Embed Size (px)
Citation preview

Unlocking Tn3-family transposase activity in vitrounveils an asymetric pathway fortransposome assemblyEmilien Nicolasa,1,2, Cédric A. Ogera,1, Nathan Nguyena, Michaël Lambina,3, Amandine Draimea, Sébastien C. Letermea,Michael Chandlerb, and Bernard F. J. Halleta,4
aInstitut des Sciences de la Vie, Université Catholique de Louvain, B-1348 Louvain-la-Neuve, Belgium; and bLaboratoire de Microbiologie et GénétiqueMoléculaires, CNRS UMR5100, F-31062 Toulouse, France
Edited by Kiyoshi Mizuuchi, National Institutes of Health, Bethesda, MD, and approved December 13, 2016 (received for review July 21, 2016)
The Tn3 family is a widespread group of replicative transposons thatare notorious for their contribution to the dissemination of antibioticresistance and the emergence of multiresistant pathogens worldwide.The TnpA transposase of these elements catalyzes DNA breakage andrejoining reactions required for transposition. It also is responsible fortarget immunity, a phenomenon that prevents multiple insertions ofthe transposon into the same genomic region. However, the molec-ular mechanisms whereby TnpA acts in both processes remainunknown. Here, we have developed sensitive biochemical assays forthe TnpA transposase of the Tn3-family transposon Tn4430 and usedthese assays to characterize previously isolated TnpAmutants that areselectively affected in immunity. Compared with wild-type TnpA,these mutants exhibit deregulated activities. They spontaneouslyassemble a unique asymmetric synaptic complex in which one TnpAmolecule simultaneously binds two transposon ends. In this complex,TnpA is in an activated state competent for DNA cleavage and strandtransfer. Wild-type TnpA can form this complex only on precleavedends mimicking the initial step of transposition. The data suggest thattransposition is controlled at an early stage of transpososomeassembly, before DNA cleavage, and that mutations affectingimmunity have unlocked TnpA by stabilizing the protein in amonomeric activated synaptic configuration. We propose an asym-metric pathway for coupling active transpososome assembly withproper target recruitment and discuss this model with respect topossible immunity mechanisms.
DDE/D transposase | DNA transposition | target immunity | Tn3 |transpososome
DNA transposition generically refers to the movement of abroad diversity of genetic elements from one location to
another within and between genomes. This movement is medi-ated by specialized recombination enzymes termed “trans-posases” that catalyze the required DNA cleavage and joiningreactions (1, 2). Transposase-encoding genes are by far the mostabundant and most ubiquitous genes in nature, reflecting theimportance of transposable elements in shaping and organizinggenetic material in all domains of life (3, 4).Tn4430 from Bacillus thuringiensis is a member of the Tn3
family, a widespread group of bacterial transposons (5, 6). Mem-bers of this family were among the first transposable elements tobe identified in bacteria. They are notorious for the number andvariety of accessory genes they transport and their prominentcontribution to antibiotic resistance dissemination (6–8). Notably,Tn3-family members are involved in the rising prevalence of re-sistance to carbapenems, often considered as the “last chance”antibiotics, causing a serious public health threat worldwide (9–11). A key to the evolutionary success of Tn3-family transposons istheir “copy-in” replicative mode of transposition, generating a newcopy of themselves every time they move from one place in thegenome to another one (6). This mechanism provides a powerfulway to mediate rapid and efficient dispersion of phenotypic traitsamong bacterial populations (6, 11). Paradoxically, very little is
known regarding the molecular aspects of this mechanism; currentreplicative transposition models are essentially based on bio-chemical studies performed with the unrelated transposable ele-ment, bacteriophage Mu (12–14).Transposase TnpA of Tn4430 belongs to a large family of
proteins, the DDE/D transposase superfamily, that share a motifof three conserved acidic residues intimately involved in catalysis(2, 15). The active site is included in an RNaseH fold structuraldomain that is characteristic of a variety of polynucleotide hy-drolases and transferases (Fig. 1A) (16, 17). In addition to medi-ating transposition, TnpA is responsible for target immunity, along-range regulatory phenomenon whereby transposons avoidinserting more than once into the same DNA region (18, 19).Other systems with target immunity, such as phage Mu or Tn7,require an adaptor protein (MuB and TnsC, respectively) thatcontrols integration by directing the transpososome to appropriatetargets (20–24). The Tn3-family TnpA is the only transposon-encoded protein directly involved in immunity and is the onlypossible determinant to account for the specificity whereby eachelement confers immunity to itself but not to other family mem-bers (18, 25). We have recently identified Tn4430 TnpA mutantsthat are proficient in transposition (T+) but impaired in targetimmunity (I−), demonstrating that the two functions can beuncoupled (25). The corresponding T+/I−mutations were found to
Significance
The Tn3 family of transposons, discovered in the early 1970s,represents a serious threat to human health because of itsprevalence in the dissemination of antibiotic resistance and,indirectly, because of its involvement in xenobiotic metabolismand in the transmission of plant pathogenicity determinants.Astonishingly, their transposition mechanism has yet to beelucidated. We have started to unravel this mechanism byreconstituting the transposition reaction of the Tn3-familytransposon Tn4430 in a cell-free in vitro system. The assays alsowere used to characterize transposase mutants affected intarget immunity, a phenomenon whereby a given transposonavoids inserting more than once into the same DNA target. Thedata support a tentative model linking target immunity withtransposition complex assembly and activation.
Author contributions: E.N., C.A.O., M.L., and B.F.J.H. designed research; E.N., C.A.O., N.N.,M.L., A.D., S.C.L., and B.F.J.H. performed research; E.N., C.A.O., N.N., M.L., A.D., S.C.L.,M.C., and B.F.J.H. analyzed data; and E.N. and B.F.J.H. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1E.N. and C.A.O. contributed equally to this work.2Present address: Fast-track Diagnostics, L-4354 Esch-sur-Alzette, Luxembourg.3Present address: GSK Biologicals, B-1300 Wavre, Belgium.4To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1611701114/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1611701114 PNAS | Published online January 17, 2017 | E669–E678
BIOCH
EMISTR
YPN
ASPL
US
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022
map across multiple TnpA structural and functional domains (Fig.1A), but their impact on the molecular mechanism of trans-position had yet to be investigated.Here, we have reconstituted consecutive steps of Tn4430
transposition in vitro, providing a more detailed understanding ofthe transposition mechanism used by the Tn3 family. We comparedwild-type TnpA (TnpAWT) activities with those of representativeT+/I− TnpA mutants and show that the T+/I− mutations have“unlocked” TnpA to different extents. This unlocking correlateswith the formation of an unusual paired-end complex (PEC) inwhich one molecule of TnpA appears to bind two transposon endssimultaneously and cooperatively. The data suggest that thisasymmetric synaptic structure represents an activated intermediatein which TnpA is competent for catalysis. We propose that for-mation of the active complex normally requires unlocking inter-actions with the target and that this requirement provides a meansto discriminate between permissive and nonpermissive targets. Wediscuss this model with respect to plausible mechanisms fortransposition immunity.
