Embed Size (px)
Citation preview

DOI: 10.1126/scitranslmed.3008661, 236ra62 (2014);6 Sci Transl Med
et al.Tatiana PerovaHigh-Risk Precursor B Cell Acute Lymphoblastic LeukemiaTherapeutic Potential of Spleen Tyrosine Kinase Inhibition for Treating
Editor's Summary
inhibitors may be promising agents for treating poor-prognosis and relapsed B-ALL.signal transduction pathways of abnormal growth in multiple subtypes of B-ALL, suggesting that small-molecule SYKanimals with SYK inhibitors reduced human leukemia burden in these tissues. Thus, SYK activation regulates key engraftment and dissemination to spleen, liver, and central nervous system. In vivo treatment of the transplantedpoor-prognosis primary human B-ALL samples into immunodeficient mice resulted in extensive bone marrow attenuated by in vitro treatment of the human leukemic cells with SYK inhibitors. Xenotransplantation ofsamples exhibited basal SYK activation. Phosphorylation of SYK and its targets and leukemic cell proliferation were inhibitors of SYK signaling. Like the mouse model, the authors show that primary pediatric and adult human B-ALL(SYK) was required for leukemic cell growth. SYK pathway activity and B-ALL cell survival were sensitive to two
. demonstrate that aberrant activation of the spleen tyrosine kinaseet almouse model of spontaneous B-ALL, Perova discovery of aberrant signaling pathways in cancers has spurred development of targeted kinase inhibitors. In achildren and achieves remission in adults, but patients in both groups relapse, resulting in low survival rates. The
Intensive chemotherapy in B cell acute lymphoblastic leukemia (B-ALL) provides improved outcomes for
Targeting the ''SYK-ness'' in B-ALL
http://stm.sciencemag.org/content/6/236/236ra62.full.htmlcan be found at:
and other services, including high-resolution figures,A complete electronic version of this article
http://stm.sciencemag.org/content/suppl/2014/05/12/6.236.236ra62.DC1.html can be found in the online version of this article at: Supplementary Material
http://www.sciencemag.org/about/permissions.dtl in whole or in part can be found at: article
permission to reproduce this of this article or about obtaining reprintsInformation about obtaining
is a registered trademark of AAAS. Science Translational Medicinerights reserved. The title NW, Washington, DC 20005. Copyright 2014 by the American Association for the Advancement of Science; alllast week in December, by the American Association for the Advancement of Science, 1200 New York Avenue
(print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except theScience Translational Medicine
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

LEUKEM IA
Therapeutic Potential of Spleen Tyrosine KinaseInhibition for Treating High-Risk Precursor B Cell AcuteLymphoblastic LeukemiaTatiana Perova,1,2 Ildiko Grandal,1 Lauryl M. J. Nutter,1 Eniko Papp,1,2 Irina R. Matei,1,2
Joseph Beyene,3 Paul E. Kowalski,1 Johann K. Hitzler,1,4 Mark D. Minden,2,5
Cynthia J. Guidos,1,6 Jayne S. Danska2,7*
Intensified and central nervous system (CNS)–directed chemotherapy has improved outcomes for pediatric B cellacute lymphoblastic leukemia (B-ALL) but confers treatment-relatedmorbidities. Moreover, many patients sufferrelapses, underscoring the need to develop new molecular targeted B-ALL therapies. Using a mouse model, weshow that leukemic B cells require pre–B cell receptor (pre-BCR)–independent spleen tyrosine kinase (SYK)signaling in vivo for survival and proliferation. In diagnostic samples from human pediatric and adult B-ALL pa-tients, SYK and downstream targets were phosphorylated regardless of pre-BCR expression or genetic subtype.Two small-molecule SYK inhibitors, fostamatinib and BAY61-3606, attenuated the growth of 69 B-ALL samples invitro, including high-risk (HR) subtypes. Orally administered fostamatinib reduced heavy disease burden afterxenotransplantation of HR B-ALL samples into immunodeficient mice and decreased leukemia dissemination intospleen, liver, kidneys, and the CNS of recipient mice. Thus, SYK activation sustains the growth of multiple HR B-ALLsubtypes, suggesting that SYK inhibitors may improve outcomes for HR and relapsed B-ALL.
INTRODUCTION
B cell acute lymphoblastic leukemia (B-ALL) is the most commonmalignancy of childhood. Use of intensified systemic and centralnervous system (CNS)–directed chemotherapy now yields survivalrates of 75 to 80% (1), but these treatments cause acute and long-termtreatment-related complications (2).However,~20%ofpediatric andmorethan 60% of adult patients fail frontline therapies and relapse with apoor prognosis (3–5), highlighting the ineffectiveness of currenttreatments. The most aggressive regimens are reserved for children(~20% of cases) deemed to be at high risk (HR) for treatment failureor relapse based on the presence of particular cytogenetic markers,clinical features, or minimal residual disease levels after inductiontherapy (6, 7). However, relapses also occur in standard-risk (SR)patients who present with favorable clinical or cytogenetic prog-nostic criteria. In both adults and children, relapses often originatefrom leukemic blasts that have disseminated to extramedullarysites, such as the CNS, and are associated with low survival rates(8). Thus, there is an unmet need to develop therapies that specifi-cally target signaling pathways that promote survival and dissemi-nation of HR B-ALL to improve survival and quality of life for bothchildren and adults.
The discovery of aberrant protein kinase activation in manycancers has spurred development of targeted kinase inhibitors (9). Ad-
dition of lesion-specific tyrosine kinase inhibitors to conventionalchemotherapy has greatly improved outcomes for HR B-ALL casesharboring BCR-ABL1 translocations (10). Mutations and/or aberrantactivation of cytokine and Janus kinase (JAK)/signal transducer andactivator of transcription (STAT) signaling pathways have recentlybeen identified in subsets of HR B-ALL patients (11, 12). Moreover,preclinical studies suggest that JAK2 ormammalian target of rapamy-cin (mTOR) inhibitors may improve outcomes for certain BCR-ABL1!
HR B-ALL patients (13). However, these cases contain diverse muta-tions that affect several different signaling pathways, suggesting theneed to develop multiple inhibitors for this poor-prognosis group.Therapeutic targeting of signaling pathways active in many HR sub-types of B-ALL would provide an ideal solution to this problem, butthis has not yet been achieved.
B-ALL results from abnormal accumulation of transformed pro-genitor B (pro-B) or precursor B (pre-B) lymphocytes. During B celldevelopment, pro-B cells are programmed to die unless they successful-ly rearrange and express the immunoglobulin m (Igm) heavy chain, whichinteracts with l5, VpreB, and CD79A/CD79B dimers to form a cell sur-face pre–B cell receptor (BCR) signaling complex (14). Pre-BCR expres-sion activates SRC-family tyrosine kinases (SFKs) to phosphorylateimmunoreceptor tyrosine-based activation motifs (ITAMs) in the cyto-plasmic tails of CD79A/CD79B, recruiting the paired N-terminal SH2domains of spleen tyrosine kinase (SYK) and causing its activation.Pre-BCR–induced SYK signaling is required for the survival, proliferation,and differentiation of pro-B cells into pre-B cells. Consequently, B cell de-velopment is arrested at the pro-B cell stage inmice with genetic defects inDNA repair that impair Igm expression or pre-BCR signaling (15). How-ever, combined disruption of DNA repair and p53-mediated DNA dam-age checkpoints causes mice to spontaneously develop early B-ALLthat lacks pre-BCR expression (16, 17). Moreover, nearly 70% of pedi-atric and adult B-ALL lack cytoplasmic Igm (cIgm) and, consequently, donot express a pre-BCR (18). Thus, B cell leukemogenesis in mice and
1Program in Developmental and Stem Cell Biology, The Hospital for Sick Children,Toronto, Ontario M5G1L7, Canada. 2Department of Medical Biophysics, University ofToronto, Toronto, Ontario M5S1A8, Canada. 3Department of Clinical Epidemiologyand Biostatistics, McMaster University, Hamilton, Ontario L8S4L8, Canada. 4Division ofHaematology/Oncology, The Hospital for Sick Children, Toronto, Ontario M5G1L7, Canada.5Ontario Cancer Institute and Princess Margaret Hospital, University Health Network,Toronto, Ontario M5T2M9, Canada. 6Department of Immunology, University of Toronto,Toronto, Ontario M5S1A8, Canada. 7Program in Genetics & Genome Biology, TheHospital for Sick Children, Toronto, Ontario M5G1L7, Canada.*Corresponding author. E-mail: [email protected]
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 1
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

humans frequently involves subversion of the pro-B developmentalcheckpoint, enabling B cell progenitors to survive and proliferate in apre-BCR–independent fashion.
These considerations prompted us to explore mechanisms of pre-BCR–independent growth in B-ALL to identify new therapeutic targets.Here, we report that pre-BCR–independent SYK signaling enabled ab-errant B-ALL survival and proliferation in p53!/!; Prkdcscid/scid double-mutant mice. Furthermore, we found that diagnostic human B-ALLsamples displayed pre-BCR–independent SYK activation, and thatSYK inhibitors potently attenuated the proliferation of a large cohortof B-ALL samples belonging to multiple HR subgroups in vitro. Weshow that oral administration of fostamatinib, a small-molecule SYKinhibitor (19), potently reduced disease burden for several HR geneticsubtypes of B-ALL in xenotransplant studies. In particular, fostamatinibreduced B-ALL dissemination from bonemarrow (BM) to spleen, liver,kidneys, and the CNS. These results document pre-BCR–independentSYKactivation acrossmany genetic subtypes of B-ALL andprovide pre-clinical in vivo evidence that SYK inhibitionmay improve outcomes forHR B-ALL.
RESULTS
Pre-BCR–independent SYK activation in a p53!/!; Prkdcscid/scid
mouse model of early B-ALLDouble-mutant p53!/!; Prkdcscid/scid mice spontaneously developearly B-ALL with 100% incidence and a median latency of 8 weeks(16, 20). Consistent with their defect in DNA repair caused by aninactivating mutation in Prkdc (Prkdcscid/scid), leukemias in p53!/!;Prkdcscid/scid mice lack Igm expression (16). We therefore expectedB-ALL blasts of the double-mutant mice to be developmentallyarrested at the pro-B stage. However, there were substantial differ-ences between the global gene expression profiles of double-mutantmouse B-ALL blasts and normal pro-B cells (Fig. 1A). Interesting-ly, 39% (398 of 1028) of these genes were also differentially expressedbetween normal pro-B and pre-B cells, suggesting that double-mutant mouse B-ALL blasts exhibit a pre-B–like expression profile.For example, genes normally down-regulated during the pro-B topre-B transition, such as Dntt, Vpreb1, Igll1 (also known as l5), andPrkch (21), were underexpressed in double-mutant mouse leuke-mias compared to pro-B cells. Reciprocally, genes normally up-regulated during this developmental transition, such as Cd2, Cd22,MhcII, Ighm, and Igl (21), were overexpressed in double-mutantmouse B-ALL blasts relative to normal pro-B cells. We confirmed thatdouble-mutant mouse leukemias expressed many pre-B cell markersusing flow cytometry (fig. S1A). Thus, despite their failure to expressthe pre-BCR, double-mutant mouse leukemias exhibited many char-acteristics of normal pre-B cells, suggesting aberrant progressionthrough the pro-B developmental checkpoint in a pre-BCR–independentfashion.
