Embed Size (px)
Citation preview

174
The Spectrum of Chronic Lymphoproliferative Disorders in Chinese People An Analysis of 64 Cases
Y. L. Kwong, M.R.C.Path.,* K. F. Wong, M.R.C.Path.,t L. C. Chan, M.R.C.Path.,S R. H. S. Liang, M.D.,*J. K. C. Chan, M.R.C.Path.,tD. Wei, F.R.A.C.P.,S E. K. W. Chiu, M.R.C.P.,* C. H. Chan, F.R.C.P.,fj D. Todd, M.D.,* and T. K. Chan, M.D.*
Background. Chronic lymphoproliferative disorders are considered rare in Oriental patients and are thought to constitute only 2% of all leukemias in these patients, compared to 20-30% in Western patients. We conducted a retrospective analysis of Chinese patients with chronic lymphoproliferative disorders to define the frequency and spectrum of these disorders.
Methods. A consecutive series of Chinese patients with leukemia and lymphoproliferative disorders seen at two regional hospitals in Hong Kong were analyzed ret- rospectively. The diagnosis of chronic lymphoprolifera- tive disorders was based on morphologic and immuno- logic criteria proposed by the French-American-British Cooperative Study Group.
Results. Sixty-four Chinese patients with chronic lymphoproliferative disorders were identified, and these patients constituted 19% of a total of 342 cases of leuke- mia diagnosed in 3 years. Chronic lymphocytic leukemia was the most common form, occurring at a frequency of 12.5% of all leukemias. The clinicopathologic features of these patients were similar to those of Western patients, except that Chinese patients tended to present with more advanced (Rai’s Stages I11 and IV; Binet’s Stage C) and bulky (splenomegaly >9 cm) disease, and expressed h light chain about six times more frequently. Other
From the University Departments of *Medicine and *Pathology, Queen Mary Hospital, and the Departments of tPathology and @Medicine, Queen Elizabeth Hospital, Hong Kong.
The authors thank Dr. H. W. Liu, Dr. S. Y. Ha, Dr. A. Lie, Dr. Y. C. Chu, and Mr. C. P. Lee for helping in the diagnosis and manage- ment of these patients, and Dr. D. Posnett and Dr. s. Poppema for the antibodies HC-2 and Bly-7, respectively.
Address for reprints: Y. L. Kwong, M.R.C.Path., University De- partment of Medicine, Professorial Block, Queen Mary Hospital, Pok- fulam Road, Hong Kong.
Received January 18,1994; accepted March 15,1994.
chronic lymphoproliferative disorders identified in this study included prolymphocytic leukemia, mantle zone lymphoma, hairy cell leukemia, splenic lymphoma with villous lymphocytes, large granular lymphocyte leuke- mia, and Sezary syndrome. The authors did not identify any case of human T-cell lymphotropic virus-I-related lymphoproliferative disorders within the study period.
Conclusion. In addition to providing the frequencies of various chronic lymphoproliferative disorders in southern Chinese people, this study also showed that these disorders no longer should be considered rare in this population. Inherent biologic differences between lymphoproliferative disorders in Chinese and Western patients also may exist. Cancer 1994; 74:174-81.
Key words: chronic lymphoproliferative disorders, Chi- nese people, chronic lymphocytic leukemia, large granu- lar lymphocytic leukemia.
Chronic lymphoproliferative disorders are thought to be rare in the East as compared to the West. Among these disorders, chronic lymphocytic leukemia (CLL) is the most common, accounting for 25-38% of all leuke- mias in the This is about 12-20 times higher than the incidence of 1.5-2% in mainland Chine~e,~ Japane~e,~ and Singapore Chinese6 people previously reported in the literature. Studies of Japanese migrants in the United States have shown that the incidence re- mained ~nchanged,~ suggesting that genetic factors might account for this difference. Information on the incidence and clinical behavior of types of chronic lym- phoproliferative disorders other than CLL in the East is lacking. It has been suggested that chronic T-cell lym- phoproliferative disorders might be more prevalent in the Orient.’ These incidence figures were all obtained

Chronic Lymphoproliferative Disorders in Chinese/Kwong et al. 175
from early reports, however, and have not been up- dated by recent studies.
Recently, the French-American-British Coopera- tive Group proposed a classification scheme for chronic B- and T-lymphoid leukemia^.^,'^ We conducted a ret- rospective analysis of a consecutive series of Chinese patients with chronic lymphoproliferative disorders di- agnosed according these criteria to define the frequen- cies and clinicopathologic features of these disorders in our population.
