Embed Size (px)
Citation preview

222 nm Photo-induced radical reactions in silazanes. A combined
laser photolysis, EPR, GC-MS and QC Studyw
Wolfgang Knolle,*a Luise Wennrich,a Sergej Naumov,a Konstanze Czihal,a
Lutz Prager,aDaniel Decker
band Michael R. Buchmeiser
c
Received 10th September 2009, Accepted 15th December 2009
First published as an Advance Article on the web 19th January 2010
DOI: 10.1039/b918814b
The initiation mechanism of the VUV-induced conversion of polyorganosilazanes into
methyl–Si–O–Si networks was studied by means of model disilazane compounds. A combined
experimental approach was chosen to determine the primary radicals and their properties
(lifetimes, spectra) as well as the major final products. It was verified that both Si–N and Si–CH3
cleavage occur in the condensed phase, the former with higher yield. The lifetime of the primary
Si- and N-centred radicals in de-oxygenated n-hexane solution is less than r10 ms. N-centred
radicals transform into amines by H abstraction, the availability of weakly bonded H as in the
case of tetramethyldisilazane accelerates the reaction considerably. In rigid matrix (frozen
solutions) �CH3, silyl radicals and methylene radicals �CH2R are trapped. In the presence
of oxygen, peroxyl radicals are formed and serve as precursors of the subsequent oxidative
conversion. Product analysis by GC-MS reveals linear R–(Si–O)n– chains rather than branched
compounds as the initial products of the oxidative conversion of tetramethyldisilazane.
It was shown that reactive silylene intermediates do not play a role in the conversion process.
Quantum chemical calculations assist in the interpretation.
Introduction
Polysilazanes are widely investigated and practically used as
precursors for SiOx gas barrier layers in packaging industry,1–3
for flexible solar cells,4,5 for applications in electronics,6 for
vacuum-insulated panels,7 and for anticorrosive protecting
coatings.8,9 Especially the inorganic oligomer perhydropoly-
silazane (PHPS, Fig. 1A) consisting of –SiH2–NH– repeat
units has been reported as a precursor for dense and stable
SiOx networks.10–14 The most conventional pathway for the
realization of such SiOx networks is the hydrolysis of the
Si–NH bonds and the subsequent condensation of the generated
silanols forming Si–O–Si bonds.15,16 At room temperature,
this process proceeds slowly. With the aim to avoid the slow
condensation process, alternatives using UV-irradiation as the
initiating step have been applied.17–20 Employing such a
technology, improved barrier properties on polymer foils as
characterized by lower oxygen and water vapour transmission
rates (OTR and WVTR, respectively) could be achieved in a
roll-to-roll process, which was also found to be suitable for
industrial applications.19 Some limitations on the application
of such coatings as the brittleness of the formed SiOx layers,
their thermoelastic properties (elongation at break, stiffness)
and the hydrophilic character of the surface can be overcome
by the use of organosilazanes, i.e. the incorporation of methyl
groups into the Si–NH–Si network.19 For these purposes,
the vacuum-UV (VUV, l o 200 nm) induced conversion
of polyorganosilazanes like poly(1,1-dimethylsilazane-co-1-
methylsilazane) (P(DMS-co-MS)) (Fig. 1B) into methyl–Si–O–Si
networks with enhanced barrier properties was studied.21
First-order kinetics were found by FTIR in the time scale of
some 10 ms for the photolytically induced decomposition
of the methyl–Si–NH–Si network, the subsequent formation
of the methyl–Si–O–Si network and the concomitant degradation
of the Si–CH3 bond. The release of ammonia and methane
accompanied the conversion process.
Thus it was concluded that the excitation of polysilazanes
leads to the direct cleavage of the Si–N bond19,21 and, in case
Fig. 1 Structures of PHPS (A), P(DMS-co-MS) (B), TMDSz (C),
HMDSz (D), and 7MDSz (E).
a Leibniz-Institut fur Oberflachenmodifizierung, Permoserstraße 15,D-04318 Leipzig, Germany. E-mail: [email protected];Fax: +49 341 2352584; Tel: +49 341 2353607
bClariant Produkte (Deutschland) GmbH, Am Unisys-Park 1,D-65843 Sulzbach a. T., Germany
c Institut fur Polymerchemie, Universitat Stuttgart, Pfaffenwaldring 55,D-70550 Stuttgart, Germanyw Electronic supplementary information (ESI) available: UV-VISspectra of silazanes, EPR spectra of intermediates between 77 and130 K, quantum chemical calculations on probable radical structures:heat of formation, hfs coupling constants and UV transitions. SeeDOI: 10.1039/b918814b
2380 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
BView Online / Journal Homepage / Table of Contents for this issue

of polyorganosilazanes, also of Si–CH3 bonds.21 In contrast to
the earlier work of Naganuma,18 who reported an essential
effect on the oxidative conversion of PHPS due to oxygen
radicals O(1D) and/or ozone (O3), both formed when
oxygen in the surrounding gas absorbs high energy photons
(172 nm, [O2] up to 20%), such processes can not occur
in the present case (222 nm photons, low O2 concentration).
It should be noted that Si–N and Si–CH3 cleavages were
also concluded from product analysis after high-power laser
irradiation of gaseous tetra- and hexamethyldisilazanes.22,23
Therefore this work focuses on the deeper elucidation of the
initiation mechanism of the VUV-induced conversion by
investigating model compounds (Fig. 1). Special attention is
devoted to the determination of the primary species and their
properties (lifetimes, spectra) and to the first reaction steps
following the absorption of photons, thus verifying the
mechanism proposed in our recent paper.21
A combined experimental approach was chosen for the
direct study of fast processes (kinetic measurements by laser
flash photolysis), to investigate intermediate species by
trapping (low temperature matrix-EPR), and to identify the
final products by GC-MS following steady state photolysis.
Quantum chemical calculations support the assignment of UV
and EPR spectra and are used to distinguish between different
reaction pathways.
Experimental
Materials
1,1,1,3,3,3-Hexamethyldisilazane (HMDSz), 1,1,3,3-tetra-
methyldisilazane (TMDSz) and heptamethyldisilazane
(7MDSz) were purchased from ABCR (Karlsruhe, Germany).
Acetonitrile (Ultra gradient HPLC grade, J.T. Baker) and
n-hexane (Uvasol, Merck) were used as received. If necessary,
n-hexane was dried by distillation from sodium/benzophenone
and stored under argon. Triethylsilane (Merck) was used as
received.
Low-temperature EPR spectroscopy. Silazanes were
dissolved in acetonitrile or n-hexane at molar solute ratios
typically between about 1 : 20 to 1 : 1000 (approx.
2 � 10�2–1 mol dm�3 in acetonitrile and 7 � 10�3–4 �10�1 mol dm�3 in hexane) and carefully degassed by freeze-
thaw technique. The solutions were irradiated at 77 K in liquid
nitrogen with 222 nm light of a KrCl* excimer lamp (intensity
at sample position 10 mW cm�2) in intervals between 2 and
180 s and transferred to the EPR spectrometer. The first
spectrum was taken as soon as possible (within about 2.5 min)
after irradiation.
Some irradiation experiments were performed directly in the
cavity of the spectrometer at 100 K with a low-pressure
mercury lamp (HgLP) of low intensity (185 and 254 nm
photons). Deuterated acetonitrile ACN-d3 was used as a
solvent in order to verify that the methyl radicals formed in
dilute solution are not the result of a reaction with the solvent.
All measurements were performed using a Bruker ELEXSYS
E500 spectrometer (9.5 GHz, 100 kHz modulation) equipped
with either a finger Dewar (77 K) or a variable temperature
control unit (ER 4121 VT, at Z 95 K). Spectra were recorded
at a microwave power of 0.1 mW and a modulation amplitude
up to 0.1 mT. Isotropic and anisotropic spectra simulations
were performed using theWinSim24 and SimFonia (BRUKERr)
software, respectively.
Laser flash photolysis experiments. The laser photolysis
set-up comprised of a 222 nm KrCl*-excimer laser (RDC-
EXC-100, Radiant Dyes, pulse width B15 ns, pulse energy up
to 10 mJ) as excitation source and a pulsed xenon short-arc
lamp (XBO 1000, Osram; power supply LP-1000, lamp pulser
MSP 05, both Muller-Elektronik, Moosinning) supplying the
analyzing light. The transient recording electronics, consisting
of a photomultiplier (R928, Hamamatsu, operated at 850 V),
a power supply (PS310, Stanford Research Systems) and a
500 MHz, 2.5 GS s�1 digitizing storage oscilloscope
(TD5034B, Tektronix) guarantee for a time resolution within
the limits set by the excitation pulse. Further details have been
published elsewhere.25,26
All experiments have been carried out in flow-through
cuvettes with 5 � 3 mm2 cross-section. The concentrations
of the solutions were adjusted to have ground state absorbances
of less than 0.3 at the excitation wavelength in order to
provide acceptable absorption conditions. Oxygen-free
solutions were prepared by purging a proper amount of
solvent for 10 min with N2 prior to adding the solute
followed by subsequent purging for 10 min. In case a certain
concentration of oxygen in the solution was to be used,
only the second purging step was performed with the
N2/O2 gas mixture. Purging was sustained during the whole
experiment.
GC-MS analysis of the irradiation products. Diluted solu-
tions of TMDSz in dry hexane (3 ml, 1–4 � 10�3 mol dm�3)
were irradiated in UV cells (Suprasil, 10 � 10 � 35 mm,
Hellma, Mullheim, Germany), modified for purging to remove
the oxygen prior to irradiation. After purging with N2
(10 min), the samples were irradiated with 222 nm photons
with an intensity of 2 mW cm�2 under continuous stirring.