Given the critical role of SYK in transducing pre-BCR signalsduring normal pre-B cell development (22), we asked if double-mutant mouse B-ALL cells exhibited pre-BCR–independent SYKactivation, which is accompanied by phosphorylation of two tyro-sine (Y) residues (Y348 and Y352) in the linker region, separating thepaired SH2 domains from the kinase domain (23). We detectedphospho-SYK (Y352) [pSYK(Y352)] by Western blotting ex vivo indouble-mutant mouse leukemia cells but could not detect it in BM
cells of p53-proficient littermate control Prkdcscid/scid mice (fig.S1B). This phospho-specific antibody for SYK also recognizes therelated kinase ZAP70. However, ZAP70 levels were low to un-detectable in double-mutant mouse B-ALL samples (fig. S1C), sug-gesting its minimal contribution to the pSYK(Y352) signals wedetected. The lack of detectable SYK levels in p53+/+; Rag2+/+;Prkcdscid/scid littermate controls could have reflected the low fre-quency of pro-B cells in these mice (fig. S1A). Thus, we also com-pared SYK abundance in double-mutant B-ALL to normal pro-Band pre-B cells by flow cytometry. Although total SYK levels weresimilar across all samples (fig. S1D), single-cell analysis usingphospho-specific flow cytometry (24) revealed robust pSYK(Y352)levels only in p53!/!; Prkdcscid/scid B-ALL samples (fig. S1E). Wefurther examined basal (stimulus-independent) phosphorylationof SYK (pSYK) and its downstream mediators BLNK and PLCg2in ex vivo p53!/!; Prkdcscid/scid leukemias because these proteins aredefined targets of pre-BCR– or BCR-dependent signaling cascades(14). We optimized and validated our phospho-flow protocol usingIgm+ Ramos B cell lymphoma cells, which rapidly and robustlyincreased the phosphorylation of SRC, SYK, BLNK, and PLCg2 in re-sponse to anti-IgM cross-linking. Furthermore, in human pre–B-ALLcell lines, phosphorylation of SYK and several other proteins wassubstantially reduced by a 2-hour pretreatment with two adenosinetriphosphate (ATP)–competitive SYK inhibitors, fostamatinib andBAY61-3606 (BAY61) (fig. S2A), suggesting basal SYK activationin these cells.
Staining with antibodies specific for pSYK(Y348) and pSYK(Y352)was significantly higher in all p53!/!; Prkdcscid/scidmouse leukemias thanin fluorescence minus one (FMO) staining controls (Fig. 1B). FMOcontrols contained all surface markers but without phospho-specificantibodies and were used to establish background fluorescence forpSYK antibodies. Furthermore, levels of pBLNK and pPLCg2 werehigher than the FMO background in p53!/!; Prkdcscid/scid mouse leuke-mic cells (Fig. 1B). Phosphorylation of all four epitopes was decreasedby a 2-hour pretreatment with fostamatinib and BAY61 (Fig. 1B). Fur-thermore, the proliferation of B-ALL cells isolated frommoribund double-mutant mice was significantly inhibited by both fostamatinib andBAY61 at clinically achievable concentrations (Fig. 1C). Fostamatiniband BAY61 displayed no effect on apoptosis in double-mutant mouseB-ALL samples (fig. S3), suggesting that SYK inhibition was not cyto-toxic in these samples. Collectively, these data suggest that pre-BCR–independent SYK signaling promotes the proliferation of double-mutantmouse leukemic B cells.
Fostamatinib has been clinically evaluated as a SYK-targetedtherapy for rheumatoid arthritis and B cell lymphoma (25). There-fore, we used fostamatinib to test the SYK dependence of growth af-ter transplantation of CD19+CD22+ B cells sorted from leukemicdouble-mutant mice into irradiated B6.CD45.1-Rag2!/! recipientmice. Fostamatinib-treated recipients exhibited lower spleen weightsand reduced engraftment of leukemic CD19+CD22+ B cells in BMand spleen compared to vehicle-treated recipients (Fig. 1D). To-gether, these data suggest that fostamatinib-sensitive SYK activityis required for double-mutant mouse leukemia survival and/or pro-liferation in vivo.
Pre-BCR–independent SYK activation in human B-ALLWe next asked if cIgm! B-ALL samples from SR andHR pediatric andadult patients exhibit evidence of SYK activation (table S1). All
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 2
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
samples expressed high levels of SYK and pSYK(Y348) but variablelevels of the pre-BCR signaling mediators CD79A, CD79B, BLNK,and PLCg2 (Fig. 2, A to C). Variable BLNK and CD79A/CD79B ex-pression in B-ALL has been previously reported (26, 27). We usedphospho-flow cytometry protocol to measure the impact of fostamatiniband BAY61 on basal levels of pSYK and downstream targets in leu-kemic blasts from diagnostic B-ALL samples. Because SYK is oftenactivated by SFKs (22), we also measured levels of autophosphory-lated SRC(Y418) using an antibody that detects several related SFKs.
The gating strategy used to positively identify live, single CD45low
leukemic blasts is shown (fig. S2B). Occasional samples containeddiscrete subsets with differing levels of the phospho-proteins, butmost displayed unimodal staining patterns, suggesting limited cel-lular heterogeneity (Fig. 2D). We observed variable levels of pSRC,pSYK, and pPLCg2 in six HR (BCR-ABL1!) cIgm! B-ALL samples(Fig. 2, D and E). Consistent with our pre–B-ALL cell line data (fig.S2A), pretreatment with 10 mM fostamatinib or 10 mMBAY61 acute-ly reduced phosphorylation of these proteins in all samples (Fig. 2, D
A
0.3 1 0
20
40
60
80
100
0.3 1 0
20
40
60
80
100
C
Fosta (µM)
BAY61 (µM)
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
0
100
200
300N
o. C
D19
+ /C
D22
+ (
106 )
Wei
ght (
mg)
D
***
0
10
20
30
40
**
FostaVehicle0
20
40
60
80
100 ***
FostaVehicleFostaVehicle
BMSpleen Spleen
0
300
600
*********
0
300
600***
*
0
300
600
900
*********
0
300
600
*********
pSYK(Y348)
pBLNK(Y84)
B
MFI
pPLC 2(Y759)
MFI
MFI
FMO Vehicle Fosta BAY61 FMO Fosta
FMO Fosta BAY61 FMO Fosta
MFI
pSYK(Y352)
BAY61
BAY61
Vehicle Vehicle
Vehicle
Pro-B:Pre-B Pro-B:DM
1274 398 630
2302
1672 1028
Myl10Vpreb1Socs2Socs3Csf2rbEngCd9SpnIgll1DnttPrkch
H2-A9H2-Eb1H2-Ab1H2-DMb2Cd22Cd2IghmTyrobpCd40Igl
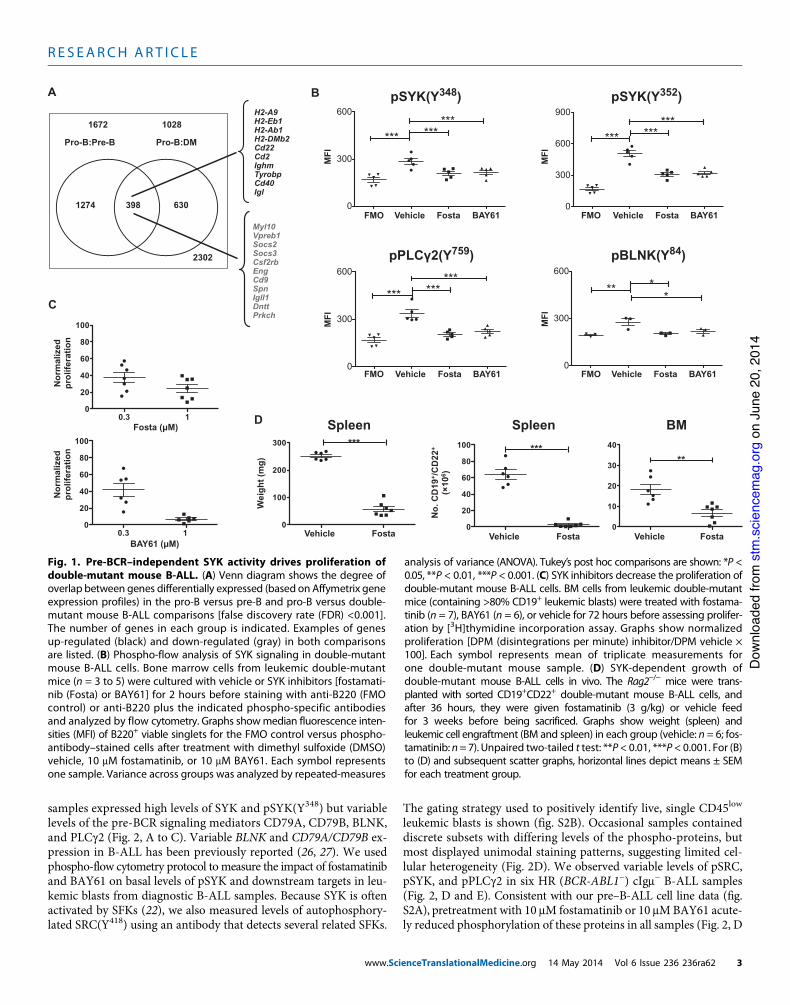
Fig. 1. Pre-BCR–independent SYK activity drives proliferation ofdouble-mutant mouse B-ALL. (A) Venn diagram shows the degree ofoverlap between genes differentially expressed (based on Affymetrix geneexpression profiles) in the pro-B versus pre-B and pro-B versus double-mutant mouse B-ALL comparisons [false discovery rate (FDR) <0.001].The number of genes in each group is indicated. Examples of genesup-regulated (black) and down-regulated (gray) in both comparisonsare listed. (B) Phospho-flow analysis of SYK signaling in double-mutantmouse B-ALL cells. Bone marrow cells from leukemic double-mutantmice (n = 3 to 5) were cultured with vehicle or SYK inhibitors [fostamati-nib (Fosta) or BAY61] for 2 hours before staining with anti-B220 (FMOcontrol) or anti-B220 plus the indicated phospho-specific antibodiesand analyzed by flow cytometry. Graphs showmedian fluorescence inten-sities (MFI) of B220+ viable singlets for the FMO control versus phospho-antibody–stained cells after treatment with dimethyl sulfoxide (DMSO)vehicle, 10 mM fostamatinib, or 10 mM BAY61. Each symbol representsone sample. Variance across groups was analyzed by repeated-measures
analysis of variance (ANOVA). Tukey’s post hoc comparisons are shown: *P <0.05, **P < 0.01, ***P < 0.001. (C) SYK inhibitors decrease the proliferation ofdouble-mutant mouse B-ALL cells. BM cells from leukemic double-mutantmice (containing >80% CD19+ leukemic blasts) were treated with fostama-tinib (n = 7), BAY61 (n = 6), or vehicle for 72 hours before assessing prolifer-ation by [3H]thymidine incorporation assay. Graphs show normalizedproliferation [DPM (disintegrations per minute) inhibitor/DPM vehicle !100]. Each symbol represents mean of triplicate measurements forone double-mutant mouse sample. (D) SYK-dependent growth ofdouble-mutant mouse B-ALL cells in vivo. The Rag2!/! mice were trans-planted with sorted CD19+CD22+ double-mutant mouse B-ALL cells, andafter 36 hours, they were given fostamatinib (3 g/kg) or vehicle feedfor 3 weeks before being sacrificed. Graphs show weight (spleen) andleukemic cell engraftment (BM and spleen) in each group (vehicle: n = 6; fos-tamatinib: n=7). Unpaired two-tailed t test: **P < 0.01, ***P < 0.001. For (B)to (D) and subsequent scatter graphs, horizontal lines depict means ± SEMfor each treatment group.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 3
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
and E), suggesting SYK dependence. These effects of SYK inhibitorswere also evident at lower doses (0.3 to 3 mM) in cell lines (fig. S4)and diagnostic samples (fig. S5), providing further evidence of SYK-dependent effects of these two inhibitors on these markers. Althoughfostamatinib reduced pBLNK levels in some samples, overall the re-ductions were not statistically significant (Fig. 2E), likely due to lowBLNK levels in some samples (26) (Fig. 2, B and C). In addition, basalSYK phosphorylation was evident across multiple samples (n = 62)regardless of immunophenotype (fig. S6), suggesting broad SYK ac-tivity in B-ALL. Collectively, these studies demonstrate that basalSYK signaling promotes phosphorylation of several pre-BCR signal-ing proteins in human B-ALL.
B-ALL survival and proliferationWe next tested the SYK dependence of survival and proliferation for alarge cohort of SR and HR B-ALL samples in vitro. Fostamatinib andBAY61 both robustly attenuated the proliferation of pediatric and adultcIgm!B-ALL samples in a dose-dependentmanner (Fig. 3, A to C). IC50
values calculated using nonlinear regression revealed that effective fos-tamatinib concentrations in this assay (fig. S7, A and B) were withinplasma concentrations achieved in clinical trials (28). Both inhibitorsalso markedly decreased the proliferation of cIgm+ B-ALL samples ina dose-dependent manner (fig. S7C). These antiproliferative effects ofSYK inhibitors were evident across many B-ALL samples and werenot correlated with variations in basal pSYK(Y348) levels (fig. S8).
