Embed Size (px)
Citation preview

1
Sustainably Green Composites of Thermoplastic Starch 1
and Cellulose Fibers – a Review 2
Amnuay Wattanakornsiri1*
, Sampan Tongnunui2 3
1Department of Agriculture and Environment, Faculty of Science and Technology, 4
Surindra Rajabhat University, Muang District, Surin Province, 32000, THAILAND 5
2Division of Biological and Natural Resource Science (Conservation Biology Program), 6
Mahidol University, Kanchanaburi Campus, Sai Yok District, 7
Kanchanaburi Province, 71150, THAILAND 8
* Corresponding author. Email: [email protected] 9
10
Abstract 11
Green composites have resulted in a renewed interest in environmentally friendly 12
materials issues for sustainable development as biodegradable renewable resource. This 13
review overviews recent advances in green composites based on thermoplastic starch (TPS) 14
and cellulose fibers. It includes information about compositions, preparations and properties 15
of starch, cellulose fibers, TPS, and green composites based on TPS and cellulose fibers. 16
Introduction and production of these composites into the material market would be important 17
for environmental sustainability in decreasing the volume of waste dumps and being cheap, 18
abundant and recyclable, and continuously being developed. 19
Keywords: Green composites/ Thermoplastic starch/ Cellulose fibers 20
21
22

2
1. Introduction 23
Worldwide environmental problems and the approaching depletion resulted from 24
petroleum-derived plastics are urging the sustainable development of green composites so-25
called environmentally friendly materials (Wattanakornsiri et al., 2011). Green composites 26
comprise biodegradable crop-derived polymers as matrixes and biodegradable plant-derived 27
fibers as fillers. When these integral parts are biodegradable, the composites are anticipated 28
to be biodegradable (Averous and Boquillon, 2004). They are considered as very promising 29
materials for environmental sustainability for a variety of reasons: 1) substitution of 30
renewable resources for depletable petrochemical feedstocks, 2) possibility of lower 31
greenhouse gas (GHG) emissions by sequestering carbon dioxide (CO2) from the atmosphere 32
in polymers and other organic chemicals, thereby closing the biogeneous carbon loop, and 33
instead of net addition of fossil carbon to the atmosphere in the case of petrochemicals 34
(Mohanty et al., 2005), and 3) closure of the loop of organic carbon and nutrients that green 35
composites can be returned to the soil by composting or biological cycling (Patel and 36
Narayan, 2005). Then, they are novel materials of the twenty-first century and would be 37
important for the environmental materials world (Mohanty et al., 2002). 38
Starch is a very attractive source and a promising raw material for the development of 39
green composites because it is naturally renewable, cheap and abundant (Angellier et al., 40
2006; Teixeira et al., 2009). Nevertheless, before thermally processable as for thermoplastic 41
polymers, starch must be converted to thermoplastic starch (TPS) by the addition of 42
plasticizers in the presence of high temperature and shear force (Curvelo et al., 2001; 43
Angellier et al., 2006). As traditional plasticizers, water (Kalichevsky and Blanshard, 1993; 44
Teixeira et al., 2009) and/or polyol plasticizers such as glycerol (Angellier et al., 2006; 45
Teixeira et al., 2009; Wattanakornsiri et al., 2011) and sorbitol (Teixeira et al., 2009) have 46
been used. However, the main plasticizer used in TPS was glycerol owing to providing the 47

3
best result in decreasing the friction between starch molecules (Janssen and Moscicki, 2006). 48
Furthermore, glycerol is a by-product generated in large amounts in the biofuel industry, and 49
is becoming nowadays a waste product that must be disposed with additional costs (Yazdani 50
and Gonzalez, 2007). 51
Despite the above clear advantages of the use of starch-based plastics for a sustainable 52
development, the applications of TPS are still restricted because of low mechanical properties 53
and high moisture absorption that are considered as the main drawbacks when compared to 54
conventional plastics (Averous and Boquillon, 2004; Teixeira et al., 2009). As an alternative 55
method to improve the properties of TPS is the reinforcement of cellulose fibers. Green 56
composites of TPS and cellulose fibers were prepared using various sources of starch, 57
including corn starch (Curvelo et al., 2001; Ma et al., 2005; Wattanakornsiri et al., 2011), 58
tapioca starch (Teixeira et al., 2009; Wattanakornsiri et al., 2012), rice starch 59
(Prachayawarakorn et al., 2010), potato starch (Thuwall et al., 2006), and wheat starch 60
(Rodriguex-Gonzalez et al., 2004), and different types of cellulose fibers, including flax and 61
ramie fibers (Wollerdorfer and Bader, 1998), potato pulp fibers (Dufresne and Vignon, 1998; 62
Dufresne et al., 2000), bleached leafwood fibers (Averous et al., 2001), bleached eucalyptus 63
pulp fibers (Curvelo et al., 2001), wood pulp fibers (Carvalho et al., 2002), cassava bagasse 64
fibers (Teixeira et al., 2009), and recycled paper cellulose fibers (Wattanakornsiri et al., 65
2012). 66
These researches have shown that tensile strength and elastic modulus of the 67
composites increased since cellulose fibers were mixed with TPS due to a good compatibility 68
between both polysaccharides, i.e. starch and cellulose fibers (Curvelo et al., 2001; Averous 69
and Boquillon, 2004). Besides, water resistance of the composites apparently increased 70
(Dufresne et al., 2000; Ma et al., 2008) as a consequence of the addition of the less 71
hydrophilic fibrous fillers (Averous and Boquillon, 2004; Ma et al., 2005). The constraint 72

4
exerted by the cellulose fibers at the interface on the matrix could perhaps also work in order 73
to reduce the swelling (Wattanakornsiri et al., 2011). In the present review, the compositions, 74
preparations and properties of starch, cellulose fibers, TPS, and green composites of TPS and 75
cellulose fibers, would be addressed that the use of green composites is attaining increased 76
importance and the world’s material manufacturers seek to replace dwindling petroleum-77
based feedstock with green composites including their advantages, i.e. biodegradability, low 78
cost, abundance and renewability (Espigule et al., 2012). 79
80
2. Starch 81
Starch is a polysaccharide polymer of D-anhydroglucose (C6H10O5) repeating units 82
comprising two main constituents, i.e. amylose and amylopectin. Amylose is an essential 83
linear polymer consisting of mainly α-1,4-D-glucosidic bond and slightly branched α-1,6-D-84
glucosidic bond as shown in Figure 1, and is soluble in water and forms a helical structure 85
(Lu et al., 2009). Besides, amylopectin is composted of α-1,4-D-glucosidic bond units 86
interlinked by α-1,6-D-glucosidic bond units to form a multiply branched structure as shown 87
in Figure 2 (Souza and Andrade, 2001; Szegda, 2009), and is able to form helical structures 88
which crystallize. Generally, the amylose and amylopectin contents are about 10 to 20% and 89
80 to 90%, respectively, depending on the type of starches (Lu et al., 2009). Starch is totally 90
biodegradable in a wide variety of environments. It can be hydrolyzed into glucose by 91
microorganisms or enzymes, and then metabolized into carbon dioxide and water 92
(Wattanakornsiri et al., 2012). 93
94
Figure 1 Chemical structure of amylose (Chiou et al., 2005) 95
96

5
Figure 2 Chemical structure of amylopectin (Chiou et al., 2005) 97
98
2.1 Starch gelatinization 99
Starch is partially in the crystalline form. When dry starch granules are heated, 100
thermal degradation occurs before the crystalline granule melting point is reached. As a 101
result, starch cannot be processed in its native form (Prachayawarakorn et al., 2013). In order 102
to meltingly process native starch, the hydrogen bonds holding the starch molecules together 103
have to be destructed and accordingly reduced. The reduction of starch hydrogen bonds can 104
be achieved in the presence of plasticizers, e.g. water, glycerol and sorbitol (Kim et al., 105
1997). Consequently, the plasticizer interacts with the starch hydroxyl groups; thus, reduces 106
the hydrogen bonds among the starch molecules. This allows individual chains to move freely 107
relative to each other (Willett and Doane, 2002). 108
When starch granules are heated in a plasticizer, their native crystalline structure is 109
disrupted and they swell irreversibly to many times of their original size. This process is 110
called “gelatinization” (Kim et al., 1997). Gelatinization gives rises not only to swelling but 111
also to loss of original crystals and solubility in the plasticizer (Park et al., 2000). During the 112
swelling amylose leaches out the plasticizer, but amylopectin forms gel (Ke and Sun, 2001). 113
The temperature at which starch begins to undergo this process is called “gelatinization 114
temperature”. Due to not all granules of a given starch begin to gelatinize at exactly the same 115
temperature; the gelatinization temperature therefore is suitably defined as a narrow 116
temperature range instead of one specific temperature. The temperature ranges also vary 117
according the source of starch (Szegda, 2009). 118
119
120