The analysis of the irradiation products in diluted solution
was performed using a GC-MS system 6890N/5973N (Agilent
Technologies, Palo Alto, CA, USA) with split/splitless
injector. The GC parameters were the following: column:
HP-5 ms (Agilent, 30 m, 0.25 mm id, 0.25 mm film thickness),
injector: split mode, 250 1C, carrier gas: helium, 1 ml min�1
(constant flow), temperature program: 40 1C held for 2 min,
then with 5 1C min�1 to 45 1C, after this with 20 1C min�1 to
250 1C, and held for 2 min. The transfer line to the MS ion
source was set to 250 1C. The MS was used in the scan mode
(scan range 12 to 600 amu). In some cases a special column for
volatile compounds (CP-Volamine, Varian, 30 m, 0.32 mm id,
0.45 mm film thickness) was used.
The irradiation experiments with undiluted TMDSz were
realized in a UV cell (Suprasil, 10 � 10 � 35 mm, Hellma,
Mullheim, Germany) with septum screwing. After optional
purging of the irradiation cell with nitrogen typically 200 mlof the prepared organosilazane was injected by a syringe.
Samples were handled by standard Schlenk-techniques and
oxygen was removed from the silazane prior to injection by
3 freeze-and-thaw cycles. UV irradiations were performed with
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2381
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
the KrCl* excimer lamp (intensity 11 mW cm�2) for 1 to 5 min
from top on a horizontally oriented cell, which was gently
shaken during irradiation to exchange the surface layer
continuously. After analysing the headspace of the samples,
the liquid phase was taken up with 1 ml of dry hexane and also
measured.
In order to analyse the volatile irradiation products and to
ensure that none of such volatile compounds is masked by the
solvent peak these were enriched by inserting a solid-phase
microextraction (SPME) fibre (PDMS/DVB, Supelco, 65 mmfilm thickness) in the headspace of the irradiation cell for 1 min
at ambient temperature and by subsequent thermodesorption
of the accumulated compounds in the GC injector (splitless
mode) at 250 1C. The other GC-MS conditions were the same
as mentioned above.
Results and discussion
In order to separate unimolecular decay processes from
bimolecular reactions, experiments in dilute solution were
performed. 222 nm excitation was chosen as a compromise
between the small absorption coefficients of the silazanes
below 230 nm (vide infra), and the limiting availability of
solvents transparent at such excitation wavelengths. In our
previous paper,21 it was shown that irradiation with photons
of different wavelengths, i.e. 172, 185 and 222 nm, had no
effect on the kinetics of the photolytical degradation of
P(DMScoMS), if the dose actually absorbed in the thin layers
is properly taken into account.21 To verify that this is also true
for the model disilazanes in the present case, a number
of comparative matrix EPR experiments were performed
using both a KrCl* excimer (222 nm) and an HgLP
lamp (emission at 185 and 254 nm). Whereas 254 nm
irradiation alone, selected by a 225 nm edge filter, did not
produce any significant amount of radicals, irradiation
with 185 nm gave the same radical species stemming from
the silazanes as where produced by 222 nm photons.
If solutions of silazanes were irradiated by 185 nm photons
the EPR spectra were complicated by additional radicals
formed by direct photolysis of the solvents, which was not
the case with 222 nm light. Acetonitrile and n-hexane were
chosen as solvents, being mostly transparent at 222 nm
(absorbances of o0.01 and o0.05 at 1 cm path length vs.
water). Especially the former is considered to be inert in
photolytical reactions.
Laser flash photolysis with 222 nm light
Laser flash photolysis experiments with B10 ns time resolution
have been carried out to investigate the luminescence properties
and the photochemically induced transformations of the
different disilazanes. All three silazanes have low absorption
coefficients at the excitation wavelength (e = 16, 120 and
615 dm3 mol�1 cm�1 for TMDSz, HMDSz and 7MDSz,
respectively, the ground state absorption spectra are given in
the ESI, Fig. 1S),w thus concentrations between 1 � 10�3–4 �10�3 mol dm�3 were used in the laser flash experiments in
order to keep the absorbance of the solution across the laser
path length (3 mm) below 0.3.
Emission measurements. Luminescence spectra of the
three silazanes were similar with a maximum emission at
280–290 nm (see ESI, Fig. 2S)w and a lifetime of B25 ns.
These spectra can most likely be assigned to the triplet state,
which is quenched by oxygen in air saturated solutions. (decay
rate increases to 1 � 108 s�1, corresponding to a bimolecular
rate constant of k(T* + O2) B 2 � 1010 dm3 mol�1 s�1, no
correction for laser pulse duration was made).
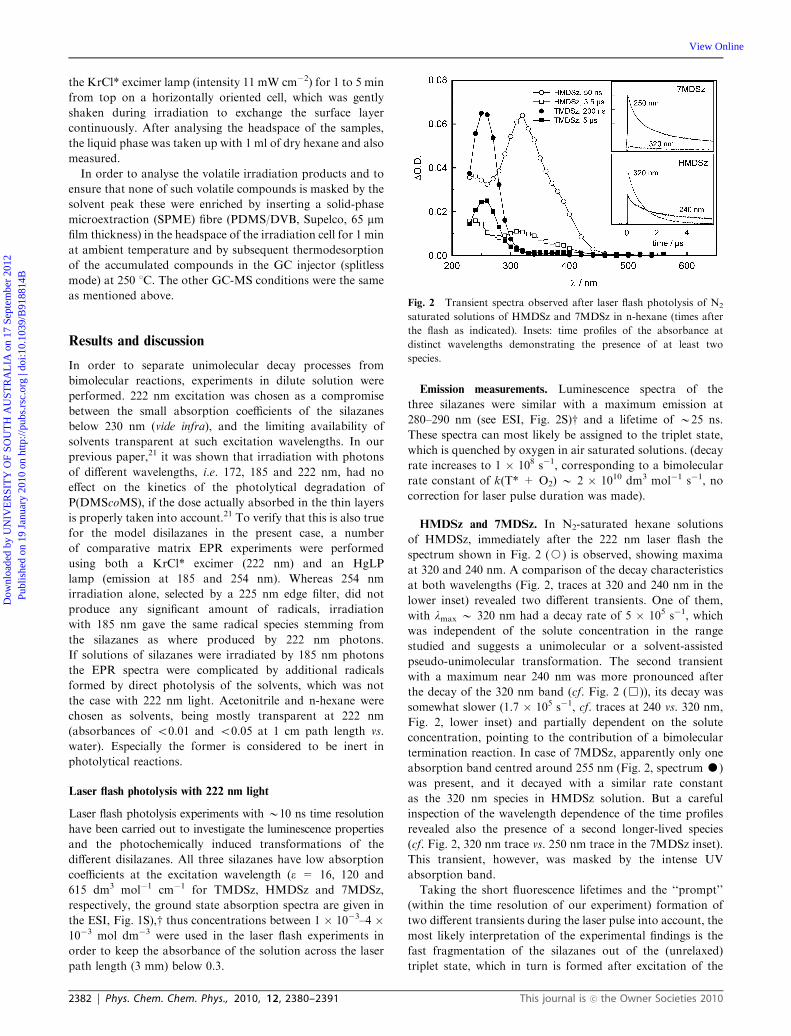
HMDSz and 7MDSz. In N2-saturated hexane solutions
of HMDSz, immediately after the 222 nm laser flash the
spectrum shown in Fig. 2 (J) is observed, showing maxima
at 320 and 240 nm. A comparison of the decay characteristics
at both wavelengths (Fig. 2, traces at 320 and 240 nm in the
lower inset) revealed two different transients. One of them,
with lmax B 320 nm had a decay rate of 5 � 105 s�1, which
was independent of the solute concentration in the range
studied and suggests a unimolecular or a solvent-assisted
pseudo-unimolecular transformation. The second transient
with a maximum near 240 nm was more pronounced after
the decay of the 320 nm band (cf. Fig. 2 (&)), its decay was
somewhat slower (1.7 � 105 s�1, cf. traces at 240 vs. 320 nm,
Fig. 2, lower inset) and partially dependent on the solute
concentration, pointing to the contribution of a bimolecular
termination reaction. In case of 7MDSz, apparently only one
absorption band centred around 255 nm (Fig. 2, spectrum K)
was present, and it decayed with a similar rate constant
as the 320 nm species in HMDSz solution. But a careful
inspection of the wavelength dependence of the time profiles
revealed also the presence of a second longer-lived species
(cf. Fig. 2, 320 nm trace vs. 250 nm trace in the 7MDSz inset).
This transient, however, was masked by the intense UV
absorption band.
Taking the short fluorescence lifetimes and the ‘‘prompt’’
(within the time resolution of our experiment) formation of
two different transients during the laser pulse into account, the
most likely interpretation of the experimental findings is the
fast fragmentation of the silazanes out of the (unrelaxed)
triplet state, which in turn is formed after excitation of the
Fig. 2 Transient spectra observed after laser flash photolysis of N2
saturated solutions of HMDSz and 7MDSz in n-hexane (times after
the flash as indicated). Insets: time profiles of the absorbance at
distinct wavelengths demonstrating the presence of at least two
species.
2382 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
silazanes into their excited singlet state followed by inter-
system crossing (reaction (1)).