0
2
4
6
8
A
SYK
-Actin
BLNK
-Actin
B c
ells
PB
MC
s
Ram
osPediatricB-ALL samples
B
SYK
-Actin
BLNK
-Actin
PLC 2
-Actin
B c
ells
PB
MC
s
Ram
osAdultB-ALL samples
C
CD79A
GAPDH
CD79B
GAPDH
Ram
os
B c
ells
D EpSRC pSYK pBLNK pPLC 2
Log2 (Treatment/FMO)
–3.5 3.50
VehicleFMO
FostaBAY61
VehicleFMO
FostaBAY61
VehicleFMO
FostaBAY61
VehicleFMO
FostaBAY61
VehicleFMO
FostaBAY61
VehicleFMO
FostaBAY61
PediatricB-ALL
AdultB-ALL
pSYK176 106 193 145 87 109 89
0
2
4
6
8 ****
MFI
FC
MFI
FC
MFI
FC
MFI
FC
pSRC
pBLNK
0
5
10
15***
***
0
2
4
6
8***
**
**
pPLC 2
pSYK
Vehicle Fosta BAY61
%Vehicle100 24 47 62 48 20 45
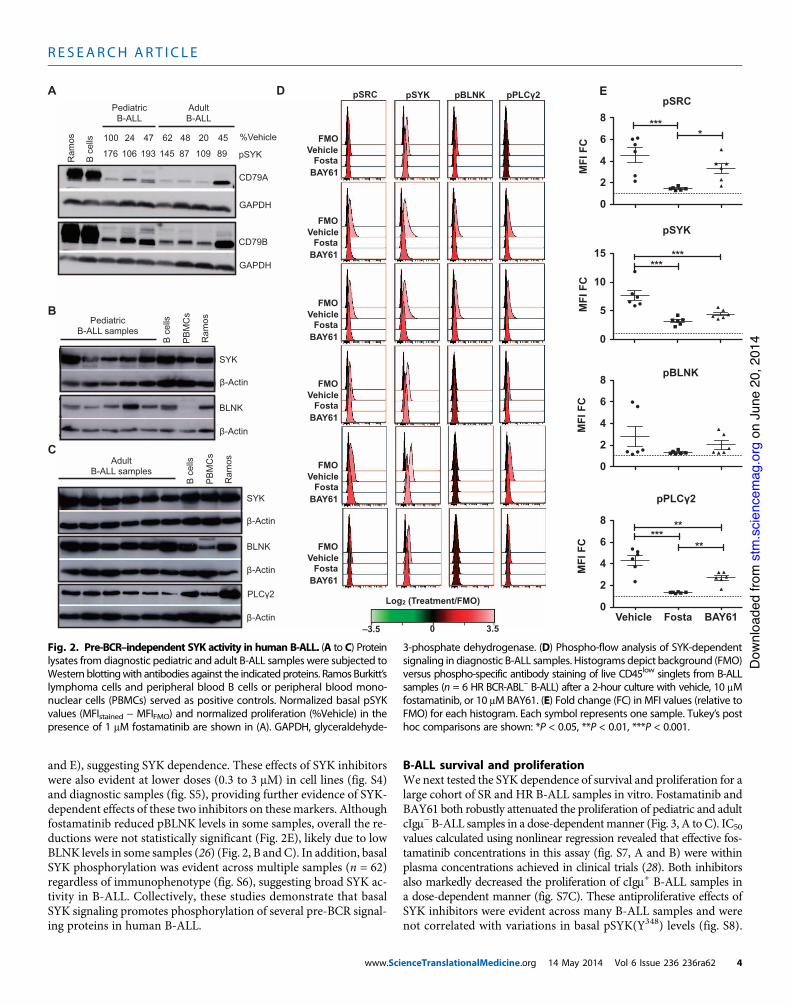
Fig. 2. Pre-BCR–independent SYK activity in human B-ALL. (A to C) Proteinlysates from diagnostic pediatric and adult B-ALL samples were subjected toWestern blottingwith antibodies against the indicated proteins. Ramos Burkitt’slymphoma cells and peripheral blood B cells or peripheral blood mono-nuclear cells (PBMCs) served as positive controls. Normalized basal pSYKvalues (MFIstained ! MFIFMO) and normalized proliferation (%Vehicle) in thepresence of 1 mM fostamatinib are shown in (A). GAPDH, glyceraldehyde-
3-phosphate dehydrogenase. (D) Phospho-flow analysis of SYK-dependentsignaling in diagnostic B-ALL samples. Histograms depict background (FMO)versus phospho-specific antibody staining of live CD45low singlets from B-ALLsamples (n = 6 HR BCR-ABL! B-ALL) after a 2-hour culture with vehicle, 10 mMfostamatinib, or 10 mM BAY61. (E) Fold change (FC) in MFI values (relative toFMO) for each histogram. Each symbol represents one sample. Tukey’s posthoc comparisons are shown: *P < 0.05, **P < 0.01, ***P < 0.001.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 4
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
The fostamatinib effects on B-ALL proliferation were not due toapoptosis induction, because fostamatinib did not increase the abun-dance of cleaved (activated) caspase-3 (Fig. 3D), similar to our obser-vations in double-mutant mouse B-ALL (fig. S3). Collectively, thesedata suggest that, similar to murine double-mutant mouse B-ALL,human B-ALL samples (both cIgm! and cIgm+) depend on SYK activa-tion for proliferation and survival in vitro.
Kinase specificity of SYK inhibitor effectsLike many small-molecule kinase inhibitors, fostamatinib inhibitsother targets, particularly FLT3 and SFKs (19). Although aberrantFLT3 activation is rare in B-ALL (29), pre-BCR–mediated SFK activa-tion plays a critical role in promoting survival in some Igm+ B-ALLsamples (30). Indeed, a 2-hour pretreatment with 10 mM fostamatinibacutely reduced pSRC levels in most samples tested (Fig. 2, D and E),
suggesting that this drug also targets SFKs. In contrast, BAY61 has amuch more restricted target spectrum that does not include FLT3 orSFKs (31). Nonetheless, we asked whether FLT3 inhibition byAGL2043 (32) or ABL/SFK inhibition by dasatinib (33) could impairB-ALL proliferation as potently as SYK inhibition by fostamatinib andBAY61. Although treatment with 1 mM AGL2043 potently inhibitedthe proliferation of MV4;11 acute myeloid leukemia (AML) cellsharboring activated mutant FLT3 (Fig. 4A), 3- to 10-fold higher dosesdid not impair the proliferation of B-ALL samples (Fig. 4B).Moreover,10 nM dasatinib potently inhibited the proliferation of 13 of 13 BCR-ABL+B-ALL samples, but only 3 of 30 BCR-ABL!B-ALL samples weresensitive to this dose (Fig. 4C). However, dasatinib inhibited basal pSRClevels in BCR-ABL! samples at concentrations as low as 10 nM, dem-onstrating the expected impact of this treatment (fig. S9). These datasuggest that ABL/SFK activation does not drive proliferation in most
A
Caspase-3
Vehicle Fosta 1 µM StaurosporineD
0.3 1 3 100
50
100
150
0.3 1 3 100
50
100
150
0.3 1 3 100
50
100
150
0.3 1 3 100
50
100
150
0.3 1 3 100
50
100
150
0.3 1 3 100
50
100
150
Fosta (µM) Fosta (µM) Fosta (µM)
C
BAY61 (µM) BAY61 (µM) BAY61 (µM)
B
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
SR pediatric HR pediatric HR adult
Vehicle Fosta, 1 µM0
10
20
30
40
% C
aspa
se-3
-pos
itive
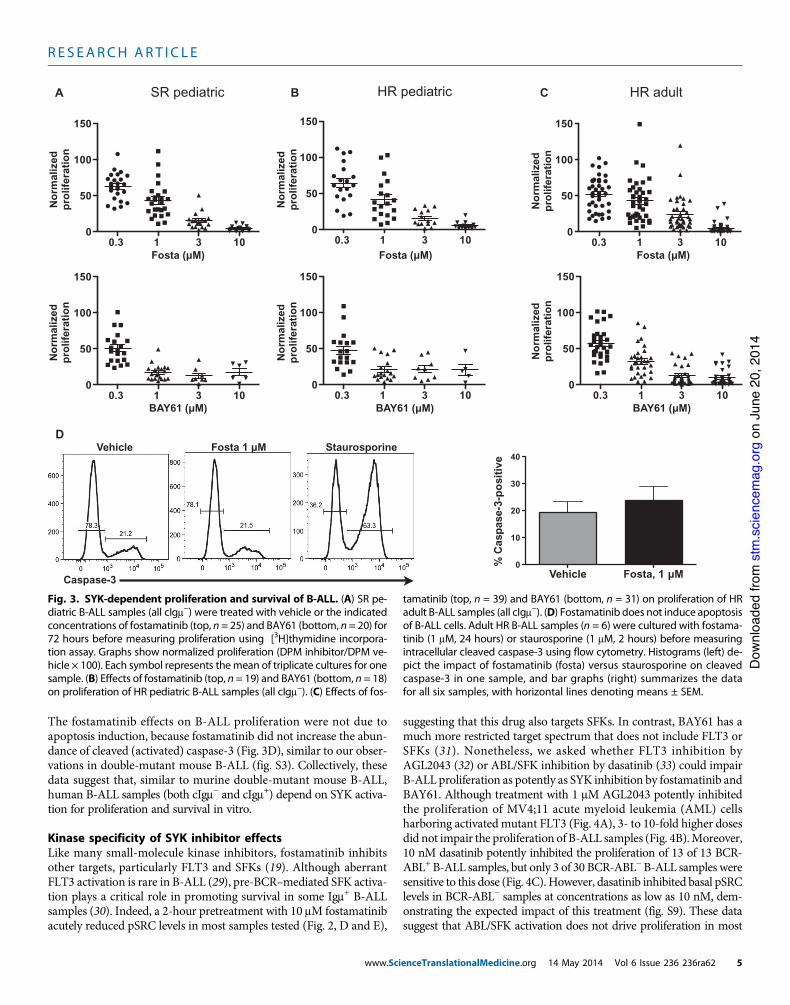
Fig. 3. SYK-dependent proliferation and survival of B-ALL. (A) SR pe-diatric B-ALL samples (all cIgm!) were treated with vehicle or the indicatedconcentrations of fostamatinib (top, n = 25) and BAY61 (bottom, n = 20) for72 hours before measuring proliferation using [3H]thymidine incorpora-tion assay. Graphs show normalized proliferation (DPM inhibitor/DPM ve-hicle ! 100). Each symbol represents themean of triplicate cultures for onesample. (B) Effects of fostamatinib (top, n = 19) and BAY61 (bottom, n = 18)on proliferation of HR pediatric B-ALL samples (all cIgm!). (C) Effects of fos-
tamatinib (top, n = 39) and BAY61 (bottom, n = 31) on proliferation of HRadult B-ALL samples (all cIgm!). (D) Fostamatinib does not induce apoptosisof B-ALL cells. Adult HR B-ALL samples (n = 6) were cultured with fostama-tinib (1 mM, 24 hours) or staurosporine (1 mM, 2 hours) before measuringintracellular cleaved caspase-3 using flow cytometry. Histograms (left) de-pict the impact of fostamatinib (fosta) versus staurosporine on cleavedcaspase-3 in one sample, and bar graphs (right) summarizes the datafor all six samples, with horizontal lines denoting means ± SEM.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 5
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
cases of BCR-ABL! B-ALL. It will be interesting to determine whetherthe three dasatinib-sensitive BCR-ABL! samples harbor other activat-ing mutations of the ABL kinase (11). Collectively, these data suggestthat SYK, rather than FLT3 or SFKs, is the primary target of fostama-tinib and BAY61 in SR and HR B-ALL samples.
Fostamatinib limits B-ALL growth after xenotransplantationWe used the well-established NOD.Prkdcscid/scidIl2rgtm1Wjl/SzJ (NSG)xenotransplantation assay to test the fostamatinib effects on B-ALLgrowth and dissemination in vivo (34). B-ALL samples were depletedof CD3+ cells and injected into the right femurs of NSG recipients tomaximize engraftment in BMniches and allow evaluation of leukemiadissemination to other bones and tissues (35). These studies focusedon B-ALL samples from HR subgroups (table S2), where there is
greatest clinical need for new therapies. SRpediatricsamples were also included because some patientsin this category also relapse. Rapid engraftment ki-netics after xenotransplantation has been shownto be an independent prognosticator of poor out-come for B-ALL patients (36).
