Embed Size (px)
Citation preview

Giles Kisby GE Y1 Genetics
CMS, Immunology and Cancer:
Spring Term:
LECTURES:10/01/14: DNA damage and its repair: Andy PorterLos (from booklet):
Lecture 3. DNA Damage and Repair Describe the ways in which DNA can be damaged by endogenous and environmental factors. Summarise the main natural DNA repair mechanisms: BER, NER, MMR and DSB repair (NHEJ & HR) Describe the DNA damage response pathways and key proteins involved Outline some inherited disease that result from mutations in genes involved in DNA repair or the DDR Identify key proteins involved in each of the above processes and give examples of diagnostic/therapeutic benefits resulting form such knowledge
Notes:
- Overview:o • Types of DNA damage o • DNA Repair mechanisms (inc sensing)o • DNA damage response (ie other than repair)o • Disorders of DNA repair
1

Giles Kisby GE Y1 Genetics
- Types of DNA damage:o Ss breako Ds breako Bulky adducts (unwanted covalent bonding between bases or to chemicals)o Mismatcheso Insertions / deletions (“in/dels”)o Base alkylation
- What can damage DNA:o Environmental damage
Chemicals (carcinogens) Radiation
o Endogenous Damage Reactive oxygen species Spontaneous deamination Replication errors
- Mutations can cause:o cancer, o other diseases (eg neurodegeneration, Immune Dysfunction, Sterility)o ageing
- Chemical attack of DNA:
2

Giles Kisby GE Y1 Genetics
o Oxidative damage Free Radicals from normal metabolism or Insulin resistance:
Attack purine and pyrimidine rings Cause strand breaks eg Guanine to 8-hydroxyguanine
o • Pairs with A instead of C eg Loss of base planar ring structure
o • Non-coding bases produced eg Helical distortion
o • Additional covalent bonding between base and sugarphosphate backbone
o Hydrolytic attack [are frequent events in the body which must be repaired] Depurination [purine bases cleaved from sugars and removed; are more
susceptible than pyrimidines] Corrected by insertion of a replacement nucleotide
Deamination • Cytosine to Uracil
o Common evento If dna replication occurs before is repaired then uracil will be
retained with adenine on other strand (ie C-G U-A); this is a disruption to the code
• Adenine to Hypoxanthine • Guanine to Xanthine [also can change to Inosine] • 5-Methylcytosine to Thymine
o DNA methylation involoves the addition of a methyl group to the cytosine or adenine DNA nucleotides. DNA methylation reduces the expression of genes in cells as cells divide and differentiate. The resulting change is normally permanent and unidirectional ie differentiation occurs
o Unmethylated CpGs are often grouped in clusters called CpG islands, which are present in the 5' regulatory regions of many genes. In many disease processes, such as cancer, gene promoter CpG islands acquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division.
3

Giles Kisby GE Y1 Genetics
o NB is prob only in Bac that the methylation plays the role of indicating which strand is the parent strand in DNA repair; prob don’t worry if any such system in euk too. In euk mainly for epigenetic gene regulation
o 5-Methylcytosine is common and already shows similarities to Thymine; deamination completes the switch
o CG TA transitions therefore a very common mutation
Strand breaks Ie breaks at the sugar phosphate backbone (= ss breaks)
o Alkylation Methylation
is one form of Alkylation commonly triggered by env agents can distort the structure of DNA eg guanine methyl-guanine corrected by dealkylation
- Non – Chemical DNA damage:o Nucleotide insertions/deletions looping out of DNAo Incorporation of wrong nucleotide mismatches/improper coding/DNA distortion
Due to replication errors Eg A-G mismatch; gives distortion; corrected by removal of the base in the
newly synth strando Incorporation of damaged nucleotides / nucleotides damaged in situ
Cyclobutane Pyrimidine Dimers (CPDs) Adjacent Thymine bases become covalently bound to each other
(via a cyclobutane group) Caused by UV/ultraviolet radiation Distorts the DNA duplex Corrected by excision of the two T residues or direct repair (cf)
4

Giles Kisby GE Y1 Genetics
- Types of DNA repair:
o • Direct repair – Reverses the DNA damage directly (ie direct reversal of the DNA damage
process) Eg Photoreactivation of CPD
Non-enzymic: 200-300nm irradiation Enzymic: Involves photolyases (occurs in Bacteria and yeast, not
mammals) Eg Reversal of Alkylation damage
O6-methylguanine-DNA methyltransferase Removes methyl groups however Enzyme is not reversibly methylated (suicide enzyme) so only single use per enzyme; is infrequently used
o • Base Excision Repair (BER) [“BAPT”]
5

Giles Kisby GE Y1 Genetics
– Removes single damaged bases; ie Removes bases damaged by hydrolysis, ROS etc.
1) DNA glycosylases identify and remove the damaged base 2) AP endonuclease and T-phosphodiesterase cut the
sugarphosphate backbone 3) Gap is filled by DNA polymerase β 4) Nick is sealed by DNA ligase
– Main protector against metabolism damage
o • Nucleotide Excision Repair (NER) – Removes Thymine dimers – Removes large chemical adducts “Global NER”:
1) Damaged bases are detected 2) Helicases and nucleases act to open up and cut either side of the
mutated base; o Incisions made 5’ by ERCC1/XP-F and 3’ by XP-G of damage;
[nb Xeroderma pigmentosa from XP-A muts etc]o 24-32bp oligonucleotide is removed
3) Gap is repaired by DNA polymerase ε or δ 4) Nick is sealed by DNA ligase
“Transcription-coupled NER”:
6

Giles Kisby GE Y1 Genetics
Coupled to the transcription process: Transcribed regions are repaired better than untranscribed regions / Preferential repair of transcribed DNA strand (5-10 fold)
XPC not required [ie is another involved in the global process] Damage recognised by stalled RNA pol Additional proteins required (that are not used in global NER): esp
CSA and CSB
o • Mismatch Repair (MMR) – Removes mismatched base pairs and insertion/deletion loops • Human mismatch repair system resembles the mutator MutHLS systems
in bacteria Bacterial MMR
o MutS recognises mismatch o MutL binds MutS and activates MutHo MutH nicks unmethylated (newly replicated) strand o Intervening DNA excised and resynthesisedo Thus repairs correct strand
Mammalian MMRo Essentially same as bacterial system o No MutH
Nicks used are thought to be those occurring normally during DNA replication i.e. between Okazaki fragments, or 3’ end of leading strand.
o HNPCC relevance
o DSB Repair: • Homologous recombination repair (HRR)
– DS break repair – Crosslink repair Process:
o • Requires intact homology template (sister chromatid) o • Operates in G2 [ie sister chromatid present]o • Many proteins involved (e.g. Rad51, BRCA2) o • Highly accurate o BRCA relavance
• Non-homologous end joining (NHEJ) – DS break repair • No homology template required • Operates in G1/S/G2 • Error-prone
7

Giles Kisby GE Y1 Genetics
[Don’t learn all this diagram detail; just to show the key differences between the two dsB processes]
- DNA Damage Response:o Cell cycle checkpoint activation
allows time for DNA repair or apoptosis CHK1/2 mediate cell cycle arrest to prevent amplification of cells with the
problem/mutationo Transcriptional program activation
Expression of DNA repair genes, or relocation e.g. Regulator: Rad51 stimulates repair genes; relocates to nucleus eg Post-translational regulation proteins associated with DNA repair
machineryo Final results:
DNA repair (good) OR Apoptosis (good) OR Incorrect/failed repair (BAD)
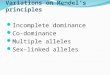
Transcription/translation giving aberrant proteins Carcinogenesis if critical targets are mutated:
o oncogenes, Uncontrolled cell proliferationo tumour suppressor genes: [gene examples then detailed
below]: 1) Brakes on cell cycle mutated
Uncontrolled cell proliferation
8