6
2.2 Starch as a material 121
The use of starch as a plastic material has been recorded since 1950s (Chadehumbe, 122
2006). Since then there have been a lot of researches done on different starches, but starch 123
has gained limited applications as general material, e.g. packaging material. The main 124
advantages of starch as a material are naturally renewable, cheap, abundant, and 125
biodegradable (Angellier et al., 2006; Teixeira et al., 2009). Nevertheless, when compared 126
with synthetic polymeric material starch has two main disadvantages as the followings. 127
- Starch contains hydroxyl groups, which indicate hydrophilic properties to starch. 128
Amylose dissolves in water and amylopectin swells in the presence of water (Lu 129
et al., 2009). This means that starch disintegrates in water and loses its properties 130
when exposed to moisture (Carvalho et al., 2001). 131
- Starch in its native form is not thermoplastic. When it is heated, pyrolysis occurs 132
before the crystalline melting point of starch is reached; then, it cannot be melt-133
processed by using conventional plastic equipments (Chadehumbe, 2006). 134
There are various techniques given for supporting starch using as a suitable material 135
such as destructuring starch as TPS, filling synthetic polymers with starch, blending starch 136
with other thermoplastic polymers, and making starch based composites. This review would 137
provide the technique of destructuring starch as TPS. 138
139
3. Thermoplastic starch 140
TPS is processed through the destructuring of native starch granules by heating at 141
relatively high temperature, under high shear conditions (Forssell et al., 1997; Hulleman et 142
al., 1998; Ma et al., 2005) and with limited amounts of liquid so-called plasticizer 143
(Prachayawarakorn et al., 2010). The plasticizer swells starch granules and reduces hydrogen 144

7
bonding and crystal in the granules. This causes an increment of molecular mobility and 145
renders it possible to melt-process native starch below its degradation temperature. The TPS 146
with different properties can be made by altering plasticizer contents and electric mixing, 147
followed by hot-press molding, extrusion or injection molding parameters (Bikiaris et al., 148
1998). 149
The amount of plasticizer used in combination of the chosen temperature has a 150
significant effect on starch conversion that can be achieved in two ways. First, all crystals in 151
starch are pulled apart by swelling, leaving none of them to be melted at higher temperature, 152
under an excess plasticizer condition. Second, the conversion can be achieved under a limited 153
plasticizer condition, which is the usual condition during electric mixing, extrusion or 154
injection. For the latter process, swelling forces are less significant and crystals melt at 155
temperatures much higher than the gelatinization temperature in the excess plasticizer 156
condition (Yu and Christie, 2001). 157
For example, during extrusion starch is affected by relatively high pressure up to 103 158
psi, heat during 90 to 180oC (Yu et al., 2005) and mechanical shear forces, resulting in 159
gelatinization, melting and fragmentation. Starch extrusion is carried out at lower moisture 160
contents from about 12 to 16%, which is below the amount of plasticizer necessary for 161
gelatinization (Chadehumbe, 2006). The starch granules are physically torn apart by 162
mechanical shear forces that influence on faster transfer of plasticizer into the starch 163
molecules. This results in the interruption of molecular bonds and loss of crystals, which lead 164
to high molecular mobility, causing the starch ability to be processed below its degradation 165
temperature (Averous et al., 2001). This clarifies that a mixture of small amounts of 166
gelatinized and melted states of starch as well as fragments exists simultaneously during 167
extrusion. Gelatinization is influenced by various variables such as moisture content, screw 168

8
speed, temperature, feed composition (ratio of amylose to amylopectin), and residence time 169
(Yu and Christie, 2001). 170
3.1 Plasticizer 171
Plasticizer is a material used to incorporate into a plastic material in order to increase 172
flexibility and workability. Plasticizer molecules penetrate the starch granules and destruct 173
the inner hydrogen bonds of the starch under high temperature, high shear force (Hulleman et 174
al., 1998; Ma et al., 2005), and high pressure (Chadehumbe, 2006). This eliminates starch-175
starch interactions; hence, they are replaced by starch-plasticizer interactions. Due to the 176
plasticizer molecules are smaller and more mobile than the starch molecules, the starch 177
network can be easily deformed without rupture (Yu et al., 1998). 178
During the TPS process, plasticizers play an importantly indispensable role 179
(Hulleman et al., 1998) because they can form hydrogen bonds with starch. This is because it 180
is a multi-hydroxyl polymer with three hydroxyl groups per monomer and there are a lot of 181
intermolecular and intramolecular hydrogen bonds in the starch. When the plasticizers form 182
hydrogen bonds with the starch, the original hydrogen bonds between hydroxyl groups of 183
starch molecules are destroyed, thus enabling the starch to display the plasticization (Ma et 184
al., 2005). 185
Hydrophilic liquids used as plasticizers for TPS are water, glycerol, sorbitol, glycol, 186
urea, and so on (Rodriguez-Gonzalez et al., 2004; Yu et al., 2005; Prachayawarakorn et al., 187
2010). Water is the most common solvent or plasticizer used with starch. The use of water as 188
a plasticizer is not preferable because the resulting TPS products are brittle when equilibrated 189
with ambient humidity (Forssell et al., 1997). The use of plasticizers such as glycerol and 190
sorbitol results in a rubbery material with better properties than the TPS plasticized by water 191
in various applications (Sugih, 2008). For the two most promising plasticizers of polyols, 192

9
glycerol and sorbitol, glycerol provides the best results in decreasing the friction between 193
starch molecules (Burgt Van Der et al., 1996; Janssen and Moscicki, 2006) and the brittleness 194
of resulting materials, and creating the easy manipulation after TPS processing (Janssen and 195
Moscicki, 2006; Teixeira et al., 2009). 196
Glycerol is a chemical compound being a sugar or sweet-tasting alcohol, which is a 197
colorless, odorless and viscous liquid and its molecular structure is C3H8O3. Generally, 198
glycerol has three hydrophilic hydroxyl groups that are responsible for its solubility in water 199
and its hygroscopic nature (Yazdani & Gonzalez, 2007). Moreover, glycerol is generated in 200
large amounts by biofuel industry, simultaneously with the increasing demand for biofuels 201
(Odling-Smee, 2007). Especially during the biodiesel production, glycerol is representing as a 202
“co-product” and rapidly becoming a waste product with a disposal cost attributed to it. 203
About 10% of crude glycerol is generated from the total amount of biodiesel produced by the 204
transesterification of vegetable oils or animal fats (Yazdani and Gonzalez, 2007). Therefore, 205
glycerol should be considered as a valuable by-product using as a plasticizer for TPS. 206
Janssen and Moscicki (2006) prepared TPS by blending potato starch plasticized with 207
varying glycerol contents of 20 to 30 wt% in two main processing steps. First, the starch was 208
gelatinized and thoroughly mixed with glycerol in an extruder; after that, the blending 209
materials were pelletized to form TPS pellets. Second, these pellets are fed to an injection 210
molding machine to produce TPS specimens. The results showed that the tensile strengths of 211
TPS were changed from varying starch to glycerol ratios and varying injection temperatures 212
that were 100 to 180oC. An increase of the glycerol content from 20 to 22 wt% led to a 213
fivefold decrease of the tensile strength from 20 to 4 MPa. The appropriate temperature of 214
molding was 140oC, indicating the highest tensile strength being 20 MPa. This value could be 215
reached the tensile strength that is comparable with commercially available polystyrene. 216

10
According to the studies of Curvelo et al. (2001) and Wattanakornsiri et al. (2012), 217
their preliminary experiments performed that the glycerol content should be in the ranges of 218
20 to 40% and 20 to 35% without added water, respectively. Lower and higher glycerol 219
content led to samples that were too much brittle or to exudation phenomena of glycerol, 220
respectively. 221
222
4. Cellulose fibers 223
Cellulose fibers are derived from plants, e.g. bast, leaf, seed and wood. They are a 224
class of hair-like materials being continuous filaments and their molecular chains are very 225
long and strong (Kaushik et al., 2010). Besides, they can be used as a component of 226
composite materials and matted into sheets for making products such as paper or felt. 227
Cellulose fibers are aligned along the length of the fibers that provide maximum tensile and 228
flexural strengths as well as support rigidity. Mechanical properties are mainly determined by 229
the cellulose content, degree of polymerization (DP), and fibrillar angle. Typically, the 230
reinforcing efficiency of cellulose fibers depends on the cellulose nature and their 231
crystallinity. Importantly, a high cellulose content and low fibrillar angle are desired 232
properties of fiber to be used as reinforcement for biological composites (John and Thomas, 233
2008). 234
Cellulose is a polysaccharide and natural linear crystalline polymer comprising D-235
anhydroglucose (C6H10O5) repeating units linked together by β-1,4-D-glucosidic bond 236
(Rowell et al., 1997) as shown in Figures 3 and 4. Each repeating unit contains three 237
hydroxyl groups. These hydroxyl groups and their ability of hydrogen bonding play an 238
important role in directing the crystalline packing and control the physical properties of 239
cellulose (Bismarck et al., 2005; John and Thomas, 2008). Solid cellulose forms a 240