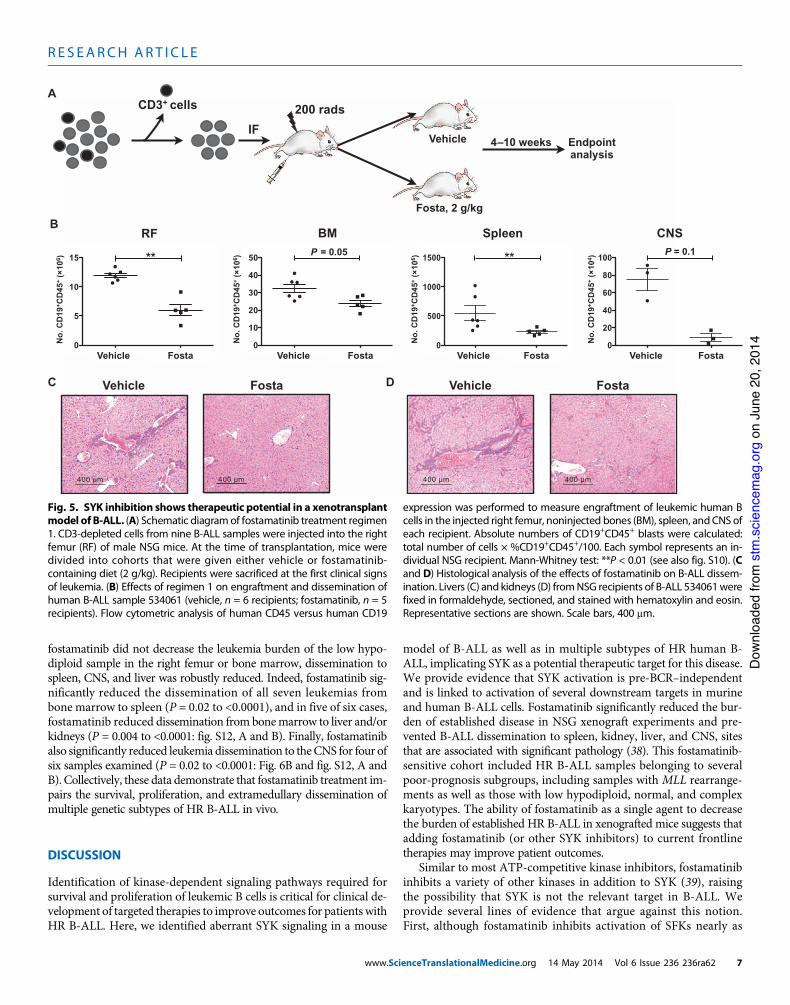
In pilot experiments, NSG recipients were giv-en vehicle or fostamatinib-impregnated feed im-mediately after transplantation, and analyzed 4to 10 weeks later for B-ALL engraftment in hema-topoietic organs (right femur, bone marrow fromnoninjected bones, and spleen) (Fig. 5A). Fosta-matinib reduced leukemia engraftment for all ninesamples in at least one tissue, as manifested by asignificant decrease in the number of engraftedleukemic B cells (P = 0.004 to 0.009: Fig. 5B, andP = 0.03 to <0.0001: fig. S10, A and B). Fostama-tinib treatment reduced disease burden in two tothree tissues for four of nine samples examined.Leukemia dissemination into nonhematopoietictissues (liver and kidney) caused marked morbid-ity, and fostamatinib also decreased disease bur-den to these sites (Fig. 5, C and D). Leukemiadissemination to the CNS is a serious complica-tion and an impediment to disease-free remissionin both pediatric and adult B-ALL (3, 37). In thesestudies, we detected CNS infiltration of B-ALLcells in three of nine samples. Fostamatinib de-monstrated a trend toward reduction of CNS in-filtration of these samples (Fig. 5B and fig. S10B).Together, these results suggested that fostamatinibtreatment can reduce B-ALL survival and expan-sion in vivo and limit dissemination and survivalin hematopoietic and nonhematopoietic tissues,including the CNS.
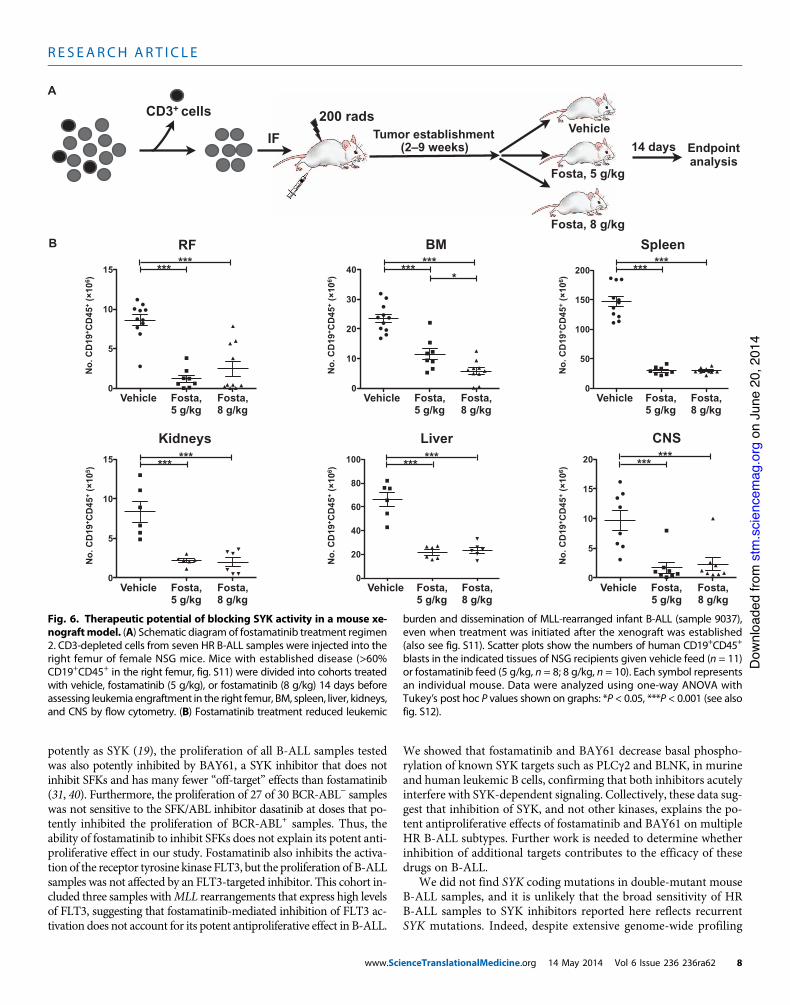
Next, we asked if fostamatinib could reduce theburden of well-established leukemic grafts, a moreclinically relevant scenario (Fig. 6A). For this sec-ond regimen, seven aggressive, fast-engraftingsamples were injected into NSGmice (25 to 31 re-cipients per sample). This HR cohort included oneBCR-ABL+ adult case, oneMLL rearranged infantcase, one low hypodiploid adult case, and four caseswith normal or complex karyotypes (table S2). To
ensure that disease was well established before initiating treatment, weassessed engraftment in sentinel recipients by timed sacrifice 2 to 9 weeksafter injection. Once sentinels displayed >60%CD19+CD45+ blasts in theright femur and robust leukemia dissemination to other bones and/orspleen (fig. S11), the remaining cohort was placed on vehicle (n = 85)or fostamatinib-containing feed (n = 108, two different concentrations).Recipients were evaluated for disease burden 14 days later.
Strikingly, even the lower fostamatinib dose significantly reducedleukemia burden in all tissues for the MLL rearranged sample 9037,with a clear dose-dependent reduction of dissemination to nonin-jected bones (P = 0.04 to <0.0001: Fig. 6B). In addition, fostamatinibsignificantly reduced leukemia burden in the injected right femurand/or other bones for four of the remaining six samples, and dose-dependent responses were also evident (fig. S12, A and B). Although
Dasatinib (nM)
C
0
50
100
150 BCR-ABL– (n = 30)BCR-ABL+ (n = 13)
10 30 100
Nor
mal
ized
prol
ifera
tion
0.3 1 3 100
50
100
150
200
250
0.3 1 3 100
50
100
150
200
250
AGL2043 (µM) AGL2043 (µM)
B
A
Pediatric Adult N
orm
aliz
edpr
olife
ratio
n
0.01 0.03 0.1 0.3 1 3 100
30
60
90
120
AGL2043 (µM)
Nor
mal
ized
prol
ifera
tion
Nor
mal
ized
prol
ifera
tion
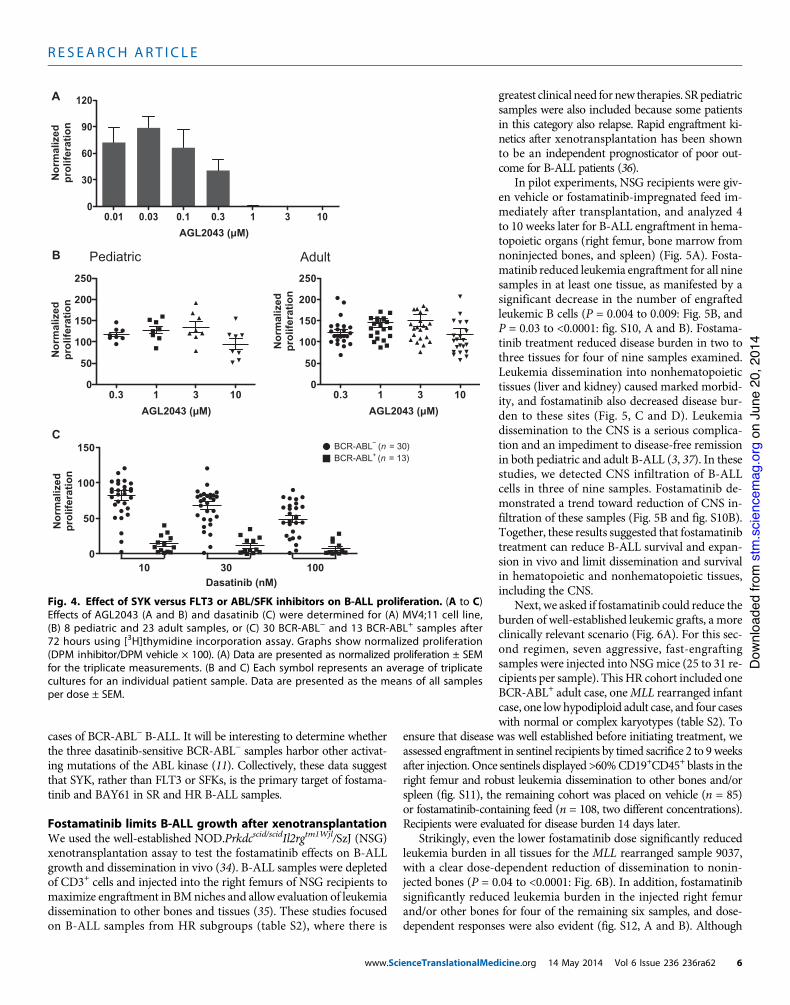
Fig. 4. Effect of SYK versus FLT3 or ABL/SFK inhibitors on B-ALL proliferation. (A to C)Effects of AGL2043 (A and B) and dasatinib (C) were determined for (A) MV4;11 cell line,(B) 8 pediatric and 23 adult samples, or (C) 30 BCR-ABL! and 13 BCR-ABL+ samples after72 hours using [3H]thymidine incorporation assay. Graphs show normalized proliferation(DPM inhibitor/DPM vehicle ! 100). (A) Data are presented as normalized proliferation ± SEMfor the triplicate measurements. (B and C) Each symbol represents an average of triplicatecultures for an individual patient sample. Data are presented as the means of all samplesper dose ± SEM.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 6
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
fostamatinib did not decrease the leukemia burden of the low hypo-diploid sample in the right femur or bone marrow, dissemination tospleen, CNS, and liver was robustly reduced. Indeed, fostamatinib sig-nificantly reduced the dissemination of all seven leukemias frombone marrow to spleen (P = 0.02 to <0.0001), and in five of six cases,fostamatinib reduced dissemination from bonemarrow to liver and/orkidneys (P = 0.004 to <0.0001: fig. S12, A and B). Finally, fostamatinibalso significantly reduced leukemia dissemination to theCNS for four ofsix samples examined (P = 0.02 to <0.0001: Fig. 6B and fig. S12, A andB). Collectively, these data demonstrate that fostamatinib treatment im-pairs the survival, proliferation, and extramedullary dissemination ofmultiple genetic subtypes of HR B-ALL in vivo.