ð1Þ
This interpretation is strongly supported by quantum chemical
calculations (vide infra). It was discussed earlier,21 that the
energy optimization of the triplet state geometry was practically
impossible, as a strong bond elongation of the Si–N bond (to
be taken as a hint towards molecular fragmentation) occurred
immediately, when triplet optimization was started from
singlet state geometry. Moreover, calculations of the electronic
absorption spectra of the fragments resulting from the Si–N
scission predict strong absorption bands at 302 nm, 275 nm
and in the 250–270 nm range for the (CH3)3SiN�(H),
(CH3)3SiN�(CH3) and (CH3)3Si
� radicals, respectively (see
ESI, Table 1S),w which is in good agreement with the experi-
mental values. Thus, the strong shorter-lived absorption bands
observed experimentally at 320 and 255 nm can be reasonably
assigned to the N-centred fragments, and the longer-lived
absorption at 240–260 is attributed to the (CH3)3Si� radical,
which is calculated to show a relatively broad band (main
transitions at 247 and 270 nm). The spectrum of the (CH3)3Si�
radical (same fragment for HMDSz and 7MDSz) strongly
overlaps with the more intense spectrum at 320 nm (HMDSz)
and is nearly completely masked by the 255 nm band in case
of 7MDSz.
In case of HMDSz, the influence of oxygen on the decay
rate was investigated. With increasing concentrations of O2 in
solution, the decay of the 320 nm transient (R–N�H, spectrum
J in Fig. 2) becomes slightly faster and a bimolecular rate
constant of B1 � 108 dm3 mol�1 s�1 was derived from a
Stern-Volmar-type plot (see ESI Fig. 3S)w taking the saturation
concentration of O2 in hexane as 1.5 � 10�2 mol dm�3.
Concomitant to the faster decay of the 320 nm band an increasing
absorption band resulting from peroxyl radicals with its
maximum around 260–270 nm was observed and nearly the
same rate constant was derived from the build-up at 270 nm
(reaction (2)). Unfortunately, the peroxyl radical band strongly
overlapped the spectrum of the primary 240 nm transient, thus
the O2-dependence of the latter could not be measured.
ð2Þ
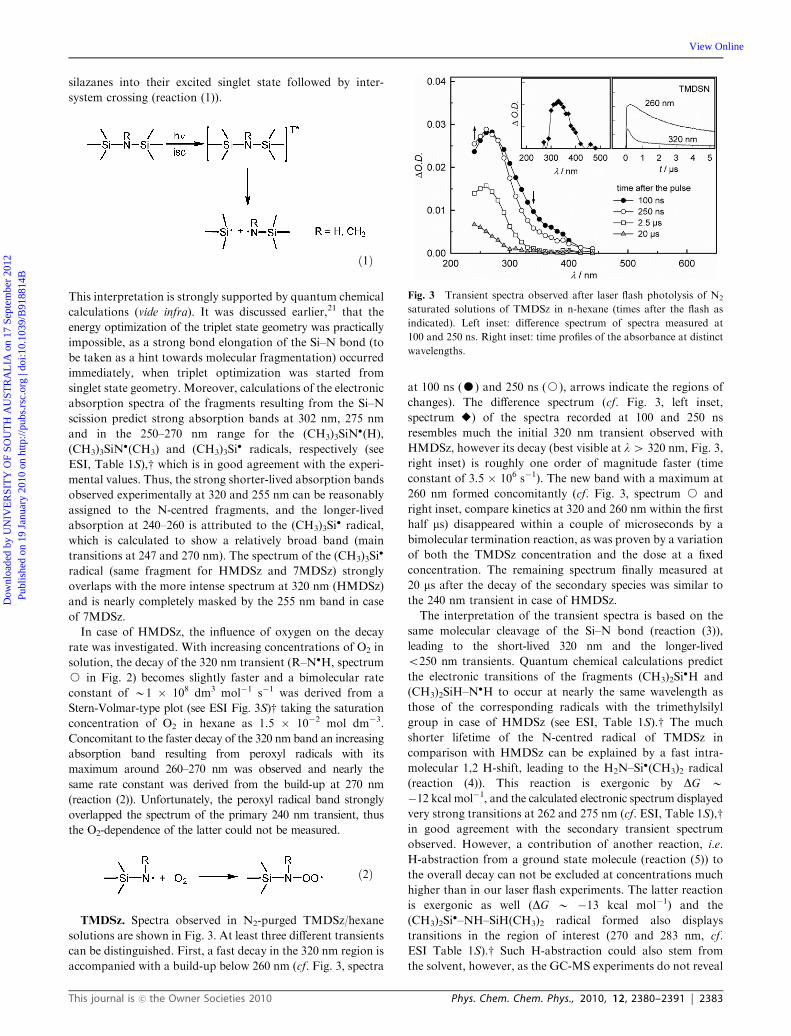
TMDSz. Spectra observed in N2-purged TMDSz/hexane
solutions are shown in Fig. 3. At least three different transients
can be distinguished. First, a fast decay in the 320 nm region is
accompanied with a build-up below 260 nm (cf. Fig. 3, spectra
at 100 ns (K) and 250 ns (J), arrows indicate the regions of
changes). The difference spectrum (cf. Fig. 3, left inset,
spectrum E) of the spectra recorded at 100 and 250 ns
resembles much the initial 320 nm transient observed with
HMDSz, however its decay (best visible at l4 320 nm, Fig. 3,
right inset) is roughly one order of magnitude faster (time
constant of 3.5 � 106 s�1). The new band with a maximum at
260 nm formed concomitantly (cf. Fig. 3, spectrum J and
right inset, compare kinetics at 320 and 260 nm within the first
half ms) disappeared within a couple of microseconds by a
bimolecular termination reaction, as was proven by a variation
of both the TMDSz concentration and the dose at a fixed
concentration. The remaining spectrum finally measured at
20 ms after the decay of the secondary species was similar to
the 240 nm transient in case of HMDSz.
The interpretation of the transient spectra is based on the
same molecular cleavage of the Si–N bond (reaction (3)),
leading to the short-lived 320 nm and the longer-lived
o250 nm transients. Quantum chemical calculations predict
the electronic transitions of the fragments (CH3)2Si�H and
(CH3)2SiH–N�H to occur at nearly the same wavelength as
those of the corresponding radicals with the trimethylsilyl
group in case of HMDSz (see ESI, Table 1S).w The much
shorter lifetime of the N-centred radical of TMDSz in
comparison with HMDSz can be explained by a fast intra-
molecular 1,2 H-shift, leading to the H2N–Si�(CH3)2 radical
(reaction (4)). This reaction is exergonic by DG B�12 kcal mol�1, and the calculated electronic spectrum displayed
very strong transitions at 262 and 275 nm (cf. ESI, Table 1S),win good agreement with the secondary transient spectrum
observed. However, a contribution of another reaction, i.e.
H-abstraction from a ground state molecule (reaction (5)) to
the overall decay can not be excluded at concentrations much
higher than in our laser flash experiments. The latter reaction
is exergonic as well (DG B �13 kcal mol�1) and the
(CH3)2Si�–NH–SiH(CH3)2 radical formed also displays
transitions in the region of interest (270 and 283 nm, cf.
ESI Table 1S).w Such H-abstraction could also stem from
the solvent, however, as the GC-MS experiments do not reveal
Fig. 3 Transient spectra observed after laser flash photolysis of N2
saturated solutions of TMDSz in n-hexane (times after the flash as
indicated). Left inset: difference spectrum of spectra measured at
100 and 250 ns. Right inset: time profiles of the absorbance at distinct
wavelengths.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2383
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
recombination products involving hexane fragments, this can
only be a minor pathway.
ð3Þ
ð4Þ
ð5Þ
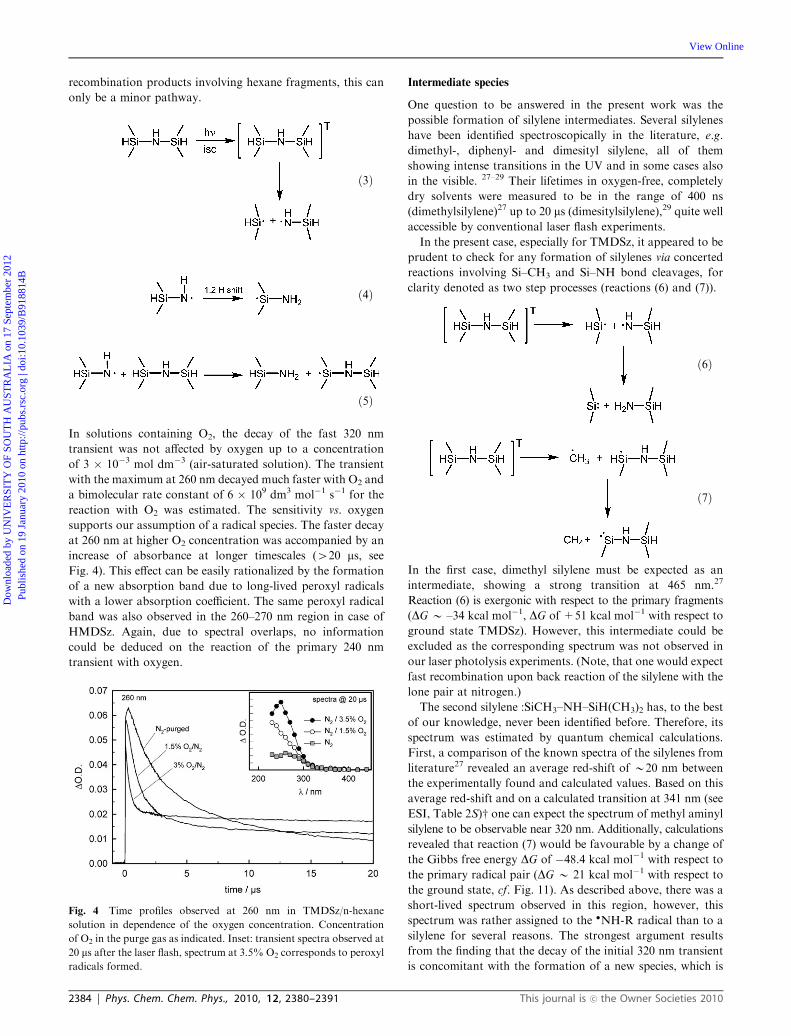
In solutions containing O2, the decay of the fast 320 nm
transient was not affected by oxygen up to a concentration
of 3 � 10�3 mol dm�3 (air-saturated solution). The transient
with the maximum at 260 nm decayed much faster with O2 and
a bimolecular rate constant of 6 � 109 dm3 mol�1 s�1 for the
reaction with O2 was estimated. The sensitivity vs. oxygen
supports our assumption of a radical species. The faster decay
at 260 nm at higher O2 concentration was accompanied by an
increase of absorbance at longer timescales (420 ms, see
Fig. 4). This effect can be easily rationalized by the formation
of a new absorption band due to long-lived peroxyl radicals
with a lower absorption coefficient. The same peroxyl radical
band was also observed in the 260–270 nm region in case of
HMDSz. Again, due to spectral overlaps, no information
could be deduced on the reaction of the primary 240 nm
transient with oxygen.
