Embed Size (px)
DESCRIPTION
计算机科学与生命科学( 9 ) 生物信息学基础 2013 年秋季学期通选课程 上课时间:周一 18:30 点 上课地点:软件园 4 区 502d 主讲人:魏天迪 讲义网址: http://www.mbtech.sdu.edu.cn/biocomp/. 分子进化. 美国人 Linus Pauling 于 1964 年提出了分子进化的理论。 在分子水平上( DNA 、 RNA 或蛋白质序列)而不是物种的外在特征,来研究进化过程。 基于某一个特定的分子在不同物种中的序列差异来构建进化树。 - PowerPoint PPT Presentation
Citation preview

计算机科学与生命科学( 9)生物信息学基础
2013 年秋季学期通选课程上课时间:周一 18:30 点 上课地点:软件园 4 区 502d主讲人:魏天迪讲义网址: http://www.mbtech.sdu.edu.cn/biocomp/

分子进化
• 美国人 Linus Pauling 于 1964 年提出了分子进化的理论。• 在分子水平上( DNA 、 RNA 或蛋白质序列)而不是物种的外在特征,来研究进化过程。• 基于某一个特定的分子在不同物种中的序列差异来构建进化树。• 基本假设:( 1 ) DNA 、 RNA 或蛋白质序列包含了物种的所有进化史的信息;( 2 )分子钟理论:一个特定蛋白质的进化变异(不同碱基或氨基酸的个数)的速度在不同物种中是基本恒定的。即两个蛋白质的序列越相近,他们距离共同祖先就越近。

DNA序列
>nameCTCCTGACCTCAGGCGATTCGCCCGCCTCGGCCTCCCAAAGTGCTAGGATTACAGGCGTGAGCCACCACGCCCGGCCACACTAACTTTTTAAGAGCCAAGAGTTCGATCGGTAGCGGGAGCGGAGAGCGGACCCCAGAGAGCCCTGAGCAGCCCCACCACCACCGCTGGCCTAGCTACCATCACACCCCGGGAGGAGCCGCAGCTGCCGCAGCCGGCCCCAGTCACCATCACCACAACCTTGAGCAGCGAGGCCGAGACCCAGCAGCCGCCCGCCGCTTGCCGCTCGCCGCCCCCCGCCCTCAGCGCCGGTGACACCACGCCCGGCACTACGGGCAGCGGCACAGGAAACGGTGGCCCGGGAGGCTTCACATCAGCAGCACCTGCCGGCGGGGACAAGAAGGTCATCGCAACGAAGGT
由 4 个不同的字母(碱基)排列组合而成。
FASTA 格式:第一行:大于号加名称或其它注释;第二行以后:每行 60 个字母。

蛋白质序列
>nameMHHHHHHSSGRENLYFQGKLPEPQFYAEPHTYEEPGRAGRSFTREIEASRIHIEKIIGSGDSGEVCYGRLRVPGQRDVPVAIKALKAGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTRGRLAMIVTEYMENGSLDTFLRTHDGQFTIMQLVGMLRGVGAGMRYLSDLGYVHRDLAARNVLVDSNLVCKVSDFGLSRVLEDDPDAAXTTTGGKIPIRWTAPEAIAFRTFSSASDVWSFGVVMWEVLAYGERPYWNMTNRDVISSVEEGYRLPAPMGCPHALHQLMLDCWHKDRAQRPRFSQIVSVLDALIRSPESLRATATVS
由 20 个不同的字母(氨基酸)排列组合而成。
FASTA 格式:第一行:大于号加名称或其它注释;第二行以后:每行 60 个字母。

创建进化树
http://www.ebi.ac.uk/Tools/phylogeny/clustalw2_phylogeny/
输入文件: sequences.txt
输出树:

看三个与进化有关的短片

基本序列算法
1. 精确子字符串搜索如:在一个 DNA 序列中搜索起始子 ATG 。• 在文本文件或 WORD 文件中直接用搜索功能。• 使用简单的计算机语言编程,如: PERL
open (FH,"<dna.txt");@get=<FH>;close FH;$content=join("", @get);$n=0;$find=index($content, "ATG", $n);while($find!=-1){print "$find\n";$n=$find+1;$find=index($content, "ATG", $n);}

