Embed Size (px)
Citation preview

pubs.acs.org/Organometallics Published on Web 09/11/2009 r 2009 American Chemical Society
5968 Organometallics 2009, 28, 5968–5981
DOI: 10.1021/om9006888
The Mechanism of the Catalytic Functionalization of Haloalkanes by
Carbene Insertion: An Experimental and Theoretical Study
Juan Urbano,‡ Ataualpa A. C. Braga,† Feliu Maseras,*,† Eleuterio �Alvarez,§
M. Mar Dıaz-Requejo,*,‡ and Pedro J. P�erez*,‡
‡Laboratorio de Cat�alisis Homog�enea, Departamento de Quımica y Ciencia de los Materiales, UnidadAsociada al CSIC, Campus de El Carmen s/n, Universidad de Huelva, 21007-Huelva, Spain, †Institute
of Chemical Research of Catalonia (ICIQ), 43007 Tarragona, Catalonia, Spain, and §Instituto deInvestigaciones Quımicas, CSIC-Universidad de Sevilla, Avenida Am�erico Vespucio 49, 41092 Sevilla, Spain
Received August 4, 2009
Carbon-halogen (C-X) bonds (X=Cl, Br) can be easily functionalized with ethyl diazoacetate(N2CHCO2Et) in the presence of silver-based catalysts containing the TpxAg core (Tpx=hydro-trispyrazolylborate ligand). Polyhalomethanes are converted into products derived from the formalinsertion of the carbene CHCO2Et units into the C-X bond. In the case of monohaloalkanes(C4-C6), cleavage of the C-X bond is observed, with formation of XCH2CO2Et and thecorresponding olefin. Experimental evidence and theoretical calculations have led to the proposalof a novel mechanism to account for these transformations, in which the metal participates along thepathway in all the reaction steps. Among the experimental data, the first example of ametal-induced,asymmetric functionalization of a C-Cl bond by carbene insertion is included (ee=14 ( 2%).
Introduction
The activation of carbon-chlorine (C-Cl) bonds is atransformation of interest since they constitute part of theskeleton of non-environmental friendly substances such aspolyvinyl chloride (PVC) and hydrochlorofluorocarbons(HCFCs).1 Several heterogeneous systems have been de-scribed for the functionalization of C-Cl bonds.2 In thehomogeneous phase, most efforts have been directed to arylhalides, a common reagent in many metal-mediated C-Nbond formation reactions.3 This is in contrast with chlor-oalkanes, where the C-Cl bond resembles those existing inHCFC and PVC, among others. Although the activation ofthis unit with transition metal complexes has been achieved,very often throughanoxidative addition step,4 the subsequent
functionalization and extraction of the modified substrate isstill rare. Alternativemethods are based on the use of a strongbase, in processes lacking regio- or stereochemical control.5
Very recently, Yorimitsu, Oshima. and co-workers havereported a cobalt-based system for the regioselective dehydro-halogenation with Grignard’s reagents.6 However, the use ofan efficient procedure that would result in the removal of thehalide from the hydrocarbon chainwith high atomic economyis still of interest and remains to be developed.Half a century ago, seminal work by Urry et al.7 described
the photochemical reaction of diazomethane or methyldiazoacetate with polyhalomethanes (Scheme 1a), leadingto products derived from the insertion of the :CHRunits intoone or more C-X bonds (X=Cl, Br). Reaction of ethyldiazoacetate with monohaloalkanes also proceeded underirradiation conditions, although when a β-hydrogen atom(with respect to the halogen) is present, the reaction affords amixture of the haloacetate and an olefin (Scheme 1b).8 In theabsence of that hydrogen, the reaction proceeds throughinsertion into the C-X bond. The main drawback of thesetransformations is that they occur through the intermediacyof photolytically generated free carbenes, and therefore nocontrol of the selectivity can be exerted.
*Corresponding authors. E-mail: [email protected];[email protected]; [email protected].(1) (a) Wiersma, A.; van de Sant, E. J. A. X.; Makkee, M.; van
Bekkum, H.; Moulijn, J. A. Stud. Surf. Sci. Catal. 1996, 101, 369.(b) Manzer, L. E.; Rao, V. N. M. Adv. Catal. 1993, 39, 329.(2) See for example: (a) Park, K. H.; Jung, I. G.; Chung, Y. K.; Han,
J.W.Adv. Synth. Catal. 2007, 349, 411. (b) York, S. C.; Cox, D. F. J. Phys.Chem. B 2003, 107, 5182. (c) Zhou, G.; Gellman, A. J. J. Catal. 2002, 194,233.(3) (a) Buchwald, S. L.; Mauger, C.; Mignani, G.; Scholz, U. Adv.
Synth. Catal. 2006, 348, 23. (b) Ley, S. V.; Thomas, A. W. Angew. Chem.,Int. Ed. 2003, 42, 5400.(4) (a) Vetter, A. J.; Rieth, R. D.; Brennessel, W. W.; Jones, W. D.
J. Am. Chem. Soc. 2009, ASAP doi: 10.1021/ja9002316. (b) Lobana, T. S.;Isobe, K.; Kitayama, H.; Nishioka, T.; Kinoshita, I. Angew. Chem., Int. Ed.2004, 43, 213. (c) Aballay, A.; Godoy, F.; Buono-Core, G. E.; Klahn, A. H.;Oelckers, B.; Garland, M. T.; Mu~noz, J. C. J. Organomet. Chem. 2003, 688,168. (d) Dorta, R.; Shimon, L. J. W.; Rozenberg, H.; Milstein, D. Eur. J.Inorg. Chem. 2002, 1827. (e) St€ohr, F.; Sturmayr, D.; Kickelbick, G.;Schubert, U. Eur. J. Inorg. Chem. 2002, 2305. (f) Haarman, H. F.; Ernsting,J. M.; Kranenburg, M.; Kooijman, H.; Veldman, N.; Spek, A. L.; vanLeeuwen, P. W. N. M.; Vrieze, K. Organometallics 1997, 16, 887.
(5) (a) Bartsch,R.A.; Za0vada, J.Chem.Rev. 1980, 80, 453. (b)Ma, Y.;Ramirez, A.; Singh, K. J.; Keresztes, I.; Collum, D. B. J. Am. Chem. Soc.2006, 128, 15399.
(6) Kobayashi, T.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. J. Am.Chem. Soc. 2008, 130, 11276.
(7) (a) Urry, W. H.; Eiszner, J. R. J. Am. Chem. Soc. 1951, 73, 2977.(b) Urry, W. H.; Eiszner, J. R. J. Am. Chem. Soc. 1952, 74, 5822. (c) Urry,W. H.; Wilt, J. W. J. Am. Chem. Soc. 1954, 76, 2594. (d) Urry, W. H.;Eiszner, J. R.; Wilt, J. W. J. Am. Chem. Soc. 1957, 79, 918.
(8) (a) Marchand, A. P.; Brockway, N. M. Chem. Rev. 1974, 74, 441.(b) Marchand, A. P.; Brockway, N. M. J. Am. Chem. Soc. 1970, 92, 5801.

