Embed Size (px)
Citation preview

3. Review of Literature
3.1. DRUG PROFILE OF KETOROLAC TROMETHAMINE
Chemical Structure:
Chemical name:5-(benzoyl)-2,3-dihydro-1H-pyrrolizine-1-carboxylicacid
Molecular formula: C15H13NO3
Molecular weight: 255.268
Description:
Melting point: 165-1670C
State: Solid
Category: Anti-Inflammatory Agent, Non-Steroidal, Cyclooxygenase inhibitors
Solubility in Water: 200 g/l
Dose: 10 mg given three to four times a day orally
Storage: Store in a cool and dry place
Pharmacology: Ketorolactromethamine (KT), an anti-inflammatory agent with analgesic
and anti-pyretic properties, is used to treat osteoarthritis and control acute pain. It is a
peripherally acting analgesic. The biological activity of ketorolac tromethamine is
associated with the S-form. Ketorolac tromethamine possesses no sedative or anxiolytic
properties.
Indications: For the short-term management of moderately severe acute pain that
requires analgesia at the opioid level, usually in a postoperative setting.

Contraindications: Ketorolac (KT) is contraindicated in patients with a
previously demonstrated hypersensitivity to ketorolac and in patients with the complete
or partial syndrome of nasal polyps, angioedema, bronchospastic reactivity or other
allergic manifestations to aspirin or other non-steroidal anti-inflammatory drugs (due to
possibility of severe anaphylaxis). Ketorolac should be avoided with all NSAID’s in
patients with renal dysfunction. Further it use in patients at highest risk was detrained
especially in elderly and those with fluid imbalances or with compromised renal function
(e.g., heart failure, diuretic use, cirrhosis, dehydration, and renal insufficiency).
Mechanism of action: The anti-inflammatory effects of KT was due to inhibition
of both cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) pathways. It leads
to inhibition of prostaglandin synthesis with decreased formation of precursors of
prostaglandins and thromboxanes from arachidonic acid. The resultant reduction in
prostaglandin synthesis and activity may be at least partially responsible for many of the
adverse and therapeutic, effects. Analgesia is probably produced peripherally due to
blockade of pain impulse generation results from decreased prostaglandinactivity.
Table 3.1.1. Pharmacokinetic properties of ketorolac tromethamine

PARAMETER DATA
Absorption Rapidly and completely absorbed after oral
administration
Bioavailability 100% (All routes)
Site of absorption Throughout the GIT
Plasma protein binding 99%
Biotransformation Primarily hepatic. Less than 50% of a dose is
metabolized. The major metabolites are a
glucuronate conjugate, which may also be
formed in the kidney, and p-hydroxy
ketorolac. Neither metabolite has significant
analgesic activity.
Route of excretion Renal: 91.4% (mean)
Biliary: 6.1% (mean)
Biological half life 2.5-5 hours
BCS class Class I
Table 3.1.2. Toxic effects of ketorolac tromethamine
BODY SYSTEMS EFFECTS
General Edema Less frequentlyhypersensitivity reactions (such as
anaphylaxis, bronchospasm, laryngeal edema, tongue edema,
hypotension), flushing, weight gain, or fever. Very infrequently,
asthenia
Cardiovascular Hypertension. Less frequently, palpitation, pallor, or fainting
(syncope
Dermatalogic Rash or pruritus. Less frequently, Lyell's syndrome, Stevens-
Johnson syndrome, musculo-papularrash, exfoliative dermatitis.
Gastrointestinal Less frequently, peptic ulceration, gastrointestinal hemorrhage,
gastrointestinal perforation, melena, rectal bleeding, gastritis.
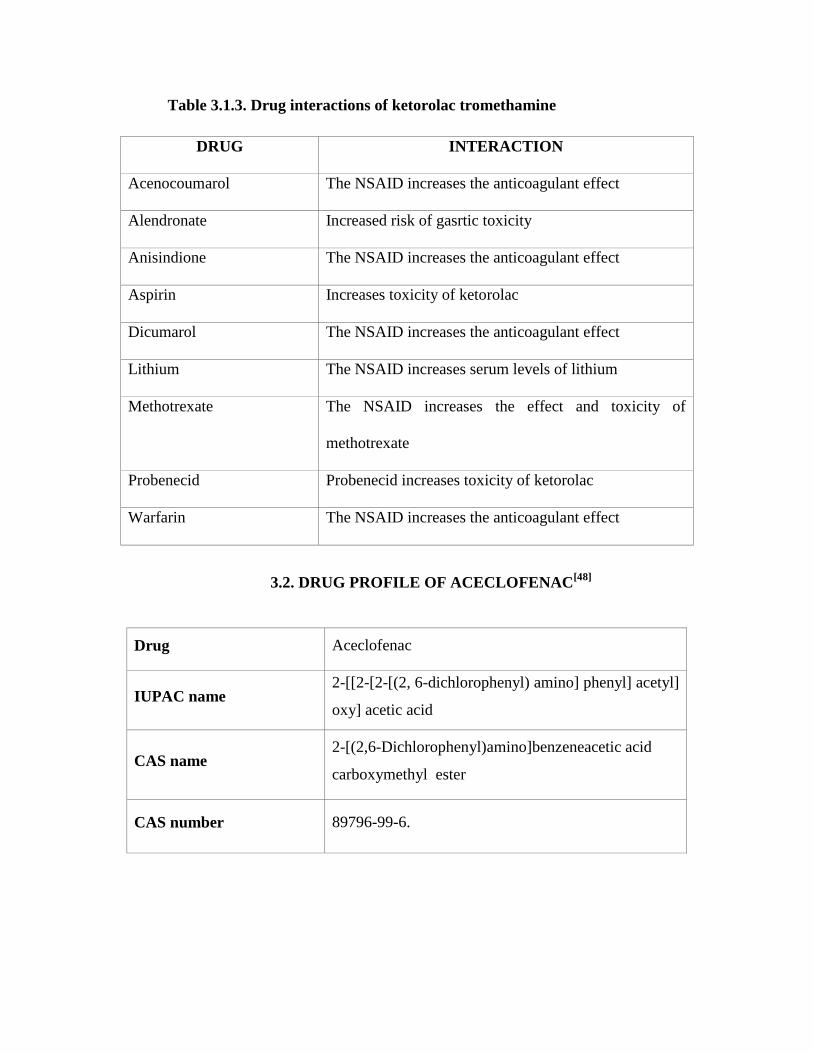
Table 3.1.3. Drug interactions of ketorolac tromethamine
DRUG INTERACTION
Acenocoumarol The NSAID increases the anticoagulant effect
Alendronate Increased risk of gasrtic toxicity
Anisindione The NSAID increases the anticoagulant effect
Aspirin Increases toxicity of ketorolac
Dicumarol The NSAID increases the anticoagulant effect
Lithium The NSAID increases serum levels of lithium
Methotrexate The NSAID increases the effect and toxicity of
methotrexate
Probenecid Probenecid increases toxicity of ketorolac
Warfarin The NSAID increases the anticoagulant effect
3.2. DRUG PROFILE OF ACECLOFENAC[48]
Drug Aceclofenac
IUPAC name 2-[[2-[2-[(2, 6-dichlorophenyl) amino] phenyl] acetyl]
oxy] acetic acid
CAS name 2-[(2,6-Dichlorophenyl)amino]benzeneacetic acid
carboxymethyl ester
CAS number 89796-99-6.