基本序列算法
2. 模式匹配搜索(正则表达式)如:在一个蛋白质序列中搜索 LxxLxLxxNxL 其中 L 代表 L, I , F 或 V , x 代表任意氨基酸。使用简单的计算机语言编程,如: PERL
open (FH,"<protein.txt");@get=<FH>;close FH;$content=join("", @get);while($content=~/[LIFV]\w{2}[LIFV]\w{1}[LIFV]\w{2}N\w{1}[LIFV]/g){print "$&\n";}
模式匹配搜索也可以实现上一张幻灯片里的精确子字符串搜索:while($content=~/ATG/g){print "$&\n";}

基本序列算法
3. 后缀树
序列: SDSDFSDFG
1: SDSDFSDFG$2: DSDFSDFG$3: SDFSDFG$4: DFSDFG$5: FSDFG$6: SDFG$7: DFG$8: FG$9: G$

基本序列算法
3. 后缀树
SDSDFSDFG功能: 1. 查找字符串 s 是否在字符串 S 中 : 从树根开始,与 s 的字符逐一比对。 s1: DFSD (在) ; s2: SDFD (不在)

基本序列算法
3. 后缀树
SDSDFSDFG功能: 2. 指定字符串 s 在字符串 S 中的重复次数:从树根开始,按照功能1 的办法找到 s ,然后看 s 之后有几片树叶。 s1: SD ( 3 次) ; s2: DF( 2 次)

