic or phosphinic acid derivatives have, in this paper, been divided into four groups depending on which of the bonds is formed as the last one at the C� atom as follows (Scheme 1):
- formation of the C�-P bond
- formation of the C�-N bond - direct formation of the C�=C� bond - formation of the C�-C� bond followed by its transformation to
C�=C� bond.
2.1. Formation of the C�-P Bond
This group of methods is used most frequently for the synthesis of �,�-dehydro-�-aminophosphonates or �-aminophosphinates. The synthetic pathway involves the replacement of the �-halogen atom of �,�-dihaloamide with phosphite or phosphonite by the Michaelis-Arbuzov reaction and further hydrogen halide elimination (Scheme 1).
NHCOR3
P
O
R4
OR1
R2
NHCOR3
P
O
R4
OR1
OR1
NHCOR3
C
R4
OOR1
�,�-dehydro-�-amino acid derivatives
�,�-dehydro-�-aminophosphinic acid derivatives
�,�-dehydro-�-aminophosphonic acid derivatives
R5
R5 R5R2 = H, Alk or Ar
Fig. (1).
1875-6271/13 $58.00+.00 © 2013 Bentham Science Publishers

412 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
Drach et al. developed a method for the synthesis of diethyl 1-(N-acylamino)-2,2-dichloroethenephosphonates via the Michaelis-Arbuzov reaction of easily accessible N-(1,2,2,2-tetrachloroethyl) acylamide (1) with triethyl phosphite (Scheme 2, R1= Et) which yielded �-aminophosphonic acid derivatives 2. The latter com-pounds underwent hydrogen chloride elimination, upon treatment with triethylamine, to give diethyl 1-(N-acylamino)-2,2-dichloro-ethenephosphonates (3) in a yield of 70–91% (Scheme 2) [11]. This procedure was later applied by Scheidecker et al. for the synthesis of 1-(N-formylamino)-2,2-dichloroethenephosphonates (Scheme 2,R = H, R1= Me, Et, i-Pr) using potassium tert-butoxide or ammonia aqueous solution for hydrogen chloride elimination [12].
In a similar way 2,2-dichloroethenephosphonate derivatives were obtained by Köckritz and Schnell starting from diethyl 2,2,2-
trichloro-1-isocyanatoethanephosphonate (5), readily prepared viathe Michaelis-Arbuzov reaction of 1,2,2,2-tetrachloroethaneis-ocyanate (4) with triethyl phosphite. The urea derivatives 6, formed as a result of the addition of a variety of aliphatic and aromatic amines to compound 5, were treated in situ after 2 h with mor-pholine, thus providing 1-carbamoylamino-2,2-dichloroethene-phosphonate derivatives 7 after 1 h (Scheme 3) [13].
Stukalo et al. developed a method for the synthesis of diethyl 1-(N-tert-butoxycarbonylamino)ethenephosphonate (11) starting from 1,2-dihalogenethylisocyanate (8) via the Michaelis-Arbuzov reaction. Subsequent hydrogen halide elimination from 2-bromo-1-isocyanatoethanephosphonate (9) under the influence of triethy-lamine and conversion of the resulting compound 10 with tert-
NHCOR3
PO
R
OR1
R2
R1OP
OR1
R2
X
NHCOR3
X
R
P
NHCOR3
X
O OR1
R2
R
- HXX = Cl, Br
O
PO OR1
R2
NH2COR3
or NH2OHNCOR3
PO OR1
R2
RR
P CH
NHCOR3
O
R1OR1O
P CR
OH
R2 = OR1
OOR1
R2
Wadsworth-Emmons reaction
CP
OOR1
R2
Y
H
H
Michaelis-Arbuzov reaction
Y = N=C or N=C=S
CR
OH
t-BuOK
R2 = OR1 R2 = OR1
Scheme 1. General approaches to the synthesis of �,�-dehydro-�-aminophosphonates and phosphinates
NHCl
ClO
Cl R
Cl
R1OP
OR1
OR1 NHCl
Cl
P
Cl
O
base- HCl
R1 = Me, Et, i-Pr
OR1
OR1
O
R NH
PO OR1
OR1
O
R
Cl
Cl
R = H, Me, Pr, Ph
1 2 3
Scheme 2.
NClCl
Cl
Cl
EtOP
OEt
OEt NClCl
P
Cl
O OEtOEt
C O C O + R1R2NH NHClCl
P
Cl
O OEtOEt
N
O
R2
R1
+ morpholine- HCl
NHCl
P
Cl
O OEtOEt
N
O
R2
R1
R1 = Me, Et, Ph, 4-Me-C6H4, 4-NO2-C6H4
R2 = H, Me, Et
4 5 6
7
Scheme 3.
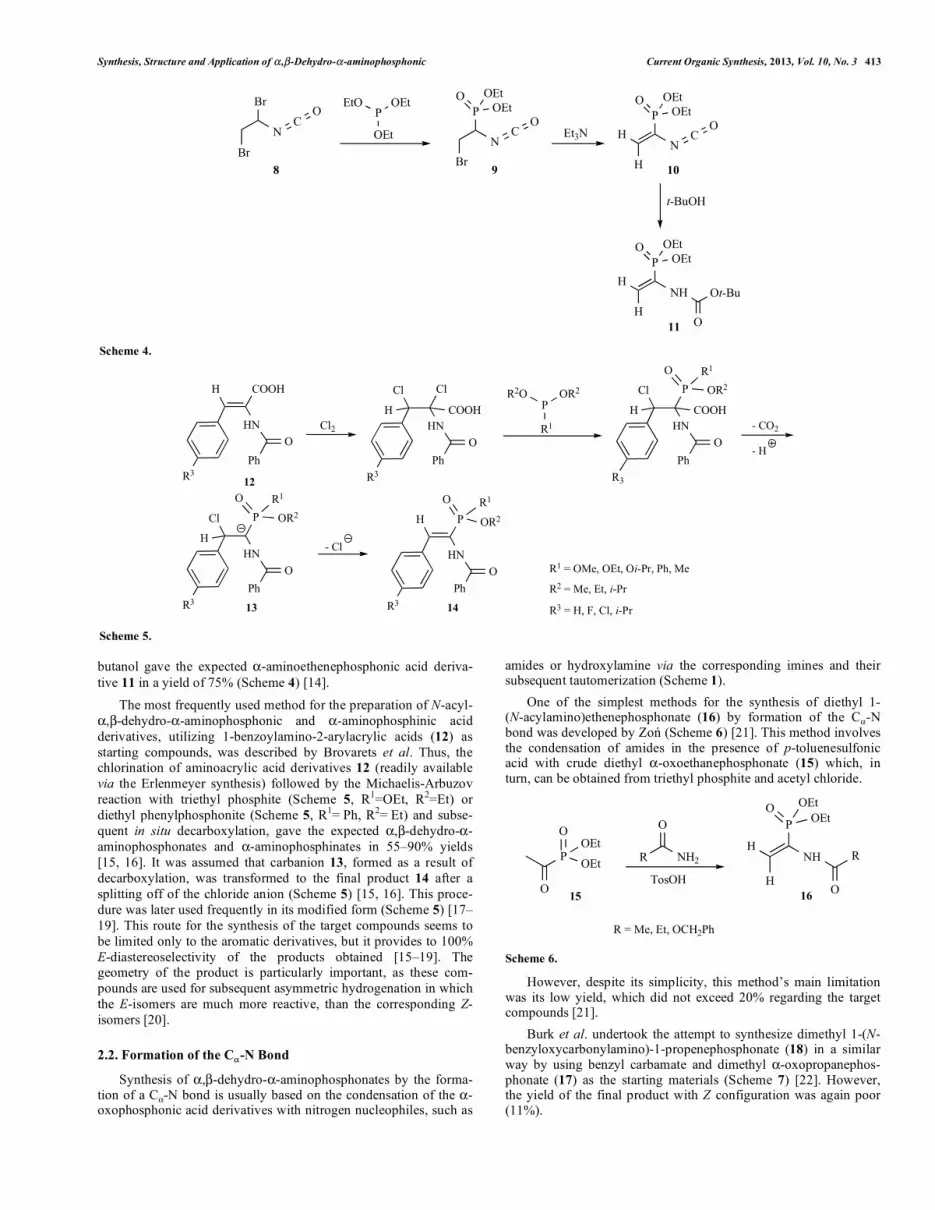
Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 413
butanol gave the expected �-aminoethenephosphonic acid deriva-tive 11 in a yield of 75% (Scheme 4) [14].