Giles Kisby GE Y1 Genetics
e.g. Rb [nb is only involved in G1 S phase step [TRUE]], p16
2) Signalling DNA damage mutated Uncontrolled cell proliferation AND
further gene mutations e.g. p53, ATM
3) Genes for Repair of DNA damage mutated further gene mutations e.g. XP-A, NBS1
- Retinoblastoma o Malignant cancer of developing retinal cellso Sporadic disease usually involves one eye. o Hereditary cases can be unilateral or bilateral and multifocal.o Due to mutation of the RB1 tumour suppressor gene on chromosome 13q14o RB1 encodes a nuclear protein that is involved in the regulation of the cell cycle
- p53 and Cancer o • Tumour suppressor gene
– Recessive – “two-hit mechanism” – 17p13
o • loss or mutation of p53 – the most common single genetic change in cancer – > 50% Tumours
o • Cancers – Breast cancer – Colon cancer – Li-Fraumeni syndrome (germ-line p53 mutation)
autosomal dominant muts have a dominant-negative effect over alternate wild type
alleles; ie the second hit must still occur but due to mature of first hit the second is v likely [the mutant protein can work to inactivate the normal protein]
- Ataxia telangectasia (AT)o Due to ATM mutations:
recruited and activated by DNA double-strand breaks
9

Giles Kisby GE Y1 Genetics
– Autosomal recessive
– Degeneration of cerebellum - ataxia – Immune dysfunction – Sterility – Sensitivity to radiation – Increased risk cancer
- Familial breast cancero • Accounts for about 5% of breast cancer.o • More than 50% associated with damage to chromosomes 17q and 13q.o • BRCA1gene mutations (17q) associated with breast, ovarian, prostate and colon
cancers.o • BRCA2gene mutations (13q) associated with breast, ovary, prostate, and
endometrial cancer.o • BRCA1 and BRCA2 function as regulators of cell cycle and genomic integrity e.g.
BRCA1/2 involved in HRRo Germline mutations in BRCA1 and BRCA2 are inherited in an autosomal dominant
manner but does not mean all are affected: The vast majority of individuals with a BRCA1 or BRCA2 mutation have inherited it from a parent. However, because of incomplete penetrance, variable age of cancer development, cancer risk reduction resulting from prophylactic surgery, or early death, not all individuals with a BRCA1 or BRCA2 mutation have a parent affected with cancer.
- Xeroderma pigmentosao • Caused by mutations in NER genes e.g. XPA-G. o • Autosomal recessiveo • Sensitivity to UV light o • Skin cancer
o • Neurodegeneration
- Cockayne’s syndrome:o – Mutations in NER gene CSA, CSB o – autosomal recessive
o – neurological dysfunction (demyelination) o – no skin cancer o – cachexia and contractions
10

Giles Kisby GE Y1 Genetics
11

Giles Kisby GE Y1 Genetics
07/02/14: Tools and mutations, pedigrees and patterns: Dr Claire Shovlin
Los (from Booklet):GENETICS 1: Tools and mutations, pedigrees and patterns Prof. Alex Blakemore Understand the contribution of genetics to human disease Understand the potential molecular effects of changing DNA sequence - for coding DNA: single nucleotide: missense; stop codons; insertions & deletions; in frame; out of frame; - for non coding DNA: very basic principles 2) Define polymorphisms and mutations 3) Define genetic markers, and explain how these are used to follow inheritance 4) Understand what is meant by loci, alleles, and phase Have the tools to describe and record inheritance - be able to draw a multigeneration family tree Recognise, describe and explain recessive, dominant and X-linked inheritance; give an example of each, and discuss broad treatment implications Understand non Mendelian segregation patterns and causes
Notes:
- Variety:o crossing over occurs during meiosis so all four haploid cells will be differento mutations can occur during meiosis
missense mutation = code changes to being for one AA to being for a different AA instead
nonsense mutation = code changes to being for one AA to being for a stop codon instead
INDELS can occur = insertions or deletions May be in frame if change is by a multiple of 3 nucleotides May be out of frame if not by a multiple of 3 nucleotides An extensive / long INDEL would be termed a “structural variant”
Strictly: [though often not used in this way]: “polymorphism” = variant with frequency >1% in pop [in reality is
the term used for non coding mutations or non – disease causing changes to proteins]
12
Giles Kisby GE Y1 Genetics
o Ie note that just because the gene relevant to a disease is mutated it may not be dysfunctional
“mutation” = variant with frequency <1% in pop [in reality is used when the mutation is causing a disease]
- GWAS: genome-wide association study o is an examination of many common genetic variants in different individuals to see if
any variant is associated with a trait. o just concerned with when genetic traits occur together with certain genetic varientso SNPs can be used; eg below are prob using SNP specific cleavage to give allele
specific fragments; allele 6 is clearly causing the disease:
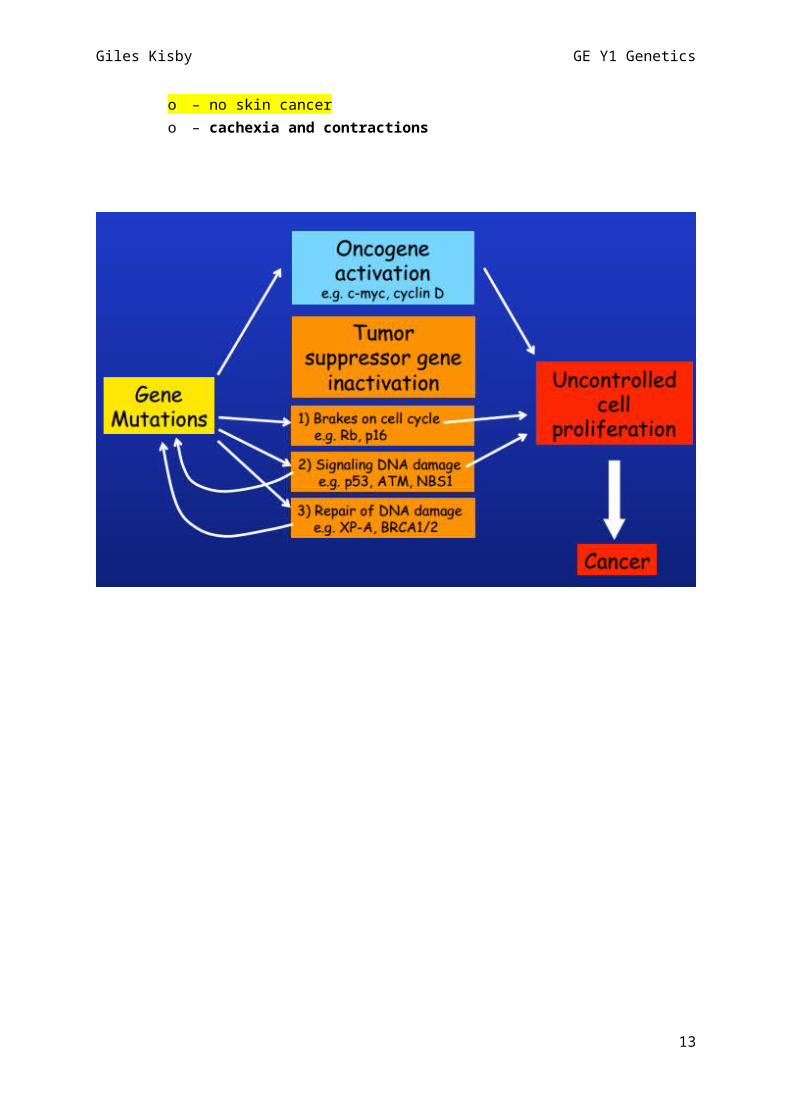
- Linkage analysis:o Linkage is the tendency of genes that are located proximal to each other on a
chromosome to be inherited together during meiosis. “genetic linkage” is usually due to “physical linkage” [ie close proximity on
the chromosome]o Linkage analysis therefore refers to tracking and analysing the inheritance of alleles
through families down generations to discern relative positions on genome to each other
o Historically minisatellites of 10-500bp were used analysed for this then microsatellites of 2-5 nuc were used. Now SNPs are used [v stable, many in the genome, easy to assay, usually only one alternative (ie are polymorphisms so are just that there is eg A instead of T at one point in > 1% of pop)]
o Allelle 1 “in phase” with allele B [ie changes in unison] but “not in phase” with allele A:
Ie sequence change at one alle can be used to infer sequence change at a disease causing gene if the two are truly in phase (prob means on same chromosome and are v close to each other)
13

