Embed Size (px)
Citation preview

European J. Biochem. 11 (1969) 405-412
y-Glutamyl Phosphate Attached to Glutamine-Specific tRNA A Precursor of Glutaminyl-tRNA in Bacillus subtilis
Michael WILCOX
Laboratory of Biochemical Genetics, National Heart Institute, National Institutes of Health, Bethesda, Maryland
(Received August 18, 1969)
B. subtilis Gln-tRNA formation, in vitro, involves the initial acceptance of glutamic acid by tRNAGln to form a missense Glu-tRNAGln intermediate which is converted to Gln-tRNA by a subsequent amidation step, catalyzed by a specific amido-transferase, and requiring divalent cations, ATP, and L-glutamine or L-asparagine as amide donor. This reaction is associated with stoichiometric cleavage of glutamine and ATP to yield glutamic acid and Pi, respectively. Amidation proceeds via the formation of an activated intermediate which is shown to be y-phos- pho-Glu-tRNAGln (P-y-Glu-tRNAGln). P-y-Glu-tRNAGln, bound to amido-transferase, is detected following incubation in the absence of amide donor. Subsequent addition of L-glutamine to the system leads to the rapid loss of the phosphate moiety from the intermediate concomitantly with the formation of Gln-tRNA, which is released from the enzyme. Consequences of the pathway, if it is duplicated in vivo, are the separation of the synthesis of glutamine destined for protein from that of free glutamine and, further, the coupling of the synthesis of the amino acid with that of protein. These implications are discussed with regard to their bearing on possible functions of the pathway.
The formation of glutaminyl-tRNA via the direct acceptance of L-glutamine by the corresponding species of tRNA has been demonstrated with extracts from gram-negative bacteria [I-31, yeast [4], a fungus [3], and mammalian [3-51 and avian [3] tissues. A second pathway, found initially in extracts from Bacillus megaterium and B. subtilis [S] and later in extracts from each of three other gram-positive organisms examined [3], involves the acceptance of L-glutamic acid by tRNAGln to form missense Glu- tRNAGln which is subsequently converted to Gln- tRNA in the presence of a specific enzyme, Glu- tRNAG1n amido-transferase, divalent cations, ATP, and L-glutamine or L-asparagine as amide donor. The amidation reaction can be summarized :
Amido-transferase Mn++ or Mg++ [14C]Glu-tRNAGln+ATP+~-glutamine --f
[l4C]G1n-tRNAGln+ ADP + Pi + glutamic acid.
The potential regulatory functions of the path- way [S] prompted further investigations which are reported here.
Unusual Abbreviations. tRNAGln, the transfer RNA re- sponding to glutamine codewords; Glu-tRNAGln, tRNAGln with glutamic acid covalently linked; P-y-Glu-tRNAG'n, Glu- tRNAG1n with a phosphate group at the y-carboxyl of the glutamyl moiety; Pyr-tRNA, tRNA covalently linked to pyrrolidone carboxylic acid; Ado-5'-P-CH2-P-P and Ado-5'- P-P-CH,-P, the C C , ~ and P,y-methylene analogues respectively of ATP; A,,, unit, the quantity of material contained in 1 ml of a solution which has an absorbance of 1 a t 260 nm when measured in a 1 om path length cell.
27 European J. Bioehem., Vol.11
MATERIALS AND METHODS Radioisotopes were obtained from the Companies
shown in parentheses : ~-[~~C,]glutamic acid, 249 mC/ mmole (Nuclear Chicago Corporation) ; ~- [3 ,4 -~H] - glutamic acid, 2 C/mmole, and ~-[l~C,]glutamine, 235 mC/mmole (Schwarz Bio-research) ; [ c ( - ~ ~ P ] - and [Y-~~PIATP, 4.8 and 17-21 C/mmole (ICN).
[Y-~~PIATP was purified by a modification of a procedure described by Khym [7]. 0.5 mC [ Y - ~ ~ P ] - ATP dissolved in 1 mlO.5 M sodium acetate, pH 3.7, containing I mM potassium phosphate, was extracted with 1 ml of a solution of 0.1 M Primene (JM-T, Rohm and Haas Co.) in iso-amyl acetate (the organic solution had been equilibrated to pH 3.7). ATP, but not Pi, was extracted into the organic layer and was recovered by re-extraction into 0.5 M NH4HC0,. The resulting solution was freed of NH,+ ions by addition of Dowex-50 beads.
Enzymes All manipulations were carried out a t 3". Glu-tRNAGln Amido-Transferase. B. subtilis (Mar-
burg) mid-log cells (obtained from General JSio- chemicals Inc.), suspended in buffer A (0.04 M Tris- C1, pH 7.2; 0.01 M 2-mercaptoethanol; I mM MgCl,, and 0.5mM MnCl,), were lysed in a French pressure cell a t 18000 lb./in.2 and the lysate centrifuged for 2 h at 1500OOxg. Protein precipitated from the 150000 x g supernatant by the addition of (NH,),SO, to 60°/, of saturation was pelleted, resuspended in

