Embed Size (px)
Citation preview


Differences between glycoproteins and proteoglycans
Functions and structures of glycoproteins and proteoglycans
Synthesis and degradation of glycoproteins and proteoglycans
Pathology related to glycoproteins and proteoglycans

Medical Biochemistry, third edition, edited by Baynes and Dominiczak.
Chapter 26, pages 351-366 on glycoproteins
Chapter 28, pages 384-388 on proteoglycans

Glycoproteins
Proteoglycans
Proteins conjugated tosaccharides lacking aserial repeat unit
Proteins conjugated topolysaccharides withserial repeat units
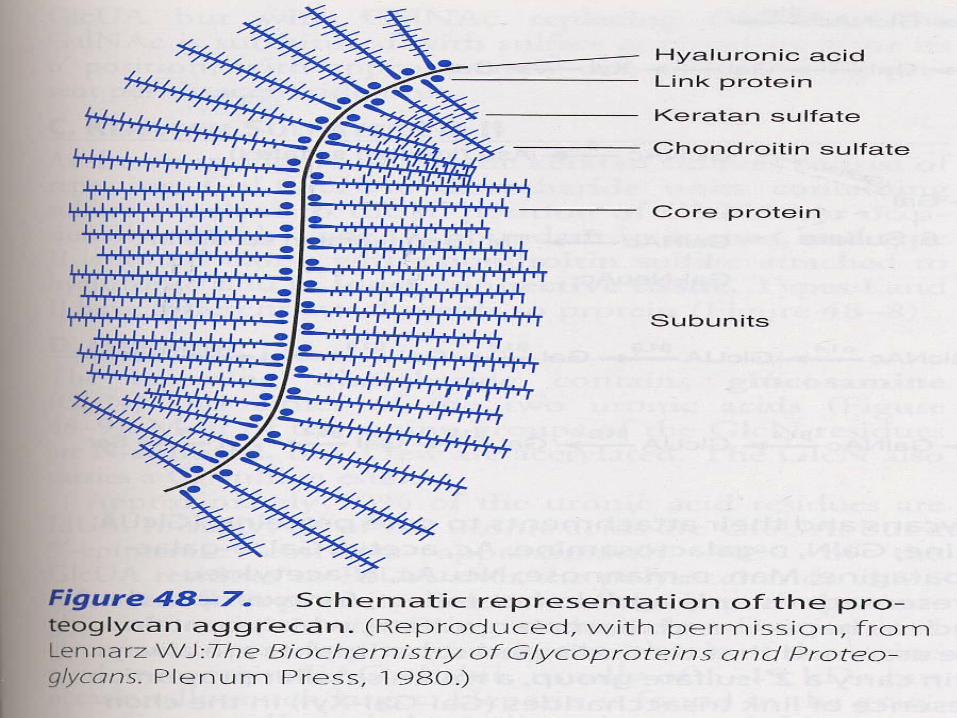
GlycosaminoglycansMucopolysaccharides
Protein>>carbohydrate
Carbohydrate>>protein
Repeat unitHexN and HexUA
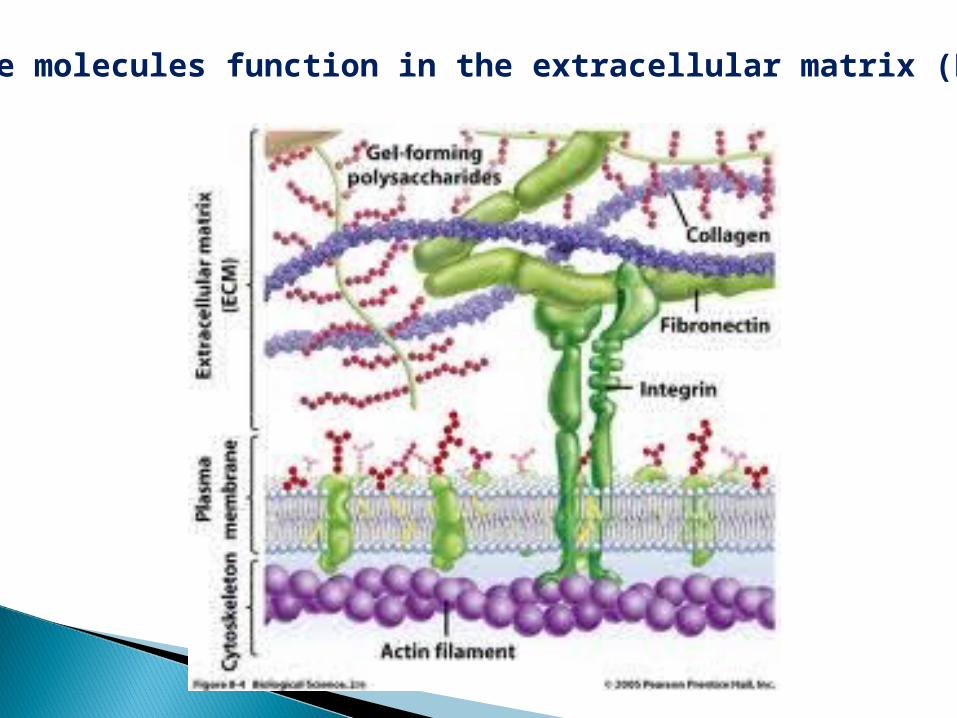
These molecules function in the extracellular matrix (ECM)