Giles Kisby GE Y1 Genetics
[also: diamond used if gender is unknown; triangle if miscarriage; dot in center if incomplete penetrance]
- Autosomal dominant:o Examples:
Huntingdons Achondroplasia [dwarfism] Retinitis pigmentosa
14

Giles Kisby GE Y1 Genetics
HHT: Hereditary hemorrhagic telangiectasia [causes nosebleeds]o May be haploinsufficiency Autosomal dominanto May be an allele with active negative function causing the disease Autosomal
dominanto In general means for a person to be effected there must have also been a parent
affected; ie no skipped generations In fact non penetrance is possible [refers to the proportion of individuals
with the mutation who exhibit clinical symptoms]: eg late onset conditions [these are sources of non mendelian genetic results]
[YES!!]
- Autosomal recessive inheritance:o Eg cystic fibrosiso Both parents carriers = children: 0.25 affected, 0.5 carriers, 0.25 unaffectedo Typically only one generation affected, not the parents
- Sex linkedo Is a source of non mendelian genetic resultso Rare for it to be Y linked as there is very little on the Y chromosome except testes
determining factoro In women one X chromosome is shut off in all their somatic cells: prob is random
whether one with the mutation is shut off or not but still compensates for the X loss of function by the other X chromosome
15

Giles Kisby GE Y1 Genetics
o Females are carriers (rare to inherit two X mut chromosomes): disease to 0.5 sons, carriers to 0.5 of their daughters
o Affected males: 100% carrier daughters, 0% sons carriers / affected (ie unless from mother)
- Co dominant:o Is a source of non mendelian genetic resultso Blood groups (antigens on RBCs): A & B tested for: means some uncertainty in the
blood group of certain individuals:
o Nb single nucleotide deletion is responsible for the absence of the antigens on O blood type allele products
o A and B are codominant and both dominant over the O type
16
Giles Kisby GE Y1 Genetics
- Mitochondrial:o Is a source of non mendelian genetic resultso Maternal onlyo 1000’s of mt per cell so “degree of heterozygosity” used to describe how many have
the mutationo Uneven transmission from mother can break clinical boundary and result in
symptoms from child (transmission from females unpredictable though)
17

Giles Kisby GE Y1 Genetics
07/02/14: Communication and calculations: Dr Claire Shovlin
Los (from Booklet):GENETICS 2: Calculations and communication (and worksheet 1) Prof. Alex Blakemore CONFIRM YOUR ABILITY TO PERFORM SIMPLE CALCULATIONS BASED ON MENDELIAN PRINCIPLES. Please note that certain topics lend themselves better to exam questions than others. You should expect to be examined on the principles of Mendelian inheritance, and if by the end of the course you do cannot produce the Worksheet 1 answers by yourself, then this topic needs further study. Appreciate issues regarding communication of genetics principles - the potential impact of the information you impart; - ways that can improve your ability to communicate information.
Notes:
- Worksheet questions:o Ensure are comfortable with such questions; will be in examo Quizlet and clinical biochemistry prob best for this learning / practice
18

Giles Kisby GE Y1 Genetics
19

Giles Kisby GE Y1 Genetics
20

Giles Kisby GE Y1 Genetics
14/02/14: Progress in the genetics of obesity: Dr Alexandra Blakemore
Los (from Booklet):
Explain the concept of genetic susceptibility to common disease. Explain how we can estimate the heritability of a common complex disease Describe the susceptibility/threshold model Discuss genetic heterogeneity in complex disease Discuss one particular disease as an example of complex inheritance Describe how genome-wide SNP association studies are designed and their contribution to our understanding of common diseases with examples Discuss the implications of genetics for clinical management of common disease
Notes: ie a lot of diseases are a combo of genetic and environemental factors; obesity is one example but ideas here are cammon to many other diseases:
- Summary:o Introduction to genetics of obesity
Syndromic
21

Giles Kisby GE Y1 Genetics
Monogenic Common obesity
o Identification of GSVs in “obesity-plus” patientso Implications for common obesityo Prospects for personalised medicine
- Fat functions:o Storage of food and watero Insulation o Support and protection of vital organs o Source of hormones – leptin (regulator of eating and reproduction / sexual
signalling)o Regulator and fuel provider of the immune system o Source of new immune cells o Aids wound healing
- Effects of a lack of fat:o Infertilityo Miscarriageo Death from infectionso Higher suicide rateo Osteoporosis
- BMIo Obesity defined as BMI ≥30 kg/m2
o Morbid obesity BMI ≥40 kg/m2
o Types of obesity: Syndromic obesity monogenic obesity common obesity
o The problem with BMI Body weight is affected by muscle/fat ratio as muscle is heavier than fat Eg Asians have a lower BMI but higher percent body fat compared to white
subjects
- Common obesity:o Env and genetic effect:
Env: High density calorie diet (genetics also feeds into this; see below) Lack of Physical Activity (genetics also feeds into this; see below) Stress [via cortisol stim of stress response; reduces thyroid function
etc]
22

Giles Kisby GE Y1 Genetics
Genetic: Gene Variations affecting behaviours:
o appetite [to gain 1lb every year requires only around 10 extra calories a day]
ALL MONOGENC FORMS OF OBESITY KNOWN SO FAR AFFECT APPETITE REGULATION
o Physical activity Eg spontaneous physical activity (fidgeting etc) Eg possible genetic influences on the drive to be
active Gene variations affecting physiology
o resting metabolism eg respiratory quotient (energy expended from
breathing) eg resting metabolic rate
o energy expenditure when activeo Heritability of obesity estimated at 0.77% [ie mainly genetic! So involuntary control
is the dominant determinant!; nb slight differences to value between studies] Established by identical twin vs non identical twin studies where both
children live separately and away from parents; show on average 77% similarity with real parents due to genetics and 23% similarity with adopted parents due to env
i.e. 77% of a child’s obesity can be explained by their genetics Genome-Wide Association Studies (GWAS)
[ie “Common disease, common variant” investigations: Looked at millions of single nucleotide polymorphisms throughout the genome (comparing obese vs non-obese people)]
Try to identify the specific genes contributing to this 77% Is polygenic though; only some of those that influence have been
found; therefore cannot be used predictively as any positive indicators for obesity may be overwhelmed by effect of the genes not known about
o Current discoveries’ contribution to the genetic component of BMI is estimated to be low (<5%) ie more work is needed
Examples: o FTO [the gene whose mutations are most strongly
associated with common obesity]o MC4R gives monogenic obesity if deleted but if just altered
with reduced function are seen as contributors to common obesity
Copy number variants: Research also shows these events are linked to obesity; ie certain
genes are present in variable numbers of copies in the genome; inheriting less copies may give iterative increase in risk of obesity
23

