Embed Size (px)
Citation preview

Temperature Regulation and the Pathogenesis of Fever
The oldest known written reference to fever exists in Akkadian cuneiform inscriptions from the 6th
century BC, most likely derived from an ancient Sumerian pictogram of a flaming brazier used to
symbolize both fever and the local warmth of inflammation.[1] Theoretical constructs of the
pathogenesis of fever did not emerge until several centuries later, when hippocratic physicians
proposed that body temperature, and physiologic harmony in general, involved a delicate balance
between four corporal humors, blood, phlegm, black bile, and yellow bile.[2] Fever was then
believed to result from an excess of yellow bile, a concept in concert with the fact that many
infections of that era caused both fever and jaundice. During the Middle Ages, demonic possession
was added to the list of mechanisms thought to be responsible for fever. By the 18th century,
Harvey’s discovery of the circulation of blood and the birth of clinical chemistry led iatrophysicists
and iatrochemists to hypothesize alternatively that body heat and fever resulted from friction
associated with the flow of blood through the vascular system and that they resulted from
fermentation and putrefaction occurring in the blood and intestines.[3] Ultimately, as a result of the
work of Claude Bernard, the metabolic processes occurring within the body came to be recognized
as the true source of body heat. Subsequent work established that body temperature is tightly
controlled within a narrow range by mechanisms regulating the rate at which such heat is allowed
to dissipate from the body.
The origin of the practice of monitoring body temperature as an aid to diagnosis is uncertain. The
oldest known references to devices used to measure temperature date to the 1st or 2nd century BC,
when Philo of Byzantium and Hero of Alexandria are believed to have invented several such
devices.[4] It is reasonably certain that Galileo manufactured a primitive (air) thermometer at about
the time he assumed the chair in mathematics at Padua in 1592.[5] However, thermometry was not
fully assimilated into medical practice until 1868, when Carl Reinhold August Wunderlich published
a magnum opus entitled Das Verhalten der Eigenw?rme in Krankenheiten (The Course of
Temperature in Diseases).[6]
Through Das Verhalten der Eigenw?rme in Krankenheiten, Wunderlich gave 37° C (98.6° F)
special significance with respect to normal body temperature.[7] He described the diurnal variation
of body temperature and, in the process, alerted clinicians to the fact that “normal body
temperature” is actually a temperature range, rather than a specific temperature. In an analysis of a
series of clinical thermometric measurements, the size of which has never been equaled
(estimated to have included some 1 million observations in as many as 25,000 subjects),
Wunderlich posited 38° C (100.4° F) as the upper limit of the normal range and in so doing,
proffered one of the first quantitative definitions of fever.
In spite of the fact that Wunderlich’s work was published over a century ago and was based
primarily on axillary measurements generally taken no more often than twice daily, it has survived
almost verbatim in modern concepts of clinical thermometry. Interestingly, recent tests conducted
with one of Wunderlich’s thermometers suggest that his instruments may have been calibrated by
as much as 1.4° to 2.2° C (2.6° to 4.0° F) higher than today’s instruments.[7] As a result, at least

some of Wunderlich’s cherished dictums regarding body temperature (e.g., the special significance
of 37° C [98.6° F]) have had to be revised.[8]
TERMINOLOGY
Of the many definitions of fever promulgated over the centuries, the one proposed by the
International Union of Physiological Sciences Commission for Thermal Physiology in 2001[9] is the
one most consistent with current concepts. It defines fever as “a state of elevated core temperature,
which is often, but not necessarily, part of the defensive responses of multicellular organisms (host)
to the invasion of live (microorganisms) or inanimate matter recognized as pathogenic or alien by
the host.” The febrile response (of which the temperature rise is a component) is a complex
physiologic reaction to disease, involving not only a cytokine-mediated rise in core temperature but
also the generation of acute-phase reactants, and the activation of numerous physiologic,
endocrinologic, and immunologic systems. The rise in temperature during fever is to be
distinguished from that occurring during episodes of hyperthermia. Unlike fever, hyperthermia
involves an unregulated rise in body temperature, in which pyrogenic cytokines are not directly
involved and against which standard antipyretics are generally ineffective. Only in the most
extreme cases, complicated by gut-derived endotoxemia, do pyrogenic cytokines appear to play a
role. In contrast to fever, hyperthermia represents a failure of thermoregulatory homeostasis, in
which there is either uncontrolled heat production, inadequate heat dissipation, or defective
thermoregulation.
In the clinical setting, fever is typically defined as a pyrogenmediated rise in body temperature
above the normal range. Although consistent with the public’s perception of fever, the definition
ignores the fact that a rise in body temperature is but one component of this multifaceted response.
This standard clinical definition is further flawed, because it implies that “body temperature” is a
single entity when, in fact, it is a pastiche of many different temperatures, each representative of a
particular body part, and each varying throughout the day in response to both activities of daily
living and the influence of endogenous diurnal rhythms.
CLINICAL THERMOMETRY
For over a century, the thermometer has been preeminent among clinical instruments used to
distinguish health from disease and to monitor the course of illness. Unfortunately, thermometric
measurements are influenced by a host of variables, all too frequently ignored when interpreting
the significance of clinical temperature readings.
Observer Variability
Thermometric measurements are generally simple to perform but involve a number of technical
details that, if not attended to, can invalidate estimates of body temperature. Few physicians, for
example, ever take the time to ensure the reliability or proper calibration of thermometers used in
clinical examinations. And yet Abbey and colleagues[10] found that a quarter of mercury-in-glass
thermometers obtained from four different manufacturers were inaccurate after 8 months of use or
storage.

Likewise, proper positioning of the temperature probe at the anatomic site employed is all too often
given less than careful attention. Erickson has reported that oral thermometric readings vary by as
much as 0.95° C (1.7° F) from the rear sublingual pocket to the area beneath the frenum in the
anterior floor of the mouth.[11] Interestingly, regional differences in readings obtained within the oral
cavity were more pronounced with electronic than with mercury thermometers, perhaps because of
differences in the dwell times required when taking measurements with the two types of
thermometers. It is also pertinent, in this regard, that studies performed earlier in the 20th century
indicated that from the anus inward, the temperature gradually rises, reaching its zenith at a depth
of approximately 2.5 inches (6.4 cm), and then gradually falls as the probe is advanced beyond 6
inches (15.2 cm).[12] More recent studies, however, found no significant differences between
temperature readings obtained with rectal probes inserted to depths of 5, 9, and 13 cm.[13]
With today’s electronic thermometers, equilibration times are relatively brief and, hence, thought
not to influence the results of clinical thermometric measurements. As noted earlier, this conclusion
is not necessarily justified. With mercury thermometers, opinions regarding proper placement times
have varied from 2 to 12 minutes for axillary recordings and 1 to 9 minutes for rectal readings.[14][15]
In a series of studies, Nichols and Kucha determined that 1 to 12 minutes are required for
equilibration of mercury-in-glass thermometers during measurements of oral temperature.[16] In
these studies, only 13% of the temperature readings reached their maximum after 3 minutes,
whereas 90% did so after 8 minutes.
Anatomic Variability
Although clinicians frequently regard temperature readings from various anatomic sites as
equivalent approximations of “body temperature,”[1] no one temperature characterizes the thermal
status of the human body. This is because the body has many different temperatures, each
representative of a particular body part. Nevertheless, within the body, there are two basic thermal
compartments worthy of special consideration—the core and the shell.
The shell, which consists of skin and subcutaneous fat, insulates the core from the external
environment. The core, of which the viscera and muscles are major components, although
insulated by the shell, has temperature gradients of its own, resulting from differences in the
metabolic rates and blood flow patterns of the various organs contained therein. Even during
baseline conditions, organs with higher metabolic rates have slightly higher temperatures than
those with lower metabolic rates; in general, tissues close to the skin have lower temperatures than
those at deeper locations.[17] Although such differences are normally small, during vigorous
exercise, muscle temperatures rise markedly in comparison with those of less metabolically active
organs. During shock and under extreme environmental conditions, regional anatomic variations in
temperature may also be exaggerated.
Rectal measurements have long been regarded as the most practical and accurate means of
obtaining routine estimates of core temperature. Benzinger and Benzinger, however, have pointed
out that no known thermoregulatory system exists at this particular anatomic site.[17] Rectal
temperature readings are consistently higher than those obtained at other sites (even pulmonary
artery blood), which some authorities have suggested might be due to heat generated as a result of
the metabolic activity of fecal bacteria.[18] However, an early study showed no significant decrease

in the rectal temperature after a reduction in the colonic bacterial content.[18] There is also concern
that stool in the rectum acts as a heat sink to delay or mitigate changes in the rectal temperature,
particularly so if the thermometer is inserted directly into stool.[19] During shock, perfusion of the
rectum may be markedly impaired, causing the rectal temperature to lag significantly behind a
rapidly rising or falling core temperature.[20] For this reason, Houdas and Ring have concluded that
the rectal temperature provides a reliable approximation of the core temperature only if the patient
is in thermal balance.[21] In neonates, even in the absence of shock, the rectal temperature
(measured by standard technique) has been reported to correlate poorly with the core temperature
(as measured by a deep rectal probe).[22] Although generally safe, such measurements are
associated with a small risk for rectal perforation—especially in neonates and very young
infants[23][24]—and if proper infection control measures are not followed, may be a source of
nosocomial infection.[25]
Of the three sites most commonly used for clinical thermometric measurement (rectum, mouth, and
tympanic membrane), the mouth is usually preferred, because it is accessible, responds promptly
to changes in the core temperature, and has a long tradition of use in monitoring body temperature
in clinical practice. The temperature of the sublingual pocket may be especially relevant clinically,
because its main artery is a branch of the external carotid artery and, like its parent artery,
responds quickly to changes in the core temperature.[18] However, because oral temperature
measurements require the cooperation of the subject being examined, not all patients (e.g., young
children, uncooperative adults, and intubated individuals) are amenable to such measurements.
It has long been suspected that the ingestion of hot or cold food or beverages and smoking
influence oral temperature readings. In a study of 22 healthy young adults, Rabinowitz and
associates showed that mastication and smoking cause both significant and persistent increases in
the oral temperature, whereas drinking ice water causes a significant but much more transient
decrease in the oral temperature.[26]
There is controversy regarding the effect of tachypnea on the accuracy of oral thermometric
readings. In studies employing electronic thermometers, Tandberg and Sklar obtained average
rectal temperature readings that were 0.96° F (0.6° C) higher than simultaneous oral temperatures
in patients with respiratory rates of 20 per minute or less, compared with 1.67° F (1.0° C) higher in
patients with respiratory rates of greater than 20 per minute.[27] A more recent study of 78 subjects
by Neff and co-workers that controlled for open- and closed-mouth breathing and used tympanic
rather than rectal temperature as a reference concluded that sublingual temperature changes do
not correlate with the respiratory rate or depth but do depend on whether the mouth is open or
closed.[28] As noted earlier, the probe location and equilibration time are two additional variables
that can alter the results of oral temperature measurements.
The right atrium is the ideal site for measuring core temperature, because it is the nexus at which
venous blood from all anatomic regions joins. However, because it is relatively inaccessible, the
temperatures of other sites are more often used as approximations of core temperature. The
tympanic membrane (TM) temperature is felt by some to be particularly useful in this regard,
because the TM is perfused by a tributary of the artery that supplies the body’s thermoregulatory
center.[29] This fact, and the ease with which TM measurements can be obtained using modern

