Embed Size (px)
Citation preview

The Combination of Alisertib, an Investigational Aurora Kinase
A Inhibitor, and Docetaxel Promotes Cell Death and Reduces
Tumor Growth in Preclinical Cell Models of Upper
Gastrointestinal Adenocarcinomas
Vikas Sehdev, PhD1; Ahmed Katsha, PhD1; Jeffrey Ecsedy, MD2; Alexander Zaika, PhD1,3;
Abbes Belkhiri, PhD1; and Wael El-Rifai, MD, PhD1,3
BACKGROUND: Upper gastrointestinal adenocarcinomas (UGCs) respond poorly to current chemotherapeutic regimes. The authors
and others have previously reported frequent Aurora kinase A (AURKA) gene amplification and mRNA and protein overexpression in
UGCs. The objective of the current study was to determine the therapeutic potential of alisertib (MLN8237) alone and in combination
with docetaxel in UGCs. METHODS: After treatment with alisertib and/or docetaxel, clonogenic cell survival, cell cycle analyses, West-
ern blot analyses, and tumor xenograft growth assays were carried out to measure cell survival, cell cycle progression, apoptotic pro-
tein expression, and tumor xenograft volumes, respectively. RESULTS: By using the AGS, FLO-1, and OE33 UGC cell lines, which have
constitutive AURKA overexpression and variable tumor protein 53 (p53) status, significantly enhanced inhibition of cancer cell sur-
vival was observed with alisertib and docetaxel treatment in combination (P < .001), compared with single-agent treatments. Cell
cycle analyses, after 48 hours of treatment with alisertib, produced a significant increase in the percentage of polyploidy in UGC cells
(P < .01) that was further enhanced by docetaxel (P < .001). In addition, an increase in the percentage of cells in sub-G1-phase
observed with alisertib (P < .01) was significantly enhanced with the combination treatment (P < .001). Western blot analysis demon-
strated higher induction of cleaved caspase 3 protein expression with the combined treatment compared with single-agent treat-
ments. In addition, FLO-1 and OE33 cell xenograft models demonstrated enhanced antitumor activity for the alisertib and docetaxel
combination compared with single-agent treatments (P < .001). CONCLUSIONS: The current study demonstrated that alisertib com-
bined with docetaxel can mediate a better therapeutic outcome in UGC cell lines. Cancer 2012;000:000–000. VC 2012 American Can-
cer Society.
KEYWORDS: Aurora kinase A, alisertib, docetaxel, stomach, esophagus, cancer, mitosis, apoptosis..
INTRODUCTIONUpper gastrointestinal adenocarcinomas (UGCs) (ie, adenocarcinomas of the stomach and esophagus) are associated withpoor patient survival rate because of inherent resistance to current therapeutic regimens.1,2 Global epidemiologic dataindicate that approximately 1.4 million new UGCs are diagnosed annually, resulting in approximately 1.1 milliondeaths.3 Over the past several decades, the incidence rates for distal gastric cancers have declined; however, incidence ratesfor adenocarcinoma of the gastric cardia, gastroesophageal (GE) junction, and esophagus continue to rise.3,4
Multiple prosurvival and drug-resistant genes, such as the epidermal growth factor receptor (EGFR), human epi-dermal growth factor receptor 2 (HER-2), v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-RAS), and Aurorakinase A (AURKA), mediate oncogenic signaling pathways in gastric and esophageal adenocarcinomas.5-9 Despite theavailability of several adjuvant and neoadjuvant chemotherapeutic treatment strategies, the survival rate for patientswith UGCs have only marginally improved.10 Therefore, laboratory investigations and preclinical studies specificallyaimed at developing novel targeted therapies and chemotherapeutic combinations with potent antitumor activity aredesperately needed to treat patients with UGCs.
We and others previously reported the amplification of the region of band 13 on the long arm of chromosome 20(20q13) in UGC.11,12 The 20q13 chromosomal region harbors the AURKA gene, which is frequently amplified and/oroverexpressed in several malignancies, including cancers of the bladder, breast, colon, liver, ovaries, pancreas, stomach,and esophagus.8,13 AURKA is a key cell cycle regulator that is critical for mitotic events.14,15 However, when
DOI: 10.1002/cncr.27801, Received: May 1, 2012; Revised: July 2, 2012; Accepted: August 1, 2012, Published online in Wiley Online Library
(wileyonlinelibrary.com)
Corresponding author: Wael El-Rifai, MD, PhD, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, 1255 Light Hall, 2215 Garland Avenue,
Nashville, TN 37232; Fax: (615) 322-7852; [email protected]
1Department of Surgery, Vanderbilt University Medical Center, Nashville, Tennessee; 2Translational Medicine, Millennium Pharmaceuticals, Inc., Cambridge, Massa-
chusetts; 3Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, Tennessee
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or Van-
derbilt University.