Giles Kisby GE Y1 Genetics
Eg 16p11 region genes: [“many skinny numbers but copies give obesity”]
o SH2B1: associated with obesity beginning early in lifeo 16p11.2: associated with obesity beginning later in life
- Syndromic obesityo In Syndromic obesity by far the dominant factor in the weight of the person is their
genetics; the genetic abnormality is the cause of the obesity/underweight hereo usually accompanied by mental retardation and particular dysmorphic or clinical
featureso Examples:
Prader-Wili syndrome is the most common Syndromic obesity seven genes (including snoRNAs) on chromosome 15 are deleted or
unexpressed on the paternal chromosome; the maternal copy is usually imprinted (and thus is silenced), while the mutated paternal copy is not functional.
floppy at birth, hypogonadism, short stature, body thinks is starving obesity
WAGR syndrome: [“same number of letters as BDNF”] Wilms tumour (a tumour of the kidneys associated with WT-1 TSG
gene), Aniridia (absence of the coloured part of the eye, the iris), Genitourinary anomalies, and Retardation
BDNF mutated Patients obese
Bardet-Beidl syndrome Gives underweight people Due to cilia defect
- Monogenic obesity [ALL MONOGENC FORMS OF OBESITY KNOWN SO FAR AFFECT APPETITE REGULATION ie “adipostat components”]:
o Dominant or recessive single gene disorders Dominant :
MC4Ro The most common single-gene form of obesity (2-6% of
morbidly obese children have this disorder)o gives monogenic obesity if deleted but if just altered with
reduced function are seen as contributors to common obesity
24

Giles Kisby GE Y1 Genetics
Recessive : Leptin:
o eg Ob/Ob humans (ie don’t produce leptin)o eg deficiency of leptin receptoro Effects of deficiency of leptin signalling:
Hunger Obesity Poor growth Low thyroid Immune problems No puberty: Infertility
[leptin feeds in to gonadotropin and gonadal steroid signalling to indicate sufficient reserves for sexual development and spermatogenesis/egg production for pregnancy]
PC1 (=PCSK1) LEP LEPR POMC
- Implications for personalised medicine:o Choice of medications that might cause weight gain (especially where there is
neurocognitive dysfunction ie drugs vs this can have obesogenic effects) o Potential for intensive lifelong preventative interventiono New drug development (eg regarding MC4R muts etc)o Choice of obesity surgery type (eg may just continue to eat same amount due to
hunger drive may cause gut/stomach damage)
- Key pointso Common diseases may have a range of causes, some very strongly genetico Progress in genetics is very fasto Genetic cause does not imply that there’s nothing that can be doneo Do not suspend evidence-based medicine because of stigmatisation of particular
patients
25

Giles Kisby GE Y1 Genetics
14/02/14: The Genetic Basis of Disease: Dr Andrew Walley
Los (from Booklet):
Explain with examples the concept of aneuploidy Draw a diagram showing possible meiotic products from a balanced translocation Describe how 3 different chromosome aberrations lead to Down syndrome Explain why sex determination is not solely based on sex chromosome karyotype Explain the classification of congenital defects. Explain how non-genetic factors lead to congenital abnormalities Give two examples of inborn errors of metabolism currently included in UK national neonatal screening programmes, including clinical features and therapeutic management of each condition Notes:
- Karyotyping o By FISH: ie each will result in dif coloured patternso By chromosome banding:
Process: Chromosomes are only visible during nuclear division
(mitosis/meiosis) Colchicine arrests cells at metaphase Chromosomes are then stained using Giemsa: gives bands
depending on the density of the DNA at that region Structural subsets of chromosomes:
Metacentric: central centromere [one arm still assigned the label “long” and the other “short”
Submetacentric: centromere toward one end Acrocentric: centromere at one end
Short arm = “p” [“petite”] Long arm = “q”
Bands are labelled as follows:
26

Giles Kisby GE Y1 Genetics
o Chromosome, Arm, band Number o eg 12q21
- Chromosome abnormalities are present in:o 7.5% of all conceptionso 60% of early spontaneous miscarriageso 4-5% of still birthso 0.6% of live births
- Chromosome abnormality types:o Numerical – ie aneuploidy: loss or gain
Loss of a chromosome gives a reduction of 50% of all fully expressed gene products (from 100% to 50%): high % change poorly tolerated
Nb the remaining chromosome will be unable to compensate as its genes often already maximally expressed
Monosomy - loss of a chromosome is almost always lethalo Exception: Monosomy X: Turner’s syndrome; cf
Gain of one chromosome gives an increase of 33% of all fully expressed gene products (from 100% to 150%): ie lower % change better tolerated
Trisomy - gain of one chromosome can be tolerated Tetrasomy - gain of two chromosomes can be tolerated
o Structural – translocations, deletions, insertions, inversions, rings Translocations:
First gen translocation will likely be asymptomatic (unless gene split at boundary of translocation) as all genes still present, just have moved between the chromosomes = balanced translocation
27

Giles Kisby GE Y1 Genetics
Second gen translocations will likely be symptomatic as the new chromosomes from above will be distributed in meiosis to give gametes with incomplete allele sets
o Mosaicism – different cell lines which develop together as a mix to form the
organism
- Down’s syndrome: o Birth incidence is 1 in 1000o Risk increases with increasing maternal ageo IQ scores ranging from 25-75o Increased risks of leukaemia and Alzheimer’so Effects:
Newborn period - severe hypotonia, sleepy, excess nuchal skin (back of neck)
Craniofacial – macroglossia (hypertrophy of the tongue), small ears, epicanthic folds, upward sloping palpebral fissures (gap between upper and lower eyelids), Brushfield spots (white spots in iris)
Limbs – single palmar crease, wide gap between first and second toes Cardiac - Atrial and Ventral septal defects Other - short stature, duodenal atresia (abnormally closed)
o Causes of DS: Trisomy 21
95% of all Down’s cases
28

Giles Kisby GE Y1 Genetics
90% maternal origin of extra chromosome Non-disjunction in meiosis I (75%) or II (25%)
Translocations 4% of all Down’s cases 2/3 de novo translocation in child 1/3 of parents are carriers of translocation Robertsonian Translocation occurs: breakage of acrocentric
chromosomes (eg 13,14,15,21,22) and fusion of their long arms High risk of further Down’s syndrome babies compared to if cause
was trisomy 21 Mosaicism
1% of all Down’s cases Children less severely affected Caused by mitotic non-disjunction in early embryo
- Sex Chromosome Aneuploidieso Turner’s syndrome
Monosomy Xo For sex chromosomes, only one X is required so the other is
randomly switched off in females (X-inactivation); is random between cells ie not the same X switched off in every cell of bodyN.B. It is not completely switched off - some genes (are also present on the Y) require expression from both copies of the X in women; ie demonstrate haploinsufficiency
1 in 3000 live female births Normal intelligence In young:
Generalised oedema and swelling in neck region can be detected in 2nd trimester (nuchal translucency)
Often no obvious problem at birth or may have oedematous extremities
In adults: short stature - 145 cm without GH treatment ovarian failure - primary amenorrhoea and infertility
Treatment oestrogen replacement for secondary sexual characteristics and
prevention of osteoporosis Genetic basis/cause:
80% due to loss of X or Y chromosome by e.g. non-disjunction in paternal meiosis
Also ring chromosome, single arm deletion, mosaicismo Polysomy X in females
1:1000 have 47,XXX karyotype 10-20 point decrease in IQ No physical abnormalities
29