Overview of glycoproteins:--carbohydrate chain short--no serial repeats--often branched, not linear--variable amounts of carbs--wide range of functions

Some Functions of Glycoproteins_________________________________________________
Function Glycoprotein_________________________________________________1. Structural molecule Collagens2. Lubricant Mucins3. Transport molecule e.g. Transferrin,
Ceruloplasmin4. Immune system Immunoglobulins,
Histocompatibility antigens,Blood group determinants
5. Hormone e.g. HCG, TSH6. Enzymes e.g. Alkaline phosphatase7. Blood clotting e.g. Fibrinogen8. Cell surface recognitionLectins__________________________________________________

One or more carbohydrate chains--covalently linked to a protein.The chains may be neutral or negatively charged. They are frequently branched.
There are two types of glycosidic links:1. O-glycosidic linkO-glycosidic link between galactose or glucose and the hydroxyl group of hydroxylysine (i.e. collagen).Other O-linked glycoproteins have a glycosidic link between N-acetyl galactosamine and either serine or threonine (i.e. blood group substances and salivary mucins).2. N-glycosidic linkN-glycosidic links exist between N-acetylglucosamine and asparagine. There are two types:A. High mannoseB. Complex. For example, in addition to mannose they may contain N-acetylglucosamine, galactose, fucose and N-acetylneuraminic acid (sialic acid)
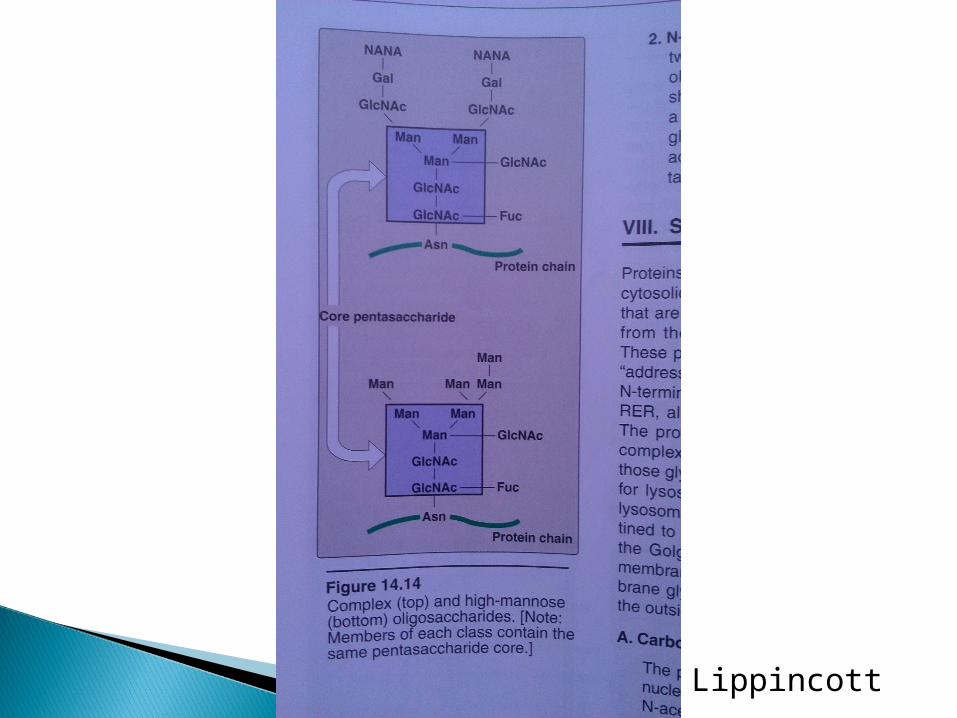
Lippincott