406 B. subtilis Glutaminyl-tRNA Synthesis European J. Biochem.
1/5 volume buffer A, and dialyzed against the same buffer. Protein was fractionally precipitated from the resulting solution by the addition of increasing amounts of (NH,),SO,. After each addition, precipi- tated protein was pelleted and resuspended in 1/5 volume buffer B (0.02 M imidazole HC1, pH 6.5; 6 mM 2-mercaptoethanol; 0.5 mM MnC1,; and loo/, glycerol). The 46-60°/, (NH,),SO, cut, containing the bulk of the amido-transferase activity, was applied to a 1 4 0 ~ 3 cm Biogel-A (0.5 m exclusion limit; Biorad) column equilibrated and run with buffer B, a t a flow rate of 25 ml/h. Tubes containing activity were pooled, concentrated by dialysis against buffer B containing 15O/, polyethylene glycol-6000, and, after the addition of KC1 to 0.01 M, applied to a granular DEAE-cellulose column (7 x 0.9 cm) equili- brated with buffer B containing 0.01 M KC1. Protein was eluted at 48 ml/h with 200 ml of buffer B that contained a linear KC1 gradient (0.01-0.40 M). Amido-transferase activity eluted as a single peak a t approximately 0.21 M KC1. Tubes containing the bulk of the activity were pooled, dialyzed, and con- centrated as described above, and stored at - 170". Protein was measured by the technique of Lowry et al. [8], and amido-transferase activity by means of the amido-transferase assay. One unit of activity is defined as equivalent t o the conversion of 1 nmole of Glu-tRNAGln to Gln-tRNAG1n per min a t 24". The purification of Glu-tRNAG1n amido-transferase a- chieved bythese procedures was25-fold withrespect to the activity of the 150000 x g supernatant fraction.
Aminoacyl-tRNA Synthetases. Aminoacyl-tRNA synthetases from B. subtilis were prepared according to the procedure of Muench and Berg 191 with modi- fications described earlier [6].
Trans f erBNA tRNAG1n was prepared by benzoylated DEAE-
cellulose chromatography as described previously 161. [l4CC]- and [3H]G1~-tRNAGln and [l4C]G1n-tRNAG1n were prepared as described earlier [6] except that 18 unlabelled L-amino acids were omitted (L-['~C]- glutamine was included for the synthesis of [14C]Gln- tRNAG1"). The 14C-labelled product released from [l4C]Gln-tRNAG1n by mild basic hydrolysis was shown by paper chromatography (System A) and paper electrophoresis a t pH 3.6 to be a mixture of 85-90°/, [14C]glutamine and 10- 15O/, [14C]glutamic acid.
Amido-Transferase Assay Complete reactions contained, in a final volume of
0.05 ml: 0.05 M potassium cacodylate, pH 6.5; 6 mM 2-mercaptoethanol; 0.35 mM MnC1,; 0.1 mM ATP; 0.2 mM ~-[~~C]glutamine; [l4C]G1u-tRNAGln (7 pmo- les ; 0.03 A,,, units), and amido-transferase protein (0.03-0.04pg). Incubation was at 24" for 5min.
Reactions were terminated, aminoacyl-tRNA de- acylated, and formation of [14C]glutamine measured as described previously [6].
Characterization of Products
Descending paper chromatography was carried out using the four systems (A)-(D) detailed earlier [6], and in addition, systems (E) the water poor phase of t-amyl alcohol-formic acid-H,O (90: 30: go), and (F) phenol saturated with 10°/, Na,CO,. Paper electrophoresis was carried out a t 3.5 kV for 1-2 h using acetic acid-pyridine-H,O (10: 1 :289), pH 3.6 and 0.55 M pyridinium acetate, pH 4.2. 14C-labelled products migrated with authentic unlabelled mate- rials in all cases.
Release of [32P]Pi from [y-32P]ATP was assayed by the procedure of Conway and Lipmann 1101.
RESULTS
The B. subtilis Glu-tRNAGln amido-transferase preparation used for these studies was purified 25-fold by the procedures described in Methods, The final step of the purification, DEAE-cellulose chromato- graphy, yielded a single peak of amido-transferase activity clearly separated from Glu-tRNA synthe- tase ; DEAE-cellulose fractionation of B. megaterium enzymes, using a modified procedure, had given two peaks of amido-transferase activity 161. The require- ments and specificity of the B. subtilis amido-trans- ferase, however, were essentially identical to those reported for the B. megaterium fractions 161.
The assay for the conversion of [l4C]G1u-tRNAG1n to [l4C]Gln-tRNAG1n is described under Methods ; the reaction components differ slightly from those given earlier 16). The requirements for amidation, catalyzed by the B. subtilis amido-transferase, are summarized in the Introduction.
Amidation is associated with the stoichiometric cleavage of both glutamine and ATP. The conversion of [3H]Glu-tRNAG1n to [3H]Gln-tRNAGln was found to be accompanied by the release of an equivalent amount of [14C]glutamic acid from ~-[~~C]glutamine, indicating that transfer of the glutamine amide nitrogen probably occurs. A phosphoryl cleavage of ATP was implied by the finding that, in the amida- tion reaction, ATP can be replaced by Ado-5'-P- CH,-P-P (the a$-methylene analogue of ATP) but not by Ado-5'-P-P-CH2-P (the p,y-methylene analo- gue). Confirmation of this came from a demonstra- tion that during the amidation of [l4C]Glu-tRNAG'n a stoichiometric amount of [32P]Pi was released from [y-32P]ATP 131. Although no stoichiometric determination has been performed, studies carried out for other purposes have shown that ADP is also released from ATP during amidation.
Vol.11, N0.3, 1969 M. WILCOX 407
Detection of Phosphorylated Glu-tRNAGLn It had been suggested [B] that amidation may
proceed via activation of the y-carboxyl group of Glu-cc-tRNAG1". Evidence in support of this came from data shown in Table 1. I n the presence of Glu- tRNAGln and amido-transferase (100-200 times more enzyme than is used for amidation, cf. Methods), radioactivity from [Y-~~PIATP but not from [ c ( - ~ ~ P ] - ATP (or, in other experiments, from ring-labelled ATP) is incorporated into cold (3") trichloroacetic acid precipitable material. No incorporation is observed in the presence of the amide donor, L-glut- amine, or when Glu-tRNAG1n is replaced by tRNAGln.