Giles Kisby GE Y1 Genetics
95% have an extra maternal X arising in meiosis I Normal fertility 48,XXXX and 49,XXXXX karyotypes show more severe mental retardation
o Polysomy X in males Klinefelter’s syndrome (47,XXY); X chromosome from either parent 1 in 1000 male live births: ONLY FOR MALES!! Abnormalities;
clumsiness, verbal learning disability 10-20 pts taller than average (long lower limbs) 30% - moderately severe gynaecomastia: enlargement of the male
breast tissue Infertile increased risk of leg ulcers, osteoporosis and breast carcinoma in
adult life 48,XXXY and 49,XXXXY are rare
o Chromosomal Sex and Gender it is possible to be chromosomally one gender and phenotypically the
opposite via SRY translocations: see diagram below loss from Y: phenotypically female [nb will not suffer turner’s
symptoms because the Y can still supply the complementary genes to avoid haploinsufficiency]
o The SRY gene is lost and leads to a female who is infertile [ie while Turner’s is avoided there is still some haploinsufficiency in genes required for fertility]
addition to X: phenotypically maleo Translocation of the SRY male determining gene from Y
chromosome to an X chromosome. Phenotypically male, testes develop, but sterile because some genes on Y chromosome needed for spermatogenesis.
SRY gene = Sex determining Region on the Y = gives male characteristics SRY gene activated at 6 wks post-conception for signalling the development
of the testes
30

Giles Kisby GE Y1 Genetics
- Inherited abnormalities:o Adult presenting abnormalities
Eg Huntingdon’so Congenital Abnormalities [NOT ALL MUTUALLY EXCLUSIVE!; e.g. a primary
malformation of the kidneys can lead to Potter’s sequence] Malformation
A primary structural defect e.g. atrial septal defects, cleft lip. Usually involves a single organ showing multifactorial inheritance (ie
polygenic with environmental influence). Disruption
A secondary abnormal structure of an organ or tissue: Caused by ischaemia, infection, trauma.
Not genetic, but genetic factors can predispose. e.g. amniotic band causing digital amputation
Deformation An abnormal mechanical force distorting a structure e.g. club foot,
hip dislocation. Often occurs late in pregnancy and can have a good prognosis as the
underlying structure is normal (addressed orthopaedically) Syndrome
A consistent pattern of abnormalities with a specific underlying cause,
o e.g. Down’s syndrome.o Eg Chromosomal abnormalities
Sequence
31

Giles Kisby GE Y1 Genetics
A sequence is a chain of events leading to a primary factor that causes multiple abnormalities
Eg Potter’s sequence and oligohydramnios [low fluid in the womb]: is not a syndrome as there are many causes of this.
Could have a genetic component contributing to primary factor. Not a syndrome as multiple causes result in the same primary factor
Dysplasia An abnormal organisation of cells into tissue e.g. Thanatophoric Dysplasia
o Caused by a single gene defect (FGFR3)o High recurrence risk for siblings/ offspringo 1:60,000 incidenceo Short flat bones, small thorax, large head
Association An unexplained but non-random occurrence of abnormalities. Cause is typically unknown. e.g. VATER/VACTERL association.
o Vertebral, Anal, Tracheo-Esophageal, Renalo Vertebral, Anal, Cardiac, Tracheo-Esophageal, Renal, Limb
- The UK Newborn Screening Programmeo Phenylketonuriao Congenital Hypothyroidismo Sickle Cell Diseaseo Cystic Fibrosiso Medium-Chain Acyl-CoA Dehydrogenase Deficiency
- The UK Newborn Screening Programme [tests are for relavant metabolites; not the genetics directly]:
o Phenylketonuria Severe mental retardation and convulsions Blond hair/blue eyes Eczema Phenylalanine hydroxylase deficiency Phenylalanine accumulates and is converted to phenylpyruvic acid -
excreted in urine Leads to tyrosine deficiency - reduced melanin [explains the blue eyes and
blond hair] Early detection allows for no mental retardation Remove phenylalanine from diet but is Difficult diet and Pregnancy requires
strict dieto Congenital Hypothyroidismo Sickle Cell Diseaseo Cystic Fibrosis
32

Giles Kisby GE Y1 Genetics
o Medium-Chain Acyl-CoA Dehydrogenase Deficiency MCAD is Medium-Chain Acyl-CoA Dehydrogenase MCADD gene is called ACADM Commonest disorder of fatty-acid oxidation Episodic hypoketotic hypoglycaemia means brain will not get adequate
energy supply if starving coma / encephalopathy Can present as coma, encephalopathy, metabolic acidosis (lactic acidosis) Sudden death can occur Maintenance of adequate calorie intake to prevent switch to fatty acid
oxidation Avoidance of fasting (<12 hrs in adults), which can be difficult in children
who become ill – teaspoon of honey or glucose drip
33

Giles Kisby GE Y1 Genetics
24/02/14: The future of genomic medicine: Dr Jess Buxton
Los (from Booklet):
Describe the basic principles of and ethical considerations in pre-implantation genetic testing Explain how next generation sequencing is being used to determine the molecular basis of monogenic diseases Compare and contrast examples of direct-to-consumer genetic testing services and discuss their relative merits and problems Give examples of how advances in genomic medicine may lead to personalised medicine
Notes:
- Outline o Advances in genomic medicine
Pharmacogenetics Risk of common, complex diseases ‘Direct-to-consumer’ genetic testing High-throughput DNA sequencing Finding the causes of monogenic disease
o Stratified medicine (‘precision’ medicine) Future perspectives Ethical issues
o Embryo testing Preimplantation genetic diagnosis (PGD) Uses and limitations Ethical issues
- Genomic medicine:o Monogenic disease; directly test for specific genes to determine if is mutated:
TPMT: Variants in TPMT gene reduce metabolism of 6-mercaptopurine,
6MP (a common chemotherapy drug) Ie Pharmacogenetics relevance
MODY [Maturity onset diabetes of the young] Can be misdiagnosed as T1D but is actually very similar to T2D but
occurs in children Ie Pharmacogenetics relevance; will need to treat with diff drugs
34

Giles Kisby GE Y1 Genetics
o Polygenic disease: Genome Wide Association Studies = GWAS Each person has ~3 million Single Nucleotide Polymorphisms (SNPs) Many sets of SNP genetic variants affect risk of common, complex traits and
diseases: Eg. heart disease, diabetes, arthritis, schizophrenia, bipolar, depression,
Crohns disease, macular degeneration, obesity, blood pressure, birthweight Problems include that not all the relevant genetic influences are being
considered and does not account for env influences on disease risko Research; DNA sequencing:
Identify novel gene mutations in monogenic disease
Eg allowed the discovery of the genetic basis of Miller syndrome: [“M looks like toes”]
o Multiple malformation syndrome
o Characteristic facial features, including ‘cupped’ ears and
absent toeso Caused by mutations in DHODH gene
Eg allowed the discovery of the genetic basis of Schinzel-Giedion syndrome:
o Severe mental retardationo Multiple congenital abnormalities o Characteristic facial featureso Life-limiting o Caused by mutations in SETBP1 gene
35