11
microcrystalline structure with regions of crystalline and amorphous. Besides, cellulose is 241
also formed of slender rod like crystalline microfibers (John and Thomas, 2008; Mo et al., 242
2010). Cellulose has received more attention for green composites since it is attacked by a 243
wide variety of microorganisms and represented an appreciable fraction of waste products 244
and composites that make up sewage and refuse (Lu et al., 2009). 245
246
Figure 3 Chemical structure of cellulose (Bismarck et al., 2005) 247
248
Figure 4 Configuration of cellulose (Bismarck et al., 2005) 249
250
4.1 Chemically treated botanical cellulose fibers 251
There have been many researches developing the utilization of botanical fibers as 252
reinforcement of plastics; however, these botanical fibers are mainly composed of cellulose, 253
hemicellulose, and lignin including lignocellulose. In order to use botanical cellulose fibers 254
for green composites, they would be chemically treated. This review would provide only the 255
alkalization treatment of the botanical cellulose fibers because it is one of the most 256
appropriate treatments, effectively changing the surface topologies of the fibers, i.e. hemp, 257
sisal, jute and kapok, and their crystallographic structures (Mwaikambo and Ansell, 2002). In 258
addition, cellulose fibers are resistant to strong alkali treatment up to 17.5 wt% but are easily 259
hydrolyzed by acid treatment to water-soluble sugars (John and Thomas, 2008). 260
Alkalization treatment is one of the most used chemical treatments for cellulose fibers 261
when used to reinforce thermoplastics. The important modification done by the alkalization 262
treatment is the disruption of hydrogen bonding in the network structure, thereby increasing 263

12
surface roughness (Li, 2008). Addition of sodium hydroxide (NaOH) to botanical cellulose 264
fibers promotes the ionization of hydroxyl group to the alkoxide as shown in the following 265
chemical equation 1. Therefore, the alkalization treatment directly influences on the cellulose 266
fibers, DP, and extraction of lignin and hemicellulose compounds (Jahn, 2002). 267
268
Fiber-OH + NaOH Fiber-O-Na + H2O (1) 269
270
In alkalization treatment, botanical fibers are immersed in NaOH solution for a given 271
period of time. A solution of 5% NaOH had been used to treat jute and sisal fibers for 2 to 72 272
h at room temperature (Mishra et al., 2001; Ray et al., 2001). Jacob et al. (2004) examined 273
the effect of NaOH concentrations ranging from 0.5 to 10% in treating sisal fiber-reinforced 274
composites and concluded that maximum tensile strength resulted from the 4% NaOH 275
treatment at room temperature. Mishra et al. (2002) investigated that sisal fiber-reinforced 276
composite treated with 5% NaOH had better tensile strength than treated with 10% NaOH. 277
Moreover, the alkalization treatment also significantly improves the mechanical, impact 278
fatigue, and dynamic mechanical behaviors of fiber-reinforced composites (Sarkar and Ray, 279
2004). 280
Bisanda (2000) investigated the effect of alkali treatment on the wetting ability and 281
coherence of sisal-epoxy composites. Treatment of sisal fiber in NaOH solution resulted in 282
more rigid composites with lower porosity and hence higher density. Additionally, the 283
treatment was shown to improve the adhesion characteristics due to an increase in surface 284
tension and roughness. The composites showed the improvements in the compressive 285
strength and water resistance. The suggestion was that the removal of intracrystalline and 286
intercrystalline lignin and other surface waxy substances by alkalinization substantially 287

13
increases a possibility for mechanical interlocking and chemical bonding. Besides, Ray and 288
Sarkar (2000) investigated the characterization of alkali-treated jute fibers for physical and 289
mechanical properties. The alkali treatment of jute fibers with 5% NaOH solution showed 290
that most of the changes occurred within 2 to 4 h of treatment. The weight loss, due to losing 291
their cementing capacity in the fiber structure, separating the fibers from the strands and 292
dissolution of hemicellulose, was maximized at these treatment hours. 293
Mwaikambo and Ansell (2002) recovered that alkalization of plant fibers, i.e. hemp, 294
sisal, jute, and kapok, effectively changes the surface morphologies of the fibers and their 295
crystallographic structures. However, the concentration of NaOH for alkalization has to be 296
taken into consideration. Besides, removal of surface impurities on plant fibers may be an 297
advantage for fiber to matrix adhesion. This may provide both mechanical interlocking and 298
bonding reaction because of the exposure of fiber to chemicals, e.g. resins and dyes. 299
Kaushik et al. (2010) expressed that the alkaline steam explosion of wheat straw 300
fibers with NaOH in an autoclave at pressure around 15 lb for 4 h resulted to substantial 301
breakdown of lignocellulosic strucuture, partial hydrolysis of hemicellulosic fraction, and 302
depolymerization of lignin components. Additionally, Liu and Huang (2013) represented that 303
the treatment of rice straw fibers by alkalization changed surface properties, improved 304
wettability and improved mechanical properties of rice straw fibers. 305
Moreover, the effect of fiber treatment on the mechanical properties of unidirectional 306
sisal reinforced epoxy composites was investigated by Rong et al. (2001). The treatments of 307
alkalization and heating were carried out to modify the fiber surface and its internal structure. 308
The results showed that chemical methods generally led to an active surface by introducing 309
some reactive groups, and provided the fibers with higher extensibility through partial 310
removal of lignin and hemicellulose. On the other hand, thermal treatment of the fibers 311
resulted in higher fiber stiffness due to the increased crystallinity of hard cellulose. The 312

14
treatments of sisal fiber, which increased the fiber strength and the adhesion between the 313
fiber bundles and the matrix, would favor an overall improvement of mechanical properties, 314
especially tensile property, of the laminated sisal fibers. 315
316
5. Green composites of thermoplastic starch and cellulose fibers 317
TPS can be reinforced with cellulose fibers in order to improve its low resistance to 318
mechanical stresses and moisture (Averous and Boquillon, 2004; Teixeira et al., 2009). TPS 319
is first introduced as matrix and cellulose fiber is then reinforced as biodegradable filler to 320
preserve their biodegradability. There have been many studies carried out on different starch 321
types of TPS and varied types of cellulose fibers. Wollerdorfer and Bader (1998) first 322
reported that the reinforced TPS prepared by wheat starch and flax and ramie cellulose fibers 323
was four times better (37 N/mm2) than the pure TPS. The reinforcement of cellulose fibers 324
and starch blends caused a stress increase of 52% (55 N/mm2) and 64% (25 N/mm
2), 325
respectively. 326
Curvelo et al. (2001) applied cellulose fibers from Eucalyptus urograndis pulp as the 327
reinforcement material for TPS in order to improve its mechanical properties. The green 328
composites were prepared from regular corn starch plasticized with glycerol and reinforced 329
with short cellulose fibers (16% wt/wt) from bleached pulp. The cellulose fibers were added 330
directly to the TPS in an intensive batch mixer at 170oC. The mixture was hot-pressed in 2 to 331
3 mm-thick plate and then cut to prepare the specimens for mechanical tests. The composites 332
showed an increase of 100% in tensile strength and more than 50% in modulus with respect 333
to the pure TPS. Scanning electron microscopy (SEM) of fractured surfaces revealed a very 334
good adhesion between the matrix and the fibers. 335

