Embed Size (px)
Citation preview

(M)- and (P)‑Bicelaphanol A, Dimeric Trinorditerpenes with PromisingNeuroprotective Activity from Celastrus orbiculatusLuo-Yi Wang, Jian Wu, Zhuo Yang, Xu-Jie Wang, Yan Fu, Shuang-Zhu Liu, Hong-Min Wang,Wei-Liang Zhu, Hai-Yan Zhang,* and Wei-Min Zhao*
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203,People’s Republic of China
*S Supporting Information
ABSTRACT: (M)-Bicelaphanol A (1) and (P)-bicelaphanolA (2), two unprecedented dimeric trinorditerpenes existing asatropisomers, together with their monomer celaphanol A (3),were isolated from the root bark of Celastrus orbiculatus. Thestructures and absolute configurations of 1 and 2 weredetermined by spectroscopic and single-crystal X-ray diffrac-tion analyses. Compound 1 exhibited a significant in vitroneuroprotective effect against a hydrogen peroxide-inducedcell viability decrease in PC12 cells at 1 μM, while compounds2 and 3 showed such effects at 10 μM.
Dimeric natural products, a special class of moleculesusually possessing complex structures and significant
bioactivities, have been attracting continuous attention fromboth the chemical and biogenetic communities.1 Because of thedevelopment of two-dimensional (2D) nuclear magneticresonance (NMR) and mass spectrometry (MS) techniques,publications about new dimeric compounds have increasedremarkably in number since the 1970s.2 Information gatheredwith dimeric chemical probes is providing crucial pharmaco-logical data about many different biological pathways and hasbeen inspiring medicinal chemists in their quest to providebetter medicines.3
Celastrus orbiculatus Thunb. (Celastraceae) has been used asa traditional Chinese medicine for the treatment of rheumatoidarthritis, vomiting, abdominal pain, and snakebite.4 Previousphytochemical investigations of this plant have resulted in theidentification of sesquiterpene esters,5 flavan-3-ols,6 alkaloids,7
carotenoids,8 phenolic and terpenic glycosides,9 norditerpenes,and triterpenes and their dimers.10 In this study, twounprecedented dimeric trinorditerpenes existing as atro-pisomers, (M)-bicelaphanol A (1) and (P)-bicelaphanol A(2), together with their monomer celaphanol A (3) (Figure 1),were isolated from the root bark of C. orbiculatus. Among them,1 exhibited potent in vitro neuroprotective activity at 1 μM.(M)-Bicelaphanol A (1), obtained as red crystals, has a
molecular formula of C34H38O7 as determined by HRESIMS atm/z 557.2545 [M − H]− (calcd for C34H37O7, 557.2539),indicating 16 degrees of unsaturation. Its 1H NMR spectrumdisplayed three aromatic or olefinic proton singlets at δH 7.25,6.43, and 6.19 (each 1H), a methine proton singlet at δH 3.19,and six methyl singlets at δH 1.55, 1.42, 1.42, 1.38, 1.30, and1.07 (each 3H) (Table 1). The 13C NMR spectrum revealed 34carbon signals corresponding to six methyl, six methylene, four
methine, and 18 quaternary carbons (including three carbonylcarbons). Further analysis of its 2D NMR data suggested thepresence of two podocarpane-type trinorditerpene moieties,indicating that 1 should be an adduct of these twotrinorditerpene moieties.Comparison of the NMR data with those of celaphanol A
(3)11 allowed one of the moieties to be established as a C-14′-substituted celaphanol A, which was confirmed by the HMBCcorrelations of H-11′ (δH 7.25)/C-8′ (δC 119.7), C-10′ (δC41.4), and C-13′ (δC 143.3) (Figure 2). The remaining
Received: November 22, 2012Published: February 19, 2013
Figure 1. Structures of compounds 1−3.
