Embed Size (px)
DESCRIPTION
Quantitative LCMS, HiLo, Multiplex LCMS, LCMSe
Citation preview

Quantitative Proteomic Analysis by Accurate MassRetention Time Pairs
Jeffrey C. Silva,*,† Richard Denny,§ Craig A. Dorschel,† Marc Gorenstein,† Ignatius J. Kass,‡Guo-Zhong Li,† Therese McKenna,§ Michael J. Nold,‡ Keith Richardson,§ Phillip Young,§ andScott Geromanos†
Waters Corporation, 34 Maple Street, Milford, Massachusetts 01757-3696, Waters Corporation, 100 Cummings Center,Beverly, Massachusetts 01915, and Waters Corporation, Atlas Park, Simons Way, M22 5PP, Manchester, Great Britain
Current methodologies for protein quantitation include2-dimensional gel electrophoresis techniques, metaboliclabeling, and stable isotope labeling methods to name onlya few. The current literature illustrates both pros and consfor each of the previously mentioned methodologies.Keeping with the teachings of William of Ockham, “withall things being equal the simplest solution tends tobe correct”, a simple LC/MS based methodology ispresented that allows relative changes in abundance ofproteins in highly complex mixtures to be determined.Utilizing a reproducible chromatographic separationssystem along with the high mass resolution and massaccuracy of an orthogonal time-of-flight mass spectrom-eter, the quantitative comparison of tens of thousands ofions emanating from identically prepared control andexperimental samples can be made. Using this configu-ration, we can determine the change in relative abundanceof a small number of ions between the two conditionssolely by accurate mass and retention time. Employingstandard operating procedures for both sample prepara-tion and ESI-mass spectrometry, one typically obtainsunder 5 ppm mass precision and quantitative variationsbetween 10 and 15%. The principal focus of this paperwill demonstrate the quantitative aspects of the methodol-ogy and continue with a discussion of the associated,complementary qualitative capabilities.
Quantitative proteomics has been chartered as the technologywhich will serve as a major contributor in studies aimed atuncovering disease pathways, biomarker discovery, and providingnew insights into biological processes for drug discovery. In theseexperiments, mass spectrometry is used to determine the relativeamounts of protein among different biological samples to char-acterize a variety of physiological conditions. In addition, furthercharacterization of the physiological perturbation may require thatthe relative degrees of posttranslational modifications associatedwith the proteins of interest be determined. However, compre-hensive quantitative proteomics remains technically challenging
due to the issues associated with sample complexity, samplepreparation, and the wide dynamic range of protein abundance.1,2
Many approaches to quantitative proteomics have involved thecombination of stable-isotope labeling methods for sample prepa-ration with automated liquid chromatography coupled to a tandemmass spectrometer (LC/MS/MS).3-12 Stable isotopes are generallyintroduced into proteins or peptides by chemical modification,3-6
metabolic labeling,7-10 or enzymatic derivatization.11,12 The speci-ficity of these isotopic labeling techniques is contingent uponobserving different mass shifts, which can be generated by usinga variety of available labeling reagents.
In two recent articles, Wang and co-workers,13 as well asRadulovic and co-workers,14 introduced quantitative, label-free LC/MS strategies for global profiling of complex protein mixtures.Both publications illustrate their specific algorithms for iondetection, clustering and quantitation. The lower resolutioninstrument employed in the studies presented by Radulovic andcolleagues requires that their data reduction scheme condenseall detections into nominal mass bins. Though the data presentedare compelling, the data reduction strategy involving nominal massbins may result in significant errors when dealing with highlycomplex mixtures. As an example, in a simple proteome such asEscherichia coli, there are ∼105 000 tryptic peptides, including one
* Corresponding author. Phone: 978-482-3005. Fax: 508-482-2055. E-mail:[email protected].
† Milford, Massachusetts.‡ Beverly, Massachusetts.§ Manchester, Great Britain.
(1) Hamdan, M.; Righetti, P. G. Mass Spectrom. Rev. 2002, 21, 287-302.(2) Lill, J. Mass Spectrom. Rev. 2003, 22, 182-194.(3) Gygi, S. P.; Rist, B.; Gerber, S. A.; Turecek, F.; Gelb, M. H.; Aebersold, R.
Nat. Biotechnol. 1999, 17, 994-999.(4) Zhou, H. L.; Ranish, J. A.; Watts, J. D.; Aebersold, R. Nat. Biotechnol. 2002,
19, 512-515.(5) Griffin, T. J.; Gygi, S. P.; Rist, B.; Aebersold, R. Anal. Chem. 2001, 73,
978-986.(6) Chakraboorty, A.; Regnier, F. J. Chromatogr., A 2002, 949, 173-184.(7) Veenstra, T. D.; Martinovic, S.; Anderson, G. A.; Pasa-Tolic, L.; Smith, R. D.
J. Am. Soc. Mass Spectrom. 2000, 11, 78-82.(8) Ong, S. E.; Kratchmarova, I.; Mann, M. J. Proteome Res. 2003, 2, 173-181.(9) Krijgsveld, J.; Ketting, R. F.; Mahmoudi, T.; Johansen, J.; Artal-Sanz, M.;
Verrijzer, C. P.; Plasterk, R. H. A.; Heck, A. J. R. Nat. Biotechnol. 2003, 21,927-931.
(10) Oda, Y.; Huang, K.; Cross, F. R.; Cowburn, D.; Chait, B. T. PNAS 1999,96, 6591-6596.
(11) Yao, X. D.; Freas, A.; Ramirez, J.; Demirev, P. A.; Fenselau, C. Anal. Chem.2001, 73, 2836-2842.
(12) Stewart, I. I.; Thomson, T.; Figeys, D. Rapid Commun. Mass Spectrom. 2001,15, 2456-2465.
(13) Wang, W.; Zhou, H.; Lin, H.; Roy, S.; Shaler, T. A.; Hill, L. R.; Norton, S.;Kumar, P.; Anderle, M.; Becker, C. H. Anal. Chem. 2003, 75, 4818-4826.
(14) Radulovic, D.; Jelveh, S.; Ryu, S.; Hamilton, T. G.; Foss, E.; Mao, Y.; Emili,A. Mol. Cell. Proteomics 2004, 3, 984-997.
Anal. Chem. 2005, 77, 2187-2200
10.1021/ac048455k CCC: $30.25 © 2005 American Chemical Society Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2187Published on Web 03/02/2005

missed cleavage between 700 and 2481 molecular mass. Anaverage of 7 tryptic peptides of the 105 000 are found within amass tolerance of 5 ppm of itself. If the mass tolerance is increasedto within 1 Da, the average number of tryptic peptides is increasedto 165. Using this logic, the opportunity to have more than onepeptide eluting within a nominal mass bin can be up to 23 timesmore likely if the data are reduced from accurate mass measure-ments to nominal mass. As a result, nominal mass binning of massspectrometric, LC/MS data may lead to problems in subsequentclustering of replicate analyzes and to variability in the corre-sponding quantitative analysis. Radulovic and co-workers reportthat their quantitative results exhibited an acceptable measure ofvariance of 2-fold or less deviation in the observed signalintensities. In addition to presenting data from an identicalinstrument platform, Wang and colleagues also illustrated LC/MS data collected on a time-of-flight mass spectrometer. In thiswork, the authors indicated that the higher resolution and massaccuracy of the TOF system was found to be advantageous fortracking and quantifying large numbers of mass spectral peaks.The results obtained from these studies provided acceptablecoefficients of variation (∼25%) across integrated peak intensities.The data acquisition platform used by Radulovic was configuredto collect two parallel LC/MS experiments in a single LC/MS runfor simultaneous quantitative and qualitative analysis. In analternating fashion, the instrument measures the masses of elutingpeptide components in MS mode in one function and then carriesout a data-dependent CID for a subset of detected precursormasses in MS/MS mode in a second function. However, theauthors affirm that considerably more peptide peaks are detectablein full-scan MS mode than can be identified in the same time frameusing the collision-induced dissociation process. This level ofinefficiency requires that additional MS/MS experiments wouldbe needed for thorough identifications to be made in a given study.
The use of MS technology in high-throughput proteomics facesseveral challenges in order to accurately compare differentiallyexpressed proteins from corresponding peptide component infor-mation, such as retention time, mass, and signal response.Included among these challenges, software solutions for peakdetection, chromatographic spectral alignment, charge-state re-duction, and deisotoping need to be implemented in order toreduce the complexity of the continuum MS data and successfullycompare differences among samples. The Expression Informaticssoftware, introduced in this study, has been developed to carryout these functionalities for comprehensive, quantitative, dif-ferential expression analysis.
Although it has been observed that electrospray ionization(ESI) provides signal responses that correlate linearly withincreasing analyte concentration,15-17 historically, there have beenconcerns regarding nonlinearity of signal response and ionsuppression effects18-21 which have prevented the implementationof a simple LC/MS solution for quantitative proteomics. We outline
a quantitative proteomics strategy which employs an LC/MSmethod as the basis for the analytical strategy for quantifyingproteome profile data for differential expression analysis. Thismethod relies on the changes in the peptide analyte signalresponse from each accurate mass measurement and correspond-ing retention time (AMRT) component, and to directly reflect theirconcentrations in one sample relative to another. This methoddoes not require the use of any stable-isotope labeling method orenrichment strategy; however, it does require that the samplepreparation conditions are carefully controlled for optimal, quan-titative performance. Regardless of the analytical technique, theprotein samples must be prepared in a fashion that ensures anefficient and reproducible separation, with concurrent eliminationof undesirable artifacts.