SYNTHESIS OF GLYCOPROTEINS
Synthesized on ribosomes attached to the RER, then transported via vesicles to the Golgi for sorting
The units in the saccharide chains are added fromUDP-glucuronic acid, UDP-N-acetylgalactosamine and GDP-mannose. Sialic acid in glycoproteins is added from CMP-NANA. These additions are catalyzed by specific glycosyltransferases.
For synthesis of O-linked glycoproteins, addition is direct. For N-linked glycoproteins, the chain is formed on dolichol pyrophosphate and then transferred to the protein.

DEGRADATION OF GLYCOPROTEINS
Degradation of the saccharide chains is achieved by hydrolytic enzymes present in lysosomes. The enzymes act on the ends of the chains on a last-on-first-off basis.
Defects can lead to a number of diseases/disorders

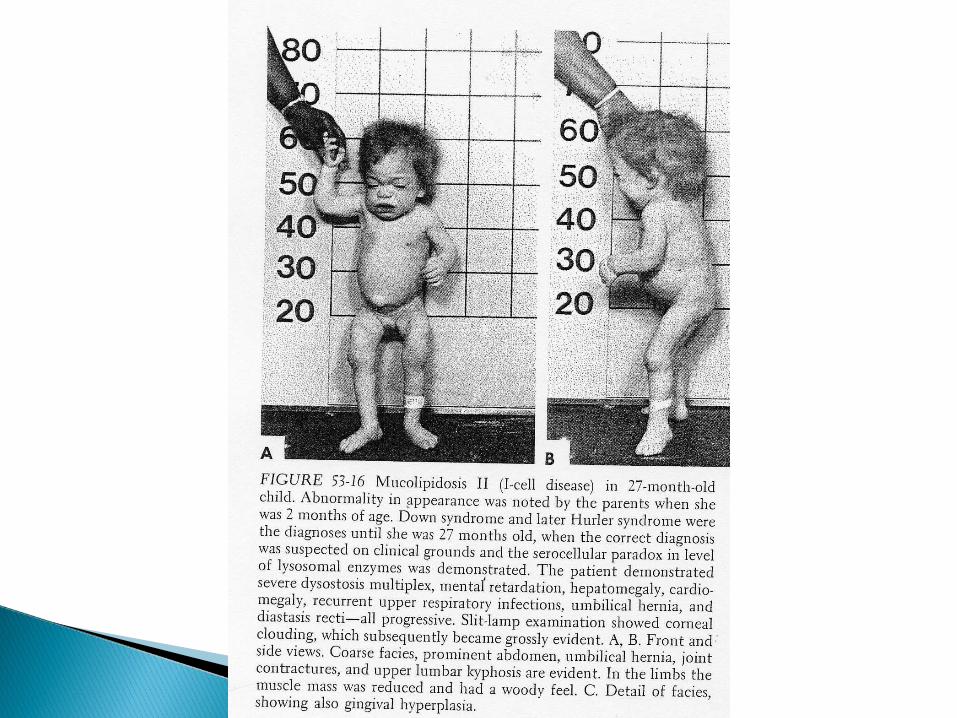
I-cell disease results from an enzyme deficiency so that lysosomal enzymes do not aquire the targeting signal, mannose 6-phosphate.
Fibroblasts in this disease have dense inclusion bodies (I-cells) and are deficient in many lysosomal enzymes.
The lysosomes become engorged with indigestible substrates, leading to death in infancy.

Proteoglycans are usually structural components of the extracellular matrix; some have a lubricant role. --bind large amounts of water--cell/cell signalling and adehsion roles
Heparin is normally intracellular and it inhibits blood clotting.