Materials and Methods
Patients
All patients were ethnic Chinese seen consecutively at initial diagnosis in two Hong Kong regional Hospitals (Queen Mary Hospital and Queen Elizabeth Hospital), which served a population of 3 million. These hospitals served as both regional hospitals and referral centers, where blood counts were done routinely for every pa- tient, and abnormal blood films were inspected by a hematopathologist. Between 1989 and 1992,342 newly dlagnosed cases of de novo leukemias were seen. Sixty- four cases (19%) were chronic lymphoid leukemias and were included in this study. Informed consent for bone marrow biopsy was obtained from all the patients in- cluded in this study.
Morphology and Immunophenotyping
Peripheral blood and bone marrow smears were exam- ined after Wright-Giemsa staining, and classified ac- cording to the French-British-American criteria pro- posed for chronic lymphoid leukemia^.^,'^ Immunophe- notyping of bone marrow or peripheral blood mononuclear cells was performed by the alkaline phos- phatase antialkaline phosphatase method for 5 1 cases, and by flow cytometry using a Coulter EPICS Profile I1 flow cytometer (Coulter, Hialeah, FL) equipped with a 15 mW (488 nm) argon laser, gating on cells with mini- mal side scatter and forward scatter characteristic of lymphocytes, for 13 cases. Monoclonal antibodies against the following surface antigens were used: CD19, CD10, CD21, CD22 (B lineage associated), CD7, CD5, CD2, CDl (T lineage associated), CD33, CD13, CD14 (myeloid lineage associated), CD16, CD56, CD57 (natural killer cell associated), HLA-DR, FMC-7, HC-2 and Bly-7. Positivity was defined by the presence of 20% or more cells expressing the antigen.
According to morphologic and immunophenotypic criteria, these cases were classified into the following categories: B-cell CLL, B- and T-cell prolymphocytic
leukemia, hairy cell leukemia, splenic lymphoma with villous lymphocytes, leukemic phase of mantle zone lymphoma, large granular lymphocyte leukemia, and Sezary syndrome. The leukemic phase of other non- Hodgkin’s lymphomas was excluded from the study.
Results
B-cell Chronic Lymphocytic Leukemia
There were 43 cases of CLL (Figure 1, top left). Their respective clinicopathologic features are summarized in Table 1. Seven patients had absolute lymphocyte counts of less than 10 X 109/1, but the monoclonal na- ture of the lymphocytes was demonstrated by mono- clonal immunoglobulin light chain expression as rec- ommended by the International Workshop on Chronic Lymphocytic Leukemia” and were included in the study. The male-to-female ratio was 1.9:1, with the modal age of presentation in the sixth decade. Spleno- megaly was present in 72% (31 of 43) of the cases, with gross splenomegaly (29 cm below costal margin) pres- ent in 26%. About 30% of patients presented with a hemoglobin of <10 g/dl or a platelet count of <lo0 X 109/L. Forty-one percent of the patients presented with a leukocyte count of >50 X 109/l. Patients tended to present with advanced disease. Staging was done by both the Rai” and the BinetI3 staging systems. Sixty percent (26 of 43) of cases were had disease of Rai’s Stages 111 and IV, and 46% had disease of Binet’s Stage C. Immunophenotyping was performed fully in 38 cases, which showed the following immunophenotypic profile: HLA-DR positive (65%), CD19 positive (loo%), CD5 positive (79’/0), CD25 positive (34%). The ratio of K to X monotypic light chain expression was 1:2.2. In the remaining five cases, B lineage was shown by the expression of CD19 without demonstration of mono- typic light chain expression.
Single-agent treatment with chlorambucil was given in 32 patients, and combination chemotherapy with cyclophosphamide, vincristine, and prednisone was administered to 11 patients who had gross spleno- megaly. The mode of therapy did not appear to affect clinical outcome. Eighteen patients died (survival in months, mean * 2 SDs = 15 k 24, range 3-105, n = 18), with infection being the cause of death in all but one, who died of myocardial infarction.