In this investigation, we prepared a tryptic digest of humanserum spiked with increasing amounts of a standard proteinmixture and observed the linear behavior in the signal fromdigested peptides corresponding to the experimentally configuredprotein concentrations. The methodology presented in this workmaximizes the duty cycle of a quadrupole-time-of-flight (Q-TOF)mass spectrometer to yield extensive quantitative and qualitativeinformation by systematically and simultaneously analyzing thepeptide components from large sets of protein mixtures.22,23
Although this work involves the analysis of human serum, thismethodology is applicable to any number of biological samples(plasma, urine, whole-cell lysate, organelle, tissue, or microbial).
MATERIALS AND METHODSSample Preparation. Six aliquots of human serum (HS,
Sigma source) were dispensed into separate eppindorf tubes(∼200 ug). An equimolar stock solution of exogenous proteins(yeast enolase and alcohol dehydrogenase, rabbit glycogenphosphorylase, and bovine serum albumin and hemoglobin,MPDS proteins) was prepared such that each protein was presentat 5 pmol/µL in 50 mM ammonium bicarbonate (pH 8.5). Theexogenous proteins were added to each of the six aliquots ofhuman serum such that the final concentration of equimolarproteins was 0.500, 0.250, 0.100, 0.050, 0.025, and 0.010 pmol/µL(final volume of 200 µL), respectively. To avoid working underthe specified limits of the pipettor, appropriate dilutions of thestock solution were made to ensure that at least 10-20 µL of stockprotein solution, from a calibrated 20-µL pipettor, was added toachieve the desired final exogenous protein concentration. Thevolumes of the samples were adjusted to 100 µL with 50 mMammonium bicarbonate (pH 8.5) containing 0.05% RapiGest.25
Protein was reduced in the presence of 10 mM dithiothreitol at60 °C for 30 min. The protein was alkylated in the dark, in thepresence of 50 mM iodoacetamide, at room temperature for 30min. Proteolytic digestion was initiated by adding modified trypsin(Promega) at a concentration of 75:1 (total protein to trypsin, by
(15) Purves, R. W.; Gabryelski, L. L. Rapid Commun. Mass Spectrom. 1998, 12,695-700.
(16) Voyksner, R. D.; Lee, H. Rapid Commun. Mass Spectrom. 1999, 13, 1427-1437.
(17) Chelius, D.; Bondarenko, P. J. Proteome Res. 2002, 1, 317-323.(18) Muller, C.; Schafer, P.; Stortzel, M.; Vogt, S.; Weinmann, W. J. Chromatogr.,
B 2002, 773, 47-52.(19) Matuszewski, B. K.; Constanzer, M. L.; Chavez-Eng, C. M. Anal. Chem.
1998, 70, 882-889.
(20) Sangster, T.; Spence, M.; Sinclair, P.; Payne, R.; Smith, C. Rapid Commun.Mass Spectrom. 2004, 18, 1361-1364.
(21) Mei, H.; Hsieh, Y.; Nardo, C.; Xu, X.; Wang, S.; Ng, K.; Korfmacher, W. A.Rapid Commun. Mass Spectrom. 2003, 17, 97-103.
(22) Bateman, R. H.; Hoyes, J. B. U.K. Patent 2,364,168A, 2002.(23) Purvine, S.; Eppel, J. T.; Yi, E. C.; Goodlett, D. R. Proteomics 2003, 3, 847-
850.(24) Geromanos, S.; Dongre, A.; Opiteck, G.; Silva, J. C. U.K. Patent 2,385,918A,
2003.(25) Yu, Y. Q.; Gilar, M.; Lee, P. J.; Bouvier, E. S. P.; Gebler, J. C. Anal. Chem.
2003, 75, 6023-6028.
2188 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005

weight) and incubated at 37 °C overnight. Each digestion mixturewas diluted to a final volume of 200 µL with 50 mM ammoniumbicarbonate (pH 8.5) to reduce the concentration of RapiGestdetergent to 0.025%. The tryptic peptide solution was centrifugedat 13 000 rpm for 10 min, and the supernatant was transferredinto an autosampler vial for peptide analysis via LC/MS. Eachsample was analyzed in triplicate. The LC/MS analysis wasperformed using 10 µL of the final tryptic digest.
HPLC Configuration. Capillary liquid chromatography (Ca-pLC) of tryptic peptides was performed with a Waters CapLC/Waters CapLC autosampler, equipped with a Waters NanoEaseAtlantis C18, 300 µm × 15 cm reversed-phase column. The aqueousmobile phase (mobile phase A) contained 1% acetonitrile in waterwith 0.1% formic acid. The organic mobile phase (mobile phaseB) contained 80% acetonitrile in water with 0.1% formic acid.Peptides were loaded onto the column with 6% mobile phase B.Peptides were eluted from the column with a gradient of 6-40%mobile phase B over 100 min at 4.4 µL/min, followed by a 10-minrinse of 99% of mobile phase B. The column was immediatelyreequilibrated at initial conditions (6% mobile phase B) for 20 min.The lock mass, [Glu1]-fibrinopeptide at 100 fmol/µL (GFP), wasdelivered from the auxiliary pump of the CapLC at 1 µL/min tothe reference sprayer of the NanoLockSpray source.
Mass Spectrometer Configuration. Mass spectrometry analy-sis of tryptic peptides was performed using a modified Waters/Micromass Q-Tof Ultima API to provide enhanced mass accuracy.Detection events were acquired at 4 GHz. For all measurements,the mass spectrometer was operated in V mode with a typicalresolving power of at least 10 000. The spectrum integration timewas 1.8 s with an interscan delay time of 0.2 s. All analyses wereperformed using positive-mode ESI using a NanoLockSpraysource. The lock mass channel was sampled every 30 s. The massspectrometer was calibrated with a GFP solution (100 fmol/µL)delivered through the reference sprayer of the NanoLockSpraysource. The doubly charged ion ([M + 2H]2+) was used for initialsingle point calibration (Lteff), and MS/MS fragment ions of GFPwere used to obtain the final instrument calibration. Data acquisi-tion was operated in the exact neutral loss mode, without aninclude list. Accurate mass LC/MS and LC/MSE data werecollected using 10 eV for MS and 28-35 eV for MSE acquisitionsuch that one cycle of MS and MSE data was acquired every 4.0s. The RF offset was adjusted such that the LC/MS data wereeffectively acquired from m/z 300 to 2000, which ensured thatany masses observed in the LC/MSE data less than m/z 300 wereknown to arise from dissociations in the collision cell.
RESULTS AND DISCUSSIONIon Detection. The ion detection algorithm of the Expression
Informatics software uses a maximum likelihood algorithm todeisotope and charge-state-reduce the m/z detections to thecorresponding monoisotopic m/z (MH+) for each scan of thecontinuum LC/MS data.26 The algorithm also calculates theobserved mass and intensity measurement deviation for everydetected component. The chromatographic area associated witheach component is calculated using an integration algorithmsimilar to the ApexTrack peak integration algorithm provided inthe MassLynx software. If a particular component exists in more
than one charge-state, the corresponding area for any givenmonoisotopic ion is reported as the summed area from allcontributing charge states. The retention time is determined foreach reported monoisotopic ion at the moment it reaches itsmaximum intensity (apex). Each detected component is referredto as an AMRT (accurate-mass, retention time) component. AnAMRT is extracted from the continuum data only if it exceeds auser-defined, minimum detection threshold. The software is alsocapable of processing the data using an autothreshold capabilitywhich automatically adjusts the ion detection threshold over timeas a function of the dynamic range within the mass spectrometricdata. The culmination of this process produces an AMRTcomponent list. This list contains many experimentally derivedattributes for each of the recorded AMRT components (AMRTs).Included in this output are the weight-averaged monoisotopic massand charge state, the calculated mass deviation, the deisotopedand charge-state-reduced sum intensity (centered by area), thechromatographic area, the calculated intensity deviation, theobserved apex retention time (centered by area), and the observedstart and stop time for the ion detection of the correspondingAMRT.
Clustering Peptide Components by Mass and RetentionTime. One of the key operations required for the comparativeanalyses of peptide mixtures is clustering chemically identicalcomponents together from replicate injections of the same sampleas well as among multiple samples. The clustering algorithmperforms multiple binary comparisons to conduct the overallclustering strategy for a complete experiment.27,28 AMRT compo-nents from each injection are clustered to align identical compo-nents to one another on the basis of a mass precision and aretention time deviation threshold. In an initial binary comparison,a subset of the AMRTs from two separate injections is comparedto establish the experimental retention time deviation behaviorof identical AMRTs between the two samples. The subset ofAMRTs considered in the initial comparison is typically thoseabove the median intensity for the entire data set. In the initialcomparison, a coarse threshold of typically 5 min is applied toconsider all potential paired candidates. Often, peptides may notreproducibly elute at exactly the same time throughout a replicateanalysis. However, one generally observes a consistent shift inretention-time, whereby the observed retention time of a givenset of peptides will deviate systematically, although not necessarilyby the same magnitude. Due to the complexity of the data, thereoften exist conditions under which an AMRT in one condition orreplicate will match within the threshold criterion to multipleAMRTs in a different replicate or condition. This, of course, isnot desirable, since an AMRT from one condition or replicateshould only match its identical companion in any other condition.To address these situations, the clustering algorithm calculatesthe delta retention time for all matched AMRTs and plots theretention time for each AMRT against the retention time differenceobserved among the corresponding matched components (Figure1A). In doing so, the algorithm can determine the expected
(26) Skilling, J.; Bryan, R. K. Mon. Not. R. Astron. Soc. 1984, 211, 111-124.
(27) Li, G.-Z.; Gorenstein, M.; Geromanos, S.; Silva, J. C.; Dorschel, C. A.; RileyT. Proc. 52nd ASMS Conf. Mass Spectrom. Allied Top. 2004, TPY 354,Nashville, TN.
