Embed Size (px)
Citation preview

Brief UltraRapid Communication
Viral Gene Transfer Rescues Arrhythmogenic Phenotypeand Ultrastructural Abnormalities in Adult
Calsequestrin-Null Mice With Inherited ArrhythmiasMarco Denegri*, Jose Everardo Avelino-Cruz*, Simona Boncompagni, Stefano Andrea De Simone,
Alberto Auricchio, Laura Villani, Pompeo Volpe, Feliciano Protasi, Carlo Napolitano, Silvia Giuliana Priori
Rationale: Catecholaminergic polymorphic ventricular tachycardia is an inherited disease that predisposes tocardiac arrest and sudden death. The disease is associated with mutations in the genes encoding for the cardiacryanodine receptor (RyR2) and cardiac calsequestrin (CASQ2). CASQ2 mutations lead to a major loss of CASQ2monomers, possibly because of enhanced degradation of the mutant protein. The decrease of CASQ2 is associatedwith a reduction in the levels of Triadin (TrD) and Junctin (JnC), two proteins that form, with CASQ2 and RyR2,a macromolecular complex devoted to control of calcium release from the sarcoplasmic reticulum.
Objective: We intended to evaluate whether viral gene transfer of wild-type CASQ2 may rescue the broadspectrum of abnormalities caused by mutant CASQ2.
Methods and Results: We used an adeno-associated serotype 9 viral vector to express a green fluorescentprotein-tagged CASQ2 construct. Twenty weeks after intraperitoneal injection of the vector in neonate CASQ2KO mice, we observed normalization of the levels of calsequestrin, triadin, and junctin, rescue of electrophys-iological and ultrastructural abnormalities caused by CASQ2 ablation, and lack of life-threatening arrhythmias.
Conclusions: We have proven the concept that induction of CASQ2 expression in knockout mice reverts themolecular, structural, and electric abnormalities and prevents life-threatening arrhythmias in CASQ2-defectivecatecholaminergic polymorphic ventricular tachycardia mice. These data support the view that development ofCASQ2 viral gene transfer could have clinical application. (Circ Res. 2012;110:663-668.)
Key Words: arrhythmias � gene therapy � genetics � sudden death
Catecholaminergic polymorphic ventricular tachycardia(CPVT) is an inherited disease that predisposes to cardiac
arrest and to sudden death.1,2 In 2001, we showed that dominantCPVT is caused by mutations in the gene encoding for thecardiac ryanodine receptor (RyR2).3 Soon after, Lahat et al4
identified mutations in the gene encoding for the cardiac calse-questrin (CASQ2) in families affected by recessive CPVT.
In This Issue, see p 651CASQ2 acts as a calcium buffer inside the sarcoplasmic
reticulum and plays a critical role in the physiology of
calcium release. Data from knock-in mice5,6 showed thateven a single amino acid replacement leads to a major lossof CASQ2 monomers, possibly because of enhanced deg-radation of the mutant protein.6 CASQ2 decrease leads toa reduction in the levels of triadin (TrD) and junctin (JnC),two proteins that form, with CASQ2 and RyR2, a macro-molecular complex devoted to control calcium releasefrom SR.
Here, we test the hypothesis that delivery of exogenousCASQ2 by viral gene transfer to CASQ2 knockout
Original received December 28, 2011; revision received January 18, 2012; accepted January 23, 2012. In December 2011, the average time fromsubmission to first decision for all original research papers submitted to Circulation Research was 14.29 days.
Brief UltraRapid Communications (BURCs) are designed to be a format for manuscripts that are of outstanding interest to the readership, reportdefinitive observations, but have a relatively narrow scope. Less comprehensive than Regular Articles but still scientifically rigorous, BURCs presentseminal findings that have the potential to open up new avenues of research. A decision on BURCs is rendered within 7 days of submission.
From the Molecular Cardiology (M.D., J.E., S.A.D.S., C.N., S.G.P.), IRCCS Fondazione Salvatore Maugeri, Pavia, Italy; Cardiovascular GeneticsProgram (C.N., S.G.P.), The Leon H. Charney Division of Cardiology, New York University School of Medicine, New York, NY; Department ofMolecular Medicine (S.G.P.), University of Pavia, Pavia, Italy; CeSI-Center for Research on Ageing & DNI-Department of Neuroscience and Imaging(S.B., F.P.), University G. d’Annunzio, Chieti, Italy; Telethon Institute of Genetics and Medicine (A.A.), Naples, Italy; Medical Genetics (A.A.),Department of Pediatrics, “Federico II” University, Naples, Italy; Pathology Division (L.V.), IRCCS Fondazione Salvatore Maugeri, Pavia, Italy;Department of Experimental Biomedical Sciences (P.V.), University of Padova, Italy.
This manuscript was sent to Gordon Tomaselli, Consulting Editor, for review by expert referees, editorial decision, and final disposition.*These authors contributed equally to this work.The online-only Data Supplement is available with this article at http://circres.ahajournals.org/content/suppl/doi:10.1161/CIRCRESAHA.111.
263939/-/DC1.Correspondence to Silvia G. Priori, MD, PhD, Division of Cardiology and Molecular Cardiology, Maugeri Foundation–University of Pavia, Via
Maugeri 10/10A, 27100 Pavia, Italy. E-mail [email protected]© 2012 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.111.263939
663 by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from