Chemical structure
Molecular formula C16H13Cl2NO4
Molecular weight 354.18472 g/mol
Description A white or almost white crystalline powder
Melting point 149 - 153˚C
Solubility Insoluble in water, freely soluble in acetone, soluble
in 96% ethanol.
Dose The usual dose is 100 mg given twice daily by
mouth, one tablet in the morning and one in the
evening.
Specifications
Loss on drying : ≤0.5%
Heavy metals : ≤10ppm
Sulphated ash : ≤0.1%
Assay : 99.0%-101.0%

Pharmacokinetics
Aceclofenac is rapidly and completely absorbed after
oral administration, peak plasma concentrations are
reached 1 to 3 hours after an oral dose. Aceclofenac is
metabolized to a major metabolite, 4'-
hydroxyaceclofenac and to a number of other
metabolites including 5-hydroxyaceclofenac, 4'-
hydroxydiclofenac, diclofenac and 5-
hydroxydiclofenac.
Oral bioavailability : nearly 100%
Effect of food : Rate of absorption will
be reduced
Protein binding : 99 %
Volume of distribution : approximately 25 litres.
Peak plasma concentration : 1-3 hours.
Plasma elimination half-life: 2-4 hours.
Metabolism : 20% primary
Steady state concentration : 3 days.
Urinary clearance : 20 %
Fecal clearance : 20 %
PHARMACODYNAMICS
Anti – inflammatory
activity
Aceclofenac relieves pain and inflammation through a
variety of mechanisms and in addition exerts
stimulatory effects on cartilage matrix synthesis.
The anti-inflammatory effects of aceclofenac have
been shown in both acute and chronic inflammation.
It inhibits various mediators of pain and inflammation
including:
PGE2 via cyclooxygenase inhibition after
intracellular metabolism to 4’ hydroxy-
aceclofenac and diclofenac in human
rheumatoid synovial cells and other
inflammatory cells.

IL-1β, IL-6 and tumor necrosis factor in
human osteoarthritic synovial cells and
human articular chondrocytes.
Reactive oxygen species has also been
observed in patients with osteoarthritis of
knee.
Expression of cell adhesion molecules has
also been shown in human neutrophils.
Drug interactions
It may increase plasma concentrations of lithium,
digoxin and methotrexate, increase the activity of
anticoagulant, inhibits the activity of diuretics,
enhance cyclosporin nephrotoxicity and precipitate
convulsions when co-administered with quinolone
antibiotics.
Adverse drug reactions
Most common events include dyspepsia, abdominal
pain, nausea, diarrhoea, flatulence, gastritis,
constipation, vomiting, ulcerative stomatitis,
pancreatitis. Other adverse effects, which are not
common, are dizziness, vertigo, and tremor.
Clinical efficacy
In osteoarthritis
In large trials of 2 to 6 months duration, aceclofenac
significantly reduced pain and improves functional
capacity and mobility relative to baseline in patients
with osteoarthritis, rheumatoid arthritis or ankylosing
spondylitis and reduces inflammation in patients with
rheumatoid arthritis.
In patients with osteoarthritis of the knee, aceclofenac
decreases pain, reduces disease severity and improves
the functional capacity of the knee to a similar extent
to diclofenac, piroxicam, and naproxen.

In rheumatoid arthritis
In ankylosing spondylitis
In dental pain
In postoperative pain
The anti-inflammatory and analgesic efficacy of
aceclofenac is similar to that of ketoprofen,
indomethacin, tenoxicam and diclofenac in patients
with rheumatoid arthritis.
The duration of morning stiffness and pain intensity
are reduced and spinal mobility improved, by
aceclofenac in patients with ankylosing spondylitis
The analgesic efficacy as single doses of aceclofenac
has been assessed in patients with moderate to severe
tooth pain and in extraction of impacted third molars.
Aceclofenac 100 mg was superior to paracetamol 650
mg in providing relief from postepisiotomy pain,
particularly 3 to 5 hours after ingestion.

3.3. PROFILE OF HYDROXY PROPYL METHYL CELLULOSE
Nonproprietarynames
BP : Hypromellose
JP : Hydroxypropylmethylcellulose
PhEur : Hypromellosum
USP : Hypromellose
Synonyms
Benecel MHPC, hydroxypropyl methylcellulose, HPMC,
methocel, methylcellulose propylene glycol ether, metolose.
Chemical name
Cellulose hydroxypropyl methyl ether
CAS registry number
[9004-65-3]
Structural formula
Description
Hypromellose is an odorless and tasteless, white or creamy
white fibrous or granular powder.

Functionalcategory
Coating agent, film-former, rate-controlling polymer for
sustained release, stabilizing agent, suspending agent, tablet
binder, viscosity-increasing agent.
Typicalproperties
Acidity/alkalinity: pH = 5.5–8.0 for a 1% w/w aqueous solution.
Ash: 1.5–3.0%, depending upon the grade and viscosity.
Auto ignition temperature: 360oC
Density (bulk): 0.341 g/cm3
Density (tapped): 0.557 g/cm3
Density (true): 1.326 g/cm3
Melting point: browns at 190–200oC; chars at 225–230
oC.
Glass transition temperature: 170–180oC.
Solubility
Soluble in cold water, forming a viscous colloidal solution,
practically insoluble in chloroform, ethanol (95%), and ether,
but soluble in mixtures of ethanol and dichloromethane,
mixtures of methanol and dichloromethane, and mixtures of
water and alcohol.
Specific gravity: 1.26 g/cm3
Viscosity (dynamic)
A wide range of viscosity types are commercially available.
Aqueous solutions are most commonly prepared. Solutions
prepared using organic solvents tend to be more viscous,
increasing concentrations also produces more viscous solutions.
Applications
Hypromellose is widely used in oral, ophthalmic, topical and
pharmaceutical formulations. In oral products, it is primarily
used as a tablet binder, in film-coating, and as a matrix former
for use in extended-release tablet formulations, as a suspending
and thickening agent in topical formulations, emulsifier, and
stabilizing agent in topical gels and ointments.
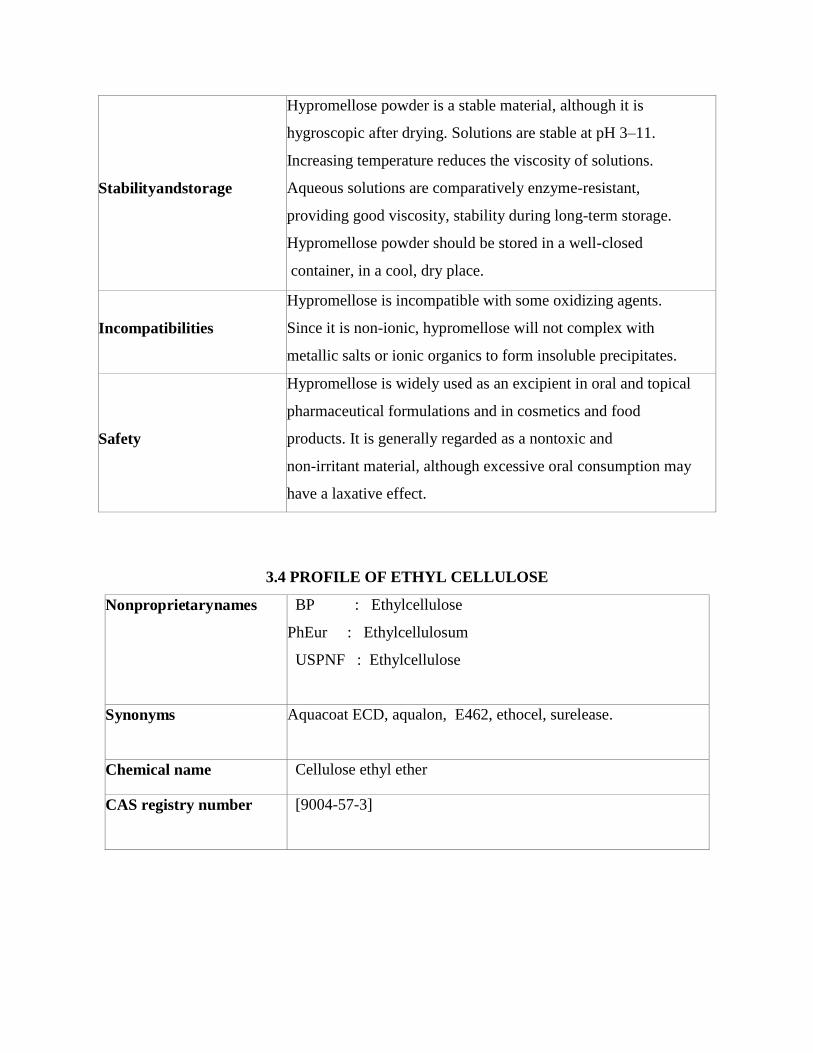
Stabilityandstorage
Hypromellose powder is a stable material, although it is
hygroscopic after drying. Solutions are stable at pH 3–11.
Increasing temperature reduces the viscosity of solutions.
Aqueous solutions are comparatively enzyme-resistant,
providing good viscosity, stability during long-term storage.
Hypromellose powder should be stored in a well-closed
container, in a cool, dry place.
Incompatibilities
Hypromellose is incompatible with some oxidizing agents.
Since it is non-ionic, hypromellose will not complex with
metallic salts or ionic organics to form insoluble precipitates.
Safety
Hypromellose is widely used as an excipient in oral and topical
pharmaceutical formulations and in cosmetics and food
products. It is generally regarded as a nontoxic and
non-irritant material, although excessive oral consumption may
have a laxative effect.
3.4 PROFILE OF ETHYL CELLULOSE
Nonproprietarynames BP : Ethylcellulose
PhEur : Ethylcellulosum
USPNF : Ethylcellulose
Synonyms Aquacoat ECD, aqualon, E462, ethocel, surelease.
Chemical name Cellulose ethyl ether
CAS registry number
[9004-57-3]