(28) Gorenstein, M.; Li, G.-Z.; Geromanos, S.; Silva, J. C.; Dorschel, C. A.; Plumb,R. S.; Stumpf, C. L.; Riley, T. Proc. 52nd ASMS Conf. Mass Spectrom. AlliedTop. 2004, WPJ 161, Nashville, TN.
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2189
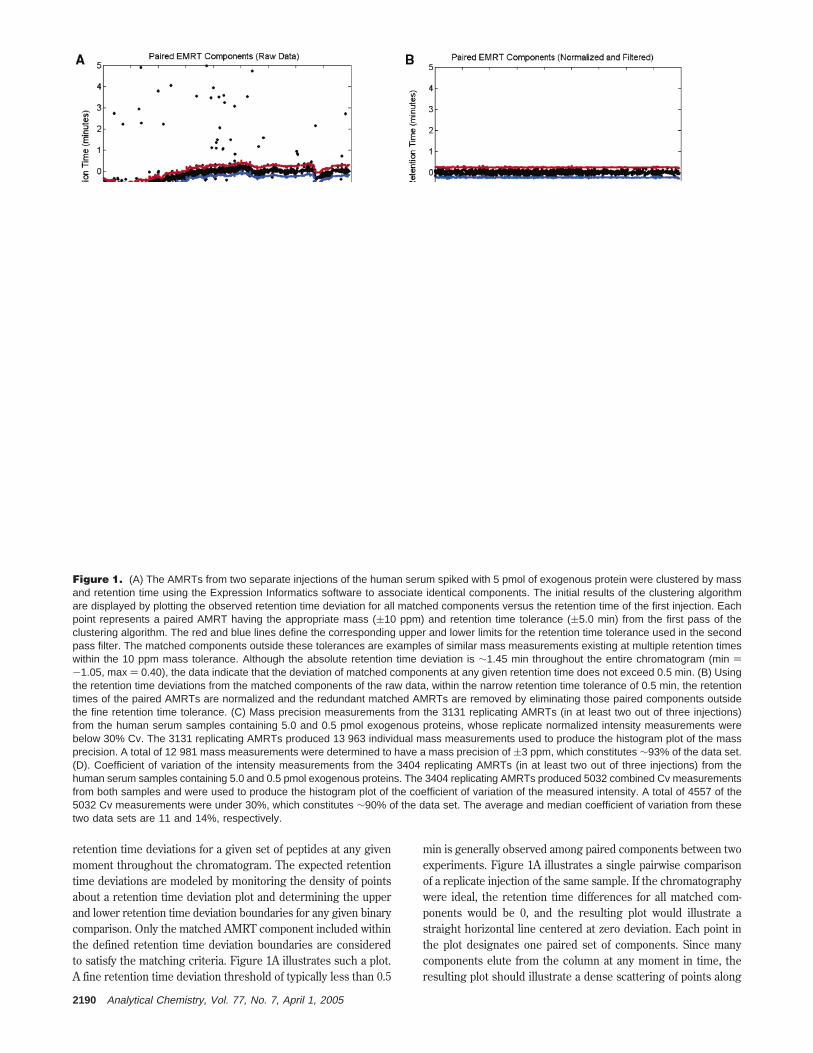
retention time deviations for a given set of peptides at any givenmoment throughout the chromatogram. The expected retentiontime deviations are modeled by monitoring the density of pointsabout a retention time deviation plot and determining the upperand lower retention time deviation boundaries for any given binarycomparison. Only the matched AMRT component included withinthe defined retention time deviation boundaries are consideredto satisfy the matching criteria. Figure 1A illustrates such a plot.A fine retention time deviation threshold of typically less than 0.5
min is generally observed among paired components between twoexperiments. Figure 1A illustrates a single pairwise comparisonof a replicate injection of the same sample. If the chromatographywere ideal, the retention time differences for all matched com-ponents would be 0, and the resulting plot would illustrate astraight horizontal line centered at zero deviation. Each point inthe plot designates one paired set of components. Since manycomponents elute from the column at any moment in time, theresulting plot should illustrate a dense scattering of points along
Figure 1. (A) The AMRTs from two separate injections of the human serum spiked with 5 pmol of exogenous protein were clustered by massand retention time using the Expression Informatics software to associate identical components. The initial results of the clustering algorithmare displayed by plotting the observed retention time deviation for all matched components versus the retention time of the first injection. Eachpoint represents a paired AMRT having the appropriate mass ((10 ppm) and retention time tolerance ((5.0 min) from the first pass of theclustering algorithm. The red and blue lines define the corresponding upper and lower limits for the retention time tolerance used in the secondpass filter. The matched components outside these tolerances are examples of similar mass measurements existing at multiple retention timeswithin the 10 ppm mass tolerance. Although the absolute retention time deviation is ∼1.45 min throughout the entire chromatogram (min )-1.05, max ) 0.40), the data indicate that the deviation of matched components at any given retention time does not exceed 0.5 min. (B) Usingthe retention time deviations from the matched components of the raw data, within the narrow retention time tolerance of 0.5 min, the retentiontimes of the paired AMRTs are normalized and the redundant matched AMRTs are removed by eliminating those paired components outsidethe fine retention time tolerance. (C) Mass precision measurements from the 3131 replicating AMRTs (in at least two out of three injections)from the human serum samples containing 5.0 and 0.5 pmol exogenous proteins, whose replicate normalized intensity measurements werebelow 30% Cv. The 3131 replicating AMRTs produced 13 963 individual mass measurements used to produce the histogram plot of the massprecision. A total of 12 981 mass measurements were determined to have a mass precision of (3 ppm, which constitutes ∼93% of the data set.(D). Coefficient of variation of the intensity measurements from the 3404 replicating AMRTs (in at least two out of three injections) from thehuman serum samples containing 5.0 and 0.5 pmol exogenous proteins. The 3404 replicating AMRTs produced 5032 combined Cv measurementsfrom both samples and were used to produce the histogram plot of the coefficient of variation of the measured intensity. A total of 4557 of the5032 Cv measurements were under 30%, which constitutes ∼90% of the data set. The average and median coefficient of variation from thesetwo data sets are 11 and 14%, respectively.
2190 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005

the retention time coordinate. Figure 1A illustrates that thereproducibility of the chromatographic peptide separation is ∼0.25min, with an overall chromatographic deviation of 1.0 min. Thepairwise comparison is performed for each of the replicateinjections, as well as across the multiple experiments. Theretention time deviations observed between the AMRTs of twoinjections serve as multiple internal standards and are used todetermine an appropriate retention time offset for AMRTs elutingat any moment. The retention time offsets are used to normalizethe observed retention time for every AMRT component. Theeffects of the retention time normalization are illustrated in Figure1B. The output that is generated from the clustering routine is alarge matrix, whereby identical components are aligned in eachrow for subsequent quantitative and statistical analysis. Theassembled matrix will not only contain AMRTs which appear ineach of the conditions for each of the replicate injections, but mayalso include those AMRTs which appear reproducibly in one ormore of the six conditions.
To illustrate the level of specificity one is capable of obtainingwith mass accuracy and retention time reproducibility, theprocessed data can be queried at different retention time and massprecision tolerances. As an example, injection 2 of the humanserum with 2 pmol of MPDS protein produced 2582 AMRTs. The2582 AMRTs were queried to determine how many were withina (1-min retention time window and a 10 ppm mass tolerance.Using these tolerances, a total of 36 AMRTs (1.4%) were found tocoexist within these parameters. Therefore, these 36 AMRTs couldpotentially add ambiguity during the clustering process and leadto incorrect clustering of the data. If the mass tolerance is allowedto expand to a 100-mDa error, the ambiguity is increases to a totalof 76 AMRTs (2.9%). At 1 Da, nominal mass, the ambiguityincreases to a total of 657 AMRTs (25.4%). These errors arecompounded if the tolerances of both the retention time and massprecision are allowed to expand. If the retention time tolerance isallowed to be within (5 min, then the following statistics aregenerated from the single data file: 293 AMRTs (11.3%) at 10 ppmmass tolerance, 441 AMRTs (17.1%) at 100-mDa mass tolerance,and 1112 AMRTs (43.1%) at 1-Da tolerance. These results arebased on a single injection of a single sample. If one were tocompare replicates among many different samples, this could leadto a significant number of AMRTs being clustered incorrectly andthereby produce highly irreproducible results. Having an LC/MSinstrumentation platform that is capable of providing reproduciblemass precision and accuracy along with reproducible chromatog-raphy will significantly increase the quality of the clustered dataand will provide a more robust quantitative proteomics platform.
Data Normalization and Statistical Analysis. Once theAMRT data have been clustered, the clustering algorithm per-forms a number of mathematical and statistical calculations forthe entire data set. To correct for injection variability and totalprotein load across samples, the intensity measurements for theentire data set are normalized. The intensity measurements of alldetected AMRTs from each injection are normalized to a set ofAMRTs (endogenous or exogenous) that are known not to havechanged among the different samples. The internal AMRTstandards used for normalization purposes were required to bepresent in all six experiments. Although the Expression Infor-matics software is capable of correcting the mass and intensity
measurements that are in dead time, there is a limit to its abilityto accurately correct for those measurements.29,30 With this inmind, the internal AMRT standards selected for normalizationwere well below dead time and existed in all replicates of eachsample. The average monoisotopic masses of the AMRTs usedfor normalization were 1273.6547, 1706.7746, and 2171.1138, withcorresponding elution times of approximately 42.60, 53.60, and101.80 min, respectively. These AMRT components were endog-enous to human serum and were determined to originate fromtransferrin (data not shown).31 Next, the algorithm calculates thereplication rate of each AMRT within and among all conditions.The algorithm also calculates the average mass, intensity, area,combined charge-state, and retention-time for each AMRT for allconditions. In addition, a standard deviation and coefficient ofvariation is determined for each of these measured attributes.Using this information, the software annotates those AMRTscommon and unique to each condition. Last, the algorithmperforms binary comparisons for each of the conditions togenerate an average normalized intensity ratio (log) for allmatched AMRTs and also performs a Student’s t-test for eachbinary comparison. The final results of the clustering algorithmcan be exported as a comma-delimited text file containing all ofthe mass spectrometric and chromatographic attributes for eachAMRT, along with all of the mathematical and statistical calcula-tions generated after the clustering process. This clustered datafile can be further manipulated or visualized in any of a numberof commercially available software packages, such as MicrosoftExcel or Spotfire Decision Site.