(CASQ2KO) mice is able to restore levels of CASQ2, TrD,and JnC, to revert electrophysiological and morphologicalabnormalities caused by CASQ2 ablation, and to preventlife-threatening arrhythmias. If confirmed, then our hypothe-sis will prove the concept that viral gene transfer may haveclinical application in recessive CPVT.
MethodsAn expanded Methods section is provided in the Online Supplement.
Viral ConstructWe generated an adeno-associated serotype-9 vector containing thecDNA of the murine CASQ2 gene (Online Figure IA). The cDNAwas cloned into a bicistronic (pIRES) eukaryotic expression vectorand subcloned into the multiple cloning site of pAAV2.1-CMV-eGFP containing the cytomegalovirus promoter and the greenfluorescent protein (eGFP) as reporter gene.
Reverse-Transcription Polymerase Chain ReactionTotal RNA was extracted and purified from myocytes, liver, lung,skeletal muscle, testis, and ovary of wild-type (WT) mice and ofCASQ2 KO mice infected with the AAV9-CASQ2 construct taggedwith eGFP (CASQ2INF-KO). Reverse-transcription polymerase chainreaction was performed using tissue-specific RNA (iScript cDNA
Synthesis kit; Bio-Rad Laboratories) and samples were analyzed onagarose gel.
Immunoblotting was performed as previously described6 using thefollowing antibodies: anti-CASQ2 (PA1-913; ABR), anti-Triadin(sc-33393; Santa Cruz), anti-junctin (kindly provided by Dr B.C.Knollmann), and anti-actin (sc-1616-R; Santa Cruz) as referenceprotein. Confocal microscopy was used for indirect immunofluores-cent labeling of cardiac myocytes from WT, CASQ2KO, andCASQ2INF-KO mice was performed with primary antibodies anti-RyR2, anti-CASQ2, anti-Junctin, or anti-Triadin, as previouslydescribed.6 Secondary antibodies were Alexa Fluor 405 goat anti-mouse and Alexa Fluor 546 goat anti-rabbit IgG. Images wereobtained in a Leica TCS-SP5 II microscope.
Electron MicroscopyFixed hearts were embedded in an epoxy resin and ultrathin sectionswere cut, stained, and analyzed as previously described.6
In Vivo and In Vitro ElectrophysiologyVentricular myocytes were enzymatically dissociated using aorticretrograde perfusion. Action potentials were recorded with patch-clamp pipettes in current-clamp configuration and analyzed withpCLAMP 9.2 (Axon Instruments). ECG was recorded using subcu-taneous telemetric recorders (DSI).
StatisticsStatistical analysis was performed using SPSS version 18. Data arereported as mean�SEM. Continuous variables were analyzed byunpaired t test or analysis of variance using Bonferroni as a post hoctest, as appropriate. Categorical variables were analyzed by contin-gency tables with Fisher exact test. P�0.05 was consideredstatistically significant.
ResultsMice were infected at birth by intraperitoneal injection ofAAV9-CASQ2 and studied at 20 weeks of age. Cardiacmyocytes isolated from CASQ2INF-KO mice showed that 50%to 60% of cells expressed the transgene as indicated by eGFPfluorescence (Figure 1C). Infection was not associated withhistological abnormalities of the heart (Online Figure IB).
Non-standard Abbreviations and Acronyms
bpm beats per minute
CASQ2INF-KO CASQ2 knockout mouse infected with the AAV9-CASQ2-GFP construct
CASQ2KO CASQ2 knockout mouse
CASQ2WT CASQ2 wild-type mouse
jSR junctional sarcoplasmic reticulum
KO knockout
SR sarcoplasmic reticulum
Figure 1. Analysis of calciumrelease-unit proteins expres-sion and efficiency of adenoassociated virus infection.Western blots (A) and quantifi-cation of protein expression(B). Protein homogenates wereobtained from isolated cardio-myocytes. Re-expression ofexogenous CASQ2 and resto-ration of endogenous TrD(gray) and JnC (white) areshown. CASQ2 knockout (KO)and infected CASQ2 knockout(INF) proteins are normalized toactin and to their respectivelevels of CASQ2, TrD or JnC inCASQ2 wild-type (WT) mice(red bar), n�3 mice/group. C,evaluation of the infection effi-ciency of CASQ2INF-KO (INF):phase contrast (PhC) andgreen fluorescent protein (GFP)signal (GFP) of CASQ2INF-KO
myocytes. D, Real Time poly-merase chain reaction quantification of expression of CASQ2 (black), TrD (gray) and JnC (white) in myocytes isolated from CASQ2KO
(KO), CASQ2INF-KO (INF) and CASQ2WT (WT) mice. Data are normalized to GAPDH and to WT (red bar) CASQ2, Triadin or Junctin levels(n�3 mice/group).
664 Circulation Research March 2, 2012
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from

