Embed Size (px)
Citation preview

88
4.設計検証及び妥当性確認文書の概要
4.1 一般情報
4.1.1 規格への適合宣言
本申請品目は、下記の基準へ適合する。 (添付資料:ニ-1)
1.薬事法第 41 条第 3 項の規定により厚生労働大臣が定める医療機器の基準
(平成 17 年 3 月 29 日 厚生労働省告示第 122 号)
2.医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令
(平成 16 年 12 月 17 日 厚生労働省令第 169 号)

89
4.2 機器の設計の妥当性確認の概要
4.2.1 機器の安全性を裏付ける試験
4.2.1.1 物理的、化学的特性
本品は、高分子材料配合成分等の特性が医療機器の本質として係るものではない。
従って本項の記載を省略する。
4.2.1.2 電気的安全性及び電磁両立性
本品は、電気的安全性及び電磁両立性に係る機能を有する医療機器ではない。
従って本項の記載を省略する。
90
4.2.1.3 生物学的安全性
添付資料:ホ-1-1
総 括
本品において、血液体液に接触する構成品である、バルーン付中心静脈カテーテルについて生物
学的安全性を確認した。既承認品の販売名「サーモガードシステム」(承認番号 22400BZI00010000)
のカテーテルと本品の原材料の差分は、バルーンと接着剤、先端チップであるため、本品の Icy カ
テーテル(モデル:IC-3893)を検体とし、ISO 10993-1: Biological evaluation of medical devices --
Part 1: Evaluation and testing に従い試験を実施した。
(1)カテーテル(コーティング含む)の生物学的安全性
Icy 及び Quattro カテーテルにおいて原材料は同一であり、評価が必要となる既承認品(サーモガ
ードシステムのカテーテル)との差分(バルーンと接着剤、先端チップ)も同一である。よって本
品の Icy カテーテル(モデル:IC-3893)を代表製品としてカテーテル全体(「Applause」ヘパリン
コーティング含む)の生物学的安全性試験の評価を行うことは妥当であると判断して、販売名「サ
ーモガードシステム」(承認番号 22400BZI00010000)平成 25 年 9 月 6 日付外国製造医療機器製造
販売承認事項一部変更承認書に生物学的安全性試験を添付し、生物学的安全性を確認済みである。
しかし、亜急性全身毒性試験は添付していなかったため、本申請において亜急性毒性試験の評価
を行った。試験の概略を表 4.2.1.3-1 に示す。
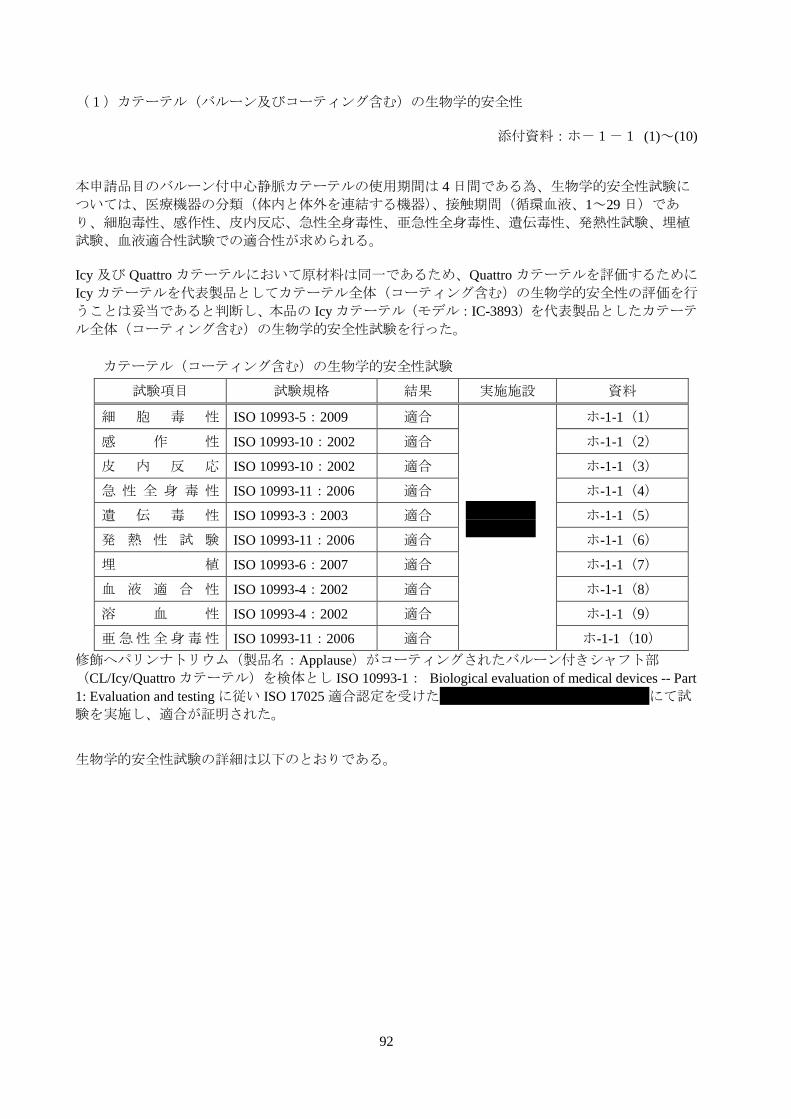
表 4.2.1.3-1:カテーテル(コーティング含む)の生物学的安全性試験概略
試験項目 試験規格 結果 実施施設 資料
細 胞 毒 性 ISO 10993-5:2009 適合
*****
*****
ホ-1-1(1)
感 作 性 ISO 10993-10:2002 適合 ホ-1-1(2)
皮 内 反 応 ISO 10993-10:2002 適合 ホ-1-1(3)
急 性 全 身 毒 性 ISO 10993-11:2006 適合 ホ-1-1(4)
遺 伝 毒 性 ISO 10993-3:2003 適合 ホ-1-1(5)
発 熱 性 試 験 ISO 10993-11:2006 適合 ホ-1-1(6)
埋 植 ISO 10993-6:2007 適合 ホ-1-1(7)
血 液 適 合 性 ISO 10993-4:2002 適合 ホ-1-1(8)
溶 血 性 ISO 10993-4:2002 適合 ホ-1-1(9)
亜急性全身毒性 ISO 10993-11:2006 適合 ホ-1-1(10)
カテーテル以外の本品の構成品の内、ダイレータの使用期間は短時間のため、医療機器の分類(体
内と体外を連結する機器)、接触期間(循環血液、24 時間以内)であり、細胞毒性、感作性、皮内
反応、急性全身毒性、発熱性、血液適合性試験での適合性が求められる。ダイレータの原材料は、
既承認品目(販売名「サーモガードシステム」、承認番号 22400BZI00010000、平成 25 年 3 月 29 日
付外国製造医療機器製造販売承認事項一部変更承認書)と同一であり、生物学的安全性が確認さ
れている原材料である。
また、その他の構成品は承認又は認証を取得しており、生物学的安全性は担保されている。
バルーン付中心静脈カテーテルの製造工程中に混入の危険性があるエンドトキシンの有無につい
て、ISO 17025 適合認定を受けた**********************において米国薬

91
局法(USP)エンドトキシン試験法に従い試験を実施し、適合証明書が発行された。本証明書は、
既承認品(サーモガードシステムの Cool Line カテーテル)を代表製品として評価しているが、既
承認品のカテーテルと Icy 及び Quattro カテーテルは、基本デザインは同一でシャフト長及びバル
ーン数が違うのみであり、製造工程において特段の差異はないため、製造工程中に混入するエン
ドトキシンの差異もない。販売名「サーモガードシステム」(承認番号 22400BZI00010000)平成 24
年 6 月 25 日付外国製造医療機器製造販売承認書に添付した適合証明書にて、本品のエンドトキシ
ンの混入が無く、適合が証明されていると判断した。
【考 察】
(1)カテーテル(コーティング含む)の生物学的安全性
今回、本品の Icy カテーテル(モデル:IC-3893)を代表製品としてカテーテル全体の生物学的
安全性試験について上記表 4.2.1.3-1 に示すとおり、必須の評価項目は全て実施し適合した。
以上より、本品の ICY 及び Quattro カテーテルの生物学的安全性が確認できた。
カテーテル以外の構成品についても、生物学的安全性への適合が確認されている。
以上より、カテーテル及び構成品について、ISO10993 に適合していると確認できた。
以上より、本申請品目は生物学的安全性に問題はない機器であるといえる。
92
(1)カテーテル(バルーン及びコーティング含む)の生物学的安全性
添付資料:ホ-1-1 (1)~(10)
本申請品目のバルーン付中心静脈カテーテルの使用期間は 4 日間である為、生物学的安全性試験に
ついては、医療機器の分類(体内と体外を連結する機器)、接触期間(循環血液、1~29 日)であ
り、細胞毒性、感作性、皮内反応、急性全身毒性、亜急性全身毒性、遺伝毒性、発熱性試験、埋植
試験、血液適合性試験での適合性が求められる。
Icy 及び Quattro カテーテルにおいて原材料は同一であるため、Quattro カテーテルを評価するために
Icy カテーテルを代表製品としてカテーテル全体(コーティング含む)の生物学的安全性の評価を行
うことは妥当であると判断し、本品の Icy カテーテル(モデル:IC-3893)を代表製品としたカテーテ
ル全体(コーティング含む)の生物学的安全性試験を行った。
カテーテル(コーティング含む)の生物学的安全性試験
試験項目 試験規格 結果 実施施設 資料
細 胞 毒 性 ISO 10993-5:2009 適合
*****
*****
ホ-1-1(1)
感 作 性 ISO 10993-10:2002 適合 ホ-1-1(2)
皮 内 反 応 ISO 10993-10:2002 適合 ホ-1-1(3)
急 性 全 身 毒 性 ISO 10993-11:2006 適合 ホ-1-1(4)
遺 伝 毒 性 ISO 10993-3:2003 適合 ホ-1-1(5)
発 熱 性 試 験 ISO 10993-11:2006 適合 ホ-1-1(6)
埋 植 ISO 10993-6:2007 適合 ホ-1-1(7)
血 液 適 合 性 ISO 10993-4:2002 適合 ホ-1-1(8)
溶 血 性 ISO 10993-4:2002 適合 ホ-1-1(9)
亜急性全身毒性 ISO 10993-11:2006 適合 ホ-1-1(10)
修飾ヘパリンナトリウム(製品名:Applause)がコーティングされたバルーン付きシャフト部
(CL/Icy/Quattro カテーテル)を検体とし ISO 10993-1: Biological evaluation of medical devices -- Part
1: Evaluation and testing に従い ISO 17025 適合認定を受けた***************にて試
験を実施し、適合が証明された。
生物学的安全性試験の詳細は以下のとおりである。

93
1)細胞毒性 添付資料:ホ-1-1(1)
試験物質に対する哺乳類細胞培養(L929)の生物学的反応性を判定。本試験は細胞の生存率を代謝
活性によって測定することに基づいている。
【規格及び試験方法】
・ISO 10993-5, 2009, Biological Evaluation of Medical Devices - Part 5: Tests for In Vitro Cytotoxicity.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration Laboratories.
【実施施設】 ********
【検 体】ZOLL/Alsius 社製 Cool line Family、ICY Family および Quattro カテーテル(SurModics 親
水性ヘパリンコーティング処理済み) *************
【対照物質】
陽性対照材料:天然ゴム *****************
陰性対照材料:陰性対照高密度ポリエチレン(陰性対照プラスチック)
*****************
未処理対照(抽出培地):血清添加(完全)最小必須培地(MEM)
*****************
添加物(培地の最終濃度):10%ウシ胎児血清、100 U/mL ペニシリン、0.1 mg/mL
ストレプトマイシン、2 mM L-グルタミン
【試験系】マウス線維芽細胞 L929
【評価基準】
生存細胞の数が減少すると、サンプルの代謝活性が低下する。この低下は直接、570 nm の吸光度(=OD)
で観察したときの青紫色ホルマザンの量に相関する。被験抽出物に曝露した細胞の生存率低下は、ブ
ランク(抽出培地に曝露した細胞、未処理)と比較するとき以下の式を用いて計算される。
生存率(%) = 100 * OD570e/OD570b
ここで、
OD570eは被験抽出物に曝露した細胞の光学濃度平均値である。
OD570bはブランク(抽出培地に曝露した細胞=未処理)の光学濃度平均値である。
生存率(%)が低下すればするほど、被験物質の細胞毒性は増加する。
生存率がブランクの<70%に低下すれば、その被験物質には細胞毒性があると考えられる。
本試験およびそのデザインは、測定の不確定度を最小限にし、データの収集および解析に対するバイ
アスを抑える方法を採用している。
【結 果】
94
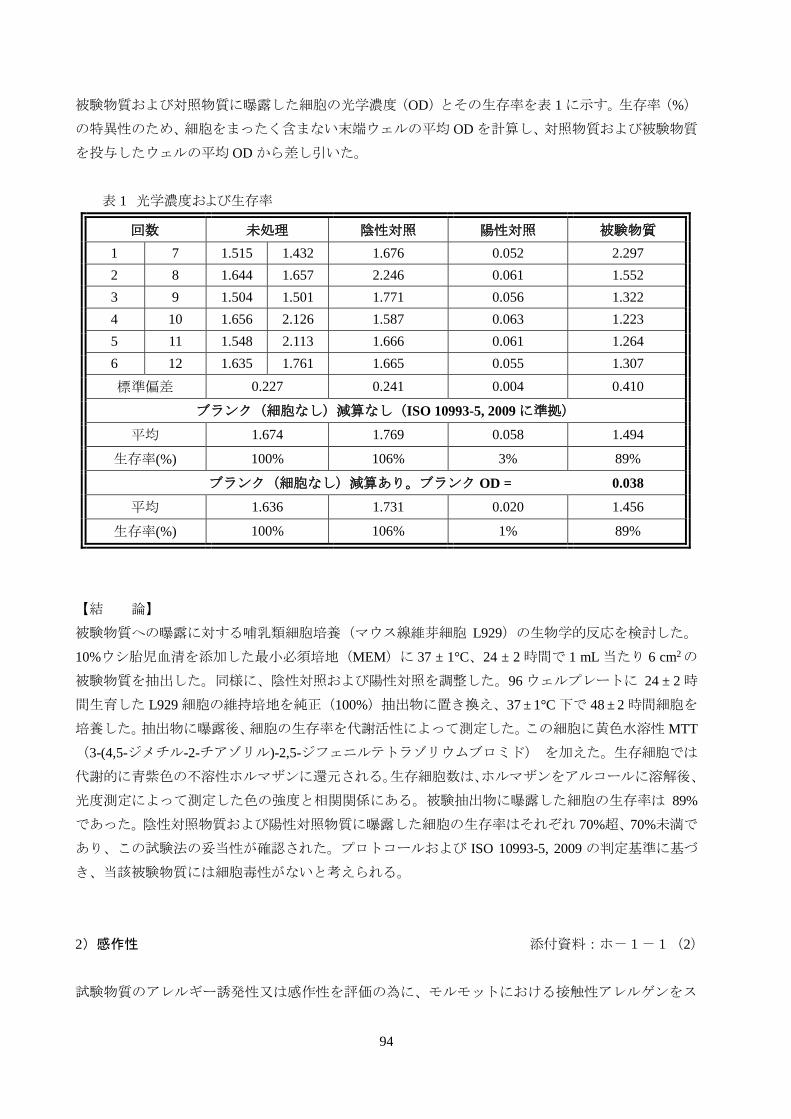
被験物質および対照物質に曝露した細胞の光学濃度(OD)とその生存率を表 1 に示す。生存率(%)
の特異性のため、細胞をまったく含まない末端ウェルの平均 OD を計算し、対照物質および被験物質
を投与したウェルの平均 OD から差し引いた。
表 1 光学濃度および生存率
回数 未処理 陰性対照 陽性対照 被験物質
1 7 1.515 1.432 1.676 0.052 2.297
2 8 1.644 1.657 2.246 0.061 1.552
3 9 1.504 1.501 1.771 0.056 1.322
4 10 1.656 2.126 1.587 0.063 1.223
5 11 1.548 2.113 1.666 0.061 1.264
6 12 1.635 1.761 1.665 0.055 1.307
標準偏差 0.227 0.241 0.004 0.410
ブランク(細胞なし)減算なし(ISO 10993-5, 2009 に準拠)
平均 1.674 1.769 0.058 1.494
生存率(%) 100% 106% 3% 89%
ブランク(細胞なし)減算あり。ブランク OD = 0.038
平均 1.636 1.731 0.020 1.456
生存率(%) 100% 106% 1% 89%
【結 論】
被験物質への曝露に対する哺乳類細胞培養(マウス線維芽細胞 L929)の生物学的反応を検討した。
10%ウシ胎児血清を添加した最小必須培地(MEM)に 37 ± 1°C、24 ± 2 時間で 1 mL 当たり 6 cm2の
被験物質を抽出した。同様に、陰性対照および陽性対照を調整した。96 ウェルプレートに 24 ± 2 時
間生育した L929 細胞の維持培地を純正(100%)抽出物に置き換え、37 ± 1°C 下で 48 ± 2 時間細胞を
培養した。抽出物に曝露後、細胞の生存率を代謝活性によって測定した。この細胞に黄色水溶性 MTT
(3-(4,5-ジメチル-2-チアゾリル)-2,5-ジフェニルテトラゾリウムブロミド) を加えた。生存細胞では
代謝的に青紫色の不溶性ホルマザンに還元される。生存細胞数は、ホルマザンをアルコールに溶解後、
光度測定によって測定した色の強度と相関関係にある。被験抽出物に曝露した細胞の生存率は 89%
であった。陰性対照物質および陽性対照物質に曝露した細胞の生存率はそれぞれ 70%超、70%未満で
あり、この試験法の妥当性が確認された。プロトコールおよび ISO 10993-5, 2009 の判定基準に基づ
き、当該被験物質には細胞毒性がないと考えられる。
2)感作性 添付資料:ホ-1-1(2)
試験物質のアレルギー誘発性又は感作性を評価の為に、モルモットにおける接触性アレルゲンをス

95
クリーニングした。
【規格及び試験方法】
・ISO 10993-10, 2002, Biological Evaluation of Medical Devices - Part 10: Tests for Irritation and
Delayed-Type Hypersensitivity, as amended 2006.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】ZOLL/Alsius 社製 Cool line Family、ICY Family および Quattro カテーテル(SurModics 親
水性ヘパリンコーティング処理済み) *************
【対照物質】
陽性対照材料:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
対照材料:綿実油(CSO)*****************
陰性対照材料:ジニトロクロロベンゼン(DNCB)*****************
【被験動物】ハートレイ系モルモット(Cavia porcellus)35 匹
【評価基準】
Kligman のスコアリングシステムを用いて、試験物質のアレルギー誘発性を次のように分類。観察
された感作率を基に試験の結果を判定した。
表 1 Magnusson-Kligman スケール
反応 グレード判定スケール
目に見える変化なし 0
分散またはパッチ部の紅斑 1*
中等度および集密的な紅斑 2*
高度紅斑および腫脹 3*
*陽性反応を示す
表 2 感作分類
感作率(%) グレード クラス
0~8 Ⅰ 軽微
9~28 Ⅱ 軽度
29~64 Ⅲ 中等度
65~80 Ⅳ 高度
81~100 Ⅴ 極度

96
【結 果】
・動物の体重:すべての動物の体重が増加した。
・臨床観察所見:実験動物および対照動物のいずれにも全身性の毒性徴候は認められなかった。
・感作:惹起時、実験動物(NaCl または CSO 抽出物)および陰性対照動物のいずれも、何ら反応
を誘発しなかった(感作率 0%)。陽性対照物質では全動物で中等度ないし強度の反応を誘発した
(感作率 100%)。
【結 論】
試験物質の USP 0.9%塩化ナトリウム注射液(NaCl)および綿実油(CSO)の抽出物では、誘導期後
の惹起時に何らの反応も誘発されなかった(感作率 0%)。そのため、Kligman のスコアリングシステ
ムに規定されているように、グレード I の反応であり、被験物質はアレルギー性が弱いものと分類さ
れる。プロトコールの基準に基づき、グレード I は重要ではないと考えられ、被験物質が ISO 10993-
10 ガイドラインの要求事項を満たしていることがわかる。
3)皮内反応 添付資料:ホ-1-1(3)
ニュージーランド白色ウサギの皮内に注射し、溶液と被験物質抽出液の刺激作用についてスクリー
ニングした。
【規格及び試験方法】
・ISO 10993-10, 2002, Biological Evaluation of Medical Devices – Part 10: Tests for Irritation and
Delayed-Type Hypersensitivity(2006 年改訂)
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices – Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 *********
【検 体】ZOLL/Alsius 社製 Cool line Family、ICY Family、Quattro カテーテル(SurModics 親水性
ヘパリンコーティング処理済み) *************
【対照物質】
陰性対照材料:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
対照材料:綿実油(CSO) *****************
【被験動物】ニュージーランド白色ウサギ(Oryctolagus cuniculus)2 匹
【評価基準】
72 時間後の評価後、それぞれの被験物質および溶媒対照について別々に、紅斑と腫脹の全等級を合

97
算した。各合算値を 12(動物 2 匹×3 等級期間×2 等級カテゴリー)で除し、それぞれの被験物質と
これに対応する溶媒対照の総平均スコアを求めた。被験物質と溶媒対照の平均スコアの差が 1.0 以下
の場合、本試験の要求事項は満たされている。いずれの観察期間でも、被験物質に対する平均反応が
溶媒対照に対する平均反応より疑わしい大きさを示した場合には、ウサギを新たに 3 匹用いて試験
を繰り返す。
【結 果】
・動物の体重:試験動物 2 匹すべての体重が増加した。
・臨床観察:いずれの観察時点においても、明らかな毒性徴候を示した動物はなかった。
・被験物質を注射した部位の方が、対照物質の注射部位より生物学的反応が有意に大きいとの結果
は示されなかった。
・被験物質および対照物質の総平均スコアの差は 0.0 であった。
【結 論】
被験物質の USP 0.9%塩化ナトリウム注射液(NaCl)および綿実油(CSO)による抽出物をニュー
ジーランドホワイトウサギに皮内注射し、刺激産生を評価した。被験物質の適用部位の方が、対照
物質の注射部位より生物学的反応が有意に大きいとの結果は示されなかった。被験物質はプロト
コールの基準に基づき、ISO 10993-10 ガイドラインの要求事項を満たすものである。
4)全身性毒性(全身性急性毒性) 添付資料:ホ-1-1(4)
マウスを対象に、単回投与による全身注射によって惹き起こされる毒性作用を検討するため、溶液お
よび被験物質抽出物をスクリーニングした。
【規格及び試験方法】
・ISO 10993-11, 2006, Biological Evaluation of Medical Devices - Part 11: Tests for Systemic Toxicity.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング) *************
【対照物質】
陽性対照材料:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
陰性対照材料:綿実油(CSO)*****************
【被験動物】動物種及び数:アルビノマウス(Mus musculus)20 匹
【観察期間】注入後 24、48、及び 72±2 時間

98
【評価基準】
被験物質を注射した動物のいずれもが、対照物質を投与した動物より生物学的反応が有意に大きい
値を示さなかった場合には、本試験を陰性と考える。
マウスが 2 匹以上死亡または痙攣や衰弱などの毒性徴候を示すか、あるいは 3 匹以上に 10%超の体
重減がみられる場合には、被験物質は本試験の要求事項を満たさないものとする。被験物質を投与し
た動物がわずかな生物学的反応のみを示したり、肉眼的な生物学的反応の徴候を示すか死亡したも
のが最大 1 匹である場合には、マウス 10 匹を用いて再度試験を実施しなければならない。再試験で
は、10 匹いずれもが、対照物質を投与した動物より有意に大きな生物学的反応を示してはならない。
【結 果】
・動物の体重:試験群及び対照群の全動物の体重が増加した。
・臨床的観察:いかなる観測時点においても、試験群又は対照群のいずれの動物にも明白な毒性徴
候は認められなかった。
・被験物質の抽出物を注射した動物のいずれもが、対照物質を投与した動物より生物学的反応が有
意に大きい値を示さなかったため、本試験を陰性と考える。
【結 論】
アルビノマウスを用いた試験では、被験物質の USP 0.9%塩化ナトリウム注射液(NaCl)および綿実
油(CSO)による抽出物の方が、対照抽出物より生物学的反応が有意に大きいとの結果は示されなか
った。本試験は、ISO 10993-11 に規定の基準に基づき陰性と考える。
5)遺伝毒性(復帰突然変異試験) 添付資料:ホ-1-1(5)
Salmonella typhimurium 及び Escherichia coli に対してそれぞれ、ヒスチジン遺伝子(his-から his+)およ
びトリプトファン遺伝子(tryp-から tryp+)に復帰突然変異を誘発するかどうかを評価した。直接プレー
ト法による本試験は、外因性哺乳類代謝活性化系の存在下および非存在下に、S. typhimurium を 4 株と
E. coli を 1 株用いて実施した。
【規格及び試験方法】
・ ISO 10993-3, 2003, Biological Evaluation of Medical Devices - Part 3: Tests for Genotoxicity,
Carcinogenicity and Reproductive Toxicity.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.

99
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング処理済み) *************
【対照物質】
陰性対照材料:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
陰性対照材料:綿実油(CSO)*****************
陽性対照材料:2-アミノアントラセン *************************
***********************************
****************
陽性対象材料:アジ化ナトリウム **************************
*********
2-ニトロフルオレン *****************
9-アミノアクリジン *****************
4-ニトロキノリン 1-オキシド *****************
【試験系】 S. typhimurium 菌株 TA98、TA100、TA1535 及び TA1537
E. coli 菌株 WP2 uvrA
【評価基準】
復帰突然変異試験の評価基準:
GraphPad Prism 統計プログラムや ANOVA(分散分析)のほか、一対比較の確認のため Newman-Keuls
検定などの統計プログラムを用いて試験成績を解析した。この統計学的方法によって、被験物質抽出
物の方が対照物質より突然変異の頻度を有意に増加させる(p ≤ 0.05)かどうかが明らかにされる。
これにより、正の用量反応関係があるかどうかがわかる。
陽性反応:
陽性対照物質試験は、直接作用変異原物質および代謝性生体内変化を必要とする突然変異原から成
る。本試験系が既知の突然変異原に関して機能することを示すには、いずれの陽性対照でも対応する
陰性対照より突然変異体の数に統計的に有意な増加が認められる必要がある。陰性対照物質のプレ
ートから、それぞれの菌株に対して試験データと比較するための基準点が得られる。
陽性対照物質が変異原性反応を示さない場合には菌株の成績を棄却する。
一般に、陰性対照物質の 2 倍の反応を示す場合に、変異原性反応と判断する。
陰性対照物質より突然変異体の数を統計的に有意に増加させ(p ≤ 0.05)、それに再現性がある場合に
は、被験物質の抽出物が本試験の陽性反応を惹き起こしたと考える。
【結 果】
・被験物質抽出物、陰性対照物質および陽性対照物質に関して、1 プレート当たりの復帰変異体の平
均数を計算した。
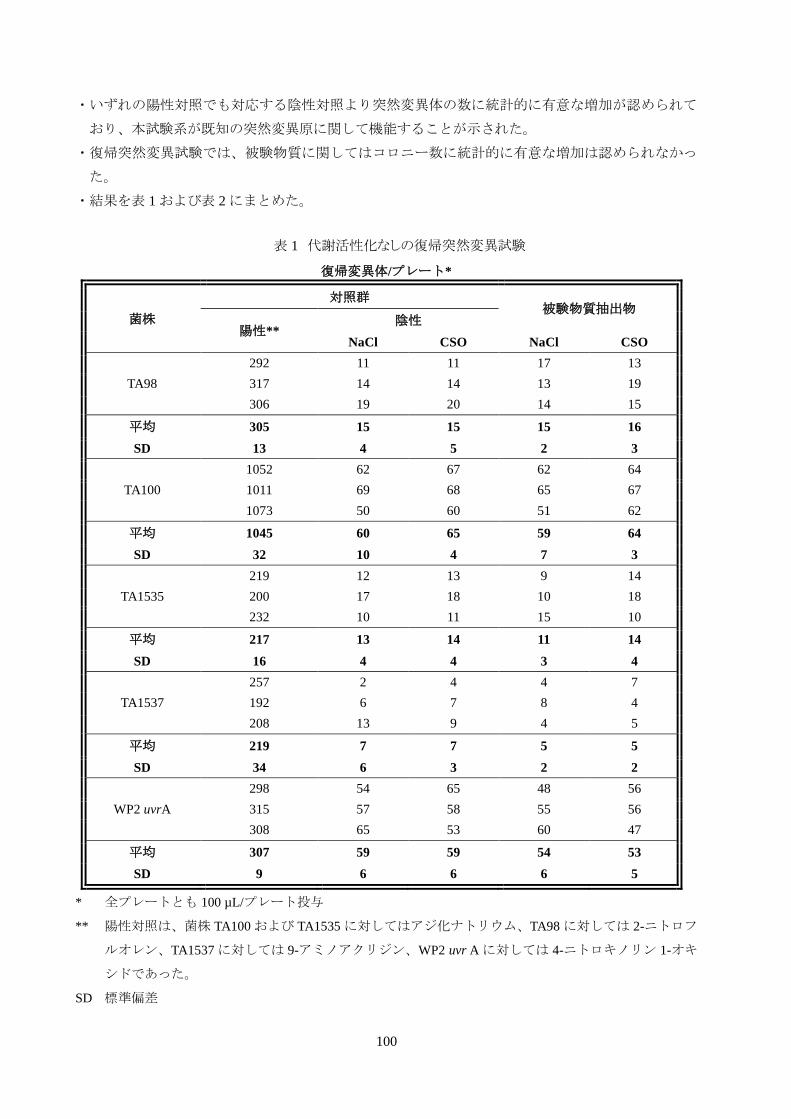
100
・いずれの陽性対照でも対応する陰性対照より突然変異体の数に統計的に有意な増加が認められて
おり、本試験系が既知の突然変異原に関して機能することが示された。
・復帰突然変異試験では、被験物質に関してはコロニー数に統計的に有意な増加は認められなかっ
た。
・結果を表 1 および表 2 にまとめた。
表 1 代謝活性化なしの復帰突然変異試験
復帰変異体/プレート*
菌株
対照群 被験物質抽出物
陽性** 陰性
NaCl CSO NaCl CSO
TA98
292 11 11 17 13
317 14 14 13 19
306 19 20 14 15
平均 305 15 15 15 16
SD 13 4 5 2 3
TA100
1052 62 67 62 64
1011 69 68 65 67
1073 50 60 51 62
平均 1045 60 65 59 64
SD 32 10 4 7 3
TA1535
219 12 13 9 14
200 17 18 10 18
232 10 11 15 10
平均 217 13 14 11 14
SD 16 4 4 3 4
TA1537
257 2 4 4 7
192 6 7 8 4
208 13 9 4 5
平均 219 7 7 5 5
SD 34 6 3 2 2
WP2 uvrA
298 54 65 48 56
315 57 58 55 56
308 65 53 60 47
平均 307 59 59 54 53
SD 9 6 6 6 5
* 全プレートとも 100 µL/プレート投与
** 陽性対照は、菌株 TA100 および TA1535 に対してはアジ化ナトリウム、TA98 に対しては 2-ニトロフ
ルオレン、TA1537 に対しては 9-アミノアクリジン、WP2 uvr A に対しては 4-ニトロキノリン 1-オキ
シドであった。
SD 標準偏差
101
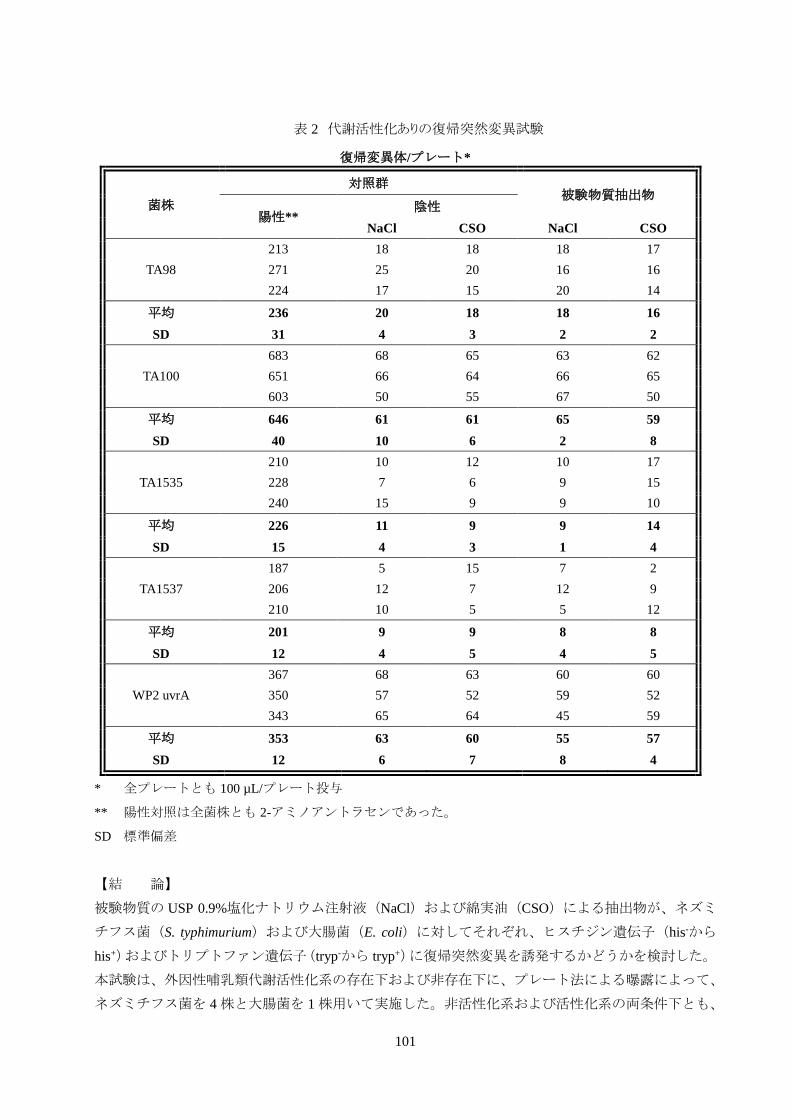
表 2 代謝活性化ありの復帰突然変異試験
復帰変異体/プレート*
菌株
対照群 被験物質抽出物
陽性** 陰性
NaCl CSO NaCl CSO
TA98
213 18 18 18 17
271 25 20 16 16
224 17 15 20 14
平均 236 20 18 18 16
SD 31 4 3 2 2
TA100
683 68 65 63 62
651 66 64 66 65
603 50 55 67 50
平均 646 61 61 65 59
SD 40 10 6 2 8
TA1535
210 10 12 10 17
228 7 6 9 15
240 15 9 9 10
平均 226 11 9 9 14
SD 15 4 3 1 4
TA1537
187 5 15 7 2
206 12 7 12 9
210 10 5 5 12
平均 201 9 9 8 8
SD 12 4 5 4 5
WP2 uvrA
367 68 63 60 60
350 57 52 59 52
343 65 64 45 59
平均 353 63 60 55 57
SD 12 6 7 8 4
* 全プレートとも 100 µL/プレート投与
** 陽性対照は全菌株とも 2-アミノアントラセンであった。
SD 標準偏差
【結 論】
被験物質の USP 0.9%塩化ナトリウム注射液(NaCl)および綿実油(CSO)による抽出物が、ネズミ
チフス菌(S. typhimurium)および大腸菌(E. coli)に対してそれぞれ、ヒスチジン遺伝子(his-から
his+)およびトリプトファン遺伝子(tryp-から tryp+)に復帰突然変異を誘発するかどうかを検討した。
本試験は、外因性哺乳類代謝活性化系の存在下および非存在下に、プレート法による曝露によって、
ネズミチフス菌を 4 株と大腸菌を 1 株用いて実施した。非活性化系および活性化系の両条件下とも、

102
陰性対照と比較して、いずれの被験物質の抽出物でも復帰変異コロニー数に統計的に有意な増加は
みられなかった。非活性化系および活性化系の両条件下とも、対応する陰性対照と比較して、いずれ
の陽性対照でも復帰変異コロニー数に統計的に有意な増加がみられたことから、この試験機能の妥
当性が確認された。試験プロトコールの基準に基づき、被験物質である Zoll/Alsius 社製Cool line family、
ICY Family および Quattro カテーテル(Sur Modics 親水性ヘパリンコーティング処理済み)は、採用
した実験条件下の試験種では変異原性を示さないと考えられる。
6)発熱性試験 添付資料:ホ-1-1(6)
患者に製品を投与した後にみられる熱性反応のリスクを許容値に抑えるため、固形材料の抽出液に
みる化学的発熱性物質の存在を明らかにすることである。本試験では、被験物質抽出物をニュージー
ランドホワイトウサギに静注し、体温の上昇を測定した。本製品は、実験対象のウサギに 1 kg あた
り 10 mL 以下の用量を最長 10 分間投与したとき、耐用性が示されるようデザインされている。
【規格及び試験方法】
・ISO 10993-11, 2006, Biological Evaluation of Medical Devices - Part 11: Tests for Systemic Toxicity.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング処理済み) *******************
【対照物質】
陰性対照材料:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
【被験動物】ニュージーランド白色ウサギ(Oryctolagus cuniculus)4 匹
【評価基準】
体温の低下を 0 上昇とした。
どのウサギも試験開始時の体温から 0.5°C 以上の上昇がみられない場合には、当該被験物質は発熱性
物質が認められない場合の要求事項に適合する。
いずれかのウサギに 0.5°C 以上の体温上昇がみられた場合には、新たに 5 匹の実験ウサギを用いて再
試験を実施する(追加費用は試験委託者が負担)。0.5°C 以上の体温上昇が認められるのが試験ウサギ
8 匹中最大でも 3 匹か、8 匹の体温上昇の合計値が最大で 3.3°C 以下の場合には、当該被験物質は発
熱性物質が認められない場合の ISO 要求事項に適合する。
【結 果】

103
実験動物の体温上昇は 0.2、0.0 および 0.0°C であった。上昇値は、個々の体温上昇の最大限界値を超
えるものではなかった。対照動物の体温上昇は 0.1°C であった。
【結 論】
被験物質の USP 0.9%塩化ナトリウム注射液(NaCl)による抽出液をニュージーランドホワイトウサ
ギに投与したときの発熱反応を評価した。プロトコールの基準に基づき、当該被験物質は非発熱性と
考えられ、発熱性物質試験の要求事項 ISO 10993-11 ガイドラインに適合するものである。
7)埋植試験 添付資料:ホ-1-1(7)
アルビノウサギの筋組織に被験物質を埋植し、局所組織反応および局所毒性作用を誘発する可能性
に関して被験物質を評価する。
【規格及び試験方法】
・ISO 10993-6, 2007, Biological Evaluation of Medical Devices - Part 6: Tests for Local Effects After
Implantation.
・ASTM F981-04, Standard Practice for Assessment of Compatibility of Biomaterials for Surgical Implants
with Respect to Effect of Materials on Muscle and Bone, 2004.
・ASTM F763-04, Standard Practice for Short Term Screening of Implant Materials, 2004.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング処理済み) *************
【対照物質】
陰性対照材料:陰性対照プラスチック *****************
【被験動物】ニュージーランド白色ウサギ(Oryctolagus cuniculus)3 匹
【評価基準】
標準スコア算出:
それぞれの埋植部位に対して合計スコアを測定した。各部位に対して炎症反応を合計し、因子 2 によ
って加重した。治癒反応は別々に合計した。炎症反応と治癒反応を合わせ、各部位に対して合計スコ
アを算出した。それぞれの動物に対して試験部位の平均スコアを対照部位の平均スコアと比較する。
全動物に対して試験部位と対照部位の間の平均差を計算し、次のように生体反応評点を割り当てる。
0.0~2.9 反応なし*
104
3.0~8.9 わずかな反応
9.0~15.0 中等度の反応
> 15 重度の反応
* 計算結果が負の場合には 0 と報告。
評点の修正:
病理評価者が算出した反応性の値を見直す。(たとえば相対的な大きさ、反応のパターン、炎症性と
その消失など)あらゆる因子の観察結果に基づいて、病理評価者が生体反応評点を修正する可能性が
ある。説明的報告のなかで、評点に対する修正の妥当性を示している。
病理評価者が、被験物質の生体適合性に関して説明的報告を提供している。
【結 果】
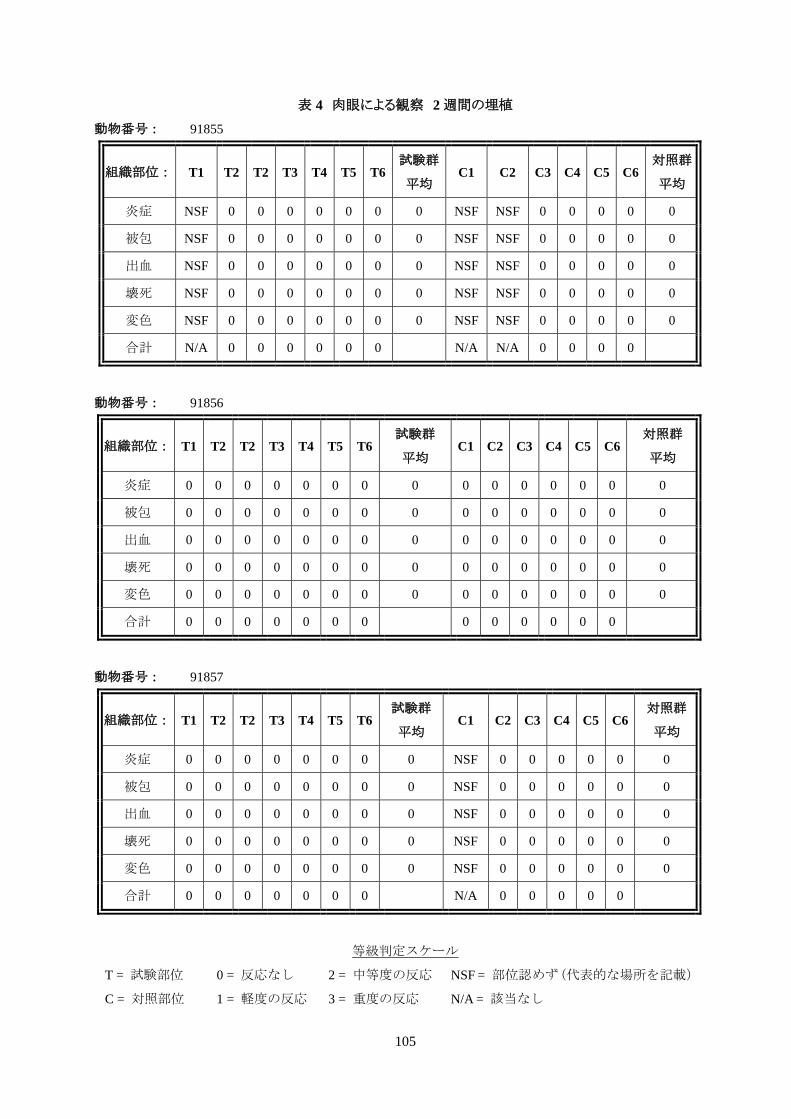
・動物の体重:3 匹の試験動物すべてで体重増加が示された。
・臨床観察所見(表 1):試験の全期間にわたって、実験動物のいずれにも毒性の徴候は認められな
かった。
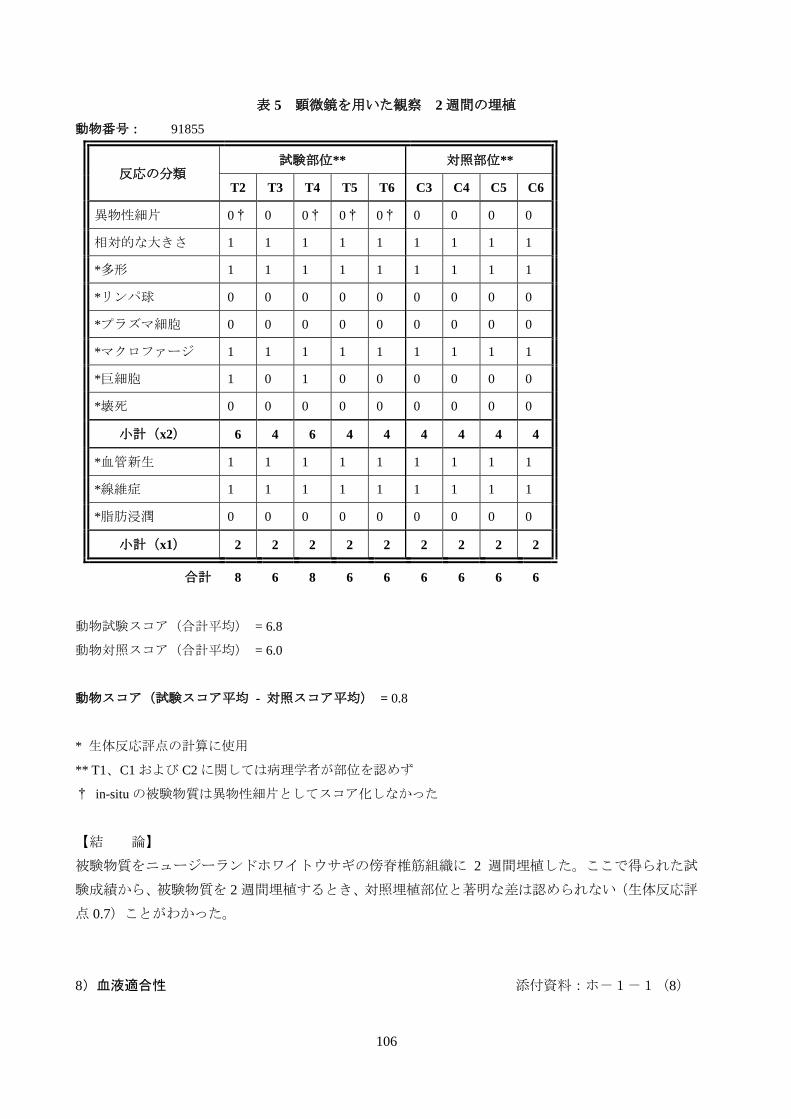
・埋植部位の観察所見(肉眼)(表 2):被験物質埋植部位の肉眼観察による評価から、2 週間の試験
期間で、炎症、被包、出血、壊死および変色の有意な徴候はみられないことがわかった。
・埋植部位の観察所見(顕微鏡)(表 3):被験物質埋植部位の顕微鏡を用いた評価から、対照物質を
埋植した部位と比較して界面に著しい差は認められないことがわかった。被験物質が in-situ のまま
であったため、界面にはわずかに肉芽腫性浸潤が認められた。被験物質の性質を考慮に入れれば、こ
の反応は予想されるものである。2 週間の試験期間での生体反応評点(平均 3 動物)は 0.7 であり、
対照埋植部位と比較すると反応なしであることがわかった。
表 1 動物の体重及び臨床観察 2 週間の埋植
動物番号 性別
体重(kg)
毒性の徴候* 第 0 日 第 14 日 体重の変化
**** ****
91855 雄 3.47 3.61 0.14 なし
91856 雌 3.09 3.30 0.21 なし
91857 雄 3.70 3.84 0.14 なし
* 第 0 日から第 14 日までの臨床観察結果の概要
105
表 4 肉眼による観察 2週間の埋植
動物番号: 91855
組織部位: T1 T2 T2 T3 T4 T5 T6 試験群
平均 C1 C2 C3 C4 C5 C6
対照群
平均
炎症 NSF 0 0 0 0 0 0 0 NSF NSF 0 0 0 0 0
被包 NSF 0 0 0 0 0 0 0 NSF NSF 0 0 0 0 0
出血 NSF 0 0 0 0 0 0 0 NSF NSF 0 0 0 0 0
壊死 NSF 0 0 0 0 0 0 0 NSF NSF 0 0 0 0 0
変色 NSF 0 0 0 0 0 0 0 NSF NSF 0 0 0 0 0
合計 N/A 0 0 0 0 0 0 N/A N/A 0 0 0 0
動物番号: 91856
組織部位: T1 T2 T2 T3 T4 T5 T6 試験群
平均 C1 C2 C3 C4 C5 C6
対照群
平均
炎症 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
被包 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
出血 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
壊死 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
変色 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
合計 0 0 0 0 0 0 0 0 0 0 0 0 0
動物番号: 91857
組織部位: T1 T2 T2 T3 T4 T5 T6 試験群
平均 C1 C2 C3 C4 C5 C6
対照群
平均
炎症 0 0 0 0 0 0 0 0 NSF 0 0 0 0 0 0
被包 0 0 0 0 0 0 0 0 NSF 0 0 0 0 0 0
出血 0 0 0 0 0 0 0 0 NSF 0 0 0 0 0 0
壊死 0 0 0 0 0 0 0 0 NSF 0 0 0 0 0 0
変色 0 0 0 0 0 0 0 0 NSF 0 0 0 0 0 0
合計 0 0 0 0 0 0 0 N/A 0 0 0 0 0
等級判定スケール
T = 試験部位 0 = 反応なし 2 = 中等度の反応 NSF = 部位認めず(代表的な場所を記載)
C = 対照部位 1 = 軽度の反応 3 = 重度の反応 N/A = 該当なし
106
表 5 顕微鏡を用いた観察 2 週間の埋植
動物番号: 91855
反応の分類 試験部位** 対照部位**
T2 T3 T4 T5 T6 C3 C4 C5 C6
異物性細片 0† 0 0† 0† 0† 0 0 0 0
相対的な大きさ 1 1 1 1 1 1 1 1 1
*多形 1 1 1 1 1 1 1 1 1
*リンパ球 0 0 0 0 0 0 0 0 0
*プラズマ細胞 0 0 0 0 0 0 0 0 0
*マクロファージ 1 1 1 1 1 1 1 1 1
*巨細胞 1 0 1 0 0 0 0 0 0
*壊死 0 0 0 0 0 0 0 0 0
小計(x2) 6 4 6 4 4 4 4 4 4
*血管新生 1 1 1 1 1 1 1 1 1
*線維症 1 1 1 1 1 1 1 1 1
*脂肪浸潤 0 0 0 0 0 0 0 0 0
小計(x1) 2 2 2 2 2 2 2 2 2
合計 8 6 8 6 6 6 6 6 6
動物試験スコア(合計平均) = 6.8
動物対照スコア(合計平均) = 6.0
動物スコア(試験スコア平均 - 対照スコア平均) = 0.8
* 生体反応評点の計算に使用
** T1、C1 および C2 に関しては病理学者が部位を認めず
† in-situ の被験物質は異物性細片としてスコア化しなかった
【結 論】
被験物質をニュージーランドホワイトウサギの傍脊椎筋組織に 2 週間埋植した。ここで得られた試
験成績から、被験物質を 2 週間埋植するとき、対照埋植部位と著明な差は認められない(生体反応評
点 0.7)ことがわかった。
8)血液適合性 添付資料:ホ-1-1(8)

107
被験物質がヒト血液の選択的血液パラメータに有害な作用を及ぼさないことを明らかにすることで
ある。試験対象とした血液パラメータは、血小板、ヘマトクリットおよび赤血球指数を含めた全血球
算定である。
【規格及び試験方法】
・ISO 10993-4, 2002, Biological Evaluation of Medical Devices - Part 4: Selection of Tests for Interactions
with Blood, as amended 2006.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング処理済み) *************
【対照物質】
陰性対照材料:陰性対照プラスチック *****************
陰性対照物質:USP 0.9%塩化ナトリウム注射液(NaCl)*****************
【試験系】ヒト血液
【評価基準】
それぞれの試験試料および陰性対照の試験成績に対して、平均偏差および標準偏差を計算した。試験
平均を陰性対照と比較する。Analytical Software, Inc.(Analyzing Data with Graph Pad Prism®, Harvey
Motulsky)の Graph Pad Prism ソフトウェアで、分散分析(ANOVA)をはじめとする統計学的手法を
実施することによって、群間に統計学的な有意差が確認された。偶然によって生じる差の確率が 5%
以下(p ≤ 0.05)の場合にのみ、試験群および対照群の間の差が統計学的に有意であると考えられる。
それぞれの血液パラメータに関して、その差が統計学的にかつ有害な方向に有意に両陰性対照との
差を示すものである場合には、被験物質は不適合であると判断する。この評価のなかで、生物学的お
よび統計学的な有意性が考慮される。
【結 果】
2 種の対照物質である未処置対照(抽出溶媒 NaCl に曝露した血液)及び陰性対照材料(陰性対照プ
ラスチック)の NaCl 抽出液に曝露した血液と比較して、試験物質である Cool Line II P/N CL2085B の
USP 0.9%塩化ナトリウム注射液(NaCl)抽出液がヒト血中選択血液パラメータに悪影響を及ぼす可
能性を評価した。評価した血液パラメータは全血球数、ヘマトクリット、赤血球指数、血小板数であ
った。被験抽出物は、試験した血液パラメータに悪影響を及ぼさなかった。
【結 論】
被験物質の USP 0.9%塩化ナトリウム注射液(NaCl)による抽出液が、非処置対照(抽出溶媒すなわ
ち NaCl に曝露した血液)および陰性対照物質の NaCl 抽出液に曝露した血液(陰性対照プラスチッ