15
Averous and Boquillon (2004) prepared the green composites from wheat starch with 336
glycerol with and without water, and incorporated with natural cellulose fibers varying 337
lengths of 60 to 900 µm from leafwood. Particularly, TPS1 and TPS2 matrixes were prepared 338
from the ratios of dried wheat starch/glycerol/water as 70:18:12 and 65:35:0, respectively; 339
besides, all fibers were supplied from companies. After extrusion and injection molding, 340
mechanical, thermo-mechanical and thermal properties of the composites were analyzed. 341
Dynamic thermal mechanical analysis (DMTA) showed important variations of main 342
relaxation temperature, which can be linked both resulting interactions in a decrease of starch 343
chain mobility and regular reinforcing effects. The results were consistent with the static 344
mechanical behavior, which varied according to the filler content as well as fibers’ nature and 345
length. In addition, the results showed that the addition of cellulose fibers improves the 346
thermal resistance of these green composites. 347
Muller et al. (2009) investigated the effect of the addition of cellulose fibers on the 348
mechanical and physical properties of TPS films plasticized with glycerol. The green 349
composites were prepared from solutions with 3 wt% of cassava starch plasticized by 350
glycerol (0.3 g/g of starch; 23 wt%) with the addition of 0.1 to 0.5 g of eucalyptus cellulose 351
fibers (about 1.2 mm length) per gram of starch. The mechanical properties of green 352
composites conditioned at differently relative humidity (RH) values were determined through 353
tensile and stress relaxation tests. SEM micrographs of the TPS films showed a homogeneous 354
and random distribution of the cellulose fibers, without pores or cracks. The TPS films with 355
cellulose fibers were more crystalline and had higher tensile strength and rigidity, but lower 356
elongation capacity. In contrast, the addition of cellulose fibers raised the stability of TPS 357
films subjected to RH variations in relative air humidity, solving a classical drawback 358
associated with these films. Hence, the addition of cellulose fibers to TPS films is a 359
promising way to prepare stronger and more stable TPS films. 360

16
In addition, Teixeira et al. (2009) prepared the green composites based on tapioca 361
starch with either glycerol or glycerol/sorbitol (1:1) as the plasticizers and tapioca bagasse 362
cellulose nanofibers from a by-product of tapioca starch industry. The cellulose nanofibers 363
displayed a relatively low crystallinity and were found to be about 2-11 nm thick and 360 to 364
1,700 nm long. The reinforcing effect of the cellulose nanofibers evaluated by DMTA and 365
tensile tests was found to depend on the nature of the employed plasticizer. Their results 366
showed the decrease of glass transition temperature of the starch after the incorporation of 367
nanofibers, and the increase of elongation at break in tensile test. Besides, the incorporation 368
of cellulose nanofibers in the TPS matrix resulted in a decrease of its hydrophilic property 369
and capacity of water uptake, especially for the glycerol plasticized samples. 370
Wattanakornsiri et al. (2012) used different cellulose fibers from used office paper 371
and newspaper as reinforcement for TPS in order to improve their poor mechanical, thermal 372
and water resistance properties. These green composites were prepared using tapioca starch 373
plasticized by glycerol at 30% wt/wt of glycerol to starch as matrix reinforced by the 374
extracted cellulose fibers with the contents ranging from 0 to 8% wt/wt of fibers to matrix. 375
The results showed that the introduction of either office paper or newspaper cellulose fibers 376
caused the improvement of tensile strength and elastic modulus, thermal stability, and water 377
resistance for composites when compared to the pure TPS. SEM showed a good adhesion 378
between matrix and fibers. Moreover, the green composites biological degraded completely 379
after 8 weeks but required a longer time compared to the pure TPS. 380
To summarize, the previous researches of green composites prepared from TPS 381
reinforced with cellulose fibers in the different types of starch, plasticizer, plasticizer ratio, 382
fiber and fiber ratio are shown in Table 1. 383
384

17
Table 1 Previous researches of green composites prepared from TPS reinforced with 385
cellulose fibers 386
387
5.1 Preparation of green composites 388
TPS reinforced with cellulose fibers processing is similar to most conventional 389
synthetic thermoplastic processing (Curvelo et al., 2001; Janssen and Moscicki, 2006). Most 390
thermoplastic operations involve heating and forming into desired shapes, and then cooling 391
(Li, 2008). Processing techniques used on thermoplastics can also be used in the TPS 392
reinforced with cellulose fibers. These include extrusion, injection molding, internal mixing, 393
compression molding, etc. 394
Henson (1997) indicated that an extrusion is a basically thermoplastic processing, 395
comprising continuously shaping a plastic or polymer through the orifice of an appropriate 396
mold, and subsequently solidifying it into a product. It is the most efficient and widely used 397
process for melting plastic resin and mixing reinforcements into the molten plastic, leading to 398
high production volumes. Temperature in an extruder should be high enough to ensure the 399
plastic fully melted and low enough to avoid burning fibers. Typically, the extrusion is 400
necessary for injection molded composite products before injection molding because 401
injection molding machines and screws are much shorter than extruders. Therefore, the ratio 402
of length to diameter for injection molding screws is lower than for extruders. The lower 403
length to diameter ratio of the screw in injection machine makes it less efficient in mixing 404
and non-homogenous melt comparison with extruders. The reason is that if the composite is 405
processed by injection molding, prior extrusion compounding is necessary for materials (Li, 406
2008). 407

18
Advani and Sozer (2002) suggested that an injection molding is an important plastic 408
processing method with the characteristics of rapid production rates and high volume 409
production. It can manufacture geometrically complex components with accurate dimensions 410
and its process is automated. In contrast, there is limitation on fiber fraction and fiber length 411
when using the injection molding to process fiber-reinforced biological composites because 412
higher natural fiber fraction and longer fiber length will make molding difficult. Then, this 413
research involves the processing techniques of internal mixer for homogenous mixing 414
between the TPS matrix and cellulose fibers and compression molding machine for hot 415
pressing the non-reinforce TPS and composites to thick sheets. 416
Curvelo et al. (2001) prepared the TPS samples, produced by corn starch plasticized 417
by glycerol as the matrix, reinforced by Eucalyptus urograndis fibers. The composites were 418
prepared in an internal mixer connected to a torque rheometer equipped with roller rotors at 419
170 oC operating at 80 rpm for 8 min. Then, the resulting materials were compression molded 420
at 160 oC to produce 10x10 cm sheets with a 2.5 mm thickness. 421
According to the research of Corradini et al. (2007) the TPS were produced from 422
starch and zein plasticized by glycerol. These materials were mixed in an internal mixer 423
connected to a torque rheometer operating at 50 rpm for 6 min. After that, these mixtures 424
were hot pressed for 5 min at 160 oC to produce 150x120x2.5 cm molded sheets. 425
Additionally, Teixeira et al. (2009) prepared the TPS composites from cassava starch 426
plasticized using either glycerol or a mixture of glycerol and sorbitol. These matrixes were 427
reinforced by cellulose cassava bagasse nano-fibers. These mixtures were processed at 428
140±10 oC in an internal mixer equipped with roller rotors rotating at 60 rpm for 6 min. There 429
processed materials were then compression molded 140 oC into 1 and 2 mm thick plates. 430
431

19
6. Properties of green composites 432
Properties of green composites based on TPS and cellulose fibers are represented as 433
the followings. 434
6.1 Mechanical properties 435
The increase of the mechanical properties, i.e. ultimate tensile strength (UTS) and 436
elastic modulus (E), of green composites when compared with pure TPS confirms the 437
interfacial adhesion and the strong interaction between matrixes and cellulose fibers (Martins 438
et al., 2009) as shown in Figure 5 in the case of green composites prepared from corn starch 439
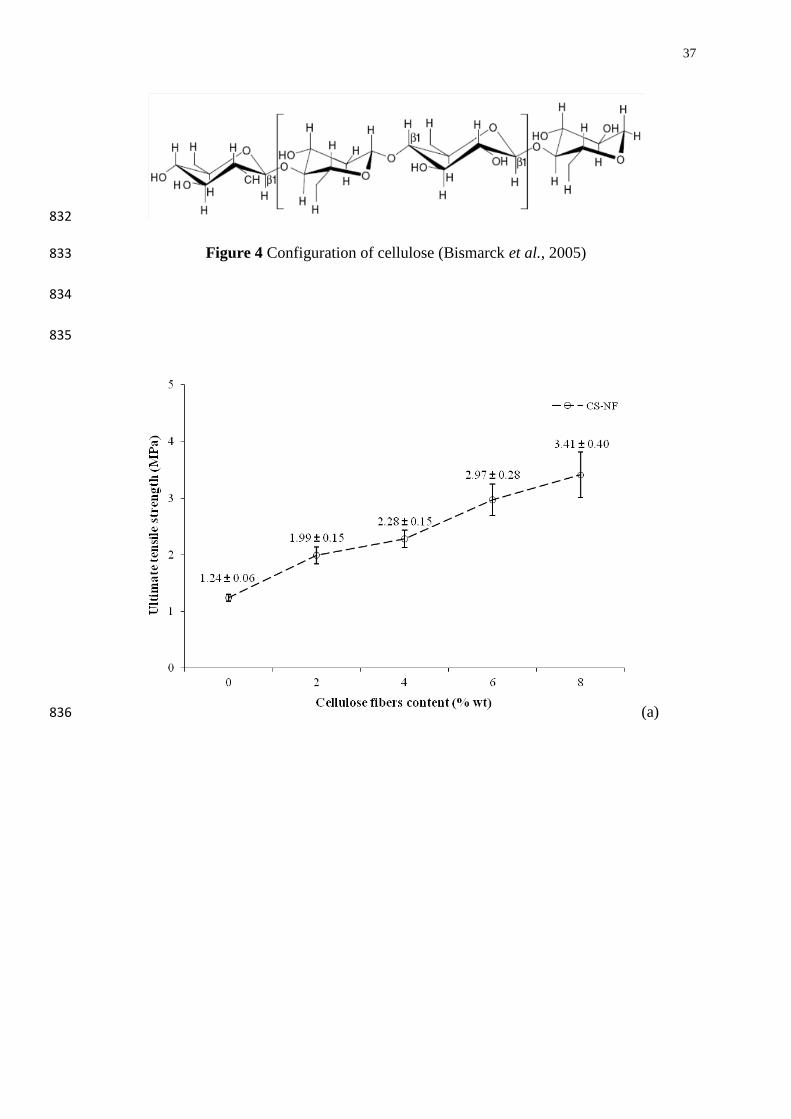
(CS) plasticized by glycerol (30% wt/wt of glycerol to starch) as matrix that was reinforced 440
with recycled paper cellulose fibers (NF) with newspaper fibers contents ranging from 0-8% 441
(wt/wt of fibers to matrix) studied by Wattanakornsiri et al. (2011). These behavior results 442
are favored by the chemical similarities between starch and cellulose fibers (Averous and 443
Boquillon, 2004; Ma et al., 2005). However, the percent elongation at break decreased with 444
respect to those of pure TPS. These results could be due to the high crystallinity of the 445
cellulose fibers, then providing higher stiffness of the green composites when compared to 446
the pure TPS (Averous and Boquillon, 2004; Martins et al., 2009; Prachayawarakorn et al., 447
2010). 448
449
Figure 5 Effect of cellulose fibers content on mechanical properties, (a) ultimate tensile 450
strength and (b) elastic modulus, of non-reinforced TPS and green composites 451
(Wattanakornsiri et al., 2011) 452
453