Giles Kisby GE Y1 Genetics
Identify genetic changes in cancer cellso Patient; DNA sequencing:
Benefits: Disease risk prediction personalised/stratified medicine possible greater role of pharmacogenetics in future
Must remember are not just finding info on the patient but also their family who may not want to know
Currently not mainstream but 100,000 patients over the next five years will be trialled in the context of research on rare diseases, susceptibility to infectious diseases and cancer
o PGD (preimplantation genetic diagnosis) = embryo testing of IVF babies: PGD is a genetic test carried out on IVF embryos, usually to ensure that only
embryos free from a particular genetic condition are returned to the woman's womb
PGD is an option for some families at risk of having a child affected by a serious genetic condition, offered as an alternative to prenatal testing: Examples:
Severe early onset genetic disease, eg. Tay Sachs Severe late onset conditions, eg. Huntington disease Disease with incomplete penetrance, eg. hereditary breast cancer
(BRCA1/2 mutations) To choose a tissue-matched baby that can provide umbilical cord
blood to treat a sick sibling Types of genetic test
Fluorescent in situ hybridisation (FISH) – to detect chromosomal conditions, eg. Down syndrome
Polymerase chain reaction (PCR) and DNA sequencing – to detect mutations in single genes
36

Giles Kisby GE Y1 Genetics
13/03/14: Final thoughts- the mainstreaming of genetics: Prof. Alex Blakemore
Los (from Booklet): In this session we will discuss the emerging requirements for genetics and genomics to be incorporated into all medical specialities, with further examples of the knowledge base required.
You will need to be able to give examples of where a genetic disease: displays more than one inheritance pattern; is caused by mutations in more than one gene; is caused by mutations in the same gene as a different disease phenotype is altered by a mutation in another gene
We will also discuss the second worksheet, and other issues arising from the course
Notes:
- Monogenic disease:o A mutated gene required to cause this specific diseaseo One mutated gene for the family (multiple mutations possible)o Protein or gene product from mutated allele is defective o May be dominant or recessive
- Complex genetic traits:o Usually attributed to an external agent (e.g. virus trauma)o Often some genetic predisposition:
No single gene causes disease; Gene can be promoting susceptibility to disease
e.g. resistance to HIV: homozygous mutation in CCR5 Threshold effect; a certain number of genetic factors may be needed Low penetrance : may have the mutation(s) but not the disease due
to certain environmental inducers being required eg viral/diet Interactions between genes (epistatsis) [“eg curly wings gene and wingless
gene”] Polymorphic variants: may function slightly differently (better/worse)
o Complex disease tend to be more common than monogenic diseases
37

Giles Kisby GE Y1 Genetics
Hypertension, cancer, IHD more common than CF, DMD for example
- Twin studieso Proband = first affected family member who seeks medical attention for a genetic
disordero high risk in relative of probands compared to general population
Shared environment as well as shared geneso λs = risk of disease in sibling of patients / risk of disease in general populationo λs values are high for single gene disorders, much lower for complex traits
- Investigation of complex diseases:o Linkage analysis:
Following markers of the disease as passes through a family / generationso Family studies
Look at twins / siblings with the disease (ie usually same environment, ethnicity, age, gender, etc already) and look for markers that correlate with disease incidence
Ie look for markers that are shared more often than random for affected sibling pairs
o Population studies / association studies: Compare diseased people for common markers relative to undiseased
controls (here must match for ethnicity etc)
- Los:- You will need to be able to give examples of where a genetic disease:
o displays more than one inheritance pattern; Muscular dystrophies
o is caused by mutations in more than one gene; neurofibromatosis Ocular albinism Albinism Ocular albinism
o is caused by mutations in the same gene as a different disease SMAD4 muts
Causes Juvenile polyposis Causes Hereditary haemorrhagic telangiectasia
o phenotype is altered by a mutation in another gene factor V leiden and haemophilia
- Genetic diseases may have:o Different patterns of inheritance
38

Giles Kisby GE Y1 Genetics
Eg Muscular dystrophies: autosomal dominant; recessive; x linked
o Phenocopies: Mutations in many different genes giving the same phenotype neurofibromatosis (autosomal dominant) Ocular albinism Albinism (autosomal recessive) Ocular albinism
o Mutations in one disease can compensate for another e.g. factor V leiden and haemophilia: the Leiden variant (form) of factor V
cannot be inactivated
o Different mutations in the same gene may give different disorders Eg SMAD4 muts
Juvenile polyposis: inherited GI pre-malignant state OR Causes Hereditary haemorrhagic telangiectasia: inherited
vascular disease, usually no cancer predisposition [associated with nosebleeds]
o Disease may be different depending on which parent has transmitted the disease allele (see tutorial below)
- Genetic imprinting:o As part of normal regulation some genes are expressed preferentially from either
the maternal or paternal DNA [therefore must have the allele from the relavant parent present to avoid the disease]
o The mechanism occurs by (reversible) DNA methylation in primordial germ cellso Eg imprint occurs on chromsome 11 and 15o Loss of imprinted 15q11-13 region on chromosome 15 case study:
Multiple genes present at this region: Loss of these genes wil have different effects according to which chromsome is affected:
paternally expressed gene SNRPN, NDN maternally expressed UBE3A [nb Point mutations in the maternal
UBE3A can also cause Angelman] Prader-Willi (if paternal copy not present) and Angelman syndrome (if
maternal copy not present)o The specific parental copy may not be present due to a deletion or inheritance of
both CH15’s from the same parent = “uniparental disomy” Paternally derived deletion: Prader Willi Syndrome = obesity;
hypogonadism; hypotonia, short stature Maternally derived deletion: Angelman syndrome = epilepsy, tremors and a
permanent smiling facial expression Maternal UPD - PW Paternal UPD - Angelman Syndrome
- Genetic tests:o Embryo testing:
Non invasive
39

Giles Kisby GE Y1 Genetics
Maternal blood tests Non invasive imaging
Invasive (risk of miscarriage increased but better estimation of disease risk: may want to terminate or initiate fetal medicine intervention)
Aminocentesis (ie test foetal cells shed into amniotic fluid) Chroionic villus sample (ie sampling of the chorionic villus =
placental tissue)o Neonatal testing: [ie are considered genetic tests even if genetic not looked at
directly; just means are identifying if has genetic disorder or not] Diseases tested:
PKU CF Hypothyroidism MCADD Sickle cell disease
o Potential benefits from genetic tests: Knowledge regarding diagnosis and prognosis Targeted avoidance of high risk states such as: diet (MCADD, PKU), infection,
life style Altered management of high risk states: infection (sickle cell), pregnancy Screening and early treatment for complication - aortic (Marfans), vascular
(HHT: Hereditary Hemorrhagic Telangiectasia) Dedicated treatment regimens: enhanced susceptibility to existing drugs
(Marfans, HHT) gene therapy
40