ResultsWild-Type TnpAWT and the T+/I− TnpA Mutants Assemble DifferentComplexes with Tn4430 Ends in Vitro. We recently developed asimple thermal shift-induction method for overexpression andpurification of a soluble form of Tn4430 TnpA (Fig. S1) (25). Weshow here that the purified protein is biochemically active andcapable of catalyzing key steps of the transposition reaction invitro. The procedure also was used to purify representative T+/I−
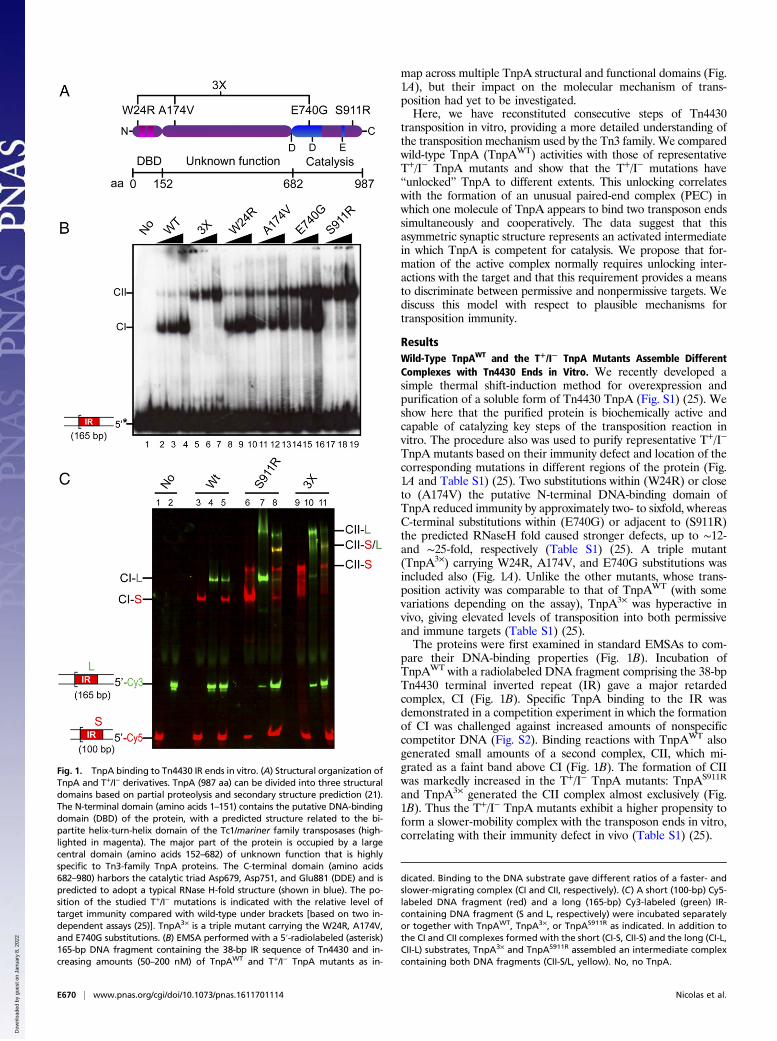
TnpA mutants based on their immunity defect and location of thecorresponding mutations in different regions of the protein (Fig.1A and Table S1) (25). Two substitutions within (W24R) or closeto (A174V) the putative N-terminal DNA-binding domain ofTnpA reduced immunity by approximately two- to sixfold, whereasC-terminal substitutions within (E740G) or adjacent to (S911R)the predicted RNaseH fold caused stronger defects, up to ∼12-and ∼25-fold, respectively (Table S1) (25). A triple mutant(TnpA3×) carrying W24R, A174V, and E740G substitutions wasincluded also (Fig. 1A). Unlike the other mutants, whose trans-position activity was comparable to that of TnpAWT (with somevariations depending on the assay), TnpA3× was hyperactive invivo, giving elevated levels of transposition into both permissiveand immune targets (Table S1) (25).The proteins were first examined in standard EMSAs to com-
pare their DNA-binding properties (Fig. 1B). Incubation ofTnpAWT with a radiolabeled DNA fragment comprising the 38-bpTn4430 terminal inverted repeat (IR) gave a major retardedcomplex, CI (Fig. 1B). Specific TnpA binding to the IR wasdemonstrated in a competition experiment in which the formationof CI was challenged against increased amounts of nonspecificcompetitor DNA (Fig. S2). Binding reactions with TnpAWT alsogenerated small amounts of a second complex, CII, which mi-grated as a faint band above CI (Fig. 1B). The formation of CIIwas markedly increased in the T+/I− TnpA mutants: TnpAS911R
and TnpA3× generated the CII complex almost exclusively (Fig.1B). Thus the T+/I− TnpA mutants exhibit a higher propensity toform a slower-mobility complex with the transposon ends in vitro,correlating with their immunity defect in vivo (Table S1) (25).
Fig. 1. TnpA binding to Tn4430 IR ends in vitro. (A) Structural organization ofTnpA and T+/I− derivatives. TnpA (987 aa) can be divided into three structuraldomains based on partial proteolysis and secondary structure prediction (21).The N-terminal domain (amino acids 1–151) contains the putative DNA-bindingdomain (DBD) of the protein, with a predicted structure related to the bi-partite helix-turn-helix domain of the Tc1/mariner family transposases (high-lighted in magenta). The major part of the protein is occupied by a largecentral domain (amino acids 152–682) of unknown function that is highlyspecific to Tn3-family TnpA proteins. The C-terminal domain (amino acids682–980) harbors the catalytic triad Asp679, Asp751, and Glu881 (DDE) and ispredicted to adopt a typical RNase H-fold structure (shown in blue). The po-sition of the studied T+/I− mutations is indicated with the relative level oftarget immunity compared with wild-type under brackets [based on two in-dependent assays (25)]. TnpA3× is a triple mutant carrying the W24R, A174V,and E740G substitutions. (B) EMSA performed with a 5′-radiolabeled (asterisk)165-bp DNA fragment containing the 38-bp IR sequence of Tn4430 and in-creasing amounts (50–200 nM) of TnpAWT and T+/I− TnpA mutants as in-
dicated. Binding to the DNA substrate gave different ratios of a faster- andslower-migrating complex (CI and CII, respectively). (C) A short (100-bp) Cy5-labeled DNA fragment (red) and a long (165-bp) Cy3-labeled (green) IR-containing DNA fragment (S and L, respectively) were incubated separatelyor together with TnpAWT, TnpA3×, or TnpAS911R as indicated. In addition tothe CI and CII complexes formed with the short (CI-S, CII-S) and the long (CI-L,CII-L) substrates, TnpA3× and TnpAS911R assembled an intermediate complexcontaining both DNA fragments (CII-S/L, yellow). No, no TnpA.
E670 | www.pnas.org/cgi/doi/10.1073/pnas.1611701114 Nicolas et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022

The Slower-Mobility CII Complex Is a PEC. For further insight into theidentity of CI and CII, we performed a short–long EMSA exper-iment coupled to differential fluorescent DNA labeling to esti-mate the number of DNA fragments in each complex. Proteinswere incubated with a short (100 bp; S) and a long (165 bp; L) IRfragment separately or together in the same reaction. The frag-ments were fluorescently labeled with cyanidin5 (Cy5) and cya-nidin3 (Cy3) fluorophores to assign them unambiguously tospecific complexes after fluorescence scanning of the gel (Fig. 1C).With TnpAWT, two differently labeled complexes were observed
in the mixed reaction (Fig. 1C, lane 5). They correspond to the CIcomplexes formed on the short and long DNA substrates sepa-rately (Fig. 1C, lanes 3 and 4). This pattern is consistent with CIbeing a single-end complex (CI/SEC) in which one IR is bound byTnpA. Binding of the TnpAS911R and TnpA3×mutants to the shortand long fragments gave a mixture of CI and CII (Fig. 1C, lanes 6and 7 and lanes 9 and 10). An additional intermediate complex(CII-S/L) also was observed in the coincubation reaction (Fig. 1C,lanes 8 and 11). This complex results from the association of ashort and long DNA substrate as demonstrated by the presence ofboth fluorophores in the corresponding band (appearing yellow inFig. 1C). This hybrid complex does not form in short–long ex-periments in which an IR-containing fragment is coincubated witha nonspecific DNA fragment (Fig. S3A). We therefore concludethat the slower-migrating CII complex is a PEC (CII/PEC) inwhich TnpA specifically binds to two IRs, and that this complexlikely corresponds to a specific synaptic intermediate in Tn4430transposition.
CII/PEC Formation Involves Local DNA Alterations Around theCleavage Sites of the Transposon. To determine the DNA re-gions of Tn4430 ends contacted by TnpA in CI/SEC and CII/PEC, both complexes were separated by standard EMSA andsubjected to in gel DNA footprinting using 1,10-phenanthroline-copper [(OP)2-Cu
+] (Fig. 2). This analysis was performedwith TnpAWT and TnpA3×, which form a majority of CI/SECs andCII/PECs, respectively, and with TnpAE740G, which produces a mixof both complexes (Fig. 1B).
TnpA binding protected a conserved 26- to 31-bp inner regionof the IR corresponding to putative recognition motifs for thetransposase (Fig. 2) (6). CI/SEC and CII/PEC gave similar foot-prints. However, on closer examination, CII/PEC patterns arecharacterized by discrete changes in the distal part of the trans-poson end, with enhanced [(OP)2-Cu
+] cleavage at the most ex-ternal IR positions and additional weak protection in the flankingDNA (Fig. 2; also see also enlargement in Fig. S4). The outermost5′-GGGG-3′ sequence of the IR is another highly conserved motifthought to be important for catalysis (12, 18). The altered foot-print pattern observed in this region suggests that the formation ofCII/PEC induces some local distortions at the very end of thetransposon, presumably making positions close to the putativeTnpA cleavage site more sensitive to [(OP)2-Cu
+]. CII/PEC thusmay correspond to an activated form of the complex in which thetransposon end is committed for catalysis.