20
In addition, amount and structure of amylose and amylopectin molecules of the 454
various starch sources play an important role in the UTS, E and percent elongation at break 455
including the formed network of TPS (Van Soest and Borger, 1997). With higher 456
amylopectin contents the UTS and E increased but the percent elongation at break decreased. 457
These can be explained that the large content of amylopectin with the branched structure is 458
less ordered and therefore has a greater degree of entanglement, which is physical 459
interlocking of polymer chains being a direct consequence of chain overlap (Shenoy et al., 460
2005), causing higher stress and lower elongation (Janssen, 2009). Or, the higher content of 461
linear amylose molecules makes that the entanglement between matrix chains is not strong; 462
they then will slide easily along each other with lower stress and higher elongation (Graff et 463
al., 2003). Besides, glycerol gave the chains more mobility and the interactions between the 464
chains of linear amylose molecules are lowered (Janssen, 2009). 465
6.2 Thermal properties 466
Generally, two glass transitions (Tg) are detected in the green composites (Forssell 467
et al., 1997; Averous et al., 2000; Shi et al., 2006) as for Figure 6 represents differential 468
scanning calorimetry (DSC) thermal traces in the case of green composites studied by 469
Wattanakornsiri et al. (2011) that the terms CS-NF0, and CS-NF4 and 8 are used to define 470
the non-reinforced TPS, and green composites containing 4 and 8% wt/wt of fibers to matrix. 471
The two glass transitions were related to phase separation phenomena that can take place in 472
starch-glycerol system with glycerol/starch ratio larger than 0.2 (Lourdin et al., 1997). The 473
lower transition temperature (Tt1) is clearly attributed to starch-poor phase and hence related 474
to the glycerol glass transition (Averous et al., 2000), whereas the higher one (Tt2) is 475
attributed to starch-rich-phase and hence referred to the TPS glass transition (Forssell et al., 476
1997; Ma et al., 2008). Depending on the type of TPS/cellulose fiber composites, the Tt1 477
values occur in the range of -50 to -70 oC (Averous and Boquillon, 2004) that is closed to 478

21
glycerol glass transition, which is about -75 oC (Averous et al., 2000; Teixeira et al., 2009). 479
Similarity, the Tt2 values are characterized by the broad temperature transition range of 60 to 480
100 oC that is the expected values for starch conditioned at 23
oC and 50% RH (Kalichevsky 481
et al., 1992). 482
483
Figure 6 DSC scans for non-reinforced TPS and composites (Wattanakornsiri et al., 2011) 484
485
The higher amylopectin TPS composites have higher Tg values than those of lower 486
amylopectin composites. The lower molar weight of amylose and its lack of branches result 487
in a larger free volume of the lower amylopectin TPS composites, so parts of polymer chains 488
move more easily (Graff et al., 2003). This can be ascribed for the lower Tg of amylose in 489
relations to the branched amylopectin; then, the green composites with lower amylose 490
contents gave higher Tg values (Janssen, 2009). 491
Generally, thermogravimetric analysis (TGA) is used to study thermal degradation 492
of green composites as for Figure 7 represents TGA results in the case of green composites 493
studied by Wattanakornsiri et al. (2011). The behavior of TGA mass loss curves was similar 494
in the non-reinforced TPS and green composites and the weight loss gradually decreased with 495
raising of fibers contents. The degradation temperatures increase with the presence of 496
cellulose fibers in green composites. These are described by the higher thermal stability of 497
fibers compared to starch, and especially the good compatibility of both polysaccharides 498
(Martins et al., 2009). The degradation temperatures of composites are between the values of 499
matrixes and fibers with an additional effect by following the rule of matrix (Averous and 500
Boquillon, 2004). And, in general, the degradation temperatures of crystalline cellulose fibers 501
occur at higher values in comparison to TPS matrixes (Teixeira et al., 2009). 502

22
503
Figure 7 TGA scans for cellulose fibers, non-reinforced TPS and composites 504
(Wattanakornsiri et al., 2011) 505
506
Moreover, the percentage weight losses decrease with the addition of cellulose 507
fibers. This is explained that at equilibrium the composites had lower water content when 508
compared to the pure matrixes, and the fibers crystallinity decreased their polar character 509
(Averous et al., 2001; Averous & Boquillon, 2004). Hence, the presence of fibers in the 510
matrixes decreased the inside water content and the diverse interactions brought by the fibers 511
took original water site of TPS matrixes (Averous & Boquillon, 2004). The addition of 512
cellulose fibers improve the thermal resistance of the pure TPS due to the good thermal 513
stability of crystalline structure for cellulose fibers and the good interaction between TPS 514
matrixes and cellulose fibers (Ma et al., 2008). 515
6.3 Water absorption properties 516
Low water resistance is a major drawback of TPS for many practical applications. 517
In fact, TPS could absorb an amount of water from the environmental humidity; as a result, 518
the mechanical properties could drastically drop down (Kalichevsky and Blanshard, 1993). 519
The presence of cellulose fibers decreased the amount of water absorption. These can be 520
mainly ascribed as the addition of the cellulose fibers; in fact, they are less hydrophilic in 521
comparison to starch (Averous and Boquillon, 2004; Ma et al., 2005) and can absorb the part 522
of glycerol with a reduction of hydrophilic behavior of TPS (Curvelo et al., 2001). Besides, 523
the presence of less hydrophilic cellulose fibers significantly reduced the water absorption of 524
TPS probably also because of the constraint exerted by the fibers at the interface on the 525

23
matrix swelling (Wattanakornsiri et al., 2011). Besides, amylopectin-rich TPS are more 526
sensitive to water absorption than amylose-rich TPS (Van Soest and Essers, 1997). 527
6.4 Ageing properties 528
Ageing property (AP) is an important issue for TPS after processing (Averous et al., 529
2000). The green composites are tested following the variation of mechanical properties 530
during several weeks after molding (Averous and Boquillon, 2004; Shi et al., 2006). The E 531
was used to estimate the ageing properties as TPS stabilization that is the ratio of E at 6 532
weeks divided by the E at 2 weeks (Averous and Boquillon, 2004; Wattanakornsiri, 2012). 533
The presence of higher cellulose fibers contents in the green composites decreases or 534
increases the ageing values that tends into the ageing stabilization value equal to one 535
depending on the type of TPS and cellulose fiber composites (Wattanakornsiri, 2012). This is 536
because of the fiber-matrix interactions that provide a kind of stabilizing three-dimensional 537
network based on low intermolecular bonds (Averous and Boquillon, 2004) and due to the 538
difference of re-crystallization or post-crystallization of starch chains between amylose and 539
amylopectin molecules (Averous et al., 2000). 540
Generally, in the TPS retrogradation taken place after cooling of gelatinized starch, 541
while amylose re-crystallization is irreversible, amylopectin re-crystallizes reversibly (Parker 542
and Ring, 1995). The crystalline structure of the higher amylose content composites is rather 543
relatively stable (Yu and Christie, 2001), then provided the higher ageing values. 544
Concurrently, during ageing the amylose and amylopectin also co-crystallize to form cross-545
links between amylose and/or amylopectin that these cross-links can also increase the E 546
(Kalichevsky and Blanshard, 1993; Yu and Christie, 2001). Besides, the re-crystallization of 547
amylopectin could contribute to the ageing or the life time of TPS, being relatively short. 548