基本序列算法
3. 后缀树
SDSDFSDFG功能: 3. 字符串 S 中的最长重复子串:找到从树根到所有节点(非叶片)的子字符串,从中找到最长的。 SDF

基本序列算法
3. 后缀树
$ 的作用:如果某一个后缀是另一个后缀的前缀,那么需要用 $ 标识出一个独立的叶片。

基本序列算法
3. 后缀树
$ 的作用:如果某一个后缀是另一个后缀的前缀,那么需要用 $ 标识出一个独立的叶片。假如第 n 个后缀是 SDSD :
$n

基本序列算法
程序自动创建 sufixtree: http://www.allisons.org/ll/AlgDS/Tree/Suffix/
基本序列算法
4. 最高分子序列问题
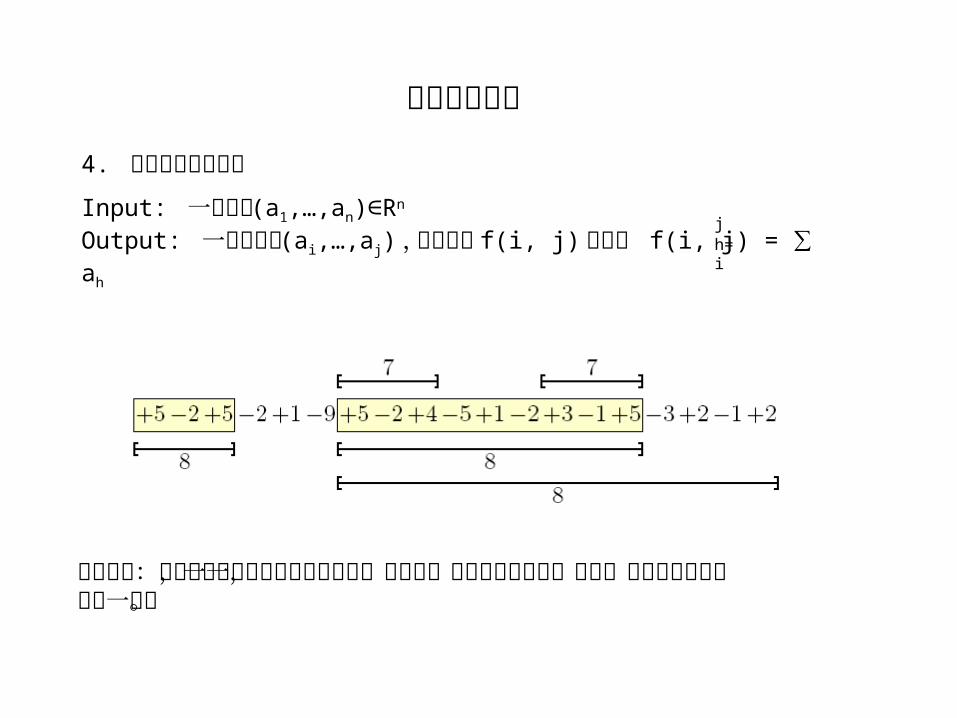
