Embed Size (px)
Citation preview

J. gen. Virol. (1986), 67, 1361-1371. Printed in Great Britain
Key words: MPS V/retrovirus vectors/ transformation/v-mos
1361
The Myeloproliferative Sarcoma Virus Retains Transforming Functions after Introduction of a Dominant Selectable Marker Gene
By W O L F R A M O S T E R T A G , 1. B A R B A R A S E L I G E R , t'2 R E G I N E K O L L E K , 1 C A R O L S T O C K I N G , 1 U L L A B E R G H O L Z ~ AND
F L O R E N C E S M A D J A - J O F F E 3
t Heinrich-Pette-Institutfi~r Experimentelle Virologie und Immunologie an der Universitiit Hamburg, Martinistrasse 52, 2000 Hamburg 20, F.R.G., 2Klinische Arbeitsgruppe 'Biologische Regulation der Wirt-Tumor-Interaktion" der Max-Planck-Gesellschaft, Gosslerstrasse lOd, 3400
Gifttingen, F.R.G. and 3 Unitk 268 I N S E R M , Oncog~nbse Appliqu~e, H6pital Paul Brousse, 14 et 16 avenue Paul Vaillant Couturier, 94800 Villejuif, France
(Accepted 20 March 1986)
SUMMARY
The dominant neomycin resistance gene (neo R) was introduced into the genome of the myeloproliferative sarcoma virus (MPSV), a replication-defective retrovirus carrying the mos oncogene. The resulting selectable neoR-MPSV virus did not lose its acute transforming property, unlike the results of attempts by other groups to insert marker genes into oncogenic viruses. NeoR-MPSV DNA was used to generate infectious virus by transfection followed by rescue with Friend or Moloney murine leukaemia virus. Infection of fibroblasts with this virus resulted in morphologically transformed cells which were resistant to the neomycin analogue G418. Segregation of the two functions (transformation and G418 resistance) was not observed in more than 500 independent viral transfers to fibroblasts. Furthermore, neoR-MPSV retained the leukaemogenesis-inducing properties of the wild-type virus. Myeloproliferation and G418-resistance transfer did not segregate after passage in mice.
INTRODUCTION
Retroviruses are naturally occurring eukaryotic cell transducing vectors, and thus provide an ideal way of introducing foreign genes into eukaryotic cells (Shimotohno & Temin, 1981 ; Tabin et al., 1982; Perkins et al., 1983; Lewis et al., 1984; Miller et al., 1984; Williams et al., 1984). Their ability to integrate stably into host chromosomal DNA and their efficient transmission to a wide variety of cell types make them a gene transfer system which is superior to the calcium phosphate method of transfection (Graham & van der Eb, 1973; Wigler et al., 1977, 1978). Furthermore, virus-introduced genes express 10- to 50-fold higher levels of transcription compared to those introduced by DNA-mediated gene transfer (Hwang & Gilboa, 1984).
The introduction of selectable markers into the retrovirus genome greatly extends the utility of such a transducing vector, facilitating the screening and isolation of infected cells. Several groups have reported the generation of selectable, infectious retroviruses by recombination with a marker gene, either regulated by its own promoter or by the functional promoter within the retrovirus long terminal repeats (LTRs). When highly transforming retroviruses were used, however, the resultant recombinant virus often had lost its transforming activity (Joyner & Bernstein, 1983a, b; Tarpley & Temin, 1984). Only two viral oncogenes, Ha-ras (Wei et al., 1981) and src (Tarpley & Temin, 1984), have been combined with a selectable recessive marker gene, the herpes simplex virus thymidine kinase gene (HSV-tk), to generate vectors which stably co-express the transforming function and the selectable marker gene.
We describe in this work the construction of a dominant, selectable transforming retrovirus vector by the introduction of the transposon 5 (Tn5) neomycin resistance (neo g) gene into the defective pol gene of the myeloproliferative sarcoma virus (MPSV). MPSV is a unique member
0000-6872 © 1986 SGM

1362 W. OSTERTAG AND OTHERS
of the Moloney murine sarcoma virus (Mo-MSV) family which not only transforms fibroblasts in vitro and induces sarcomas in adult mice, but also causes extensive changes in the haematopoietic system (Ostertag et al . , 1980). The molecular analysis of the MPSV genome has shown that it is composed entirely of Moloney murine leukaemia virus (Mo-MuLV) and cellular mos oncogene (c-mos) related sequences, as is the Mo-MSV (Pragnell et al. , 1981 ; Kollek et al . , 1984; Stacey et al. , 1984). The unique leukaemogenesis-inducing activity of MPSV requires cooperative interaction of the MPSV 3' LTR region, which differs slightly but signifcantly from those of either Mo-MuLV and Mo-MSV, and the mos oncogene (Stocking et al . , 1985).
Transfected cell lines or established non-producer cell lines were superinfected with helper virus, resulting in release of infectious pseudotypes that confer resistance to G418, a neomycin analogue, to infected fibroblasts without loss of mos expression. A series of experiments was designed to estimate the segregation frequency of the two genes on passage of the modified virus to fibroblasts and to haematopoietic cells of the mouse.
METHODS
Cell lines. Cell lines used in these studies and referred to in the figure were as follows: RAT1, NIH 3T3 and NRK (normal rat kidney) cells were used for transfection and infection experiments. RAT1 and NRK are rat fibroblast cell lines (Ostertag et al., 1980; Topp, 1981). N IH 3T3 cells were obtained from Dr R. A. Weinberg. Cell line 643/22N is a clone of SCI cells, which releases high titres of twice-cloned Friend helper virus (F-MuLV) (Bilello et at., 1980; Ostertag et al., 1980). Cell line Mo-MuLV CI 2 c, provided by R. Jaenisch, is a NIH 3T3 cell clone that releases high titres of cloned Mov-3 Mo-MuLV. All cell lines were grown in modified Eagle's medium supplemented with 10~o foetal calf serum.