Table 1. Formation of [32P]P-Glu-tRNAG1n from Clu- tRNAGln and [y-32P]ATP
Complete reactions (0.05 ml) contained: 0.05 M potassium cacodylate pH 6.5; 6 mM 2-mercaptoethanol; 0.35 mM MnCl,; 10 pM ATP containing [ C ~ - ~ ~ P ) - or [y-s2P]ATP (2 x lo5 counts/min, about 380 counts x min-l x pmole-l) ; 4 pM potassium phosphate; [3H]Gl~-tRNAGln (4.0 pmoles; 0.013 A,,, units) and 8.0 pg amido-transferase protein. Where stated Glu-tRNAGln was replaced by tRNAGln (0.02 A,, units) or [14C]Gln-tRNAGln (7.2 pmole; 0.14 A,,, units). Incubation was a t 24" for 5 min and reactions were terminated by the addition of 0.05 ml of a 1 : 1 mixture of 0.1 M ATP and 0.1 M potassium phosphate, and 1 ml of loo/, trichloroacetic acid at 3'. Precipitates were washed on nitrocellulose filters with 5O/, trichloroacetic acid at 3", dried and radioactivity determined in a liquid scintillation
counter
[ srP]P- Glu-t RNAQln formed
from from [Y-~'PIATP [a-"PJATP
pmoles pmoles
Modification
Complete 1.72 0.36 - Enzyme 0.22 0.25 + L-Glutamine 0.33 0.31 - Glu-tRNAGln 0.17 0.21 - Glu-tRNAGln + tRNAGln 0.21 - - Glu-tRNAG'n + Gln-tRNAG1n 0.53 -
A low level of incorporation obtained in the presence of Gln-tRNAG1n can be ascribed, in light of later results, to the 10-15°/o Glu-tRNAGln contaminant present in Gln-tRNAGln preparations (cf. Methods).
In anticipation of studies, outlined below, which have shown that this initial observation reflects a mono-phosphorylation of the glutamyl moiety of Glu-tRNAGln, the cold acid precipitable product is referred to from this point as P-Glu-tRNAGln.
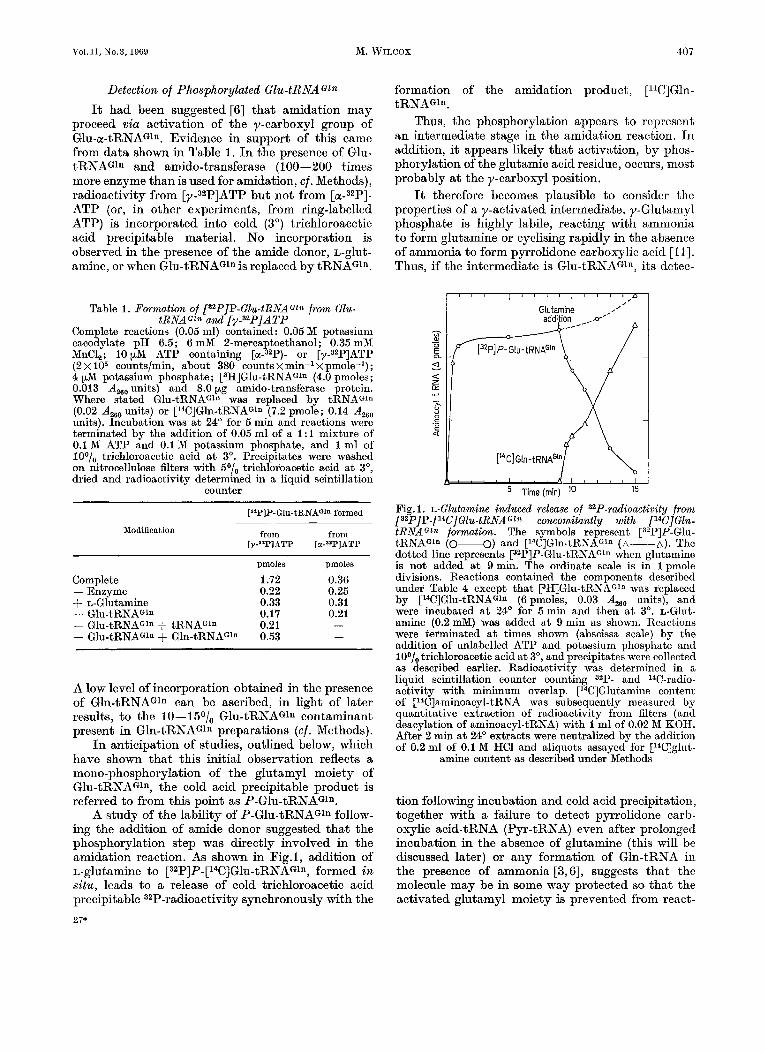
A study of the lability of P-Glu-tRNAG1n follow- ing the addition of amide donor suggested that the phosphorylation step was directly involved in the amidation reaction. As shown in Fig.1, addition of L-glutamine to [32P]P-[14C]Glu-tRNAGln, formed in situ, leads to a release of cold trichloroacetic acid precipitable 32P-radioactivity synchronously with the 27"
formation of the amidation product, [14C]Gln- tRNAGln.
Thus, the phosphorylation appears to represent an intermediate stage in the amidation reaction. In addition, it appears likely that activation, by phos- phorylation of the glutamic acid residue, occurs, most probably a t the y-carboxyl position.