Prolymphocytic Leukemia
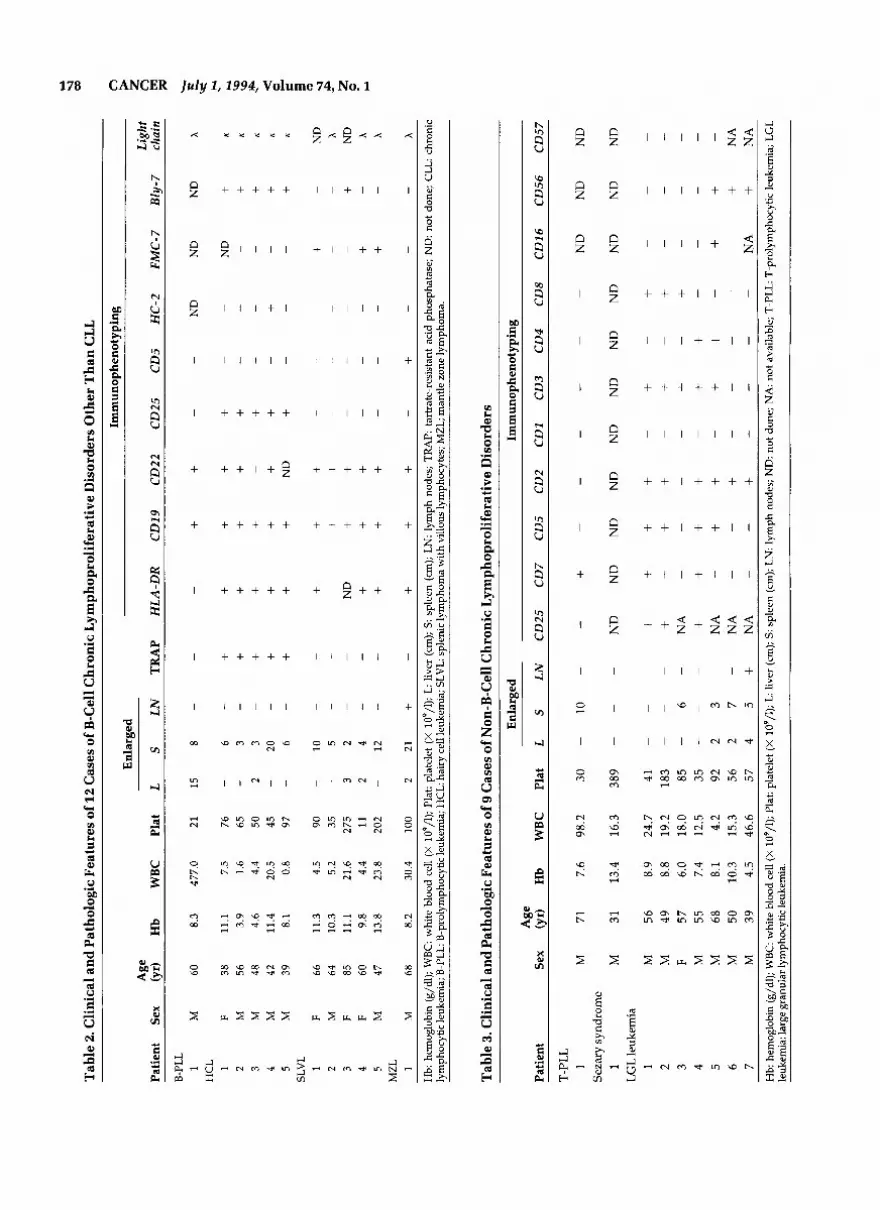
Two cases of prolymphocytic leukemia were found. The first case was a B-cell prolymphocytic leukemia (Fig. 1, top right) (Table 2) and showed an immunophenotype

176 CANCER July 1,1994, Volume 74, No. 1
Figure 1. (Top left) Chronic lymphocytic leukemia, with mature, small lymphocytes (Wright-Giemsa, original magnification XlOOO). (Top right) B-cell prolymphocytic leukemia. Note the large central nucleolus (Wright-Giemsa, original magnification XlOOO). (Middle left) T-cell prolymphocytic leukemia. Some cytoplasmic vacuolation is seen together with a prominent central nucleolus (Wright-Giemsa, original magnification X 1000). (Middle right) Hairy cell leukemia cell, with typical hairy cytoplasmic projections (Wright-Giemsa, original magnification XlOOO). (Bottom left) Splenic lymphoma with villous lymphocytes. Note the short and stubby cytoplasmic projections (Wright-Giemsa, original magnification XlOOO). (Bottom right) Large granular lymphocyte leukemia cell. Note the prominent cytoplasmic granules (Wright-Giemsa, original magnification XlOOO).
of CD19+CD22+. The second case was a T-cell prolym- phocytic leukemia (Fig. 1, middle left) (Table 3) and ex- pressed only CD7, being negative for other T-cell anti- gens tested. Both patients died (one at 3 months after diagnosis and the other at 6 months after diagnosis) de- spite combination chemotherapy with cyclophospha- mide, vincristine, and prednisone.
