Embed Size (px)
Citation preview

Catalysis Communications 17 (2012) 118–125
Contents lists available at SciVerse ScienceDirect
Catalysis Communications
j ourna l homepage: www.e lsev ie r .com/ locate /catcom
Short Communication
Total oxidation of selected mono-carbon VOCs over hydrotalcite originated metaloxide catalysts
Lucjan Chmielarz a,⁎, Zofia Piwowarska a, Małgorzata Rutkowska a, Magdalena Wojciechowska a,Barbara Dudek a, Stefan Witkowski a, Marek Michalik b
a Jagiellonian University, Faculty of Chemistry, Ingardena 3, 30-060 Kraków, Polandb Jagiellonian University, Institute of Geological Sciences, Oleandry 2a, 30-063 Kraków, Poland
⁎ Corresponding author. Tel.: +48 126632006; fax: +E-mail address: [email protected] (L. Chmi
1566-7367/$ – see front matter © 2011 Elsevier B.V. Alldoi:10.1016/j.catcom.2011.10.030
a b s t r a c t
a r t i c l e i n f oArticle history:Received 8 September 2011Received in revised form 20 October 2011Accepted 26 October 2011Available online 4 November 2011
Keywords:HydrotalcitesMixed metal oxidesCatalysisVOCsTotal oxidation
Hydrotalcite originated Cu–Mg–Al, Co–Mg–Al and Cu–Co–Mg–Al oxide systems were tested as catalysts forthe total oxidation of mono-carbon VOCs (methane, methanol, and formic acid). Both calcination tempera-ture of the hydrotalcite precursors as well as doping of the catalysts with potassium promoter influencedtheir catalytic activity. Increased calcination temperature, which resulted in a decrease of the surface areaof the samples and formation of the spinel phases, activated the Co–Mg–Al catalyst, while the opposite effectwas observed for the Cu–Mg–Al and Cu–Co–Mg–Al catalysts. On the other hand doping of the catalysts withpotassium promoter significantly activated the Cu–Mg–Al and Cu–Co–Mg–Al catalysts in the processes ofmethanol and formic acid conversion, while only slightly influenced the catalytic performance of the Co–Mg–Al sample.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
Hydrotalcite-like material, called also layered double hydroxides(LDHs), were reported to be precursors of active catalysts for variousprocesses, including among other DeNOx [1], ammonia oxidation [2]and N2O decomposition [3]. Recently, there is an increased interest inapplication of hydrotalcite-like materials as precursors of the catalystsfor total oxidation for volatile organic compounds (VOCs). Much atten-tion was paid for the cobalt containing oxide systems. Gennequin et al.[4,5] used Mg–Al hydrotalcites, calcined at various temperatures, assupports for the deposition of cobalt. Such catalysts were tested in theprocess of toluene incineration. The best effectiveness in this processwas obtained for cobalt supported on hydrotalcite calcined at 700 °C.Calcination of Mg–Al hydrotalcite at lower and higher temperaturesresulted in the lower activity of the catalysts. These authors [5] showedalso that the significantly more active catalysts for the total toluene ox-idation can be obtained from theCo–Mg–Al hydrotalcite-like precursorscalcined at 500 °C [6]. They related the increased activity of the secondgroup of the catalysts to better dispersion of cobalt species. Pérez et al.[7] reported that the ultrasound assisted synthesis of the Co–Mg–Alhydrotalcite-like materials is much faster comparing to the traditionalmethod and their calcination results in very active catalysts for thetotal oxidation of butanol. Authors related high activity of the catalysts
48 126340515.elarz).
rights reserved.
to the increased surface area and porosity of the samples produced bythe ultrasound assisted method. Kovanda at al. [8] reported the benefi-cial effect of using the Co–Mg–Mn–Al hydrotalcite precursors for thepreparation of the catalysts for the ethanol incineration. The best resultswere obtained for the cobalt reach Co–Mn–Al oxide system, which assuggested authors, has an optimum content of the reducible compo-nents. Recently, the promoting effect of the potassium doping on activ-ity of these catalysts in the total oxidation of toluene was reported [9].There is also a lot of papers reporting successful application of the cop-per containing catalysts for VOCs oxidation [e.g. 10–13], however onlylimited number those related to using the hydrotalcite originated cata-lysts. Kovanda et al. [14] reported high catalytic activity of the hydrotal-cite originated Cu–Mg–Al oxide systems (calcination at 450 °C) in thetoluene combustion. The activity of the studied catalysts increasedwith an increase of the copper loading and, as it was suggested by theauthors, is related to the contribution of the easy reducible copper spe-cies. Also the hydrotalcite-like materials containing apart from copperalso other transition metal were reported to be precursors of the activecatalysts for the VOCs combustion. Bahranowski et al. [15] showed thehigh catalytic activity of the Cu–Cr–Al hydrotalcite-like materials cal-cined at 600 °C in the complete oxidation of toluene and ethanol. Alsothe Cu–Mn–Al and Cu–Zn–Al oxide systems obtained from thehydrotalcite-like materials were found to be active and selective cata-lysts of the toluene combustion [16]. It was shown that the samples cal-cined at lower temperature (450 °C) were more active than thosecalcined at higher temperature (600 °C). Recently, Kovanda et al. [17],who studied a broad range of the hydrotalcite-like materials containingvarious transition metals as precursors of the metal oxide catalysts for

119L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
the ethanol combustion, reported very high activity of the Cu–Co–Mn,Cu–Ni–Mn and Cu–Co–Al oxide systems.
The present paper reports the studies of the hydrotalcite originatedCu–Mg–Al, Co–Mg–Al and Cu–Co–Mg–Al metal oxide catalysts for thecomplete oxidation ofmethane,methanol and formic acid. An influenceof the calcination temperature of the hydrotalcite precursors and dop-ing of the samples with potassium promoter on their catalytic activityis discussed.
Table 1Composition, calcination temperature and surface area of the hydrotalcite basedsamples.