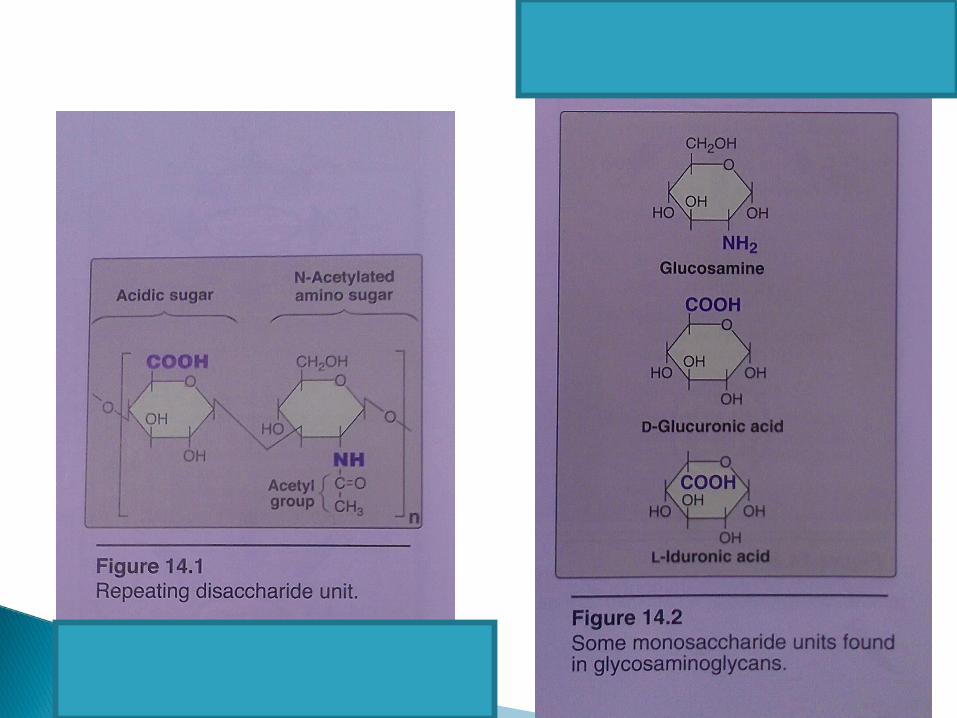
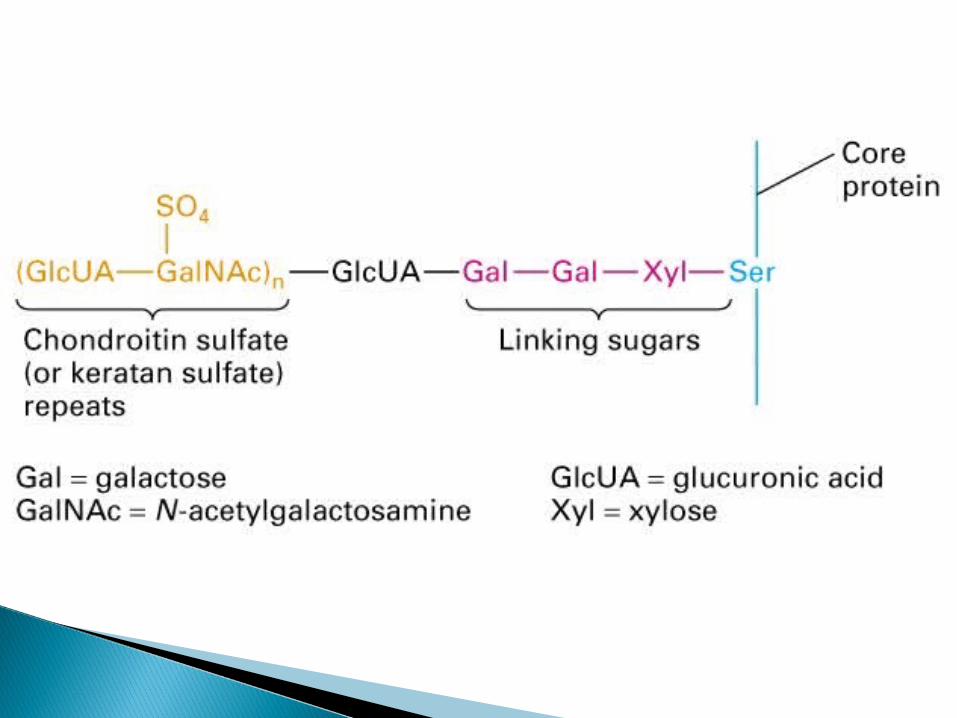
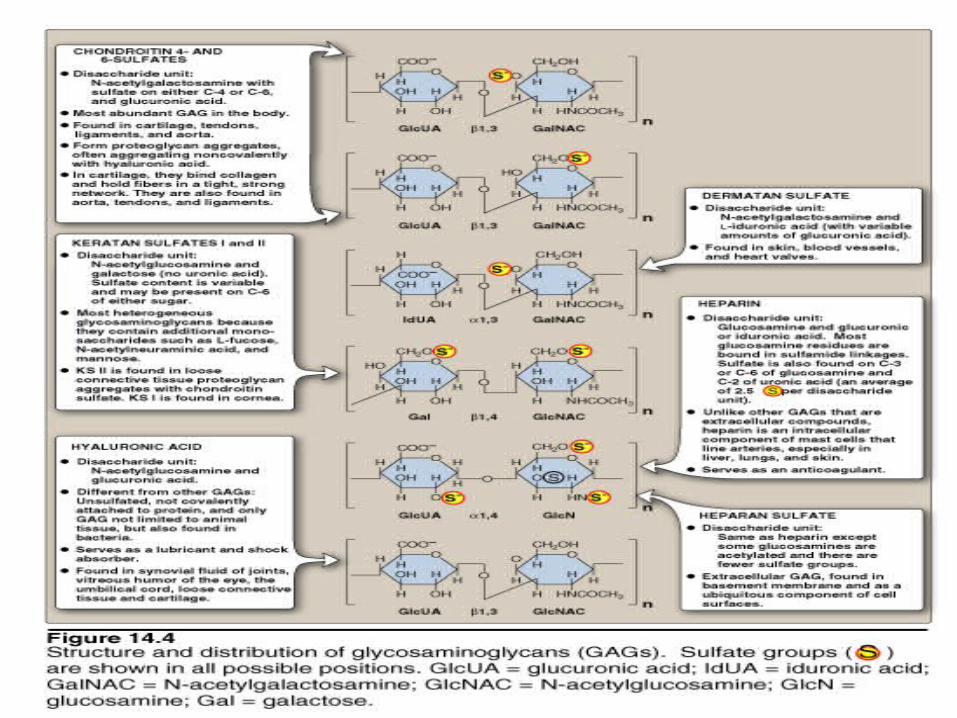