Intermediate species
One question to be answered in the present work was the
possible formation of silylene intermediates. Several silylenes
have been identified spectroscopically in the literature, e.g.
dimethyl-, diphenyl- and dimesityl silylene, all of them
showing intense transitions in the UV and in some cases also
in the visible. 27–29 Their lifetimes in oxygen-free, completely
dry solvents were measured to be in the range of 400 ns
(dimethylsilylene)27 up to 20 ms (dimesitylsilylene),29 quite well
accessible by conventional laser flash experiments.
In the present case, especially for TMDSz, it appeared to be
prudent to check for any formation of silylenes via concerted
reactions involving Si–CH3 and Si–NH bond cleavages, for
clarity denoted as two step processes (reactions (6) and (7)).
ð6Þ
ð7Þ
In the first case, dimethyl silylene must be expected as an
intermediate, showing a strong transition at 465 nm.27
Reaction (6) is exergonic with respect to the primary fragments
(DG B –34 kcal mol�1, DG of +51 kcal mol�1 with respect to
ground state TMDSz). However, this intermediate could be
excluded as the corresponding spectrum was not observed in
our laser photolysis experiments. (Note, that one would expect
fast recombination upon back reaction of the silylene with the
lone pair at nitrogen.)
The second silylene :SiCH3–NH–SiH(CH3)2 has, to the best
of our knowledge, never been identified before. Therefore, its
spectrum was estimated by quantum chemical calculations.
First, a comparison of the known spectra of the silylenes from
literature27 revealed an average red-shift of B20 nm between
the experimentally found and calculated values. Based on this
average red-shift and on a calculated transition at 341 nm (see
ESI, Table 2S)w one can expect the spectrum of methyl aminyl
silylene to be observable near 320 nm. Additionally, calculations
revealed that reaction (7) would be favourable by a change of
the Gibbs free energy DG of �48.4 kcal mol�1 with respect to
the primary radical pair (DG B 21 kcal mol�1 with respect to
the ground state, cf. Fig. 11). As described above, there was a
short-lived spectrum observed in this region, however, this
spectrum was rather assigned to the �NH-R radical than to a
silylene for several reasons. The strongest argument results
from the finding that the decay of the initial 320 nm transient
is concomitant with the formation of a new species, which is
Fig. 4 Time profiles observed at 260 nm in TMDSz/n-hexane
solution in dependence of the oxygen concentration. Concentration
of O2 in the purge gas as indicated. Inset: transient spectra observed at
20 ms after the laser flash, spectrum at 3.5% O2 corresponds to peroxyl
radicals formed.
2384 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
definitely a radical due to its reactivity with oxygen. Such a
reaction (leading to a radical) would not be possible with a
silylene as precursor. Moreover, the insensitivity of the
primary 320 nm transient towards O2 also rules out silylene
as a candidate for the assignment.
It is therefore concluded that reactive silylene intermediates
are unlikely formed or should be extremely short-lived to
escape direct observation. In order to corroborate this con-
clusion, trapping experiments at high scavenger concentration
were conducted (vide infra).
Low-temperature EPR
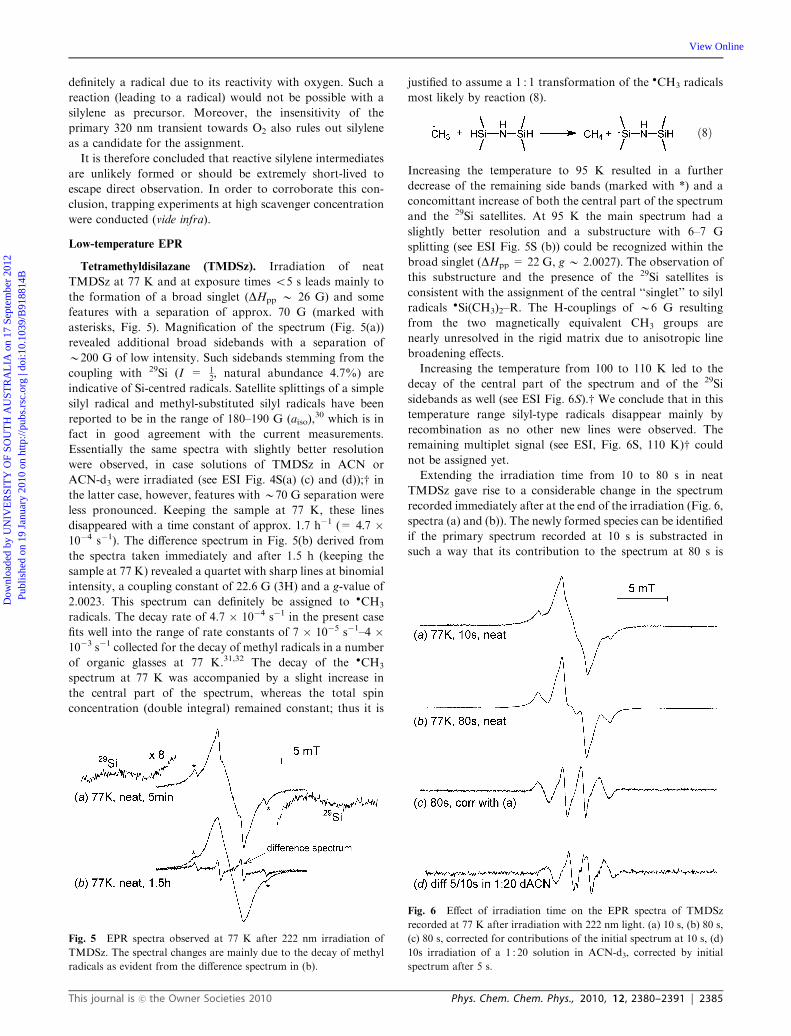
Tetramethyldisilazane (TMDSz). Irradiation of neat
TMDSz at 77 K and at exposure times o5 s leads mainly to
the formation of a broad singlet (DHpp B 26 G) and some
features with a separation of approx. 70 G (marked with
asterisks, Fig. 5). Magnification of the spectrum (Fig. 5(a))
revealed additional broad sidebands with a separation of
B200 G of low intensity. Such sidebands stemming from the
coupling with 29Si (I = 12, natural abundance 4.7%) are
indicative of Si-centred radicals. Satellite splittings of a simple
silyl radical and methyl-substituted silyl radicals have been
reported to be in the range of 180–190 G (aiso),30 which is in
fact in good agreement with the current measurements.
Essentially the same spectra with slightly better resolution
were observed, in case solutions of TMDSz in ACN or
ACN-d3 were irradiated (see ESI Fig. 4S(a) (c) and (d));w in
the latter case, however, features with B70 G separation were
less pronounced. Keeping the sample at 77 K, these lines
disappeared with a time constant of approx. 1.7 h�1 (= 4.7 �10�4 s�1). The difference spectrum in Fig. 5(b) derived from
the spectra taken immediately and after 1.5 h (keeping the
sample at 77 K) revealed a quartet with sharp lines at binomial
intensity, a coupling constant of 22.6 G (3H) and a g-value of
2.0023. This spectrum can definitely be assigned to �CH3
radicals. The decay rate of 4.7 � 10�4 s�1 in the present case
fits well into the range of rate constants of 7 � 10�5 s�1–4 �10�3 s�1 collected for the decay of methyl radicals in a number
of organic glasses at 77 K.31,32 The decay of the �CH3
spectrum at 77 K was accompanied by a slight increase in
the central part of the spectrum, whereas the total spin
concentration (double integral) remained constant; thus it is
justified to assume a 1 : 1 transformation of the �CH3 radicals
most likely by reaction (8).
ð8Þ
Increasing the temperature to 95 K resulted in a further
decrease of the remaining side bands (marked with *) and a
concomittant increase of both the central part of the spectrum
and the 29Si satellites. At 95 K the main spectrum had a
slightly better resolution and a substructure with 6–7 G
splitting (see ESI Fig. 5S (b)) could be recognized within the
broad singlet (DHpp = 22 G, g B 2.0027). The observation of
this substructure and the presence of the 29Si satellites is
consistent with the assignment of the central ‘‘singlet’’ to silyl
radicals �Si(CH3)2–R. The H-couplings of B6 G resulting
from the two magnetically equivalent CH3 groups are
nearly unresolved in the rigid matrix due to anisotropic line
broadening effects.
Increasing the temperature from 100 to 110 K led to the
decay of the central part of the spectrum and of the 29Si
sidebands as well (see ESI Fig. 6S).w We conclude that in this
temperature range silyl-type radicals disappear mainly by
recombination as no other new lines were observed. The
remaining multiplet signal (see ESI, Fig. 6S, 110 K)w could
not be assigned yet.