Structural formula
Description Ethylcellulose is a tasteless, free-flowing, white to light tan
colored powder.
Solubility Ethylcellulose is practically insoluble in glycerin,
propylene glycol, and water.
Functionalcategory Coating agent, flavoring fixative, tablet binder, tablet filler,
viscosity-increasing agent.
Typicalproperties Density (bulk): 0.4 g/cm3
Glass transition temperature: 129–1330C
Specific gravity 1.12–1.15 g/cm3
Viscosity The viscosity of ethylcellulose is measured typically at
250C using 5% w/v ethylcellulose dissolved in a solvent blend
of 80% toluene : 20% ethanol (w/w).
Uses Use Concentration (%)
Microencapsulation 10.0–20.0
Sustained release tablet coating 3.0–20.0
Tablet coating 1.0–3.0

Tablet granulation 1.0–3.0
Applications
It is widely used in oral and topical pharmaceutical
formulations, hydrophobic coating agent for tablets and
granules, to modify the release of a drug, to mask an unpleasant
taste, or to improve the stability of a formulation. Higher
viscosity grades tend to produce stronger and more durable
films.
Safety Ethylcellulose is not metabolized following oral consumption
and is therefore a non-calorific substance. Parenteral use may be
harmful to the kidneys.
Incompatibilities
Incompatible with paraffin wax and microcrystalline wax.
Stabilityandstorage Ethylcellulose is a stable, slightly hygroscopic material. It is
chemically resistant to alkalis, both dilute and concentrated, and
to salt solutions, although it is more sensitive to acidic materials
than are cellulose esters.
It should be stored at a temperature not exceeding 320C in a dry
area away from all sources of heat. It should not be stored next
to peroxides or other oxidizing agents.

3.5 PROFILE OF POLY VINYL PYRROLIDONE
Nonproprietarynames BP : Pyrrolidone
PhEur : Pyrrolidone
Synonyms γ-Aminobutyric acid lactam, 4-aminobutyric acid lactam,
gaminobutyric lactam, γ -aminobutyrolactam, γ -butyrolactam.
Butyrolactam, 2-ketopyrrolidine, 2-oxopyrrolidine, 2-Pyrol.
Chemical name 2-Pyrrolidinone
CAS registry number
[616-45-5]
Empiricalformula C4H7NO
Molecular weight 85.11 g/mol
Structural formula
Description N-Methylpyrrolidone occurs as a clear, hygroscopic
liquid with a mild amine odor.
Solubility Miscible with ethanol (95%), propan-2-ol, and water.
Also miscible with other organic solvents like benzene, carbon
disulfide, chloroform, ether, and ethyl acetate.
Specific gravity 1.11 at 25oC
Viscosity (dynamic) 13.3 mPa s at 25oC
Functionalcategory Penetration agent, plasticizer, solvent, solubilizing agent.
Typicalproperties Acidity/alkalinity pH = 8.2 – 10.8 for a 10% v/v aqueous
solution.
Boiling point : 245oC
Dipole moment : 2.3 Debye at 25oC
Flash point : 129oC

Applications
Pyrrolidone and N-methylpyrrolidone are mainly used as
solvents in veterinary injections, a better solubilizer than
glycerin, propylene glycol, or ethanol, as effective penetration
enhancers in topical applications.
Stability and storage Pyrrolidone is chemically stable and, if it is kept in unopened
original containers, the shelf-life is approximately one year. It
should be stored in a well-closed container protected from light
and oxidation, at temperatures below 20oC.
Incompatibilities
Incompatible with oxidizing agents and strong acids.
Safety Mainly used in veterinary injections and have also been
suggested for use in human oral, topical, and parenteral
pharmaceutical formulations.
3.6 PROFILE OF DIBUTYL PHTHALLATE
Nonproprietarynames BP : Dibutyl Phthalate
PhEur : Dibutyl Phthalate
USP-NF : Dibutyl Phthalate
Synonyms Araldite 502, benzenedicarboxylic acid, butyl phthalate,
celluflex DBP, dibutyl benzene 1,2-dicarboxylate, dibutyl
ester of 1,2-benzenedicarboxylic acid, dibutylisphthalas,
dibutyl-o-phthalate, Polycizer DBP, PX 104, RC Plasticizer
DBP.
Chemical name Dibutyl benzene-1,2-dicarboxylate
CAS registry number
[84-74-2]

Empiricalformula C16H22O4
Molecular weight 278.34 g/mol
Structural Formula
Description Dibutyl phthalate occurs as an odorless, oily, colorless, or
very slightly yellow-colored, viscous liquid.
Solubility Very soluble in acetone, benzene, ethanol (95%), and ether;
soluble 1 in 2500 of water at 20oC.
Functionalcategory Film-forming agent, plasticizer, solvent.
Typicalproperties Boiling point : 34oC
Flash point : 171oC
Melting point : 35oC
Incompatibilities
Dibutylphthallate reacts violently with chlorine. It also
reacts with oxidizing agents, acids, bases, and nitrates.
Applications
Dibutyl phthalate is used in pharmaceutical formulations as a
plasticizer in film-coatings, evaluated as a pore-forming
agent in novel delivery systems, extensively as a solvent,
particularly in cosmetic formulations such as antiperspirants,
hair shampoos, and hair sprays, as an insect repellent,
although it is not as effective as dimethyl phthalate.