24
Thus, TPS should be composted of high amylose content due to the effect of retrodegradation 549
(Van Soest and Knooren, 1997; Ma et al., 2005). 550
6.5 Functional groups 551
Fourier transform infrared spectroscopy (FT-IR) is a powerful technique for 552
identifying types of chemical bonds of polymer composites in a molecule by producing 553
infrared absorption spectrum that is like a molecule finger print. FT-IR spectra in the case of 554
green composites investigated by Wattanakornsiri et al. (2012) display the typical profiles of 555
polysaccharide as illustrated in Figure 8 that the terms CS-NF0, and NF4 and 8 are defined to 556
the non-reinforced TPS, and green composites containing 4 and 8% wt/wt of fibers to matrix. 557
The peaks in the range of 1,026-1,027 and 1,079-1,155 cm-1
are attributed to C-O stretching 558
of C-O-C group in the anhydroglucose ring and of C-O-H group, respectively. The wave 559
numbers in the range of 1,414-1,454 cm-1
are designed for O-H bonding (Prachayawarakorn 560
et al., 2011). The peak positions in the range of 1,638-1,639 cm-1
are owing to the bound 561
water present in the non-reinforced TPS and composites. The bands of 2,931 cm-1
are 562
associated with C-H stretching. Besides, the bands belonging to hydrogen bonded hydroxyl 563
(O-H) group appear in the range of 3,414-3,420 cm-1
that are attributed to the complex 564
vibrational stretching, associated with free, inter and intra molecular bound hydroxyl groups 565
(Wu et al., 2009; Glicia-Garcia et al., 2011). 566
Especially in the last case, the bands slightly shifted to lower wave numbers by the 567
presence of cellulose fibers; referring that the increase of intermolecular hydrogen bonding 568
by the addition of cellulose fibers. This phenomenon is ascribed that when polymers are 569
compatible, a distinct interaction, i.e. hydrogen bonding or dipolar interaction, exists between 570
the chains of TPS matrix and cellulose fibers, providing the changes of FT-IR spectra on the 571
composites, e.g. band shifts and broadening (Prachayawarakorn et al., 2010). 572

25
573
Figure 8 FT-IR spectra for cellulose fibers, non-reinforced TPS and composites 574
(Wattanakornsiri et al., 2012) 575
576
6.6 Morphology 577
Scanning electron microscopy (SEM) is one of the most worldwide using techniques 578
for studying green composites’ morphologies and compatibilities. SEM micrograph of the 579
fractured surface of green composites studied by Wattanakornsiri et al. (2011) is illustrated in 580
Figure 9. Not only the cellulose fibers appear to be embedded in the matrix and good 581
adhesion features but also, in fact, the TPS matrix remains tightly jointed to the fibers even 582
after a cryo-fracture test without any evident debonding phenomena (Curvelo et al., 2001). 583
Moreover, the absence of fiber pullout indicates their good interfacial adhesion (Ma et al., 584
2005; Wattanakornsiri et al., 2011) 585
586
Figure 9 SEM micrograph of fragile fractured surface of green composites (Wattanakornsiri 587
et al., 2011) 588
589
6.7 Biodegradation 590
Starch is a nutrient for many microorganisms and once water is present in the starch 591
structure of TPS it is readily biodegraded. Starch easily absorbs water, resulting in 592
disintegration of green composites by partial solubility. Partially solubilised starch is even 593
more readily biodegraded by enzymes principally from microorganisms (Shanks and Kong, 594
2012). The green composites prepared by TPS and cellulose fibers were fully biodegraded. 595

26
Biodegradation rate showed that when fiber content increased the green composites degraded 596
slower when compared to the pure TPS (Prachayawarakorn et al., 2011; Wattanakornsiri et 597
al., 2012). The more difficult biodegradability is related to the more hydrophobic cellulose 598
fibers when compared to starch. Besides, this occurrence is due to the phase compatibility of 599
TPS matrix and cellulose fibers (Prachayawarakorn et al., 2011). 600
601
7. Concluding remarks 602
Green composites have rapidly evolved over the last decade due to the approaching 603
depletion of fossil fuels and worldwide environmental problems resulted from petroleum-604
derived plastics. The main advantage of green composites is their biological decomposition 605
with organic wastes and returned to enrich the soil. Their use would be useful to the 606
environment not only reduce injuries to wild animals but also lessen the labor cost for 607
removal plastic wastes. In addition, their decomposition would help to increase the longevity 608
and stability of landfills by reducing the volume of garbage as well as they could be recycled 609
to useful monomers and oligomers by microbial activities. This review outlines the 610
significance of research and development of green composites based on thermoplastic starch 611
(TPS) and cellulose fibers derived from naturally renewable resources. These composites 612
could be used as commodity plastics like biodegradable artifacts, e.g. organic waste bags and 613
seeding grow bags, being cheap, abundant and recyclable. However, the future growth and 614
sustainability of green composites is reliant to continued research, in particularly 615
improvement of hydrophobic character, surface modification and advanced processing 616
technique. These should be more understood that they are expected to replace petroleum-617
derived plastics. 618
619

27
7. References 620
Advani, S.G. and Sozer, E.M. 2002. Process Modeling in Composites Manufacturing, Marcel 621
Dekker, New York, U.S.A. 622
Angellier, H., Molina-Boisseau, S., Dole, P. and Dufresne, A. 2006. Thermoplastic-waxy 623
maize starch nanocrystals nanocomposites. Biomacromolecules 7, 531-539. 624
Averous, L. and Boquillon, N. 2004. Biocomposites based on plasticized starch: thermal and 625
mechanical behaviours. Carbohydrate Polymers 56, 111-122. 626
Averous, L., Fringant, C. and Moro, L. 2001. Plasticized starch-cellulose interactions in 627
polysaccharide composites. Polymer 42, 6571-6578. 628
Bikiaris, D., Pavlidou, E., Prinos, J., Aburto, J., Alric, I., Borredon, E. and Panayiotou, C. 629
1998. Biodegradation of octanoated starch and its blends with LDPE. Polymer 630
Degradation and Stability 60, 437-447. 631
Bisanda, E.T.N. 2000. The effect of alkali treatment on the adhesion characteristics of sisal 632
fibres. Applied Composite Materials 7, 331-339. 633
Bismarck, A., Mishra, S. and Lampke, T. 2005. Plant fibers as reinforcement for green 634
composites. In Natural Fibers, Biopolymers and Biocomposites, A.K. Mohanty, M. 635
Misra and L.T. Drazl, editors. CRC Press (Taylor & Francis Group), New York, pp 636
37-108. 637
Burgt Van Der, M.C., Woude Van Der M.E. and Janssen L.P.M.B. 1996. The influence of 638
plasticizer on extruded thermoplastic starch. Journal Vinyl Additive and Technology 639
2, 170-174. 640

28
Carvalho, A.J.F.D., Curvelo, A.A.S. and Agnelli, J.A.M. 2002. Wood pulp reinforced 641
thermoplastic starch composites. International Journal of Polymeric Materials 51, 642
647-660. 643
Corradini, E., Carvalho, A.J.F.D. and Curvelo, A.A.D.S. 2007. Preparation and 644
characterization of thermoplastic starch/zein blends. Materials Research 10, 227-231. 645
Curvelo, A.A.S., Carvalho, A.J.F.D. and Angelli, J.A.M. 2001. Thermoplastic starch-646
cellulosic fibers composites: preliminary results. Carbohydrate Polymers 45, 183-188. 647
Chadehumbe, C. 2006. Tensile properties of thermoplastic starch and its blends with 648
polyvinyl butyral and polyamides. Ph.D. dissertation, University of Pretoria. 649
Chiou, B., Glenn, G.M., Imam, S.H., Inglesby, M.K., Wood, D.F. and Orts, W.J. 2005. 650
Starch polymer: Chemistry, engineering, and novel products. In Natural Fibers, 651
Biopolymers and Biocomposites, A.K. Mohanty, M. Misra and L.T. Drazl, editors. 652
CRC Press (Taylor & Francis Group), New York, pp 639-669. 653
Dufresne, A. and Vignon, M.R. 1998. Improvement of starch film performance using 654
cellulose microfibrils. Macromolecules 31, 2693-2696. 655
Dufresne, A., Dupeyre, D. and Vignon, M.R. 2000. Cellulose microfibrils from potato tuber 656
cells: processing and characterization of starch cellulose microfibril composites. 657
Journal of Applied Polymer Science 76, 2080-2092. 658
Espigule, E., Puigvert, X., Vilaseca, F., Mendez, J.A., Mutje, P. and Girones, J. 2013. 659
Thermoplastic starch-based composites reinforced with rape fibers: water uptake and 660
thermomechanical properties. Bioresources 8, 2620-2630. 661