It therefore becomes plausible to consider the properties of a y-activated intermediate. y-Glutamyl phosphate is highly labile, reacting with ammonia to form glutamine or cyclising rapidly in the absence of ammonia to form pyrrolidone carboxylic acid [ill. Thus, if the intermediate is Glu-tRNAGln, its detec-
- - M E a a
a - z rx c - > 0 0 ._ E a
15 Time (min) lo
Fig. 1. L-Clutamine induced release of 3zP-radioactivity from [32P]P-[14C]Glu-tRNAG1n concomitantly with [l*C]Cln- tRNAGzn formation. The symbols represent [32P]P-Glu- tRNAGIn (0-0) and [14C]Gln-tRNAG1n (A-A). The dotted line represents [32P]P-Glu-tRNAGln when glutamine is not added a t 9 min. The ordinate scale is in 1 pmole divisions. Reactions contained the components describcd under Table 4 except that [3H]Glu-tRNAGln was replaced by [14C]Glu-tRNAGln (6 pmoles, 0.03 Aaao units), and were incubated a t 24' for 5min and then a t 3". L-Glut- amine (0.2 mM) was added a t 9 min as shown. Reactions were terminated a t times shown (abscissa scale) by the addition of unlabelled ATP and potassium phosphate and loo/, trichloroacetic acid a t 3", and precipitates were collected as described earlier. Radioactivity was determined in a liquid scintillation counter counting 32P- and 14C-radio- activity with minimum overlap. [WIGlutamine content of [14C]aminoacyl-tRNA was subsequently measured by quantitative extraction of radioactivity from filters (and deacylation of aminoacyl-tRNA) with 1 ml of 0.02 M KOH. After 2 min at 24' extracts were neutralized by the addition of 0.2 ml of 0.1 M HCl and aliquots assayed for [14C]glut-
amine content as described under Methods
tion following incubation and cold acid precipitation, together with a failure to detect pyrrolidone carb- oxylic acid-tRNA (Pyr-tRNA) even after prolonged incubation in the absence of glutamine (this will be discussed later) or any formation of Gln-tRNA in the presence of ammonia [3,6], suggests that the molecule may be in some way protected so that the activated glutamyl moiety is prevented from react-

408 B. subtilis Glutaminyl-tRNA Synthesis European J. Biochem.
Table 2. Protection of [W]Glu-tRNAG$n from degradation by RNase A
Complete reactions (0.05 ml) contained potassium cacodylate, 2-mercaptoethanol, and MnCl, as described under Table 1, and in addition 0.1 mM ATP, [14C]Glu-tRNAG1n (7.5 pmoles, 0.035 A,,, units) and 8.0 pg amido-transferase protein. Modifications are described in the Table. Where stated 0.2 mM L-glutamine was added. Incubation was for 8 min at 24'; except where stated, 0.1 pg RNase A was added after 5 min. Reactions were terminated by the addition of 1 ml of loo/, trichloroacetic acid a t 3'. Precipitates were washed on nitrocellulose filters with 5"/, trichloroacetic acid a t 3', dried, and radioactivity determined in a liquid scintillation counter. Other experiments showed that, under these conditions, the amount of RNase resistant Glu- tRNAGln obtained in the complete reaction remained essentially constant from 1.5 to 8min after the point of RNase addition, whereas, in the absence of enzyme or ATP (or in the presence of glutamine), degradation was complete
by 1.5 rnin
ILNase resistant aminoacyl-tRNA
114C1G1u-tRNAG~n I'VlGln-tRNAG'n Modification
~
pmoles pmoles
- RNase A 7.51 7.84 Complete 3.61 0.50 - Enzyme 0.06 0.06 - ATP 0.44 0.45 + L-Glutamine 0.13 0.06
Table 3. Formation of [l4C]Pyr-tRNAG1n from [14C]Glu- tRNAGln
The protection of [WIGlu-tRNAGln from degradation by RNase A was assayed as described under Table 2. For assay of [14C]Pyr-tRNAG1n formation, 0.05 ml reactions contained potassium cacodylate, 2-mercaptoethanol, MnCl,, ATP, [ ~ ~ C I G ~ U - ~ R N A G ~ ~ and amido-transferase as described in the legend t o Table 2. After a 5 min incubation at 24", 0.05 ml of 0.1 N HC1 was added t o reactions which were subsequently heated for 2 min a t 75-80'. Reactions were cooled and aminoacyl-tRNA deacylated by the addition of 0.5 ml of 0.02 M KOH. After 2 rnin a t 24' each reaction was neutralized by the addition of 0.05 ml of 0.1 M HCI and applied to a 2 ~ 0 . 5 om column of Dowex-BOW (X-8, Hf form). Under these conditions glutamic acid and glutamine but not pyrrolidone carboxylic acid are retained by the column. Each column was washed with 2 x 0.5 ml H,O and the effluant was collected and counted in a liquid scintilla- tion counter. For characterization, [14C]pyrrolidone carb- oxylic acid was released from isolated [14C]aminoacyl- tRNAG1n by mild alkaline hydrolysis and analyzed by paper chromatography (System D) and by paper electrophoresis
(PH 4.2) ~~~~ ~ ~
RNase resistant [WIPyr- tRNAG'n Reaction [ "C] Glu-
tRNAG'n formed pmoles pmoles
Comdete 3.24 3.45 - Ekzyme - ATP
0.06 0.16 0.38 0,22 + L-Glutamine 0.15 0149
ing. Evidence for protection of the intermediate by interaction with amido-transferase was provided by data in Table 2. Following incubation under condi- tions where P-Glu-tRNAGln is formed, a part of [14C]Glu-tRNAG1n, but not of [l4C]Gln-tRNAG1n, is protected from degradation by RNase A, as deter- mined by cold trichloroacetic acid precipitation. No protection is observed in the absence of ATY or the presence of L-glutamine. When unlabelled ATP is replaced by [Y-~~PIATP, approximately equivalent amounts of 32P-radioactivity and [14C]Glu-tRNAG1n were found to be resistant to RNase (data not shown).