Hairy Cell Leukemia
There were five cases of hairy cell leukemia (Fig. 1, mid- dle right), and their clinical features are summarized in Table 2. There were four men and one woman, whose ages ranged from 39 years to 56 years (mean, 45). Sple- nomegaly was a constant feature. Tartrate-resistant acid
phosphatase was shown in all of them. The common immunophenotypic profile was CD19+CD22+CD25 +Bly-7+, and all cases expressed the K light chain. HC-2 was expressed in only one case. All patients responded to a 12-18-month course of a-interferon (3 megaunits 3 times per week) with clinical remission, which had lasted from 4 months to 18 months. One patient (Case 1, Table 2) relapsed 12 months after being taken off therapy and has responded to another course of a-in- terferon.
Splenic Lymphoma with Villous Lymphocytes
There were five cases of splenic lymphoma with villous lymphocytes (Fig. 1, bottom left) (Table 2). The modal

Chronic Lyrnphoproliferative Disorders in Chinese/Kwong et al . 177
Table 1. Clinicopathologic Features of 43 Chinese CLL Patients
No. of patients
Male 28 Female 15
Age < 40 yr 1 41-50 yr 2 51-60 yr 11 61-70 yr 18 71-80 yr 8 81-90 yr 2 > 90 yr 1
Splenomegaiy 31 1-3 cm 6 3-6 cm 9 6-9 cm 5 9-12 cm 8 > 12cm 3
1-3 cm 16 3-6 cm 5 > 6 c m 1
> 10 g/dl 30 < 10 g/dl 13
Hepa tomegaly 22
Hemoglobin
Leukocyte count (X lo*/]) > I00 51-100 10-50 < 10
> 100 < 100
Platelet count (X 1o9/1)
Immunophenot p i n g
HLA-Dr CD 19 CD 20 CD 5 CD 25 CD 14 I[ light chain X light chain
Staging
+ 27 38 13 34 15 6
12 26
Rai
0 = 1 I = 1
I I = 15 111 = 15 IV = 11
5 0 8 4 9
11
11 7
18 7
29 14
ND
11 5
22 5
19 26 5 -
Binet
A = 11 B = 12 c = 20
CLL chronic lymphocytic leukemia; ND: not done.
age of presentation was in the sixth decade, with sple- nomegaly present in all patients. The circulating villous lymphocytes ranged from 3970 to 76%. None of the
cases showed tartrate-resistant acid phosphatase. In ad- dition to B-cell antigens CD19 and CD22, three cases expressed FMC-7, and one case expressed Bly-7. In three cases tested, the X light chain was expressed. Two cases were treated with chlorambucil and were alive, one at 10 months after diagnosis and the other at 12 months after diagnosis. One patient received combina- tion chemotherapy with cyclophosphamide, vincris- tine, procarbazine, and prednisone, and his disease was in apparent remission 6 months after diagnosis. One patient died of unrelated carcinoma of the lung, while the last one refused therapy and defaulted follow-up.
Mantle Zone Lymphoma
One patient presented with the leukemic phase of man- tle zone lymphoma (Table 2). Surface coexpression of immunoglobulins M and D was found. He received combination chemotherapy with cyclophosphamide, vincristine, procarbazine, and prednisone, and showed a partial clinical response but died 12 months after di- agnosis because of infection.
Large Granular Lymphocyte Leukemia
Seven cases of large granular lymphocyte leukemia were found (Figure 1, bottom right) (Table 3). Accord- ing to immunophenotyping, three subgroups could be defined. The first group consisted of three cases that were CD2+CD3+CD4TD8+. All patients received chlorambucil and were alive 33-40 months after diag- nosis. The second group of two cases were CD2+C- D3+CD4+CD8-. One patient (Case 4) received combi- nation chemotherapy with cyclophosphamide, vincris- tine, and prednisone, and was alive 30 months after diagnosis. The other patient died 1 month after diagno- sis because of infection. The third group of two cases were CD2+CD3-CD4-CD8-CD56'. Both cases died within 1 month of diagnosis because of uncontrolled disease. Details of these cases will be reported sepa- rately (unpublished data, Y.L. Kwong et al., 1994).
Sezary Syndrome
One case of Sezary syndrome was identified (Table 3). He presented with mycosis fungoides 3 years before the development of Sezary syndrome. Hemi-body irradia- tion was given and he was alive 26 months after diag- nosis.