Samplecode
Composition Atomicratio
Calcinationtemperature[°C]
SBET of calcinedhydrotalcite[m2/g]
HT700-Cu15 Cu/Mg/Al 15/56/29 700 193HT800-Cu15 Cu/Mg/Al 15/56/29 800 88HT700-Co15 Co/Mg/Al 15/56/29 700 217HT800-Co15 Co/Mg/Al 15/56/29 800 61HT700-CuCo15 Cu/Co/Mg/Al 15/15/41/29 700 107HT800-CuCo15 Cu/Co/Mg/Al 15/15/41/29 800 60
2. Experimental
Cu(II)Mg(II)Al(III), Co(II)Mg(II)Al(III) and Cu(II)Co(II)Mg(II)Al(III)hydrotalcite-like materials were prepared by the co-precipitationmethod using aqueous solutions of the following metal nitrates:Mg(NO3)2·6H2O (Sigma), Al(NO3)3·9H2O (Fluka), Cu(NO3)2·3H2O(Merck) and Co(NO3)2·6H2O (POCh). A solution of NaOH (POCh)was used as a precipitating agent. The mixture of metal nitrate solu-tions was slowly added to a vigorously stirred aqueous solution con-taining a slight excess of Na2CO3 (POCh). The pH was maintainedconstant at 10.0±0.2 by dropwise addition of NaOH solution. Pre-cipitates were aged in a suspension at 60 °C for 30 min under vigor-ous stirring. In the next step the suspension was filtered, washedwith distilled water and dried overnight at 120 °C. Finally, the pre-pared hydrotalcite-like materials were calcined at 700 or 800 °Cfor 16 h. The hydrotalcites calcined at 800 °Cweremodifiedwith po-tassium using the incipient wetness impregnation method. Aqueoussolutions of KNO3 (POCh) were used for the deposition of potassium(0.9 wt.%). The samples impregnated with potassium nitrate werecalcined at 600 °C. A detailed description of the catalysts was pre-sented in our previous paper [3].
The surface area of hydrotalcite-like materials calcined at differenttemperatures was determined by the BET method. The measurementswere performed using ASAP 2010 (Micromeritics). Prior to the nitrogenadsorption at −196 °C the samples were outgassed under vacuum at350 °C for 12 h. The X-ray diffraction (XRD) patterns of the fresh andcalcined hydrotalcite-like materials were obtained with a Philips X'PertAPD diffractometer using CuK∝ radiation (λ=1.54178 Å). The TPRed(temperature-programmed reduction) of the samples (30 mg)was car-ried out from room temperature to 1100 °Cwith a linear heating rate of5 °C/min. Measurements were performed in a fixed-bed flow micro-reactor. Prior to the TPRed experiment, the catalysts were outgassedat 600 °C for 12 h. The TPRed runs were carried out in a flow of10 vol.% of H2 diluted in Ar with a total flow rate of gas mixture of6 mL/min. Evolving water was removed from effluent gas by means ofa cold trap. The evolution of hydrogen was detected by microvolumeTCD (Valco). More detailed characteristic of the samples was presentedin our previous paper [3].
Calcined hydrotalites were tested as the catalysts of methane,meth-anol and formic acid incineration. The catalytic experiments have beenperformed under atmospheric pressure in a fixed-bed flow microreac-tor system. The reactant concentrations were continuously measuredusing a quadruplemass spectrometer RGA 200 (PREVAC) connected di-rectly to the reactor outlet. Prior to the catalytic test each sample of thecatalyst (100 mg) was outgassed in a flow of pure helium at 500 °C for1 h. The isothermal saturator with a constant flow of helium was usedfor supply of methanol and formic acid into the reactionmixture. Meth-anewas supplied from gas cylinder containingmixture of 1 vol.% of CH4
diluted in pure helium. The composition of gas mixture at the reactorinlet was [CH4]=0.5 vol.% (or alternatively methanol or formic acid),[O2]=4.5 vol.% and [He]=95 vol.%. The reaction was studied in therange from 100 to 450 °C (or 650 °C for methane incineration) withthe linear temperature increase of 10 °C/min. The signal of the heliumline served as an internal standard to compensate possible small fluctu-ations of the operating pressure. The sensitivity factors of analysed lineswere calibrated using commercial mixtures of gases.
3. Results and discussion
The chemical composition and BET surface area of hydrotalcitescalcined at 700 and 800 °C are presented in Table 1. In a series ofhydrotalcites calcined at 700 °C, the samples containing only onetype of transition metal (HT700-Co15 or HT-700-Cu15) have signif-icantly higher surface area than the sample containing both copperand cobalt (HT700-CuCo15). An increase in the calcination tempera-ture from 700 °C to 800 °C dramatically reduced surface area of thesamples.
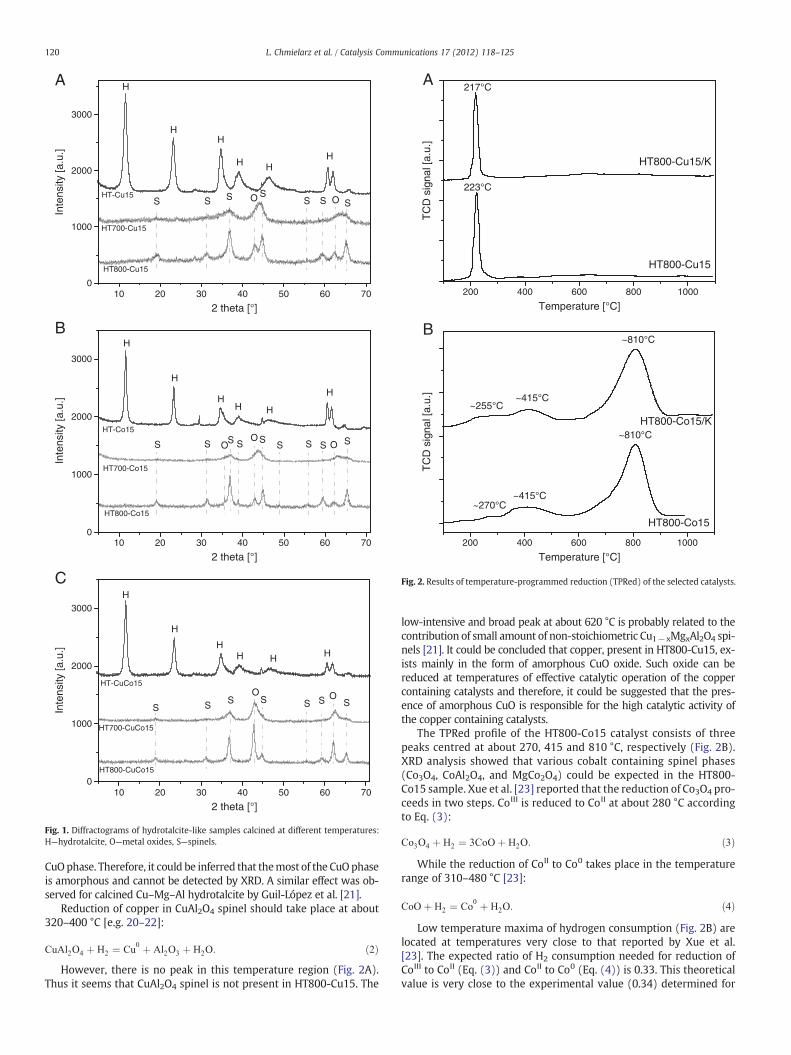
The XRD patterns of the as-prepared and calcined hydrotalcite-likematerials are shown in Fig. 1. The reflections typical of the hydrotalcitestructure (marked as H in Fig. 1, JCPDS 37-0630) were present in diffrac-tograms of the non-calcined samples [18]. It should be noted that inten-sity of XRD reflections, which is related to crystallinity of hydrotalcitephases, is higher for the HT-Cu15 sample than for HT-Co15 and HT-Cu15Co15. Calcination of the samples at 700 °C resulted in a disappear-ance of diffraction lines typical of the hydrotalcite structure and forma-tion of new broad reflections. Two of them, positioned at about 42° and62°, are characteristic of the MgO phase (marked as O in Fig. 1, JCPDS4–0829), whereas the band at about 37° could be attributed to the pres-ence of the following spinel phases: MgAl2O4 (JCPDS 21-1152), CuAl2O4
(JCPDS-33-0448), CoAl2O4 (JCPDS 82-2246), MgCo2O4 (JCPDS 02-1073),Co3O4 (JCPDS 74-2120) and possible non-stoichiometric spinel phasescontaining Cu, Cu, Mg and Al cations. The spinel phases are marked as Sin Fig. 1. Any intensive peaks related to the presence of CuO (JCPDS 41-0254) were not found [18,19]. The low-intensive reflections at about19°, 31°, 59° and 65° are related to formation of the above-mentionedspinel phases. An increase in the calcination temperature to 800 °Cresulted in an increase of the intensity of these reflections, suggesting asignificant increase of the crystallinity of the spinel phases. Therefore, itcould be concluded that calcination of the hydrotalite-like samples re-sults in their thermal decomposition to the spinel and metal oxidephases. An increase in the calcination temperature favours formation ofthe low-surface area spinel phases. The detailed studies of thermal de-composition hydrotalcite-like materials into metal oxide systems werepresented in our previous paper [3].