Plasmids. The construction of the plasmid vector neoR-MPSV used for transfection experiments is described in Results. The neomycin resistance gene from pACYC neo R (Chang & Cohen, 1978) was ligated into the wild-type MPSV plasmid p18-663 (Kollek et al., 1984). Plasmid pAG60 (Colb~re-Garapin et al., 1981), which contains the HSV-tk transcription controls and the neo R coding region, was used as control for transfection experiments (see below), as well as for a nick-translated probe. Plasmid pmSI (Oskarsson et al., 1980) containing the c-mos gene was used to detect mos homologous sequences. Large-scale plasmid preparations were purified by CsCl-ethidium bromide density gradient centrifugation.
TransJbction experiments. These were performed as described previously (Wigler et al., 1978; Kollek et al., 1984). A calcium phosphate precipitate prepared with 0.1 ~tg to 1-0 ~tg MPSV plasmid DNA in the presence of 30 p,g of high molecular weight DNA from calf thymus was added to 5 x 105 cells/T75 flask. For selection ofneo R colonies, cells were grown in medium containing 400 ~tg/ml G418 (Gibco). Cells resistant to the antibiotic appeared 2 to 4 weeks after DNA-mediated gene transfer.
Virus rescue and virus assays. The neo R colonies were superinfected with F-MuLV or Mo-MuLV and the titre of infectious neoR-MPSV virus was determined by measuring reverse transcriptase activity, fibroblast focus formation (as £f.u.) and spleen focus formation (s.f.f.u.). The infectious virus was obtained either directly from spleens or from cell culture supernatants (Ostertag et al., 1980). In some experiments virus from tissue culture supernatants was concentrated 10- to 100-fold with a DC hollow fibre apparatus followed by a second step of concentration and dialysis with an Amicon type XM300 membrane (Ostertag et al., 1980).
The reverse transcriptase activity was determined using a simplified procedure described earlier (Pragnell et al., 1977), and corrected for variations by using viral supernatant of Friend cells as a standard. The fibroblast focus formation assay was carried out with N RK or RAT1 fibroblasts (Ostertag et at., 1980). Spleen focus formation was assayed by injecting the filtered viral supernatant into the lateral tail vein of DBA/2J mice. Serial viral dilutions were made and three mice were injected with 0-5 ml of viral suspension for each virus concentration. Spleens were removed, fixed, and spleen foci were counted 16 and 21 days later (Ostertag et al., 1980).
Southern and Northern blot analysis. Cellular DNA was extracted as described previously (Miller et al., 1984): DNA samples were digested with restriction endonucleases (Bethesda Research Laboratories) under the conditions recommended by the supplier and separated on a 0.8 ~ agarose gel in TEA buffer (40 mM-Tris-OH, 20 raM-sodium acetate, 20 mM-NaCI, 2 mM-EDTA, pH 8. l). The gels were treated and the DNA was transferred to Gene Screen (New England Nuclear) filters (Southern, 1975). The filters were hybridized to 3zp-labelled nick- translated mos or neo R probes (see Plasmids).
Total cellular RNA was extracted (Auffray & Rougeon, 1980), glyoxylated and subjected to agarose gel electrophoresis (20 ~tg/slot) (McMaster & Carmichael, 1977). The RNA was transferred to Gene Screen as recommended by the supplier and hybridized to 32p-labelled mos and neo R probes.
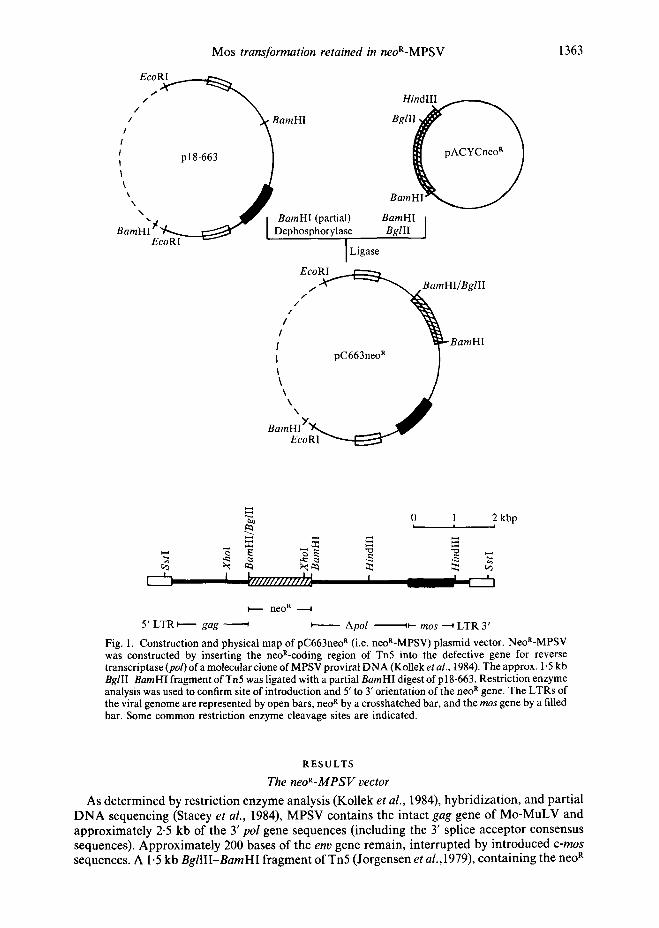
Mos transformation retained in neoR-MPSV 1363
EcoRI
/ /
/
I I
I I
\
k
Ban
p18-663
EcoRI
H i n d l ~ - - ~
• BamHl B g l I ( p A C Y C n e o ~
I I Dephosphorylase BgllI
I Ligase
E c o R I j ~ . . { ~ . _ HI/BglII
i/////"~ -
I ~ B a m H I
'~ 0 1 2 kbp
I I t : : / / # / / / / / , , / ) z l . I I I t l
v--- neo R
5' LTR I gag ~ : Apol ~ mos ~ LTR 3'
Fig. 1. Construction and physical map of pC663neo rt (i.e. neoR-MPSV) plasmid vector• NeoR-MPSV was constructed by inserting the neoR-coding region of Tn5 into the defective gene for reverse transcriptase (pol) of a molecular clone of MPSV proviral DNA (Kollek et al., 1984). The approx. 1.5 kb BglII-BamHI fragment of Tn5 was ligated with a partial Barn HI digest of p l 8-663. Restriction enzyme analysis was used to confirm site of introduction and 5' to 3' orientation of the neo R gene. The LTRs of the viral genome are represented by open bars, neo R by a crosshatched bar, and the mos gene by a filled bar. Some common restriction enzyme cleavage sites are indicated.