Cancer Month 00, 2012 1
Original Article

overexpressed, AURKA is a bona fide oncogene andresults in genetic instability, dedifferentiated morphol-ogy, and a poor prognosis in patients with UGCs.6,16
The overexpression of AURKA promotes cancer cellgrowth and resistance to chemotherapy by up-regulatingoncogenic signaling pathways and suppressing cell-deathmechanisms, respectively.13 In addition, AURKA over-expression induces growth-promoting and survival-promoting oncogenic signaling pathways, such as thephosphoinositide 3-kinase/protein kinase B (PI3K/AKT)and b-catenin pathways, in UGC cancer cells.13 Thedocetaxel, cisplatin, and 5-fluorouracil (DCF) chemo-therapy regimen is 1 of the most efficacious chemothera-peutic regimens against advanced gastric cancer.17 It isnoteworthy that AURKA overexpression reportedlymediates resistance against paclitaxel and cisplatin-induced cell death.18,19 The cell-death mechanisms,which are regulated by the tumor protein 53 (p53) familyof proapoptotic proteins, are activated in cancer cells aftertreatment with chemotherapeutic agents. However,mutations in the p53 function are frequently observed inUGCs. In p53-mutant cancers, the related tumor protein73 (p73) can mediate p53-like apoptotic functions andactivate apoptotic pathways after treatment with chemo-therapeutic agents.20 Also noteworthy is the fact thatAURKA overexpression in cancer suppresses p53 andp73 protein expression and function.21,22 These findingssuggest that p53-mutant UGCs with constitutively higherlevels of AURKA expression can respond poorly to chem-otherapy. Therefore, given the poor response of UGCs tocurrent therapeutic regimens; novel therapeutic strategiesthat take into account the molecular make-up of tumorsto activate cell death response are critically needed tocombat UGCs.
Alisertib (MLN8237) is an investigational small mol-ecule inhibitor developed by Millennium Pharmaceuticals,Inc. (Cambridge, Mass.) that has demonstrated the abilityto selectively inhibit AURKA and thereby induce cell cyclearrest, aneuploidy, polyploidy, mitotic catastrophe, and celldeath.8,23 Currently, alisertib is being tested in variousphase 1, 2, and 3 clinical trials for advanced solid tumorsand hematologic malignancies (http://clinicaltrials.gov/;Accessed August 26, 2012). In addition, multiple clinicaltrials have indicated that docetaxel and its combinationwith cisplatin and 5-fluorouracil have significant antitumoractivity in UGCs.17 Docetaxel is a microtubule-polymeriz-ing chemotherapeutic agent that disrupts microtubule dy-namics by binding to the b-subunit of tubulin andpromoting its polymerization.24 Consequently, docetaxelcauses cell cycle arrest in mitosis and subsequently induces
apoptosis. In the current study, we investigated the poten-tial therapeutic benefit of alisertib alone and in combina-tion with docetaxel using in vitro and in vivo cell models ofUGC with variable p53 status. We hypothesized that thealisertib and docetaxel combination treatment would causeenhanced cell cycle arrest, resulting in polyploidy and sub-sequent induction of apoptosis in UGC cancer cells, irre-spective of their p53 status.
MATERIALS AND METHODS
Cell Culture and Pharmacologic Reagents
The AGS (p53 wild type) gastric adenocarcinoma cell lineand the FLO-1/OE33 (p53 mutant) esophageal adenocar-cinoma cell lines were maintained as a monolayer culturein Dulbecco modified Eagle medium (DMEM) (Gibco,Carlsbad, Calif) cell culture medium supplementedwith 10% (volume/volume) fetal bovine serum (FBS)(Gibco).25 All cell lines were evaluated weekly to ascertainconformity to the appropriate in vitro morphologiccharacteristics.26 MLN8237 (alisertib) was provided byMillennium Pharmaceuticals, Inc., and alisertib stocksolutions for in vitro and in vivo studies were preparedaccording to our previously reported methods.8 Docetaxel(Sanofi Aventis, Bridgewater, NJ) stock solution (11.6mM) prepared in 13% ethanol (volume/volume) was pro-vided by the TVC Outpatient Pharmacy at VanderbiltUniversity Medical Center (Nashville, Tenn).
Clonogenic Cell Survival Assay
AGS, FLO-1 and OE33 cells were seeded at 5000 cellsper well, respectively, onto 6-well plates overnight andthen were treated with various concentrations of alisertib(0.25 lM, 0.5 lM, 1.0 lM, 2.0 lM, and 5.0 lM) for 24hours. After treatment, UGC cell survival was determinedaccording to our previously reported protocol.8 Briefly,after treatment, the cells were incubated in drug-free cellculture medium for 10 days. Subsequently, the cells werefixed with 2% paraformaldehyde solution, stained withcrystal violet dye solution, and cell survival was quantifiedby measuring the dye signal in each well using ImageJanalysis software (National Institutes of Health, Bethesda,Md). In addition, we selected an alisertib dose close to the50% inhibitory concentration (0.5 lM) and treated thecells with alisertib (0.5 lM) and/or with docetaxel (0.5nM, 1.0 nM, or 5.0 nM) for 24 hours.
Cell Cycle Analysis
AGS and FLO-1 cells were treated with alisertib (0.1lM) and/or docetaxel (0.5 nM) in cell culture medium(2.5% FBS) for 24 hours and 48 hours, respectively.OE33 cells were treated with alisertib (0.5 lM) and/or
Original Article
2 Cancer Month 00, 2012

docetaxel (1.0 nM) in cell culture medium (2.5% FBS)for 24 hours and 48 hours, respectively. After treatment,supernatant media was collected, and adherent cells weretrypsinized. The supernatant and trypsinized cells werecentrifuged together at �2000g at 4�C for 10 minutes.Then, the cells were resuspended in 1 mL propidiumiodide (PI) solution (PI 50 lg/mL and RNase 1 lg/mLin 1 � phosphate-buffered saline) and incubated at roomtemperature in the dark for 30 minutes. Subsequently,the cells were analyzed with the BD LSR III flowcytometer (BD Biosciences, San Jose, Calif), and thedata were processed with BD FACS Diva software (BDBiosciences) at the VMC Flow Cytometry SharedResource, Vanderbilt Ingram Cancer Center.
