Embed Size (px)
Citation preview

Archives of Biochemistry and Biophysics 475 (2008) 18–24
Contents lists available at ScienceDirect
Archives of Biochemistry and Biophysics
journal homepage: www.elsevier .com/ locate /yabbi
Tyramine oxidation by copper/TPQ amine oxidase and peroxidase fromEuphorbia characias latex
Anna Mura, Francesca Pintus, Antonella Fais, Simona Porcu, Marcella Corda, Delia Spanò, Rosaria Medda,Giovanni Floris *
Department of Applied Sciences in Biosystems, University of Cagliari, I-09042 Monserrato, CA, Italy
a r t i c l e i n f o a b s t r a c t
Article history:Received 21 February 2008and in revised form 19 March 2008Available online 7 April 2008
Keywords:Euphorbia characiasAmine oxidasePeroxidaseTyramine
0003-9861/$ - see front matter � 2008 Elsevier Inc. Adoi:10.1016/j.abb.2008.03.034
* Corresponding author. Fax: +39 070 6754523.E-mail address: [email protected] (G. Floris).
1 Abbreviations used: ELAO, Euphorbia latex amineperoxidase; TPQ, 6-hydroxydopa diquinone; pHA, pdi-pHA, di-p-hydroxyphenylacetaldehyde.
Tyramine, an important plant intermediate, was found to be a substrate for two proteins, a copper amineoxidase and a peroxidase from Euphorbia characias latex. The oxidation of tyramine took place by two dif-ferent mechanisms: oxidative deamination to p-hydroxyphenylacetaldehyde by the amine oxidase andformation of di-tyramine by the peroxidase. The di-tyramine was further oxidized at the two aminogroups by the amino oxidase, whereas p-hydroxyphenylacetaldehyde was transformed to di-p-hydroxy-phenylacetaldehyde by the peroxidase. Data obtained in this study indicate a new interesting scenario inthe metabolism of tyramine.
� 2008 Elsevier Inc. All rights reserved.
Plant latex is a milky sap contained in the laticifers, specializedcells forming vessel-like structures that permeate various aerialtissues of about 20 plant families and present in all Euphorbiaceae[1,2]. Latex is a complex environment with a diverse and not thor-oughly investigated composition that includes alkaloids, terpenoidcompounds, and a number of proteins [3]. These substances arecollectively believed to provide an important contribution to plantdefense mechanisms by repelling and killing phytopathogens andsealing wounded areas [4]. In the latex of the Mediterranean shrubEuphorbia characias two enzymes has been purified and character-ized, a copper amine oxidase (ELAO)1 and a class III peroxidase(ELP).
ELAO (amine oxygen oxidoreductase, deaminating, copper/TPQcontaining; EC 1.4.3.6) [5] is a soluble homodimeric protein andeach subunit (MW ffi 74 kDa) contains an active site with a tightlybound CuII ion and an organic cofactor, the 6-hydroxydopa diqui-none, known as Topaquinone (TPQ or TOPA), derived from thepost-translational modification of a Tyr residue in the polypeptidechain [6]. Due to the presence of TPQ, the oxidized form of ELAOhas a distinctive pink color and shows, in addition to the proteinabsorbance maximum at 278 nm, a broad absorption band in thevisible region at 490 nm. ELAO catalyzes the oxidative deaminationof primary amines to the corresponding aldehydes concomitantly
ll rights reserved.
oxidase; ELP, Euphorbia latex-hydroxyphenylacetaldehyde;
with a two-electron reduction of dioxygen to hydrogen peroxide,and could have an important role in the lignification of cell walland in the defensive oxidative burst.
ELP (EC 1.11.1.7, donor: hydrogen peroxide oxidoreductase),belonging to the superfamily of heme-containing peroxidases, is atypical class III secreted peroxidase [7]. This enzyme has, at variancewith other known plant peroxidases, low specific activity for classi-cal peroxidase substrates and its activity seems to be finely regulatedby the Ca2+/calmodulin system. After addition of Ca2+, the kcat in-creased 100 times with a decrease of KM for H2O2 [8]. ELP presumablyparticipates in plant defense mechanisms based on the production ofH2O2-signaling pathways with associated responses against invad-ing pathogens and environmental stresses [9,10].
Tyramine is widely considered an important compound in theplant kingdom for its involvement in a variety of physiological con-ditions such as the biosynthesis of some benzylisoquinoline alka-loids, which have been suggested to function as herbivoredeterrents, and in the protection against pathogens. Moreovertyramine is also involved in the formation of the hydroxycinna-moyl amines, N-feruloyltyramine and 4-N-coumaroyltyramine[11,12], widely distributed metabolites that, incorporated into cellwalls and secreted into the cell culture medium [13], are consid-ered to be integral components of plant defence responses topathogens [14,15].