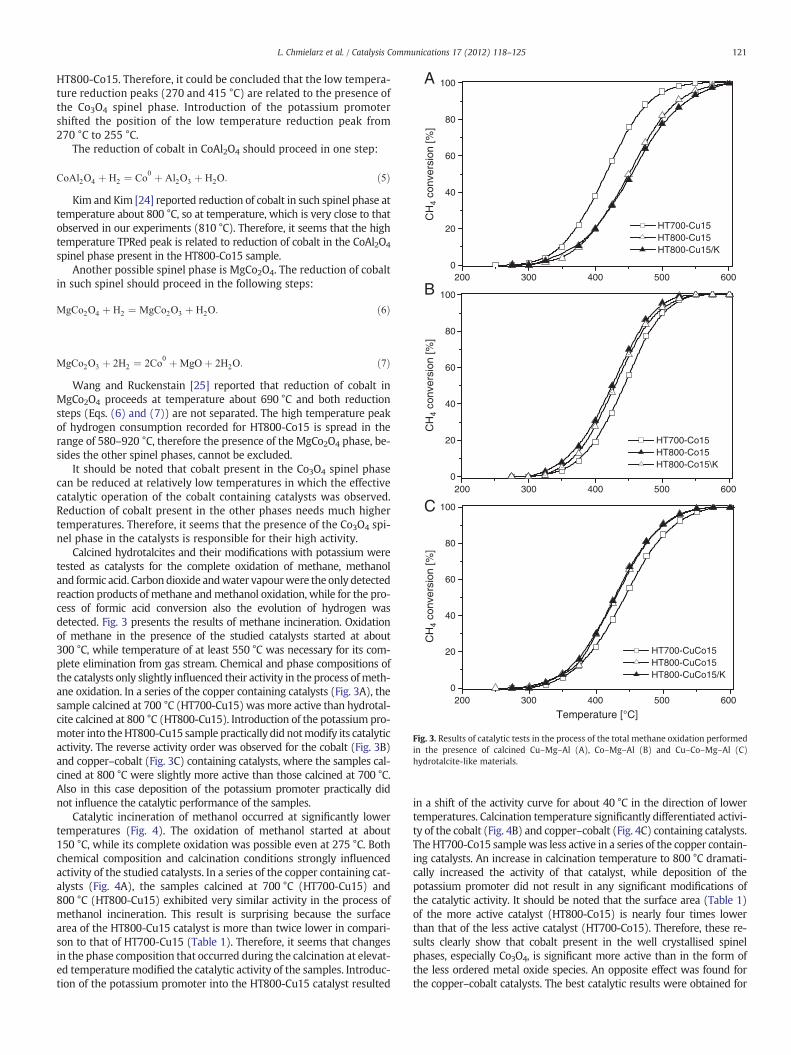
Fig. 2 presents the results of TPRed studies of HT800-Cu15 andHT800-Co15 and their modifications with potassium promoter. Thereduction of copper oxide species [14] present in the HT800-Cu15sample took place in the range of 120–305 °C with a sharp maximumlocated at 223 °C (Fig. 2A). The promotion of that catalyst with potas-sium (HT-800-Cu15/K) shifted the reduction maximum into 217 °C.Additionally, low-intensive and broad peak of hydrogen consump-tion, centred at about 620 °C, was detected for the samples promotedand non-promoted with potassium. According to the literature re-ports [e.g. 20–22] the reduction of CuO to Cu0 occurs at 200–300 °Cdepending on the size and nature of CuO crystals.
CuO þ H2 ¼ Cu0 þ H2O: ð1Þ
The results of TPRed are not supported by XRD studies, in whichthere were not found any intensive peaks related to the presence of
0
1000
2000
3000
O O SSSSSSS
HHH
HH
HT800-Cu15
HT700-Cu15
Inte
nsity
[a.u
.]In
tens
ity [a
.u.]
2 theta [°]
HT-Cu15
A
B
C
H
0
1000
2000
3000
O OO SSSS
SSSSS
H
HHH
H
HT800-Co15
HT700-Co15
HT-Co15
H
10 20 30 40 50 60 70
2 theta [°]10 20 30 40 50 60 70
2 theta [°]10 20 30 40 50 60 70
0
1000
2000
3000
H
S SO O
SSSS
H HH
H
HT800-CuCo15
HT700-CuCo15
Inte
nsity
[a.u
.]
HT-CuCo15
H
S
Fig. 1. Diffractograms of hydrotalcite-like samples calcined at different temperatures:H—hydrotalcite, O—metal oxides, S—spinels.
200 400 600 800 1000
217°C
HT800-Cu15/K
TC
D s
igna
l [a.
u.]
Temperature [°C]
200 400 600 800 1000
Temperature [°C]
HT800-Cu15
223°C
A
B
~270°C
~255°C
~415°C
~415°C
~810°CHT800-Co15/K
TC
D s
igna
l [a.
u.]