infrared TM thermometers, have made these instruments the thermometers of choice in many
clinics and intensive care units. There are two basic types of infrared TM thermometers. One type
detects radiant energy emitted from the TM and portions of the ear canal, processes the
information, and then displays a value representing tissue temperature in the ear canal (unadjusted
mode).[30] The other displays an (adjusted) estimate of the core temperature (e.g., pulmonary
arterial blood temperature) based on comparison data obtained from selected study samples.
Readings obtained using the former type of TM thermometer tend to be lower than simultaneously
obtained oral readings, whereas those obtained with the latter type are generally higher.[29]
Unfortunately, numerous studies of many different TM thermometers have shown that although
convenient, such instruments tend to give highly variable readings that correlate poorly with
simultaneously obtained oral or rectal readings.[26][30][31][32][33]
Although Wunderlich’s monumental treatise on clinical thermometry was based primarily on axillary
measurements, recent experience indicates that although the axillary temperature provides a
reasonable approximation of body temperature in the neonate, it does not in the older child or adult.
In studies of core (deep rectal), anus, axillary, and skin temperature measurements in the newborn,
Mayfield and associates observed that axillary measurements obtained with a mercury
thermometer are as reliable as rectal temperature measurements.[22] Buntain and colleagues have
also reported that in the neonate, axillary temperature measurements are sufficiently accurate to
replace rectal measurements if a mercury-in-glass thermometer is used with an axillary dwell time
of 5 minutes or more.[34] Schiffman has shown a similarly good correlation between axillary and
rectal temperature measurements taken with mercury thermometers.[35]
Loudon, on the other hand, has shown that in older children, axillary temperature measurements
with mercury thermometers vary from 1.6° C (2.6° F) lower to 0.6° C (1° F) higher than
simultaneous oral measurements.[36] Nichols and colleagues have reported that in adults, even
with 12-minute dwell times, axillary temperatures exhibit differences of 0° to 2.5° C (0° to 4.2° F)
compared with oral temperature readings.[37] Furthermore, they noted that whereas 90% of rectal
temperature readings reached their zenith at 4 minutes and 28% of oral readings reached their
zenith at 5 minutes, only 18% of axillary readings reached a maximum at 5 minutes. Following a
similar experience in children, Ogren has concluded that axillary temperature readings may be
misleading and should be abandoned in the outpatient setting.[38]
Several studies have shown that monitoring the skin temperature using temperature-sensitive
crystals incorporated into plastic strips placed on the forehead is an insensitive technique for
detecting elevations in the core temperature.[39][40] The detection of fever by palpation is similarly
insensitive. Bergeson and Stienfeld found that 42% of 138 febrile children (as defined by a “body
temperature” of 38° C or greater) were judged to be afebrile by nurse assistants using palpation to
detect fever.[41] Only 1.8% of over 1000 afebrile children were judged to be febrile using this same
technique. In an evaluation of a mother’s ability to assess the temperature of her child by palpation,
Banco and Veltri found mothers to have a sensitivity of 73.9% and a specificity of 85.6% for
detecting fever of greater than 38° C (100.4° F).[42] Thus, palpation by mothers was more sensitive
than that by nurse assistants but was less specific for detecting the febrile state. Finally, Bonadio
and co-workers have reported that among infants younger than 2 months of age presenting to the
emergency room with a history of fever, those in whom fever had been documented at home by

rectal thermometer were twice as likely to be febrile on presentation or during hospitalization than
those whose fever had been documented by palpation alone (92% versus 46%; P < .001).[43]
Because the temperature of the rectum, mouth, and tympanic membrane are related but not
identical, it would be useful to have a reliable formula for converting data from one site to that of
another. In a study of healthy young adults, Rabinowitz and associates determined that on average,
rectal readings exceed concurrent oral readings by 0.4° C (0.8° F) and exceed TM readings
(obtained with the IVAC Core) by 0.8° C (1.6° F).[26] However, these relationships were extremely
variable. Their findings concerning the relationship between rectal and oral readings were in
agreement with those of several earlier investigations.[44][45] Their findings with respect to the
relationship between oral and TM readings, however, differed from earlier reports,[19] which had
generally shown TM readings to be higher than simultaneously obtained oral measurements. This
discrepancy most likely reflected the fact that unadjusted-mode TM thermometers—for example,
the IVAC Core—generally give lower readings than adjusted-mode TM thermometers, such as
those used in earlier studies.[30]
Togawa has reported that on average, in a resting healthy adult, the core temperature (pulmonary
artery) is 0.4° C (0.7° F) higher than the oral temperature and 0.2° C (0.4° F) lower than the rectal
temperature—however, again with considerable individual variability.[46] Anagnostakis and
co-workers have concluded in studies comparing rectal and axillary temperatures in infants that
because of a similarly high degree of variability, no standard factor can be developed for converting
axillary to rectal temperatures.[47] Thus, using a temperature reading from one anatomic site to
predict the temperature at another must be done with caution.
Physiologic Variables
Wunderlich and Seguin[48] believed that “old” people have lower body temperatures than younger
persons, and their views in this regard were corroborated by Howell in a report published in Lancet
in 1948.[49] There is also a substantial body of data suggesting that thermoregulation is impaired in
older persons because of various effects of aging on the autonomic nervous system.[50]
Nevertheless, more recent work has not shown lower average core temperatures among healthy
older subjects (mean age, 80.3 years; range, 62 to 99 years) than among healthy younger
subjects.[51] Comparisons of simultaneous oral, axillary, and rectal temperature readings from such
subjects have shown lower average oral and axillary readings in older persons but comparable
average rectal temperatures in older and young subjects.
It has long been known that women exhibit increases in body temperature of about 0.5° C (0.9° F)
at the time of ovulation.[21] Wunderlich and Seguin also maintained that women have slightly higher
normal temperatures than men overall and often show greater and more sudden changes in
temperature.[48] Two more recent studies have corroborated Wunderlich and Seguin’s former but
not latter observation.[8][52]
Body temperature, like most physiologic functions, exhibits circadian rhythmicity that is linked to
the sleep-wake cycle.[53] During normal sleep-wake cycles (i.e., asleep during the night and awake
during the day), the core temperature reaches its zenith in the late afternoon or early evening and
its nadir in the early morning.[8] Adaptation to night-shift work causes a reversal of this pattern.

Thermoregulation has also been reported to be altered in patients with neuropsychiatric disorders
such as chronic depression.[54] Therefore, when interpreting clinical thermometric measurements, it
is important to consider not only the time of the measurement and the site at which the temperature
was taken, but also the sleep-wake cycle and mental health of the subject being studied.
In addition to these physiologic variables, exercise, digestion, and underlying disorders such as
chronic renal failure, shock, and local inflammation at the site of the thermometric measurement
(e.g., proctitis, external otitis, or stomatitis) may alter thermoregulatory responses or local
temperatures, or both. It has, for example, been shown that the core temperature varies by as
much as 3° C (36° to 39° C) in states ranging from sleep to moderately high levels of sustained
exercise, and this continuum of body temperature is related to a continuum of activity.[55] Ambient
temperature and humidity have been shown experimentally to affect both human sleep stages and
body temperature,[56] suggesting that body temperature might also vary according to the time of
year and local climate. It is pertinent in this regard that Cheng and Partridge have shown that
bundling and warm environments can elevate rectal temperatures of newborns to the febrile
range.[57]
Normal “Body” Temperature
A survey of physicians’ perceptions of body temperature published in 1995 indicated widespread
confusion regarding key features of the human body temperature.[58] Seventy-five percent of 268
physicians and medical students surveyed gave 37° C (98.6° F) as their definition of normal body
temperature. An additional 13% defined the normal temperature as a narrow range of
temperatures about a mean of 37° C (98.6° F). Only 10 (4%) subjects in the group as a whole
specified a particular body site (e.g., oral or rectal) for temperature measurements in their definition.
Ninety-eight percent believed that the normal temperature varies during the day, with quantitative
estimates of such diurnal variability ranging from 0.2° C (0.4° F) to 2.8° C (5° F) (mean ± SD, 0.8° ±
0.4° C [1.6° ± 0.8° F]). Estimates of the lower and upper ends of the normal temperature range
varied between 32.8° C (91° F) and 37.2° C (99° F) and between 36.7° C (98.0° F) and 39° C
(102.2° F), respectively. Temperatures used to define fever (i.e., the lower end of the febrile range)
varied between 36.9° C (98.5° F) and 40° C (104° F). Seventy-nine percent of the subjects
believed that body temperature normally reaches its zenith in the evening and its nadir in the
morning. However, fewer medical students (72%) than graduate physicians (85%) believed this to
be the case (P = .01).
The origin of these perceptions of body temperature is uncertain, but in all likelihood it lies in Carl
Wunderlich’s 1868 book on clinical thermometry (mentioned previously), which many regard to this
day as the definitive work on the subject.[6] Unfortunately, several of Wunderlich’s dictums
concerning body temperature, like the perceptions of modern-day physicians, appear to be in error.
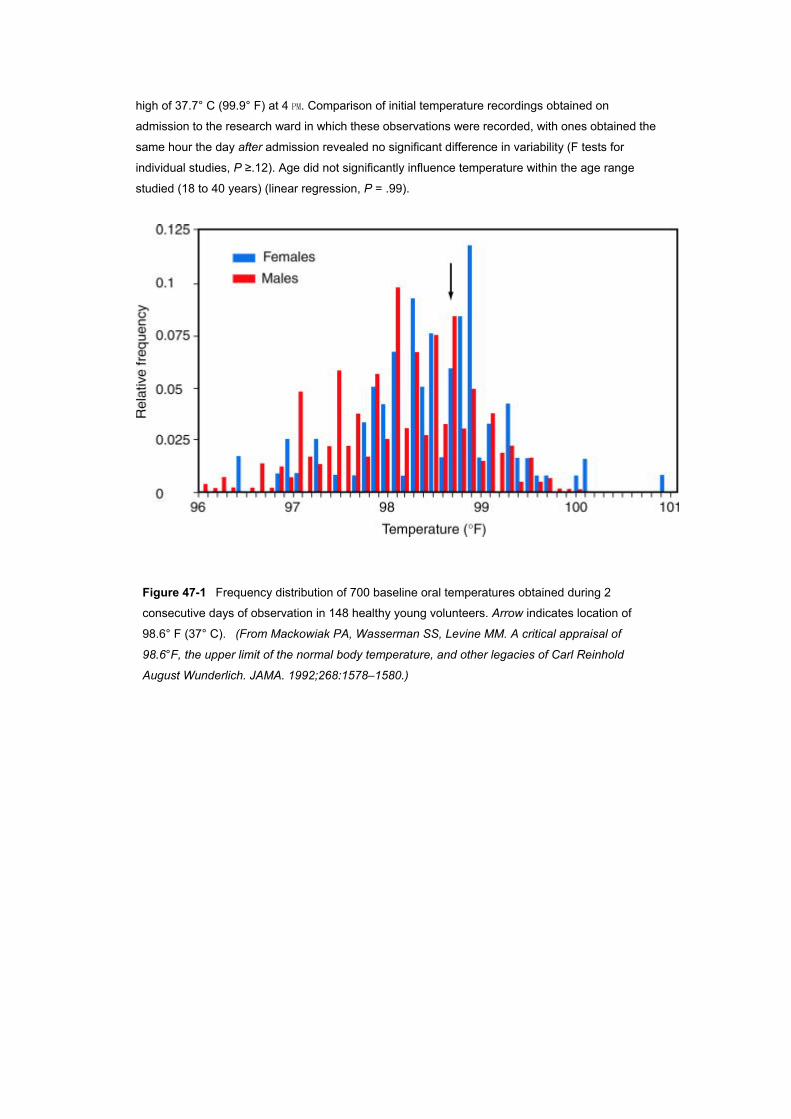
A 1992 descriptive analysis of 700 baseline oral temperature observations from 148 healthy men
and women found a range of 35.6° C (96.0° F) to 38.2° C (100.8° F), an overall mean of 36.8° ±0.4°
C (98.2° (±0.7° F), a median of 36.8° C (98.2° F), and a mode of 36.7° C (98.0° F); 37° C (98.6° F)
accounted for only 56 (8%) of the 700 oral temperature observations recorded ( Fig. 47–1 ).[8] The
mean temperature varied diurnally, with a 6 AM nadir and a 4 to 6 PM peak ( Fig. 47–2 ). The maximal
temperature (as reflected by the 99th percentile) varied from a low of 37.2° C (98.9° F) at 6 AM to a
high of 37.7° C (99.9° F) at 4 PM. Comparison of initial temperature recordings obtained on
admission to the research ward in which these observations were recorded, with ones obtained the
same hour the day after admission revealed no significant difference in variability (F tests for
individual studies, P ≥.12). Age did not significantly influence temperature within the age range
studied (18 to 40 years) (linear regression, P = .99).
Figure 47-1 Frequency distribution of 700 baseline oral temperatures obtained during 2
consecutive days of observation in 148 healthy young volunteers. Arrow indicates location of
98.6° F (37° C). (From Mackowiak PA, Wasserman SS, Levine MM. A critical appraisal of
98.6°F, the upper limit of the normal body temperature, and other legacies of Carl Reinhold
August Wunderlich. JAMA. 1992;268:1578–1580.)
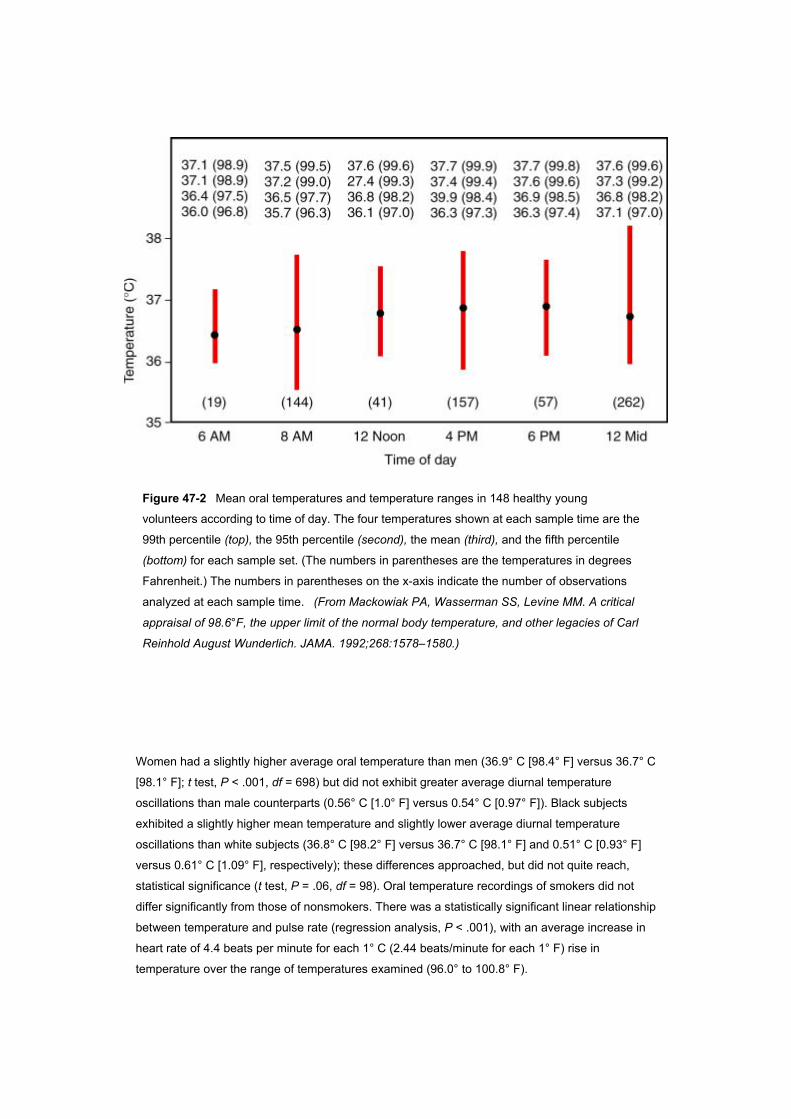
Figure 47-2 Mean oral temperatures and temperature ranges in 148 healthy young
volunteers according to time of day. The four temperatures shown at each sample time are the
99th percentile (top), the 95th percentile (second), the mean (third), and the fifth percentile
(bottom) for each sample set. (The numbers in parentheses are the temperatures in degrees
Fahrenheit.) The numbers in parentheses on the x-axis indicate the number of observations
analyzed at each sample time. (From Mackowiak PA, Wasserman SS, Levine MM. A critical
appraisal of 98.6°F, the upper limit of the normal body temperature, and other legacies of Carl
Reinhold August Wunderlich. JAMA. 1992;268:1578–1580.)
Women had a slightly higher average oral temperature than men (36.9° C [98.4° F] versus 36.7° C
[98.1° F]; t test, P < .001, df = 698) but did not exhibit greater average diurnal temperature
oscillations than male counterparts (0.56° C [1.0° F] versus 0.54° C [0.97° F]). Black subjects
exhibited a slightly higher mean temperature and slightly lower average diurnal temperature
oscillations than white subjects (36.8° C [98.2° F] versus 36.7° C [98.1° F] and 0.51° C [0.93° F]
versus 0.61° C [1.09° F], respectively); these differences approached, but did not quite reach,
statistical significance (t test, P = .06, df = 98). Oral temperature recordings of smokers did not
differ significantly from those of nonsmokers. There was a statistically significant linear relationship
between temperature and pulse rate (regression analysis, P < .001), with an average increase in
heart rate of 4.4 beats per minute for each 1° C (2.44 beats/minute for each 1° F) rise in
temperature over the range of temperatures examined (96.0° to 100.8° F).