Thus, the requirements for the protection of Glu- tRNAGln (or, a t least, a part of the molecule including the CpCpA-amino acid, 3' end) from degradation by RNase A, and for the production of an equivalent amount of P-Glu-tRNAGln, are identical, and indicate the formation of P-Glu-tRNAGln bound to amido- transferase as an intermediate step in amidation. It should be noted that not all [14C]Glu-tRNAG1n was resistant to RNase; maximum level of protection achieved in other experiments, under a variety of conditions, was 50-6Qo/,. This finding remains to be clarified.
Characterisation of the Intermediate As anticipated in view of the already mentioned
lability of the y-glutamyl phosphate moiety, at- tempts t o 'isolate [32P]P-Gl~-tRNAGln, following phenol extraction and ethanol precipitation, were
unsuccessful. Characterisation was achieved, however, by identification of the expected products of reaction of the intermediate, as follows.
Experiments to determine the position of phos- phorylation were based on the assumption that the stability of the postulated y-activated intermediate results from its interaction with amido-transferase ; thus, under conditions (e.g. low pH) where the com- plex would be expected to dissociate subsequent cyclisation of the intermediate to form Pyr-tRNA should occur. As shown in Table 3, when P-[14C]Glu- tRNAGln, formed in situ, is treated briefly with acid a t 75-80' and the aminoacyl bond then hydro- lyzed by base treatment, [14C]pyrrolidone carboxylic acid is released. An equimolar amount of [14C]G1u- tRNAGln is protected from degradation by RNase under the same conditions. In other experiments, 14C-labelled aminoacyl-tRNA was re-isolated following acid treatment ; subsequent base treatment yielded the same product indicating that Pyr-tRNA, rather than free pyrrolidone carboxylic acid, was initially formed. [14C]Pyr-tRNAGln formation is dependent, as expected, upon amido-transferase and ATP. No Pyr-tRNAGln is formed in the presence of L-glut- amine. I n experiments not shown here it was demon- strated that cyclisation occurs rapidly a t room temperature in the presence of mild acid or base.
To determine the nature of the 32P-labelled residue, trichloroacetic acid precipitated RNase A resistant [32P] P-Glu-tRNAG1n was collected on a
Vol. 11, No. 3, 1969 1\1. WILCOX 409
Table 4. Formation of y-NHOH-(14C]Glu-tRiVAG~n The reaction (0.50 ml) contained potassium cacodylate, 2-mercaptoethanol, MnCl, and ATP as described in the legend to Table 2 and, in addition, [l4C]G1u-tRNAG1n (75 pmole; 0.35 A,,, units) and amido-transferase protein (80 pg). After incubation for 5 min a t 24O, an aliquot (0.05 ml) was removed and assayed for RNase A resistant [14C]amino- acyl-tRNA as described earlier, and the remainder of the reaction was treated with 0.5 M hydroxylamine HC1,pH 6.5, for 2 min a t 24". [14C]Aminoacyl-tRNA was then precipitated by addition of 3 ml of loo/, trichloroacetic acid a t 3"; the precipitate was collected on a nitrocellulose filter, washed with 5 O / , trichloroacetic acid a t 3" and with 95O/, ethanol a t - lo", and dried. The filter was immersed in 0.7 ml of 0.1 M Tris-C1, pH 9.0, and [14C]aminoacyl-tRNA was extracted and deacylated. The extract was applied to a 3 x 2 cm Dowex-1 column and the effluant, together with 2 x 2 ml H,O washings, was lyophilized. The residue was taken up in H,O, an aliquot counted to determine 14C-radioactivity, and the remainder analyzed by paper chromatography
(System F) and paper electrophoresis (pH 4.2)
nNase resistant '~C-label,ed ''C-labelled products from Dowex-1 ["C]Glu- product eluted tRNAG'n from Dowex-1 r-"4c1G1u- 14c]pyr other NHOR
~~ ~
pmoles pmoles "1. "0 "0
22.5 24.3 83 11 6
nitrocellulose filter, washed with 5OI0 trichloroacetic acid a t 3" and then with 9501, ethanol. Radioactivity was extracted quantitatively from the filter into 0.1 M Tris-C1 pH 9.0 a t 3". Subsequent analysis revealed that, by this treatment, more than goo/, of 32P-radioactivity was released as [32P]Pi (assayed by the procedure of Conway and Lipmann [lo] and characterized by paper chromatography (System E)) ; no [32P]PPi was detected. As a control, [y-32P]ATP was carried through the same manipulations; no [32P]Pi was released.
Thus it was confirmed that mono-phosphoryla- tion of the glutamic acid residue of Glu-tRNAG1n occurs ; the formation of Pyr-tRNA argues strongly that the intermediate is P-y-Glu-tRNAGln. However, to rule out phosphorylation elsewhere (e.9. of the a-amino group) the experiment shown in Table 4 was performed. P-[l4C]Glu-tRNAG1n, formed in situ, was treated with 0.5 M hydroxylamine (with 32P-[14C]- Glu-tRNAG1n this led to a rapid loss of 32P but not of 14C radioactivity from cold trichloroacetic acid precipitable material). Hydroxylamine treatment led, after deacylation, to the formation of y- [14C]glutamyl- hydroxamate in an amount approximately equal to that of [14C]Glu-tRNAG1n rendered resistant to RNase under the same conditions. The small amount of [14C]pyrrolidone carboxylic acid detected probably resulted from cyclisation of the hydroxamate during the work up procedures [Ill.
This experiment provided final proof that the intermediate was y-phospho-Glu-tRNAGln (P-y-Glu- cr-tRNAGln).