The most frequently used method for the preparation of N-acyl-�,�-dehydro-�-aminophosphonic and �-aminophosphinic acid derivatives, utilizing 1-benzoylamino-2-arylacrylic acids (12) as starting compounds, was described by Brovarets et al. Thus, the chlorination of aminoacrylic acid derivatives 12 (readily available via the Erlenmeyer synthesis) followed by the Michaelis-Arbuzov reaction with triethyl phosphite (Scheme 5, R1=OEt, R2=Et) or diethyl phenylphosphonite (Scheme 5, R1= Ph, R2= Et) and subse-quent in situ decarboxylation, gave the expected �,�-dehydro-�-aminophosphonates and �-aminophosphinates in 55–90% yields [15, 16]. It was assumed that carbanion 13, formed as a result of decarboxylation, was transformed to the final product 14 after a splitting off of the chloride anion (Scheme 5) [15, 16]. This proce-dure was later used frequently in its modified form (Scheme 5) [17–19]. This route for the synthesis of the target compounds seems to be limited only to the aromatic derivatives, but it provides to 100% E-diastereoselectivity of the products obtained [15–19]. The geometry of the product is particularly important, as these com-pounds are used for subsequent asymmetric hydrogenation in which the E-isomers are much more reactive, than the corresponding Z-isomers [20].
2.2. Formation of the C�-N Bond
Synthesis of �,�-dehydro-�-aminophosphonates by the forma-tion of a C�-N bond is usually based on the condensation of the �-oxophosphonic acid derivatives with nitrogen nucleophiles, such as
amides or hydroxylamine via the corresponding imines and their subsequent tautomerization (Scheme 1).
One of the simplest methods for the synthesis of diethyl 1-(N-acylamino)ethenephosphonate (16) by formation of the C�-N bond was developed by Zo� (Scheme 6) [21]. This method involves the condensation of amides in the presence of p-toluenesulfonic acid with crude diethyl �-oxoethanephosphonate (15) which, in turn, can be obtained from triethyl phosphite and acetyl chloride.
O
P
OOEt
OEt R
O
NH2
TosOH
NH
PO OEt
OEt
O
R
H
H
R = Me, Et, OCH2Ph
15 16
Scheme 6.
However, despite its simplicity, this method’s main limitation was its low yield, which did not exceed 20% regarding the target compounds [21].
Burk et al. undertook the attempt to synthesize dimethyl 1-(N-benzyloxycarbonylamino)-1-propenephosphonate (18) in a similar way by using benzyl carbamate and dimethyl �-oxopropanephos-phonate (17) as the starting materials (Scheme 7) [22]. However, the yield of the final product with Z configuration was again poor (11%).
N
Br
BrC
OEtO
POEt
OEt NBr
PC
O
O OEtOEt
Et3NN
PC
O
O OEtOEt
H
H
t-BuOH
NH
PO OEt
OEt
H
H
O
Ot-Bu
8 9 10
11
Scheme 4.
COOH
HN
H
OPh
Cl2
R2OP
OR2
R1
Cl
HN
Cl
OPh
H COOH
R3 R3
P
HN
H
OPh
R3
O R1
OR2
R1 = OMe, OEt, Oi-Pr, Ph, Me
R2 = Me, Et, i-Pr
R3 = H, F, Cl, i-Pr
P
HN
Cl
OPh
H COOH
R3
O R1
OR2
- CO2
- H
P
HN
Cl
OPh
H
R3
O R1
OR2
- Cl
12
13 14
Scheme 5.

414 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
Another interesting method for the synthesis of dimethyl N-acetyl-�,�-dehydro-�-aminophosphonates starting from �-oxophosphonates was developed by Zard et al. �-Oxophosphonates 19 can be readily prepared via the Michaelis-Arbuzov reaction of an acid chloride with trialkyl phosphite. Treatment of the �-oxophosphonates (19) with hydroxylamine and then with acetic anhydride provided the corresponding oxime acetates 20. Subse-quent reduction of these compounds by the double electron transfer from iron powder in acetic acid at 50–60°C led to the formation of a corresponding iminyl radical 21 and then iminyl anion 22 as a result of the rupture of the N-O bond. Trapping the intermediate iminyl anion (22) by acetic anhydride gave the desired 1-(N-acetylamino)-1-alkenephosphonates (23) as an equilibrium mixture of Z- and E-isomers in a ratio of 1:1 in a yield of 71–89% (Scheme 8) [23].
Quite a different approach to the preparation of �,�-dehydro-�-aminophosphonate derivatives was claimed in the US patent by Talley. The multi-step synthesis of dimethyl 1-(N-benzyl-oxycarbonylamino)-2-(4-benzyloxyphenyl)ethenephosphonate (28)was based on the Curtius rearrangement of the corresponding acyl azide 27, followed by trapping the isocyanate intermediate with benzyl alcohol. The �-(dimethoxyphosphoryl)propenic acid deriva-tive (25) was prepared in Knovenagel condensation of dimethoxy-phosphorylacetate (24) with 4-(benzyloxy)benzaldehyde. Subse-quent treatment of the condensation product 25 with trifluoroacetic acid removed the protecting tert-butoxy group and led to a mixture of Z- and E-isomers of compound 26. Its reaction with diphenyl-phosphoryl azide in the presence of triethylamine provided the expected acyl azide 27, which then underwent the above-mentioned Curtius rearrangement to produce the target compound 28 as a single E-isomer in a yield of 56% (Scheme 9) [24].
2.3. Direct Formation of the C�=C� Bond Synthesis of �,�-dehydro-�-aminophosphonates by the direct
formation of the double C�=C� bond involves the Wadsworth-Emmons reaction of �-aminomethanebisphosphonic acid deriva-tives with aldehydes (Scheme 1).
There are only two known procedures for the synthesis of �,�-dehydro-�-aminophosponates via the Wadsworth-Emmons reac-
O
P
OOMe
OMeO
O
NH2
NH
PO OMe
OMe
O
O
H
PhPh
17 18
Scheme 7.
RO
Cl
MeOP
OMe
OMe PO O
OMeOMe
NH2OH PHON O
OMeOMe
Ac2O / AcOH
PAcON O
OMeOMe
Fe
1e
PN O
OMeOMe
R
R R
R
1e PN O
OMeOMe
R
Ac2O
NHAc
P
O OMe
OMe
R
19
20 21 22 23
R = Ph, 4-F-C6H4, 3-MeO-C6H4, CH2Ph, 1-Np, i-Pr, Bu
Scheme 8.
O
+P
O OMe
OMe
O
Ot-Bu
TiCl4
NMM
P
OOMeOMeH
O
Ot-Bu
O
H
O
TFA
P
OOMeOMe
H
O
OH
O
DPPAEt3N
P
OOMeOMe
H
O
N
O
N NPh
P
OOMeOMeH
NH
OO
O Curtiusrearrangement
Ph
PhPh
PhPh
OH
24 25 26
2728
Ph
Scheme 9.

Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 415
tion. The first procedure consists in the conversion of ami-nomethanebisphosphonic acid (29) with trimethyl orthoformate into tetramethyl �-(N-formylamino)methanebisphosphonate (30), fol-lowed by its reaction with aldehydes in the presence of sodium methanolate, which results in the expected 1-(N-formylamino)-1-alkenephosponates (31) in reasonable yields (45–92%) as a mixture of E- and Z-isomers (Scheme 10) [25].
Wu and Tishler described an analogous synthesis of dimethyl 1-(N-formylamino)-2-(4-imidazolyl)ethenephosphonate (35) starting from 4-formylimidazole. Commercially available 4-imidazolyl-methanol, after N-protection of the imidazole ring by tritylation, was oxidized to the corresponding aldehyde 32 and then subjected to the Wadsworth-Emmons reaction with �-(N-formylamino)- methanebisphosphonate (33) to provide the imidazolylethenephos-phonate (34) as a single E-isomer in a yield of 50%. Its subsequent selective detritylation with 50% formic acid gave the deprotected product 35 in a yield of 62% (Scheme 11) [26]. After catalytic hydrogenation of this compound, the phosphonic analogue of his-tidine, which is responsible for a variety of physiological and patho-logical processes [27], was obtained.
2.4. Formation of the C�-C� Followed by its Transformation to a C�=C� Bond
This group of methods for the synthesis of �,�-dehydro-�-aminophosphonic acid derivatives is based on the condensation of the phosphonic acid derivatives possessing active hydrogen atoms in the � position with carbonyl compounds. Further elimination or a ring opening reaction results in the formation of the C�=C� bond of the target compounds (Scheme 1).