According to Wunderlich and Seguin, “When the organism (man) is in a normal condition, the
general temperature of the body maintains itself at the physiologic point: 37° C = 98.6° F.”[48]
Although several subsequent investigations have recorded mean temperatures of normal adult
populations closer to 36.6° C (98.0° F),[59] Wunderlich’s intimation that 37° C (98.6° F) is the most
normal of temperatures[60] persists to this day in lay thinking, although to a lessening extent in the
thinking of health care workers. The special significance formerly accorded 37° C (98.6° F) is
perhaps best illustrated by the 1990 edition of Stedman’s Medical Dictionary, which defines fever
as “a body temperature above the normal of 37° C (98.6° F).”[61] In the 2000 edition, fever is
defined as “A complex physiologic response to disease mediated by pyrogenic cytokines and
characterized by a rise in core temperature, generation of acute-phase reactants and activation of
immunological systems.”[62]
The data reviewed earlier suggest that 37° C (98.6° F) has no special significance vis-à-vis body
temperature in healthy young adults when such temperature is measured orally using modern
thermometers. In the population examined, 37° C (98.6° F) was not the overall mean temperature,
the mean temperature of any of the time periods studied, the median temperature, or the single
most frequent temperature recorded. Furthermore, it did not fall within the 99.9% confidence limits
for the sample mean (36.7° to 36.8° C; 98.1° to 98.2° F).
Wunderlich identified 38.0° C (100.4° F) as the upper limit of normal body temperature in his
patient population and, therefore, regarded any temperature greater than 38.0° C (100.4° F) as
fever.[48] However, the upper limit of normal body temperature varies among individuals, thereby
limiting the applicability of mean values derived from population studies (even those as large as
Wunderlich’s) to individual subjects. However, the maximal temperature, like the mean
temperature, exhibited by a population varies according to the time of day and the site at which
temperature measurements are taken. Because of such variability, no single temperature can be
designated as the upper limit of normal. In the study population considered earlier, 37.2° C (98.9° F)
was the maximal oral temperature (i.e., the 99th percentile) recorded at 6 AM, whereas at 4 PM, the
maximal oral temperature observed reached 37.7° C (99.9° F). Thus, these data suggest that when
modern thermometers are used to monitor oral temperature in young or middle-aged adults, fever
is roughly defined as an early-morning temperature of 37.2° C (99.0° F) or greater or a temperature
of 37.8° C (100° F) or greater at any time during the day.
Wunderlich wrote in 1868 that “[temperature] oscillates even in healthy persons according to time
of day by 0.5° C = 0.9° F.” The next year, Wunderlich and Reeve wrote, “The lowest point is
reached in the morning hours between two and eight, and the highest in the afternoon between
four and nine.”[63] Modern authorities have generally concurred with these observations. However,
Tauber has suggested that the amplitude of diurnal variation might be as high as 1° C (1.8° F).[64]
The data described earlier are more consistent with the views of Wunderlich and colleagues.
Nevertheless, the subjects examined in that study exhibited considerable individual variability,
some having daily temperature oscillations as wide as 1.3° C (2.4° F) and others having
oscillations as narrow as 0.1° C (0.2° F).
According to Wunderlich and Seguin, women have slightly higher normal temperatures than men
and often show greater and more sudden changes of temperature.[48] In a study of nine healthy

young adults (six male and three female), Dinarello and Wolff corroborated both observations.[52]
The investigation described earlier, which did not control for the effects of ovulation on thermal
observations, was able to corroborate only the former observation of Wunderlich and Seguin.[8]
It has been maintained for over a century that older persons have lower body temperatures than
younger persons.[48] Howell’s 1948 study (mentioned previously) seemed to substantiate this
belief.[49] Although there are considerable data suggesting that thermoregulation is impaired in
older persons because of various effects of aging on the autonomic system,[50] as noted previously,
more recent investigation has not shown lower average core temperatures among healthy older
persons than among healthy young people.[51]
Some authors believe that the first temperature reading obtained on admission to a hospital can be
falsely elevated, because stress, in the broadest sense, has the capacity to elevate body
temperature.[48][65] The study by Mackowiak and colleagues described previously did not find
evidence that the first temperature reading obtained after admission to a research study unit was
any more likely to be elevated than measurements obtained at later times.[8] The Maryland
investigators, however, could not be certain that stress levels at the time of admission to their unit
were comparable to levels of stress experienced by patients at the time of admission to a hospital.
As a result of work conducted earlier this century,[45][66] it is widely believed that the heart rate
increases 10 beats per minute for each 1° F rise in body temperature. More recent data (presented
earlier) indicate that the heart rate increases only 2.44 beats per minute for each 1° F rise in
temperature.[8] The difference between the earlier and more recent investigations most likely
reflects the fact that in the latter instance, subjects were afebrile and were examined seated,
whereas those examined in earlier investigations were mostly febrile and rested reclining on a
couch for 20 minutes before examination.
The normal range of body temperature in children is not well delineated. Lorin has written that the
range is higher in children than in adults and that a decrease toward adult levels begins at about 1
year of age, continues through puberty, and stabilizes at 13 to 14 years of age in girls and at 17 to
18 years of age in boys.[67] He offers as documentation of his views on the matter a 1937
publication by Bayley and Stolz.[68] Unfortunately, these early investigators did not control for
variables such as the time of day, bundling, and the thermometer dwell time, each of which might
have significantly affected the results of their survey. It has also been maintained that the circadian
rhythm that characterizes body temperature in the adult is less evident in the first few months of life,
is well established by the second birthday, and tends to be more pronounced during childhood than
during adulthood.[67] This concept, like many concerned with the normal temperature of children, is
difficult to substantiate with published data.
THERMOREGULATION
Heat is derived from biochemical reactions occurring in all living cells.[69] At the mitochondrial level,
energy derived from the catabolism of metabolites such as glucose is used in oxidative
phosphorylation to convert adenosine diphosphate to adenosine triphosphate (ATP). At rest, more
than half of the body’s heat is generated as a result of the inefficiency of the biochemical processes
that convert food energy into the free energy pool (e.g., ATP). Even if no external work is being