RESULTS
The neoR-MPSV vector
As de te rmined by restr ict ion enzyme analysis (Kollek et al., 1984), hybr id iza t ion , and par t ia l D N A sequencing (Stacey et al., 1984), MPSV conta ins the in tact gag gene of M o - M u L V and approximate ly 2.5 kb of the 3' pol gene sequences ( inc luding the 3' splice acceptor consensus sequences). Approx imate ly 200 bases of the env gene remain , in te r rupted by in t roduced c-mos sequences. A 1.5 kb BgllII-BamHI f ragment of Tn5 (Jorgensen et al., 1979), con t a in ing the neo R

1364 W. OSTERTAG AND OTHERS
coding region but not the eukaryotic transcription signals from the plasmid pACYC neo R, was inserted into the first BamHI site of the defective MPSV gene for reverse transcriptase (pol) (Fig. 1). The neo R gene confers resistance to the antibiotic G418 in eukaryotes (Chang & Cohen, 1978). Under conditions in which transcription of the introduced neo R gene is initiated from the viral LTR, we postulated that translation of the protein would be possible in a manner similar to that of thepolgene of Mo-MuLV (Weiss et al., 1982). It was further assumed that an insert in the already defective pol gene would not interfere with the expression of the mos oncogene.
Transfer o f the neoR-MPSV DNA to fibroblasts and virus rescue
Experiments were carried out to answer the following questions. (i) Is the neo R gene expressed in the frame of the MPSV genome? (ii) Is MPSV still expressing mos fibroblast-transforming functions? (iii) Are haematopoietic cell-transforming functions still being expressed ? (iv) Do the two genes (mos and neo R) segregate and what is the frequency of segregation?
Unlike other spleen focus-forming virus isolates (Ostertag & Pragnell, 1982), MPSV also transforms fibroblasts, thus allowing expression of both neo R and the mos oncogene to be tested directly in fibroblasts. RAT1 and NIH 3T3 fibroblasts were utilized for transfection experiments with either neoR-MPSV plasmid DNA or pAG60 DNA as a parallel control. Selection was applied 2 days after transfection by adding 400 ~tg/ml G418 to the culture medium. Colonies of resistant cells were counted 3 weeks after initiation of selection. Similar numbers of resistant colonies were obtained at equal molarities of each plasmid DNA (approx. 200 colonies/0.1pmol), indicating that the potential for neo R expression was not altered by its inclusion in the MPSV genome. Furthermore, all resistant colonies obtained on transfection with neoR-MPSV DNA appeared transformed, in contrast to the resistant colonies obtained from control pAG60 DNA transfer experiments. This was to be expected if viral transforming functions (mos gene expression) and G418 resistance are expressed coordinately in the same viral genome.
Several primary and independently isolated neoR-MPSV-transfected RAT1 clones were infected with F-MuLV. Released virus was tested for focus formation on N R K fibroblasts. Clone RAT1-4Mneo had the highest titre of focus-forming virus (2 × 105 to 2 × 106 f.f.u./ml of tissue culture supernatant) and also proved to be unusually stable. Virus of Ratl-4Mneo cells was therefore used exclusively to obtain secondary non-producer cell lines.
Generation o f non-producer cell lines: transformed cells are resistant to G418
Cell lines transfected by neoR-MPSV were very often unstable and contained a variable copy number and size of the newly introduced genes (Kollek et al., 1984; and see below). To obtain cell lines with a single, stably integrated viral copy, virus from RAT1-4Mneo cells at limiting dilutions was used to infect N R K cell lines. Foci were picked and cloned in agar. Cloned cell lines were tested for virus release and non-producer cell lines were retained. All transformed non-producer clones proved to be resistant to G418.
The clonability of transformed cloned cell lines in the presence of G418 was determined in order to test whether cells selected on the basis of their transformed phenotype expressed the neo R gene at sufficient levels to enable growth under G418 selection or whether variant cells were subsequently generated in which the expression of the neo R gene had been suppressed when selective pressure was not employed. Because normal N R K cells cannot be cloned efficiently in agar, the transformed cell clone N R K no. 10 Ag2 obtained by infection with cloned wild-type MPSV virus (Kollek et al., 1984) was used as control. In the presence of 400 ~tg G418/ml medium, no resistant colonies were obtained with the control cell line, whereas all of the non-producer cell clones obtained by infection with neoR-MPSV proved to be resistant. The cloning efficiency with G418 selection in medium was comparable to that without G418 selection in agar (Table 1). These data thus indicate that the two functions, G418 resistance and mos-mediated transformation, were not segregating.

(a) 1 2 3
Mos transformation retained in neoR-MPSV
4 5 6 7 (b) 1 2 3 4 5 6
1365
7
8.1
6.1
4.5
2.6
Fig. 2. Southern blot analysis of neoR-MPSV cell lines. Genomic DNAs were digested with SstI restriction enzyme, separated on a 0.8 ~ agarose gel, and transferred to a nitrocellulose filter. The filter was probed with (a) mos-specific and (b) neoR-specific 32p-labelled sequences. Cell lines analysed were RAT1 (lane 1), RAT1-4Mneo (a transfected cell line) (lane 2) and five NRK non-producer cell clones obtained by infection with virus from cell clone 1-4Mneo: clone no. 10 (lane 3), clone no. 12 (lane 4), clone no. 21 (lane 5), clone no. 23 (lane 6) and clone no. 32 (lane 7).
Table 1. Expression o f the transformed phenotype and the selectable dominant marker neo R o f neoR-MPSV non-producer clones
Cloning efficiency (~o) in* A
Cell line MEM Agar MEM + G4181" Agar + G418t NRK 56 12 0 0 NRK no. 10 Ag2:~ ND§ 30-5 0 0 neoR-MPSV NRK no. 10 56 70 42 41 neoR-MPSV NRK no. 12 39 36 37 35 neoR-MPSV NRK no. 21 31 24.5 28 34-5 neoR-MPSV NRK no. 23 42 30 38 31 neoR-MPSV NRK no. 32 28 26.5 26 34
* The cloning efficiency was calculated from at least two separate experiments. t 400 ~tg/ml G418. :~ NRK no. 10 Ag2 is an agar-derived non-producer clone of molecularly cloned wild-type MPSV. § ND, Not done.