108
ク)のふたつの比較対照と比べて、ヒト血液の選択的血液パラメータに有害な作用を及ぼす可能性が
あるかどうかを評価した。評価した血液パラメータは、全血球算定、ヘマトクリット、赤血球指数お
よび血小板数であった。被験抽出物は、試験対象とした血液パラメータにいかなる有害作用も及ぼさ
なかった。試験プロトコールの評価基準に基づいて、採用した実験条件下では、被験物質は in vitro
の血液適合性試験に合格している。
9)溶血性 添付資料:ホ-1-1(9)
ウサギ血液を用いた間接接触法により、被験物質の溶血活性を評価した。
【規格及び試験方法】
・ASTM F756-08, Standard Practice for Assessment of Hemolytic Properties of Materials, 2008.
・ISO 10993-4, 2002, Biological Evaluation of Medical Devices - Part 4: Selection of Tests for Interactions
with Blood, as amended 2006.
・ISO 10993-12, 2007, Biological Evaluation of Medical Devices - Part 12: Sample Preparation and
Reference Materials.
・ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration
Laboratories.
【実施施設】 ********
【検 体】Zoll/Alsius 社製 Cool line family、ICY Family および Quattro カテーテル(Sur Modics 親水
性ヘパリンコーティング処理済み) *************
【対照物質】
陰性対照材料:陰性対照プラスチック *****************
陽性対照材料:ブナ天然ゴム *****************
媒体対照の名称:マグネシウムおよびカルシウムを含まないリン酸緩衝生理食塩水(PBS)
*****************
【試験系】クエン酸添加ウサギ血液
【動物種及び数】ニュージーランド白色ウサギ(Oryctolagus cuniculus)3 匹
【評価基準】
・上清のヘモグロビン濃度を測定するために吸光度を使用する。被験物質の溶血率(%)は、ブラン
クおよび陰性対照の値を引くことによって得られる。
・本試験が有効であるためには、陰性対照の溶血率(%)が 0~2%の範囲に収まる必要がある。陰性
対照が 2%超の場合、ASTM F756-08 に従って本試験を再度実施しなければならない。
・被験物質の溶血率(%)が 0~2%以下の場合、被験物質は採用した実験条件下において非溶血性と
考える。溶血率が 2~5%の間の場合には、被験物質はわずかに溶血性と考えられ、5%以上の場合
には溶血性と判断を下すことができる。
・反復試験試料の平均値が<5%であるが、複数の反復試料が>5%の溶血指数を示した場合、その反復
試料の 2 倍の数だけ試験を繰り返す。
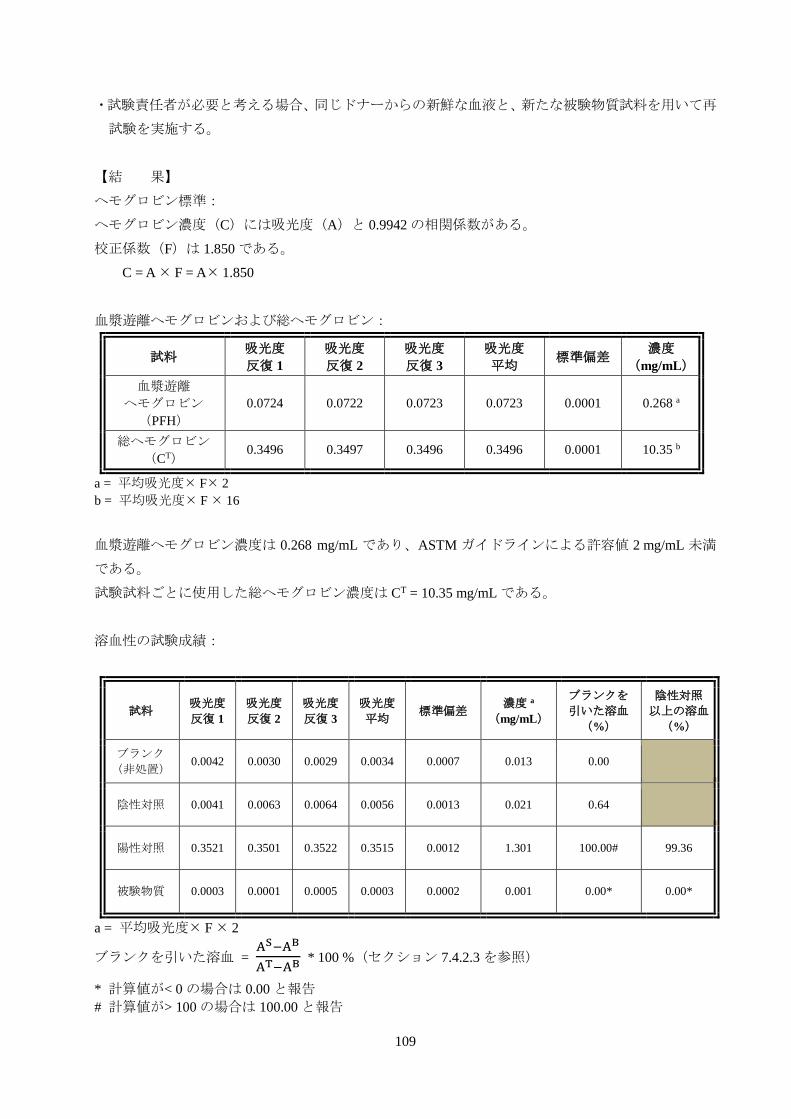
109
・試験責任者が必要と考える場合、同じドナーからの新鮮な血液と、新たな被験物質試料を用いて再
試験を実施する。
【結 果】
ヘモグロビン標準:
ヘモグロビン濃度(C)には吸光度(A)と 0.9942 の相関係数がある。
校正係数(F)は 1.850 である。
C = A F = A 1.850
血漿遊離ヘモグロビンおよび総ヘモグロビン:
試料 吸光度
反復 1
吸光度
反復 2
吸光度
反復 3
吸光度
平均 標準偏差
濃度
(mg/mL)
血漿遊離
ヘモグロビン
(PFH)
0.0724 0.0722 0.0723 0.0723 0.0001 0.268 a
総ヘモグロビン
(CT) 0.3496 0.3497 0.3496 0.3496 0.0001 10.35 b
a = 平均吸光度 F 2
b = 平均吸光度 F 16
血漿遊離ヘモグロビン濃度は 0.268 mg/mL であり、ASTM ガイドラインによる許容値 2 mg/mL 未満
である。
試験試料ごとに使用した総ヘモグロビン濃度は CT = 10.35 mg/mL である。
溶血性の試験成績:
試料 吸光度
反復 1
吸光度
反復 2
吸光度
反復 3
吸光度
平均 標準偏差
濃度 a
(mg/mL)
ブランクを
引いた溶血
(%)
陰性対照
以上の溶血
(%)
ブランク
(非処置) 0.0042 0.0030 0.0029 0.0034 0.0007 0.013 0.00
陰性対照 0.0041 0.0063 0.0064 0.0056 0.0013 0.021 0.64
陽性対照 0.3521 0.3501 0.3522 0.3515 0.0012 1.301 100.00# 99.36
被験物質 0.0003 0.0001 0.0005 0.0003 0.0002 0.001 0.00* 0.00*
a = 平均吸光度 F 2
ブランクを引いた溶血 = AS−AB
AT−AB * 100 %(セクション 7.4.2.3 を参照)
* 計算値が< 0 の場合は 0.00 と報告
# 計算値が> 100 の場合は 100.00 と報告

110
【結 論】
ASTM F756-08 を用いた間接接触により、被験物質である Zoll/Alsius 社製 Cool line family、ICY Family
および Quattro カテーテル(Sur Modics 親水性ヘパリンコーティング処理済み)の溶血活性を評価し
た。被験物質は 0.00%の溶血性を示し、間接接触法により陰性対照が示した溶血性の水準以上であっ
た。被験物質は、採用した実験条件下において非溶血性と考える。
10)亜急性全身毒性 添付資料:ホ-1-1(10)
被験物質(ZOLL/Alsius が提供する、SurModics の親水性被膜又はヘパリンでコーティングされた Cool
Line Family/ICY Family/Quattro カテーテル)を、ラットの筋肉組織に 28 日間埋め込み、被験物質に関
連する毒性の局所兆候及び全身兆候を評価した。被験物質及び対照物質は、それぞれ、10 体の動物
(雌雄各 5 体)に埋め込んだ。観察には、体重観察、臨床観察、血液学、臨床化学、凝固パラメータ、
臓器重量/相対臓器重量、並びに埋め込み部位及び選択した臓器の巨視的評価及び微視的評価(顕微
鏡的評価)を含めた。
【規格及び試験方法】
・ISO 10993-11, 2006, Biological Evaluation of Medical Devices - Part 11: Tests for Systemic Toxicity.
・ISO 10993-6, 2007, Biological Evaluation of Medical Devices - Part 6: Tests for Local Effects After
Implantation
.
【実施施設】********
【試験試料】SurModics 親水性ヘパリンでコーティングされた Cool Line Family/ICY Family/Quattro
カテーテル(ZOLL/Alsius 提供)
*************
試験試料は事前にカットした(1 X EO sterile)
被験物質の測定サイズは、およそ、直径 3 mm、長さ 2 cm であった。
【対照物質】陰性対照高密度ポリエチレン********
*****************
対照物質は、およそ、直径 1 mm、長さ 8 mm にカットし、70%のエタノールに浸して
滅菌した。
【被験動物】Sprague Dawley ラット 20 体(Rattus norvegicus)
試験群: オス 5 体及びメス 5 体
対照群: オス 5 体及びメス 5 体
生後 5 週間以上の成体で体重 227.4 - 376.8g*******************
*****
最低 5 日間馴化し、埋植前に体重を測定した。
身体の大きなものから選別し、好ましくない臨床兆候が見られないことを確認した。手
術前の鎮痛を除き、試験のどの段階でも治療薬は使用しなかった。
本試験でラットを使用した理由は、ラットが長期にわたって安全性評価試験に使用さ
れてきたこと、及びガイドラインに別の(動物を使用しない)方法の記載がないことに
よる。動物は、皮下埋め込みにより処理した。動物種、数、及び試験材料の投与経路は、
ISO 10993-11 及び ISO 10993-6 のガイドライン、並びに試験依頼者が推奨するものであ
った。

111
【埋植】 各ラットには、被験物質又は対照物質のいずれかを埋め込んだ。正中線から約 2 cm の
位置に切り込みを入れ、皮下組織に生じた小さなくぼみに、動物当たり 1 つの部位に 4
つの被験物質(T1 - T4)を埋め込んだ。
【観察期間】埋植後、28 日間維持管理し、毎日臨床兆候を観察した。
部検前後に体重測定し、部検による総括的観察、全血・血清・血漿による血液学的検査
(血液学、臨床化学、凝固)、臓器重量測定、埋植部位の巨視的観察、摘出後固定した
組織による組織病理学的検査を行った。
【評価基準】
生物反応性評点(埋め込み部位):
埋め込まれた部位ごとに、総得点を求める。炎症反応の場合は、埋め込み部位ごとに合計を出し、
因数 2 で重みを付ける(合計×2)。治癒反応の場合は、重み付けなしに合計を出す。2 つの合計を
共に埋め込み部位の得点に追加する。試験動物の全ての試験部位の平均点を対照動物の全ての部
位の平均点と比較する。試験群と対照群との差を計算し、最初の生物反応性評点を以下のように割
り当てる。
0-2.9 無反応*
> 2.9-8.9 軽度の反応
>8.9-15.0 中等度の反応
>15.0 重度の反応
* 計算結果が負の場合はゼロ(0)として報告する。
体重などの定量的データは適切な統計学的処理によって解析する。有意差がある場合は、生物学的
関連性を文献データや病歴データと比較してさらに評価する。対照動物と被験動物との間に統計
学的有意差があると見なせるのは、偶然に起因する差の発生確率が 5%(p ≤ 0.05)以下の場合に限
定される。対応のない t-検定(性別でグループ化される)は、体重の解析と臨床病理の連続値の解
析に使用する。統計解析は、GraphPad Software 社(San Diego, California, USA)の Windows 用 GraphPad
Prism Version 3.02 を使用して実施する。
<巨視的観察>
各部位は、炎症、包嚢形成、出血、壊死、及び変色の兆候について、以下の基準を用いて検査した。
0 = 正常
1 = 軽度
2 = 中等度
3 = 重度
<病理学的評価>
(埋め込み部位)
埋め込み/組織境界面から、正常な組織および血管状態の特性を示す、影響を受けなかった領域ま
での幅を評価して、影響を受けた領域の相対的サイズを採点した。関係する領域の相対的サイズは、
以下の基準を使用して採点した。
0 = 0 mm、部位なし
1 = 0.5 mm 以下、微少
2 = 0.6-1.0 mm、軽度
3 = 1.1 - 2.0 mm、中等度
4 = 2.0 mm より大、重度
(非埋め込み部位)
非埋め込み組織の評価は、容認される病理学的枠組みを使用して実施した。
0 = 正常、1 = 微少、2 = 軽度、3 = 中等度、4 = 重度

112
<血液学パラメータ>
1. 白血球数 6. 平均赤血球ヘモグロビン
2. 赤血球数 7. 平均赤血球ヘモグロビン濃度
3. ヘモグロビン 8. 血小板数
4. ヘマトクリット値 9. 白血球分画
5. 平均細胞体積
<臨床化学パラメータ>
1. アルブミン 10. グルコース
2. アルブミン/グロブリン比 11. 総ビリルビン
3. クレアチニン 12. 総タンパク
4. 血中尿素窒素(BUN) 13. トリグリセリド
5. グロブリン 14. ナトリウム
6. アラニンアミノトランスフェラーゼ(ALT) 15. カルシウム
7. アスパラギン酸アミノトランスフェラーゼ(AST) 16. 塩素
8. アルカリホスファターゼ(ALK) 17. リン
9. コレステロール 18. カリウム
<凝固パラメータ>
プロトロンビン時間(PT) 活性化部分トロンボプラスチン時間(APTT)
【結 果】
(1) 体重
体重又は全般的な体重変化において、試験群と対照群との間に統計学的な有意差はなかった。試験
動物と対照動物の全ては、試験の過程を通して体重が増加した、または不変であった。
(2) 臨床観察
試験動物又は対照動物はいずれも、試験の過程を通して毒性の兆候を示さなかった。
(3) 剖検
#4 の試験動物(オス)の頭蓋骨内側表面に突起物の増殖を確認した。過度の骨の成長に伴って脳
内に直径 2mm 未満の小さな腔が認められた。#17 の対照動物(メス)の右卵巣付近の脂肪組織内
に腫瘤を確認した。試験動物又は対照動物のいずれにも、剖検において肉眼で確認できる毒性を
示す観察又は有意な兆候はなかった。
(4) 血液学
剖検直前に採取した血液に対して血液学的分析を実施した。全てのパラメータに関し、試験動物
と対照動物との間に有意差は観察されなかった。
(5) 臨床化学
剖検直前の動物から採取した血液の血清分画に対して、臨床化学プロファイルの分析を実施した。
対照群と比較したとき、試験群のオスに、かなり高い血清塩化物値(p=0.0486)を確認した。塩化
物の値は、この種のラット系統の過去の基準値の範囲内(93.7-107.7 mmol/l)にあるため、この差
は、生物学的に有意とは見なさなかった。全てのパラメータに関し、試験群と対照群との間に他
の有意差は観察されなかった。
(6) 凝固
剖検直前に採取した血液の血漿分画に対して凝固パラメータの分析を実施した。全てのパラメー
タに関して、試験群と対照群との間に有意差はなかった。

113
(7) 臓器重量/相対的臓器重量
いずれの性別でも、試験群と対照群の間には臓器重量/相対的臓器重量に有意差はなかった。
(8) 巨視的所見 - 埋め込み部位
被験物質の埋め込み部位の巨視的評価では、28 日の期間を通し、炎症、包嚢形成、出血、壊死又
は変色について生物学的に有意な兆候はなかった。
(9) 顕微鏡的所見 - 埋め込み部位
埋め込み部位組織には、被験物質に関連すると考えられる顕微鏡下での異常はなかった。ラット
の筋肉への 28 日間の埋め込みに対する生物反応性評点は 0.1 であり、局所的な組織反応性がない
ことを示した。
(10) 顕微鏡的所見 - 非埋め込み組織
試験群オス#4 の突出した骨は、頭蓋骨に形成される異常性のない新しい骨並びに下層の脳軟膜へ
の単核細胞浸潤、色素(おそらくヘモジデリン)及び大脳の圧力面付近のうっ血に関連するもの
であった。対照群メス#17 の右卵巣付近の腫瘤は、顕微鏡下で観察したところ、卵巣とは無関係の
大網脂肪の脂肪肉芽腫に関連するものであった。被験物質に関連すると考えられる顕微鏡下の組
織異常はなかった。試験動物と対照動物の両方の心臓、肝臓、肺及び腎臓に認められた偶発性の
顕微鏡的所見は、この年齢のラット系統には正常な背景所見であると見なした。
【結 論】
全てのパラメータに対して、対照物質を埋め込んだ動物と比較した結果、被験物質を埋め込んだ動
物は、被験物質に起因する変化を示さなかった。従って、被験物質は、指示書に従ってラットに 28
日間埋め込んだとき、毒性の局所兆候または全身兆候を示さなかった。
4 週間の生物反応性評点は 0.1 であった。これは、対照部位と比較して、無反応であることを示し
ている。

114
4.2.1.4 放射線に関する安全性
本品は放射線を利用しない医用電気機器である。
従って本項の記載を省略する。
4.2.1.5 機械的安全性
本品は、機械的安全性に係る機能を有する医療機器ではない。
従って本項の記載を省略する。
115
4.2.1.6 安定性及び耐久性
添付資料:ハ-1-1~ハ-1-2
総 括
本品のバルーン付中心静脈カテーテルは単回使用の滅菌品であり、安定性及び耐久性を確認する
ため、加速エージングおよび実時間寿命性能試験を実施した。試験の概略を表 4.2.1.6-1 及び表
4.2.1.6-2 に示す。
(1)バルーン付中心静脈カテーテル(Icy バルーン)の安定性及び耐久性試験の概略
(添付資料:ハ-1-1)
本品カテーテルの旧モデル(IC-3585)(「Duraflo」ヘパリンコーティング)を検体として検証し
た結果を表 4.2.1.6-1 に示す。ICY 及び Quattro カテーテルの原材料は同一であり、必要とされる
有効期間、貯蔵及び使用に係る条件も同一であるため、ICY カテーテルを代表製品として検証
を行った。ICY カテーテルの旧モデル(IC-3585)から形状を変更(注入ルーメン数を 1 個から
3 個に、シャフト径を 8.5Fr から 9.3Fr に変更)した現モデル(IC-3893)は、変更した以外の形
状は同等であり、原材料の変更がない(下表参照)。安定性試験は、原材料に対して評価を行っ
ているため、旧モデルを使用した安定性試験は本品にも外挿できるとして、510(k)(K052443)
を取得しているため、本試験を本申請品の安定性及び耐久性試験に外挿できると判断した。
試験検体のカテーテルとの比較
販売名
(構成品名)
本邦未承認
(旧モデルの ICY カテーテル)
本品
(ICY カテーテル)
本品
(Quattro カテーテル)
バルーン個数 3 個 3 個 4 個
有効長 380 mm 380 mm 450 mm
シャフト径 8.5 Fr 9.3 Fr 9.3 Fr
ルーメン数 3 ルーメン
(1infusion, 1inflow, 1outflow)
5 ルーメン
(3infusion, 1inflow,
1outflow)
5 ルーメン
(3infusion, 1inflow,
1outflow)
ヘパリンコー
ティング剤
Duraflo
(Edwards Lifesciences 社製)
Applause
(SurModics 社製)
Applause
(SurModics 社製)
原材料 ヘパリンコーティング剤以外同一
表 4.2.1.6-1:バルーン付中心静脈カテーテル(Icy バルーン)の安定性及び耐久性試験の概略
試 験 目 的 バルーン付中心静脈カテーテルの安定性及び耐久性の評価。
実 施 施 設 ALSIUS Corporation 社試験室
対 象 バルーン付中心静脈カテーテル(モデル IC-3585)
(「Duraflo」ヘパリンコーティング)
加 速 条 件
アレニウスモデルに基づき、各有効期間のバルーン付中心静脈カテー
テル IC-3585 を調整するため、以下の条件にて実施した。(本品の有効
期間は 2 年である。)
貯蔵時間 昇温 加速された曝露時間
2 年 *℃ *日
試験項目 試験方法又は試験規格 結果
1. 目視検査 EN ISO 10555-1 Section 4.3.に従い評価。 適合
2. 寸法検査 各部寸法を測定し、規格範囲内であるとことを確認。 適合
3. ガイドワイヤ
試験
直径 0.81mm と 0.89mm のガイドワイヤをディスタルルーメ
ンに通し、ルーメン内を通ることを確認。
適合
4. パージング/プラ
イミング試験
インフロールーメンから生理食塩水を注入し、バルーン等の
流路内のエア抜きが出来ることを確認。
適合

116
(つづき)
試験項目 試験方法又は試験規格 結果
5. 流量測定 EN ISO 10555-3(Section 4.6. and Annex A)に従い、ディスタルル
ーメンの流量を測定。
適合
6. 熱交換能試験 シミュレートされた模擬患者モデルを用い、ポンプ流量
200mL/min で、********************
以下の項目を測定、算出。
適合
7. カテーテル破壊
試験(陰圧)
37℃の恒温槽に保持し、各ルーメン内をシリンジで空気を完
全に除き(陰圧にし)、ルーメンを観察。
適合
8. カテーテル寿命
試験
インフロールーメン、アウトフロールーメンにチューブセッ
トを接続し、37℃の恒温槽に保持し、240mL/min の流量で、水
を 4 日間、循環させ、バルーンからの漏れを観察。
適合
9. カテーテル破壊
及びバルーン破裂
試験(陽圧)
37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi
ずつ加圧し、破裂時の圧力を測定。
適合
10. 曲げ耐性試験 チップから**mm の位置を**回繰り返し**°屈曲させ、
観察。
適合
11. 破断強度試験 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.5,Annex B)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.7.1)に従い評価。
適合
注)ALSIUS Corporation は、現 ZOLLCirculation
(2)カテーテルの追加検証;加速エージング後の安定性 (添付資料:ハ-1-2)
510(k)(K052443)を取得した後、唯一コーティング剤の原材料を「Applause」ヘパリンコーティン
グに変更したため、コーティング剤が影響を与えると考えられる試験について追加検証を行った。
ICY 及び Quattro カテーテルの原材料及び包装は同一であり、必要とされる有効期間、貯蔵及び使
用に係る条件も同一であるため、本試験は ICY カテーテル(モデル:IC-3893)を代表製品とし、
カテーテル全体を検体として評価できるとして検証を行った。
表 4.2.1.6-2:バルーン付中心静脈カテーテル(Icy バルーン)の安定性及び耐久性試験の概略
試 験 目 的 「Applause」ヘパリンコーティングを有するバルーン付中心静脈カテー
テルの安定性及び耐久性の評価。
実 施 施 設 ZOLL Circulation
対 象 バルーン付中心静脈カテーテル(ICY 3 カテーテル/IC-3893)
(「Applause」ヘパリンコーティング)

117
(つづき)
加 速 条 件
アレニウスモデルに基づき、以下の条件にて実施した。(本品の有効期間
は 2 年である。)
貯蔵時間 昇温 加速された曝露時間
12 ヶ月 **℃ **日間
24 ヶ月 **℃ **日間
試験項目 試験方法又は試験規格 結果
目視検査 異常なきこと。 適合
熱交換能試験 シミュレートされた模擬患者モデルを用い、ポンプ流量
200mL/min で、*********************
以下の項目を測定、算出。
適合
カテーテル寿命試
験
インフロールーメン、アウトフロールーメンにチューブセット
を接続し、36.5℃の恒温槽に保持し、200mL/min の流量で、水
を 7 日間、循環させ、バルーンからの漏れを観察。
適合
カテーテル破壊及
びバルーン破裂試
験(陽圧)
37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi ず
つ加圧し、破裂時の圧力を測定。
適合
【考 察】
(1)バルーン付中心静脈カテーテル(Icy バルーン)の安定性及び耐久性試験の概略
(添付資料ハ-1-1)
アレニウスの式(ファントホッフの法則に基づく)に従って加速試験を実施し、それぞれの試験
の結果は、有効期間 2 年を担保するものであった。
(2)カテーテルの追加検証;加速エージング後の安定性 (添付資料ハ-1-2)
本品カテーテルの原材料においては、温度による劣化が大きな影響をもつと推測され、アレニ
ウスの式(ファントホッフの法則に基づく)に従って上昇した保存温度と加速暴露時間を決定
し、加速エージング後に性能試験を実施した。本試験においては、EtO 滅菌サイクルに 2 回暴露
したサンプルで試験を行った。それぞれの試験の結果は、有効期間 2 年を担保するものであっ
た。
以上より、バルーン付中心静脈カテーテル及びスタートアップキットにおいて、滅菌後 2 年間の
条件にて本品の性能を維持しており、有効期間 2 年間の安定性及び耐久性に問題はないといえる。

118
(1)バルーン付中心静脈カテーテル(Icy バルーン)の安定性及び耐久性試験の概略
(添付資料ハ-1-1)
サンプルは、通常の EtO 滅菌サイクルの後、2 年の加速エージングにて試験された。
【目 的】バルーン付中心静脈カテーテルの安定性及び耐久性の評価。
【実施施設】・ALSIUS Corporation 社試験室
【サンプル】バルーン付中心静脈カテーテル(モデル IC-3585)
(「Duraflo」ヘパリンコーティング)
【加速条件】
アレニウスモデルに基づき、各有効期間のバルーン付中心静脈カテーテル IC-3835 を調整するた
め、以下の条件にて実施した。
貯蔵時間 昇温 加速された曝露時間
2 年 *℃ *日
【結 果】
試 験 項 目 目視検査
試 験 方 法 EN ISO 10555-1 Section4.3.に従い評価。
結 果 汚染や表面の傷を 2.5 倍の倍率で検査し、全てのサンプルに異常がなかった。
試 験 項 目 寸法検査
試 験 方 法 各部寸法を測定し、規格範囲内であるとことを確認。
結 果 すべての寸法仕様に合格した。
試 験 項 目 ガイドワイヤ試験
試 験 方 法 直径 0.81mm と 0.89mm のガイドワイヤをディスタルルーメンに通し、ルーメ
ン内を通ることを確認。
結 果 22 個中 1 個が通らなかったが、ルアー材料に空洞が認められた。バックローデ
ィング中にこの空洞にディスタルルーメンの引っかかりが生じて、試験に合格
しなかった。そのため、16 個の追加試験を行い、ルーメン内を通ることを確認
した。38 個中 1 個が通らなかったが、90%信頼性基準及び 90%信頼度であるた
め、判定基準に準じて要求事項に適合したと判断した。
試 験 項 目 パージング/プライミング試験
試 験 方 法 インフロールーメンから生理食塩水を注入し、バルーン等の流路内のエア抜き
が出来ることを確認。
結 果 冷却膜内に残留したエア容量は極少量で、臨床的に問題なく製品の安全性や性
能に影響を与えないと判断した。

119
試験項目 流量測定
試験方法 EN ISO 10555-3(Section4.6. andAnnex A)に従い、ディスタルルーメンの流量を測
定。
結果 近位注入用ルーメンでの仕様値:最小平均流量 2200mL/時であり、平均流量
**mL/時であったが、90%信頼性基準及び 90%信頼度であるため、要求事項
に適合したと判断した。
試験項目 熱交換能試験
試験方法 モデルを用い、ポンプ流量 200mL/min で、****************
*****以下の項目を測定、算出。
結果 仕様値は、カテーテル流量 200mL/分において、最小値 125W である。平均値は
**W であり、信頼性基準/信頼度が 90/90 で仕様値に合格した。
試験項目 カテーテル破壊試験(陰圧)
試験方法 37℃の恒温槽に保持し、各ルーメン内をシリンジで空気を完全に除き(陰圧に
し)、ルーメンを観察。
結果 遠位注入用ルーメン及び近位注入用ルーメンは、*分間 真空に耐えて、内腔は
つぶれなかった。
試験項目 カテーテル寿命試験
試験方法 インフロールーメン、アウトフロールーメンにチューブセットを接続し、37℃
の恒温槽に保持し、240mL/min の流量で、水を4日間、循環させ、バルーンか
らの漏れを観察。
結果 59 個のうち 2 個にバルーンの不具合があった。1 個は近位バルーンの孔、1 個
は遠位バルーンの亀裂であった。そのため、4 個の追加試験を行い、総数 63 個
のうち 2 個が不具合となったが、その他のカテーテルに割れその他の故障は見
られなかったので、90%信頼性基準及び 95%信頼度であるため、要求事項に適
合したと判断した。
試験項目 カテーテル破壊及びバルーン破裂試験(陽圧)
試験方法 37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi ずつ加圧し、破裂
時の圧力を測定。
結果 各ルーメンの破裂圧力は、仕様値:最小値 100psi に対してすべてのサンプルが
合格した。

120
試験項目 曲げ耐性試験
試験方法 チップから**mm の位置を**回繰り返し**°屈曲させ、観察。
結果 破損はなく、また、カテーテル先端部とシャフト材料との間で、剥がれも生じ
なかった。
試験項目 破断強度試験
試験方法 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.7.1)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.5,Annex B )に従い評価。
結果 延長チューブ接合部のルアーコネクター、延長チューブとマニフォールド接合
部、及び複数ルーメンシャフトとマニフォールド接合部、そして、カテーテル
先端部と複数ルーメン接合部間の引張り強度は仕様に合格した。
【結 論】
旧モデルのカテーテル(モデル IC-3585)は、全てのプロトコルの要求事項に適合し、安定性耐久
性に問題はない。
旧モデル(IC-3585)から形状を変更(ルーメン数を 1 個から 3 個に、シャフト径を 8.5Fr から 9.3Fr
に変更)した現モデル(IC-3893)は、変更した以外の形状は同等であり、原材料の変更がない。
旧モデルを使用した安定性試験は本品にも外挿できるとして、510(k)(K052443)を取得している
ことも含め、本試験を本申請品の安定性及び耐久性試験に外挿できると判断した。
(2)カテーテルの追加検証;加速時間後の安定性 添付資料:ハ-1-2
唯一コーティング剤の原材料が変更になったため、コーティング剤が影響を与えると考えられる試
験について、安定性及び耐久性について追加検証を行った。本試験は ICY カテーテルを代表製品と
し、カテーテル全体を検体としている。
サンプルは、EtO 滅菌サイクル 2 回暴露の後、1 年、2 年の加速エージングを行い、実時間にて試験
された。EtO 滅菌 2 回暴露及び実時間 7 日間の寿命試験(本品の使用期間は 4 日間)は、過酷条件と
なるため、本品に外挿できると考えた。
本試験においては、ヘパリンコーティングの安定性についても確認しているが、当該安定性試験は既
承認品目(販売名「サーモガードシステム」、承認番号 22400BZI00010000、平成 24 年 6 月 25 日付外
国製造医療機器製造販売承認、平成 25 年 9 月 6 日付一部変更承認)にて確認の通りであるため、記
載を省略する。
【目 的】SurModics 社ヘパリンコーティング(製品名「Applause」)を使用するカテーテルの重要
な安全及び性能特性について、24 ヶ月の加速劣化寿命を検証する。
【実施施設】ZOLL Circulation
【サンプル】バルーン付中心静脈カテーテル(ICY 3 カテーテル/IC-3893)
ICY カテーテルモデル#IC-3893 のサンプルは、Alsius ストレートバルーンカテーテルの
すべての材料とプロセスの分散を代表し、SurModics 親水性/ヘパリンコーティング実施
の検証と立証に使用される。
【加速条件】アレニウスの式(ファント ホッフの公式)を使用し、このプロトコルでは標準室温 22°C、
および、通常の加速係数 2.0 を使用して、以下の条件とした。
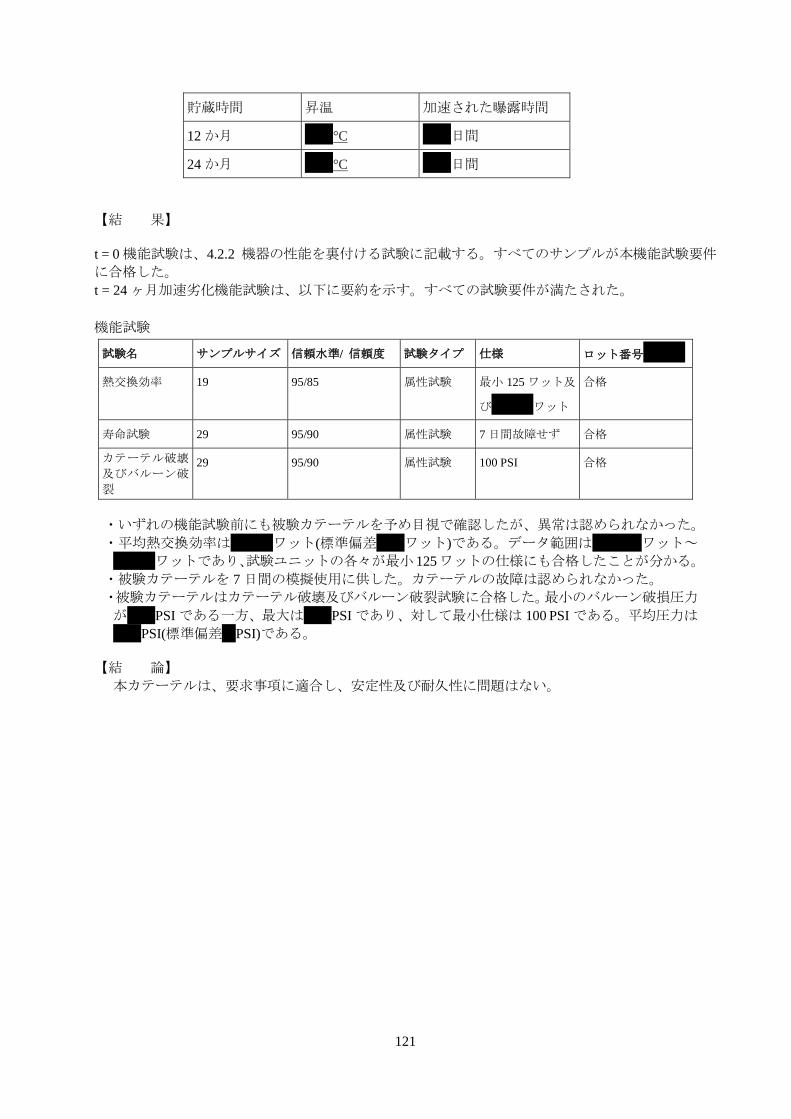
121
貯蔵時間 昇温 加速された曝露時間
12 か月 **°C **日間
24 か月 **°C **日間
【結 果】
t = 0 機能試験は、4.2.2 機器の性能を裏付ける試験に記載する。すべてのサンプルが本機能試験要件
に合格した。
t = 24 ヶ月加速劣化機能試験は、以下に要約を示す。すべての試験要件が満たされた。
機能試験
試験名 サンプルサイズ 信頼水準/ 信頼度 試験タイプ 仕様 ロット番号***
熱交換効率 19 95/85 属性試験 最小 125 ワット及
び***ワット
合格
寿命試験 29 95/90 属性試験 7 日間故障せず 合格
カテーテル破壊
及びバルーン破
裂
29 95/90 属性試験 100 PSI 合格
・いずれの機能試験前にも被験カテーテルを予め目視で確認したが、異常は認められなかった。
・平均熱交換効率は***ワット(標準偏差**ワット)である。データ範囲は ***ワット~
***ワットであり、試験ユニットの各々が最小 125ワットの仕様にも合格したことが分かる。
・被験カテーテルを 7 日間の模擬使用に供した。カテーテルの故障は認められなかった。
・被験カテーテルはカテーテル破壊及びバルーン破裂試験に合格した。最小のバルーン破損圧力
が**PSI である一方、最大は**PSI であり、対して最小仕様は 100 PSI である。平均圧力は
**PSI(標準偏差*PSI)である。
【結 論】
本カテーテルは、要求事項に適合し、安定性及び耐久性に問題はない。

122
4.2.2 機器の性能を裏付ける試験
添付資料:ホ-1-3~ホ-1-5
総 括
本品の構成は、バルーン付中心静脈カテーテル、スタートアップキット、及びカテーテルイントロ
デューサキットより成る。
バルーン付中心静脈カテーテルにおいては、本品は 2 つの異なるタイプを有する。それぞれの性
能を裏付ける試験を実施し適合を確認した。また、カテーテルイントロデューサキット構成品の
内、ダイレータついて性能を裏付ける試験にて適合を確認した。
スタートアップキット及びその他の構成品については、既承認品目(販売名「サーモガードシステ
ム」、承認番号 22400BZI00010000、平成 24 年 6 月 25 日付外国製造医療機器製造販売承認)にて確
認の通りである。
(1)バルーン付中心静脈カテーテルに関する試験1
(添付資料ホ-1-3(1)、添付資料ホ-1-3(2))
本品カテーテルの性能を評価する為に検証試験を行った。ICY カテーテル(モデル:IC-3893)及
び Quattro カテーテル(モデル:IC-4593)の検証試験の概略を表 4.2.2-1 及び表 4.2.2-2 に示す。
本試験に使用されたカテーテルのコーティング剤は製品名「Duraflo」であり、コーティング剤に
係る以外の試験は評価できると判断した。また、本申請品目のコーティング剤は製品名
「Applause」であるが、当該コーティング剤はどちらも既承認品目である販売名「サーモガード
システム」(承認番号 22400BZI00010000、平成 24 年 6 月 25 日付承認)において代替品として承
認を受けているものであり(平成 25 年 9 月 6 日付一部変更承認)、(2)及び(3)にて別途評
価を行っているため、問題はないと判断した。
(2)バルーン付中心静脈カテーテルに関する試験2 (添付資料ホ-1-3(3))
本品カテーテルのバルーンのコーティング剤の変更に伴い、コーティング剤が影響を与える試験
に係る追加検証で評価できると判断し、バルーン部(コーティング剤「Applause」含む)の性能
を評価する為に、ICY カテーテル(モデル:IC-3893)を代表製品として追加検証を行った。ICY
及び Quattro カテーテルにおいて原材料は同一であり、上記性能試験(1)から確認が必要とな
る差分も同一である。よって ICY カテーテルを代表製品として評価した。概略を表 4.2.2-3 に示
す。
なお本試験は 4.2.1.6 安定性及び耐久性 (1)カテーテルの追加検証;加速の安定性における、エ
ージング前に相当する試験である。

123
表 4.2.2-1:ICY カテーテルにおける試験の概略
試 験 項 目 試 験 方 法 又 は 試 験 規 格 結 果 実施施設 資 料
目視検査 EN ISO 10555-1 Section 4.3.に従い評価。 適合
社内試験
ZOLL
Circulation
ホ-1-3
(1)
寸法検査 各部寸法を測定し、規格範囲内であるとことを
確認。
適合
ガイドワイヤ
試験
直径 0.81mm のガイドワイヤをディスタルルー
メンに通し、ルーメン内を通ることを確認。
適合
パージング/プ
ライミング試
験
全てのルーメンについて、プライミング時のエ
ア抜きが出来ることを確認し、プライミング量
を測定。
適合
流量測定 EN ISO 10555-3(Section 4.6. and Annex A)に従い、
プロキシマルルーメン、メディアルルーメン、
ディスタルルーメンの流量を測定。
適合
熱交換能試験 模擬大静脈を用いた実験形を用い、ポンプ流量
200mL/min で、**************
******以下の項目を測定、算出。
適合
カテーテル破
壊試験(陰圧)
37℃の恒温槽に保持し、各ルーメン内をシリン
ジで空気を完全に除き(陰圧にし)、ルーメンを
観察。
適合
カテーテル寿
命試験
インフロールーメン、アウトフロールーメンに
チューブセットを接続し、36.5℃の恒温槽に保
持し、200~240mL/min の流量で、水を 7 日間、
循環させ、バルーンからの漏れを観察。
適合
カテーテル破
壊及びバルー
ン破裂試験(陽
圧)
37℃の恒温槽に保持し、加圧デバイスで、各ル
ーメンを*psi ずつ加圧し、破裂時の圧力を測定。
適合
曲げ耐性試験 カテーテル先端部を**回繰り返し*°屈曲さ
せ、観察。
適合
破断強度試験 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.7.1)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.5,Annex B )に従い評
価。
適合
注)ALSIUS Corporation は、現 ZOLLCirculation

124
表 4.2.2-2:Quattro カテーテルにおける試験の概略
試 験 項 目 試 験 方 法 又 は 試 験 規 格 結 果 実施施設 資 料
目視検査 EN ISO 10555-1 Section 4.3.に従い評価。 適合
社内試験
ZOLL
Circulation
ホ-1-3
(2)
寸法検査 各部寸法を測定し、規格範囲内であるとことを
確認。
適合
ガイドワイヤ
試験
直径 0.81mm のガイドワイヤをディスタルルー
メンに通し、ルーメン内を通ることを確認。
適合
パージング/プ
ライミング試
験
全てのルーメンについて、プライミング時のエ
ア抜きが出来ることを確認し、プライミング量
を測定。
適合
流量測定 EN ISO 10555-3(Section 4.6. and Annex A)に従い、
プロキシマルルーメン、メディアルルーメン、
ディスタルルーメンの流量を測定。
適合
熱交換能試験 模擬大静脈を用いた実験形を用い、ポンプ流量
200mL/min で、**************
******以下の項目を測定、算出。
適合
カテーテル破
壊試験(陰圧)
37℃の恒温槽に保持し、各ルーメン内をシリン
ジで空気を完全に除き(陰圧にし)、ルーメンを
観察。
適合
カテーテル寿
命試験
インフロールーメン、アウトフロールーメンに
チューブセットを接続し、36.5℃の恒温槽に保
持し、200mL/min の流量で、水を 7 日間、循環
させ、バルーンからの漏れを観察。
適合
カテーテル破
壊及びバルー
ン破裂試験(陽
圧)
37℃の恒温槽に保持し、加圧デバイスで、各ル
ーメンを*psi ずつ加圧し、破裂時の圧力を測定。
適合
曲げ耐性試験 カテーテル先端部を**回繰り返し*°屈曲さ
せ、観察。
適合
破断強度試験 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.7.1)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.5,Annex B)に従い評
価。
適合
注)ALSIUS Corporation は、現 ZOLLCirculation

125
表 4.2.2-3:カテーテルの追加検証(コーティング剤「Applause」含む)の概略
添付資料:ホ-1-3 (3)
試 験 目 的 「Applause」ヘパリンコーティングを有するバルーン付中心静脈カテー
テルの設計検証。
実 施 施 設 ZOLL Circulation
対 象 バルーン付中心静脈カテーテル(ICY 3 カテーテル/IC-3893)
目視検査 異常なきこと。 適合
熱交換能試験 シミュレートされた模擬患者モデルを用い、ポンプ流量
200mL/min で、********************
以下の項目を測定、算出。
適合
カテーテル寿命試
験
インフロールーメン、アウトフロールーメンにチューブセッ
トを接続し、36.5℃の恒温槽に保持し、200mL/min の流量で、
水を 7 日間、循環させ、バルーンからの漏れを観察。
適合
カテーテル破壊及
びバルーン破裂試
験(陽圧)
37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi
ずつ加圧し、破裂時の圧力を測定。
適合
(3)カテーテルコーティング剤(修飾ヘパリンナトリウム)に関する試験
(添付資料ホ-1-4)
本申請品目のカテーテルコーティング剤(製品名「Applause」)の性能を評価する為に、カテーテ
ル血栓評価試験を行った。ICY 及び Quattro カテーテルにおいて原材料は同一でありコーティング
剤に必要とされる性能も同一である。よって ICY カテーテル(モデル:IC-3893)を代表製品とし
て評価した。概略を表 4.2.2-4 に示す。
表 4.2.2-4:カテーテル血栓評価試験概略 (添付資料:ホ-1-4)
目 的 SurModics 社のヘパリンコーティング(製品名:Applause)の血栓形成性評
価
実 施 施 設 ******************
被 験 機 器 被験カテーテル:コーティングを施した ICY カテーテル
対照カテーテル:コーティング施していない ICY カテーテル
試 験 概 略 ウシ新鮮血を用いた in-vitro フローモデルによる被験カテーテル(コーティ
ングカテーテル)と対照カテーテル(非コーティングカテーテル)におけ
る 7 日間の持続試験後の血栓形成性の比較。
評 価 方 法 放射性標識血小板を用いて、試験終了時の血栓を定量する。
判 定 基 準 被験カテーテルは、対照カテーテルと比較し、顕著には血栓を生じてはな
らない。
結 果 被験カテーテルは、対照カテーテルと比較して血栓の付着に統計的に有意
な減少が認められた。

126
(4)流速シミュレーション試験 (添付資料ホ-1-5)
Computational Fluid Dynamics (CFD)を用いて、中心静脈内に留置した際の本品カテーテル及び血液
の挙動について、シミュレーションを実施した。
形状構造において、ICY 及び Quattro カテーテルはバルーン数が異なるが、バルーンを直列に配し
ておりバルーンの基本デザインは同一であり、バルーン周囲における血流も同じ挙動を示すと考
えられるため、ICY カテーテル(モデル:IC-3893)を代表製品として評価した。
本シミュレーションは、血管内にカテーテルを挿入した際の血流速度と血液温度を示したもので
ある。バルーン部とバルーン部の間の血液の流れのシミュレーションにおいて、バルーン部とコ
ンタクトのない外側の血流に比べ、血流の速度は遅いが、血液が流れてくる側のバルーン部に渦
が発生し、血液を留めることなく血流を作りだしている様子を確認した。
【考 察】
(1)バルーン付中心静脈カテーテルに関する試験1
(添付資料ホ-1-3(1)、添付資料ホ-1-3(2))
本品のカテーテルは、中心静脈カテーテルとして薬剤を注入することが可能なカテーテルであ
る。中心静脈カテーテルの機能を兼ね備えていることから、本邦の中心静脈カテーテル承認基
準(薬食発第 0107 第2号(平成 25 年1月7日))を担保する必要がある。本試験においては、
表面、流量、破断強度、気密性を、中心静脈基準 ISO 10555-1:Sterile, single-use intravascular catheters
-- Part 1: General requirements 及び、ISO 10555-3:Sterile, single-use intravascular catheters -- Part 3:
General requirements に従い確認した。よって、本邦の中心静脈カテーテル承認基準を満たすもの
であるといえる。
(2)バルーン付中心静脈カテーテルに関する試験2 (添付資料ホ-1-3(3))
本品カテーテルのコーティング剤の変更に伴い、バルーン(コーティング剤「Applause」含む)
について、上記(1)カテーテル性能試験と同様の試験をおこなった。バルーンに関する品目仕
様への適合を確認し、本品で使用される原材料にて問題がないことが確認できた。
(3)カテーテルコーティング剤(修飾ヘパリンナトリウム)に関する試験
(添付資料ホ-1-4)
本試験モデル(ウシ新鮮血を用いた in-vitro フローモデル)は、医療機器の血栓形成性を評価す
るために以前に発表され既にバリデートされた方法を用いたものである。
本試験の各段階において本品カテーテルのバルーンは「拡張させた状態」で試験を実施した。
放射性標識血小板を用いて定量的に比較を行った結果、被験カテーテルは、対照カテーテルと比
較して血栓の付着に統計的に有意な減少が認められたことから、SurModics 社のヘパリンコーテ
ィング剤(製品名:Applause)を使用した場合においても血栓形成を抑制することが確認でき、
本申請品目のカテーテルのコーティング剤としての抗血栓性能を確保していると判断した。
(4)流速シミュレーション試験 (添付資料ホ-1-5)
Computational Fluid Dynamics (CFD)を用いて、中心静脈内に留置した際の血液の挙動シミュレー
ションを実施したところ、バルーン部とバルーン部の間の血液は、血流を留めることなく渦を発
生させていることが確認できた。血液の滞留は血栓のリスクを増大すると考えられるが、シミュ
レーション結果においては、少なくとも、滞留箇所はないと判断した。
ダイレータ性能については、ダイレータ供給業者が行った設計バリデーション試験を行った(販
売名「サーモガードシステム」(承認番号 22400BZI00010000)平成 25 年 3 月 29 日付外国製造医
療機器製造販売承認事項一部変更承認書に添付)。
本品のダイレータは径が 10.5F(厚み 1.28mm)のダイレータであり、径が大きいほど物理的強度
は強くなること、及びダイレータの厚みがあるほど物理的強度は強くことから、径が 10F(厚み
1.2mm)の製品を代表品として評価を行い、カテーテル拡張器の国際規格である ISO 11070:

127
1998_Sterile, single-use intravascular catheter Introducers に準拠し実施した。なお、ISO 11070:1998
はカテーテル拡張器規格(JIS T3260:2007)の基規格である。ISO 規格より厳しい試験条件に従
って実施しており(ISO 11070 の引張りレート:20mm/min/mm、実施した試験の引張りレート:
** mm/min/mm 又は**mm/min/mm)、全ての試験に合格していることから、本邦における規
格に準拠したものであるといえる。
その他のカテーテルイントロデューサキット構成品は、承認又は認証を取得しており性能は担
保できている。
以上、本申請品目の品目仕様に掲げる特性及び性能を担保できており、品目仕様の設定に問題は
ないといえる。