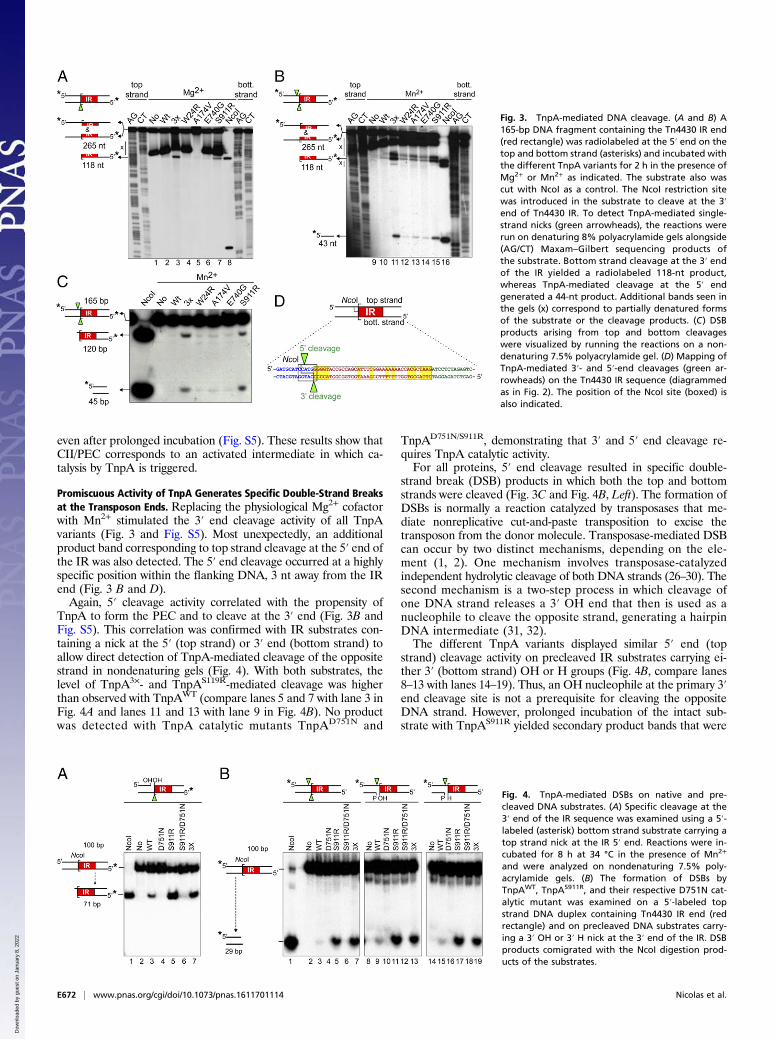
CII/PEC Formation Correlates with Catalytic Activation. DNA cleav-age at the transposon ends was examined by incubating wild-typeand mutant TnpAs with top- and bottom-strand radiolabeled IRsubstrates and by analyzing the reactions on denaturing poly-acrylamide gels (Fig. 3 and Fig. S5). An NcoI restriction site wasintroduced at the end of the IR sequence to generate a substratethat mimics TnpA-mediated 3′ end cleavage. Reactions wereperformed with protein concentrations giving equivalent levels ofDNA binding to allow direct comparison of specific activities.No cleavage was observed in the presence of EDTA or if
Asp751 of the DDE catalytic triad was replaced by Asn, asexpected for the DDE/D transposase superfamily (Fig. S5) (15–17). Incubation with Mg2+ generated a radiolabeled 120-ntproduct that comigrated with the bottom strand cleavage productof NcoI, demonstrating that TnpA introduces specific nicks at thetransposon 3′ ends as predicted from current models of copy-inreplicative transposition (Fig. 3 A and D) (6). The amount ofcleavage was proportional to the amount of CII/PEC produced bythe different TnpA variants (compare Fig. 1B with Fig. 3A andFig. S5A), with TnpA3× and TnpAS911R showing the highest ac-tivity and cleavage mediated by TnpAWT being barely detected
Fig. 2. DNA footprint analysis of the CI/SEC and CII/PEC. (Upper) The 165-bp DNA fragment containingthe IR end of Tn4430 (red box) was 5′-radiolabeledon the bottom (Left) or top (Right) strand and in-cubated with TnpAWT, TnpAE740G, or TnpA3×. Theunbound DNA substrates and the resulting CI/SECand CII/PEC complexes were separated by standardEMSA and subjected to in-gel [(OP)2-Cu
+] DNAcleavage. The footprint reactions were analyzedon denaturing 8% polyacrylamide gels alongsideMaxam–Gilbert DNA sequencing reactions (AG/CT)performed on the substrates. A densitometry graphcomparing the TnpAE740G SEC (purple trace) and PEC(orange trace) cleavage patterns is shown to theright of the gels. For the top strand-labeled sub-strate, the signal given by the unbound DNA (dashedline) in the bottom of the gel is out of range becauseof overdigestion with [(OP)2-Cu
+]. Symbols showingprotected regions (solid bars and circles) and [(OP)2-Cu+] hypersensitive sites (triangles) in the SEC andPEC are colored purple and orange, respectively.(Lower) The data are summarized on the DNA se-quence of the substrate. The 38-bp IR sequence ofTn4430 is shown in red with the conserved motifs(Boxes A, B1, and B2) highlighted in yellow (6). “In”and “Out” refer to Tn4430 inner and outer se-quences, respectively.
Nicolas et al. PNAS | Published online January 17, 2017 | E671
BIOCH
EMISTR
YPN
ASPL
US
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022
even after prolonged incubation (Fig. S5). These results show thatCII/PEC corresponds to an activated intermediate in which ca-talysis by TnpA is triggered.
Promiscuous Activity of TnpA Generates Specific Double-Strand Breaksat the Transposon Ends. Replacing the physiological Mg2+ cofactorwith Mn2+ stimulated the 3′ end cleavage activity of all TnpAvariants (Fig. 3 and Fig. S5). Most unexpectedly, an additionalproduct band corresponding to top strand cleavage at the 5′ end ofthe IR was also detected. The 5′ end cleavage occurred at a highlyspecific position within the flanking DNA, 3 nt away from the IRend (Fig. 3 B and D).Again, 5′ cleavage activity correlated with the propensity of
TnpA to form the PEC and to cleave at the 3′ end (Fig. 3B andFig. S5). This correlation was confirmed with IR substrates con-taining a nick at the 5′ (top strand) or 3′ end (bottom strand) toallow direct detection of TnpA-mediated cleavage of the oppositestrand in nondenaturing gels (Fig. 4). With both substrates, thelevel of TnpA3×- and TnpAS119R-mediated cleavage was higherthan observed with TnpAWT (compare lanes 5 and 7 with lane 3 inFig. 4A and lanes 11 and 13 with lane 9 in Fig. 4B). No productwas detected with TnpA catalytic mutants TnpAD751N and
TnpAD751N/S911R, demonstrating that 3′ and 5′ end cleavage re-quires TnpA catalytic activity.For all proteins, 5′ end cleavage resulted in specific double-
strand break (DSB) products in which both the top and bottomstrands were cleaved (Fig. 3C and Fig. 4B, Left). The formation ofDSBs is normally a reaction catalyzed by transposases that me-diate nonreplicative cut-and-paste transposition to excise thetransposon from the donor molecule. Transposase-mediated DSBcan occur by two distinct mechanisms, depending on the ele-ment (1, 2). One mechanism involves transposase-catalyzedindependent hydrolytic cleavage of both DNA strands (26–30). Thesecond mechanism is a two-step process in which cleavage ofone DNA strand releases a 3′ OH end that then is used as anucleophile to cleave the opposite strand, generating a hairpinDNA intermediate (31, 32).The different TnpA variants displayed similar 5′ end (top
strand) cleavage activity on precleaved IR substrates carrying ei-ther 3′ (bottom strand) OH or H groups (Fig. 4B, compare lanes8–13 with lanes 14–19). Thus, an OH nucleophile at the primary 3′end cleavage site is not a prerequisite for cleaving the oppositeDNA strand. However, prolonged incubation of the intact sub-strate with TnpAS911R yielded secondary product bands that were
Fig. 3. TnpA-mediated DNA cleavage. (A and B) A165-bp DNA fragment containing the Tn4430 IR end(red rectangle) was radiolabeled at the 5′ end on thetop and bottom strand (asterisks) and incubated withthe different TnpA variants for 2 h in the presence ofMg2+ or Mn2+ as indicated. The substrate also wascut with NcoI as a control. The NcoI restriction sitewas introduced in the substrate to cleave at the 3′end of Tn4430 IR. To detect TnpA-mediated single-strand nicks (green arrowheads), the reactions wererun on denaturing 8% polyacrylamide gels alongside(AG/CT) Maxam–Gilbert sequencing products ofthe substrate. Bottom strand cleavage at the 3′ endof the IR yielded a radiolabeled 118-nt product,whereas TnpA-mediated cleavage at the 5′ endgenerated a 44-nt product. Additional bands seen inthe gels (x) correspond to partially denatured formsof the substrate or the cleavage products. (C) DSBproducts arising from top and bottom cleavageswere visualized by running the reactions on a non-denaturing 7.5% polyacrylamide gel. (D) Mapping ofTnpA-mediated 3′- and 5′-end cleavages (green ar-rowheads) on the Tn4430 IR sequence (diagrammedas in Fig. 2). The position of the NcoI site (boxed) isalso indicated.
Fig. 4. TnpA-mediated DSBs on native and pre-cleaved DNA substrates. (A) Specific cleavage at the3′ end of the IR sequence was examined using a 5′-labeled (asterisk) bottom strand substrate carrying atop strand nick at the IR 5′ end. Reactions were in-cubated for 8 h at 34 °C in the presence of Mn2+
and were analyzed on nondenaturing 7.5% poly-acrylamide gels. (B) The formation of DSBs byTnpAWT, TnpAS911R, and their respective D751N cat-alytic mutant was examined on a 5′-labeled topstrand DNA duplex containing Tn4430 IR end (redrectangle) and on precleaved DNA substrates carry-ing a 3′ OH or 3′ H nick at the 3′ end of the IR. DSBproducts comigrated with the NcoI digestion prod-ucts of the substrates.
E672 | www.pnas.org/cgi/doi/10.1073/pnas.1611701114 Nicolas et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022

not seen with the precleaved 3′H substrate (Fig. S6). One of thesebands (labeled “y” in Fig. S6) migrated above the substrate andthus may correspond to strand transfer product. Together, theseobservations suggest that cleavage at the 5′ end takes place pri-marily by direct hydrolysis, although some hairpin-dependentmechanism may act in the formation of secondary products as wasreported for other transposase families (26).