Southern and Northern blot analysis o f non-producer cell lines transfected or infected by neoR-MPS V
High molecular weight D N A of the clone RAT 1-4Mneo obtained by transfection and of five neoR-MPSV non-producer N R K cell lines obtained by infection with virus from cell clone RAT1-4Mneo was analysed by Southern blot hybridization to determine copy number and size of integrated neoR-MPSV genomes. The restriction enzyme SstI cut within the retrovirus LTR, generating a neoR-MPSV-specific fragment of 8.1 kb (Fig. 1 and 2). An SstI digest of clone RAT1-4Mneo showed multiple full-length as well as altered copies of the neoR-MPSV genomes, whereas all clones obtained by infection contained a single unaltered copy. Clone no. 12 contained an additional altered copy. All full-length bands hybridized to both the mos and neo probes.
Total cellular R N A from RAT1-4Mneo clone no. 23 was isolated and subjected to Northern blot analysis. The filter was hybridized to a mos probe and subsequently re-hybridized to a neo R probe. A full-length band of approx. 8-0 kb was detected with both probes and two additional
1366 w. OSTERTAG AND OTHERS
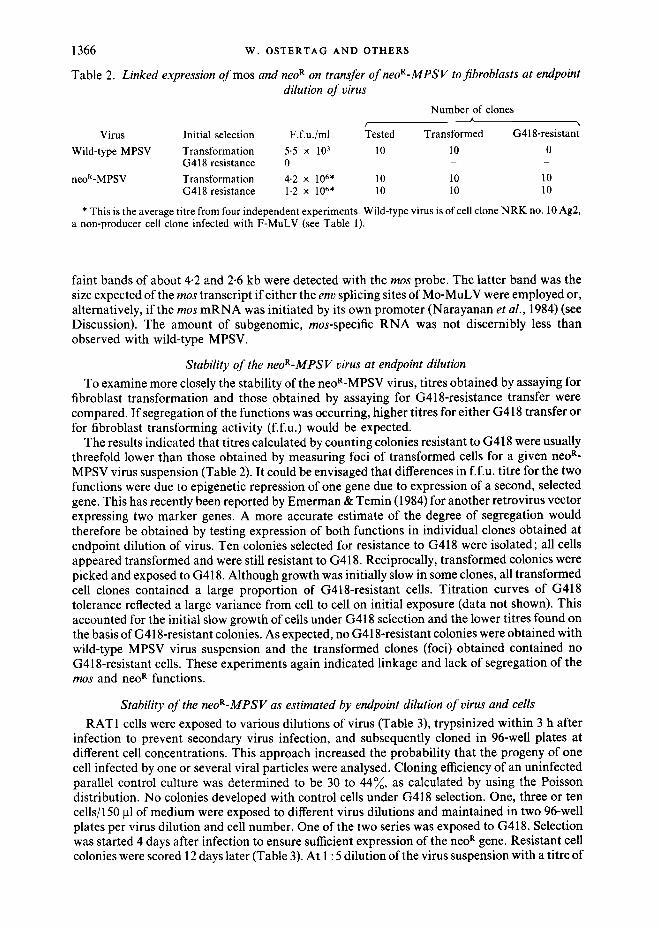
Table 2. Linked expression ofmos and neo R on transfer o f neoR-MPSV to fibroblasts at endpoint dilution o f virus
Number of clones A
Virus Initial selection F.f.u./ml Tested Transformed G418-resistant Wild-type MPSV Transformation 5.5 x 103 10 10 0
G418 resistance 0 - - - neoR-MPSV Transformation 4.2 x 106* 10 10 10
G418 resistance 1.2 x 106* 10 10 10
* This is the average titre from four independent experiments. Wild-type virus is of cell clone NRK no. 10 Ag2, a non-producer cell clone infected with F-MuLV (see Table 1).
faint bands of about 4.2 and 2.6 kb were detected with the mos probe. The latter band was the size expected of the mos transcript if either the env splicing sites of Mo-MuLV were employed or, alternatively, if the mos m R N A was initiated by its own promoter (Narayanan et al., 1984) (see Discussion). The amount of subgenomic, mos-specific R N A was not discernibly less than observed with wild-type MPSV.
Stability o f the neoR-MPSV virus at endpoint dilution
To examine more closely the stability of the neoR-MPSV virus, titres obtained by assaying for fibroblast transformation and those obtained by assaying for G418-resistance transfer were compared. I f segregation of the functions was occurring, higher titres for either G418 transfer or for fibroblast transforming activity (f.f.u.) would be expected.
The results indicated that titres calculated by counting colonies resistant to G418 were usually threefold lower than those obtained by measuring foci of transformed cells for a given neo R- MPSV virus suspension (Table 2). It could be envisaged that differences in f.f.u, titre for the two functions were due to epigenetic repression of one gene due to expression of a second, selected gene. This has recently been reported by Emerman & Temin (1984) for another retrovirus vector expressing two marker genes. A more accurate estimate of the degree of segregation would therefore be obtained by testing expression of both functions in individual clones obtained at endpoint dilution of virus. Ten colonies selected for resistance to G418 were isolated; all cells appeared transformed and were still resistant to G418. Reciprocally, transformed colonies were picked and exposed to G418. Although growth was initially slow in some clones, all transformed cell clones contained a large proportion of G418-resistant cells. Titration curves of G418 tolerance reflected a large variance from cell to cell on initial exposure (data not shown). This accounted for the initial slow growth of cells under G418 selection and the lower titres found on the basis of G418-resistant colonies. As expected, no G418-resistant colonies were obtained with wild-type MPSV virus suspension and the transformed clones (foci) obtained contained no G418-resistant cells. These experiments again indicated linkage and lack of segregation of the mos and neo R functions.