Stabilityandstorage Should be stored in a well-closed container in a cool, dry,
location. Containers may be hazardous when empty since
they can contain product residues such as vapors and liquids.
Safety Dibutyl phthalate is generally regarded as a relatively
nontoxic material, although it has occasionally been reported
to cause hypersensitivity reactions. It is widely used in
topical cosmetic and some oral pharmaceutical formulations.
3.7. PROFILE OF TRIETHYL CITRATE
Nonproprietarynames BP : Triethyl citrate
PhEur : Triethyl citrate
USP-NF : Triethyl citrate
Synonyms Citric acid ethyl ester, citroflex 2, citrofol AI, E1505, ethyl
citrate, hydagen CAT, 1,2,3-propanetricarboxylic acid, 2
hydroxy-, triethyl ester , TEC, triethyliscitras.ester of
1,2-benzenedicarboxylic acid, dibutylisphthalas,
dibutyl-o-phthalate, di-n-butyl phthalate.
Chemical name 2-Hydroxy-1,2,3-propanetricarboxylic acid triethyl ester
CAS registry number
[77-93-0]
Empiricalformula C12H20O7
Molecular weight 276.29 g/mol

Structural Formula
Description Triethyl citrate is a clear, viscous, odorless, and practically
colorless, hygroscopic liquid.
Solubility Soluble 1 in 125 of peanut oil, 1 in 15 of water.
Miscible with ethanol (95%), acetone, and propan-2-ol.
Viscosity (dynamic) 35.2 mPa s at 250C
Functionalcategory Plasticizer, solvent.
Typicalproperties Acid value : 0.02
Boiling point : 294oC
Flash point : 155oC
Pour point : 45oC
Applications
Citrate, and acetyltributyl citrate are used to plasticize
polymers in formulated pharmaceutical coatings. The coating
applications include capsules, tablets, beads, and granules for
taste masking, immediate release, sustained-release, and
enteric formulations. It is also used as a direct food additive
for flavoring, for solvency, and as a surface active agent.
Stabilityandstorage Triethyl citrate should be stored in a closed container in a
cool, dry location. When stored in accordance with these
conditions, triethyl citrate is a stable product.
Incompatibilities
Incompatible with strong alkalis and oxidizing materials.

Safety Triethyl citrate is used in oral pharmaceutical formulations
and as a direct food additive. It is generally regarded as a
nontoxic and nonirritant material. However, ingestion of large
quantities may be harmful.
3.8 PROFILE OF TALC
Nonproprietarynames BP : Purified Talc
JP : Talc
PhEur : Talc
USP : Talc
Synonyms Altalc, E553b, hydrous magnesium calcium silicate, hydrous
magnesium silicate, magnesium hydrogen meta silicate,
magsilosmanthus, powdered talc, purified French chalk,
purtalc,soapstone, steatite, superiore, talcum.
Chemical name Talc
CAS registry number
[14807-96-6]
Description Talc is a very fine, white to grayish-white, odorless,
impalpable, unctuous, crystalline powder. It adheres readily to
the skin and is soft to the touch and free from grittiness.
Solubility Practically insoluble in dilute acids and alkalis, organic
solvents, and water.
Specific gravity 2.7–2.8 g/cm3
Functionalcategory Anti-caking agent, glidant, tablet and capsule diluents, tablet
and capsule lubricant.

Typicalproperties Acidity/alkalinity pH = 7–10 for a 20% w/v aqueous
dispersion.
Hardness (Mohs) 1.0–1.5
Moisture content : Talc absorbs insignificant amounts of water
at 250C and relative humidity’s up to about 90%.
Applications
Talc was once widely used in oral solid dosage formulations
as a lubricant and diluent, as a dissolution retardant in the
development of controlled-release products, as an adsorbant.
In topical preparations, talc is used as a dusting powder. Talc
is additionally used to clarify liquids and is also used in
cosmetics and food products, mainly for its lubricant
properties.
Stabilityandstorage Talc is a stable material and may be sterilized by heating at
1600C for not less than 1 hour. It may also be sterilized by
exposure to ethylene oxide or gamma irradiation. Talc should
be stored in a well-closed container in a cool, dry place.
Incompatibilities
Incompatible with quaternary ammonium compounds
Safety Talc is used mainly in tablet and capsule formulations. Talc is
not absorbed systemically following oral ingestion and is
therefore regarded as an essentially nontoxic material.

3.9.1. PAST WORK CARRIED OUT ON KETOROLAC
TROMETHAMINE
Bytul Rahman et al., prepared and compared ketorolac tromethamine tablets.
Formulated KT tablets gave higher dissolution rates than marketed product. Direct
compression method was adopted for preparation of tablet using different excipients
namely; microcrystalline cellulose, spray dried lactose and starch 1500. The effect of
excipients on the drug release from prepared tablets was also studied. All the tablet
quality control tests were studied. All formulations showed good mechanical properties
and complied with the USP 28 pharmacopoeial standard requirements for content
uniformity of dosage units and friability [49]
.
Rukmani et al., prepared matrix type formulations with dicalcium phosphate
dihydrate (DCPD) using a polymeric binder (Eudragit RSPO) to obtain controlled release
of Ketorolac tromethamine (KT).The drug, DCPD and Eudragit RSPO were granulated
by using isopropyl alcohol with and without a plasticizer (Diethyl phthalate, DEP).
Addition of Eudragit appear to affect the release profile. However, addition of a
plasticizer had a significant effect on the rate of release. The release appeared to follow
first order kinetics and the rate constant decreased linearly with increasing DEP
concentration. A directly compressible mixture was also formulated by coating DCPD
particles with DEP with and without Eudragit RSPO[50]
.
Sakshikanth et al., formulated gastro retentive multiparticulate drug delivery
systems to improve the absorption and bioavailability of ketorolac trometamol by

retaining the system in the stomach for prolonged period of time. The floating drug
delivery system of ketorolac trometamol was prepared by emulsion solvent diffusion
method by using ethyl cellulose, HPMC K4M, Eudragit R 100, Eudragit S 100 polymers
in varying concentration. Formulations were evaluated for percent yield, particle size,
entrapment efficiency, in vitro buoyancy and in vitro release studies. The percentage
yield of all the formulated batches of microspheres was more than 70 %. The optimized
formulations showed good buoyancy and invitro controlled release of ketorolac
trometamol. In vitro dissolution studies in 0.1N HC1 and in phosphate buffer pH 7.4
showed pH independent release of ketorolac trometamol[51]
.
Samir Roy et al., worked on transdermal delivery of ketorolac tromethamine, a
potent non-narcotic analgesic, through human skin in vitro and in vivo. In order to
enhance and sustain the flux of ketorolac through human skin, various compositions of
isopropyl alcohol (IPA), water, and isopropyl myristate (IPM) were evaluated. The
solubility of ketorolac acid in an IPA/water binary vehicle mixture increased as the
volume fraction of IPA increased from 0 to 90%. The solubility of ketorolac acid in an
IPA/water/IPM (saturated) ternary vehicle mixture was practically the same as in the
IPA/water binary vehicle mixture. The permeation of ketorolac acid through cadaver skin
was evaluated using modified franz diffusion cells. The skin flux increased as the IPA
volume fraction was increased from 0 to 50% and then leveled off beyond 80% IPA
loading. When IPM was added to the IPA/water binary vehicle mixture, a significant
increase in the skin flux of ketorolac was observed. The skin flux decreased exponentially
as the donor solution pH was raised from 3.5 to 7.0. The permeability of ketorolac