Input: 一个序列 (a1,…,an) R∈ n
Output: 一个子序列 (ai,…,aj) ,使得函数 f(i, j) 最大, f(i, j) = ∑ ah
jh=i
最短原则:在几个子序列同时拥有最高分时,如果某一个完全包含在另一之内,则只返回被包含的那一个。
基本序列算法
4. 最高分子序列问题
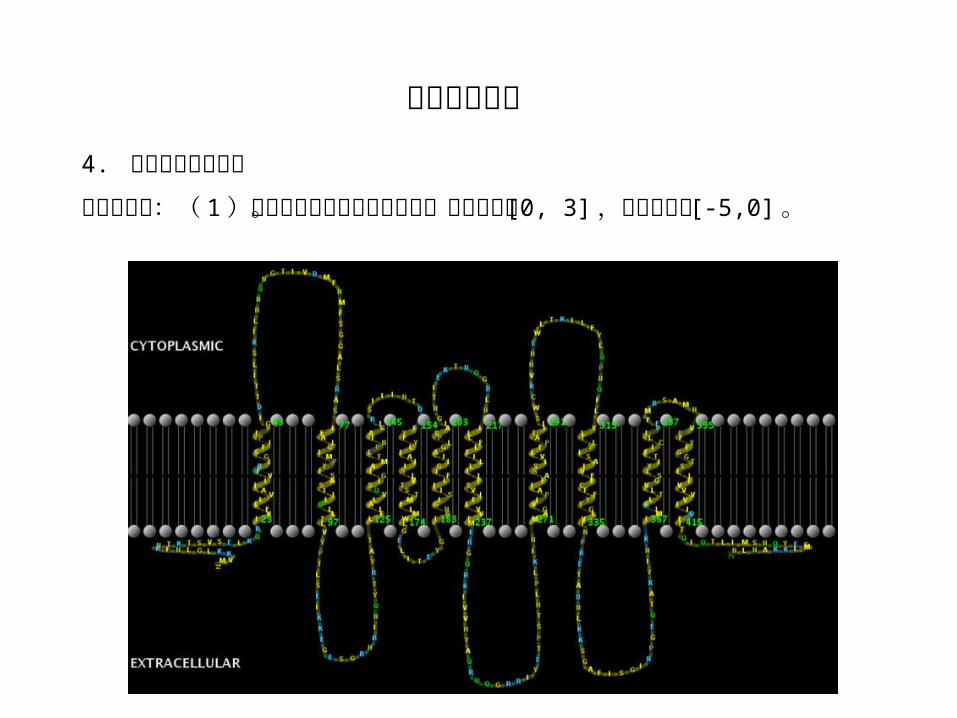
生物学应用:( 1 )蛋白质序列跨膜区域的预测。疏水氨基酸 [0, 3] ,亲水氨基酸 [-5,0] 。
基本序列算法
4. 最高分子序列问题
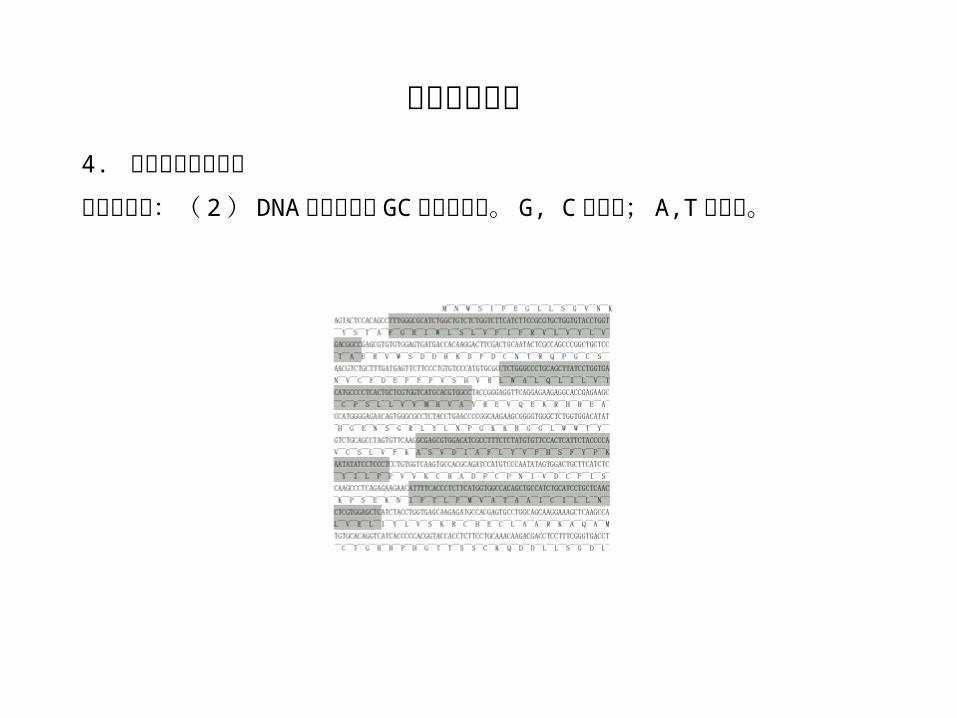
生物学应用:( 2 ) DNA 序列中富含 GC 区域的预测。 G, C 给正分; A,T 给负分。