Article Organometallics, Vol. 28, No. 20, 2009 5969
Late transition metal complexes are known to promotediazo compound decomposition and subsequent transfer ofthe carbene moiety to unsaturated or saturated substrates.9 Inthe early 1990s, Pirrung and co-workers studied the behaviorof Rh2(OAc)4 as the catalyst for dipolar cycloaddition reac-tions using cyclic diazo compounds. During their studies theyobserved10 that the reaction of 2-diazo-1,3-cyclohexanedionewith furan could be performed in several solvents such asTHF, benzene, or fluorobenzene; however, in the case ofmethylene or ethylene chloride as the solvent no reactionoccurred due to the formation of another product that waslater characterized11 as the formal HCl abstraction product(Scheme 2a). To explain the formation of this compound, theauthors invoked the rhodium-catalyzed formation of a chlor-onium ylide that undergoes loss of chlorine-containing moi-eties. Further work byM€uller and co-workers12 with the samesystem led to the observation of different compounds in whicha CH2Cl group migrates from the chloronium to the vicinalcarbonyl O-atom (Scheme 2b). Migration of ArCH2 was alsoobserved13 by Lee and co-workers with a related system usingaryl halides ArCH2Cl as the substrate in the reaction with2-diazo-1,3-cyclohexanedione.A few years ago Dias, Lovely, et al. reported14 the first
example based on this methodology of a metal-catalyzedfunctionalization of a C-X bond (X=Cl, Br) using simpleethyl diazoacetate (EDA) as the carbene source.Theyobserveda similar behavior to that previously reported under irradia-tion: polyhalomethanes underwent formal insertion of thecarbene group, whereas in the case of halocyclohexane, ethylhaloacetate and cyclohexene were obtained (Scheme 3).
In spite of the low number of contributions regarding thereaction of haloalkanes and diazocompounds, this is anextremely interesting transformation, which could find apotential application for the degradation of polyvinyl chlo-ride, PVC. Equation 1 shows a hypothetical decompositionof such material using the methodology of carbene transferfrom ethyl diazoacetate in the presence of a metal-basedcatalyst. The PVC polymer chain would undergo a formaldehydrochlorination process in such a way that an unsatu-rated polyolefin and ethyl chloroacetate would be the finalproducts. The former could be reused in different ways,whereas the latter, ClCH2CO2Et, is a rawmaterial employedin chemical industry in several processes. However, thedesign of the appropriate catalysts requires the knowledgeof the mechanism that governs this transformation. Duringthe past decade, our group has been involved in the use ofgroup 11 metal-based catalysts for the functionalization ofhydrocarbons, either saturated or
unsaturated, with ethyl diazoacetate.15 We have been parti-cularly interested in the conversion of simple hydrocarbonswith EDA, in a reaction inwhich the :CHCO2Et group (fromEDA) formally inserts into the C-H bond of the alkane.16-18
Among the catalysts developed for such transformation, the[TpBr3Ag]2 (1) complexwas found18 to promote high conversions
Scheme 1. Photochemical Functionalization of C-X (X=halogen)with Diazo Compounds
Scheme 2. First Examples of Metal-Catalyzed Carbene
Insertion into C-Cl Bonds
Figure 1. Trispyrazolylborate ligands (Tpx) relevant to thiswork.
Scheme 3. Silver-CatalyzedFunctionalization ofC-X (X=halogen)with Diazo Compounds
(9) Doyle, M. P.; McKervey, M. A.; Ye, T. Modern CatalyticMethods for Organic Synthesis with Diazo Compounds; John Wiley &Sons: New York 1998.(10) Pirrung,M.C.; Zhang, J.;McPhail, A. T. J. Org. Chem. 1991, 56,
6269.(11) Pirrung, M. C.; Zhang, J.; Lackey, J.; Sternbach, D. D.; Brown,
F. J. Org. Chem. 1995, 60, 2112.(12) M€uller, P.; Allenbach, Y. F.; Bernardinelli, G.Helv. Chim. Acta
2003, 86, 3164.(13) Lee, Y. R.; Cho, B. S.; Kwon, H. J. Tetrahedron 2003, 59, 9333.(14) Dias, H. V. R.; Browning, R. G.; Polach, S. A.; Diyabalange,
H. V. K.; Lovely, C. J. J. Am. Chem. Soc. 2003, 125, 9270.
(15) (a) Dıaz-Requejo, M. M.; P�erez, P. J. Chem. Rev. 2008, 108,3379. (b) Díaz-Requejo,M.M.; P�erez, P. J. J. Organomet. Chem. 2005, 690,5441. (c) Díaz-Requejo, M.M.; P�erez, P. J. J. Organomet. Chem. 2001, 617,110.
(16) (a) Dıaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.;Trofimenko, S.; P�erez, P. J. J. Am. Chem. Soc. 2002, 124, 896.(b) Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.;Trofimenko, S.; P�erez, P. J. J. Am. Chem. Soc. 2003, 125, 1446.(c) Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.;Trofimenko, S.; P�erez, P. J. Organometallics 2003, 22, 4145.
(17) (a) Fructos, M. R.; Belderrain, T. R.; de Fr�emont, P.; Scott,N. M.; Nolan, S. P.; Dıaz-Requejo, M. M.; P�erez, P. J. Angew. Chem.,Int. Ed. 2005, 44, 5284. (b) Fructos, M. R.; de Fr�emont, P.; Scott, N. M.;Nolan, S. P.; Díaz-Requejo, M. M.; P�erez, P. J. Organometallics 2006, 25,2237.
(18) Urbano, J.; Belderraın, T. R.; Nicasio, M. C.; Trofimenko, S.;Dıaz-Requejo, M. M.; P�erez, P. J. Organometallics 2005, 24, 1528.

5970 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
when employed with several plain alkanes (pentane, hexane,2-methylbutane, 2,3-dimethylbutane, amongothers), using thealkane as the reaction solvent. Given the similarity of thiscomplex to the Dias-Lovely catalyst14 (complex Tp(CF3)2
Ag(thf); seeFigure 1 for Tpx ligands), we decided to investigatethe behavior of our [TpBr3Ag]2 catalyst in the presence ofcarbon-halogen bonds. In this contribution we present theresults obtained with these silver complexes as catalysts for theabove transformations, including the first example of anasymmetric insertion of a carbene group into the C-Cl bond.Experimental and theoretical data have allowed the proposalof a novel reaction mechanism that will serve as the basis forthe design of useful catalysts for these processes.
Results and Discussion
Catalytic Functionalization of Polyhalomethanes and
Monohaloalkanes with [TpBr3Ag]2 as the Catalyst Precursor.A first series of experiments was carried out with [TpBr3Ag]2as the catalyst in the reaction of ethyl diazoacetate andseveral polyhalomethanes as the substrate (and solvent). A1:20 [catalyst]:[EDA] ratio was employed in all cases(considering TpxAg monomeric units). As shown inScheme 4, chloro and bromo derivatives were converted intothe corresponding products derived from the formal inser-tion of the :CHCO2Et unit into theC-Xbond. In no case didthe iodo analogues verify any reaction, all the initial EDAbeing converted into a mixture of diethyl fumarate andmaleate after extended reaction times. Some decompositionwas observed, and therefore the formation of the olefinscould be due to the presence of some naked silver in themixture. We believe that the iodo-containing substratesinduce a certain blocking of the silver center, avoidingfurther catalytic reaction. In the case of Cl- and Br-contain-ing substrates, the conversions observed are quite similarto those reported14 with Tp(CF3)2Ag(thf), as it could beexpected, since both catalysts have also been reported toinduce a similar activity in the C-H bond functionalizationby carbene insertion from EDA.18,19 Competition experi-ments (Scheme 5) have also been carried out to determine therelative reactivity of C-Cl and C-Br bonds. Thus, when
equimolar mixtures of dichloro- and dibromomethane werereacted with EDA, a 7-fold excess of the product derivedfrom the bromo derivative was observed. This trend is inaccord with the well-known order of bond dissociationenergies (BDE) for C-Cl and C-Br bonds, the latter beingca. 16 kcal/mol lower than the former. In another competi-tion experiment with dichloromethane and chloroform, onlythe product formed from the latter was obtained.