through various membranes such as a micro porous membrane and pressure-sensitive
adhesive was evaluated. While a micro porous membrane offered practically no diffusion
resistance, the in vitro flux of ketorolac through cadaver skin decreased substantially
upon lamination of pressure-sensitive adhesive onto a micro porous membrane [52]
.
Ibrahim et al., investigated permeation of ketorolac across excised rabbit skin
from various proniosome gel formulations using franz diffusion cells. Each of the
prepared proniosomes significantly improved drug permeation and reduced the lag time.
Proniosomes prepared with span 60 provided a higher ketorolac flux across the skin
thanthose prepared with Tween 20. A change in the cholesterol content did not affected
the efficiency of the proniosomes, and the reduction in the lecithin content did not
significantly decrease the flux. The encapsulation efficiency and size of niosomal vesicles
formed by proniosome hydration were also characterized by specific high performance
liquid chromatography method and scanning electron microscopy. Each of the prepared
niosomes achieved about 99% drug encapsulation. Vesicle size was markedly dependent
on the composition of the proniosomal formulations [53]
.
Aysegul et al., prepared occular inserts of water-soluble Ketorolac tromethamine
and water- insoluble indomethacin using hydrogels such as poly (butyl methacrylate),
poly (2-hydroxyethyl methacrylate), and poly (2-hydroxypropyl methacrylate), and a
plasticizer such as polyethylene glycol 300 by film casting method. Swelling properties
of these inserts was determined and they were irradiated with an absorbed dose of 1.2 M
rad by means of a Co- 60 source. The effects of these parameters on the drug release were
examined. The mechanism of drug release was identified by means of the semi-empirical

equation developed by korsmeyer and peppas. Water–soluble ketorolac showed higher
release than water–insoluble indomethacin. The release of ketorolac mainly fitted to the
fickian diffusion mechanism whereas the drug release of indomethacin fitted to non-
fickian release mechanism as per the ‘n’ exponent values [54]
.
Sanatkumar et al., formulated microspheres of ketorolac tromethamine for oral
delivery by complex coacervation and simple coacervation methods without the use of
chemical cross–linking agent (glutaraldehyde) to avoid the toxic reactions and other
undesirable effects of the chemical cross-linking agents. Alternatively, ionotropic
gelation was employed by using sodium-tripolyphosphate as a cross linking agent.
Chitosan and gelatin B were used as polymer and copolymer respectively. The release
study was carried out in pH 1.2 buffer for 2 hours andpH 7.4 phosphate buffer medium
for 8hours. It was observed that the rate of release decreased as the concentration of the
carrier was increased. This may be due to low permeability of polymer to the drug [56]
.
Lutfi gen et al., prepared enteric-coated film tablets of Ketorolac by the spray
technique. Different members of the Eudragit series may be employed for taste-masking
or as enteric-coating agents or dissolution rate-controlling membranes in sustained-
release dosage forms.Eudragit S-100 and L-100 were selected as coating materials.
Polyethylene glycol (PEG) 4000 was used as a plasticizer agent. Core tablets of ketorolac
were prepared by the direct compression technique. Tablet specifications were
determined and evaluated statistically [57]
.

Somanikumar et al., fabricated controlled release microcapsules of ketorolac by
coacervation phase separation and solvent evaporation technique. Microcapsules were
formulated using Eudragit RL100, Eudragit RS100 and Ethyl cellulose, bearing core:
polymer ratio of (1:1) and (1:2). The effects of various formulation and process variables
on the internal and external particle morphology, micromeritic properties, physical state
of the incorporated drug, drug loading and in vitro drug release were studied [58]
.
Sam Mathew et al., prepared albumin microspheres of ketorolac tromethamine by
emulsion cross-linking method. Selected formulations were characterized for their
entrapment efficiency, particle size, surface morphology, and release behavior. The
release pattern was biphasic, characterized by an initial burst effect followed by a slow
release. All selected microspheres, except those having less polymer proportion exhibited
a prolonged release for almost 24 hours. The release mechanism was regulated by drug:
polymer ratio and amount of cross-linking agent. From the experimental data obtained
with respect to particle size and extent of drug release, it was concluded that the prepared
microspheres are useful for once-a-day intramuscular administration of ketorolac
tromethamine[55]
.
Mohammad Etman et al., prepared ketorolac pellets using nonpareil seeds as an
inert carrier. Nonpareil seeds were first pan coated successively with EudragitRS 100
solution in isopropyl alcohol (12.5%) until they became visually insoluble in water. KT
was then applied onto nonpareil coated pellets in a conventional stainless steel pan.
Subsequently, the prepared KT pellets were sieved (2 mm) and coated by four coating

formulations using various proportions of Eudragit RL100 and Eudragit RS100 and the
drug release studies of the prepared pellets were studied [55]
.
Esmail Mohammed et al., prepared Ketorolac tromethamine loaded mucoadhesive
liquid suppository as a site-specific mucoadhesive rectal dosage form. Poloxamer mixture
formed of 21% P407 and 9% P188 were used as liquid suppository base. Invitro release
rate of KT from liquid suppository was studied and compared to that from conventional
suppository. The safety of the prepared suppository on GIT was conducted,
hepatotoxicity of KT after 5 days of administration of liquid suppository was evaluated
histologically and biochemically [60]
.
3.9.2. PAST WORK DONE ON ACECLOFENAC
Raghavendra Rao et al. ,developedaceclofenacloaded chitosan microparticles by
ionotropic gelation method using binary polymer mixture and by surface coating method.
Drug loadingefficiency (DLE) of microparticles was found to be between 62.20 to 92.93
% and depended up on the formulation variables. An increase in the
tripolyphosphateconcentration and cross-linking time decreased the drug release. The
particle size decreased with increase in cross-linking time and found between therange of
1194.1 to 1568.9 µm. Microparticles swelled and showed burst effect when shifted from
acid buffer pH 1.2 to phosphate buffer pH 7.4 andwere having poor mechanical strength.
Surface coating with alginate, alginate-pectin, chemical cross-linking with glutaraldehyde
and addition ofnon-ionizing polymer HPMC, were proved to be effective in controlling
the burst effect, prolonging the drug release with improved particle strength in both the

media. The research finally concluded that tripolyphosphate-chitosan microparticles
developed with binary polymer mixture and by surface coating method may
becomepotential delivery system to prolong the release of drug aceclofenac[61]
.
Jignesh Modi et al., investigated the potential of a nanoemulsion formulation for
topical delivery of aceclofenac. Various oil-in-water nanoemulsions were prepared by the
spontaneous emulsification method. The nanoemulsion area was identified by
constructing pseudoternary phase diagrams. Topical permeation of aceclofenac through
rat abdominal skin was determined. The in vitro skin permeation profile of optimized
formulations was compared with that of aceclofenac conventional gel and Nanoemulsion
gel. The anti-inflammatory effects of formulation showed a significant increase percent
inhibition value after 24 hours when compared with aceclofenac conventional gel and
nanoemulsion gel on carrageenan-induced paw edema in rats. These results suggested
that nanoemulsions are potential vehicles for improved transdermal delivery of
aceclofenac[62]
.
Pavan Kumar et al., investigated the use of biodegradable polymers for
microencapsulation of aceclofenac using solvent evaporation technique. Poly lactic acid
was selected as a retardant polymer because of its advantage over otherbiodegradable
polymers. Four different batches of microspheres were prepared by varying the
concentration of polymer from50% to 80% w/w. It was observed that the increase in
concentration of the polymer had increased the meanparticle size of the microspheres.It
can be seen that by increasing the polymer concentration, the rate of drug releasefrom the

