Embed Size (px)
Citation preview

8/10/2019 0003-9985%281999%29123-0358%3Apqc-2%2E0%2Eco%3B2.pdf
http://slidepdf.com/reader/full/0003-998528199929123-03583apqc-22e02eco3b2pdf 1/3
358 Arch Pathol Lab Med—Vol 123, April 1999 Pathologic Quiz Case —Malone et al
Residents’ Page
Pathologic Quiz Case
Janine C. Malone, MD; Kelly Z. Brown, MD; Joseph C. Parker, Jr, MD; Louisville, KY
The patient was a 46-year-old white man with a historyof hypertension for 15 years (most recently treated
with lisinopril), peptic ulcer disease, arthritis, chronic ob-structive pulmonary disease, and hepatitis A 4 years be-fore presentation. He also had a 3-pack-per-day smokinghistory for the past 30 years (having quit 1 year before
presentation) and a history of 6 to 8 beers per day for thepast 30 years. Twenty years ago, he had been diagnosedwith and treated for pulmonary tuberculosis.
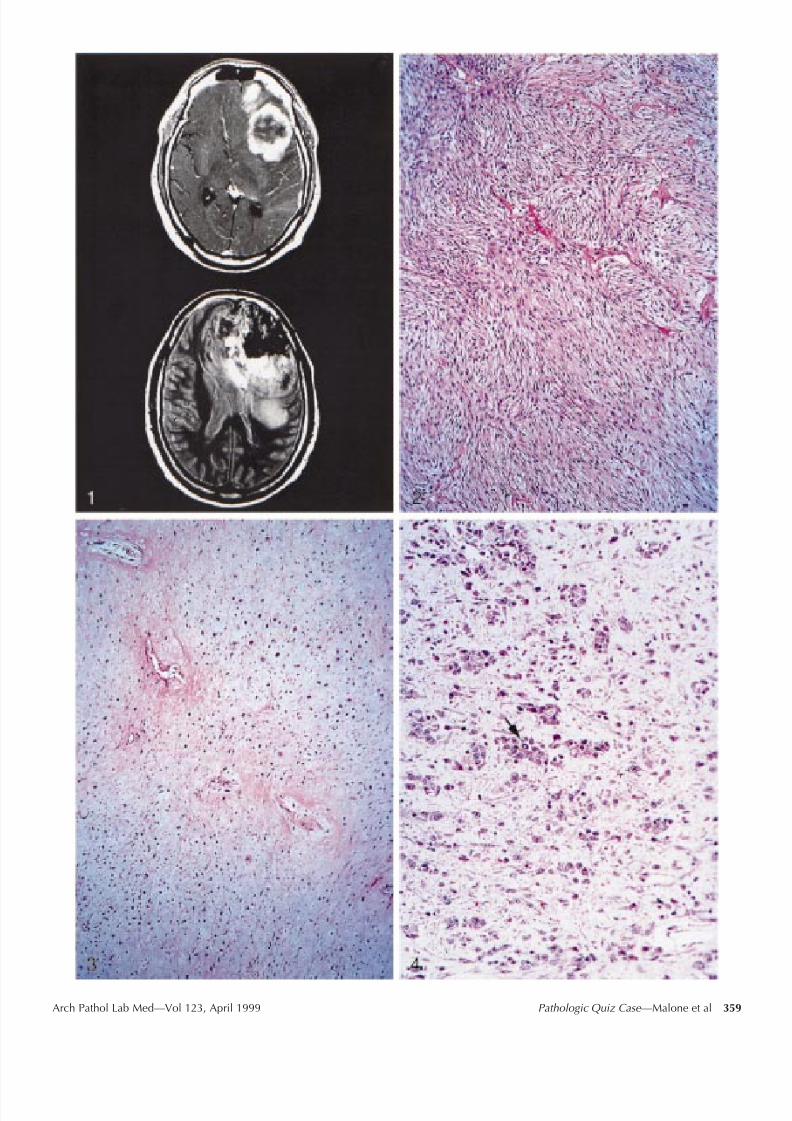
Radiologic studies included computed tomographicscan of the head, which demonstrated an inhomogeneous,calcified left frontal lobe mass crossing the corpus callos-um and producing a shift of midline structures. Subse-quent magnetic resonance imaging demonstrated a 9 6-cm, irregular, rim-enhancing left frontal lobe mass withlow signal intensity corresponding to calcification andseveral unenhancing areas suggestive of necrosis. Shift of midline structures and subfalcine herniation were alsonoted (Figure 1).