Once it was demonstrated that the TpBr3-containing silvercomplex was also capable of inducing the functionalizationof C-Xbonds by this methodology, we explored this featuretoward monohaloalkanes. Chloro- and bromobutane or-hexane were employed as the substrates, again with a 1:20ratio of [catalyst]:[EDA]. In all cases, mixtures of severalproducts were obtained. For example, the use of 1-chlor-ohexane as the substrate afforded a mixture of ethyl chlor-oacetate, 1-hexene, and several compounds derived from theinsertion of the carbene unit into the C-H bonds of1-chlorohexane (Scheme 6). This is a notable variance withthe results reported by Dias, Lovely, et al., which did notmention the functionalization of C-H bond with haloalk-anes as the substrates: only the formation of the ethylhaloacetate was mentioned,14 in spite of the reported cata-lytic capabilities of their Tp(CF3)2-containing complex forthis transformation.19 Back to our system, the four haloalk-anes studied displayed the same behavior using [TpBr3Ag]2 asthe catalyst (Scheme 6), the respective amounts of productsderived from C-X cleavage and the C-H functionalizationbeing nearly similar. It could seem that the bromoalkanes areless reactive than the chloroalkanes, but these results justreflect the relative reactivity of each haloalkane comparedwith EDA dimerization. Intermolecular competition experi-ments provided intractable mixtures of compounds, so wedecided to evaluate the intramolecular competition using1-chloro-4-bromobutane as the substrate (Scheme 7). Pro-ducts derived from the cleavage of both the C-Cl and theC-Br bonds were observed, the ratio of products indicatingthat the C-Br bond is more prone to be activated, in goodaccord with its lower value of BDE. Chloro- and bromo-cyclohexane have also been studied in this transformation(Scheme 7). They converted mainly into a mixture of ethylhaloacetate and cyclohexene, the bromo derivative beingmore active toward the functionalization. Small amounts ofthe products derived from C-H functionalization werealso detected (<10%) by GC and characterized by GC-MS(see Supporting Information).
Therefore, at this stage we can conclude that the[TpBr3Ag]2 complex catalyzes the functionalization of halo-alkanes with a dual reactivity: polyhalomethanes CXnH4-n
(X=Cl, Br; n=2-4) can be converted into products derivedfrom the insertion of the carbene unit into the C-X bonds.
Scheme 4. Functionalization of Polyhalomethanes with EDA
Using [TpBr3Ag]2 as the Catalyst Precursora
aDiethyl fumarate andmaleate account for the remaining initial EDA.
Scheme 5. Competition Experiments with Polyhalomethanes
(19) (a) Dias, H. V. R.; Browning, R. G.; Richey, S. A.; Lovely, C. J.Organometallics 2005, 24, 5784. (b) Dias, H. V. R.; Browning, R. G.;Richey, S. A.; Lovely, C. J. Organometallics 2004, 23, 1200. (c) Dias,H. V. R.; Lovely, C. J. Chem. Rev. 2008, 108, 3233.

Article Organometallics, Vol. 28, No. 20, 2009 5971
In the case of monohalogenated substrates, with availableβ-hydrogen atoms, the overall reaction supposes the cleavage ofthe carbon-halogenbondand the formationof ethylhaloacetateand an olefin. This reaction competes with the functionalizationof theC-Hbondof the startingmaterial by carbene insertion. Inall cases, the reactions are performed at room temperature, andthe yields are moderate. It is worth mentioning that we have notoptimized the conversions, at this stage, but focused in a generalscreening with the aim of studying the reaction mechanism.
Mechanistic Studies
Nature of the Catalytic Species. The above results haveshown that the complex [TpBr3Ag]2 (1) can be employed as amodel to study the catalytic functionalization of haloalk-anes. The catalyst precursor was first isolated and character-ized with the composition [TpBr3Ag]2 3 1/2(acetone).
18 Wehave now fully characterized this complex by X-ray studies,the structure of the molecule being shown in Figure 2. The[TpBr3Ag]2 group exists in the solid state as a consequence ofthe coordination of each silver ion to two pyrazolyl rings ofone TpBr3 ligand and a third pyrazolyl ring of a second TpBr3
ligand. This structure is very similar to that already reportedfor [Tp*Cu]2.
20 It is only worth mentioning that the distanceAg-Ag of 2.89 A is above the sum of the covalent radii21
(1.33 A þ 1.33 A=2.66 A). The related [Tp*Ag]2 complexdisplayed a 2.78 A distance,22 although with a distinctligand distribution around the two silver centers. This
Ag-Ag distance falls in the range expected for the so-calledargentophilic interactions,23 weak bonding forces that aresimilar in strength to hydrogen bonding. In good accordwiththis, determination of themolecular weight in thf solution bySigner’s method24 has shown that an equilibrium betweenthe dinuclear and monomeric species exists in solution atroom temperature, favoring the latter by a factor of 3.
Two other complexes containing the core TpxAg, with theTp*,Br and TpMs ligands, respectively, previously prepared inour laboratory,18,25 corresponded with the formulations [Tp*,BrAg]2 (2) and [Tp
Ms2Ag3][NO3] (3). The structure of the former
is similar to that of 1 (Figure 3), with a 2.80 A separationbetween both silver centers. In the case of the TpMs derivative, atrinuclear silver structure is sustained by two TpMs ligands, theoverall positive charge being balanced with a nitrate counterion(Figure 3). Each silver center is coordinated to two pyrazolylrings of different TpMs ligands. This μ3-η
1:η1:η1 trinucleatingbridgingmodeof theTpMs structure is rare; aprecedenthasbeenreported for the complex [TpAn2Ag3][ClO4] (An= anisyl).26
Scheme 6. Functionalization of 1-Halohexane with EDA Using [TpBr3Ag]2 as the Catalyst Precursora
aDiethyl fumarate and maleate account for the remaining initial EDA.
Scheme 7. Intramolecular Competition Experiments with
1-Chloro-4-butane and Functionalization of Halocyclohexanes
Figure 2. X-ray structure of the molecules of the complex[TpBr3Ag]2, 1.
(20) Mealli, C.; Arcus, C. S.; Wilkinson, J. L.; Marks, T. J.; Ibers,J. A. J. Am. Chem. Soc. 1976, 98, 711.(21) Bayler, A.; Schier, A.; Bowmaker, G. A.; Schmidbaur, H. J. Am.
Chem. Soc. 1976, 98, 711.(22) Humphrey, E. R.; Reeves, Z.; Jeffery, J. C.; McCleverty, J. A.;
Ward, M. D. Polyhedron 1999, 18, 1335.(23) Mohamed, A. A.; P�erez, L. M.; Fackler, J. P., Jr. Inorg. Chim.
Acta 2005, 358, 1657.
(24) Zoellner, R. W. J. Chem. Educ. 1990, 67, 714.(25) G�omez-Emeterio, B. P.; Urbano, J.; Dıaz-Requejo, M. M.;
P�erez, P. J. Organometallics 2008, 27, 4126.(26) Humphrey, E. R.; Harden, N. C.; Rees, L. H.; Jeffery, J. C.;
McCleverty, J. A.;Ward,M.D. J. Chem. Soc., Dalton Trans. 1998, 3353.

5972 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
In spite of the oligonuclear structures of 2 and 3, molecularweight studies in solution by Signer’s method have shown theexistence of a dissociation equilibrium, similar to that reportedfor complex 1. Therefore, in all cases, the oligomeric complexes1-3 deliver in solution a certain amount of the unsaturatedfragments TpxAg that are available to react with EDA.Kinetic and Isotopic Experiments. It is well established that
the reaction of ethyl diazoacetate with late transition metalcomplexes leads to the formation of highly reactive, electro-philic metallocarbene intermediates,27 usually this step beingrate-determining.28 In addition, previous theoretical workfrom our laboratories has led to the knowledge of thegeometries and energies of the TpBr3MdC(H)CO2Me inter-mediates (M=Cu, Ag).29 We have first carried out kineticmeasurements of the nitrogen evolution in the reactions ofEDA and dihalomethanes CH2X2 (X=Cl, Br) to assess this
proposal. First-order kinetics are observed for N2 evolution,e.g., for EDA consumption (each molecule of EDA con-sumed originates one molecule of N2). The kobs values forCH2Br2 and CH2Cl2 were nearly identical (1.95 � 10-3 and1.93� 10-3 s-1, Figure 4). This is in good agreement with theabove statement for the metallocarbene formation as therate-determining step, the effect of the halomethane beingnull in the reaction rate. It is worth mentioning that thekinetic experiment is very sensitive to changes in the tem-perature of (1 �C, due to its effect on the pressure of theevolved nitrogen.