It is well known that tyramine, obtained by the decarboxylationof tyrosine, can be a substrate for amine oxidases and peroxidases,leading to the formation of p-hydroxyphenylacetaldehyde (pHA)and di-tyramine, respectively [16–19]. In view of the importance

Table 1Kinetic parameters of Euphorbia amine oxidase
Compound KM (mM) kcata (s�1) kcat/KM (mM�1 s�1)
Putrescine 0.2 ± 0.02 34 ± 1.2 142Tyramine 0.2 ± 0.03 3 ± 0.13 15Di-tyramine 14 ± 1.6 15.30 ± 1.7 1.09
The buffer used was 100 mM Tris/HCl buffer, pH 7.0, at 37 �C. Data represent meansof at least five different measurements.
a kcat: (mol of substrate consumed) � (mol of enzyme active sites)�1 � s�1.
A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24 19
of the possible dual role of tyramine in Euphorbia latex, the reac-tion between Euphorbia amine oxidase and Euphorbia peroxidasein the presence of tyramine as substrate was investigated. Wefound that ELAO catalyzes the oxidative deamination of tyramineto pHA whereas ELP catalyzes the oxidation of tyramine to di-tyra-mine. The di-tyramine was further oxidized by ELAO in both thetwo amino groups, whereas pHA was transformed to di-p-hydroxy-phenylacetaldehyde (di-pHA) by ELP. Data obtained in this studyindicate a new interesting scenario in the complex metabolism oftyramine.
Materials and methods
Reagents
Ascorbic acid, 2,20-azinobis(3-ethylbenzo-6-thiazolinesulfonic acid) (ABTS), 1,4-diaminobutane (putrescine) and p-hydroxyphenylethylamine hydrochloride (tyra-mine) were purchased from Sigma (St. Louis, MO, USA). Hydrogen peroxide wasfrom Merck (Darmstadt, Germany) and an e240 = 43.6 M�1 cm�1 was used to deter-mine its concentration. p-Hydroxyphenylacetaldehyde and di-tyramine were ob-tained as previously reported [16–18]. All chemicals were pure commercialproducts and used without further purification.
Enzymes
ELP (RZ value A401/A278 = 2.7) was purified as previously described [7]. The en-zyme concentration was determined spectrophotometrically using ane401 = 130.7 mM�1 cm�1.
ELAO was purified according to the procedure reported [5]. An e490 of6 mM�1 cm�1 or an e278 of 378 mM�1 cm�1 for the purified enzyme (2 copper ionsand a Mr of 150,000) was used to estimate enzyme concentration.
The purified enzymes showed a single band in SDS–PAGE with a molecularmass of 72 ± 1 kDa for ELAO and 46 ± 1 kDa for ELP, in good agreement with thedata reported [5,7].
Peroxidase activityActivity measurements were always made in 100 mM Tris/HCl buffer, pH 7.0, at
25 �C, in the presence of 10 mM Ca2+, using 25 mM hydrogen peroxide and 10 mMof the reducing substrate ABTS following the increase in absorbance at 415 nmresulting from the formation of the ABTS cation radical product(e415 = 36 mM�1 cm�1). Catalytic center activity (kcat) was defined as (mol of sub-strate consumed)/(mol of enzyme active sites) � s1.
The value of KM for ELP using varying tyramine concentrations at saturatingconcentrations of hydrogen peroxide (63 lM), or varying concentrations of hydro-gen peroxide at saturating concentrations of reducing substrate (9 mM tyramine),was calculated from initial velocity data fitted to the Michaelis–Menten equationby nonlinear regression and by double reciprocal plots by Michaelis–Menten anal-ysis in 100 mM Tris/HCl buffer, pH 7.0, at 25 �C. The kcat/KM value was also used as amore useful measure of substrate specificity.
Amine oxidase activityThe KM value for tyramine was obtained from a double reciprocal plot in
100 mM Tris/HCl buffer, pH 7.0. Catalytic activity (kcat) and the kcat/KM values werecompared to those of the best substrate (putrescine). Kinetic parameters were cal-culated as the mean of at least five different measurements.
Oxygen uptake was determined polarographically at 37 �C by an oxygraph(Hansatech Instruments LTD., Norfolk, UK) equipped with a Clark electrode. Thestandard reaction mixture (1 mL) contained the enzyme in 100 mM Tris/HCl buffer,pH 7.0. The reaction was started by addition of substrate after at least 10 min pre-incubation.
Kinetics of inactivation
The time dependence of the inhibition was determined by preincubatingELAO at 37 �C in the presence of various concentrations of tyramine. Periodi-cally aliquots of the mixture were withdrawn and tested polarographically forthe initial rates of putrescine oxidation under standard assay conditions. Theequation used for a single exponential decay is y = A0 � e�kt, i.e. the observedvalues decay with time, t, from an initial value, A0 at t = 0, to 0 at t =1. Therate constant for the decay is k; the half-life for the process, t½, is given byloge 2/k. The data were plotted and fitted to a first-order exponential decay toobtain values of kapp. The rate constant of inactivation (kinh) and dissociationconstant Ki were calculated from double reciprocal plot of kapp against tyramineconcentrations [20].