I , I I I I
Void AA-tRNA Amino acid
Ct Glutamine ::k 0 5 0 20 40 60
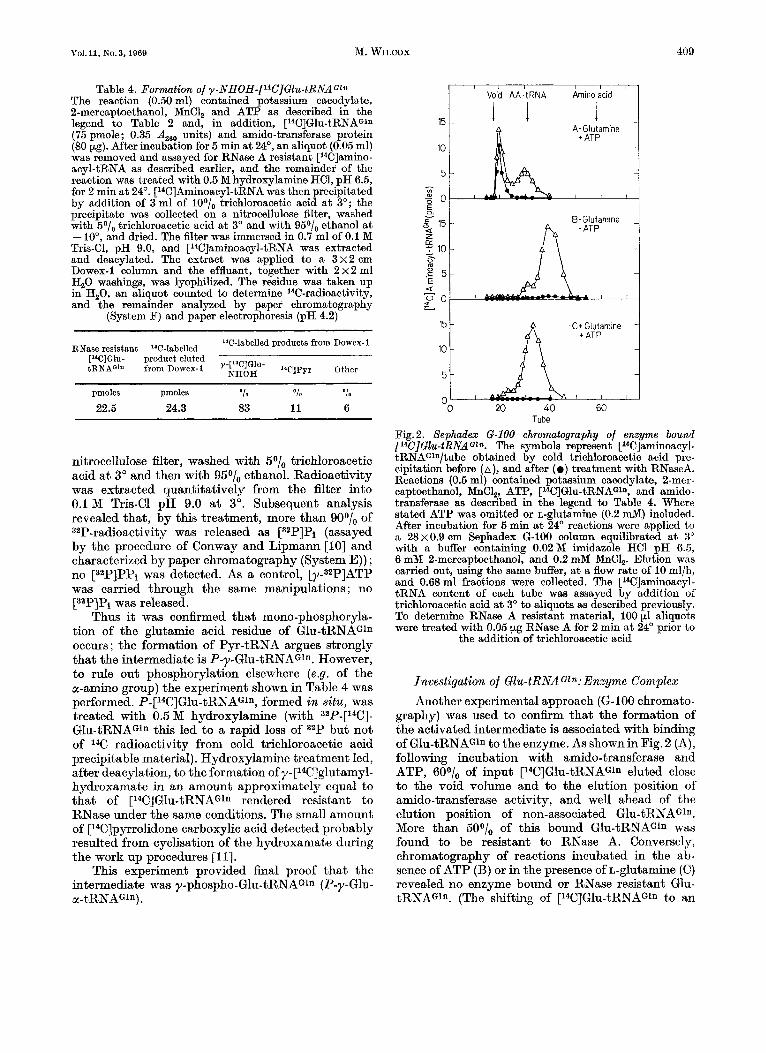
Tube Fig. 2. Sephadex (2-100 chromatography of enzyme bound [14C]Gl~-tRNAG~*. The symbols represent [14C]aminoacyl- tRNAG'n/tube obtained by cold trichloroacetic acid pre- cipitation before (A), and after ( 0 ) treatment with RNaseA. Reactions (0.5 ml) contained potassium cacodylate, 2-mer- captoethanol, MnCl, ATP, [14C]G1~-tRNAG1n, and amido- transferase as described in the legend to Table 4. Where stated ATP was omitted or L-glutamine (0.2 mM) included. After incubation for 5 min a t 24" reactions were applied to a 28x0.9 cm Sephadex 6-100 column equilibrated a t 3" with a buffer containing 0.02M imidazole HCl pH 6.5, 6 mM 2-mercaptoethanol, and 0.2 mM MnCl,. Elution was carried out, using the same buffer, a t a flow rate of 10 ml/h, and 0.68 ml fractions were collected. The [14C]aminoacyl- tRNA content of each tube was assayed by addition of trichloroacetic acid a t 3' to aliquots as described previously. To determine RNase A resistant material, 100 pl aliquots were treated with 0.05 pg RNase A for 2 min a t 24" prior to
the addition of trichloroacetic acid
Investigation of Glu-tRNAGln: Enzyme Complex Another experimental approach (G-I00 chromato-
graphy) was used to confirm that the formation of the activated intermediate is associated with binding of Glu-tRNAGln to the enzyme. As shownin Fig.2 (A), following incubation with amido-transferase and ATP, 60°/, of input [14C]Gl~-tRNAGln eluted close to the void volume and to the elution position of amido-transferase activity, and well ahead of the elution position of non-associated Glu-tRNAGln. More than 50°/0 of this bound Glu-tRNAGln was found to be resistant to RNase A. Conversely, chromatography of reactions incubated in the ab- sence of ATP (B) or in the presence of L-glutamine (C) revealed no enzyme bound or RNase resistant Glu- tRNAGln. (The shifting of [WIGlu-tRNAGln to an

410 B. subtilis Glutami nyl-tRNA Synthesis European J. Biocliein.
apparent lower molecular weight following incuba- tion in the absence of ATP is not explained.)
Surprisingly, when unlabelled ATP was replaced by [Y-~~P]ATP, it was found that the eluted amido- transferase bound [14C]G1~-tRNAG1n was no longer phoephorylated. Basic hydrolysis of the of the com- plex yielded only [14C]glutamic acid; that is, the phosphate group had been lost during chromato- graphy by direct hydrolysis, rather than as a result of cyclisation to form Pyr-tRNAGln. An explanation for this was provided by findings that can be briefly summarized as follows.