DNA Cleavage at the 3′ End of Tn4430 Stabilizes PEC Formation. Theresults reported above show that mutations affecting target im-munity in vivo appear to activate TnpA in vitro, correlating with ahigher level of CII/PEC formation. As a corollary, CII/PEC as-sembly may impose an activation barrier that must be overcomeduring normal transposition to trigger catalysis. To test this idea,complex formation was examined on precleaved DNA substratesto reproduce conditions under which specific requirements forinitiating transposition have been bypassed.Precleaved substrates were first generated by cutting the IR-
containing DNA fragment with NcoI to mimic 3′ end cleavage byTnpA (Fig. 5; also see Fig. 3). Binding of all TnpA variants (in-cluding TnpAWT) to this substrate yielded a single retarded bandthat migrated at the same position within the gel (Fig. 5A). Ashort–long experiment performed with TnpAWT and TnpAS911R
on Cy3- and Cy5-labeled substrates revealed that this band cor-responds to a cleaved paired-end complex (CII/PECC) containingtwo cleaved ends (Fig. 5B). As for the intact (uncleaved) ends (Fig.S3A), CII/PECC does not form in reactions where specific pre-cleaved IR ends are mixed with equivalent nonspecific substrates(Fig. S3B), further demonstrating that PEC formation requiresspecific interactions between TnpA and two transposon ends.Enhanced CII/PEC formation was also observed with DNA
substrates containing a single-strand nick at the 3′ end of the IRsequence. Binding of TnpAWT (and its catalytic mutant derivativeTnpAD751N) to this substrate gave significant amounts of CII/PECC which was undetectable or barely detectable with theunnicked substrate (Fig. S7, compare B and C with A). In addition,binding of two ends displayed some kind of cooperativity, with ahigher CII/PEC-to-CI/SEC ratio at low TnpA concentrations thanat higher concentrations (Fig. S7). In the case of TnpAS911R (andTnpAD751N/S911R), CI/SEC was detected only at protein concen-trations reaching substrate saturation for both the nicked andunnicked IR (Fig. S7 A–C). These results support the conclusionthat CII/PEC formation involves a conformational change that
enables TnpA to bind two transposon ends with a higher affinitythan a single one. This conformational change is facilitated byactivating T+/I− TnpA mutations and/or by bypassing the initialcleavage step of transposition.
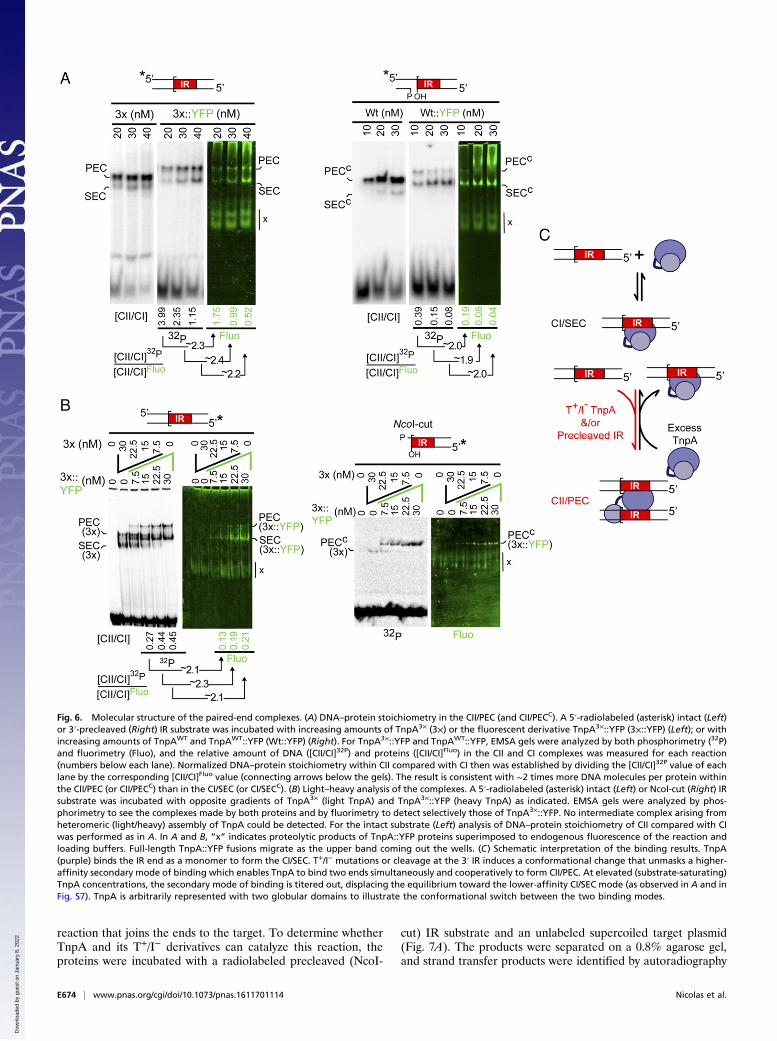
CII/PEC Has a Noncanonical Asymmetrical Architecture with a SingleTnpA Molecule Tethering Two Transposon Ends. For further insightsinto CI/SEC and CII/PEC architecture, we sought to establish theprotein–DNA stoichiometry in both complexes. Size-exclusionchromatography showed that TnpA exists as a monomer in solu-tion, eluting as a single peak corresponding to an apparent mo-lecular mass of ∼143 kDa (Fig. S8A). This result is a slightoverestimation compared with the calculated mass (116 kDa),likely because of the multidomain structure of TnpA (Fig. 1A)(25). Also consistent with this conclusion, dynamic light scattering(DLS) measurements indicate that TnpA has an average diameterof ∼11.4 nm, which is closer to the expected value for a monomer(10.2 nm) than for a dimer (14.1 nm) (Fig. S8B).To establish the protein/DNA ratio within the complexes,
fluorescent derivatives of TnpAWT and TnpA3× with C-terminalYFP Venus tags were used. The binding properties of TnpAWT::YFP and TnpA3×::YFP were similar to those of their native (i.e.,unfused) counterparts. In particular, TnpA3×::YFP gave a mixtureof CI/SEC and CII/PEC on an intact IR, whereas TnpAWT::YFPgave detectable levels of the CII/PEC only on a precleaved IRcarrying a nick at the 3′-end (Fig. 6A). Note however that for-mation of CII/PEC was more sensitive to increasing amounts ofthe fusion proteins than the native proteins, suggesting that thepresence of YFP at the TnpA C terminus destabilizes CII/PEC infavor of CI/SEC. For both proteins, we measured the relativeamount of radioactivity (DNA) and fluorescence (protein) withinthe CII/PEC and CI/SEC bands and found that the DNA/proteinratio of the CII/PEC is twice that measured for CI/SEC (Fig. 6A).Thus, the same numbers of TnpA molecules bind to a single endwithin CI/SEC and to two synapsed ends within CII/PEC.To determine the oligomeric state of TnpA within both
complexes, we performed a light–heavy experiment, the proteinequivalent of the short–long assay described above for the DNA.Incubation of an IR end with different ratios of unfused TnpA3×
(light TnpA) and TnpA3×::YFP (heavy TnpA) yielded a mixedpattern corresponding to the CI/SECs and CII/PECs producedby both proteins separately (Fig. 6B). No intermediate band wasdetected, as would be expected for the formation of heteromeric(light–heavy) TnpA/DNA complexes. The presence of a singleTnpA molecule in the CI/SEC and CII/PEC was demonstratedfurther by performing the binding reactions in the presence ofincreasing amounts of a monoclonal antibody raised against thepurification cMyc-His6-tag (Fig. S9). For both CI/SEC and CII/PEC, a single supershifted band was detected, indicating that theantibody binds to a single TnpA molecule in both complexes.Together, these results show that TnpA binds to the transposon
end as a monomer and that under specific conditions that favorCII/PEC formation (i.e., with T+/I− activating TnpA mutations oron precleaved DNA ends), the protein can adopt an alternativeconfiguration that enables it to bind two ends simultaneously. Thisability therefore implies that TnpA has separate modes of DNAbinding: a primary mode to bind one end and a secondary mode,that, when unmasked, allows the protein to capture a second end(Fig. 6C). This conclusion also clarifies the binding pattern ob-served at high protein/DNA ratios for the T+/I− TnpA mutantsand/or the precleaved ends (Fig. 6A and Fig. S7). At substrate-saturating TnpA concentrations, the primary mode of bindingoutcompetes the secondary mode, promoting CII/PEC dissocia-tion into CI/SEC (Fig. 6C).
Tn4430 Transposition Is Regulated at the Level of TranspososomeAssembly and DNA Cleavage. During transposition, DNA cleav-age at the ends of the transposon is followed by a strand transfer
Fig. 5. TnpA binding on precleaved ends. (A) A 5′-radiolabeled (asterisk)NcoI-cleaved IR end was incubated with different TnpA variants as indicated,and the reactions were analyzed by standard EMSA. TnpA binding to theprecleaved end gave a single retarded complex with all variants. (B) A shortCy5-labeled (S, 58 bp) and a long Cy3-labeled (L, 120 bp) precleaved endwere incubated together or separately with TnpAWT and TnpAS911R as in-dicated. The presence of an intermediate yellow band in the mixed reactions(lanes 3 and 6) indicates that the complex formed on the short (lanes 1 and 5)or long (lanes 4 and 7) substrates is a PECC containing two transposon ends.