29
Forssell, P.M., Mikkila, J.M., Moates, G.K. and Parker, R. 1997. Phase and glass transition 662
behavior of concentrated barley starch-glycerol-water mixtures, a model for 663
thermoplastic starch. Carbohydrate Polymers 34, 275-282. 664
Galicia-Garcia, T., Martinez-Brutos, F., Jimenez-Arevalo, O., Martinez, A.B., Ibarra-Gomez, 665
R., Gaytan-Martinez, M. and Mendoza-Duarte, M. 2011. Thermal and microstructural 666
characterization of biodegradable films prepared by extrusion-calendering process. 667
Carbohydrate Polymers 83, 354-361. 668
Graff, R.A.D, Karman, A.P. and Janssen, L.P.B.M. 2003. Material properties and glass 669
transition temperatures of different thermoplastic starches after extrusion processing. 670
Starch/Starke 55, 80-86. 671
Henson, F. 1997. Plastic Extrusion Technology (2nd
ed.), Hanser, New York, U.S.A. 672
Hulleman, S.H.D., Jansen, F.H.P. and Feli, H. 1998. The role of water during the 673
plasticization of native starches. Polymer 39, 2034-2048. 674
Jacob, M., Thomas, S. and Varughese, K.T. 2004. Mechanical properties of sisal/oil palm 675
hybrid fiber reinforced natural rubber composites. Composites Science and 676
Technology 64, 955-965. 677
Janssen, L.P.B.M. 2009. Influence of process condtions on the physical properties of TPS. In 678
Thermoplastic Starch, L.P.B.M. Janssen and L. Moscicki, editors. WILEY-VCH 679
Verlag GmbH & Co. KGaA, Weinheim, pp 159-171. 680
Janssen, L.P.B.M. and Moscicki, L. 2006. Thermoplastic starch as packaging material. Acta 681
Sci. Pol., Technica Agraria 5, 19-25. 682

30
Jahn, A. 2002. Characterization of alkali treated flax fibers by means of FT raman 683
spectroscopy and environmental scanning electron microscopy. Spectrochimica Acta 684
Part A: Molecular and Biomolecular Spectroscopy 58, 2271-2279. 685
John, M.J. and Thomas, S. 2008. Biofibres and biocomposites. Carbohydrate Polymers 71, 686
343-364. 687
Kalichevsky, M.T. and Blanshard, J.M.V. 1993. The effect of fructose and water on the glass 688
transition of amylopectin. Carbohydrate Polymers 20, 107-113. 689
Kalichevsky, M.T., Jaroszkiewicz, E.M., Ablett, S., Blanshard, J.M.V. and Lillford, P.J. 690
1992. The glass transition of amylopectin measured by DSC, DMTA and NMR. 691
Carbohydrate Polymers 18, 77-88. 692
Kaushik, A., Singh, M. and Verma, G. 2010. Green nanocomposites based on thermoplastic 693
starch and steam exploded cellulose nanofibrils from wheat straw. Carbohydrate 694
Polymers 82, 337-345. 695
Ke, T. and Sun, X. 2001. Effect of moisture content and heat treatment on the physical 696
properties of starch and poly (lactic acid) blends. Journal of Applied Polymer Science 697
81, 3069-3082. 698
Kim, J., Kim, W. and Shin, M.A. 1997. Comparative study on the retrogradation of rice 699
starch gels by DSC, X-ray and α-amylose methods. Starch/Starke 49, 71-75. 700
Li, X. 2008. Development of flax fiber-reinforced polyethylene biocomposites by injection 701
molding. Ph.D. dissertation, University of Saskatchewan. 702
Liu, J. and Huang, C. 2013. Biodegradable composites from rice straw and cornstarch 703
adhesives. Advance Journal of Food Science and Technology 5, 41-45. 704

31
Lourdin, D., Bizot, H. and Colonna, P. 1997. “Antiplasticization” in starch-glycerol films?. 705
Journal of Applied Polymer Science 63, 1047-1053. 706
Lu, D.R., Xiao, C.M. and Xu, S.J. 2009. Starch-based completely biodegradable polymer 707
materials. Express Polymer Letter 6, 366-375. 708
Ma, X., Chang, P.R. and Yu, J. 2008. Properties of biodegradable thermoplastic pea 709
starch/carboxymethyl cellulose and pea starch/microcrystalline cellulose composites. 710
Carbohydrate Polymers 72, 369-375. 711
Ma, X., Yu, J. and Kennedy, J.F. 2005. Studies on the properties of natural fibers-reinforced 712
thermoplastic starch composites. Carbohydrate Polymers 62, 19-24. 713
Martins, I.M.G., Magina, S.P., Oliveira, L., Freire, C.S.R., Silvestre, A.J.D., Neto, C.P. and 714
Gandini, A. (2009). New biocomposites based on thermoplastic starch and bacterial 715
cellulose. Composites Science and Technology 69, 2163-2168. 716
Mishra, S., Misra, M., Tripathy, S.S., Nayak, S.K. and Mohanty, A.K. 2001. Graft 717
copolymerization of acrylonitrile on chemically modified sisal fibers. Macromolecular 718
Materials and Engineering 286, 107-113. 719
Mishra, S., Misra, M., Tripathy, S.S., Nayak, S.K. and Mohanty, A.K. 2002. The influence of 720
chemical surface modification on the performance of sisal polyester biocomposites. 721
Polymer Composites 23, 164-170. 722
Mo, X., Zhong, Y., Liang, C. and Yu, S. 2010. Studies on the properties of banana fibers-723
reinforced thermoplastic cassava starch composites: preliminary results. Advanced 724
Materials Research 87-88, 439-444. 725

32
Mohanty, A.K., Misra, M. and Drzal, L.T. 2002. Sustainable bio-composites from renewable 726
resources: opportunities and challenges in the green materials world. Journal of 727
Polymers and the Environment 10, 19-26. 728
Mohanty, A.K., Misra, M., Drzal, L.T., Selke, S.E., Harte, B.R. and Hinrichsen, G. 2005. 729
Natural fibers, biopolymers, and biocomposites: an introduction. In Natural Fibers, 730
Biopolymers and Biocomposites, A.K. Mohanty, M. Misra and L.T. Drazl, editors. 731
CRC Press (Taylor & Francis Group), New York, pp 1-36. 732
Muller, C.M.O., Laurindo, J.B. and Yamashita, F. 2009. Effect of cellulose fibers on the 733
crystallinity and mechanical properties of starch-based films at different relative 734
humidity values. Carbohydrate Polymers 77, 293-299. 735
Mwaikambo, L.Y. and Ansell, M.P. 2002. Chemical modification of hemp, sisal, jute, and 736
kapok fibers by alkalization. Journal of Applied Polymer Science 84: 2222-2234. 737
Odling-Smee, L. 2007. Biofules bandwagon hits a rut. Nature 446, 483. 738
Park, J.W., Im, S.S., Kim, S.H. and Kim, Y.H. 2000. Biodegradable polymer blends of poly 739
(L-lactic acid) and gelatinized starch. Polymer Engineering and Science 40, 2539-740
2550. 741
Patel, M. and Narayan, R. 2005. How sustainable are biopolymers and biobased products? 742
The hope, the doubts, and the reality. In Natural Fibers, Biopolymers and 743
Biocomposites, A.K. Mohanty, M. Misra and L.T. Drazl, editors. CRC Press (Taylor 744
& Francis Group), New York, pp 833-853. 745
Prachayawarakorn, J., Chaiwatyothin, S., Mueangta, S. and Hanchana, A. 2013. Effect of jute 746
and kapok fibers on properties of thermoplastic cassava starch composites. Materials 747
and Design 47, 309-315. 748