Extending the irradiation time from 10 to 80 s in neat
TMDSz gave rise to a considerable change in the spectrum
recorded immediately after at the end of the irradiation (Fig. 6,
spectra (a) and (b)). The newly formed species can be identified
if the primary spectrum recorded at 10 s is substracted in
such a way that its contribution to the spectrum at 80 s is
Fig. 5 EPR spectra observed at 77 K after 222 nm irradiation of
TMDSz. The spectral changes are mainly due to the decay of methyl
radicals as evident from the difference spectrum in (b).
Fig. 6 Effect of irradiation time on the EPR spectra of TMDSz
recorded at 77 K after irradiation with 222 nm light. (a) 10 s, (b) 80 s,
(c) 80 s, corrected for contributions of the initial spectrum at 10 s, (d)
10s irradiation of a 1 : 20 solution in ACN-d3, corrected by initial
spectrum after 5 s.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2385
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
minimized. As one can see in Fig. 6(c), a nearly binomial
quartet is observed with a(3H)B 18.5 G and gB 2.0026. This
quartet is also formed in solution (Fig. 6 (d), 1 : 20 in ACN-d3)
and it seems to be already present at short irradiation times
(cf. ESI Fig. 7S (b) 1 : 99 ACN solution).w In case a neat
system was irradiated at 120 K, only one species was observed,
which is obviously again the same four line spectrum with
somewhat better resolution.
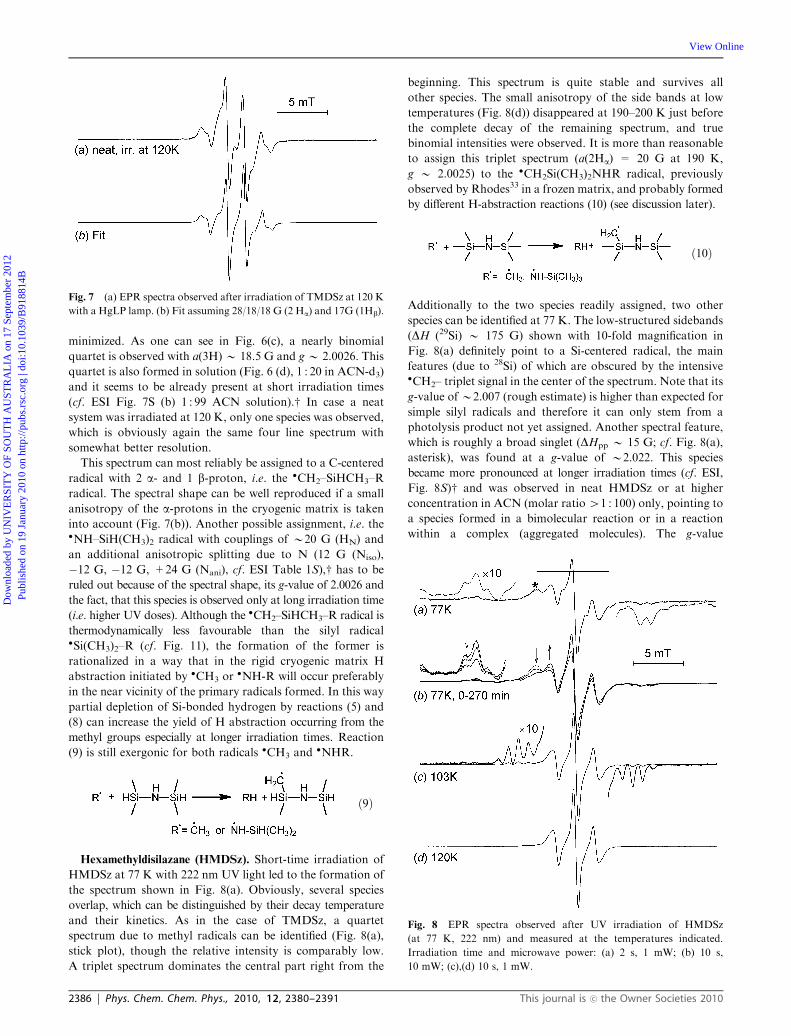
This spectrum can most reliably be assigned to a C-centered
radical with 2 a- and 1 b-proton, i.e. the �CH2–SiHCH3–R
radical. The spectral shape can be well reproduced if a small
anisotropy of the a-protons in the cryogenic matrix is taken
into account (Fig. 7(b)). Another possible assignment, i.e. the�NH–SiH(CH3)2 radical with couplings of B20 G (HN) and
an additional anisotropic splitting due to N (12 G (Niso),
�12 G, �12 G, +24 G (Nani), cf. ESI Table 1S),w has to be
ruled out because of the spectral shape, its g-value of 2.0026 and
the fact, that this species is observed only at long irradiation time
(i.e. higher UV doses). Although the �CH2–SiHCH3–R radical is
thermodynamically less favourable than the silyl radical�Si(CH3)2–R (cf. Fig. 11), the formation of the former is
rationalized in a way that in the rigid cryogenic matrix H
abstraction initiated by �CH3 or�NH-R will occur preferably
in the near vicinity of the primary radicals formed. In this way
partial depletion of Si-bonded hydrogen by reactions (5) and
(8) can increase the yield of H abstraction occurring from the
methyl groups especially at longer irradiation times. Reaction
(9) is still exergonic for both radicals �CH3 and�NHR.
ð9Þ
Hexamethyldisilazane (HMDSz). Short-time irradiation of
HMDSz at 77 K with 222 nm UV light led to the formation of
the spectrum shown in Fig. 8(a). Obviously, several species
overlap, which can be distinguished by their decay temperature
and their kinetics. As in the case of TMDSz, a quartet
spectrum due to methyl radicals can be identified (Fig. 8(a),
stick plot), though the relative intensity is comparably low.
A triplet spectrum dominates the central part right from the
beginning. This spectrum is quite stable and survives all
other species. The small anisotropy of the side bands at low
temperatures (Fig. 8(d)) disappeared at 190–200 K just before
the complete decay of the remaining spectrum, and true
binomial intensities were observed. It is more than reasonable
to assign this triplet spectrum (a(2Ha) = 20 G at 190 K,
g B 2.0025) to the �CH2Si(CH3)2NHR radical, previously
observed by Rhodes33 in a frozen matrix, and probably formed
by different H-abstraction reactions (10) (see discussion later).
ð10Þ
Additionally to the two species readily assigned, two other
species can be identified at 77 K. The low-structured sidebands
(DH (29Si) B 175 G) shown with 10-fold magnification in
Fig. 8(a) definitely point to a Si-centered radical, the main
features (due to 28Si) of which are obscured by the intensive�CH2– triplet signal in the center of the spectrum. Note that its
g-value ofB2.007 (rough estimate) is higher than expected for
simple silyl radicals and therefore it can only stem from a
photolysis product not yet assigned. Another spectral feature,
which is roughly a broad singlet (DHpp B 15 G; cf. Fig. 8(a),
asterisk), was found at a g-value of B2.022. This species
became more pronounced at longer irradiation times (cf. ESI,
Fig. 8S)w and was observed in neat HMDSz or at higher
concentration in ACN (molar ratio41 : 100) only, pointing to
a species formed in a bimolecular reaction or in a reaction
within a complex (aggregated molecules). The g-value
Fig. 7 (a) EPR spectra observed after irradiation of TMDSz at 120 K
with a HgLP lamp. (b) Fit assuming 28/18/18 G (2 Ha) and 17G (1Hb).
Fig. 8 EPR spectra observed after UV irradiation of HMDSz
(at 77 K, 222 nm) and measured at the temperatures indicated.
Irradiation time and microwave power: (a) 2 s, 1 mW; (b) 10 s,
10 mW; (c),(d) 10 s, 1 mW.
2386 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online
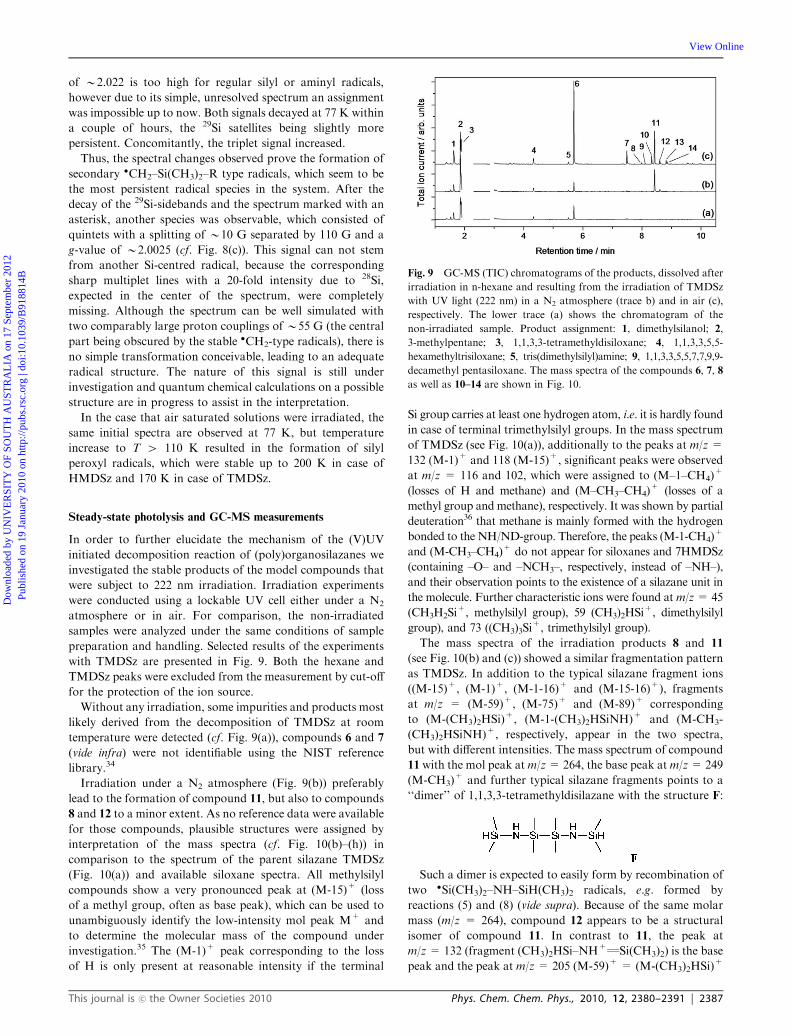
of B2.022 is too high for regular silyl or aminyl radicals,
however due to its simple, unresolved spectrum an assignment
was impossible up to now. Both signals decayed at 77 K within
a couple of hours, the 29Si satellites being slightly more
persistent. Concomitantly, the triplet signal increased.