128
(1)バルーン付中心静脈カテーテルに関する試験1
添付資料:ホ-1-3 (1)
<ICY カテーテルにおける試験>
試 験 項 目 目視検査
試 験 方 法 EN ISO 10555-1 Section4.3.に従い評価。
結 果 目視検査と触知検査の結果、要求条件に合格
試 験 項 目 寸法検査
試 験 方 法 各部寸法を測定し、規格範囲内であるとことを確認。
結 果 すべての寸法仕様に合格した。
試 験 項 目 ガイドワイヤ試験
試 験 方 法 直径 0.81mm のガイドワイヤをディスタルルーメンに通し、ルーメン内を通る
ことを確認。
結 果 直径 0.81mm のガイドワイヤが通過し、合格。
試 験 項 目 パージング/プライミング試験
試 験 方 法 全てのルーメンについて、プライミング時のエア抜きが出来ることを確認し、
プライミング量を測定。
結 果 冷却膜内に残留したエア容量は極少量で、臨床的に問題なく製品の安全性や性
能に影響を与えない。
試験項目 流量測定
試験方法 EN ISO 10555-3(Section4.6. andAnnex A)に従い、プロキシマルルーメン、ディス
タルルーメンの流量を測定。
結果 近位注入用ルーメンでの仕様値:最小平均流量 1300mL/時、中位注入用ルーメ
ンでの仕様値:最小平均流量 1000mL/時、ガイドワイヤルーメンでの仕様値:
最小平均流量 1900mL/時であり、各ルーメンとも 95%信頼水準で合格であっ
た。
試験項目 熱交換能試験
試験方法 模擬大静脈を用いた実験形を用い、ポンプ流量 200mL/min で、*******
*************以下の項目を測定、算出。
結果 仕様値は、カテーテル流量 200mL/分において、最小値 125W であるが、95%信
頼水準で仕様値に合格しており、平均値は**Wであった。
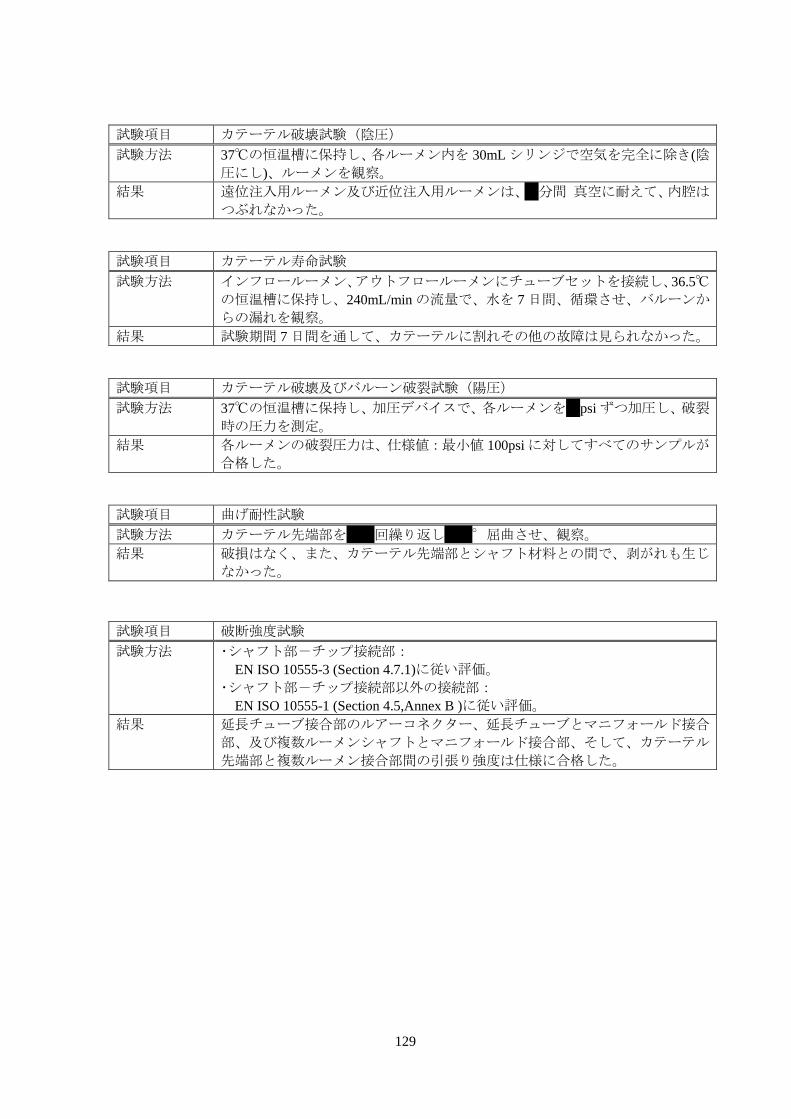
129
試験項目 カテーテル破壊試験(陰圧)
試験方法 37℃の恒温槽に保持し、各ルーメン内を 30mL シリンジで空気を完全に除き(陰
圧にし)、ルーメンを観察。
結果 遠位注入用ルーメン及び近位注入用ルーメンは、*分間 真空に耐えて、内腔は
つぶれなかった。
試験項目 カテーテル寿命試験
試験方法 インフロールーメン、アウトフロールーメンにチューブセットを接続し、36.5℃
の恒温槽に保持し、240mL/min の流量で、水を 7 日間、循環させ、バルーンか
らの漏れを観察。
結果 試験期間 7 日間を通して、カテーテルに割れその他の故障は見られなかった。
試験項目 カテーテル破壊及びバルーン破裂試験(陽圧)
試験方法 37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi ずつ加圧し、破裂
時の圧力を測定。
結果 各ルーメンの破裂圧力は、仕様値:最小値 100psi に対してすべてのサンプルが
合格した。
試験項目 曲げ耐性試験
試験方法 カテーテル先端部を**回繰り返し**°屈曲させ、観察。
結果 破損はなく、また、カテーテル先端部とシャフト材料との間で、剥がれも生じ
なかった。
試験項目 破断強度試験
試験方法 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.7.1)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.5,Annex B )に従い評価。
結果 延長チューブ接合部のルアーコネクター、延長チューブとマニフォールド接合
部、及び複数ルーメンシャフトとマニフォールド接合部、そして、カテーテル
先端部と複数ルーメン接合部間の引張り強度は仕様に合格した。

130
<Quattro カテーテルにおける試験> 添付資料:ホ-1-3 (2)
試験項目 目視検査
試験方法 EN ISO 10555-1 Section4.3.に従い評価。
結果 目視検査と触知検査の結果、要求条件に合格
試験項目 寸法検査
試験方法 各部寸法を測定し、規格範囲内であるとことを確認。
結果 すべての寸法仕様に合格した。
試験項目 ガイドワイヤ試験
試験方法 直径 0.81mm のガイドワイヤをディスタルルーメンに通し、ルーメン内を通る
ことを確認。
結果 直径 0.81mm のガイドワイヤが通過し、合格。
試験項目 パージング/プライミング試験
試験方法 全てのルーメンについて、プライミング時のエア抜きが出来ることを確認し、
プライミング量を測定。
結果 冷却膜内に残留したエア容量は極少量で、臨床的に問題なく製品の安全性や性
能に影響を与えない。
試験項目 流量測定
試験方法 EN ISO 10555-3(Section4.6. andAnnex A)に従い、プロキシマルルーメン、ディス
タルルーメンの流量を測定。
結果 近位注入用ルーメンでの仕様値:最小平均流量 1300mL/時、中位注入用ルーメ
ンでの仕様値:最小平均流量 1000mL/時、ガイドワイヤルーメンでの仕様値:
最小平均流量 1900mL/時であり、各ルーメンとも 95%信頼水準で合格であっ
た。
試験項目 熱交換能試験
試験方法 模擬大静脈を用いた実験形を用い、ポンプ流量 200mL/min で、*******
*************以下の項目を測定、算出。
結果 仕様値は、カテーテル流量 200mL/分において、最小値 170W であるが、95%信
頼水準で仕様値に合格しており、平均値は**Wであった。

131
試験項目 カテーテル破壊試験(陰圧)
試験方法 37℃の恒温槽に保持し、各ルーメン内を 30mL シリンジで空気を完全に除き(陰
圧にし)、ルーメンを観察。
結果 遠位注入用ルーメン、中位注入用ルーメン及び近位注入用ルーメンは、*分間
真空に耐えて、内腔はつぶれなかった。
試験項目 カテーテル寿命試験
試験方法 インフロールーメン、アウトフロールーメンにチューブセットを接続し、36.5℃
の恒温槽に保持し、200mL/min の流量で、水を 7 日間、循環させ、バルーンか
らの漏れを観察。
結果 試験期間 7 日間を通して、カテーテルに割れその他の故障は見られなかった。
試験項目 カテーテル破壊及びバルーン破裂試験(陽圧)
試験方法 37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi ずつ加圧し、破裂
時の圧力を測定。
結果 各ルーメンの破裂圧力は、仕様値:最小値 100psi に対してすべてのサンプルが
合格した。
試験項目 曲げ耐性試験
試験方法 カテーテル先端部を**回繰り返し**°屈曲させ、観察。
結果 破損はなく、また、カテーテル先端部とシャフト材料との間で、分離領域は認
められなかった。
試験項目 破断強度試験
試験方法 ・シャフト部-チップ接続部:
EN ISO 10555-3 (Section 4.7.1)に従い評価。
・シャフト部-チップ接続部以外の接続部:
EN ISO 10555-1 (Section 4.5,Annex B)に従い評価。
結果 ルアー - 延長チューブ接合部、延長チューブ - マニフォールド接続部、及び
複数ルーメンシャフト -マニフォールド接続部は、すべて仕様に合格。また、
カテーテル先端部 – 複数ルーメンシャフト接続部も仕様に合格した。
【考 察】
各カテーテルモデルにおいて、設計仕様における重要な安全性特性と性能特性の要求仕様或いは合
格基準に適合するといえる。

132
(2)バルーン付中心静脈カテーテルに関する試験2
添付資料:ホ-1-3 (3)
本試験は、ICY カテーテルを代表製品としてコーティング剤の変更に際してバルーン部の設計検証
のため行った追加検証である。試験の概略は以下のとおりである。
なお本試験は 4.2.1.6 安定性及び耐久性 (1)カテーテルの追加検証;加速および実時間の安定性に
おける、エージング前に相当する試験である。
試験項目 目視検査
試験方法 異常なきこと。
結果 すべての試験サンプルで異常は認められなかった。
試験項目 熱交換能試験
試験方法 模擬大静脈を用いた実験形を用い、ポンプ流量 200mL/min で、*******
*************以下の項目を測定、算出。
結果 仕様値は、カテーテル流量 200mL/分において、最小値 125W であるが、すべて
のユニットが仕様値に合格しており、平均値は***Wであった。
試験項目 カテーテル寿命試験
試験方法 インフロールーメン、アウトフロールーメンにチューブセットを接続し、36.5℃
の恒温槽に保持し、200mL/min の流量で、水を 7 日間、循環させ、バルーンか
らの漏れを観察。
結果 試験期間 7 日間を通して、カテーテルに割れその他の故障は見られなかった。
試験項目 カテーテル破壊及びバルーン破裂試験(陽圧)
試験方法 37℃の恒温槽に保持し、加圧デバイスで、各ルーメンを*psi ずつ加圧し、破裂
時の圧力を測定。
結果 各ルーメンの破裂圧力は、仕様値:最小値 100psi に対してすべてのサンプルが
合格した。

133
繰り返し実施しなかった試験項目に対して妥当性の説明を以下の表に示す。
試験項目 妥当性説明
ガイドワイヤ試験 カテーテルのルーメンはコーティングで覆われないため、ヘパリン
コーティングの変更はガイドワイヤの通過に影響しない。
パージング試験 インフロールーメン・アウトフロールーメン又は流路の内側はコー
ティングで覆われないため、ヘパリンコーティングの変更はエア抜
きに影響しない。
流量測定 測定されるルーメンはコーティングで覆われないため、ヘパリンコ
ーティングの変更は流量の測定に影響しない。
曲げ耐性試験 ヘパリンコーティングの種類又は有無はカテーテル曲げ耐性に影響
しないため、ヘパリンコーティングの変更はカテーテル曲げ耐性に
影響しない。
破断強度試験 ヘパリンコーティングの種類又は有無はカテーテル破断に影響しな
いため、ヘパリンコーティングはカテーテル破断に影響しない。
カテーテル破壊試験(陰
圧)
ヘパリンコーティングは外面に薄い層として塗布されているので、
シャフト、バルーン及びアッセンブリの構造に影響しない。
無菌バリア性試験 ヘパリンコーティングの変更は無菌バリア自体に影響しない。
【考 察】
すべての被験カテーテルが、すべての試験に関する判定基準に合格した。SurModics親水性/ヘパリンを施した
現行の ICY系統、CL系統及びQuattroカテーテル設計の機能的性能が実証されたといえる。

134
(3)カテーテルコーティング剤(修飾ヘパリンナトリウム)に関する試験
添付資料:ホ-1-4
ウシ新鮮血を用いた in-vitro フローモデルによる被験カテーテル(コーティングカテーテル)と対照
カテーテル(非コーティングカテーテル)における 7 日間の持続試験後の血栓形成性の比較。
【目的】SurModics 社のヘパリンコーティング剤(製品名:Applause)の血栓形成性評価
【実施施設】******************
【被験機器】
製品番号 説明 個数
601028-001
ロット番号***
コーティングを施した ICY カテーテル
(被験カテーテル) 12 本
500647-001 Rev C
ロット番号***
コーティングしていない ICY カテーテル
(対照カテーテル) 12 本
【準 備】
①被験カテーテル 6 本と対照カテーテル 6 本を、7 日間、リン酸緩衝化生理食塩水(PBS)にて洗
浄処理する。流速は、典型的な大腿静脈にみられる流れ(方向および流速)に類似させる(500mL
/分)。各カテーテルをポリ塩化ビニール製(PVC)導管内に留置し、順番につなげる。蠕動ポ
ンプにより、所定の速度で PBS を(37℃に維持して)流動させる。PBS の容器は、新鮮な PBS
に毎日交換する。
②24 本のカテーテルすべてを、流動ループ内で活性化血小板を含むウシの血液に曝露させ、以下
の 4 群に分ける。
被験カテーテルで、7 日間 PBS 洗浄したもの:6 本
被験テーテルで、そのままのもの(洗浄しなかったもの):6 本
対照カテーテルで、7 日間 PBS 洗浄したもの:6 本
対照カテーテルで、そのままのもの(洗浄しなかったもの):6 本
【試験モデル】
【試験手順】

135
【評価方法】
放射性標識血小板を用いて、実験終了時の血栓を定量する。
① 実験開始前にウシの自家血液からの分注液を酸性クエン酸ブドウ糖液(ACD)中に採取し、350g
で 12 分間遠沈し、赤血球を取り除く。得られた血小板を多く含む血漿を 1000g で 15 分間遠沈
し、血小板を沈降させる。
② 上澄みの液を静かに移すことにより当該血漿を除去後、沈降させた血小板を 5 mL の ACD 生
理食塩水中に再懸濁させ、50~100μCi のインジウム 111 オキシン*************
**********を添加し、37℃で 30 分間保温する。
③ 実験開始前に、インジウム 111 標識血小板を当該血液に加える。実験終了時に、管の箇所をカ
テーテルから取り外し、ガンマ線測定装置により、当該カテーテル上の血栓に関連する放射活
性を定量する。
④ 当該カテーテル上の血栓の撮影も行い、選択した標本を SEM 下での検査用に Karnovsky 固定
液中に保存する。
【分析方法】
カテーテル上の血栓の有無を視覚的に評価し、当該カテーテル除去後のカテーテル表面の血栓を
すべて撮影する。代表的標本を SEM 下での検査用に Karnovsky 固定液中に保存する。上記のよう
に、血液標本を採取し、全血球算定(CBC)、血小板数および血漿遊離ヘモグロビンの測定用に提
出する。コーティングを施したカテーテル群とコーティングしていないカテーテル群間で各血球
数の値を分散分析(ANOVA)により比較。
【判定基準】被験カテーテルは、対照カテーテルと比較し、顕著には血栓を生じてはならない。
【結果】
① 放射活性の定量による血栓付着評価
対照カテーテルと比較して被験カテーテルは、PBS により処置したものと未処置のも双方につい
て、血栓の付着に統計的に有意な減少が認められた(p<0.05, ボンフェローニ補正なしの片側検定
による対応のある t 検定)。結果を図 4.2.2-5 及び図 4.2.2-6 に示す。

136
図 4.2.2-5:放射活性の定量による血栓付着評価(PBS 未処理群)
図 4.2.2-6:放射活性の定量による血栓付着評価(PBS 処理群)
放射活性の定量による血栓付着評価は、対照カテーテルに対する割合で表した(平均値±標準誤差)。
被験カテーテルにおいて PBS 処置なし(図 4.2.2-5)と PBS 処置あり(図 4.2.2-6)の両方に、各々の
対照と比較して血栓付着の有意な減少が認められた。
注:被験カテーテルのそれぞれで、四分位範囲法に基づき、外れ値として 1 つのデータポイントを本
解析から除外。
② 外観による血栓付着評価
当該カテーテルの外観は、放射活性データを反映していた。
【結 論】

137
ウシ新鮮血を用いた in-vitro フローモデルにおいて、被験カテーテルは、PBS 処置したものと未処置
の両方で、各々の対照カテーテルと比較して、血栓の付着で統計的に有意な減少が認められた。
【考 察】
本試験は、in-vitro で行った試験である。in-vitro モデルは、血流、抗凝固などの条件を in-vivo モデル
の場合よりも比較的管理しやすく、in-vivo での評価を混乱させる可能性のある血管サイズ、動物の
活動性、可変性の止血およびホメオスタシス、感染などの副次的パラメータは、in-vitro モデルでは
除外できる。このことにより、他のパラメータを比較的一定に保ち、1 つの特定の変数(たとえば、
界面化学に関するもの)に集中して評価することが可能である。さらに、当該機器を同じ条件下に設
定してあるため、このような in-vitro モデルにより、当該機器の相対的な血栓形成性の直接比較が可
能となる。
本試験モデルは、医療機器の血栓形成性を評価するために以前に発表され既にバリデートされた方
法を用いたものである。一般的に、ウシ新鮮血の in-vitro フローモデルを用いたカテーテルへの血栓
付着に関する試験では、適切な信頼区間(95%)と検出力(80%超)を得るためには、6 回以上の反
復測定が必要であることが示されている。本試験では、6 回反復測定し、外れ値を判定する四分位
範囲の統計学的基準に基づき、コーティングを施した各カテーテルについて、1 つのデータポイン
トを外れ値として除外したが、違いについての信頼性は維持されたと言える。
添付資料ホ-3-1 において、旧コーティング剤である Duraflo でヘパリンコーティングされた本品カテ
ーテルをヒツジの中心静脈に長期留置した際の血栓付着を評価している。被験群として Duraflo コー
ティングが施された旧コーティング品を用い、対照群としてヘパリンコーティングが施されていな
い市販の中心静脈カテーテルを用いて、4 日間後の血栓付着を定性的に評価した。外観検査及び組織
病理学検査の結果に基づき、4 日間留置後に肺塞栓のエビデンスが認められなかったことより、旧ヘ
パリンコーティングのカテーテルの安全性は対照群に比べて非劣性であることが示された。
一方、本試験においては、現コーティング剤である Applause でヘパリンコーティングされた ZOLL
製カテーテルに無菌リン酸緩衝化生理食塩水(PBS)の 7 日間洗浄処理を行い、長期留置を模擬した
際の血栓付着を評価した。被験群として Applause でコーティングを施されたカテーテルを用い、対
照群としてコーティングを施さないカテーテルを用いて、7 日間洗浄処理後の血栓付着を定量的に評
価した。インジウムの放射性マーカー計数値に基づき、本品の Applause でコーティングを施された
カテーテルは対照群に比べて有意に血栓付着が低下することが示された。なお、本品カテーテルの使
用期間は 4 日間であり、7 日間の試験は過酷条件となる。
両被験群の Duraflo コーティング(変更前)及び Applause コーティング(変更後)を施されたカテー
テルは、ヘパリンコーティングを施されていない対照群に比べて、抗血栓性能が同等もしくは優れて
いることが確認された。両試験結果はどちらも本品カテーテル及び両コーティング剤の製造元から
独立している試験施設により実施され保証されている。これらの結果を踏まえると、Applause コーテ
ィングは変更前の Duraflo コーティングと比較して劣っておらず、同様に血栓形成を抑制することが
できると考えられる。
本試験の結果、被験カテーテルは、対照カテーテルと比較して血栓の付着に統計的に有意な減少が
認められた。このことから SurModics 社のヘパリンコーティング剤(製品名:Applause)を本品カ
テーテルのコーティング剤に使用した場合においても血栓形成を抑制することが確認できた。これ
によって、本コーティング剤を利用しても、性能を示すことは確保できると判断した。

138
(4)流速シミュレーション試験
添付資料:ホ-1-5
Computational Fluid Dynamics (CFD)を用いて、臨床使用を模擬したモデルとして ICY カテーテルを中
心静脈内に留置した際の本品及び血液の挙動について、初期の血液温度 37℃、バルーンを流れる生
理食塩水の初期温度 4℃の条件下における流速シミュレーションを実施した。本シミュレーションは、
血管内にカテーテルを挿入した際の血流速度と血液温度を示したものである。
図 4.2.4-7 は、カテーテル内を流れる生理食塩水の温度と周辺の血液温度を示したものである。
********************************************
********************************************
********************************************
********************************************
****************************
図 4.2.4-7:生理食塩水の温度と周辺の血液温度
図 4.2.4-8 は、バルーン部とバルーン部の間の血液の流れをシミュレーションしたものである。**
*********************************
********************************************
********************************************
********************************************
***************
図 4.2.4-8:バルーン間の血液の流れ
①
② ⑪
①
139
4.2.3 効能効果を裏付ける試験
添付資料ホ-2-1
総 括
ヒツジを用い、中心静脈内にカテーテルを留置して、本品の体温調節機器としての有効性を確認
し、またカテーテル留置による血行及び組織への安全性確認を行った。試験にはモデル ICY のカ
テーテル(米国モデル Icy-2、モデル:3585)を使用した。
本試験に使用されたカテーテルのコーティング剤は製品名「Duraflo」であり、本申請品目のコー
ティング剤は製品名「Applause」であるが、当該コーティング剤はどちらも既承認品目である販
売名「サーモガードシステム」(承認番号 22400BZI00010000、平成 24 年 6 月 25 日付承認)にお
いて代替品として承認を受けているものであり(平成 25 年 9 月 6 日付一部変更承認)、コーティ
ング剤は熱交換能に影響を与えないため、「Duraflo」で行った試験を本品に外挿できると判断し
た。
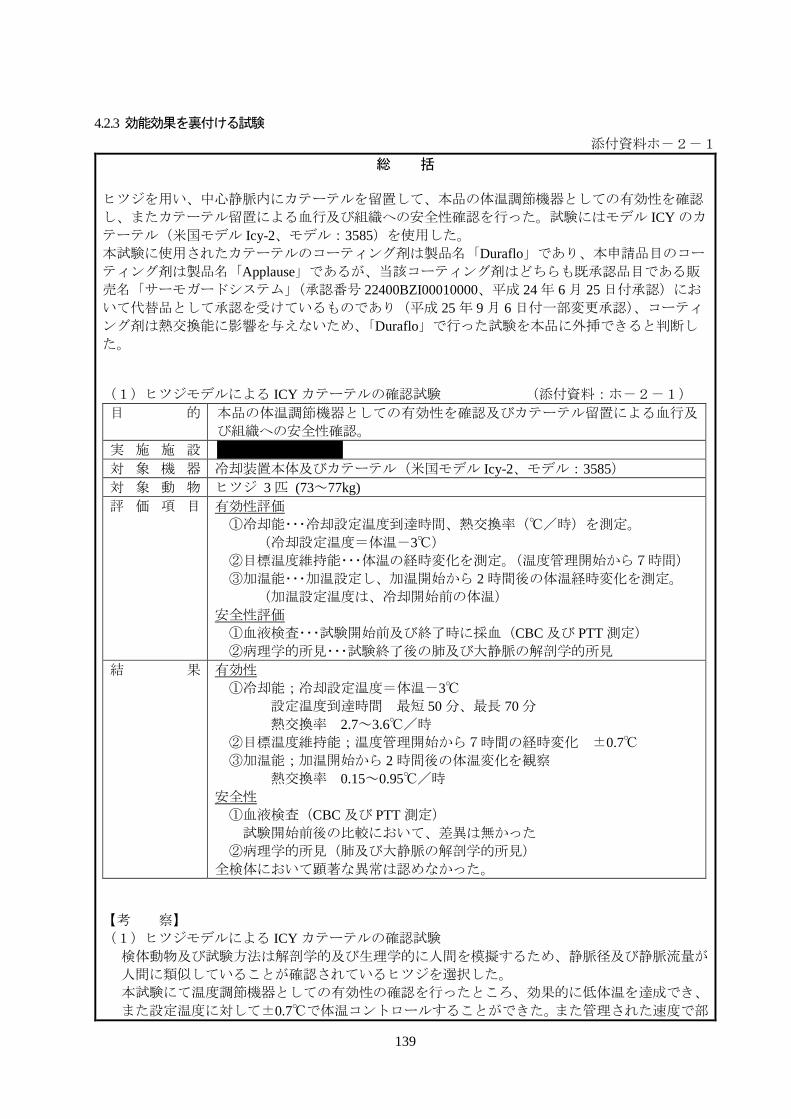
(1)ヒツジモデルによる ICY カテーテルの確認試験 (添付資料:ホ-2-1)
目 的 本品の体温調節機器としての有効性を確認及びカテーテル留置による血行及
び組織への安全性確認。
実 施 施 設 *********
対 象 機 器 冷却装置本体及びカテーテル(米国モデル Icy-2、モデル:3585)
対 象 動 物 ヒツジ 3 匹 (73~77kg)
評 価 項 目 有効性評価
①冷却能・・・冷却設定温度到達時間、熱交換率(℃/時)を測定。
(冷却設定温度=体温-3℃)
②目標温度維持能・・・体温の経時変化を測定。(温度管理開始から7時間)
③加温能・・・加温設定し、加温開始から 2 時間後の体温経時変化を測定。
(加温設定温度は、冷却開始前の体温)
安全性評価
①血液検査・・・試験開始前及び終了時に採血(CBC 及び PTT 測定)
②病理学的所見・・・試験終了後の肺及び大静脈の解剖学的所見
結 果 有効性
①冷却能;冷却設定温度=体温-3℃
設定温度到達時間 最短 50 分、最長 70 分
熱交換率 2.7~3.6℃/時
②目標温度維持能;温度管理開始から7時間の経時変化 ±0.7℃
③加温能;加温開始から 2 時間後の体温変化を観察
熱交換率 0.15~0.95℃/時
安全性
①血液検査(CBC 及び PTT 測定)
試験開始前後の比較において、差異は無かった
②病理学的所見(肺及び大静脈の解剖学的所見)
全検体において顕著な異常は認めなかった。
【考 察】
(1)ヒツジモデルによる ICY カテーテルの確認試験
検体動物及び試験方法は解剖学的及び生理学的に人間を模擬するため、静脈径及び静脈流量が
人間に類似していることが確認されているヒツジを選択した。
本試験にて温度調節機器としての有効性の確認を行ったところ、効果的に低体温を達成でき、
また設定温度に対して±0.7℃で体温コントロールすることができた。また管理された速度で部

140
分的再加温も可能であったことから、温度調節機器としての有効性が確認できた。また安全性
において、血液検査の結果から、手技前後の値において、データの傾向は認められず、また解
剖学的所見においても、全検体において顕著な異常は認めなかった。
以上より、本品の効能効果に必要とされる性能及び安全性は確保されているといえる。
(1)ヒツジモデルによる ICY カテーテルの確認試験 添付資料:ホ-2-1
【試験目的】 本品の体温調節機器としての有効性を確認及びカテーテル留置による血行及び組織
への安全性確認。
【実施施設】 *********
【対象機器】 冷却装置本体及びカテーテル(米国モデル Icy-2)
【対象動物】 ヒツジ 3 匹 (73~77kg)
【評価項目】
(1)有効性評価
①冷却能・・・冷却設定温度到達時間、熱交換率(℃/時)を測定。
(冷却設定温度=体温-3℃)
②設定温度維持能・・・体温の経時変化を測定。(温度管理開始から7時間)
③加温能・・・加温設定し、加温開始から 2 時間後の体温経時変化を測定。
(加温設定温度は、冷却開始前の体温)
(2)安全性評価
①血液検査・・・試験開始前及び終了時に採血(CBC 及び PTT 測定)
②病理学的所見・・・試験終了後の肺及び大静脈の解剖学的所見
【試験方法】
(1)処 置
・ヒツジは麻酔投与し、試験開始前、最低 60 分間以上、体温変動が 0.3℃以上にならない安定し
た体温状態を保った。(試験中、ブランケットで覆った。)
・カテーテルは、直径 0.81mm のガイドワイヤを介し、大腿静脈から挿入し、IVC に留置。
(2)温度コントロール
・本体のポンプ流量を 200±5mL/min とし、生理食塩水をカテーテル内に循環させた。
・目標温度を体温より 3℃低い温度と設定し、7時間温度コントロールを行った。
・7時間温度コントロールを行った後、新たな目標温度を設定(試験開始前の体温)し、2 時間
実施した。
・体温は、食道温を経時的に計測。

141
【結 果】
(1)有効性 ; 体温測定結果を下表に示す。
項 目 検体番号
21 23 29
冷却開始前体温(℃)
冷却設定温度(℃)
冷却設定温度到達時間(min)
熱交換率(℃/hr);冷却能
(冷却設定温度以下)最大値
体温維持 最小体温測定値
最大体温測定値
熱交換率;加温
能(℃/hr)
加温後 0~1 時間
加温後 1~2 時間
試験終了後体温(℃)
①冷却能;冷却設定温度=体温-3℃
設定温度到達時間 最短 50 分、最長 70 分
熱交換率 2.7~3.6℃/時
②目標温度維持能;温度管理開始から7時間の経時変化 ±0.7℃
③加温能;加温開始から 2 時間後の体温変化を観察
熱交換率 0.15~0.95℃/時
(2)安全性
①血液検査結果
手技前後の値において、データの傾向は認められなかった。血液検査結果を下表に示す。
検体番号 21 23 29
ベースライン 手技後 ベースライン 手技後 ベースライン 手技後
赤血球(×100 万/μL)
白血球(×1000/μL)
血小板数(×1000/μL)
PTT(秒)
pH
ヘマトクリット値(%)
血漿遊離ヘモグロビン(mg/d
L)
Na+(mEq/L)
K+(mEq/L)
Ca+(mEq/L)
②病理学的所見(肺及び大静脈の解剖学的所見)
解剖学的所見は下表のとおりであり、全検体において顕著な異常は認めなかった。
検体番号 肺の所見 大静脈の所見
21 顕著な異常なし 顕著な異常なし
23 顕著な異常なし 顕著な異常なし
29 顕著な異常なし 顕著な異常なし

142
【考 察】
ヒツジモデルにおいて、ICY カテーテル(米国モデル Icy-2 を使用)の温度調節機器の有効性の確
認を行ったところ、効果的に低体温を達成でき、また設定温度に対して±0.7℃で体温コントロー
ルすることができた。また管理された速度で部分的再加温も可能であったことから、温度調節補助
装置としての有効性が確認できた。また安全性において、血液検査の結果から、手技前後の値にお
いて、データの傾向は認められず、また解剖学的所見においても、全検体において顕著な異常は認
めなかった。またカテーテル抜去後、カテーテルに血栓は認められなかったため、血栓形成のリス
クは低いと考えられることから、安全性に問題はないと言える。

143
4.2.4 使用方法を裏付ける試験
添付資料:ホ-3-1
総 括
使用方法を裏付ける試験として、ヒツジモデルを用い、本品カテーテルを中心静脈に長期留置し
た際の安全性を評価した。試験にはモデル ICY のカテーテル(米国モデル Icy-2、モデル:IC-
3585)を使用し、市販されている中心静脈カテーテル(**************)を比較
対象として使用した。
本試験に使用されたカテーテルのコーティング剤は製品名「Duraflo」であり、本申請品目のコー
ティング剤は製品名「Appulause」であるが、当該コーティング剤はどちらも既承認品目である販
売名「サーモガードシステム」(承認番号 22400BZI00010000、平成 24 年 6 月 25 日付承認)にお
いて代替品として承認を受けているものであり(平成 25 年 9 月 6 日付一部変更承認)、「4.2.2 機
器の性能を裏付ける試験」に示したように別途評価試験を行っているため、本品に外挿すること
ができると判断した。
本申請における ICY と Quattro はシャフト長及びバルーン数の違いのみであり、当該カテーテル
は患者の体格の大きさに応じて使い分けるものであり(患者体形の目安は操作方法又は使用方法
欄に記載済み)、使用方法は ICY も Quattro も一般的な中心静脈カテーテルと同一であることか
ら、ICY を代表モデルとして評価することと判断した。
(1)ヒツジモデルによる ICY カテーテルの4日間挿入試験 (添付資料:ホ-3-1)
目 的 ICY カテーテル(Icy-2)を中心静脈に長期留置した際の血栓形成を評価する。
実 施 施 設 *********
対 象 機 器 被験群:冷却装置本体及びカテーテル(米国モデル Icy-2、モデル:3585)
コントロール:中心静脈カテーテル*******
対 象 動 物 被験群:ヒツジ 3 匹 (73~91kg)
コントロール:ヒツジ 3 匹 (86~102kg)
評 価 項 目 4 日間留置した後、カテーテルを抜去。抜去後の血栓の有無を確認。
①カテーテルの目視検査
②下大静脈及び肺の解剖学的所見
結 果 ①カテーテルの目視検査
抜去後のカテーテルに血栓を認めたが、コントロールとの差異は無かった。
②下大静脈及び肺の解剖学的所見
被験群、コントロール群共、肺切片に塞栓は認められなかった。
被験群、コントロール群共に静脈組織において、出血性壁在血栓を認めた。
結 論 ヒツジモデルにおいて、ICY カテーテル(Icy-2)の 4 日間留置の安全性が確認
された。
【考 察】
(1)ヒツジモデルによる ICY カテーテルの4日間挿入試験
被験群及びコントロール群のいずれも肺血栓は認められなかった。静脈組織においては、壁在性
の血栓が認められたが、コントロールにおいても同様であったことから、本品のカテーテルの安
全性は、中心静脈カテーテルの留置における安全性と同等であるといえる。
以上より、本品の使用方法において必要とされる安全性は確保されているといえる。

144
(1)ヒツジモデルによる ICY カテーテルの4日間挿入試験
添付資料:ホ-3-1
【試験目的】 ICY カテーテル(Icy-2)を中心静脈に長期留置した際の血栓形成を評価する。
【実施施設】 *********
【対象機器】 被験群:ICY カテーテル(米国モデル Icy-2)
コントロール群:中心静脈カテーテル*******
コントロールに使用した中心静脈カテーテルは、****************
*********************であり、国内においては、本中心静脈
カテーテルの抗菌コートされていないタイプが承認されている(販売名:*****
***********、承認番号:*********)。
【対象動物】 被験群:ヒツジ 3 匹 (73~91kg)
コントロール群:ヒツジ 3 匹 (86~102kg)
【評価項目】
4 日間カテーテルを留置した後、カテーテルを抜去。抜去後の血栓の有無を確認。
①カテーテルの外観検査
②下大静脈及び肺の解剖学的所見
【試験方法】
①ヒツジに麻酔剤を投与し、鎮静状態にした。
②カテーテルを直径 0.81mm のガイドワイヤを介して、大腿静脈から挿入し、下大静脈に留置。
③カテーテル留置期間は、水を自由に与えた。また、カテーテル挿入部位に感染の兆候が現れた場
合、軟膏を塗布した。
④テーテル挿入から 5 日後、抜去。抜去後のカテーテルは生理食塩水で軽く洗い流し、10%ホルマ
リンに浸漬。(カテーテルに血塊が付着している場合は、それを除去しない)
⑤20,000U のヘパリンを投与し、安楽死の後、解剖。
【結 果】
①カテーテルの外観検査
<被験群>
検体番号 抜去後のカテーテル外観
24 遠位バルーン及び中位バルーンの部位において血栓を認めた。
22 近位バルーンの遠位部において血栓を認めた。
27 顕著な血栓を認めなかった。
<コントロール群>
検体番号 抜去後のカテーテル外観
25 顕著な血栓を認めなかった。
26 挿入部位に繊維性血栓が認めらた。
28 顕著な血栓を認めなかった。
②下大静脈及び肺の解剖学的所見
肺の所見において、いずれも血栓は、認められなかった。
静脈組織においては、被験群及びコントロール群のすべてにおいて、出血性壁在血栓及び血栓
に伴う血管壁の変化が認められた。

145
【考 察】
被験群及びコントロール群のいずれも肺血栓は認められなかった。静脈組織においては、壁在性の
血栓が認められたが、コントロールにおいても同様であったことから、本品のカテーテルの安全性
は、中心静脈カテーテルの留置における安全性と同等であるといえる。
146
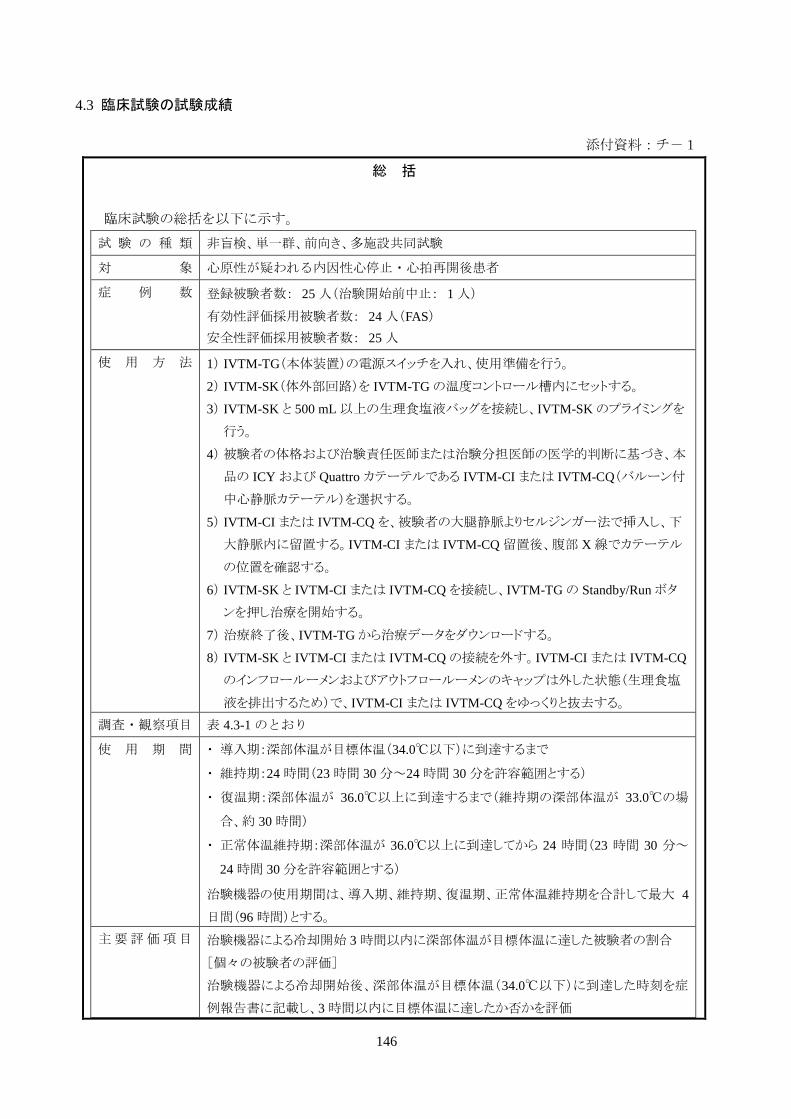
4.3 臨床試験の試験成績
添付資料:チ-1
総 括
臨床試験の総括を以下に示す。
試 験 の 種 類 非盲検、単一群、前向き、多施設共同試験
対 象 心原性が疑われる内因性心停止・心拍再開後患者
症 例 数 登録被験者数: 25 人(治験開始前中止: 1 人)
有効性評価採用被験者数: 24 人(FAS)
安全性評価採用被験者数: 25 人
使 用 方 法 1) IVTM-TG(本体装置)の電源スイッチを入れ、使用準備を行う。
2) IVTM-SK(体外部回路)を IVTM-TGの温度コントロール槽内にセットする。
3) IVTM-SK と 500 mL以上の生理食塩液バッグを接続し、IVTM-SKのプライミングを
行う。
4) 被験者の体格および治験責任医師または治験分担医師の医学的判断に基づき、本
品の ICY および Quattro カテーテルである IVTM-CI または IVTM-CQ(バルーン付
中心静脈カテーテル)を選択する。
5) IVTM-CI または IVTM-CQ を、被験者の大腿静脈よりセルジンガー法で挿入し、下
大静脈内に留置する。IVTM-CI または IVTM-CQ留置後、腹部 X線でカテーテル
の位置を確認する。
6) IVTM-SK と IVTM-CI または IVTM-CQを接続し、IVTM-TG の Standby/Runボタ
ンを押し治療を開始する。
7) 治療終了後、IVTM-TGから治療データをダウンロードする。
8) IVTM-SK と IVTM-CI または IVTM-CQの接続を外す。IVTM-CI または IVTM-CQ
のインフロールーメンおよびアウトフロールーメンのキャップは外した状態(生理食塩
液を排出するため)で、IVTM-CI または IVTM-CQをゆっくりと抜去する。
調査・観察項目 表 4.3-1 のとおり
使 用 期 間 ・ 導入期:深部体温が目標体温(34.0℃以下)に到達するまで
・ 維持期:24 時間(23 時間 30 分~24 時間 30 分を許容範囲とする)
・ 復温期:深部体温が 36.0℃以上に到達するまで(維持期の深部体温が 33.0℃の場
合、約 30 時間)
・ 正常体温維持期:深部体温が 36.0℃以上に到達してから 24 時間(23 時間 30 分~
24 時間 30 分を許容範囲とする)
治験機器の使用期間は、導入期、維持期、復温期、正常体温維持期を合計して最大 4
日間(96 時間)とする。
主要評価項目 治験機器による冷却開始 3 時間以内に深部体温が目標体温に達した被験者の割合
[個々の被験者の評価]
治験機器による冷却開始後、深部体温が目標体温(34.0℃以下)に到達した時刻を症
例報告書に記載し、3 時間以内に目標体温に達したか否かを評価

147
副次的評価項目 1)治験機器使用開始 14 日後の Cerebral Performance Categories(CPC)
2)治験機器使用開始 14 日後の modified Rankin Scale(mRS)
3)治験機器使用中の体温の推移
4)単位時間あたりの体温変化量(低下速度)
安全性評価項目 1)有害事象(臨床検査値の異常変動を含む)
2)治験機器の不具合(体温のオーバーシュートを含む)
治 験 期 間 2013 年*月 ~ 2014 年*月
施 設 名 *************************
*************************
*************************
*************************
*************************
*************************
*************************
*************************
*************************
************************* 全 10 施設
【結果及び考察】
本治験の同意を取得した被験者は 25人、治験を中止した被験者は 3人だった。治験の中止は、1人が治
験機器による治療開始前中止、残る 2 人は治療開始後の中止だった。治験の中止理由は、全員が「有害
事象の発現により治験の継続が困難と治験責任医師または治験分担医師が判断した場合」であった。3
人とも有害事象と治験機器との因果関係はないと判断された。
<主要評価項目>
治験機器による治療を開始した 24人全員が 3時間以内に目標体温に達し、目標達成率は 100%(95%
CI 85.8-100%)だった。治療開始時の深部体温は 36.20±1.11℃で、冷却開始から目標体温到達まで
に要した時間は 54.63±37.46分(IQR 33.0-73.3分)だった。
<副次的評価項目>
1)治験機器使用開始 14日後の Cerebral Performance Categories(CPC)
治験機器使用開始から 14日後の CPCは、良好な神経学的転帰を示す CPC 1または 2の被験者が 12
人(50%)だった。
2)治験機器使用開始 14日後の modified Rankin Scale(mRS)
治験機器使用開始から 14 日後のmRS は、良好な神経学的転帰を示す mRS 0~2 の被験者が 11 人
(45.9%)だった。
3)治験機器使用中の体温の推移
維持期(設定温度 33.0℃)の平均体温は 33.09±0.06℃、95%信頼区間は 33.08-33.10℃であった。体
温測定回数 242 回中、±0.2℃を超えたのは 2 回(0.8%)で±0.5℃および±1.0℃を超えた変動はなか

148
った。
正常体温維持期(設定温度 36.5℃)の平均体温は 36.64±0.17℃、95%信頼区間は 36.60-36.69℃であ
った。体温測定回数 188 回中、±0.2℃を超えたのは 30 回(16.0%)、±0.5℃を超えたのは 6 回
(3.2%)、±1.0℃を超えたのは 1回(0.5%)であった。
維持期から正常体温維持期に移行する復温期は 0.1℃/hr で復温することとした。維持期と正常体温到
達時の体温差は 3.0℃であり、復温時間は 28.69±4.03時間(IQR 27.9-30.0)(FAS)だった。
4)単位時間あたりの体温変化量(低下速度)
導入期の体温冷却速度は、3.08±1.45℃/hr(中央値 2.70℃/hr、IQR 2.3-3.7℃/hr)だった。
<安全性評価項目>
本治験の同意を取得した全被験者を対象に安全性を評価した。
1)有害事象
少なくとも 1 件以上の有害事象(重篤、非重篤を問わない、また因果関係も問わない)が報告された被
験者数(有害事象発現率)(95%CI)は 25人中 19 人(76.0%)(54.9-90.6%)だった。治験機器との因果
関係が否定できない有害事象の発現被験者数は 4 人(16.0%)(4.5-36.1%)であり、いずれも非重篤だ
った。重篤な有害事象の発現被験者数は 5 人(20%)(6.8-40.7%)であり、いずれも治験機器との因果
関係はなかった。治験機器との因果関係が否定できない重篤な有害事象は発現しなかった。
2)治験機器の不具合
治験機器の不具合は 2 件報告された。内訳は、IVTM-SK のチューブ亀裂による破損 1 件、IVTM-CQ
のキット内に同梱されているガイドワイヤの折れ曲がり 1件だった。どちらの事象も、不具合に伴う有害事
象はなかった。
<考察>
治験機器 IVTM(IVTM-TG、IVTM-SK、IVTK-CI または IVTM-CQ)を用いた血管内体温管理のよる低
体温療法は、心原生が疑われる内因性心停止・心拍再開後患者に対し安全に実施できる。また、本治験機
器は目標体温への到達、低体温の管理・維持、および復温と正常体温の管理・維持が治験実施計画書どお
り有効に実施できた。本治験機器による低体温療法が安全および有効に実施できることが示された本治験結
果から、中心静脈カテーテルが留置可能な心停止・心拍再開後患者に対して、本治験機器は対象集団の体
温管理に用いる医療機器として適切であると考える。
149
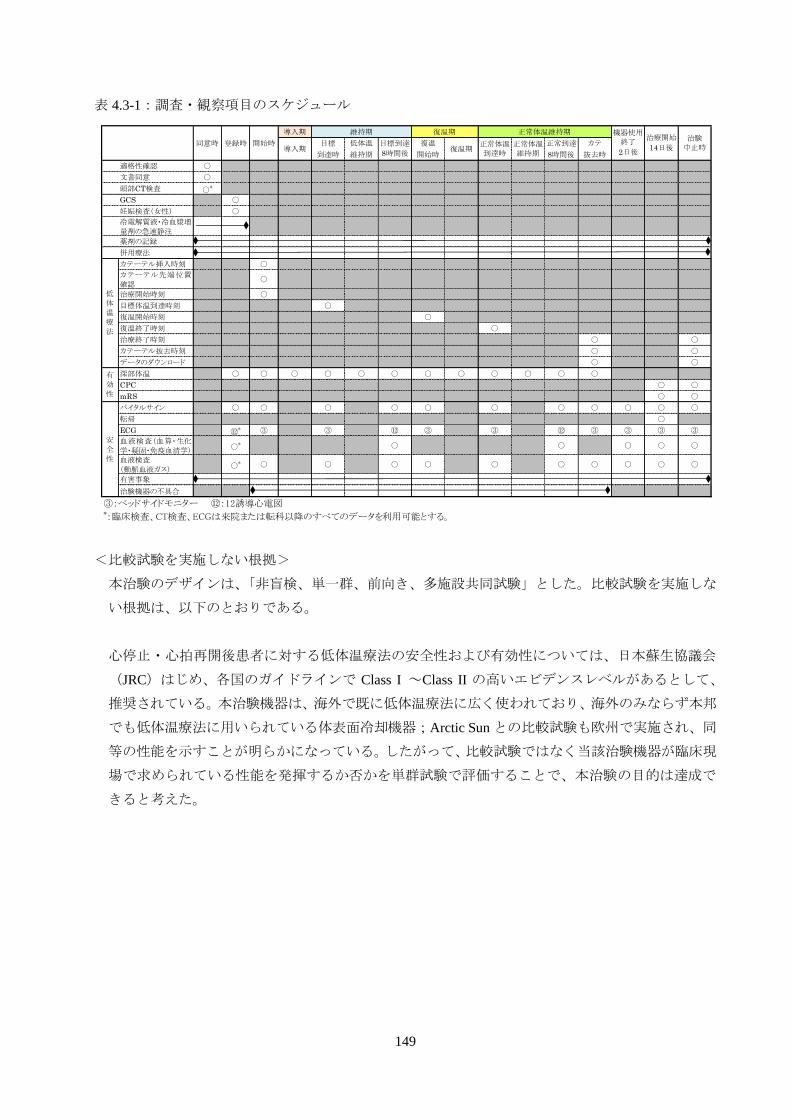
表 4.3-1:調査・観察項目のスケジュール
<比較試験を実施しない根拠>
本治験のデザインは、「非盲検、単一群、前向き、多施設共同試験」とした。比較試験を実施しな
い根拠は、以下のとおりである。
心停止・心拍再開後患者に対する低体温療法の安全性および有効性については、日本蘇生協議会
(JRC)はじめ、各国のガイドラインで Class I ~Class II の高いエビデンスレベルがあるとして、
推奨されている。本治験機器は、海外で既に低体温療法に広く使われており、海外のみならず本邦
でも低体温療法に用いられている体表面冷却機器;Arctic Sun との比較試験も欧州で実施され、同
等の性能を示すことが明らかになっている。したがって、比較試験ではなく当該治験機器が臨床現
場で求められている性能を発揮するか否かを単群試験で評価することで、本治験の目的は達成で
きると考えた。
導入期
目標 低体温 復温 正常到達 カテ
到達時 維持期 開始時 8時間後 抜去時
適格性確認 ○
文書同意 ○
頭部CT検査 ○*
GCS ○
妊娠検査(女性) ○
冷電解質液・冷血漿増量剤の急速静注
薬剤の記録
併用療法
カテーテル挿入時刻 ○
カテーテル先端位置確認
○
治療開始時刻 ○
目標体温到達時刻 ○
復温開始時刻 ○
復温終了時刻 ○
治療終了時刻 ○ 〇
カテーテル抜去時刻 ○ 〇
データのダウンロード ○ 〇
深部体温 ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○
CPC ○ 〇
mRS ○ 〇
バイタルサイン ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ 〇
転帰 〇
ECG ⑫* ③ ③ ⑫ ③ ③ ⑫ ③ ③ ③ ③
血液検査(血算・生化学・凝固・免疫血清学) ○* ○ ○ ○ ○ 〇
血液検査(動脈血液ガス)
○* ○ ○ ○ ○ ○ ○ ○ ○ ○ 〇
有害事象
治験機器の不具合
③:ベッドサイドモニター ⑫:12誘導心電図*:臨床検査、CT検査、ECGは来院または転科以降のすべてのデータを利用可能とする。
治験中止時
治療開始
14日後導入期目標到達
8時間後復温期
正常体温到達時
正常体温維持期
維持期 復温期
低体温療法
有効性
安全性
正常体温維持期 機器使用終了
2日後同意時 登録時 開始時