Several procedures for the preparation of N-acyl-�,�-dehydro-�-aminophosponates by the formation of the C�-C� bond and its subsequent transformation into the double bond have been de-scribed. One of them is an efficient route which utilizes 5-substituted (3-thio-4,2-oxazolidine)phosphonic acid diethyl esters
(37). The substituted oxazolidine-2-thiones, used as the starting materials, were obtained in a diastereoselective manner from metal-lated diethyl (isothiocyanatomethane)phosphonate (36) and alde-hydes and were transformed into N-Boc derivatives 38. The elimi-nation of carbon oxysulfide, performed at �78ºC in the presence of 2 equivalents of potassium tert-butoxide and completed within half an hour, provided the desired diethyl 1-(N-tert-butoxycarbonylamino)-1-alkenephosponates (39) in high yields (69–94%) and stereospecificity (Scheme 12) [28]. Thus, (Z)-N-Boc-1-aminoalkenephosphonates (39a) were formed from cis-oxazolidine-2-thiones (37a), whereas the diastereomeric trans-oxazolidine-2-thiones (37b) afforded E-isomers of the correspond-ing 1-aminoalkenephosphonates (39b).
A similar procedure for the synthesis of N-formyl-�,�-dehydro-�-aminophosphonates from diethyl (isocyanomethane)phosphonate via oxazolines was developed by Schöllkopf et al. The starting compound 42 was prepared from trimethyl N-(formylamino)- methylammonium halide (40) and triethyl phosphite followed by the reaction of the resulting N-(formylamino)methanephosphonate (41) with phosphoryl chloride in the presence of triethylamine. Diethyl (isocyanomethane)phosphonate (42) in the presence of catalytic amounts of sodium cyanide in ethanol was transformed into its �-metallated form 43. Condensation of this compound with carbonyl compounds gave diethyl (1,5-dihydro-4,2-oxazole)phos-phonates (46) as a result of the irreversible protonation of the corre-sponding sodium oxazoline 45, which was in equilibrium with adduct 44. (1,5-Dihydro-4,2-oxazole)phosphonates, upon treatment with potassium tert-butoxide, underwent deprotonation, providing thus the cyclic anion 47. Subsequent ring opening in electrocyclic reaction and trapping of the anion (48) so formed by protonation gave the expected diethyl 1-(N-formylamino)-1-alkenephospho-nates (49) in a yield of 78–92% as a mixture of E- and Z-isomers (Scheme 13) [29, 30].
P POO
OHOH
HOHO
NH2 HOMe
OMeOMe
HP P
OO
OMeOMe
MeOMeO
NH
O
H
RO
H
PO
OMeOMe
NH
O
H
H
R
R = H, Me, Hexyl, Ph, CH=CHPh
NaOMe
29 30 31
Scheme 10.
N
NH HCl.
OHPh3CCl
Et3N
N
NCPh3
OHMnO2
N
NCPh3
HO
NaOMeN
NCPh3
H P
HN
O OMe
OMe
H
Oaq. 50% HCOOH N
NH
H P
HN
O OMe
OMe
H
OP P
OO
OMeOMe
MeOMeO
NH
O
H
32
32
33 34 35
Scheme 11.

416 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
2.4. Miscellaneous Methods The utility of N-acyl-�,�-dehydro-�-aminophosphonic and
�-aminophosphinic acid species has been derived from their appli-cation in asymmetric catalytic hydrogenation, leading thus to opti-cally active �-aminophosphonic and �-aminophosphinic acids. The N-protecting group plays an essential role in this process. One of the most frequently used protection of an amino group of �-amino acids is the N-benzoyl group. N-Benzoyl-protected �,�-dehydro-�-amino acids are frequently the precursors of the corresponding �,�-dehydro-�-aminophosphonic and phosphinic acid derivatives. However, removal of this kind of N-protection by acidic hydrolysis is often connected with racemization of optically active products. This problem may be overcome by replacing the N-benzoylamino group with the N-alkoxycarbonylamino group under mild condi-tions. Thus, the one-pot transformation of N-benzoyl-�,�-dehydro-�-aminophosphonates and �-aminophosphinates to imides 51
comprises the reaction of compounds 50 with di-tert-butyl dicar-bonate in the presence of DMAP, as was claimed in the German patent by Dwars et al. Removal of the N-benzoyl group after the introduction of the N-Boc group was achieved upon treatment with hydrazine (Scheme 14) [5].
It has also been shown that it is possible to obtain �,�-dehydro-�-aminophosphonate derivatives by trapping phosphorus-stabilized allyl carbanions 55 (derived from �,�-dehydro-�-aminophosphonic acid) with common electrophiles, such as proton and methyl iodide. In order to generate the required stabilized allyl anion, dimethyl 1-(N-acetyl-N-isopropylamino)-3-phenyl-2-propenephosphonate (54) was prepared in the reaction of imine 52 with dimethyl phosphite in an overall yield of 63%, followed by the acetylation of the resulting aminoalkenephosphonate 53 using acetyl chloride and DMAP/pyridine. Further treatment of compound 54 with sodium hydride and quenching the generated anion 55 with water provided
P N
OEtO
EtO
CS
+
R
O
H
NHO
S
R P
O
OEtOEt
NHO
S
R P
O
OEtOEt
Boc2O
DMAP
Boc2O
DMAP
NBocO
S
R P
O
OEtOEt
P
NH
OOEt
OEt
Boc
R
t-BuOK
NBocO
S
R P
O
OEtOEt
P
NH
OOEt
OEt
Boc
t-BuOKR
R = i-Pr, t-Bu, Ph, 2-Furyl, PhCH=CH
H
H
36 37a
37b
38a 39a
38a 39a
Scheme 12.
EtOP
OEt
OEt NHN
O
H X+
P
O
EtOEtO NH
H
OCl3P O
Et3N
P
O
EtOEtO N
C10% mol. NaCN
EtOHP
O
EtOEtO N
CNa
+ OR1
R2
O PR1
R2H
N
OEtOEtNa
NO
PHR2
R1
OOEtOEt
Na
NO
PHR2
R1
OOEtOEt
H
+ EtOH- EtONa
t-BuOK NO
PR2R1 O
OEtOEt
H
NO
P
O OEtOEt
H
R1
R2
NHO
P
O OEtOEt
H
R1
R2
+ H
X = Br, I
R1 = Me, i-Pr, PhR2 = H, Me
40 41
42 43
43
44 45
46 47 48 49
O
C
Scheme 13.

Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 417
a mixture of starting material 54 and its isomer 56 in a ratio of 13:87, respectively. Methyl iodide as a trapping agent gave �-methylated product 57 as a mixture of the E- and Z-isomers (Scheme 15) [31].
3. STRUCTURE OF �,�-DEHYDRO-�-AMINOPHOS-PHONIC AND PHOSPHINIC ACID DERIVATIVES
The stereochemical result of hydrogenation of �,�-dehydro-�-aminophosphonic and �-aminophosphinic acids depends on the configuration at the double bond of the substrate. Determining this configuration is, therefore, an important issue.
Determining the configuration at the double bond in �,�-dehydro-�-aminophosphonic or �-aminophosphinic acid deriva-tives is based on an analysis of the values of vinyl proton–phosphorus (3JHP) and carbon–phosphorus (3JCP) coupling constants (Scheme 16, Table 1). Thus, according to literature, the values of the vicinal coupling constants (3JHP) for the E-isomers (58) of
1-(N-acylamino)-1-alkenephosphonates [18, 25, 26, 28] or phosphi-nates [19] are in the range of 14–24 Hz, while for Z-isomers (59)these values are significantly larger (J = 39–44 Hz) [25, 28]. But when it comes to 3JCP coupling constants, an inverse relationship can be noticed (Table 1). For Z-isomers these values of the coupling constants are smaller (J = 4–7 Hz) than those characteristic of the E-isomers (J = 14-22 Hz) [28].
P
HN
H
O
Ph
Ph
O R1
OEt
t-BuOO
O
Ot-BuO
DMAP
P
N
H
Ph
O R1
OEt
Ot-BuO
Ph
OH2N NH2
MeOH
P
NH
H
Ph
O R1
OEt
Ot-BuO
R1 = Ph, OEt
50 51
Scheme 14.
H
NP
O
OMeHOMe
P
OCl
Me
DMAP/Pyridine
NaH
HN
OOMe
OMe
P
N
OOMe
OMe
O
P
N
OOMe
OMe
O
P
N
OOMe
OMe
O
P
N
OOMe
OMe
O
P
N
OOMe
OMe
O
+
P
N
OOMe
OMe
O
MeIH2O
52 53
54 55
54 57
56
Scheme 15.
NHCOR3
P
O
R
H
3JHP = 39-44 Hz3JCP = 4-7 Hz
3JHP = 14-24 Hz3JCP = 14-22 Hz
d = 12-26 ppm31P NMR NHCOR3
P
O
H
ROR1
R2OR1
R2
58 59
Scheme 16.

418 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
Table 1. 31P NMR Chemical Shifts (�) and Vicinal Coupling Constants (J) of Selected 1-(N-acylamino)-1-alkenephosphonates.
Isomer R R1 R2 R3 31P NMR, � (ppm) 1H NMR 3JHP (Hz)13C NMR 3JCP
(Hz) Ref.