Assays of products
Reaction of ELAO and tyramineAmmonia production was determined from the amount of NADH consumed in
the presence of glutamate dehydrogenase. Hydrogen peroxide was determined withthe peroxidase/4-hydroxy-3-methoxyphenylacetic acid method [21]. The aldehydereleased was determined as follow: after incubation of ELAO (10 lM) with 1 mMtyramine, in 1 mL of 100 mM Tris/HCl buffer, pH 7.0, at 37 �C, the reaction productswere separated from protein by Centricon filters (Millipore Corporation, Bedford,MA) with a cut-off of approximately 30 kDa. The aldehyde was separated by HPLCchromatography Agilent 1100 series equipped with Zorbax 300 SB-C18 5-lm parti-cle size, 250 � 4.6 mm ID column. The assignment of the peaks to the correspondingcompounds was done by comparing the retention time (Rt) with that of genuinealdehyde sample as standard. In this system pHA (Rt = 25.3 min) was easily distin-guishable from tyramine (Rt = 14.7 min).
Reaction of ELP and tyramineAt 1 mL of 100 mM Tris/HCl buffer, pH 7.0, at 25 �C, containing ELP (0.1 lM),
tyramine (1 mM) and 10 mM Ca2+, 4 aliquots of 300 nmol H2O2 (each 5 lL) wereadded. At the end of the incubation, the reaction products were separated from pro-teins by Centricon filters and analyzed by HPLC as above reported. In this system di-tyramine showed an Rt = 18.4 min.
Spectroscopic methods
Absorption spectra and data from all activity assays were obtained with anUltrospec 2100 spectrophotometer (Biochrom Ltd., Cambridge, UK) using cells witha 1 cm path length. Anaerobic experiments were made in a Thunberg-type spectro-photometer cuvette (Soffieria Vetro, Sassari, Italy) after several cycles of evacuationfollowed by flushing with O2-free argon. Anaerobic additions of various reagents tothe cuvette were made through a rubber cap with a syringe. Fluorescence spectrawere obtained using a Perkin-Elmer LS-3 spectrofluorimeter (Perkin-Elmer Ltd.,Beaconsfield, UK).
Results
Reaction of native ELAO with tyramine
Tyramine turned out to be a substrate for ELAO. The stoichiom-etry of the products, obtained after oxidation of tyramine withELAO, showed that 1 mol of oxygen was consumed and 1 mol ofammonia, 1 mol of pHA and 1 mol of hydrogen peroxide wereformed (not shown). Thus a 1:1:1 stoichiometry of the reactionproducts was found. In 100 mM Tris/HCl buffer, pH 7.0, the KM va-lue for tyramine (0.2 mM) was found to be very similar to that ofthe best substrate, putrescine (Table 1). ELAO showed to give non-linear double reciprocal plots if extended ranges of substrate con-centration were used. The enzyme showed the highest Vmax valueat 30 mM and 1.5 mM for putrescine and tyramine concentrations,respectively, and the inhibition by excess of substrate was shownat 40 and 2.5 mM, respectively. At 2.3 mM concentration of tyra-mine, native ELAO showed a kcat value of approximately 3 s�1, amarkedly lower value on respect to putrescine (34 s�1), making itcatalytically low efficient substrate (Table 1).
HPLC analysis
After incubation of ELAO with 1 mM tyramine for 30 min, whenno more tyramine oxidation occurred, the reaction products wereanalyzed as reported in Materials and methods. A peak with reten-tion time at about 25.3 min was identified as pHA by comparison

Fig. 2. Inactivation of Euphorbia amine oxidase by tyramine. Time courses deter-mined with 10 nM ELAO pre-incubated with the indicated concentrations of tyra-mine in 100 Tris/HCl buffer, pH 7.0, at 37 �C, then assayed with putrescine assubstrate. Concentrations of tyramine were: (s) 1.25 mM; (d) 2.5 mM; (h) 5 mM;(j) 10 mM. Inset: Double reciprocal plot of apparent first-order rate constants ofinactivation (k app) versus tyramine concentrations. The kapp values were obtainedfrom the data in (A) (see Materials and methods: kinetics of inactivation).
20 A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24
with a synthetic pHA standard obtained as reported ([16–17];Fig. 1).
Mechanism of inhibition of ELAO
It is well known that the quantitative analysis of the productsobtained after oxidation of a good substrate (putrescine) by ELAOshows that the amount of O2 consumed is stoichiometric to theamount of products released (ammonia, aldehyde, and hydrogenperoxide). In the presence of tyramine as substrate, when the reac-tion time was extended from 0.5 to 15 min, the enzyme graduallybecame inactivated. The time course of inactivation of ELAO byvarious concentrations of tyramine is shown in Fig. 2. The rate ofenzyme inactivation increased linearly with tyramine concentra-tions, the inactivation following apparent first-order kinetics(Fig. 2). This inhibition was found to be irreversible: after dialysisagainst 100 mM Tris/HCl buffer, pH 7.0, or filtration through aSephacryl S-200 column, the activity of the native enzyme wasnot restored. A double reciprocal plot of the inactivation rate con-stants against tyramine concentrations showed a linear concentra-tion dependence and a positive intercept on the y-axis (Fig. 2,inset), corresponding to 1/kinh [20]. An inactivation rate constantkinh (1 ± 0.02 � s�1) and an apparent inhibition constant Ki of4.9 ± 0.4 mM were determined from the plot.