Western Blot Analysis
AGS, FLO-1, and OE33 cancer cells were plated over-night at 30% confluence in cell culture medium (10%FBS). AGS cells were treated with alisertib (0.25 lM)and/or docetaxel (1.0 nM), FLO-1 cells were treated withalisertib (0.1 lM) and/or docetaxel (0.5 nM), and OE33cells were treated with alisertib (0.5 lM) and/or docetaxel(1.0 nM) for 48 hours in cell culture medium supple-mented with 2.5% FBS. After treatment, cell lysates wereprepared and evaluated for total and phosphorylated (p-)proteins; p-AURKA (threonine 288), AURKA, cleavedcaspase 3, and b-actin (Cell Signaling Technology, Bev-erly, Mass), according to standard protocols.27
In Vivo Tumor Xenograft Inhibition
Four million FLO-1 or OE33 cells suspended in a 200-lL DMEM-Matrigel mixture (50% DMEM supple-mented with 10% FBS and 50% Matrigel) were injectedinto the flank regions of female athymic nude-Foxn1 nu/nu mice (Harlan Laboratories Inc., Indianapolis, Ind).The tumors were allowed to grow until they measured200 mm3 in size before the treatment was started withdaily alisertib (30 mg/kg, orally) and/or once-weeklydocetaxel (10 mg/kg as an intraperitoneal injection) for 3weeks. Tumor xenografts were measured every alternateday, and tumor size was calculated according to the fol-lowing formula: Tvol ¼ L�W2 � 0.5, in which Tvol is tu-mor volume, L is tumor length, andW is tumor width.23
Immunohistochemistry
After 21 days of animal treatment, the tumors were iso-lated, and an immunohistochemical analysis was carriedout to measure Ki-67 and cleaved caspase 3 proteinexpression levels, as previously reported.8 Protein expres-sion was scored using a composite expression score (CES)that was determined by using the formula ‘‘CES ¼ 4(in-tensity � 1) þ frequency,’’ as described previously, with
intensity measured on a scale from 0 to 3 and frequencymeasured on a scale from 0 to 4.28
Statistical Analysis
Data are presented as means � standard error of themean (SEM). All in vitro experiments were performed intriplicate. A 1-way analysis of variance (ANOVA) withTukey post hoc analysis was used to demonstrate statisti-cal differences between control groups and treatmentgroups at the treatment endpoints. A 2way ANOVA withBonferroni post hoc analysis was used to demonstrate sta-tistical differences between various treatment groups andcell cycle stages. For tumor xenograft data, a 2-wayANOVA (time point-matched analysis) with Bonferronipost-test was used to compare the ‘‘mean tumor size’’ of atreatment group on any given treatment day with the‘‘mean tumor size’’ of another other treatment groups onthe corresponding treatment day. All statistical analysesdescribed above were carried out using GraphPad Prism5 software (GraphPad Software Inc., La Jolla, Calif). AllP values � .05 were considered statistically significantand are marked in the figures, in which a single asteriskindicates P < .05, and double asterisks indicate P < .01.
RESULTS
Alisertib Significantly EnhancedDocetaxel-Mediated Inhibition of Cell Survival
Both FLO1 and OE33 cell lines exhibited gene amplifica-tion and overexpression of AURKA at the mRNA andprotein levels.8,29 Similarly, AGS cells exhibited anincrease in AURKA DNA copy number (2.26-fold) andmRNA level (4.98-fold; data not shown). Therefore, thesecell models mimic the previously reported in vivo data onAURKA overexpression in primary UGCs.30 The clono-genic cell-survival assay data indicated that alisertib (0.5lM) or docetaxel (1.0 nM) as single-agent treatmentsdecreased the percentage of AGS cells that survived (aliser-tib 0.5 lM: 45.5% � 4.6% AGS cell survival [mean �SEM]; P < .01; docetaxel 1.0 nM, 53.6% � 1.8% AGScell survival; P < .01) (Fig. 1A), FLO-1 cells (alisertib 0.5lM: 45.5% � 4.6% FLO-1 cell survival; P < .01; doce-taxel 1.0 nM: 70.1% � 5.6% FLO-1 cell survival; P <
.05) (Fig. 1B), and OE33 cells (alisertib 0.5 lM: 45.5%� 4.6% OE33 cell survival; P < .01; docetaxel 1.0 nM:32.4% � 3.5% OE33 cell survival; P < .01) (Fig. 1C).Treatment with the alisertib (0.5 lM) and docetaxel (1.0nM) combination led to a significantly enhanced inhibi-tion of the percentage of surviving AGS cells (alisertib 0.5lM plus docetaxel 1.0 nM: 5.5% � 0.7% AGS cell sur-vival; P < .01) (Fig. 1A), FLO-1 cells (alisertib 0.5 lM
Alisertib & Docetaxel Inhibit Tumor Growth/Sehdev et al
Cancer Month 00, 2012 3

plus docetaxel 1.0 nM: 4.5%� 0.5% FLO-1 cell survival;P < .01) (Fig. 1B), and OE33 cells (alisertib 0.5 lM plusdocetaxel 1.0 nM: 13% � 1.8% OE33 cell survival; P <
.01) (Fig. 1C). These results suggest that the combinationof alisertib with docetaxel may have a significantly greaterinhibitory effect on UGC cell survival.
Alisertib-Enhanced Docetaxel InducedPolyploidy and Apoptosis in UpperGastrointestinal Adenocarcinoma Cells
By using the AGS, FLO-1, and OE33 cell lines as in vitromodels of UGC to study the effect of alisertib and doce-taxel on cell cycle progression, treatment with alisertib
Figure 1. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) significantly inhibits cell survival in 3 celllines with constitutive Aurora kinase A overexpression and variable p53 status. (A-C) Cell survival assay data indicated significantcell survival inhibition in the cell lines (A) AGS, (B) FLO-1, and (C) OE33 after combined treatment with MLN and DOCE. The AGS,FLO-1, and OE33 cell lines were treated with MLN 0.5 lM (MLN 0.5) and/or DOCE 0.5 nM (DOCE 0.5), 1.0 nM (DOCE 1.0), or 5.0nM (DOCE 5.0) for 24 hours and incubated in drug-free medium for 10 days. Combined treatment with MLN 0.5 lM and DOCE1.0 nM significantly suppressed the survival of all 3 cell lines. CV indicates control vehicle; �, negative; þ, positive. A single aster-isk indicates P < .05; double asterisks; P < .01. CV indicates control vehicle.