Z Ph Et EtO t-BuO 13.45 41.20 5.84 [28]
E Ph Et EtO t-BuO 15.00 16.15 19.00 [28]
Z 2-Furyl Et EtO t-BuO 13.20 39.20 6.95 [28]
E 2-Furyl Et EtO t-BuO 14.76 15.20 21.95 [28]
Z PhCH=CH Et EtO t-BuO 14.17 40.00 6.92 [28]
E PhCH=CH Et EtO t-BuO 14.89 � a 17.86 [28]
E t-Bu Et EtO t-BuO 15.71 16.75 15.65 [28]
Z i-Pr Et EtO t-BuO 15.17 42.50 4.80 [28]
E i-Pr Et EtO t-BuO 14.96 14.75 13.90 [28]
Z Me Me MeO H 16.77 b 42.5 b 5.4 b [20]
Z Me Me MeO H 14.06 c 40.5 c 4.1 c [20]
E Me Me MeO H 16.35 b 14.2 b 15.6 b [20]
E Me Me MeO H 15.25 c 14.2 c 14.2 c [20]
Z Ph Me MeO H 15.82 b 41.5 b 6.0 d [20]
Z Ph Me MeO H 13.88 c 38.5 c 6.0 d [20]
E Ph Me MeO H 15.43 b 16.6 b 17.4 b [20]
E Ph Me MeO H 17.08 c � 18.8 c [20]
Z Me Me MeO H � 44 � [25]
Z Hexyl Me MeO H � 43 � [25]
E Hexyl Me MeO H � 24 � [25]
Z Ph Me MeO H 16.5 � � [25]
E Ph Me MeO H 14.99 16 � [25]
Z PhCH=CH Me MeO H � 44 � [25]
E 4-imidazolyl Me MeO H � 15.5 � [26]
E Ph Me MeO Ph � 16.3 18.5 [18]
E Ph Et EtO Ph � 16.6 18.8 [18]
E Ph i-Pr i-PrO Ph 12.7 � a 19.0 [18]
E 2-FC6H4 Me MeO Ph 16.8 16.7 19.0 [18]
E 4-FC6H4 Me MeO Ph 17.6 16.3 19.2 [18]
E 4-ClC6H4 Me MeO Ph 17.3 16.0 19.1 [18]
E 4-i-PrC6H4 Me MeO Ph 18.0 16.5 18.8 [18]
E 4-NO2C6H4 Me MeO Ph 16.2 16.7 21.0 [18]
E 4-CF3C6H4 Me MeO Ph 26.6 16.4 19.0 [18]
a Overlapped with other signals; b major rotamer; c minor rotamer; d the value was not assigned to the specific rotamer
4. APPLICATIONS OF �,�-DEHYDRO-�-AMINOPHOS-PHONIC AND PHOSPHINIC ACID DERIVATIVES 4.1. Enantioselective Hydrogenation of �,�-Dehydro-�-amino-phosphonates and Phosphinates
Since N-acyl-�-aminoalkenephosphonates and phosphinates can be regarded as the analogues of �,�-dehydro-�-amino acid esters, they are valuable synthons in stereoselective reduction to the corresponding �-aminophosphonates and �-aminophosphinates (Scheme 17). The asymmetric catalytic hydrogenation of �,�-dehydro-�-aminophosphonates and phosphinates is one of the most
important routes that allows access to homochiral �-aminophosphonates [3, 4, 32-36] and �-aminophosphinates [5, 19, 37].
�-Aminophosphonic and �-aminophosphinic acids are struc-tural analogues of �-aminocarboxylic acids. The great interest in �-aminophosphonic acids and their related derivatives results from their unique biological activities [38-40] as potent antibiotics [41, 42], herbicides [43], fungicides [44], as well as antibacterial [42, 45, 46], antiviral [47] and anti-tumor agents [48]. Their wide range of applications also includes proteolytic enzyme inhibitors [49, 50],
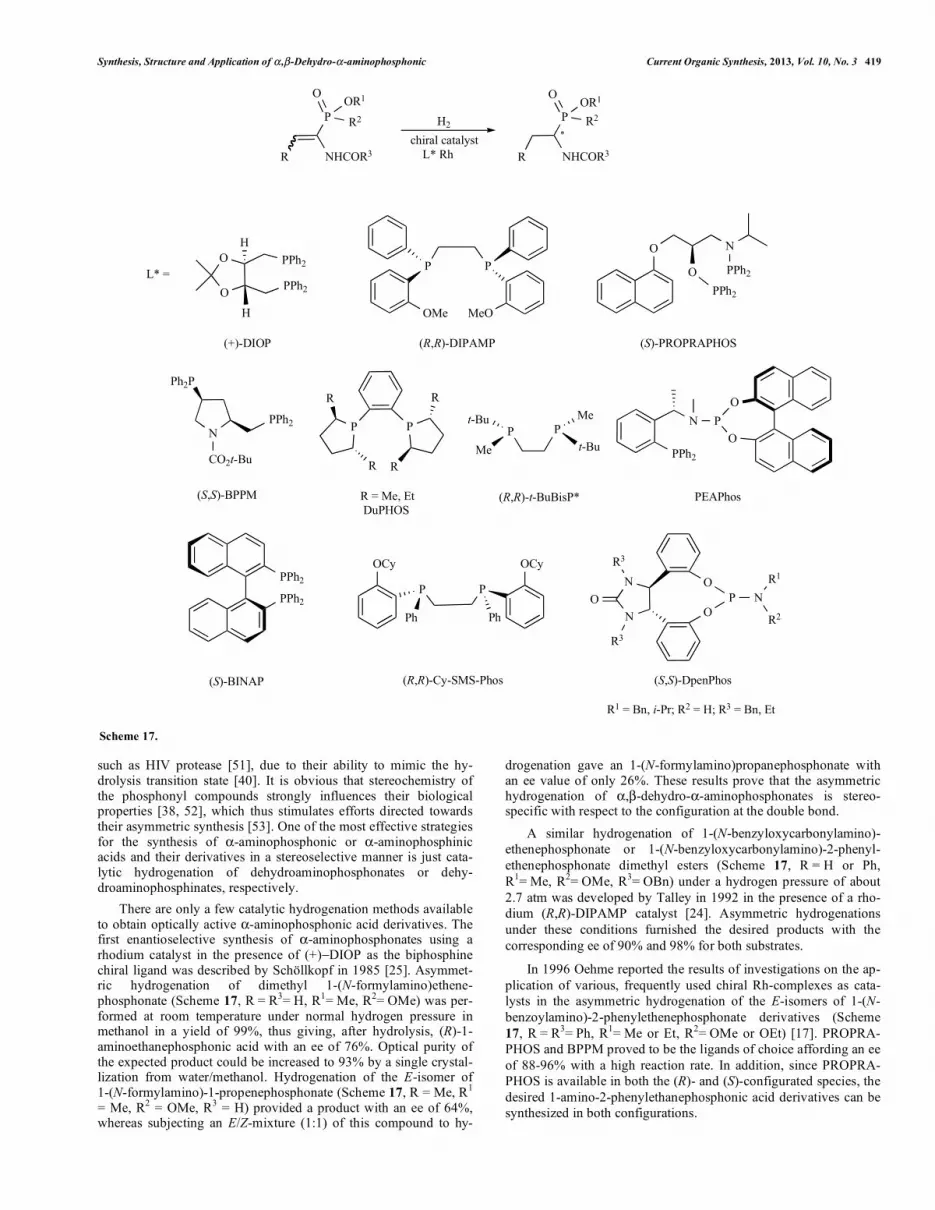
Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 419
such as HIV protease [51], due to their ability to mimic the hy-drolysis transition state [40]. It is obvious that stereochemistry of the phosphonyl compounds strongly influences their biological properties [38, 52], which thus stimulates efforts directed towards their asymmetric synthesis [53]. One of the most effective strategies for the synthesis of �-aminophosphonic or �-aminophosphinic acids and their derivatives in a stereoselective manner is just cata-lytic hydrogenation of dehydroaminophosphonates or dehy-droaminophosphinates, respectively.