Evaluation of different organs showed the absence of eGFP inlung, ovary, and testis, whereas a weak positive signal wasdetected in liver and skeletal muscle (Online Figures IIA,IIB). Expression analysis of CASQ2, TrD and JnC wasperformed by real-time polymerase chain reaction (Figure1D) and Western blot (Figure 1A, 1B) in CASQ2WT (WT),CASQ2KO (KO), and CASQ2INF-KO mice; on infection,expressions of the three proteins were restored to levelscomparable to those of WT mice.
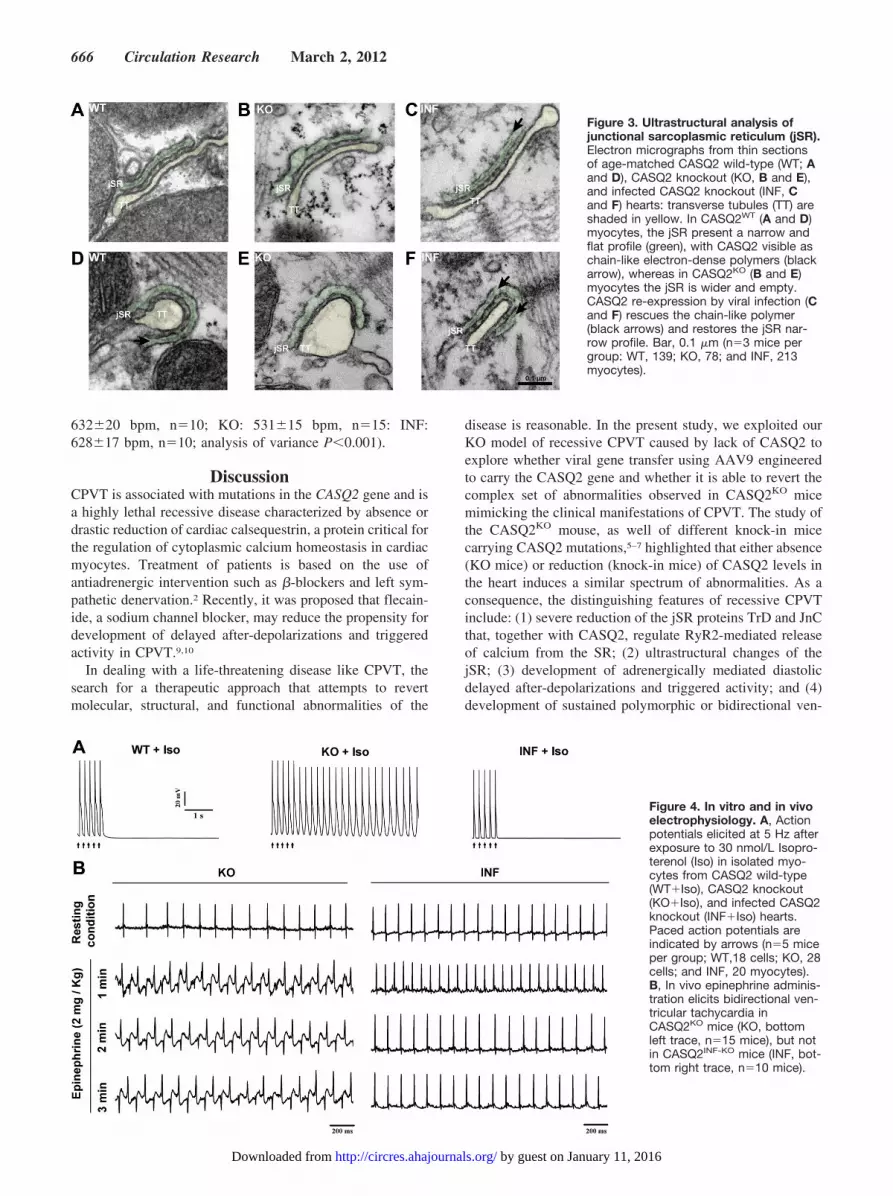
Immunofluorescence studies showed that exogenousCASQ2, TrD, and JnC were properly localized in myocytesderived from CASQ2INF-KO mice (Figure 2C, Online FigureIII); confocal microscopy showed that virally inducedCASQ2 colocalized with endogenous RyR2 as occurs in WTmice (Figure 2). By electron microscopy, we investigatedwhether ultrastructural abnormalities of calcium release unitsin CASQ2KO mice were also reverted by CASQ2 genetransfer (Figure 3, Online Figure IV). Calcium-release unitsare formed by the close apposition of T tubules to junctionalsarcoplasmic reticulum (jSR). In electron microscopy images,CASQ2 appears as electrodense chain-like polymers that holdtogether the opposite sides of the jSR membranes, conferringthem the typical narrow and flat profile seen in CASQ2WT
mice (Figure 3A, 3D). It has been reported that absence ofCASQ2 in CASQ2KO myocytes induces disappearance of thechain-like polymers and widening of the jSR lumen.7 Ourexperiments confirm those findings; the width of jSR lumenincreased (Figure 3D, 3E) from 26�0.3 nm to 37�1.2 nm inCASQ2WT and CASQ2KO myocytes (P�0.001), respectively,and returned to normal size in CASQ2INF-KO myocytes(21�0.3 nm; analysis of variance P�0.001; Figure 3C, 3F).This observation is important because it shows that toxicityinduced by CASQ2 overexpression in CASQ2WT mice andthe peculiar jSR swelling do not develop when CASQ2overexpression is performed on the null background.
Action potential recording was performed in CASQ2KO
and in CASQ2INF-KO myocytes to test susceptibility to devel-opment of delayed after-depolarizations and triggered activityduring �-adrenergic stimulation. A remarkable reduction ofisoproterenol (30 nmol/L) induced triggered action potentialswas observed in CASQ2INF-KO (Figure 4A) as compared withCASQ2KO (from 70% in KO to 5% in CASQ2INF-KO myo-cytes; P�0.001) or CASQ2empty-vector and GFP-negativeCASQ2INF-KO myocytes (Online Figures V, VI).
Arrhythmias susceptibility was investigated in vivo in miceinstrumented with ECG telemetry. A striking reduction ofventricular tachycardia during epinephrine (2 mg/kg) stimu-lation was observed. Ventricular tachycardia occurrence de-creased from 15/15 in CASQ2KO mice to 1/10 amongCASQ2INF-KO mice (P�0.001; Figure 4B). Interestingly,CASQ2INF-KO mice also showed normalization of heart ratethat increased to values similar to those of WT mice (WT:
Figure 2. Subcellular localization of adeno associated virus9-CASQ2. Immunolabeling of CASQ2 and RyR2 in CASQ2WT
(A), CASQ2KO (B), and CASQ2INF-KO (C) myocytes. Mergedimages are shown at low and high magnifications. Colocaliza-tion of CASQ2 with RyR2 is present in CASQ2 wild-type (WT)heart, whereas in CASQ2 knockout (CASQ2KO; KO) hearts onlyRyR2 is visualized; in infected CASQ2 knockout (INF) myocytes,
colocalization of CASQ2 and RyR2 is restored. On the bottomleft side of each panel, graphs show the distribution of the fluo-rescent signals (blue, RyR2; red, CASQ2) along the white line(white arrows). Scale bars: 15 �m. Images are representative ofthree independent experiments for each group.
Denegri et al Gene Therapy for CPVT 665
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from
632�20 bpm, n�10; KO: 531�15 bpm, n�15: INF:628�17 bpm, n�10; analysis of variance P�0.001).
DiscussionCPVT is associated with mutations in the CASQ2 gene and isa highly lethal recessive disease characterized by absence ordrastic reduction of cardiac calsequestrin, a protein critical forthe regulation of cytoplasmic calcium homeostasis in cardiacmyocytes. Treatment of patients is based on the use ofantiadrenergic intervention such as �-blockers and left sym-pathetic denervation.2 Recently, it was proposed that flecain-ide, a sodium channel blocker, may reduce the propensity fordevelopment of delayed after-depolarizations and triggeredactivity in CPVT.9,10
In dealing with a life-threatening disease like CPVT, thesearch for a therapeutic approach that attempts to revertmolecular, structural, and functional abnormalities of the
disease is reasonable. In the present study, we exploited ourKO model of recessive CPVT caused by lack of CASQ2 toexplore whether viral gene transfer using AAV9 engineeredto carry the CASQ2 gene and whether it is able to revert thecomplex set of abnormalities observed in CASQ2KO micemimicking the clinical manifestations of CPVT. The study ofthe CASQ2KO mouse, as well of different knock-in micecarrying CASQ2 mutations,5–7 highlighted that either absence(KO mice) or reduction (knock-in mice) of CASQ2 levels inthe heart induces a similar spectrum of abnormalities. As aconsequence, the distinguishing features of recessive CPVTinclude: (1) severe reduction of the jSR proteins TrD and JnCthat, together with CASQ2, regulate RyR2-mediated releaseof calcium from the SR; (2) ultrastructural changes of thejSR; (3) development of adrenergically mediated diastolicdelayed after-depolarizations and triggered activity; and (4)development of sustained polymorphic or bidirectional ven-
Figure 3. Ultrastructural analysis ofjunctional sarcoplasmic reticulum (jSR).Electron micrographs from thin sectionsof age-matched CASQ2 wild-type (WT; Aand D), CASQ2 knockout (KO, B and E),and infected CASQ2 knockout (INF, Cand F) hearts: transverse tubules (TT) areshaded in yellow. In CASQ2WT (A and D)myocytes, the jSR present a narrow andflat profile (green), with CASQ2 visible aschain-like electron-dense polymers (blackarrow), whereas in CASQ2KO (B and E)myocytes the jSR is wider and empty.CASQ2 re-expression by viral infection (Cand F) rescues the chain-like polymer(black arrows) and restores the jSR nar-row profile. Bar, 0.1 �m (n�3 mice pergroup: WT, 139; KO, 78; and INF, 213myocytes).
Figure 4. In vitro and in vivoelectrophysiology. A, Actionpotentials elicited at 5 Hz afterexposure to 30 nmol/L Isopro-terenol (Iso) in isolated myo-cytes from CASQ2 wild-type(WT�Iso), CASQ2 knockout(KO�Iso), and infected CASQ2knockout (INF�Iso) hearts.Paced action potentials areindicated by arrows (n�5 miceper group; WT,18 cells; KO, 28cells; and INF, 20 myocytes).B, In vivo epinephrine adminis-tration elicits bidirectional ven-tricular tachycardia inCASQ2KO mice (KO, bottomleft trace, n�15 mice), but notin CASQ2INF-KO mice (INF, bot-tom right trace, n�10 mice).
666 Circulation Research March 2, 2012
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from