Nicolas et al. PNAS | Published online January 17, 2017 | E673
BIOCH
EMISTR
YPN
ASPL
US
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022
reaction that joins the ends to the target. To determine whetherTnpA and its T+/I− derivatives can catalyze this reaction, theproteins were incubated with a radiolabeled precleaved (NcoI-
cut) IR substrate and an unlabeled supercoiled target plasmid(Fig. 7A). The products were separated on a 0.8% agarose gel,and strand transfer products were identified by autoradiography
Fig. 6. Molecular structure of the paired-end complexes. (A) DNA–protein stoichiometry in the CII/PEC (and CII/PECC). A 5′-radiolabeled (asterisk) intact (Left)or 3′-precleaved (Right) IR substrate was incubated with increasing amounts of TnpA3× (3×) or the fluorescent derivative TnpA3×::YFP (3×::YFP) (Left); or withincreasing amounts of TnpAWT and TnpAWT::YFP (Wt::YFP) (Right). For TnpA3×::YFP and TnpAWT::YFP, EMSA gels were analyzed by both phosphorimetry (32P)and fluorimetry (Fluo), and the relative amount of DNA ([CII/CI]32P) and proteins ([CII/CI]Fluo) in the CII and CI complexes was measured for each reaction(numbers below each lane). Normalized DNA–protein stoichiometry within CII compared with CI then was established by dividing the [CII/CI]32P value of eachlane by the corresponding [CII/CI]Fluo value (connecting arrows below the gels). The result is consistent with ∼2 times more DNA molecules per protein withinthe CII/PEC (or CII/PECC) than in the CI/SEC (or CI/SECC). (B) Light–heavy analysis of the complexes. A 5′-radiolabeled (asterisk) intact (Left) or NcoI-cut (Right) IRsubstrate was incubated with opposite gradients of TnpA3× (light TnpA) and TnpA3×::YFP (heavy TnpA) as indicated. EMSA gels were analyzed by phos-phorimetry to see the complexes made by both proteins and by fluorimetry to detect selectively those of TnpA3×::YFP. No intermediate complex arising fromheteromeric (light/heavy) assembly of TnpA could be detected. For the intact substrate (Left) analysis of DNA–protein stoichiometry of CII compared with CIwas performed as in A. In A and B, “x” indicates proteolytic products of TnpA::YFP proteins superimposed to endogenous fluorescence of the reaction andloading buffers. Full-length TnpA::YFP fusions migrate as the upper band coming out the wells. (C) Schematic interpretation of the binding results. TnpA(purple) binds the IR end as a monomer to form the CI/SEC. T+/I− mutations or cleavage at the 3′ IR induces a conformational change that unmasks a higher-affinity secondary mode of binding which enables TnpA to bind two ends simultaneously and cooperatively to form CII/PEC. At elevated (substrate-saturating)TnpA concentrations, the secondary mode of binding is titered out, displacing the equilibrium toward the lower-affinity CI/SEC mode (as observed in A and inFig. S7). TnpA is arbitrarily represented with two globular domains to illustrate the conformational switch between the two binding modes.
E674 | www.pnas.org/cgi/doi/10.1073/pnas.1611701114 Nicolas et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022
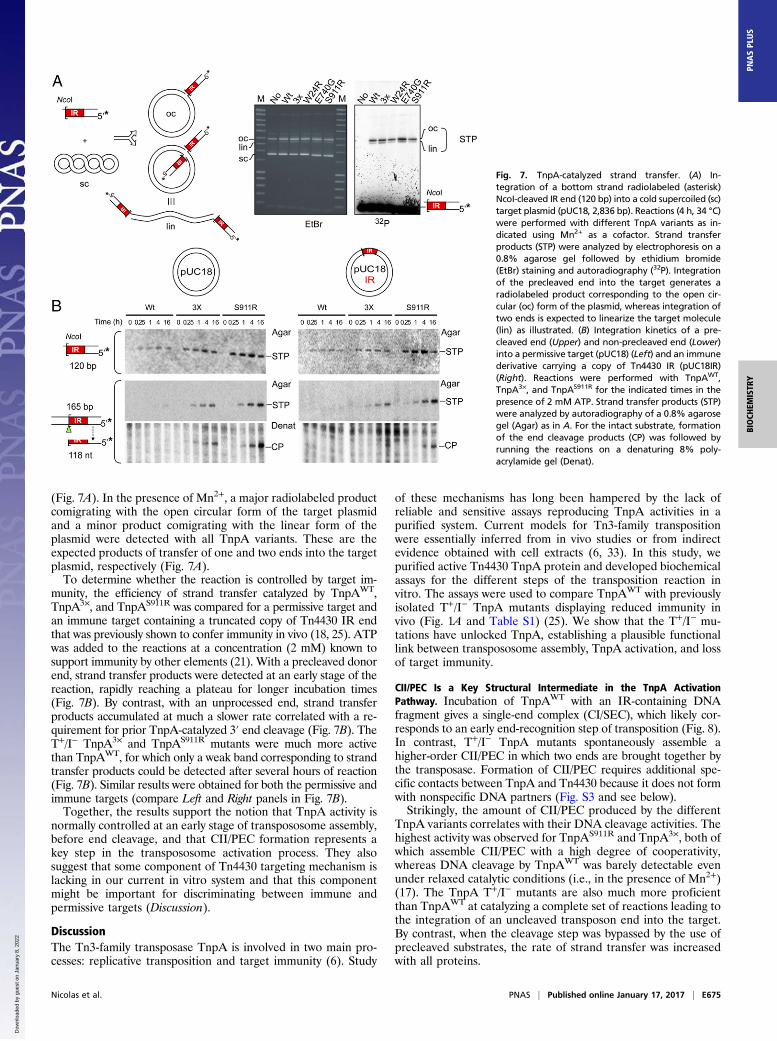
(Fig. 7A). In the presence of Mn2+, a major radiolabeled productcomigrating with the open circular form of the target plasmidand a minor product comigrating with the linear form of theplasmid were detected with all TnpA variants. These are theexpected products of transfer of one and two ends into the targetplasmid, respectively (Fig. 7A).To determine whether the reaction is controlled by target im-
munity, the efficiency of strand transfer catalyzed by TnpAWT,TnpA3×, and TnpAS911R was compared for a permissive target andan immune target containing a truncated copy of Tn4430 IR endthat was previously shown to confer immunity in vivo (18, 25). ATPwas added to the reactions at a concentration (2 mM) known tosupport immunity by other elements (21). With a precleaved donorend, strand transfer products were detected at an early stage of thereaction, rapidly reaching a plateau for longer incubation times(Fig. 7B). By contrast, with an unprocessed end, strand transferproducts accumulated at much a slower rate correlated with a re-quirement for prior TnpA-catalyzed 3′ end cleavage (Fig. 7B). TheT+/I− TnpA3× and TnpAS911R mutants were much more activethan TnpAWT, for which only a weak band corresponding to strandtransfer products could be detected after several hours of reaction(Fig. 7B). Similar results were obtained for both the permissive andimmune targets (compare Left and Right panels in Fig. 7B).Together, the results support the notion that TnpA activity is
normally controlled at an early stage of transpososome assembly,before end cleavage, and that CII/PEC formation represents akey step in the transpososome activation process. They alsosuggest that some component of Tn4430 targeting mechanism islacking in our current in vitro system and that this componentmight be important for discriminating between immune andpermissive targets (Discussion).
DiscussionThe Tn3-family transposase TnpA is involved in two main pro-cesses: replicative transposition and target immunity (6). Study
of these mechanisms has long been hampered by the lack ofreliable and sensitive assays reproducing TnpA activities in apurified system. Current models for Tn3-family transpositionwere essentially inferred from in vivo studies or from indirectevidence obtained with cell extracts (6, 33). In this study, wepurified active Tn4430 TnpA protein and developed biochemicalassays for the different steps of the transposition reaction invitro. The assays were used to compare TnpAWT with previouslyisolated T+/I− TnpA mutants displaying reduced immunity invivo (Fig. 1A and Table S1) (25). We show that the T+/I− mu-tations have unlocked TnpA, establishing a plausible functionallink between transpososome assembly, TnpA activation, and lossof target immunity.
CII/PEC Is a Key Structural Intermediate in the TnpA ActivationPathway. Incubation of TnpAWT with an IR-containing DNAfragment gives a single-end complex (CI/SEC), which likely cor-responds to an early end-recognition step of transposition (Fig. 8).In contrast, T+/I− TnpA mutants spontaneously assemble ahigher-order CII/PEC in which two ends are brought together bythe transposase. Formation of CII/PEC requires additional spe-cific contacts between TnpA and Tn4430 because it does not formwith nonspecific DNA partners (Fig. S3 and see below).Strikingly, the amount of CII/PEC produced by the different
TnpA variants correlates with their DNA cleavage activities. Thehighest activity was observed for TnpAS911R and TnpA3×, both ofwhich assemble CII/PEC with a high degree of cooperativity,whereas DNA cleavage by TnpAWT was barely detectable evenunder relaxed catalytic conditions (i.e., in the presence of Mn2+)(17). The TnpA T+/I− mutants are also much more proficientthan TnpAWT at catalyzing a complete set of reactions leading tothe integration of an uncleaved transposon end into the target.By contrast, when the cleavage step was bypassed by the use ofprecleaved substrates, the rate of strand transfer was increasedwith all proteins.