SYNTHESIS OF PROTEOGLYCANS
Synthesized in Golgi
The units in the saccharide chains are elongated in alternating acidic/amino sugars, donated from UDP derivatives. Last step is sulfation of some amino sugars.
For glycosaminoglycan synthesis and synthesis of O-linked glycoproteins, the addition is direct. For N-linked glycoproteins, the chain is formed on dolichol pyrophosphate and then transferred to the protein.
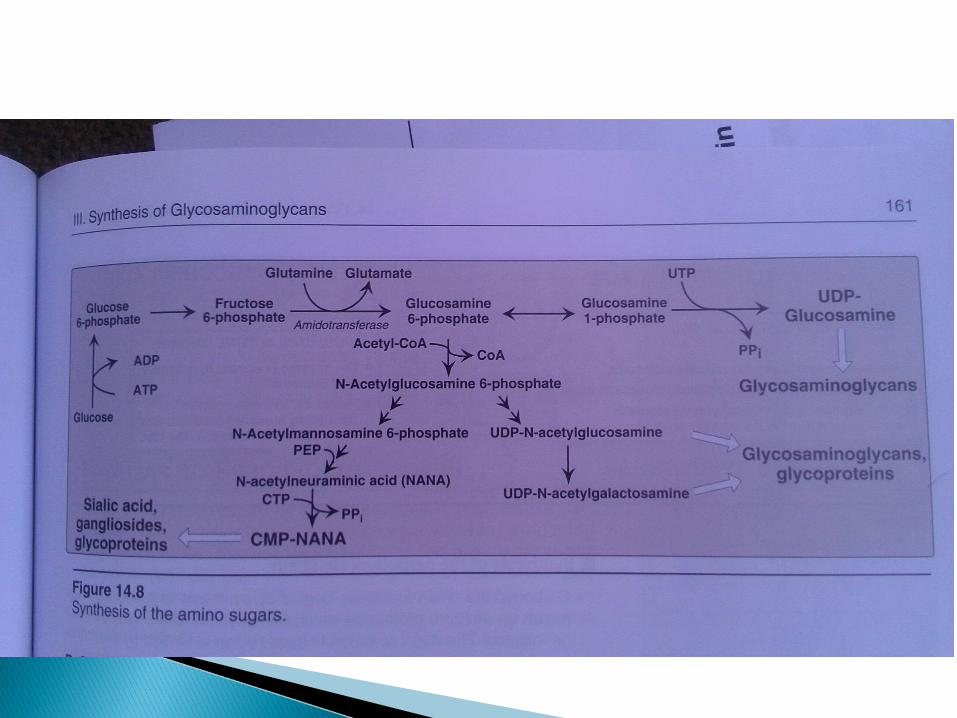