Stability o f the neoR-MPSV as estimated by endpoint dilution o f virus and cells
RAT1 cells were exposed to various dilutions of virus (Table 3), trypsinized within 3 h after infection to prevent secondary virus infection, and subsequently cloned in 96-well plates at different cell concentrations. This approach increased the probability that the progeny of one cell infected by one or several viral particles were analysed. Cloning efficiency of an uninfected parallel control culture was determined to be 30 to 44~ , as calculated by using the Poisson distribution. No colonies developed with control cells under G418 selection. One, three or ten cells/150 ~tl of medium were exposed to different virus dilutions and maintained in two 96-well plates per virus dilution and cell number. One of the two series was exposed to G418. Selection was started 4 days after infection to ensure sufficient expression of the neo R gene. Resistant cell colonies were scored 12 days later (Table 3). At 1 : 5 dilution of the virus suspension with a titre of
Mos transformation retained in neoR-MPSV
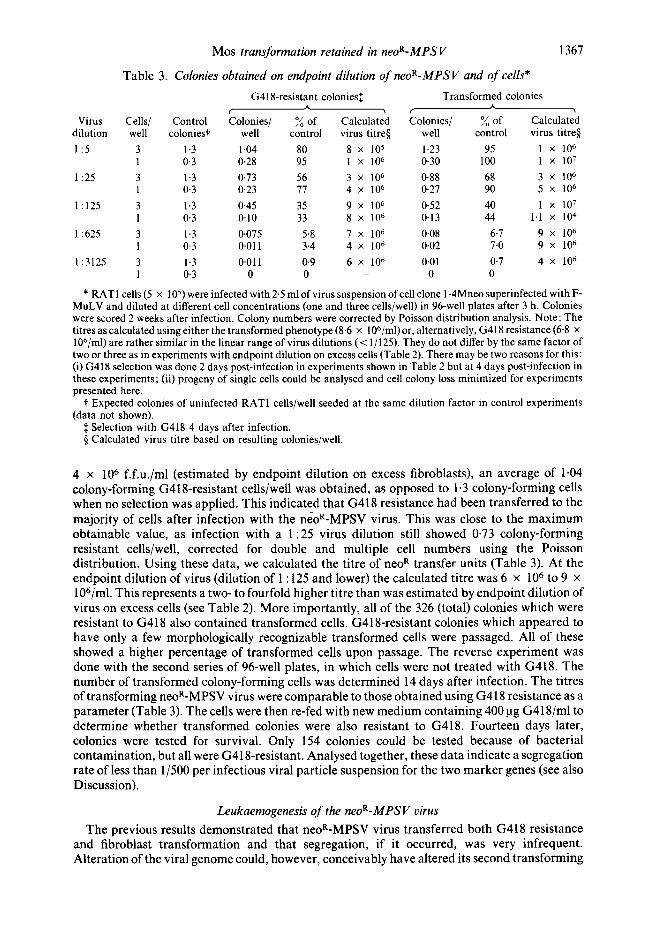
Table 3. Colonies obtained on endpo&t dilution of neoR-MPSV and of cells*
1367
G418-resistant colonies:~ Transformed colonies A A
Virus C e l l s / Control Colonies/ ~ of Calculated Colonies/ ~ of Calculated dilution well colonies? we l l control virus titre§ well control virus titre§ 1:5 3 1.3 1.04 80 8 × 105 1.23 95 1 x 106
1 0-3 0.28 95 1 × 106 0.30 100 1 × 107 1:25 3 1-3 0.73 56 3 × 106 0.88 68 3 × 106
1 0.3 0.23 77 4 × 106 0.27 90 5 × 106 1:125 3 1.3 0.45 35 9 x 106 0.52 40 1 x 107
1 0.3 0.10 33 8 x 106 0.13 44 1-1 × 104
1:625 3 1.3 0-075 5.8 7 x 106 0.08 6.7 9 × 106 1 0.3 0.011 3.4 4 × 106 0.02 7.0 9 × 106
1:3125 3 1-3 0.011 0.9 6 × 106 0.01 0.7 4 × 106 1 0.3 0 0 0 0
* RAT1 cells (5 × 105) were infected with 2.5 mlofvirussuspensionofcellclone 1-4Mneosuperinfected withF- MuLV and diluted at different cell concentrations (one and three cells/well) in 96-well plates after 3 h. Colonies were scored 2 weeks after infection. Colony numbers were corrected by Poisson distribution analysis. Note: The titres as calculated using either the transformed phenotype (8.6 × 106/ml)or, alternatively, G418 resistance (6"8 x 106/ml) are rather similar in the linear range of virus dilutions (< 1/125). They do not differ by the same factor of two or three as in experiments with endpoint dilution on excess cells (Table 2). There may be two reasons for this: (i) G418 selection was done 2 days post-infection in experiments shown in Table 2 but at 4 days post-infection in these experiments; (ii) progeny of single cells could be analysed and cell colony loss minimized for experiments presented here.
t Expected colonies of uninfected RAT1 cells/well seeded at the same dilution factor in control experiments (data not shown).
:~ Selection with G418 4 days after infection. § Calculated virus titre based on resulting colonies/well.
4 x 106 f.f.u./ml (estimated by endpoint dilution on excess fibroblasts), an average of 1-04 colony-forming G418-resistant cells/well was obtained, as opposed to 1.3 colony-forming cells when no selection was applied. This indicated that G418 resistance had been transferred to the majority of cells after infection with the neoR-MPSV virus. This was close to the maximum obtainable value, as infection with a 1:25 virus dilution still showed 0-73 colony-forming resistant cells/well, corrected for double and multiple cell numbers using the Poisson distribution. Using these data, we calculated the titre of neo R transfer units (Table 3). At the endpoint dilution of virus (dilution of 1:125 and lower) the calculated titre was 6 x 106 to 9 x 106/ml. This represents a two- to fourfold higher titre than was estimated by endpoint dilution of virus on excess cells (see Table 2). More importantly, all of the 326 (total) colonies which were resistant to G418 also contained transformed cells. G418-resistant colonies which appeared to have only a few morphologically recognizable transformed ceils were passaged. All of these showed a higher percentage of transformed cells upon passage. The reverse experiment was done with the second series of 96-well plates, in which cells were not treated with G418. The number of transformed colony-forming cells was determined 14 days after infection. The titres of transforming neoR-MPSV virus were comparable to those obtained using G418 resistance as a parameter (Table 3). The cells were then re-fed with new medium containing 400 ~tg G418/ml to determine whether transformed colonies were also resistant to G418. Fourteen days later, colonies were tested for survival. Only 154 colonies could be tested because of bacterial contamination, but all were G418-resistant. Analysed together, these data indicate a segregation rate of less than 1/500 per infectious viral particle suspension for the two marker genes (see also Discussion).