DISCUSSION
Identification of kinase-dependent signaling pathways required forsurvival and proliferation of leukemic B cells is critical for clinical de-velopment of targeted therapies to improve outcomes for patients withHR B-ALL. Here, we identified aberrant SYK signaling in a mouse
model of B-ALL as well as in multiple subtypes of HR human B-ALL, implicating SYK as a potential therapeutic target for this disease.We provide evidence that SYK activation is pre-BCR–independentand is linked to activation of several downstream targets in murineand human B-ALL cells. Fostamatinib significantly reduced the bur-den of established disease in NSG xenograft experiments and pre-vented B-ALL dissemination to spleen, kidney, liver, and CNS, sitesthat are associated with significant pathology (38). This fostamatinib-sensitive cohort included HR B-ALL samples belonging to severalpoor-prognosis subgroups, including samples with MLL rearrange-ments as well as those with low hypodiploid, normal, and complexkaryotypes. The ability of fostamatinib as a single agent to decreasethe burden of established HR B-ALL in xenografted mice suggests thatadding fostamatinib (or other SYK inhibitors) to current frontlinetherapies may improve patient outcomes.
Similar to most ATP-competitive kinase inhibitors, fostamatinibinhibits a variety of other kinases in addition to SYK (39), raisingthe possibility that SYK is not the relevant target in B-ALL. Weprovide several lines of evidence that argue against this notion.First, although fostamatinib inhibits activation of SFKs nearly as
0
20
40
60
80
100
0
5
10
15
200 radsCD3+ cells
IFEndpoint analysis
Vehicle
Fosta, 2 g/kg
4–10 weeks
RF BM Spleen CNS
A
B
0
10
20
30
40
50
0
500
1000
1500
No.
CD
19+ C
D45
+ (
106 )
Vehicle Fosta Vehicle Vehicle VehicleFosta Fosta Fosta
**
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
104 )
C Vehicle Fosta D Vehicle Fosta
** P = 0.1P = 0.05
400 µm400 µm400 µm400 µm
Fig. 5. SYK inhibition shows therapeutic potential in a xenotransplantmodel of B-ALL. (A) Schematic diagramof fostamatinib treatment regimen1. CD3-depleted cells from nine B-ALL samples were injected into the rightfemur (RF) of male NSG mice. At the time of transplantation, mice weredivided into cohorts that were given either vehicle or fostamatinib-containing diet (2 g/kg). Recipients were sacrificed at the first clinical signsof leukemia. (B) Effects of regimen 1 on engraftment and dissemination ofhuman B-ALL sample 534061 (vehicle, n = 6 recipients; fostamatinib, n = 5recipients). Flow cytometric analysis of human CD45 versus human CD19
expression was performed to measure engraftment of leukemic human Bcells in the injected right femur, noninjected bones (BM), spleen, andCNS ofeach recipient. Absolute numbers of CD19+CD45+ blasts were calculated:total number of cells ! %CD19+CD45+/100. Each symbol represents an in-dividual NSG recipient. Mann-Whitney test: **P < 0.01 (see also fig. S10). (Cand D) Histological analysis of the effects of fostamatinib on B-ALL dissem-ination. Livers (C) and kidneys (D) fromNSG recipients of B-ALL 534061werefixed in formaldehyde, sectioned, and stained with hematoxylin and eosin.Representative sections are shown. Scale bars, 400 mm.
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 7
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
potently as SYK (19), the proliferation of all B-ALL samples testedwas also potently inhibited by BAY61, a SYK inhibitor that does notinhibit SFKs and has many fewer “off-target” effects than fostamatinib(31, 40). Furthermore, the proliferation of 27 of 30 BCR-ABL! sampleswas not sensitive to the SFK/ABL inhibitor dasatinib at doses that po-tently inhibited the proliferation of BCR-ABL+ samples. Thus, theability of fostamatinib to inhibit SFKs does not explain its potent anti-proliferative effect in our study. Fostamatinib also inhibits the activa-tion of the receptor tyrosine kinase FLT3, but the proliferation of B-ALLsamples was not affected by an FLT3-targeted inhibitor. This cohort in-cluded three samples withMLL rearrangements that express high levelsof FLT3, suggesting that fostamatinib-mediated inhibition of FLT3 ac-tivation does not account for its potent antiproliferative effect in B-ALL.
We showed that fostamatinib and BAY61 decrease basal phospho-rylation of known SYK targets such as PLCg2 and BLNK, in murineand human leukemic B cells, confirming that both inhibitors acutelyinterfere with SYK-dependent signaling. Collectively, these data sug-gest that inhibition of SYK, and not other kinases, explains the po-tent antiproliferative effects of fostamatinib and BAY61 on multipleHR B-ALL subtypes. Further work is needed to determine whetherinhibition of additional targets contributes to the efficacy of thesedrugs on B-ALL.
We did not find SYK coding mutations in double-mutant mouseB-ALL samples, and it is unlikely that the broad sensitivity of HRB-ALL samples to SYK inhibitors reported here reflects recurrentSYK mutations. Indeed, despite extensive genome-wide profiling
0
5
10
15
20
0
20
40
60
80
100
0
5
10
15
0
50
100
150
200
0
10
20
30
40
0
5
10
15
200 radsTumor establishment
(2–9 weeks)
CD3+ cells
IFVehicle
Fosta, 5 g/kg
Fosta, 8 g/kg
14 days
A
RF BM Spleen
CNSLiverKidneys
B
Endpoint analysis
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
105 )
No.
CD
19+ C
D45
+ (
106 )
No.
CD
19+ C
D45
+ (
106 )
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
Vehicle Fosta,5 g/kg
Fosta,8 g/kg
*** *** *** *** *** ***
*** *** *** *** *** ***
*
Fig. 6. Therapeutic potential of blocking SYK activity in a mouse xe-nograftmodel. (A) Schematic diagram of fostamatinib treatment regimen2. CD3-depleted cells from seven HR B-ALL samples were injected into theright femur of female NSG mice. Mice with established disease (>60%CD19+CD45+ in the right femur, fig. S11) were divided into cohorts treatedwith vehicle, fostamatinib (5 g/kg), or fostamatinib (8 g/kg) 14 days beforeassessing leukemia engraftment in the right femur, BM, spleen, liver, kidneys,and CNS by flow cytometry. (B) Fostamatinib treatment reduced leukemic
burden and dissemination of MLL-rearranged infant B-ALL (sample 9037),even when treatment was initiated after the xenograft was established(also see fig. S11). Scatter plots show the numbers of human CD19+CD45+
blasts in the indicated tissues of NSG recipients given vehicle feed (n = 11)or fostamatinib feed (5 g/kg, n = 8; 8 g/kg, n = 10). Each symbol representsan individual mouse. Data were analyzed using one-way ANOVA withTukey’s post hoc P values shown on graphs: *P < 0.05, ***P < 0.001 (see alsofig. S12).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 8
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

studies, SYK mutations have not been reported in HR B-ALL (41).Furthermore, SYK is not a frequent or recurrent target of point muta-tions or copy number aberrations in epithelial and other solid tumors(http://www.cbioportal.org; http://cancer.sanger.ac.uk/cosmic/gene/analysis), although epigenetic dysregulation of SYK expression leadingto lineage-inappropriate SYK dependence has been reported in retino-blastoma (42). Chronically active SYK-dependent BCR signaling, like-ly driven by self-antigens in a cell-autonomous fashion, is essential forsurvival of B cell chronic lymphocytic leukemia (43), as well as onesubtype of diffuse large B cell lymphoma (DLBCL) (44). Although20% of these DLBCL cases harbored gain-of-function mutations inCD79A or CD79B ITAMs, mutational mechanisms that promoteBCR signaling have not been identified in most cases. Similarly, someAMLcases express active SYKand are sensitive to SYK inhibition (45),but SYK mutations have not been identified in this disease (46). It isimportant to note that SYK can be activated by many receptorsexpressed on B cells, including Fc receptors, integrins, C-type lectins,and cytokines (22, 47). Although many of these receptors containpaired or single ITAMs, ITAM-independent mechanisms for SYK ac-tivation have also been described (22). Furthermore, like many ki-nases, SYK is regulated by phosphatases and ubiquitin ligases (22,48). Thus, although we cannot exclude the possibility that some HRB-ALLs harbormutations in SYK orCD79A/CD79B (44), we favor thenotion that nonmutational mechanisms that induce states akin toligand-independent “tonic” signaling promote widespread SYK acti-vation in HR B-ALL.
Evidence for the role of SYK in early B-ALL has been contro-versial. SYK deficiency was reported in an early study of pediatricB-ALL, suggesting that SYK functions as a tumor suppressor (49),and down-regulation of B cell signaling genes was identified as a sig-nature associatedwith IKAROSdeletion in adult B-ALL (50). AlthoughSYK deletion was reported in 1 of 22 cases of BCR-ABL+ B-ALL (27), 8of 9 of the BCR-ABL+ cases we have profiled expressed fostamatinib/BAY61-sensitive pSYK(Y348) and pPLCg2 epitopes (Perova et al., un-published), suggesting that SYK signaling is active in most cases ofBCR-ABL+ B-ALL. In contrast, SYK served as a proto-oncogene in amodel of transduction-mediated TEL-SYK B-ALL, supporting a tu-morigenic role of SYK signaling (51). Our findings provide direct evi-dence that pre-BCR–independent SYK activation is a common event inhuman B-ALL. Moreover, SYK inhibition reduced the proliferation ofsamples, among which 70% came from poor-prognosis groups at HRfor relapse. Further studies are needed to define the upstream mech-anisms of pre-BCR–independent SYK activation in B-ALL because thismay reveal additional targets for therapeutic development. Regardless ofthemechanisms of activation, SYK appears to regulate key signal trans-duction pathways of abnormal proliferation and survival in B-ALL.Thus, small-molecule SYK inhibitors represent promising agents fortreating poor-prognosis and relapsed B-ALL.
CNS involvement with ALL is associated with poor prognosisand increased risk of ALL relapse (3). There has been little innova-tion in CNS-directed therapy since the introduction of intrathecalchemotherapy and cranial irradiation, which contemporary treat-ment seeks to avoid because of their debilitating side effects (52).Our findings have two important implications for B-ALL in theCNS. First, we demonstrated the utility of xenografted NSG miceto model CNS disease, providing a valuable screen for preclinicaltesting of known or new therapeutic agents. Second, we describethat SYK inhibition by small-molecule therapeutics can impair dis-
semination and/or survival of leukemic blasts in the CNS. Fostama-tinib substantially reduced CNS dissemination of four HR B-ALLsamples after engraftment into NSG mice, using an experimentalprotocol designed to mimic treatment of patients presenting witha significant leukemia cell burden. This cohort included an MLL-rearranged infant case with CNS involvement at diagnosis as wellas other poor-prognosis samples. Our data suggest that SYK inhi-bitors may provide an appropriate treatment option in poor-prog-nosis B-ALL patients with CNS involvement.
MATERIALS AND METHODS
Study designThis study used 91 diagnostic B-ALL samples (table S1) to gain in-sights into the signaling abnormalities that regulate aberrant prolif-eration and survival of B-ALL cells and to identify new therapeutictargets in this malignancy. The sample size was based on the availa-bility of B-ALL samples from viable tissue biobanks at the Hospitalfor Sick Children and University Health Network (Toronto, Canada).For all in vivo experiments, mice were randomly assigned to eitherthe vehicle or the fostamatinib treatment group. The sample selectionfor in vivo studies was based on our preliminary experiments, wherewe prescreened each sample to ensure that they engrafted robustly(>80% human leukemic cells in NSG BM) and caused leukemia-related morbidities within 10 weeks. The numbers of samples andexperimental replicates for each experiment are indicated in the figurelegends.
Micep53!/!; Prkdcscid/scid double-mutant mice and Rag2!/! mice werehoused in specific pathogen–free (SPF) conditions at the Hospitalfor Sick Children (Toronto, Canada), as previously described (16, 20).NSG mice were housed in SPF conditions at the Max Bell ResearchCentre facility (Toronto, Canada). All experiments were conductedin accordance with and approval from the Institutional Animal CareCommittees.
