Embed Size (px)
Citation preview

HIPERPLASIA SUPRARRENAL CONGÉNITA
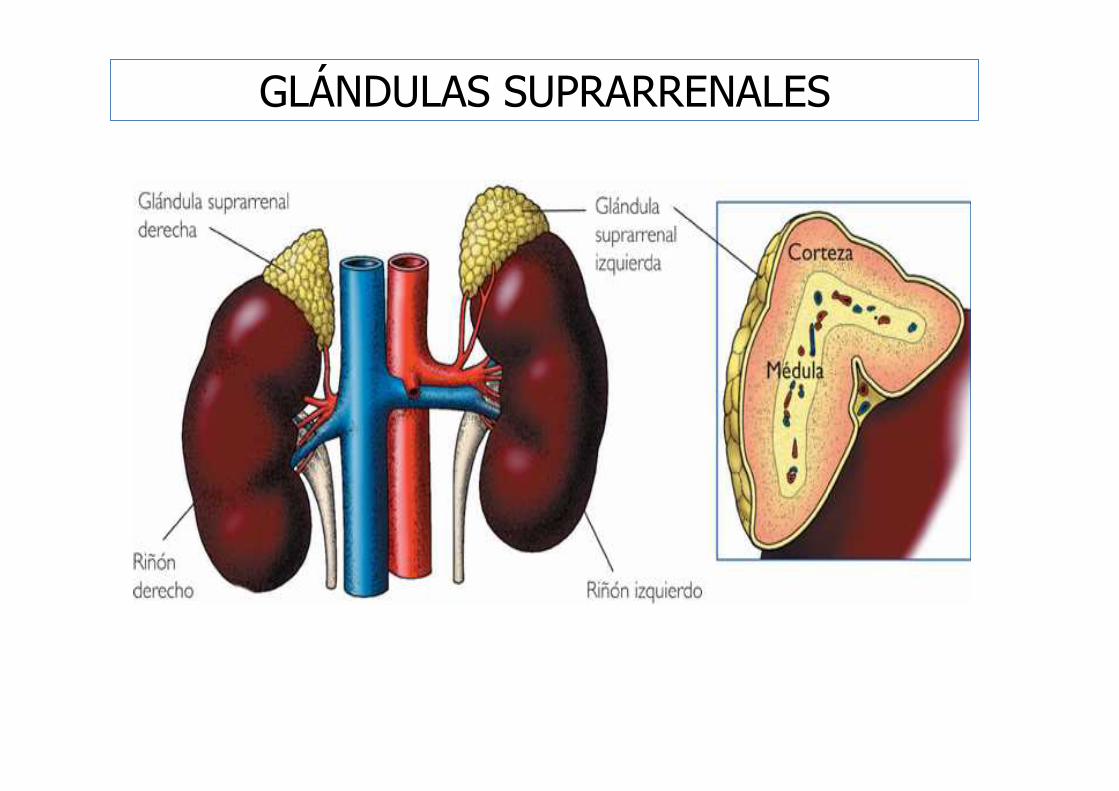
GLÁNDULAS SUPRARRENALES
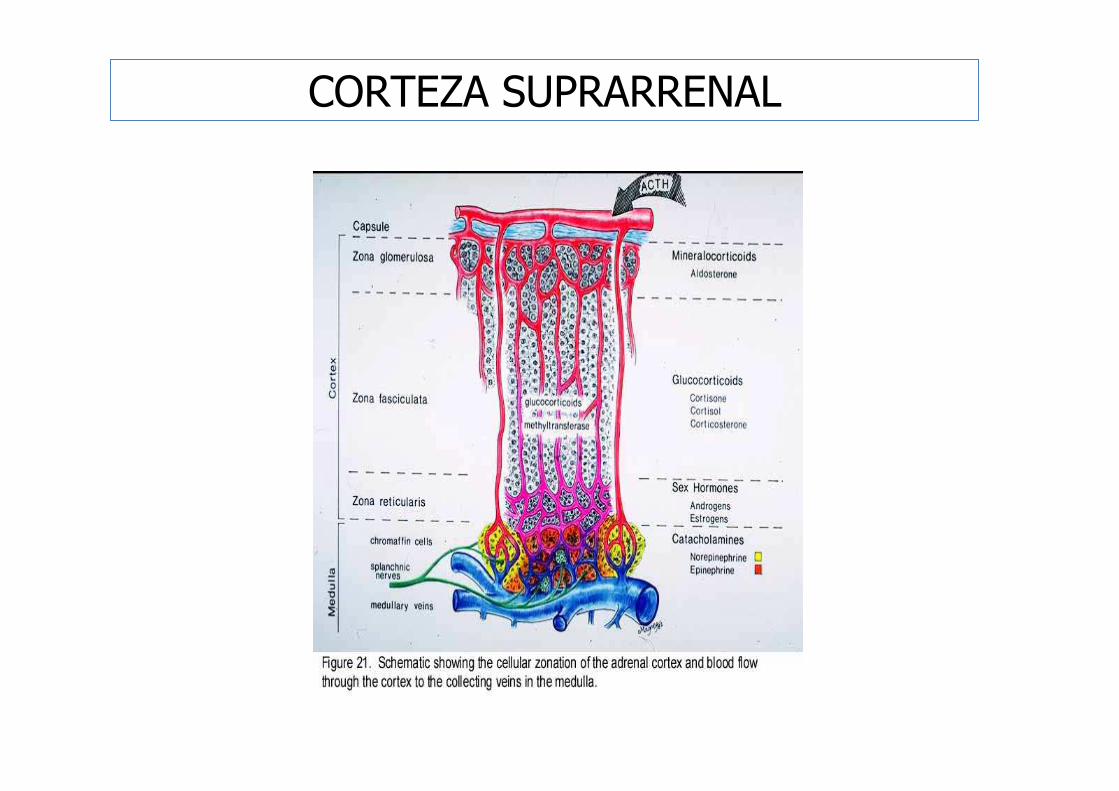
CORTEZA SUPRARRENAL
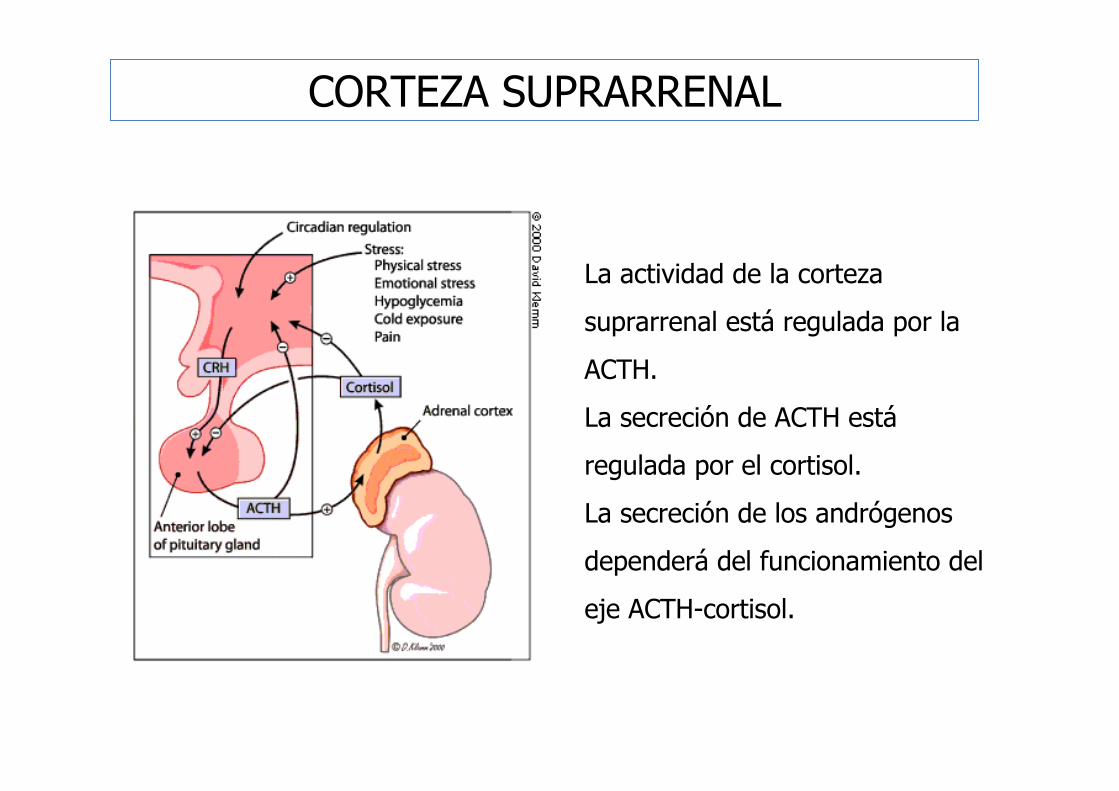
CORTEZA SUPRARRENAL
La actividad de la corteza
suprarrenal está regulada por la
ACTH.
La secreción de ACTH está
regulada por el cortisol.
La secreción de los andrógenos
dependerá del funcionamiento del
eje ACTH-cortisol.