tricular tachycardia elicited by stress or emotion. This com-plex phenotype results from the absence of CASQ2 duringembryonic development; therefore, it challenges the view thatrestoration of the missing protein in adults may revertphenotypic abnormalities. The mere quantitative restorationof CASQ2, in fact, does not imply that the protein willproperly localize and will re-establish physiological interac-tions with TrD and JnC in the appropriate stochiometry.8
Likewise, it is difficult to predict whether overexpressedCASQ2 will revert the ultrastructural abnormalities of thecalcium-release units, leading to an antiarrhythmic effect.With these challenges in mind, we designed an experimentalprotocol that verifies the rescue of all the key defects thatcharacterize the KO model of CPVT. Results showed that50% of myocytes express the transgene, suggesting that theheart of such infected mice resembles that of heterozygousKO mice that are asymptomatic and do not present cardiacarrhythmias,6 just like the heterozygous parents of affectedhomozygous individuals. Interestingly, in our experiments,the CASQ2 increase was accompanied by restoration of thelevels of TrD and JnC and by normalization of ultrastructuralabnormalities. One of our concerns in the design of the studywas the possibility that viral gene transfer would induceoverexpression of CASQ2, leading to the loss of the physio-logical ratio between CASQ2 and JnC/TrD, leading to the“toxic” effects observed in transgenic mice overexpressingCASQ28 and characterized by jSR swelling. Quite on thecontrary, long-term gene delivery in KO mice was able tonormalize ultrastructural defects of calcium-release units inthe majority of myocytes. By electron microscopy, onlyapproximately 2% of myocytes displayed overswelling of thejSR; this observation (Online Table I and Online Figure VII)supports the view that there is heterogeneity in the amount ofprotein that is produced in response to infection in each cell.Although 50% of cells appear to be infected as judged byeGFP labeling (green color), the actual production of thetransfected CASQ2 protein is probably more than an average50% increase, as inferred by the Western blot data (Figure1A, 1B). Interestingly, despite heterogeneity of the infectionrate and its efficiency, the relevant consequence of viral genetransfer is normalization of jSR morphology in most infectedcells (Online Table I) and suppression of electrophysiologicalabnormalities and arrhythmias in vivo. Online Figure VIIshows that re-expression of CASQ2 by viral infection maycause morphological changes in the jSR correlated withdifferent levels of CASQ2 spanning from absence of CASQ2(Online Table I) to presence of diffuse CASQ2 (OnlineFigure VIIC) to restoration of the physiological polymer-likestructure (Figure 3) to CASQ2 overexpression causing over-swelling of the SR (Online Figure VIIB). After observing thenormalization of the molecular and ultrastructural abnormal-ities of CASQ2KO mice, we anticipated that the substrate fordevelopment of triggered activity probably had been elimi-nated. Accordingly, in vitro and in vivo studies documentedthat electrophysiological abnormalities and arrhythmogenesisin vivo were completely suppressed in the KO mice infectedwith the AAV9-CASQ2, supporting the view that arrhythmo-genesis is the consequence of the complex set of abnormal-ities that occurs in response to CASQ2 decrease or absence.
Our data prove that systemic administration of cardiac-specific viral vectors to deliver CASQ2 to the heart ofCASQ2KO mice is able to revert all the abnormalities reportedin this CPVT mouse model. Considering that recently theCUPID trial11 demonstrated the safety of AAV-mediatedgene transfer in humans, we suggest that development ofCASQ2 viral gene transfer could have clinical application forseverely affected recessive CPVT resistant to conventionaltherapy. As part of our commitment to bring gene therapyto the clinical arena, we now need to investigate the durabilityof the AAV approach and whether the viral gene transfer willwork in adult mice as well as in newborns.
AcknowledgmentsThe authors thank Patrizia Vaghi and “Centro Grandi Strumenti” ofthe University of Pavia for technical assistance provided to theConfocal Microscopy facility.
Sources of FundingTelethon grants GGP11141 (to S.G.P. and P.V.), Telethon grantGGP08153 (to F.P.), Foundation Leducq research grant 0812 CVD01 (to S.G.P.), CARIPLO pr.2008.2275 (to S.G.P.), MIUR GR2009-1606636 (to S.G.P.), and Veronesi Foundation grant “Modellicellulari e terapia sperimentale dei difetti cardiaci associati a patolo-gie aritmogene ereditarie” (to S.G.P.).
DisclosuresNone.
References1. Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum
Ca2� handling and arrhythmogenesis. Circ Res. 2011;108:871–883.2. Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M,
DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G,Benatar A, DeLogu A. Clinical and molecular characterization of patientswith catecholaminergic polymorphic ventricular tachycardia. Circulation.2002;106:69–74.
3. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R,Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptorgene (hRyR2) underlie catecholaminergic polymorphic ventriculartachycardia. Circulation. 2001;103:196–200.
4. Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-NissenbaumE, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutationin a highly conserved region of CASQ2 is associated with autosomal recessivecatecholamine-induced polymorphic ventricular tachycardia in Bedouin familiesfrom Israel. Am J Hum Genet. 2001;69:1378–1384.
5. Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, Berul CI,Eldar M, Seidman CE, Seidman JG. Calsequestrin 2 (CASQ2) mutationsincrease expression of calreticulin and ryanodine receptors, causing cat-echolaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007;117:1814–1823.
6. Rizzi N, Liu N, Napolitano C, et al. Unexpected structural and functionalconsequences of the R33Q homozygous mutation in cardiac calse-questrin: a complex arrhythmogenic cascade in a knock in mouse model.Circ Res. 2008;103:298–306.
7. Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, KnollmannBE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR,Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulumvolume increase, premature Ca2� release, and catecholaminergic polymorphicventricular tachycardia. J Clin Invest. 2006;116:2510–2520.
8. Franzini-Armstrong C. Architecture and regulation of the Ca2� deliverysystem in muscle cells. Appl Physiol Nutr Metab. 2009;34:323–327.
9. Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, DuffHJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catechol-aminergic polymorphic ventricular tachycardia in mice and humans. NatMed. 2009;15:380–383.
10. Liu N, Denegri M, Ruan Y, Avelino-Cruz JE, Perissi A, Negri S,Napolitano C, Coetzee WA, Boyden PA, Priori SG. Short communi-cation: flecainide exerts an antiarrhythmic effect in a mouse model of
Denegri et al Gene Therapy for CPVT 667
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from