33
Prachayawarakorn, J., Ruttanabus, P. and Boonsom, P. 2011. Effect of cotton fiber contents 749
and lengths on properties of thermoplastic starch composites prepared from rice and 750
waxy rice starches. Journal of Polymers and the Environment, 274-282. 751
Prachayawarakorn, J., Sangnitidej, P. and Boonpasith, P. 2010. Properties of thermoplastic 752
rice starch composites reinforced by cotton fiber or low-density polyethylene. 753
Carbohydrate Polymers 81, 425-433. 754
Ray, D. and Sarkar, B.K. (2000). Characterization of alkali-treated jute fibers for physical 755
and mechanical properties. Journal of Applied Polymer Science 80, 1013-1020. 756
Ray, D., Sarkar, B.K., Rana, A.K. and Bose, N.R. 2001. Effect of alkali treated jute fibers on 757
composite properties. Bulletin of Material Sciences 24, 129-135. 758
Rodriguez-Gonzalez, F.J., Ramsay, B.A. and Favis, B.D. 2010. Rheological and thermal 759
properties of thermoplastic starch with high glycerol content. Carbohydrate Polymers 760
58, 139-147. 761
Rong, M.X., Zhang, M.Q., Liu, Y., Yang, G.C. and Zeng, H.M. 2001. The effect of fiber 762
treatment on the mechanical properties of unidirectial sisal-reinforced epoxy 763
composites. Composites Science and Technology 61, 1437-1447. 764
Rowell, R.M., Young, R.A. and Rowell, J.K. 1997. Paper and Composites from Agro-based 765
Resources, CRC Lewis, Boca Raton, FL. 766
Shanks, R. and Kong, I. 2012. Thermoplastic starch. In Thermoplastic Elastomers, A. El-767
Sonbati, editor. InTech Europe, Rijeka, pp 95-116. 768
Sarkar, B.K. and Ray, D. 2004. Effect of the defect concentration on the impact fatigue 769
endurance of untreated and alkali treated jute-vinylester composites under normal and 770
liquid nitrogen atmosphere. Composites Science and Technology 64, 2213-2219. 771

34
Shenoy, S.L., Bates, W.D., Frisch, H.L. and Wnek, G.E. 2005. Role of chain entanglements 772
on fiber formation during electrospinning of polymer solutions: good solvent, non-773
specific polymer-polymer interaction limit. Polymer 46, 3372-3384. 774
Shi, R., Liu, Q., Ding, T., Han, Y., Zhang, L., Chen, D. and Tian, W. 2006. Ageing of soft 775
thermoplastic starch with high glycerol content. Journal of Applied Polymer Science 776
103, 574-586. 777
Souza, R.C.R. and Andrade, C.T. 2001. Processing and properties of thermoplastic starch and 778
its blends with sodium alginate. Journal of Applied Polymer Science 81, 412-420. 779
Sugih, A.K. 2008. Synthesis and properties of starch based biomaterials. Ph.D. dissertation, 780
University of Groningen. 781
Szegda, D. 2009. Experimental investigation and computational modeling of the 782
thermoforming process of thermoplastic starch. Ph.D. dissertation, Brunel University. 783
Teixeira, E.D.M., Pasquini, D., Curvelo, A.A.S., Corradini, E., Belgacem, M.N. and 784
Dufresne, A. 2009. Cassava bagasse cellulose nanofibrils reinforced thermoplastic 785
cassava starch. Carbohydrate Polymers 78, 422-431. 786
Thuwall, M., Boldizar, A. and Rigdahl, M. 2006. Extrusion processing of high amylase 787
potato starch materials. Carbohydrate Polymers 65, 441-446. 788
Van Soest, J.J.G. and Borger, D.B. 1997. Structure and properties of compression-molded 789
thermoplastic starch materials from normal and high-amylose maize starches. Journal 790
of Applied Polymer Science 64, 631-644. 791
Van Soest, J.J.G. and Essers, P. 1997. Influence of amylose-amylopectin ratio on properties 792
of extruded starch plastic sheets. Journal of Macromolecular Science, Part A 34, 793
1665-1689. 794

35
Van Soest, J.J.G. and Knooren, N. 1997. Influence of glycerol and water content on the 795
structure and properties of extruded starch plastic sheets during aging. Journal of 796
Applied Polymer Science 64, 1411-1422. 797
Wattanakornsiri, A. 2012. Thermoplastic starch reinforced with recycled paper cellulose 798
fibers. Ph.D. dissertation, Burapha University. 799
Wattanakornsiri, A., Pachana, K., Kaewpirom, S., Sawangwong, P. and Migliaresi, C. 2011. 800
Green composites of thermoplastic corn starch and recycled paper cellulose fibers. 801
Songklanakarin Journal of Science and Technology 33, 461-467. 802
Wattanakornsiri, A., Pachana, K., Kaewpirom, S., Traina, M. and Migliaresi, C. 2012. 803
Preparation and properties of green composites based on tapioca starch and differently 804
recycled paper cellulose fibers. Journal of Polymers and the Environment 20, 801-805
809. 806
Willett, J.L. and Doane, W.M. 2002. Effect of moisture content on the tensile properties of 807
starch/poly (hydroxyester ether) composite materials. Polymer 43, 4413-4420. 808
Wollerdorfer, M. and Bader, H. 1998. Influence on natural fibers on the mechanical 809
properties of biodegradable polymers. Industrial Crops and Products 8, 105-112. 810
Wu, R.L., Wang, X.L., Li, F., Li, H.Z., and Wang, Y.Z. 2009. Green composite films 811
prepared from cellulose, starch and lignin in room-temperature ionic liquid. 812
Bioresource Technology 100, 2569-2574. 813
Yazdani, S.S. and Gonzalez, R. 2007. Aerobic fermentation of glycerol: a path to economic 814
viability for the biofuels industry. Current Opinion in Biotechnology 18, 213-219. 815
Yu, J., Chen, S., Gao, J., Huawu, Z., Zheng, H., Zhang, J. and Lin, T.A. 1998. Study on the 816
properties of starch/glycerine blend. Starch/Starke 50, 246-250. 817

36
Yu, J., Wang, N. and Ma, X. 2005. The effects of citric acid on the properties of 818
thermoplastic starch plasticized by glycerol. Starch/Starke 57, 494-504. 819
Yu, L. and Christie, G. 2001. Measurement of starch thermal transition using differential 820
scanning calorimetry. Carbohydrate Polymers 46, 179-184. 821
Figures 822
823
Figure 1 Chemical structure of amylose (Chiou et al., 2005) 824
825
826
Figure 2 Chemical structure of amylopectin (Chiou et al., 2005) 827
828
829
Figure 3 Chemical structure of cellulose (Bismarck et al., 2005) 830
831
37
832
Figure 4 Configuration of cellulose (Bismarck et al., 2005) 833
834
835
(a) 836

38
(b) 837
Figure 5 Effect of cellulose fibers content on mechanical properties, (a) ultimate tensile 838
strength and (b) elastic modulus, of non-reinforced TPS and green composites 839
(Wattanakornsiri et al., 2011) 840
841
842
Figure 6 DSC scans for non-reinforced TPS and composites (Wattanakornsiri et al., 2011) 843
844

39
845
Figure 7 TGA scans for cellulose fibers, non-reinforced TPS and composites 846
(Wattanakornsiri et al., 2011) 847
848
Figure 8 FT-IR spectra for cellulose fibers, non-reinforced TPS and composites 849
(Wattanakornsiri et al., 2012) 850
851

40
852
Figure 9 SEM micrograph of fragile fractured surface of green composites 853
(Wattanakornsiri et al., 2011) 854
855
856
857
858
859
860
861
862
863
864
865
Table 866
Table 1 Previous researches of green composites prepared from TPS reinforced with 867
cellulose fibers 868

41
Researchers Starch
type
Plasticizer ratio
(% wt/wt of
plasticizer to
starch)
Fiber type Fiber ratio
(% wt/wt of
fibers to
matrix*)
Curvelo et al.
(2001)
Corn 30 wt% (glycerol) Eucalyptus
urograndis
0 and16
Averous and
Boquillon
(2004)
Wheat TPS1: 18 wt%
(glycerol), and 12
wt% (water)
TPS2: 35 wt%
(glycerol)
Leafwood, and
paper
pulpfibers from
broad-leaved
species
TPS1: 0, 15,
and 30
TPS2: 0, 4, 8,
10, 12, 16, and
20
Ma et al.
(2005)
Corn 15.4 wt% (urea), and
7.7 wt%
(formamide)
Winceyette 0, 5, 10, 15,
and 20
Muller et al.
(2009)
Cassava 23 wt% (glycerol) Eucalyptus 0, 7, 19, and 28
Teixeira et al.
(2009)
Cassava
(tapioca)
TPS1: 30 wt%
(glycerol)
TPS2: 15 wt%
(glycerol), and 15
wt% (sorbitol)
Cassava
bagasse
TPS1 and TPS2:
0, 5, 10, and 20
Wattanakornsiri
et al. (2012)
Tapioca 30 wt% (glycerol) Used office
paper and
newspaper
0, 2, 4, 6 and 8
Remark: * Starch plus plasticizer 869
870





