Note
pubs.acs.org/jnp
© 2013 American Chemical Society andAmerican Society of Pharmacognosy 745 dx.doi.org/10.1021/np3008182 | J. Nat. Prod. 2013, 76, 745−749
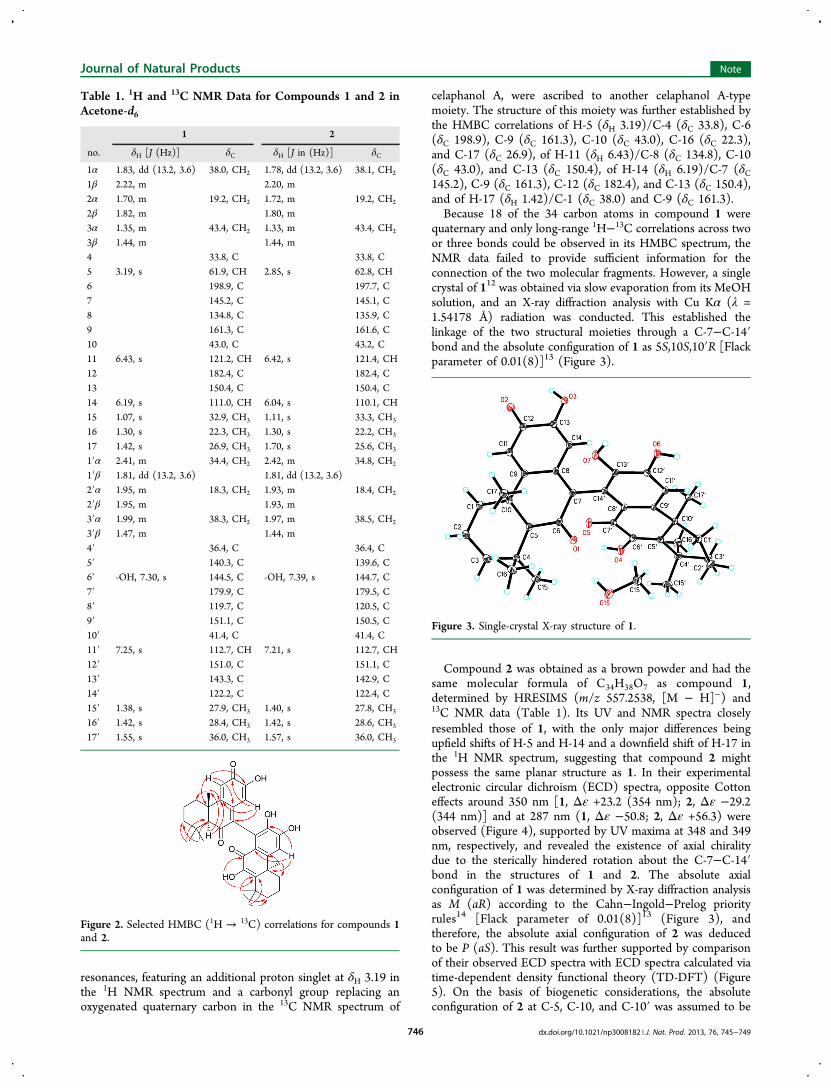
resonances, featuring an additional proton singlet at δH 3.19 inthe 1H NMR spectrum and a carbonyl group replacing anoxygenated quaternary carbon in the 13C NMR spectrum of
celaphanol A, were ascribed to another celaphanol A-typemoiety. The structure of this moiety was further established bythe HMBC correlations of H-5 (δH 3.19)/C-4 (δC 33.8), C-6(δC 198.9), C-9 (δC 161.3), C-10 (δC 43.0), C-16 (δC 22.3),and C-17 (δC 26.9), of H-11 (δH 6.43)/C-8 (δC 134.8), C-10(δC 43.0), and C-13 (δC 150.4), of H-14 (δH 6.19)/C-7 (δC145.2), C-9 (δC 161.3), C-12 (δC 182.4), and C-13 (δC 150.4),and of H-17 (δH 1.42)/C-1 (δC 38.0) and C-9 (δC 161.3).Because 18 of the 34 carbon atoms in compound 1 were
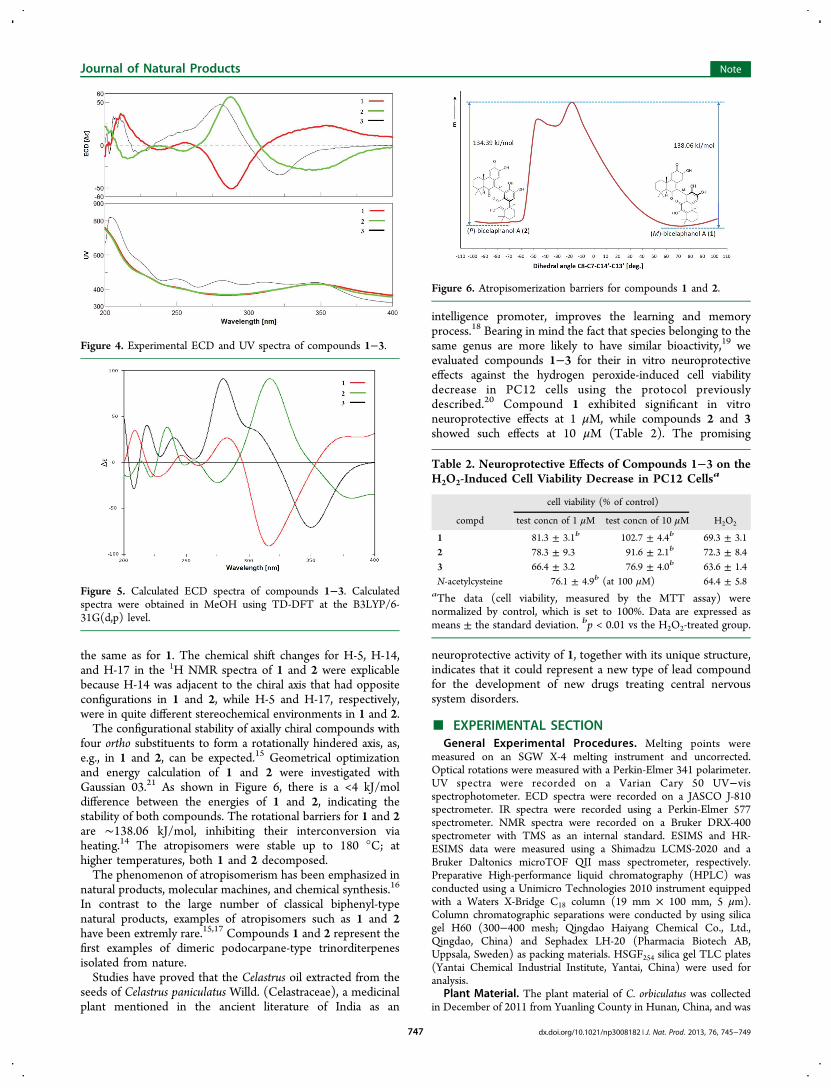
quaternary and only long-range 1H−13C correlations across twoor three bonds could be observed in its HMBC spectrum, theNMR data failed to provide sufficient information for theconnection of the two molecular fragments. However, a singlecrystal of 112 was obtained via slow evaporation from its MeOHsolution, and an X-ray diffraction analysis with Cu Kα (λ =1.54178 Å) radiation was conducted. This established thelinkage of the two structural moieties through a C-7−C-14′bond and the absolute configuration of 1 as 5S,10S,10′R [Flackparameter of 0.01(8)]13 (Figure 3).