catecholaminergic polymorphic ventricular tachycardia by increasingthe threshold for triggered activity. Circ Res. 2011;109:291–295.
11. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B,Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ, Calcium Upregulation
by Percutaneous Administration of Gene Therapy in Cardiac Disease(CUPID) Investigators. A phase 2 trial of intracoronary gene therapy ofsarcoplasmic reticulum Ca2�-ATPase in patients with advanced heartfailure. Circulation. 2011;124:304–313.
Novelty and Significance
What Is Known?
● Recessive catecholaminergic polymorphic ventricular tachycardia (CPVT)is an inherited disease that predisposes to cardiac arrest and suddendeath, and it is caused by mutations in the calsequestrin2 (CASQ2)gene, leading to CASQ2 reduction or disappearance.
● CASQ2 reduction decreases levels of triadin (TrD) and junctin (JnC),which are important components of the sarcoplasmic reticulum(SR) calcium-release channel complex leading to impaired SRcalcium release, junctional sarcoplasmic reticulum (jSR) structuralabnormalities, and life-threatening arrhythmias.
What New Information Does This Article Contribute?
● Administration of the CASQ2 gene by in vivo viral transfer infects 50%of myocytes and increases the tissue levels of CASQ2, TrD, andJnC to 80% to 90% of control.
● Exogenous CASQ2 rescues ultrastructural abnormalities, revertselectrophysiological instability of myocytes, and abolishes ventric-ular tachycardia.
The development of novel treatments to correct abnormalities ofinherited diseases is a top priority in translational research. Inthe present study, we demonstrate that an increase in CASQ2 isable to revert the arrhythmogenic substrate as well as theultrastructural abnormalities present in CASQ2 knockout mice,even though the infection of myocytes was nonuniform. Wefound that an average increase of CASQ2 to 80% of the levelpresent in wild-type animals increased the amount of JnC andTrD, thereby preventing triggered activity and life-threateningarrhythmias. These findings could help in designing effectivestrategies for reverting structural and functional abnormalities inthe setting of inherited deficiencies of CASQ2.
668 Circulation Research March 2, 2012
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from

Circ Res AHA/2011/263939/R2 - online supplemental material
1
Denegri et al. Gene therapy for CPVT - Supplemental Material
Material and Methods
Generation of Knock-out of CASQ2 in Mouse Model
The CASQ2 knock-out strain was generated by homologous recombination of the targeting
vector with 129Sv/J embryonic stem cells genome. The linearized targeting vector was electroporated
into 129Sv/J embryonic stem cells. The clone selected with G418 and gancyclovir was injected into
C57BL/6NCrL blastocyts and transferred to pseudopregnant CD-1 females. Genotype was determined
by sequencing of DNA extracted from tail biopsy specimens (DNeasy Tissue Kit, Qiagen).
Cloning procedures and Virus production
The WT murine CASQ2 coding sequence (NM_009814.2), without 5’ and 3’-UTR sequences,
was cloned into pGEM-T-Easy vector (Promega). By enzymatic digestion, EcoRI, the insert
corresponding to the WT-CASQ2 was excised from the pGEM vector and subcloned in bis-cistronic
pIRES vector (BD Biosciences, Clontech Palo Alto CA, USA). Indeed, the fragment corresponding to
the WT-CASQ2 followed by the IRES sequence was subcloned via PCR amplification using specific
primers (Forward: 5’-CACAGCGGCCGCACAATGAAGAGGATTTACCTGCTCATGG-3’ and
Reverse 5’-CGAAGCATTAACCCTCACTAAAGGG-3’ containing the Not I enzymatic site. The
amplicon was inserted into the backbone vector pAAV-2.1-eGFP for AAV production 1, provided by
the Adeno-Associated Virus (AAV) vector Core facility (Tigem, Napoli, Italy), by the enzymatic
digestion with Not I. All the plasmids were entirely sequenced. The AAV particles production was
carried out by the Tigem AAV Vector core. The AAV was produced using a transient transfection of 3
plasmids in 293 cells: pAd helper, pAAV rep-cap (packaging, containing the cap sequence from
AAV9), pAAV Cis (including our insert, WT-mCASQ2-IRES, cloned in the pAAV2.1-CMV-eGFP
plasmid). The vector was purified from cellular contaminants by two rounds of CsCl2
ultracentrifugation and dialyzed against PBS. The viral titer was 2.60 x e12 GC/ml for the AAV9-
CASQ2-eGFP and 4.3 x e12 GC/ml for the CASQ2empty vector.