There are only a few catalytic hydrogenation methods available to obtain optically active �-aminophosphonic acid derivatives. The first enantioselective synthesis of �-aminophosphonates using a rhodium catalyst in the presence of (+)�DIOP as the biphosphine chiral ligand was described by Schöllkopf in 1985 [25]. Asymmet-ric hydrogenation of dimethyl 1-(N-formylamino)ethene-phosphonate (Scheme 17, R = R3= H, R1= Me, R2= OMe) was per-formed at room temperature under normal hydrogen pressure in methanol in a yield of 99%, thus giving, after hydrolysis, (R)-1-aminoethanephosphonic acid with an ee of 76%. Optical purity of the expected product could be increased to 93% by a single crystal-lization from water/methanol. Hydrogenation of the E-isomer of 1-(N-formylamino)-1-propenephosphonate (Scheme 17, R = Me, R1
= Me, R2 = OMe, R3 = H) provided a product with an ee of 64%, whereas subjecting an E/Z-mixture (1:1) of this compound to hy-
drogenation gave an 1-(N-formylamino)propanephosphonate with an ee value of only 26%. These results prove that the asymmetric hydrogenation of �,�-dehydro-�-aminophosphonates is stereo-specific with respect to the configuration at the double bond.
A similar hydrogenation of 1-(N-benzyloxycarbonylamino)- ethenephosphonate or 1-(N-benzyloxycarbonylamino)-2-phenyl-ethenephosphonate dimethyl esters (Scheme 17, R = H or Ph, R1= Me, R2= OMe, R3= OBn) under a hydrogen pressure of about 2.7 atm was developed by Talley in 1992 in the presence of a rho-dium (R,R)-DIPAMP catalyst [24]. Asymmetric hydrogenations under these conditions furnished the desired products with the corresponding ee of 90% and 98% for both substrates.
In 1996 Oehme reported the results of investigations on the ap-plication of various, frequently used chiral Rh-complexes as cata-lysts in the asymmetric hydrogenation of the E-isomers of 1-(N-benzoylamino)-2-phenylethenephosphonate derivatives (Scheme 17, R = R3= Ph, R1= Me or Et, R2= OMe or OEt) [17]. PROPRA-PHOS and BPPM proved to be the ligands of choice affording an ee of 88-96% with a high reaction rate. In addition, since PROPRA-PHOS is available in both the (R)- and (S)-configurated species, the desired 1-amino-2-phenylethanephosphonic acid derivatives can be synthesized in both configurations.
NHCOR3
P
O
Rchiral catalyst L* Rh
H2*
NHCOR3
P
O
R
O
O
H
HPPh2
PPh2L* =
(+)-DIOP
P P
OMe MeO
(R,R)-DIPAMP
O N
OPPh2
PPh2
(S)-PROPRAPHOS
N
Ph2P
PPh2
CO2t-Bu
(S,S)-BPPM
P P
R
R
R
R
R = Me, Et DuPHOS
P PMe
t-Bu
t-Bu
Me
(R,R)-t-BuBisP*
OR1
R2
OR1
R2
PPh2
PPh2
(S)-BINAP
PP
PhPh
OCy OCy
(R,R)-Cy-SMS-Phos
N
NO
R3
R3
O
OP N
R1
R2
(S,S)-DpenPhos
R1 = Bn, i-Pr; R2 = H; R3 = Bn, Et
PPh2
N
PEAPhos
PO
O
Scheme 17.

420 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
Two years later the same ligands were used by Oehme’s group for the catalytic hydrogenation of (E)-1-(N-benzoylamino)-2-phenylethenephosphinates (Scheme 17, R = R3= Ph, R1= Me or Et, R2= Ph or Me), performed at room temperature under normal hy-drogen pressure [19]. Their investigations on the catalytic activity of chiral rhodium complexes containing these ligands showed lower activity than for the hydrogenations of the aminophosphonic acid precursors. Greater ee values were obtained with BPPM-Rh (79–98% ee) than with PROPRAPHOS-Rh (31–79% ee). Strong solvent dependence on the enantioselectivities was observed in the case of the Rh-complex possessing PROPRAPHOS as the chiral ligand (MeOH: 75% ee, C6H6: 31% ee, CH2Cl2: 79% ee).
Noyori et al. investigated the asymmetric hydrogenation of both isomers of dimethyl 1-(N-formylamino)-1-alkenephosphonates (Scheme 17, R = H, Me, or Ph, R1 = Me, R2 = OMe, R3 = H) with rhodium complexes of BINAP at 30ºC under a hydrogen pressure of 4 atm in methanol [20]. Use of the (S)-BINAP catalysts resulted in the formation of predominantly (R)-configurated products, while the (R)-BINAP complexes afforded the (S)-products. The elabo-rated procedure allowed the synthesis of 1-aminoethanephosphonic acid, 1-aminopropanephosphonic acid and 1-amino-2-phenyl-ethanephosphonic acid with an enantiomeric purity above 97% ee, and in quantitative yields when the substrates used for hydrogena-tion were the single E-isomers. Dependence of the reactivity on geometry of substrate was observed. Thus, the E-isomers turned out to be more reactive than the corresponding Z-isomers by a factor of 100. Because of the lower reactivity of the Z-configurated sub-strates, an E/Z mixture could be used directly for catalytic hydro-genation, without separation of the stereoisomers and providing, after hydrolysis and recrystallization, (R)-1-aminopropane-phosphonic acid and (R)-1-amino-2-phenylethanephosphonic acid in a yield of 62% and 78%, respectively.
DuPHOS was another chiral ligand tested for the enantioselec-tive hydrogenation of N-acetyl- and N-benzyloxycarbonyl-�,�-dehydro-�-aminophosphonates (Scheme 17, R1= Me, R2= OMe, R3= Me or OBn) by Burk and co-workers [22]. The reaction was performed with an initial hydrogen pressure of 4 atm at room tem-perature in methanol, which resulted in an ee of 95% using the alkyl derivatives of dehydroaminophosphonates (R = H or i-Pr) as sub-strates, and Et-DuPHOS-Rh as the catalyst. By comparison, hydro-genation of (E)-phenyl-substituted and (Z)-methyl-substituted 1-aminoethenephosphonate derivatives (R = Ph or Me, R3 = OBn) with Me-DuPHOS-Rh gave lower enantioselectivities of 76% and 71%, respectively.
Imamoto and co-workers reported that the catalytic hydrogena-tion of diethyl 1-(N-acetylamino)ethenephosphonate (Scheme 17,R = H, R1= Et, R2= OEt, R3= Me) with a rhodium complex of (R,R)-t-Bu-BisP* proceeded at a hydrogen pressure of 4 atm in methanol, thus providing an (R)-configurated product with an ee of 90% [54].
In 2008 Hu et al. developed an Rh-complex of the chiral phosphine-aminophosphine ligand (PEAphos) derived from (S)-1-phenylethylamine as an effective catalyst for the asymmetric hy-drogenation of �,�-dehydro-�-aminophosphonates under a hydro-gen pressure of 10 atm in methylene chloride [55]. Both 1-(N-benzyloxycarbonylamino)ethenephosphonate and the E-isomer of 1-(N-benzyloxycarbonylamino)-2-phenylethenephosphonate (Scheme 17, R = H or Ph, R1= Me, R2= OMe, R3= OBn) were reduced completely to give the corresponding (S)-1-amino-phosphonates with 96% ee.
The use of an Rh-(Cy-SMS-Phos) catalyst in the asymmetric hydrogenation of diethyl 1-(N-acetylamino)ethenephosphonate (Scheme 17, R = H, R1= Et, R2= OEt, R3= Me) was disclosed by Stephan et al. [56]. The reaction was carried out at room tempera-ture under a hydrogen pressure of 1 atm in methanol resulting in an (R)-configurated product with an excellent ee of 99.9%. Such an
extremely effective catalyst and simple reaction conditions are of industrial relevance.
Recently, chiral monodentate phosphoramidite DpenPhos ligands have been found to be highly efficient in the rhodium cata-lyzed asymmetric hydrogenation of a wide variety of �-aryl-, �-heteroaryl-, and �-alkyl-substituted �,�-dehydro-�-aminophos-phonates (Scheme 17, R = H, Ph, 2-naphthyl, 2-thienyl, styryl, i-Pr, t-Bu or Cy, R1= Me or Et, R2= OMe or OEt, R3= Me, Ph or OBn) [57]. Regardless of the structure of the substrate to be reduced, all of the reactions were completed within 1 hour at room temperature with an ambient pressure of hydrogen, providing the expected chiral �-aminophosphonates in quantitative conversion and in most cases with excellent ee values (97-99%). The effect of the solvent on enantioselectivity and reactivity has been demonstrated. Thus, using CH2Cl2, ClCH2CH2Cl, toluene or i-PrOH for the asymmetric hydrogenation of dimethyl 1-(N-benzoylamino)-2-phenylethene-phosphonate yielded full conversions with high ee values above 96%, whereas using methanol resulted in poor conversion (29%) as well as in a poor ee value (40%). Similarly, the influence of sub-strate geometry on enantioselectivity in the model hydrogenation reaction with the E- or Z-isomeric substrate has also been investi-gated. After subjecting to hydrogenation both pure E- and Z-isomers of dimethyl 1-(N-acetylamino)-2-phenylethenephos-phonate, the corresponding �-aminophosphonate ester was obtained in 99 and 88% ee, respectively, upon full conversion of the starting compounds. For the asymmetric hydrogenation of an E/Z-isomeric mixture (2.88:1), a satisfactory ee value (95%) of the product was accomplished along with complete conversion of the substrate, which demonstrates high efficiency and versatility of the catalyst used.