HT800-Co15
~810°C
Fig. 2. Results of temperature-programmed reduction (TPRed) of the selected catalysts.
120 L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
CuOphase. Therefore, it could be inferred that themost of the CuOphaseis amorphous and cannot be detected by XRD. A similar effect was ob-served for calcined Cu–Mg–Al hydrotalcite by Guil-López et al. [21].
Reduction of copper in CuAl2O4 spinel should take place at about320–400 °C [e.g. 20–22]:
CuAl2O4 þ H2 ¼ Cu0 þ Al2O3 þ H2O: ð2Þ
However, there is no peak in this temperature region (Fig. 2A).Thus it seems that CuAl2O4 spinel is not present in HT800-Cu15. The
low-intensive and broad peak at about 620 °C is probably related to thecontribution of small amount of non-stoichiometric Cu1−xMgxAl2O4 spi-nels [21]. It could be concluded that copper, present in HT800-Cu15, ex-ists mainly in the form of amorphous CuO oxide. Such oxide can bereduced at temperatures of effective catalytic operation of the coppercontaining catalysts and therefore, it could be suggested that the pres-ence of amorphous CuO is responsible for the high catalytic activity ofthe copper containing catalysts.
The TPRed profile of the HT800-Co15 catalyst consists of threepeaks centred at about 270, 415 and 810 °C, respectively (Fig. 2B).XRD analysis showed that various cobalt containing spinel phases(Co3O4, CoAl2O4, and MgCo2O4) could be expected in the HT800-Co15 sample. Xue et al. [23] reported that the reduction of Co3O4 pro-ceeds in two steps. CoIII is reduced to CoII at about 280 °C accordingto Eq. (3):
Co3O4 þ H2 ¼ 3CoO þ H2O: ð3ÞWhile the reduction of CoII to Co0 takes place in the temperature
range of 310–480 °C [23]:
CoO þ H2 ¼ Co0 þ H2O: ð4Þ
Low temperature maxima of hydrogen consumption (Fig. 2B) arelocated at temperatures very close to that reported by Xue et al.[23]. The expected ratio of H2 consumption needed for reduction ofCoIII to CoII (Eq. (3)) and CoII to Co0 (Eq. (4)) is 0.33. This theoreticalvalue is very close to the experimental value (0.34) determined for
200 300 400 500 600
200 300 400 500 600
200 300 400 500 600
0
20
40
60
80
100
0
20
40
60
80
100
0
20
40
60
80
100
CH
4 co
nver
sion
[%]
CH
4 co
nver
sion
[%]
CH
4 co
nver
sion
[%]
HT700-Cu15 HT800-Cu15 HT800-Cu15/K
A
Temperature [°C]
HT700-CuCo15 HT800-CuCo15 HT800-CuCo15/K
C
HT700-Co15 HT800-Co15 HT800-Co15\K
B
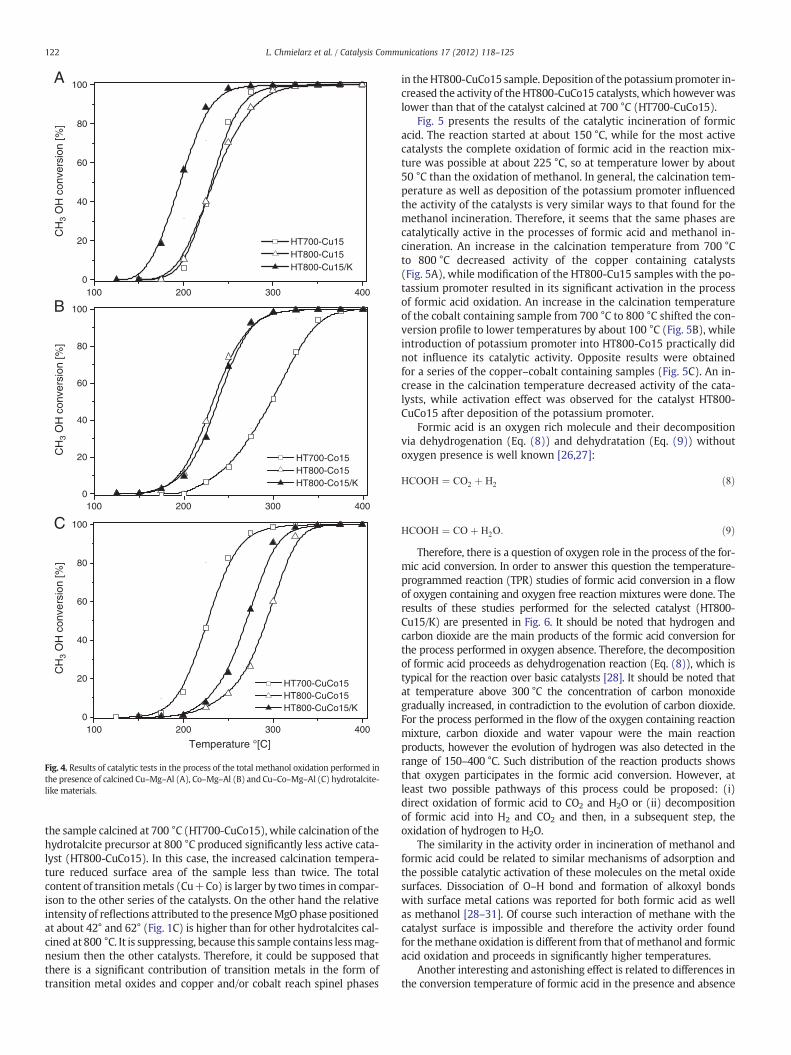
Fig. 3. Results of catalytic tests in the process of the total methane oxidation performedin the presence of calcined Cu–Mg–Al (A), Co–Mg–Al (B) and Cu–Co–Mg–Al (C)hydrotalcite-like materials.
121L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
HT800-Co15. Therefore, it could be concluded that the low tempera-ture reduction peaks (270 and 415 °C) are related to the presence ofthe Co3O4 spinel phase. Introduction of the potassium promotershifted the position of the low temperature reduction peak from270 °C to 255 °C.
The reduction of cobalt in CoAl2O4 should proceed in one step:
CoAl2O4 þ H2 ¼ Co0 þ Al2O3 þ H2O: ð5Þ
Kim and Kim [24] reported reduction of cobalt in such spinel phase attemperature about 800 °C, so at temperature, which is very close to thatobserved in our experiments (810 °C). Therefore, it seems that the hightemperature TPRed peak is related to reduction of cobalt in the CoAl2O4
spinel phase present in the HT800-Co15 sample.Another possible spinel phase is MgCo2O4. The reduction of cobalt
in such spinel should proceed in the following steps:
MgCo2O4 þ H2 ¼ MgCo2O3 þ H2O: ð6Þ
MgCo2O3 þ 2H2 ¼ 2Co0 þMgO þ 2H2O: ð7Þ
Wang and Ruckenstain [25] reported that reduction of cobalt inMgCo2O4 proceeds at temperature about 690 °C and both reductionsteps (Eqs. (6) and (7)) are not separated. The high temperature peakof hydrogen consumption recorded for HT800-Co15 is spread in therange of 580–920 °C, therefore the presence of the MgCo2O4 phase, be-sides the other spinel phases, cannot be excluded.