Thus, the spectral changes observed prove the formation of
secondary �CH2–Si(CH3)2–R type radicals, which seem to be
the most persistent radical species in the system. After the
decay of the 29Si-sidebands and the spectrum marked with an
asterisk, another species was observable, which consisted of
quintets with a splitting of B10 G separated by 110 G and a
g-value of B2.0025 (cf. Fig. 8(c)). This signal can not stem
from another Si-centred radical, because the corresponding
sharp multiplet lines with a 20-fold intensity due to 28Si,
expected in the center of the spectrum, were completely
missing. Although the spectrum can be well simulated with
two comparably large proton couplings of B55 G (the central
part being obscured by the stable �CH2-type radicals), there is
no simple transformation conceivable, leading to an adequate
radical structure. The nature of this signal is still under
investigation and quantum chemical calculations on a possible
structure are in progress to assist in the interpretation.
In the case that air saturated solutions were irradiated, the
same initial spectra are observed at 77 K, but temperature
increase to T 4 110 K resulted in the formation of silyl
peroxyl radicals, which were stable up to 200 K in case of
HMDSz and 170 K in case of TMDSz.
Steady-state photolysis and GC-MS measurements
In order to further elucidate the mechanism of the (V)UV
initiated decomposition reaction of (poly)organosilazanes we
investigated the stable products of the model compounds that
were subject to 222 nm irradiation. Irradiation experiments
were conducted using a lockable UV cell either under a N2
atmosphere or in air. For comparison, the non-irradiated
samples were analyzed under the same conditions of sample
preparation and handling. Selected results of the experiments
with TMDSz are presented in Fig. 9. Both the hexane and
TMDSz peaks were excluded from the measurement by cut-off
for the protection of the ion source.
Without any irradiation, some impurities and products most
likely derived from the decomposition of TMDSz at room
temperature were detected (cf. Fig. 9(a)), compounds 6 and 7
(vide infra) were not identifiable using the NIST reference
library.34
Irradiation under a N2 atmosphere (Fig. 9(b)) preferably
lead to the formation of compound 11, but also to compounds
8 and 12 to a minor extent. As no reference data were available
for those compounds, plausible structures were assigned by
interpretation of the mass spectra (cf. Fig. 10(b)–(h)) in
comparison to the spectrum of the parent silazane TMDSz
(Fig. 10(a)) and available siloxane spectra. All methylsilyl
compounds show a very pronounced peak at (M-15)+ (loss
of a methyl group, often as base peak), which can be used to
unambiguously identify the low-intensity mol peak M+ and
to determine the molecular mass of the compound under
investigation.35 The (M-1)+ peak corresponding to the loss
of H is only present at reasonable intensity if the terminal
Si group carries at least one hydrogen atom, i.e. it is hardly found
in case of terminal trimethylsilyl groups. In the mass spectrum
of TMDSz (see Fig. 10(a)), additionally to the peaks at m/z =
132 (M-1)+ and 118 (M-15)+, significant peaks were observed
at m/z = 116 and 102, which were assigned to (M–1–CH4)+
(losses of H and methane) and (M–CH3–CH4)+ (losses of a
methyl group and methane), respectively. It was shown by partial
deuteration36 that methane is mainly formed with the hydrogen
bonded to the NH/ND-group. Therefore, the peaks (M-1-CH4)+
and (M-CH3–CH4)+ do not appear for siloxanes and 7HMDSz
(containing –O– and –NCH3–, respectively, instead of –NH–),
and their observation points to the existence of a silazane unit in
the molecule. Further characteristic ions were found atm/z= 45
(CH3H2Si+, methylsilyl group), 59 (CH3)2HSi+, dimethylsilyl
group), and 73 ((CH3)3Si+, trimethylsilyl group).
The mass spectra of the irradiation products 8 and 11
(see Fig. 10(b) and (c)) showed a similar fragmentation pattern
as TMDSz. In addition to the typical silazane fragment ions
((M-15)+, (M-1)+, (M-1-16)+ and (M-15-16)+), fragments
at m/z = (M-59)+, (M-75)+ and (M-89)+ corresponding
to (M-(CH3)2HSi)+, (M-1-(CH3)2HSiNH)+ and (M-CH3-
(CH3)2HSiNH)+, respectively, appear in the two spectra,
but with different intensities. The mass spectrum of compound
11 with the mol peak atm/z=264, the base peak atm/z=249
(M-CH3)+ and further typical silazane fragments points to a
‘‘dimer’’ of 1,1,3,3-tetramethyldisilazane with the structure F:
Such a dimer is expected to easily form by recombination of
two �Si(CH3)2–NH–SiH(CH3)2 radicals, e.g. formed by
reactions (5) and (8) (vide supra). Because of the same molar
mass (m/z = 264), compound 12 appears to be a structural
isomer of compound 11. In contrast to 11, the peak at
m/z= 132 (fragment (CH3)2HSi–NH+QSi(CH3)2) is the base
peak and the peak at m/z= 205 (M-59)+ = (M-(CH3)2HSi)+
Fig. 9 GC-MS (TIC) chromatograms of the products, dissolved after
irradiation in n-hexane and resulting from the irradiation of TMDSz
with UV light (222 nm) in a N2 atmosphere (trace b) and in air (c),
respectively. The lower trace (a) shows the chromatogram of the
non-irradiated sample. Product assignment: 1, dimethylsilanol; 2,
3-methylpentane; 3, 1,1,3,3-tetramethyldisiloxane; 4, 1,1,3,3,5,5-
hexamethyltrisiloxane; 5, tris(dimethylsilyl)amine; 9, 1,1,3,3,5,5,7,7,9,9-
decamethyl pentasiloxane. The mass spectra of the compounds 6, 7, 8
as well as 10–14 are shown in Fig. 10.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2387
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online

has significant intensity, the latter pointing to a preferable loss
of the dimethylsilyl group. We tentatively assign the
mass spectrum 12 to the branched structure G, which is
formed either by recombination of secondary radicals�Si(CH3)2–NH–SiH(CH3)2 and SiH(CH3)2–N
�–SiH(CH3)2 or
by an intramolecular rearrangement of F to its energetically
more stable isomer G (DG B 16 kcal mol�1)
Compound 8 also shows the typical silazane fragmentation
pattern and a mol peak at m/z = 250. In comparison to 11,
low molecular fragments up to m/z = 130 appear at the same
m/z values with quite similar intensities, whereas the masses of
the higher molecular fragments at m/z = 161, 175, 191, 219,
235 and 249 are all shifted by 14 units towards lower masses.
This strongly indicates that 8 should have an analogous
structure to 11, however, with one methyl group substituted
by a hydrogen. In this case, recombination of a secondary�Si(CH3)2–NH–SiH(CH3)2 radical with a primary �SiHCH3–
NH–SiH(CH3)2 radical photolytically formed by Si–CH3
cleavage was likely to form structure H.
In the presence of oxygen (Fig. 9(c)), considerable amounts
of 6 and 7 were generated. The mass spectrum of 6 with the
base peak at m/z = 192 (M-15)+, a mol peak at m/z = 207
and typical silazane fragments points to an oxygen-containing
compound with a hybrid structure consisting of both a
silazane and a siloxane unit (structure I).
This assignment is further supported by comparison
with the spectrum of 1,1,3,3,5,5-hexamethyltrisiloxane.34 The
siloxane displays a similar fragmentation pattern as 6 with the
main difference that all significant ion peaks with m/z Z 132
are shifted by 1 unit towards higher masses, as was expected
from the replacement of the –NH– group (m/z = 15) by
oxygen (m/z = 16), if the –O–SiH(CH3)2– fragment is the
primary leaving group in both cases. In the mass spectrum
of 7, significant ions similar to 6 with m/z = (M-15)+,
(M-75)+and (M-89)+ are present, all shifted towards higher
masses by m/z = 74, corresponding to an additional
–(CH3)2Si–O– unit. Again, the spectrum is quite similar to
the analogous 1,1,3,3,5,5,7,7-octamethyltetrasiloxane34 with
the difference of 1 unit for peaks with m/z Z 132 (vide supra).
It is more than reasonable to assign 7 to a structure with one
silazane and two siloxane fragments, probably as structure J.
The peaks 13 and 14 show both similar mass spectra (cf. 13
in Fig. 10(h)) and retention times. This suggests that the
chemical structures of these isomers differ only marginally.
Comparing the spectrum of 13 with that of compound 7 one
can easily rationalize that the former differs by an additional
group with 74 mass units at an otherwise similar pattern. Thus
we assign compound 13 to structure K with an additional
–(CH3)2Si–O– unit in comparison to J.