This result is consistent with a mechanism-based enzyme inac-tivation [22] and the kinetic scheme of inactivation is well de-scribed by the following equation:
Fig. 1. Reverse-phase HPLC separation of tyramine and p-hydroxyphenylacetalde-hyde. After 15 min incubation of 1 mM tyramine with ELAO in 1 mL of 100 mM Tris/HCl buffer, pH 7, at 37 �C, the reaction products were separated from protein bycentrifugal filtration. Products were subsequently analyzed by injection (20 lL) ofreaction mixture onto a reverse-phase HPLC column as described under ‘‘Materialsand methods”. The pHA (Rt = 25.3 min) was easy distinguishable from tyramine (Rt
= 14.7 min). The full disappearance of tyramine was observed after 30 min wherepHA reached its maximum. Synthetic pHA was used as reference standard.
Since hydrogen peroxide and ammonia were also produced in theenzyme-catalyzed oxidation of putrescine, as in the oxidation ofothers substrates, and since the inhibition was observed in the pres-ence and in absence of catalase (data not shown), the pHA was thelikely inhibitory product. To test this hypothesis, the oxidation oftyramine was measured after addition of authentic pHA to the sam-ple: the enzyme was inactivated much faster (not shown). Inhibi-tion was not observed in the presence of the good substrateputrescine indicating that a substrate with a similar KM but a muchhigher Vmax may protect the enzyme.
Spectroscopic features of ELAO
The visible spectrum of resting ELAO has a typical broad absorp-tion centered around 490 nm (Fig. 3). The addition of tyramine to a1 mL solution containing 17 lM ELAO, in the absence of air, causedthe immediately disappearance of the 490 nm absorbance band
Fig. 3. Absorption spectra of ELAO upon reaction with tyramine. Continuous line:spectrum of native enzyme (17 lM) in 1 mL of 100 mM air-saturated Tris/HCl bu-ffer, pH 7.0; dashed line: 6 h after the addition of 1 mM tyramine.

A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24 21
indicating the rapid conversion of the TPQ cofactor to a bleachedspecies, a CuII–aminoquinol [23] (not shown). Afterwards newabsorbance bands centered at 360, 434 and 464 nm appeared con-ferring a yellow color to the solution (not shown). This absorptionspectrum was due to the generation of a CuI–semiquinolamine rad-ical species [24] confirming that the oxidation of tyramine fol-lowed the same mechanistic pathway already reported for othersubstrates. When putrescine was used as substrates, oxygenationrestored the pink-red color of the native, active enzyme. However,when tyramine was used as substrate, after admission of oxygenthe reaction mixture turned blue within 4 h and a new band ap-peared in the spectrum at approximately 648 nm (Fig. 3). No fur-ther changes were observed in the spectrum, while the enzymewas completely inactivated. Even after exhaustive dialysis the en-zyme neither recovered the original absorption, nor the activity.
Reaction of native ELP with tyramine
Tyramine was also shown to be a substrate for ELP. In 100 mMTris/HCl buffer, pH 7.0, and 10 mM Ca2+, the value of KM for tyra-mine, at 63 lM concentration of hydrogen peroxide, was shownto be 5 mM, whereas the KM for hydrogen peroxide, at saturatingconcentrations of tyramine (9 mM), was calculated to be 6.6 lM.ELP showed to give nonlinear double reciprocal plots if extendedranges of hydrogen peroxide concentration are used. The inhibitionby excess of hydrogen peroxide, as previously reported using ABTSas one of the best substrate for ELP [25] was shown at 70 lMhydrogen peroxide and 10 mM tyramine concentrations (notshown). At maxima concentrations of hydrogen peroxide andtyramine, in which no inhibition by excess of substrates was seen,native ELP showed a kcat value of approximately 0.13 s�1, a verylow value when compared with the substrate ABTS (kcat value of270 s�1).
Spectroscopic features of ELP
The electronic absorption spectrum of ELP (PrIXFeIII) showedmaxima at 278, 401, 498 and 637 nm in 100 mM Tris/HCl buffer,pH 7.0 (Fig. 4). The reaction of hydrogen peroxide with ELP gener-ates the green enzyme intermediate compound I (PrIX�+FeIV = O2�),with characteristic absorption maxima at 278, 398, 544, 577 and
Fig. 4. Absorption spectra of 2.5 lM Euphorbia peroxidase. (a) Native ELP. (b) Co-mpound I is formed after addition of equimolar amount of hydrogen peroxide to thenative enzyme. (c) Compound II is formed after addition of 1 equivalent of tyramineto compound I. (d and e) One and three minutes after addition of 1 equivalent oftyramine to compound II showing the re-formation of native ELP, which is totallyrecovered after 10 min incubation (not shown). Conditions: 100 mM Tris/HCl buf-fer, pH 7.0, 25 �C and 10 mM Ca2+.