Discussion
Chronic lymphoproliferative disorders generally are considered rare in Oriental patients. Most of the data in
Tab
le 2
. Clin
ical
and
Pat
holo
gic
Fea
ture
s of 1
2 C
ases
of B
-Cel
l Chr
onic
Lym
phop
rolif
erat
ive
Dis
orde
rs O
ther
Th
an C
LL
~ ~~
~
Imm
unop
heno
typi
ng
Enl
arge
d A
ge
Light
Patie
nt
Sex
(yr)
H
b W
BC
Pl
at
L S
LN
TRA
P HL
A-DR
CD
19
CD22
CD
25
CD5
HC-2
FM
C-7
Bly-7
chain
B-PL
L +
+ -
ND
N
D
ND
A
1 M
60
8
3
4770
21
15
8
-
ND
+
1 F
38
11 1
7
5
76
- 6
- +
+ +
+ +
K
+ 2
M
56
39
1
6
65
- 3
- +
+ +
+ +
K
+ +
3 M
48
4
6
44
50
2
3 -
+ +
+ K
+ +
4 M
42
1
14
20
5 45
-
20
-
+ +
+ +
+ K
+ 5
M
39
81
0
8
97
-
6 -
+ +
+ N
D
+ K
1 F
66
11 3
4
5
90
-
10
-
+ +
+ +
+ 2
M
64
10
3
52
35
-
5 -
3 F
85
11 1
21
6
275
3 2
-
ND
+
+ 4
F 60
9
8
44
11
2
4 -
+ +
+ +
+ +
M
47
13
8
23
8
202
- 12
-
- 5
MZL
1
M
68
82
3
04
10
0 2
21
+ -
-
-
-
HC
L -
-
-
- -
- -
- -
- -
-
-
-
SLVL
-
-
-
ND
x
+ N
D
-
+ -
-
-
-
-
-
-
-
-
-
-
-
-
x x -
+ + -
-
-
- -
-
- -
x -
-
-
+ -
+ +
+ H
b h
emog
lobi
n (g
/dl);
WBC
: w
hite
blo
od c
ell (
X 1
09/l)
; Pla
t: p
late
let
(X 1
09/l)
; L l
iver
(cm
); S
: spl
een
(cm
); L
N: l
ymph
nod
es;
TR
AP:
tart
rate
-res
istan
t ac
id p
hosp
hata
se; N
D:
not
done
; CL
L ch
roni
c ly
mph
ocyt
ic le
ukem
ia; B
-PLL
: B-p
roly
mph
ocyt
ic le
ukem
ia; H
CL:
hai
ry c
ell l
euke
mia
; SLV
L: sp
leni
c lym
phom
a w
ith v
illou
s lym
phoc
ytes
; MZL
; man
tle zo
ne ly
mph
oma.
Tab
le 3
. Clin
ical
and
Pat
holo
gic
Fea
ture
s of 9
Cas
es o
f Non
-B-C
ell C
hron
ic L
ymph
opro
lifer
ativ
e D
isor
ders
Enl
arge
d Im
mun
ophe
not y
ping
A
ge
Patie
nt
Sex
(yr)
Hb
WB
C
Plat
L
S LN
CD25
CD
7 CD
5 CD
2 CD
1 CD
3 CD
4 CD
8 CD
16
CD56
CD
57
T-PL
L
Sez
ary
synd
rom
e
LGL
leuk
emia
-
ND
N
D
ND
-
-
-
-
-
+ -
1 M
71
7.
6 98
.2
30
-
10
-
1 M
31
13
.4
16.3
38
9 -
-
-
ND
N
D
ND
N
D
ND
N
D
ND
N
D
ND
N
D
ND
+ 1
M
56
8.9
24.7
41
-
-
+ +
+ +
2 M
49
8.
8 19
.2
183
-
-
+ +
+ +
+ +
3 F
57
6.0
18.0
85
-
6 -
NA
4
M
55
7.4
12.5
35
-
~ +
+ +
+ +
+ 5
M
68
8.1
4.2
92
2 3
-
NA
+
+ +
+ +
+ 6
M
50
10.3
15
.3
56
2 7
-
NA
M
39
4.
5 46
.6
57
4 5
+ N
A
7
-
-
-
-
-
+ -
~ ~
-
-
-
-
-
-
-
-
-
-
-
-
-
+ -
-
-
-
-
-
-
-
-
-
~
+ N
A
-
-
NA
+
NA
-
-
-
-
-
-
+ + -
-
-
-
Hb:
hem
oglo
bin
(g/d
l); W
BC: w
hite
blo
od ce
ll (X
109
/l); P
lat:
pla
tele
t (X
109
/l); L
liv
er (c
m);
S: s
plee
n (c
m);
LN
: lym
ph n
odes
; ND
: not
don
e; N
A: n
ot a
vaila
ble;
T-P
LL: T
-pro
lym
phm
ytic
leuk
emia
; LG
L le
ukem
ia: l
arge
gra
nula
r lym
phoc
ytic
leuk
emia
.
n
4
w r,
CI c z ?