performed, heat is generated as a result of both internal work (e.g., peristalsis, myocardial
contractions, and the circulation of blood) and biochemical reactions involved in maintaining the
structural and functional integrity of the various organ systems (i.e., the utilization and resynthesis
of ATP). When external work is performed, additional heat is generated as a byproduct of skeletal
muscle contractions.
In adult humans and most other large mammals, shivering is the primary means by which heat
production is enhanced. Nonshivering thermogenesis is more important in smaller mammals,
newborns (including humans), and cold-acclimated mammals.[69][70] Although several tissues (e.g.,
the heart, respiratory muscles, and adipose tissue) contribute to the process, brown adipose tissue
has been most closely associated with nonshivering thermogenesis. This highly specialized form of
adipose tissue located near the shoulder blades, neck, adrenals, and deep blood vessels (adjacent
to vital organs) is characterized by its brownish color, a profuse vascular system, and an
abundance of mitochondria.[69][71]
Heat generated primarily in vital organs lying deep within the body core, is distributed throughout
the body via the circulatory system. In response to input from the nervous system, the circulatory
system determines both the temperature of the various body parts and the rate at which heat is lost
from body surfaces to the environment (by conduction, convection, radiation, and evaporation).[72]
In a warm environment, or in response to an elevation in the core temperature resulting from
exercise, cutaneous blood flow increases so that heat is transported from the core to be dissipated
at the skin surface. Simultaneous activation of sweating enhances such heat loss via evaporation.
In anesthetized animals, increases in cutaneous blood flow in response to hypothalamic warming
are offset by concomitant reductions in gastrointestinal blood flow.[73] In a cold environment or in
response to a reduction in core temperature, cutaneous blood flow normally decreases as a means
of conserving heat within the body core.
Thermoregulation is a process that involves a continuum of neural structures and connections
extending to and from the hypothalamus and limbic system through the lower brain stem and
reticular formation to the spinal cord and sympathetic ganglia ( Fig. 47–3 ).[69] Nevertheless, an
area of the brain located in and near the rostral hypothalamus appears to be especially important to
the process of thermoregulation. Although generally referred to as the “preoptic area,” it actually
includes the medial and lateral aspects of the preoptic area, anterior hypothalamus, and septum.
Numerous studies extending over 60 years have established that neurons located in this area are
thermosensitive and exert at least partial control over physiologic and behavioral thermoregulatory
responses.[72][74]
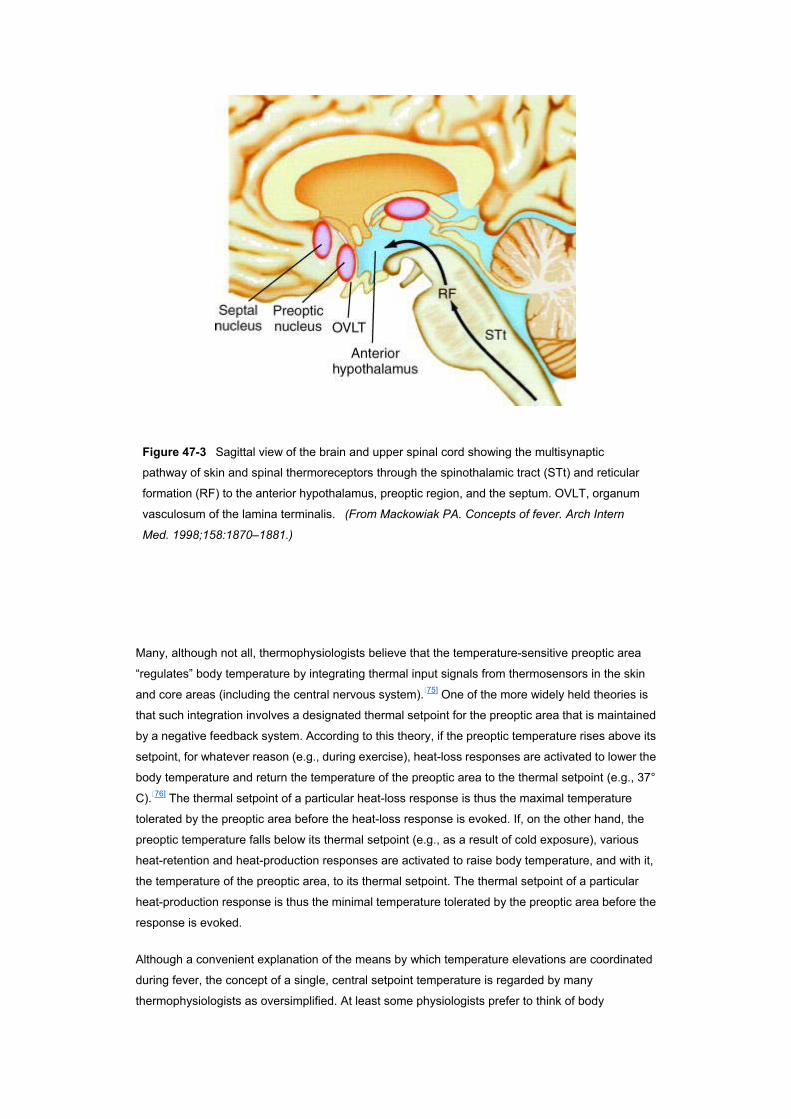
Figure 47-3 Sagittal view of the brain and upper spinal cord showing the multisynaptic
pathway of skin and spinal thermoreceptors through the spinothalamic tract (STt) and reticular
formation (RF) to the anterior hypothalamus, preoptic region, and the septum. OVLT, organum
vasculosum of the lamina terminalis. (From Mackowiak PA. Concepts of fever. Arch Intern
Med. 1998;158:1870–1881.)
Many, although not all, thermophysiologists believe that the temperature-sensitive preoptic area
“regulates” body temperature by integrating thermal input signals from thermosensors in the skin
and core areas (including the central nervous system).[75] One of the more widely held theories is
that such integration involves a designated thermal setpoint for the preoptic area that is maintained
by a negative feedback system. According to this theory, if the preoptic temperature rises above its
setpoint, for whatever reason (e.g., during exercise), heat-loss responses are activated to lower the
body temperature and return the temperature of the preoptic area to the thermal setpoint (e.g., 37°
C).[76] The thermal setpoint of a particular heat-loss response is thus the maximal temperature
tolerated by the preoptic area before the heat-loss response is evoked. If, on the other hand, the
preoptic temperature falls below its thermal setpoint (e.g., as a result of cold exposure), various
heat-retention and heat-production responses are activated to raise body temperature, and with it,
the temperature of the preoptic area, to its thermal setpoint. The thermal setpoint of a particular
heat-production response is thus the minimal temperature tolerated by the preoptic area before the
response is evoked.
Although a convenient explanation of the means by which temperature elevations are coordinated
during fever, the concept of a single, central setpoint temperature is regarded by many
thermophysiologists as oversimplified. At least some physiologists prefer to think of body

temperature as regulated within a narrow range of temperatures by a composite setpoint of several
thermosensitive areas and several different thermoregulatory responses.[77][78][79]
A variety of endogenous substances and drugs appear to affect temperature regulation by altering
the activity of hypothalamic neurons. Perhaps the best examples of such substances are the
pyrogenic cytokines discussed later. These are released by mononuclear phagocytes in response
to a wide array of stimuli and have the capacity to raise the thermoregulatory center’s thermal
setpoint. Whether they cross the blood-brain barrier to do so[80][81] or act by evoking the release of
other mediators (e.g., prostaglandin E2 [PGE2]) in circumventricular organs, such as the organum
vasculosum laminae terminalis,[80] is uncertain. Whatever the precise endogenous mediators of
fever, their primary effect appears to be to decrease the firing rate of preoptic warm-sensitive
neurons, leading to the activation of responses designed to decrease heat loss and increase heat
production.
ENDOGENOUS PYROGENS
Pyrogens have traditionally been divided into two general categories: those that originate outside
the body (exogenous pyrogens) and those that are derived from host cells (endogenous pyrogens).
Exogenous pyrogens are, for the most part, microorganisms and toxins or other products of
microbial origin, whereas endogenous pyrogens are host cell–derived (pyrogenic) cytokines that
are the principal central mediators of the febrile response.[82] According to traditional concepts,
exogenous pyrogens, regardless of their physicochemical structure, initiate fever by inducing host
cells (primarily macrophages) to produce endogenous pyrogens. Such concepts notwithstanding,
certain endogenous molecules also have the capacity to induce endogenous pyrogens. These
include, among others, antigen-antibody complexes in the presence of complement,[83][84] certain
androgenic steroid metabolites,[85][86][87] inflammatory bile acids,[88] complement,[89] and various
lymphocyte-derived molecules.[90][91] Likewise, data recently obtained in studies employing guinea
pigs suggest that bacterial lipopolysaccharide (LPS) induces fever directly (rather than indirectly
through the induction of pyrogenic cytokines) by interacting with Kupffer’s cells, thereby initiating
pyrogenic signals that are transmitted to the preoptic area of the hypothalamus via the hepatic
branch of the vagus nerve.[92] Thus, the distinction between endogenous and exogenous pyrogens
is artificial at best.
Complete understanding of the function of individual pyrogenic cytokines has been hampered by
the fact that one cytokine often influences the expression of other cytokines or their receptors, or
both, and may also induce more distal co-mediators of cytokine-related bioactivities (e.g.,
prostaglandins and platelet-activating factor).[93] In short, cytokines function within a complex
regulatory network in which information is conveyed to cells by combinations, and perhaps by
sequences, of a host of cytokines and other hormones.[94] Like the words of human communication,
individual cytokines are basic units of information. On occasion, a single cytokine, like a single
word, may communicate a complete message. More often, however, complete messages received
by cells probably resemble sentences, in which combinations and sequences of cytokines convey
information. Because of such interactions, it has been difficult to ascertain the direct in vivo
bioactivities of particular cytokines. Nevertheless, several cytokines have in common the capacity