4.2. Miscellaneous Applications of � ,�-Dehydro-�-aminophos-phonic and Phosphinic Acid Derivatives
Dimethyl 1-(N-formylamino)ethenephosphonate (60) and dia-zoalkanes undergo [2+3] dipolar cycloaddition reactions regio-specifically to afford 5-substituted 3-(formylamino)-4,5-dihydro-3H-pyrazol-3-phosphonates (61) in high yields (88-94%). The obtained adducts 61 can serve as versatile synthons for some impor-tant compounds. Thus, their aromatization under acidic conditions results in 3-phosphonylated pyrazoles 62, while thermal decompo-sition of 61 followed by hydrolysis leads to 2-substituted 1-aminocyclopropanephosphonic acids 63 (Scheme 18) [58]. The latter compounds can be incorporated into peptide chains, which is a prominent route to conformationally constrained peptidomimetics [59, 60]. Furthermore, the 1-aminocyclopropanephosphonic acid proved to be a potent inhibitor of 1-aminocyclopropanecarboxylate (ACC) deaminase from Pseudomonas sp. and alanine racemase from Bacillus stearothermophilus [61]. According to recent litera-ture data, several types of oligopeptides containing the 1-aminocyclopropanephosphonic moiety were found to be Hepatitis C virus (HCV) NS3 protease inhibitors [62, 63].
Stevens and Vanderhoydonck disclosed the application of ethenephosphonate derivatives in a Michael type addition reaction during their research on the ring opening chemistry of 1-vinyl-2-phosphonoaziridines 64. Thus, the regioselective aziridine ring opening at C-3 with methyl chloroformate afforded 1-(N-acylamino)-2-chloroethanephosphonates 65, which upon treatment with ammonia in methanol underwent elimination of HCl and gave rise to the corresponding ethenephosphonates 66 in a yield of 51-61%. The subsequent Michael type addition of these compounds with several primary amines followed by in situ cyclization yielded the expected phosphonylated 2-imidazolidinones 67 in 33-52% (Scheme 19) [64]. It is worth mentioning that interest in the synthesis of heterocyclic aminophosphonates of this type is due to their potential pharmaceutical and agrochemical applications [38, 47].

Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 421
1-(1,3,4-Oxadiazol-2-yl)-1-aminomethanephosphonic acids 71are a similar group of heterocyclic aminophosphonic acid deriva-tives that are tested for potential use in various fields of medicine and agriculture. These compounds were synthesized by the reaction of diethyl 1-[N-(4-methylbenzoyl)amino]-2,2-dichloroethene-phosphonate (68) with hydrazine hydrate followed by treatment of the 5-hydrazino-1,3-oxazole derivative 69 with chlorides of various heterocyclic carboxylic acids in the presence of N,N-dimethyl-aniline. As a result new oxazole derivatives 70 were formed which upon heating in acetic acid underwent recyclization and ester bond rupture, providing thus the corresponding 1-(1,3,4-oxadiazol-2-yl)-1-aminomethanephosphonic acids (71) in a yield of 47-86% (Scheme 20) [65].
Another example of the practical application of �,�-dehydro-�-aminophosphonates is the use of diethyl (E)-1-(N,N-dimethylamino)-2-(tiophen-2-yl)ethenephosphonate derivatives (74) as intermediates in the multi-stage synthesis of 1, 3 and 4
substituted azetidin-2-ones 76. These novel 3-(thienyl) �-lactam derivatives (76) exhibiting antiproliferative and tubulin-binding activity are useful scaffolds for the development of tubulin-targeting drugs in cancer therapy [66, 67]. The above-mentioned ethenephosphonates 74 were prepared by the Wadsworth-Emmons reaction of tetraethyl dimethylaminomethanediphosphonate (72)with substituted 2-thienyl aldehydes 73 and then hydrolyzed with strong acid to produce acetic acid derivatives 75 (Scheme 21) [68]. It should be stressed that the described experiment shows that the 1-aminoalkenephosphonyl moiety can be considered as a masked form of a carboxyl group.
�,�-Dehydro-�-aminophosphonates also exhibit coordination properties. The conjugated C=C bond with the amino group has high affinity to late transition metals. Such a skeleton was presented as coordinated in a diiron complex [69]. On the other hand, Senge et al. have shown a nickel complex where the dehydroaminophos-
NHO
P
O OMeOMe
H
RCHN2, Et2O, r.t. N N
RP
HN
O OMeOMe
O
H
1. toluene, reflux, 3 h2. 6N HClaq, reflux, 6 h3. propylene oxide, MeOH
NH2
P
O OHOH
R
MeOH, cat Me3SiCl - HC(O)NH2
HN N
R
P
O
OMeOMe
R = H, Me, Ph
60 61
62 63
H
H
Scheme 18.
NR1
R1 P
OOEt
OEt ClCO2MeN
P
OOEt
OEt
R1 R1
MeOO
Cl
MeO
O
N
R1
R1
P
O
OEt
OEt
Cl
NH3 (7N in MeOH) toluene
CH2Cl2
MeO
O
N
R1
R1
P
O
OEt
OEt6 equiv. NH2R
MeOH, K2CO3
MeO
O
N
R1
R1
P
O
OEt
OEt
NHR
R1
R1
N
NO
R
P
O
OEtOEt
R = allyl, Pr, i-Bu, BnR1 = Et, (CH2)5
64 65
6667
Scheme 19.

422 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
phonate moiety was shielded from direct coordination by a flat porphyrin chelate [70].
Furthermore, �,�-dehydro-�-aminophosphonates are used as plant growth regulators, in particular for growth inhibition in the crops of soya and sunflower in order to improve stability and in-crease the yield [71].
5. OUTLOOK In summary, the literature describes several routes to �,�-
dehydro-�-aminophosphonic and �,�-dehydro-�-aminophosphinic acid derivatives which allow the synthesis of these compounds from various substrates. Different synthetic paths can be considered depending on the desired model of the target compound and available synthons. The most important application of the above-mentioned compounds is their use in catalytic asymmetric hydrogenation, which leads to homochiral �-aminophosphonic and �-aminophosphinic acid derivatives exhibiting a wide spectrum of biological activity. Due to the appealing application of these products of hydrogenation, there is a need for further research on the synthesis of �,�-dehydro-�-aminophosphonates and �,�-dehydro-�-aminophosphinates.
CONFLICT OF INTEREST The author(s) confirm that this article content has no conflict of
interest.
ACKNOWLEDGEMENTS
The financial help of the Ministry of Science and Higher Edu-cation of Poland (Grant No. N N204 165636) is gratefully acknowl-edged.
ABBREVIATIONS
Bn = Benzyl Boc = tert-Butoxycarbonyl DMAP = 4-N,N-dimethylaminopyridine DPPA = Diphenylphosphoryl azide NMM = N-Methylmorpholine 1-Np = (1-Naphthyl) TFA = Trifluoroacetic acid
O
NHP
ClCl
O OEtOEt
MeNH2 NH2
. H2O
N
O
PO OEt
OEt
NH
NH2
Me
Ht
O
ClPhNMe2
N
O
PO OEt
OEt
NH
NH
Me O
Ht AcOHO
NH P
O OHOH
N
NO
Ht
Me
Ht =O S
NOPh
O O O
O
68 69
70 71
Scheme 20.
P P
OO
OEtOEt
EtOEtO
N
+ R1O
H
NaH
R1
NP
OOEtOEt
R1
OOH
NO
OMe
OMe
OMe
OMe
R1
R1 =S S
Me
72 73 74
75 76
10 M HCl
Scheme 21.

Synthesis, Structure and Application of � ,�-Dehydro-�-aminophosphonic Current Organic Synthesis, 2013, Vol. 10, No. 3 423
REFERENCES[1] Kreuzfeld, H.J.; Michalik, M.; D�bler, C. Unusual amino acids VIII. Asym-
metric hydrogenation of some heteroaryl-N-CBZ and N-BOC aminocinnamic acid derivatives. Amino Acids, 1999, 16, 21-27.
[2] Kreuzfeld, H.J.; D�bler, C.; Schmidt, U.; Krause, H.W. Synthesis of non-proteinogenic (D)- or (L)-amino acids by asymmetric hydrogenation. Amino Acids, 1996, 11, 269-282.
[3] Ordó�ez, M.; Rojas-Cabrera, H.; Cativiela, C. An overview of stereoselec-tive synthesis of �-aminophosphonic acids and derivatives. Tetrahedron,2009, 65, 17-49.