The precision of the extracted mass measurements of theclustered components from the replicate injections of all sampleswere typically within (5 ppm of the mean mass measurement.These data are illustrated in Figure 1C and demonstrate therobustness of the ion extraction software and the stability of themass measurement instrumentation. In fact, 90% of the totalnumber of replicated components were measured with a precisionof (3 ppm. The reproducibility of the quantitative intensitymeasurements from the Expression Informatics software issummarized in Figure 1D. These results indicate that the coef-ficient of variation (Cv) among the replicate injections and acrossmultiple samples were typically less than 15%, with a majority ofthe quantitative variation lying between 11 and 14% Cv. Theseobservations are typically expected from the Expression Infor-matics software when using standard protocols for efficient samplepreparation.32
Expression Analysis of AMRT Components. The purposeof these experiments was to demonstrate that the ExpressionInformatics software could ascertain the relative change inabundance of a small subset of proteins (MPDS proteins) spikedinto a complex protein background (human serum). The MPDS
(29) Rockwood, A. L.; Fabbi, J. C.; Harris, L.; Davis, L.; Lee, E. D.; Ogden, C.;Tolley, H.; Gunsay, M.; Sin, J. C. N.; Lee, H. G. Proc. 45th ASMS Conf. MassSpectrom. Allied Top. 1997, WOE 0250, Palm Springs, CA.
(30) Barbacci, D. C.; Russel, D. H.; Schultz, J. A.; Holocek, J.; Ulrich, S.; Burton,W.; Van Stipdonk, M. J. Am. Soc. Mass Spectrom. 1998, 9, 1328-1333.
(31) Silva, J. C.; Richardson, K.; Young, P.; Denny, R.; Neeson, K.; McKenna,T.; Dorschel, C. A.; Li, G.-L.; Gorenstein, M.; Riley, T.; Geromanos, S. Proc.52nd ASMS Conf. Mass Spectrom. Allied Top. 2004, MPX 452, Nashville,TN.
(32) Dorschel, C. A.; Gorenstein, M.; Li, G.-Z.; Silva, J. C.; Geromanos, S.; Riley,T. Proc. 52nd Ann. ASMS Conf. Mass Spectrom. Allied Top. 2004, TPY 458,Nashville, TN.
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2191

proteins were spiked at levels well below that of the most abundantproteins in the complex background. Six samples were preparedto reflect a dilution series of the MPDS proteins ranging from 10to 500 fmol/µL. The samples were digested with trypsin asdescribed in the Material and Methods Section, and the resultingpolypeptide mixtures were analyzed in triplicate by LC/MS.22-24
To demonstrate that the quantitative information relating to theMPDS proteins was available in the acquired LC/MS data, amanual analysis was performed on a previously characterizedAMRT (m/z 724.41 at 69.5 min). Figure 2A depicts six total ionchromatograms (TICs) obtained from the LC/MS acquisitions.For the sake of space, only one replicate TIC is illustrated foreach of the six different samples. The TICs illustrate a high degreeof similarity among the six different samples, despite an overall50-fold change in the relative levels of MPDS peptides throughoutthe six samples. Figure 2B illustrates the selected ion chromato-grams (SICs) for the m/z 724.41 (z ) 2, MH2+) ion at ∼69.5 minand the associated integrated peak areas, as determined byMassLynx. The identity of this peptide was validated by DDA touse as a proof-of-concept model for the subsequent quantitativecomparison (data not shown, VVGLSTLPEIYEK peptide fromyeast ADH). Figure 2C illustrates the six individual MS spectraobtained from each sample at the chromatographic apex of theSIC in Figure 2B (m/z 724.41). Each spectrum presented in Figure
2C is normalized to the highest ion in the spectrum to illustratethe dilution of the 724.41 MH2+ ion over the six differentconcentrations. The data presented in each spectrum illustrate avery high degree of similarity with respect to the other coelutingpeptides in the background of human serum. This similarity isreflected not only in the number of ions present in each scan butalso in the correlation among their respective intensities andrelative intensity ratios. The degree of chromatographic reproduc-ibility is further supported, at the global level, from the ExpressionInformatics processing and analysis of the clustered AMRTsobtained from each of the replicate analyses, as will be illustratedlater. Figure 2D depicts each spectrum after it has been smoothed(Savistky-Golay smoothing, three channels, two smoothes),centered (three channels, 80% of the centroid top, centered byarea), and lock-mass corrected against the monoisotopic ion ofGFP (m/z 785.8426). Comparison of the lock-mass-corrected massmeasurements obtained from the six individual samples (m/z724.41, MH2+) reflects the level of mass precision obtained fromthis methodology. It also establishes that one can use an LC/MS-based approach for relative quantitation of peptide componentsin a complex protein sample, provided that sufficient mass andretention time reproducibility are obtained. Table 1 outlines theresults obtained from the manual interrogation of the raw datausing the commercially available MassLynx software. The inte-
Figure 2. (A) The base peak intensity (BPI) of human serum with five equimolar exogenous proteins spiked at decreasing levels (5.00, 2.00,1.00, 0.50, 0.25, and 0.10 pmol), (B) the selected ion chromatogram (SIC) of the doubly charged peptide ion, 724.34 ((0.05 m/z). Thecorresponding SICs were integrated using MassLynx processing software between 68.00 and 71 min. Processing parameters were set forautomatic noise measurement, Savitzky-Golay smoothing (three channels, two smoothes), and ApexTrack peak integration. (C) The continuummass spectrum at the apex of the corresponding 724.34 selected ion chromatogram in panel B (from 600 to 825 m/z). (D) The lock-mass-corrected, centroided mass spectrum of the 724.34 isotope cluster (between 722 and 729 m/z) from panel C (smoothing: Savitzky-Golay,three channels, two smoothes; centering: three channels, centroid top 80%, centered by area) and lock-mass-corrected against the monoisotopicion of Glu-Fib, 785.8426 m/z).
2192 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005
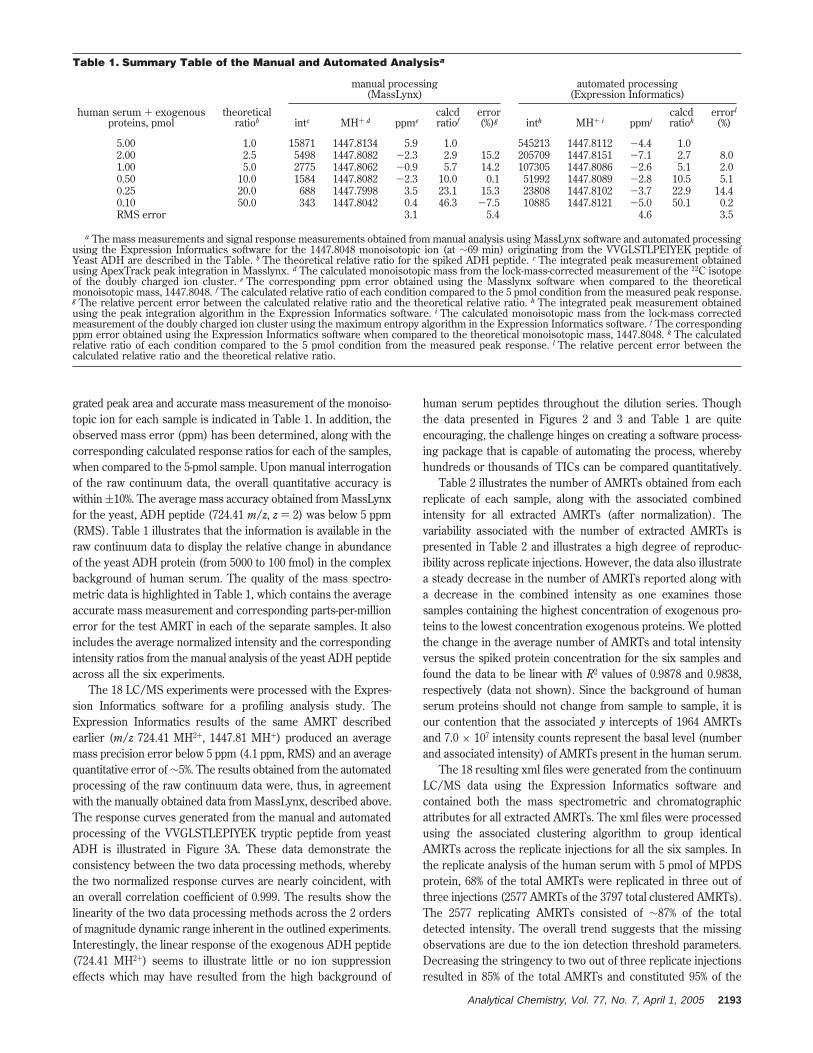
grated peak area and accurate mass measurement of the monoiso-topic ion for each sample is indicated in Table 1. In addition, theobserved mass error (ppm) has been determined, along with thecorresponding calculated response ratios for each of the samples,when compared to the 5-pmol sample. Upon manual interrogationof the raw continuum data, the overall quantitative accuracy iswithin (10%. The average mass accuracy obtained from MassLynxfor the yeast, ADH peptide (724.41 m/z, z ) 2) was below 5 ppm(RMS). Table 1 illustrates that the information is available in theraw continuum data to display the relative change in abundanceof the yeast ADH protein (from 5000 to 100 fmol) in the complexbackground of human serum. The quality of the mass spectro-metric data is highlighted in Table 1, which contains the averageaccurate mass measurement and corresponding parts-per-millionerror for the test AMRT in each of the separate samples. It alsoincludes the average normalized intensity and the correspondingintensity ratios from the manual analysis of the yeast ADH peptideacross all the six experiments.