基本序列算法
4. 最高分子序列问题
Input: 一个序列 (a1,…,an) R∈ n
Output: 一个子序列 (ai,…,aj) ,使得函数 f(i, j) 最大, f(i, j) = ∑ ah
jh=i
Naïve 算法:对于所有 i<=j [1, n]∈ ,计算 f(i, j) ,在找出最大值对应的 (i, j) 。
所有可能的 (i, j) 组合的数量,即计算 f(i, j) 的次数:
(n-1)+(n-2)+…+1 = n*(n-1)/2 = O(n2)
计算一次 f(i, j) 所需的步骤: O(n)
=> Naïve 算法的总运算步骤为 O(n3)
一个算法的运算步骤: k -> log(n) -> n -> n*log(n) -> n2

基本序列算法
4. 最高分子序列问题
Input: 一个序列 (a1,…,an) R∈ n
Output: 一个子序列 (ai,…,aj) ,使得函数 f(i, j) 最大, f(i, j) = ∑ ah
jh=i
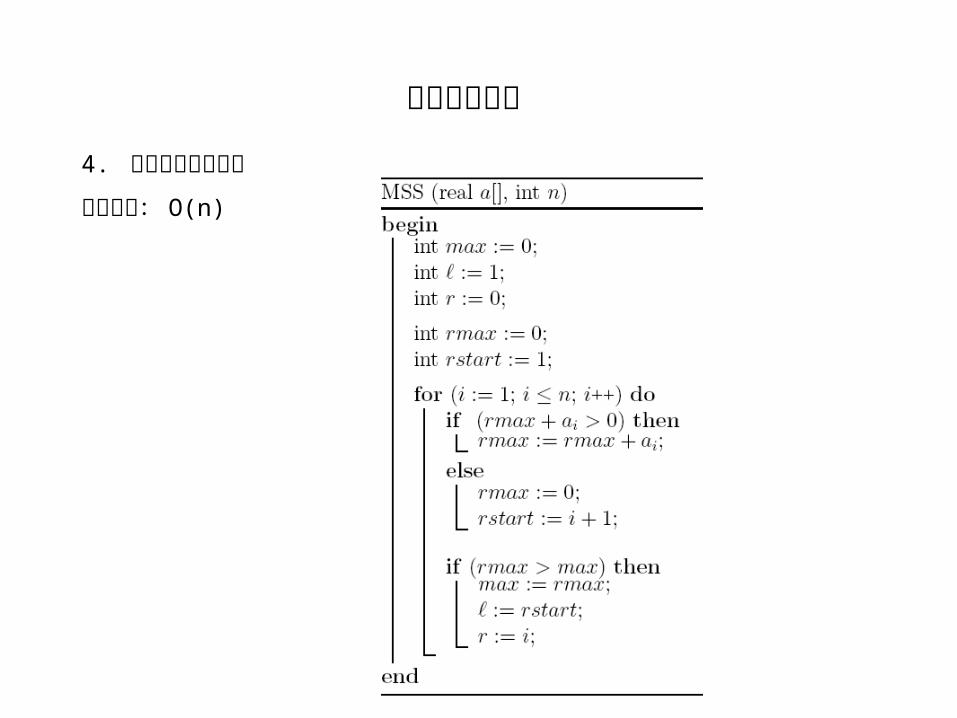
动态算法: O(n2)
分而治之算法: O(n*log(n))
聪明算法: O(n)
基本序列算法
4. 最高分子序列问题
聪明算法: O(n)