[4] Ma, J.A. Catalytic asymmetric synthesis of �- and �-amino phosphonic acid derivatives. Chem. Soc. Rev., 2006, 35, 630–636.
[5] Dwars, T.; Schmidt, U.; Krause, H.; Oehme, G. One-pot preparation of N-alkoxycarbonyl-protected acrylate, phosphonate or phosphinate esters com-prises reacting N-acyl-protected esters with dicarbonate ester and then hydra-zine. DE Patent 19,840,861 A1, March 2, 2000.
[6] Blaskovich, M.A. Handbook on Syntheses of Amino Acids; University Press: New York, 2010.
[7] Hughes, A.B. Amino Acids, Peptides and Proteins in Organic Chemistry;Wiley-VCH: Weinheim, 2009.
[8] Bonauer, C.; Walenzyk, T.; König, B. �,�-Dehydroamino acids. Synthesis,2006, 1-20.
[9] Humphrey, J.M.; Chamberlin, A.R. Chemical synthesis of natural product peptides: coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev., 1997, 97, 2243-2266.
[10] Schmidt, U.; Lieberknecht, A.; Wild, J. Didehydroamino acids (DDAA) and didehydropeptides (DDP). Synthesis, 1988, 159-172.
[11] Drach, B.S.; Sviridov, E.P.; Shaturskii, Y.P. Reaction of diethyl 1-acylamido-2,2-dichlorovinylphosphonates with primary and secondary amines. Zh. Obshch. Khim., 1974, 44, 1712-1715.
[12] Scheidecker, S.; K�ckritz, A.; Schnell, M. �-Substituted phosphonates. 56. Synthesis and reactions of 1-formylamino-2,2,2-trichloroethanephos-phonates. J. Prakt. Chem., 1990, 332, 968-976.
[13] K�ckritz, A.; Schnell, M. �-Substituted phosphonates 68. �-Aminophosphonates and phosphono-substituted heterocycles from diethyl [2,2,2-trichloro-1-isocyanatoethyl]phosphonate. Phosphorus, Sulfur Silicon Relat. Elem., 1993, 83, 125-133.
[14] Stukalo, E.A.; Yureva, E.M.; Markovskii, L.N. 1-Dihalophosphonyl-vinylisocyanates. Zh. Obshch. Khim., 1980, 50, 343-347.
[15] Brovarets, V.S.; Zyuz, K.V.; Budnik, L.V.; Solodenko, V.A.; Drach, B.S. New approach to the synthesis of [1-(acylamino)alkenyl]phosphonic acids, their analogs, and their derivatives. Zh. Obshch. Khim., 1993, 63, 1259-1265.
[16] Brovarets, V.S.; Budnik, L.V.; Drach, B.S. Substitution of a carbonyl group by a phosphoryl group in unsaturated azlactones. Zh. Obshch. Khim., 1992,62, 707-708.
[17] Schmidt, U.; Oehme, G.; Krause, H. Catalytic stereoselective synthesis of �-amino phosphonic acid derivatives by asymmetric hydrogenation. Synth. Commun., 1996, 26, 777-781.
[18] Schmidt, U.; Krause, H.W.; Oehme, G.; Michalik, M.; Fischer, C. Enantiose-lective synthesis of �-aminophosphonic acid derivatives by hydrogenation. Chirality, 1998, 10, 564-572.
[19] Dwars, T.; Schmidt, U.; Fischer, C.; Grassert, I.; Kempe, R.; Fr�hlich, R.; Drauz, K.; Oehme, G. Synthesis of optically active �-aminophosphinic acids by catalytic asymmetric hydrogenation in organic solvents and aqueous mi-cellar media. Angew. Chem. Int. Ed., 1998, 37, 2851-2853.
[20] Kitamura, M.; Yoshimura, M.; Tsukamoto, M.; Noyori, R. Synthesis of �-amino phosphonic acids by asymmetric hydrogenation. Enantiomer, 1996, 1,281-303.
[21] Zo�, J. A simple preparation of diethyl 1-acylamino-1-ethenephosphonates. Synthesis, 1981, 324.
[22] Burk, M.J.; Stammers, T.A.; Straub, J.A. Enantioselective synthesis of �-hydroxy and �-amino phosphonates via catalytic asymmetric hydrogenation. Org. Lett., 1999, 1, 387-390.
[23] Quiclet-Sire, B.; Zard, S.Z.; Zhang, H. A practical access to �-phosphonoenamides. J. Organomet. Chem., 2002, 643, 404-408.
[24] Talley, J.J. Process for making chiral �-amino phosphonates selected novel chiral �-amino phosphonates. U.S. Patent 5,321,153, June 14, 1994.
[25] Sch�llkopf, U.; Hoppe, I.; Thiele, A. Asymmetric synthesis of �-aminophosphonic acids, I. Enantioselective synthesis of L-(1-aminoethyl)phosphonic acid by asymmetric catalytic hydrogenation of N-[1-(dimethoxyphosphoryl)ethenyl]-formamide. Liebigs Ann. Chem., 1985, 555-559.
[26] Wu, Y.L.; Tishler, M. Synthesis of �-amino-�-(4-imidazolyl)ethylphos-phonic acid, the phosphonoisostere of histidine. Chinese Chem. Lett., 1991,2, 95-98.
[27] Merrett, J.H.; Spurden, W.C.; Thomas, W.A.; Tong, B.P.; Whitcombe, I.W.A. The synthesis and rotational isomerism of 1-amino-2-imidazol-4-ylethylphosphonic acid [phosphonohistidine, His (P)] and 1-amino-2-imidazol-2-ylethylphosphonic acid [phosphonoisohistidine, Isohis (P)]. J. Chem. Soc., Perkin Trans. 1, 1988, 61-67.
[28] Baewska, K.; Gajda, T. A concise synthesis of diethyl 1-(tert-butoxycarbonylamino)-1-alkenylphosphonates. Tetrahedron, 2004, 60,11701-11707.
[29] Sch�llkopf, U.; Schr�der, R. Synthesen mit �-metallierten isocyaniden, XXI. Umsetzungen von �-metalliertem isocyanmethan-phosphonsäure-diäthylester mit carbonylverbindungen. Tetrahedron Lett., 1973, 14, 633-636.
[30] Sch�llkopf, U.; Schr�der, R.; Stafforst, D. Syntheses with �-metalated isocyanides, XXVII. Reactions of �-metalated diethyl isocyanomethyl- and �-isocyanobenzylphosphonates with carbonyl compounds. Liebigs Ann. Chem., 1974, 44-53.
[31] Van Speybroeck, V.; Moonen, K.; Hemelsoet, K.; Stevens, C.V.; Waroquier, M. Unexpected four-membered over six-membered ring formation during the synthesis of azaheterocyclic phosphonates: experimental and theoretical evaluation. J. Am. Chem. Soc., 2006, 128, 8468-8478.
[32] Grassert, I.; Schmidt, U.; Ziegler, S.; Fischer, C.; Oehme, G. Use of rhodium complexes with amphiphilic and nonamphiphilic ligands for the preparation of chiral �-aminophosphonic acid esters by hydrogenation in micellar media. Tetrahedron: Asymmetry, 1998, 9, 4193-4202.
[33] Holz, J.; Quirmbach, M.; Schmidt, U.; Heller, D.; Stürmer, R.; B�rner, A. Synthesis of a new class of functionalized chiral bisphospholane ligands and the application in enantioselective hydrogenations. J. Org. Chem., 1998, 63,8031-8034.
[34] Holz, J.; Stürmer, R.; Schmidt, U.; Drexler, H.J.; Heller, D.; Krimmer, H.P.; B�rner, A. Synthesis of chiral 2,5-bis(oxymethyl)-functionalized bis(phospholanes) and their application in Rh- and Ru-catalyzed enantiose-lective hydrogenations. Eur. J. Org. Chem., 2001, 4615-4624.
[35] Wassenaar, J.; Reek, J.N.H. Asymmetric hydrogenation of enamides, �-enol and �-enamido ester phosphonates catalyzed by IndolPhos-Rh complexes. J. Org. Chem., 2009, 74, 8403-8406.
[36] Wassenaar, J.; Kuil, M.; Lutz, M.; Spek, A.L.; Reek, J.N.H. Asymmetric hydrogenation with highly active IndolPhos-Rh catalysts: kinetics and reac-tion mechanism. Chem. Eur. J., 2010, 16, 6509-6517.
[37] Schmidt, U.; Dwars, T.; Grassert, I.; Oehme, G.; Fischer, C. Preparation of phosphinic and phosphonic acid derivatives potentially useful as herbicides or as antiviral or antibacterial. DE Patent 19,826,211 A1, December 16, 1999.
[38] Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Wiley & Sons: Chichester, 2000.