Compound 2 was obtained as a brown powder and had thesame molecular formula of C34H38O7 as compound 1,determined by HRESIMS (m/z 557.2538, [M − H]−) and13C NMR data (Table 1). Its UV and NMR spectra closelyresembled those of 1, with the only major differences beingupfield shifts of H-5 and H-14 and a downfield shift of H-17 inthe 1H NMR spectrum, suggesting that compound 2 mightpossess the same planar structure as 1. In their experimentalelectronic circular dichroism (ECD) spectra, opposite Cottoneffects around 350 nm [1, Δε +23.2 (354 nm); 2, Δε −29.2(344 nm)] and at 287 nm (1, Δε −50.8; 2, Δε +56.3) wereobserved (Figure 4), supported by UV maxima at 348 and 349nm, respectively, and revealed the existence of axial chiralitydue to the sterically hindered rotation about the C-7−C-14′bond in the structures of 1 and 2. The absolute axialconfiguration of 1 was determined by X-ray diffraction analysisas M (aR) according to the Cahn−Ingold−Prelog priorityrules14 [Flack parameter of 0.01(8)]13 (Figure 3), andtherefore, the absolute axial configuration of 2 was deducedto be P (aS). This result was further supported by comparisonof their observed ECD spectra with ECD spectra calculated viatime-dependent density functional theory (TD-DFT) (Figure5). On the basis of biogenetic considerations, the absoluteconfiguration of 2 at C-5, C-10, and C-10′ was assumed to be
Table 1. 1H and 13C NMR Data for Compounds 1 and 2 inAcetone-d6
1 2
no. δH [J (Hz)] δC δH [J in (Hz)] δC
1α 1.83, dd (13.2, 3.6) 38.0, CH2 1.78, dd (13.2, 3.6) 38.1, CH2
1β 2.22, m 2.20, m2α 1.70, m 19.2, CH2 1.72, m 19.2, CH2
2β 1.82, m 1.80, m3α 1.35, m 43.4, CH2 1.33, m 43.4, CH2
3β 1.44, m 1.44, m4 33.8, C 33.8, C5 3.19, s 61.9, CH 2.85, s 62.8, CH6 198.9, C 197.7, C7 145.2, C 145.1, C8 134.8, C 135.9, C9 161.3, C 161.6, C10 43.0, C 43.2, C11 6.43, s 121.2, CH 6.42, s 121.4, CH12 182.4, C 182.4, C13 150.4, C 150.4, C14 6.19, s 111.0, CH 6.04, s 110.1, CH15 1.07, s 32.9, CH3 1.11, s 33.3, CH3
16 1.30, s 22.3, CH3 1.30, s 22.2, CH3
17 1.42, s 26.9, CH3 1.70, s 25.6, CH3
1′α 2.41, m 34.4, CH2 2.42, m 34.8, CH2
1′β 1.81, dd (13.2, 3.6) 1.81, dd (13.2, 3.6)2′α 1.95, m 18.3, CH2 1.93, m 18.4, CH2
2′β 1.95, m 1.93, m3′α 1.99, m 38.3, CH2 1.97, m 38.5, CH2
3′β 1.47, m 1.44, m4′ 36.4, C 36.4, C5′ 140.3, C 139.6, C6′ -OH, 7.30, s 144.5, C -OH, 7.39, s 144.7, C7′ 179.9, C 179.5, C8′ 119.7, C 120.5, C9′ 151.1, C 150.5, C10′ 41.4, C 41.4, C11′ 7.25, s 112.7, CH 7.21, s 112.7, CH12′ 151.0, C 151.1, C13′ 143.3, C 142.9, C14′ 122.2, C 122.4, C15′ 1.38, s 27.9, CH3 1.40, s 27.8, CH3
16′ 1.42, s 28.4, CH3 1.42, s 28.6, CH3
17′ 1.55, s 36.0, CH3 1.57, s 36.0, CH3
Figure 2. Selected HMBC (1H → 13C) correlations for compounds 1and 2.
Figure 3. Single-crystal X-ray structure of 1.