数据库中的序列相似性搜索
在麻将连连看中,你需要用眼睛从一推麻将牌中找出一对相同的麻将牌。
序列相似性

数据库中的序列相似性搜索
对于一个蛋白质或 DNA 序列,你需要从序列数据库中找到与它相同或相似的序列。不可能再用眼睛去比较每一对序列,因为数据库中有太多序列,甚至用眼睛比较一对序列都是不可能做到的。
…… > 100,000
BLAST
序列相似性
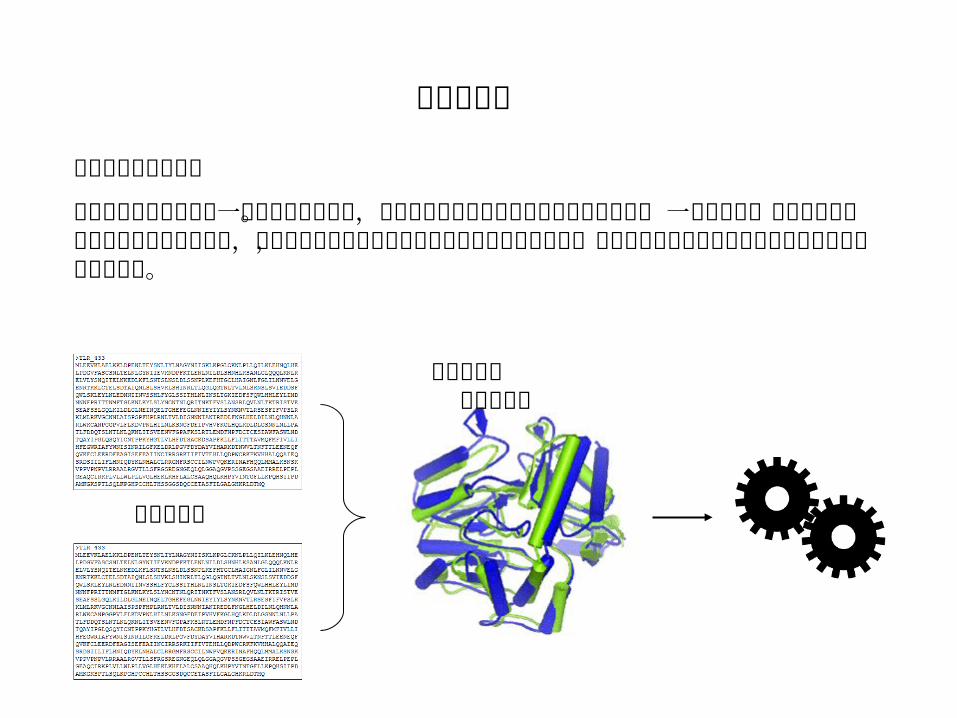
序列相似性的重要性
相似的序列往往起源于一个共同的祖先序列。它们很可能有相似的空间结构和生物学功能 ,因此对于一个已知序列但未知结构和功能的蛋白质,如果与它序列相似的某些蛋白质的结构和功能已知,则可以推测这个未知结构和功能的蛋白质的结构和功能。
相似的序列
相似的结构 相似的功能
序列相似性
结构相似?功能相似?
序列相似性
序列相似性的重要性
相似的序列往往起源于一个共同的祖先序列。它们很可能有相似的空间结构和生物学功能 ,因此对于一个已知序列但未知结构和功能的蛋白质,如果与它序列相似的某些蛋白质的结构和功能已知,则可以推测这个未知结构和功能的蛋白质的结构和功能。
序列相似性
序列相似性的重要性
相似的序列往往起源于一个共同的祖先序列。它们很可能有相似的空间结构和生物学功能 ,因此对于一个已知序列但未知结构和功能的蛋白质,如果与它序列相似的某些蛋白质的结构和功能已知,则可以推测这个未知结构和功能的蛋白质的结构和功能。
结构相似?功能相似?
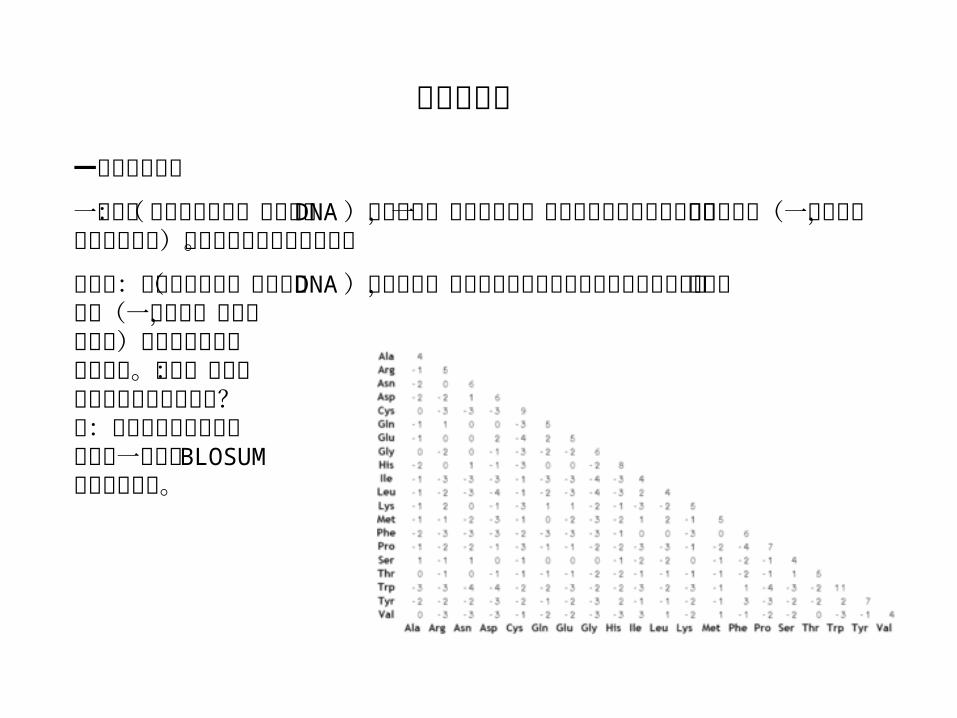
一致度与相似度
一致度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的一致度定义为他们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
相似度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的相似度定义为他们对应位置上相似的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。问题:哪个残基与哪个残基算作相似?答:残基两两相似的量化关系被一个称为 BLOSUM的矩阵所定义。
序列相似性