microspheres dramatically decreased. The kinetics of drug release from F1 and F2
microspheres predominantly followsHiguchi pattern followed by zero order and then first
order. The release kinetics of F3 and F4 predominantly follows zeroorder followed by
Higuchi and then first order[63]
.
Hindustan Abdul Ahad et al., developed matrix tablets of aceclofenac with
Prosophisjulifloragum and studied its functionality as a matrix forming agent for once
daily sustained release tablet formulations. Various formulations of
aceclofenacProsophisjulifloragum were prepared and the formulated tablets found to
have better uniformity of weight and drug content with low standard deviation values.
The swelling behavior and release rate characteristics were studied. The dissolution study
proved that the driedProsophisjulifloragum can be used as a matrix forming material for
making once daily sustained release matrix tablets.This result has shown thatas the
proportion of Prosophisjulifloragum increased, theoverall time of release of the drug
from the matrix tabletwas also increased[64]
.
Tank Chintankumar et al., prepared aceclofenac loaded maltodextrin based
proniosomes by slurry method with different surfactantto cholesterol ratio. The niosomal
suspensions were evaluated for entrapment efficiency, invitro release study, release
kinetic dataanalysis, stability study, and invivo anti-inflammatory study. The result from
SEM analysis has showed smooth surface ofproniosome. The optimized formulation
showed higher entrapment efficiency of 83.24 ± 1.34 and in-vitro releases of 97.122%at
the end of 24hrs was found to be best among the other formulations. Release was best

explained by zero order kinetics.Kinetic analysis shows that the drug release follows
super case II transport diffusion. Proniosome formulation has showedappropriate stability
for 90 days by storing the formulation at refrigerator condition[65]
.
Yadav et al., designed the work to investigate the effect of size enlargement
methods like melt granulation and liquisolid technique on the physicochemical properties
such as solubility and dissolution rate of a poorly water-soluble drugaceclofenac.
Initially, the granules were prepared by melt granulation technique using melt binders,
diluent and super-disintegrants. The liquisolid systems were also prepared by using non-
volatile organic solvent, coating polymer, diluents and super disintegrants. The size
enlarged granules were prepared by both techniques and they exhibited improvement in
solubility, dissolution rate, wettability and flowability compared to aceclofenac. The melt
granules containing poloxamer with sodium starch glycolate and liquisolid systems,
containing micro crystalline cellulose with croscaramellose sodium showed higher
solubility and dissolution rate compared to other melt-granules andliquisolid systems.
The XRD and FTIR studies revealed acharacteristic decrease in thecrystallinity of the
granules[66]
.
Gopal Venkatesh Shavi et al., developed an enteric coated multi-unit dosage form
containing aceclofenac, a non-steroidal anti-inflammatory drug. The pellets were
prepared by using extrusion/spheronization method, and the core pellets were coated with
a pH sensitive poly methacrylate copolymer to achieve site specific drug release. The
formulated pellets were characterized for percentage yield, size distribution, surface

morphology studies, drug content and flow properties. In vitro dissolution test was used
for comparison of drug release profiles of various coated pellets and the practical yield
was found to be 90-95%[67]
.
PAST WORK CARRIED OUT ON ETHYL CELLULOSE
Ravikumaran et al,. prepared surfactant-coated, nimesulide-free, and nimesulide-
loaded ethylcellulose or methylcellulose nanoparticles by varying drug concentration,
polymer concentration, and surfactant concentration. Ethyl cellulose and methyl cellulose
nanoparticles prepared by desolvation method produced discrete particles and they were
characterized by SEM, AFM, and FTIR studies. The encapsulation efficiency decreased
with the increase of nimesulide concentration with respect to polymer concentration [68]
.
Satit Prasertmanakit et al., developed ethyl cellulose microcapsules for use as a
drug-delivery device for protecting folic acid from release and degradation in the
undesirable environmental conditions of the stomach, whilst allowing its release in the
intestinal tract to make it available for absorption. The controlled release folic acid-
loaded ethyl cellulose microcapsules were prepared by oil-in-oil emulsion solvent
evaporation using a mixed solvent system, consisting of a 9:1 (v/v) ratio of acetone:
methanol and light liquid paraffin as the dispersed and continuous phase.
Samuelov et al., prepared cast films composed of different ratios of polyethylene
glycol and ethyl cellulose containing salicylic acid, caffeine, and tripelennamine as model
dispersed drugs were for sustained release. The drug content of the film declined at an

apparent first-order rate initially, whereas the drug quantity released was proportional to
the square root of time. The release rates were independent of film thickness and
proportional to drug concentration in pure ethyl cellulose films; in polyethylene glycol-
ethyl cellulose films, a positive deviation from linearity was observed [69]
.
Parthasarathy et al., reported the development of a sustained-release system of
sparfloxacin for use in the treatment of periodontal disease. A sustained-release
sparfloxacin device was formulated, based on ethyl cellulose (EC) 10 cps, polyethylene
glycol (PEG) 4000, and diethyl phthalate (DEP). It will hereafter be called the
sparfloxacin chip. The chip has dimensions of 10 mm length, 2 mm width, and 0.5 mm
thickness. The in vitro drug release pattern and clinical evaluation of the formulations
were studied [70]
.
Emeje et al., investigated the effect of some commonly used release enhancers on
the compaction characteristics of EC. The wet granulation method of massing and
screening was used, and compacts were produced by compressing granules for 60
seconds at various compression pressures.He also studied the effects of four commonly
used hydrophilic additives such as polyethylene glycol (PEG) 4000, PEG 10, 000,
sorbitol, and mannitolon the compaction characteristics of ethyl cellulose using density
measurements and the Heckel equation. It can be concluded that additives such as PEG
4000, PEG 10 000, sorbitol, and mannitol, which are often used as channeling agents in
sustained-release formulations containing hydrophobic matrix formers, affect the

deformation characteristics of EC, with the extent and nature of the effect dependent on
the nature of the additive[71]
.
Baria et al., prepared sustained release (SR) suppositories containing aceclofenac
microspheres. In the first part of the study, aceclofenac microspheres were prepared by
solvent evaporation method employing ethyl cellulose as a microsphere forming polymer.
The effect of drug: polymer ratio and stirring rate on microspheres formation, average
particle size, incorporation efficiency, micromeritic properties and in vitroaceclofenac
release were investigated [72]
.
Muhammad Khan Sarfraz et al., developed sustained released matrix tablets of
naproxen with ethyl cellulose, a hydrophobic polymer. Matrix tablets were prepared by
incorporating various proportions of ethyl cellulose in the matrix system using wet
granulation technique [73]
.
Golomb et al., prepared cast films of ethyl cellulose with or without polyethylene
glycol, containing metronidazole, and they exhibited sustained release. The
microbiological results proved that embedding metronidazole in ethyl cellulose film does
not inhibit the biological activity. The release kinetics of in vivowere correlated with in
vitro results, exhibiting a sustained release of metronidazole over a period of three days
[74].
Hemangi Patil et al., prepared floating microspheres of acyclovir using different
viscosities of ethyl cellulose to achieve an extended retention in upper GIT which may be
resulted in enhanced absorption and thereby improves bioavailability. The floating