It should be noted that cobalt present in the Co3O4 spinel phasecan be reduced at relatively low temperatures in which the effectivecatalytic operation of the cobalt containing catalysts was observed.Reduction of cobalt present in the other phases needs much highertemperatures. Therefore, it seems that the presence of the Co3O4 spi-nel phase in the catalysts is responsible for their high activity.
Calcined hydrotalcites and their modifications with potassium weretested as catalysts for the complete oxidation of methane, methanoland formic acid. Carbon dioxide andwater vapourwere the only detectedreaction products of methane andmethanol oxidation, while for the pro-cess of formic acid conversion also the evolution of hydrogen wasdetected. Fig. 3 presents the results of methane incineration. Oxidationof methane in the presence of the studied catalysts started at about300 °C, while temperature of at least 550 °C was necessary for its com-plete elimination from gas stream. Chemical and phase compositions ofthe catalysts only slightly influenced their activity in the process ofmeth-ane oxidation. In a series of the copper containing catalysts (Fig. 3A), thesample calcined at 700 °C (HT700-Cu15) wasmore active than hydrotal-cite calcined at 800 °C (HT800-Cu15). Introduction of the potassium pro-moter into theHT800-Cu15 sample practically did notmodify its catalyticactivity. The reverse activity order was observed for the cobalt (Fig. 3B)and copper–cobalt (Fig. 3C) containing catalysts, where the samples cal-cined at 800 °C were slightly more active than those calcined at 700 °C.Also in this case deposition of the potassium promoter practically didnot influence the catalytic performance of the samples.
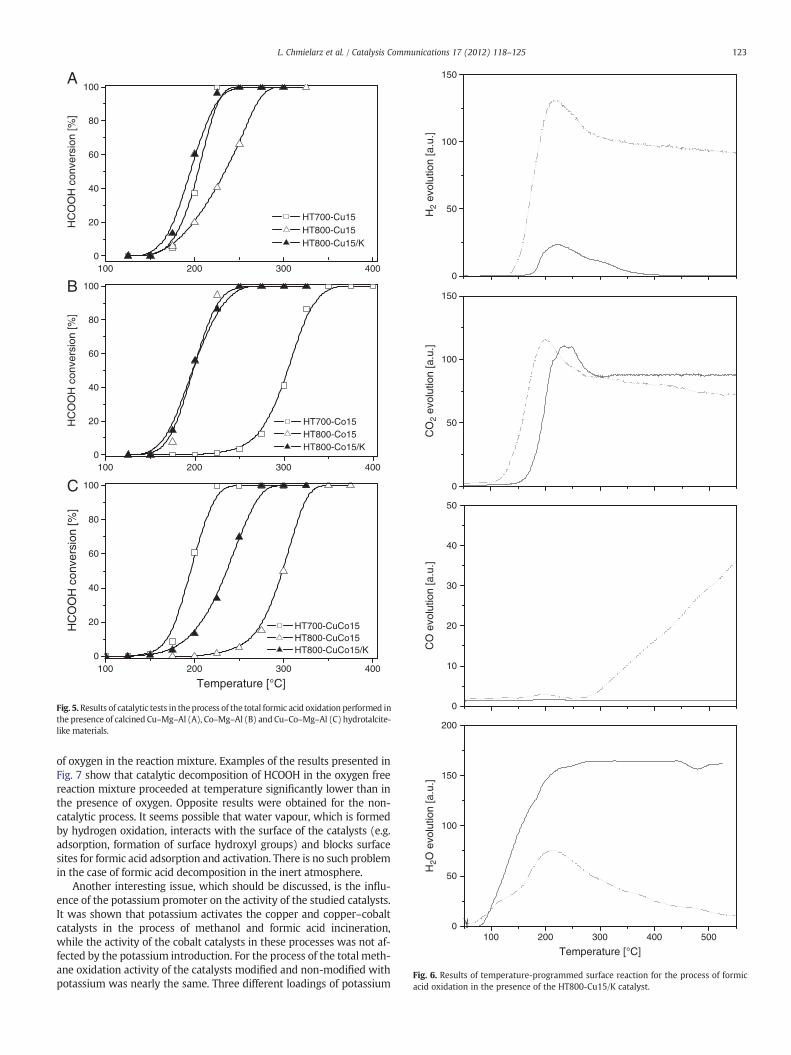
Catalytic incineration of methanol occurred at significantly lowertemperatures (Fig. 4). The oxidation of methanol started at about150 °C, while its complete oxidation was possible even at 275 °C. Bothchemical composition and calcination conditions strongly influencedactivity of the studied catalysts. In a series of the copper containing cat-alysts (Fig. 4A), the samples calcined at 700 °C (HT700-Cu15) and800 °C (HT800-Cu15) exhibited very similar activity in the process ofmethanol incineration. This result is surprising because the surfacearea of the HT800-Cu15 catalyst is more than twice lower in compari-son to that of HT700-Cu15 (Table 1). Therefore, it seems that changesin the phase composition that occurred during the calcination at elevat-ed temperature modified the catalytic activity of the samples. Introduc-tion of the potassium promoter into the HT800-Cu15 catalyst resulted
in a shift of the activity curve for about 40 °C in the direction of lowertemperatures. Calcination temperature significantly differentiated activi-ty of the cobalt (Fig. 4B) and copper–cobalt (Fig. 4C) containing catalysts.The HT700-Co15 samplewas less active in a series of the copper contain-ing catalysts. An increase in calcination temperature to 800 °C dramati-cally increased the activity of that catalyst, while deposition of thepotassium promoter did not result in any significant modifications ofthe catalytic activity. It should be noted that the surface area (Table 1)of the more active catalyst (HT800-Co15) is nearly four times lowerthan that of the less active catalyst (HT700-Co15). Therefore, these re-sults clearly show that cobalt present in the well crystallised spinelphases, especially Co3O4, is significant more active than in the form ofthe less ordered metal oxide species. An opposite effect was found forthe copper–cobalt catalysts. The best catalytic results were obtained for
0
20
40
60
80
100
0
20
40
60
80
100
100 200 300 400
100 200 300 400
100 200 300 400
0
20
40
60
80
100
Temperature °[C]
HT700-CuCo15 HT800-CuCo15 HT800-CuCo15/K
C
HT700-Co15 HT800-Co15 HT800-Co15/K
B
CH
3 O
H c
onve
rsio
n [%
] C
H3
OH
con
vers
ion
[%]
CH
3 O
H c
onve
rsio
n [%
]
HT700-Cu15 HT800-Cu15 HT800-Cu15/K
A
Fig. 4. Results of catalytic tests in the process of the total methanol oxidation performed inthe presence of calcined Cu–Mg–Al (A), Co–Mg–Al (B) and Cu–Co–Mg–Al (C) hydrotalcite-like materials.