Journal of Natural Products Note
dx.doi.org/10.1021/np3008182 | J. Nat. Prod. 2013, 76, 745−749746
the same as for 1. The chemical shift changes for H-5, H-14,and H-17 in the 1H NMR spectra of 1 and 2 were explicablebecause H-14 was adjacent to the chiral axis that had oppositeconfigurations in 1 and 2, while H-5 and H-17, respectively,were in quite different stereochemical environments in 1 and 2.The configurational stability of axially chiral compounds with
four ortho substituents to form a rotationally hindered axis, as,e.g., in 1 and 2, can be expected.15 Geometrical optimizationand energy calculation of 1 and 2 were investigated withGaussian 03.21 As shown in Figure 6, there is a <4 kJ/moldifference between the energies of 1 and 2, indicating thestability of both compounds. The rotational barriers for 1 and 2are ∼138.06 kJ/mol, inhibiting their interconversion viaheating.14 The atropisomers were stable up to 180 °C; athigher temperatures, both 1 and 2 decomposed.The phenomenon of atropisomerism has been emphasized in
natural products, molecular machines, and chemical synthesis.16
In contrast to the large number of classical biphenyl-typenatural products, examples of atropisomers such as 1 and 2have been extremly rare.15,17 Compounds 1 and 2 represent thefirst examples of dimeric podocarpane-type trinorditerpenesisolated from nature.Studies have proved that the Celastrus oil extracted from the
seeds of Celastrus paniculatus Willd. (Celastraceae), a medicinalplant mentioned in the ancient literature of India as an
intelligence promoter, improves the learning and memoryprocess.18 Bearing in mind the fact that species belonging to thesame genus are more likely to have similar bioactivity,19 weevaluated compounds 1−3 for their in vitro neuroprotectiveeffects against the hydrogen peroxide-induced cell viabilitydecrease in PC12 cells using the protocol previouslydescribed.20 Compound 1 exhibited significant in vitroneuroprotective effects at 1 μM, while compounds 2 and 3showed such effects at 10 μM (Table 2). The promising
neuroprotective activity of 1, together with its unique structure,indicates that it could represent a new type of lead compoundfor the development of new drugs treating central nervoussystem disorders.
■ EXPERIMENTAL SECTIONGeneral Experimental Procedures. Melting points were
measured on an SGW X-4 melting instrument and uncorrected.Optical rotations were measured with a Perkin-Elmer 341 polarimeter.UV spectra were recorded on a Varian Cary 50 UV−visspectrophotometer. ECD spectra were recorded on a JASCO J-810spectrometer. IR spectra were recorded using a Perkin-Elmer 577spectrometer. NMR spectra were recorded on a Bruker DRX-400spectrometer with TMS as an internal standard. ESIMS and HR-ESIMS data were measured using a Shimadzu LCMS-2020 and aBruker Daltonics microTOF QII mass spectrometer, respectively.Preparative High-performance liquid chromatography (HPLC) wasconducted using a Unimicro Technologies 2010 instrument equippedwith a Waters X-Bridge C18 column (19 mm × 100 mm, 5 μm).Column chromatographic separations were conducted by using silicagel H60 (300−400 mesh; Qingdao Haiyang Chemical Co., Ltd.,Qingdao, China) and Sephadex LH-20 (Pharmacia Biotech AB,Uppsala, Sweden) as packing materials. HSGF254 silica gel TLC plates(Yantai Chemical Industrial Institute, Yantai, China) were used foranalysis.
Plant Material. The plant material of C. orbiculatus was collectedin December of 2011 from Yuanling County in Hunan, China, and was
Figure 4. Experimental ECD and UV spectra of compounds 1−3.
Figure 5. Calculated ECD spectra of compounds 1−3. Calculatedspectra were obtained in MeOH using TD-DFT at the B3LYP/6-31G(d,p) level.
Figure 6. Atropisomerization barriers for compounds 1 and 2.
Table 2. Neuroprotective Effects of Compounds 1−3 on theH2O2-Induced Cell Viability Decrease in PC12 Cellsa
cell viability (% of control)
compd test concn of 1 μM test concn of 10 μM H2O2
1 81.3 ± 3.1b 102.7 ± 4.4b 69.3 ± 3.12 78.3 ± 9.3 91.6 ± 2.1b 72.3 ± 8.43 66.4 ± 3.2 76.9 ± 4.0b 63.6 ± 1.4N-acetylcysteine 76.1 ± 4.9b (at 100 μM) 64.4 ± 5.8aThe data (cell viability, measured by the MTT assay) werenormalized by control, which is set to 100%. Data are expressed asmeans ± the standard deviation. bp < 0.01 vs the H2O2-treated group.