The spectrum of 10 shows a mol peak at m/z = 280 and
typical silazane fragments ((M-15)+, (M-31)+, (M-59)+,
(M-89)+). Additionally, a distinct (M-17)+ peak was observed
with much higher relative intensity than found for TMDSz due
to the subsequent losses of H and methane (M-1-CH4)+. Also,
as the precursor ion (M-1)+ for the CH4 loss did not appear,
we concluded that a direct loss of a fragment with 17 mass
units was observed, which can be assigned to the loss of
NH3.35 This fact points to the presence of a terminal amino
group and a tentative structure L is supposable, formed by a
Fig. 10 Mass spectra of compounds resulting from the irradiation of
TMDSz. For comparison, the spectrum of TMDSz is given at the top
(a). (b)–(d) Irradiation under N2 atmosphere, (e)–(h) air atmosphere.
2388 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online

recombination reaction involving a secondary radical�Si(CH3)2–NH2 (cf. reaction (4)).
In addition to the irradiation products in the liquid phase
(cf. Fig. 9), the volatile compounds in the headspace above the
liquid were analyzed. These were accumulated using an SPME
fibre, then thermo-desorbed in the hot GC injector and
subsequently analyzed by means of GC-MS. The components
detected in both the gaseous and the liquid phases were
essentially the same. Although the sensitivity using SPME is
generally higher for volatile compounds as compared to
standard GC-MS with diluted samples, one should note that
quantification in SPME is difficult because of the different
volatilities of the analytes. Peak 3 corresponding to tetra-
methyldisiloxane, which was partially clipped by the solvent
cut-off in the conventional GC-MS, is clearly present in the
SPME measurements. However, no dependency on the
irradiation was observed whether under N2 or O2/N2 conditions,
therefore TMDSO is likely to be an impurity in TMDSz.
Using SPME and a special GC column for the separation of
more volatile compounds, we could additionally observe
dimethylsilane and traces of trimethylsilane. The amount of
dimethylsilanol in the chromatograms (peak 1) varied from
sample to sample on a low level. It was confirmed that it is
formed by hydrolysis due to residual moisture introduced during
sample handling. As thermal decomposition at room temperature
is assumed to be slow,37 it is likely to occur in the injector and
transfer line of the GC-MS. If a TMDSz/hexane solution was
intentionally saturated with H2O, the yield of dimethylsilanol
strongly increased already for the non-irradiated reference sample,
however, its concentration further increased with increasing
irradiation time. This shows that thermally activated hydrolysis
occurs with an enhanced rate under photolytic conditions.
Additionally to the experiments with pure TMDSz and
with TMDSz/hexane solutions, respectively, irradiation was
performed using 1 : 1 mixtures of TMDSz and triethylsilane
with the aim to scavenge any highly reactive silylene inter-
mediates according to the literature26 (reaction (11)):
ð11Þ
The corresponding compound could not be detected. Thus, the
GC-MS results further support our hypothesis that silylenes
do not play a significant role (if any) in the entire reaction
mechanism.
Quantum chemical investigations
Recently, we reported quantum chemical calculations on the
irradiation-induced oxidative conversion of perhydropoly-
silazane19 and poly(1,1-dimethylsilazane-co-1-methylsilazane)21
into Si–O–Si or methyl–Si–O–Si networks, respectively.
Parameters such as the electronic absorption spectra, energy
levels of the excited states and the dissociation Gibbs energies
(DGdiss) were calculated on appropriate model molecules
showing structural resemblance to the silazane precursor
networks such as perhydro-, tetramethyl- and octamethyl
cyclotetrasilazanes. The calculations supported substantially
the discussion of the experimental results. Similar calculations
(see ESI, Table 1S)w were made here on TMDSz, HMDSz and
7MDSz using the DFT B3LYP method with 6-31G(d)
and 6-31+G(d,p) basis sets for geometry optimization and
calculation of the excitation energies, respectively, as implemented
in the Gaussian 03 (Rev. B.02) package.38 Frequency calculations
were done at the same level of theory to characterize the
stationary points on the potential surface and to obtain the
Gibbs free energy (G) at a standard temperature of 298.15 K
and a pressure of 1 atm using unscaled vibrations. The electronic
transition spectra were calculated using the time-dependent
(TD) B3LYP/6-31+G(d,p) method.39As an example, the
results obtained on the fragmentation of tetramethyldisilazane
(TMDSz) are summarized in Fig. 11 (the dissociation Gibbs
energy DGdiss is more reliable to characterise the probability of
Fig. 11 Quantum chemical calculations on the photolytic excitation of TMDSz and possible fragmentation pathways.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2389
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online

the fragmentation and the relative order of the different path-
ways than the bond dissociation enthalpy).
Calculation of the energy of the first excited singlet state S1
(129 kcal mol�1) showed that photons with an energy of
E 4 5.6 eV (l o 222 nm) are required in order to excite
the molecule. The absorption thresholds of the hexa- and hepta-
methylated disilazane compounds shifted towards lower energies
with respect to TMDSz and accounted for 5.4 eV (230 nm) and
5.1eV (243 nm) for HMDSz and 7MDSz, respectively.
Fast vibrational relaxation shall lead to the S1 state, which
serves as a precursor for the subsequent intersystem crossing
(ISC) into the excited triplet state T*. The energy of the lowest
triplet state T1 could not be calculated. Attempts to perform
geometry optimization for the triplet state were unsuccessful.
Simulations running from the singlet state geometry as starting
point did not converge into a stable triplet conformation.
Instead, immediate bond elongation of the Si–N bond was
observed. Thus, the QC calculations suggest that bond scission
maybe faster than the relaxation of the excited triplet state T*
into its T1 ground state, or at least, that any triplet state is
expected to be rather short-lived. In fact, the laser flash experi-
ments reveal a short-lived luminescence band at 285 nm, being
oxygen sensitive, which was assigned to the short-lived triplet
state. Taking the lmax of 285 nm as a rough measure of the triplet
energy, this wavelength corresponds to B100 kcal mol�1.
In Fig. 11, the dissociation Gibbs energies (DGdiss) are
calculated as differences of Gibbs free energies of the corres-
ponding fragments. As can be seen, all types of bonds present in
the molecule may be subject to dissociation with respect to the
excited singlet state, however, taking the estimate for the triplet
energy of 100 kcal mol�1 as a threshold, H cleavages from the
methyl groups and from the amine group are less likely.
By comparing the HOMO and LUMO orbitals (the latter can
be taken as a first approximation of the electron distribution in
the excited state) given in Fig. 11, it is obvious that excitation
leads to a strong intramolecular electron redistribution, resulting
in a reduced Coulomb attraction between these atoms, and
therefore gives rise to weakening of the corresponding –Si–NH–
bond. Thus, the partial charges in the ground state of +0.73e
and �0.85e at Si and N, respectively, change to +0.41e (Si) and
�0.32e (N) in the excited state. From the analysis of the electron
distribution from MOs shown in Fig. 11 it can also be seen that
the n-electron from nitrogen is delocalized in the ground state
onto the Si–C s-bonds due to a favourable symmetry. After
excitation this s-electron density will be removed from the Si–C
bonds resulting in weakening the bond, facilitating the cleavage.
Thus, according to the calculations the reaction pathway may be
based on both Si–NH bond scission and on the degradation of
methyl groups (Si–CH3 scission).
Conclusion
Summarizing the time-resolved, matrix-assisted and steady-
state experiments, two main photolytically-induced pathways
are confirmed, i.e. the cleavage of the Si–N and of the Si–CH3
bonds. Unlike the gas-phase photolysis of HMDSz, where
Si–CH3 cleavage with subsequent formation of methane was
found to be the favourable reaction pathway (54% CH4 vs.
14% (CH3)3SiH due to cleavages of Si–C and Si–N bonds,
respectively),40 in the condensed phase Si–N cleavage
dominates the overall process. Si–CH3 scission in fact takes
place, as approved by the observation of CH3� radicals by
low-temperature EPR, and as earlier suggested21 by the
observation of small amounts of CH4 (gas-phase IR) and by
the decay of Si–CH3 vibrations (ATR-FTIR). Even though the
smallest dissociation Gibbs energy was calculated for the
Si–CH3 bond cleavage, Si–NH bond scission is found to be
the preferred photochemically induced reaction pathway in
condensed media, which can be rationalised by the favourable
change of electron distribution along the Si–NH–Si bonds
upon excitation. (The higher relative yield of methane
observed in the gas phase photolysis is likely due to a higher
probability of the small �CH3 fragment to escape radical pair
recombination in comparison with the larger �SiH(CH3)2 and�NH–SiH(CH3)2 fragments.)
The cleavage of the excited silazane was expected to be fast,
as the excited triplet state was calculated to be dissociative.
Indeed, only short-lived fluorescence spectra were observed.
On the basis of the current results it can not be completely
excluded that the cleavage of the silazanes may also occur
from the excited singlet state. In order to finally distinguish
between excited singlet and triplet state processes, additional
ultra-fast kinetic measurements have to be performed, which
were beyond the scope of the present paper. Nevertheless, the
origin of the primary radical pairs has no impact on the
subsequent reactions discussed here.
Aminyl radicals �NH–R could not be trapped in the matrix
EPR experiments in the present case (although silylaminyl
radicals were readily observed under stationary conditions)41–43
and there was no indication of stable products bearing the amino
group in the GC-MS experiments under oxygen-free conditions.
However, aminyl radicals were observed by laser flash photolysis
and they disappear rapidly, most probably by H-transfer
reactions leading to terminal amino groups R-NH2. The faster
decay of the aminyl radicals in case of TMDSz in comparison
with HMDSz and 7MDSz can be explained by a facile intra-
molecular 1,2 H-shift (reaction (4), DG = �12 kcal mol�1)
leading to �Si(CH3)2–NH2, and in undiluted TMDSz also by
H-abstraction from another molecule (reaction (5), DG B �13kcal mol�1) with the formation of the �Si(CH3)2–NH–SiH(CH3)2radical and dimethylsilylamine. Although the �NH-R radical
is longer-lived in case of HMDSz, it still reacts fast enough
to escape trapping in the matrix EPR. In this case
H-abstraction will occur from one of the six methyl groups
(at a lower rate than from SiH in case of TMDSz), and indeed�CH2–Si(CH3)2–NH–R radicals are observed as the dominating
species in the EPR spectra of irradiated HMDSz (Fig. 8).