Circ Res AHA/2011/263939/R2 - online supplemental material
2
AAV- CASQ2 infection procedure in CASQ2KO mice
Animals were bred and raised at the Charles River Laboratories in Calco, Italy, and transferred to
the Maugeri Foundation for the characterization of the phenotype. Infection of homozygous CASQ2KO
mice with AAV9 viral particles (100 µl) was performed by intraperitoneal injection.
Isolation of Adult Mice Ventricular Myocytes
Ventricular myocytes were isolated using an established enzymatic digestion protocol2 from
homozygous CASQ2KO, CASQ2INF-KO, CASQ2empty vector and CASQ2WT mice (20 weeks) of either sex.
RT PCR
Total RNA from the myocytes of CASQ2INF-KO and CASQ2WT mice and organs (liver, lung,
muscle, testis and ovary) derived from infected mice (INF) and not infected mice (WT) were purified
with RNeasy mini kit (Qiagen), 0.5 µg RNA per reaction was used for RT-PCR assay using iScript
cDNA Synthesis kit (Bio-Rad Laboratories, Inc., USA). PCR was performed using standard conditions.
The following primers were used: GFP; Forward: 5’-TATATCATGGCCGACAAGCA-3’ and
Reverse: 5’-GAACTCCAGCAGGACCATGT-3’3 and β-Actin; Forward: 5’-
TGGAATCCTGTGGCATCCATGAAA-3’ and Reverse: 5’-
TAAAACGCAGCTCAGTAACAGTCCG-3’.4 Samples were analyzed by 1% agarose gel
electrophoresis in presence of Ethidium Bromide.
Quantitative Real Time PCR
Total RNA from the adult myocytes CASQ2INF-KO and CASQ2WT (n = 3 mice) and organs (liver,
lung, muscle, testis and ovary) derived from infected mice (INF) were purified with RNeasy mini kit
(Qiagen). A total amount of 0.25 μg template RNA per reaction was taken for RT-PCR assay
performed with iScript cDNA Synthesis kit (Bio-Rad Laboratories, Inc., USA). Real Time
quantification of mRNAs of the targets genes (CASQ2, Triadin, Junctin, GFP) and the reference gene
(GAPDH) was performed in optical 96-well plates using CFX96 detection module (Bio-Rad
Laboratories, Inc.). All samples were analyzed in triplicate with SsoFast EvaGreen Supermix using
specific primer mix and 100 ng of cDNA template with previously described Primers.5 Real-Time PCR
was performed using the CFX96 Real-Time PCR Detection System and analyzed using the Bio-Rad

Circ Res AHA/2011/263939/R2 - online supplemental material
3
CFX Manager software package (Bio-Rad Laboratories, Inc., USA). The mean values were normalized
against the GAPDH. Values for Fold Expression, based on ΔΔC(t) methods, were generated
automatically by the Bio-Rad CFX Manager software 1.5.
Electron Microscopy
Hearts from CASQ2KO, CASQ2INF-KO and CASQ2WT mice of 20 weeks of age were fixed by
retrograde aortic perfusion with 3.5% glutaraldehyde in 0.1 mol/L NaCaCo buffer (pH 7.2) and hearts
were fixed and analyzed. Small bundles of cells were post-fixed in 2% OsO4 in NaCaCo buffer for 2
hours and then block-stained in saturated uranyl acetate. After dehydration, specimens were embedded
in an epoxy resin (Epon 812). Ultrathin sections were cut in a Leica Ultracut R microtome (Leica
Microsystem, Austria) using a Diatome diamond knife (Diatome Ltd. CH-2501 Biel, Switzerland) and
double stained with uranyl acetate and lead citrate. All sections were examined with an FP 505
Morgagni Series 268D electron microscope (FEI Company, Brno, Czech Republic), equipped with
Megaview III digital camera. The average width of jSR terminal cisternae were measured in random
images at 56.000X using the Soft Imaging System.
Action potential recordings in Isolated Ventricular Myocytes
Isolated ventricular myocytes were seeded on a glass bottom perfusion chamber mounted on the
stage of an inverted microscope. After 5 minutes, the myocytes were bathed in the following solution at
35°C (in mmol/L): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 5 glucose, pH 7.4, with NaOH.
Action potential recording was performed using patch electrodes made of borosilicated (resistance of 2
to 3 MΩ) and filled with a solution containing: (in mmol/L): 120 potassium aspartate, 20 KCl, 1
MgCl2, 4 Na2ATP, 0.1 GTP, 10 HEPES, 10 glucose, pH 7.2, with NaOH. Signals were acquired at 10
kHz (Digidata 1322A, Axon Instruments) and analyzed with pCLAMP version 9.2 software (Axon
Instruments). Quiescent, calcium-tolerant, rod-shaped cells with clear cross striations and a resting
potential < –80 mV were electrically stimulated using depolarizing pulses with duration of 3 ms and
amplitude of 1.5 fold the minimal current needed to evoke and action potential. Correction for the
liquid junction potential between pipette and bath solution was performed.