Chronic Lymphoproliferative Disorders in Chinese/Kwong et al. 179
the literature and standard textbooks were on CLL in Orientals, which quoted a frequency of 1.5-2% of all leukemias, A recent study of more than 4000 Chinese leukemic patients cited an incidence of CLL of 4.6%,14 which was in fact very close to that of 4.2% reported in Hawaiian Japanese pe0p1e.l~ This has led to the conclu- sion that inherent genetic factors might account for the low incidence of CLL in Oriental populations.I6
The present study, however, has shown a high fre- quency of CLL of 12.5% of all leukemias in Hong Kong Chinese people. If we excluded those cases presenting with a leukocyte count of less than 10 X lo9 cells/l, the frequency is about 10%. This underscores the impor- tance of immunophenotypic demonstration of mono- clonal light chain restriction in persistent lymphocytosis for the diagnosis of CLL with less than 10 X lo9 cells/l, as recognized by the International Workshop on Chronic Lymphocytic Leukemia.” This high frequency is not due to a relative rarity in other types of leuke- mia-the overall incidence of leukemia per 100,000 people in this study is 3.6 (similar to the overall inci- dence of leukemia in Hong Kong, at 4 per 100,000 peo- ple17), which is comparable to that of 2.4 previously re- ported in Chinese people,” 2.2 in Indians,” and 3.4 in Japanese people.” This finding is intriguing. Less phy- sician awareness and inadequate diagnostic facilities that were standard when these past studies were con- ducted might account in part for the difference, al- though recent studies in migrant Hawaiian Japanese people in the United States still showed a very low inci- dence of CLL.” One therefore might speculate that fac- tors other than genetic ones might have accounted for the relatively higher frequency of CLL observed in our population, and further series of CLL patients in Orien- tal populations are needed to validate this observation. The frequency, however, is still lower than that of West- ern populations.
The clinicopathologic features of CLL patients in this study show a number of similarities and differences when compared to other typical series of Western pa- tients.21,22 Although the male-to-female ratio, modal age of presentation, and the range of initial leukocyte counts are similar, the following differences are noticed: 26% of the patients in our study had gross splenomeg- aly (vs. 14% in Western patients2’*22), 46% (Binet’s Stage C) to 60% (Rai’s Stages I11 and IV) of our patients had advanced disease (vs. 16-25% in Western pa- tients21f22), and the K-to-X light chain ratio was 1:2.2 (vs. 2.8:l in Western series21,22). Although the small number of patients in this series precludes meaningful statistical analysis, it appears that Chinese patients are more likely to present with late stage and bulky diseases. The much higher frequency of expression of the A light chain
(about six times that seen in Western patients) might reflect that cases of CLL in Chinese patients have inher- ently different biologic characteristics. Of interest in this respect is that a previous study on non-Hodgkin’s lym- phoma of the Hong Kong Chinese people also demon- strated X light chain predominance ( K : X = 1:1.9) in the B-cell lymphomas.23
Besides CLL, other chronic lymphoproliferative disorders that occurred in our Chinese population also were identified in this study. Prolymphocytic leukemia was apparently rare, accounting for only 3% of cases in this study, which is similar to the rate reported in West- ern patientsz4 Five cases of splenic lymphoma with vil- lous lymphocytes were seen. The clinicopathologic fea- tures, including the mean age of presentation (64 years), leukocyte count (4.4-23.8 X 109/1), and splenomegaly (2-12 cm), immunophenotypic profile (HLD-DR’CDI - 9+CD22+FMC-7’), absence of lymphadenopathy, and indolent clinical course, were all very similar to those of Western patient^.'^ One West African study of splenic lymphoma with villous lymphocytes showed certain different clinical features, including female predomi- nance and high lymphocyte counts, and suggested that this might imply different etiologic links.26 This possible geographic difference was not observed in this study.
There are no data on hairy cell leukemia in Chinese patients in the literature. The clinicopathologic features, including male predominance, immunophenotyping, and response to therapy in the five patients analyzed in this study were quite similar to those reported for West- em patients.27 In contrast to our cases of CLL, however, expression of the K light chain was predominant, whereas in Western patients, the K light chain is ex- pressed as frequently as the X light chain is.” Machii et aLZ9 recently reported a variant form of hairy cell leuke- mia in Japanese patients, characterized by hairy cells with prominent nucleolus, weak to negative tartrate-re- sistant acid phosphatase, weak surface immunoglobu- lin expression, a K-to-X expression of 7:1, and no expres- sion of CD25. Careful review of our cases, however, did not show such a variant, except that we found the same predominance of K light chain expression.