Human B-ALL samples and cell linesThis study had research ethics board approvals at the Hospital forSick Children and University Health Network (Toronto, Canada).BM or peripheral blood samples from newly diagnosed patientswere obtained with informed consent. Mononuclear cells wereisolated with Ficoll-Paque density gradient separation (GE Health-care) according to the manufacturer’s instructions. Mononuclearcells were viably frozen in 90% (v/v) fetal bovine serum containing10% (v/v) DMSO and stored long-term in a vapor phase of liquidnitrogen. Pre–B-ALL cell lines were purchased from the GermanCollection of Microorganisms and Cell Culture (KOPN-8 andNALM-6) or American Type Culture Collection (MV4;11 andRS4;11) and maintained as per suppliers’ instructions.
Kinase inhibitorsAstraZeneca provided two fostamatinib formulations: R406, ametabolic active form, was used for the in vitro experiments, andR788, an orally bioavailable form, was impregnated into AIN-76Arodent diet (2 to 8 g of R788 per kilogram of feed; Research Diets) forthe in vivo experiments. Both forms are referred to as “fostamatinib.”
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 9
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

BAY61 and AGL2043 were purchased from EMD Chemicals. Dasatinibwas purchased from Toronto Research Chemicals.
Gene expression microarraysRNA was isolated from BM or lymph node cell suspensions in TRI-zol reagent (Invitrogen), as per the manufacturer’s instructions,and cleaned with the RNeasy isolation kit (Qiagen). Biotin-labeledcomplementary RNA (cRNA) probes were prepared (The Centrefor Applied Genomics at the Hospital for Sick Children) as recom-mended by the manufacturer (Affymetrix). Biotin-labeled cRNAwas hybridized to the Affymetrix Mouse Genome 430 2.0 array,as recommended by the manufacturer, and washed with an auto-mated fluidics workstation, and arrays were scanned on a GeneChipScanner 3000 (Affymetrix). All microarray data were deposited in theNational Center for Biotechnology Information Gene Expression Om-nibus database (GSE56345).
Statistical and graphical analyses were carried out in R forWindows, version 2.4. One-way ANOVA was implemented withthe linear models for microarray analysis (LIMMA) package (53),part of the Bioconductor project (http://www.Bioconductor.org) (54).Affymetrix CEL files were preprocessed with Robust MultiChip Anal-ysis (55, 56). Statistical analysis was performed on log2-transformeddata, and differential expression of genes was determined with an em-pirical Bayes approach and moderated F statistics within LIMMA(53), with the Benjamini-Hochberg FDR method to correct for multi-ple testing (57). For the Venn analysis, probe sets were selected on thebasis of differential expression in the pro-B versus pre-B and pro-Bversus p53!/!; Prkdcscid/scid B-ALL comparisons (FDR "0.001). Poorlyannotated probe sets (for example, with Riken and LOC descriptions)were filtered out. For genes represented by multiple probe sets, weselected the probe set with the lowest (most significant) P value. Theresulting two nonredundant gene lists were then subjected to Vennanalysis to identify the degree of overlap between them.
Western blot analysisA B Cell Isolation Kit (BD Biosciences) was used to enrich for Bcells from peripheral blood (purity >98%). Protein extracts wereprepared from single-cell suspensions in modified radioimmuno-precipitation assay (RIPA) buffer [50 mM tris-Cl (pH 7.4), 1%NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA]with protease inhibitor (Roche) and Halt phosphatase inhibitorcocktails (Pierce) and separated by SDS–polyacrylamide gel elec-trophoresis. Proteins were transferred onto polyvinylidene difluoridemembranes before Western blotting using standard techniques withantibodies against CD79a, BLNK, PLCg2, pSYK(Y352), GAPDH, b-actin (Cell Signaling), CD79b (Santa Cruz Biotechnology), total SYK(SYK-01, Monosan), and total ZAP70 (Upstate Biotechnologies Inc.),followed by species-specific horseradish peroxidase–conjugated anti-bodies (Santa Cruz Biotechnology and Cell Signaling) for 45 min. De-tection was performed with ECL reagents (GE Healthcare).
Flow cytometry and adoptive transfer of murine cellsSingle-cell suspensions were prepared from BM, spleen, or lymph nodesof moribund p53!/!; Prkdcscid/scid double-mutant mice, and flow cyto-metric analyses were performed as previously described (16, 58). All sam-ples contained >85% CD19+ leukemic blasts. In some experiments, totalBM from littermate control mice (p53+/+ or p53+/!; Prkdcscid/scid), contain-ing 5 to 10% CD19+ cells, was used as a control.
p53!/!; Prkdcscid/scid B-ALL cells were stained with fluorochrome-conjugated antibodies, specific for CD19-PE (phycoerythrin) (1D3),CD43-FITC (fluorescein isothiocyanate) (S7), CD22-biotin andCD22-PE (Cy34.1), CD2-biotin andCD2-PE (RM2-5), andmajor his-tocompatibility complex (MHC) II–PE (M5/114.15.2), all purchasedfrom BD Biosciences. Biotinylated primary antibodies were revealedby streptavidin–Alexa Fluor 633 (Molecular Probes). Data werecollected on a FACSCalibur flow cytometer (BDBiosciences) and ana-lyzed with FlowJo software v 9.1 (Tree Star).
To purify B-ALL cells for adoptive transfer experiments, BM cellsuspensions from moribund p53!/!; Prkdcscid/scid mice were stainedwith antibodies specific for murine CD19-PE-Cy7, CD43-FITC,CD22-PE, and CD11b-APC (allophycocyanin) (M1/70). ViableCD19+CD11b!CD43!CD22+ cells were sorted (purity >97%) andinjected intravenously (103 per mouse) into irradiated (650 cGy)Rag2!/!mice. Recipientmice received AIN-76A rodent diet (ResearchDiets) impregnated with fostamatinib (3 g/kg) or vehicle 36 hours lat-er. Mice were sacrificed 3 weeks later to analyze leukemia engraftmentin BM and spleen by stainingwithmurine CD19-APC (1D3), CD22-PE,and CD45.2-FITC (104). Viable cell counts were determined by trypanblue exclusion, and absolute numbers of CD19+CD22+CD45.2+ donorcells were calculated using the following formula: total number of livecells ! % CD19+CD45+/100. Fluorescence compensation was performedusing anti-mouse, anti-rat, or anti-hamster Igk and negative control com-pensationparticles (BDBiosciences), stainedwith fluorochrome-conjugatedantibody.
Lymphoblast isolationLiver, kidneys, brain, and spinal cord were removed from mice.Cell suspensions were prepared in staining medium [Hanks’ balancedsalt solution, 10 mM Hepes (pH 7.2), 2% calf serum]. Isolation of leu-kemic cells from CNS tissue was performed with Percoll gradients[70%/37%/30% Percoll (GE Healthcare)]. After centrifugation (400g,20 min), cells were collected from the 37%/70% interface, washed,and resuspended in RPMI, supplemented with 10% calf serum. Isola-tion of leukemic cells from liver and kidneys was performed by resus-pending cells in 33% Percoll gradient. After centrifugation (400g,20 min), cell pellets were washed and resuspended in RPMI, supple-mented with 10% calf serum.
Flow cytometric analyses of human samplesFor immunophenotyping and engraftment analysis of human leuke-mias, cells were stained with antibodies specific for human CD19-PE(4G7), human CD45-FITC (2D1), human CD10-APC (HI10a), hu-man CD20–peridinin chlorophyll protein (PerCP)–Cy5.5 (H1 FB1),human CD34-PE-Cy7 (8G12), and human CD38-APC (HB7), allfrom BD Biosciences. Live cells were discriminated by staining with4!,6-diamidino-2-phenylindole (100 ng/ml) (Molecular Probes) orFixable Blue viability dye (1:1000; Molecular Probes), as per the man-ufacturer’s instruction. For apoptosis assays, B-ALL samples werecultured with fostamatinib (1 to 3 mM) or DMSO (0.1%, v/v) for24 hours. Treatment with staurosporine was included as a positive con-trol for caspase-3 activation. Fixable Blue viability stain was added30 min before fixing and permeabilizing cells with Cytofix/CytopermFixation and Permeabilization Solution (BD Biosciences). Cells werethen stained with an FITC-conjugated antibody specific for activecaspase-3 (Active Caspase-3 Apoptosis kit, BD Biosciences), accord-ing to the manufacturer’s instructions. The ArC Amine Reactive
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 10
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

Compensation Bead Kit (Molecular Probes) was used for FixableBlue stain compensation. Cellular fluorescence was measured on anLSRII or LSRFortessa flow cytometer (BD Biosciences).
Phospho-specific flow cytometryPhospho-specific antibodies were purchased fromBDBiosciences andincluded SRC(Y418)–Alexa Fluor 488 (K98-37), SYK(Y352)/ZAP70(Y319)–Alexa Fluor 488 (17a), SYK(Y348)–Alexa Fluor 488/PE (I120-722), BLNK(Y84)–Alexa Fluor 647 (J117-1278), and PLCg2(Y759)–Alexa Fluor 488 (K86-689.37). CD45-APC-Cy7 (BD Biosciences,2D1) antibody was used to discriminate human leukemic cells.Murine B-ALL samples were stained with anti-B220–PE–Cy7 (BDBiosciences, RA3-6B2). Before phospho-flow analysis, B-ALL celllines were serum-deprived for 24 hours in serum- and phenol red–freeRPMI 1640, supplemented with 25 mM Hepes (pH 7.2), 1 mM sodi-um pyruvate, 2 mM L-glutamine, and 0.1 mM nonessential aminoacids [serum-free medium (SFM)]. BM cells from murine B-ALLand patient samples were rapidly thawed and allowed to recover for1 hour in SFM. Cells were then cultured with fostamatinib (0.3 to10 mM), BAY61 (0.3 to 10 mM), or DMSO (0.1%, v/v) vehicle in SFMfor 2 hours at 37°C. Fixable Blue viability dye (1:1000) was added for thelast 30 min of culture. Cells were fixed with BD Cytofix buffer (10 min,37°C) and permeabilized on ice (30 min) with Perm Buffer III (BDBiosciences). Cellswere rehydrated andwashedwithphosphate-bufferedsaline (PBS) containing 1% (w/v) bovine serum albumin (BSA), collectedby centrifugation, and stained with predetermined optimal concen-trations of antibodies (30 min). Cells were washed and resuspendedin PBS/BSA for flow cytometric analysis on an LSRII flow cytometer.FCS3.0 data files were analyzed with FlowJo v 9.6 and Cytobank(http://cytobank.org/) (59).
Proliferation assayPatient samples were thawed and incubated at 37°C overnight inStemSpanmedium (Stem Cell Technologies) containing 25 mMHepes(pH 7.2), 1 mM sodium pyruvate, 1 mM L-glutamine, and 1 mM non-essential amino acids. Cells were cultured in triplicate (1.5 ! 105 perwell) in 96-well flat-bottom plates with vehicle or the indicated inhibi-tors for 72 hours. [methyl-3H]Thymidine (1 mCi per well) was added16 hours before harvesting (Inotech Cell Harvester). DPMs were mea-sured (Beckman LS 6500) by scintillation counting.
B-ALL xenograft assaysB-ALL samples were depleted of CD3+ cells with CD3 MicroBeads(Miltenyi Biotec) and an AutoMACS Pro Separator (Miltenyi Biotec),as per the manufacturer’s protocols. Purity of eluted cells was assessedby flow cytometry with anti-CD3 antibody (BD Biosciences, SK7)and was >99% in all cases. NSGmale and female mice (8 to 12 weeksold) were sublethally irradiated (200 cGy) 24 hours before intrafe-moral injection of CD3-depleted leukemia cells (3 ! 105 to 5 ! 106
in 30 ml of PBS, depending on leukemia sample) as previously de-scribed (60). For fostamatinib regimen 1, animals were treated withcontrol or fostamatinib-impregnated (2 g/kg) AIN-76A rodent dietstarting on the day of B-ALL injection. For fostamatinib regimen 2,B-ALL engraftment was monitored by flow cytometry for 2 to 8 weeksafter injection. After xenografts were well established, mice were givencontrol AIN-76A diet, fostamatinib feed with 5 kg/kg, or fostamatinibfeed with 8 g/kg (Research Diets). Mice were monitored daily for signsof morbidity and sacrificed 4 to 12 weeks after transplantation. The
injected right femur, noninjected bones (BM), spleen, brain, liver, andkidneys were analyzed for human leukemia cells by flow cytometrywith human-specific CD45 and CD19 antibodies (see “Flow cyto-metric analyses of human samples”). Viable cell counts were deter-mined by trypan blue exclusion, and absolute numbers of CD19+CD45+
blasts were calculated using the following formula: total number oflive cells ! % CD19+CD45+/100.