Leukaemogenesis of the neoR-MPSV virus
The previous results demonstrated that neoR-MPSV virus transferred both G418 resistance and fibroblast transformation and that segregation, if it occurred, was very infrequent. Alteration of the viral genome could, however, conceivably have altered its second transforming

1368 W. OSTERTAG AND OTHERS
Table 4. Leukaemogenic action o f neoR-MPSV
Ratio S.f.f.u./ml* F.f.u./ml s.f.f.u. : f.f.u.
Wild-type MPSV 5 x 102 6 × 103 0.08 neoR-MPSV 4.7 x 102 6.6 X 10 3 0"07
* Spleen foci were counted 14 to 16 days post-injection with wild-type MPSV and 18 to 21 days post-injection with neoR-MPSV.
action, that on haematopoietic cells. Therefore, six DBA/2J mice were injected intravenously for each dilution of a fivefold serial dilution of neoR-MPSV virus. Mice infected at high virus concentrations developed the typical myeloproliferative syndrome with excessive proliferation of all cells of the myeloid linkage. The disease developed, however, more slowly than with wild- type virus. Spleen foci induced by neoa-MPSV were therefore counted at days 18 to 21 post- injection, compared to wild-type MPSV virus where foci were counted 14 to 16 days post- injection. The ratio of spleen focus-forming units to fibroblast focus-forming units was only slightly lower with the neoR-MPSV virus than with the molecularly cloned wild-type virus (Table 4).
The integrity of the neoR-MPSV virus after passage through mice was determined. Virus of spleen homogenates from mice was used to infect N R K cells and titres were counted when selection with G418 was made (0.5 x 106) and compared with the titres obtained after selection for the transformed phenotype (4.2 x 106) . Once again, individual clones were analysed to obtain a more accurate estimate of segregation. Ten transformed clones were picked and were grown in the presence of G418; conversely, ten G418-resistant clones were tested for transformation. All clones checked expressed both phenotypes.
DISCUSSION
Retrovirus oncogenes (v-onc) were originally considered to be peculiar features of oncogenic retroviruses, and of only theoretical interest for the study of malignant transformation in humans. This conclusion has changed radically during the last few years and it is now generally accepted that activated or mutated c-onc genes are prerequisites for generating 'spontaneously' arising transformed cells in humans or vertebrates (Der et al., 1982; Parada et al., 1982; Santos et al., 1982).
Direct studies of the transformation process of the oncogenes have been hampered by many obstacles: first, it is difficult to culture and identify target cells; second, introduced genes are not expressed in all cells; third, inherent differences exist between the oncogenes found in the cellular genome and in the transforming retrovirus, making interpretations difficult. We have characterized here a retrovirus vector, neoR-MPSV, that overcomes some of these difficulties.
The use of selectable dominant markers in conjunction with an oncogene-carrying retrovirus vector allows selection of infected cells immediately after transfer of the oncogene by elimination of cells that have not incorporated the transduced genes. With a few exceptions (Wei et al., 1981 ; Tarpley & Temin, 1984), the alteration in the genome of any transforming retrovirus by introduction of marker genes was connected with unexpected difficulties: most such constructs are either highly unstable or coordinate expression of the oncogene and selectable marker gene could not be obtained (Varmus et al., 1981 ; Chen & Temin, 1982; Joyner & Bernstein, 1983 b). Insertion of the marker gene in the region that functions as an intron for the mRNA of the oncogene often inhibits expression of the transforming gene.
The neoR-MPSV vector was, however, able to transmit and express both the mos oncogene and the neo R gene on transfection and following viral infection. More than 500 independently isolated cell lines, selected either by their transformed phenotype or by G418 resistance after viral transfer, proved to express both transduced genes. These results indicate that not only can MPSV be used as a neoR-transducing retrovirus vector, but inclusion of the selectable gene into the genome does not disrupt mos expression. It has been postulated that the mos gene in the Mo- MSV genome is expressed via splicing, utilizing the env splice acceptor sites 5' to the mos gene

Mos transformation retained in neoR-MPSV 1369
(Weiss et al., 1982; Nash et al., 1984). Although recent work has also indicated the importance of intron sequences in the pol gene for sufficient splicing of env in Mo-MuLV (Hwang et al., 1984), the varying pol deletions found to date in Mo-MSV variants (Weiss et al., 1982; Kollek et al., 1984) seem to indicate that sufficient mos expression is independent of some rearrangements and deletions in the intron sequences. Alternatively, some reports suggest that the transcription of the mos gene in the Mo-MSV genome is initiated by its own promoter and enhanced by sequences in the 3' LTR (Narayanan et al., 1984). If a similar promoter is used for expression of mos in the MPSV genome, the enhancement of mos transcription would not be affected by the insertion of the neo R gene 5' to the putative promoter.
Earlier work has shown that MPSV is an exceptional member of the Mo-MSV family in having a dual transforming potential in both fibroblasts and cells of the haematopoietic compartment (Ostertag et al., 1980; Ostertag & Pragnell, 1982), due to unique properties of the MPSV LTR that have altered the target cell specificity (Stocking et al., 1985). MPSV, as compared to the conventional Mo-MSV and to Mo-MuLV, could possibly confer the advantage of an increased target cell range, as compared to vectors using Mo-MuLV or Mo-MSV LTRs, and hence expression of the transduced gene in a wider range of cell types. The neoR-MPSV virus not only transforms fibroblasts as efficiently as the wild-type virus (see above), but also causes leukaemogenic transformation in adult mice as shown by spleen focus formation (Table 4), spleen enlargement, induction of anaemia, and myeloproliferation (data not shown). The delayed onset of leukaemic symptoms after infection with neoR-MPSV into mice is at present unexplained, although it could reflect a somewhat lower expression of mos not discernible with Northern analysis. Virus derived from leukaemic animals was similarly stable and segregation had not occurred, indicating that loss or alteration of the neo R function is not necessary for induction of leukaemogenesis. Moreover, infection of haematopoietic stem cell cultures by neo R- MPSV and selection of neoR-resistant colonies shows release of intact transforming virus in all of 30 such colonies which were screened (G. Johnson & W. Ostertag, unpublished).