We identified a total of seven large granular lym- phocyte leukemias in this study. Five of these seven cases expressed multiple T-cell antigens, and two ex- pressed variably natural killer cell antigens. The former group showed a fairly indolent course, similar to that observed in Western patient^,^^,^^ while the disease in the latter group was aggressive. This is different from the experience in Western patients, where the expres- sion of natural killer antigens was not associated with a poor p r o g n o s i ~ . ~ ~ - ~ ~
Besides large granular lymphocyte leukemia, we

180 CANCER July 1,1994, Volume 74, No. 1
have identified only one case of Sezary syndrome. No cases of human T-cell lymphotropic virus-I-related lymphoproliferative disorders were identified within the study period, which is a reflection of the fact that, other than in Japan and certain parts of Taiwan,33 hu- man T-cell lymphotropic virus4 infection is rare in Asia.
This study has clarified a number of points and be- liefs noted in the literature on chronic lymphoprolifera- tive disorders in the East. First, the finding of a relatively high frequency and a broad spectrum of chronic lym- phoproliferative diseases means that these disorders no longer should be considered rare in the Chinese. Sec- ond, the frequency of chronic lymphoproliferative dis- orders of non-B-cell origin is only 12% (8 of 64). There- fore, the belief that T-cell chronic lymphoproliferative disorders are the predominant form in Asia" appears to be unwarranted in the Chinese. Third, the patterns of light chain expression (predominant K light chain in hairy cell leukemia and X light chain in other lympho- proliferative disorders) in our patients appear very different from those in the West. Finally, these different clinicopathologic features suggest that biologic differ- ences might exist. To support this, we recently have de- termined by fluorescence in situ hybridization that the incidence of trisomy 12 in Chinese CLL cases was only one third that of the 30% incidence in Western pa- t i e n t ~ . ~ ~ ' ~ ~ Further clinical and molecular studies of chronic lymphoproliferative diseases in the Orient thus may give useful information for our understanding of these disorders.
References
1 .
2.
3.
4.
5.
6.
7.
Young JL, Percy CL, Asire AJ. Surveillance, epidemiology and end results: incidence and mortality data, 1973-77. National Cancer Institute monograph 57. Department of Health and Hu- man Services Publication: National Institute of Health, 1981:81- 2330. Brinker H. Population-based age- and sex-specific incidence rates in the 4 main types of leukaemia. Scand J Haematol 1982; 29:241-9. Rai KR, Sawitsky A. Diagnosis and treatment of chronic lym- phocytic leukemia. In: Wiemik PH, Canellos GP, Kyle RA, Schiffer CA, editors. Neoplastic diseases of the blood. New York: Churchill Livingstone, 1991:97- 109. Miller RW. Cancer epidemics in the People's Republic of China. I N a t l Cancerlnst 1978; 60:1195-1203. Weiss NS. Geographical variation in the incidence of the leuke- mias and lymphomas. In: Henderson BE, editor. Second sympo- sium on epidemiology and cancer registries in the Pacific basin. National Cancer Institute monograph 53. Bethesda, MD: Na- tional lnstitutes of Health, 1978:79-1864. Wells R, Lau KS. lncidence of leukemia in Singapore and rarity of chronic lymphocytic leukemia in Chinese. Br Med ] 1960; 1: 759. Haenszel W, Kurihara M. Studies of Japanese migrants. 1: mor-
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
tality from cancer and other diseases among Japanese in the United States. JNat l Cancer Inst 1968; 40:43-68. Gallo RC. Human T-cell leukemia-lymphoma virus and T-cell malignancies in adults. Cancer Sun, 1984; 3:113-59. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, and the French-American-British (FAB) Cooperative Group. Proposals for the classification of chronic (mature) B and T lymphoid leukemias. J Clin Pathol
Bennett JM, Juliusson G, Mecucci G. A conference on the mor- phologic, immunologic, and cytogenetic classification of the chronic (mature) B and T-lymphoid leukemias (MIV IV). Hema- fol Pafhol 1990; 4:161-2. International Workshop on Chronic Lymphocytic Leukemia. Chronic lymphocytic leukemia: recommendations for diagnosis, staging, and response criteria. Ann Intern Med 1989; 110:236-38. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pas- ternack BS. Clinical staging of chronic lymphocytic leukemia.