microspheres were prepared by emulsion solvent diffusion technique and triethyl citrate
was used as a plasticizer [75]
.
Bhupendra .Prajapati et al., formulated matrix tablets of nicorandil by the polymer
blend to get desirable release profile. In the present study, HPMC K200M, which was
used in hydrophilic matrix drug delivery systems have been employed to formulate
sustained-release tablets of nicorandil but alone it did not give good results. So it was
used in combination with hydrophobic polymer like Eudragit RSPO and Ethyl cellulose
[76].
Hosseinali Tabandeh et al., fabricated matrix aspirin tablets with ethylcellulose
(EC), Eudragit RS100, and Eudragit S100 were prepared by direct compression. EC with
an amount as little as 10 percent in formulation could make sustained-release aspirin
tablets in which the release profile is not sensitive to moderate changes in hardness.
Anjali et al., studied the applicability of fine particle ethyl cellulose to produce
matrix tablets by wet granulation technique. Tablets were prepared by wet granulation of
drug and fine particle ethyl cellulose in an appropriate mass ratio. As its content and the
hardness of the tablets were increased, the release rate of the drug was decreased [77]
.
Phutaneet et al., developed microspheres by emulsion solvent diffusion-
evaporation technique by using the modified ethanol,-dichloromethane co-solvent
system. The polymer mixture of ethyl cellulose and Eudragit® S100 was used in different

ratios (1:0, 1:1, 2:3, 1:4 and 0:1) to formulate batches. The formulated microspheres were
discrete, spherical with relatively smooth surface, and with good flow properties. Among
different formulations, the fabricated microspheres of batch F3 had shown the optimum
percent drug encapsulation of microspheres and the sustained release of the Glipizide for
about 12 h.The data obtained thus suggest that a microparticulate system can be
successfully designed for sustained delivery of Glipizide and to improve dosage form
characteristics for easy formulation.[78]
.
Sandip Tiwari et al., studied the effect of concentration of hydrophilic
(hydroxypropyl methylcellulose [HPMC]) and hydrophobic polymers (hydrogenated
castor oil [HCO], ethylcellulose) on the release rate of tramadol. Hydrophilic matrix
tablets were prepared by wet granulation technique, while hydrophobic (wax) matrix
tablets were prepared by melt granulation technique and in vitro dissolution studies were
performed. Hydrophobic matrix tablets resulted in sustained in vitro drug release (>20
hours) as compared with hydrophilic matrix tablets (<14 hours) [79]
.
Akhil Sharma et al., formulated and evaluated salbutamol sulphate sustained
release tablets using different polymers as release retarding agents. Preformulation study
was done initially and the results were directed for further course of formulation. Based
on preformulation studies, different batches of salbutamol sulphate were prepared using
xanthan gum, carbopol and ethyl cellulose due to their different hydrophilic properties to
calculate the sustained release properties [80]
.

Vatsaraj et al., formulated a sustained-release tablet of Ketorolac tromethamine,
which is a nonsteroidal anti-inflammatory agent. A 23 full factorial design (8 runs) was
selected. The variables studied were the amount of drug (30 and 40 mg), ratio of
hydroxypropyl methylcellulose (HPMC)/sodium carboxymethylcellulose (NaCMC)
(240/40 and 140/140 mg), and amount of ethylcellulose (140 and 180 mg). Swelling-
controlled matrix tablets were manufactured by direct compression of formulation
ingredients using a Stokes single punch tablet press [81]
.
Giunchedi et al., studied on Ketoprofen which was a non-steroidal anti-
inflammatory drug. It has been incorporated into polymeric micro matrices
(microspheres) made of cellulose acetate trimellitate (CAT)/ethylcellulose (EC) blends
by spray drying process. Drug loaded microspheres were obtained by spray-drying
organic solutions of the two polymers and the drug. Characterization of the
microparticles (morphology, particle size distribution, drug content, yield of production,
surface properties, and solvent residues) was carried out and invitro release behavior was
measured. The release rate of the drug was diminished as the proportion of EC was
raised[82]
.
Hitesh Gevariya et al., formulated ocular inserts of levofloxacin and evaluated
their potential for sustained ocular delivery. Matrix-type of ocular inserts were prepared
by the film casting technique in Teflon-coated Petri dishes and in vitrocharacterization
was done by drug release studies using a flow-through apparatus that simulated the eye
conditions. Total 9 formulations were developed, which differed in the ratio of polymers

such as polyethylene oxide (PEO), hydroxypropyl cellulose (HPC) and ethyl cellulose
(EC). It was also observed that increasing the proportion of PEO and HPC to EC
increases the rate of release of Levofloxacin [83]
.
Abu HenaMostafa Kamal et al., prepared Indomethacin microcapsules using ethyl
cellulose (EC) and hydroxy propyl methyl cellulose phthalate (HPMCP) by o/w
emulsification-solvent evaporation technique. The rate was increased exponentially with
the addition of HPMCP in EC. IM rate was observed highest with the highest
concentration of HPMCP (3:7 ratio of EC: HPMCP), used in the present studies. On the
other hand, IM rate was lowest when EC and HPMCP combination was used at the ratio
of 10:0 [84]
.
Verhoeven et al., developedmini-matrices (multiple-unit dosage form) with
release-sustaining properties by means of hot-melt extrusion using ibuprofen as the
model drug and ethylcellulose as sustained-release agent. Xanthan gum, a hydrophilic
polymer, was added to the formulation to increase the drug release since ibuprofen
release from the ibuprofen/ethylcellulose matrices (60/40, w/w) was too slow (20% in
24 h). Drug release from the mini-matrices was mainly diffusion controlled, but swelling
played an important role to obtain complete drug release within 24 hours[85]
.

PAST WORK CARRIED OUT ON PELLETS
Eskandari has developed an extended release pellet formulation of indomethacin
by the centrifugation (rotary fluid bed granulation) or powder layering method. Layered,
nonpareil pellets composed of sugar, Avicel PH 101 and lactose were prepared using
FREUND CFgranulator and were treated by a binder solution (HPC-L) applied by spray
gun. A conical designed powder-feeding unit applies the drug powder. Drug content of
pellets was determined by HPLC method. Eudragit NE 30 D was used for coating the
prepared pellets. The results showed that increasing the amount of Eudragit NE 30 D,
Opadray and SDS in coating solution adjusts the release rate of the pellets [86]
.
Cronlein et al., investigated the enteric performance of aqueous enteric-coated
multiparticulate formulations containing lansoprazole in bio-relevant media which better
simulates the gastric environment of a patient on a multiple dose regimen of PPI’s. A
secondary objective of this study was to characterize the stability of the finished dosage
form under room temperature and accelerated storage conditions [87]
.
Sonali Naikwade et al., developed enteric-coated pellets of piroxicam in order to
avoid local gastrointestinal irritation which is one of the major side effects of non-
steroidal anti-inflammatory drugs after oral ingestion. Three polymers namely Eudragit
L100, cellulose acetate phthalate and hydroxyl propyl methyl cellulose phthalate were
used. Pellets were made by an extrusion–spheronization process. Two approaches were
used viz. enteric matrix and enteric polymer coating. Matrix and coated pellets were
evaluated for physicochemical properties. Influence of excipients on in vitro release of