Original Article
4 Cancer Month 00, 2012

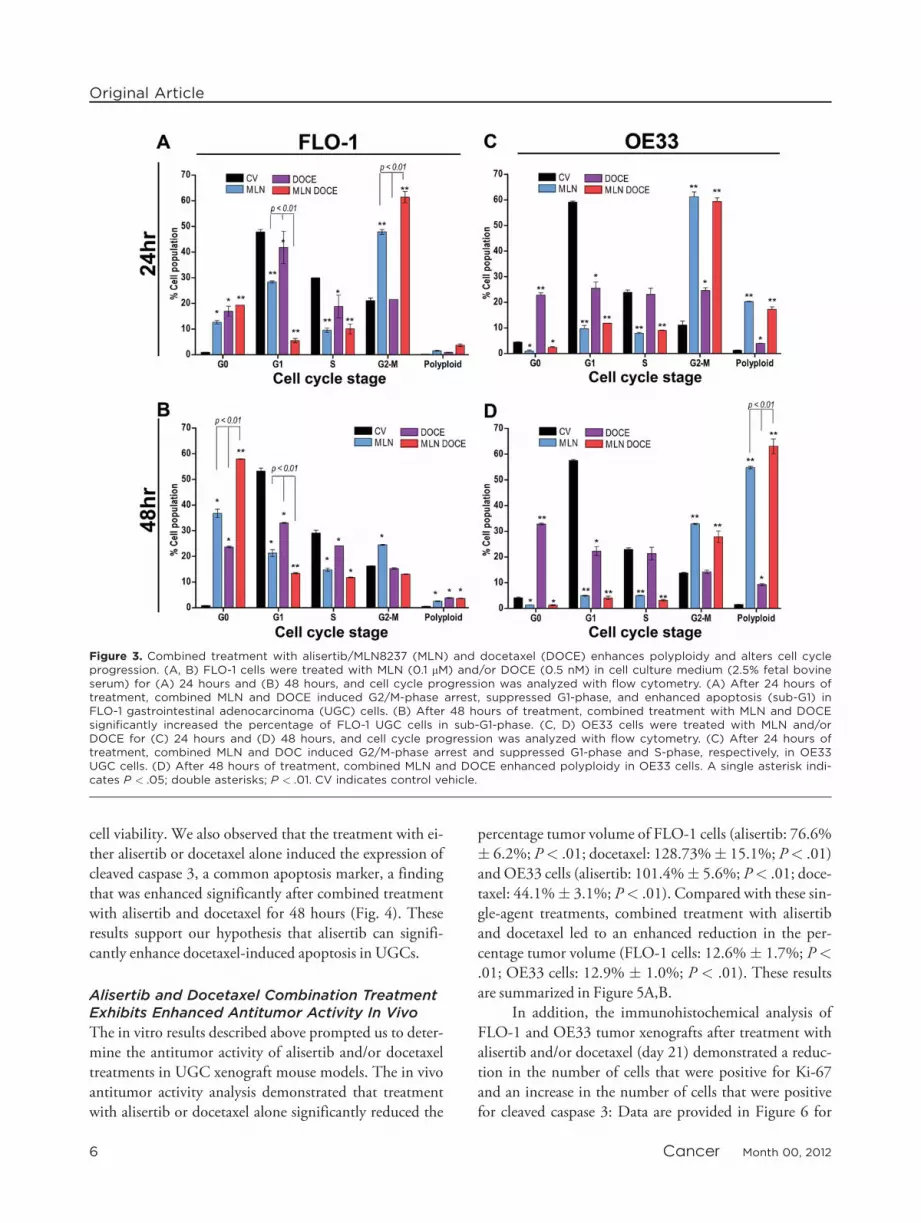
alone or in combination with docetaxel for 24 hours sig-nificantly reduced the percentage of cells in G1-phase andS-phase and induced a significant delay in the transitionfrom G2-phase to M-phase in AGS and OE33 cells(Figs. 2A, 3C). The 24-hour treatment with alisertib alonehad a similar effect on G1-phase, S-phase, and the G2-phase to M-phase transition in FLO-1 cells; however, thiseffect was significantly more pronounced after 24 hours ofcombined treatment with combined alisertib and doce-taxel (Fig. 3A). In addition, treatment with alisertib alonefor 24 hours significantly increased the percentage of pol-yploid cells, which was enhanced further by combined ali-sertib and docetaxel treatment in AGS and FLO-1 cells(Figs. 2A, 3A). At the 48-hour time point, FLO-1 cellswith that were treated with either alisertib or docetaxel asa single-agent demonstrated an increase in the percentageof cells in sub-G1-phase (P < .05) (Fig. 3B). This effectwas significantly enhanced in FLO-1 cells that received
the combined treatment (P < .01) (Fig. 3B). The cellcycle analyses indicate that FLO-1 cells were more sensi-tive to alisertib and/or docetaxel treatments, as evidencedby an increase in the number of cells in sub-G1-phase af-ter 24-hour and 48-hour treatment. In addition, 48-hourtreatment with alisertib and docetaxel enhanced poly-ploidy in AGS and OE33 UGC cells. These findings arein agreement with previously published reports, in whichinvestigators observed that docetaxel-induced microtu-bule stabilization impaired mitosis, generating aneuploidand tetraploid cells that subsequently underwent apopto-tic cell death.24,31,32 Our data suggest that the combina-tion treatment promotes polyploidy early on and,depending on the cell line, polyploidy likely leads to celldeath (sub-G1) either early on (FLO-1 cells) or at latertime points (AGS and OE33 cells). These findings pro-vide a plausible explanation and support for the clono-genic cell survival assay results that measured long-term
Figure 2. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) enhances polyploidy and alters cell cycleprogression. (A, B) AGS cells were treated with MLN (0.1 lM) and/or DOCE (0.5 nM) for (A) 24 hours and (B) 48 hours, and cellcycle progression was analyzed with flow cytometry. After treatment for (A) 24 hours and (B) 48 hours, MLN (0.1 lM) in combi-nation with DOCE (0.5 nM) significantly enhanced polyploidy in AGS cells. A single asterisk indicates P < .05; double asterisks; P< .01. CV indicates control vehicle; PI-A, indicates propidium iodide.