The 18 LC/MS experiments were processed with the Expres-sion Informatics software for a profiling analysis study. TheExpression Informatics results of the same AMRT describedearlier (m/z 724.41 MH2+, 1447.81 MH+) produced an averagemass precision error below 5 ppm (4.1 ppm, RMS) and an averagequantitative error of ∼5%. The results obtained from the automatedprocessing of the raw continuum data were, thus, in agreementwith the manually obtained data from MassLynx, described above.The response curves generated from the manual and automatedprocessing of the VVGLSTLEPIYEK tryptic peptide from yeastADH is illustrated in Figure 3A. These data demonstrate theconsistency between the two data processing methods, wherebythe two normalized response curves are nearly coincident, withan overall correlation coefficient of 0.999. The results show thelinearity of the two data processing methods across the 2 ordersof magnitude dynamic range inherent in the outlined experiments.Interestingly, the linear response of the exogenous ADH peptide(724.41 MH2+) seems to illustrate little or no ion suppressioneffects which may have resulted from the high background of
human serum peptides throughout the dilution series. Thoughthe data presented in Figures 2 and 3 and Table 1 are quiteencouraging, the challenge hinges on creating a software process-ing package that is capable of automating the process, wherebyhundreds or thousands of TICs can be compared quantitatively.
Table 2 illustrates the number of AMRTs obtained from eachreplicate of each sample, along with the associated combinedintensity for all extracted AMRTs (after normalization). Thevariability associated with the number of extracted AMRTs ispresented in Table 2 and illustrates a high degree of reproduc-ibility across replicate injections. However, the data also illustratea steady decrease in the number of AMRTs reported along witha decrease in the combined intensity as one examines thosesamples containing the highest concentration of exogenous pro-teins to the lowest concentration exogenous proteins. We plottedthe change in the average number of AMRTs and total intensityversus the spiked protein concentration for the six samples andfound the data to be linear with R2 values of 0.9878 and 0.9838,respectively (data not shown). Since the background of humanserum proteins should not change from sample to sample, it isour contention that the associated y intercepts of 1964 AMRTsand 7.0 × 107 intensity counts represent the basal level (numberand associated intensity) of AMRTs present in the human serum.
The 18 resulting xml files were generated from the continuumLC/MS data using the Expression Informatics software andcontained both the mass spectrometric and chromatographicattributes for all extracted AMRTs. The xml files were processedusing the associated clustering algorithm to group identicalAMRTs across the replicate injections for all the six samples. Inthe replicate analysis of the human serum with 5 pmol of MPDSprotein, 68% of the total AMRTs were replicated in three out ofthree injections (2577 AMRTs of the 3797 total clustered AMRTs).The 2577 replicating AMRTs consisted of ∼87% of the totaldetected intensity. The overall trend suggests that the missingobservations are due to the ion detection threshold parameters.Decreasing the stringency to two out of three replicate injectionsresulted in 85% of the total AMRTs and constituted 95% of the
Table 1. Summary Table of the Manual and Automated Analysisa
manual processing(MassLynx)
automated processing(Expression Informatics)
human serum + exogenousproteins, pmol
theoreticalratiob intc MH+ d ppme
calcdratiof
error(%)g inth MH+ i ppmj
calcdratiok
errorl
(%)
5.00 1.0 15871 1447.8134 5.9 1.0 545213 1447.8112 -4.4 1.02.00 2.5 5498 1447.8082 -2.3 2.9 15.2 205709 1447.8151 -7.1 2.7 8.01.00 5.0 2775 1447.8062 -0.9 5.7 14.2 107305 1447.8086 -2.6 5.1 2.00.50 10.0 1584 1447.8082 -2.3 10.0 0.1 51992 1447.8089 -2.8 10.5 5.10.25 20.0 688 1447.7998 3.5 23.1 15.3 23808 1447.8102 -3.7 22.9 14.40.10 50.0 343 1447.8042 0.4 46.3 -7.5 10885 1447.8121 -5.0 50.1 0.2RMS error 3.1 5.4 4.6 3.5
a The mass measurements and signal response measurements obtained from manual analysis using MassLynx software and automated processingusing the Expression Informatics software for the 1447.8048 monoisotopic ion (at ∼69 min) originating from the VVGLSTLPEIYEK peptide ofYeast ADH are described in the Table. b The theoretical relative ratio for the spiked ADH peptide. c The integrated peak measurement obtainedusing ApexTrack peak integration in Masslynx. d The calculated monoisotopic mass from the lock-mass-corrected measurement of the 12C isotopeof the doubly charged ion cluster. e The corresponding ppm error obtained using the Masslynx software when compared to the theoreticalmonoisotopic mass, 1447.8048. f The calculated relative ratio of each condition compared to the 5 pmol condition from the measured peak response.g The relative percent error between the calculated relative ratio and the theoretical relative ratio. h The integrated peak measurement obtainedusing the peak integration algorithm in the Expression Informatics software. i The calculated monoisotopic mass from the lock-mass correctedmeasurement of the doubly charged ion cluster using the maximum entropy algorithm in the Expression Informatics software. j The correspondingppm error obtained using the Expression Informatics software when compared to the theoretical monoisotopic mass, 1447.8048. k The calculatedrelative ratio of each condition compared to the 5 pmol condition from the measured peak response. l The relative percent error between thecalculated relative ratio and the theoretical relative ratio.
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2193
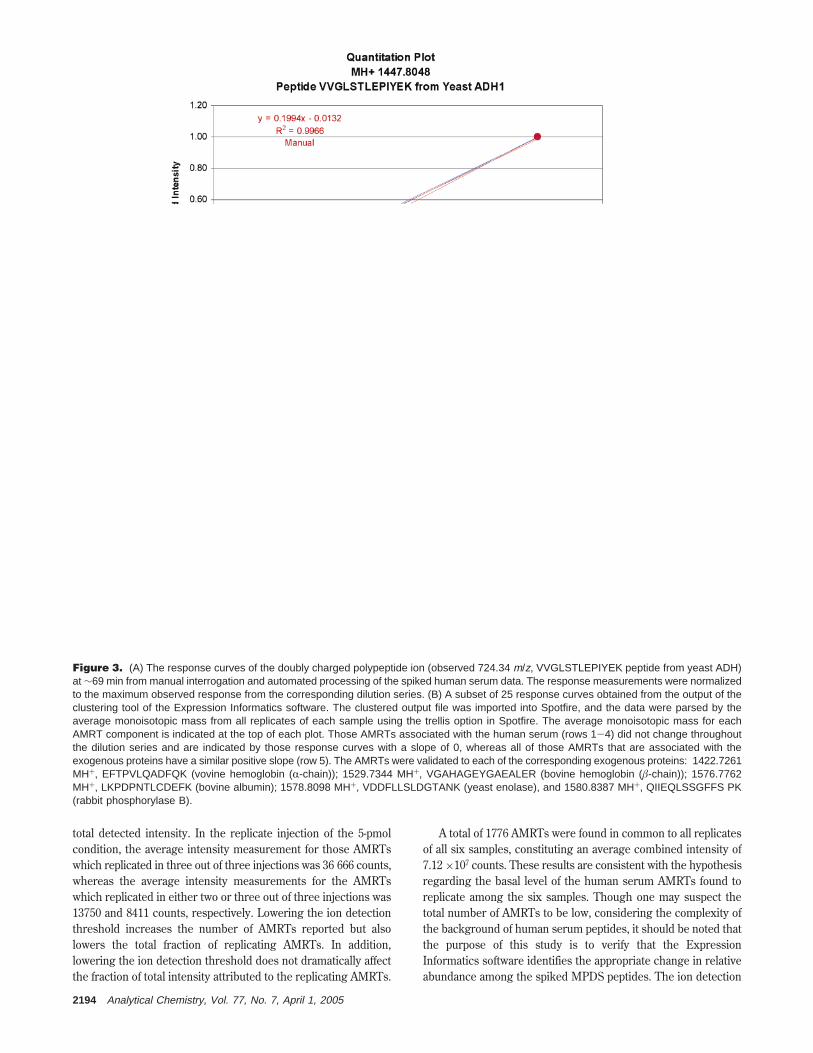
total detected intensity. In the replicate injection of the 5-pmolcondition, the average intensity measurement for those AMRTswhich replicated in three out of three injections was 36 666 counts,whereas the average intensity measurements for the AMRTswhich replicated in either two or three out of three injections was13750 and 8411 counts, respectively. Lowering the ion detectionthreshold increases the number of AMRTs reported but alsolowers the total fraction of replicating AMRTs. In addition,lowering the ion detection threshold does not dramatically affectthe fraction of total intensity attributed to the replicating AMRTs.