651 nm (Fig. 4). After addition of tyramine, as observed with otherreducing substrate molecules, the red compound II (PrIXFeIV = O),with absorption maxima at 278, 406, 522 and 555 nm, is produced(Fig. 4) by the first electron transfer from tyramine to compound I.Compound II then reverts to the resting state by a successive one-electron reaction transfer from tyramine.
Tyramine shows a maximum in the UV region at 274 nm(e274 = 1325 M�1 cm�1). To 1 mL of 100 mM Tris/HCl buffer, pH7.0, containing 0.1 lM ELP, 1 mM tyramine and 10 mM Ca2+, ali-quots of 5 lL freshly prepared hydrogen peroxide (300 nmol) wereadded stepwise. It resulted in a UV/visible spectrum characterizedby the formation of a shoulder at 306–347 nm with a maximum at323 nm (Fig. 5), indicative of the formation of di-tyramine [18],whereas the absorption at 274 nm was quite unchanged. Asshowed in Fig. 5, an increase in absorbance at 245 nm with a shiftat 259 nm was also observed. The end point for maximum spectralchange at each addition of H2O2 was 30 min and about 4 additionsof H2O2 (corresponding at about 1.2 lmol) were required to obtainthe maximal spectral change. As reported [26], the molar absorp-tion coefficient at 323 nm of 2200 M�1 cm�1 for di-tyramine wascalculated.
Fluorimetric methods for dimer identification
Di-tyramine was measured fluorimetrically by excitation at 274and 323 nm and emission at 305 and 403 nm, respectively [18].Tyramine showed a fluorescence spectrum at 305 nm whenexcited at 274 nm, but it did not show fluorescence detectable inthe emission band 350–450 nm when excited at 323 nm. Whentyramine was incubated with ELP and hydrogen peroxide, a newfluorescence peak was observed at 403 nm when excited at323 nm (not shown). Again, this was indicative of the formationof di-tyramine.
HPLC analysis
After incubation of ELP with 1 mM tyramine, 10 mM Ca2+ andH2O2 for 2 h (for details see Materials and methods), a peak with
Fig. 5. Absorption spectra of tyramine in the presence of ELP. Reaction mixturecontained, in 1 mL, 100 mM Tris/HCl buffer, pH 7.0, 0.1 lM Euphorbia peroxidase,10 mM Ca2+, and 1 mM tyramine (dashed line). After addition of 300 nmol H2O2
spectra were recorded from 5 to 30 min (continuous line). Arrows indicate theincrease in absorbance at 323 and 245 nm. Inset: Reverse-phase HPLC separation oftyramine and di-tyramine. After 10 min incubation of tyramine and ELP in 1 mL of100 mM Tris/HCl buffer, pH 7.0, at 37 �C, in the presence of Ca2+, the reaction pr-oducts were separated as in Fig. 1. The formation of di-tyramine (Rt = 18.4 min)was showed. The full disappearance of tyramine was observed after 120 min wheredi-tyramine reached its maximum. The synthetic di-tyramine was used as referencestandard.
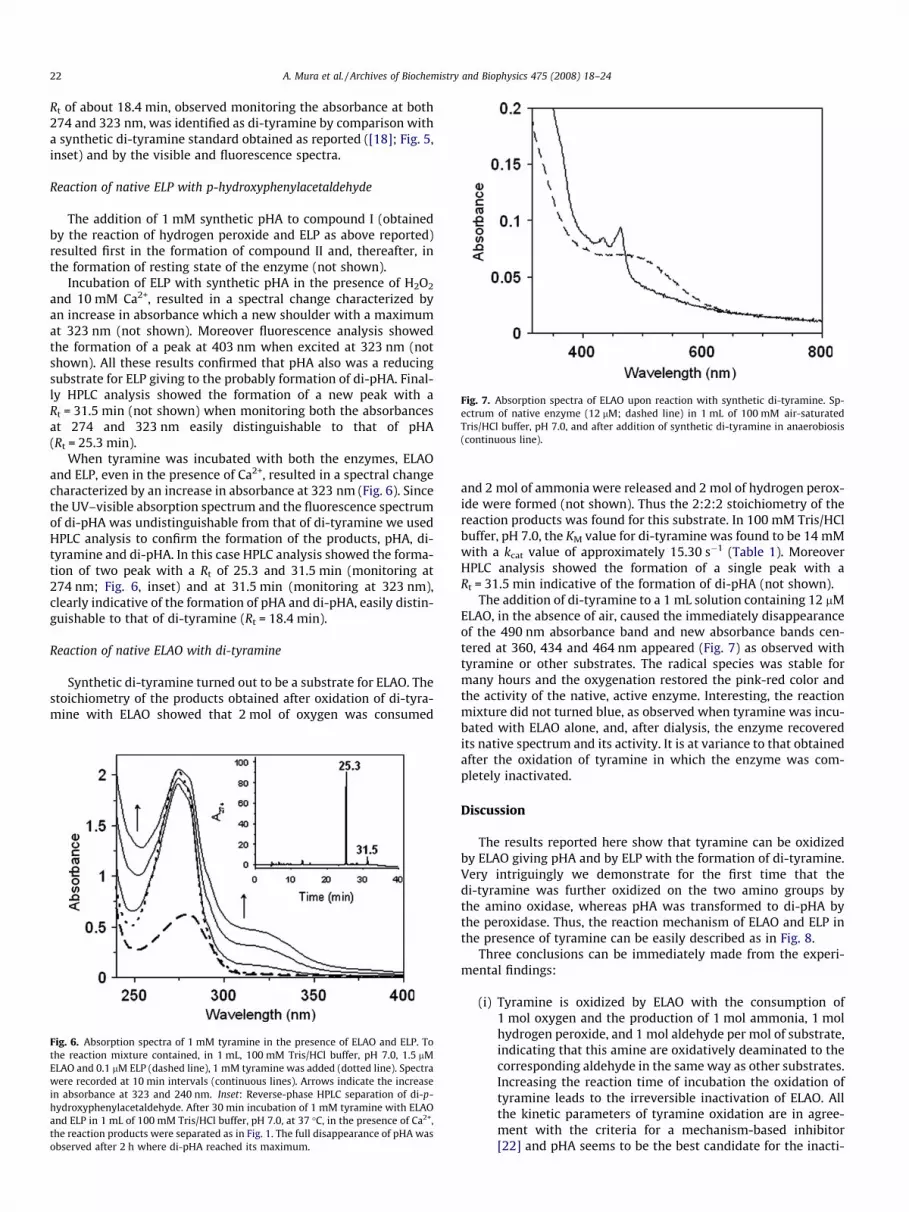
Fig. 7. Absorption spectra of ELAO upon reaction with synthetic di-tyramine. Sp-ectrum of native enzyme (12 lM; dashed line) in 1 mL of 100 mM air-saturatedTris/HCl buffer, pH 7.0, and after addition of synthetic di-tyramine in anaerobiosis(continuous line).
22 A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24