Fig. 7. TnpA-catalyzed strand transfer. (A) In-tegration of a bottom strand radiolabeled (asterisk)NcoI-cleaved IR end (120 bp) into a cold supercoiled (sc)target plasmid (pUC18, 2,836 bp). Reactions (4 h, 34 °C)were performed with different TnpA variants as in-dicated using Mn2+ as a cofactor. Strand transferproducts (STP) were analyzed by electrophoresis on a0.8% agarose gel followed by ethidium bromide(EtBr) staining and autoradiography (32P). Integrationof the precleaved end into the target generates aradiolabeled product corresponding to the open cir-cular (oc) form of the plasmid, whereas integration oftwo ends is expected to linearize the target molecule(lin) as illustrated. (B) Integration kinetics of a pre-cleaved end (Upper) and non-precleaved end (Lower)into a permissive target (pUC18) (Left) and an immunederivative carrying a copy of Tn4430 IR (pUC18IR)(Right). Reactions were performed with TnpAWT,TnpA3×, and TnpAS911R for the indicated times in thepresence of 2 mM ATP. Strand transfer products (STP)were analyzed by autoradiography of a 0.8% agarosegel (Agar) as in A. For the intact substrate, formationof the end cleavage products (CP) was followed byrunning the reactions on a denaturing 8% poly-acrylamide gel (Denat).
Nicolas et al. PNAS | Published online January 17, 2017 | E675
BIOCH
EMISTR
YPN
ASPL
US
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022

These results indicate that transposition is controlled at anearly stage of the process, before end cleavage and that CII/PECformation represents a limiting step in the pathway that normallyactivates TnpA. We propose that T+/I− mutations reduce theactivation barrier, enabling TnpA to bind two transposon ends ina cleavage-competent configuration. This conformational changeis also facilitated by 3′ end cleavage, which may stabilize thecomplex for subsequent steps of transposition.
Activation of TnpA Involves a Substantial Rearrangement of its DNA-Binding Interface. An intriguing and surprising finding is that CII/PEC formation engages a single TnpA monomer with two trans-poson ends. The structural transition between CI/SEC and CII/PEC involves a switch from a noncooperative mode of DNAbinding to a cooperative mode allowing TnpA to capture two endswith a higher affinity than a single end. Concomitant unlocking ofTnpA within CII/PEC also involves specific structural changes tocommit the protein to catalysis. Therefore, it is likely that transitionfrom CI/SEC to CII/PEC requires a substantial reorganization ofthe DNA-bound TnpA molecule. This requirement may explainwhy mutations in different TnpA domains all result in enhancedPEC formation, each domain contributing to the structural rear-rangement either directly or indirectly.DNA footprint analysis revealed that in both CI/SEC and CII/
PEC TnpA protects an inner IR region containing specific protein-recognition motifs (6). However, compared with CI/SEC, CII/PECexhibits extended contacts between TnpA and DNA adjacent tothe end cleavage site. This pattern supports a scenario in whichTnpA interacts primarily with the inner part of one IR (e.g.,through its putative N-terminal DNA binding domain) and thenexposes an additional binding interface (likely connected to the
C-terminal catalytic domain) to contact the cleavage site of asecond end (Fig. 8). A similar arrangement of the synaptic complexin which a transposase molecule primarily bound in cis to one endcleaves the opposite end in trans has been reported for othermembers of the DDE/D-recombinase superfamily and is viewedas a convergent mechanism to ensure concerted chemistry at bothtransposon ends (34–37). It remains to be determined whetherthis arrangement is also true for Tn4430 and other Tn3-familytransposons.DNA distortions in the 5′-GGGG-3′ motif at the IR tip, as
revealed by [(OP)2-Cu+] hypersensitive sites, may be required to
engage the scissile phosphate for catalysis. However, the pres-ence or absence of metal ion cofactors (Mn2+ or Mg2+) or theD751N catalytic mutation seem not to affect the relative amountof CI/SEC and CII/PEC observed with different TnpA variants(compare Fig. S7A with Fig. S7 D and E). This finding suggeststhat CII/PEC formation does not rely simply on a functionalconfiguration of the active site in which all the components re-quired to bind and activate the scissile phosphate are present;rather, additional contacts between the protein and DNA arelikely to be involved. These contacts may be important to distortDNA locally and to promote the rearrangement of the flankingsequences with respect to the transposon ends, as is required forthe activation of Mu and other transposable elements (38). Sup-porting this possibility is the finding that precleaving the Tn4430IR end enhances CII/PEC formation, even with TnpAWT. Thisenhancement was even more pronounced for the NcoI-cutIR (from which all flanking DNA has been resected) than forsubstrates containing a single nick at the 3′ IR end (Fig. 5 andFig. S7). Removing the flanking DNA or increasing DNA flexi-bility at the cleavage site thus may relieve conformational strainswithin the complex, stabilizing the active configuration of TnpA.The primary cleavage activity of TnpA is to introduce a nick at
the 3′ end of the IR, as expected from classical models for copy-inreplicative transposition (1, 2). However, TnpA also can catalyzespecific DSBs, a property that is normally associated with trans-posases that mediate nonreplicative cut-and-paste transposition.This activity was unveiled under conditions that stimulate cataly-sis (i.e., in the presence of Mn2+) and was most prominent for theT+/I− TnpA mutants. Although some hairpins may arise as sec-ondary products of the reaction, the data suggest that DSBs occurprimarily by a sequential mechanism in which TnpA first cleavesthe 3′ end and then repositions onto the complementary strand tocleave the 5′ end. This promiscuous activity further illustrates thestructural flexibility that allows TnpA to adopt different geome-tries of the active site with respect to the other protein domains.The repositioning of the TnpA active site is also reminiscent ofthat proposed for Tc1/mariner family transposons. For the latter,however, the transposase first cleaves the nontransferred strandsat the 5′ ends of the transposon before cleaving the transferredstrands at the 3′ ends (26, 28). The reversed order of cleavagedisplayed by TnpA thus provides an additional example of thefunctional versatility of DDE(D) enzymes.
An Asymmetric Pathway for Active Transpososome Assembly andPossible Molecular Models for Target Immunity. What promotesTnpA unlocking during normal transposition? Although it rep-resents an activated intermediate, the monomeric organizationof CII/PEC has never been reported for other studied elements,suggesting a unique pathway for transposition complex assembly.This structure also suggests that perhaps CII/PEC is only part ofthe assembly process and that formation of the fully activetranspososome has additional requirements. Consistent with thispossibility, strand transfer catalyzed by both wild-type and T+/I−
TnpAs was relatively weak (detected only in the presence ofMn2+) and essentially led to single-end integration products. Wepropose that assembly of the fully functional transpososome is
Fig. 8. An asymmetric model for Tn4430 transpososome assembly and acti-vation. TnpA (purple) binds Tn4430 IR end (red box) as a monomer giving aCI/SEC in which the protein is in an inactive, locked configuration (Left).Unlocking TnpA requires an activating conformational change that unmasks asecondary binding mode, enabling the transposase to connect both transposonends and become competent for DNA cleavage as observed in the CII/PEC(Center). This conformational transition is completely effective in the contextof a fully assembled transpososome comprising both transposon ends and anappropriate (permissive, green) target (Right). In the broadest interpretation ofthe model, formation of the active ternary complex requires specific and re-ciprocal interactions between end-bound and target-linked TnpA subunits. Onan immune (nonpermissive, pink) target, interactions involving the lockedconfiguration of TnpA are nonproductive (and may promote the dissociationof the target-bound TnpA subunits or prevent their association). By contrast,T+/I− mutations have a lower energy barrier for SEC to PEC transition, makingTnpA less discriminant between permissive and immune targets. TnpA is ar-bitrarily drawn with two globular domains to illustrate the conformationalswitch between the CI/SEC and CII/PEC configurations.