to induce fever. On the basis of this characteristic, they have been codified together as so-called
pyrogenic cytokines.
The list of currently recognized pyrogenic cytokines includes, among others, interleukin-1 (IL-1
[IL-1α and IL-β]), tumor necrosis factor-α (TNF-α), IL-6, ciliary neurotropic factor (CNF), and
interferon (IFN).[95][96][97][98][99][100][101][102][103] Even among these few cytokines, complex relationships
exist, with certain members upregulating the expression of other members or their receptors in
certain situations and downregulating them in others.[93] The four major pyrogenic cytokines have
monomeric molecular masses that range from 17 to 30 kDa. Undetectable under basal conditions
in healthy subjects, they are produced by many different tissues in response to appropriate stimuli.
Once released, pyrogenic cytokines have short intravascular half-lives. They are pleiotropic, in that
they interact with receptors present on many different host cells. They are active in picomolar
quantities, induce maximal cellular responses even at low receptor occupancy, and exert local
(autocrine-paracrine) as well as systemic (endocrine) effects.[93]
It has long been suspected that interactions between pyrogenic cytokines and their receptors in the
preoptic region of the anterior hypothalamus activate phospholipase A2, liberating plasma
membrane arachidonic acid as a substrate for the cyclooxygenase pathway. Some cytokines
appear to do so by increasing cyclooxygenase expression directly, causing liberation of the
arachidonate metabolite PGE2. Because this small lipid molecule easily diffuses across the
blood-brain barrier, it is thought by some to be the local mediator that actually activates
thermosensitive neurons. Although it is not yet widely accepted, additional studies indicate that the
C5a component of the complement cascade is integral to LPS-induced fever[104] and that in some
situations, thermal information involved in the febrile response is transmitted from the periphery to
the thermoregulatory center via vagal pathways (see earlier).[105]
Figure 47–4 depicts the modern, hypothetical model for the febrile response,[106] in which
pyrogenic cytokines released by phagocytic leukocytes into the blood stream in response to
exogenous pyrogens find their way to the organum vasculosum of the lamina terminalis (OVLT),
where they induce synthesis of prostaglandins mediating the febrile response. The model has
several shortcomings that have caused thermophysiologists to suspect that multiple pathways
might be involved in the induction of fever (e.g., the vagal pathways referred to earlier, local
production of pyrogenic cytokines in the hypothalamus itself, and participation of membrane-bound
cytokines as mediators), with different pathways or combinations of pathways being responsible for
fever in different situations.[105][107] All of the models proposed to date have been concerned with
mechanisms responsible for the induction phase of fever. None have considered the plateau or
ascending phases of fever or explained why a disorder such as endocarditis, in which exogenous
pyrogens (i.e., bacteria) are present continuously in the blood, is associated with a remittent rather
than a continuous fever pattern. As a consequence, our understanding of the febrile response
remains incomplete and largely speculative. As indicated previously, it is not yet clear whether
circulating cytokines cross the blood-brain barrier or have to be produced within the central
nervous system in order to activate thermosensitive neurons; or if each of the pyrogenic cytokines
is capable of raising the thermoregulatory setpoint independently or must exert this effect through
some final, common pathway (see Fig. 47–4 ); or if PGE2 or other local mediators are a sine qua
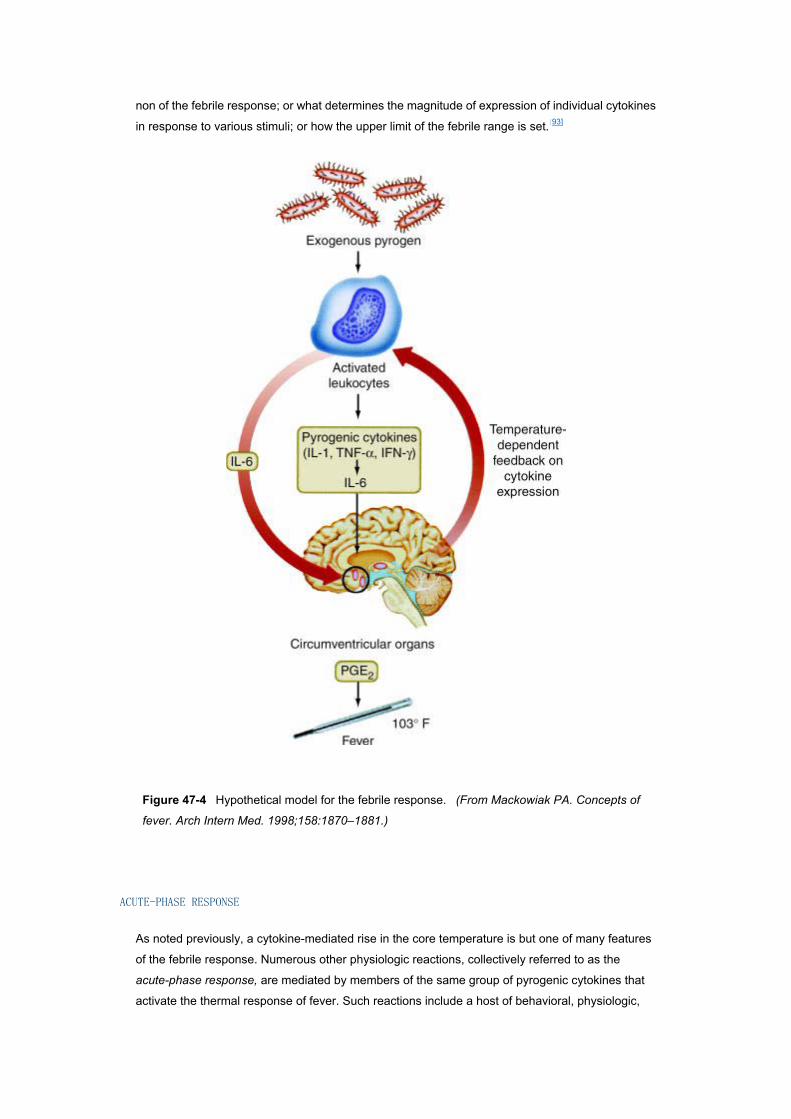
non of the febrile response; or what determines the magnitude of expression of individual cytokines
in response to various stimuli; or how the upper limit of the febrile range is set.[93]
Figure 47-4 Hypothetical model for the febrile response. (From Mackowiak PA. Concepts of
fever. Arch Intern Med. 1998;158:1870–1881.)
ACUTE-PHASE RESPONSE
As noted previously, a cytokine-mediated rise in the core temperature is but one of many features
of the febrile response. Numerous other physiologic reactions, collectively referred to as the
acute-phase response, are mediated by members of the same group of pyrogenic cytokines that
activate the thermal response of fever. Such reactions include a host of behavioral, physiologic,
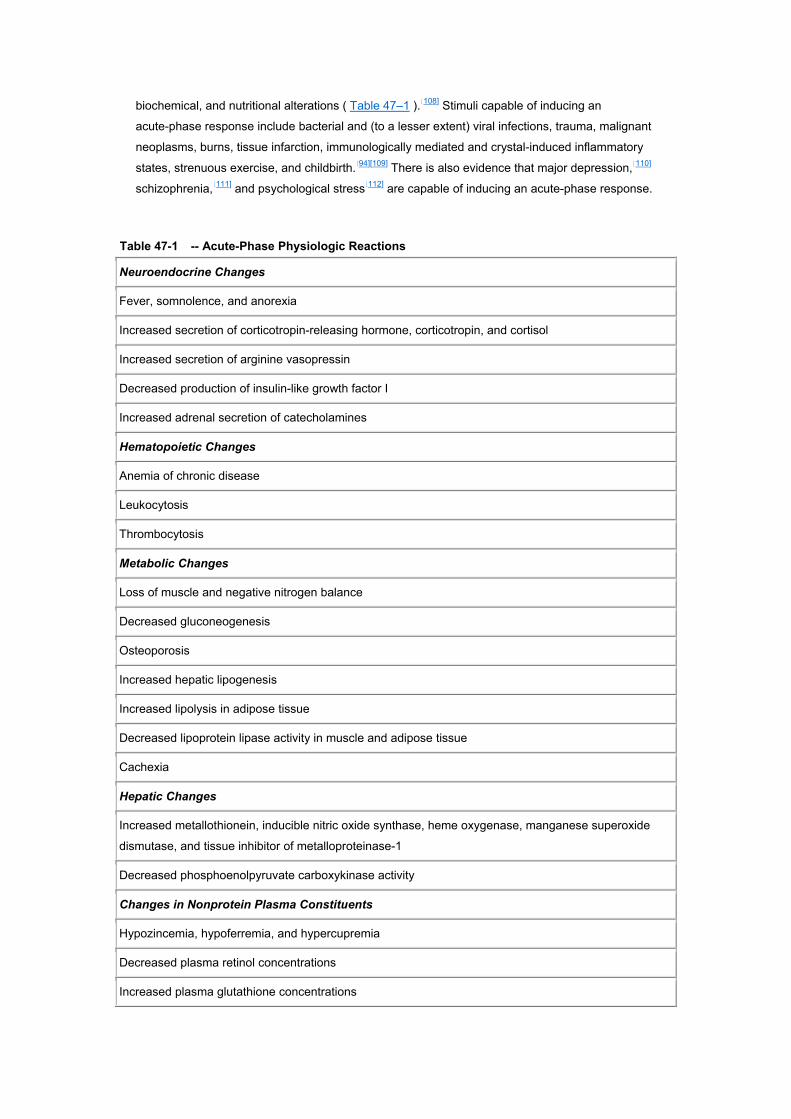
biochemical, and nutritional alterations ( Table 47–1 ).[108] Stimuli capable of inducing an
acute-phase response include bacterial and (to a lesser extent) viral infections, trauma, malignant
neoplasms, burns, tissue infarction, immunologically mediated and crystal-induced inflammatory
states, strenuous exercise, and childbirth.[94][109] There is also evidence that major depression,[110]
schizophrenia,[111] and psychological stress[112] are capable of inducing an acute-phase response.
Table 47-1 -- Acute-Phase Physiologic Reactions
Neuroendocrine Changes
Fever, somnolence, and anorexia
Increased secretion of corticotropin-releasing hormone, corticotropin, and cortisol
Increased secretion of arginine vasopressin
Decreased production of insulin-like growth factor I
Increased adrenal secretion of catecholamines
Hematopoietic Changes
Anemia of chronic disease
Leukocytosis
Thrombocytosis
Metabolic Changes
Loss of muscle and negative nitrogen balance
Decreased gluconeogenesis
Osteoporosis
Increased hepatic lipogenesis
Increased lipolysis in adipose tissue
Decreased lipoprotein lipase activity in muscle and adipose tissue
Cachexia
Hepatic Changes
Increased metallothionein, inducible nitric oxide synthase, heme oxygenase, manganese superoxide
dismutase, and tissue inhibitor of metalloproteinase-1
Decreased phosphoenolpyruvate carboxykinase activity
Changes in Nonprotein Plasma Constituents
Hypozincemia, hypoferremia, and hypercupremia
Decreased plasma retinol concentrations
Increased plasma glutathione concentrations

From Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J
Med. 1999;340:448–454. Copyright ? 1999 Massachusetts Medical Society. All rights reserved.
Traditionally, the phrase acute-phase response has been used to denote changes in plasma
concentrations of a number of secretory proteins derived from hepatocytes. Acute-phase proteins,
of which there are many ( Table 47–2 ),[108] exhibit either increased synthesis (positive
acute-phase proteins) or decreased synthesis (negative acute-phase proteins) during the
acute-phase response. IL-6 is the chief stimulator of the production of most acute-phase proteins.
Other pyrogenic cytokines, however, also influence the production of various subgroups of such
proteins.[108]
Table 47-2 -- Human Acute-Phase Proteins
Proteins Whose Plasma Concentrations Increase
Complement system
C3
C4
C5
C9
MAC
Factor B
C1 inhibitor
C4b-binding protein
Mannose-binding lectin
Coagulation and fibrinolytic system
Fibrinogen
Plasminogen
Tissue plasminogen activator
Urokinase
Protein S
Vitronectin
Plasminogen-activator inhibitor I
Kininogen
Antiproteases
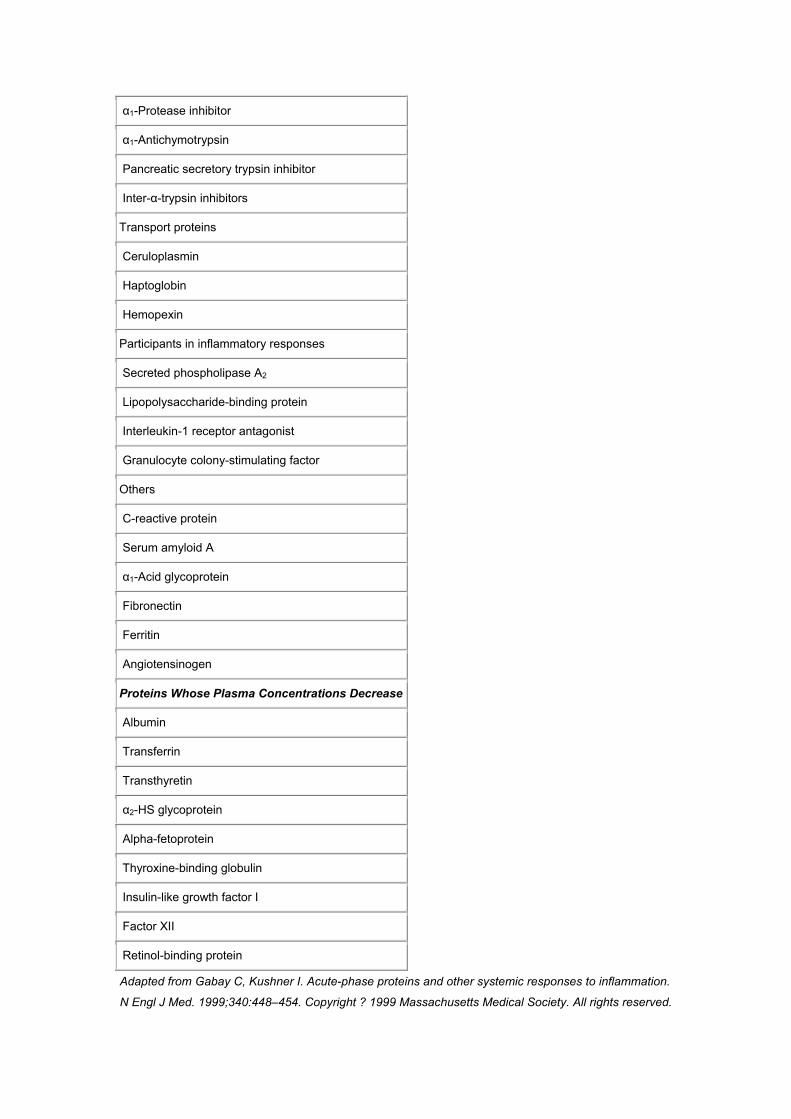
α1-Protease inhibitor
α1-Antichymotrypsin
Pancreatic secretory trypsin inhibitor
Inter-α-trypsin inhibitors
Transport proteins
Ceruloplasmin
Haptoglobin
Hemopexin
Participants in inflammatory responses
Secreted phospholipase A2
Lipopolysaccharide-binding protein
Interleukin-1 receptor antagonist
Granulocyte colony-stimulating factor
Others
C-reactive protein
Serum amyloid A
α1-Acid glycoprotein
Fibronectin
Ferritin
Angiotensinogen
Proteins Whose Plasma Concentrations Decrease
Albumin
Transferrin
Transthyretin
α2-HS glycoprotein
Alpha-fetoprotein
Thyroxine-binding globulin
Insulin-like growth factor I
Factor XII
Retinol-binding protein
Adapted from Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation.
N Engl J Med. 1999;340:448–454. Copyright ? 1999 Massachusetts Medical Society. All rights reserved.