drug was evaluated. Piroxicam release from uncoated pellets was measured in phosphate
buffer (pH 6.8) using paddle dissolution method(USP XXIII). Enteric-coated pellets were
tested in 0.1 N HCl and phosphate buffer, pH 6.8. Optimum formulations were subjected
to stability studies under accelerated conditions. Enteric pellets prepared using eudragit
L100 gave promising results for matrix pellets [88]
.
Cara Young et al., worked on Lansoprazoledelayed-release pellets which have
been shown to have better absorption properties than a delayed-release tablet. However,
binder solutions for drug layering and extrusion-spheronization excipients were
incompatible with lansoprazole. In contrast, dry powder layering technology has been
reported to provide a more stable manufacturing method for acid-labile drugs. The
objective of this study was to evaluate the performance of lansoprazole powder layered
pellets coated with a fully formulated aqueous enteric system (Acryl-EZE®
93F19255) in
various media [89]
.
Radke et al., cinnarizine pellets were prepared by powder-layering technique,
using cinnarizine as active ingredient and Eudragit RS-100, Eudragit RL-100, Ethyl
cellulose as coating agents, propylene glycol as plasticizer and PVPK-30 as binder. Drug
content was estimated spectrophotometrically at 254 nm. The prepared pellets were
further evaluated for surface texture by scanning electron microscopy, uniformity of
diameter, thickness and weight,in vitro drug release pattern and short-term stability.
Pellets coated with 10% Eudragit RS-100 showed promising results releasing more than
95% of drug up to 12 hours. This study concluded that the powder layering technique can

be used for designing sustained release drug delivery systems providing drug release over
a period of 12 hours [90]
.
Mustafa Sinan Kaynak has prepared the pellet formulations by pan coating
method. They have prepared formulations in order to decrease the dosage regimen which
is twice daily for conventional tablet. Formulation of Glipizide pellets were aimed to
maintain the necessary blood Glipizide concentration for the treatment. The in
vitrocharacterization as well as microscopic investigation of the pellet formulations was
evaluated[91]
.
Alireza Ghaffari et al., prepared pellets containing theophylline as a model drug
and microcrystalline cellulose, in a ratio of 6:4, by the extrusion-spheronization method.
The pellets were coated with Eudragit RS aqueous dispersions, containing various
amounts of pectin-chitosan complex and different coating mass gains, using a fluidized-
bed apparatus. Twelve formulations were developed, which differed in two factors:
coating mass gain (10, 15 and 20%, w/w) and the amount of pectin-chitosan complex (5,
10, 15 and 20%, w/w). Drug release studies were conducted using the USP apparatus I
(basket) in dissolution media, mimicking the conditions prevailing in the stomach, small
intestine or colon. Studies have shown that the drug release rate and pattern were
dependent on both of the two mentioned factors. Some formulations showed bimodal and
burst drug release, being triggered in the colonic medium bythe action of
pectinolyticenzymes [92]
.

Gopal Venktesh Shavi et al., developed an enteric-coated multi-unit dosage form
containing aceclofenac, a nonsteroidal anti-inflammatory drug (NSAID). The pellets
were prepared by using extrusion/spheronization method, and the core pellets were
coated with a pH-sensitive poly (meth acrylate) copolymer (Eudragit L100-55) to achieve
site-specific drug release. The formulated pellets were characterized for percentage yield,
size distribution, surface morphology studies, drug content, and flow properties. In vitro
dissolution test was used for the comparison of drug release profiles of various coated
pellets. The practical yield was found to be 90–95% [93]
.
Halle Bechgaard et al., conducted a clinical trial investigating the safety and
efficacy of two multiple unit controlled release esomeprazole capsule formulations
containing enteric coated pellets bioequivalent to a standard capsule in respect of extent
of bioavailability in a cross over study with normal human subjects. However, drug
absorption from the enteric coated pellet formulations was slower when compared to that
from the reference capsule, the standard reference capsule releases 85% of its drug
content in vitroduring 10min at pH 6.5 and 98% during 1hr at pH 7.5. The data indicated
that multiple units of controlled release formulations represent a reliable and reproducible
source of esomeprazole, which by avoiding extremes of local or systemic drug
concentration[94]
.
Akelesh et al., developeda pharmaceutically equivalent, stable, cost effective and
quality improved formulation of venlafaxine HCl pellets in the form of sustained release
capsule. Pellets containing different polymers have been investigated with the intention

of gaining a deeper understanding the formulation factors. The optimized formulation
gave satisfactory dissolution profile and releases more than 98.29% of drug within 24 hrs.
Drug release rate was more when compared with the innovator sample and its dissolution
profile matches with the innovatordissolution profile and follows first order kinetics[95]
.
Audity Ganguly Rupesh, et al,. designed an oral controlled onset of dosage form
intended to approximate the chronobiology ofrheumatoid arthritis which was proposed
for colonic targeting. The multiparticulate systemcomprising of non-pareil seeds coated
with eudragit S100 was designed for chronotherapeuticdelivery of valdecoxib. The drug
was coated onto non-pareil seeds by powder layeringtechnique using the conventional
coating pan. The in vitro dissolution tests showed that the release of valdecoxib from the
coatedpellets depended on the pH of the dissolution fluid and the coat weights applied.
All theformulations exhibited no release of drug in the pH 1.2 and pH 4 buffers, drug
release has taken place in phosphate buffer of pH 7.4. Further intactness of the drug in the
formulation and theuniformity of the polymer coating were checked by the infrared study
and scanning electronmicroscopy. All the above results showed thatthe formulation could
be highlyadvantageous in the chronotherapy of rheumatoid arthritis withappreciable drug
release and physiochemical properties[96]
.
Lian-Dong Hu et al., prepared metformin hydrochloride sustained-release pellets
by centrifugal granulation. Experimental results indicated that talc modification made a
decisive contribution to control the drugrelease by avoiding dose dumping.The relative
bioavailability of the sustainedreleasepellets was studied in 12 healthy volunteers after

oral administration in a fast state using a commercially available immediate release
tablet. Restricted delivery of metformin hydrochloride to the small intestine from
differently coated pellets resulted in increased relativebioavailability and a sustained
release effect. So the adoption of several different pH dissolution media established a
better relationshipbetween the in vitro release and in vivo absorption of the sustained-
release pellets[97]
.
Heinämäki evaluated the tackiness of aqueous chitosan film coatings and effects
of anti-sticking agents on sticking tendency. High molecularweight chitosan plasticized
with glycerol was used as a film-forming agent.Film coatings were performed in
aminiaturized top-spray coater. The incorporation of anti-sticking agents led to a clear
decrease in tackiness of the chitosan films. Film-coated pellets containing magnesium
stearate as an anti-sticking agent were very easily fluidized and were thusclassified as
the best flowing and the least sticking samples.Thus the research gave an idea on the
determination of the experimental minimum fluidization velocity in a fluidized bed,
which is a usefuland sensitive method of measuring the tackiness tendency of film-coated
pellets[98]
.
Rajesh et al., prepared pellets loaded withaceclofenac sodium (ACS) as model
drug through pelletization technique by using theblend of sodium alginate (SA) and
glycerylpalmito stearate (GPS) as hydrophilic andhydrophobic carriers, along with
methyl crystalline cellulose (MCC) as spheronizerenhancer in various concentrations and
examined the influence of various processparameters on drug containing pellets. The
research showed that oral controlled release aceclofenac sodium pellets were able to

prolong the drug release,minimizing the drug related adverse effects and improve
bioavailability in different GItractconditions. Formulated drug loaded pellets were
investigated for physicochemicalproperties and drug release potential. The release of
drug was controlled for more than 24 h. The rate of drug releasefollowed first order
kinetics and the mechanism of drug release followed fickiandiffusion. Thus they finally
concluded that the drug release performance was greatly affected by the materials used
inpellets preparations, which allows absorption in the intestinal tract by a
controlledmanner[99]
.
Fatemeh Sadeghi et al., investigated the release of metoclopramide hydrochloride
(a water-soluble cationic drug) and diclofenac sodium (a sparingly soluble anionic drug)
from pellets coated with ethylcellulose from an aqueous ethylcellulose dispersion
(Surelease®) at different coating loads. The release rates of each drug decreased as the
coating load of Surelease® increased. [100]





