一致度与相似度
一致度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的一致度定义为他们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
相似度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的一致度定义为他们对应位置上相似的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。问题:哪个残基与哪个残基算作相似?答:残基两两相似的量化关系被一个称为 BLOSUM的矩阵所定义。
序列相似性
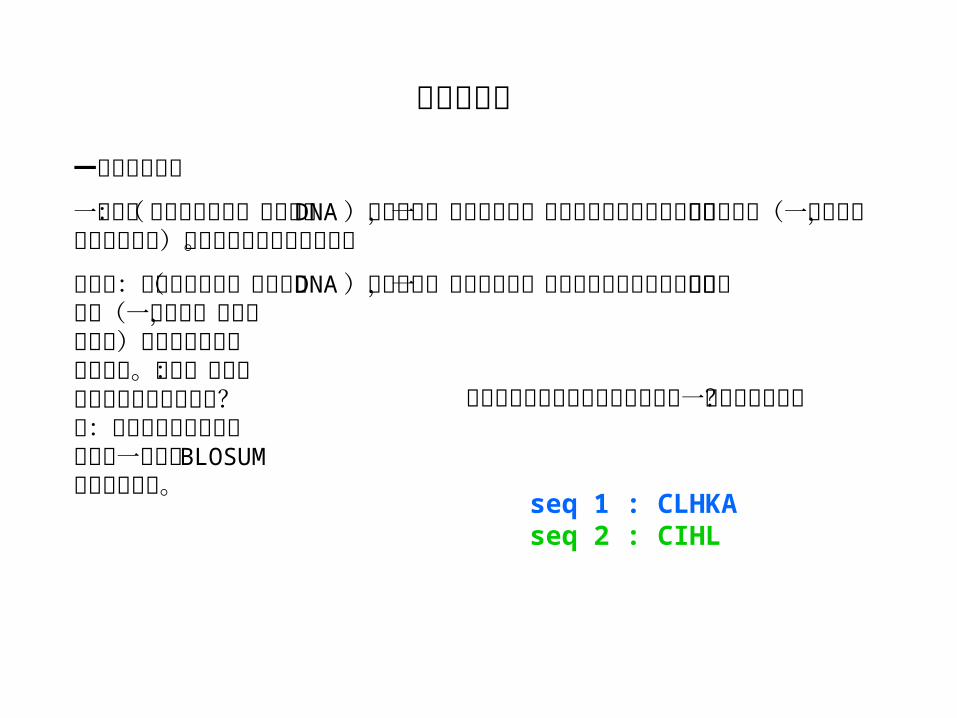
序列 1 : CLHK序列 2 : CIHL
一致度 = 2/4 = 50%
相似度 = 3/4 = 75%
一致度与相似度
一致度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的一致度定义为他们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
相似度:如果两个序列(蛋白质或 DNA )长度相同,那么它们的一致度定义为他们对应位置上相似的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。问题:哪个残基与哪个残基算作相似?答:残基两两相似的量化关系被一个称为 BLOSUM的矩阵所定义。
序列相似性
如果像个序列的长度不同怎么计算一致度与相似度?