Many of the acute-phase proteins are believed to modulate inflammation and tissue repair.[113] A
major function of C-reactive protein (CRP), for example, is presumed to involve binding of
phosphocholine on pathogenic microorganisms, as well as phospholipid constituents on damaged
or necrotic host cells. Through such binding, CRP might both activate the complement system and
promote phagocyte adherence, thereby initiating the process by which pathogenic microbes or
necrotic cells are cleared from the host. Such activities are most likely potentiated by CRP-induced
production of inflammatory cytokines[114] and tissue factor[115] by monocytes. Nevertheless, the
ultimate function of CRP is uncertain, in that several in vivo studies have shown it to have
anti-inflammatory properties.[116][117][118]
Another major human acute-phase protein, serum amyloid A, has been reported to potentiate
adhesiveness and chemotaxis of phagocytic cells and lymphocytes.[119] There is also evidence that
macrophages bear specific binding sites for serum amyloid A, that serum amyloid A–rich,
high-density lipoproteins mediate the transfer of cholesterol to macrophages at sites of
inflammation,[120] and that serum amyloid A enhances low-density lipoprotein oxidation in arterial
walls.[121]
Complement components, many of which are acute-phase reactants, induce pyrogenic cytokines
and PGE2; modulate chemotaxis, opsonization, vascular permeability, and vascular dilation; and
have cytotoxic effects as well.[108] Haptoglobin, hemopexin, and ceruloplasmin are all antioxidants.
It is, therefore, reasonable to assume that, like the antiproteases α1-antichymotrypsin and
C1-esterase inhibitor, they play important roles in modulating inflammation. However, the
functional capacity of such proteins is broad. There is also a growing literature concerned with the
acute-phase protein LPS-binding protein, which appears both to enhance and to neutralize the
biologic activity of LPS (through its interaction with the CD14 receptor on macrophages).[122]
Although closely associated with fever, the acute-phase response is not an invariable component
of the febrile response.[108] Some febrile patients (e.g., those with certain viral infections) have
normal blood levels of CRP. Moreover, patients with elevated blood levels of CRP are not always
febrile. The acute-phase response, like the febrile response, is a complex response consisting of
numerous integrated, though separately regulated, components. The particular components
expressed in response to a given disease process more than likely reflect the specific cytokines
induced by the disease.
ENDOGENOUS CRYOGENS
Hippocrates maintained that “heat is the immortal substance of life endowed with intelligence….
However, heat must also be refrigerated by respiration and kept within bounds if the source or
principle of life is to persist; for if refrigeration is not provided, the heat will consume itself.”[123]
Modern-day clinicians also generally subscribe to the notion that the febrile range has an upper
limit but do not agree on a precise temperature defining this limit.[57] The lack of a consensus in this
regard is understandable, because “body” temperature profiles exhibit considerable individual,
anatomic, and diurnal variability. For this reason, the upper limit of the febrile range cannot be
defined as a single temperature applicable to all body sites of all people at all times during the day.

Nevertheless, the febrile response is a regulated physiologic response, in which the temperature is
maintained within a specific range, the upper limit of which virtually never exceeds 41° C in adult
humans, regardless of the cause of the fever or the site at which the temperature measurements
are taken.[124] The physiologic necessity of this upper limit is supported by considerable
experimental data demonstrating adverse physiologic consequences of core temperatures of
greater than 41° to 42° C (107.6° F).[125]
The mechanisms regulating fever’s upper limit have yet to be fully elucidated. They could lie with
the intrinsic properties of the neurons themselves or involve the release of endogenous antipyretic
substances that antagonize the effects of pyrogens on thermosensitive neurons. With regard to the
former possibility, plots of the firing rates of neurons coordinating thermoregulatory responses and
heat production tend to converge at 42° C (107.6° F) ( Fig. 47–5 ).[125] At this temperature, the
sustained firing rates of warm-sensitive neurons reach their zenith and cannot be increased further
in response to higher temperatures. Similarly, the firing rates of cold-sensitive neurons reach their
nadir at 42° C (107.6° F) and cannot decrease further, even if the temperature continues to
increase. Thus, regardless of the pyrogen concentration, thermosensitive neurons appear to be
incapable of providing additional thermoregulatory signals once the temperature reaches 42° C
(107.6° F).
Figure 47-5 Model showing responses (A and B) of neuronal firing rates (FR) in the preoptic
region and anterior hypothalamus and whole-body metabolic heat production (C) during
changes in hypothalamic temperature (Th). Thermosensitivity is reflected by the slope of each
plot. The letters inside the cells indicate a warm-sensitive (w) neuron and a cold-sensitive (c)
neuron. With increases in Th, warmsensitive neurons raise their FRs, and heat production
decreases. Pyrogens inhibit (-) the FRs of warm-sensitive neurons, thereby resulting in

accelerated FRs of cold-sensitive neurons and increased heat production. The plots show FR
and heat production responses during normal conditions in the absence of pyrogens (N) and in
the presence of low concentrations (P1) and high concentrations (P2) of pyrogens. (From
Mackowiak PA, Boulant JA. Fever’s glass ceiling. Clin Infect Dis. 1996;22:525–536.)
These same thermosensitive neurons are influenced by a variety of endogenous substances, at
least some of which appear to function as endogenous cryogens.[125] One such substance is
arginine vasopressin. Studies from several laboratories employing a variety of animal models have
established that arginine vasopressin is present in the fibers and terminals of the ventral septal
area of the hypothalamus, is released into the ventral septal area during fever, reduces fever by its
action at type 1 vasopressin receptors when introduced into the ventral septal area, and, when
inhibited, prolongs fever.[126][127][128]
α-Melanocyte-stimulating hormone (α-MSH) is another neuropeptide exhibiting endogenous
antipyretic activity.[129] Unlike some of the other antipyretic peptides, α-MSH has not been identified
in fibers projecting into the dorsolateral septal area.[130] It does, nevertheless, reduce
pyrogen-induced fever when administered to experimental animals in doses below those affecting
the basal body temperature.[131][132][133][134][135] When given centrally, α-MSH is more than 25,000
times more potent as an antipyretic than acetaminophen.[129] Repeated central administration of
α-MSH does not induce tolerance to its antipyretic effect.[136] In addition, injection of anti-α-MSH
antiserum into the cerebral ventricles augments the febrile response of experimental animals to
IL-1.[137]
Glucocorticoids and their inducers (corticotropin-releasing hormone and corticotropin) inhibit the
synthesis of pyrogenic cytokines such as IL-6 and TNF-α.[138][139][140] Through such effects, they are
believed to exert inhibitory feedback on LPS-induced fever.[141] Lipocortin-1, a putative mediator of
glucocorticoid function, has also been shown to inhibit the pyrogenic actions of IL-1 and IFN.[142]
Injection of corticotropin-releasing hormone (CRH) into the third ventricle of experimental animals
produces similar antipyretic effects.[143]
Thyrotropin-releasing hormone,[144] gastric-inhibitory peptide,[145] neuropeptide Y,[146] nitric
oxide,[147] carbon monoxide,[148] and bombesin[149] likewise exhibit cryogenic properties under
certain conditions. Of these, bombesin has exhibited the highest potency, in that it consistently
produces hypothermia associated with changes in heat dissipation and heat production when
injected into the preoptic area or anterior hypothalamus of conscious goats and rabbits.[149][150][151]
Bombesin is believed to exert its hypothermic effect by decreasing the sensitivity of warm-sensitive
neurons.[151]
Pyrogenic cytokines, the mediators of the febrile response, might themselves have a role in
determining fever’s upper limit. There is, for instance, experimental evidence indicating that under
certain conditions (e.g., with intracerebral injection of recombinant human TNF-α in Zucker rats),

TNF-α acts to lower, rather than to raise, body temperature,[152][153] although only in the presence of
LPS. Thus, it is possible that at certain concentrations or in the appropriate physiologic milieu,
pyrogenic cytokines function paradoxically as endogenous cryogens.
A growing body of literature indicates that the release of pyrogenic cytokines such as IL-1 is
followed by increased shedding of soluble receptors for such cytokines, which function as
endogenous scavengers of these pyrogens.[154] In the case of IL-1, a 22- to 25-kDa molecule
identified in supernates of human monocytes blocks binding of IL-1 to its receptors.[155] The IL-1
receptor antagonist is structurally related to IL-1α and IL-1β[156] and binds to both type I and type II
receptors on various target cells without inducing a specific biologic response.[157][158] Shedding of
soluble receptors of TNF-α that bind to circulating TNF-α and thereby inhibit binding to
cell-associated receptors has also been described.[159][160][161][162][163] The precise biologic function
of such circulating receptor antagonists and soluble receptors is not known. However, it is possible
that one function is to serve as a natural braking system for the febrile response.
RISK-TO-BENEFIT CONSIDERATIONS
Questions concerning fever’s risk-to-benefit quotient have generated considerable controversy.[164]
The controversy arises because of data indicating both potentiating and inhibitory effects of the
response on resistance to infection. As a result, there is as yet no consensus as to the appropriate
clinical situations (if any) in which fever or its mediators should be suppressed.
Data illustrating fever’s beneficial effects originate from several sources. Studies of the phylogeny
of fever have shown the response to be widespread within the animal kingdom.[165] With few
exceptions, mammals, reptiles, amphibians, and fish, as well as several invertebrate species, have
been shown to elevate the core temperature in response to a challenge with microorganisms or
other known pyrogens ( Fig. 47–6 ). It has been assumed, although not established conclusively,
that such elevations in temperature are the poikilothermic corollary of fever. The prevalence of
such “febrile responses” has been offered as some of the strongest evidence that fever is an
adaptive response, on the basis of the argument that the metabolically expensive increase in body
temperature that accompanies the febrile response would not have evolved and been so faithfully
preserved in the animal kingdom unless fever had some net benefit to the host.
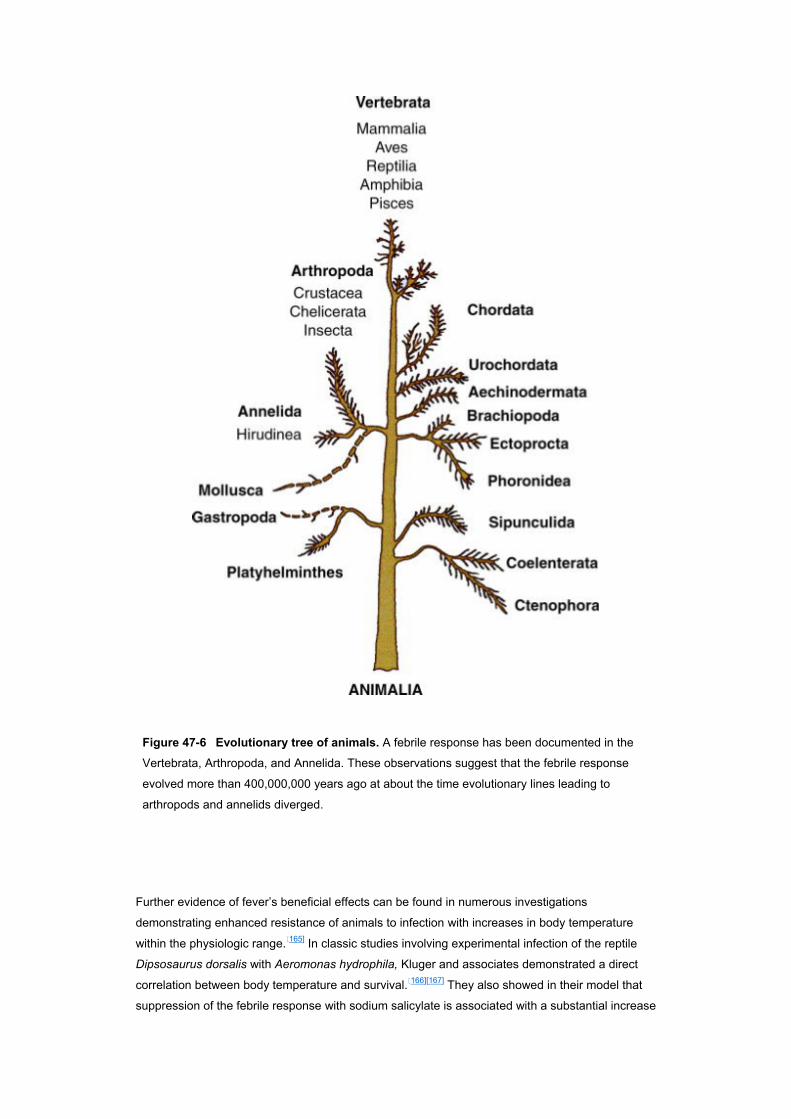
Figure 47-6 Evolutionary tree of animals. A febrile response has been documented in the
Vertebrata, Arthropoda, and Annelida. These observations suggest that the febrile response
evolved more than 400,000,000 years ago at about the time evolutionary lines leading to
arthropods and annelids diverged.
Further evidence of fever’s beneficial effects can be found in numerous investigations
demonstrating enhanced resistance of animals to infection with increases in body temperature
within the physiologic range.[165] In classic studies involving experimental infection of the reptile
Dipsosaurus dorsalis with Aeromonas hydrophila, Kluger and associates demonstrated a direct
correlation between body temperature and survival.[166][167] They also showed in their model that
suppression of the febrile response with sodium salicylate is associated with a substantial increase