150
4.3.1 臨床試験成績
4.3.1.1 臨床試験の概要
臨床試験の概略を以下に示す。
目的 臨床試験の治験機器(IVTM)を用いて、心原性が疑われる内因性心停止・心拍再開後
患者の体温を適切に管理でき、低体温療法が行えることを検証すること。
試験デザイン 非盲検、単一群、前向き、多施設共同試験
症例数 登録被験者数:25 人(治療開始前中止 1 人)
有効性評価採用被験者数: 24 人(FAS)
安全性評価採用被験者数: 25 人
対象患者 心原性が疑われる内因性心停止・心拍再開後患者
選択基準:
以下のすべてを満たす患者
1) 非外傷性の院内または院外心停止患者
2) 目撃されない心停止(心室細動)または目撃された心停止(心室細動、無
脈性電気活動、発症後 15 分以内の心静止)
3) 同意取得時に 20 歳以上 80 歳未満の患者
4) 口頭指示に従うことができないと治験責任医師または治験分担医師が判断
した患者
5) 自己心拍再開(Return of Spontaneous Circulation;ROSC)から 6 時間以内
に治験機器による冷却を開始できる患者
6) 代諾者からの文書同意が取得できる患者
除外基準:
以下のいずれかに該当する患者
1) 外傷性心停止患者(鈍的外傷、鋭的外傷、熱傷、失血、窒息、煙の吸引、
感電、溺水等)
2) 深部体温が 35.0℃未満の偶発性低体温症患者
3) 妊娠または妊娠している可能性がある患者
4) DNR を意思表示している患者
5) 手術歴、既往歴、解剖学的構造等により大腿静脈アクセスが禁忌の患者
6) 下大静脈フィルターを留置している患者
7) 重度の出血(肺出血、消化管出血等)がある患者
8) CT 検査により頭蓋内出血が確認された患者
9) 昇圧薬や強心薬の使用にもかかわらず循環動態が不安定な患者
10) ヘパリン過敏症の患者
11) 重篤な全身性感染症(敗血症等)患者
12) 血小板 30,000/mm3未満の患者
151
13) 重篤な肝機能障害のある患者
14) 重篤な腎機能障害のある患者
15) 経皮的心肺補助装置(Percutaneous Cardio Pulmonary Support;PCPS)を使
用している患者
16) 持続的血液透析濾過療法(Continuous Hemodiafiltration;CHDF)を施行し
ている患者
17) ROSC から治験開始時までに他の低体温療法を実施した患者(ブドウ糖を
含まない冷電解質液またはブドウ糖を含まない冷血漿増量剤の急速静注
を除く)
18) 深部体温モニタリングができない患者
19) 過去 6 ヶ月以内または現在他の治験に参加している患者
20) その他、治験責任医師または治験分担医師が本治験実施を不適当と判断
した患者
使用方法 1) IVTM-TG(本体装置)の電源スイッチを入れ、使用準備を行う。
2) IVTM-SK(体外部回路)を IVTM-TGの温度コントロール槽内にセットする。
3) IVTM-SK と 500 mL 以上の生理食塩液バッグを接続し、IVTM-SK のプライミングを
行う。
4) 被験者の体格および治験責任医師または治験分担医師の医学的判断に基づき、本
品の ICY および Quattro カテーテルである IVTM-CI または IVTM-CQ(バルーン付
中心静脈カテーテル)を選択する。
5) IVTM-CI または IVTM-CQ を、被験者の大腿静脈よりセルジンガー法で挿入し、下
大静脈内に留置する。 IVTM-CI または IVTM-CQ 留置後、腹部 X 線でカテーテル
の位置を確認する。
6) IVTM-SK と IVTM-CIまたは IVTM-CQを接続し、IVTM-TGの Standby/Runボタン
を押し治療を開始する。
7) 治療終了後、IVTM-TGから治療データをダウンロードする。
8) IVTM-SK と IVTM-CI または IVTM-CQ の接続を外す。IVTM-CI または IVTM-CQ
のインフロールーメンおよびアウトフロールーメンのキャップは外した状態(生理食塩液
を排出するため)で、IVTM-CI または IVTM-CQをゆっくりと抜去する。
調査・観察項目 表 4.3-1 のとおり
使用期間 ・導入期:深部体温が目標体温(34.0℃以下)に到達するまで
・維持期:深部体温が 34.0℃以下に到達してから 24時間(23時間 30分~24時間 30分
を許容範囲とする)
・復温期:深部体温が 36.0℃以上に到達するまで
・正常体温維持期:深部体温が 36.0℃以上に到達してから 24 時間(23 時間 30 分~24
時間 30 分を許容範囲とする)
機器の使用期間は、導入期、維持期、復温期、正常体温維持期を合計して最大 4 日間
(96 時間)とする。
主要評価項目 治験機器による冷却開始 3 時間以内に深部体温が目標体温に達した被験者の割合

152
[個々の被験者の評価]
治験機器による冷却開始後、深部体温が目標体温(34.0℃以下)に到達した時刻を症例
報告書に記載し、3 時間以内に目標体温に達したか否かを評価する。
副次的評価項目 1) 治験機器使用開始 14 日後の Cerebral Performance Categories(CPC)
2) 治験機器使用開始 14 日後の modified Rankin Scale(mRS)
3) 治験機器使用中の体温の推移
4) 単位時間あたりの体温変化量(冷却速度)
安全性評価項目 1) 有害事象(臨床検査値の異常変動を含む)
2) 治験機器の不具合(体温のオーバーシュートを含む)
治験調整医師/
医学専門家
*************************
*************************
施設名 *************************
*************************
*************************
*************************
*************************
*************************
*************************
*************************
*************************
************************* 全 10 施設
治験期間 2013 年*月~2014 年*月
試験成績 本治験の同意を取得した被験者は 25人、治験を中止した被験者は 3人だった。治験の
中止は、1人が治験機器による治療開始前中止、残る 2人は治療開始後の中止だった。
治験の中止理由は、全員が「有害事象の発現により治験の継続が困難と治験責任医師
または治験分担医師が判断した場合」であった。3 人とも有害事象と治験機器との因果
関係はないと判断された。
<被験者背景>
被験者の背景は、男性 22 人(88.0%)、女性 3 人(12.0%)、年齢 56.1±15.4 歳(27-
78 歳)(平均値±標準偏差、カッコ内は最小値-最大値;以下同様)、身長 168.5±8.8
cm(148-181 cm)、体重 69.0±13.2 kg(39-100 kg)、BMI 24.2±3.5 kg/m2(16.8-31.1
kg/m2)、BSA 1.737±0.193 m2(1.24-2.15 m2)だった。
全員が院外心停止で、22 人(88.0%)が目撃ありだった。初期波形は 25 人中 19 人
(76.0%)が心室細動で、残り 6 人(24.0%)が無脈性電気活動だった。心静止の被験
者はいなかった。
心停止から ROSCまでの時間は 24.6±14.5分、ROSCから治験機器使用開始までの
時間は 262.3±73.5 分で、全員が治験機器による冷却開始前に冷電解質液が投与さ
れた。
治療に使用したカテーテルは、IVTM-CI が 7 人(29.2%)、IVTM-CQ が 17 人
(70.8%)だった。
153
<主要評価項目>
治験機器による治療を開始した 24 人全員が 3 時間以内に目標体温に達し、目標達
成率は 100%(95%CI 85.8-100%)だった。冷却開始時の深部体温は 36.20±1.11℃
で、冷却開始から目標体温到達までに要した時間は 54.63±37.46 分(中央値 45.00
分、IQR 33.0-73.5 分)だった。
<副次的評価項目>
1)治験機器使用開始 14 日後の Cerebral Performance Categories(CPC)
治験機器使用開始から 14 日後の CPC は、良好な神経学的転帰を示す CPC 1 また
は 2 の被験者が 12 人(50%)だった。
2)治験機器使用開始 14 日後の modified Rankin Scale(mRS)
治験機器使用開始から 14日後のmRSは、良好な神経学的転帰を示す mRS 0~2の
被験者が 11 人(45.9%)だった。
3)治験機器使用中の体温の推移
維持期(設定温度 33.0℃)の平均体温は 33.09±0.06℃、95%信頼区間は 33.08-
33.10℃であった。体温測定回数 242 回中、±0.2℃を超えたのは 2 回(0.8%)で±
0.5℃および±1.0℃を超えた変動はなかった。
正常体温維持期(設定温度 36.5℃)の平均体温は 36.64±0.17℃、95%信頼区間は
36.60-36.69℃であった。体温測定回数 188 回中、±0.2℃を超えたのは 30 回
(16.0%)、±0.5℃を超えたのは 6 回(3.2%)、±1.0℃を超えたのは 1 回(0.5%)であ
った。
維持期から正常体温維持期に移行する復温期は 0.1℃/hrで復温することとした。維持
期と正常体温到達時の体温差は 3.0℃であり、復温時間は 28.69±4.03 時間(IQR
27.9-30.0)(FAS)だった。
4)単位時間あたりの体温変化量(低下速度)
導入期の体温冷却速度は、3.08±1.45℃/hr(中央値 2.70℃/hr、IQR 2.3-3.7℃/hr)だ
った。
<安全性評価項目>
本治験の同意を取得した全被験者を対象に安全性を評価した。
1)有害事象
少なくとも 1 件以上の有害事象(重篤、非重篤を問わない、また因果関係も問わない)
が報告された被験者数(有害事象発現率)(95%CI)は 25 人中 19 人(76.0%)(54.9-
90.6%)だった。治験機器との因果関係が否定できない有害事象の発現被験者数は4
人(16.0%)(4.5-36.1%)であり、いずれも非重篤だった。重篤な有害事象の発現被験
者数は 5 人(20%)(6.8-40.7%)であり、いずれも治験機器との因果関係はなかった。
治験機器との因果関係が否定できない重篤な有害事象は発現しなかった。

154
発現率の高かった重篤な有害事象は心室細動で、3 人(12.0%)に発現した。3 件とも
重症度は高度で治験機器との因果関係は「関連がない」だった。転帰は 2 人が軽快、
1 人が死亡だった。
発現率の高かった非重篤な有害事象は、肺炎 6 人(24.0%)、高血圧 3 人(12.0%)だ
った。
重症度は肺炎(6 人)が軽度 3 人、中等度 3 人、高血圧(3 人)が軽度 3 人だった。
治験機器との因果関係が否定できない有害事象は、肺炎、徐脈、大静脈血栓症、血
中クレアチンホスホキナーゼ増加、血圧低下がそれぞれ 1件発現した。重症度はいず
れも中等度だった。なお、徐脈については、低体温療法を行う場合、心臓脈の減少
は通常起こりえることであり、可逆的で予期される効果であると考える。
発現時期は、大静脈血栓症のみ治療終了後に発現した。残りの治験機器との因果関
係が否定できない有害事象は、治療期間中に発現したが、治験機器の使用状況は
「変更なし(治療継続)」だった。転帰は、いずれも「回復」だった。治験機器との因果
関係が否定できない有害事象は、いずれも治験機器による治療継続が可能であった
ことや、処置により回復していることから被験者の安全性には問題がないと考えられ
る。
本治験の登録被験者 25 人のうち、最終確認時の転帰が生存だったのは 23 人
(92%)、死亡は 2人(8.0%)であった。生存率の 95%信頼区間は 71.6-97.9%だった。
死亡した被験者の死因は 2 人とも原疾患の悪化で、死亡と治験機器との因果関係は
関係ないと治験責任医師が判断した。いずれの被験者も治験中止後の死亡だった。
治験期間中の死亡はなかった。死亡以外の重篤な有害事象は 5 人 5 件(心室細動 3
件、心停止 1 件、急性呼吸窮迫症候群 1 件)が報告された。すべての重篤な有害事
象は治験機器との因果関係がないと判断された。重篤な有害事象のうち、心室細動 1
件と心停止 1 件は治験機器の使用前に発現した有害事象だった。
臨床検査値に関する有害事象は、血中クレアチンホスホキナーゼ増加の 2件だった。
心電図の異常所見に関する有害事象は報告されなかった。
バイタルサイン(収縮期/拡張期血圧、心拍数、呼吸数、深部温および末梢温)では、
目標体温到達時から 8 時間後および 24 時間後に心拍数の減少が認められた。登録
時、目標体温到達時から 8時間後および 24時間後の心拍数は、108.4±22.0回/分、
76.1±14.6 回/分、76.1±17.1 回/分であり、著明な徐脈は認められなかった。
2) 治験機器の不具合
治験機器の不具合は 2件報告された。内訳は、IVTM-SKのチューブ亀裂による破損
1 件、IVTM-CQ のキット内に同梱されているガイドワイヤの折れ曲がり 1 件だった。ど
ちらの事象も、不具合に伴う有害事象はなかった。

155
(1)選択基準の設定根拠
1)JRC ガイドライン 2010[1]に基づく。
2)JRC ガイドライン 2010[1]に基づく。
3)倫理的配慮から設定した。
4)心停止・心拍再開後患者のうち、意識障害がある患者が低体温療法の対象となることから設定し
た。
5)J-PULSE-HYPO[2]では、低体温療法の Window of Opportunity は 6 時間と考えられていたことから
設定した。
6)被験者の利益と治験の倫理性確保のために設定した。
(2)除外基準の設定根拠
1)本治験の対象ではないと考えられるため除外した。
2)本治験の対象ではないと考えられるため除外した。
3)妊娠中の使用に関して安全性が確立されていないため除外した。
4)倫理的配慮から除外した。
5)安全性確保の観点から除外した。
6)安全性確保の観点から除外した。
7)安全性確保の観点から除外した。
8)本治験の対象ではないと考えられるため除外した。
9)安全性確保の観点から除外した。
10)安全性確保の観点から除外した。
11)安全性確保の観点から除外した。
12)安全性確保の観点から除外した。
13)安全性確保の観点から除外した。
14)安全性確保の観点から除外した。
15)有効性および安全性評価に影響を及ぼすと考えられるため除外した。
16)有効性および安全性評価に影響を及ぼすと考えられるため除外した。
17)有効性および安全性評価に影響を及ぼすと考えられたため除外した。
18)安全性確保の観点から除外した。
19)有効性および安全性評価に影響を及ぼす可能性があること、安全性確保の観点から除外した。
20)上記の項目以外にも、本治験の有効性および安全性の評価に影響を及ぼす可能性がある場合、ま
た、治験責任医師または治験分担医師の判断で被験者の安全性の確保が困難であると判断した
場合には除外するよう設定した。
(3)評価項目の設定根拠
1)主要評価項目の設定根拠
低体温療法は低体温導入、低体温維持、復温、正常体温維持の行程からなるため、この 4 つの行程
で治験機器が体温を適切に管理できることを、信頼性の担保された臨床試験で示す必要がある。こ
の 4 つの行程で、現在最も医学的コンセンサスが得られている部分は、32℃~34 ℃で一定時間(24

156
時間程度)低体温を維持することと、低体温導入時の目標体温への早期到達である。前者は、一定
の温度幅で低体温を維持することが重要と考えられているが、これまでの臨床研究で管理温度幅
と神経学的予後に関する考察はほとんどない。一方後者は、自己心拍再開後一定時間内に目標体温
へ到達させることで、生存率あるいは神経学的予後に良好な結果をもたらすことがいくつかの報
告で示されており、治験機器を用いた低体温療法で、最低体温への早期到達が神経細胞死と関連の
深い神経特異的エノラーゼの血清中濃度を低値に抑え、神経予後が良好なことが報告されている
[3]。
臨床的に重要な意味を持つ低体温到達時間の閾値は、現在のところ明確なエビデンスが得られて
いないため、これまでに報告された治験機器の臨床性能をもとに評価時間を決定した。その結果、
低体温療法の臨床実績がある体表面冷却機器(Arctic Sun)と治験機器の臨床比較をした Tømte ら
の報告[4]で、いずれの機器も目標体温達成の中央値が 3 時間前後であることから、本治験による主
要評価項目を「治験機器による冷却開始 3 時間以内に深部体温が目標体温に達した被験者の割合」
と定めた。なお、本邦の J-PULSE-HYPO 臨床試験[2]での冷却導入時間も中央値は 3 時間であり、
この目標時間は本邦での低体温療法の実態にも合致すると考えられた。
2)副次的評価項目の設定根拠
本治験は、心原性が疑われる内因性心停止・心拍再開後患者で意識障害を有する患者を対象にして
いることから、低体温療法実施後の被験者の意識障害の程度を評価するために CPC、mRS を設定
した。また、体温が適切に管理されているかを評価するために治験機器使用中の体温の推移を、治
験機器による冷却開始時の体温には個人差があり、3 時間での目標達成の有無だけでは本治験機器
の冷却性能は評価できないと考えたことから単位時間あたりの体温変化量(冷却速度)を設定した。
3)安全性評価項目の設定根拠
(1) 有害事象(臨床検査値の異常変動を含む)
本治験機器を用いた低体温療法による有害事象を検討するために設定した。
(2) 治験機器の不具合(体温のオーバーシュートを含む)
本治験機器の「製品の仕様上の問題」、「不良品」、「故障」、「治験機器に添付している文書の不十
分な記載」を検討するために設定した。また、体温のオーバーシュートは、本治験機器に設定体
温の許容範囲を逸脱する現象が発現するかを検討するため設定した。
(4)被験者数の設定根拠
本治験機器は、既に欧州で低体温療法の適応で CE マークを取得しているため、国内外での適応の
乖離は医療現場での混乱を招く懸念があり、これを解消するために臨床試験を迅速に実施し、低体
温療法への適応拡大申請を早期に行う必要がある。したがって、迅速な治験実施のためには被験者
登録期間*年間、実施医療機関数 10 医療機関以上は困難であると判断した。
本治験の目的は、「治験機器による冷却開始 3 時間以内に深部体温が目標体温に達した被験者の割
合」であり、Tømte[4]らの報告などから本治験機器の目標体温達成時間の中央値または平均値がお

157
よそ 3 時間であることが示されているため、本治験での 3 時間以内の目標達成率は 50%と想定し
た。
一方、エビデンスレベルが高いとされる欧州 HACA グループの試験[5]では、低体温療法開始後か
ら目標体温達成までの時間は示されていないものの、ROSC から目標温度(32℃~34℃)に到達す
るまでの時間は平均 8 時間、25 パーセンタイル値は 4 時間であることが示されている。さらに当
該試験では、ROSC から低体温療法を開始するまでの平均時間は 105 分、25 パーセンタイル値は
61 分であると報告されているので、低体温療法開始後から 3 時間後に目標体温を達成する被験者
の割合はおよそ 25%と推察した。
以上のことから、低体温療法開始後 3 時間で目標体温(32℃~34℃)に到達する被験者の割合が
25%以上を達成できれば臨床的に意義があると考え、目標達成率 50%と想定した本治験機器の
95%信頼区間の下限値が、目標達成率 25%を上回ることを検出力 80%で検証するために必要な被
験者数を算出したところ、23 人であった。
本治験の対象はきわめて重症な患者であり、同様の対象を扱った種々の臨床試験で、退院時までの
生存率は 50%~60%である。したがって、本治験期間である 2 週間を完遂できる被験者数は登録
被験者数の 60%~70%と想定されるが、主要評価項目である治験機器による冷却開始後 3 時間ま
での評価可能被験者数は登録被験者数の 80%以上と見込んだ。
以上のことから、登録被験者数 30 人を目標とした場合、主要評価項目が評価可能な被験者数は 24
人以上となり、必要被験者数の 23 人を上回ることから上記の目標を達成することは可能であると
考えた。
(5)使用方法の設定根拠
低体温維持期は JRC ガイドライン 2010[1]に従い 24 時間、32.0℃~34.0℃とし、これを実現するた
め設定温度を 33.0℃とした。ガイドライン[1]では、復温速度および復温後の正常体温維持時間の
推奨条件は示されていないが Kokubu N らの報告[6]によれば、J-PULSE-HYPO レジストリにおける
復温速度を≧2.0℃/day の F 群、1.0-1.9℃/day の M 群、<1.0℃/day の S 群の 3 群に分けて解析する
と、30 日生存率は M 群が有意に高かったが、生存者だけの CPC1(神経予後良好)の比率は F 群で
有意に高く予後良好であった。このため、復温速度は 0.1℃/hr に設定した。低体温療法後の発熱を
避けるため、復温後 36.0℃~37.0℃の正常体温を 24 時間強制的に持続することとした。なお、本治
験機器の使用期間は最大 4 日であり、その範囲内で治療を終了することとした。
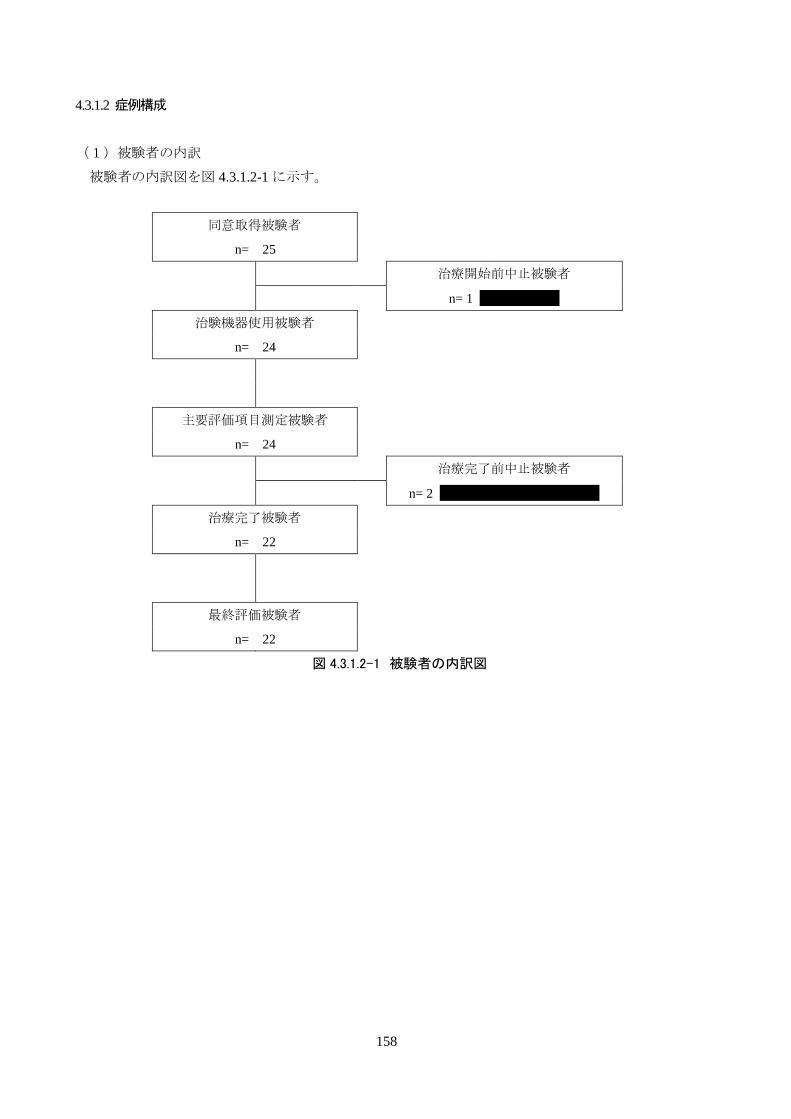
158
4.3.1.2 症例構成
(1)被験者の内訳
被験者の内訳図を図 4.3.1.2-1 に示す。
同意取得被験者
n= 25
治療開始前中止被験者
n= 1 ******
治験機器使用被験者
n= 24
主要評価項目測定被験者
n= 24
治療完了前中止被験者
n= 2 ************
治療完了被験者
n= 22
最終評価被験者
n= 22
図 4.3.1.2-1 被験者の内訳図

159
(2)解析対象
1)有効性解析対象集団
有効性解析対象集団は、「最大の解析集団(FAS)」および「治験実施計画書に適合した解析集団(PPS)」
の 2 集団を設定する。有効性の主たる解析対象集団は FAS とし、必要に応じて PPS による解析も
実施する。
(1) FAS(Full Analysis Sets)
本治験を実施した被験者のうち、下記のいずれにも該当しない被験者;
・治験機器による治療を施行していない被験者
・主要評価項目の評価が存在しない被験者
・GCP を逸脱した被験者。
(2) PPS(Per Protocol Sets)
FAS から下記に該当する被験者を除いた集団;
・治験機器の使用開始から 14 日後の評価が存在しない被験者
・治験実施計画書からの逸脱が認められ、症例検討により解析の対象外とされた被験者
2)安全性解析対象集団(SAF)
安全性解析対象被験者は、本治験の同意を取得した全被験者とした。ただし、治験機器の不具合の
解析対象は、IVTM-TG は被験者に使用した(治療に至った)回数を、IVTM-SK、IVTM-CI および
IVTM-CQ は治療開始前の準備も含め開封した数を母数とした。
要約表の作成および有害事象の集計にあたり、症例報告書に記載された有害事象名を MedDRA/J
(Version 17.0)でコーディング、分類した。
有害事象の発現率は被験者数を用いて算出した。なお、1 人の被験者に同一名称の有害事象が複数
回発現した場合は、より重篤な、またはより重症な事象を採用した。

160
4.3.1.3 中止・脱落・プロトコル逸脱
治験を中止した被験者の内訳を表 4.3.1.3-1 に、治験実施計画書からの重要な逸脱の内訳を表 4.3.1.3-
2 に、解析対象被験者数と内訳を表 4.3.1.3-3 に、解析から除外された被験者一覧を表 4.3.1.3-4 に示
す。
被験者の内訳は、同意取得被験者 25 人、治験を中止した被験者 3 人であった。
治験を中止した被験者 3 人の内訳は、治験機器による治療開始前に治験を中止した被験者 1 人、治験
機器による治療開始後に治験を中止した被験者 2 人であった。
治験機器による治療開始後に治験を中止した被験者は、2 人とも維持期(治験機器使用中;主要評価
項目の評価後)に治験を中止した。
治験を中止した被験者 3 人の中止理由は、全員が「有害事象の発現により治験の継続が困難と治験責任
医師または治験分担医師が判断した場合」であった。3 人とも有害事象と治験機器との因果関係はないと判
断された。
治験機器による治療を完遂した 22 人は、全員が治験機器使用開始から 14 日後の評価を実施した。
治験実施計画書からのすべての逸脱は、症例検討会(***年*月*日実施)に報告し、審議の上取
扱いを決定した。逸脱のうち、「GCP 違反」、「患者選択」に該当する逸脱、有効性に影響を及ぼす逸
脱を重要な逸脱として取扱った。重要な逸脱は 4 人に認められた。内訳は、「必須療法」2 人、「契約」
1 人、「選択基準」1 人であった。
いずれの逸脱も被験者の安全性およびデータの信頼性は損なわれていないと考えられ、本治験の有
効性および安全性の評価に影響を与えるものではないと判断した。
本治験の同意を取得した 25 人のうち、FAS は 24 人(除外 1 人)、PPS は 17 人(除外 8 人)であっ
た。FAS からの除外理由は、「治験機器による治療を施行していない」と「主要評価項目の評価が存
在しない」であった。PPS からの除外理由で複数人が該当した理由は、「治験機器の使用開始から 14
日後の評価が存在しない」3 人、「正常体温維持期の時間がアローアンス(23.5~24.5 hr)から外れて
いる」2 人、「導入期または維持期に鎮痛・鎮静・シバリング抑制プロトコルに従った薬剤を使用し
ていない」2 人であった。
本治験では、治験実施計画書で許容した範囲外に調査・測定されたデータがあった被験者や、データ
欠測があった被験者は不完全例と定義し、集計は次のとおりデータを取り扱った。
アローアンス外に調査・測定されたデータは集計に含めた。ただし、被験者識別コード******
の治験中止時のバイタルサイン:体温(腋下温)は膀胱温で測定されており、一覧表には参考値とし
てデータを表示したが集計からは除外した。また、被験者識別コード******の治験機器使用終
了日の 2 日後の臨床検査:血液生化学的検査:LDH は溶血のため高値を示しており、一覧表には参
考値としてデータを表示したが集計からは除外した。
欠測値の取り扱いは統計解析計画書で定めたとおり、心停止発生日時、来院・転科日時および GCS
判定日時の「分」データが欠測の場合を除いて、調査・観察項目の欠測値の補完は行わなかった(デ

161
ータの取扱いは、添付資料チ-1(1)「6.11.2. 統計解析の方法」参照)。症例検討会(**年*月*
日実施)で確認した結果、本治験の有効性評価に係るすべての時刻データおよび治療開始から終了ま
での深部体温データに欠測はなかった。
表 4.3.1.3-1 治験を中止した被験者の内訳
番号 被験者
識別コード 性別 実施医療機関名 中止時期 中止理由※1
1 ****** 男 ************ 治療開始前 1※2
2 ****** 男 ************ 治療開始後
(維持期)
1※2
3 ****** 男 ************ 治療開始後
(維持期)
1※2
※1:中止理由の詳細
1. 有害事象の発現により、治験の継続が困難と治験責任医師または治験分担医師が判断した場合
2. 低体温療法の実施が困難と治験責任医師または治験分担医師が判断した場合
3. 症状の悪化等により、治験の継続が困難と治験責任医師または治験分担医師が判断した場合
4. 治験機器による低体温療法開始後 6 時間を超えても目標体温に到達せず、治験責任医師または治験分担
医師が本治験機器単独での目標体温到達が困難と判断した場合
5. 代諾者および/もしくは被験者が治験の中止を申し出た場合
6. その他、治験の中止が必要と治験責任医師または治験分担医師が判断した場合(不適格の場合を含む)
※2:発現した有害事象と治験機器との因果関係はないと判断された。

162
表 4.3.1.3-2 治験実施計画書からの重要な逸脱の内訳
項目 該当被験者 詳細 取扱い
【契約】
治験責任医師または治
験分担医師に指名され
ていない者による治験
行為があった。
****** カテーテル挿入のための一連
の治験業務(使用するカテー
テルの選択、カテーテル挿
入、カテーテル挿入長の確
認、被験者の安全確保)のう
ち、カテーテル挿入を治験分
担医師・治験協力者リストに記
載のない医師が実施した。
治験分担医師の指導・監督の
下にカテーテルを挿入し、被験
者の安全性確保に対する配慮
は適切にされていた。また、カテ
ーテル挿入長の確認は治験分
担医師が行い、治験データの信
頼性も損なわれていないと考え
られることから、FAS 採用、PPS
不採用、SAF 採用とする。
【選択基準】
ROSC から 6 時間以内
に治験機器による冷却
を開始できない(できな
かった)患者
****** ROSC から治療開始までの時
間が 6 時間 5 分であった。
被験者の適格性確認時は
ROSCから 6時間以内に治療開
始される見込みで選択基準を満
たすと判断されていたため、
FAS 採用、PPS 不採用、SAF 採
用とする。
【除外基準】
除外基準に関する違反
該当なし
【中止基準】
中止基準に関する違反
該当なし
【設定温度】
導入期の設定温度違反
該当なし
【必須療法】
ROSC から治験機器に
よる治療開始まで:冷電
解質液または冷血漿増
量剤を投与していない。
****** ROSCの 7分前に冷電解質液
の投与を開始した。
ROSC から治験機器による治療
開始の間にも投与が継続されて
いるため、FAS採用、PPS採用、
SAF採用とする。
****** ROSCの 3分前に冷電解質液
の投与を開始した。
【禁止療法】
禁止療法に関する違反
該当なし
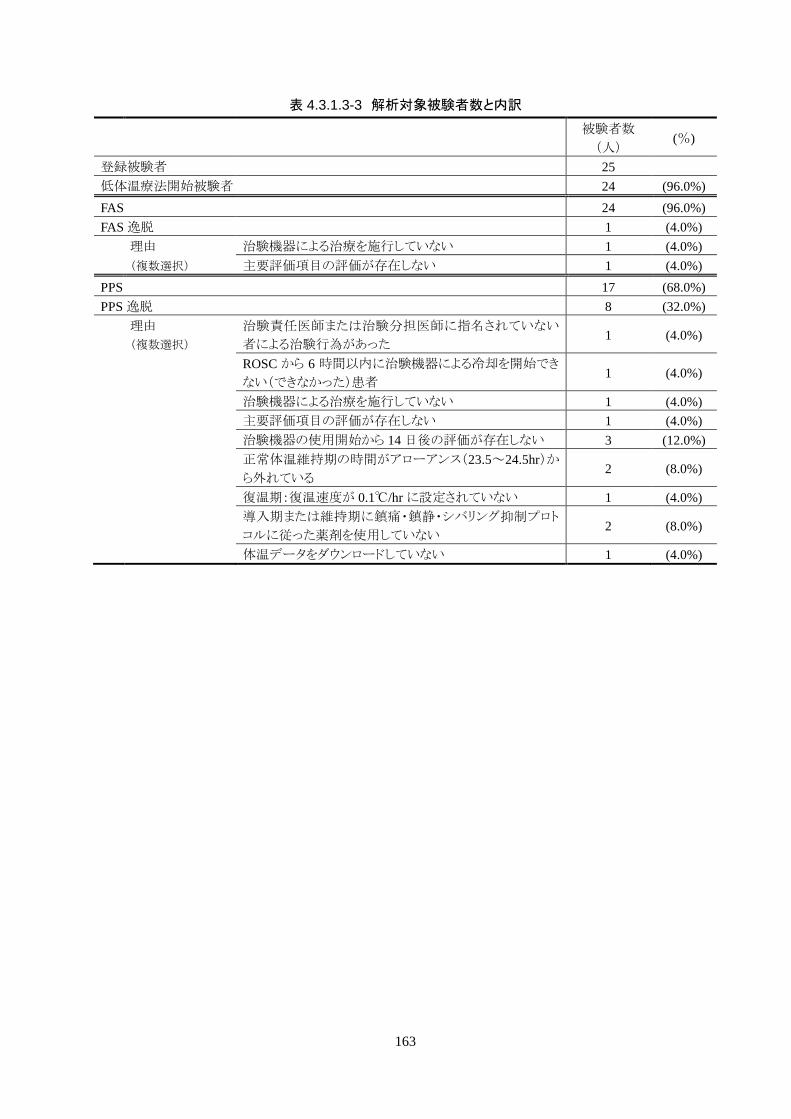
163
表 4.3.1.3-3 解析対象被験者数と内訳
被験者数
(人) (%)
登録被験者 25
低体温療法開始被験者 24 (96.0%)
FAS 24 (96.0%)
FAS逸脱 1 (4.0%)
理由 治験機器による治療を施行していない 1 (4.0%)
(複数選択) 主要評価項目の評価が存在しない 1 (4.0%)
PPS 17 (68.0%)
PPS逸脱 8 (32.0%)
理由
(複数選択)
治験責任医師または治験分担医師に指名されていない
者による治験行為があった 1 (4.0%)
ROSC から 6 時間以内に治験機器による冷却を開始でき
ない(できなかった)患者 1 (4.0%)
治験機器による治療を施行していない 1 (4.0%)
主要評価項目の評価が存在しない 1 (4.0%)
治験機器の使用開始から 14日後の評価が存在しない 3 (12.0%)
正常体温維持期の時間がアローアンス(23.5~24.5hr)か
ら外れている 2 (8.0%)
復温期:復温速度が 0.1℃/hr に設定されていない 1 (4.0%)
導入期または維持期に鎮痛・鎮静・シバリング抑制プロト
コルに従った薬剤を使用していない 2 (8.0%)
体温データをダウンロードしていない 1 (4.0%)
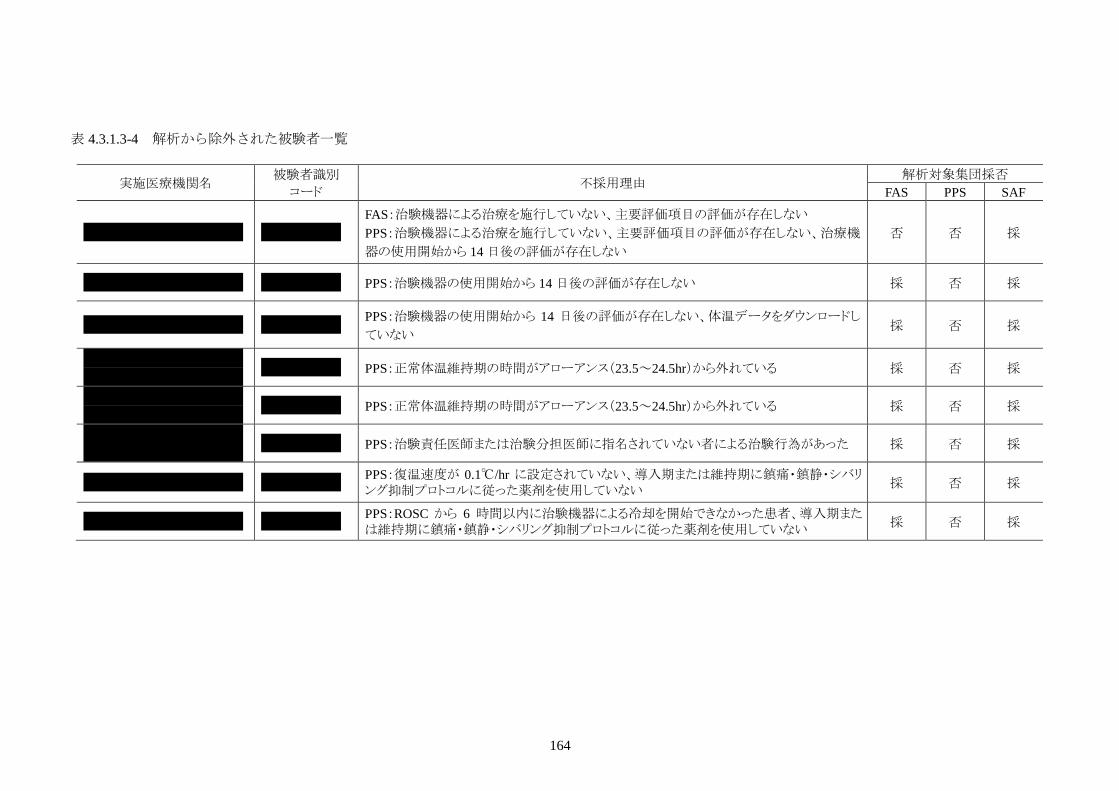
164
表 4.3.1.3-4 解析から除外された被験者一覧
実施医療機関名 被験者識別
コード 不採用理由
解析対象集団採否
FAS PPS SAF
************ ******
FAS:治験機器による治療を施行していない、主要評価項目の評価が存在しない
PPS:治験機器による治療を施行していない、主要評価項目の評価が存在しない、治療機
器の使用開始から 14 日後の評価が存在しない
否 否 採
************ ****** PPS:治験機器の使用開始から 14 日後の評価が存在しない 採 否 採
************ ****** PPS:治験機器の使用開始から 14 日後の評価が存在しない、体温データをダウンロードし
ていない 採 否 採
************
************ ****** PPS:正常体温維持期の時間がアローアンス(23.5~24.5hr)から外れている 採 否 採
************
************ ****** PPS:正常体温維持期の時間がアローアンス(23.5~24.5hr)から外れている 採 否 採
************
************ ****** PPS:治験責任医師または治験分担医師に指名されていない者による治験行為があった 採 否 採
************ ****** PPS:復温速度が 0.1℃/hr に設定されていない、導入期または維持期に鎮痛・鎮静・シバリング抑制プロトコルに従った薬剤を使用していない
採 否 採
************ ****** PPS:ROSC から 6 時間以内に治験機器による冷却を開始できなかった患者、導入期または維持期に鎮痛・鎮静・シバリング抑制プロトコルに従った薬剤を使用していない
採 否 採

165
4.3.1.4 患者背景
(1)人工統計学的変数および他の基準値の特性
人口統計学的変数の解析結果を表 4.3.1.4.1-1に、心停止の原因疾患の内訳を表 4.3.1.4.1-2 に、既往
歴の内訳を表 4.3.1.4.1-3 に、合併症の内訳を表 4.3.1.4.1-4 に示す。
要約表の作成および集計にあたり、症例報告書に記載された心停止の原因疾患、既往歴、合併症を
MedDRA/J(Version 17.0)でコーディング、分類した。
被験者背景は、男性 22 人(88.0%)、女性 3 人(12.0%)、年齢 56.1±15.4 歳(27-78 歳)(平均値±
標準偏差、カッコ内は最小値-最大値;以下同様)、身長 168.5±8.8 cm(148-181 cm)、体重 69.0±
13.2 kg(39-100 kg)、BMI 24.2±3.5 kg/m2(16.8-31.1 kg/m2)、BSA 1.737±0.193 m2(1.24-2.15 m2)
であった。
心停止の原因疾患で多かったのは、急性心筋梗塞 5 人(20%)、異型狭心症 3 人(12.0%)、急性冠
症候群 3 人(12.0%)などであった。
既往歴は、なし 20 人(80%)、あり 5 人(20%)だった。心臓障害に分類される既往歴は、急性心
筋梗塞、狭心症、心筋梗塞、洞不全症候群、ブルガダ症候群がそれぞれ 1 人ずつだった。
合併症は、なし 5 人(20%)、あり 20 人(80%)だった。合併症で多かったのは、高血圧 9 人(36.0%)、
誤嚥性肺炎 7 人(28.0%)、心房細動 5 人(20.0%)、低酸素性虚血性脳症、心不全、大動脈弁置換
がそれぞれ 3 人(12.0%)などであった。
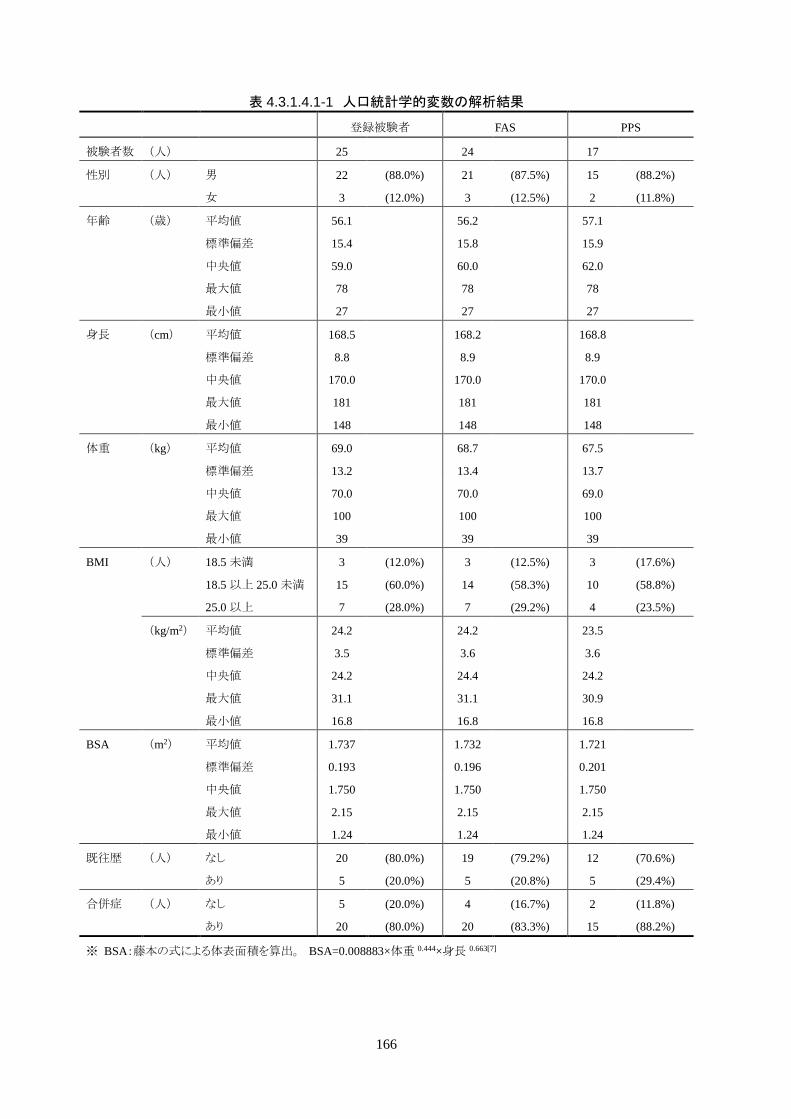
166
表 4.3.1.4.1-1 人口統計学的変数の解析結果
登録被験者 FAS PPS
被験者数 (人) 25 24 17
性別 (人) 男 22 (88.0%) 21 (87.5%) 15 (88.2%)
女 3 (12.0%) 3 (12.5%) 2 (11.8%)
年齢 (歳) 平均値 56.1 56.2 57.1
標準偏差 15.4 15.8 15.9
中央値 59.0 60.0 62.0
最大値 78 78 78
最小値 27 27 27
身長 (cm) 平均値 168.5 168.2 168.8
標準偏差 8.8 8.9 8.9
中央値 170.0 170.0 170.0
最大値 181 181 181
最小値 148 148 148
体重 (kg) 平均値 69.0 68.7 67.5
標準偏差 13.2 13.4 13.7
中央値 70.0 70.0 69.0
最大値 100 100 100
最小値 39 39 39
BMI (人) 18.5 未満 3 (12.0%) 3 (12.5%) 3 (17.6%)
18.5 以上 25.0 未満 15 (60.0%) 14 (58.3%) 10 (58.8%)
25.0 以上 7 (28.0%) 7 (29.2%) 4 (23.5%)
(kg/m2) 平均値 24.2 24.2 23.5
標準偏差 3.5 3.6 3.6
中央値 24.2 24.4 24.2
最大値 31.1 31.1 30.9
最小値 16.8 16.8 16.8
BSA (m2) 平均値 1.737 1.732 1.721
標準偏差 0.193 0.196 0.201
中央値 1.750 1.750 1.750
最大値 2.15 2.15 2.15
最小値 1.24 1.24 1.24
既往歴 (人) なし 20 (80.0%) 19 (79.2%) 12 (70.6%)
あり 5 (20.0%) 5 (20.8%) 5 (29.4%)
合併症 (人) なし 5 (20.0%) 4 (16.7%) 2 (11.8%)
あり 20 (80.0%) 20 (83.3%) 15 (88.2%)
※ BSA:藤本の式による体表面積を算出。 BSA=0.008883×体重 0.444×身長 0.663[7]
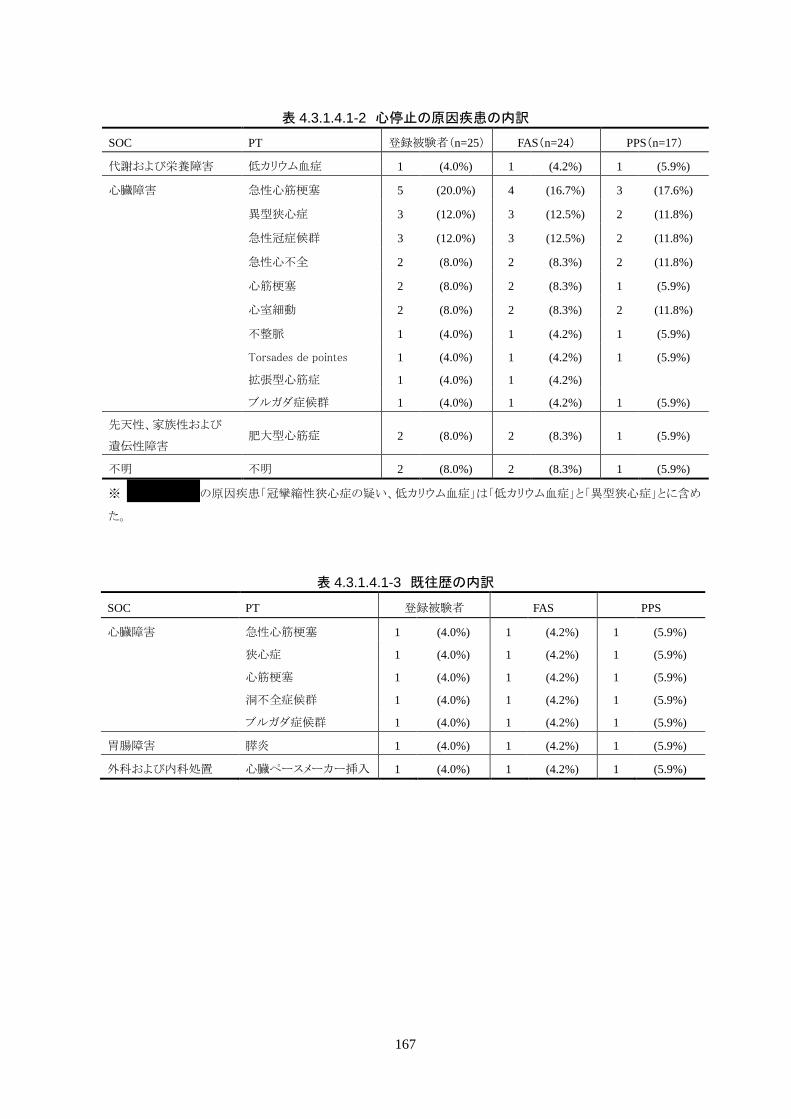
167
表 4.3.1.4.1-2 心停止の原因疾患の内訳
SOC PT 登録被験者(n=25) FAS(n=24) PPS(n=17)
代謝および栄養障害 低カリウム血症 1 (4.0%) 1 (4.2%) 1 (5.9%)
心臓障害 急性心筋梗塞 5 (20.0%) 4 (16.7%) 3 (17.6%)
異型狭心症 3 (12.0%) 3 (12.5%) 2 (11.8%)
急性冠症候群 3 (12.0%) 3 (12.5%) 2 (11.8%)
急性心不全 2 (8.0%) 2 (8.3%) 2 (11.8%)
心筋梗塞 2 (8.0%) 2 (8.3%) 1 (5.9%)
心室細動 2 (8.0%) 2 (8.3%) 2 (11.8%)
不整脈 1 (4.0%) 1 (4.2%) 1 (5.9%)
Torsades de pointes 1 (4.0%) 1 (4.2%) 1 (5.9%)
拡張型心筋症 1 (4.0%) 1 (4.2%)
ブルガダ症候群 1 (4.0%) 1 (4.2%) 1 (5.9%)
先天性、家族性および
遺伝性障害 肥大型心筋症 2 (8.0%) 2 (8.3%) 1 (5.9%)
不明 不明 2 (8.0%) 2 (8.3%) 1 (5.9%)
※ ******の原因疾患「冠攣縮性狭心症の疑い、低カリウム血症」は「低カリウム血症」と「異型狭心症」とに含め
た。
表 4.3.1.4.1-3 既往歴の内訳
SOC PT 登録被験者 FAS PPS
心臓障害 急性心筋梗塞 1 (4.0%) 1 (4.2%) 1 (5.9%)
狭心症 1 (4.0%) 1 (4.2%) 1 (5.9%)
心筋梗塞 1 (4.0%) 1 (4.2%) 1 (5.9%)
洞不全症候群 1 (4.0%) 1 (4.2%) 1 (5.9%)
ブルガダ症候群 1 (4.0%) 1 (4.2%) 1 (5.9%)
胃腸障害 膵炎 1 (4.0%) 1 (4.2%) 1 (5.9%)
外科および内科処置 心臓ペースメーカー挿入 1 (4.0%) 1 (4.2%) 1 (5.9%)
168
表 4.3.1.4.1-4 合併症の内訳
SOC PT 登録被験者 FAS PPS
感染症および寄生虫症 慢性C型肝炎 1 (4.0%) 1 (4.2%) 1 (5.9%)
C型肝炎 1 (4.0%) 1 (4.2%) 1 (5.9%)
白癬感染 1 (4.0%) 1 (4.2%) 1 (5.9%)
良性、悪性および詳細不
明の新生物(嚢胞および
ポリープを含む)
前立腺癌 1 (4.0%) 1 (4.2%)
血液およびリンパ系障害 貧血 1 (4.0%) 1 (4.2%) 1 (5.9%)
播種性血管内凝固 1 (4.0%) 1 (4.2%) 1 (5.9%)
内分泌障害 バセドウ病 1 (4.0%) 1 (4.2%)
甲状腺炎 1 (4.0%) 1 (4.2%) 1 (5.9%)
代謝および栄養障害 糖尿病 2 (8.0%) 2 (8.3%) 1 (5.9%)
高尿酸血症 1 (4.0%) 1 (4.2%) 1 (5.9%)
低カリウム血症 1 (4.0%) 1 (4.2%) 1 (5.9%)
脂質異常症 2 (8.0%) 2 (8.3%) 1 (5.9%)
高脂血症 2 (8.0%) 2 (8.3%) 2 (11.8%)
精神障害 不眠症 1 (4.0%) 1 (4.2%) 1 (5.9%)
神経系障害 低酸素性虚血性脳症 3 (12.0%) 3 (12.5%) 2 (11.8%)
眼障害 結膜出血 1 (4.0%) 1 (4.2%) 1 (5.9%)
ブドウ膜炎 1 (4.0%) 1 (4.2%) 1 (5.9%)
心臓障害 大動脈弁閉鎖不全症 1 (4.0%) 1 (4.2%) 1 (5.9%)
心房細動 5 (20.0%) 5 (20.8%) 5 (29.4%)
心不全 3 (12.0%) 3 (12.5%) 3 (17.6%)
慢性心不全 1 (4.0%) 1 (4.2%) 1 (5.9%)
心筋梗塞 1 (4.0%) 1 (4.2%) 1 (5.9%)
血管障害 高血圧 9 (36.0%) 9 (37.5%) 6 (35.3%)
鎖骨下動脈狭窄 1 (4.0%) 1 (4.2%) 1 (5.9%)
呼吸器、胸郭および縦隔 肺気腫 1 (4.0%) 1 (4.2%)
障害 鼻出血 1 (4.0%) 1 (4.2%) 1 (5.9%)
誤嚥性肺炎 7 (28.0%) 7 (29.2%) 5 (29.4%)
肺水腫 1 (4.0%) 1 (4.2%)
胃腸障害 腹部腫瘤 1 (4.0%) 1 (4.2%) 1 (5.9%)
胃腸障害 1 (4.0%) 1 (4.2%) 1 (5.9%)
肝胆道系障害 アルコール性肝疾患 1 (4.0%) 1 (4.2%) 1 (5.9%)
脂肪肝 1 (4.0%) 1 (4.2%) 1 (5.9%)
生殖系および乳房障害 良性前立腺肥大症 1 (4.0%) 1 (4.2%) 1 (5.9%)
傷害、中毒および処置合
併症 胸骨骨折 1 (4.0%) 1 (4.2%) 1 (5.9%)
外科および内科処置 大動脈弁置換 3 (12.0%) 3 (12.5%) 3 (17.6%)
僧帽弁置換 2 (8.0%) 2 (8.3%) 2 (11.8%)
大動脈吻合 1 (4.0%) 1 (4.2%) 1 (5.9%)