An initial finding that 32P-radioactivity was rapidly lost from [32P]P-G1~-tRNAG1n after the addition of unlabelled ATP, was clarified by a study of the Glu-tRNAGln directed release of Pi from ATP under conditions where P-Glu-tRNAGln formation occurs. Glu-tRNAGln stimulated the rate of release of [32P]Pi from [Y-~~P]ATP 1.6-fold over that observed with enzyme alone. No stimulation was observed when Glu-tRNAGln and L-glutamine were both present. The amount of [32P]Pi released in response to the addition of Glu-tRNAGln was %fold higher (after 5 min) and 20-fold higher (after 20 min) than the constant level of phosphorylation of Glu-tRNAGln achieved under the same conditions. Conversely, although the amido-transferase preparation catalyzed an exchange between ADP and ATP, no sti- mulation of the exchange was observed after the addition of Glu-tRNAGl*.
These data are consistent with the concept that the y-phosphate group of enzyme bound P-Glu- tRNAG1n is highly susceptible t o chemical hydrolysis and that a continuous process of hydrolysis and re- phosphorylation occurs.
The loss of the y-phosphate moiety from the P-Glu-tRNAG1n : enzyme complex does not affect appreciably the stability of the complex (as demon- strated by G-100 chromatography). Hence it appeared likely that a distinction could be drawn between the binding and phosphorylation steps.This wasconfirmed by a demonstration (Table 5 ) that both Ado-5'-P- CH,-P-P and Ado-5'-P-P-CH,-P partially replace ATP in directing the binding of Glu-tRNAGln to amido-transferase (as assayed by the protection of Glu-tRNAGln from degradation by RNase A). With Ado-5'-P-CH2-P-P, where phosphorylation would be expected to occur, addition of glutamine to the system removed all protection, and acid treatment led to the formation of the expected amount of Pyr- tRNA.
With Ado-5'-P-P-CH,-P, where a phosphoryl cleavage is not possible, the level of RNase resistant Glu-tRNAG'n, in the presence of glutamine, increased to SOo/, of that obtained with ATP (suggesting a possible allosteric role for the amide donor), while acid treatment produced no Pyr-tRNA indicating that activation did not occur.
Table 5. Protection of (14C]Gl~-tRNAGl* against RNase by ATP analogues
Protection of [14C]Gl~-tRNAGl* from degradation by RNase A, and formation of [14C]Pyr-tRNAG1n were assayed as described in the legends to Tables 2 and 3, respectively, with the exception that in each case nucleotides (nucleotide modifications are given in the Table) were present a t a
concentration of 40 pM
Reaction [ 'YWyr- RNase resistant
[ 'dC]Glu-tRNAG~n tRNAG'n --Cln +G,n formation
A pmoles
Complete 2.00 0.09 2.03 - ATP 0.17 - 0.17
Ado-5'-P-P-CHZ-P 0.78 1.60 0.16 - Al'P + - ATP +
Ado-5'-P-CH2-P-P 1.26 0 I .24
Thus, although binding of Glu-tRNAG1n to amido- transferase in the presence of ATP yields P-Glu- tRNAGln, an initial role for ATP may be to direct the association of Glu-tRNAGln and amido-trans- ferase, phosphorylation occurring either coincidently with or after this step.
Another question of interest is whether the highly reactive P-Glu-tRNAG1n intermediate might be released from the enzyme. The data presented to this point, particularly that demonstrating the stability of the Glu-tRNAGIn :enzyme complex, argue against this. I n other experiments, summarized here, prolonged incubation (30-60 min) of [l*C]- Glu-tRNAG1n and ATP with catalytic amounts of amido-transferase in the absence of glutamine pro- duced no [14C]Pyr-tRNAGln or [l4C]G1n-tRNAGln (the latter could have derived from reaction of free P-Glu-tRNAGln with any ammonia in the system). When high concentrations of amido-transferase werc used, short or prolonged incubation (3-60 min) yielded identical amounts of Pyr-tRNA (derived from the constant amount of P-Glu-tRNAGln formed under these conditions). Preliminary experiments suggest, in addition, that enzyme bound P-Glu- tRNAG1n does not exchange with free Glu-tRNAG1n.
Hence, it is clear that no free P-Glu-tRNAGln is produced in the system in vitro.
Cellular Levels of Amido-Transferase and tRNAGln Amido-transferase and tRNAGln levels were
determined in extracts prepared from identical batches of B. subtilis cells. Whole cell nucleic acid, obtained as described earlier for B. megaterium [6], contained tRNAGln accepting 670- 1100 pmoles of L-glutamic acid/g of cells. A 150000 x g supernatant prepared as described under Methods, contained

Vol.11, No.3, 1969 M. WILCOX 411
20 units of amido-transferase activitylg of cells (units are defined in the experimental section) ; this quantity of enzyme, after purification, bound 740 to 1040pmoles of Glu-tRNAGln, as measured by the RNase assay.
Thus, the cell contains sufficient amido-trans- ferase to interact with all potentially available Glu- tRNAG1n.
DISCUSSION
The amidation of Glu-tRNAGln, the final step of B. subtilis Gln-tRNA formation, proceeds, in vitro, by a mechanism which may be broadly summarized as follows :
Glu-tRNAGln + enzyme + ATP N*++ *
* P-y-Glu-tRNAGln :enzyme + ADP.
P-y-Glu-tRNAG1” : enzyme + L-glutamine M E + + Gln-tRNAGln + enzyme + Pi + glutamic acid.
Other findings suggest that ATP may play an allo- steric role in the formation of a stable complex of Glu-tRNAGln and amido-transferase. Following the phosphorylation step ADP may remain bound to the enzyme, its presence may be required for the integrity of the complex. Consistent with this view are the findings that ADP is required for the de- amidation of Gln-tRNAGl” [3] which occurs under arsenolysis conditions [12], and that ADP is a competitive inhibitor of the amidation reaction [3].