Journal of Natural Products Note
dx.doi.org/10.1021/np3008182 | J. Nat. Prod. 2013, 76, 745−749747

identified by Prof. Jin-Gui Shen (Department of Natural ProductsChemistry, Shanghai Institute of Materia Medica, Chinese Academy ofSciences). A voucher specimen (NST-20120220) has been depositedin Shanghai Institute of Materia Medica.Extraction and Isolation. The air-dried and powdered root bark
of C. orbiculatus (20 kg) were percolated with 95% EtOH (3 × 25 L)at room temperature to give a crude extract, which was suspended inwater (2 L) and partitioned with EtOAc (3 × 3 L). The EtOAc extractwas subjected to CC over silica gel (200−300 mesh) and eluted with apetroleum ether/EtOAc gradient (from 10:1 to 1:1, v/v) to yieldfractions 1−5. Fraction 3 was separated over RP-18 CC eluted with60% aqueous CH3CN, affording compound 3 (560 mg). Fraction 5was chromatographed over RP-18 CC (MeOH/H2O, 4:6 to 10:0, v/v), followed by Sephadex LH-20 gel (95% EtOH) and RP-18preparative HPLC (CH3CN/H2O, 4:1, v/v) to give compounds 1 (55mg) and 2 (39 mg).(M)-Bicelaphanol A (1): red crystals; mp 270−271 °C; [α]22D +527
(c 0.9, acetone); UV (MeOH) λmax (log ε) 348 nm (4.46); ECD(MeOH) 211 nm (Δε +36.8), 236 nm (Δε −5.5), 257 nm (Δε +3.0),287 nm (Δε −50.8), 354 nm (Δε +23.2); IR (film) νmax 3534, 3377,3188, 2933, 1678, 1614, 1583, 1525, 1448, 1306, 1228, 1201, 1146,887, 789, 656 cm−1; see Table 1 for 1H NMR (acetone-d6, 400 MHz)and 13C NMR (acetone-d6, 100 MHz) data; HRESIMS m/z 557.2545[M − H]− (calcd for C34H37O7, 557.2539).(P)-Bicelaphanol A (2): brown powder; [α]22D −294 (c 0.7,
acetone); UV (MeOH) λmax (log ε) 349 nm (4.47); ECD (MeOH)216 nm (Δε −15.8), 238 nm (Δε −0.7), 254 nm (Δε −7.0), 287 nm(Δε +56.3), 344 nm (Δε −29.2); IR (film) νmax 3357, 2931, 2870,1680, 1622, 1539, 1435, 1379, 1309, 1228, 1203, 1178, 1146, 879, 598cm−1; see Table 1 for 1H NMR (acetone-d6, 400 MHz) and 13C NMR(acetone-d6, 100 MHz) data; HRESIMS m/z 557.2538 [M − H]−
(calcd for C34H37O7, 557.2539).Neuroprotective Activity Assay. PC12 neuroblastoma cells were
digested with trypsin and suspended in DMEM medium (low sugar)containing 10% new bovine serum (Gibco). Cells were seeded into 96-well plates (Greiner) at a density of 8 × 104 cells/mL, 100 μL/well,maintained at 37 °C in a constant-temperature incubator containing5% CO2. Serum-free DMEM medium was used to substitute theoriginal medium 24 h after cells had been seeded.Appropriate concentrations of H2O2 were prepared on the day of
application to cultures. The PC12 cells were preincubated with thedifferent compounds (1 or 10 μM) 2 h before H2O2 (300 μM) wasadded, and the assays for cell viability were performed 24 h after H2O2
was added. Cell survival was evaluated by reduction of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma]. Thevalues of cell survival were normalized against the values for thecontrol group, which was set to 100%. Data were evaluated forstatistical significance with a one-way ANOVA followed by an LSDtest using a computerized statistical package.N-Acetylcysteine (NAC) was used as a positive control. Compared
with 100 μM NAC, 10 μM (M)-bicelaphanol A (1) showed asignificantly protective effect against H2O2 injury.Computational Methods. Geometrical optimization and energy
calculation of (M)-bicelaphanol A (1) and (P)-bicelaphanol A (2)were performed using DFT with the B3LYP functional and the 6-31G(d,p) basis set in the gas phase with Gaussian 03.21 Anatropisomerization barrier scan was performed by optimizing thestructure with restriction of the C-8−C-7−C-14′−C-13′ dihedralangle, ranging from −100° to 100°. Vibrational analysis was conductedat the same level to confirm minima. TD-DFT/B3LYP/6-31G(d,p) inMeOH using the SCRF (self-consistent reaction field) method withthe CPCM (conductor-like polarizable continuum) model wasemployed to calculate excitation energy (denoted by the wavelengthin nanometers) and rotatory strength R in dipole velocity (Rvel) anddipole length (Rvel) forms. ECD curves were calculated on the basis ofrotatory strengths under a half-bandwidth of 0.2 eV using SpecDisversion 1.51.22 The spectra were combined after Boltzmann weightingaccording to their population contribution.