A total of 1776 AMRTs were found in common to all replicatesof all six samples, constituting an average combined intensity of7.12 ×107 counts. These results are consistent with the hypothesisregarding the basal level of the human serum AMRTs found toreplicate among the six samples. Though one may suspect thetotal number of AMRTs to be low, considering the complexity ofthe background of human serum peptides, it should be noted thatthe purpose of this study is to verify that the ExpressionInformatics software identifies the appropriate change in relativeabundance among the spiked MPDS peptides. The ion detection
Figure 3. (A) The response curves of the doubly charged polypeptide ion (observed 724.34 m/z, VVGLSTLEPIYEK peptide from yeast ADH)at ∼69 min from manual interrogation and automated processing of the spiked human serum data. The response measurements were normalizedto the maximum observed response from the corresponding dilution series. (B) A subset of 25 response curves obtained from the output of theclustering tool of the Expression Informatics software. The clustered output file was imported into Spotfire, and the data were parsed by theaverage monoisotopic mass from all replicates of each sample using the trellis option in Spotfire. The average monoisotopic mass for eachAMRT component is indicated at the top of each plot. Those AMRTs associated with the human serum (rows 1-4) did not change throughoutthe dilution series and are indicated by those response curves with a slope of 0, whereas all of those AMRTs that are associated with theexogenous proteins have a similar positive slope (row 5). The AMRTs were validated to each of the corresponding exogenous proteins: 1422.7261MH+, EFTPVLQADFQK (vovine hemoglobin (R-chain)); 1529.7344 MH+, VGAHAGEYGAEALER (bovine hemoglobin (â-chain)); 1576.7762MH+, LKPDPNTLCDEFK (bovine albumin); 1578.8098 MH+, VDDFLLSLDGTANK (yeast enolase), and 1580.8387 MH+, QIIEQLSSGFFS PK(rabbit phosphorylase B).
2194 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005

threshold was set to generate AMRTs which spanned 3-4 ordersof magnitude dynamic range within a given sample. The MPDSproteins were spiked into the human serum at levels such thattheir intensities were within this window of dynamic range. Byapplying these threshold parameters, we were able to demonstratethe appropriate response with the ADH peptide and, therefore,continue with the analysis to characterize the remaining AMRTs.
The clustering results were exported from the ExpressionInformatics software and imported directly into Spotfire forevaluation. With identical components clustered across thereplicate injections of the six samples (dilution series), one canreadily obtain response curves for each of the clustered compo-nents. Figure 3B illustrates response curves for a subset ofclustered AMRTs, in which the average normalized intensity isplotted as a function of the quantity (femtomole) of spiked MPDSproteins. The bottom five plots represent an individual peptidefrom four of the remaining five exogenous proteins. All of theseresponse curves have a similar slope that is indicative of theconfigured serial dilution. The response curves in Figure 3Bcorrespond to extracted AMRTs that replicated in all six samplesof human serum with the exogenous proteins. The AMRTs withthe experimentally determined monoisotopic m/z of 1422.7261,1529.7344, 1576.7762, 1578.8098, and 1580.8387 represent peptidesfrom bovine hemoglobin (â-chain), bovine hemoglobin (R-chain),bovine albumin, yeast enolase, and rabbit phosphorylase B,respectively. The mass accuracies associated with these corre-sponding peptides are all within ( 5 ppm of the theoretical trypticpeptide mass. All of the plots for the remaining AMRTs have aslope of 0 and, therefore, correspond to background serumpeptides that do not change in relative concentration across thesix individual samples. For the point of this illustration, the x axiscorresponds to the concentration of spiked exogenous proteins.In a biomarker discovery study, the concentration dependencecould easily be replaced by a time course or different perturba-tions, such as drug dosage or environmental conditions. The abilityto display these response curves (or conditional profiles) for allmatched AMRTs enables one to perform comprehensive globalcomparisons rather than multiple binary comparisons. Using thisapproach, the AMRTs can be rapidly screened and characterizedon the basis of their collective behavior across the multipleconditions. Self-organizing maps (SOMs) or k-means clusteringtechniques can be used to associate AMRTs that exhibit the same
behavior, and by extension, may be related to the same protein,metabolic, or regulatory pathway(s).33,34
Figure 4A illustrates a diagonal plot of the log of the averagenormalized intensity for matched AMRTs from the 5-pmol mixture(x axis) versus the 2-pmol mixture (y axis). The data illustratetwo distinct clusters of ions spanning close to 4 orders ofmagnitude dynamic range in ion detection and share 2997matched AMRT component pairs between the two conditions. Thedata points are colored by their respective t-test score of thenormalized intensities for all replicate injections between the twoconditions to illustrate that the variance between the two condi-tions is statistically significant. The yellow data points illustratethose matched AMRT components with a t-test score of <0.01,indicating that there is less than a 1% chance that the observedchange is not due to the applied perturbation. Although moresophisticated multicomponent statistical methods could be per-formed on these data, all the comparisons in this work wereperformed using a binary Student’s t-test. The t-test was performedon only the highly reproducible AMRTs which were found to bein the majority of the replicates for each of the two test conditions(at least two out of three). In the presentation of this work, therewas no attempt to correct for missing data. If an AMRT occurredin only one out of three replicate injections in either of the twoconditions, the AMRT was ignored in the subsequent quantitativeprocessing. Since this approach does not require the use ofenrichment techniques, there is quite a bit of peptide redundancyfor each representative protein in the sample. By not limiting thenumber of peptides per protein, we can afford to use a conservativeapproach to our data reduction scheme and propogate the highestquality data into the quantitative processing without jeopardizingthe number of proteins that can be quantified and subsequentlyidentified. In Figure 4A-E, the blue data points are those AMRTsthat did not exhibit any change due to the applied perturbationas defined by the Student’s t-test (>0.01). The red data pointhighlights the AMRT described in Table 1, for the purpose of themanual analysis and comparison to the automated processing.
(33) Mirkin, B. Mathematical Classification and Clustering, Nonconvex Optimiza-tion and Its Applications; Pardalos, P., Horst, R., Eds.; Kluwer AcademicPublishers: The Netherlands, 1996, Chapter 11.
(34) MacQueen, J. Some Methods for Classification and Analysis of MultivariateObservations. In Proceedings of the Fifth Berkeley Symposium on MathematicalStatistics and Probability; Le Cam, L. M., Neyman, J., Eds.; University ofCalifornia Press: Berkeley and Los Angeles, CA; Vol 1, pp 281-297.
Table 2. Summary Table of the Ion Detection Resultsa
sample inj 1 inj 2 inj 3 CV, % inj 1 inj 2 inj 3 CV, %
5 pm ProStds HsSera 2 pm ProStds HsSeraAMRTs 3142 3231 3212 1.47 2382 2582 2758 7.31normalized intensity 1.04 × 108 1.03 × 108 9.90 × 107 2.59 8.40 × 107 8.58 × 107 8.85 × 107
2.661 pm ProStds HsSera 0.5 pm ProStds HsSera
AMRTs 2383 2087 2244 6.62 2005 2062 2106 2.46normalized intensity 7.61 × 107 8.22 × 107 8.18 × 107 4.27 7.56 × 107 7.46 × 107 7.97 × 107
3.570.25 pm ProStds HsSera 0.1 pm ProStds HsSera
AMRTs 2012 1939 2058 3.00 1972 2002 1923 2.03normalized intensity 8.00 × 107 7.27 × 107 8.12 × 107 5.88 7.79 × 107 7.08 × 107 7.38 × 107 4.81
a The total number of AMRTs is indicated for each replicate analysis of the six human serum samples. The sum of the normalized intensity foreach replicate injection is listed below each of the corresponding total AMRT values. The coefficient of variation of the extracted AMRTs and theirassociated normalized intensity is calculated for each replicate injection. The ion detection parameters were set up to extract those multiply chargedions (charge states between 2 and 6) which exceeded 200 counts (center by area, after deisotoping).
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2195

Figure 4. Diagonal plots of the normalized log intensity. (A) Comparison of clustered AMRTs between human serum with 5.0 pmol of exogenousprotein mixture versus human serum with 2.0 pmol of exogenous protein mixture. For each matched AMRT component, the average log intensity fromeach condition is plotted along each of the two axes. The data are presented without applying any statistical filters, which are obtained from the clustereddata set. (B) Same comparison as illustrated in Panel A; however, the data have been filtered using a number of the available statistical measuresobtained from the clustering tool of the Expression Informatics software. The data have been filtered to show only those matched AMRTs which werefound to have a coefficient of variation of the normalized intensity of e30% among the replicate injections, (minimum two out of three replicates percondition), as well as an observed mass precision of e10 ppm among the replicate injections. (C) Comparison of clustered AMRTs between humanserum with 5 pmol of exogenous protein mixture versus human serum with 0.1 pmol of exogenous protein mixture after applying the statistical filterdescribed above. (D) Comparison of clustered AMRTs between human serum with 5.0 pmol of exogenous protein mixture and human serum with 1.0pmol of exogenous protein mixture after applying the statistical filter described above. (E) Comparison of clustered AMRTs between human serum with0.50 pmol of exogenous protein mixture and human serum with 0.25 pmol of exogenous protein mixture after applying the statistical filter describedabove. The data presented in all panels are colored by binned probability score (p score) from a binary Student’s t-test. Those AMRTs which had aprobability score of e0.01 are yellow, whereas those that are >0.01 are blue. The red data point corresponds to the monoisotopic ion of 1447.8048, whichoriginates from the VVGLSTLPEIYEK peptide of yeast ADH. The interpolated black line corresponds to the expected fold change for each binary comparison.