� La secreción de glucocorticoides y andrógenos
está regulada fundamentalmente por factores
hipotalámicos e hipofisarios.
� La producción de aldosterona está controlada por
un sistema cuyos componentes más importantes son
el sistema renina-angiotensina y la concentración
extracelular de potasio.
CORTEZA SUPRARRENAL
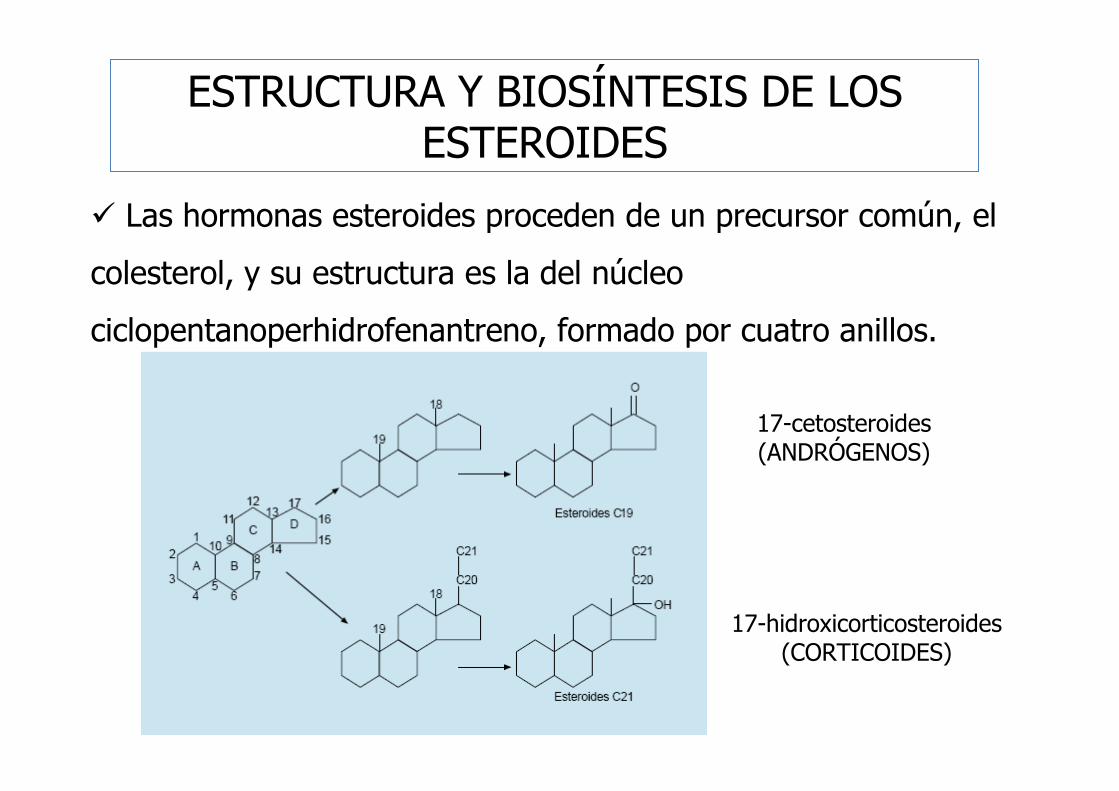
ESTRUCTURA Y BIOSÍNTESIS DE LOS ESTEROIDES
� Las hormonas esteroides proceden de un precursor común, el
colesterol, y su estructura es la del núcleo
ciclopentanoperhidrofenantreno, formado por cuatro anillos.
17-cetosteroides(ANDRÓGENOS)
17-hidroxicorticosteroides(CORTICOIDES)
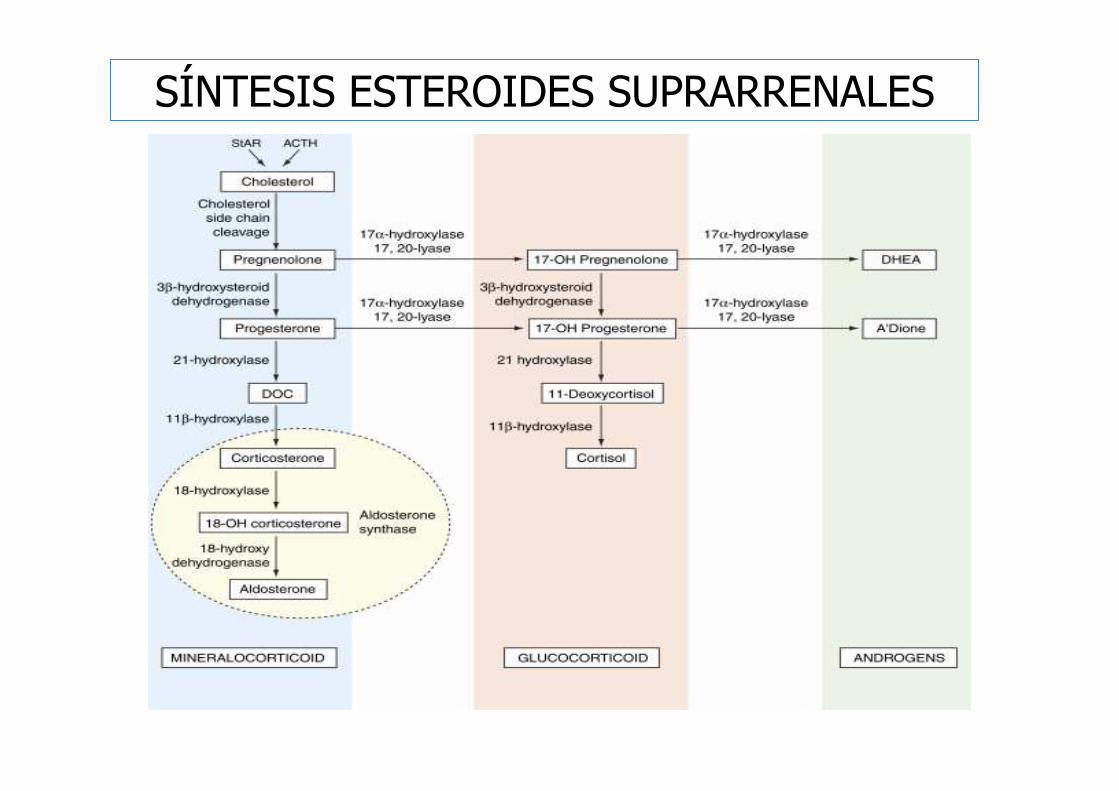
SÍNTESIS ESTEROIDES SUPRARRENALES