169
(2)心停止から治験機器による低体温療法までの状況
心停止・自己心拍再開の状況を表 4.3.1.4.2-1 に、低体温療法の状況を表 4.3.1.4.2-2 に、深部体温モ
ニタリングプローブ留置部位を表 4.3.1.4.2-3 に示す。
心停止の場所は、全員(25 人)が院外心停止だった。目撃者の有無は、目撃者あり 22 人(88.0%)、
目撃者なし 3 人(12.0%)、心停止の状態(初期波形)は心室細動 19 人(76.0%)、無脈性電気活動
6 人(24.0%)、心静止 0 人(0.0%)だった。
心停止から ROSC までの時間は 24.6±14.5 分で、全体の 56.0%が 20 分以内だった。
心停止から来院までの時間は 63.5±58.8 分(中央値 31.0分、範囲 15-195分)で、他院からの転院等
により来院までに 61 分以上要した被験者も 8 人(32.0%)いた。
来院時の GCS(トータルスコア)は 4.3±1.9 点で、最頻値は 3 点(15 人)だった。
心停止から冷却(治験機器による治療)開始までの時間は 287.6±75.3 分(中央値 273.5 分、範囲
133-413 分)で、120 分未満の被験者はいなかった。また、361 分以上要した被験者が 5 人(20.8%)
いた。
ROSC から冷却(治験機器による治療)開始までの時間は 262.3±73.5 分(中央値 248.5 分、範囲
101-365 分)だった。
治療に使用したカテーテルは IVTM-CI が 7 人(29.2%)、IVTM-CQ が 17 人(70.8%)だった。
治験機器による治療開始前の必須療法だった冷却輸液は、全員が冷電解質液を使用し、投与量は
20.2±6.6 mL/kg(中央値 20.5 mL/kg、範囲 10-30 mL/kg)だった。
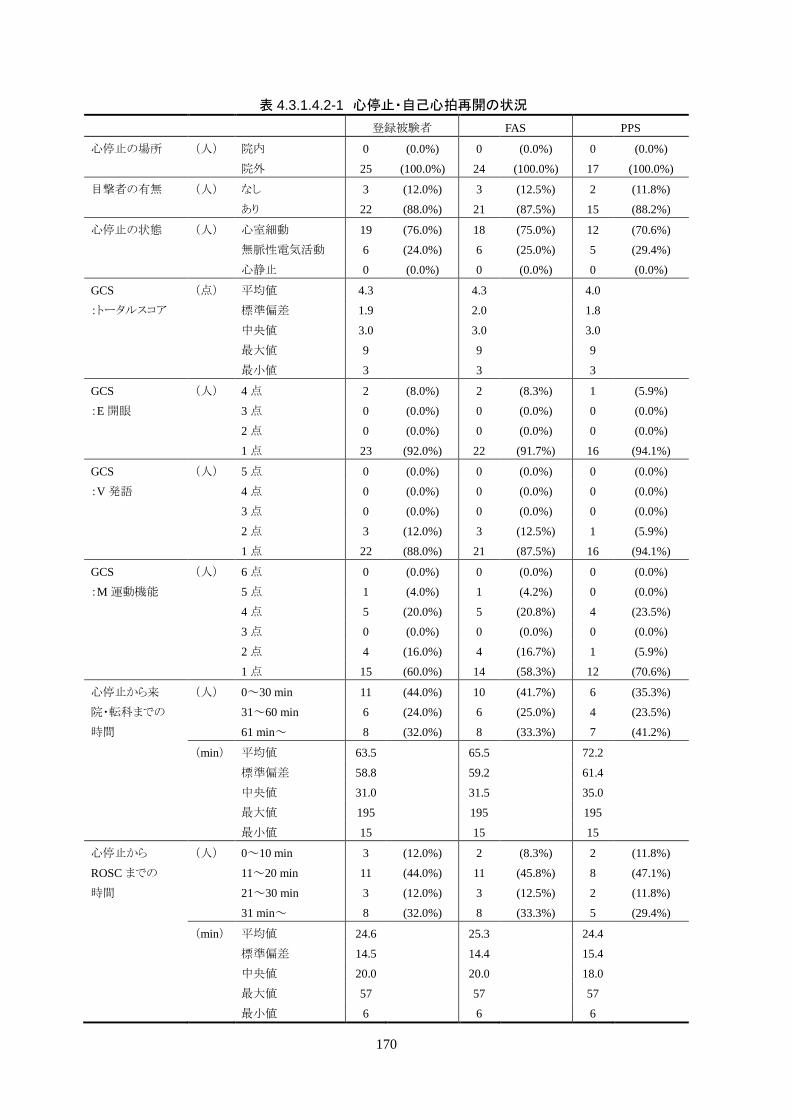
170
表 4.3.1.4.2-1 心停止・自己心拍再開の状況
登録被験者 FAS PPS
心停止の場所 (人) 院内 0 (0.0%) 0 (0.0%) 0 (0.0%)
院外 25 (100.0%) 24 (100.0%) 17 (100.0%)
目撃者の有無 (人) なし 3 (12.0%) 3 (12.5%) 2 (11.8%)
あり 22 (88.0%) 21 (87.5%) 15 (88.2%)
心停止の状態 (人) 心室細動 19 (76.0%) 18 (75.0%) 12 (70.6%)
無脈性電気活動 6 (24.0%) 6 (25.0%) 5 (29.4%)
心静止 0 (0.0%) 0 (0.0%) 0 (0.0%)
GCS (点) 平均値 4.3 4.3 4.0
:トータルスコア 標準偏差 1.9 2.0 1.8
中央値 3.0 3.0 3.0
最大値 9 9 9
最小値 3 3 3
GCS (人) 4 点 2 (8.0%) 2 (8.3%) 1 (5.9%)
:E 開眼 3 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
2 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
1 点 23 (92.0%) 22 (91.7%) 16 (94.1%)
GCS (人) 5 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
:V 発語 4 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
3 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
2 点 3 (12.0%) 3 (12.5%) 1 (5.9%)
1 点 22 (88.0%) 21 (87.5%) 16 (94.1%)
GCS (人) 6 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
:M 運動機能 5 点 1 (4.0%) 1 (4.2%) 0 (0.0%)
4 点 5 (20.0%) 5 (20.8%) 4 (23.5%)
3 点 0 (0.0%) 0 (0.0%) 0 (0.0%)
2 点 4 (16.0%) 4 (16.7%) 1 (5.9%)
1 点 15 (60.0%) 14 (58.3%) 12 (70.6%)
心停止から来 (人) 0~30 min 11 (44.0%) 10 (41.7%) 6 (35.3%)
院・転科までの 31~60 min 6 (24.0%) 6 (25.0%) 4 (23.5%)
時間 61 min~ 8 (32.0%) 8 (33.3%) 7 (41.2%)
(min) 平均値 63.5 65.5 72.2
標準偏差 58.8 59.2 61.4
中央値 31.0 31.5 35.0
最大値 195 195 195
最小値 15 15 15
心停止から (人) 0~10 min 3 (12.0%) 2 (8.3%) 2 (11.8%)
ROSC までの 11~20 min 11 (44.0%) 11 (45.8%) 8 (47.1%)
時間 21~30 min 3 (12.0%) 3 (12.5%) 2 (11.8%)
31 min~ 8 (32.0%) 8 (33.3%) 5 (29.4%)
(min) 平均値 24.6 25.3 24.4
標準偏差 14.5 14.4 15.4
中央値 20.0 20.0 18.0
最大値 57 57 57
最小値 6 6 6
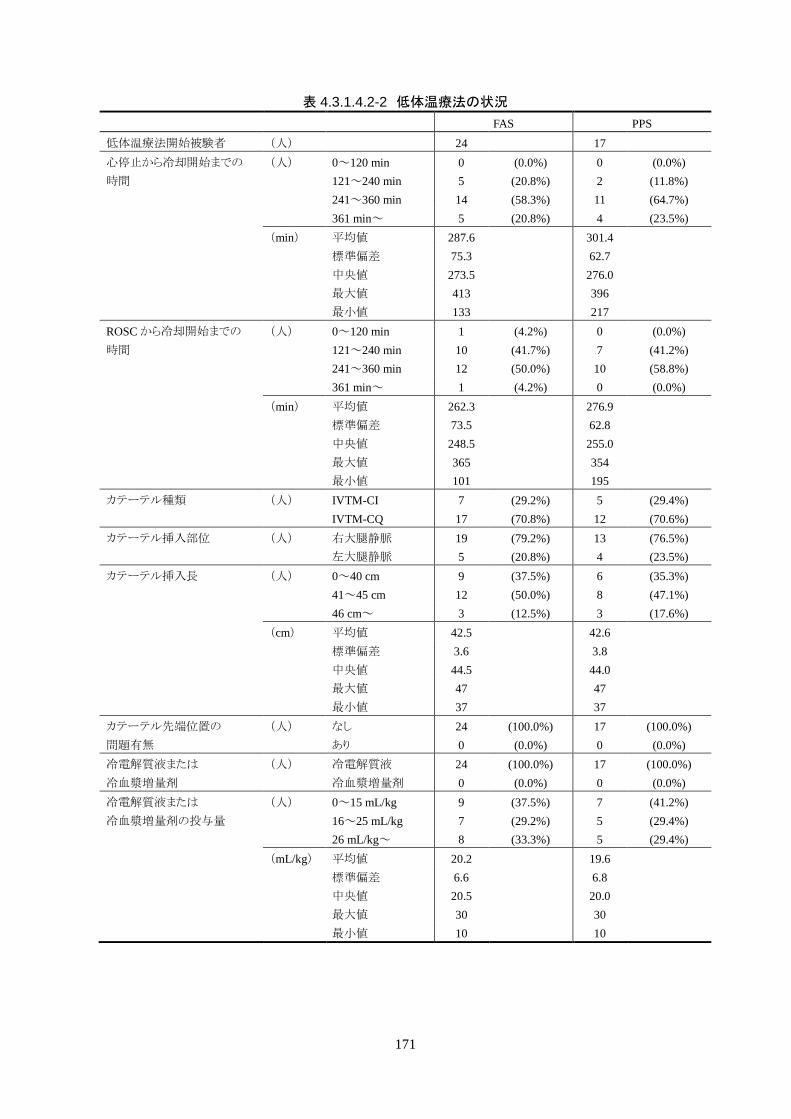
171
表 4.3.1.4.2-2 低体温療法の状況
FAS PPS
低体温療法開始被験者 (人) 24 17
心停止から冷却開始までの (人) 0~120 min 0 (0.0%) 0 (0.0%)
時間 121~240 min 5 (20.8%) 2 (11.8%)
241~360 min 14 (58.3%) 11 (64.7%)
361 min~ 5 (20.8%) 4 (23.5%)
(min) 平均値 287.6 301.4
標準偏差 75.3 62.7
中央値 273.5 276.0
最大値 413 396
最小値 133 217
ROSCから冷却開始までの (人) 0~120 min 1 (4.2%) 0 (0.0%)
時間 121~240 min 10 (41.7%) 7 (41.2%)
241~360 min 12 (50.0%) 10 (58.8%)
361 min~ 1 (4.2%) 0 (0.0%)
(min) 平均値 262.3 276.9
標準偏差 73.5 62.8
中央値 248.5 255.0
最大値 365 354
最小値 101 195
カテーテル種類 (人) IVTM-CI 7 (29.2%) 5 (29.4%)
IVTM-CQ 17 (70.8%) 12 (70.6%)
カテーテル挿入部位 (人) 右大腿静脈 19 (79.2%) 13 (76.5%)
左大腿静脈 5 (20.8%) 4 (23.5%)
カテーテル挿入長 (人) 0~40 cm 9 (37.5%) 6 (35.3%)
41~45 cm 12 (50.0%) 8 (47.1%)
46 cm~ 3 (12.5%) 3 (17.6%)
(cm) 平均値 42.5 42.6
標準偏差 3.6 3.8
中央値 44.5 44.0
最大値 47 47
最小値 37 37
カテーテル先端位置の (人) なし 24 (100.0%) 17 (100.0%)
問題有無 あり 0 (0.0%) 0 (0.0%)
冷電解質液または (人) 冷電解質液 24 (100.0%) 17 (100.0%)
冷血漿増量剤 冷血漿増量剤 0 (0.0%) 0 (0.0%)
冷電解質液または (人) 0~15 mL/kg 9 (37.5%) 7 (41.2%)
冷血漿増量剤の投与量 16~25 mL/kg 7 (29.2%) 5 (29.4%)
26 mL/kg~ 8 (33.3%) 5 (29.4%)
(mL/kg) 平均値 20.2 19.6
標準偏差 6.6 6.8
中央値 20.5 20.0
最大値 30 30
最小値 10 10
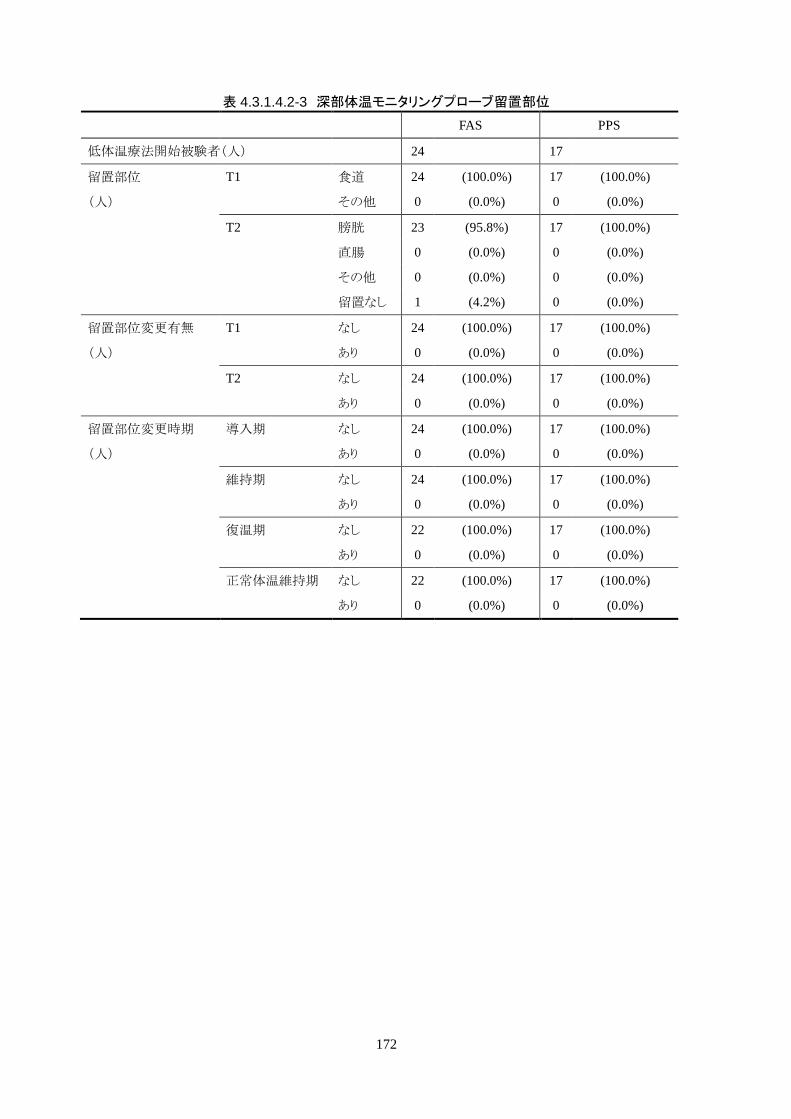
172
表 4.3.1.4.2-3 深部体温モニタリングプローブ留置部位
FAS PPS
低体温療法開始被験者(人) 24 17
留置部位 T1 食道 24 (100.0%) 17 (100.0%)
(人) その他 0 (0.0%) 0 (0.0%)
T2 膀胱 23 (95.8%) 17 (100.0%)
直腸 0 (0.0%) 0 (0.0%)
その他 0 (0.0%) 0 (0.0%)
留置なし 1 (4.2%) 0 (0.0%)
留置部位変更有無 T1 なし 24 (100.0%) 17 (100.0%)
(人) あり 0 (0.0%) 0 (0.0%)
T2 なし 24 (100.0%) 17 (100.0%)
あり 0 (0.0%) 0 (0.0%)
留置部位変更時期 導入期 なし 24 (100.0%) 17 (100.0%)
(人) あり 0 (0.0%) 0 (0.0%)
維持期 なし 24 (100.0%) 17 (100.0%)
あり 0 (0.0%) 0 (0.0%)
復温期 なし 22 (100.0%) 17 (100.0%)
あり 0 (0.0%) 0 (0.0%)
正常体温維持期 なし 22 (100.0%) 17 (100.0%)
あり 0 (0.0%) 0 (0.0%)