122 L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
the sample calcined at 700 °C (HT700-CuCo15), while calcination of thehydrotalcite precursor at 800 °C produced significantly less active cata-lyst (HT800-CuCo15). In this case, the increased calcination tempera-ture reduced surface area of the sample less than twice. The totalcontent of transitionmetals (Cu+Co) is larger by two times in compar-ison to the other series of the catalysts. On the other hand the relativeintensity of reflections attributed to the presenceMgO phase positionedat about 42° and 62° (Fig. 1C) is higher than for other hydrotalcites cal-cined at 800 °C. It is suppressing, because this sample contains lessmag-nesium then the other catalysts. Therefore, it could be supposed thatthere is a significant contribution of transition metals in the form oftransition metal oxides and copper and/or cobalt reach spinel phases
in theHT800-CuCo15 sample. Deposition of the potassiumpromoter in-creased the activity of theHT800-CuCo15 catalysts, which howeverwaslower than that of the catalyst calcined at 700 °C (HT700-CuCo15).
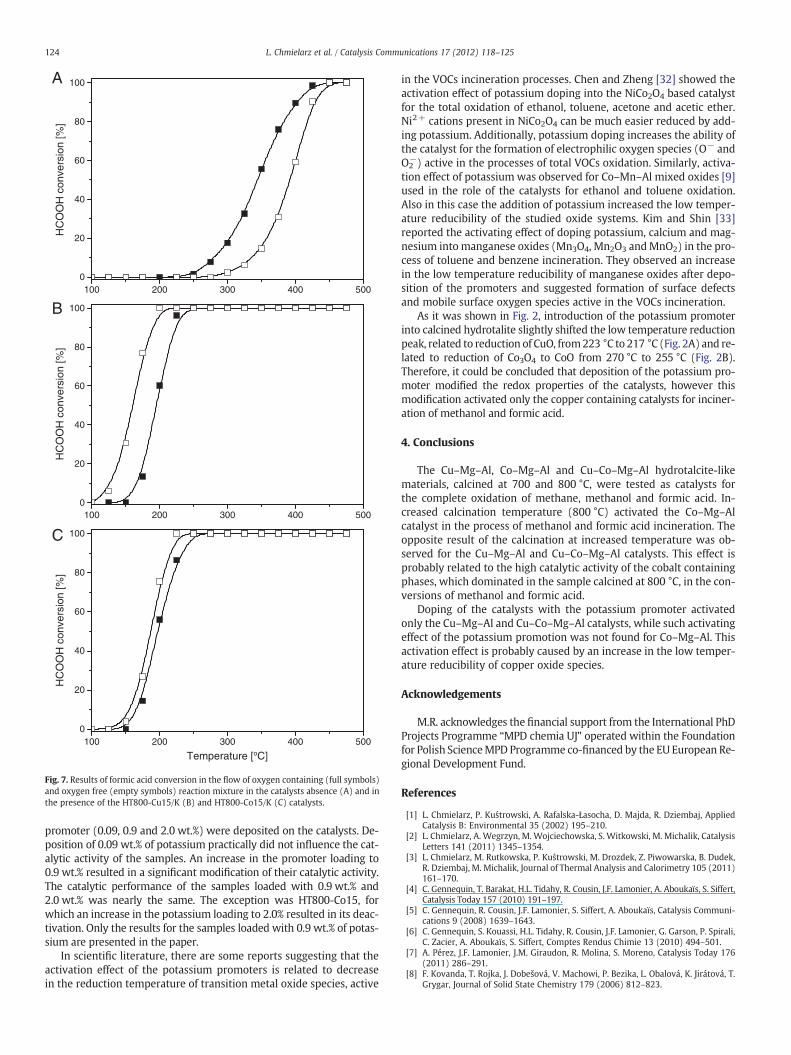
Fig. 5 presents the results of the catalytic incineration of formicacid. The reaction started at about 150 °C, while for the most activecatalysts the complete oxidation of formic acid in the reaction mix-ture was possible at about 225 °C, so at temperature lower by about50 °C than the oxidation of methanol. In general, the calcination tem-perature as well as deposition of the potassium promoter influencedthe activity of the catalysts is very similar ways to that found for themethanol incineration. Therefore, it seems that the same phases arecatalytically active in the processes of formic acid and methanol in-cineration. An increase in the calcination temperature from 700 °Cto 800 °C decreased activity of the copper containing catalysts(Fig. 5A), while modification of the HT800-Cu15 samples with the po-tassium promoter resulted in its significant activation in the processof formic acid oxidation. An increase in the calcination temperatureof the cobalt containing sample from 700 °C to 800 °C shifted the con-version profile to lower temperatures by about 100 °C (Fig. 5B), whileintroduction of potassium promoter into HT800-Co15 practically didnot influence its catalytic activity. Opposite results were obtainedfor a series of the copper–cobalt containing samples (Fig. 5C). An in-crease in the calcination temperature decreased activity of the cata-lysts, while activation effect was observed for the catalyst HT800-CuCo15 after deposition of the potassium promoter.
Formic acid is an oxygen rich molecule and their decompositionvia dehydrogenation (Eq. (8)) and dehydratation (Eq. (9)) withoutoxygen presence is well known [26,27]:
HCOOH ¼ CO2 þ H2 ð8Þ
HCOOH ¼ COþ H2O: ð9ÞTherefore, there is a question of oxygen role in the process of the for-
mic acid conversion. In order to answer this question the temperature-programmed reaction (TPR) studies of formic acid conversion in a flowof oxygen containing and oxygen free reaction mixtures were done. Theresults of these studies performed for the selected catalyst (HT800-Cu15/K) are presented in Fig. 6. It should be noted that hydrogen andcarbon dioxide are the main products of the formic acid conversion forthe process performed in oxygen absence. Therefore, the decompositionof formic acid proceeds as dehydrogenation reaction (Eq. (8)), which istypical for the reaction over basic catalysts [28]. It should be noted thatat temperature above 300 °C the concentration of carbon monoxidegradually increased, in contradiction to the evolution of carbon dioxide.For the process performed in the flow of the oxygen containing reactionmixture, carbon dioxide and water vapour were the main reactionproducts, however the evolution of hydrogen was also detected in therange of 150–400 °C. Such distribution of the reaction products showsthat oxygen participates in the formic acid conversion. However, atleast two possible pathways of this process could be proposed: (i)direct oxidation of formic acid to CO2 and H2O or (ii) decompositionof formic acid into H2 and CO2 and then, in a subsequent step, theoxidation of hydrogen to H2O.