Statistical analysisAll data are presented as means ± SEM. Gaussian distribution wasassessed by the D’Agostino-Pearson normality test. Two-group com-parisons were performed with paired Student’s t test orMann-Whitneytest (for data with non-Gaussian distribution). One-way ANOVA withTukey’s post hoc test was used to perform three-group comparisons,whereas repeated-measures ANOVA was used to compare matchedgroups. Correlations were assessed by two-tailed Spearman correlation.Statistical differences with two-tailed probability values of P < 0.05 wereconsidered significant. All data were analyzed with GraphPad Prismsoftware, version 5.0, for Mac OS X.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/236/236ra62/DC1Fig. S1. p53!/!; Prkdcscid/scid mouse leukemias display a pre-BCR–independent pro-B to pre-Bcell transition, related to Fig. 1.Fig. S2. Phospho-flow analysis of SYK-dependent signaling in B-ALL, related to Fig. 2.Fig. S3. Assessment of apoptosis in double-mutant B-ALL after treatment with SYK inhibitors,related to Fig. 1.Fig. S4. Dose-response effects of SYK inhibitors on SYK-dependent signaling in cell lines, re-lated to Fig. 2.Fig. S5. Dose-response effects of fostamatinib and BAY61 on SYK-dependent signaling in B-ALL, related to Fig. 2.Fig. S6. Relationship between immunophenotype and SYK activation in B-ALL, related to Fig. 2.Fig. S7. IC50 values for fostamatinib and BAY61 effects on B-ALL proliferation, related to Fig. 3.Fig. S8. Relationship between SYK phosphorylation and activity of SYK inhibitors in B-ALL,related to Fig. 3.Fig. S9. Relationship between pSRC inhibition and activity of dasatinib in B-ALL, related to Fig. 4.Fig. S10. Fostamatinib reduces B-ALL tumor burden after xenografting into NSG mice, relatedto Fig. 5.Fig. S11. Tumor burden in NSG mice at the start of fostamatinib regimen 2, related to Fig. 6.Fig. S12. Fostamatinib decreases the burden of well-engrafted B-ALL, related to Fig. 6.Table S1. Clinical characteristics of B-ALL samples used in phospho-flow and in vitro prolifer-ation assays (Figs. 1 to 4).Table S2. Clinical characteristics of 13 B-ALL samples used in xenotransplant studies (Figs. 5and 6).
REFERENCES AND NOTES
1. C. H. Pui, L. L. Robison, A. T. Look, Acute lymphoblastic leukaemia. Lancet 371, 1030–1043(2008).
2. C. H. Pui, C. Cheng, W. Leung, S. N. Rai, G. K. Rivera, J. T. Sandlund, R. C. Ribeiro, M. V. Relling,L. E. Kun, W. E. Evans, M. M. Hudson, Extended follow-up of long-term survivors of childhoodacute lymphoblastic leukemia. N. Engl. J. Med. 349, 640–649 (2003).
3. F. Locatelli, M. Schrappe, M. E. Bernardo, S. Rutella, How I treat relapsed childhood acutelymphoblastic leukemia. Blood 120, 2807–2816 (2012).
4. E. Liew, E. G. Atenafu, A. D. Schimmer, K. W. Yee, A. C. Schuh, M. D. Minden, V. Gupta,J. M. Brandwein, Outcomes of adult patients with relapsed acute lymphoblastic leukemiafollowing frontline treatment with a pediatric regimen. Leuk. Res. 36, 1517–1520 (2012).
5. C. H. Pui, W. E. Evans, Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354,166–178 (2006).
6. M. J. Borowitz, M. Devidas, S. P. Hunger, W. P. Bowman, A. J. Carroll, W. L. Carroll, S. Linda,P. L. Martin, D. J. Pullen, D. Viswanatha, C. L. Willman, N. Winick, B. M. Camitta; Children’sOncology Group, Clinical significance of minimal residual disease in childhood acute lym-
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 11
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

phoblastic leukemia and its relationship to other prognostic factors: A Children’s OncologyGroup study. Blood 111, 5477–5485 (2008).
7. K. R. Schultz, D. J. Pullen, H. N. Sather, J. J. Shuster, M. Devidas, M. J. Borowitz, A. J. Carroll,N. A. Heerema, J. E. Rubnitz, M. L. Loh, E. A. Raetz, N. J. Winick, S. P. Hunger, W. L. Carroll,P. S. Gaynon, B. M. Camitta, Risk- and response-based classification of childhood B-precursoracute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediat-ric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood 109, 926–935 (2007).
8. C. Domenech, M. Mercier, E. Plouvier, M. Puraveau, P. Bordigoni, G. Michel, Y. Benoit,G. Leverger, A. Baruchel, Y. Bertrand, First isolated extramedullary relapse in childrenwith B-cell precursor acute lymphoblastic leukaemia: Results of the Cooprall-97 study.Eur. J. Cancer 44, 2461–2469 (2008).
9. D. S. Krause, R. A. Van Etten, Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med.353, 172–187 (2005).
10. B. J. Druker, C. L. Sawyers, H. Kantarjian, D. J. Resta, S. F. Reese, J. M. Ford, R. Capdeville,M. Talpaz, Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis ofchronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chro-mosome. N. Engl. J. Med. 344, 1038–1042 (2001).
11. K. G. Roberts, R. D. Morin, J. Zhang, M. Hirst, Y. Zhao, X. Su, S. C. Chen, D. Payne-Turner,M. L. Churchman, R. C. Harvey, X. Chen, C. Kasap, C. Yan, J. Becksfort, R. P. Finney, D. T. Teachey,S. L. Maude, K. Tse, R. Moore, S. Jones, K. Mungall, I. Birol, M. N. Edmonson, Y. Hu, K. E. Buetow,I.M. Chen, W. L. Carroll, L. Wei, J. Ma, M. Kleppe, R. L. Levine, G. Garcia-Manero, E. Larsen,N. P. Shah, M. Devidas, G. Reaman, M. Smith, S. W. Paugh, W. E. Evans, S. A. Grupp, S. Jeha,C. H. Pui, D. S. Gerhard, J. R. Downing, C. L. Willman, M. Loh, S. P. Hunger, M. A. Marra,C. G. Mullighan, Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22, 153–166 (2012).
12. S . K . Tas ian , M. Y . Dora l , M. J . Borowitz , B . L . Wood, I . M. Chen, R . C . Harvey ,J . M. Gastier-Foster, C. L. Willman, S. P. Hunger, C. G. Mullighan, M. L. Loh, Aberrant STAT5and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acutelymphoblastic leukemia. Blood 120, 833–842 (2012).
13. S. L. Maude, S. K. Tasian, T. Vincent, J. W. Hall, C. Sheen, K. G. Roberts, A. E. Seif, D. M. Barrett,I. M. Chen, J. R. Collins, C. G. Mullighan, S. P. Hunger, R. C. Harvey, C. L. Willman, J. S. Fridman,M. L. Loh, S. A. Grupp, D. T. Teachey, Targeting JAK1/2 and mTOR in murine xenograftmodels of Ph-like acute lymphoblastic leukemia. Blood 120, 3510–3518 (2012).
14. S. Herzog, M. Reth, H. Jumaa, Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol. 9, 195–205 (2009).
15. I. R. Matei, C. J. Guidos, J. S. Danska, ATM-dependent DNA damage surveillance in T-celldevelopment and leukemogenesis: The DSB connection. Immunol. Rev. 209, 142–158 (2006).
16. C. J. Guidos, C. J. Williams, I. Grandal, G. Knowles, M. T. Huang, J. S. Danska, V(D)J recom-bination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precur-sors. Genes Dev. 10, 2038–2054 (1996).
17. M. Nacht, A. Strasser, Y. R. Chan, A. W. Harris, M. Schlissel, R. T. Bronson, T. Jacks, Mutationsin the p53 and SCID genes cooperate in tumorigenesis. Genes Dev. 10, 2055–2066 (1996).
18. C. H. Pui, M. V. Relling, D. Campana, W. E. Evans, Childhood acute lymphoblastic leukemia.Rev. Clin. Exp. Hematol. 6, 161–180 (2002).
19. S. Braselmann, V. Taylor, H. Zhao, S. Wang, C. Sylvain, M. Baluom, K. Qu, E. Herlaar, A. Lau,C. Young, B. R. Wong, S. Lovell, T. Sun, G. Park, A. Argade, S. Jurcevic, P. Pine, R. Singh,E. B. Grossbard, D. G. Payan, E. S. Masuda, R406, an orally available spleen tyrosine kinaseinhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation.J. Pharmacol. Exp. Ther. 319, 998–1008 (2006).
20. R. A. Gladdy, M. D. Taylor, C. J. Williams, I. Grandal, J. Karaskova, J. A. Squire, J. T. Rutka,C. J. Guidos, J. S. Danska, The RAG-1/2 endonuclease causes genomic instability and controls CNScomplications of lymphoblastic leukemia in p53/Prkdc-deficientmice. Cancer Cell 3, 37–50 (2003).
21. R. R. Hardy, K. Hayakawa, B cell development pathways, Annu. Rev. Immunol. 19, 595–621 (2001).22. A. Mócsai, J. Ruland, V. L. Tybulewicz, The SYK tyrosine kinase: A crucial player in diverse
biological functions. Nat. Rev. Immunol. 10, 387–402 (2010).23. M. T. Furlong, A. M. Mahrenholz, K. H. Kim, C. L. Ashendel, M. L. Harrison, R. L. Geahlen,
Identification of the major sites of autophosphorylation of the murine protein-tyrosinekinase Syk. Biochim. Biophys. Acta 1355, 177–190 (1997).
24. P. O. Krutzik, J. M. Irish, G. P. Nolan, O. D. Perez, Analysis of protein phosphorylation and cellularsignaling events by flow cytometry: Techniques and clinical applications. Clin. Immunol. 110,206–221 (2004).
25. D. G. Efremov, L. Laurenti, The Syk kinase as a therapeutic target in leukemia and lymphoma.Expert Opin. Investig. Drugs 20, 623–636 (2011).
26. H. Jumaa, L. Bossaller, K. Portugal, B. Storch, M. Lotz, A. Flemming, M. Schrappe, V. Postila,P. Riikonen, J. Pelkonen, C. M. Niemeyer, M. Reth, Deficiency of the adaptor SLP-65 in pre-B-cell acute lymphoblastic leukaemia. Nature 423, 452–456 (2003).
27. D. Trageser, I. Iacobucci, R. Nahar, C. Duy, G. von Levetzow, L. Klemm, E. Park, W. Schuh,T. Gruber, S. Herzog, Y. M. Kim, W. K. Hofmann, A. Li, C. T. Storlazzi, H. M. Jäck, J. Groffen,G. Martinelli, N. Heisterkamp, H. Jumaa, M. Müschen, Pre–B cell receptor–mediated cell cyclearrest in Philadelphia chromosome–positive acute lymphoblastic leukemia requires IKAROSfunction. J. Exp. Med. 206, 1739–1753 (2009).
28. J. W. Friedberg, J. Sharman, J. Sweetenham, P. B. Johnston, J. M. Vose, A. Lacasce,J. Schaefer-Cutillo, S. De Vos, R. Sinha, J. P. Leonard, L. D. Cripe, S. A. Gregory, M. P. Sterba,A. M. Lowe, R. Levy, M. A. Shipp, Inhibition of Syk with fostamatinib disodium has signif-icant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood115, 2578–2585 (2010).
29. S. A. Armstrong, M. E. Mabon, L. B. Silverman, A. Li, J. G. Gribben, E. A. Fox, S. E. Sallan,S. J. Korsmeyer, FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 103,3544!3546 (2004).
30. V. T. Bicocca, B. H. Chang, B. K. Masouleh, M. Muschen, M. M. Loriaux, B. J. Druker, J. W. Tyner,Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lym-phoblastic leukemia. Cancer Cell 22, 656!667 (2012).
31. N. Yamamoto, K. Takeshita, M. Shichijo, T. Kokubo,M. Sato, K. Nakashima, M. Ishimori, H. Nagai,Y. F. Li, T. Yura, K. B. Bacon, The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY61-3606) blocks antigen-induced airway inflammation in rodents. J. Pharmacol. Exp. Ther.306, 1174!1181 (2003).
32. A. Gazit, K. Yee, A. Uecker, F. D. Böhmer, T. Sjöblom, A. Ostman, J. Waltenberger, G. Golomb,S. Banai, M. C. Heinrich, A. Levitzki, Tricyclic quinoxalines as potent kinase inhibitors ofPDGFR kinase, Flt3 and Kit. Bioorg. Med. Chem. 11, 2007!2018 (2003).