Circ Res AHA/2011/263939/R2 - online supplemental material
4
In vivo exercise stress test and pharmacological challenge
In vivo ECG recordings was performed using subcutaneous devices (Data Sciences International).
Phenotypical characterization was performed as follows: DAY 1: baseline 24 hours ECG recording was
collected. DAY 2: ECG recording during exercise on a treadmill until exhaustion (15-20 min). DAY 3:
ECG recording after epinephrine injection (2 mg/Kg i.p.). All the animals were studied according to the
protocols approved by the Animal Care and Use committee at the Maugeri Foundation, Pavia.
Legends for Online Figures
Online Figure I
AAV9 construct and histological assay. (A) diagram of the pAAV2.1-CMV-CASQ2-IRES-
eGFP construct. (B) histological sections from hearts of CASQ2WT (WT) and CASQ2INF-KO (INF)
mice; from left: first and second panels are stained with hematoxylin-eosin; third and fourth panels are
stained with Masson staining.
Online Figure II
RT-PCR and Real Time analysis. (A) GFP expression in CASQ2WT (WT), CASQ2KO (KO) and
CASQ2INF-KO (INF) mice; Ctrl = negative control. (B) RealTime PCR quantification of GFP expression
in different organs from CASQ2INF-KO (INF) mice. Isolated myocytes from CASQ2INF-KO (INF) and
CASQ2WT (WT) mice were used as positive and negative controls, respectively. Data were collected
from 3 independent experiments. Real Time PCR data are given as mean ± SEM and are expressed as
fold expression normalized to GAPDH and to the positive control.
Online Figure III
Sub-cellular localization of CRU proteins. Left panels: phase contrast (PhC) image of GFP
positive myocytes isolated from CASQ2INF-KO mice. GFP signal is depicted in middle panels. Right
panels from top to bottom depict staining with anti-TrD (A) and anti-JnC (B) antibodies.
Immunolabeling indicates the correct localization of restored TrD and JnC along the Z lines (top and
middle right panels). Myocyte stained with the secondary antibody is the negative control (C, bottom
right panel). (n = 3 animals/group).

Circ Res AHA/2011/263939/R2 - online supplemental material
5
Online Figure IV
Ultrastructural analysis of jSR. (A) Electron micrographs from thin sections of age-matched
CASQ2WT, CASQ2KO (B) and CASQ2INF-KO (C) hearts: yellow labeling marks the position of T-tubules
in correspondence of junctions with the jSR. In CASQ2WT myocytes (A) jST/T-tubule junctions are
mostly disposed in correspondence of the Z-lines, but longitudinal dyads are also present between
myofibrils. In CASQ2KO and CASQ2INF-KO (B and C) myocytes dyads are also disposed in
correspondence of the Z-line between myofibrils. Insets display high magnification images of jSR/T-
tubule couplons: jSR is narrow in CASQ2WT, wide in CASQ2KO and narrow again in CASQ2INF-KO (see
Figure 3 for more details). Scale bars: A, B and C = 0.5 μm; insets= 0.1 μm, (n = 3 animals/group).
Online Figure V
In vitro electrophysiology of CASQ2empty-vector mice. (A) Action potentials and TA induced
with pacing at 5 Hz during exposure to 30 nmol/L Isoproterenol (Iso) in isolated myocytes from
hearts infected with an empty viral vector (Empty vector+Iso). Paced action potentials are indicated
by arrows. (B) comparison of the incidence of TA in myocytes derived from the heart of a knock out
mouse infected with CASQ2 (INF) and in myocytes derived from the heart of a knock out mouse
infected with an empty vector (Empty Vector) . We studied 8 cells in each group; *** p>0.0001.
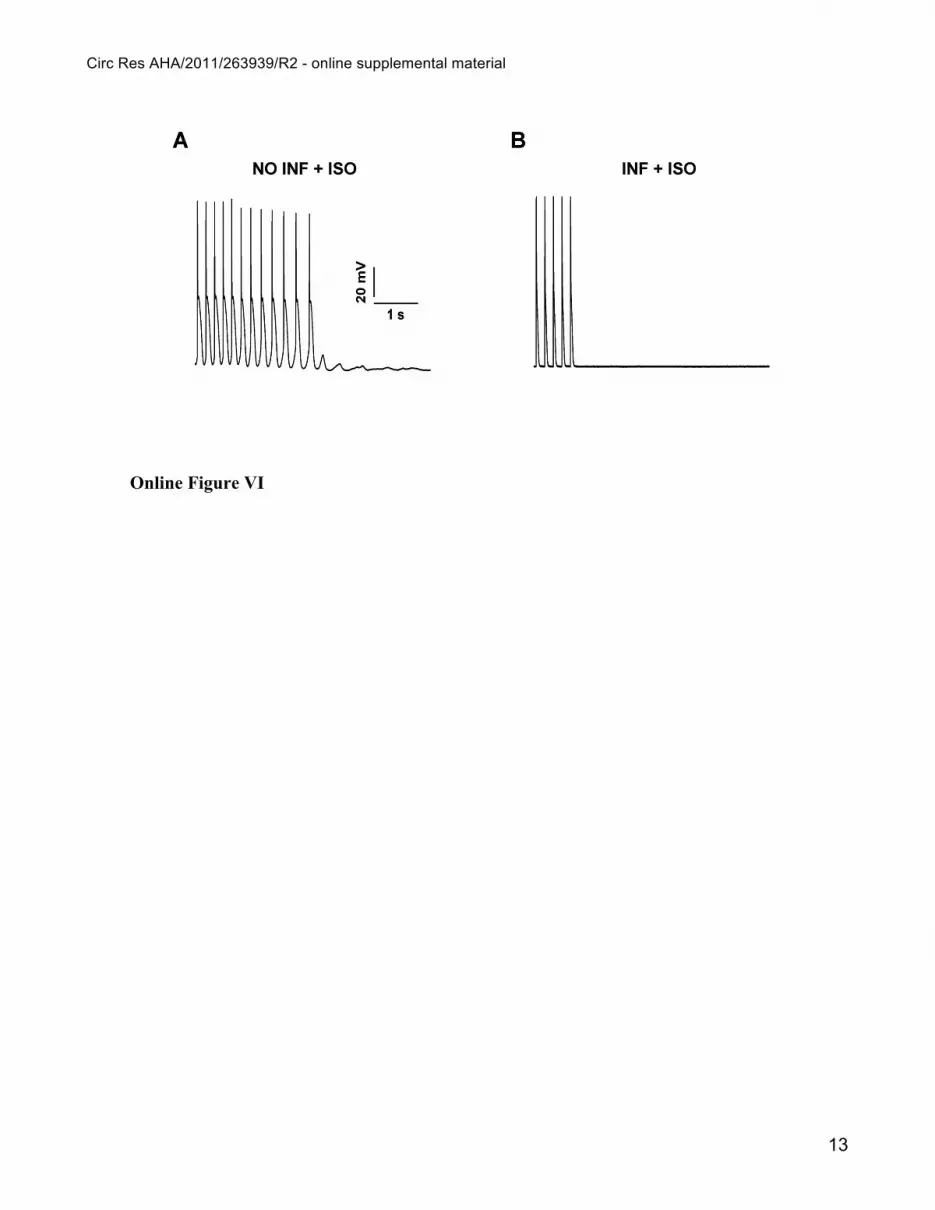
Online Figure VI
In vitro electrophysiology of GFP negative CASQ2INF-KO mice. Action potentials and TA
induced with pacing at 5 Hz after exposure to 30 nmol/L Isoproterenol (ISO) in isolated myocytes
derived from the heart of a CASQ2 knock out mouse infected with a viral vector containing wild type
CASQ2 labeled with eGFP (CASQ2INF-KO). Panel A shows recording from a myocyte that did not
present the GFP signal and therefore had not been infected (NO INF +ISO); Panel B Shows recording
from a myocyte that presented the GFP signal and therefore had been infected and expresses the
CASQ2 protein. (A) Representative recording of a GFP negative CASQ2INF-KO myocytes (NO-
INF+Iso) that presents TA during Iso administration; (B) A representative trace of a GFP positive
myocytes ( INF+Iso) that does not develop TA. The experimental protocol included 11 GFP negative
cells and 19 GFP positive cells.