The similarity in the activity order in incineration of methanol andformic acid could be related to similar mechanisms of adsorption andthe possible catalytic activation of these molecules on the metal oxidesurfaces. Dissociation of O–H bond and formation of alkoxyl bondswith surface metal cations was reported for both formic acid as wellas methanol [28–31]. Of course such interaction of methane with thecatalyst surface is impossible and therefore the activity order foundfor themethane oxidation is different from that ofmethanol and formicacid oxidation and proceeds in significantly higher temperatures.
Another interesting and astonishing effect is related to differences inthe conversion temperature of formic acid in the presence and absence
0
20
40
60
80
100
0
20
40
60
80
100
100 200 300 400
100 200 300 400
100 200 300 400
0
20
40
60
80
100
HC
OO
H c
onve
rsio
n [%
]
Temperature [°C]
HT700-CuCo15 HT800-CuCo15 HT800-CuCo15/K
C
HC
OO
H c
onve
rsio
n [%
]
HT700-Co15 HT800-Co15 HT800-Co15/K
B
HC
OO
H c
onve
rsio
n [%
]
HT700-Cu15 HT800-Cu15 HT800-Cu15/K
A
Fig. 5. Results of catalytic tests in theprocess of the total formic acid oxidation performed inthe presence of calcined Cu–Mg–Al (A), Co–Mg–Al (B) and Cu–Co–Mg–Al (C) hydrotalcite-like materials.
0
50
100
150
H2
evol
utio
n [a
.u.]
0
50
100
150
CO
2 ev
olut
ion
[a.u
.]
0
10
20
30
40
50
CO
evo
lutio
n [a
.u.]
100 200 300 400 5000
50
100
150
200
H2O
evo
lutio
n [a
.u.]
Temperature [°C]
Fig. 6. Results of temperature-programmed surface reaction for the process of formicacid oxidation in the presence of the HT800-Cu15/K catalyst.
123L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
of oxygen in the reaction mixture. Examples of the results presented inFig. 7 show that catalytic decomposition of HCOOH in the oxygen freereaction mixture proceeded at temperature significantly lower than inthe presence of oxygen. Opposite results were obtained for the non-catalytic process. It seems possible that water vapour, which is formedby hydrogen oxidation, interacts with the surface of the catalysts (e.g.adsorption, formation of surface hydroxyl groups) and blocks surfacesites for formic acid adsorption and activation. There is no such problemin the case of formic acid decomposition in the inert atmosphere.
Another interesting issue, which should be discussed, is the influ-ence of the potassium promoter on the activity of the studied catalysts.It was shown that potassium activates the copper and copper–cobaltcatalysts in the process of methanol and formic acid incineration,while the activity of the cobalt catalysts in these processes was not af-fected by the potassium introduction. For the process of the total meth-ane oxidation activity of the catalysts modified and non-modified withpotassium was nearly the same. Three different loadings of potassium
0
20
40
60
80
100
HC
OO
H c
onve
rsio
n [%
]
B
0
20
40
60
80
100
Temperature [oC]
HC
OO
H c
onve
rsio
n [%
]
C
100 200 300 400 500
100 200 300 400 500
100 200 300 400 500
0
20
40
60
80
100
HC
OO
H c
onve
rsio
n [%
]
A
Fig. 7. Results of formic acid conversion in the flow of oxygen containing (full symbols)and oxygen free (empty symbols) reaction mixture in the catalysts absence (A) and inthe presence of the HT800-Cu15/K (B) and HT800-Co15/K (C) catalysts.
124 L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
promoter (0.09, 0.9 and 2.0 wt.%) were deposited on the catalysts. De-position of 0.09 wt.% of potassium practically did not influence the cat-alytic activity of the samples. An increase in the promoter loading to0.9 wt.% resulted in a significant modification of their catalytic activity.The catalytic performance of the samples loaded with 0.9 wt.% and2.0 wt.% was nearly the same. The exception was HT800-Co15, forwhich an increase in the potassium loading to 2.0% resulted in its deac-tivation. Only the results for the samples loaded with 0.9 wt.% of potas-sium are presented in the paper.
In scientific literature, there are some reports suggesting that theactivation effect of the potassium promoters is related to decreasein the reduction temperature of transition metal oxide species, active
in the VOCs incineration processes. Chen and Zheng [32] showed theactivation effect of potassium doping into the NiCo2O4 based catalystfor the total oxidation of ethanol, toluene, acetone and acetic ether.Ni2+ cations present in NiCo2O4 can be much easier reduced by add-ing potassium. Additionally, potassium doping increases the ability ofthe catalyst for the formation of electrophilic oxygen species (O− andO2−) active in the processes of total VOCs oxidation. Similarly, activa-
tion effect of potassium was observed for Co–Mn–Al mixed oxides [9]used in the role of the catalysts for ethanol and toluene oxidation.Also in this case the addition of potassium increased the low temper-ature reducibility of the studied oxide systems. Kim and Shin [33]reported the activating effect of doping potassium, calcium and mag-nesium into manganese oxides (Mn3O4, Mn2O3 andMnO2) in the pro-cess of toluene and benzene incineration. They observed an increasein the low temperature reducibility of manganese oxides after depo-sition of the promoters and suggested formation of surface defectsand mobile surface oxygen species active in the VOCs incineration.
As it was shown in Fig. 2, introduction of the potassium promoterinto calcined hydrotalite slightly shifted the low temperature reductionpeak, related to reduction of CuO, from223 °C to 217 °C (Fig. 2A) and re-lated to reduction of Co3O4 to CoO from 270 °C to 255 °C (Fig. 2B).Therefore, it could be concluded that deposition of the potassium pro-moter modified the redox properties of the catalysts, however thismodification activated only the copper containing catalysts for inciner-ation of methanol and formic acid.
4. Conclusions
The Cu–Mg–Al, Co–Mg–Al and Cu–Co–Mg–Al hydrotalcite-likematerials, calcined at 700 and 800 °C, were tested as catalysts forthe complete oxidation of methane, methanol and formic acid. In-creased calcination temperature (800 °C) activated the Co–Mg–Alcatalyst in the process of methanol and formic acid incineration. Theopposite result of the calcination at increased temperature was ob-served for the Cu–Mg–Al and Cu–Co–Mg–Al catalysts. This effect isprobably related to the high catalytic activity of the cobalt containingphases, which dominated in the sample calcined at 800 °C, in the con-versions of methanol and formic acid.