Giles Kisby GE Y1 Genetics
17/02/14: Cancer in families and individuals: Alistair Reid [genetics]
Los (from Booklet):
Explain the difference between somatic and germline mutations Understand why genetic changes cause cancer and describe the 2 main classes of cancer
gene Understand the contribution of chromosome rearrangements to the formation of gene
fusions and their contribution to oncogenesis. Discuss how inherited mutations in BRCA1 and BRCA2 genes influence risk of breast and
ovarian cancer Outline how defects in cell division or DNA repair influence risk of colorectal cancer Explain, using an example, how chromosome translocations are used to quantify residual
disease in some leukaemias. Explain with examples what is meant by a “pharmacogenomic marker”
Notes:
- Summaryo Cancer caused by abnormalities of tumour suppressors or oncogeneso TS genes usually require “2 hits” the second of which is often a deletion (manifesting
as LOH)o Usually sporadic but can be inherited – some genetic causes with obvious mendelian
inheritance are well characterisedo Somatic abnormalities, particularly those involving a novel fusion gene can be used
to monitor therapy (if successful therapy available)o Certain somatic abnormalities confer resistance or response to new targeted
therapies – “Pharmagogenomics”
- Some facts about cancero One in three people will develop cancer during their lifetimeo One in eight people will die from cancero Cancer can develop at any age, but three-quarters of cases occur in people aged 60
or oldero Encompasses >100 different diseases from most of the cell types and organs of the
body
41

Giles Kisby GE Y1 Genetics
- Tumour suppressor vs oncogene:o Normal (proto)oncogene functions (favour the halt of cell cycle)
Growth and proliferation eg
Growth factors Transcription factors Tyrosine kinases
o Normal functions of TS genes (favour prolif but in a controlled way) Regulating cell division DNA damage checkpoints (damage=no division) Apoptosis DNA repair
- Mutation progression:
o Mechanisms for TSG inactivation / protooncogene activation: TSG inactivation
Typically “two hits” required but some TSGs demonstrate “haploinsufficiency”
Mut types:o mutation in promotoro mutation in coding regiono Genomic deletion or whole chromosome loss
Most common seq of events:o 1st hit is a point mutation germline mutation
42

Giles Kisby GE Y1 Genetics
1% of cases of cancer are initiated by the inheritance from one parent (or occasionally both) of a mutation in “germline” tissue, usually in a tumour suppressor “(hit 1”)
Ie 99% of cancers are sporadic (non-inherited) and for those that are the cancer wasn’t guaranteed; just confer a “high risk” of cancer due to the 1st hit
o 2nd hit is a deletion in somatic cell Such deletions can be detected as a loss of
hetrozygosity; SNPs for a region of the genome would be expected to show some hetrozygosity due to the diff options that could exist but deletion means that only one of each possibility of each demonstrated for that region when interrogated
protooncogene activation Mut types:
o mutation in promotoro mutation in coding regiono Genomic amplification or whole chromosome gaino Gene fusion via chromosome rearrangement
Examples:
43

Giles Kisby GE Y1 Genetics
- Breast cancer:o BRCA 1/2:
Are TSGs: Normal function of BRCA1 and BRCA2 is DNA repair, specifically a process called homologous recombination. A truncated/non-functional protein causes impaired DNA repair
2-4% of breast cancer cases are caused by germline mutation of BRCA1 or BRCA2 genes (“hit 1”) [ie 2-4% of BC due to the Inherited predisposition to breast and ovarian cancer]
Mutations in BRCA1 and BRCA2 are uncommon, and breast cancer is relatively common, so these mutations consequently account for a small percentage of all breast cancer cases in women
Effect of having mutation: 60% risk of developing breast cancer by age of 90, earlier average age of onset
There must be other genes linked to breast cancer!: 10-20% of cases have some evidence of family history with no BRCA1/2 mutations
- Colorectal cancer:o FAP (familial adenomatous polyposis): APC TSG
Characterised by the growth of 1000s of intestinal polyps, one or more of which is likely to become cancerous >1% of all colorectal cancers AUTOSOMAL DOMINANT Mutation of APC (adenomatous polyposis coli) gene APC controls cell division Virtually 100% lifetime risk of cancer
o Hereditary non-polyposis colorectal cancer (HNPCC = Lynch syndrome): MLH1 & MSH2 TSGs [Microsatellite Instability - High (“MSI-H”)
3% of all colorectal cancer cases, AUTOSOMAL DOMINANT Most common inherited form (90% of familial cases) Mutation of MLH1 [MutL homolog 1] or MSH2 [MutS homolog 2] (DNA
“mismatch repair” genes) Lifetime risk of cancer 80%
- Leukaemia:o Chronic Myeloid Leukaemia:
A clonal myeloproliferative disorder of the pluripotent haematopoeic stem cell leading to an overproduction of mature granulocytes [ie high rate of BM stem cells mature granulocytes, hence is termed chronic]
Triphasic : indolent chronic phase, accelerated and terminal acute stage
44

Giles Kisby GE Y1 Genetics
Caused b y: translocation between chromosomes 9 abl gene and 22 bcr gene resulting in a fusion gene called BCR-ABL1 that functions as a
tyrosine kinase[nb is still referred to as an oncogene] Treatment : use TKIs; tyrosine kinase inhibitors:
eg Imatinib = Glivec which blocks the ATP binding pocket; however note that the fusion prot sometimes undergoes a further
mutation to become impervious to TKIs: BCR-ABL1 “T315I” = unlikely to respond to TKIs
also: resistance to TKIs can develop Following efficacy of treatment : [want to know the amount/concentration
of cells in blood that still have the fusion gene mutation]; In order of increasing sensitivity:
Cytogenetics (ie karyotyping using banding) [absent bands looked for]
FISH [colocalisation of colours looked for] RQ-PCR (reverse transcriptase quantitative PCR) [primers that can
only amplify the fusion prot are used]o Pathology
Malignancy of granulocytes → increased myeloid precursor production Philadelphia chromosome: result of Bcr-abl fusion gene: constituitively active
tyrosine kinase o Risk factors
Ionising radiation + benzene exposure o Symptoms + Signs
Anaemia, weight loss, lassitude, anorexia, sweating, gout, bleeding [prob due to thrombocytopenia], priapism, low grade fever [prob due to neutrophillia]
Bruising, petechiae, hepatosplenomegaly
o Acute promyelocytic leukemia Abnormal accumulation of immature granulocytes called promyelocytes Characterized by a chromosomal translocation forming a fusion protein:
45

Giles Kisby GE Y1 Genetics
The proteins involved are the retinoic acid receptor alpha (RARα or
RARA) gene on chromosome 17 and the promyelocytic
leukemia gene (PML) on chromosome 15, a translocation denoted as t(15;17)(q22;q12); ie RARα::::PML
Normal form : RARα is a member of the nuclear family of receptors; its ligand, retinoic acid is a form of Vitamin A and acts as a regulator of DNA transcription
Pathogenic form : The translocation product is the PML-RARα fusion protein – binds too strongly to DNA via enhanced interaction with co-repressor molecules, blocking transcription (“dominant negative mutant”)
o acute promyelocytic leukemia disseminated intravascular coagulation
o Is in AML M3 subset
o Treatment : APML responds to all trans retinoic acid (ATRA) therapy,
a vitamin A derivative. ATRA binds, degrades and cleaves PML-RARα dissociating co-repressors allowing normal transcription and cell differentiation
Following efficacy of treatment : Like CML, APML is monitored by cytogenetics and/or FISH and/or RQ-PCR
- Pharmacogenomics and cancero In cancer treatment, pharmacogenomic tests are used to identify which patients are
most likely to respond to certain cancer drugs based on the presence or absence of particular somatic mutations.
o Examples include: KRAS test with cetuximab for colorectal cancer
KRAS mutation = less likelihood of response EGFR test with gefitinib for nonsmall-cell lung cancer
EGFR mutation = greater likelihood of response BCR-ABL1 “T315I” test with dasatinib for chronic myeloid leukaemia
BCR-ABL1 T315I mutation = unlikely to respond to TKIs [was mentioned above]
46