The patient presented with his family, who described
recurrent sudden ‘‘blackouts’’ in the patient during theprevious 2 weeks. Physical examination revealed a slightlydepressed and lethargic obese man with a blood pressureof 152/98 mm Hg and a pulse of 85 beats per minute. Hewas disoriented to place and time and demonstrated slightpronator drift of the right upper extremity. Following im-aging studies, the patient underwent left fronto-temporo-
parietal craniotomy with near-complete tumor resection.Following surgery, he received 65 Gy of external beamradiation. Approximately 3 months after surgery, his fam-ily reported increased confusion and right-sided weak-ness. A repeat computed tomographic scan revealed alarge recurrent tumor mass. The patient died a few weeks
later, nearly 4 months after his initial presentation. Per-mission for autopsy was denied.At the time of surgical resection, a large left frontal neo-
plasm with dural attachment was noted. The meningeal sur-face was ossified. Pathologic evaluation revealed 190 g of fragmented, tan to pale yellow, soft to rubbery tissue withassociated white chondroosseous portions. Gross hemor-rhage and necrosis were absent. Microscopically, the tumordemonstrated several patterns. Most prominent were highlycellular areas composed of malignant spindle cells with astoriform pattern (Figure 2). These areas demonstrated pos-itive immunoreactivity for CD68 and negative immunore-activity for glial fibrillary acidic protein. Separate distinct ar-eas of malignant, focally necrotic cartilage and malignant
osseous tissue were also identified (Figure 3). These com-ponents exhibited increased mitotic activity with 2 to 5 mi-toses per high-power field. Atypical to frankly malignantastrocytes were present (Figure 4) and demonstrated positivestaining for glial fibrillary acidic protein. Bizarre mitotic fig-ures were occasionally seen (Figure 4, arrow).
What is your diagnosis?
8/10/2019 0003-9985%281999%29123-0358%3Apqc-2%2E0%2Eco%3B2.pdf
http://slidepdf.com/reader/full/0003-998528199929123-03583apqc-22e02eco3b2pdf 2/3
Arch Pathol Lab Med—Vol 123, April 1999 Pathologic Quiz Case —Malone et al 359

8/10/2019 0003-9985%281999%29123-0358%3Apqc-2%2E0%2Eco%3B2.pdf
http://slidepdf.com/reader/full/0003-998528199929123-03583apqc-22e02eco3b2pdf 3/3
360 Arch Pathol Lab Med—Vol 123, April 1999 Pathologic Quiz Case —Malone et al
Clinicopathologic Data Compiled From 63 Cases of Gliosarcoma(1,2,5)
Number Percent
Age,4040–5960
82728
134344
SexMale
Female
35
28
56
44Location*
FrontalTemporalParietalOccipitalOther
183026
73
213631
84
* Tumors located in overlapping lobes are scored in all areas whichapply.
Pathologic Diagnosis
Gliosarcoma Containing Malignant Fibrohistiocytic,Osseous, and Chondroid Elements
Gliosarcoma is a biphasic neoplasm that contains bothmalignant glial and mesenchymal elements. In most le-sions, the malignant glial component is glioblastoma mul-tiforme (GBM), although an oligodendroglial componenthas occasionally been described. These tumors are rare,
representing between 1.8% and 8% of malignant glial neo-plasms.1,2
Feigin and Gross3 described 3 patients with tumorscomposed of mixed glial and sarcomatous elements. Theseauthors postulated that the sarcomatous component arosefrom neoplastic transformation of vascular endotheliumwithin a primary GBM. Many studies utilizing immuno-histochemical and ultrastructural markers for vascular or-igin have both supported and refuted this hypothesis.Still, these tumors are often referred to as Feigin tumors.Later investigators classified mixed glial and sarcomatousneoplasms according to their presumed primary compo-nent. In 1979, Lalitha and Rubinstein4 suggested the termsarcoglioma for malignant mesenchymal brain tumors con-taining ‘‘secondary’’ glial elements. They described le-sions with a central meningioma or sarcoma surrounded by gliomatous elements ranging from reactive astrocytosisto GBM. Based on the tumor arrangement, the authorsproposed that the sarcoma induced the development of glioma.