Doping of the catalysts with the potassium promoter activatedonly the Cu–Mg–Al and Cu–Co–Mg–Al catalysts, while such activatingeffect of the potassium promotion was not found for Co–Mg–Al. Thisactivation effect is probably caused by an increase in the low temper-ature reducibility of copper oxide species.
Acknowledgements
M.R. acknowledges the financial support from the International PhDProjects Programme “MPD chemia UJ” operated within the Foundationfor Polish ScienceMPD Programme co-financed by the EU European Re-gional Development Fund.
References
[1] L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha, D. Majda, R. Dziembaj, AppliedCatalysis B: Environmental 35 (2002) 195–210.
[2] L. Chmielarz, A. Wegrzyn, M. Wojciechowska, S. Witkowski, M. Michalik, CatalysisLetters 141 (2011) 1345–1354.
[3] L. Chmielarz, M. Rutkowska, P. Kuśtrowski, M. Drozdek, Z. Piwowarska, B. Dudek,R. Dziembaj, M. Michalik, Journal of Thermal Analysis and Calorimetry 105 (2011)161–170.
[4] C. Gennequin, T. Barakat, H.L. Tidahy, R. Cousin, J.F. Lamonier, A. Aboukaïs, S. Siffert,Catalysis Today 157 (2010) 191–197.
[5] C. Gennequin, R. Cousin, J.F. Lamonier, S. Siffert, A. Aboukaïs, Catalysis Communi-cations 9 (2008) 1639–1643.
[6] C. Gennequin, S. Kouassi, H.L. Tidahy, R. Cousin, J.F. Lamonier, G. Garson, P. Spirali,C. Zacier, A. Aboukaïs, S. Siffert, Comptes Rendus Chimie 13 (2010) 494–501.
[7] A. Pérez, J.F. Lamonier, J.M. Giraudon, R. Molina, S. Moreno, Catalysis Today 176(2011) 286–291.
[8] F. Kovanda, T. Rojka, J. Dobešová, V. Machowi, P. Bezika, L. Obalová, K. Jirátová, T.Grygar, Journal of Solid State Chemistry 179 (2006) 812–823.

125L. Chmielarz et al. / Catalysis Communications 17 (2012) 118–125
[9] K. Jirátová, J. Mikulová, J. Klempa, T. Grygar, Z. Bastil, F. Kovanda, Applied CatalysisA: General 361 (2009) 106–116.
[10] D. Delimaris, T. Ioannides, Applied Catalysis B: Environmental 89 (2009) 295–302.[11] V.H. Vu, J. Belkouch, A. Ould-Dris, B. Taouk, Journal of Hazardous Materials 169
(2009) 758–765.[12] R. Dziembaj,M.Molenda, L. Chmielarz,M.M. Zaitz, Z. Piwowarska, A. Rafalska-Łasocha,
Catalysis Today 169 (2011) 112–117.[13] R. Dziembaj, M. Molenda, L. Chmielarz, M. Drozdek, M.M. Zaitz, B. Dudek, A.
Rafalska-Łasocha, Z. Piwowarska, Catalysis Letters 135 (2010) 68–75.[14] F. Kovanda, R. Jirátová, J. Rymeš, D. Koloušek, Applied Clay Science 18 (2001) 71–80.[15] K. Bahranowski, E. Bielańska, R. Janik, T. Machej, E.M. Serwicka, Clay Minerals 34
(1999) 67–77.[16] L.A. Palacio, J. Velásquez, A. Echavarría, A. Faro, F.R. Ribeiro, M.F. Ribeiro, Journal
of Hazardous Materials 177 (2010) 407–413.[17] F. Kovanda, K. Jirátová, Applied Clay Science 53 (2011) 305–316.[18] L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha, R. Dziembaj, Thermochimica
Acta 395 (2003) 225–236.[19] D.I. Enache, B. Rebours, M. Roy-Auberger, R. Revel, Journal of Catalysis 205 (2002)
346–353.[20] Y. Tanaka, T. Utaka, R. Kikuchi, K. Sasaki, K. Eguchi, Applied Catalysis A: General 242
(2003) 287–295.
[21] R. Guil-López, R.N. Navarro, M.A. Pena, J.L.G. Fierro, International Journal of HydrogenEnergy 36 (2011) 1512–1523.
[22] M.M.V.M. Souza, K.A. Ferreira, O.R. deMacedo Neto, N.F.P. Ribero, M. Schmal, CatalysisToday 133–135 (2008) 750–754.
[23] L. Xue, C. Zhang, H. He, Y. Teraoka, Applied Catalysis B: Environmental 75 (2007)167–174.
[24] N.N. Kim, D.C. Kim, Journal of Materials Online 1 (2005) 1–11.[25] H.Y. Wang, H.C. Ruckenstein, Applied Catalysis A: General 209 (2001) 207–215.[26] X. Li, F. Shi, X. Ma, L. Lu, Y. Deng, Journal of Fuel Chemistry and Technology 38
(2010) 544–553.[27] R. Larsson, M.H. Jamróz, M.A. Borowiak, Journal of Molecular Catalysis A: Chemical
129 (1998) 41–51.[28] E.A. Ivanov, G.Y. Popova, Y.A. Chesalov, T.V. Andrushkevich, Journal of Molecular
Catalysis A: Chemical 312 (2009) 92–96.[29] M.A. Borowiak, M.H. Jamróz, R. Larsson, Journal of Molecular Catalysis A: Chemical
152 (2000) 121–132.[30] S.A. Kirillov, P.E. Tsiakaras, I.V. Romanova, Journal of Molecular Structure 651–653
(2003) 365–370.[31] R. Zhang, Y. Sun, S. Peng, Fuel 81 (2002) 1619–1624.[32] M. Chen, X.M. Zhen, Journal of Molecular Catalysis A: Chemical 221 (2004) 77–80.[33] S.C. Kim, W.G. Shim, Applied Catalysis B: Environmental 98 (2010) 180–185.
![Propuestas para una nueva Constitución (Originada en democracia) [Proposals for a new Constitution (originated in democracy)]](https://img.pdfslide.net/doc/110x75/634534b3596bdb97a908d26f/propuestas-para-una-nueva-constitucion-originada-en-democracia-proposals-for.jpg)
![Arsenate [As (V)] in serpentinites originated from the fore-arc mantle: X-ray absorption spectroscopy study](https://img.pdfslide.net/doc/110x75/63341979a1ced1126c0a2a59/arsenate-as-v-in-serpentinites-originated-from-the-fore-arc-mantle-x-ray-absorption.jpg)



