E676 | www.pnas.org/cgi/doi/10.1073/pnas.1611701114 Nicolas et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022

conditioned by the target, providing a checkpoint to discriminatebetween permissive and nonpermissive targets (Fig. 8).For other transposable elements, delivery of an appropriate
target to the transpososome is mediated by specific interactionsbetween the transposase and an element-encoded adaptor pro-tein, typically a member of the AAA+ ATPase family (20–23, 39,40). These proteins not only recruit and activate the target but alsoact as an allosteric effector by stimulating transposase activity andassisting conformational changes underlying subsequent trans-position steps. For Mu and Tn7, the target activators MuB andTnsC also provide the key for target immunity (14, 30). In bothcases, interaction between the target-bound activator and the end-bound transposase of a resident copy of the element (or an “im-mature” version of the transposition complex) stimulates ATPhydrolysis by the target activator, promoting its dissociation fromDNA and relocation to a remote site (21, 41–43).In Tn3-family members there is no transposon-encoded pro-
tein, aside from TnpA itself, that could play a role in target se-lection. TnpA also is the only possible determinant that canaccount for the self-recognition mechanism whereby each Tn3-family transposon expresses target immunity against itself butnot against other members of the family (25). The TnpA proteinthus carries all the specific information needed to identify whichtarget is permissive and which is not and to discriminate betweenboth kinds of target by catalyzing or not catalyzing transposition.Based on this dual role of TnpA and the monomeric structure ofCII/PEC, we propose an asymmetric model for transpososomeassembly in which concerted activation of the donor and thetarget depends on separate sets of TnpA interactions (Fig. 8).In the broadest version of the model, reciprocal interactions
between target- and IR-bound TnpA promote a conformationalchange that facilitates or stabilizes an unlocked protein configu-ration as observed in CII/PEC. Adopting this configuration would,in turn, be critical for functional multimerization of TnpA by in-corporating reciprocally activated target- and end-bound TnpAmolecules into the developing transpososome (Fig. 8). Note thatin a more restrictive interpretation of the model, the TnpA mol-ecule that interacts primarily with the target already may be boundto the transposon end before fully active transpososome assembly.In any case, we postulate that the conformational transition thatleads to productive complex formation (in which TnpA adoptsthe unlocked CII/PEC configuration) (Fig. 8) is normally a rate-limiting step of transposition. Interaction with end-bound TnpAmolecules in the locked configuration, as TnpAWT is in CI/SEC,would be nonproductive or would inactivate TnpA-tagged targets(e.g., by preventing their formation or promoting their dissocia-tion), thus making DNA regions already containing a copy of thetransposon refractory to further insertions. Therefore, the higherpropensity of T+/I− TnpA mutants to adopt the transposition-committed CII/PEC configuration displaces the reaction towardthe formation of the fully activated ternary complex, making thesemutants less discriminative between permissive and immune tar-gets (Fig. 8).A conceptually equivalent mechanism for coordinating trans-
posase activation with target capture has been proposed for Tn7and Mu (14, 30). For Tn7, assembly of the heteromeric trans-posase TnsAB requires the formation of a pretranspositioncomplex in which the TnsB subunits derive from the donorsynaptic complex and the TnsA subunits are delivered from apreassembled target complex also containing the target activatorTnsC (44). Assembly of the pretransposition complex is a pre-requisite for transposition, ensuring that all appropriate partnersare brought together before triggering the reaction (30, 45). ForMu, the target activator MuB is not a subunit of the MuAtransposase per se, but its interaction with MuA stimulates everystep of transposition (14). The exact mechanism whereby re-ciprocal MuA–MuB interactions switch from a dissuasive to aproductive outcome has yet to be clarified but is thought to
depend on conformational transitions during transpososomeprogression, as we propose here for Tn4430 (14, 40). Again, theability of a single polypeptide, TnpA, to mediate different rolesduring transposition likely reflects the overall size of the protein(which is more than twice that of other DDE/D transposases)and its organization into multiple domains, some of which haveno assigned functions (Fig. 1) (12, 29).If TnpA is a specific target “matchmaker,” what target feature is
recognized by the protein to mediate efficient transposition? ForMu, the target adaptor MuB binds DNA with relatively lowspecificity, preferring A/T-rich DNA stretches (23, 24, 40). Incontrast, TnsC is addressed to specific sites by the Tn7-encodedtarget selector proteins TnsD or TnsE. TnsD promotes assemblyof the target complex at a specific chromosomal site, attTn7,whereas TnsE directs transposition to DNA regions undergoingactive lagging-strand DNA synthesis (30, 46). Although Tn4430inserts in a broad variety of DNA sequences in vivo, the datasuggest that integration depends on specific host factors or cellularprocesses that locally affect DNA, directing transposition to spe-cific DNA regions (6, 18). Identifying this factor (or factors) thusconstitutes the next important step to validate our current modelfor Tn3-family transposition and target immunity mechanisms.
Materials and MethodsDNA Substrates. The Escherichia coli TOP10 strain and the plasmids used inthis study are listed in Table S2. IR substrates were generated by PCR am-plification or by annealing specific oligonucleotides as detailed in Table S3.Precleaved IR substrates were obtained by cleaving linear duplexes with NcoIunless otherwise specified. Substrates containing a nick at the 3′ or 5′ end ofthe IR were assembled by annealing three oligonucleotides as specified inTable S3. 32P 5′-labeling of PCR primers and oligonucleotides was performedwith [γ-32P]-ATP (Perkin-Elmer) and T4 polynucleotide kinase (New EnglandBiolabs). Fluorescently labeled substrates were PCR amplified using 5′ Cy5-labeled and 5′ Cy3-labeled primers (Eurogentec) (Table S3). DNA substrateswere separated on 8% polyacrylamide gels in Tris/borate/EDTA (TBE) buffer(88 mM Tris base, 88 mM boric acid, and 2 mM EDTA) and were eluted in TESbuffer [10 mM Tris HCl (pH 8.0), 1 mM EDTA, 300 mM NaCl]. DNA wasethanol precipitated and resuspended in TE buffer [10 mM Tris HCl (pH 8.0),1 mM EDTA].
TnpA Expression and Purification. Wild-type and T+/I− TnpAs were fused to acMyc-His6 epitope tag at their C termini and were expressed in E. coli TOP10cells under the control of the pAra promoter (Table S1). TnpAWT::YFP andTnpA3×::YFP are C-terminal fusions between the TnpAWT and TnpA3×
transposases and a cMyc-His6–tagged version of Venus YFP (Table S1) (47).TnpA derivatives were purified using the thermal shift-induction procedureto increase the yield of soluble proteins followed by nickel column chroma-tography (Fig. S1) (25). After purification, proteins were dialyzed against theno-buffer condition [50 mM Tris·HCl (pH 8.0), 500 mM NaCl, 20% (vol/vol)glycerol], typically giving ∼700-nM fractions.
EMSA. Binding reactions were performed in a final volume of 20 μL containingTnpA (20–150 nM), 5 mg/mL BSA, 10% (vol/vol) DMSO, 50mM Tris·HCl (pH 8.0),1 mM DTT, 5 mM EDTA (pH 8.0), 50 mM NaCl, and 1 μL of radiolabeled(100 cps/μL) or fluorescently labeled DNA substrates. For the supershift assay,0.2 and 1 μg of anti-cMyc antibody (Invitrogen) were added to the bindingreaction, and DTT was omitted. Reactions were incubated at 34 °C for 30 minand were run on 4% (wt/vol) polyacrylamide gels in TBE buffer at 4 °C. Gelswere scanned with an Ettan DIGE Imager (GE-Healthcare) for fluorescencedetection or with a Pharos FX scanner (Bio-Rad) for 32P detection.
DNA Footprinting Analysis. TnpAWT, TnpAE740G, and TnpA3× were incubatedwith a 165-bp IR fragment labeled with 32P on the 5′ end of the top orbottom strand, and the resulting complexes were separated on a 4% (wt/vol)polyacrylamide gel using standard EMSA conditions. In-gel footprinting withthe [(OP)2-Cu
+] ion was performed as described (48). Cleavage productsobtained for CI/SEC, CII/PEC, and the unbound substrate were eluted fromthe gel and run on a denaturing 8% (wt/vol) polyacrylamide gel alongsideMaxam–Gilbert AG/CT sequencing reactions (49).
Cleavage Assay. Reaction mixtures (30 μL) contained 10% (vol/vol) DMSO,50 mM Tris·HCl (pH 8.0), 5 mM DTT, 5 mg/mL BSA, 50 mM NaCl, 1 μL of
Nicolas et al. PNAS | Published online January 17, 2017 | E677
BIOCH
EMISTR
YPN
ASPL
US
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022

end-labeled DNA substrates (100 cps/μL), and 5 mM MgCl2 or MnCl2. After in-cubation at 34 °C, reactions were stopped by proteinase K treatment(600 μg/mL) (Sigma) in 0.5% SDS, 12.5 mM EDTA (pH 8.0) followed by phe-nol-chloroform extraction and ethanol precipitation. For the detection ofTnpA-mediated DSBs, reaction products were resuspended in TE buffer andseparated on a nondenaturing 7.5% polyacrylamide, 0.1% SDS gel in TBEbuffer. For mapping single-strand nicks, cleavage products (5 μL) weredenatured by adding 5 μL of formamide loading buffer [80% (vol/vol)formamide, 1 mM EDTA (pH 8.0), 10 mM NaOH, bromophenol blue] and a10-min treatment at 95 °C. Samples then were run on denaturing 6% or8% (wt/vol) polyacrylamide, 25% (vol/vol) formamide gels in TBE bufferalongside Maxam–Gilbert AG/CT sequencing reactions. NcoI digestionproducts of the substrate were used as a size marker.
Integration Assay.DNA strand transfer reactions (30 μL) contained 10% (vol/vol)DMSO, 50 mM Tris·HCl (pH 8.0), 1 mM DTT, 10 mg/mL BSA, 10% (wt/vol) PEG-6000, 1.5 μg of pUC18 or pUC18IR target plasmid (Table S1), 1 μL of radiola-beled IR substrate (100 cps/μL), and 5 mM MgCl2 or MnCl2. For testing targetimmunity, reactions also were supplemented with 2 mM ATP. Protein con-
centrations were standardized according to their DNA-binding activity.Reactions were incubated at 34 °C and were stopped by proteinase Ktreatment followed by phenol-chloroform extraction and ethanol pre-cipitation. Products were analyzed by electrophoresis on 0.8% agarose gels in TAEbuffer [40 mM Tris-acetate, 1 mM EDTA (pH 7.8)]. The gels were stained withethidium bromide and then dried for autoradiography or phosphorimaging by aPharos FX scanner (Bio-Rad).
ACKNOWLEDGMENTS. We thank Bao Ton-Hoang, Guy Duval-Valentin, andPhilippe Rousseau from the Laboratoire de Microbiologie et GénétiqueMoléculaires and all the members of the Institut des Sciences de la Vie atUniversité Catholique de Louvain (UCL) for fruitful discussions and adviceand Marie Deghorain and Patrice Soumillion for critical reading of the manu-script. This work was supported by the Fonds Spéciaux de la Recherche (FSR) atUCL and by Fonds National de la Recherche Scientifique (FRS-FNRS) Grants2.5496.04, 1.9226.04, and 1.5.225.10. M.L. and C.A.O. held fellowships fromthe Fonds de la Recherche dans l’Industrie et l’Agriculture. E.N. was a researchassistant at the FNRS. E.N. and B.F.J.H. were supported by a short-term fellow-ship from the European Molecular Biology Organization (EMBO) and an FNRStravelling grant, respectively, for 3-mo research stays in the laboratory of M.C.