We have described in this work a dominant selectable retrovirus vector facilitating the study of the mos oncogene in haematopoietic differentiation and mouse development. Unlike other v- onc genes, which have accumulated mutations making the oncogenes divergent from the original c-onc genes (Weiss et al., 1982), v-mos and c-mos are interchangeable in their transformation potential (Blair et al., 1981 ; Stocking et al., 1985). Deletion and temperature-sensitive mutants of MPSV (Kollek et al., 1984; Ostertag et al., 1984; Stocking et al., 1985) can be utilized together with the neo R marker gene to define further the role of the mos oncogene. Use of psi-2 cells or of similar cell lines (Mann et al., 1983) would eliminate the problems inherent in utilization of helper virus during transfer.
We thank Marion Ziegler and G6khan Arman for excellent technical assistance. Some of this work has been done for part of the Ph.D. thesis of B.S. We acknowledge support of the Deutsche Krebshilfe and the Deutsche Forschungsgemeinschaft. The Heinrich-Pette-Institut is supported by Freie und Hansestadt Hamburg and
• Bundesministerium f/Jr Jugend, Familie und Gesundheit.
REFERENCES AUFFRAY, C. & ROUGEON, F. (1980). Purification of mouse immunoglobulin heavy-chain messenger RNA from total
myeloma tumor RNA. European Journal of Biochemistry 107, 303-314. BILELLO, J. A., COLLETTA, G., WARNECKE, G., KOCH, G., FRISBY, D., PRAGNELL, I.B. & OSTERTAG, W. (1980). Analysis of
the expression of SFFV related RNA and gp55, a Friend and Rauscher virus specific protein. Virology 107, 331-344.
BLAIR, D. G., OSKARSSON, M., WOOD, T. G., McCLEMEN'I~, W. L., FISCHINGER, P. J. & VANDE WOUDE, G. (1981). Activation of the transforming potential of a normal cell sequence - a molecular model for oncogenesis. Science 212, 941-943.
CHANG, A. C. Y. & COHEN, S. N. (1978). Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. Journal of Bacteriology 134, 1141-1156.
CHEN, I. S. Y. & TEMIN, H. M. (1982). Substitution of 5' helper virus sequences into non-rel portion of reticuloendotheliosis virus strain T suppresses transformation of chicken spleen cells. Cell 31 111-120.
COLB~'RE-GARAPIN, F., HORODNICEANU, F., KOURILSKY, P. & GARAPIN, A. (1981). m new dominant hybrid selective marker for higher eukaryotic cells. Journal of Molecular Biology 150, 1-14.

1370 W . O S T E R T A G AND O T H E R S
DER, C., KROUTINS, T. & COOPER, G. (1982). Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proceedings of the National Academy of Sciences', U.S.A. 79, 3637-3640.
EMERMAN, M. & TEMIN, H. M. (1984). Genes with promoters in retrovirus vectors can be independently suppressed by an epigenetic mechanism. Cell 39, 459-467.
GRAHAM, F. t . & VAN DER EB, A. J. (1973). A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52, 456-467.
HWANG, L-H. S. & GILBOA, E. (1984). Expression of genes introduced into cells by retroviral infection is more efficient than that of genes introduced into cells by D N A transfection. Journal of Virology 50, 417-424.
HWANG, L-H. S., PARK, J. & GILBOA, E. (1984). Role of intron-contained sequences in formation of Moloney murine leukemia virus env m R N A . Molecular and Cellular Biology 4, 2289 2297.
JORGENSEN, R. A., ROTHSTEIN, S. J. & REZNIKOFF, W. S. (1979). A restriction enzyme cleavage map of Tn5 and location of a region encoding neomycin resistance. Molecular and General Genetics 177, 65-72.
JOYNER, A. & BERNSTEIN, A. (1983a). Retrovirus transduction: generation of infectious retroviruses expressing dominant and selectable genes is associated with in vivo recombination and deletion events. Molecular and Cellular Biology 3, 2180 2190.
JOYNER, A. & BERNSTEIN, A. (1983 b). Retrovirus transduction : segregation of the viral t ransforming function and the herpes simplex virus gene in infectious Friend spleen focus-forming virus thymidine kinase vectors. Molecular and Cellular Biology 3, 2191 2202.
KOLLEK, R., STOCKING, C., SMADJA-JOFFE, I. & OSTERTAG, W. (1984). Molecular cloning and characterization of a leukemia inducing myeloproliferative sarcoma virus and two of its temperature-sensit ive mutants . Journal of Virology 50, 717-724.
LEWIS, S., GIFFORD, A. & BALTIMORE, D. (1984). Joining of V~ to Jr gene segments in a retroviral vector introduced into lymphoid cells. Nature, London 308, 425 428.
McMASTER, G. K. & CARMICrlAEL, G. G. (1977). Analysis of single- and double-stranded nucleic acids on polyacrylamide and agarose gels by using glyoxal and acridine orange. Proceedings of the National Academy of Sciences, U.S.A. 74, 4835-4839.
MANN, R., MULLIGAN, R. C. & BALTIMORE, D. (1983). Construction of a retrovirus packaging mutan t and its use to produce helper-free defective retrovirus. Cell 33, 153-159.
MILLER, A. D., ONG, E. S., ROSENFELD, M. G., VERMA, I. M. & EVANS, R. M. (1984). Infectious and selectable retrovirus containing an inducible rat growth hormone minigene. Science 225, 993 998.
NARAYANAN, R., SRINIVASAN, A. & AARONSON, S. A. (1984). Sequences in the long terminal repeats of the Moloney murine sarcoma virus-124 genome which control t ransforming gene function. Virology 137, 32-40.
NASH, M., BROWN, N. V., WONG, J. L., ARLINGHAUS, R. B. & MURPHY, E. C., JR (1984). S1 nuclease mapping of viral R N A s from a temperature-sensitive t ransformation mutan t of murine sarcoma virus. Journal of Virology 50, 478 488.