Rt of about 18.4 min, observed monitoring the absorbance at both274 and 323 nm, was identified as di-tyramine by comparison witha synthetic di-tyramine standard obtained as reported ([18]; Fig. 5,inset) and by the visible and fluorescence spectra.
Reaction of native ELP with p-hydroxyphenylacetaldehyde
The addition of 1 mM synthetic pHA to compound I (obtainedby the reaction of hydrogen peroxide and ELP as above reported)resulted first in the formation of compound II and, thereafter, inthe formation of resting state of the enzyme (not shown).
Incubation of ELP with synthetic pHA in the presence of H2O2
and 10 mM Ca2+, resulted in a spectral change characterized byan increase in absorbance which a new shoulder with a maximumat 323 nm (not shown). Moreover fluorescence analysis showedthe formation of a peak at 403 nm when excited at 323 nm (notshown). All these results confirmed that pHA also was a reducingsubstrate for ELP giving to the probably formation of di-pHA. Final-ly HPLC analysis showed the formation of a new peak with aRt = 31.5 min (not shown) when monitoring both the absorbancesat 274 and 323 nm easily distinguishable to that of pHA(Rt = 25.3 min).
When tyramine was incubated with both the enzymes, ELAOand ELP, even in the presence of Ca2+, resulted in a spectral changecharacterized by an increase in absorbance at 323 nm (Fig. 6). Sincethe UV–visible absorption spectrum and the fluorescence spectrumof di-pHA was undistinguishable from that of di-tyramine we usedHPLC analysis to confirm the formation of the products, pHA, di-tyramine and di-pHA. In this case HPLC analysis showed the forma-tion of two peak with a Rt of 25.3 and 31.5 min (monitoring at274 nm; Fig. 6, inset) and at 31.5 min (monitoring at 323 nm),clearly indicative of the formation of pHA and di-pHA, easily distin-guishable to that of di-tyramine (Rt = 18.4 min).
Reaction of native ELAO with di-tyramine
Synthetic di-tyramine turned out to be a substrate for ELAO. Thestoichiometry of the products obtained after oxidation of di-tyra-mine with ELAO showed that 2 mol of oxygen was consumed
Fig. 6. Absorption spectra of 1 mM tyramine in the presence of ELAO and ELP. Tothe reaction mixture contained, in 1 mL, 100 mM Tris/HCl buffer, pH 7.0, 1.5 lMELAO and 0.1 lM ELP (dashed line), 1 mM tyramine was added (dotted line). Spectrawere recorded at 10 min intervals (continuous lines). Arrows indicate the increasein absorbance at 323 and 240 nm. Inset: Reverse-phase HPLC separation of di-p-hydroxyphenylacetaldehyde. After 30 min incubation of 1 mM tyramine with ELAOand ELP in 1 mL of 100 mM Tris/HCl buffer, pH 7.0, at 37 �C, in the presence of Ca2+,the reaction products were separated as in Fig. 1. The full disappearance of pHA wasobserved after 2 h where di-pHA reached its maximum.
and 2 mol of ammonia were released and 2 mol of hydrogen perox-ide were formed (not shown). Thus the 2:2:2 stoichiometry of thereaction products was found for this substrate. In 100 mM Tris/HClbuffer, pH 7.0, the KM value for di-tyramine was found to be 14 mMwith a kcat value of approximately 15.30 s�1 (Table 1). MoreoverHPLC analysis showed the formation of a single peak with aRt = 31.5 min indicative of the formation of di-pHA (not shown).