2196 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005

From this analysis, it is suggested that the yellow data pointsrepresent peptides from the MPDS proteins, whereas the bluedata points originate from peptides from human serum proteins.The information that is provided from this methodology allowsone to apply user-defined thresholds to the resulting statisticalanalysis performed on any of the experimental attributes relatingto each AMRT cluster, as well as a minimum replication rate withinand across conditions as a means to extract the highest qualitydata for subsequent quantitative analysis. Figure 4B depicts 1840(61.4%) of the matched AMRT component pairs from Figure 4Aafter applying a specific set of statistical thresholds to reveal thehighest quality data. These statistical measurements are providedby the Expression Informatics software and are included in thecorresponding output file. In this instance, the data were filteredby (1) applying a replication requirement, in which correspondingAMRTs must exist in at least two out of the three replicateinjections for each condition, (2) requiring that the coefficient ofvariation for the normalized intensities of an AMRT be e30% and(3) requiring that the mass precision of clustered AMRTs be <10ppm across all samples. After applying the statistical thresholds,1840 of the initial 2997 matched AMRTs (61.4%) remained toillustrate the two distinct sets of peptides, the unaffected humanserum peptides and the affected MPDS peptides. The breadth ofeach group of ions along the two diagonals is influenced by thedegree of variability inherent to the analytical method and willdetermine the confidence interval for a specific fold change.Interestingly, the 1840 statistically significant AMRTs represent>90% of the total average normalized intensity found in eachcondition. A total of 724 of the 2997 AMRTs were attributed toAMRTs which occurred in only one out of the three replicateinjections, an additional 384 AMRTs had coefficients of variation>30%, and 49 AMRTs had mass precision errors exceeding 10ppm. This indicates that the most variable data are due to thelower intensity AMRTs, as can be seen in Figure 5A. Figure 5Adepicts a scatter plot of the average normalized intensity of each
AMRT from the filtered data versus the observed coefficient ofvariation for the entire clustered data set. The blue data pointsare the subset of 1840 AMRTs which meet the statisticalparameters described above. As expected, the data illustrate thatthe statistical filtering process had the most significant effect onthe lowest intensity AMRTs, since they will be most influencedby coeluting AMRTs and will therefore tend to exhibit the highestvariability (Cv).
Manual inspection of the clustered output of the replicateinjections of the 5-pmol condition indicated that less than 80 ofthe AMRTs determined to be found in only one out of threereplicate injections could have been associated with an AMRTdetermined to have replicated in only two out of three replicateinjections. In this particular example, this represents a falseclustering rate of ∼2%. However, since only the AMRTs found toreplicate in only one out of three injections are eliminated fromthe quantitative processing, the information describing thesepotentially discarded AMRTs is still captured in those AMRTswhich occurred in two out of three injections.
One of the key features of this methodology is that it is anunbiased approach. The method does not require prescreeningof polypeptide pools for those peptides that contain specific aminoacids. This unbiased approach produces significantly more peptideions per protein than some other quantitative methodologies whichutilize isotope-coded affinity tags. In addition, the quantitativenature of this methodology allows the user to apply statisticalmethods to remove polypeptide ions (AMRTs) that exhibitquestionable reproducibility from further consideration withoutjeopardizing the ability to find lower level changes. Figure 5Bdepicts a histogram plot of the observed fold change for the 1840filtered AMRTs. The data presented illustrate two Gaussiandistributions about the x axis which are centered at values of 1.0and 2.5. These values correlate with the predicted results for theserum-related peptides (no change) and the spiked exogenouspeptides (2.5-fold change).
Figure 5. (A) A scatter plot of the average normalized intensity of the clustered AMRTs versus their corresponding coefficient of variationamong the replicate injections for human serum spiked with 5 pmol of exogenous protein versus human serum spiked with 2 pmol of exogenousprotein. The blue data points represent 1840 AMRTs which satisfy the statistical filters described in Figure 2B, whereas the red data pointsillustrate the 1157 AMRTs that were removed during the filtering process. (B) A histogram plot of the corresponding fold changes determinedamong the 1840 AMRTs which met the applied statistical measures.
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2197

Figure 4CD represents two additional diagonal plots of the logaverage normalized intensity of the 5-pmol mixtures versus boththe 100-fmol and 1-pmol mixtures. The results from Figure 4Cbegin to test the limits of this methodology. At 100 fmol of spikedMPDS protein, we are approaching the limit of detection for the300-µm scale chromatography selected for these series of experi-ments. This can manifest itself in the results by attenuating theexpected fold change, producing more scatter between the upperand lower limits of the expected fold change. In addition, it shouldbe noted that there are a number of peptides from the exogenousMPDS proteins that are chemically identical to a subset of thehuman serum proteins. Among these are human serum albuminand human hemoglobin. These chemically identical peptides willshow an attenuated fold change as a function of their relativeabundance over that of the endogenous peptide. Figure 4Eillustrates the 250-fmol mixture versus the 100-fmol mixture. Theseplots illustrate two distinct ion distributions of AMRTs, whichcorrelate with the relative concentration change of the MPDSproteins between the two samples as well as those unaffectedhuman serum proteins. The blue data points represent thoseAMRTs that do not show any relative change with statisticalsignificance between the two conditions (human serum proteins);the yellow data points represent those peptide components thatdo exhibit statistically significant changes between the twoconditions (MPDS proteins).
To confirm the quantitative results illustrated in Figure 4A-E, we performed a simple peptide mass fingerprinting search usingthe average mass measurement of each AMRT that was found inat least two out of three replicate injections from all six conditionswith a t-test probability score of e0.01 (67 AMRTs in all). Wesearched a Swissprot database of over 200 000 entries at 5 ppmmass accuracy with no missed cleavages and required fourminimum peptides to match. The search results accounted for59 of the 67 total AMRTs. The 59 AMRTs identified 47 proteinsby peptide mass fingerprint, which included the 5 spiked inproteins (MPDS proteins) as well as 37 isoforms of the MPDSproteins from different species, including 23 different isoforms ofglycogen phosphorylase. Last, the final five identifications wereexamples of very high molecular weight proteins (>120 kDa)which have tryptic peptides with monoisotopic masses in commonwith the MPDS proteins. The level of redundancy is not surprising,since the search was performed using a non-species-specificdatabase. In a true biomarker discovery experiment, the peptidemass fingerprint would most likely be restricted to a nonredundantdatabase of a specific organism to reduce the number of isoformsone may obtain from the homology/identity found in a cross-species database.
If we had spiked the proteins in at different concentrations,we could have used the quantitative fold change of the AMRTsas an additional filter or scoring mechanism to eliminate thewrongfully assigned high molecular weight protein assignments.We also suggest that the use of accurate mass in conjunction withfold change is a powerful strategy for MS-based protein identifica-tion. Since enzymatically digested proteins typically produce manypeptides and this methodology does not limit the number ofobserved peptides per protein through the use of any type ofaffinity capture enrichment protocol, proteins which exhibit arelative fold change in expression will produce a number of
AMRTs (tryptic peptides) that will exhibit the same change inexpression within some reasonable tolerance. It is suggested thatthe use of accurate mass in conjunction with the quantitative foldchange provides additional specificity to allow rapid screening ofcomplex protein mixtures for targeted proteins of interest whichexhibit a change in relative abundance. In instances for whichfurther validation is needed, the user has the ability to constructa targeted include list for subsequent MS/MS analysis from theaccurate mass and retention times (AMRTs) obtained from theLC/MS acquisition. However, the parallel LC/MS and LC/MSE
strategy implemented for this analysis contains not only theprecursor ion information but also the associated fragment ioninformation from all the observed precursors and allows one toidentify the precursor ions without having to perform the targetedMS/MS experiment.31 Low-energy precursor data are collectedinto function 1, while the associated elevated-energy data arecollected into the second function. The low-energy precursor ionsare associated with their corresponding high-energy fragment ionsusing the obtained chromatographic attributes. In this type ofexperiment, the software uses both the low- and elevated-energydata for qualitative assignment.20
The data presented in this manuscript illustrate that theExpression Informatics software is capable of reducing large setsof LC/MS analyses from complex protein mixtures to a simplelist of AMRT components that have undergone a change in relativeabundance due to the applied perturbation. These capabilities areprovided for by the use of the ion detection, clustering, andquantitative functionalities. Having the ability to reduce thesecomplex protein mixtures to a simple list of AMRT componentsgreatly simplifies the problem of properly identifying the proteinsaffected by the applied perturbation. In many cases, a subsequentprotein identification from such complex protein mixtures can beascertained from a simple peptide mass fingerprint of the specificAMRTs within a given fold change window. To illustrate thispowerful capability, we conducted a PMF search with only thoseAMRTs present in at least two out of the three replicate injectionsfor all conditions (5000-100 fmol MPDS proteins), with Cv’s ofthe associated replicating intensities of under 30%, with a massprecision of under 10 ppm, and illustrating a fold change with at-test score of <1% (Figure 4). The PMF search was queriedagainst a human database of 27 000 entries along with the fiveexogenous proteins and was conducted without considering anymissed cleavages and with a mass accuracy of <10 ppm. The PMFsearch returned 33 peptides from rabbit glycogen phosphorylase,18 peptides from bovine serum albumin, 14 peptides from yeastenolase, 12 peptides from yeast alcohol dehydrogenase, 4 peptidesfrom bovine hemoglobin (R), and 7 peptides from bovine hemo-globin (â). Among the set of identified exogenous proteins, thepeptide VVGLSTLEPIYEK (1447.8048 MH+) was among the 12peptides matched to yeast alcohol dehydrogenase. This peptidewas one of the most intense peptides from yeast ADH andexhibited a linear response when spiked into human serum, from100 to 5000 fmol (Figure 3A).