in mortality.[167] Covert and Reynolds corroborated these findings in an experimental model
involving goldfish.[168]
In mammalian experimental models, increasing the body temperature by artificial means has been
reported to enhance the resistance of mice to herpes simplex virus,[169] poliovirus,[170] Coxsackie B
virus,[171] rabies virus,[172] and Cryptococcus neoformans[173] but to decrease resistance to
Streptococcus pneumoniae. [174] Increased resistance of rabbits to S. pneumoniae[175] and C.
neoformans,[176] dogs to herpesvirus,[177] piglets to gastroenteritis virus,[178] and ferrets to influenza
virus[179] has also been observed after the induction of artificial fever. Unfortunately, because
raising the body temperature by artificial means does not duplicate the physiologic alterations that
occur during fever in homeotherms (and, indeed, entails a number of opposite physiologic
responses[180]), data obtained using mammalian experimental models must be interpreted with
caution when used to understand the febrile response.
Clinical data supporting an adaptive role for fever have accumulated slowly. Like animal data,
clinical data include evidence of both beneficial effects of fever and adverse effects of antipyretics
on the outcome of infections. In a retrospective analysis of 218 patients with gram-negative
bacteremia, Bryant and associates reported a positive correlation between maximal temperature
on the day bacteremia was diagnosed and survival.[181] A similar relationship has been observed in
patients with polymicrobial sepsis and mild (but not severe) underlying diseases.[182] In an
examination of factors influencing the prognosis of spontaneous bacterial peritonitis, Weinstein and
co-workers identified a positive correlation between a temperature reading of greater than 38° C
(100.4° F) and survival.[183]
It has been reported that children with chickenpox who are treated with acetaminophen have a
longer time to total crusting of lesions than placebo-treated controls.[184] Stanley and colleagues
have reported that adults infected with rhinovirus exhibit more nasal viral shedding when they
receive aspirin than when given placebo.[185] Furthermore, Graham and colleagues have reported
a trend toward a longer duration of rhinovirus shedding in association with antipyretic therapy and
have shown that the use of aspirin or acetaminophen is associated with suppression of the serum
neutralizing antibody response and with increased nasal symptoms and signs.[186] A more recent,
retrospective, observational analysis of studies of human volunteers infected with influenza A
found a relationship between antipyretic therapy and prolonged illness.[187] These data, like those
reviewed in the preceding paragraph, are subject to several interpretations and do not prove a
causal relationship between fever and improved prognosis during infection. Nevertheless, they are
consistent with such a relationship and when considered in concert with the phylogeny of the
febrile response and the animal data summarized earlier, constitute strong circumstantial evidence
that fever is an adaptive response in most situations.
Whereas many of the foregoing investigations examined the relationship between the elevation of
the core temperature and the outcome of infection, others have considered the endogenous
mediators of the febrile response. In such studies, all of the principal pyrogenic cytokines have
been shown to have immune-potentiating capabilities, which might theoretically enhance
resistance to infection.[93][188] In vitro and in vivo investigations of these cytokines have provided

evidence of a protective effect of IFN, TNF-α, or IL-1, or all of these, against Plasmodium,[189][190][191]
Toxoplasma gondii,[192] Leishmania major,[193] Trypanosoma cruzi,[194] and Cryptosporidium.[195]
Several reports have also shown enhancement of resistance to viral[196][197][198] and bacterial
infections[199][200] by pyrogenic cytokines. Treatment of normal and granulocytopenic animals with
IL-1 has been shown to prevent death in some gram-positive and gram-negative bacterial
infections.[200] However, IL-1 is effective only if administered an appreciable time (e.g., 24 hours)
before the initiation of infections having rapidly fatal courses. In less acute infections, IL-1
administration can be delayed until shortly after the infectious challenge. Such observations
suggest that those physiologic effects of the febrile response that enhance resistance to infection
might be limited to localized infections or systemic infections of only mild to moderate severity.
The febrile response’s potential for harm is reflected in a recent flurry of reports suggesting that
IL-1, TNF-α, IL-6, and IFN mediate the physiologic abnormalities of certain infections. Although
proof of an adverse effect of fever on the clinical outcome of these infections has yet to be
established, the implication is that if pyrogenic cytokines contribute to the pathophysiologic burden
of infections, both the mediators themselves and the febrile response are potentially deleterious.
The most persuasive evidence in this regard derives from studies of gram-negative bacterial
sepsis.[201] It has long been suspected that bacterial LPS is involved in the pathophysiology of the
syndrome. Purified LPS induces a spectrum of physiologic abnormalities that are similar to those
occurring in patients with gram-negative bacterial sepsis. In experimental animals, challenge with
LPS causes TNF-α and IL-1 to be released into the blood stream coincident with the appearance of
signs of sepsis.[202] Furthermore, patients with the septic syndrome have detectable levels of
circulatory TNF-α, IL-1, and IL-6 independent of culture-documented infection, and such levels
correlate inversely with survival.[203] IL-1, alone or in combination with other cytokines, induces
many of the same physiologic abnormalities (e.g., fever, hypoglycemia, shock, and death) seen
after the administration of purified LPS.[204] In a murine experimental model for septic shock, IFN
administered before or as long as 4 hours after LPS challenge increases mortality, whereas
pretreatment with anti-IFN antibody significantly reduces mortality.[205] In several investigations, the
adverse effects of gram-negative bacterial sepsis or LPS injections, or both, have been attenuated
by pretreating experimental animals with IL-1 antagonists[206][207] and monoclonal antibodies
directed against TNF-α.[208][209] Furthermore, animals rendered tolerant to TNF-α by repeated
injections of the recombinant cytokine are protected against the hypotension, hypothermia, and
lethality of gram-negative bacterial sepsis.[210]
The theory derived from these observations, that death from sepsis is the consequence of
cytokine-mediated overstimulation of the immune system, unfortunately correlates only loosely with
the clinical picture in humans, most likely because the studies cited here used large doses of
endotoxin or bacteria that induced levels of circulating pyrogenic cytokines exponentially higher
than those detected in patients with sepsis.[211] Thus, the “cytokine storm” created in such animals
most likely has only limited relevance for human sepsis. This perhaps explains why in clinical trials,
inhibition of pyrogenic cytokines in septic patients has had only modest success, improving
outcome in patients with a high risk for death but not those with a low risk.[212]
ANTIPYRETIC THERAPY

Although clinicians have long had at their disposal effective means of lowering the core
temperature in febrile patients, the actual benefit of such reductions in temperature is still uncertain.
Moreover, it has yet to be shown in humans that increases in the core temperature encountered
during fever are harmful per se. Certainly, during the course of heat stroke and other forms of
hyperthermia, the core temperature can, and frequently does, rise to levels that are inherently
harmful.[213] However, as discussed previously, such levels are almost never reached during
fever’s regulated rise in temperature, which probably never exceeds 41° C (105.8° F) in
humans.[125] Nevertheless, whereas healthy volunteers have been reported to withstand core
temperatures of 42° C (107.6° F) for periods of as long as 4 hours without apparent ill effect,[214] the
possibility remains that in certain patients, even the relatively modest increases in core
temperature encountered during fever are deleterious and should therefore be suppressed.
One such category of patients includes children—primarily between the ages of 3 months and 5
years. In such children, seizures have been reported to occur during episodes of fever at a
frequency of as high as 14% in selected populations.[215] Although most children with febrile
seizures have temperatures of 39° C (102.2° F) or more at the time of their seizure,[216] many
tolerate even higher fevers at later dates without convulsing.[217] Unfortunately, antipyretic therapy
has not been shown to protect against recurrences of febrile seizures in the few controlled trials
conducted thus far (see later).[218]
It has also been suggested that patients with underlying cardiovascular or pulmonary disorders
might be especially susceptible to the adverse effects of fever, because of metabolic demands
imposed by the elevated temperature.[219] Such demands are particularly high during the chill
phase if shivering is present, as evidenced by increases in the sympathetic tone,[180] oxygen
consumption, respiratory minute volume, and respiratory quotient.[220] As a result of the associated
increase in metabolic demand, the chill phase of fever might be expected to add to the burden of
cardiac or pulmonary disorders. Although this possibility has been offered as justification for
antipyretic therapy in patients with these disorders, the risk-to-benefit ratio of such therapy has yet
to be determined.
Antipyretic therapy might also be justified, at least in theory, if fever’s metabolic cost exceeded its
physiologic benefit, if the treatment provided symptomatic relief without adversely affecting the
course of the febrile illness, or if the toxicologic costs (side effects) of the antipyretic regimen were
appreciably lower than its beneficial effects. Unfortunately, although clinicians have long argued
the validity of each of these propositions as justification for antipyretic therapy, few scientific data
exist to support any of these arguments.
Antipyretic drugs can be grouped into three general categories on the basis of their mechanisms of
action. These include corticosteroids, aspirin and the other nonsteroidal anti-inflammatory drugs
(NSAIDs), and acetaminophen. Each exerts its effects at different points in the febrile response
pathway.
Although not generally used for antipyresis, corticosteroids suppress fever through both direct and
indirect mechanisms. They block the transcription of pyrogenic cytokines and inducible
cyclooxygenase via interactions involving the glucocorticoid receptor.[82][221] They downregulate the

synthesis of cytokine receptors,[211] and by inducing lipocortin-1, they secondarily inhibit the activity
of phospholipase A2, a critical enzyme in the prostaglandin synthetic pathway.[221]
Acetaminophen and aspirin and the other NSAIDs all block the conversion of arachidonic acid to
prostaglandins such as PGE2 by inhibiting cyclooxygenase (COX, also known as prostaglandin GH
synthetase).[221] This effect is thought to be critical to their antipyretic activity, in that production of
PGE2 at key sites within the hypothalamus is widely regarded as a crucial step in the process by
which the physiologic cascade responsible for raising the core temperature during the febrile
response is activated (see Fig. 47–4 ).[222] Cyclooxygenase has at least two distinct isoforms: a
constitutive isoform, COX-1, and a predominantly inducible isoform, COX-2, which is undetectable
in most resting cells. A third isoform, COX-3, has recently been identified.[223] It is a COX-1 variant
selectively inhibited by antipyretic drugs such as acetaminophen, phenacetin, antipyrine, and
dipyrone. Although it has been suggested that COX-3 could be the primary site of action of these
drugs, their inhibitory effect is both nonspecific and weak.[224] COX-1 initiates production of
prostacyclin, which has both antithrombogenic and cytoprotective properties, whereas COX-2 is a
principal mediator of fever and the inflammatory response. The anti-inflammatory action of NSAIDs
is believed to result from inhibition of COX-2, and the unwanted adverse effects, such as gastric
irritation, from inhibition of COX-1.[225]
The structure and catalytic activity of the COX-1 and -2 isoforms are similar, with approximately
600 amino acids, of which 63% are in identical sequence, and active sites located at the apex of a
long, narrow, hydrophobic channel. The amino acids forming the channel, as well as catalytic sites
and neighboring residues, are identical in the two isoforms with two exceptions. Valine in COX-1 is
substituted for isoleucine at positions 434 and 523 in COX-2. Aspirin acetylates serine 530 of both
isoforms. In COX-1, this blocks access of arachidonic acid to the catalytic site, causing irreversible
inhibition of the enzyme. Because of the wider hydrophobic channel of COX-2, access of
arachidonic acid to the active site persists after acetylation of serine 530 by aspirin.[225]
Physical Methods of Antipyresis
A variety of physical techniques are used to cool febrile patients. These include sponging with
various solutions (e.g., tepid water or alcohol), the application of ice packs or cooling blankets, and
exposure to circulating fans (most often in conjunction with sponging). With the latter method, helox
(80% helium, 20% oxygen) has been shown to be superior to air in lowering core temperature, at
least in experimental animals, because of the greater thermal conductivity of helium compared with
that of nitrogen.[226] In contrast to antipyretic drugs, external cooling lowers the temperature of
febrile patients by overwhelming effector mechanisms that have been evoked by an elevated
thermoregulatory setpoint, rather than by lowering that setpoint. Therefore, unless concomitant
antipyretic agents are used, or shivering is inhibited by other pharmacologic means, external
cooling is vigorously opposed in the febrile patient by thermoregulatory mechanisms endeavoring
to maintain the elevated body temperature.
Physical methods of antipyresis promote heat loss by conduction, convection, and evaporation.
Evaporative methods have traditionally been touted as the most effective physical means of
promoting heat loss in febrile patients, because such methods are deemed to be least likely to