Competition experiments using deuterated solvents havebeen carried out to determine the existence of any isotopiceffect. Thus, when equimolar amounts of chloroform anddeuterochloroform, CHCl3 and CDCl3, were employed,a ca. 1.5:1 ratio of products derived from the insertion intothe C-Cl bond was obtained favoring the protio derivative
Figure 3. X-ray structures of the molecules of the complexes [Tp*,BrAg]2, 2 (left), and [TpMs2Ag3][NO3], 3 (right).
Scheme 8. Competition Experiments Carried out with CHCl3and CDCl3
Figure 4. Monitoring of N2 evolution in the reaction of EDAand dihalomethanes using [TpBr3Ag]2 as the catalyst precursor.Experimental errors are <1%. Experiments were carried outthree times.
Scheme 9. Effect of the Tpx Ligand on the Ratio of C-Cl and
C-Br Functionalization
(27) Kirmse, W. Angew. Chem., Int. Ed. 2003, 42, 1088.(28) See: Dıaz-Requejo, M. M.; Caballero, A.; Belderrain, T. R.;
Nicasio,M. C.; Trofimenko, S.; P�erez P. J. J. Am. Chem. Soc. 2002, 124,978, and references therein.(29) Braga, A. A. C.; Maseras, F.; Urbano, J.; Caballero, A.;
Dıaz-Requejo, M. M.; P�erez, P. J. Organometallics 2006, 25, 5292.
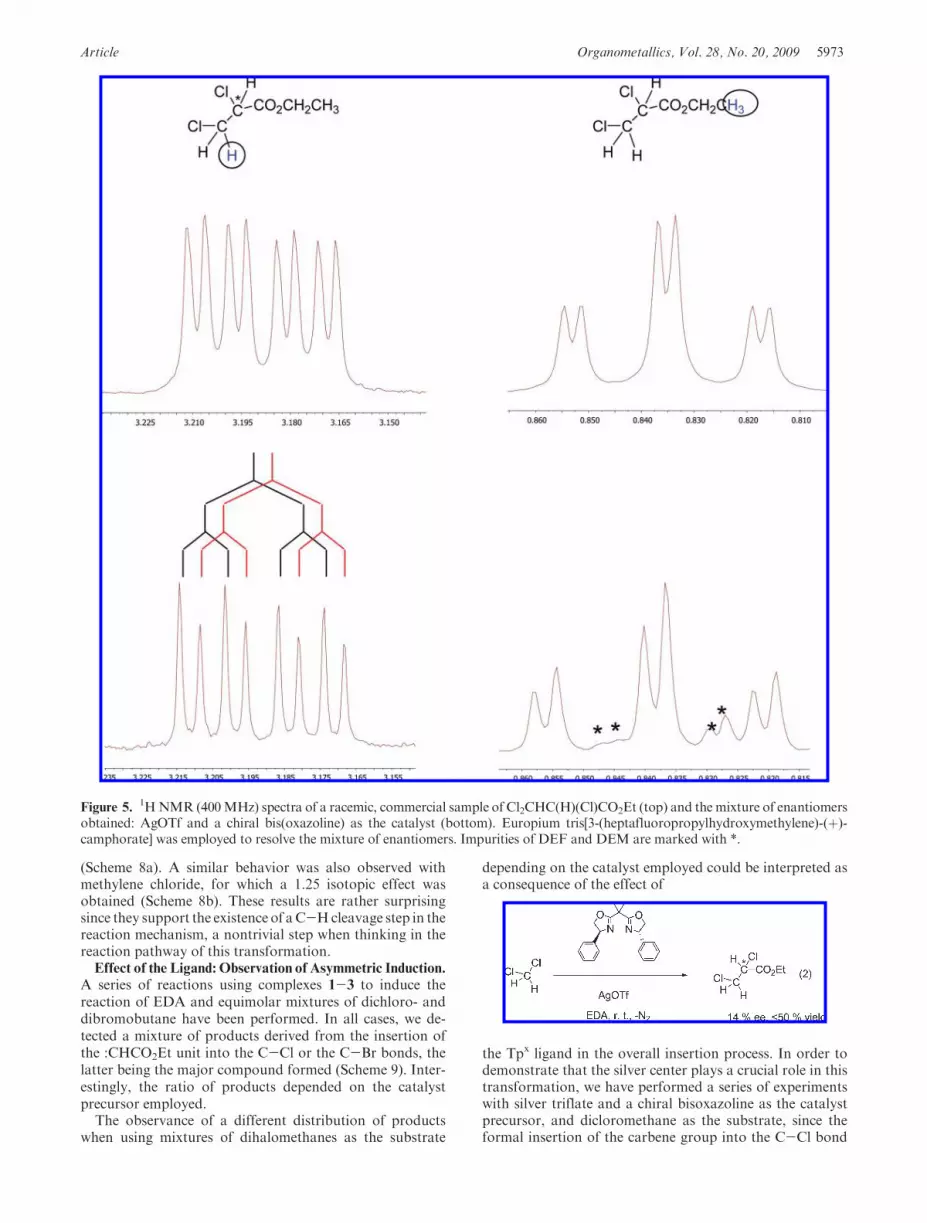
Article Organometallics, Vol. 28, No. 20, 2009 5973
(Scheme 8a). A similar behavior was also observed withmethylene chloride, for which a 1.25 isotopic effect wasobtained (Scheme 8b). These results are rather surprisingsince they support the existence of aC-Hcleavage step in thereaction mechanism, a nontrivial step when thinking in thereaction pathway of this transformation.Effect of the Ligand: Observation of Asymmetric Induction.
A series of reactions using complexes 1-3 to induce thereaction of EDA and equimolar mixtures of dichloro- anddibromobutane have been performed. In all cases, we de-tected a mixture of products derived from the insertion ofthe :CHCO2Et unit into the C-Cl or the C-Br bonds, thelatter being the major compound formed (Scheme 9). Inter-estingly, the ratio of products depended on the catalystprecursor employed.
The observance of a different distribution of productswhen using mixtures of dihalomethanes as the substrate
depending on the catalyst employed could be interpreted asa consequence of the effect of
the Tpx ligand in the overall insertion process. In order todemonstrate that the silver center plays a crucial role in thistransformation, we have performed a series of experimentswith silver triflate and a chiral bisoxazoline as the catalystprecursor, and dicloromethane as the substrate, since theformal insertion of the carbene group into the C-Cl bond
Figure 5.1HNMR (400MHz) spectra of a racemic, commercial sample of Cl2CHC(H)(Cl)CO2Et (top) and the mixture of enantiomers
obtained: AgOTf and a chiral bis(oxazoline) as the catalyst (bottom). Europium tris[3-(heptafluoropropylhydroxymethylene)-(þ)-camphorate] was employed to resolve the mixture of enantiomers. Impurities of DEF and DEM are marked with *.