DEGRADATION OF PROTEOGLYCANS
Some proteoglycans must be phagocytosized first Degradation of the saccharide chains is achieved by hydrolytic enzymes present in lysosomes. The enzymes act on the ends of the chains on a last-on-first-off basis.
Defects can lead to a number of diseases/disorders

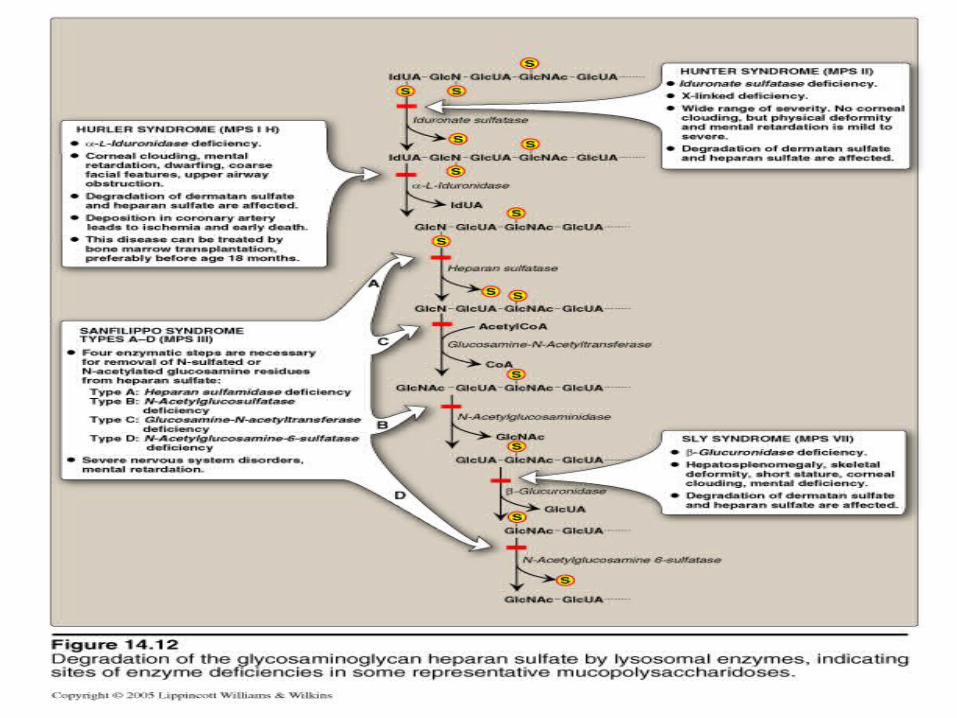
Rare inborn errors in the degradation of glycosaminoglycans result in a series of diseases called mucopolysaccharidoses;
characterized by mental retardation and/or structural defects.
MPS Type IHurler’s syndrome results from a deficiency of alpha-L-
iduronidase. Heparan sulfate and dermatan sulfate accumulate. There is growth and mental retardation with characteristic facial changes.