Circ Res AHA/2011/263939/R2 - online supplemental material
6
Online Figure VII
Low magnification analysis of jSR. (A) Low magnification image of a CASQ2INF-KO fibre
showing over-swollen SR vesicles (asterisks), which are suggestive of CASQ2 over-expression. (B)
large SR vesicles, filled with a granular electron-dense content representing CASQ2, that may or may
not be associated with T-tubules (labelled in yellow). (C) CRUs showing slightly dilated jSR
containing a diffuse, granular content suggestive of CASQ2 presence. (n = 3 animals/group). Scale
bars: A, 2 μm; B, 0.5 μm; C, 0.2 μm.
Online Table I
Evaluation of the efficiency of viral infection by EM. A detailed EM analysis of the SR in 3
hearts from CASQ2INF-KO mice showed 3 types of ultrastructural changes:
Column A: no rescue of CASQ2 levels and enlargement of jSR as seen in CASQ2KO mice.
Column B: rescue of CASQ2 with variable recovery of jSR: 80% of fibers (9 fibers in Heart 1, 8 fibers
in Heart 2 and 6 fibers in Heart 3) presented full rescue of CASQ2 with chain like polymer
configuration and normal jSR (Figure 3); 20% of cells had less complete rescue presenting diffused
CASQ2 and a slightly wider than normal jSR (Online Figure VII C).
Column C: over-swollen jSR with a diffused granular CASQ2 suggestive of CASQ2 over-expression
(Online Figure VII B).

Circ Res AHA/2011/263939/R2 - online supplemental material
7
REFERENCES
1. Auricchio A, Hildinger M, O’Connor E, Gao GP, Wilson JM.Isolation of highly
infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow
column. Hum Gene Ther 2001;12:71-76.
2. Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M,
Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in Catecholaminergic Polymorphic
Ventricular Tachycardia. Insights From a RyR2 R4496C Knock-In Mouse Model. Circulation
Research 2006;99:292-298.
3. White JD, Thesier DM, Swain JB, Katz MG, Tomasulo C, Henderson A, Wang L,
Yarnall C, Fargnoli A, Sumaroka M, Isidro A, Petrov M, Holt D, Nolen-Walston R, Koch WJ,
Stedman HH, Rabinowitz J, Bridges CR. Myocardial gene delivery using molecular cardiac
surgery with recombinant adeno-associated virus vectors in vivo. Gene Ther 2011;18:546-552.
4. Kordek R, Nerurkar VR, Liberski PP, Isaacson S, Yanagihara R, Gajdusek DC.
Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary
acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc Natl Acad Sci U S A
1996;93:9754-9758.
5. Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D,
Bigioggera M, Scelsi M, Villani L, Volpe P, Priori SG. Unexpected structural and functional
consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex
arrhythmogenic cascade in a knock in mouse model. Circulation Research 2008;103:298-306.
6. Tijskens P, Jones BL, Franzini-Armstrong C. Junctin and calsequestrin overexpression
in cardiac muscle: the role of junctin and the synthetic and delivery pathways for the two
proteins. J Mol Cell Cardiol. 2003;35:961-74

Circ Res AHA/2011/263939/R2 - online supplemental material
8
Online Figure I

Circ Res AHA/2011/263939/R2 - online supplemental material
9
Online Figure II

Circ Res AHA/2011/263939/R2 - online supplemental material
10
Online Figure III

Circ Res AHA/2011/263939/R2 - online supplemental material
11
Online Figure IV

Circ Res AHA/2011/263939/R2 - online supplemental material
12
Online Figure V
Circ Res AHA/2011/263939/R2 - online supplemental material
13
Online Figure VI

Circ Res AHA/2011/263939/R2 - online supplemental material
14
Online Figure VII

Circ Res AHA/2011/263939/R2 - online supplemental material
15
Online Table I
A B C
No. of Fibers
No. of Fibers with no rescue of jSR and no CASQ2
No. of Fibers with CASQ2 re-
expressed and variable degree of
jSR widening
No. of Fibers with CASQ2 and over-swollen
SR suggestive of over-expression
Heart 1 27 11 (41%)
9 + 5* (52%)
2** (7%)
Heart 2 23 14 (61%)
8 + 1* (39%) 0
Heart 3 21 15 (71%)
6 (29%) 0
Average 58% 40% 2%
*Fibers with dilated jSR as shown in Online Figure VII C. ** Fibers with over-swollen SR such as shown in Online Figure VII B.

and Silvia Giuliana PrioriSimone, Alberto Auricchio, Laura Villani, Pompeo Volpe, Feliciano Protasi, Carlo Napolitano
Marco Denegri, José Everardo Avelino-Cruz, Simona Boncompagni, Stefano Andrea DeAbnormalities in Adult Calsequestrin-Null Mice With Inherited ArrhythmiasViral Gene Transfer Rescues Arrhythmogenic Phenotype and Ultrastructural
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2012 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.111.2639392012;110:663-668; originally published online January 31, 2012;Circ Res.
http://circres.ahajournals.org/content/110/5/663World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2012/01/31/CIRCRESAHA.111.263939.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on January 11, 2016http://circres.ahajournals.org/Downloaded from





