5974 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
generates a stereogenic center (eq 2) following the proceduredescribed byMa and co-worker.30 Although the transforma-tion is quite slow (50% of EDA is consumed after 36 h),a ca. 14%of enantiomeric excess has been observed byNMRstudies using europium salts as discriminating reagent(Figure 5). At this stage, this result has little syntheticrelevance in terms of development of an efficient enantiose-lective process. However, it is remarkable in terms of provid-ing an unambiguous indication that the silver center, now ina chiral environment, is responsible for exerting a certainasymmetric induction. The involvement of free versus com-plexed ylides is a rather subtle question that has beenthoroughly discussed by Hodgson some years ago.31,32 Sup-posedly, if the coordinating metal carries chiral ligands, oneshould observe enantioselectivity if the ylide is complexed,but no enantioselectivity in the absence of complexation.Free Ylide versus the Metal-Based Mechanism. The pre-
vious data can be summarized in the following: (i) the firststep in this transformation is the formation of a silver-carbene intermediate, from the reaction of TpxAg and ethyldiazoacetate (EDA); (ii) the interaction of this intermediatewith the haloalkanes does not affect the reaction rate; (iii) aprocess involving C-H cleavage must occur in order toexplain the observance of kinetic effects with deuteratedsubstrates; (iv) the formation of the products must occur inthe coordination sphere of the metal complex inferred fromthe observation of different ratio products when varying theTpx ligand and, more interestingly, from the induction of acertain asymmetry when employing a silver-bisoxazolinecatalyst precursor.
Seminal work on this reaction10-12 invoked the formation ofmetal-free ylides as intermediates (Scheme 2). In the recentcontribution using the silver catalyst,14 Dias and Lovely pro-posed a similarmechanism inwhich an ylide is formed from theinteraction of the silver-carbene intermediate and the halo-methane. The free ylide subsequently undergoes a rearrange-ment that actually corresponds to a Stevens rearrangement
(Scheme 10).33 This mechanism supposes that the ylide, a metal-free intermediate, would control the formation of the products,with nopossible influence of themetal catalyst on the selectivity ofthe reaction. Such a proposal has been later applied by the sameauthors to the reaction of related allyl and propargyl halidesand diazoacetates.34 However, the aforementioned informa-tion collectedwithourmodel system is somewhat contradictoryto such a proposal. Thus, the observance of a different dis-tribution of products when using mixtures of dihalomethanesas the substrate depending on the catalysts employed or theunprecedented observation of enantiomeric excesses whenemploying the silver-bisoxazoline catalyst precursor cannotbe explained on the basis of a Stevens rearrangement of a freeylide. On the other hand, the transformation must occur alonga pathway in which the metal center exerts the appropriateinfluence to induce those values of regio- (with several Tpx
ligands) or enantioselection (with the bisox ligand). It is alsoworth mentioning that the observance of a certain deuteroeffect must be related to the existence of some C-H cleavagestep during the catalytic transformation, after the silver-car-bene intermediate is formed. This step also defies the free-ylide-based mechanism. Therefore, the overall transformation, i. e.,the formal insertion of a carbene group into aC-Xbond,musttherefore be accomplished throughout a more complex me-chanism that themere formationof an ylide10-14 or a concertedinsertion of the carbene group into such a bond, similar to therelated alkane C-H bond functionalization.29
On the basis of the available data, we believe that thetransformation described in this contribution occurs throughthe intermediacy of a silver-coordinated ylide (CY) formedfrom the silver metallocarbene (MC) and the haloalkane,either polyhalomethanes or monohaloalkanes (Scheme 11).The rate-determining step is the formation of the silver-carbene from the [LAg] core and EDA (L= Tpx, bisox).The coordinated ylide CY subsequently undergoes furtherrearrangement, to yield the observed products. Althoughthe final steps of this mechanism cannot be proposedfrom experimental data, it is worth mentioning that (i) theroutes in Scheme 11 would explain the asymmetric inductionby the metal catalyst during the formation of the productderived from the formal insertion of the CHCO2Et and (ii) themissing steps should reproduce the observation of kineticeffects in the experiment carried out with deuterated sub-strates. The low enantiomeric excess might not arise from
Scheme 10. Previously Proposed Mechanism for Haloalkane
Functionalization with Ethyl Diazoacetate (top)That Is Formally
Similar to a Stevens Rearrangement (bottom)
Scheme 11. Possible Pathway for the Silver-Catalyzed
Functionalization of C-X Bondsa
aThe metal center must participate along the pathway, with no freeylides being involved.
(30) Ma, S.; Wu, S. New J. Chem. 2001, 25, 1337.(31) Hodgson, D. M.; Pierard, F. Y. T. M.; Stupple, M. A. Chem.
Soc. Rev. 2001, 30, 50.(32) (a) Hodgson, D. M.; Stupple, M. A.; Johnstone, C. Chem.
Commun. 1999, 2185. (b) Hodgson, D. M.; Stupple, M. A.; Pierard,F. Y. T. M.; Labande, A. H.; Johnstone, C. Chem.;Eur. J. 2001, 7, 4465.(c) Hodgson, D. M.; Labande, A. H.; Pierard, F. Y. T. M. Synlett 2003, 59.(d) Hodgson, D. M.; Labande, A. H.; Pierard, F. Y. T. M.; Castro, M. A. E.J. Org. Chem. 2003, 68, 6153.
(33) Smith, M. B; March, J. Advanced Organic Chemistry, 6th ed.;Wiley-Interscience: New York.
(34) Krishnamoorthy, P.; Browning, R. G.; Singh, S.; Sivappa, R.;Lovely, C. J.; Dias, H. V. R. Chem. Commun. 2007, 731.

Article Organometallics, Vol. 28, No. 20, 2009 5975
metal involvement but from other less usual processes, forexample, selective destruction by the catalyst of one of theenantiomers of the product. In view of the novelty of themechanism that does not follow the previous proposals basedon free ylides, we decided to complete this work from atheoretical point of view in order to propose a completepathway that accounts for the experimental data. This com-putational study will provide, together with the availableexperimental evidence, a complete mechanistic picture of theprocess at hand.Theoretical Calculations. a. Polyhalomethanes. In order
to collect information relevant to the mechanism governingthe transformation described above, computational DFTstudies at the B3LYP level have been carried out on themechanism of the reactions for both the polyhalomethanesand the monohaloalkane systems. For the polyhalomethanesystems, the main purpose was to find a general mechanismthat could accommodate the experimental data presentedabove. The two simplest mechanistic alternatives would bethe Dias-Lovely mechanism, actually a Stevens rearrange-ment, outlined above (Scheme 10)14 and the direct σ bondcleavage process analogous to that previously reported forC-Hactivation (Scheme 12).29 Although only the second oneis compatiblewith the observed asymmetric induction, none ofthem can easily explain the presence of isotope effects in theCHCl3/CDCl3 andCH2Cl2/CD2Cl2 competition experiments.
We have studied the reaction of [TpBr3AgdC(H)CO2Me](R1) with the three possible polychloromethane systemsCH2Cl2 (RA), CHCl3 (RB), and CCl4 (RC). As will be shownbelow, the mechanisms are quite complex in terms of thepresence of intermediates and transition states, and becauseof this, we will use a systematic labeling system.Wewill use aunique label for each process considered,A for the particularcase of dichloromethane, and classify the structures asreactants (R), products (P), intermediates (I), or transitionstates (TS). Intermediates will be numbered consecutively asthey appear in a given mechanism, and transition states willbe labeled according to the species they connect.
Figure 6 presents schematically the overall mechanism forprocessesA, B, andC corresponding to the transition metal-catalyzed C-Xactivation in CH2Cl2 (RA), CHCl3 (RB), andCCl4 (RC), respectively. The corresponding energetics arecollected in Table 1. The overall mechanism is similar, butthe number of intermediates changes. The simplest case isthat of dichloromethane, A. The initial approach of thereactants produces an ylide intermediate, AI1, where thechlorine binds to the carbenic center, which remains attachedto the metal center. A reorganization then takes place; theylide ligand retains its general shape, but ends up attached tothe metal through its carbonylic oxygen, inAI2. For dichlor-omethane, the C-Cl activation takes place directly fromAI2, resulting in intermediate AI5, where the formal inser-tion has already taken place. The movement of the productaway from the silver center results in the products of thisprocess, [TpBr3Ag] (P1) and ClCH2CHClCO2Me (PA).