OSKARSSON, M., McCLEMENTS, W. L., BLAIR, D. G., MAIZEL, J. V. & VANDE WOUDE, G. F. (1980). Properties of a normal mouse cell D N A sequence (sarc) homologous to the src sequence of Moloney sarcoma virus. Science 207, 1222 1224.
OSTERTAG, W. & PRAGNELL, I. B. (1982). Differentiation and viral involvement in differentiation of t ransformed mouse and erythroid cells. Current Topics in Microbiology and Immunology 94/95, 143 208.
OSTERTAG, W., VEHMEYER, K., FAGG, B., PRAGNELL, I. B., PAETZ, W., LEBOUSSE, M. C., SMADJA-JOFFE, F., KLEIN, B., JASMIN, C. & EISEN, H. (1980). Myeloproliferative virus, a cloned murine sarcoma virus with spleen focus- forming properties in adult mice. Journal of Virology 33, 573-583.
OSTERTAG, W., FRESHNEY, M., VEHMEYER, K., JASMIN, C. & RUTTER, G. (1984). Action of temperature-sensit ive mutants of myeloproliferative sarcoma virus suggests that fibroblast-transforming and hematopoietic transforming viral properties are related. Journal of Virology 49, 173 181.
PARADA, L., TABIN, C., SHUH, C. & WEINBERG, R. (1982). H u m a n EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature, London 297, 474-478.
PERKINS, A. S., KIRSCHMEIER, P. T., GATTONI-CELLI, S. & WEINSTEIN, I. B. (1983). Design o f a retrovirus-derived vector for expression and transduction of exogenous genes in mammal i an cells. Molecular and Cellular Biology 3, 1123-1132.
PRAGNELL, I. B., OSTERTAG, w. & PAUL, J. (1977). The expression of viral and globin genes during differentiation of the Friend cell. Experimental Cell Research 108, 269-278.
PRAGNELL, I. B., FUSCO, A., ARBUTHNOTT, C., SMADJA-JOFFE, F., KLEIN, B., JASMIN, C. & OSTERTAG, W. (1981). Analysis of the myeloproliferation sarcoma virus genome: limited changes in the prototype lead to altered target cell specificity. Journal of Virology 38, 952-957.
SANTOS, E., TRONICK, S., AARONSON, S., PULCIANI, S. & BARBACID, M. (1982). T24 human bladder carcinoma oncogene is an activated form of the normal h u m a n homologue of Balb- and Harvey-MSV transforming genes. Nature, London 298, 343-347.
SHIMOTOHNO, K. & TEM1N, H.M. (1981). Formation of infectious progeny virus after insertion of herpes simplex thymidine kinase gene into D N A of an avian retrovirus. Cell 26, 67-77.
SOUTHERN, E. M. (1975). Dectection of specific sequences among D N A fragments separated by gel eleetrophoresis. Journal of Molecular Biology 98, 503-513.

M o s transformation retained in neoR-MPSV 1371
STACEY, A., ARBUTHNOTT, C., KOLLEK, R., COGGINS, L. & OSTERTAG, W. (1984). Comparison of myeloproliferative sarcoma virus with Moloney murine sarcoma virus variants by nucleotide sequencing and heteroduplex analysis. Journal of Virology 50, 725-732.
STOCKING, C., KOLLEK, R., BERGHOLZ, U. & OSTERTAG, W. (1985). Long t e rmina l repea t sequences i m p a r t hematopoietic transformation property to the myeloproliferative sarcoma virus. Proceedings of the National Academy oJ Sciences, U.S.A. 82, 5746 5750.
TAB/N, C~ J., HOFFMANN, J. w., GOFF, S. P. & WEINBERG, R. A. (1982). Adaptation o fa retrovirus as a eukaryotic vector transmitting the herpes simplex virus thymidine gene. Molecular and Cellular Biology 2, 426-436.
TARPLEY, w. G. & TEMIN, H. M. (1984). The location of v-src in a retrovirus vector determines whether the virus is toxic or transforming. Molecular and Cellular Biology 4, 2653 2660.
TOPP, W. C. (1981). Normal rat cell lines deficient in nuclear thymidine kinase. Virology 113, 408~411. VARMUS, H. E., QUINTRELL, N. & ORTIZ, S. (1981). Retroviruses as mutagens: insertion and excision of a
nontransforming provirus alter expression of a resident transforming provirus. Cell 25, 23 36. WEI, C.-M., GIBSON, M., SPEAR, P. G. & SCOLNICK, E. M. (1981). Construction and isolation of a transmissible
retrovirus containing the scr gene of Harvey murine sarcoma virus and the thymidine kinase gene of herpes simplex virus type 1. Journal of Virology 39, 935-944.
WEISS, R., TEICH, N. M., VARMUS, H. & COFFIN, J. (editors) (1982). RNA Tumor Viruses, (Molecular Biology of Tumor Viruses, 2nd edn.). New York: Cold Spring Harbor Laboratory.
WlGLER, M., SILVERSTEIN, S., LIH-SYNG, L., PELLICER, A., YUNG-CHI, C. & AXEL, R. (1977). T rans fe r of purif ied herpes simplex virus thymidine kinase gene to cultured mouse cells. Cell 11, 223-232.
WIGLER, M., PELLICER, A., SILVERSTEIN, S. & AXEL, R. (1978). B iochemica l t ransfer o f s ingle-copy eukaryo t i c genes using total cellular DNA as donor. Cell 14, 725-731.
WILLIAMS, D. A., LEMISCHKA, I. R., NATHAN, D. G. & MULLIGAN, R. C. (1984). In t roduc t ion of new genet ic m a t e r i a l into pluripotent haematopoietic stem cells of the mouse. Nature, London 310, 476-480.
(Received 16 September 1985)






















![[Diagnosis and treatment of BCR/ABL-negative myeloproliferative diseases]](https://img.pdfslide.net/doc/110x75/63493507f4145ce0ba031ff6/diagnosis-and-treatment-of-bcrabl-negative-myeloproliferative-diseases.jpg)