The addition of di-tyramine to a 1 mL solution containing 12 lMELAO, in the absence of air, caused the immediately disappearanceof the 490 nm absorbance band and new absorbance bands cen-tered at 360, 434 and 464 nm appeared (Fig. 7) as observed withtyramine or other substrates. The radical species was stable formany hours and the oxygenation restored the pink-red color andthe activity of the native, active enzyme. Interesting, the reactionmixture did not turned blue, as observed when tyramine was incu-bated with ELAO alone, and, after dialysis, the enzyme recoveredits native spectrum and its activity. It is at variance to that obtainedafter the oxidation of tyramine in which the enzyme was com-pletely inactivated.
Discussion
The results reported here show that tyramine can be oxidizedby ELAO giving pHA and by ELP with the formation of di-tyramine.Very intriguingly we demonstrate for the first time that thedi-tyramine was further oxidized on the two amino groups bythe amino oxidase, whereas pHA was transformed to di-pHA bythe peroxidase. Thus, the reaction mechanism of ELAO and ELP inthe presence of tyramine can be easily described as in Fig. 8.
Three conclusions can be immediately made from the experi-mental findings:
(i) Tyramine is oxidized by ELAO with the consumption of1 mol oxygen and the production of 1 mol ammonia, 1 molhydrogen peroxide, and 1 mol aldehyde per mol of substrate,indicating that this amine are oxidatively deaminated to thecorresponding aldehyde in the same way as other substrates.Increasing the reaction time of incubation the oxidation oftyramine leads to the irreversible inactivation of ELAO. Allthe kinetic parameters of tyramine oxidation are in agree-ment with the criteria for a mechanism-based inhibitor[22] and pHA seems to be the best candidate for the inacti-

Fig. 8. Oxidation of tyramine. Tyramine (A) is oxidatively deaminated by ELAO topHA (B) and ammonia. The formation of di-tyramine (C) arise after the reaction oftyramine with ELP and hydrogen peroxide. The di-tyramine is further oxidized onthe two amino groups by ELAO giving di-pHA (D). Finally pHA (B) is transformed todi-pHA (D) by ELP.
A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24 23
vation of ELAO. The radical species generated in the absenceof air and with tyramine as substrate remained stable formany hours suggesting that pHA, in anaerobic conditions,did not react directly with any of the two anaerobic interme-diates (the semiquinolamine radical and aminoquinol spe-cies). Thus the inactivation of ELAO and the formation ofthe absorption band at 648 nm requires, at least, severalcycles of reaction and the presence of oxygen. At themoment we cannot assigned at this species any possiblestructure.
(ii) Tyramine is also oxidized by ELP. The compound I, formedafter addition of H2O2 to native ELP, is converted to com-pound II by the first electron transfer from tyramine to com-pound I. Compound II then reverts to the resting state by asuccessive one-electron reaction transfer from tyramine.The reaction gives to the formation of di-tyramine, asshowed by spectrophotometric, fluorimetric and HPLCdeterminations.
(iii) The di-tyramine is oxidized at the two amino groups byELAO, whereas pHA is transformed to di-pHA by ELP. More-over, di-tyramine does not show to be a mechanism-basedinhibitor for ELAO.
It is well known that pHA can bind lysine residues in proteinsand the inhibition of lentil amine oxidase after tyramine oxidationhas been reported [19]. A mechanism involving pHA with one ormore lysine residues and a cross-link between pHA and the semi-
quinolamine radical by formation of carbon–carbon bonds wasproposed interfering with the normal function in the catalytic pro-cess. We believe that the same mechanism can be proposed for theinhibition of ELAO after the reaction with tyramine. Moreover, animportant question springs on why ELAO is not inactivated afterthe reaction with di-tyramine. A plausible explanation could bethat, probably due to the steryc hindrance, the formation of thecross-link between di-pHA and semiquinolamine radical isprevented.
When tyramine is incubated with both the enzymes, ELAO andELP, all tyramine is faster oxidized by ELAO resulting in the forma-tion of pHA and H2O2 that, acting both as substrates for ELP, give tothe formation of di-pHA. It could preserved ELAO from the mecha-nism-based inactivation by tyramine.