Figure 7 presents some selected structures involved inreaction A. The initial intermediate AI1 clearly has an ylidenature, the chlorine interacting with two carbon atoms. TheC-Cl distances are 1.870 A for the methyl carbon and 1.916A for the carbene carbon. These distances have to becompared with the 1.790 A C-Cl distance computed forfree dichloromethane. This strong interaction weakenscoordination of the carbene to silver; the Ag-C distance is2.213 A in AI1, to be compared with the 2.051 A in thestarting complex R1. AI1 is significantly different from theadducts we had previously reported between the same silvercarbene complex and alkanes.29AI1 does not directly under-go C-Cl cleavage, but evolves to another intermediate,AI2,where the ylide coordinates silver through the carbonyloxygen. AI2 has a relative energy of-1.1 kcal/mol, compar-able to that of AI1, and they are connected through thetransition state ATS1-2 with a low relative energy of4.9 kcal/mol. AI2 undergoes a C-Cl cleavage step throughtransition state ATS2-5, also shown in Figure 7. Thistransition state is quite strained and is the rate-determiningstep of the whole process (referred to the metallocarbene
Scheme 12. Operating Mechanism for Alkane C-H
Functionalization with Ethyl Diazoacetate29
Figure 6. Computed mechanism for the C-Cl activation inpolyhalomethane systems CH2Cl2, CHCl3, and CCl4.
Table 1. Computed B3LYP Energetics (kcal/mol) for SpeciesInvolved in the C-Cl Activation in Polyhalomethane Systems
CH2Cl2, CHCl3, and CCl4
reaction A reaction B reaction C
R1 þ 0.0 0.0 0.0I1 -5.3 -1.1 -3.4I2 -1.1 -6.5 -12.1I3 -12.7 -15.2I4 -5.7I5 -77.2 -77.1 -76.3P1 þ -64.1 -64.7 -63.9TS1-2 4.9 1.9 -1.9TS2-5 10.7TS2-3 6.4TS3-5 3.6 -1.6TS3-4 -2.3TS4-5 4.9

5976 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
intermediate).ATS2-5 is 10.7 kcal/mol above the reactants.In fact, it involves an inversion at the carbon center and thusgoes against the rules of orbital symmetry conservation.These rules apply however only to concerted mechanisms,and we see this process rather as an arrested dissociation; thecarbene fragment goes away from the metal and then itreturns. Its energy is still relatively low, likely because ofthe high exothermicity of the process. The following inter-mediate, AI5, is already 77.2 kcal/mol below the reactants.A computational estimation of the isotope effect associatedwith ATS2-5 produces a value of 1.35:1, in reasonableagreement with the experimental measurement of 1.25:1.No bond involving a hydrogen atom is cleaved at this step,but the movement of the CH2Cl unit in the reaction coordi-nate involves the hydrogen centers. It is difficult to give aqualitative explanation for the sign of this isotope effect, butthis is not surprising given the complexity of the transitionstate. The good agreement between experimental and theo-retical values provides in any case strong support to thecomputed mechanism.
A completely different alternative to the mechanism justdiscussed could be envisaged where the chloride coordinatesinitially at silver, and then this complex reorganizes to the
ylide proposed above, or alternatively through an intra-molecular activation and reduction elimination. This possi-bility was investigated for reaction A. A putative intermedi-ate, altAI1, where there is anAg-Cl bond (2.365 A) resultingfrom the addition of the C-Cl bond to the AgdC doublebond, is certainly quite stable, 35.2 kcal/mol below theseparated reactants. However, there is no low-energy pathconnecting it to the reactants. We could not locate thetransition state for the direct connection, but we did estimatea lower limit for the barrier through constrained geometryoptimizations. The late stages of the CH2Cl2 approach to thereactant were considered in three steps, with the Cl-Agdistance frozen at 2.6, 2.5, and 2.4 A in structures altAaux1,altAaux2, and altAaux3. These calculations proved thatapproach of a chlorine atom to the metal is not sufficientto detach the chlorine from the alkane. Even in altAaux3
(available in the Supporting Information), with a Ag-Cldistance of 2.400 A, the elongation of the C-Cl bond wasvery small (1.806 A). Evenmore significant were the energiesof altAaux1, altAaux2, and altAaux3 with respect to thereactants. They were 10.8, 18.1, and 22.2 kcal/mol, respec-tively. The initial point of this alternative approach,altAaux1, has an energy already higher than that of the
Figure 7. Computed structures for intermediate AI1 and transition state ATS2-5. Selected distances are given in A.
Figure 8. Computed structures for intermediate BI3 and transition state BTS2-3. Selected distances are given in A.

Article Organometallics, Vol. 28, No. 20, 2009 5977
rate-limiting transition state ATS2-5 of the favored path-way. It thus seems unlikely that a low-energy path can befound where the haloalkane attacks first the metal centerthrough one of its halogen atoms.
Process B involves CHCl3 (RB) and is also summarized inFigure 6 and Table 1. The key difference with process A isthat there is no direct connection between I2 and I5. Instead,there is a new intermediate, BI3, and the correspondingtransition states BTS2-3 and BTS3-5. The geometries ofBI3 andBTS2-3 are presented in Figure 8.BI3 is an unusualstructure, which may better be described as a complex with aC(O)(OMe)(CH2Cl) unit coordinated to the silver complexplus a free CCl2 carbene. This description is neverthelesssimplistic, as the CCl2 unit stays much closer (by 1.5 A) toone of the two adjacent C-Hbonds, which are thus far fromequivalent. Moreover the C-H bond further away is in a
more hindered position, therefore making scrambling un-likely. BI3 is quite stable (12.7 kcal/mol below reactants,Table 1) and is reached through transition state BTS2-3,which has the highest energy (6.4 kcal/mol) in this profile.Our calculations found out a computed isotope effect of 1.40for this step, in reasonable agreement with the experimen-tally measured value of 1.5. BI3 evolves toward productsthrough transition state BTS3-5 (energy 3.6 kcal/mol)where the CCl2 carbene unit inserts into a C-H bond.
Another difference between reactions A and B is in therelative energies of intermediates I1 and I2. I1 is 4.2 kcal/molmore stable in caseA, but the situation is inverted in cases B,with I2 being 5.4 kcal/mol more stable. The reason can beunderstood by analyzing the structures reported in theSupporting Information. The change in bond distances fromI1 to I2 indicates an increased transfer of a chloride from the
Figure 9. Computed energy profile for C-Cl activation in CH2Cl2 with (path A) and without (path D) involvement of the silvercomplex throughout the whole process.

5978 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
haloalkane to the carbene. As mentioned above,methyl-carbon and carbene-carbon distances for inter-mediate AI1 are 1.870 and 1.916 A, respectively. The valuesevolve to 2.010 and 1.686 A, respectively, inAI2. The trend isthe same for BI1 and BI2, with corresponding values of1.940, 1.876 and 2.283, 1.668 A, respectively. A chloridefragment thus moves from the haloalkane to the carbene,and the resulting haloalkyl fragment C4-xClx-2 ends up in acarbocationic form. The more substituted R-halogeno car-benium ions are known to bemore stable,35 and this explainsthe increased stability of I2 in the case of reaction B.
The results corresponding to system C, consisting of[TpBr3AgdC(H)CO2Me] plus CCl4, are also included inFigure 6 and Table 1, as well as in the Supporting Informa-tion, but will not be discussed here in detail. We will justmention that a new intermediate, CI4, appears in the pathand that the overall barrier is also low, with the highestenergy being the 4.9 kcal/mol for CTS4-5.
Reaction A was also evaluated computationally for thecases ofCH2Br2 andCH2I2. Themechanism is very similar tothat of the chlorine case, the rate-determining transitionstates being BrATS2-5 and IATS2-5. The structures aregiven in the Supporting Information. The energies ofBrATS2-5 and IATS2-5 relative to the correspondingreactants are 9.8 and 9.5 kcal/mol, respectively. Both valuesare lower than the 10.7 kcal/mol reported above for thechlorine case. This result fits well with our experimentalobservation of an easier reaction for bromoalkanes thanchloralkanes and is likely explained by the diminution in theC-X bond dissociation energies when X changes fromchlorine to bromine and to iodine.