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Engloba un grupo de trastornos enzimáticos de la
glándula suprarrenal que conlleva una alteración en
la síntesis de cortisol y aldosterona, con acúmulo de
precursores androgénicos.

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
El cortisol inhibe la síntesis de CRH y de ACTH.
En la HSC existe un déficit de cortisol, el “feed back”
negativo se pierde, por tanto, aumentan los niveles
de CRH y de ACTH.
La ACTH hiperestimula la corteza adrenal que se
hipertrofia.

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Cada déficit enzimático se caracteriza por un
fenotipo característico y, dependiendo del bloqueo,
se producirá acúmulo de sustancias que serán las
responsables de esa clínica y ayudarán al
diagnóstico.

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Así, podemos encontrar:
- Trastorno de la diferenciación sexual en el
momento del nacimiento (genitales ambiguos),
asociado o no a un síndrome adrenogenital.
- Amenorrea primaria e hipertensión arterial en la
adolescencia.

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Engloba 5 diferentes déficit enzimáticos que alteran
la síntesis de cortisol y aldosterona, junto con
acúmulo de andrógenos. Por orden de frecuencia:
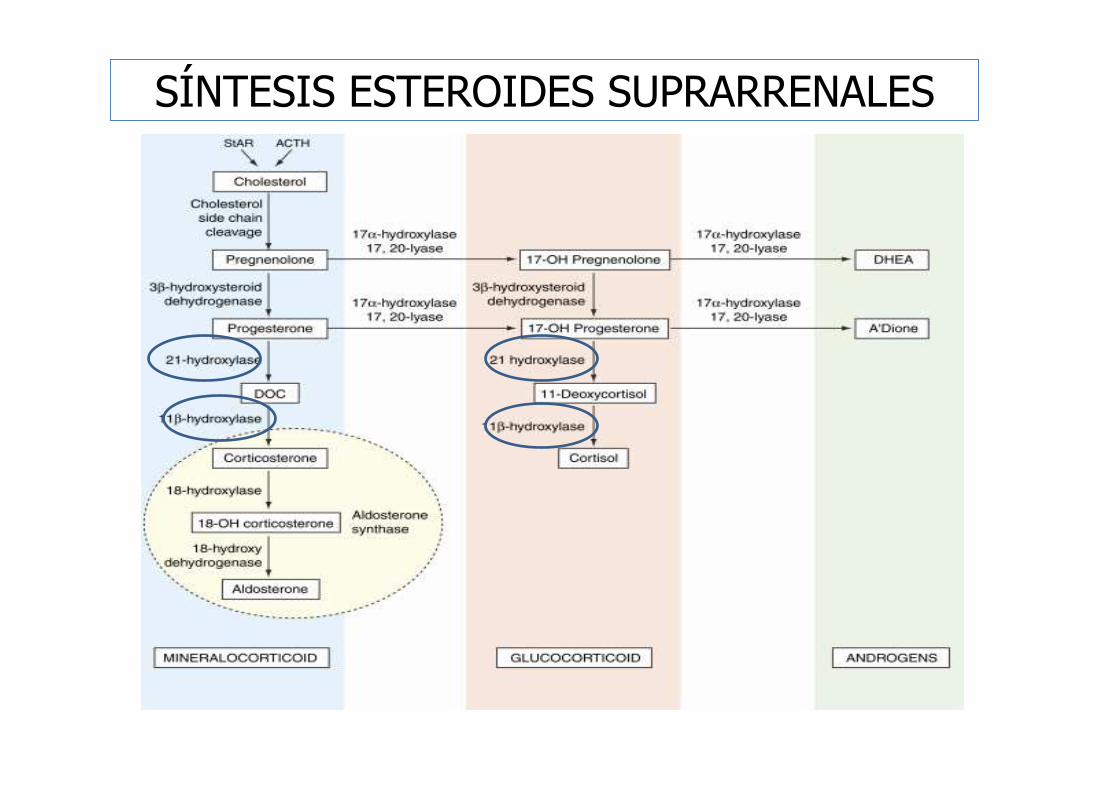
- Déficit de 21-α-hidroxilasa.
- Déficit de 11-β-hidroxilasa.
- Déficit de 3-β-hidroxiesteroide deshidrogenasa.
- Déficit de 17-α-hidroxilasa.
- Déficit de la proteína StAR.
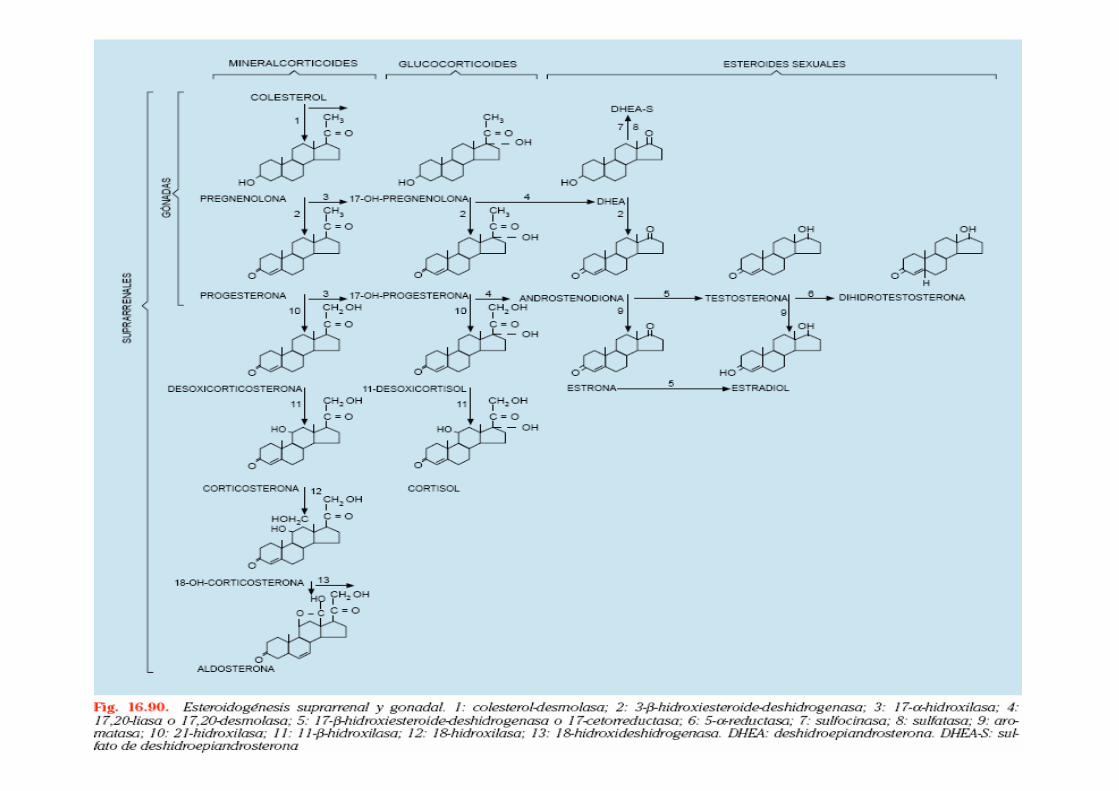
SÍNTESIS ESTEROIDES SUPRARRENALES

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Su transmisión hereditaria es autosómica recesiva,
con penetrancia variable.
Incidencia anual*: 1/15.000 recién nacidos vivos.
*Hiperplasia suprarrenal congénita. Pediatr Integral 2007;XI(7):601-610.

HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC)
Se pueden presentar como:
� formas clásicas (graves): déficit enzimático
completo. Se manifiestan clínicamente en la época
fetal.
� formas no clásicas (tardías): déficit parcial. Su
manifestación clínica puede aparecer en la infancia,
adolescencia o edad adulta.