Alisertib & Docetaxel Inhibit Tumor Growth/Sehdev et al
Cancer Month 00, 2012 5
cell viability. We also observed that the treatment with ei-ther alisertib or docetaxel alone induced the expression ofcleaved caspase 3, a common apoptosis marker, a findingthat was enhanced significantly after combined treatmentwith alisertib and docetaxel for 48 hours (Fig. 4). Theseresults support our hypothesis that alisertib can signifi-cantly enhance docetaxel-induced apoptosis in UGCs.
Alisertib and Docetaxel Combination TreatmentExhibits Enhanced Antitumor Activity In Vivo
The in vitro results described above prompted us to deter-mine the antitumor activity of alisertib and/or docetaxeltreatments in UGC xenograft mouse models. The in vivoantitumor activity analysis demonstrated that treatmentwith alisertib or docetaxel alone significantly reduced the
percentage tumor volume of FLO-1 cells (alisertib: 76.6%� 6.2%; P< .01; docetaxel: 128.73%� 15.1%; P< .01)and OE33 cells (alisertib: 101.4%� 5.6%; P< .01; doce-taxel: 44.1%� 3.1%; P< .01). Compared with these sin-gle-agent treatments, combined treatment with alisertiband docetaxel led to an enhanced reduction in the per-centage tumor volume (FLO-1 cells: 12.6%� 1.7%; P<
.01; OE33 cells: 12.9% � 1.0%; P < .01). These resultsare summarized in Figure 5A,B.
In addition, the immunohistochemical analysis ofFLO-1 and OE33 tumor xenografts after treatment withalisertib and/or docetaxel (day 21) demonstrated a reduc-tion in the number of cells that were positive for Ki-67and an increase in the number of cells that were positivefor cleaved caspase 3: Data are provided in Figure 6 for
Figure 3. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) enhances polyploidy and alters cell cycleprogression. (A, B) FLO-1 cells were treated with MLN (0.1 lM) and/or DOCE (0.5 nM) in cell culture medium (2.5% fetal bovineserum) for (A) 24 hours and (B) 48 hours, and cell cycle progression was analyzed with flow cytometry. (A) After 24 hours oftreatment, combined MLN and DOCE induced G2/M-phase arrest, suppressed G1-phase, and enhanced apoptosis (sub-G1) inFLO-1 gastrointestinal adenocarcinoma (UGC) cells. (B) After 48 hours of treatment, combined treatment with MLN and DOCEsignificantly increased the percentage of FLO-1 UGC cells in sub-G1-phase. (C, D) OE33 cells were treated with MLN and/orDOCE for (C) 24 hours and (D) 48 hours, and cell cycle progression was analyzed with flow cytometry. (C) After 24 hours oftreatment, combined MLN and DOC induced G2/M-phase arrest and suppressed G1-phase and S-phase, respectively, in OE33UGC cells. (D) After 48 hours of treatment, combined MLN and DOCE enhanced polyploidy in OE33 cells. A single asterisk indi-cates P < .05; double asterisks; P < .01. CV indicates control vehicle.
Original Article
6 Cancer Month 00, 2012

FLO-1 xenografts, and similar results were obtained inOE33 xenografts. In concordance with the tumor growthresults (Fig. 5), these immunostaining patterns were moresignificant with the combined treatment (P < .01) thanwith the single-agent treatments (P< .05) (Fig. 6). There-fore, the in vivo data indicate that combined treatmentwith alisertib and docetaxel has enhanced antitumor activ-ity in UGC tumor xenograft models.
DISCUSSIONDespite novel therapeutic advancements, improvement inthe survival rate of patients with UGC has been marginal,suggesting the presence of unique, active, intrinsic mecha-nisms that impart resistance to chemotherapeutic agentsin UGCs.33,34 AURKA is frequently overexpressed and/oramplified in various cancers, including UGCs.8,13 Recentreports suggest that AURKA can induce chemotherapeu-tic resistance and regulate several key signaling pathwaysin cancer cells, suggesting its role as a central node in can-cer cell signaling.13 Docetaxel has demonstrated signifi-cant in vitro and in vivo antitumor activity against avariety of UGC cell lines.35
In the current study, we used UGC cell lines todetermine the therapeutic response of the recently devel-oped AURKA selective inhibitor alisertib as a single agentand in combination with docetaxel. Although previous invitro studies with Aurora kinase inhibitors have demon-strated their antitumor activity in combination with other
chemotherapeutic agents, such as cisplatin, docetaxel,nilotinib, and vorinostat,8,36-38 alisertib, an investiga-tional small-molecule AURKA inhibitor currently in clin-ical development, has not been tested in combinationwith docetaxel in gastrointestinal cancer models. In thisregard, our current results suggest a potential therapeuticbenefit of combined treatment with alisertib anddocetaxel.