Due to the thick rubber composition of the latex and to thepresence of a number of compounds, actually we are unable toidentify any of tyramine oxidation products in the latex. It is dif-ficult to establish a definite physiological role for pHA, di-tyra-mine and di-pHA formed by the coupled pathway of ELAO andELP toward tyramine. Nevertheless the ability of different en-zymes to oxidize these molecules and the contemporarily pres-ence of AO and peroxidase in the latex suggest that theseactivities may not be by chance. Although data available cannotdemonstrate the physiological role of these molecules and theirimportance in plants, it has to be investigated in more detaildue to the possibility of finding new important activities. In thiscontext it should be recalled that tyramine, resulting from thedecarboxylation of tyrosine, have received much attention inthe plant kingdom for its involvement in a variety of physiologi-cal conditions such as the biosynthesis of some benzylisoquino-line alkaloids, which have been suggested to function asherbivore deterrents, and in the protection against pathogens.Moreover tyramine is also involved in the formation of thehydroxycinnamoyl amines, N-feruloyltyramine and 4-N-couma-royltyramine, widely distributed metabolites that, incorporatedinto cell walls and secreted into the cell culture medium, are con-sidered to be integral components of plant defence responses topathogens. Finally, although hydrogen peroxide could be consid-ered the main cytotoxic product on cell cultures, when formedin the reaction between ELAO and tyramine, it may be utilizedby ELP as a crucial substrate for important biochemical processes,leading a signal and a defense molecules rather than a noxiouswaste product.
Acknowledgment
This study was partially supported by PRIN 2006 (Progetti diricerca di interesse nazionale) funds.
References
[1] P. Rudall, Ann. Mo. Bot. Gard. 81 (1994) 270–282.[2] G.L. Webster, Ann. Mo. Bot. Gard. 81 (1994) 3–32.[3] J.H. Ko, K.S. Chow, K.H. Han, Plant Mol. Biol. 53 (2003) 479–492.[4] R.G.O. Kekwick, in: John Wiley & Sons, Inc. (Ed.), Encyclopedia of Life Sciences,
Latex and Laticifers, 2001. Available from: http://www.els.net.[5] A. Padiglia, R. Medda, A. Lorrai, B. Murgia, J.Z. Pedersen, A. Finazzi Agrò, G.
Floris, Plant Physiol. 117 (1998) 1363–1371.[6] S.M. Janes, D. Mu, D. Wemmer, A.J. Smith, S. Kaur, D. Maltby, A.L. Burlingame,
J.P. Klinman, Science 248 (1990) 981–987.[7] R. Medda, A. Padiglia, S. Longu, A. Bellelli, A. Arcovito, S. Cavallo, J.Z. Pedersen,
G. Floris, Biochemistry 42 (2003) 8909–8918.[8] A. Mura, R. Medda, S. Longu, G. Floris, A.C. Rinaldi, A. Padiglia, Biochemistry 44
(2005) 14120–14130.[9] T. Kawano, Plant Cell Rep. 21 (2003) 829–837.
[10] M.L. Orozco-Cardenas, J. Narvaez-Vasquez, C.A. Ryan, Plant Cell 13 (2001) 179–191.
[11] P.J. Facchini, V. De Luca, J. Biol. Chem. 269 (1994) 26684–26690.[12] P.J. Facchini, Annu. Rev. Plant Physiol. Plant Mol. Biol. 52 (2001) 29–66.

24 A. Mura et al. / Archives of Biochemistry and Biophysics 475 (2008) 18–24
[13] A. Schimidt, R. Grimm, J. Schmidt, D. Scheel, D. Strack, S. Rosahl, J. Biol. Chem.274 (1999) 4273–4280.
[14] L. Bonneau, M. Carre, J. Martintanguy, Plant Growth Regul. 15 (1994)75–82.
[15] P.J. Facchini, J. Hagel, K.G. Zulag, Can. J. Bot. 80 (2002) 577–589.[16] S.L. Hazen, F.F. Hsu, J.W. Heinecke, J. Biol. Chem. 271 (1996) 1861–1867.[17] S.L. Hazen, J.P. Gaut, F.F. Hsu, J.R. Crowley, A. d’Avignon, J.W. Heinecke, J. Biol.
Chem. 272 (1997) 16990–16998.[18] M. Valoti, J.A. Morón, A. Benocci, G. Sgaragli, M. Unzeta, Biochem. Pharmacol.
55 (1998) 37–43.[19] A. Padiglia, G. Floris, S. Longu, M.E. Schininà, J.Z. Pedersen, A. Finazzi Agrò, F. De
Angelis, R. Medda, Biol. Chem. 385 (2004) 323–329.
[20] E. Cardemil, in: J. Eyzaguirre (Ed.), Chemical Modification of Enzymes: ActiveSite Studies, Kinetics of the Chemical Modification of Enzymes, Chichester,England, 1987, pp. 23–34.
[21] G.B. Leyton, Pathology 13 (1981) 327–333.[22] C.T. Walsh, Annu. Rev. Biochem. 53 (1984) 493–535.[23] R. Medda, A. Padiglia, J.Z. Pedersen, G. Rotilio, A. Finazzi Agrò, G. Floris,
Biochemistry 34 (1995) 16375–16381.[24] R. Medda, A. Padiglia, A. Bellelli, J.Z. Pedersen, A. Finazzi Agrò, G. Floris, FEBS
Lett. 453 (1999) 1–5.[25] A. Mura, F. Pintus, P. Lai, A. Padiglia, A. Bellelli, G. Floris, R. Medda, Biol. Chem.
387 (2006) 559–567.[26] M. Valoti, K.F. Tipton, P. Sgaragli, Biochem. Pharmacol. 43 (1992) 945–951.





