HSC: Déficit 21-α-hidroxilasa
El déficit de 21-hidroxilasa es la forma clínica más
frecuente (90-95%)* de HSC.
Se debe a una mutación o deleción en el gen que lo
codifica (CYP21B). Brazo corto del cromosoma 6.
El déficit de la 21-hidroxilasa presenta dos
características fundamentales: la insuficiencia
suprarrenal y el hiperandrogenismo.
•Hiperplasia suprarrenal congénita. Pediatr Integral 2007;XI(7):601-610.
• Clinical Diagnosis and Management by Laboratory Methods, 20th ed. John Bernard Henry.
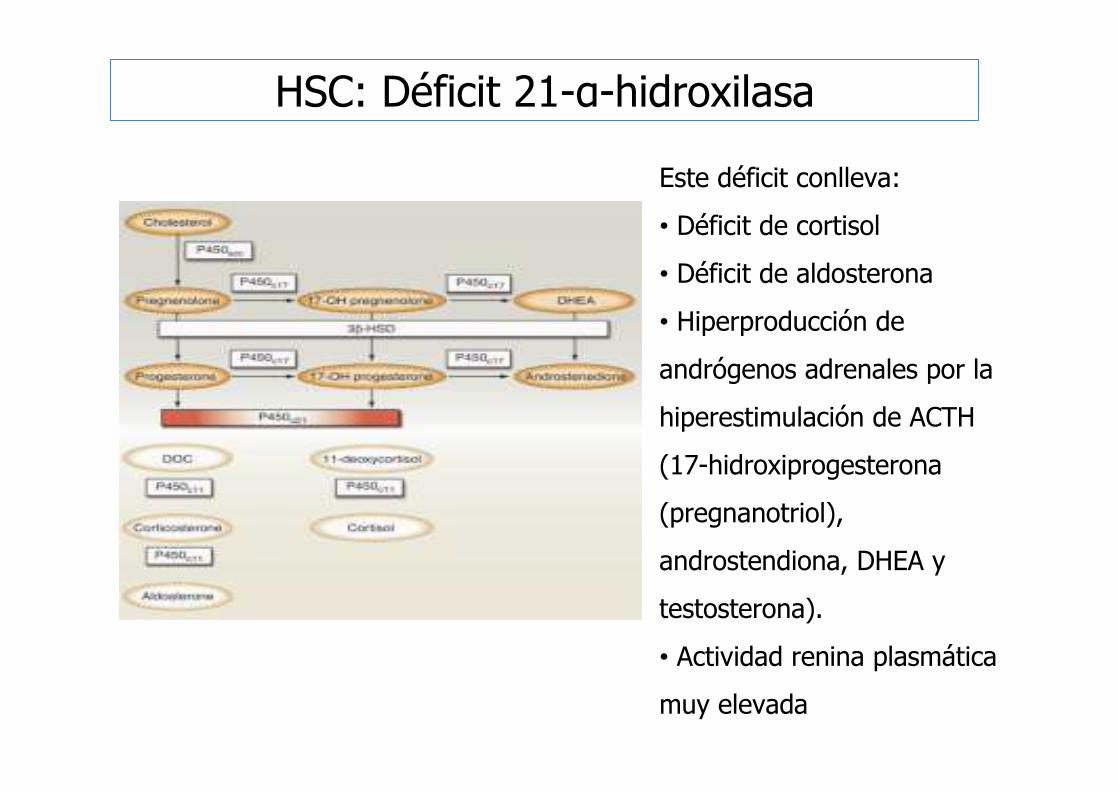
Este déficit conlleva:
• Déficit de cortisol
• Déficit de aldosterona
• Hiperproducción de
andrógenos adrenales por la
hiperestimulación de ACTH
(17-hidroxiprogesterona
(pregnanotriol),
androstendiona, DHEA y
testosterona).
• Actividad renina plasmática
muy elevada
HSC: Déficit 21-α-hidroxilasa

Existen dos formas de presentación de la forma
clásica:
Síndrome pierde sal
Forma virilizante simple
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Síndrome pierde sal): 75% de los
pacientes afectos de la forma clásica.
La mutación de la 21-α-hidroxilasa produce un grado
máximo de inactividad enzimática que conlleva un
déficit total de cortisol y aldosterona.
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Síndrome pierde sal):
Excreción excesiva de sodio por la orina.
Disminución de la eliminación urinaria de potasio.
(hiponatremia e hiperpotasemia)
La eliminación de sodio, arrastra agua y bicarbonato
(hipovolemia, hipotensión y acidosis
metabólica)
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Síndrome pierde sal):
Este síndrome se manifiesta entre el 5º-15º día de
vida por vómitos, pérdida de apetito, diarrea,
hipotensión.
Alteraciones bioquímicas: hipoglucemia,
hiponatremia, hiperpotasemia y acidosis metabólica.
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Síndrome pierde sal):
La elevación de los andrógenos (por encima del
bloqueo enzimático) desde la semana 7ª de la
gestación produce en el sexo femenino un trastorno
de la diferenciación sexual. Los niños pueden
presentar signos de virilización.
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Síndrome pierde sal):
La elevación de los niveles de 17-
hidroxiprogesterona ayudarán a establecer el
diagnóstico:
Neonatos sanos:
17-OH-progesterona basal < 20 ng/mL
Forma clásica:
17-OH-progesterona > 1000 ng/mL
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Forma virilizante simple): 25% de los
casos
La mutación de la 21-α-hidroxilasa permite producir
la cantidad de cortisol y aldosterona suficiente como
para no desarrollar un síndrome pierde sal.
El diagnóstico clínico va a depender del exceso de
andrógenos por encima del bloqueo enzimático.
HSC: Déficit 21-α-hidroxilasa

Forma clásica (Forma virilizante simple):
Niveles anormalmente elevados de 17-OH-
progesterona
Nivel basal > 100 ng/mL
HSC: Déficit 21-α-hidroxilasa