Figure 6 shows the 19 peptides matched from bovine serumalbumin via the PMF search. The average normalized intensityvalues are plotted for each of the albumin peptides for each ofthe six conditions (5000 to 100 fmol, on column). It is clear fromthis illustration that not all peptides ionized with sufficient
2198 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005
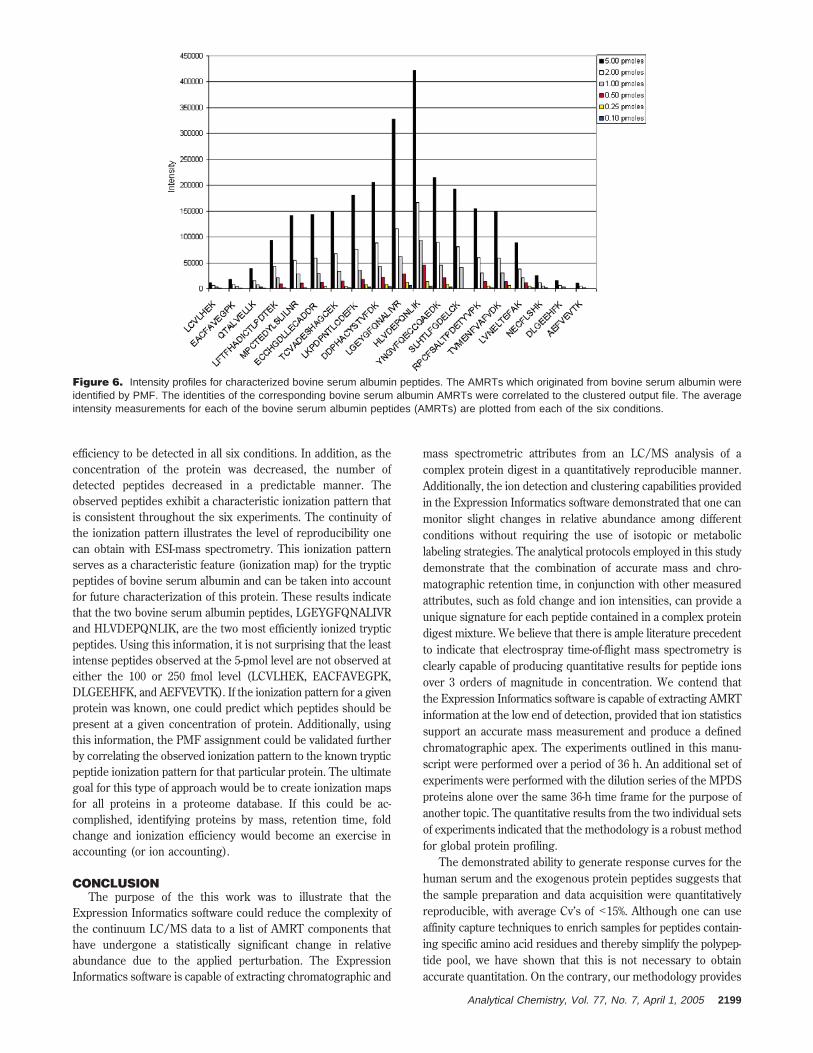
efficiency to be detected in all six conditions. In addition, as theconcentration of the protein was decreased, the number ofdetected peptides decreased in a predictable manner. Theobserved peptides exhibit a characteristic ionization pattern thatis consistent throughout the six experiments. The continuity ofthe ionization pattern illustrates the level of reproducibility onecan obtain with ESI-mass spectrometry. This ionization patternserves as a characteristic feature (ionization map) for the trypticpeptides of bovine serum albumin and can be taken into accountfor future characterization of this protein. These results indicatethat the two bovine serum albumin peptides, LGEYGFQNALIVRand HLVDEPQNLIK, are the two most efficiently ionized trypticpeptides. Using this information, it is not surprising that the leastintense peptides observed at the 5-pmol level are not observed ateither the 100 or 250 fmol level (LCVLHEK, EACFAVEGPK,DLGEEHFK, and AEFVEVTK). If the ionization pattern for a givenprotein was known, one could predict which peptides should bepresent at a given concentration of protein. Additionally, usingthis information, the PMF assignment could be validated furtherby correlating the observed ionization pattern to the known trypticpeptide ionization pattern for that particular protein. The ultimategoal for this type of approach would be to create ionization mapsfor all proteins in a proteome database. If this could be ac-complished, identifying proteins by mass, retention time, foldchange and ionization efficiency would become an exercise inaccounting (or ion accounting).
CONCLUSIONThe purpose of the this work was to illustrate that the
Expression Informatics software could reduce the complexity ofthe continuum LC/MS data to a list of AMRT components thathave undergone a statistically significant change in relativeabundance due to the applied perturbation. The ExpressionInformatics software is capable of extracting chromatographic and
mass spectrometric attributes from an LC/MS analysis of acomplex protein digest in a quantitatively reproducible manner.Additionally, the ion detection and clustering capabilities providedin the Expression Informatics software demonstrated that one canmonitor slight changes in relative abundance among differentconditions without requiring the use of isotopic or metaboliclabeling strategies. The analytical protocols employed in this studydemonstrate that the combination of accurate mass and chro-matographic retention time, in conjunction with other measuredattributes, such as fold change and ion intensities, can provide aunique signature for each peptide contained in a complex proteindigest mixture. We believe that there is ample literature precedentto indicate that electrospray time-of-flight mass spectrometry isclearly capable of producing quantitative results for peptide ionsover 3 orders of magnitude in concentration. We contend thatthe Expression Informatics software is capable of extracting AMRTinformation at the low end of detection, provided that ion statisticssupport an accurate mass measurement and produce a definedchromatographic apex. The experiments outlined in this manu-script were performed over a period of 36 h. An additional set ofexperiments were performed with the dilution series of the MPDSproteins alone over the same 36-h time frame for the purpose ofanother topic. The quantitative results from the two individual setsof experiments indicated that the methodology is a robust methodfor global protein profiling.
The demonstrated ability to generate response curves for thehuman serum and the exogenous protein peptides suggests thatthe sample preparation and data acquisition were quantitativelyreproducible, with average Cv’s of <15%. Although one can useaffinity capture techniques to enrich samples for peptides contain-ing specific amino acid residues and thereby simplify the polypep-tide pool, we have shown that this is not necessary to obtainaccurate quantitation. On the contrary, our methodology provides
Figure 6. Intensity profiles for characterized bovine serum albumin peptides. The AMRTs which originated from bovine serum albumin wereidentified by PMF. The identities of the corresponding bovine serum albumin AMRTs were correlated to the clustered output file. The averageintensity measurements for each of the bovine serum albumin peptides (AMRTs) are plotted from each of the six conditions.
Analytical Chemistry, Vol. 77, No. 7, April 1, 2005 2199

access to more peptides per protein and allows one to establishhigh confidence levels for each quantified protein.
The peptide components which exhibit significant up- or down-regulation can be further investigated by conducting a modificationof the traditional peptide mass fingerprint analysis. One canmaximize the information obtained from the clustered AMRTanalysis by recognizing that a relative change in abundance for aparticular protein will manifest itself by producing multiple peptidefragments which should exhibit the same relative change inabundance. Using the quantitative information available from theclustered AMRT analysis, the user can choose to submit for PMFidentification, only those accurate mass measurements whichexhibit the proper fold change. Organizing the AMRTs byobserved fold change for subsequent PMF identification is quiteempowering, since it provides additional stringency to the qualita-tive identification of a protein that is quantitatively consistent withthe data. For those users who require structural information forqualitative peptide/protein assignment, the list of AMRTs can beused to organize a targeted include list for subsequent peptideidentification studies by traditional methods, such as targeted MS/MS. Using the accurate mass and retention time informationobtained from the AMRT analysis, as well as the associatedquantitative and statistical analysis, one can carry out a targetedMS/MS analysis to identify only those AMRTs that have under-gone a statistically significant change in relative abundancebetween conditions. This would eliminate the accumulation of MS/MS data on proteins that are not affected in specific studies andwould allow one to maximize the efficiency of the MS/MS datacollection per unit time in a biomarker discovery setting. However,the initial LC/MS experiments could have been acquired usingthe alternate scanning methodology described in this work, inwhich the collision energy alternates between low and elevatedenergy throughout the entire LC/MS analysis to capture bothprecursor and associated fragment ion information in one experi-ment. Precursor information is captured in one function underlow-energy conditions, and the associated fragment ions arecaptured in a second function under elevated-energy conditions.Each reported precursor will have an associated set of fragment
ions that were detected in the elevated energy function. Althoughit is not described in this manuscript, the additional informationprovided in the elevated energy function affords additionalspecificity for each of the detected precursors in the low energyfunction. Although changing the chromatography column maycause a slight shift in the observed retention time, the associatedelevated energy accurate mass measurements will allow one tomanage the data properly across multiple experiments. Asdescribed in this work, the precursor information obtained in thismode is quantitative and reproducible. A more detailed explanationof the alternate scanning methodology is described in thefollowing study by Silva and co-workers31 and will be the topic offuture work.
ACKNOWLEDGMENTThe authors acknowledge the valuable contributions of Timo-
thy Riley and Bob Bateman throughout the development of thiswork. The authors also acknowledge Jeanne Li for her contribu-tions in the laboratory and throughout the editing of thismanuscript. Last, we extend our gratitude to our collaboratorswho helped develop the Expression Informatics software byembracing the methodology and applying themselves to demon-strate its utility (Stanely Hefta, Ashok Dongre, Gregory Opiteck,Martin Wiedmann, Deborah H. Smith, Arthur Moseley, KevinBlackburn, Danie Schlatzer, Craig A. Townsend, Minerva Hughes,Christopher T. Walsh, and Jun Yin).
SUPPORTING INFORMATION AVAILABLEFour posters that were presented at the 52nd ASMS Confer-
ence on Mass Spectrometry and Allied Topics, 2004, Nashville,TN (see explanations of refs 27, 28, 31, 32 in text) are availableas Supporting Information. This material is available free of chargevia the Internet at http://pubs.acs.org.
Received for review October 19, 2004. Accepted January13, 2005.
AC048455K
2200 Analytical Chemistry, Vol. 77, No. 7, April 1, 2005