MPS Type IIHunters syndrome is similar to Hurler’s syndrome but the
enzyme deficiency is for iduronate sulfatase and the inheritance is X-linked.
MPS Type IIISanfilipo’s syndrome is caused by a deficiency of one of four
enzymes of which three are hydrolases and one is an N-acetyltransferase. There is severe mental retardation but only mild structural features.
Other MPS Types are IV, VI and VII. There is no MPS Type V.
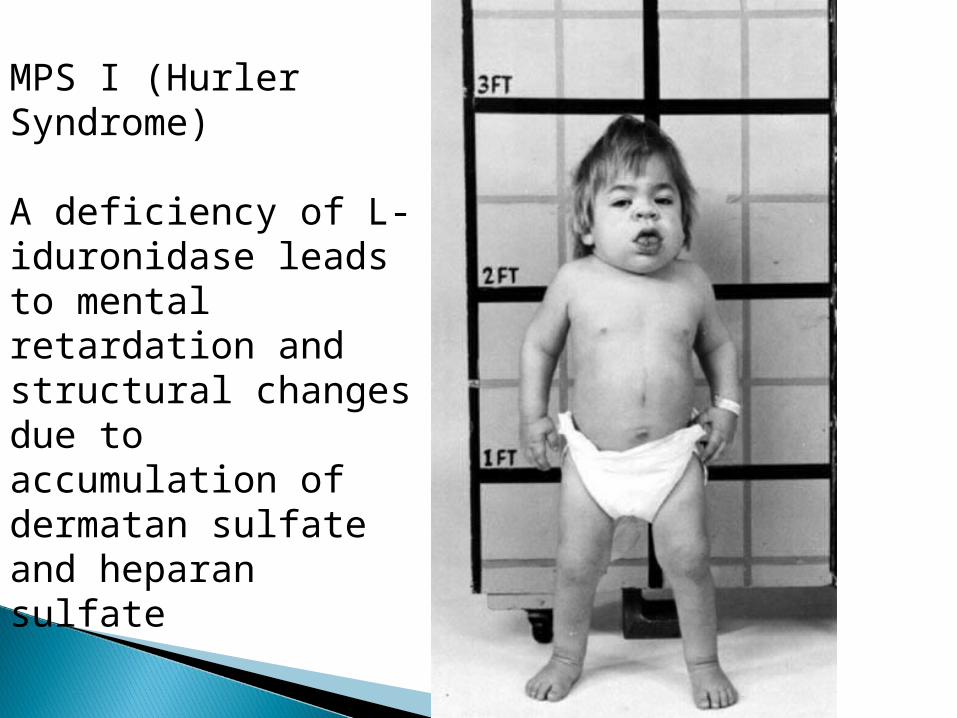
MPS I (Hurler Syndrome)
A deficiency of L-iduronidase leads to mental retardation and structural changes due to accumulation of dermatan sulfate and heparan sulfate