We completed this part of the theoretical study on thepolyhalomethane systems by analyzing the mechanismpreviously proposed by Dias and Lovely,14 where the ylideis detached from the metal complex before cleavage of theC-X bond, following a Stevens rearrangement (seeScheme 10). The study was carried out only for the CH2Cl2system, in what we label as systemD (Figure 9). The startingpoint is the same as for system A, the ylide adduct AI1, withan energy 5.3 kcal/mol below the separated reactants. After-ward, we separated the ylide from the silver complex, result-ing in speciesDI2, which rearranges through transition stateDTS2-P to produce directly the product PA. The relation-ship, from both energetic and structural points of viewbetween DI2 and DTS2-P, is very similar to that betweenAI2 and ATS2-5. In fact, the energetics favor even slightlythe D process in this regard: DTS2-P is 10.5 kcal/mol aboveDI2, while the corresponding difference for the A processwas 11.8 kcal/mol. However, this mechanism has to bediscarded because of the relative energy of the DI2 inter-mediate. It is 18.2 kcal/mol above the separate reactants and23.5 kcal/mol above intermediate AI1. This brings DTS2-P
to a prohibitively high energy of 28.7 kcal/mol above thereactants, unable to compete with the 10.7 kcal/mol ofATS2-5. The silver fragment certainly does not seem toenhance in any way the ylide rearrangement, but it isnecessary for the ylide formation, and there is simply noreason for dissociation before the reaction is completed.b. Monoalkanes. The second part of the computational
study concerns the monohaloalkane systems. In this case thepurpose was to justify the experimental observation of the
simultaneous formation of different products, coming fromeither dehydrohalogenation or C-H activation in differentpositions of the chain. The computational study was in thiscase carried out on the system [TpBr3AgdC(H)CO2Me] (R1)plus CH3CH2Cl (RE), which is the simplest possible, andavoids the conformational problems that would appear iflonger chains were considered. Three different processeswere considered for the system: dehydrohalogenation (E)and C-H cleavage leading to either MeCO2CH2CH2CH2Cl(F) or MeCO2CH2CHClCH3 (G).
The computed mechanism for dehydrohalogenation issummarized in Figure 10. The initial steps are analogous tothose reported above for the polychloromethane systems.The approach of chloroethane to the carbene complexproduces the ylide intermediate EI1, which is attached tothe metal through the carbon, and this reorganizes toisomeric form EI2, where the ylide attached to the metalthrough the carbonyl oxygen. Here the mechanisms deviate.There is a new conformational rearrangement of this ylidecomplex affecting the ethyl chain that produces intermediateEI3, which undergoes β elimination through transition stateETS3-4, which yields directly the product adduct EI4. Thiscontains already the units of each of the three products:[TpBr3Ag] (P1), methyl chloroacetate (PE1), and ethylene(PE2).
The energetics, also shown in Figure 10, deserve somecomment. The transition stateETS3-4 for β elimination hasa low relative energy of 2.1 kcal/mol below reactants, which
Figure 10. Computed mechanism for the dehydrohalogenationof CH3-CH2Cl. Relative energies are indicated in kcal/mol.
(35) Gr€utzmacher, H.; Marchand, C. M. Coord. Chem. Rev. 1997,163, 287.

Article Organometallics, Vol. 28, No. 20, 2009 5979
is in fact lower than that for the initial ylide rearrangementthrough ETS1-2 (3.2 kcal/mol), which becomes, because ofthis, the rate-determining step of the overall process (relativeto the initial metallocarbene complex). The structures ofboth ETS1-2 and ETS3-4 are presented in Figure 11.ETS1-2 corresponds to the movement of the ylide in thecoordination sphere of the metal, and ETS3-4 correspondsto the β elimination step.ETS3-4 is only 1.0 kcal/mol abovethe previous intermediate EI3. β elimination is thus veryeasy, which explains the absence of formal carbene insertionin the C-X bond when β hydrogens are available.
Processes F andG, involving insertion of the carbene intoa C-Hbond, are mechanistically simpler. They take place inone single step from the initial ylide intermediate EI1. Thecorresponding transition states, FTS1-2 and GTS1-2, areshown in Figure 12. They differ only in the carbon where theC-H insertion is taking place. It is that where chlorine is notattached in FTS1-2 and that with chlorine inGTS1-2. Themost relevant part concerning the placement of these struc-tures in the overall reactivity of the system is in the energetics.The relative energies above the reactants of FTS1-2 andGTS1-2 are 3.1 and 2.8 kcal/mol, respectively. The valuesare in fact close to that of 2.8 kcal/mol previously obtainedfor C-Hactivation in ethane.29 The similarity to the value of
3.2 kcal/mol reported above for ETS1-2 is in any casecritical for the reactivity of CH3CH2Cl.
The system [TpBr3AgdC(H)CO2Me] plusCH3CH2Cl thushas three possible reaction paths, whichwe have labeledE,F,and G, leading to different products, and their respectivebarriers are 3.2, 3.1, and 2.8 kcal/mol. This points out to amixture of the three products, and this is exactly what isexperimentally observed in these systems, with mixture ofhydrodehalogenation and C-H insertion products. Thecomputed barriers are in fact so close that it is not possibleto make a computational prediction of the exact propor-tions, which would require the use of a more realistic model(i.e., 1-chlorobutane instead of chloroethane) and the intro-duction of solvation and entropic effects.Mechanistic Proposal. The overall mechanistic picture
encompassing all considered systems is summarized inScheme 13, which could be also in principle be applied tothe reaction of other haloalkane systems with [TpBr3MdCHCOOEt] complexes. The reaction starts, in all cases, withthe formation of an ylide complex where the halogen atombinds to the carbene without losing its connection to thealkane chain. This initial ylide can evolve directly towardC-H insertion through a transition state similar to thatof the reaction of unsubstituted alkanes. An alternative
Figure 11. Computed structures for transition states ETS1-2 and ETS3-4. Selected distances are given in A.
Figure 12. Computed structures for transition states FTS1-2 and GTS1-2. Selected distances are given in A.

5980 Organometallics, Vol. 28, No. 20, 2009 Urbano et al.
pathway is that in which the initial C-bound ylide rearrangesto bind to the metal through an oxygen. This O-bound ylidecan then undergo two different processes. If β hydrogens areavailable, it undergoes a low-barrier dehydrohalogenation.If this is not possible, as is the case of halomethanes, it canundergo a C-X insertion through a multistep process,involving in some cases C-H cleavage steps.
Itmay seem surprising thatC-H insertion occurs after theylide formation. This is however consistentwith our previouscomputational study on C-H insertion by the same type ofsilver complexes in alkanes with no halogen substituents.29
The energies of the van der Waals adducts between alkaneand metal complex were between -0.7 and -1.0 kcal/moldepending on the substitution at the alkane. This is much lessstable than the ylide complexEI1 reported above (-8.4 kcal/mol), which should thus be formed quantitatively through anessentially barrierless complex.
The above proposal supports the involvement of themetalcenter in the various steps and is in contrast with thepreviously proposed mechanism that is based on the forma-tion of a free ylide.10-14 The contribution of the silver centerin this transformation is of interest when designing catalystsfor the dechlorination of the aforementioned PVC, since theappropriate tuning of the metal center will exert a certaineffect in the reaction products.
This study cannot provide a detailed mechanism forthe asymmetric induction observed in the case of thebis(oxazoline) ligands, which has been presented onlyto prove metal involvement in the reaction. The stereo-induction mechanism by this type of ligand, which willreplace the (tris)pyrazolyl in the silver coordination sphere,
has been shown in other cases to be based on subtle combi-nations of steric effects36 and is out of the scope of the presentwork.