33. L. J. Lombardo, F. Y. Lee, P. Chen, D. Norris, J. C. Barrish, K. Behnia, S. Castaneda,L. A. Cornelius, J. Das, A. M. Doweyko, C. Fairchild, J. T. Hunt, I. Inigo, K. Johnston,A. Kamath, D. Kan, H. Klei, P. Marathe, S. Pang, R. Peterson, S. Pitt, G. L. Schieven,R. J. Schmidt, J. Tokarski, M. L.Wen, J.Wityak, R.M. Borzilleri, Discovery ofN-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity inpreclinical assays. J. Med. Chem. 47, 6658–6661 (2004).
34. A. Agliano, I. Martin-Padura, P. Mancuso, P. Marighetti, C. Rabascio, G. Pruneri, L. D. Shultz,F. Bertolini, Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rg null mice generatea faster and more efficient disease compared to other NOD/scid-related strains. Int. J. Cancer123, 2222–2227 (2008).
35. J. L. McKenzie, O. I. Gan, M. Doedens, J. E. Dick, Human short-term repopulating stem cellsare efficiently detected following intrafemoral transplantation into NOD/SCID recipientsdepleted of CD122+ cells. Blood 106, 1259–1261 (2005).
36. L. H. Meyer, S. M. Eckhoff, M. Queudeville, J. M. Kraus, M. Giordan, J. Stursberg, A. Zangrando,E. Vendramini, A. Möricke, M. Zimmermann, A. Schrauder, G. Lahr, K. Holzmann,M. Schrappe, G. Basso, K. Stahnke, H. A. Kestler, G. Te Kronnie, K. M. Debatin,Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterizedby a gene signature involving survival pathways. Cancer Cell 19, 206–217 (2011).
37. J. M. Sancho, J. M. Ribera, A. Oriol, J. M. Hernandez-Rivas, C. Rivas, C. Bethencourt, R. Parody,G. Deben, J. L. Bello, E. Feliu; Programa para el Estudio y Tratamiento de Hemopatias MalignasGroup, Central nervous system recurrence in adult patients with acute lymphoblastic leuke-mia: Frequency and prognosis in 467 patients without cranial irradiation for prophylaxis.Cancer 106, 2540–2546 (2006).
38. A. Redaelli, B. L. Laskin, J. M. Stephens, M. F. Botteman, C. L. Pashos, A systematic literaturereview of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL).Eur. J. Cancer Care 14, 53–62 (2005).
39. M. I. Davis, J. P. Hunt, S. Herrgard, P. Ciceri, L. M. Wodicka, G. Pallares, M. Hocker, D. K. Treiber,P. P. Zarrinkar, Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29,1046–1051 (2011).
40. K. S. Lau, T. Zhang, K. R. Kendall, D. Lauffenburger, N. S. Gray, K. M. Haigis, BAY61-3606affects the viability of colon cancer cells in a genotype-directed manner. PLOS One 7,e41343 (2012).
41. M. L. Loh, J. Zhang, R. C. Harvey, K. Roberts, D. Payne-Turner, H. Kang, G. Wu, X. Chen,J. Becksfort, M. Edmonson, K. H. Buetow, W. L. Carroll, I. M. Chen, B. Wood, M. J. Borowitz,M. Devidas, D. S. Gerhard, P. Bowman, E. Larsen, N. Winick, E. Raetz, M. Smith, J. R. Downing,C. L. Willman, C. G. Mullighan, S. P. Hunger, Tyrosine kinome sequencing of pediatric acutelymphoblastic leukemia: A report from the Children’s Oncology Group TARGET Project. Blood121, 485–488 (2013).
42. J. Zhang, C. A. Benavente, J. McEvoy, J. Flores-Otero, L. Ding, X. Chen, A. Ulyanov, G. Wu,M. Wilson, J. Wang, R. Brennan, M. Rusch, A. L. Manning, J. Ma, J. Easton, S. Shurtleff,C. Mullighan, S. Pounds, S. Mukatira, P. Gupta, G. Neale, D. Zhao, C. Lu, R. S. Fulton,L. L. Fulton, X. Hong, D. J. Dooling, K. Ochoa, C. Naeve, N. J. Dyson, E. R. Mardis, A. Bahrami,D. Ellison, R. K. Wilson, J. R. Downing, M. A. Dyer, A novel retinoblastoma therapy fromgenomic and epigenetic analyses. Nature 481, 329–334 (2012).
43. M. Dühren-von Minden, R. Übelhart, D. Schneider, T. Wossning, M. P. Bach, M. Buchner,D. Hofmann, E. Surova, M. Follo, F. Köhler, H. Wardemann, K. Zirlik, H. Veelken, H. Jumaa,Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomoussignalling. Nature 489, 309–312 (2012).
44. R. E. Davis, V. N. Ngo, G. Lenz, P. Tolar, R. M. Young, P. B. Romesser, H. Kohlhammer, L. Lamy,H. Zhao, Y. Yang, W. Xu, A. L. Shaffer, G. Wright, W. Xiao, J. Powell, J. K. Jiang, C. J. Thomas,A. Rosenwald, G. Ott, H. K. Muller-Hermelink, R. D. Gascoyne, J. M. Connors, N. A. Johnson,
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 12
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m

L. M. Rimsza, E. Campo, E. S. Jaffe, W. H. Wilson, J. Delabie, E. B. Smeland, R. I. Fisher,R. M. Braziel, R. R. Tubbs, J. R. Cook, D. D. Weisenburger, W. C. Chan, S. K. Pierce, L. M. Staudt,Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92(2010).
45. C. K. Hahn, J. E. Berchuck, K. N. Ross, R. M. Kakoza, K. Clauser, A. C. Schinzel, L. Ross, I. Galinsky,T. N. Davis, S. J. Silver, D. E. Root, R. M. Stone, D. J. DeAngelo, M. Carroll, W. C. Hahn, S. A. Carr,T. R. Golub, A. L. Kung, K. Stegmaier, Proteomic and genetic approaches identify Syk as anAML target. Cancer Cell 16, 281–294 (2009).
46. Cancer Genome atlas Research Network, Genomic and epigenomic landscapes of adult denovo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
47. E. Schweighoffer, L. Vanes, J. Nys, D. Cantrell, S. McCleary, N. Smithers, V. L. Tybulewicz, TheBAFF receptor transduces survival signals by co-opting the B cell receptor signalingpathway. Immunity 38, 475–488 (2013).
48. L. Chen, P. Juszczynski, K. Takeyama, R. C. Aguiar, M. A. Shipp, Protein tyrosine phosphatasereceptor–type O truncated (PTPROt) regulates SYK phosphorylation, proximal B-cell–receptorsignaling, and cellular proliferation. Blood 108, 3428–3433 (2006).
49. P. A. Goodman, C. M. Wood, A. Vassilev, C. Mao, F. M. Uckun, Spleen tyrosine kinase (Syk)deficiency in childhood pro-B cell acute lymphoblastic leukemia. Oncogene 20, 3969–3978(2001).
50. I. Iacobucci, N. Iraci, M. Messina, A. Lonetti, S. Chiaretti, E. Valli, A. Ferrari, C. Papayannidis,F. Paoloni, A. Vitale, C. T. Storlazzi, E. Ottaviani, V. Guadagnuolo, S. Durante, M. Vignetti,S. Soverini, F. Pane, R. Foà, M. Baccarani, M. Müschen, G. Perini, G. Martinelli, IKAROSdeletions dictate a unique gene expression signature in patients with adult B-cell acutelymphoblastic leukemia. PLOS One 7, e40934 (2012).
51. T. Wossning, S. Herzog, F. Köhler, S. Meixlsperger, Y. Kulathu, G. Mittler, A. Abe, U. Fuchs,A. Borkhardt, H. Jumaa, Deregulated Syk inhibits differentiation and induces growthfactor–independent proliferation of pre–B cells. J. Exp. Med. 203, 2829–2840 (2006).
52. C. H. Pui, D. Campana, D. Pei, W. P. Bowman, J. T. Sandlund, S. C. Kaste, R. C. Ribeiro,J. E. Rubnitz, S. C. Raimondi, M. Onciu, E. Coustan-Smith, L. E. Kun, S. Jeha, C. Cheng,S. C. Howard, V. Simmons, A. Bayles, M. L. Metzger, J. M. Boyett, W. Leung, R. Handgretinger,J. R. Downing, W. E. Evans, M. V. Relling, Treating childhood acute lymphoblastic leukemiawithout cranial irradiation. N. Engl. J. Med. 360, 2730–2741 (2009).
53. G. K. Smyth, Linear models and empirical Bayes methods for assessing differential expres-sion in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 (2004).
54. R. C. Gentleman, V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis, L. Gautier,Y. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R. Irizarry, F. Leisch, C. Li,M. Maechler, A. J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney, J. Y. Yang, J. Zhang,Bioconductor: Open software development for computational biology and bioinformatics.Genome Biol. 5, R80.1–R80.16 (2004).
55. R. A. Irizarry, B. M. Bolstad, F. Collin, L. M. Cope, B. Hobbs, T. P. Speed, Summaries of Affy-metrix GeneChip probe level data. Nucleic Acids Res. 31, e15 (2003).
56. R. A. Irizarry, B. Hobbs, F. Collin, Y. D. Beazer-Barclay, K. J. Antonellis, U. Scherf, T. P. Speed,Exploration, normalization, and summaries of high density oligonucleotide array probelevel data. Biostatistics 4, 249–264 (2003).
57. Y. Benjamini, Y. Hochberg, Controlling the false discovery rate: A practical and powerfulapproach to multiple testing. J. R. Statist. Soc. B 57, 289–300 (1995).
58. J. S. Danska, F. Pflumio, C. J. Williams, O. Huner, J. E. Dick, C. J. Guidos, Rescue of T cell–specificV(D)J recombination in SCID mice by DNA-damaging agents. Science 266, 450–455 (1994).
59. N. Kotecha, P. O. Krutzik, J. M. Irish, Web-based analysis and publication of flow cytometryexperiments. Curr. Protoc. Cytom. Chapter 10, Unit10.17 (2010).
60. F. Mazurier, M. Doedens, O. I. Gan, J. E. Dick, Rapid myeloerythroid repopulation after in-trafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat.Med. 9, 959–963 (2003).
Funding: This work was supported by grants to C.J.G. and J.S.D. from the Cancer Stem CellConsortium with funds through Canadian Institutes for Health Research (CIHR) and GenomeCanada to the Highly Active Leukemia Therapies project, and from the Ontario Institute ofCancer Research, Program in Cancer Stem Cells. T.P. was a recipient of a CIHR doctoral researchaward. Flow cytometry was performed in The SickKids–University Health Network Flow Cytom-etry Facility, with funding from the Ontario Institute for Cancer Research, McEwen Centre forRegenerative Medicine, Canadian Foundation for Innovation, and SickKids Foundation. Authorcontributions: T.P., C.J.G., and J.S.D. designed the study, analyzed the data, and wrote the man-uscript. T.P. performed in vitro and in vivo characterization of SYK inhibitors in B-ALL, Westernblotting, and phospho-flow profiling. I.G. performed Western blotting and assisted with xeno-graft assays. L.M.J.N. performed microarrays and immunophenotyping of double-mutant mice.E.P. performed adaptive transfers of double-mutant B-ALL. I.R.M. performed phospho-flowexperiments. T.P. analyzed the data and prepared the figures. J.B. analyzed the microarray data.P.E.K. analyzed the data. J.K.H. and M.D.M. provided patient samples and edited the manuscript.Competing interests: The authors declare that they have no competing interests.
Submitted 28 January 2014Accepted 25 April 2014Published 14 May 201410.1126/scitranslmed.3008661
Citation: T. Perova, I. Grandal, L. M. J. Nutter, E. Papp, I. R. Matei, J. Beyene, P. E. Kowalski,J. K. Hitzler, M. D. Minden, C. J. Guidos, J. S. Danska, Therapeutic potential of spleentyrosine kinase inhibition for treating high-risk precursor B cell acute lymphoblasticleukemia. Sci. Transl. Med. 6, 236ra62 (2014).
R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 14 May 2014 Vol 6 Issue 236 236ra62 13
on
June
20,
201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m





