induce shivering.[227] However, carefully designed comparative trials have not yet established any
one physical method of antipyresis as superior.
Direct comparisons of pharmacologic and physical methods of antipyresis are, likewise, all but
nonexistent. In the only extant controlled study, Wenzel and Werner reported that salicylates
reduced the second phase of endotoxin-induced fever in rabbits, whereas abdominal cooling
increased heat production and did not lower the core temperature unless the animals were
simultaneously exposed to environmental hyperthermia.[228] Neither antipyretic modality abolished
the initial febrile response.
The few available clinical studies of the efficacy of physical methods of antipyresis have differed in
their conclusions. Interpretation of the results of these studies has been difficult, because
pharmacologic agents have almost invariably been administered concomitantly with external
cooling. Steele and co-workers found acetaminophen (in age-adjusted dosages ranging from 80 to
320 mg) and sponging to be equally effective in lowering fever in children admitted to a pediatric
hospital because of fever.[229] However, when combined, the two modalities produced more rapid
cooling than either alone. By contrast, Newman found that tepid-water sponging in combination
with acetaminophen (5 to 10 mg/kg) was no more effective than acetaminophen alone in lowering
the temperature of febrile children.[230] O’Donnell and colleagues have concluded that in adults,
although hypothermia blanket therapy adds little to the action of pharmacologic agents in lowering
temperature, it induces wider temperature fluctuations and more episodes of rebound
hyperthermia.[227]
Diagnostic Considerations
Numerous investigators have observed a direct correlation between the height of fevers and the
rate of serious bacterial infections in children, with the maximal incidence of such infections at
temperatures in excess of 40° C (104° F).[231][232][233][234] It has also been suggested that the
response of a fever to antipyretic therapy might have diagnostic implications, in that a drop in
temperature or improvement in the appearance of a febrile child, or both, generally indicates that
the fever is not the result of a serious illness.[235] This conclusion, however, is not supported by
numerous investigations comparing the temperature response of bacteremic and nonbacteremic
infections to antipyretic therapy in children.[236][237][238][239][240][241]
Several studies have suggested that an antipyretic response to NSAIDs can distinguish fevers of
infectious origin from those due to cancer by virtue of the fact that the latter fevers are more readily
suppressed by such agents. Naproxen was the first such agent to be studied in this regard.[242]
Subsequent randomized comparisons have shown naproxen, indomethacin, and diclofenac to be
equally effective in inhibiting cancer-induced fever,[243] although the sensitivity and specificity of the
“naproxen test” for differentiating neoplastic from infectious fevers are not yet known. Moreover,
there is no physiologic rationale to explain why NSAIDs might be more effective in reducing fever
due to cancer than that due to infection.
Benefits versus Risks

Two critical assumptions are made when prescribing antipyretic therapy. One is that fever is, at
least in part, noxious, and the other is that suppressing fever will reduce if not eliminate fever’s
noxious effects. Neither assumption has been validated experimentally. In fact, there is
considerable evidence that fever is an important defense mechanism that contributes to the host’s
ability to resist infection.[244] However, even if fever (or its mediators) does adversely affect the
course of certain disorders, as for example bacterial sepsis,[164] it does not necessarily follow that
inhibiting fever using current modes of antipyretic therapy will obviate this effect, especially if such
therapy has intrinsic toxicity of its own.
One of the reasons commonly given to justify suppressing fever is that the metabolic cost of fever
exceeds its clinical benefit. In fact, the metabolic cost of fever is substantial, especially during the
chill phase of the response with its shivering-induced increase in metabolic rate,
norepinephrine-mediated peripheral vasoconstriction, and increased arterial blood pressure.[180]
Because of the potential adverse consequences of these metabolic effects on cardiovascular and
pulmonary function, fever has been attacked with particular vigor in patients with underlying
cardiovascular and pulmonary diseases.[245] Although antipyretic therapy has theoretical merit in
this regard (if it does not induce shivering[246]), the detrimental effects of fever and the salutary
effects of antipyretic therapy have yet to be critically evaluated.
External cooling, which is widely used in critically ill patients to suppress fevers unresponsive to
antipyretic drugs, has been shown to decrease oxygen consumption by as much as 20% if
shivering is prevented by therapeutic paralysis.[246] If shivering is not inhibited, external cooling
causes a rise, rather than a fall, in oxygen consumption.[246] Perhaps more important to febrile
patients with underlying cardiovascular disease, external cooling has the capacity to cause
vasospasm of diseased coronary arteries by inducing a cold pressor response.[247][248] For all these
reasons, it has been suggested that a more rational strategy for treating fevers unresponsive to
antipyretic drugs is to warm rather than to cool selected skin surfaces, thereby reducing the
vasoconstriction and shivering thresholds dictated by the elevated hypothalamic thermal setpoint
and, in turn, effecting a decrease in the core temperature.[249]
Unfortunately, certain antipyretic drugs also appear to cause coronary vasoconstriction in patients
with coronary artery disease. Friedman and associates observed significant increases in the mean
arterial pressure, coronary vascular resistance, and myocardial arteriovenous oxygen difference
after intravenous indomethacin (0.5 mg/kg) in such patients.[250] Coronary blood flow decreased
simultaneously from 181 ± 29 to 111 ± 14 mL/min (P < .05). Thus, in this investigation, myocardial
oxygen demand increased in the face of a fall in coronary blood flow after indomethacin
administration. The authors believe that indomethacin’s vasoconstrictor effect most likely derives
from to its capacity to block the synthesis of vasodilatory prostaglandins. Perhaps even more
disturbing are recent reports suggesting that compared to other nonsteroidal anti-inflammatory
drugs, COX-2–selective NSAIDS seem to increase the risk for cardiovascular thrombotic events in
patients not taking aspirin.[251]
Antipyretic therapy is also commonly administered to enhance patient comfort.[245] General
experience with antipyretic drugs, which are for the most part also analgesic agents, seems to
support this contention. However, carefully controlled efficacy studies have not yet established the

validity of this contention. Moreover, the relative cost of such symptomatic relief, in terms of drug
toxicity and adverse effects of antipyretic agents on the course of the illness responsible for the
fever, have never been determined. The importance of such information is underscored by reports
that acetaminophen prolongs the time to crusting of lesions in children with chickenpox,[184] that
both acetaminophen and aspirin increase viral shedding and nasal signs and symptoms while
suppressing the serum neutralizing antibody response in adults with rhinovirus infections,[185][186]
and that antipyretic drugs might prolong the course of influenza A infections.[187]
Antipyretic therapy is also occasionally given to prevent febrile seizures in children, and to prevent
or to reverse fever-induced mental dysfunction in frail older patients. Beisel and co-workers have
shown that aspirin (in combination with propoxyphene) ameliorates fever-induced decrements in
mental work performance in young volunteers infected with sand fly fever virus, even in the face of
only partial relief of either the fever or other symptoms of the illness.[252] In view of these
observations, antipyretic therapy might be expected to have a beneficial effect on fever-induced
mental dysfunction in frail older patients. However, studies designed to test this hypothesis have
not yet been reported.
Unfortunately, antipyretic therapy does not appear to be effective in preventing febrile seizures.[218]
Camfield and colleagues conducted a randomized double-blind study comparing single-daily-dose
phenobarbital plus antipyretic instruction to placebo plus antipyretic instruction to prevent recurrent
seizure after an initial simple febrile seizure.[253] In children treated with both phenobarbital and
antipyretics, the febrile seizure recurrence rate was 5%, whereas in those given placebo with
antipyretics, the rate was 25%, suggesting that a single daily 5 mg/kg dose of phenobarbital is
more effective than counseling parents about antipyretic therapy in preventing recurrent febrile
seizures. More recently, acetaminophen has been given to children with fever as prophylaxis
against febrile seizure recurrences. Whether given in moderate dosage (10 mg/kg dose four times
a day)[254] or in relatively high doses (15 to 20 mg/kg dose every 4 hours),[255] acetaminophen failed
to reduce the rate of febrile seizure recurrence.
Finally, there has been mounting interest in the use of certain antipyretic drugs to modulate the
activity of pyrogenic cytokines during bacterial sepsis.[256] In some animal models of sepsis,
antipyretic drugs that inhibit cyclooxygenase confer protection when given soon after bacterial
challenge, presumably by blunting the adverse effects of TNF-α and IL-1. In a large clinical trial,
Bernard and associates reported that 48 hours of intravenous therapy with the cyclooxygenase
inhibitor ibuprofen lowered the core temperature, heart rate, oxygen consumption, and lactic acid
blood levels but did not decrease the incidence of organ failure or mortality at 30 days.[257] In a
more recent retrospective analysis of sepsis trials, Eichacker and co-workers could find evidence
of a beneficial effect of antipyretic agents only in septic patients with a high risk for death (see
above).[212] Thus, in spite of promising results obtained in some experimental models, antipyretic
agents have been shown to be of only limited value clinically in the treatment of bacterial sepsis.
Indications
Although clinicians have resorted to various forms of antipyretic therapy since time immemorial,
there is a dearth of scientific data concerning the actual benefits and relative risks of such
treatments.[258] Nevertheless, several tentative conclusions regarding antipyretic therapy seem

warranted in light of the limited data available. It is clear, for instance, that short courses of
approved doses of standard antipyretic drugs carry a low risk for toxicity. Most of these drugs have
analgesic as well as antipyretic properties. Therefore, if not otherwise contraindicated (e.g., aspirin
in young children because of the risk for Reye’s syndrome), such drugs can be prescribed to
provide symptomatic relief in febrile patients, to reduce the metabolic demands of fever in patients
with underlying cardiovascular and pulmonary disorders, and, possibly, to prevent or alleviate
fever-induced mental dysfunction in older patients. To minimize antipyretic-induced fluctuations in
temperature (as well as the risk for recurrent shivering with its associated increased metabolic
demands), antipyretic agents should be administered to febrile patients at regular intervals that
preclude abrupt recurrences of fever, rather than as needed for temperatures above some arbitrary
level. Whenever such medications are prescribed, it should also be recognized that each carries its
own risk for toxicity and might prolong the course of the illness responsible for the fever while
reducing the intensity of its symptoms.
In view of the capacity of external cooling measures to induce a cold pressor response, it is
questionable whether this form of antipyretic therapy should ever be administered to febrile
patients (much less to intensive care unit patients for whom it is so frequently prescribed). If
external cooling is used to treat fever, care must be taken to prevent shivering, because of its
associated increased oxygen consumption. Unfortunately, even if shivering is prevented, there is
no guarantee that a cold pressor response will be averted. In view of indomethacin’s capacity to
cause coronary vasoconstriction in patients with coronary artery disease and the possible
increased risk for cardiovascular thrombotic events associated with COX-2–selective NSAIDs
should be used cautiously to suppress fever in such patients.
(From:
http://www.mdconsult.com/das/book/body/110858782-4/0/1259/376.html?tocnode=51377509&fromURL
=376.html#4-u1.0-B0-443-06643-4..50050-7_1905)


















![[Micro] pathogenesis](https://img.pdfslide.net/doc/110x75/55d6fc34bb61eb0d2b8b47a6/micro-pathogenesis-55d98896d0eb8.jpg)