Conclusions
Silver complexes containing the TpxAg core can be em-ployed as catalysts in the reaction of ethyl diazoacetate andhaloalkanes. There are two different types of reactivitydepending of the nature of the substrate. For polyhalo-methanes, only the product derived from the insertion ofthe CHCO2Et unit into the C-X bond is observed. In thecase of monohaloalkanes, a more complex reaction takesplace, resulting in a mixture of an alkene and ethyl haloace-tate. Theoretical calculations and experimental studies, in-cluding the use of a chiral silver complex to induceenantiomeric excesses in the functionalization of CH2Cl2,have led to the proposal of a novel mechanism in which themetal center is involved in all the steps. Development ofrelated catalysts for PVC dechlorination is currently under-way in our laboratories.
Experimental Section
General Methods. All preparations and manipulations werecarried out under an oxygen-free nitrogen atmosphere usingconventional Schlenk techniques or inside a drybox. All thehaloalkanes and the ethyl diazoacetate were purchased fromAldrich and employed without further purification. The com-plexes TpxAg (Tpx=TpBr3, TpMs, Tp*) were prepared accordingto literature methods.18,25 NMR experiments were run in aVarian Mercury 400 MHz spectrometer. GC and GC-MSstudies were carried out using Varian 3900 and Varian Saturn2100 instruments, respectively.
General Catalytic Experiment. The silver catalyst was dis-solved (0.02mmol based onTpxAg units, 20.6mg) in 5mLof thehaloalkane, and ethyl diazoacetate (0.75 mmol) was added inone portion. The flask was covered with aluminum foil, and themixture was stirred until all the starting diazo compound wasconsumed (verified by GC). The products were identified byNMR, GC, and GC-MS, by comparison with reported data orcommercial samples (see Supporting Information). The lowboiling point of most of the products induced loss of materialduring workup. Therefore, the conversions were measured byNMR studies of the reaction mixture, using an internal stan-dard. In the case of CBr4 as the substrate, methylene chloridewas used as the solvent of this solid haloalkane.
For competition experiments the different substrates weremixed before the catalyst was added. For example, 10 mmol ofCHCl3 and 10mmol of CDCl3 were mixed at room temperatureand 0.02 mmol (290.6 mg) of TpBr3Ag was added. After 15 minof stirring, 0.1 mmol of EDAwas added in one portion, and themixture was stirred for 8 h. After that time, the mixture wasinvestigated by 1H NMR, the relative amount of insertionproducts obtained from each substrate being established byintegration.
Asymmetric Induction.The catalyst precursorwas prepared asdescribed in the literature.30 (-)-2,20-Isopropylidenebis[(4S)-4-phenyl-2-oxazoline] (0.03 mmol, 10 mg) was dissolved in 15 mLof dichloroethane, and 0.025mmol of silver triflate was added tothe stirring solution. After 1.5 h the solvent was removed undervacuum and the residue was employed as the catalyst precursorin the reaction of methylene chloride with EDA, following theabove procedure. After 36 h of stirring, GC studies revealedca. 50%of EDA consumed. Volatiles were removed at 0 �C, and
Scheme 13. Overall Mechanistic Proposal for the Reactivity of
Haloalkanes CnH2nþ2-mXm with [TpBr3AgdCHCOOEt]([Ag] = TpBr3Ag)
(36) Drudis-Sole, G.; Maseras, F.; Lledos, A.; Vallribera, A.;Moreno-Manas, M. Eur. J. Org. Chem. 2008, 5614.

Article Organometallics, Vol. 28, No. 20, 2009 5981
the residue was purified by column chromatography (SiO2, 9:1petroleum ether/diethyl ether as eluent) to give the product as anoil.
The procedure to measure ee by NMR was the following. Astock solution of europium tris[3-(heptafluoropropylhydroxy-methylene)-(þ)-camphorate] (75 mg in 0.5 mL of C6D6) wasprepared. A separate solution of the product was prepared bydissolving 5 μL of the isolated oil in 0.5 mL of C6D6. Afterseveral additions of the europium solution, the enantiomerswere resolved by adding ca. 200 μL of the Eu3þ solution to thatof the product. Integrationof the resonances gave a ca. 14(2%ee.X-ray Studies. Experimental procedures and cif files for the
determination of the structures of complexes 1, 2, and 3 are givenin the Supporting Information. CCDC 687284, 687285, and687286, respectively, contain the supplementary crystallo-graphic data for the compounds described in this paper. Thesedata can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge CrystallographicData Centre, 12 Union Road, Cambridge CB21EZ, UK; fax:þ44 1223-336-033; or [email protected]).Kinetic Studies. A solution of the catalyst (0.02 mmol) in
methylene chloride (4 mL) was placed into a 25 mL round-bottom flask connected to a computer-connected pressuregauge. Ethyl diazoacetate was added in one portion (0.5 mmol),and the evolution of nitrogen was monitored until the pressurewas maintained constant, i.e., until all EDA was consumed.Calculations. Calculations were performed at the DFT level
by means of the B3LYP functional37 using the Gaussian suite ofprograms.38 Ag and Br atoms were described using an effectivecore potential (LANL2DZ)39 for the inner electrons and its
associated valence double-ζ basis set for the outer ones. Polar-ization shells, of f and d type, were added for Ag and Br,respectively.40 The 6-31G(d) basis set was used for H, B, C, N,O, and Cl atoms.41 The structures of reactants, intermediates,transition states, and products were fully optimized with nosymmetry restrictions. Transition states were identified by hav-ing one negative eigenvalue in the Hessian matrix, and theirconnectivity to intermediates was checked through small stepgeometry optimizations from the transition state. All reportedenergies are potential energies. This method was proved to givecorrect results in a previous study by our groups,29 where it wascalibrated against CCSD(T) calculations, and entropic andsolvation effects were shown to have a minor effect.
Acknowledgment. Financial support from MICINN(GrantsCTQ2008-06866-CO2-02/BQU,CTQ2008-00042/BQU, Consolider Ingenio 2010 CSD2006-0003), theCatalan DURSI (project 2005SGR00715), the Junta deAndalucıa (Grant P07-FQM 2870), and the ICIQfoundation is acknowledged. J.U. also thanks the Juntade Andalucıa for a student fellowship. A.A.C.B. thanksthe Spanish MICINN for a “Juan de la Cierva” contract.
Supporting Information Available: Full reference for theGaussian03 program, Cartesian coordinates, and summary ofenergetics for all stationary points. Thismaterial is available freeof charge via the Internet at http://pubs.acs.org.
(37) (a) Lee, C.; Parr, R. G.; Yang, W. Phys. Rev. B 1988, 37, 785.(b) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (c) Stephens, P. J.; Devlin,F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623.(38) Frisch, M. J. Gaussian 03, Revision C.02; Gaussian, Inc.:
Wallingford, CT, 2004.(39) Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284.
(40) (a) Ehlers, A. W.; B€ohme, M.; Dapprich, S.; Gobbi, A.;H€ollwarth, A.; Jonas, V.; K€ohler, K. F.; Stegmann, R.; Veldkamp, A.;Frenking, G.Chem. Phys. Lett. 1993, 208, 111. (b) H€ollwarth, A.; B€ohme,M.; Dapprich, S.; Ehlers, A. W.; Gobbi, A.; Jonas, V.; K€ohler, K. F.;Stegmann, R.; Veldkamp, A.; Frenking, G. Chem. Phys. Lett. 1993, 208,237.
(41) Francl, M. M.; Pietro, W. J.; Hehre, W. J.; Binkley, J. S.;Gordon,M. S.; Defrees,D. J.; Pople, J. A. J. Chem. Phys. 1982, 77, 3654.





