The p53 gene is frequently mutated in variouscancers in which p53-mutant tumors exhibit inherentresistance to several chemotherapeutic drugs.39 This is ofparticular significance in UGC therapeutics; because, inaddition to AURKA overexpression, a high frequency ofdefective p53 signaling caused by mutation or deletion isobserved in UGCs, presenting a formidable clinical chal-lenge.40 In this study, we used p53-mutant (FLO-1 andOE33) and wild-type (AGS) cell lines and obtained simi-lar results. It is noteworthy that both in vitro and in vivomodels suggested a promising therapeutic potential forthe alisertib and docetaxel combination, as indicated bysuppressed cell survival in vitro (P< .001) and significantregression of tumor growth in vivo (P< .001). On the ba-sis of our findings, we suggest that AURKA-targeted ther-apy, either alone or in combination with docetaxel, iseffective independent of p53 status. Our data indicatethat the therapeutic effect is largely because of the induc-tion of aberrant mitosis, leading to polyploidy andsubsequent apoptosis. However, others factors, such as
Figure 4. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) significantly enhances apoptotic markerexpression. The cell lines (A) AGS, (B), FLO-1, and (C) OE33 were treated with MLN and/or DOCE for 48 hours. Combined treat-ment with MLN and DOCE significantly enhanced the expression of cleaved caspase 3 in all 3 cell lines. CV indicates control vehi-cle; AURKA, Aurora kinase A; p-AURKA, phosphorylated Aurora kinase A.
Alisertib & Docetaxel Inhibit Tumor Growth/Sehdev et al
Cancer Month 00, 2012 7

suppressed proliferation and nonapoptotic forms of celldeath, also may be occurring. These findings are timelygiven the finding that alisertib is being tested actively invarious phase 1, 2, and 3 clinical trials (http://clinical-trials.gov/; [accessed August 26, 2012]).
AURKA is a serine/threonine kinase that facilitatesaccurate mitosis by regulating vital cell cycle events duringvarious stages of mitosis.14,15 It has been demonstratedthat AURKA inhibition induces aneuploidy, polyploidy,and mitotic catastrophe in cells.36 After treatment withalisertib, we observed a significant increase in the percent-age of cells with polyploidy. This suggests that mitotic ca-tastrophe remains 1 of the predominant functions ofalisertib, a finding that is in agreement with previouslypublished data.8,36 Docetaxel-based combination chemo-therapeutic regimens like DCF are widely used for thetreatment of advanced gastric cancers.17 Docetaxel-induced defects in spindle formation and function (tubu-
lin polymerization) activate the spindle assembly check-point (SAC) and subsequently result in aneuploidy,polyploidy, and/or apoptosis.24 It is worth noting previ-ous reports that AURKA overexpression was able to over-ride the SAC and impart resistance to paclitaxel in HEK-293 cells, suggesting that AURKA overexpression is criti-cal for resistance to cell cycle inhibitors.19 Treatment withdocetaxel increased the percentage of cells in the sub-G1-phase, indicating late-stage cell death, which conforms tothe finding that docetaxel-induced spindle defects activateSAC, which subsequently activates apoptotic pathways.24
Treatment with alisertib alone and in combination withdocetaxel induced G2-M-phase arrest in vitro. It is knownthat AURKA regulates G2-phase to M-phase transitions,and its inhibition should result in G2/M-phase arrest.41
At low treatment concentrations, docetaxel-induced SACis transient and weak, an effect that can be easily overrid-den by overexpressed AURKA, as previously reported in
Figure 5. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) exhibits enhanced antitumor activity in vivo.FLO-1 and OE33 tumor xenografts were treated with MLN (30 mg/kg) and/or DOCE (10 mg/kg) for 21 days, and tumor size wasmeasured every other day. (A, B) The data indicate that combined treatment with MLN (30 mg/kg) and DOCE (10 mg/kg) hadsignificantly enhanced antitumor activity against FLO-1 and OE33 tumor xenografts. A single asterisk indicates P < .05; doubleasterisks; P < .01.
Original Article
8 Cancer Month 00, 2012

HeLa cells.19 However, alisertib-mediated specific inhibi-tion of AURKA may sensitize AURKA-overexpressingcells to docetaxel, resulting in prolonged activation ofSAC that is subsequently translated into accelerated mi-totic slippage, polyploidy, and cell death.31 After a short,48-hour treatment, our results demonstrated an increasein the percentage of polyploid cells in AGS and OE33cells, whereas an increase in apoptotic cells was observedin FLO-1 cells. These data suggest that FLO-1 cells arerelatively more sensitive to AURKA inhibition comparedwith AGS and OE33 cells when treated with alisertibalone, an effect that is further enhanced by docetaxel.
Although treatment of AGS and OE33 cells with doce-taxel alone for 48 hours increased the percentage of apo-ptotic sub-G1-phase cells, even higher percentages ofpolyploid cells were always observed after treatment withcombined alisertib and docetaxel. It is well documentedthat combination chemotherapy is a common and effec-tive therapeutic approach in the treatment of UGCs.17 Inour study, combined alisertib and docetaxel treatmentinduced a higher percentage of polyploidy that, subse-quently, resulted in increased apoptosis, as indicated bythe significantly enhanced expression of cleaved caspase 3protein levels in AGS, FLO-1, and OE33 cells. In
Figure 6. Combined treatment with alisertib/MLN8237 (MLN) and docetaxel (DOCE) suppresses proliferation and enhances apo-ptotic marker expression in FLO-1 tumor xenografts. FLO-1 tumor xenografts were treated with MLN (30 mg/kg) and/or DOCE(10 mg/kg) for 21 days. Subsequently, tumors were isolated and immunohistochemical analyses were done to measure Ki-67 andcleaved caspase 3 expression. (A) The data indicate that combined treatment with MLN (30 mg/kg) and DOCE (10 mg/kg) sig-nificantly inhibited Ki-67 expression in FLO-1 tumor xenografts. C indicates control. (B) Combination treatment with MLN (30mg/kg) and DOCE (10 mg/kg) exhibited enhanced cleaved caspase 3 protein expression in FLO-1 tumor xenografts. A single as-terisk indicates P < .05; double asterisks; P < .01.