seq 1 : CLHKAseq 2 : CIHL
比较两个长度不同的序列的方法:打点法、序列比对法
打点法:最简单的比较两个序列的方法,理论上可以用纸和笔来完成。
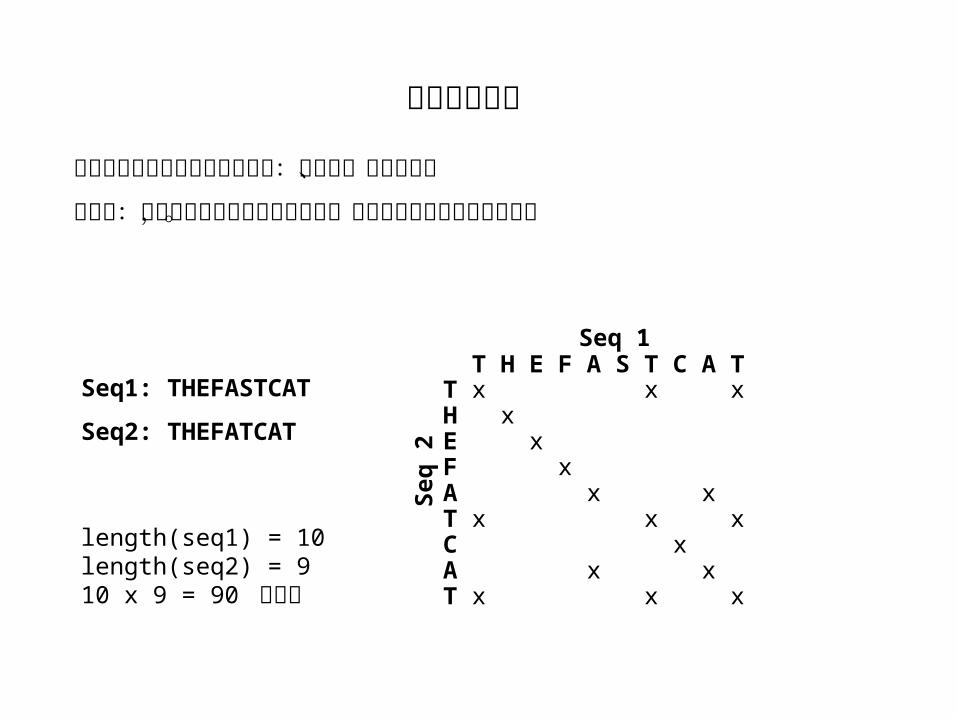
Seq1: THEFASTCAT
Seq2: THEFATCAT
T H E F A S T C A TT x x xH xE xF xA x xT x x xC xA x xT x x x
length(seq1) = 10length(seq2) = 910 x 9 = 90 次比较
序列两两比较
Seq 1
Seq 2
序列两两比较
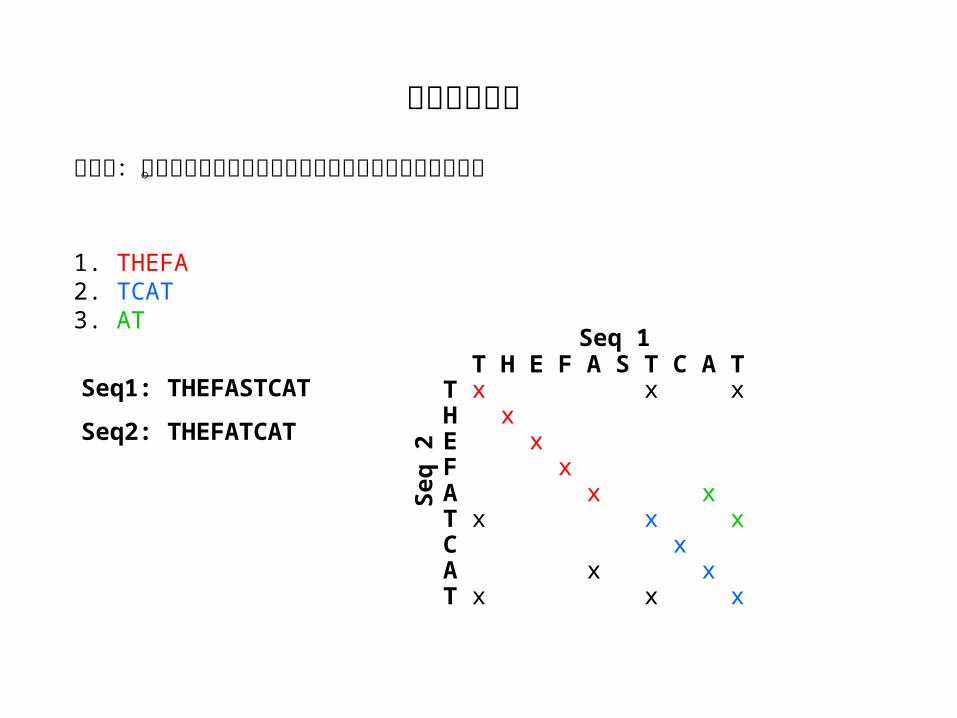
打点法:对角线及对角线的平行线代表两条序列中相同的区域。
1. THEFA2. TCAT3. AT
Seq1: THEFASTCAT
Seq2: THEFATCAT
T H E F A S T C A TT x x xH xE xF xA x xT x x xC xA x xT x x x
Seq 1
Seq 2
Seq1: THEFASTHE T H E F A S T H ET x xH x xE x xF xA xS xT x xH x xE x x
序列两两比较
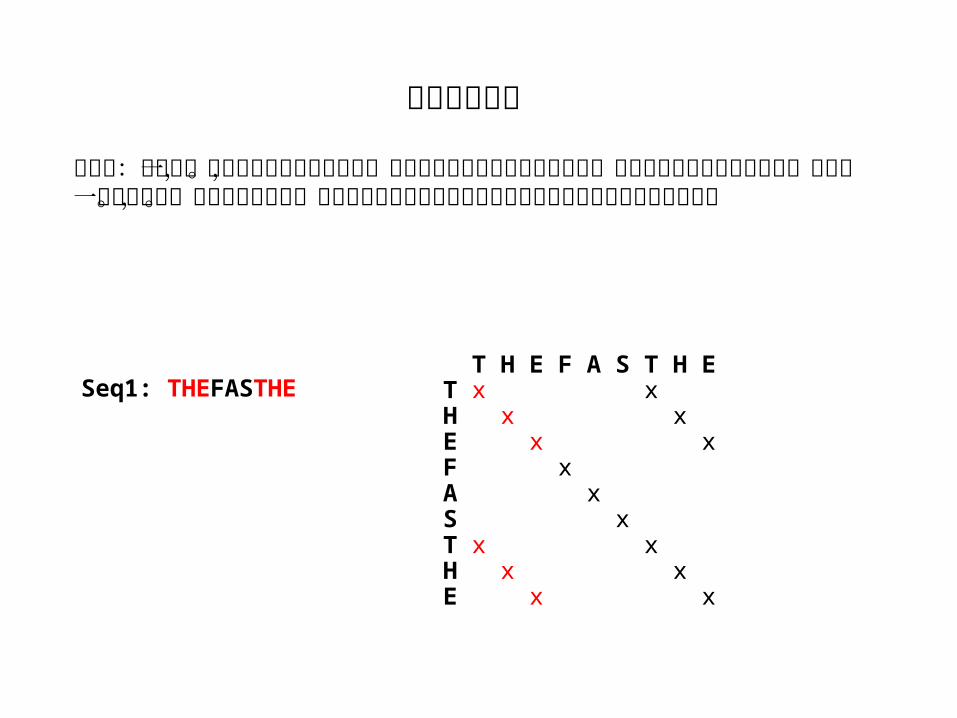
打点法:可以用一条序列自己对自己做打点,从而可以发现序列中重复的片段。这样的点矩阵必然是对称的,并且有一条主对角线。在横向或纵向上,与主对角线重叠的小对角线所对应的序列片段就是重复的部分。





