A temporal location is classically reported for gliosar-comas, and compiled data support this observation (Ta- ble). As in most gliomas, gliosarcomas are more commonin males than females. The majority occur in the aged, asis the case for most high-grade glial neoplasms. In con-trast, sarcogliomas tend to occur in children or youngadults. Gliosarcomas have shown a predilection to arise
in patients having undergone previous cerebral radiationtherapy. The mean survival of patients with gliosarcomais similar to that of patients with GBM, being less than 12months. In contrast to GBM, there is a propensity forgliosarcoma to disseminate and produce extracranial me-tastases.6
Neuroimaging reveals a hypodense enhancing massthat is superficially located within the cerebral parenchy-ma and often associated with the leptomeninges and dura.Peritumoral edema is common, and calcification and cys-tification are sometimes evident. Grossly, gliosarcomas ap-pear firm, discrete, and lobulated. Frequently, there ismeningeal attachment, and the macroscopic appearancemay suggest meningioma. The cut surface is often varie-gated with firm gritty areas alternating with softened yel-low areas. Microscopically, the tumor possesses fasciclesof sarcoma interspersed with GBM in a marbled arrange-ment. In advanced lesions, neoplastic glial cells may beonly sparsely distributed within the sarcoma. The spindlecells of sarcomatous areas typically have cytologic featuresof malignancy, including pleomorphic nuclei with vesic-ular or clumped chromatin and prominent nucleoli. Mi-totic figures are present, although they may not be abun-dant. Although fibrosarcoma is the most frequently re-ported stromal malignancy,7 other patterns resemblingmalignant fibrous histiocytoma, rhabdomyosarcoma, os-
teosarcoma, and chondrosarcoma have been described.5
Special stains help delineate the biphasic nature of thesetumors. Silver stains demonstrate the reticulin-rich stroma
of the sarcomatous component, while glial fibrillary acidicprotein immunoperoxidase techniques decorate the glialcomponent. Reactivity for histiocytic markers and myoidmarkers may be seen in the mesenchymal component of gliosarcomas.
An area of debate centers on the cell of origin of thesarcomatous component of gliosarcoma. Ultrastructuraland immunohistochemical studies have suggested a pos-sible endothelial cell origin, as Feigin and Gross3 suggest-ed, whereas other studies have refuted these results. Alsoimplicated as the cell of origin are adventitial and men-ingeal fibroblasts, histiocytes, vascular smooth musclecells, or some undifferentiated pluripotential mesenchy-mal cell.
In summary, this case illustrates an unusual malignantglioma with characteristic gross and microscopic features.Gliosarcoma should be included in the differential diag-nosis of any discrete, superficially located or meningeal- based tumor that radiographically or grossly suggests ameningioma. The diagnosis moves to the forefront if thetumor arises in the temporal lobe or demonstrates necro-sis. The biphasic nature of the tumor may be difficult todiscern if there is a paucity of glial elements or if glialcells assume a spindled configuration. Reticulin and glialfibrillary acidic protein stains are helpful in these cases.
References
1. Morantz RA, Feigin I, Ransohoff J. Clinical and pathological study of 24cases of gliosarcoma. J Neurosurg . 1976;45:398–408.
2. Meis JM, Martz KL, Nelson JS. Mixed glioblastoma multiforme and sarcoma:a clinicopathologic study of 26 radiation therapy oncology group cases. Cancer .1991;67:2342–2349.
3. Feigin I, Gross SW. Sarcoma arising in glioblastoma of the brain. Am J Pathol . 1954;31:633–649.
4. Lalitha VS, Rubinstein LJ. Reactive glioma in intracranial sarcoma: a formof mixed sarcoma and glioma (‘‘sarcoglioma’’), report of eight cases. Cancer .1979;43:246–257.
5. Sreenan JJ, Prayson RA. Gliosarcoma, a study of 13 tumors, including p53and CD34 immunohistochemistry. Arch Pathol Lab Med . 1997;121:129–133.
6. Cerame MA, Guthikonda M, Kohli CM. Extraneural metastases in gliosar-coma: a case report and review of the literature. Neurosurgery . 1985;17:413–418.
7. Burger PC, Scheithauer BW. Tumors of the Central Nervous System. Wash-ington, DC: Armed Forces Institute of Pathology; 1994. Atlas of Tumor Pathology ;3rd series, No. 10.




![· "!# $%'& " !)(* $ +" , - , . $ /1032547682968:= 0@?34BA ?3CED F G8HJIKH;@2E0>DMLON@=P=P?3CO=PQEN H?3LORS;@T.05UVNOLW? X'Y Z@[]\_^a` Y bV](https://img.pdfslide.net/doc/110x75/5f51303d42d8670c615987fc/teisenkopaperslochpdf-1032547682968.jpg)















![ADjoint: An Approach for the Rapid Development of Discrete ...adl.stanford.edu/papers/AIAA-29123-431.pdf · systems [9]. The usefulness of the adjoint method lies in the fact that](https://img.pdfslide.net/doc/110x75/5f0982447e708231d427297c/adjoint-an-approach-for-the-rapid-development-of-discrete-adl-systems-9.jpg)