173
4.3.1.5 臨床試験結果
(1)有効性の解析結果
① 主要評価項目の解析
治験機器による冷却開始 3 時間以内に目標体温に到達した被験者の割合を表 4.3.1.5.1-1 に、治験機
器による冷却開始から目標体温到達までに要した時間を表 4.3.1.5.1-2 に示す。
本治験の主要評価項目である 3 時間以内の目標達成率は 100%であり、治験機器による治療を開始
した被験者全員が 3 時間以内に目標体温に到達した。目標達成率の 95%信頼区間は 85.8-100%だ
った。
冷却開始時の深部体温は 36.20±1.11℃で、冷却開始から目標体温到達までに要した時間は、54.63
±37.46 分(中央値 45.00 分、IQR 33.0-73.5 分)であった。24 人中 16 人(66.7%)が治療開始から
60 分以内に目標体温に到達した。
導入期の深部体温と目標体温到達被験者の割合の推移(FAS および PPS)を表 4.3.1.5.1-3 および表
4.3.1.5.1-4 に、心停止から目標体温到達までに要した時間を表 4.3.1.5.1-5 に、導入期の体温(深部
温)の推移(FAS および PPS)を図 4.3.1.5.1-6 および図 4.3.1.5.1-7 に、目標体温到達被験者の割合
の推移(導入期)(FAS および PPS)を図 4.3.1.5.1-8 および図 4.3.1.5.1-9 に示す。
導入期の深部体温の目標到達被験者の割合は、治療開始 30 分後が 25.0%、60 分後が 66.7%、90 分
後が 87.5%、120 分後が 95.8%だった。
心停止から目標体温到達までに要した時間は、342.25±82.05 分(中央値 341.50 分、IQR 288.0-415.5
分)であった。
表 4.3.1.5.1-1 治験機器による冷却開始 3時間以内に目標体温に到達した被験者の割合
FAS PPS
低体温療法開始被験者(人) 24 17
3 時間以内に目標体温に到達した被験者(人) 24 17
割合(%) 100.0% 100.0%
95%信頼区間上限 100.0% 100.0%
95%信頼区間下限 85.8% 80.5%
※ 95%信頼区間は、F分布を用いた正確な出現率の信頼区間計算法により算出した。
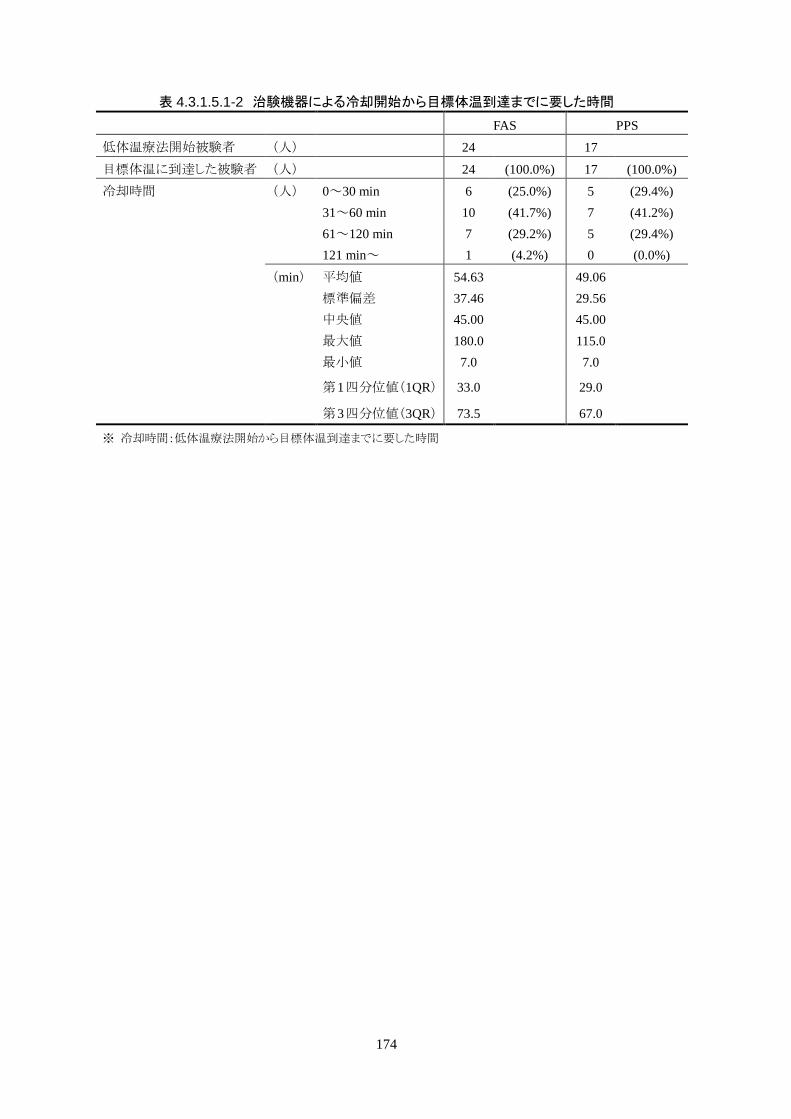
174
表 4.3.1.5.1-2 治験機器による冷却開始から目標体温到達までに要した時間
FAS PPS
低体温療法開始被験者 (人) 24 17
目標体温に到達した被験者 (人) 24 (100.0%) 17 (100.0%)
冷却時間 (人) 0~30 min 6 (25.0%) 5 (29.4%)
31~60 min 10 (41.7%) 7 (41.2%)
61~120 min 7 (29.2%) 5 (29.4%)
121 min~ 1 (4.2%) 0 (0.0%)
(min) 平均値 54.63 49.06
標準偏差 37.46 29.56
中央値 45.00 45.00
最大値 180.0 115.0
最小値 7.0 7.0
第1四分位値(1QR) 33.0 29.0
第3四分位値(3QR) 73.5 67.0
※ 冷却時間:低体温療法開始から目標体温到達までに要した時間
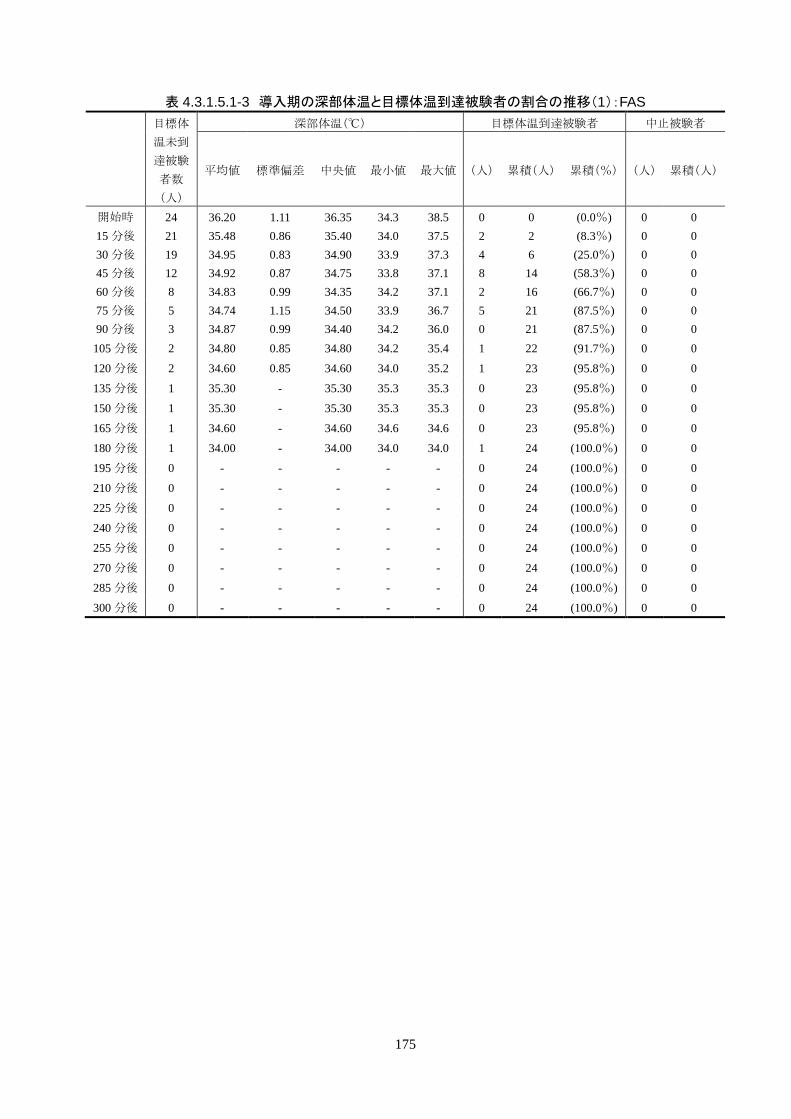
175
表 4.3.1.5.1-3 導入期の深部体温と目標体温到達被験者の割合の推移(1):FAS
目標体
温未到
達被験
者数
(人)
深部体温(℃) 目標体温到達被験者 中止被験者
平均値 標準偏差 中央値 最小値 最大値 (人) 累積(人) 累積(%) (人) 累積(人)
開始時 24 36.20 1.11 36.35 34.3 38.5 0 0 (0.0%) 0 0
15 分後 21 35.48 0.86 35.40 34.0 37.5 2 2 (8.3%) 0 0
30 分後 19 34.95 0.83 34.90 33.9 37.3 4 6 (25.0%) 0 0
45 分後 12 34.92 0.87 34.75 33.8 37.1 8 14 (58.3%) 0 0
60 分後 8 34.83 0.99 34.35 34.2 37.1 2 16 (66.7%) 0 0
75 分後 5 34.74 1.15 34.50 33.9 36.7 5 21 (87.5%) 0 0
90 分後 3 34.87 0.99 34.40 34.2 36.0 0 21 (87.5%) 0 0
105 分後 2 34.80 0.85 34.80 34.2 35.4 1 22 (91.7%) 0 0
120 分後 2 34.60 0.85 34.60 34.0 35.2 1 23 (95.8%) 0 0
135 分後 1 35.30 - 35.30 35.3 35.3 0 23 (95.8%) 0 0
150 分後 1 35.30 - 35.30 35.3 35.3 0 23 (95.8%) 0 0
165 分後 1 34.60 - 34.60 34.6 34.6 0 23 (95.8%) 0 0
180 分後 1 34.00 - 34.00 34.0 34.0 1 24 (100.0%) 0 0
195 分後 0 - - - - - 0 24 (100.0%) 0 0
210 分後 0 - - - - - 0 24 (100.0%) 0 0
225 分後 0 - - - - - 0 24 (100.0%) 0 0
240 分後 0 - - - - - 0 24 (100.0%) 0 0
255 分後 0 - - - - - 0 24 (100.0%) 0 0
270 分後 0 - - - - - 0 24 (100.0%) 0 0
285 分後 0 - - - - - 0 24 (100.0%) 0 0
300 分後 0 - - - - - 0 24 (100.0%) 0 0
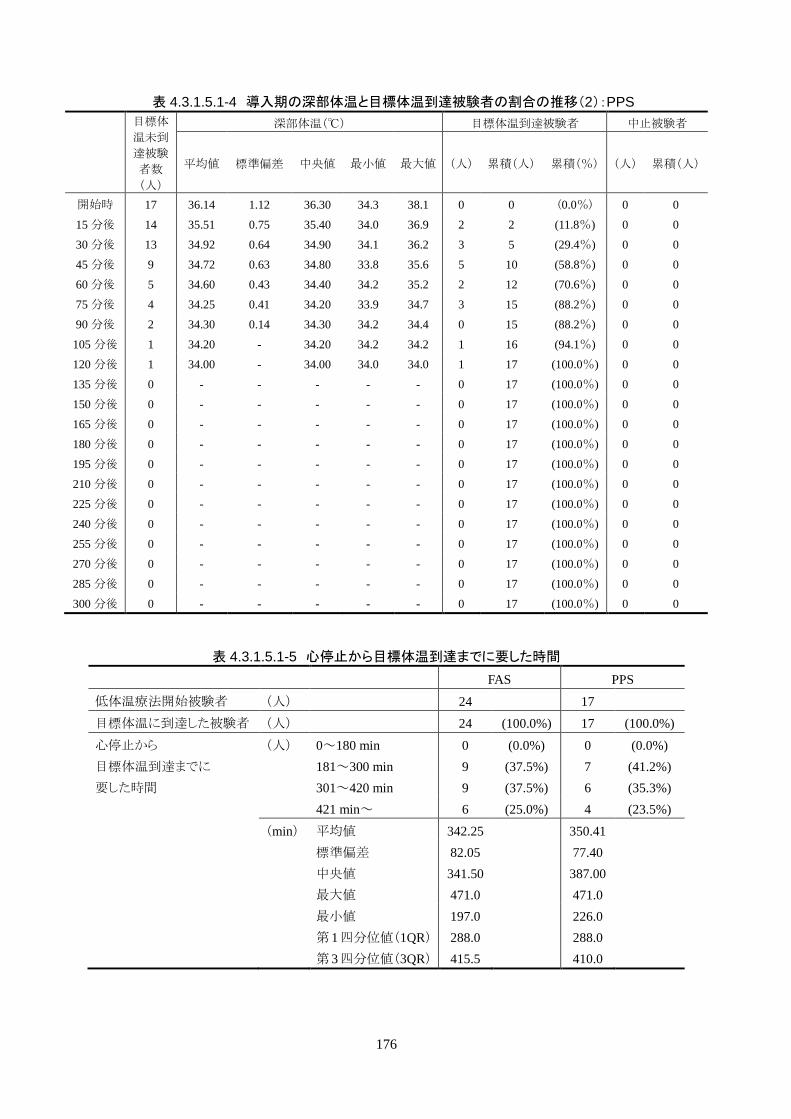
176
表 4.3.1.5.1-4 導入期の深部体温と目標体温到達被験者の割合の推移(2):PPS
目標体
温未到
達被験
者数
(人)
深部体温(℃) 目標体温到達被験者 中止被験者
平均値 標準偏差 中央値 最小値 最大値 (人) 累積(人) 累積(%) (人) 累積(人)
開始時 17 36.14 1.12 36.30 34.3 38.1 0 0 (0.0%) 0 0
15 分後 14 35.51 0.75 35.40 34.0 36.9 2 2 (11.8%) 0 0
30 分後 13 34.92 0.64 34.90 34.1 36.2 3 5 (29.4%) 0 0
45 分後 9 34.72 0.63 34.80 33.8 35.6 5 10 (58.8%) 0 0
60 分後 5 34.60 0.43 34.40 34.2 35.2 2 12 (70.6%) 0 0
75 分後 4 34.25 0.41 34.20 33.9 34.7 3 15 (88.2%) 0 0
90 分後 2 34.30 0.14 34.30 34.2 34.4 0 15 (88.2%) 0 0
105 分後 1 34.20 - 34.20 34.2 34.2 1 16 (94.1%) 0 0
120 分後 1 34.00 - 34.00 34.0 34.0 1 17 (100.0%) 0 0
135 分後 0 - - - - - 0 17 (100.0%) 0 0
150 分後 0 - - - - - 0 17 (100.0%) 0 0
165 分後 0 - - - - - 0 17 (100.0%) 0 0
180 分後 0 - - - - - 0 17 (100.0%) 0 0
195 分後 0 - - - - - 0 17 (100.0%) 0 0
210 分後 0 - - - - - 0 17 (100.0%) 0 0
225 分後 0 - - - - - 0 17 (100.0%) 0 0
240 分後 0 - - - - - 0 17 (100.0%) 0 0
255 分後 0 - - - - - 0 17 (100.0%) 0 0
270 分後 0 - - - - - 0 17 (100.0%) 0 0
285 分後 0 - - - - - 0 17 (100.0%) 0 0
300 分後 0 - - - - - 0 17 (100.0%) 0 0
表 4.3.1.5.1-5 心停止から目標体温到達までに要した時間
FAS PPS
低体温療法開始被験者 (人) 24 17
目標体温に到達した被験者 (人) 24 (100.0%) 17 (100.0%)
心停止から (人) 0~180 min 0 (0.0%) 0 (0.0%)
目標体温到達までに 181~300 min 9 (37.5%) 7 (41.2%)
要した時間 301~420 min 9 (37.5%) 6 (35.3%)
421 min~ 6 (25.0%) 4 (23.5%)
(min) 平均値 342.25 350.41
標準偏差 82.05 77.40
中央値 341.50 387.00
最大値 471.0 471.0
最小値 197.0 226.0
第 1四分位値(1QR) 288.0 288.0
第 3四分位値(3QR) 415.5 410.0
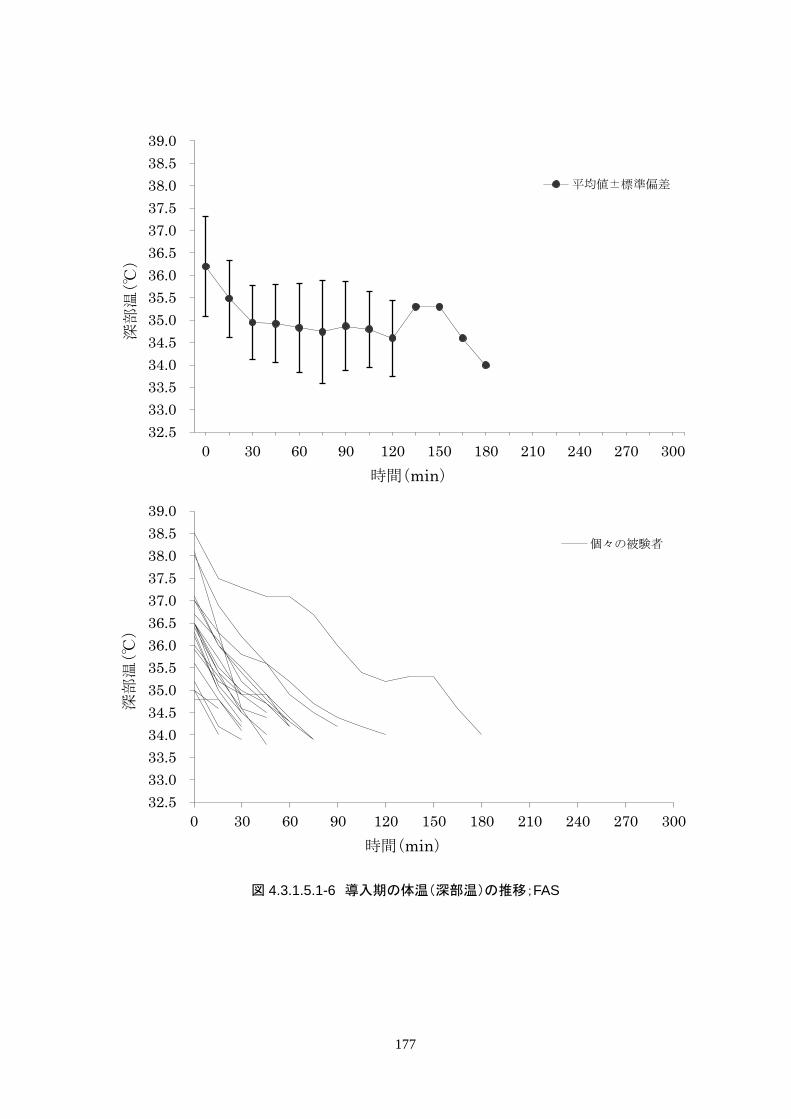
177
図 4.3.1.5.1-6 導入期の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
個々の被験者
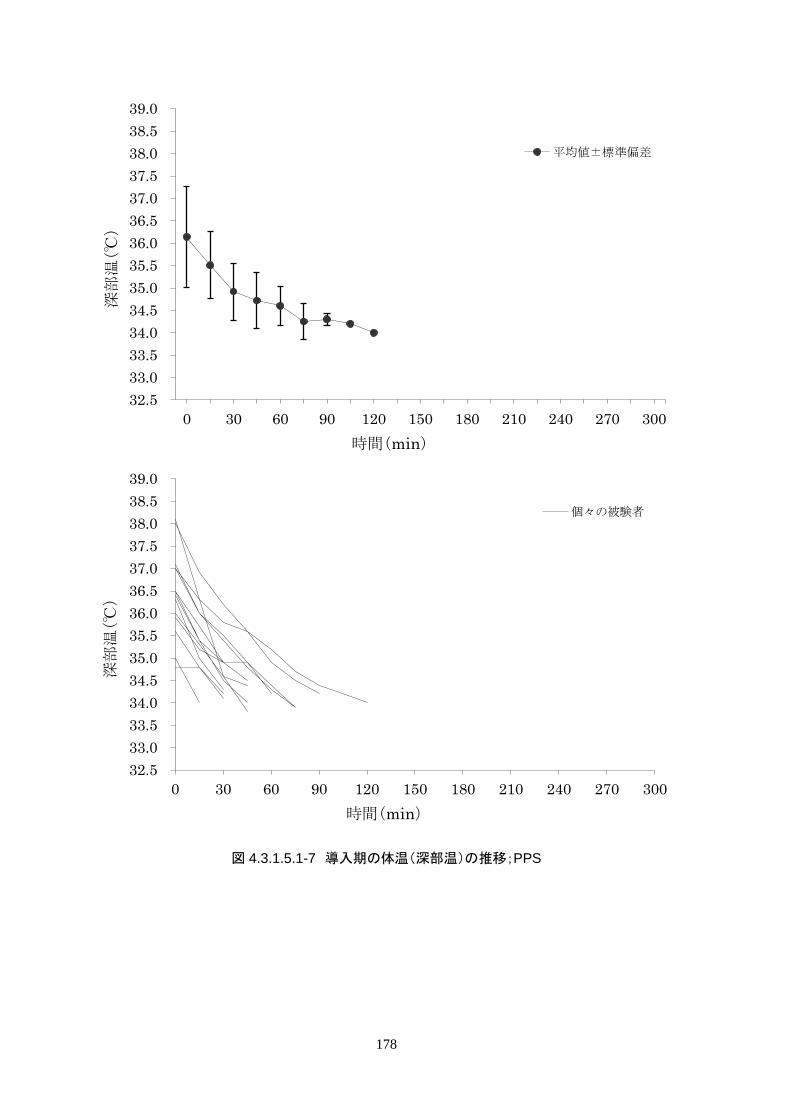
178
図 4.3.1.5.1-7 導入期の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
個々の被験者
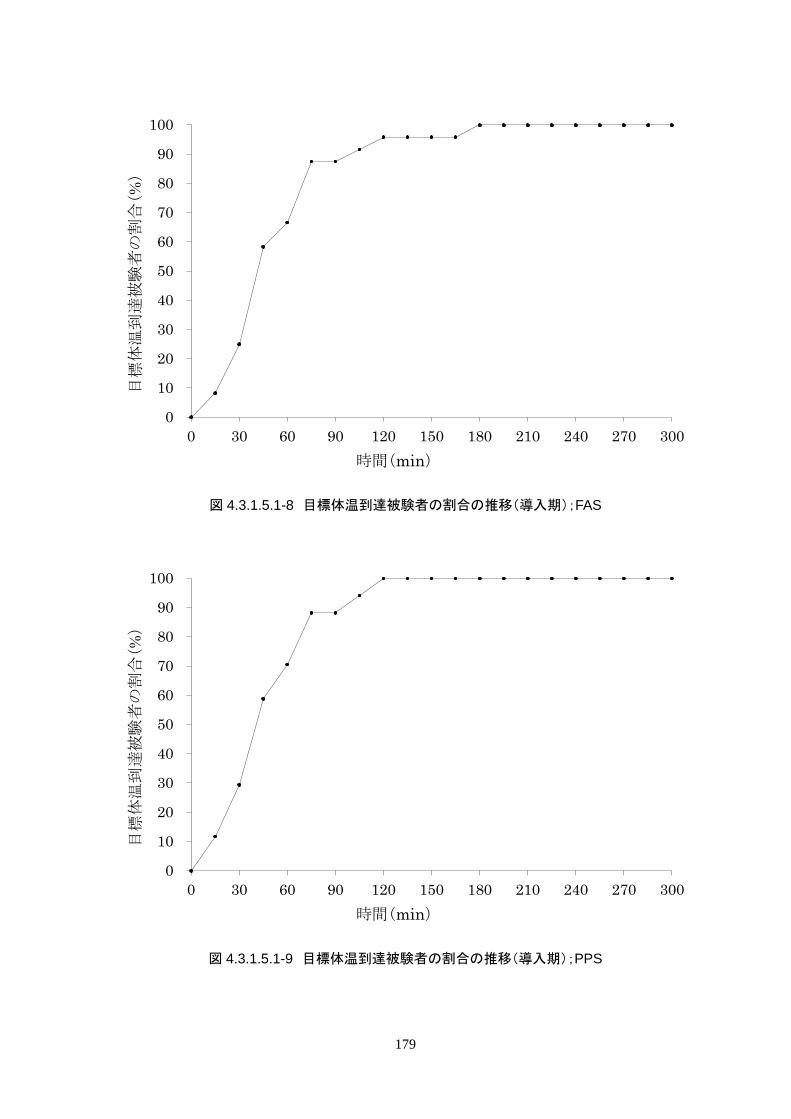
179
図 4.3.1.5.1-8 目標体温到達被験者の割合の推移(導入期);FAS
図 4.3.1.5.1-9 目標体温到達被験者の割合の推移(導入期);PPS
0
10
20
30
40
50
60
70
80
90
100
0 30 60 90 120 150 180 210 240 270 300
目標体温到達被験者の割合(%)
時間(min)
0
10
20
30
40
50
60
70
80
90
100
0 30 60 90 120 150 180 210 240 270 300
目標体温到達被験者の割合(%)
時間(min)
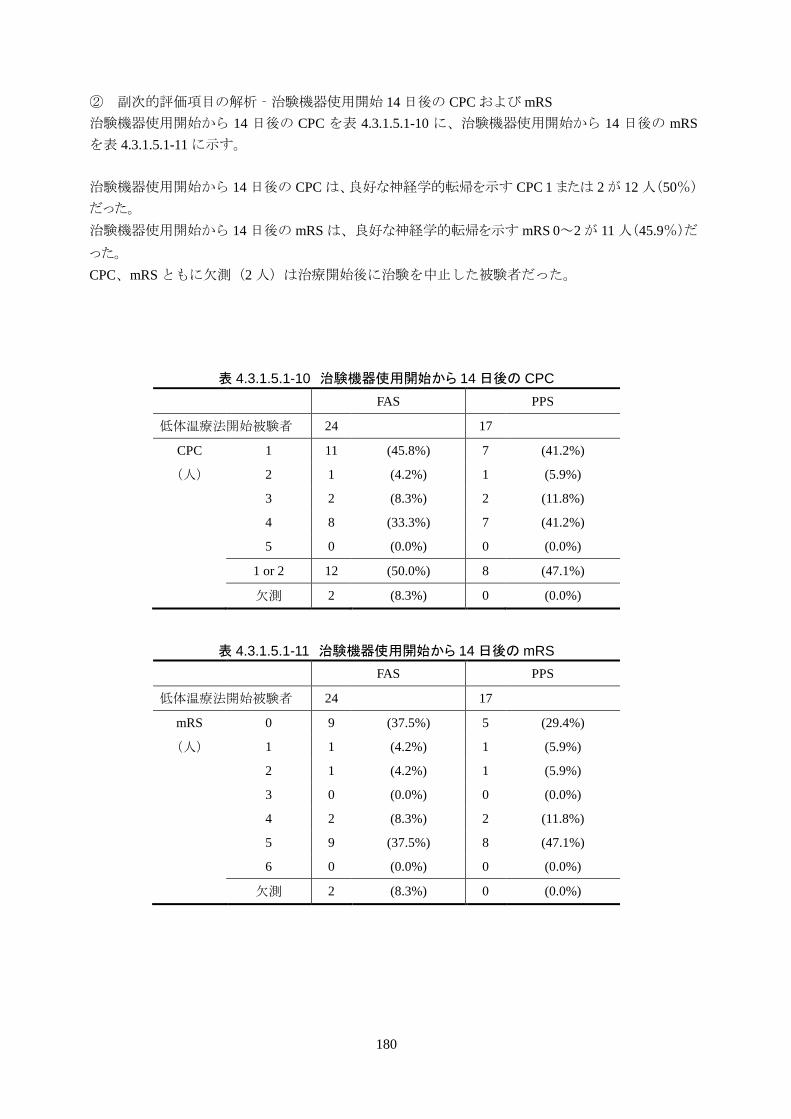
180
② 副次的評価項目の解析‐治験機器使用開始 14 日後の CPC および mRS
治験機器使用開始から 14 日後の CPC を表 4.3.1.5.1-10 に、治験機器使用開始から 14 日後の mRS
を表 4.3.1.5.1-11 に示す。
治験機器使用開始から 14 日後の CPC は、良好な神経学的転帰を示す CPC 1または 2が 12人(50%)
だった。
治験機器使用開始から 14 日後の mRS は、良好な神経学的転帰を示す mRS 0~2が 11人(45.9%)だ
った。
CPC、mRS ともに欠測(2 人)は治療開始後に治験を中止した被験者だった。
表 4.3.1.5.1-10 治験機器使用開始から 14日後の CPC
FAS PPS
低体温療法開始被験者 24 17
CPC 1 11 (45.8%) 7 (41.2%)
(人) 2 1 (4.2%) 1 (5.9%)
3 2 (8.3%) 2 (11.8%)
4 8 (33.3%) 7 (41.2%)
5 0 (0.0%) 0 (0.0%)
1 or 2 12 (50.0%) 8 (47.1%)
欠測 2 (8.3%) 0 (0.0%)
表 4.3.1.5.1-11 治験機器使用開始から 14日後のmRS
FAS PPS
低体温療法開始被験者 24 17
mRS 0 9 (37.5%) 5 (29.4%)
(人) 1 1 (4.2%) 1 (5.9%)
2 1 (4.2%) 1 (5.9%)
3 0 (0.0%) 0 (0.0%)
4 2 (8.3%) 2 (11.8%)
5 9 (37.5%) 8 (47.1%)
6 0 (0.0%) 0 (0.0%)
欠測 2 (8.3%) 0 (0.0%)
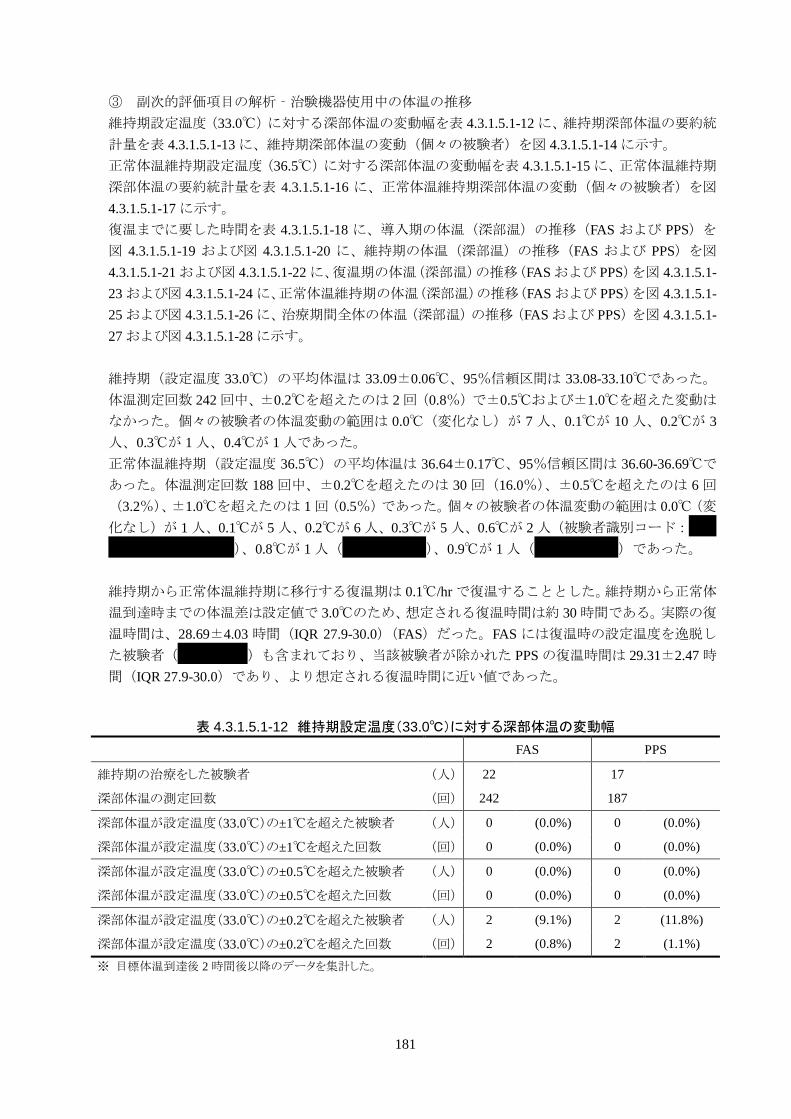
181
③ 副次的評価項目の解析‐治験機器使用中の体温の推移
維持期設定温度(33.0℃)に対する深部体温の変動幅を表 4.3.1.5.1-12 に、維持期深部体温の要約統
計量を表 4.3.1.5.1-13 に、維持期深部体温の変動(個々の被験者)を図 4.3.1.5.1-14 に示す。
正常体温維持期設定温度(36.5℃)に対する深部体温の変動幅を表 4.3.1.5.1-15 に、正常体温維持期
深部体温の要約統計量を表 4.3.1.5.1-16 に、正常体温維持期深部体温の変動(個々の被験者)を図
4.3.1.5.1-17 に示す。
復温までに要した時間を表 4.3.1.5.1-18 に、導入期の体温(深部温)の推移(FAS および PPS)を
図 4.3.1.5.1-19 および図 4.3.1.5.1-20 に、維持期の体温(深部温)の推移(FAS および PPS)を図
4.3.1.5.1-21 および図 4.3.1.5.1-22 に、復温期の体温(深部温)の推移(FAS および PPS)を図 4.3.1.5.1-
23 および図 4.3.1.5.1-24 に、正常体温維持期の体温(深部温)の推移(FAS および PPS)を図 4.3.1.5.1-
25 および図 4.3.1.5.1-26 に、治療期間全体の体温(深部温)の推移(FAS および PPS)を図 4.3.1.5.1-
27 および図 4.3.1.5.1-28 に示す。
維持期(設定温度 33.0℃)の平均体温は 33.09±0.06℃、95%信頼区間は 33.08-33.10℃であった。
体温測定回数 242 回中、±0.2℃を超えたのは 2 回(0.8%)で±0.5℃および±1.0℃を超えた変動は
なかった。個々の被験者の体温変動の範囲は 0.0℃(変化なし)が 7 人、0.1℃が 10 人、0.2℃が 3
人、0.3℃が 1 人、0.4℃が 1 人であった。
正常体温維持期(設定温度 36.5℃)の平均体温は 36.64±0.17℃、95%信頼区間は 36.60-36.69℃で
あった。体温測定回数 188 回中、±0.2℃を超えたのは 30 回(16.0%)、±0.5℃を超えたのは 6 回
(3.2%)、±1.0℃を超えたのは 1 回(0.5%)であった。個々の被験者の体温変動の範囲は 0.0℃(変
化なし)が 1 人、0.1℃が 5 人、0.2℃が 6 人、0.3℃が 5 人、0.6℃が 2 人(被験者識別コード:**
*********)、0.8℃が 1 人(******)、0.9℃が 1 人(******)であった。
維持期から正常体温維持期に移行する復温期は 0.1℃/hr で復温することとした。維持期から正常体
温到達時までの体温差は設定値で 3.0℃のため、想定される復温時間は約 30 時間である。実際の復
温時間は、28.69±4.03 時間(IQR 27.9-30.0)(FAS)だった。FAS には復温時の設定温度を逸脱し
た被験者(*****)も含まれており、当該被験者が除かれた PPS の復温時間は 29.31±2.47 時
間(IQR 27.9-30.0)であり、より想定される復温時間に近い値であった。
表 4.3.1.5.1-12 維持期設定温度(33.0℃)に対する深部体温の変動幅
FAS PPS
維持期の治療をした被験者 (人) 22 17
深部体温の測定回数 (回) 242 187
深部体温が設定温度(33.0℃)の±1℃を超えた被験者 (人) 0 (0.0%) 0 (0.0%)
深部体温が設定温度(33.0℃)の±1℃を超えた回数 (回) 0 (0.0%) 0 (0.0%)
深部体温が設定温度(33.0℃)の±0.5℃を超えた被験者 (人) 0 (0.0%) 0 (0.0%)
深部体温が設定温度(33.0℃)の±0.5℃を超えた回数 (回) 0 (0.0%) 0 (0.0%)
深部体温が設定温度(33.0℃)の±0.2℃を超えた被験者 (人) 2 (9.1%) 2 (11.8%)
深部体温が設定温度(33.0℃)の±0.2℃を超えた回数 (回) 2 (0.8%) 2 (1.1%)
※ 目標体温到達後 2 時間後以降のデータを集計した。
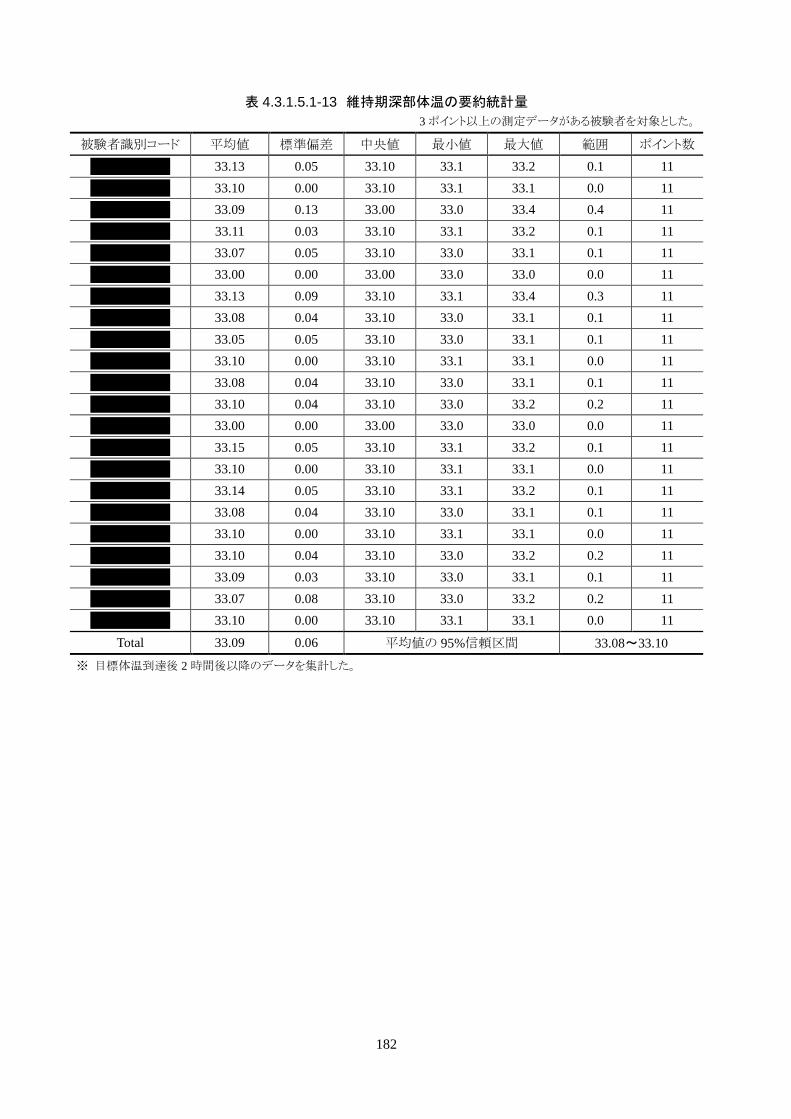
182
表 4.3.1.5.1-13 維持期深部体温の要約統計量
3 ポイント以上の測定データがある被験者を対象とした。
被験者識別コード 平均値 標準偏差 中央値 最小値 最大値 範囲 ポイント数
****** 33.13 0.05 33.10 33.1 33.2 0.1 11
****** 33.10 0.00 33.10 33.1 33.1 0.0 11
****** 33.09 0.13 33.00 33.0 33.4 0.4 11
****** 33.11 0.03 33.10 33.1 33.2 0.1 11
****** 33.07 0.05 33.10 33.0 33.1 0.1 11
****** 33.00 0.00 33.00 33.0 33.0 0.0 11
****** 33.13 0.09 33.10 33.1 33.4 0.3 11
****** 33.08 0.04 33.10 33.0 33.1 0.1 11
****** 33.05 0.05 33.10 33.0 33.1 0.1 11
****** 33.10 0.00 33.10 33.1 33.1 0.0 11
****** 33.08 0.04 33.10 33.0 33.1 0.1 11
****** 33.10 0.04 33.10 33.0 33.2 0.2 11
****** 33.00 0.00 33.00 33.0 33.0 0.0 11
****** 33.15 0.05 33.10 33.1 33.2 0.1 11
****** 33.10 0.00 33.10 33.1 33.1 0.0 11
****** 33.14 0.05 33.10 33.1 33.2 0.1 11
****** 33.08 0.04 33.10 33.0 33.1 0.1 11
****** 33.10 0.00 33.10 33.1 33.1 0.0 11
****** 33.10 0.04 33.10 33.0 33.2 0.2 11
****** 33.09 0.03 33.10 33.0 33.1 0.1 11
****** 33.07 0.08 33.10 33.0 33.2 0.2 11
****** 33.10 0.00 33.10 33.1 33.1 0.0 11
Total 33.09 0.06 平均値の 95%信頼区間 33.08~33.10
※ 目標体温到達後 2 時間後以降のデータを集計した。
183
※ 目標体温到達後 2 時間後以降のデータを集計した。
図 4.3.1.5.1-14維持期深部体温の変動(個々の被験者)
32.0
32.5
33.0
33.5
34.0
34.5
深部体温(℃)
平均値±標準偏差
To
tal
32.0
32.5
33.0
33.5
34.0
34.5
深部体温(℃)
中央値,最小値,最大値
To
tal
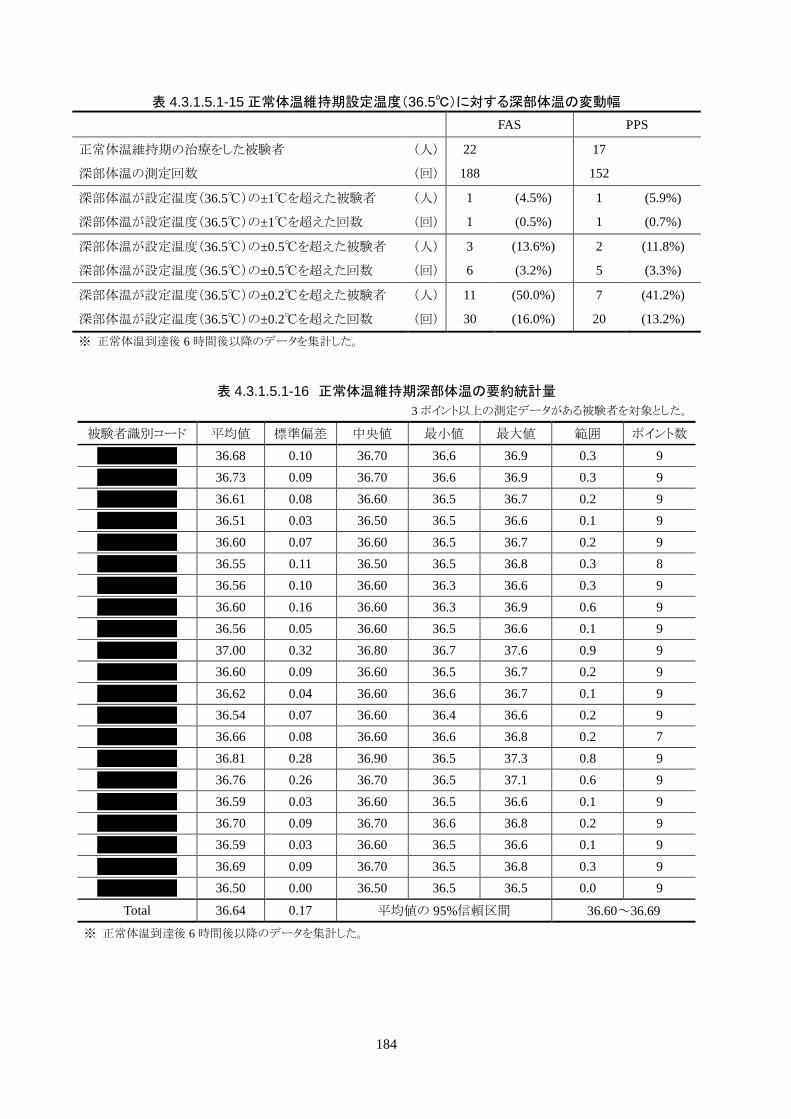
184
表 4.3.1.5.1-15正常体温維持期設定温度(36.5℃)に対する深部体温の変動幅
FAS PPS
正常体温維持期の治療をした被験者 (人) 22 17
深部体温の測定回数 (回) 188 152
深部体温が設定温度(36.5℃)の±1℃を超えた被験者 (人) 1 (4.5%) 1 (5.9%)
深部体温が設定温度(36.5℃)の±1℃を超えた回数 (回) 1 (0.5%) 1 (0.7%)
深部体温が設定温度(36.5℃)の±0.5℃を超えた被験者 (人) 3 (13.6%) 2 (11.8%)
深部体温が設定温度(36.5℃)の±0.5℃を超えた回数 (回) 6 (3.2%) 5 (3.3%)
深部体温が設定温度(36.5℃)の±0.2℃を超えた被験者 (人) 11 (50.0%) 7 (41.2%)
深部体温が設定温度(36.5℃)の±0.2℃を超えた回数 (回) 30 (16.0%) 20 (13.2%)
※ 正常体温到達後 6 時間後以降のデータを集計した。
表 4.3.1.5.1-16 正常体温維持期深部体温の要約統計量
3 ポイント以上の測定データがある被験者を対象とした。
被験者識別コード 平均値 標準偏差 中央値 最小値 最大値 範囲 ポイント数
****** 36.68 0.10 36.70 36.6 36.9 0.3 9
****** 36.73 0.09 36.70 36.6 36.9 0.3 9
****** 36.61 0.08 36.60 36.5 36.7 0.2 9
****** 36.51 0.03 36.50 36.5 36.6 0.1 9
****** 36.60 0.07 36.60 36.5 36.7 0.2 9
****** 36.55 0.11 36.50 36.5 36.8 0.3 8
****** 36.56 0.10 36.60 36.3 36.6 0.3 9
****** 36.60 0.16 36.60 36.3 36.9 0.6 9
****** 36.56 0.05 36.60 36.5 36.6 0.1 9
****** 37.00 0.32 36.80 36.7 37.6 0.9 9
****** 36.60 0.09 36.60 36.5 36.7 0.2 9
****** 36.62 0.04 36.60 36.6 36.7 0.1 9
****** 36.54 0.07 36.60 36.4 36.6 0.2 9
****** 36.66 0.08 36.60 36.6 36.8 0.2 7
****** 36.81 0.28 36.90 36.5 37.3 0.8 9
****** 36.76 0.26 36.70 36.5 37.1 0.6 9
****** 36.59 0.03 36.60 36.5 36.6 0.1 9
****** 36.70 0.09 36.70 36.6 36.8 0.2 9
****** 36.59 0.03 36.60 36.5 36.6 0.1 9
****** 36.69 0.09 36.70 36.5 36.8 0.3 9
****** 36.50 0.00 36.50 36.5 36.5 0.0 9
Total 36.64 0.17 平均値の 95%信頼区間 36.60~36.69
※ 正常体温到達後 6 時間後以降のデータを集計した。
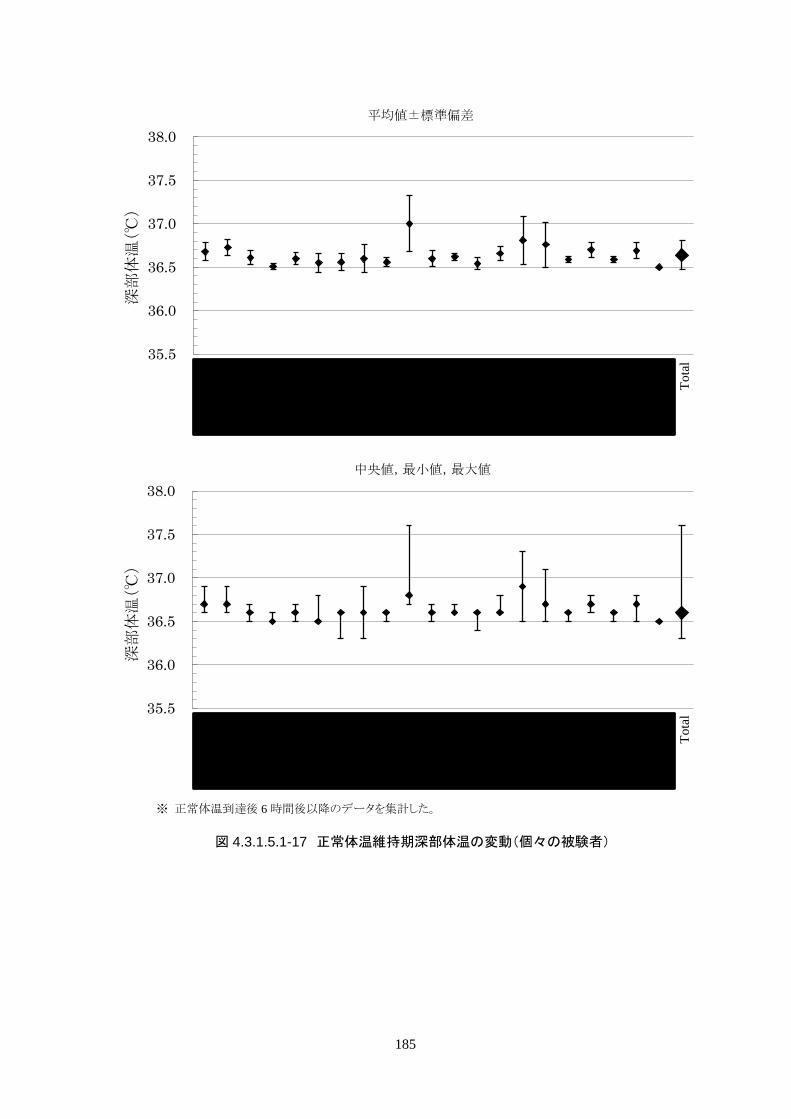
185
※ 正常体温到達後 6 時間後以降のデータを集計した。
図 4.3.1.5.1-17 正常体温維持期深部体温の変動(個々の被験者)
35.5
36.0
36.5
37.0
37.5
38.0深部体温(℃)
平均値±標準偏差
To
tal
35.5
36.0
36.5
37.0
37.5
38.0
深部体温(℃)
中央値,最小値,最大値
To
tal
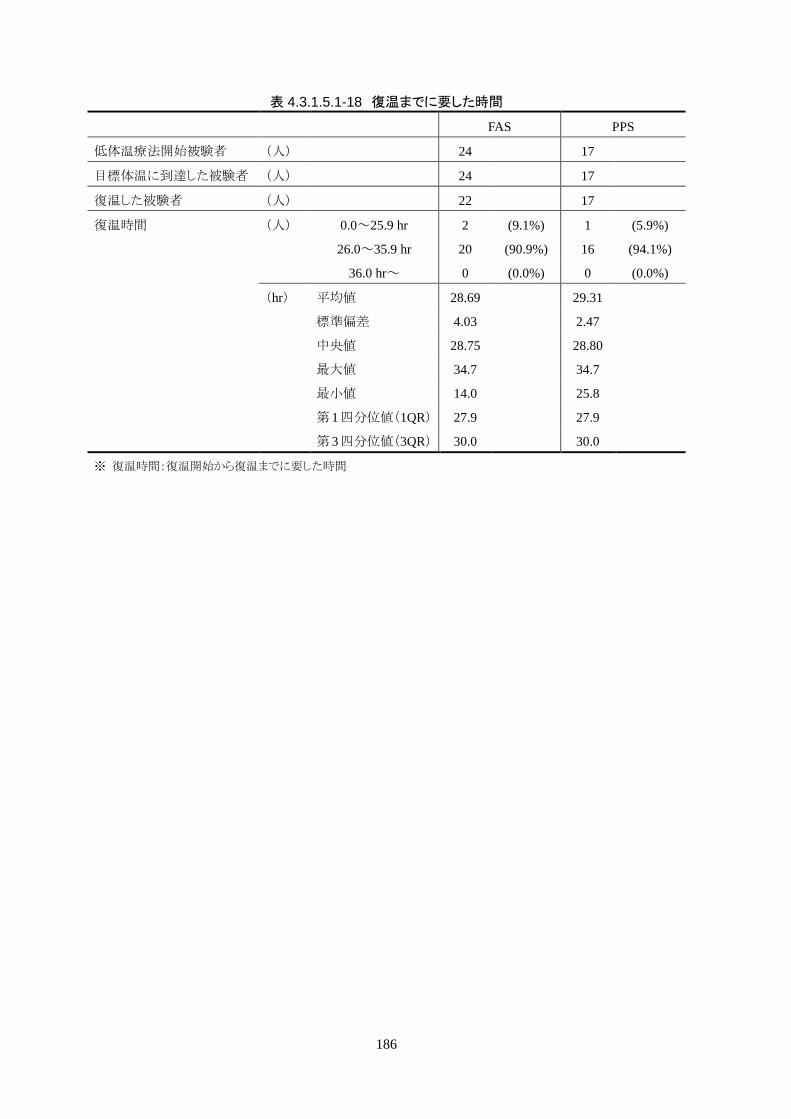
186
表 4.3.1.5.1-18 復温までに要した時間
FAS PPS
低体温療法開始被験者 (人) 24 17
目標体温に到達した被験者 (人) 24 17
復温した被験者 (人) 22 17
復温時間 (人) 0.0~25.9 hr 2 (9.1%) 1 (5.9%)
26.0~35.9 hr 20 (90.9%) 16 (94.1%)
36.0 hr~ 0 (0.0%) 0 (0.0%)
(hr) 平均値 28.69 29.31
標準偏差 4.03 2.47
中央値 28.75 28.80
最大値 34.7 34.7
最小値 14.0 25.8
第 1四分位値(1QR) 27.9 27.9
第 3四分位値(3QR) 30.0 30.0
※ 復温時間:復温開始から復温までに要した時間
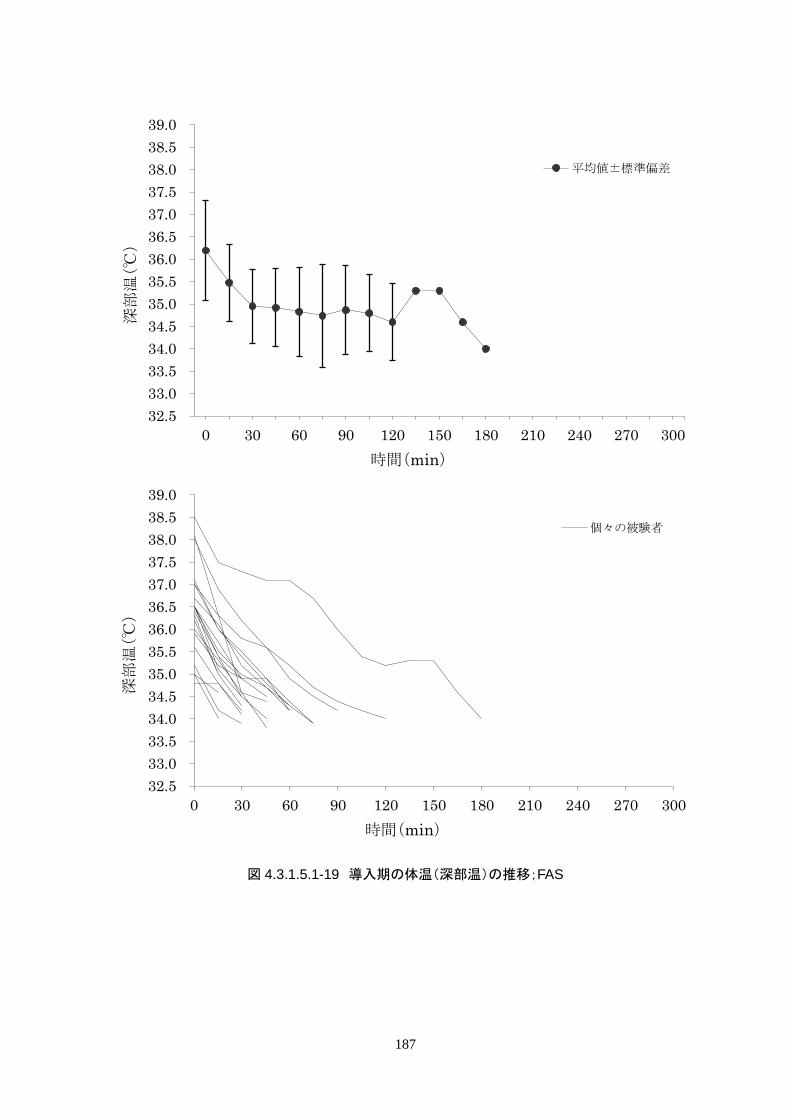
187
図 4.3.1.5.1-19 導入期の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
個々の被験者
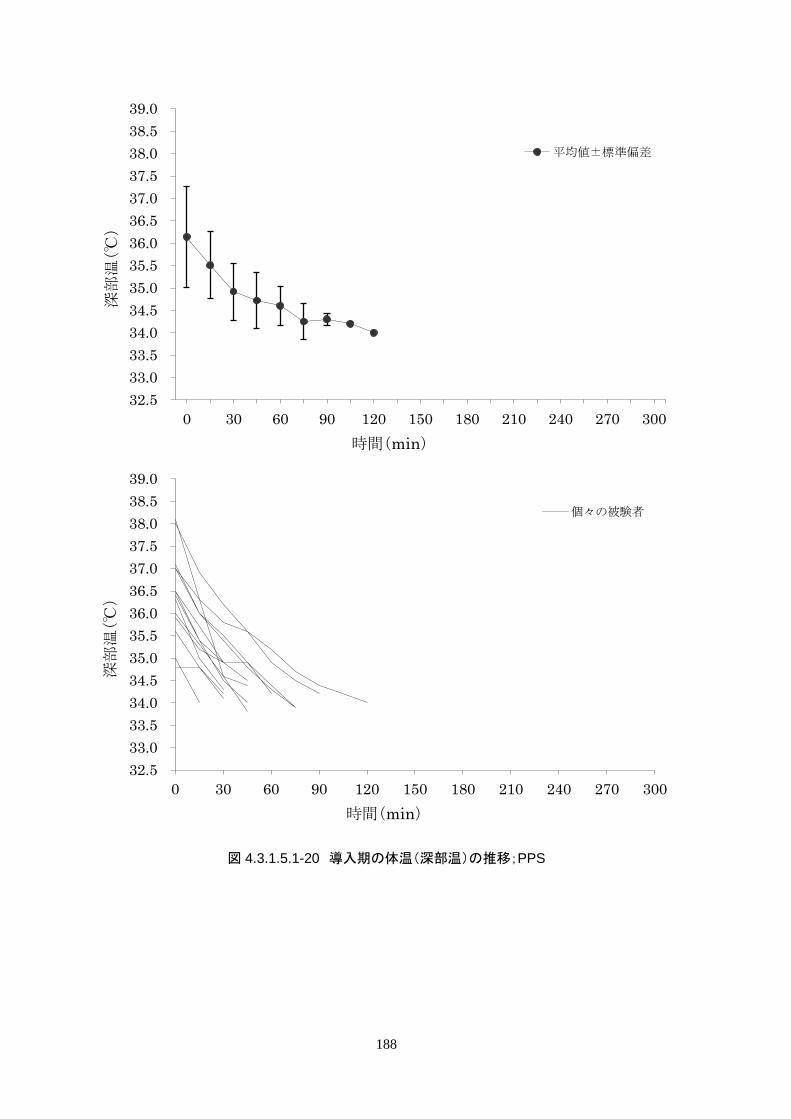
188
図 4.3.1.5.1-20 導入期の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 30 60 90 120 150 180 210 240 270 300
深部温(℃)
時間(min)
個々の被験者
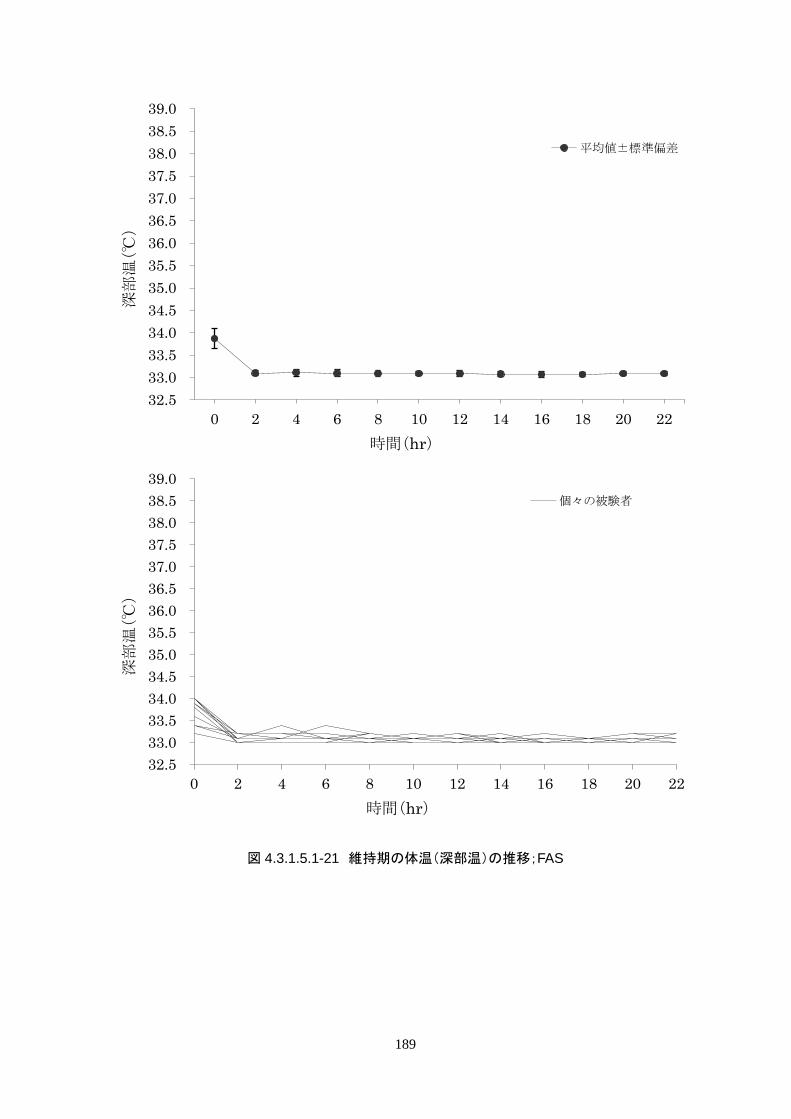
189
図 4.3.1.5.1-21 維持期の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
個々の被験者
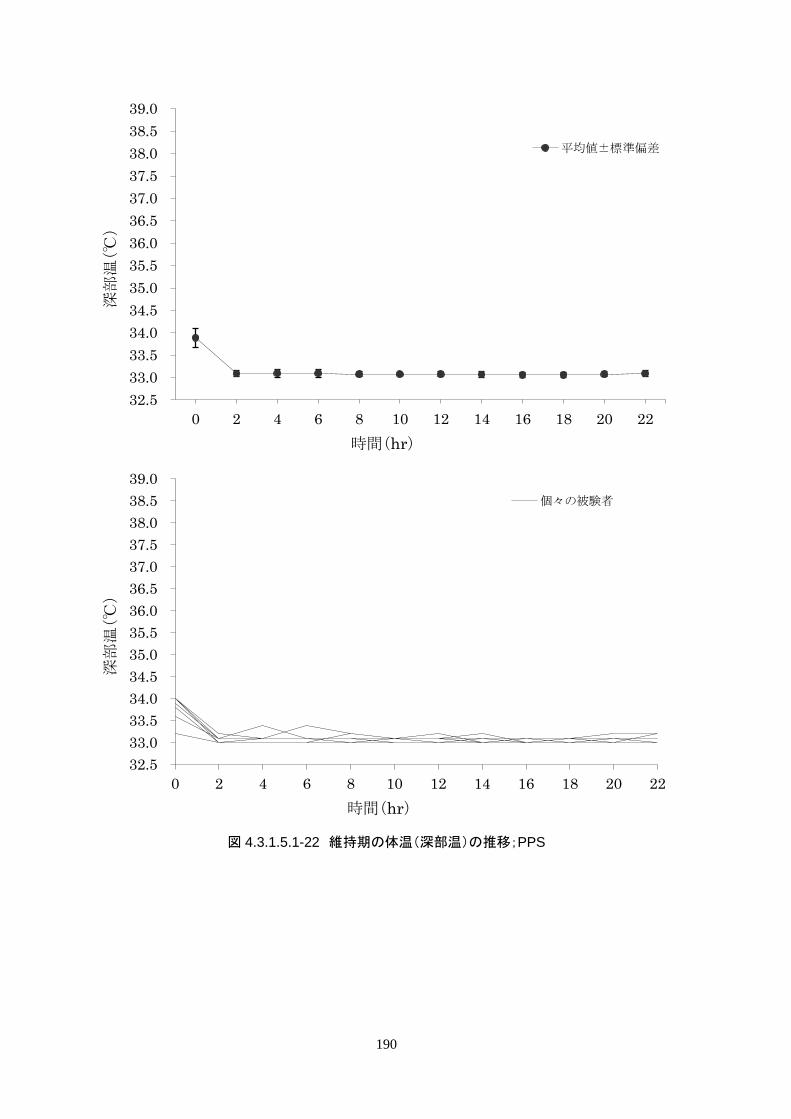
190
図 4.3.1.5.1-22 維持期の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
個々の被験者
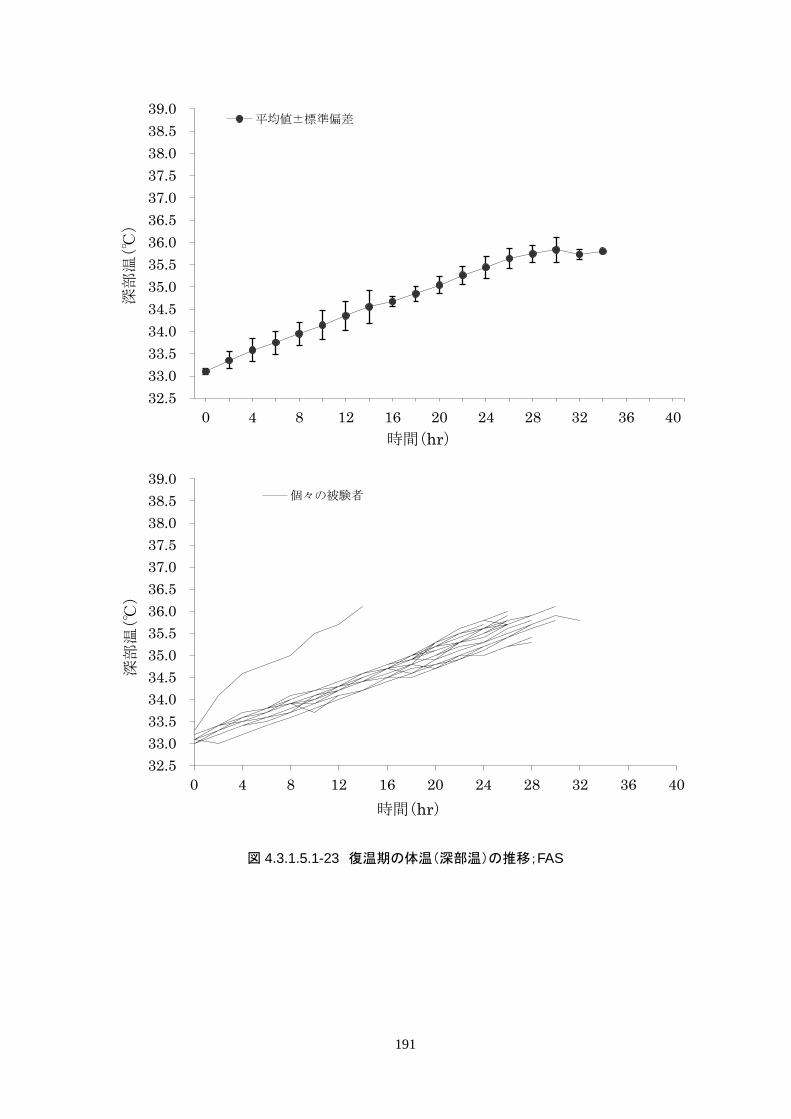
191
図 4.3.1.5.1-23 復温期の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 4 8 12 16 20 24 28 32 36 40
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 4 8 12 16 20 24 28 32 36 40
深部温(℃)
時間(hr)
個々の被験者
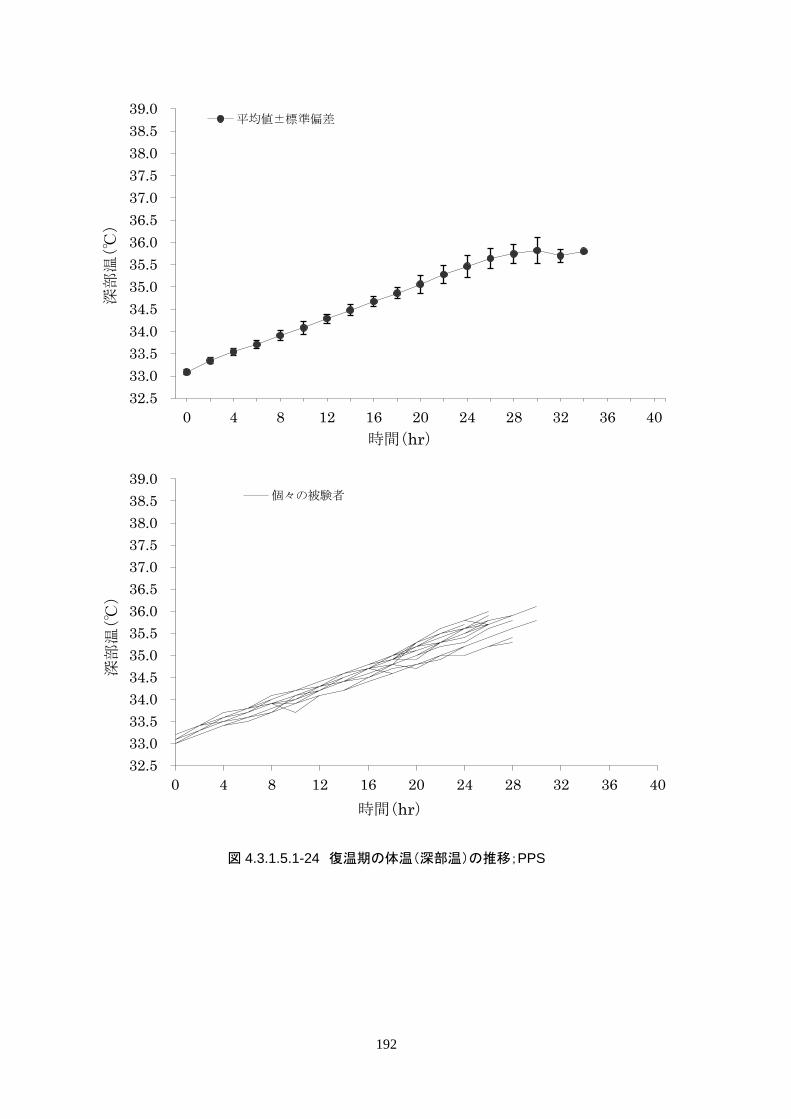
192
図 4.3.1.5.1-24 復温期の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 4 8 12 16 20 24 28 32 36 40
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 4 8 12 16 20 24 28 32 36 40
深部温(℃)
時間(hr)
個々の被験者
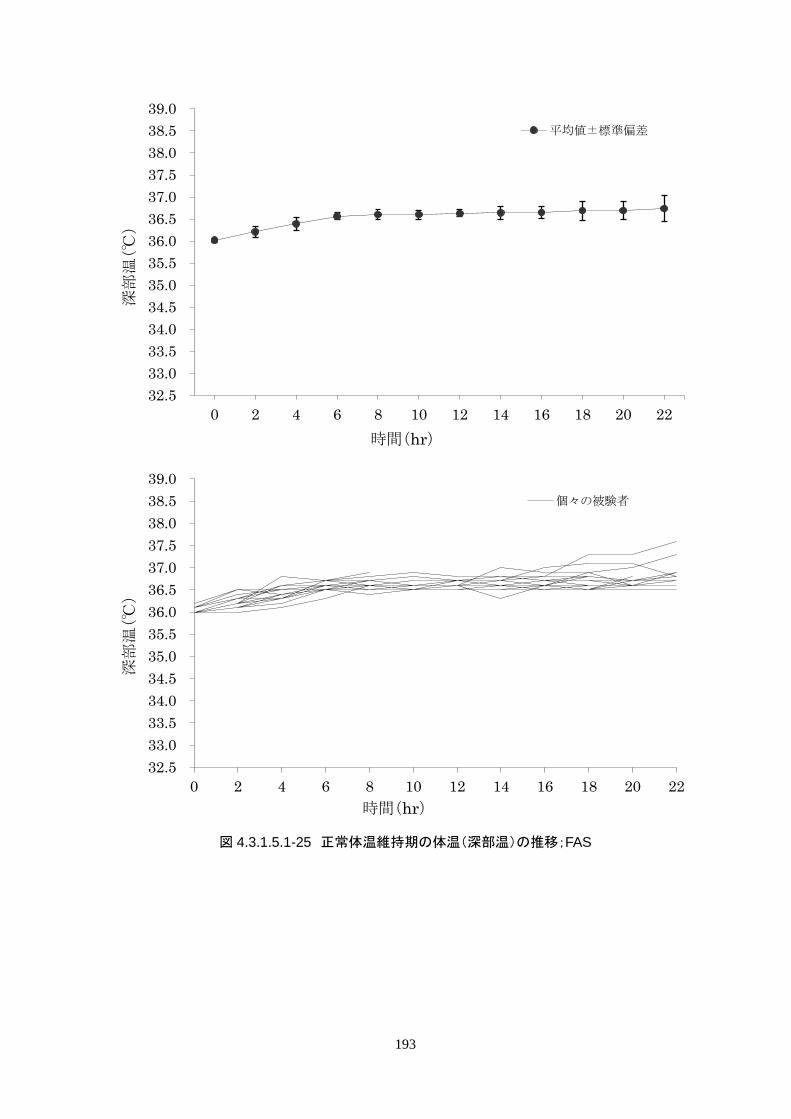
193
図 4.3.1.5.1-25 正常体温維持期の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
個々の被験者
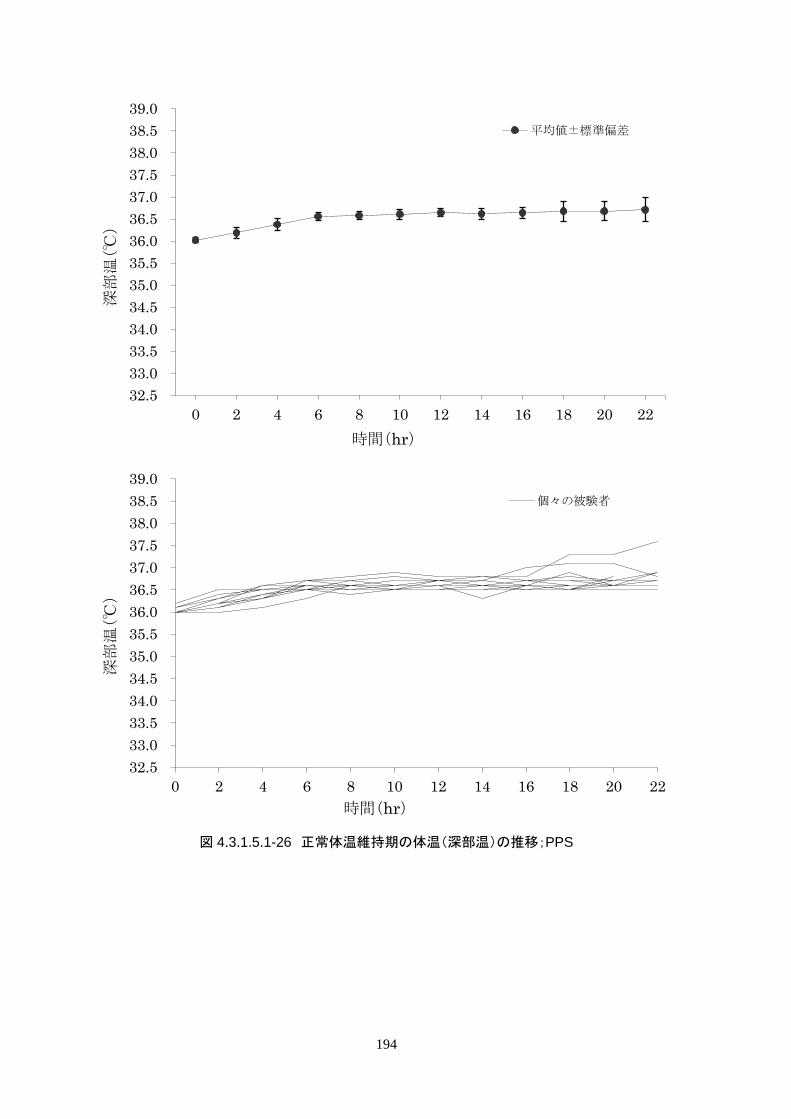
194
図 4.3.1.5.1-26 正常体温維持期の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
平均値±標準偏差
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
0 2 4 6 8 10 12 14 16 18 20 22
深部温(℃)
時間(hr)
個々の被験者
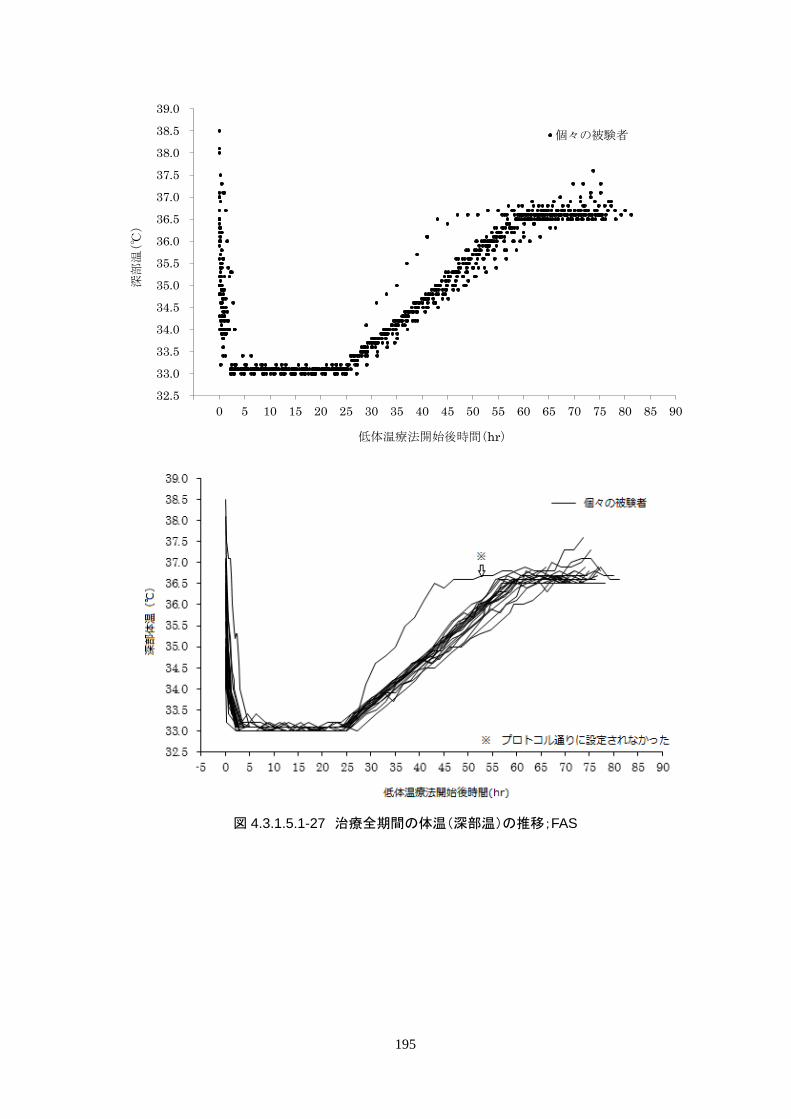
195
図 4.3.1.5.1-27 治療全期間の体温(深部温)の推移;FAS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
-5 0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
深部温(℃)
低体温療法開始後時間(hr)
個々の被験者
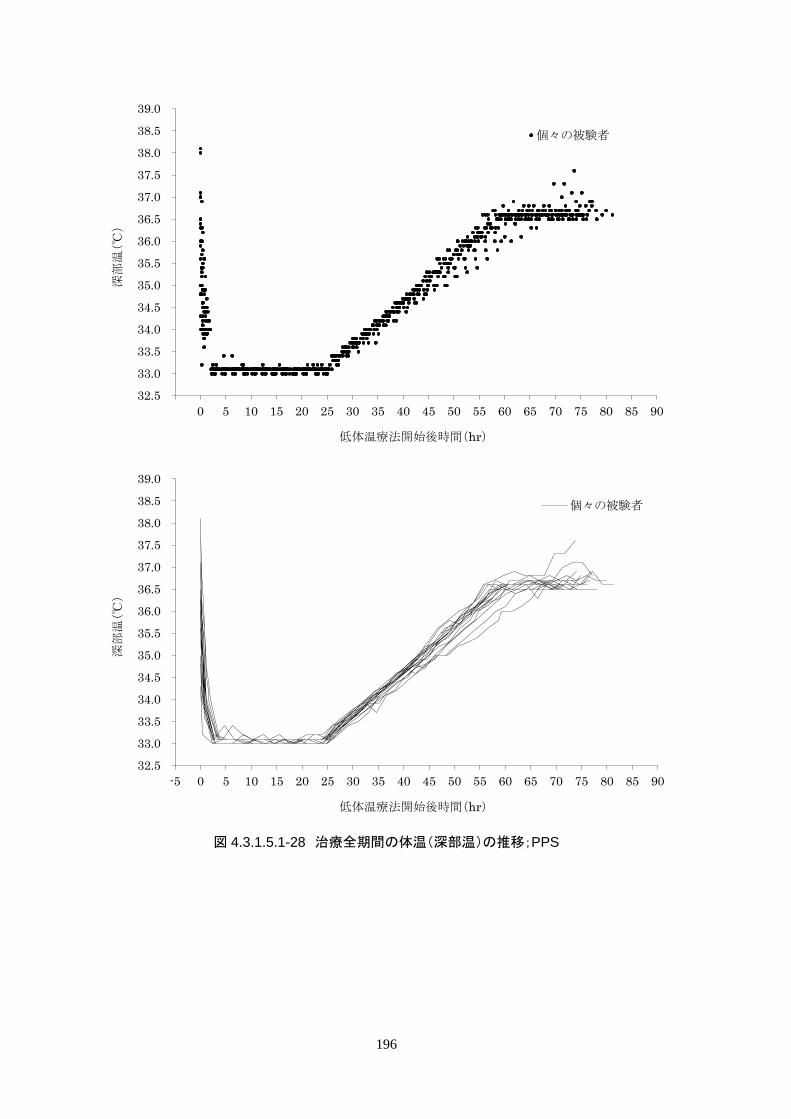
196
図 4.3.1.5.1-28 治療全期間の体温(深部温)の推移;PPS
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
-5 0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
深部温(℃)
低体温療法開始後時間(hr)
個々の被験者
32.5
33.0
33.5
34.0
34.5
35.0
35.5
36.0
36.5
37.0
37.5
38.0
38.5
39.0
-5 0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
深部温(℃)
低体温療法開始後時間(hr)
個々の被験者
197
④ 副次的評価項目の解析‐単位時間あたりの体温変化量(冷却速度)
単位時間あたりの体温変化率(冷却速度)を表 4.3.1.5.1-29 に示す。
導入期の体温冷却速度は、3.08±1.45℃(中央値 2.70℃/hr、IQR 2.3-3.7℃/hr)だった。
表 4.3.1.5.1-29 単位時間あたりの体温変化率(冷却速度)
FAS PPS
低体温療法開始被験者 (人) 24 17
目標体温に到達した被験者 (人) 24 (100.0%) 17 (100.0%)
冷却速度 (人) 0.0~2.0℃/hr 3 (12.5%) 2 (11.8%)
2.1~3.0℃/hr 12 (50.0%) 8 (47.1%)
3.1~4.0℃/hr 7 (29.2%) 5 (29.4%)
4.1℃/hr~ 2 (8.3%) 2 (11.8%)
(℃/hr) 平均値 3.08 3.27
標準偏差 1.45 1.63
中央値 2.70 2.80
最大値 8.6 8.6
最小値 1.5 1.5
第 1四分位値(1QR) 2.3 2.5
第 3四分位値(3QR) 3.7 3.7
※ 冷却速度:(冷却開始時体温-目標体温)/目標体温到達までに要した時間
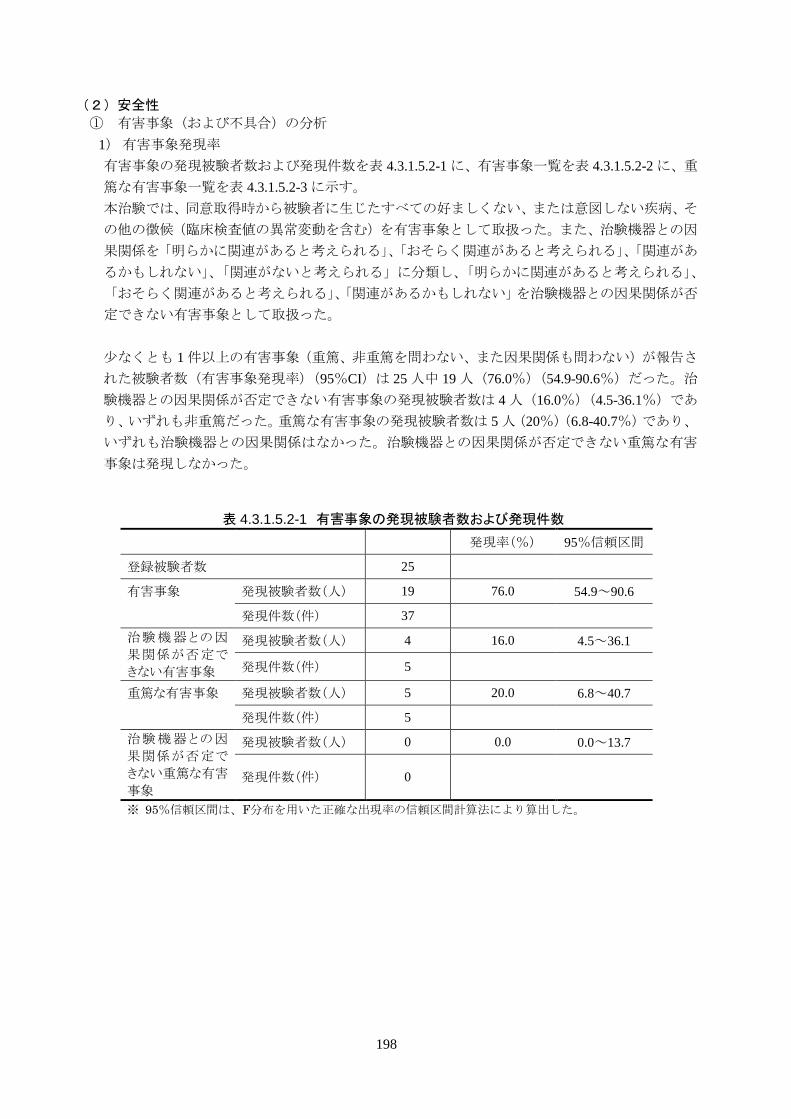
198
(2)安全性
① 有害事象(および不具合)の分析
1) 有害事象発現率
有害事象の発現被験者数および発現件数を表 4.3.1.5.2-1 に、有害事象一覧を表 4.3.1.5.2-2 に、重
篤な有害事象一覧を表 4.3.1.5.2-3 に示す。
本治験では、同意取得時から被験者に生じたすべての好ましくない、または意図しない疾病、そ
の他の徴候(臨床検査値の異常変動を含む)を有害事象として取扱った。また、治験機器との因
果関係を「明らかに関連があると考えられる」、「おそらく関連があると考えられる」、「関連があ
るかもしれない」、「関連がないと考えられる」に分類し、「明らかに関連があると考えられる」、
「おそらく関連があると考えられる」、「関連があるかもしれない」を治験機器との因果関係が否
定できない有害事象として取扱った。
少なくとも 1 件以上の有害事象(重篤、非重篤を問わない、また因果関係も問わない)が報告さ
れた被験者数(有害事象発現率)(95%CI)は 25 人中 19 人(76.0%)(54.9-90.6%)だった。治
験機器との因果関係が否定できない有害事象の発現被験者数は 4 人(16.0%)(4.5-36.1%)であ
り、いずれも非重篤だった。重篤な有害事象の発現被験者数は 5 人(20%)(6.8-40.7%)であり、
いずれも治験機器との因果関係はなかった。治験機器との因果関係が否定できない重篤な有害
事象は発現しなかった。
表 4.3.1.5.2-1 有害事象の発現被験者数および発現件数
発現率(%) 95%信頼区間
登録被験者数 25
有害事象 発現被験者数(人) 19 76.0 54.9~90.6
発現件数(件) 37
治験機器との因
果関係が否定で
きない有害事象
発現被験者数(人) 4 16.0 4.5~36.1
発現件数(件) 5
重篤な有害事象 発現被験者数(人) 5 20.0 6.8~40.7
発現件数(件) 5
治験機器との因
果関係が否定で
きない重篤な有害
事象
発現被験者数(人) 0 0.0 0.0~13.7
発現件数(件) 0
※ 95%信頼区間は、F分布を用いた正確な出現率の信頼区間計算法により算出した。
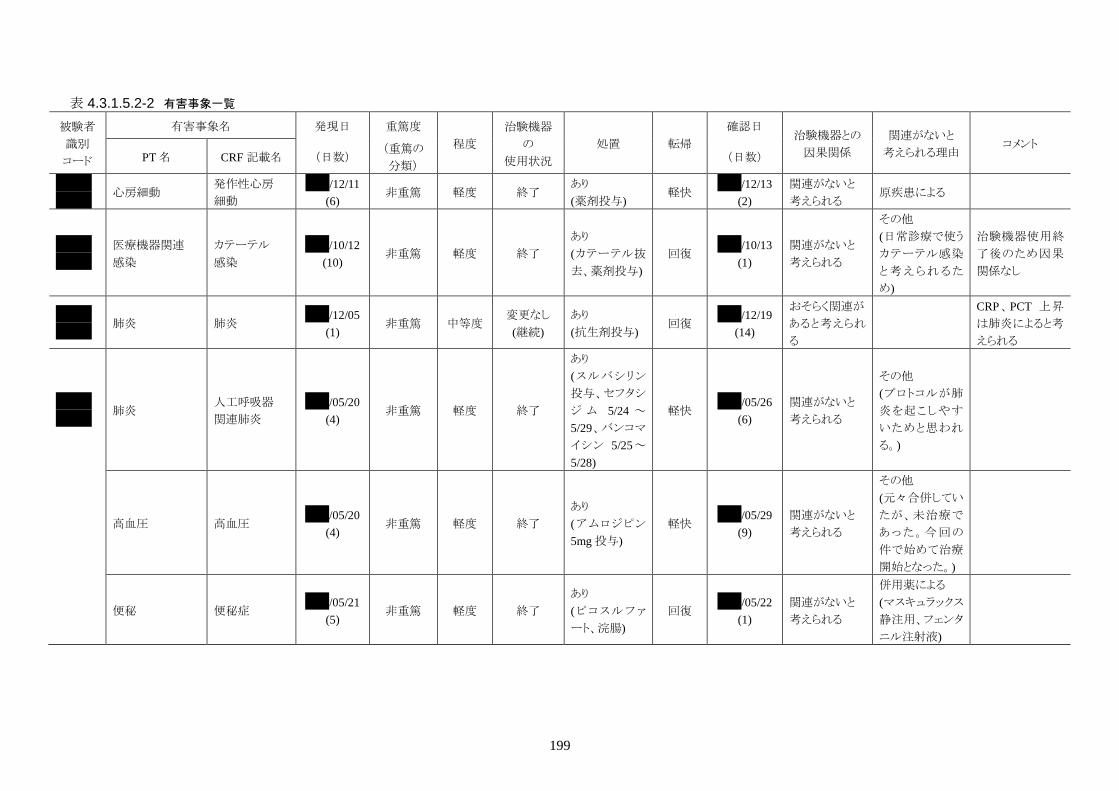
199
表 4.3.1.5.2-2 有害事象一覧
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 心房細動
発作性心房
細動
**/12/11
(6) 非重篤 軽度 終了
あり
(薬剤投与) 軽快
**/12/13
(2)
関連がないと
考えられる 原疾患による
***
***
医療機器関連
感染
カテーテル
感染
**/10/12
(10) 非重篤 軽度 終了
あり
(カテーテル抜
去、薬剤投与)
回復 **/10/13
(1)
関連がないと
考えられる
その他
(日常診療で使う
カテーテル感染
と考えられるた
め)
治験機器使用終
了後のため因果
関係なし
***
*** 肺炎 肺炎
**/12/05
(1) 非重篤 中等度
変更なし
(継続)
あり
(抗生剤投与) 回復
**/12/19
(14)
おそらく関連が
あると考えられ
る
CRP、PCT 上昇
は肺炎によると考
えられる
***
*** 肺炎
人工呼吸器
関連肺炎
**/05/20
(4) 非重篤 軽度 終了
あり
(スルバシリン
投与、セフタシ
ジ ム 5/24 ~
5/29、バンコマ
イシン 5/25~
5/28)
軽快 **/05/26
(6)
関連がないと
考えられる
その他
(プロトコルが肺
炎を起こしやす
いためと思われ
る。)
高血圧 高血圧 **/05/20
(4) 非重篤 軽度 終了
あり
(アムロジピン
5mg投与)
軽快 **/05/29
(9)
関連がないと
考えられる
その他
(元々合併してい
たが、未治療で
あった。今回の
件で始めて治療
開始となった。)
便秘 便秘症 **/05/21
(5) 非重篤 軽度 終了
あり
(ピコスルファ
ート、浣腸)
回復 **/05/22
(1)
関連がないと
考えられる
併用薬による
(マスキュラックス
静注用、フェンタ
ニル注射液)
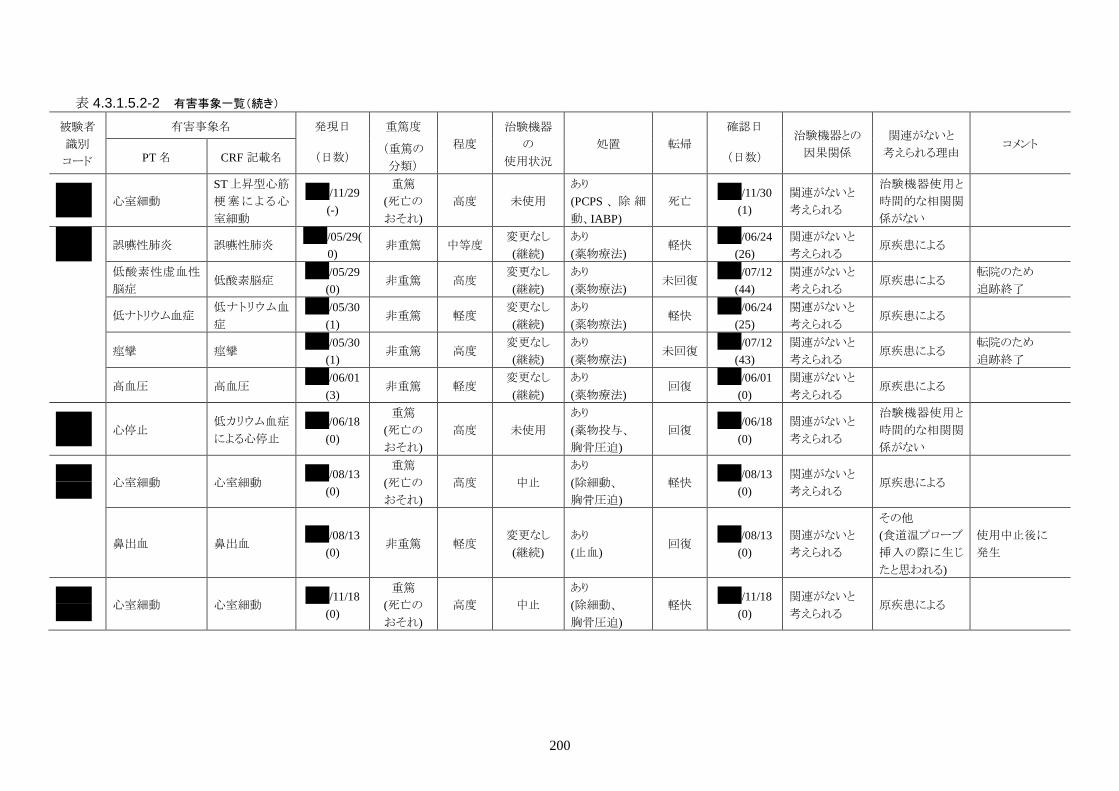
200
表 4.3.1.5.2-2 有害事象一覧(続き)
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 心室細動
ST上昇型心筋
梗塞による心
室細動
**/11/29
(-)
重篤
(死亡の
おそれ)
高度 未使用
あり
(PCPS 、 除 細
動、IABP)
死亡 **/11/30
(1)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 誤嚥性肺炎 誤嚥性肺炎
**/05/29(
0) 非重篤 中等度
変更なし
(継続)
あり
(薬物療法) 軽快
**/06/24
(26)
関連がないと
考えられる 原疾患による
低酸素性虚血性
脳症 低酸素脳症
**/05/29
(0) 非重篤 高度
変更なし
(継続)
あり
(薬物療法) 未回復
**/07/12
(44)
関連がないと
考えられる 原疾患による
転院のため
追跡終了
低ナトリウム血症 低ナトリウム血
症
**/05/30
(1) 非重篤 軽度
変更なし
(継続)
あり
(薬物療法) 軽快
**/06/24
(25)
関連がないと
考えられる 原疾患による
痙攣 痙攣 **/05/30
(1) 非重篤 高度
変更なし
(継続)
あり
(薬物療法) 未回復
**/07/12
(43)
関連がないと
考えられる 原疾患による
転院のため
追跡終了
高血圧 高血圧 **/06/01
(3) 非重篤 軽度
変更なし
(継続)
あり
(薬物療法) 回復
**/06/01
(0)
関連がないと
考えられる 原疾患による
***
*** 心停止
低カリウム血症
による心停止
**/06/18
(0)
重篤
(死亡の
おそれ)
高度 未使用
あり
(薬物投与、
胸骨圧迫)
回復 **/06/18
(0)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 心室細動 心室細動
**/08/13
(0)
重篤
(死亡の
おそれ)
高度 中止
あり
(除細動、
胸骨圧迫)
軽快 **/08/13
(0)
関連がないと
考えられる 原疾患による
鼻出血 鼻出血 **/08/13
(0) 非重篤 軽度
変更なし
(継続)
あり
(止血) 回復
**/08/13
(0)
関連がないと
考えられる
その他
(食道温プローブ
挿入の際に生じ
たと思われる)
使用中止後に
発生
***
*** 心室細動 心室細動
**/11/18
(0)
重篤
(死亡の
おそれ)
高度 中止
あり
(除細動、
胸骨圧迫)
軽快 **/11/18
(0)
関連がないと
考えられる 原疾患による
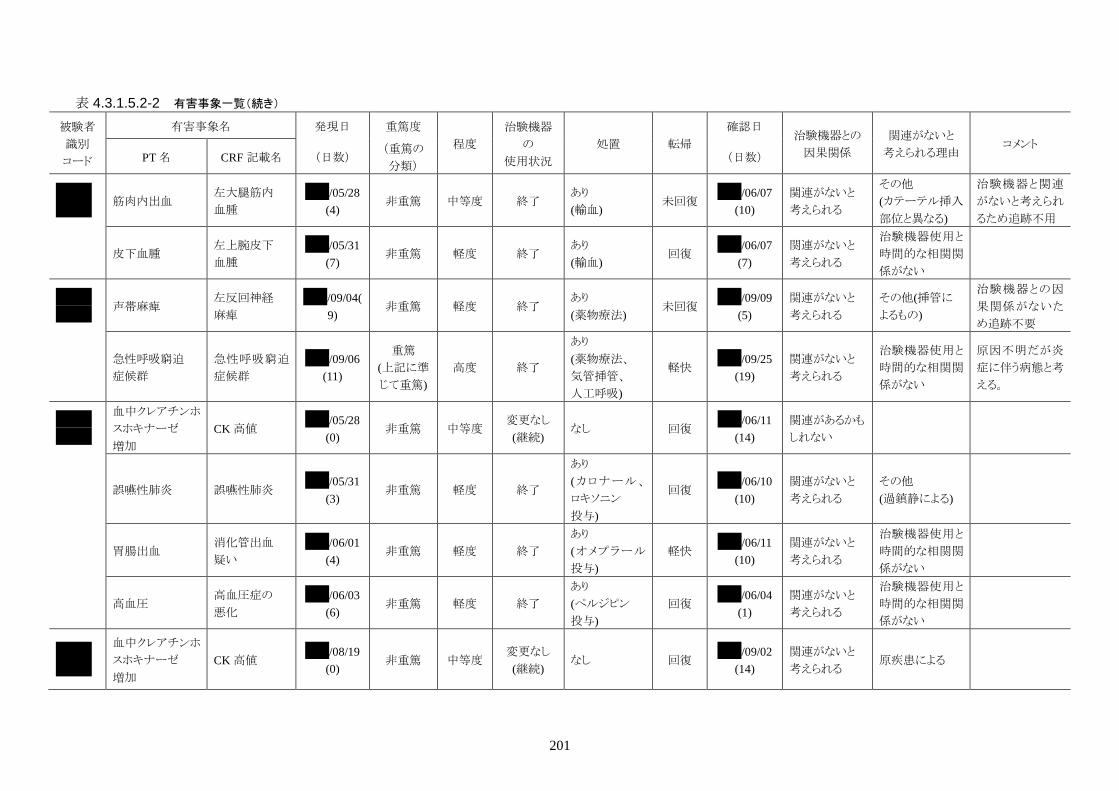
201
表 4.3.1.5.2-2 有害事象一覧(続き)
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 筋肉内出血
左大腿筋内
血腫
**/05/28
(4) 非重篤 中等度 終了
あり
(輸血) 未回復
**/06/07
(10)
関連がないと
考えられる
その他
(カテーテル挿入
部位と異なる)
治験機器と関連
がないと考えられ
るため追跡不用
皮下血腫 左上腕皮下
血腫
**/05/31
(7) 非重篤 軽度 終了
あり
(輸血) 回復
**/06/07
(7)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 声帯麻痺
左反回神経
麻痺
**/09/04(
9) 非重篤 軽度 終了
あり
(薬物療法) 未回復
**/09/09
(5)
関連がないと
考えられる
その他(挿管に
よるもの)
治験機器との因
果関係がないた
め追跡不要
急性呼吸窮迫
症候群
急性呼吸窮迫
症候群
**/09/06
(11)
重篤
(上記に準
じて重篤)
高度 終了
あり
(薬物療法、
気管挿管、
人工呼吸)
軽快 **/09/25
(19)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
原因不明だが炎
症に伴う病態と考
える。
***
***
血中クレアチンホ
スホキナーゼ
増加
CK高値 **/05/28
(0) 非重篤 中等度
変更なし
(継続) なし 回復
**/06/11
(14)
関連があるかも
しれない
誤嚥性肺炎 誤嚥性肺炎 **/05/31
(3) 非重篤 軽度 終了
あり
(カロナール、
ロキソニン
投与)
回復 **/06/10
(10)
関連がないと
考えられる
その他
(過鎮静による)
胃腸出血 消化管出血
疑い
**/06/01
(4) 非重篤 軽度 終了
あり
(オメプラール
投与)
軽快 **/06/11
(10)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
高血圧 高血圧症の
悪化
**/06/03
(6) 非重篤 軽度 終了
あり
(ペルジピン
投与)
回復 **/06/04
(1)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
***
血中クレアチンホ
スホキナーゼ
増加
CK高値 **/08/19
(0) 非重篤 中等度
変更なし
(継続) なし 回復
**/09/02
(14)
関連がないと
考えられる 原疾患による
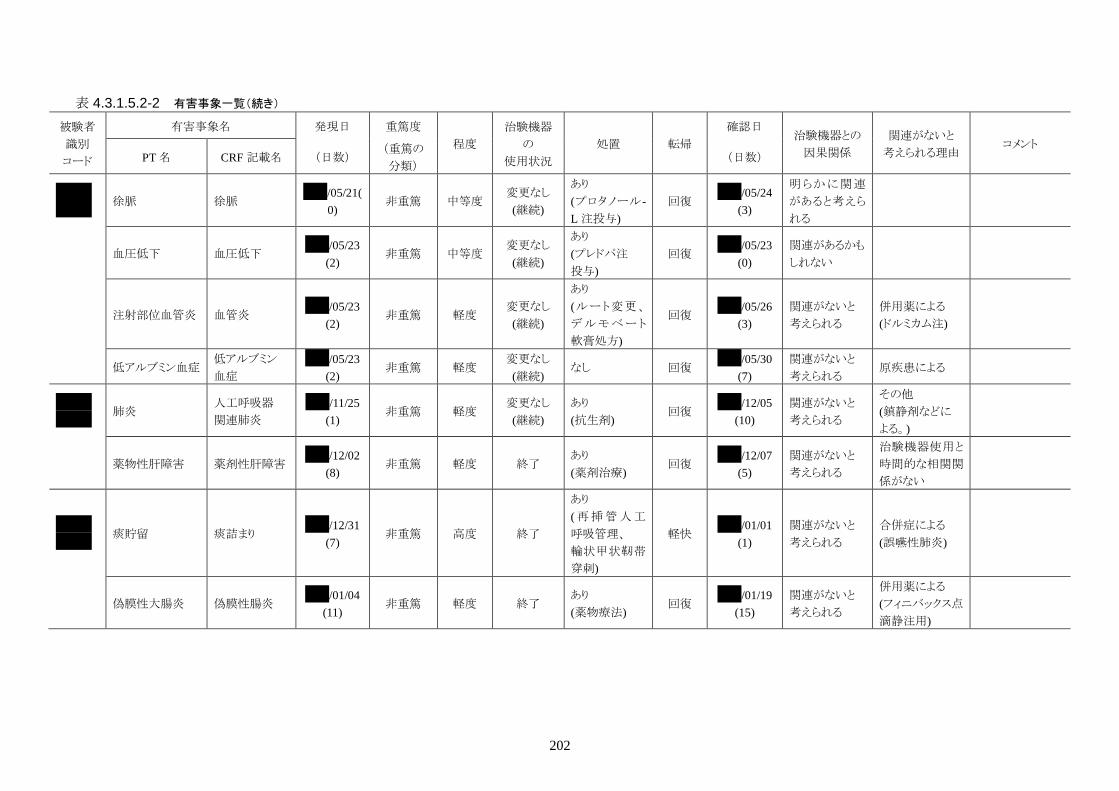
202
表 4.3.1.5.2-2 有害事象一覧(続き)
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 徐脈 徐脈
**/05/21(
0) 非重篤 中等度
変更なし
(継続)
あり
(プロタノール-
L注投与)
回復 **/05/24
(3)
明らかに関連
があると考えら
れる
血圧低下 血圧低下 **/05/23
(2) 非重篤 中等度
変更なし
(継続)
あり
(プレドパ注
投与)
回復 **/05/23
(0)
関連があるかも
しれない
注射部位血管炎 血管炎 **/05/23
(2) 非重篤 軽度
変更なし
(継続)
あり
(ルート変更、
デルモベート
軟膏処方)
回復 **/05/26
(3)
関連がないと
考えられる
併用薬による
(ドルミカム注)
低アルブミン血症 低アルブミン
血症
**/05/23
(2) 非重篤 軽度
変更なし
(継続) なし 回復
**/05/30
(7)
関連がないと
考えられる 原疾患による
***
*** 肺炎
人工呼吸器
関連肺炎
**/11/25
(1) 非重篤 軽度
変更なし
(継続)
あり
(抗生剤) 回復
**/12/05
(10)
関連がないと
考えられる
その他
(鎮静剤などに
よる。)
薬物性肝障害 薬剤性肝障害 **/12/02
(8) 非重篤 軽度 終了
あり
(薬剤治療) 回復
**/12/07
(5)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 痰貯留 痰詰まり
**/12/31
(7) 非重篤 高度 終了
あり
(再挿管人工
呼吸管理、
輪状甲状靭帯
穿刺)
軽快 **/01/01
(1)
関連がないと
考えられる
合併症による
(誤嚥性肺炎)
偽膜性大腸炎 偽膜性腸炎 **/01/04
(11) 非重篤 軽度 終了
あり
(薬物療法) 回復
**/01/19
(15)
関連がないと
考えられる
併用薬による
(フィニバックス点
滴静注用)
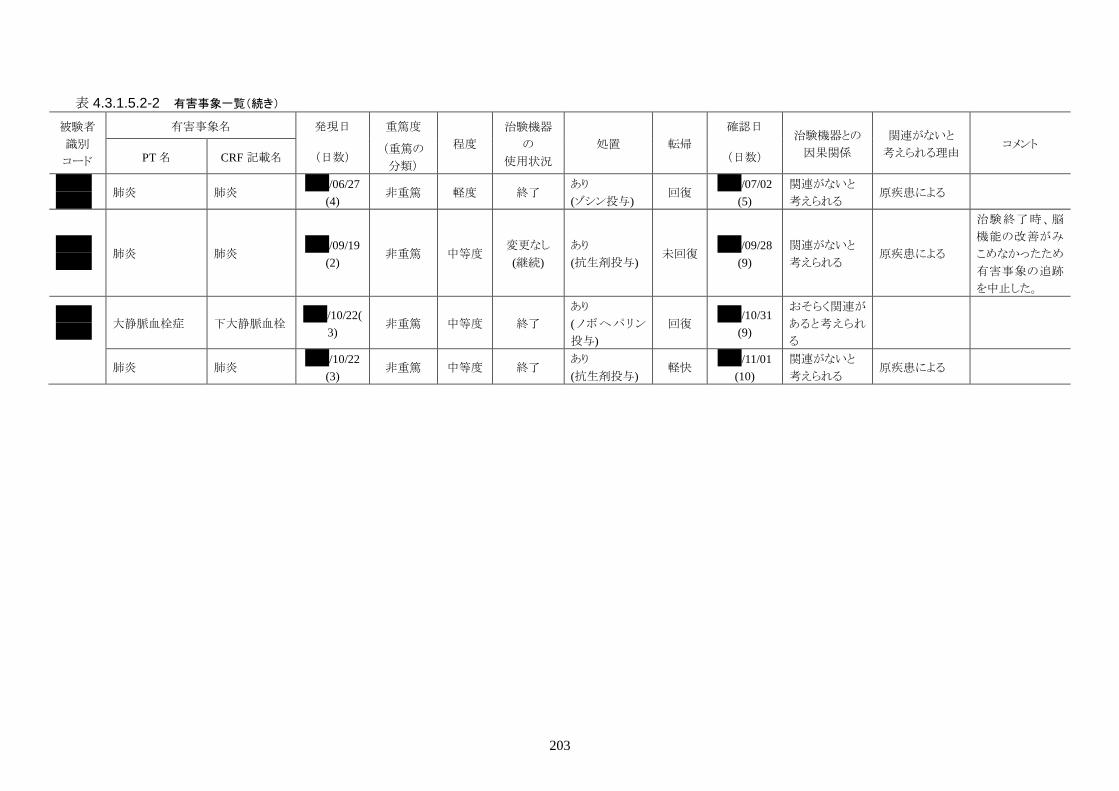
203
表 4.3.1.5.2-2 有害事象一覧(続き)
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 肺炎 肺炎
**/06/27
(4) 非重篤 軽度 終了
あり
(ゾシン投与) 回復
**/07/02
(5)
関連がないと
考えられる 原疾患による
***
*** 肺炎 肺炎
**/09/19
(2) 非重篤 中等度
変更なし
(継続)
あり
(抗生剤投与) 未回復
**/09/28
(9)
関連がないと
考えられる 原疾患による
治験終了時、脳
機能の改善がみ
こめなかったため
有害事象の追跡
を中止した。
***
*** 大静脈血栓症 下大静脈血栓
**/10/22(
3) 非重篤 中等度 終了
あり
(ノボヘパリン
投与)
回復 **/10/31
(9)
おそらく関連が
あると考えられ
る
肺炎 肺炎 **/10/22
(3) 非重篤 中等度 終了
あり
(抗生剤投与) 軽快
**/11/01
(10)
関連がないと
考えられる 原疾患による
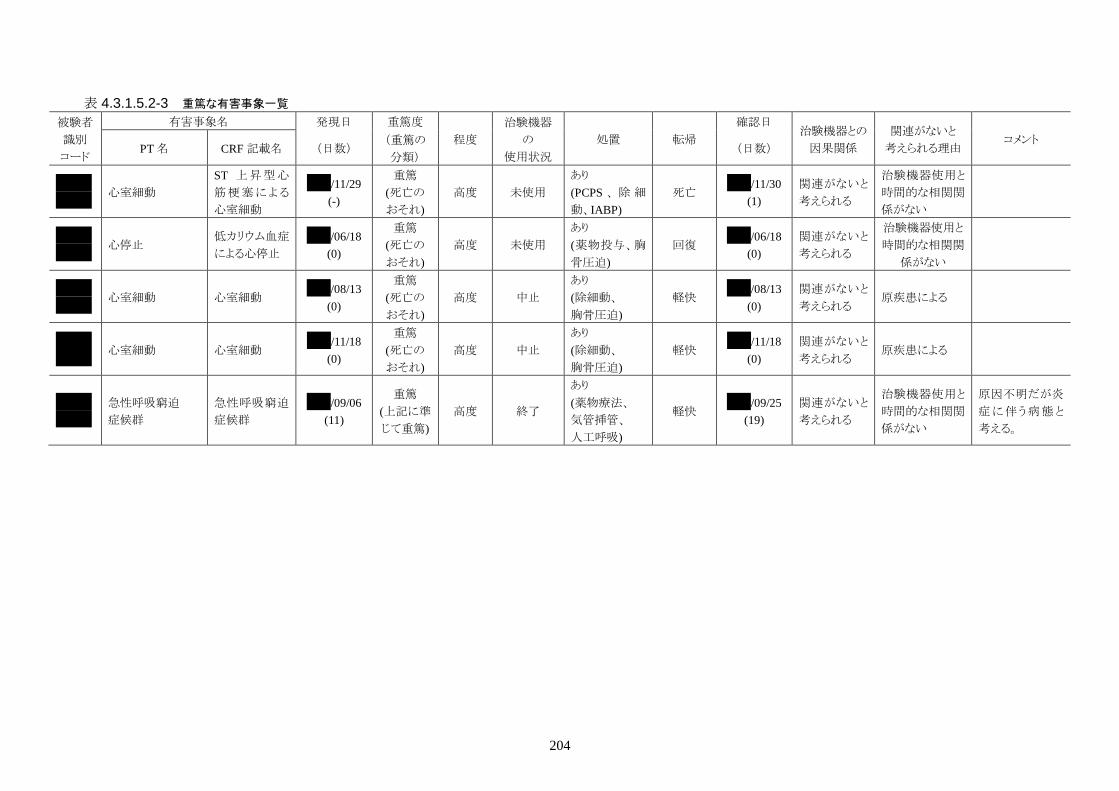
204
表 4.3.1.5.2-3 重篤な有害事象一覧
被験者
識別
コード
有害事象名 発現日 重篤度
程度
治験機器
の
使用状況
処置 転帰
確認日 治験機器との
因果関係
関連がないと
考えられる理由 コメント
PT名 CRF 記載名 (日数) (重篤の
分類) (日数)
***
*** 心室細動
ST 上昇型心
筋梗塞による
心室細動
**/11/29
(-)
重篤
(死亡の
おそれ)
高度 未使用
あり
(PCPS 、 除 細
動、IABP)
死亡 **/11/30
(1)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 心停止
低カリウム血症
による心停止
**/06/18
(0)
重篤
(死亡の
おそれ)
高度 未使用
あり
(薬物投与、胸
骨圧迫)
回復 **/06/18
(0)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
***
*** 心室細動 心室細動
**/08/13
(0)
重篤
(死亡の
おそれ)
高度 中止
あり
(除細動、
胸骨圧迫)
軽快 **/08/13
(0)
関連がないと
考えられる 原疾患による
***
*** 心室細動 心室細動
**/11/18
(0)
重篤
(死亡の
おそれ)
高度 中止
あり
(除細動、
胸骨圧迫)
軽快 **/11/18
(0)
関連がないと
考えられる 原疾患による
***
***
急性呼吸窮迫
症候群
急性呼吸窮迫
症候群
**/09/06
(11)
重篤
(上記に準
じて重篤)
高度 終了
あり
(薬物療法、
気管挿管、
人工呼吸)
軽快 **/09/25
(19)
関連がないと
考えられる
治験機器使用と
時間的な相関関
係がない
原因不明だが炎
症に伴う病態と
考える。