Alisertib & Docetaxel Inhibit Tumor Growth/Sehdev et al
Cancer Month 00, 2012 9

addition, compared with single-agent treatments, thecombination treatment significantly reduced Ki-67 levelsand enhanced cleaved caspase 3 protein levels in tumorxenografts, providing additional evidence of the improvedantitumor activity of this regimen.
In conclusion, the combination of alisertib anddocetaxel results in significantly enhanced antitumoractivity in cell line models, possibly mediated by apopto-tic pathways induced after the activation of SAC. Thus,as clinical trials for alisertib are being actively conducted,our current findings provide a credible rationale forevaluating AURKA-targeted therapy in combinationwith docetaxel as a therapeutic approach for the treat-ment of UGCs.
FUNDING SOURCESThis study was supported by grants from the National Institute ofHealth; R01CA131225 (Dr. El-Rifai), VICTR pilot project sup-port from Vanderbilt CTSA grant UL1 RR024975; VanderbiltSpecialized Programs of Research Excellence (SPORE) in Gastroin-testinal Cancer (P50 CA95103), Vanderbilt Ingram Cancer Center(P30 CA68485) and the Vanderbilt Digestive Disease ResearchCenter (DK058404).
CONFLICT OF INTEREST DISCLOSURESThe authors made no disclosures.
REFERENCES1. Hohenberger P, Gretschel S. Gastric cancer. Lancet. 2003;362:305-
315.2. Reim D, Gertler R, Novotny A, et al. Adenocarcinomas of the
esophagogastric junction are more likely to respond to preoperativechemotherapy than distal gastric cancer. Ann Surg Oncol. 2012;19:2108-2118.
3. Kamangar F, Dores GM, Anderson WF. Patterns of cancer inci-dence, mortality, and prevalence across 5 continents: defining prior-ities to reduce cancer disparities in different geographic regions ofthe world. J Clin Oncol. 2006;24:2137-2150.
4. Devesa SS, Blot WJ, Fraumeni JF. Jr Changing patterns in the inci-dence of esophageal and gastric carcinoma in the United States.Cancer. 1998;83:2049-2053.
5. Cronin J, McAdam E, Danikas A, et al. Epidermal growth factorreceptor (EGFR) is overexpressed in high-grade dysplasia and ade-nocarcinoma of the esophagus and may represent a biomarker ofhistological progression in Barrett’s esophagus (BE). Am J Gastroen-terol. 2011;106:46-56.
6. Rugge M, Fassan M, Zaninotto G, et al. Aurora kinase A in Bar-rett’s carcinogenesis. Hum Pathol. 2010;41:1380-1386.
7. Deng N, Goh LK, Wang H, et al. A comprehensive survey ofgenomic alterations in gastric cancer reveals systematic patterns ofmolecular exclusivity and co-occurrence among distinct therapeutictargets. Gut. 2012;61:673-684.
8. Sehdev V, Peng D, Soutto M, et al. The aurora kinase A inhibitorMLN8237 enhances cisplatin-induced cell death in esophageal ade-nocarcinoma Cells. Mol Cancer Ther. 2012;11:763-774.
9. Lord RV, O’Grady R, Sheehan C, Field AF, Ward RL. K-ras codon12 mutations in Barrett’s oesophagus and adenocarcinomas of theoesophagus and oesophagogastric junction. J Gastroenterol Hepatol.2000;15:730-736.
10. Matuschek C, Bolke E, Peiper M, et al. The role of neoadjuvantand adjuvant treatment for adenocarcinoma of the upper gastroin-testinal tract. Eur J Med Res. 2011;16:265-274.
11. Rygiel AM, Milano F, Ten Kate FJ, et al. Gains and amplificationsof c-myc, EGFR, and 20q13 loci in the no dysplasia-dysplasia-ade-nocarcinoma sequence of Barrett’s esophagus. Cancer Epidemiol Bio-markers Prev. 2008;17:1380-1385.
12. El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Mietti-nen M. DNA sequence copy number changes in gastrointestinalstromal tumors: tumor progression and prognostic significance.Cancer Res. 2000;60:3899-3903.
13. Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinaseinhibitors—rising stars in cancer therapeutics? Mol Cancer Ther.2010;9:268-278.
14. Berdnik D, Knoblich JA. Drosophila Aurora-A is required for cen-trosome maturation and actin-dependent asymmetric protein local-ization during mitosis. Curr Biol. 2002;12:640-647.
15. Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an inter-acting activator, the LIM protein Ajuba, are required for mitoticcommitment in human cells. Cell. 2003;114:585-598.
16. Sakakura C, Hagiwara A, Yasuoka R, et al. Tumour-amplified kinaseBTAK is amplified and overexpressed in gastric cancers with possibleinvolvement in aneuploid formation. Br J Cancer. 2001; 84:824-831.
17. Van Cutsem E, Van de Velde C, Roth A, et al. Expert opinion onmanagement of gastric and gastro-oesophageal junction adenocarci-noma on behalf of the European Organisation for Research andTreatment of Cancer (EORTC)-Gastrointestinal Cancer Group.Eur J Cancer. 2008;44:182-194.