■ ASSOCIATED CONTENT*S Supporting InformationProposed biosynthesis pathway of 1 and 2, characterization dataof 3, optimized structures and calculated energies of 1 and 2,NMR, HRESIMS, and IR spectra of compounds 1−3, and X-ray crystallographic data (CIF file) of 1. This material isavailable free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] (H.-Y.Z.) or [email protected] (W.-M.Z.). Telephone and fax: +86-21-50806052.Author ContributionsL.-Y.W., J.W., and Z.Y. contributed equally to this work.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was supported financially by the State KeyLaboratory of Drug Research (SIMM1203KF-02) and NationalScience & Technology Major Project “Key New Drug Creationand Manufacturing Program” (2011ZX09307-002-03).
■ REFERENCES(1) Xu, G.; Yang, X. W.; Wu, C. Y.; Li, X. N.; Su, J.; Deng, X.; Li, Y.;Qin, H. B.; Yang, L. X.; Zhao, Q. S. Chem. Commun. 2012, 48, 4438−4440.(2) Lian, G. Y.; Yu, B. Chem. Biodiversity 2010, 7, 2660−2691.(3) Berube, G. Curr. Med. Chem. 2006, 13, 131−154.(4) Jiangsu New Medical College. Dictionary of Chinese Herb Medicines;Shanghai Scientific and Technologic Press: Shanghai, 1986; p 1567.(5) (a) Kim, S. E.; Kim, Y. H.; Lee, J. J.; Kim, Y. C. J. Nat. Prod. 1998,61, 108−111. (b) Kim, S. E.; Kim, H. S.; Hong, Y. S.; Kim, Y. C.; Lee,J. J. J. Nat. Prod. 1999, 62, 697−700. (c) Guo, Y. Q.; Li, X.; Xu, J.; Li,N.; Meng, D. L.; Wang, J. H. Chem. Pharm. Bull. 2004, 52, 1134−1136.(d) Guo, Y. Q.; Li, X.; Xu, J.; Meng, D. L.; Li, Y. S.; Ohizumi, Y. Chem.Lett. 2005, 34, 764−765. (e) Zhu, Y. D.; Miao, Z. H.; Ding, J.; Zhao,W. M. J. Nat. Prod. 2008, 71, 1005−1010.(6) (a) Min, K. R.; Hwang, B. Y.; Lim, H.; Kang, B.; Oh, G.; Lee, J.;Kang, S.; Lee, K. S.; Ro, J. S.; Kim, Y. Planta Med. 1999, 65, 460−462.(b) Hwang, B. Y.; Kim, H. S.; Lee, J. H.; Hong, Y. S.; Ro, J. S.; Lee, K.S.; Lee, J. J. J. Nat. Prod. 2001, 64, 82−84.(7) Guo, Y. Q.; Li, X.; Wang, J. H.; Xu, J.; Li, N. Fitoterapia 2005, 76,273−275.(8) Maoka, T. Phytochemistry 2009, 70, 920−923.(9) Zhang, Y.; Tan, C. H.; Tan, J. J.; Yang, P. M.; Jiang, S. H.; Ni, X.;Zhu, D. Y. Helv. Chim. Acta 2010, 93, 1407−1412.(10) Wu, J.; Zhou, Y.; Wang, L. Y.; Zuo, J. P.; Zhao, W. M.Phytochemistry 2012, 75, 159−168.(11) Chen, B.; Duan, H. Q.; Takaishi, Y. Phytochemistry 1999, 51,683−687. See the Supporting Information for detailed data of 3.