With the obvious exception that ammonia does not function as a donor of amide nitrogen [3,6], the mechanism bears obvious similarities to that pro- posed for the glutamine synthetase catalysed forma- tion of glutamine from glutamic acid and ammonia [ill-
The pathway, if duplicated in vivo, could fulfil several functions. The failure to detect Gln-tRNA synthetase activity in B. subtilis and B. megaterum extracts under a variety of conditions [3,6] suggests that the enzyme is probably absent. Nevertheless, the wide occurrence of the pathway in gram-positive bacteria [3] makes it unlikely that it operates as a suppression mechanism compensating for a defective Gln-tRNA synthetase. If, as seems likely, the pathway is restricted to gram-positive and closely related organisms then the possibility should be considered that it has evolved either to fulfil a function required by, or as a result of, some specialised feature of the metabolism of these organisms.
Other potential regulatory roles may be men- tioned. As discussed earlier [6], the pathway serves to separate the synthesis of glutamine for insertion into protein from that of free glutamine and, in addition, to couple directly the synthesis of the
amino acid with that of protein; i.e. the rate of synthesis of glutamine for insertion into protein may depend upon the rate of glutamine incorporation into protein and upon the availability of glutamic acid and amide donor. Although glutamine synthe- tase is found in Bacillus extracts [13], the cellular pool of glutamine is very low [14]. This fact, together with their own findings, has led Rebello and Strauss [15] to postulate that the synthesis of glutamine is essentially rate limiting in B. subtilis. Such a situa- tion might necessitate a separate pathway for the synthesis of glutamine for protein, and hence place some import on the finding that asparagine can re- place glutamine as an amide donor (so that free glutamine may not be required for protein synthesis). Only one other example of asparagine acting as an amide donor, in the first step of purine biosynthesis in wheat germ, has been reported [16].
On the assumption that the P-y-Glu-tRNAGln: enzyme complex plays no role in protein synthesis, the mechanism potentially provides a high level of control over the concentration of Gln-tRNA avail- able for protein synthesis and, further, could serve to preserve the fidelity of glutamine insertion into protein (the intermediate Glu-tRNAGln responds also t o the glutamine codewords, CpApA and CpApG [17]; hence, a t low amide donor levels, the sub- stitution of glutamic acid for glutamine in protein could occur). The feasibility of such a “fidelity” mechanism, however, would hinge upon the extremely rapid binding of Glu-tRNAGln to amido-transferase or, alternatively, upon a preferred binding situation stemming, for example, from some close association of amido-transferase and Glu-tRNA synthetase. An alternative mechanism could result if Glu-tRNAGln is inactive in protein synthesis (for example, because of an inability to bind to transfer factor) and is activated, during amidation, as a result of some modification of the tRNA molecule. Further exper- imentation is required to confirm or rule out such possibilities.
The author gratefully acknowledges the aid and encou- ragement received from Dr. Marshall Nirenberg in whose laboratory this work was carried out.
1.
2.
3.
4.
5. 6.
7. 8.
REFERENCES Lazzarini, R. A., and Mehler, A. H., Biochemistry, 3
Ravel. J. &I.. Wane. S.-F.. Heinemever. C.. and Shive. (1964) 1445.
I - ,
W.,’J. Biol. Che;. 240 (1965) 432.
(1969) in press.
Biophys. 121 (1967) 614.
U. S. 61 (1968) 229.
dall, R. J., J . BioZ Chem. 193 (1951) 265.
Wilcox, M., Cold Spring Harbor Symp. Quant. Biol. 34
Lee, L. W., Ravel, J. M., and Shive, W., Arch. Biochem.
Deutscher, M. P., J . Biol. Chem. 242 (1967) 1123. Wilcox, M., and Nirenberg, M., Proc. Natl. Acad. Sci.
Khym, J. X., Biochemistry, 2 (1965) 401. Lowry, 0. H., Rosebrough, N. J., Farr, A. I,., and Ran-

412 M. WILCOX: R. subtilis Glutaminyl-tRNA Synthesis Europrsn J. Biochcm.
9. Muench, K. H., and Berg, P., Procedures in Nucleic Acid Research. (edited by G. L. Cantoni and D. R. Davies), Harper and Row, New York 1966, p. 375.
10. Conwav. T. W.. and Linmann. 3.. Proc. Natl. Acad. Sci. , I
U. g’52 (1964) 1462: 11. Meister, A.. T h e Enzwmes (edited bv P. D. Bover.
H. Lardy, and K. Myrback), AcadeGic Press, &ew York 1962, Vol. 6, p. 443.
12. Levintow, L., and Rieister, A., J . Biol. Chem. 209 (1954) 265.
13. Hubbard, J. S., and Stadtman, E. R., J . Bacteriol. 93 (1967) 1045.
14. Pfennig, N., Arch. Mikrobiol. 26 (1957) 345. 15. Rebello, J. L., and Strauss, N., J . Bacteriol. 98 (1969)
16. Kapoor, &I., and Waygood, E. R., Biochem. Biophys.
17. Wilcox, M., and Nirenberg, M., Federation Proc. (Abstr.),
683.
Res. Commun. 9 (1962) 7.
27 (1968) 341.
M. Wilcox’s present address: M. R. C . Laboratory of Molecular Biology Hills Road, Cambridge CB2 2QH, Great Britain





![RESEARCH ARTICLE Open Access Fragmentation of ... - SLU.SE · 18–46 nt pieces derived from mature tRNA or the 3 ′ end of precursor-tRNA (pre-tRNA) [14-16]. tRNA fragmenta-tion](https://img.pdfslide.net/doc/110x75/60474a078cb48655a57c0958/research-article-open-access-fragmentation-of-sluse-18a46-nt-pieces-derived.jpg)