205
2) 重篤度別の有害事象
重篤度別有害事象の発現率を表 4.3.1.5.2-4 に、重篤度別の治験機器との因果関係が否定できな
い(因果関係がある)有害事象の発現率を表 4.3.1.5.2-5 に示す。
発現率の高かった有害事象の SOC 分類は、感染症および寄生虫症 8 件(32.0%)、心臓障害 6
件(24.0%)、呼吸器、胸郭および縦隔障害 5 件(20.0%)、血管障害 4 件(16.0%)、臨床検査
3 件(12.0%)であった。
発現率が高かった重篤な有害事象は、心室細動 3 件(12.0%)だった。
発現率が高かった非重篤な有害事象は、肺炎 6 件(24.0%)、高血圧 3 件(12.0%)だった。
非重篤な治験機器との因果関係が否定できない有害事象の発現件数は 5 件(4 人 5 件)だった。
内訳は、肺炎、徐脈、大静脈血栓症、血中クレアチンホスホキナーゼ増加、血圧低下がそれぞ
れ 1 件だった。また、重篤な治験機器との因果関係が否定できない有害事象はなかった。なお、
徐脈については、低体温療法を行う場合、心臓脈の減少は通常起こりえることであり、可逆的で予
期される効果であると考える。
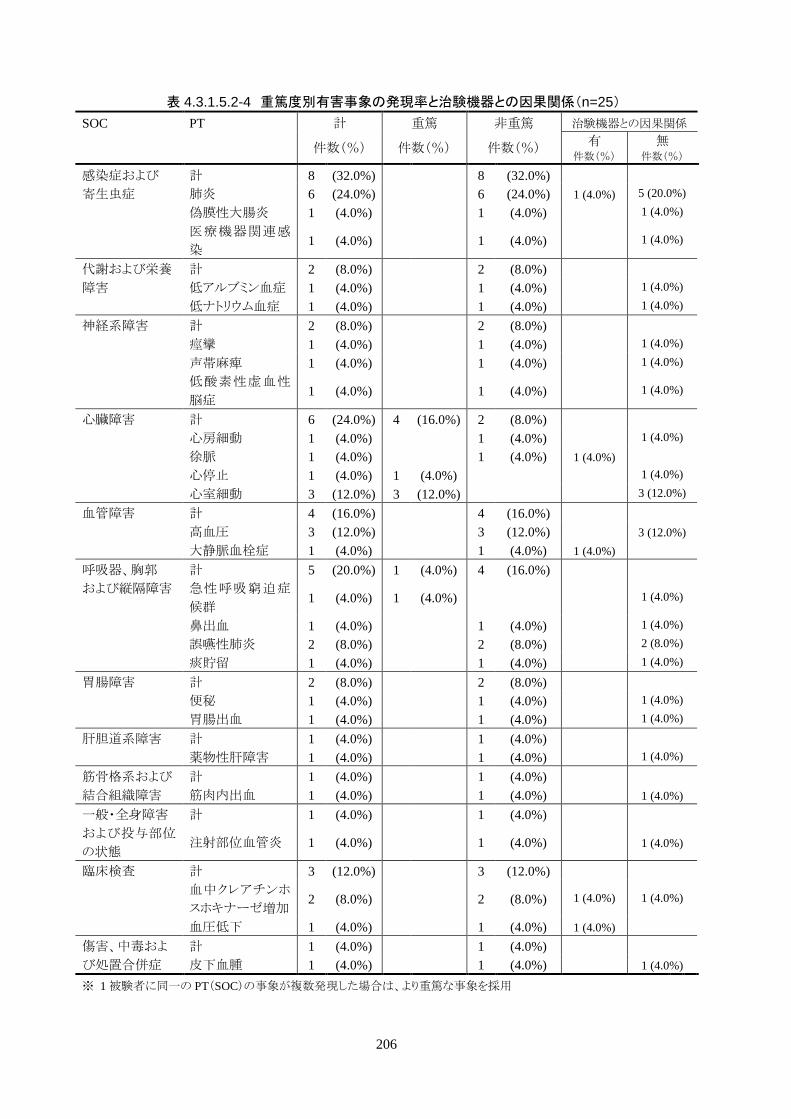
206
表 4.3.1.5.2-4 重篤度別有害事象の発現率と治験機器との因果関係(n=25)
SOC PT 計 重篤 非重篤 治験機器との因果関係
件数(%) 件数(%) 件数(%) 有
件数(%)
無 件数(%)
感染症および 計 8 (32.0%) 8 (32.0%)
寄生虫症 肺炎 6 (24.0%) 6 (24.0%) 1 (4.0%) 5 (20.0%)
偽膜性大腸炎 1 (4.0%) 1 (4.0%) 1 (4.0%)
医療機器関連感
染 1 (4.0%) 1 (4.0%)
1 (4.0%)
代謝および栄養 計 2 (8.0%) 2 (8.0%)
障害 低アルブミン血症 1 (4.0%) 1 (4.0%) 1 (4.0%)
低ナトリウム血症 1 (4.0%) 1 (4.0%) 1 (4.0%)
神経系障害 計 2 (8.0%) 2 (8.0%)
痙攣 1 (4.0%) 1 (4.0%) 1 (4.0%)
声帯麻痺 1 (4.0%) 1 (4.0%) 1 (4.0%)
低酸素性虚血性
脳症 1 (4.0%) 1 (4.0%)
1 (4.0%)
心臓障害 計 6 (24.0%) 4 (16.0%) 2 (8.0%)
心房細動 1 (4.0%) 1 (4.0%) 1 (4.0%)
徐脈 1 (4.0%) 1 (4.0%) 1 (4.0%)
心停止 1 (4.0%) 1 (4.0%) 1 (4.0%)
心室細動 3 (12.0%) 3 (12.0%) 3 (12.0%)
血管障害 計 4 (16.0%) 4 (16.0%)
高血圧 3 (12.0%) 3 (12.0%) 3 (12.0%)
大静脈血栓症 1 (4.0%) 1 (4.0%) 1 (4.0%)
呼吸器、胸郭 計 5 (20.0%) 1 (4.0%) 4 (16.0%)
および縦隔障害 急性呼吸窮迫症
候群 1 (4.0%) 1 (4.0%)
1 (4.0%)
鼻出血 1 (4.0%) 1 (4.0%) 1 (4.0%)
誤嚥性肺炎 2 (8.0%) 2 (8.0%) 2 (8.0%)
痰貯留 1 (4.0%) 1 (4.0%) 1 (4.0%)
胃腸障害 計 2 (8.0%) 2 (8.0%)
便秘 1 (4.0%) 1 (4.0%) 1 (4.0%)
胃腸出血 1 (4.0%) 1 (4.0%) 1 (4.0%)
肝胆道系障害 計 1 (4.0%) 1 (4.0%)
薬物性肝障害 1 (4.0%) 1 (4.0%) 1 (4.0%)
筋骨格系および 計 1 (4.0%) 1 (4.0%)
結合組織障害 筋肉内出血 1 (4.0%) 1 (4.0%) 1 (4.0%)
一般・全身障害 計 1 (4.0%) 1 (4.0%)
および投与部位
の状態 注射部位血管炎 1 (4.0%) 1 (4.0%)
1 (4.0%)
臨床検査 計 3 (12.0%) 3 (12.0%)
血中クレアチンホ
スホキナーゼ増加 2 (8.0%) 2 (8.0%) 1 (4.0%) 1 (4.0%)
血圧低下 1 (4.0%) 1 (4.0%) 1 (4.0%)
傷害、中毒およ 計 1 (4.0%) 1 (4.0%)
び処置合併症 皮下血腫 1 (4.0%) 1 (4.0%) 1 (4.0%)
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重篤な事象を採用
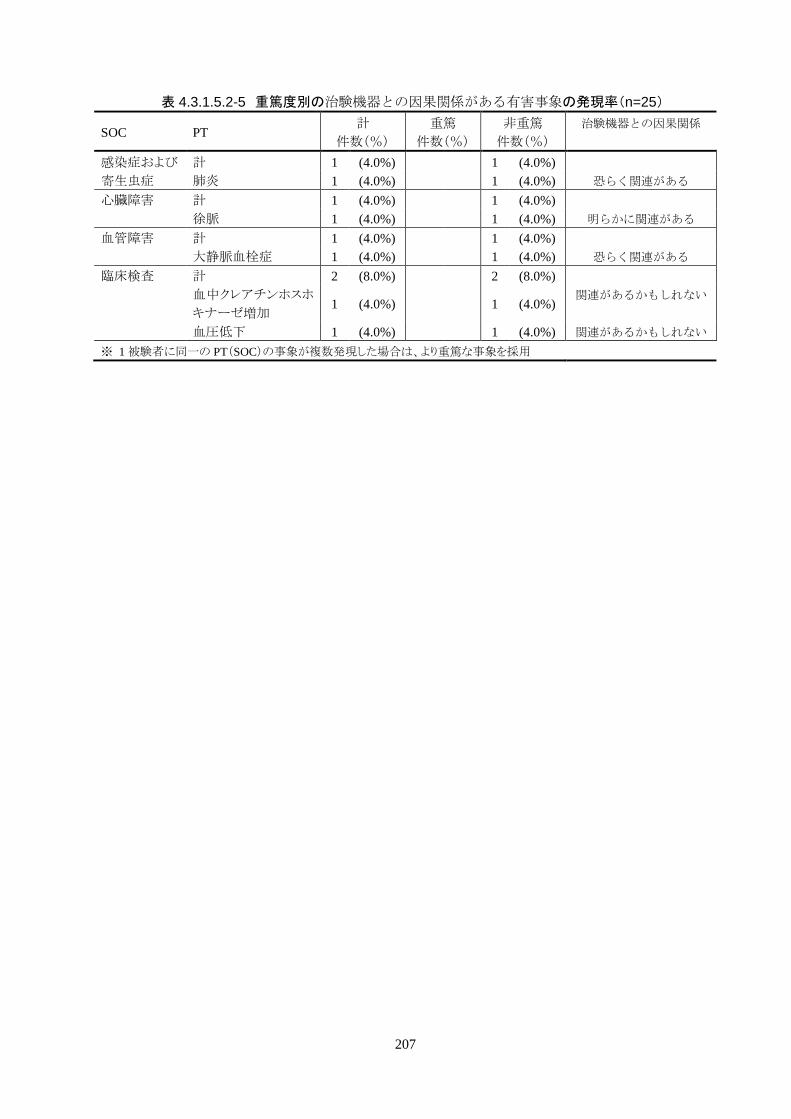
207
表 4.3.1.5.2-5 重篤度別の治験機器との因果関係がある有害事象の発現率(n=25)
SOC PT 計
件数(%)
重篤
件数(%)
非重篤
件数(%)
治験機器との因果関係
感染症および 計 1 (4.0%) 1 (4.0%)
寄生虫症 肺炎 1 (4.0%) 1 (4.0%) 恐らく関連がある
心臓障害 計 1 (4.0%) 1 (4.0%)
徐脈 1 (4.0%) 1 (4.0%) 明らかに関連がある
血管障害 計 1 (4.0%) 1 (4.0%)
大静脈血栓症 1 (4.0%) 1 (4.0%) 恐らく関連がある
臨床検査 計 2 (8.0%) 2 (8.0%)
血中クレアチンホスホ
キナーゼ増加 1 (4.0%) 1 (4.0%)
関連があるかもしれない
血圧低下 1 (4.0%) 1 (4.0%) 関連があるかもしれない
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重篤な事象を採用
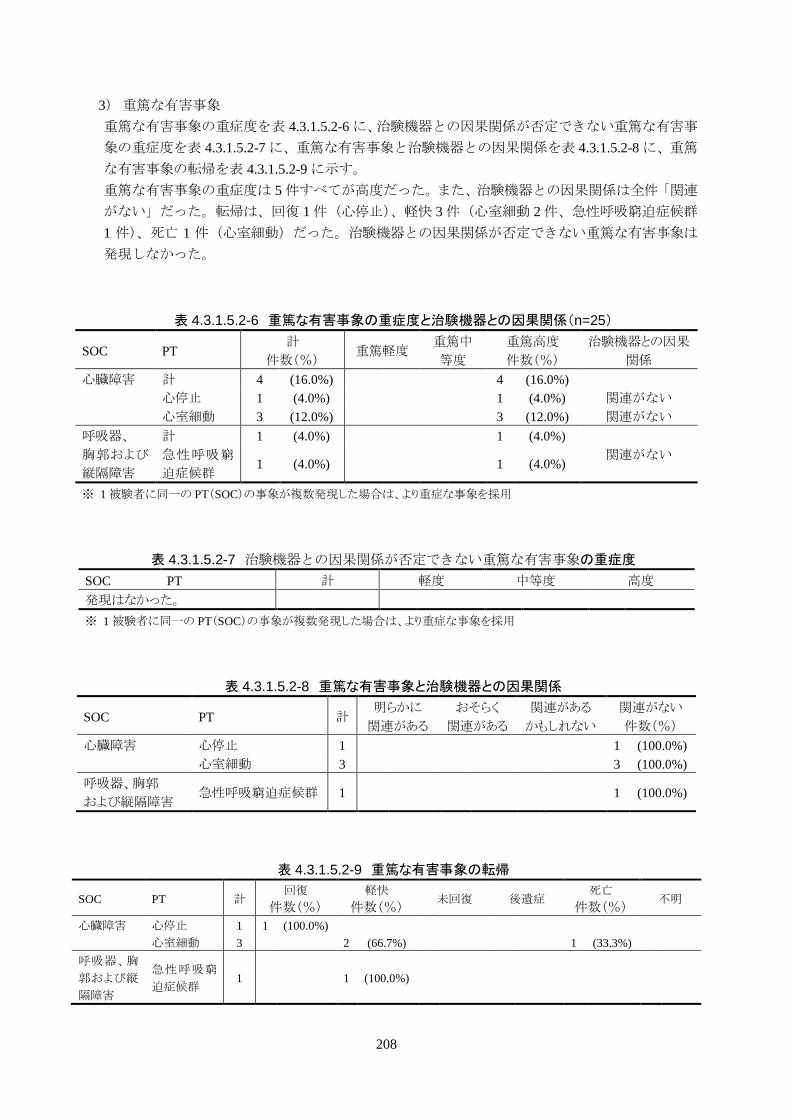
208
3) 重篤な有害事象
重篤な有害事象の重症度を表 4.3.1.5.2-6 に、治験機器との因果関係が否定できない重篤な有害事
象の重症度を表 4.3.1.5.2-7 に、重篤な有害事象と治験機器との因果関係を表 4.3.1.5.2-8 に、重篤
な有害事象の転帰を表 4.3.1.5.2-9 に示す。
重篤な有害事象の重症度は 5 件すべてが高度だった。また、治験機器との因果関係は全件「関連
がない」だった。転帰は、回復 1 件(心停止)、軽快 3 件(心室細動 2 件、急性呼吸窮迫症候群
1 件)、死亡 1 件(心室細動)だった。治験機器との因果関係が否定できない重篤な有害事象は
発現しなかった。
表 4.3.1.5.2-6 重篤な有害事象の重症度と治験機器との因果関係(n=25)
SOC PT 計
件数(%) 重篤軽度
重篤中
等度
重篤高度
件数(%)
治験機器との因果
関係
心臓障害 計 4 (16.0%) 4 (16.0%)
心停止 1 (4.0%) 1 (4.0%) 関連がない
心室細動 3 (12.0%) 3 (12.0%) 関連がない
呼吸器、 計 1 (4.0%) 1 (4.0%)
胸郭および
縦隔障害
急性呼吸窮
迫症候群 1 (4.0%) 1 (4.0%)
関連がない
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重症な事象を採用
表 4.3.1.5.2-7 治験機器との因果関係が否定できない重篤な有害事象の重症度
SOC PT 計 軽度 中等度 高度
発現はなかった。
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重症な事象を採用
表 4.3.1.5.2-8 重篤な有害事象と治験機器との因果関係
SOC PT 計 明らかに
関連がある
おそらく
関連がある
関連がある
かもしれない
関連がない
件数(%)
心臓障害 心停止 1 1 (100.0%)
心室細動 3 3 (100.0%)
呼吸器、胸郭
および縦隔障害 急性呼吸窮迫症候群 1 1 (100.0%)
表 4.3.1.5.2-9 重篤な有害事象の転帰
SOC PT 計 回復
件数(%)
軽快
件数(%) 未回復 後遺症
死亡
件数(%) 不明
心臓障害 心停止 1 1 (100.0%)
心室細動 3 2 (66.7%) 1 (33.3%)
呼吸器、胸
郭および縦
隔障害
急性呼吸窮
迫症候群 1 1 (100.0%)