(12) X-ray crystallographic data of 1: C34H38O7·CH3OH, M =590.69, monoclinic, space group P21, a = 8.4324(2) Å, b = 16.6283(3)Å, c = 10.8816(2) Å, α = 90.00°, β = 96.1740(10)°, γ = 90.00°, V =1516.93(5) Å3, Z = 2, Dcalcd = 1.293 mg/m3, F(000) = 632, μ = 0.739mm−1, T = 173(2) K. A single crystal with dimensions of 0.14 mm ×0.05 mm × 0.03 mm was used for X-ray measurements. A total of19415 reflections, collected in the θ range of 4.09−66.50°, yielded5100 unique reflections (Rint = 0.0298). Final R indices for I > 2σ(I):R1 = 0.0279, and wR2 = 0.0864. R indices for all data: R1 = 0.0282, andwR2 = 0.0870. Crystallographic data have been deposited in theCambridge Crystallographic Data Center as entry 883806. A copy ofthe data can be obtained, free of charge, on application to the Director,CCDC, 12 Union Road, Cambridge CB21EZ, U.K. [fax, +44(0)-1233−336033; e-mail, [email protected]].(13) Flack, H. D. Acta Crystallogr. 1983, A39, 876−881.
Journal of Natural Products Note
dx.doi.org/10.1021/np3008182 | J. Nat. Prod. 2013, 76, 745−749748

(14) Bringmann, G.; Mortimer, A. J. P.; Keller, P. A.; Gresser, M. J.;Garner, J.; Breuning, M. Angew. Chem., Int. Ed. 2005, 44, 5384−5427.(15) Bringmann, G.; Gulder, T.; Gulder, T. A. M.; Breuning, M.Chem. Rev. 2011, 111, 563−639.(16) Yao, S.; Tang, C. P.; Ye, Y.; Kurtan, T.; Kiss-Szikszai, A.; Antus,S.; Pescitelli, G.; Salvadori, P.; Krohn, K. Tetrahedron: Asymmetry2008, 19, 2007−2014.(17) Li, E. W.; Zhang, F.; Niu, S. B.; Liu, X. Z.; Liu, G.; Che, Y. S.Org. Lett. 2012, 14, 3320−3323.(18) Nalini, K.; Karanth, K. S.; Rao, A.; Aroor, A. R. J.Ethnopharmacol. 1995, 47, 101−108.(19) Pillon, Y.; Fogliani, B. Pac. Sci. 2009, 63, 97−103.(20) Dong, K.; Pu, J. X.; Zhang, H. Y.; Du, X.; Li, X. N.; Zou, J.;Yang, J. H.; Zhao, W.; Tang, X. C.; Sun, H. D. J. Nat. Prod. 2012, 75,249−256.(21) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.;Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A.D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.;Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford,S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.;Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.;Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.;Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez,C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong,M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.Gaussian 03; Gaussian: Wallingford, CT, 2003.(22) Bruhn, T.; Hemberger, Y.; Schaumloffel, A.; Bringmann, G.SpecDis, version 1.51; University of Wuerzburg: Wuerzburg, Germany,2011.
Journal of Natural Products Note
dx.doi.org/10.1021/np3008182 | J. Nat. Prod. 2013, 76, 745−749749





