Forma no clásica o tardía
Grado suficiente de actividad enzimática como para
producir adecuadamente cortisol y aldosterona
Producción de andrógenos no excesiva.
Hiperandrogenismo variable de manifestación en la
infancia, adolescencia o edad adulta.
HSC: Déficit 21-α-hidroxilasa

Forma no clásica o tardía
Habitualmente los recién nacidos son asintomáticos.
Niñas: adrenarquia prematura, acné, aceleración del
crecimiento y de la edad ósea.
En la adolescencia: acné, hirsutismo,
irregularidades menstruales, amenorrea.
Niños: adrenarquia prematura, acné, aceleración del
crecimiento.
HSC: Déficit 21-α-hidroxilasa

Forma no clásica o tardía
El déficit de 21-α- hidroxilasa tardío en la mujer es el
que plantea el diagnóstico diferencial con otras
formas de hiperandrogenismo (ovario poliquístico,
síndrome de Cushing, hirsutismo idiopático,
hiperandrogenismo de origen tumoral).
Para el diagnóstico de la forma tardía es necesaria la realización de un
TEST DE ESTIMULACIÓN CON ACTH.
HSC: Déficit 21-α-hidroxilasa

PRUEBA DE ESTIMULACIÓN CON ACTH
La deficiencia parcial de 21-OH puede confirmarse
comparando los niveles séricos de 17-OHP antes y 60
minutos después de la administración de 0,250 mg
de ACTH para estimular la síntesis hormonal.
Si existe una deficiencia parcial, los niveles de 17-
OHP deberían aumentar.

ACTH: ritmo circadiano.
17-OH-progesterona (fase folicular del ciclo).
Se considera prueba positiva cuando los valores
superan los 15 ng/mL tras estímulo.
PRUEBA DE ESTIMULACIÓN CON ACTH

El déficit de 11-β-hidroxilasa: responsable del 5-8%
de los casos de HSC.
Su incidencia se estima entre 1/250.000 y 1/100.000.
Disminución de la síntesis de aldosterona y cortisol,
que produce un aumento de ACTH y,
secundariamente, aumento de los precursores al
bloqueo enzimático.
HSC: Déficit 11-β-hidroxilasa