Under continuous irradiation conditions, intermediate
products with terminal NH2–groups undergo further photolysis
and the cleavage of the NH2–Si bond most likely occurs with the
subsequent formation of NH3 by H abstraction. Ammonia was
found to be a major volatile decomposition product in the VUV
irradiation of PHPS19 and polyorganosilazanes.21 The present
results suggest that the rate of photolytical decomposition of
oligosilazanes should depend on the amount of labile (reactive) H
available in the matrix (i.e. H bound to Si). This conclusion
explains at least qualitatively the experimental finding that PHPS
is much faster converted into an Si–O–Si network than the
2390 | Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 This journal is �c the Owner Societies 2010
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online

corresponding (P(DMScoMS)) into a methyl–Si–O–Si network.
P(DMScoMS) contains only 33% H bound to Si in comparison
to PHPS (Fig. 1).
No silylenes were observed in the present laser flash experi-
ments, and all attempts to trap them by high concentration of
triethylsilane (as in Moisseev et al.44) also failed. It is therefore
concluded that silylenes do not play a (significant) role in the
photochemistry of silazanes.
The main product identified by GC-MS after photolysis of
TMDSz is a ‘‘dimer’’ of the silazane, most probably formed by
recombination of two Si–N–Si� fragments. This dimer is also
formed in the presence of oxygen, and this clearly shows that
such radical recombination is fast enough to compete with the
reaction of O2 with the Si–N–Si� radical. In the presence of O2,
siloxazane compounds are the main irradiation products, formed
after several reaction steps from the secondary silylperoxyl
species. The identification of oligomers with subsequently
increasing number of –Si(CH3)2–O– units strongly points to a
reaction mechanism for the conversion based on building of
linear R–(Si–O)n– chains, which in turn may later crosslink by
further cleavage of Si–CH3 bonds along the backbone.
If traces of moisture are present in the system, a photo-
lytically-assisted hydrolysis leads to the enhanced formation of
silanol groups as byproducts.
The present work shows that the reactivity of organo-
silazanes does not depend only on the quantum yield of radical
pair formation, but also on structural parameters such as the
availability of reactive hydrogen atoms. Thus, investigations
of the influence of different substituents at the Si atom like
vinyl or acrylate groups will be performed in the future.
Acknowledgements
These studies have been supported by the Bundesministerium
fur Bildung und Forschung, BMBF, contract no. 01RI06007,
the Federal State of Germany and the Free State of Saxony.
The authors are grateful to Mrs Ingrid Reinhardt for the
measurement of the UV-VIS spectra of silazanes.
References
1 H. Chatham, Surf. Coat. Technol., 1996, 78, 1–9.2 J. Madocks, J. Rewhinkle and L. Barton, Mater. Sci. Eng., B,2005, 119, 268–273.
3 Y. Leterrier, Prog. Mater. Sci., 2003, 48, 1–55.4 F. Kessler, D. Herrmann and M. Powalla, Thin Solid Films, 2005,480–481, 491–498.
5 D. Pech, P. Steyer, A. S. Loir, J. C. Sanchez-Lopez and J. P. Millet,Surf. Coat. Technol., 2006, 201, 347–352.
6 T. N. Chen, D. S. Wuu, C. C. Wu, C. C. Chiang, Y. P. Chen andR. H. Horng, J. Electrochem. Soc., 2006, 153, F244–F248.
7 J. Fricke, H. Schwab and U. Heinemann, Int. J. Thermophys.,2006, 27, 1123–1139.
8 F. Fracassi, R. d’Agostino, F. Palumbo, E. Angelini, S. Grassiniand F. Rosalbino, Surf. Coat. Technol., 2003, 174–175, 107–111.
9 E. Angelini, S. Grassini, F. Rosalbino, F. Fracassi, S. Laera andF. Palumbo, Surf. Interface Anal., 2006, 38, 248–251.
10 K. Reichelt and X. Jiang, Thin Solid Films, 1990, 191, 91–126.11 A. Gruniger and P. Rudolf von Rohr, Thin Solid Films, 2004, 459,
308–312.12 A. G. Erlat, B. C. Wang, R. J. Spontak, Y. Tropsha, K. D. Mar,
D. B.Montgomery andE.A.Vogler, J.Mater. Res., 1999, 15, 704–717.13 S. Iwamori, Y. Gotoh and K. Moorthi, Surf. Coat. Technol., 2003,
166, 24–30.
14 T. P. Chou, C. Chandrasekaran and G. Z. Cao, J. Sol–Gel Sci.Technol., 2003, 26, 321–327.
15 H. Kriegsmann and G. Engelhardt, Z. Anorg. Allg. Chem., 1960,310, 100–109.
16 K. Kamiya, T. Tange, T. Hashimoto, H. Nasu and Y. Shimizu,Res. Rep. Fac. Eng. Mie Univ., 2001, 26, 23–31.
17 C. Kato, S. Tanaka, Y. Naganuma and T. Shindo, J. Photopolym.Sci. Tec., 2003, 16, 163–164.
18 Y. Naganuma, S. Tanaka, C. Kato and T. Shindo, J. Ceram. Soc.Jpn., 2004, 112, 599–603.
19 L. Prager, A. Dierdorf, H. Liebe, S. Naumov, S. Stojanovic,R. Heller, L. Wennrich and M. R. Buchmeiser, Chem.–Eur. J.,2007, 13, 8522–8529.
20 Eur. Pat. EP0745974B1, 1996; Jap. Pat. JP11092666AA, 1999; Jap.Pat. JP10279362AA, 1998.
21 L. Prager, L. Wennrich, R. Heller, W. Knolle, S. Naumov,A. Prager, D. Decker, H. Liebe and M. R. Buchmeiser,Chem.–Eur. J., 2009, 15, 675–683.
22 J. Pola, A. Galikova, Z. Bastl, J. Subrt, K. Vacek, J. Brus andA. Ouchi, Appl. Organomet. Chem., 2006, 20, 648–655.
23 J. Pola, A. Galikova, A. Galik, V. Blechta, Z. Bastl, J. Subrt andA. Ouchi, Chem. Mater., 2002, 14, 144–153.
24 D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110.25 J. von Sonntag and W. Knolle, J. Photochem. Photobiol., A, 2000,
132, 25–27.26 J. von Sonntag, J. Photochem. Photobiol., A, 1999, 126, 1–5.27 G. Levin, P. K. Das, C. Bilgrien and C. L. Lee, Organometallics,
1989, 8, 1206–1211.28 A.G.Moiseev andW. J. Leigh,Organometallics, 2007, 26, 6268–6276.29 R. T. Conlin, J. C. Nettoferreira, S. Zhang and J. C. Scaiano,
Organometallics, 1990, 9, 1332–1334.30 C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251.31 T. Doba, K. U. Ingold, W. Siebrand and T. A. Wildman, Faraday
Discuss. Chem. Soc., 1984, 175–191.32 W. G. French and J. E. Willard, J. Phys. Chem., 1968, 72, 4604.33 C. J. Rhodes, J. Chem. Soc., Perkin Trans. 2, 1992, 235–241.34 NIST Standard Reference Database 1A (Data Version: NIST 02),
National Institute of Standards and Technology, Gaithersburg,MD, 2002.
35 J. Silbiger, C. Lifshitz, J. Fuchs and A. Mandelba, J. Am. Chem.Soc., 1967, 89, 4308.
36 J. Tamas, M. Mak and S. Makleit, Org. Mass Spectrom., 1974, 9,847–853.
37 F. Bauer, U. Decker, A. Dierdorf, H. Ernst, R. Heller, H. Liebeand R. Mehnert, Prog. Org. Coat., 2005, 53, 183–190.
38 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, J. Montgomery, J. A., T. Vreven,K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi,V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega,G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota,R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian,J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts,R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli,J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth,P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich,A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick,A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz,Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov,G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin,D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng,A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson,W. Chen,M.W.Wong, C. Gonzalez and J. A. Pople,GAUSSIAN 03(Revision C.02), Gaussian, Inc., Wallingford, CT, 2004.
39 R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256,454–464.
40 J. Pola and R. Taylor, J. Organomet. Chem., 1993, 446, 131–134.41 J. C. Brand, M. D. Cook and B. P. Roberts, J. Chem. Soc., Perkin
Trans. 2, 1984, 1187–1196.42 J. C. Brand, B. P. Roberts and J. N. Winter, J. Chem. Soc., Perkin
Trans. 2, 1983, 261–270.43 M. D. Cook, B. P. Roberts and K. Singh, J. Chem. Soc., Perkin
Trans. 2, 1983, 635–643.44 A. G. Moiseev and W. J. Leigh, Organometallics, 2007, 26,
6277–6289.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 2380–2391 | 2391
Dow
nloa
ded
by U
NIV
ER
SIT
Y O
F SO
UT
H A
UST
RA
LIA
on
17 S
epte
mbe
r 20
12Pu
blis
hed
on 1
9 Ja
nuar
y 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/B91
8814
B
View Online







![Impacts of aerosols and clouds on photolysis frequencies and ... of aerosols and cloud… · [2] Photolysis reactions play a very important role in atmospheric chemistry. Ozone photolysis](https://img.pdfslide.net/doc/110x75/5f07e35b7e708231d41f41d6/impacts-of-aerosols-and-clouds-on-photolysis-frequencies-and-of-aerosols-and.jpg)





