18. Sumi K, Tago K, Kasahara T, Funakoshi-Tago M. Aurora kinase Acritically contributes to the resistance to anti-cancer drug cisplatinin JAK2 V617F mutant-induced transformed cells. FEBS Lett.2011;585:1884-1890.
19. Anand S, Penrhyn-Lowe S, Venkitaraman AR. Aurora-A amplifica-tion overrides the mitotic spindle assembly checkpoint, inducing re-sistance to Taxol. Cancer Cell. 2003;3:51-62.
20. Alsafadi S, Tourpin S, Andre F, Vassal G, Ahomadegbe JC. P53family: at the crossroads in cancer therapy. Curr Med Chem.2009;16:4328-4344.
21. Liu Q, Kaneko S, Yang L, et al. Aurora-A abrogation of p53 DNAbinding and transactivation activity by phosphorylation of serine215. J Biol Chem. 2004;279:52175-52182.
22. Dar AA, Belkhiri A, Ecsedy J, Zaika A, El-Rifai W. Aurora kinaseA inhibition leads to p73-dependent apoptosis in p53-deficient can-cer cells. Cancer Res. 2008;68:8998-9004.
23. Gorgun G, Calabrese E, Hideshima T, et al. A novel Aurora-Akinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrestin multiple myeloma. Blood. 2010;115:5202-5213.
24. Hernandez-Vargas H, Palacios J, Moreno-Bueno G. Telling cellshow to die: docetaxel therapy in cancer cell lines. Cell Cycle.2007;6:780-783.
25. Soussi T, Asselain B, Hamroun D, et al. Meta-analysis of the p53 muta-tion database for mutant p53 biological activity reveals a methodologi-cal bias in mutation detection. Clin Cancer Res 2006;12:62-69.
26. Boonstra JJ, van Marion R, Beer DG, et al. Verification andunmasking of widely used human esophageal adenocarcinoma celllines. J Natl Cancer Inst. 2010;102:271-274.
27. Soutto M, Belkhiri A, Piazuelo MB, et al. Loss of TFF1 is associatedwith activation of NF-kappaB-mediated inflammation and gastric ne-oplasia in mice and humans. J Clin Invest. 2011;121: 1753-1767.
28. Mukherjee K, Peng D, Brifkani Z, et al. Dopamine and cAMPregulated phosphoprotein MW 32 kDa is overexpressed in earlystages of gastric tumorigenesis. Surgery. 2010;148:354-363.
29. Fichter CD, Herz C, Munch C, Opitz OG, Werner M, LassmannS. Occurrence of multipolar mitoses and association with aurora-A/-B kinases and p53 mutations in aneuploid esophageal carcinomacells [serial online]. BMC Cell Biol. 2011;12:13.
30. Dar AA, Zaika A, Piazuelo MB, et al. Frequent overexpression of Au-rora Kinase A in upper gastrointestinal adenocarcinomas correlateswith potent antiapoptotic functions. Cancer. 2008;112: 1688-1698.
31. Wysong DR, Chakravarty A, Hoar K, Ecsedy JA. The inhibition ofAurora A abrogates the mitotic delay induced by microtubule per-turbing agents. Cell Cycle. 2009;8:876-888.
Original Article
10 Cancer Month 00, 2012

32. Weaver BA, Cleveland DW. Decoding the links between mitosis,cancer, and chemotherapy: the mitotic checkpoint, adaptation, andcell death. Cancer Cell. 2005;8:7-12.
33. Kubota E, Kataoka H, Tanaka M, et al. ERas enhances resistanceto CPT-11 in gastric cancer. Anticancer Res. 2011;31:3353-3360.
34. Hildebrandt MA, Yang H, Hung MC, et al. Genetic variations inthe PI3K/PTEN/AKT/mTOR pathway are associated with clinicaloutcomes in esophageal cancer patients treated with chemoradio-therapy. J Clin Oncol. 2009;27:857-871.
35. Tanaka M, Obata T, Sasaki T. Evaluation of antitumour effects ofdocetaxel (Taxotere) on human gastric cancers in vitro and in vivo.Eur J Cancer. 1996;32A:226-230.
36. Qi W, Cooke LS, Liu X, et al. Aurora inhibitor MLN8237 incombination with docetaxel enhances apoptosis and anti-tumoractivity in mantle cell lymphoma. Biochem Pharmacol. 2011; 81:881-890.
37. Kelly KR, Ecsedy J, Medina E, et al. The novel Aurora A kinase in-hibitor MLN8237 is active in resistant chronic myeloid leukemiaand significantly increases the efficacy of nilotinib. J Cell Mol Med.2011;15:2057-2070.
38. Kretzner L, Scuto A, Dino PM, et al. Combining histone deacety-lase inhibitor vorinostat with Aurora kinase inhibitors enhanceslymphoma cell killing with repression of c-Myc, hTERT, andmicroRNA levels. Cancer Res. 2011;71:3912-3920.
39. Koike M, Fujita F, Komori K, et al. Dependence of chemotherapyresponse on p53 mutation status in a panel of human cancer linesmaintained in nude mice. Cancer Sci. 2004;95:541-546.
40. Oki E, Zhao Y, Yoshida R, et al. The difference in p53 mutationsbetween cancers of the upper and lower gastrointestinal tract. Diges-tion. 2009;79(suppl 1):33-39.
41. Qin L, Tong T, Song Y, Xue L, Fan F, Zhan Q. Aurora-A interactswith cyclin B1 and enhances its stability. Cancer Lett. 2009;275:77-85.
Alisertib & Docetaxel Inhibit Tumor Growth/Sehdev et al
Cancer Month 00, 2012 11





