[39] Kafarski, P.; Lejczak, B. Aminophosphonic acids of potential medical importance. Curr. Med. Chem.-Anti-Cancer Agents, 2001, 1, 301-312.
[40] Kafarski, P.; Lejczak, B. Biological activity of aminophosphonic acids. Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193-215.
[41] Atherton, F.R.; Hassall, C.H.; Lambert, R.W. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl)phosphonic acid. J. Med. Chem., 1986, 29, 29-40.
[42] Lejczak, B.; Kafarski, P.; Sztajer, H.; Mastalerz, P. Antibacterial activity of phosphono dipeptides related to Alafosfalin. J. Med. Chem., 1986, 29, 2212-2217.
[43] Natchev, I.A. Synthesis, enzyme-substrate interaction, and herbicidal activity of phosphoryl analogues of glycine. Liebigs Ann. Chem., 1988, 861-867.
[44] Maier, L.; Diel, P.J. Synthesis, physical and biological properties of the phosphorus analogues of phenylalanine and related compounds. Phosphorus, Sulfur Silicon Relat. Elem., 1994, 90, 259-279.
[45] Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based de-sign, chemistry, and activity. J. Med. Chem., 2003, 46, 2641-2655.
[46] Liu, W.; Rogers, C.J.; Fisher, A.J.; Toney, M.D. Aminophosphonate inhibi-tors of dialkylglycine decarboxylase: structural basis for slow binding inhibi-tion. Biochemistry, 2002, 41, 12320-12328.
[47] Huang, J.; Chen, R. An overview of recent advances on the synthesis and biological activity of �-aminophosphonic acid derivatives. Heteroat. Chem.,2000, 11, 480-492.
[48] Lavielle, G.; Hautefaye, P.; Schaeffer, C.; Boutin, J.A.; Cudennec, C.A.; Pierre, A. New �-amino phosphonic acid derivatives of vinblastine: chemis-try and antitumor activity. J. Med. Chem., 1991, 34, 1998-2003.
[49] Allen, M.C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J.M. Renin inhibitors. Synthesis of transition-state analogue inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem., 1989, 32, 1652-1661.
[50] Bird, J.; De Mello, R.C.; Harper, G.P.; Hunter, D.J.; Karran, E.H.; Markwell, R.E.; Miles-Williams, A.J.; Rahman, S.S.; Ward, R.W. Synthesis of novel N-phosphonoalkyl dipeptide inhibitors of human collagenase. J. Med. Chem.,1994, 37, 158-169.
[51] Stowasser, B.; Budt, K.-H.; Jian-Qi, L.; Paymen, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett., 1992, 33, 6625-6628.
[52] Patel, D.V.; Rielly-Gauvin, K.; Ryono, D.E.; Free, C.A.; Rogers, W.L.; Smith, S.A.; DeForrest, J.M.; Oehl, R.S.; Petrillo, E.W.Jr. �-Hydroxy phosphinyl-based inhibitors of human renin. J. Med. Chem., 1995, 38, 4557-4569.

424 Current Organic Synthesis, 2013, Vol. 10, No. 3 Ku�nik et al.
[53] Kukhar, V.P.; Soloshonok, V.A.; Solodenko, V.A. Asymmetric synthesis of phosphorus analogs of amino acids. Phosphorus, Sulfur Silicon Relat. Elem.,1994, 92, 239-264.
[54] Gridnev, I.D.; Yasutake, M.; Imamoto, T.; Beletskaya, I.P. Asymmetric hydrogenation of �,�-unsaturated phosphonates with Rh-BisP* and Rh-MiniPHOS catalysts: scope and mechanism of the reaction. Proc. Natl. Acad. Sci. USA, 2004, 101, 5385-5390.
[55] Wang, D.Y.; Huang, J.D.; Hu, X.P.; Deng, J.; Yu, S.B.; Duan, Z.C.; Zheng, Z. Readily available chiral phosphine-aminophosphine ligands for highly ef-ficient Rh-catalyzed asymmetric hydrogenation of �-enol ester phosphonates and �-enamido phosphonates. J. Org. Chem., 2008, 73, 2011-2014.
[56] Zupancic, B.; Mohar, B.; Stephan, M. Impact on hydrogenation catalytic cycle of the R groups’ cyclic feature in “R-SMS-Phos”. Org. Lett., 2010, 12,3022-2025.
[57] Zhang, J.; Li, Y.; Wang, Z.; Ding, K. Asymmetric hydrogenation of �- and �-enamido phosphonates: rhodium(I)/monodentate phosphoramidite catalyst. Angew. Chem. Int. Ed., 2011, 50, 11743-11747.
[58] Goulioukina, N.S.; Makukhin, N.N.; Beletskaya, I.P. 1,3-Dipolar cycloaddi-tion of diazoalkanes onto dimethyl 1-(formylamino) ethylenephosphonate: a new route to 1-aminocyclopropanephosphonic acids and 3-phosphorylated pyrazoles. Tetrahedron, 2011, 67, 9535-9540.
[59] Brackmann, F.; de Meijere, A. Natural occurrence, syntheses, and applica-tions of cyclopropyl-group-containing �-amino acids. Part 1. 1-Aminocyclopropanecarboxylic acid and other 2,3-methanoamino acids. Chem. Rev., 2007, 107, 4493-4537.
[60] Pellicciari, R.; Marinozzi, M.; Camaioni, E.; del Carmen Nùnez, M.; Costan-tino, G.; Gasparini, F.; Giorgi, G.; Macchiarulo, A.; Subramanian, N. Spiro [2.2]pentane as a dissymmetric scaffold for conformationally constrained analogues of glutamic acid: focus on racemic 1-aminospiro[2.2]pentyl-1,4-dicarboxylic acids. J. Org. Chem., 2002, 67, 5497-5507.
[61] Erion, M.D.; Walsh, C.T. 1-Aminocyclopropanephosphonate: time-dependent inactivation of 1-aminocyclopropanecarboxylate deaminase and Bacillus stearothermophilus alanine racemase by slow dissociation behavior. Biochemistry, 1987, 26, 3417-3425.
[62] Pompei, M.; Francesco, M.E.D.; Koch, U.; Liverton, N.J.; Summa, V. Phosphorous acid analogs of novel P2-P4 macrocycles as inhibitors of HCV-NS3 protease. Bioorg. Med. Chem. Lett., 2009, 19, 2574-2578.
[63] Sheng, X.C.; Pyun, H.-J.; Chaudhary, K.; Wang, J.; Doerffler, E.; Fleury, M.; McMurtrie, D.; Chen, X.; Delaney, W.E.; Kim, C.U. Discovery of novel phosphonate derivatives as hepatitis C virus NS3 protease inhibitors. Bioorg. Med. Chem. Lett., 2009, 19, 3453-3457.
[64] Vanderhoydonck, B.; Stevens, C.V. Ring transformations of aziridinyl 2-phosphonates: synthesis of 5-phosphono-2-oxazolidinones and 5-phosphono-2-imidazolidinones. Tetrahedron, 2007, 63, 7679-7689.
[65] Golovchenko, A.V.; Solomyannyi, R.N.; Brovarets, V.S. Synthesis of C-heteryl-substituted aminomethylphosphonic acids derivatives. Russ. J. Gen. Chem., 2010, 80, 723-727.
[66] Carr, M.; Greene, L.M.; Knox, A.J.S.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Lead identification of conformationally restricted �-lactam type com-bretastatin analogues: synthesis, antiproliferative activity and tubulin target-ing effects. Eur. J. Med. Chem., 2010, 45, 5752-5766.
[67] O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and evaluation of azetidinone analogues of combretastatin A-4 as tubulin target-ing agents. J. Med. Chem., 2010, 53, 8569-8584.
[68] O’Boyle, N.M.; Greene, L.M.; Bergin, O.; Fichet, J.B.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, evaluation and structural stud-ies of antiproliferative tubulin-targeting azetidin-2-ones. Bioorg. Med. Chem., 2011, 19, 2306-2325.
[69] Marchetti, F.; Zacchini, S.; Zanotti, V. Unprecedented transformation of diiron bridging vinyliminium ligands into carboxyamido- and alkylphospho-nate- vinylalkylidenes. Eur. J. Inorg. Chem., 2012, 2456-2463.
[70] Locos, O.B.; Dahms, K.; Senge, M.O. Allenylporphyrins: a new motif on the porphyrin periphery. Tetrahedron Lett., 2009, 50, 2566-2569.
[71] Maier, L.; Diel, P.J. Phosphonyl-enamines as plant growth regulators. DE Patent 4,029,444 A1, March 28, 1991.
�
Received: May 02, 2012 Revised: June 21, 2012 Accepted: September 24, 2012
�





