El gen responsable se encuentra en la región Q21-22
del brazo largo del cromosoma 8 y se denomina
CYP11B1.
A pesar de estar alterada la producción de
aldosterona, estos pacientes no presentan signos ni
síntomas de insuficiencia suprarrenal, ya que existe
elevación de desoxicorticosterona (DOCA).
HSC: Déficit 11-β-hidroxilasa
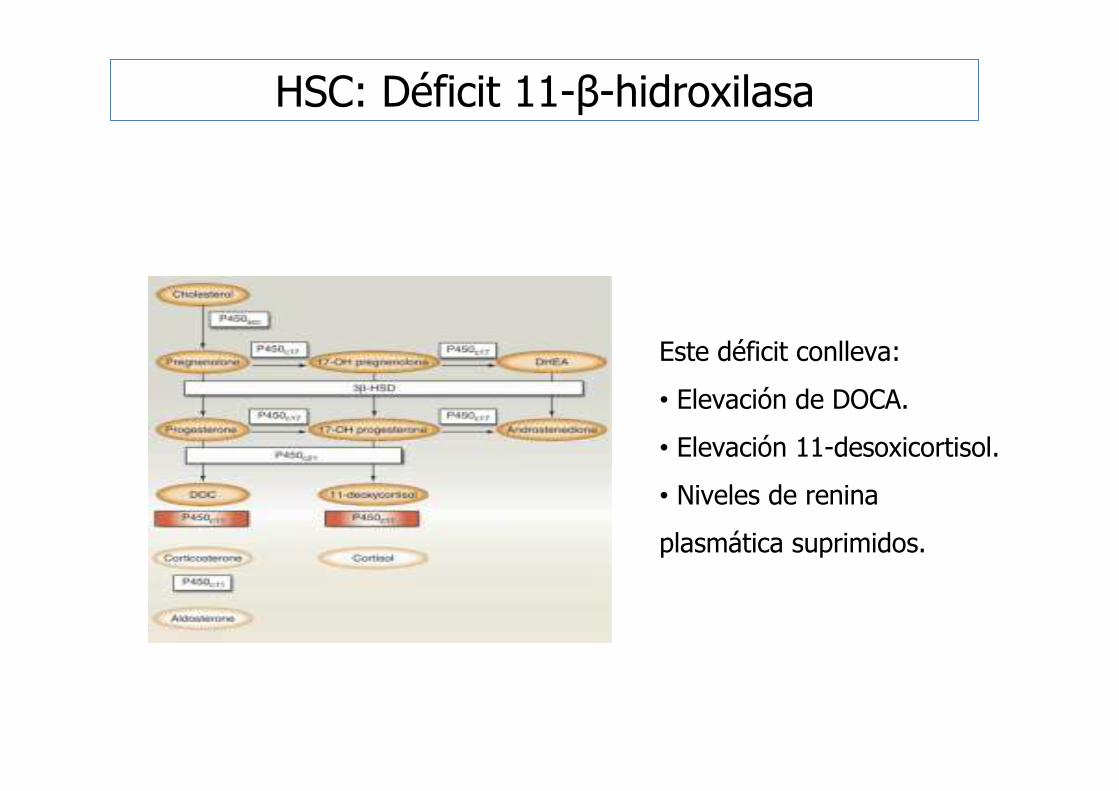
Este déficit conlleva:
• Elevación de DOCA.
• Elevación 11-desoxicortisol.
• Niveles de renina
plasmática suprimidos.
HSC: Déficit 11-β-hidroxilasa
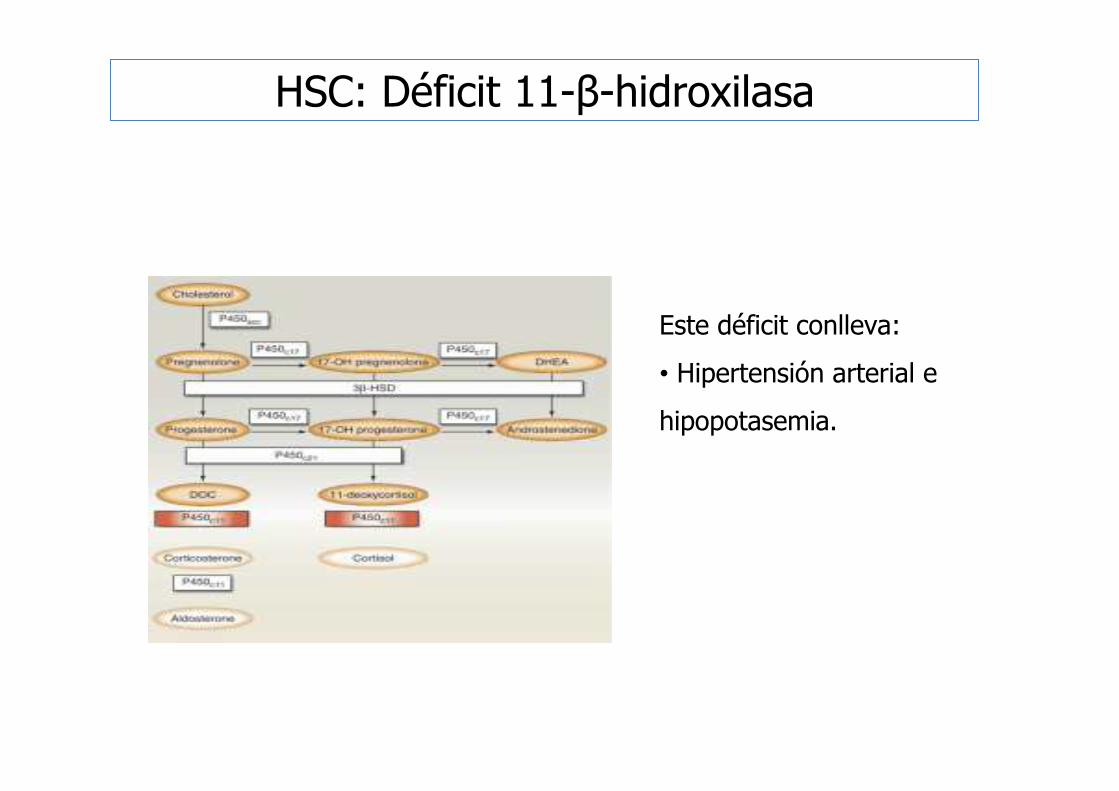
Este déficit conlleva:
• Hipertensión arterial e
hipopotasemia.
HSC: Déficit 11-β-hidroxilasa

Déficit de 11-β-hidroxilasa:
Elevación de los precursores androgénicos.
Niñas: virilización excesiva (genitales ambiguos) en el
momento del parto.
HSC: Déficit 11-β-hidroxilasa

Diagnóstico diferencial:
Determinar niveles de 11-desoxicortisol.
Encontraremos niveles de renina plasmática
suprimidos, por la elevación de DOCA, junto con
hipertensión e hipopotasemia.
HSC: Déficit 11-β-hidroxilasa

DIAGNÓSTICO
El diagnóstico definitivo:
Estudio genético de las posibles mutaciones del gen
responsable.

TRATAMIENTO
El objetivo del tratamiento sustitutivo con
glucocorticoides y mineralcorticoides en los pacientes
con deficiencia de 21-OH es:
- Niveles de 17-OHP < 2 mg/L
- Niveles de ACTH < 100 ng/L
Evitar el desvío hacia la síntesis de testosterona, lo
que queda demostrado por un nivel normal de
androstendiona.





