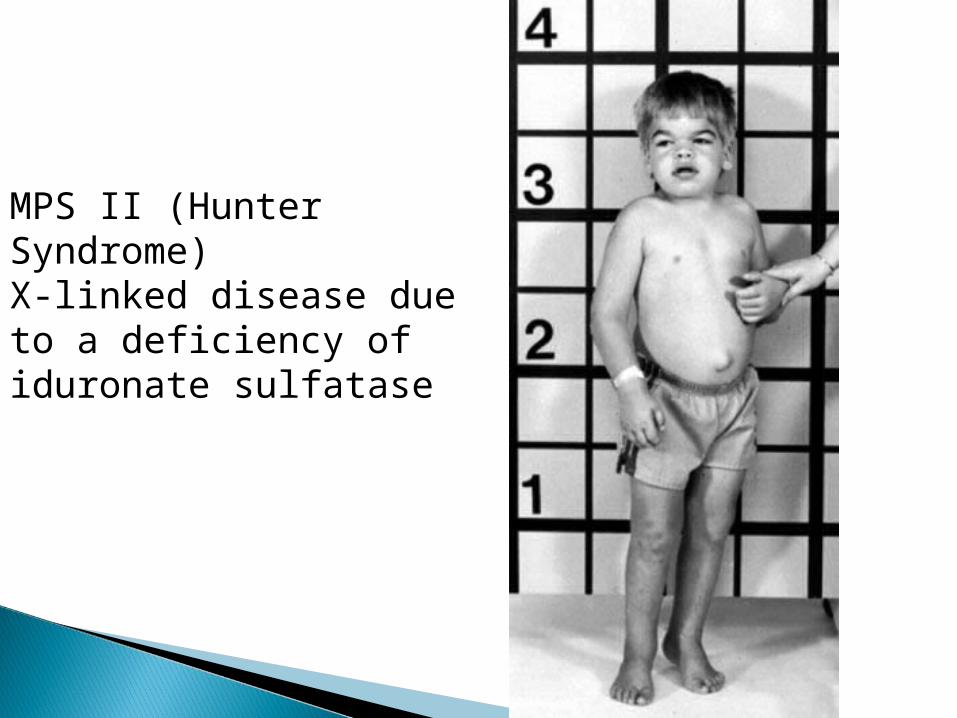
MPS II (Hunter Syndrome)X-linked disease due to a deficiency of iduronate sulfatase
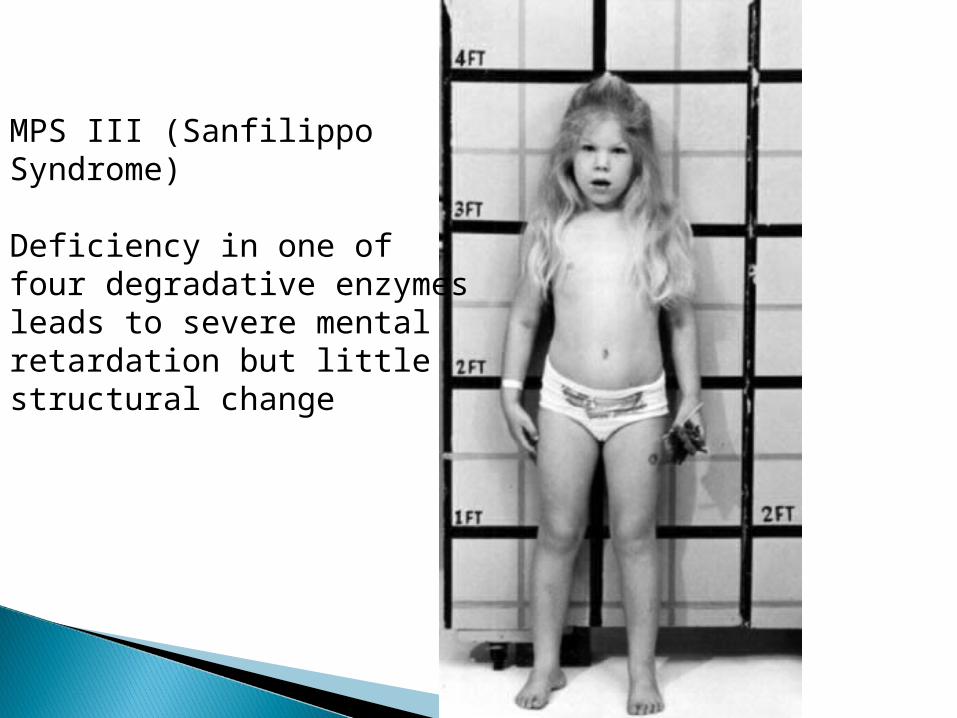
MPS III (Sanfilippo Syndrome)
Deficiency in one of four degradative enzymes leads to severe mental retardation but little structural change
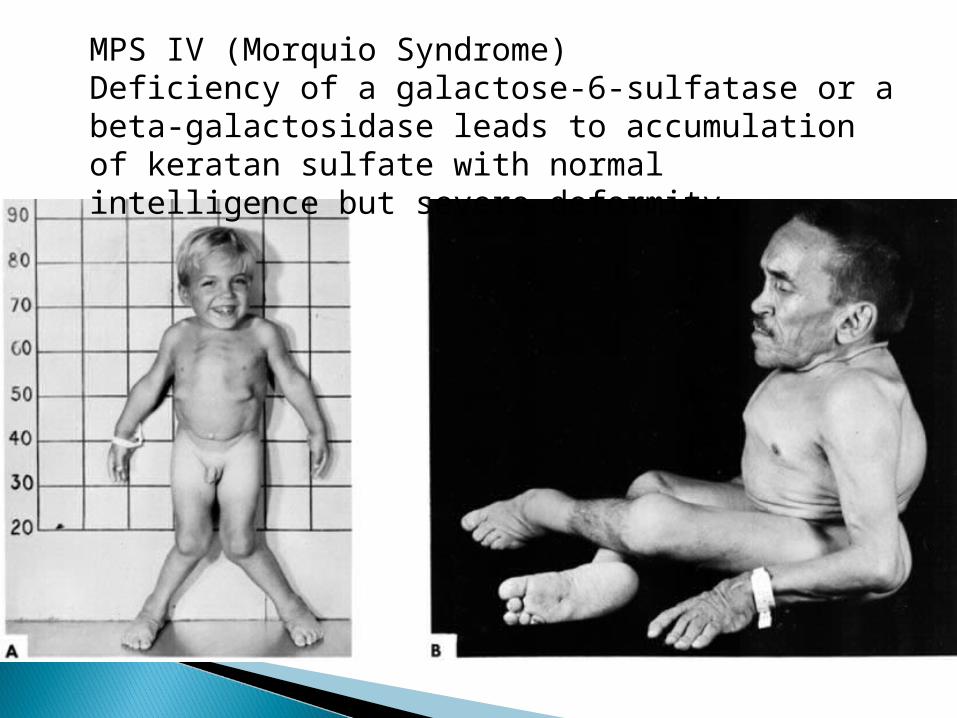
MPS IV (Morquio Syndrome)Deficiency of a galactose-6-sulfatase or a beta-galactosidase leads to accumulation of keratan sulfate with normal intelligence but severe deformity

Summary
Glycoproteins and proteoglycans are distinct:--functions/structures--synthesis/degradation--associated pathologies





