Giles Kisby GE Y1 Genetics
Tutorial:
- nb part of you was in your grandmother(!)- nb genetic testing is a test specifically for an individual vs screening where is a general test
against the whole population- nb “affected” may be referring to not just sufferers but also carriers- nb be able to distinguish autosomal dominant from a mt DNA mutation disease; look similar
but for mt mutation it will be the children of females ONLY that are affected- hardy Weinberg assumptions:
o natural selection is not actingo no new mutations or migration of peopleo infinitely large population is assumedo mating is random
- hardy Weinberg equation:o p = A = dominant / other trait; q = a = recessive / other traito ensure use fraction / decimal in population, not raw numberso consider copying in pharma notes
- balanced translocation if 1st gen; no disease unless gene at boundary; only next gen disease risk when becomes unbalanced in the 2nd gen
- unbalanced translocation gives immediate phenotypes; due to balanced translocation of previous gen
- translocation examples:o Bcr-Abl = philidelphia chromosome:
Gives CML; use imatinib = gleevec to treato Dek – can
Gives AMLo Ews – fli
Gives ‘Ewings sarcoma’
47

Giles Kisby GE Y1 Genetics
48

Giles Kisby GE Y1 Genetics
Self-directed learning objectives [clarify if the below are still part of course; the first is only on last years and the second not found anywhere; if not then why are there still the learning objectives?!]
GENETICS WORKSHEET 1You will gain more from this exercise if you perform the calculations yourself before the answers are discussed in the first practical. Demonstrate an understanding of Mendelian segregation, by being able to perform simple genetic risk calculations.
[use these to practice]:You should work through these problems on your own between the 1st and 3rd genetics Lectures to gain the most from these examples Tip: draw family trees wionalith segregation of relevant alleles.
1. A woman affected by Charcot Marie Tooth Disease (autosomal dominant) already has one affected child. What is the probability that her next child will be affected?
2. A couple have one child with cystic fibrosis (autosomal recessive) and another who is not affected. What is the probability that the one who is not affected, is homozygous normal?
3. A man with AB blood group marries a woman with B blood group. What is the probability that a child of theirs will be B blood group?
4. A couple have a son who has Duchenne Muscular Dystrophy (X-linked). What is the probability that their unaffected daughter is a carrier?
5. If a couple who both have achondroplasia (autosomal dominant) have children, what percentage of their children will be of normal stature?
TIP: Remember to consider what is likely to happen to affected homozygotes 6. A woman's nephew (her sister’s son) has Haemophilia A (X-linked). Assuming that this is not a new mutation, what is the probability of the woman being a carrier?
7. A couple are hoping to start a family. His brother has osteogenesis imperfecta (autosomal dominant; affects 1 in 20,000). Their parents are not affected. What is the probability of a child of theirs being affected by osteogenesis imperfecta?
8. A man with G6PD deficiency (X-linked) has 1 unaffected daughter. What is the probability that her sons will be affected?
a) her first son b) All of her 3 sons c) One of her 3 sons
49

Giles Kisby GE Y1 Genetics
GENETICS WORKSHEET 2You will gain more from this if you do these exercises yourself, before the discussions scheduled for the last day of the course. Be aware of online resources for information on genetic disease, including OMIM, Genecards, the National Genetics Education Development Centre and the NHS library resources on genetic conditions; Understand the basic principles of meiosis and non-disjunction; Understand the basic principles of karyotype analysis and the clinical features of trisomy 21; Be able to give named examples of genetic disorders with different modes of inheritance; Be able to give two examples where different types of mutation in the same gene cause different monogenic disorders; Understand the basic principles of DNA purification, PCR and agarose gel electrophoresis and how these might be used to investigate human diseases.
A) The cause of Huntington’s disease: affects muscle coordination and leads to cognitive decline and psychiatric problems Huntingtin gene: [nb sp true]
o Part of this gene is a repeated section called a trinucleotide repeat, which varies in length between individuals.
o When the length of this repeated section reaches a certain threshold, it produces an altered form of the protein, called mutant Huntingtin protein (mHtt).
o The Huntington's disease mutation is genetically dominant and almost fully penetrant
B) Record the mode of inheritance, gene name and main clinical features of the following conditions:
Phenylketonuria mode of inheritance
autosomal recessivegene name
phenylalanine hydroxylase (PAH)main clinical features
This enzyme is necessary to metabolize the amino acid phenylalanine (Phe) to the amino acid tyrosine
When PAH activity is reduced, phenylalanine accumulates and is converted into phenylpyruvate (also known as phenylketone), which can be detected in the urine.
Untreated PKU can lead to intellectual disability, seizures and other mental problems
Incontinentia pigmenti mode of inheritance
X-linked dominant
50

Giles Kisby GE Y1 Genetics
gene name IKBKG gene
main clinical features affects the skin, hair, teeth, nails, and central nervous system ie skin lesions, alopecia, hypodontia, abnormal tooth shape, dystrophic
nails, cognitive delays/mental retardation
MELAS: Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes –abbreviated to MELAS
mode of inheritancemitochondrial genome
gene name nothing specific: just mutations in the genes in mitochondrial DNA
main clinical featuresencephalopathy and myopathy: muscle weakness and pain, recurrent headaches, loss of appetite, vomiting, and seizures
46XY complete gonadal dysgenesis mode of inheritance
inherited male to male; ie is Y chromosome problemgene name
multiple options: eg SRY or DHH mutation: Desert hedgehog main clinical features
hypogonadism in a person whose karyotype is 46,XY The person is externally female with streak gonads, and left untreated, will
not experience puberty Congenital androgen insensitivity
mode of inheritanceInheritance is typically maternal (due to the problems it causes in men) and follows an X-linked recessive pattern
gene name human androgen receptor (AR)
main clinical features partial or complete inability of the cell to respond to androgens impair or prevent the masculinization of male genitalia in the developing
fetus, as well as impair the development of male secondary sexual characteristics at puberty
51

Giles Kisby GE Y1 Genetics
does not significantly impair female genitalia or sexual development Kennedy disease
mode of inheritanceInheritance is typically maternal (due to the problems it causes in men) and follows an X-linked recessive pattern
gene name mutation of the androgen receptor (AR) gene
main clinical featuresmuscle cramps and progressive weakness due to degeneration of motor neurons in the brain stem and spinal cord.
Cystic fibrosis mode of inheritance
autosomal recessivegene name
point mutation in the gene cystic fibrosis transmembrane conductance regulator (CFTR)
52

Giles Kisby GE Y1 Genetics
main clinical features thick, viscous secretions The name cystic fibrosis refers to the characteristic scarring (fibrosis) and
cyst formation within the pancreas Myotonic dystrophy
mode of inheritanceautosomal dominant
gene name DMPK or ZNF9
main clinical featureswasting of the muscles (muscular dystrophy)
Haemophilia B mode of inheritance
X-linked recessive trait (NB as is haemophilia A)gene name
mutation of the Factor IX genemain clinical features
increased propensity for haemorrhage
DMD gene: = dystrophin gene: X - linked different types of mutation in this gene cause different types of muscular dystrophy: Duchenne muscular dystrophy (DMD) Becker's muscular dystrophy (BMD)
53

Giles Kisby GE Y1 Genetics
54