Binet JL, Leporrier M, Dighiero G, Charron D, DAthis Ph, Vaughier G, et al. A clinical staging system for chronic lympho- cytic leukemia: prognostic significance. Cancer 1977; 40:855-64. Boggs DR, Chen SC, Zhang ZN, Zhang A. Chronic lymphocytic leukemia in China. Am JHematol 1987; 25:349-54. Elizaga FV, Oishi N. Chronic lymphocytic leukemia in Japanese in Hawaii. Hawaii M e d J 1977; 36:169-71. Linet MS, Blattner WA. The epidemiology of chronic lympho- cytic leukemia. In: Polliack A, Catovsky D, editors. Chronic lym- phocytic leukemia. Chur, Switzerland Harwood Academic Publishers, 1988:ll-32. Hong Kong Cancer Registry. 1989 Annual Statistical Report. Hong Kong: Institute of Radiology and Oncology, Hospital Au-
Chu JX. The Preliminary report of the epidemiology survey in China. In: Advances in leukemia research in China. Beijing: De- partment of Medical Information Research, Institute of Hema- tology, Chinese Academy of Medical Sciences, 1978. Fraumeni JF Jr, Hoover RN, Devesa SS, Kinlen LJ. Epidemiology of cancer. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer: principles and practice in oncology. Philadelphia: J. B. Lippincott, 1989: 196-227. Yanagihara ET, Blaisdell RD, Hayashi T, Lukes RJ. Malignant lymphoma in Hawaii-Japanese: a retrospective morphologic survey. Hemafol Oncol 1989; 7:219-32. Catovsky D, Fooks J, Richards S. Prognostic factors in chronic lymphocytic leukaemia: the importance of age, sex and response to treatment in survival: a report from the MRC CLL 1 Trial. BrJ Haematol 1989; 72:141-49. Vallespi T, Montserrat E, Sanz MA. Chronic lymphocytic leu- kaemia: prognostic value of lymphocyte morphological sub- types. A multivariate survival analysis in 146 patients. Br ] Haematoll991; 77:478-85. Ng CS, Chan JKC, Lo STH, Poon YF. Immunophenotypic anal- ysis of non-Hodgkin's lymphoma in Chinese: a study of 75 cases in Hong Kong. Pathology 1986; 18:419-25. Stone RM. Prolymphocytic leukemia. Hemafol Oncol CIin North Am 1990; 4:457-71. Melo JV, Hegde U, Parreira A, Thompson I, Lampert IA, Catov- sky D. Splenic B cell lymphoma with circulating villous lympho- cytes: differential diagnosis of B cell leukaemias with large spleens. J Clin Pathol 1987; 40:642-51. Bates I, Bedu-Addo G, Rutherford T, Bevan DH. Splenic lym-
1989; 42~567-84.
Blood 1975; 46219-34.
thority, 1993:50-1.

Chronic Lymphoproliferative Disorders in Chinese/Kwong et al. 181
27.
28.
phoma with villous lymphocytes in tropical West Africa. Lancet
Moormeier JA, Golomb HM. Diagnosis and treatment of hairy cell leukemia. In: Wiernik PH, Canellos GP, Kyle RA, editors. Neoplastic diseases of the blood. New York: Churchill Living- stone, 1991:lll-21. Melo JV, Robinson DSF, Catovsky D. The differential diagnosis between chronic lymphocytic leukemia and other B-cell lym- phoproliferative disorders: morphological and immunological studies. In: Polliack A, Catovsky D, editors. Chronic lympho- cytic leukemia. Chur, Switzerland: Harwood Academic Publish- ers, 1988:85-103.
1993; 3401575-7. 31.
32.
33.
34.
cyte expansions: differential expression of the CD8m and CD8g chains. Blood 1992; 80:1765-73. Sun T, Brody J, Koduru P, Vinciguerra V, Weiselberg L, Ma- rino J, et al. Study of the major phenotype of large granular T-cell lymphoproliferative disorder. Am 1 C f i n Pnthof 1992;
Chan WC, Gu LB, Masih A, Nicholson J, Vogler WR, Yu G, Nasr S. Large granular lymphocyte proliferation with the natural killer-cell phenotype. Am Cfin Dathol 1992; 97:353-8. Shih LY, Liang DC. Non-Hodgkin’s lymphomas in Asia. Hema- tol Oncof Clin North Am 1991; 5:983-1001. Crossen PE. Cytogenetic and molecular changes in chronic B- cell leukemia. Cancer Genet Ciitopnet 1989; 43:143-SO.
98~516-21.
“ I
29. Machii T, Tikumine Y, Inoue R, Kitani T. Predominance of a distinct subtype of hairy cell leukemia in Japan. Leukemia 1993; 7181-6. de Totero D, Tazzari PL, DiSanto JP, di Celle PF, Raspadori D, Conte R. Heterogenous immunophenotype of granular lympho-
35, K~~~~ y ~ , pang J, Ching LM, ~i~ HW, ~i~~~ RHS, Chan LC, Trisomy 12 in chronic lymphocytic leukemia: an interphase cy- togenetic study by fluorescence in situ hybridization. Cancer Genet Cytogenet 1994;72:83-5.
30.




