1. Curcio MJ, Derbyshire KM (2003) The outs and ins of transposition: From Mu tokangaroo. Nat Rev Mol Cell Biol 4(11):865–877.
2. Hickman AB, Dyda F (2016) DNA transposition at work. Chem Rev 116:12758–12784.3. Aziz RK, Breitbart M, Edwards RA (2010) Transposases are the most abundant, most
ubiquitous genes in nature. Nucleic Acids Res 38(13):4207–4217.4. Craig NL, et al. (2015) Mobile DNA III (ASM, Washington, DC).5. Mahillon J, Lereclus D (1988) Structural and functional analysis of Tn4430: Identifi-
cation of an integrase-like protein involved in the co-integrate-resolution process.EMBO J 7(5):1515–1526.
6. Nicolas E, et al. (2015) The Tn3-family of replicative transposons.Microbiol Spectr 3(4):MDNA3-0060-2014.
7. Mindlin S, Petrova M (2013) Mercury resistance transposons. Bacterial IntegrativeGenetic Elements, eds Roberts AP, Mullany P (Landes Bioscience, Austin, TX), pp33–52.
8. Partridge SR (2011) Analysis of antibiotic resistance regions in Gram-negative bacte-ria. FEMS Microbiol Rev 35(5):820–855.
9. Cuzon G, Naas T, Nordmann P (2011) Functional characterization of Tn4401, a Tn3-basedtransposon involved in blaKPC genemobilization. Antimicrob Agents Chemother 55(11):5370–5373.
10. Chen L, et al. (2014) Carbapenemase-producing Klebsiella pneumoniae: Molecularand genetic decoding. Trends Microbiol 22(12):686–696.
11. He S, et al. (2015) Insertion sequence IS26 reorganizes plasmids in clinically isolatedmultidrug-resistant bacteria by replicative transposition. MBio 6(3):e00762.
12. Mizuuchi K (1992) Transpositional recombination: Mechanistic insights from studiesof Mu and other elements. Annu Rev Biochem 61:1011–1051.
13. Chaconas G, Harshey RM (2002) Transposition of Phage Mu DNA. Mobile DNA II, edsCraig NL, Craigie R, Gellert M, Lambowitz AM (ASM, Washington, DC), pp 384–402.
14. Harshey RM (2014) Transposable phage Mu. Microbiol Spectr 2(5):MDNA3-0007-2014.15. Montaño SP, Rice PA (2011) Moving DNA around: DNA transposition and retroviral
integration. Curr Opin Struct Biol 21(3):370–378.16. Nowotny M (2009) Retroviral integrase superfamily: The structural perspective. EMBO
Rep 10(2):144–151.17. Yang W, Lee JY, Nowotny M (2006) Making and breaking nucleic acids: Two-Mg2+-ion
catalysis and substrate specificity. Mol Cell 22(1):5–13.18. Nicolas E, Lambin M, Hallet B (2010) Target immunity of the Tn3-family transposon
Tn4430 requires specific interactions between the transposase and the terminal in-verted repeats of the transposon. J Bacteriol 192(16):4233–4238.
19. Craig NL (1997) Target site selection in transposition. Annu Rev Biochem 66:437–474.20. Adzuma K, Mizuuchi K (1988) Target immunity of Mu transposition reflects a dif-
ferential distribution of Mu B protein. Cell 53(2):257–266.21. Stellwagen AE, Craig NL (1997) Avoiding self: Two Tn7-encoded proteins mediate
target immunity in Tn7 transposition. EMBO J 16(22):6823–6834.22. Stellwagen AE, Craig NL (1998) Mobile DNA elements: Controlling transposition with
ATP-dependent molecular switches. Trends Biochem Sci 23(12):486–490.23. Ge J, Lou Z, Cui H, Shang L, Harshey RM (2011) Analysis of phage Mu DNA trans-
position by whole-genome Escherichia coli tiling arrays reveals a complex relationshipto distribution of target selection protein B, transcription and chromosome archi-tectural elements. J Biosci 36(4):587–601.
24. Mizuno N, et al. (2013) MuB is an AAA+ ATPase that forms helical filaments to controltarget selection for DNA transposition. Proc Natl Acad Sci USA 110(27):E2441–E2450.
25. Lambin M, et al. (2012) Separate structural and functional domains of Tn4430transposase contribute to target immunity. Mol Microbiol 83(4):805–820.
26. Dawson A, Finnegan DJ (2003) Excision of the Drosophila mariner transposon Mos1.Comparison with bacterial transposition and V(D)J recombination. Mol Cell 11(1):225–235.
27. Feng X, Colloms SD (2007) In vitro transposition of ISY100, a bacterial insertion se-quence belonging to the Tc1/mariner family. Mol Microbiol 65(6):1432–1443.
28. Tellier M, Bouuaert CC, Chalmers R (2015) Mariner and the ITm superfamily oftransposons. Microbiol Spectr 3(2):MDNA3-0033-2014.
29. Sarnovsky RJ, May EW, Craig NL (1996) The Tn7 transposase is a heteromeric complexin which DNA breakage and joining activities are distributed between different geneproducts. EMBO J 15(22):6348–6361.
30. Peters JE (2014) Tn7. Microbiol Spectr 2(5): MDNA3-0010-2014.31. Kennedy AK, Haniford DB, Mizuuchi K (2000) Single active site catalysis of the suc-
cessive phosphoryl transfer steps by DNA transposases: Insights from phosphor-othioate stereoselectivity. Cell 101(3):295–305.
32. Zhou L, et al. (2004) Transposition of hAT elements links transposable elements andV(D)J recombination. Nature 432(7020):995–1001.
33. Maekawa T, Yanagihara K, Ohtsubo E (1996) A cell-free system of Tn3 transpositionand transposition immunity. Genes Cells 1(11):1007–1016.
34. Davies DR, Goryshin IY, Reznikoff WS, Rayment I (2000) Three-dimensional structureof the Tn5 synaptic complex transposition intermediate. Science 289(5476):77–85.
35. Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD (2009) Molecular archi-tecture of the Mos1 paired-end complex: The structural basis of DNA transposition ina eukaryote. Cell 138(6):1096–1108.
36. Maertens GN, Hare S, Cherepanov P (2010) The mechanism of retroviral integrationfrom X-ray structures of its key intermediates. Nature 468(7321):326–329.
37. Montaño SP, Pigli YZ, Rice PA (2012) The μ transpososome structure sheds light onDDE recombinase evolution. Nature 491(7424):413–417.
38. Yanagihara K, Mizuuchi K (2003) Progressive structural transitions within Mu trans-positional complexes. Mol Cell 11(1):215–224.
39. Arias-Palomo E, Berger JM (2015) An atypical AAA+ atpase assembly controls efficienttransposition through DNA remodeling and transposase recruitment. Cell 162(4):860–871.
40. Greene EC, Mizuuchi K (2004) Visualizing the assembly and disassembly mechanismsof the MuB transposition targeting complex. J Biol Chem 279(16):16736–16743.
41. Skelding Z, Queen-Baker J, Craig NL (2003) Alternative interactions between the Tn7transposase and the Tn7 target DNA binding protein regulate target immunity andtransposition. EMBO J 22(21):5904–5917.
42. Greene EC, Mizuuchi K (2002) Target immunity during Mu DNA transposition.Transpososome assembly and DNA looping enhance MuA-mediated disassembly ofthe MuB target complex. Mol Cell 10(6):1367–1378.
43. Han YW, Mizuuchi K (2010) Phage Mu transposition immunity: Protein pattern for-mation along DNA by a diffusion-ratchet mechanism. Mol Cell 39(1):48–58.
44. Skelding Z, Sarnovsky R, Craig NL (2002) Formation of a nucleoprotein complexcontaining Tn7 and its target DNA regulates transposition initiation. EMBO J 21(13):3494–3504.
45. Bainton RJ, Kubo KM, Feng JN, Craig NL (1993) Tn7 transposition: Target DNA rec-ognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system.Cell 72(6):931–943.
46. Parks AR, et al. (2009) Transposition into replicating DNA occurs through interactionwith the processivity factor. Cell 138(4):685–695.
47. Nagai T, et al. (2002) A variant of yellow fluorescent protein with fast and efficientmaturation for cell-biological applications. Nat Biotechnol 20(1):87–90.
48. Kuwabara MD, Sigman DS (1987) Footprinting DNA-protein complexes in situ fol-lowing gel retardation assays using 1,10-phenanthroline-copper ion: Escherichia coliRNA polymerase-lac promoter complexes. Biochemistry 26(23):7234–7238.
49. Maxam AM, Gilbert W (1980) Sequencing end-labeled DNA with base-specificchemical cleavages. Methods Enzymol 65(1):499–560.
E678 | www.pnas.org/cgi/doi/10.1073/pnas.1611701114 Nicolas et al.
Dow
nloa
ded
by g
uest
on
Janu
ary
8, 2
022





