209
4) 非重篤な有害事象
非重篤な有害事象の重症度を表 4.3.1.5.2-10 に、治験機器との因果関係が否定できない非重篤
な有害事象の重症度を表 4.3.1.5.2-11 に示す。
重症度が軽度で発現件数が多かった非重篤な有害事象は、肺炎 3件(12.0%)、高血圧 3 件(12.0%)
だった。中等度では、肺炎 3 件(12.0%)、血中クレアチンホスホキナーゼ増加 2 件(8.0%)が
複数件発現した有害事象だった。重症度が高度と判定された非重篤な有害事象は、痙攣、低酸
素性虚血性脳症、痰貯留がそれぞれ 1 件だった。
痙攣、低酸素性虚血性脳症は同一の被験者に発現した事象だった。いずれも原疾患(ブルガダ
症候群)により発現し、薬物療法を行ったが未回復で、転院により追跡調査を終了した。治験
機器との因果関係は関連がないと考えられた。本治験の対象となった心停止・自己心拍再開後
患者は、心停止時の虚血により低酸素脳症に至る可能性の高い被験者集団であり、集学的治療
後でも神経学的予後が不良の患者は少なくない。本治験でも、通常の経過を辿ったと判断され
た場合は、低酸素脳症を有害事象として捉えないと判断された被験者が多かった。本被験者は、
来院時の CT 検査で低酸素脳症の疑いを認め、その後、症状の悪化があったことから一連の経
過を有害事象として捉えたものだった。
痰貯留は合併症(誤嚥性肺炎)により発現した有害事象で、再挿管、人工呼吸管理等の処置に
より軽快した。治験機器との因果関係は関連がないと考えられた。
治験機器との因果関係が否定できない非重篤な有害事象の重症度は 5 件すべてが中等度だっ
た。
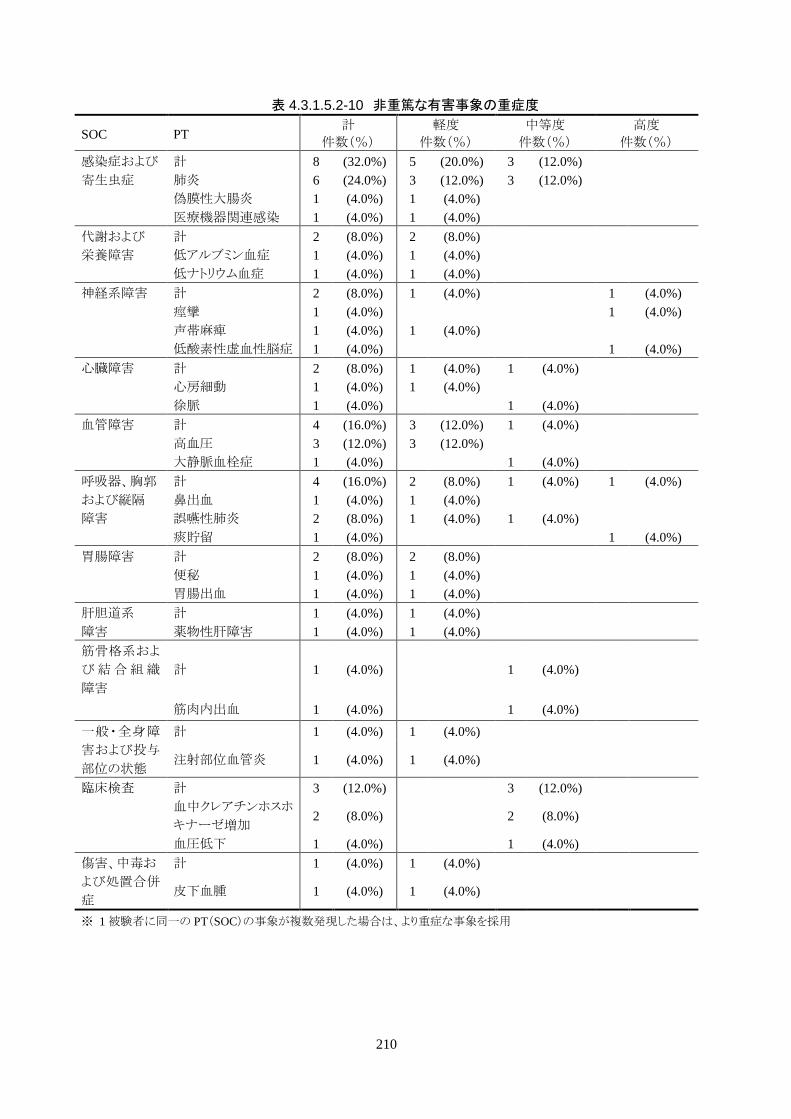
210
表 4.3.1.5.2-10 非重篤な有害事象の重症度
SOC PT 計
件数(%)
軽度
件数(%)
中等度
件数(%)
高度
件数(%)
感染症および 計 8 (32.0%) 5 (20.0%) 3 (12.0%)
寄生虫症 肺炎 6 (24.0%) 3 (12.0%) 3 (12.0%)
偽膜性大腸炎 1 (4.0%) 1 (4.0%)
医療機器関連感染 1 (4.0%) 1 (4.0%)
代謝および 計 2 (8.0%) 2 (8.0%)
栄養障害 低アルブミン血症 1 (4.0%) 1 (4.0%)
低ナトリウム血症 1 (4.0%) 1 (4.0%)
神経系障害 計 2 (8.0%) 1 (4.0%) 1 (4.0%)
痙攣 1 (4.0%) 1 (4.0%)
声帯麻痺 1 (4.0%) 1 (4.0%)
低酸素性虚血性脳症 1 (4.0%) 1 (4.0%)
心臓障害 計 2 (8.0%) 1 (4.0%) 1 (4.0%)
心房細動 1 (4.0%) 1 (4.0%)
徐脈 1 (4.0%) 1 (4.0%)
血管障害 計 4 (16.0%) 3 (12.0%) 1 (4.0%)
高血圧 3 (12.0%) 3 (12.0%)
大静脈血栓症 1 (4.0%) 1 (4.0%)
呼吸器、胸郭 計 4 (16.0%) 2 (8.0%) 1 (4.0%) 1 (4.0%)
および縦隔 鼻出血 1 (4.0%) 1 (4.0%)
障害 誤嚥性肺炎 2 (8.0%) 1 (4.0%) 1 (4.0%)
痰貯留 1 (4.0%) 1 (4.0%)
胃腸障害 計 2 (8.0%) 2 (8.0%)
便秘 1 (4.0%) 1 (4.0%)
胃腸出血 1 (4.0%) 1 (4.0%)
肝胆道系 計 1 (4.0%) 1 (4.0%)
障害 薬物性肝障害 1 (4.0%) 1 (4.0%)
筋骨格系およ
び結合組織
障害
計 1 (4.0%) 1 (4.0%)
筋肉内出血 1 (4.0%) 1 (4.0%)
一般・全身障
害および投与
部位の状態
計 1 (4.0%) 1 (4.0%)
注射部位血管炎 1 (4.0%) 1 (4.0%)
臨床検査 計 3 (12.0%) 3 (12.0%)
血中クレアチンホスホ
キナーゼ増加 2 (8.0%) 2 (8.0%)
血圧低下 1 (4.0%) 1 (4.0%)
傷害、中毒お 計 1 (4.0%) 1 (4.0%)
よび処置合併
症 皮下血腫 1 (4.0%) 1 (4.0%)
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重症な事象を採用
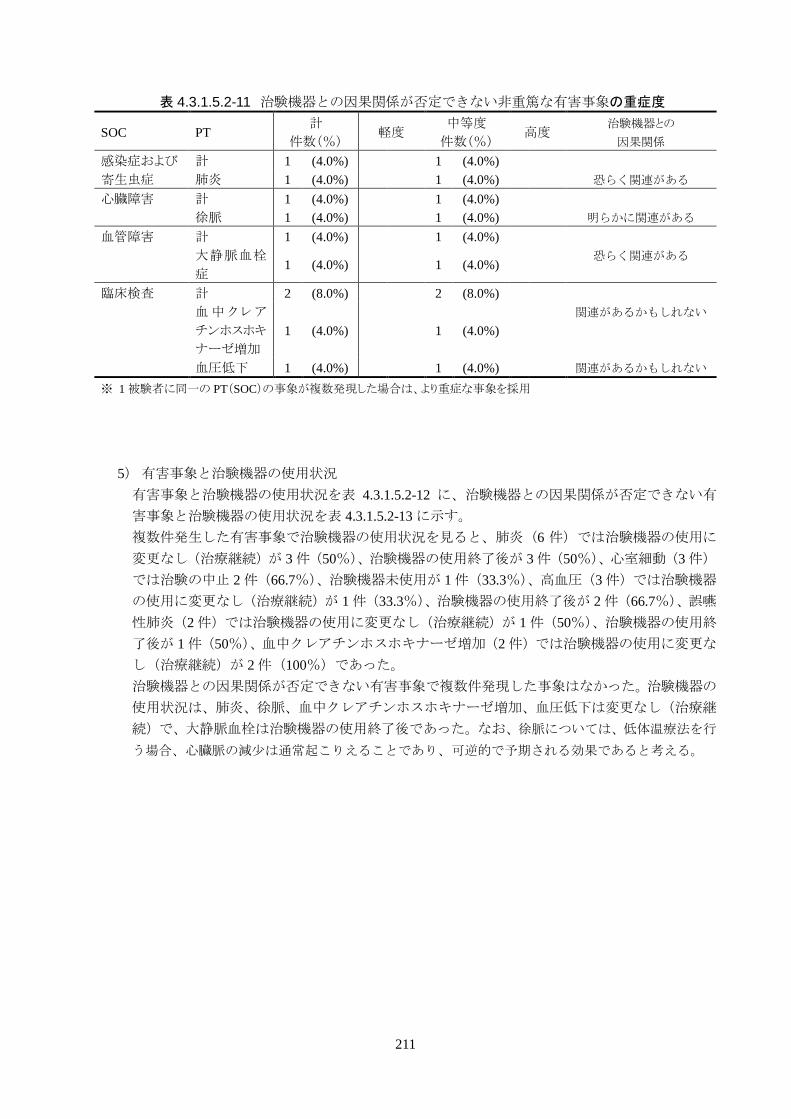
211
表 4.3.1.5.2-11 治験機器との因果関係が否定できない非重篤な有害事象の重症度
SOC PT 計
件数(%) 軽度
中等度
件数(%) 高度
治験機器との
因果関係
感染症および 計 1 (4.0%) 1 (4.0%)
寄生虫症 肺炎 1 (4.0%) 1 (4.0%) 恐らく関連がある
心臓障害 計 1 (4.0%) 1 (4.0%)
徐脈 1 (4.0%) 1 (4.0%) 明らかに関連がある
血管障害 計 1 (4.0%) 1 (4.0%)
大静脈血栓
症 1 (4.0%) 1 (4.0%)
恐らく関連がある
臨床検査 計 2 (8.0%) 2 (8.0%)
血中クレア
チンホスホキ
ナーゼ増加
1 (4.0%) 1 (4.0%)
関連があるかもしれない
血圧低下 1 (4.0%) 1 (4.0%) 関連があるかもしれない
※ 1 被験者に同一の PT(SOC)の事象が複数発現した場合は、より重症な事象を採用
5) 有害事象と治験機器の使用状況
有害事象と治験機器の使用状況を表 4.3.1.5.2-12 に、治験機器との因果関係が否定できない有
害事象と治験機器の使用状況を表 4.3.1.5.2-13 に示す。
複数件発生した有害事象で治験機器の使用状況を見ると、肺炎(6 件)では治験機器の使用に
変更なし(治療継続)が 3 件(50%)、治験機器の使用終了後が 3 件(50%)、心室細動(3 件)
では治験の中止 2 件(66.7%)、治験機器未使用が 1 件(33.3%)、高血圧(3 件)では治験機器
の使用に変更なし(治療継続)が 1 件(33.3%)、治験機器の使用終了後が 2 件(66.7%)、誤嚥
性肺炎(2 件)では治験機器の使用に変更なし(治療継続)が 1 件(50%)、治験機器の使用終
了後が 1 件(50%)、血中クレアチンホスホキナーゼ増加(2 件)では治験機器の使用に変更な
し(治療継続)が 2 件(100%)であった。
治験機器との因果関係が否定できない有害事象で複数件発現した事象はなかった。治験機器の
使用状況は、肺炎、徐脈、血中クレアチンホスホキナーゼ増加、血圧低下は変更なし(治療継
続)で、大静脈血栓は治験機器の使用終了後であった。なお、徐脈については、低体温療法を行
う場合、心臓脈の減少は通常起こりえることであり、可逆的で予期される効果であると考える。

212
表 4.3.1.5.2-12 有害事象と治験機器の使用状況
SOC PT 計 中止
件数(%)
変更なし
件数(%) 中断
終了
件数(%)
未使用
件数(%)
感染症およ
び寄生虫症
肺炎 6 3 (50.0%) 3 (50.0%)
偽膜性大腸炎 1 1 (100.0%)
医療機器関連
感染 1 1 (100.0%)
代謝および
栄養障害
低アルブミン血症 1 1 (100.0%)
低ナトリウム血症 1 1 (100.0%)
神経系障害 痙攣 1 1 (100.0%)
声帯麻痺 1 1 (100.0%)
低酸素性虚血性
脳症 1 1 (100.0%)
心臓障害 心房細動 1 1 (100.0%)
徐脈 1 1 (100.0%)
心停止 1 1 (100.0%)
心室細動 3 2 (66.7%) 1 (33.3%)
血管障害 高血圧 3 1 (33.3%) 2 (66.7%)
大静脈血栓症 1 1 (100.0%)
呼吸器、胸
郭および縦
隔障害
急性呼吸窮迫症
候群 1 1 (100.0%)
鼻出血 1 1 (100.0%)
誤嚥性肺炎 2 1 (50.0%) 1 (50.0%)
痰貯留 1 1 (100.0%)
胃腸障害 便秘 1 1 (100.0%)
胃腸出血 1 1 (100.0%)
肝胆道系障
害 薬物性肝障害 1 1 (100.0%)
筋骨格系お
よび結合組
織障害
筋肉内出血 1 1 (100.0%)
一般・全身
障害および
投与部位の
状態
注射部位血管炎 1 1 (100.0%)
臨床検査 血中クレアチンホ
スホキナーゼ増加 2 2 (100.0%)
血圧低下 1 1 (100.0%)
傷害、中毒
および処置
合併症
皮下血腫 1 1 (100.0%)
表 4.3.1.5.2-13 治験機器との因果関係が否定できない有害事象と治験機器の使用状況
SOC PT 計 中止 変更なし
件数(%) 中断
終了
件数(%) 未使用
感染症およ
び寄生虫症 肺炎 1 1 (100.0%)
心臓障害 徐脈 1 1 (100.0%)
血管障害 大静脈血栓症 1 1 (100.0%)
臨床検査 血中クレアチンホ
スホキナーゼ増加 1 1 (100.0%)
血圧低下 1 1 (100.0%)

213
6) 治験機器の不具合
治験機器の不具合の発現被験者数および発現件数を表 4.3.1.5.2-14 に、治験機器の不具合分類
(治験責任医師の評価)と有害事象の有無を表 4.3.1.5.2-15 に、治験機器の不具合一覧を表
4.3.1.5.2-16 に示す。
治験機器を使用した被験者は 24 人で、うち 2 人に治験機器の不具合が認められた。発現率は
8.3%(95%CI 1.0-27.0%)で、発現件数は 2 件だった。内訳は IVTM-SK 1 件(治験責任医師の
判断した分類:故障)、IVTM-CQ 1 件(治験責任医師の判断した分類:取り扱い説明書の不十
分な記載)で、いずれも治験機器の不具合に伴う有害事象はなかった。
なお、IVTM-CQ の不具合(カテーテルキット内のガイドワイヤの折れ曲がり)について、製造
元(治験依頼者)はカテーテル挿入時の操作方法によるもの(単発のヒューマンエラー)であ
り、取り扱いの説明書によらないものと判断した。
本治験では不具合集計とは別に、治験機器を使用した体温管理中に発生した体温のオーバーシ
ュートを集計した。低体温維持期では体温が 34.1℃以上または 31.9℃以下、正常体温維持期で
は体温が 37.6℃以上または 35.4℃以下の場合を体温のオーバーシュートと定義した。
治験機器使用中の体温のオーバーシュートは 1 件認められた。オーバーシュート時に使用して
いた治験機器の不具合はなかった。
治験機器の不具合および体温のオーバーシュートの概要を以下に示す。
<不具合>
不具合 1:IVTM-SK(被験者識別コード:******)
・発現日:**年 8 月 25 日
・治験機器適応期間:**年 8 月 22 日~**年 8 月 25 日
・不具合の状態:IVTM-SK(体外部回路)のチューブ(ローラポンプに接触する部分)の亀裂
による破損
・経過:
**年 8 月 22 日:本治験参加の文書同意を代諾者の*より取得。20 時 40 分 IVTM-CQ バ
ルーンを挿入し、レントゲンにて挿入長 43 cm を確認。21 時 10 分に低体温療法を開始。
21 時 55 分に低体温目標温度 33.8℃に到達。
8 月 23 日:5 時 55 分に低体温目標到達 8 時間後の検査を実施。22 時復温開始。
8 月 25 日:4 時に正常体温 36.2℃に到達。12 時に正常体温到達 8 時間後の検査を実施。23
時 50 分 IVTM-TG 装置本体のエアートラックアラームが鳴り出す。環流する生理食塩液が
なくなっている事に気がつき、生理食塩液を付け替えるもアラーム音は消失せず、IVTM-
CQ 側は異常がない事を確認し、治験依頼者に問い合わせの連絡を入れる。
8 月 26 日:0 時 20 分 IVTM-TG 装置本体のコンソール部を確認するとチューブ(ローラポ
ンプに接触する部分)に亀裂を発見し、治験機器の継続使用不可能と判断し正常体温維持
期 20 hr で治療終了を決定した。IVTM-CQ を抜去する(治験は継続)。抜去時後、カテー
テルに生理食塩液を注入しカテーテルのバルーンに破損がないことを確認した。0 時 30 分
カテーテル抜去時の検査を実施し、被験者の安全性に問題ないことを確認した。
・コメント:機器の不具合はチューブ(ローラポンプに接触する部分)の亀裂による破損と判
明した。カテーテル抜去後、心電図・採血・バイタルサイン測定を実施し、安全性に問題な
いことを確認した。被験者への健康被害(有害事象)はなかったが、治験機器の不具合とし
て報告する。

214
不具合 2:IVTM-CQ(被験者識別コード:******)
・発現日:**年 11 月 24 日
・治験機器適応期間:**年 11 月 24 日~**年 11 月 28 日
・不具合の状態:カテーテルキット内のガイドワイヤの折れ曲がり
・経過:
**年 11 月 24 日:本治験参加の文書同意を代諾者より取得した。右大腿静脈よりカテー
テル挿入するためのダイレーション時に、ガイドワイヤが曲がった。ガイドワイヤを抜去
し、被験者の体内に残存していないことを確認した。被験者の安全性に問題がないことを
確認した上で、新しいカテーテルキットを開封し、ガイドワイヤを交換してカテーテルを
挿入した。16 時 05 分に低体温療法を開始した。
・コメント:被験者への健康被害(有害事象)はなかったが、治験機器の不具合として報告す
る。
<体温のオーバーシュート>(被験者識別コード:******)
・発見日:**年 7 月 4 日(正常体温維持期)
・体温:37.6℃(正常体温到達から 22 時間後)
・コメント:7/4 10:50 ごろ(正常体温維持期)に定速モードに変更し、治療終了まで維持した。
筋弛緩薬投与終了や肺炎が発熱の要因と考えられる。
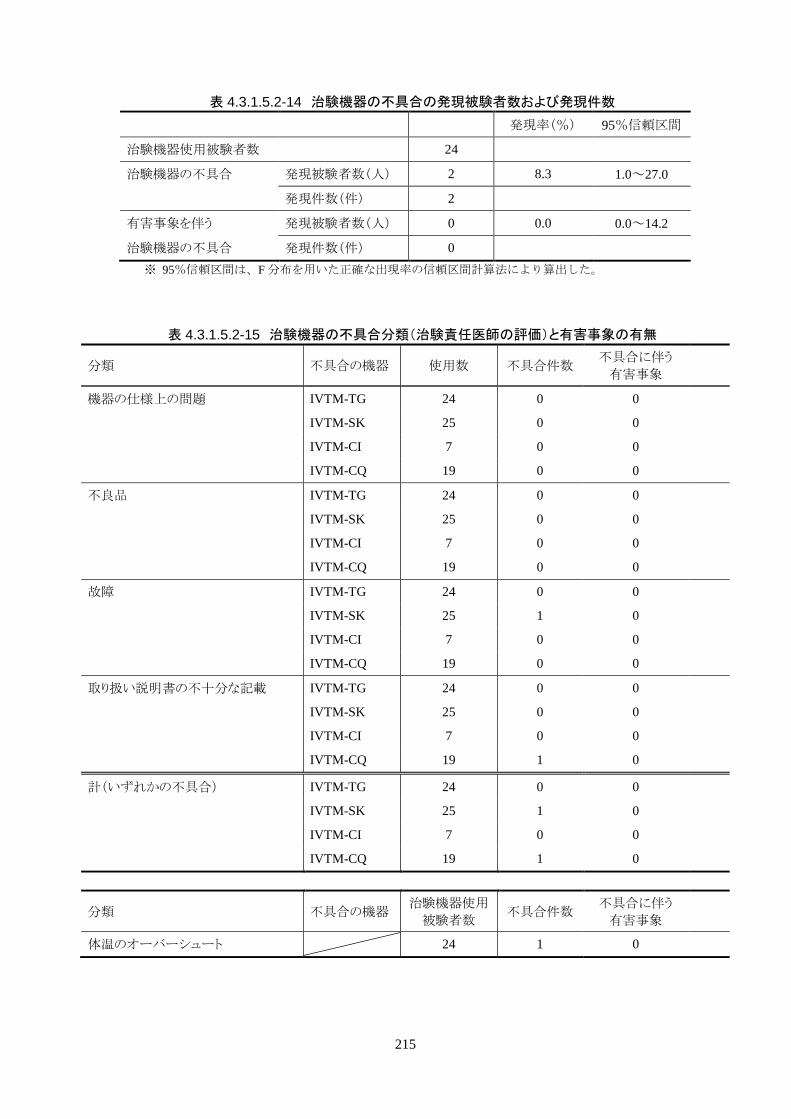
215
表 4.3.1.5.2-14 治験機器の不具合の発現被験者数および発現件数
発現率(%) 95%信頼区間
治験機器使用被験者数 24
治験機器の不具合 発現被験者数(人) 2 8.3 1.0~27.0
発現件数(件) 2
有害事象を伴う 発現被験者数(人) 0 0.0 0.0~14.2
治験機器の不具合 発現件数(件) 0
※ 95%信頼区間は、F 分布を用いた正確な出現率の信頼区間計算法により算出した。
表 4.3.1.5.2-15 治験機器の不具合分類(治験責任医師の評価)と有害事象の有無
分類 不具合の機器 使用数 不具合件数 不具合に伴う
有害事象
機器の仕様上の問題 IVTM-TG 24 0 0
IVTM-SK 25 0 0
IVTM-CI 7 0 0
IVTM-CQ 19 0 0
不良品 IVTM-TG 24 0 0
IVTM-SK 25 0 0
IVTM-CI 7 0 0
IVTM-CQ 19 0 0
故障 IVTM-TG 24 0 0
IVTM-SK 25 1 0
IVTM-CI 7 0 0
IVTM-CQ 19 0 0
取り扱い説明書の不十分な記載 IVTM-TG 24 0 0
IVTM-SK 25 0 0
IVTM-CI 7 0 0
IVTM-CQ 19 1 0
計(いずれかの不具合) IVTM-TG 24 0 0
IVTM-SK 25 1 0
IVTM-CI 7 0 0
IVTM-CQ 19 1 0
分類 不具合の機器 治験機器使用
被験者数 不具合件数
不具合に伴う
有害事象
体温のオーバーシュート 24 1 0
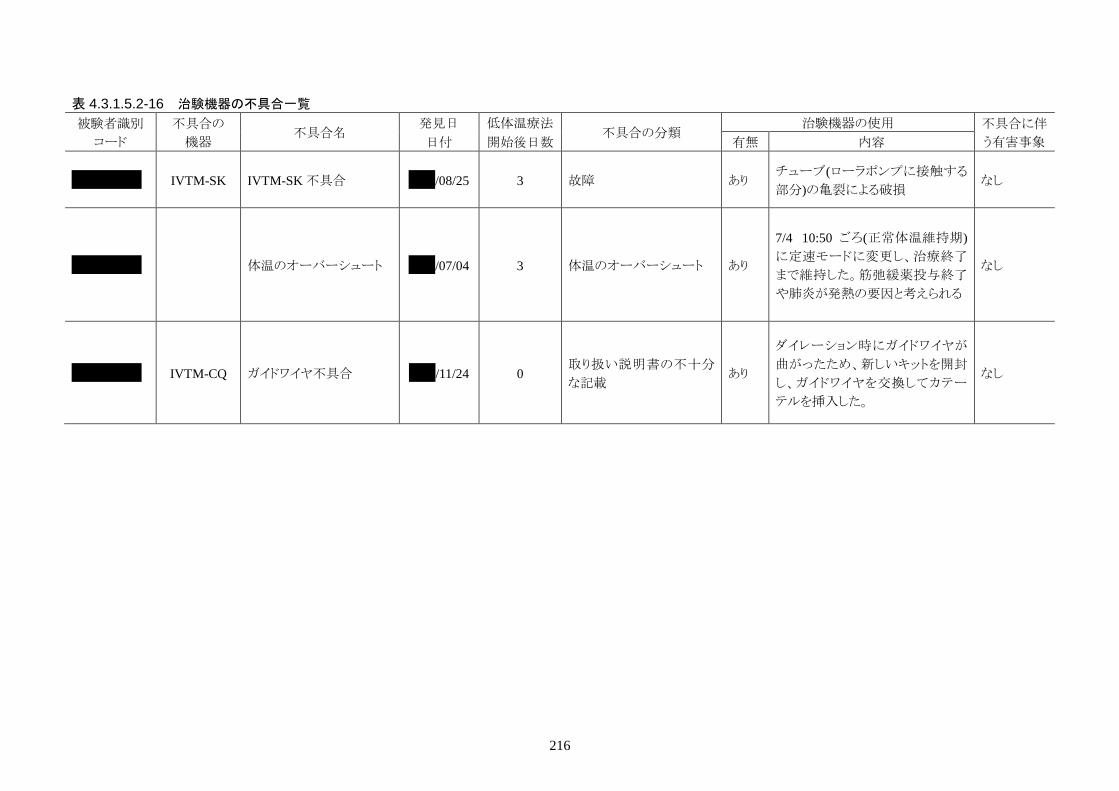
216
表 4.3.1.5.2-16 治験機器の不具合一覧
被験者識別
コード
不具合の
機器 不具合名
発見日 低体温療法 不具合の分類
治験機器の使用 不具合に伴
う有害事象 日付 開始後日数 有無 内容
***** IVTM-SK IVTM-SK不具合 **/08/25 3 故障 あり チューブ(ローラポンプに接触する
部分)の亀裂による破損 なし
***** 体温のオーバーシュート **/07/04 3 体温のオーバーシュート あり
7/4 10:50 ごろ(正常体温維持期)
に定速モードに変更し、治療終了
まで維持した。筋弛緩薬投与終了
や肺炎が発熱の要因と考えられる
なし
***** IVTM-CQ ガイドワイヤ不具合 **/11/24 0 取り扱い説明書の不十分
な記載 あり
ダイレーション時にガイドワイヤが
曲がったため、新しいキットを開封
し、ガイドワイヤを交換してカテー
テルを挿入した。
なし
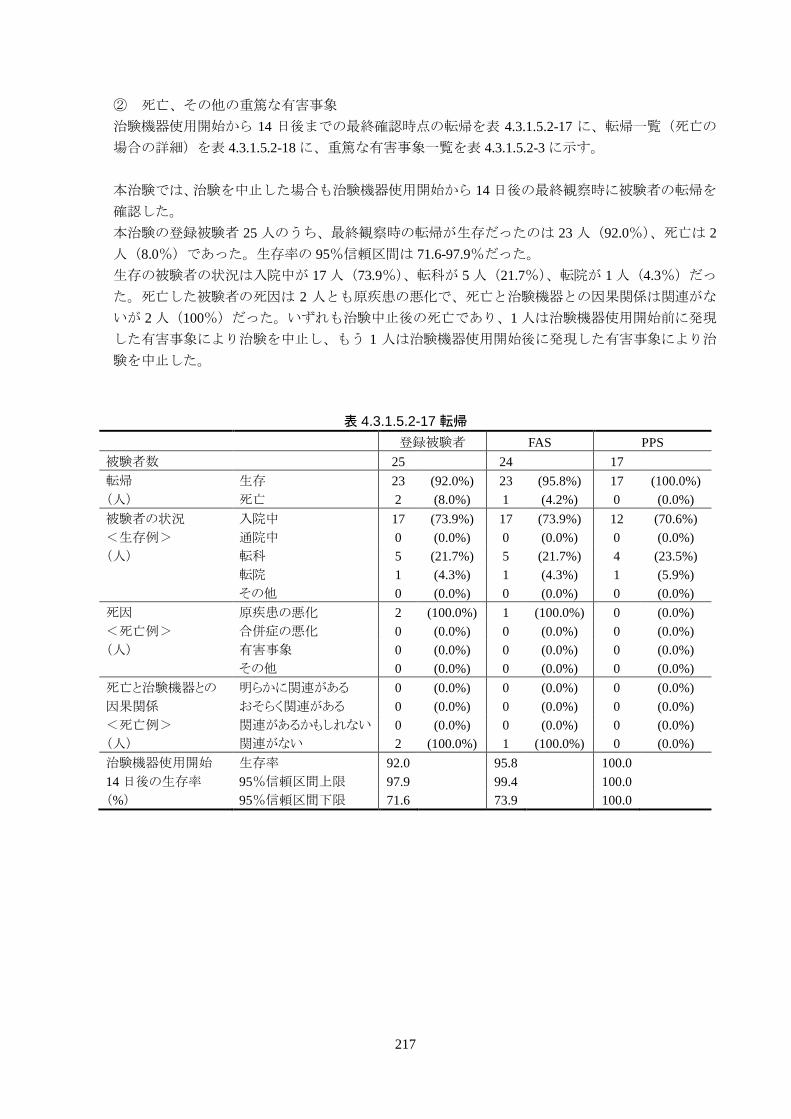
217
② 死亡、その他の重篤な有害事象
治験機器使用開始から 14 日後までの最終確認時点の転帰を表 4.3.1.5.2-17 に、転帰一覧(死亡の
場合の詳細)を表 4.3.1.5.2-18 に、重篤な有害事象一覧を表 4.3.1.5.2-3 に示す。
本治験では、治験を中止した場合も治験機器使用開始から 14 日後の最終観察時に被験者の転帰を
確認した。
本治験の登録被験者 25 人のうち、最終観察時の転帰が生存だったのは 23 人(92.0%)、死亡は 2
人(8.0%)であった。生存率の 95%信頼区間は 71.6-97.9%だった。
生存の被験者の状況は入院中が 17 人(73.9%)、転科が 5 人(21.7%)、転院が 1 人(4.3%)だっ
た。死亡した被験者の死因は 2 人とも原疾患の悪化で、死亡と治験機器との因果関係は関連がな
いが 2 人(100%)だった。いずれも治験中止後の死亡であり、1 人は治験機器使用開始前に発現
した有害事象により治験を中止し、もう 1 人は治験機器使用開始後に発現した有害事象により治
験を中止した。
表 4.3.1.5.2-17転帰
登録被験者 FAS PPS
被験者数 25 24 17
転帰 生存 23 (92.0%) 23 (95.8%) 17 (100.0%)
(人) 死亡 2 (8.0%) 1 (4.2%) 0 (0.0%)
被験者の状況 入院中 17 (73.9%) 17 (73.9%) 12 (70.6%)
<生存例> 通院中 0 (0.0%) 0 (0.0%) 0 (0.0%)
(人) 転科 5 (21.7%) 5 (21.7%) 4 (23.5%)
転院 1 (4.3%) 1 (4.3%) 1 (5.9%)
その他 0 (0.0%) 0 (0.0%) 0 (0.0%)
死因 原疾患の悪化 2 (100.0%) 1 (100.0%) 0 (0.0%)
<死亡例> 合併症の悪化 0 (0.0%) 0 (0.0%) 0 (0.0%)
(人) 有害事象 0 (0.0%) 0 (0.0%) 0 (0.0%)
その他 0 (0.0%) 0 (0.0%) 0 (0.0%)
死亡と治験機器との 明らかに関連がある 0 (0.0%) 0 (0.0%) 0 (0.0%)
因果関係 おそらく関連がある 0 (0.0%) 0 (0.0%) 0 (0.0%)
<死亡例> 関連があるかもしれない 0 (0.0%) 0 (0.0%) 0 (0.0%)
(人) 関連がない 2 (100.0%) 1 (100.0%) 0 (0.0%)
治験機器使用開始 生存率 92.0 95.8 100.0
14 日後の生存率 95%信頼区間上限 97.9 99.4 100.0
(%) 95%信頼区間下限 71.6 73.9 100.0

218
表 4.3.1.5.2-18 転帰一覧(死亡の場合の詳細)
被験者識別
コード 死亡日時 死亡時期 死因 死因の特定根拠 死亡までの経過
剖検 死亡と治験機器との因果関係
有無 所見 因果関係 因果関係の判定根拠
***** ****
***
治験機器
使用前
原 疾 患
の悪化 循環動態不全のため SAE レポート参照 なし
関連がない
と考えられる
治験機器使用前に中
止したため
***** ****
***
治 験 中
止、11 日
後
原 疾 患
の悪化
陳旧性心筋梗塞が原
因で発症した低酸素
脳症の増悪が直接の
死因と考える
*月*日に発症した心室細動により治験を中止し、
治験実施計画書の規程外の体温管理を実施した。
体温管理は問題なく施行できたが、低酸素脳症によ
る致命的なダメージが残り、死亡に至った。
なし 関連がない
と考えられる
治験開始前より低酸素
脳症を発症していたと
考えられるため

219
本治験では、「重篤な有害事象」は治験実施計画書で以下の 7 項目とした;
(1)死亡
(2)死亡のおそれ
(3)入院または入院期間の延長
(4)障害
(5)障害のおそれ
(6)上記に準じて重篤
(7)先天異常
治験期間中の被験者の死亡はなかった。死亡以外の重篤な有害事象は 5 人 5 件(心室細動 3 件、
心停止 1 件、急性呼吸窮迫症候群 1 件)が報告された。すべての重篤な有害事象は治験機器との
因果関係はないと判断された。重篤な有害事象のうち、2 人 2 件(心室細動 1 件、心停止 1 件)
は、治験機器の使用前に発現したものであった。
治験機器の使用前に発現した重篤な有害事象を除く、3 人 3 件(心室細動 2 件、急性呼吸窮迫症
候群 1 件)の重篤な有害事象の概要(重篤な有害事象及び治験機器との因果関係が否定できない
重篤な有害事象に関する報告書の最終報からの抜粋)を以下に示す。

220
1) 被験者識別コード:******(男性、193*年*月*日生まれ)
有害事象等名 心室細動(既知)
発現日 **年 8 月 13 日
重篤と判断した理由 死亡のおそれ
有害事象の転帰 軽快(転帰日**年 8 月 13 日)
治験機器適応期間 **年 8 月 13 日~8 月 13 日
有害事象発現後の措置 有(胸骨圧迫)
因果関係 有害事象と治験機器:否定できる
有害事象とその他の事項:原疾患(陳旧性心筋梗塞)
経過
**/8/13 14:32 救急車搬送にて、当院に到着。胸骨圧迫等の救命処置を施行。
14:38 心拍再開。
16:30 頃 家族より文書にて同意を取得。
全身 CT、心臓カテーテル検査等施行後、治験用カテーテル(IVTM-CI)挿
入透視下で確認。
18:10 ICU 入室。
18:43 治験機器による低体温療法を開始。
19:20 目標体温(34.0℃)に到達。
19:25 心室細動が出現 胸骨圧迫、除細動を開始。
19:27 治験機器の使用を中断。
規定された低体温療法の継続は不可能と判断し、治験を中止。
19:28 心拍再開し、転帰を『軽快』と判断。
患者の安全性を考慮し、安定した体温管理のため、治験機器を継続して使
用。
22:10 治験機器の使用を再開(34.5℃にて管理)
**/8/14 09:00 36.5℃を目標に 0.15℃/hr で復温を開始。
**/8/15 00:45 目標体温到達。
治験中止時検査を実施。
**/8/16 16:30 治験機器の使用を終了し、治験用カテーテル(IVTM-CI)を抜去。
治験中止からカテーテルを抜去するまでの間、短い不整脈は認められたが、
回数は激減し、致命的な不整脈は確認されなかった。
コメント 治験機器の不具合等は確認されず、有害事象との因果関係は否定
できる。原疾患の陳旧性心筋梗塞により、低体温療法による心室細
動が発生しやすい状況であった。

221
2) 被験者識別コード:******(男性、194*年*月*日生まれ)
有害事象等名 心室細動(既知)
発現日 **年 11 月 18 日
重篤と判断した理由 死亡のおそれ
有害事象の転帰 軽快(**年 11 月 18 日)
治験機器適応期間 **年 11 月 18 日~**年 11 月 18 日
有害事象発現後の措置 有(胸骨圧迫・除細動)
因果関係 有害事象と治験機器:否定できる
経過
**/11/18 13:30 *****************************救
急要請。
13:37 救急隊現着時 CPA、瞳孔対光反射なし。
13:40 モニター上 VF のため除細動施行、その後 PEA。エピネフリン投与。
13:53 心拍再開、BP149/109、HR130。
14:20 当院着。CT 撮影。CAG の結果、#7 の PCI 実施。
18:53 低体温療法開始。
19:33 目標体温到達。
19:40 VF 出現に対して胸骨圧迫実施。治験を一時中断。胸部圧迫にて sinus
rhythm に復帰したが、再び VF が出現し除細動にて sinus rhythm に復帰。
20:08 治験の続行は不可能と医師の判断にて中止。
治験機器は継続して使用し、36.5℃に復温。
**/11/22 11:00 カテーテルを抜去。治験機器の使用を終了し、治験用カテーテル
(IVTM-CI)を抜去。治験中止からカテーテルを抜去するまで、致死的な
不整脈は確認されなかった。
コメント 治験機器の不具合などは確認されず、原疾患の急性冠症候群のた
め低体温療法による心室細動が発生しやすい状況であった。した
がって、本治験と有害事象との因果関係は否定できる。

222
3) 被験者識別コード:******(女性、194*年*月*日生まれ)
有害事象等名 急性呼吸窮迫症候群(未知)
発現日 **年 9 月 6 日
重篤と判断した理由 上記に準じて重篤
有害事象の転帰 軽快(**年 9 月 25 日)
治験機器適応期間 **年 8 月 26 日~**年 8 月 30 日
有害事象発現後の措置 ‐
因果関係 有害事象と治験機器:否定できる
経過
**/8/26 ***********心肺停止状態で近医搬送。
心拍再開し精査・治療目的で当院転院搬送。
**/8/26 治験参加に同意され、治験機器カテーテル留置、低体温療法開始。
**/8/27 復温開始。
**/8/30 低体温療法終了、治験機器カテーテル抜去。
**/9/2 呼吸・循環動態安定。意識レベルが回復し気管内チューブ抜去(人工呼吸器
離脱)。
**/9/6 呼吸状態悪化し再挿管、人工呼吸管理再開。
呼吸窮迫症候群(ARDS)と診断されソル・メドロール開始。肺水腫あり利
尿剤投与。
**/9/9 CT 所見:ARDS、心原性肺うっ血混在。誤嚥性肺炎の合併も否定しかねる。
人工呼吸管理、治療継続。
治験機器使用開始 14 日後調査実施。
**/9/18 呼吸状態改善し再抜管。
**/9/25 経鼻 1L/分酸素投与にて血液ガス分析良好。
コメント 治験機器抜去後 6 日経過しており、時間的な相関がないため因果
関係は否定できる。
本事象の原因は不明だが、炎症に伴う病態と考えられる。

223
③ 臨床検査値の評価
臨床検査:血液学的検査を表 4.3.1.5.2-19 に、臨床検査:血液生化学的検査を表 4.3.1.5.2-20 に、臨
床検査:凝固・線溶系検査を表 4.3.1.5.2-21 に、臨床検査:動脈血液ガス分析を表 4.3.1.5.2-22 に、
臨床検査:免疫血清学的検査を表 4.3.1.5.2-23 に、臨床検査:サイトカインを表 4.3.1.5.2-24 に示
す。
臨床検査値に関する有害事象は、血中クレアチンホスホキナーゼ増加の 2 件だった。
臨床検査値で異常変動が多かったのは CRP(56.5%)、プロカルシトニン(43.5%)であった。い
ずれの検査値も登録時から徐々に上昇し、治療中または中止時にピークを迎え、その後徐々に低
下していた。
サイトカインは IL-4、IL-1β、IL-10、IL-6、HMGB1、TNF-αの 6 項目を登録時、目標体温到達時
から 8 時間後(維持期)、正常体温到達時から 8 時間後(正常体温維持期)の 3 回測定した。登録
時データから変化が認められたのは IL-10 と HMGB1 で登録時、目標体温到達時から 8 時間後、
正常体温到達時から 8 時間後のそれぞれの値(いずれも中央値)は、IL-10 が 33.100 pg/mL、5.490
pg/mL、3.635 pg/mL、HMGB1 が 10.30 ng/mL、6.25 ng/mL、5.20 ng/mL だった。
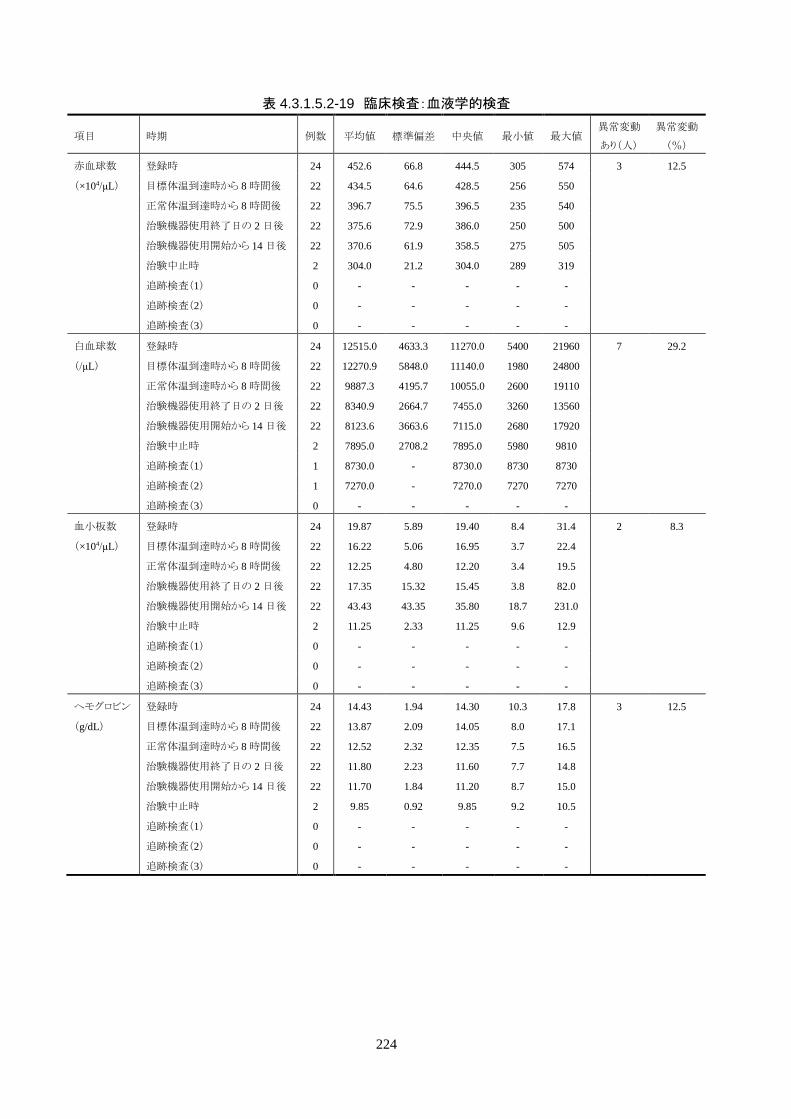
224
表 4.3.1.5.2-19 臨床検査:血液学的検査
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
赤血球数 登録時 24 452.6 66.8 444.5 305 574 3 12.5
(×104/μL) 目標体温到達時から 8 時間後 22 434.5 64.6 428.5 256 550
正常体温到達時から 8 時間後 22 396.7 75.5 396.5 235 540
治験機器使用終了日の 2 日後 22 375.6 72.9 386.0 250 500
治験機器使用開始から 14 日後 22 370.6 61.9 358.5 275 505
治験中止時 2 304.0 21.2 304.0 289 319
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
白血球数 登録時 24 12515.0 4633.3 11270.0 5400 21960 7 29.2
(/μL) 目標体温到達時から 8 時間後 22 12270.9 5848.0 11140.0 1980 24800
正常体温到達時から 8 時間後 22 9887.3 4195.7 10055.0 2600 19110
治験機器使用終了日の 2 日後 22 8340.9 2664.7 7455.0 3260 13560
治験機器使用開始から 14 日後 22 8123.6 3663.6 7115.0 2680 17920
治験中止時 2 7895.0 2708.2 7895.0 5980 9810
追跡検査(1) 1 8730.0 - 8730.0 8730 8730
追跡検査(2) 1 7270.0 - 7270.0 7270 7270
追跡検査(3) 0 - - - - -
血小板数 登録時 24 19.87 5.89 19.40 8.4 31.4 2 8.3
(×104/μL) 目標体温到達時から 8 時間後 22 16.22 5.06 16.95 3.7 22.4
正常体温到達時から 8 時間後 22 12.25 4.80 12.20 3.4 19.5
治験機器使用終了日の 2 日後 22 17.35 15.32 15.45 3.8 82.0
治験機器使用開始から 14 日後 22 43.43 43.35 35.80 18.7 231.0
治験中止時 2 11.25 2.33 11.25 9.6 12.9
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
ヘモグロビン 登録時 24 14.43 1.94 14.30 10.3 17.8 3 12.5
(g/dL) 目標体温到達時から 8 時間後 22 13.87 2.09 14.05 8.0 17.1
正常体温到達時から 8 時間後 22 12.52 2.32 12.35 7.5 16.5
治験機器使用終了日の 2 日後 22 11.80 2.23 11.60 7.7 14.8
治験機器使用開始から 14 日後 22 11.70 1.84 11.20 8.7 15.0
治験中止時 2 9.85 0.92 9.85 9.2 10.5
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
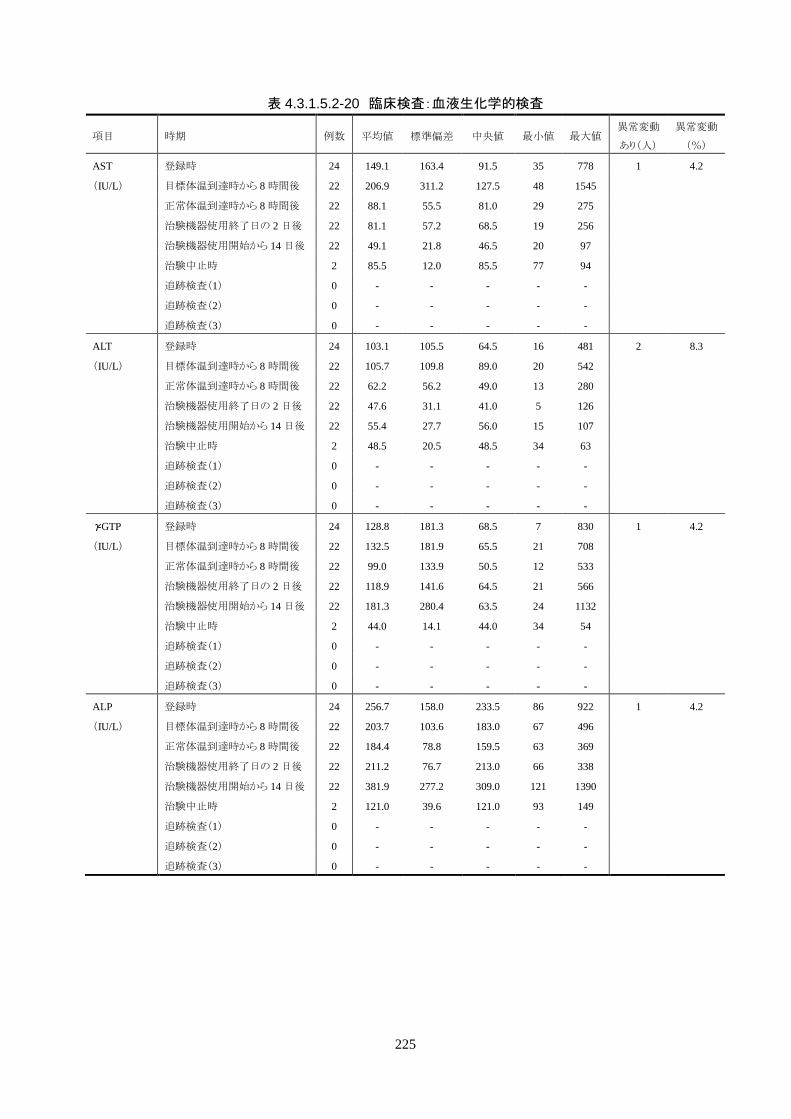
225
表 4.3.1.5.2-20 臨床検査:血液生化学的検査
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
AST 登録時 24 149.1 163.4 91.5 35 778 1 4.2
(IU/L) 目標体温到達時から 8 時間後 22 206.9 311.2 127.5 48 1545
正常体温到達時から 8 時間後 22 88.1 55.5 81.0 29 275
治験機器使用終了日の 2 日後 22 81.1 57.2 68.5 19 256
治験機器使用開始から 14 日後 22 49.1 21.8 46.5 20 97
治験中止時 2 85.5 12.0 85.5 77 94
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
ALT 登録時 24 103.1 105.5 64.5 16 481 2 8.3
(IU/L) 目標体温到達時から 8 時間後 22 105.7 109.8 89.0 20 542
正常体温到達時から 8 時間後 22 62.2 56.2 49.0 13 280
治験機器使用終了日の 2 日後 22 47.6 31.1 41.0 5 126
治験機器使用開始から 14 日後 22 55.4 27.7 56.0 15 107
治験中止時 2 48.5 20.5 48.5 34 63
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
γ-GTP 登録時 24 128.8 181.3 68.5 7 830 1 4.2
(IU/L) 目標体温到達時から 8 時間後 22 132.5 181.9 65.5 21 708
正常体温到達時から 8 時間後 22 99.0 133.9 50.5 12 533
治験機器使用終了日の 2 日後 22 118.9 141.6 64.5 21 566
治験機器使用開始から 14 日後 22 181.3 280.4 63.5 24 1132
治験中止時 2 44.0 14.1 44.0 34 54
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
ALP 登録時 24 256.7 158.0 233.5 86 922 1 4.2
(IU/L) 目標体温到達時から 8 時間後 22 203.7 103.6 183.0 67 496
正常体温到達時から 8 時間後 22 184.4 78.8 159.5 63 369
治験機器使用終了日の 2 日後 22 211.2 76.7 213.0 66 338
治験機器使用開始から 14 日後 22 381.9 277.2 309.0 121 1390
治験中止時 2 121.0 39.6 121.0 93 149
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
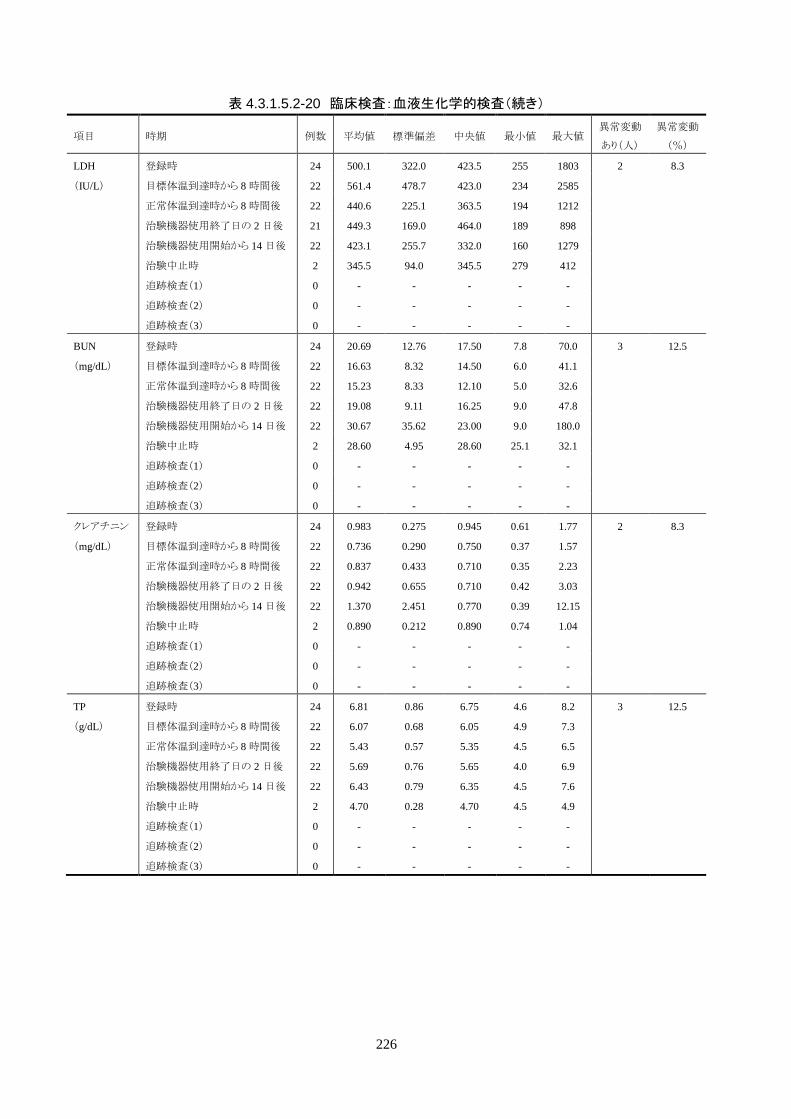
226
表 4.3.1.5.2-20 臨床検査:血液生化学的検査(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
LDH 登録時 24 500.1 322.0 423.5 255 1803 2 8.3
(IU/L) 目標体温到達時から 8 時間後 22 561.4 478.7 423.0 234 2585
正常体温到達時から 8 時間後 22 440.6 225.1 363.5 194 1212
治験機器使用終了日の 2 日後 21 449.3 169.0 464.0 189 898
治験機器使用開始から 14 日後 22 423.1 255.7 332.0 160 1279
治験中止時 2 345.5 94.0 345.5 279 412
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
BUN 登録時 24 20.69 12.76 17.50 7.8 70.0 3 12.5
(mg/dL) 目標体温到達時から 8 時間後 22 16.63 8.32 14.50 6.0 41.1
正常体温到達時から 8 時間後 22 15.23 8.33 12.10 5.0 32.6
治験機器使用終了日の 2 日後 22 19.08 9.11 16.25 9.0 47.8
治験機器使用開始から 14 日後 22 30.67 35.62 23.00 9.0 180.0
治験中止時 2 28.60 4.95 28.60 25.1 32.1
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
クレアチニン 登録時 24 0.983 0.275 0.945 0.61 1.77 2 8.3
(mg/dL) 目標体温到達時から 8 時間後 22 0.736 0.290 0.750 0.37 1.57
正常体温到達時から 8 時間後 22 0.837 0.433 0.710 0.35 2.23
治験機器使用終了日の 2 日後 22 0.942 0.655 0.710 0.42 3.03
治験機器使用開始から 14 日後 22 1.370 2.451 0.770 0.39 12.15
治験中止時 2 0.890 0.212 0.890 0.74 1.04
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
TP 登録時 24 6.81 0.86 6.75 4.6 8.2 3 12.5
(g/dL) 目標体温到達時から 8 時間後 22 6.07 0.68 6.05 4.9 7.3
正常体温到達時から 8 時間後 22 5.43 0.57 5.35 4.5 6.5
治験機器使用終了日の 2 日後 22 5.69 0.76 5.65 4.0 6.9
治験機器使用開始から 14 日後 22 6.43 0.79 6.35 4.5 7.6
治験中止時 2 4.70 0.28 4.70 4.5 4.9
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
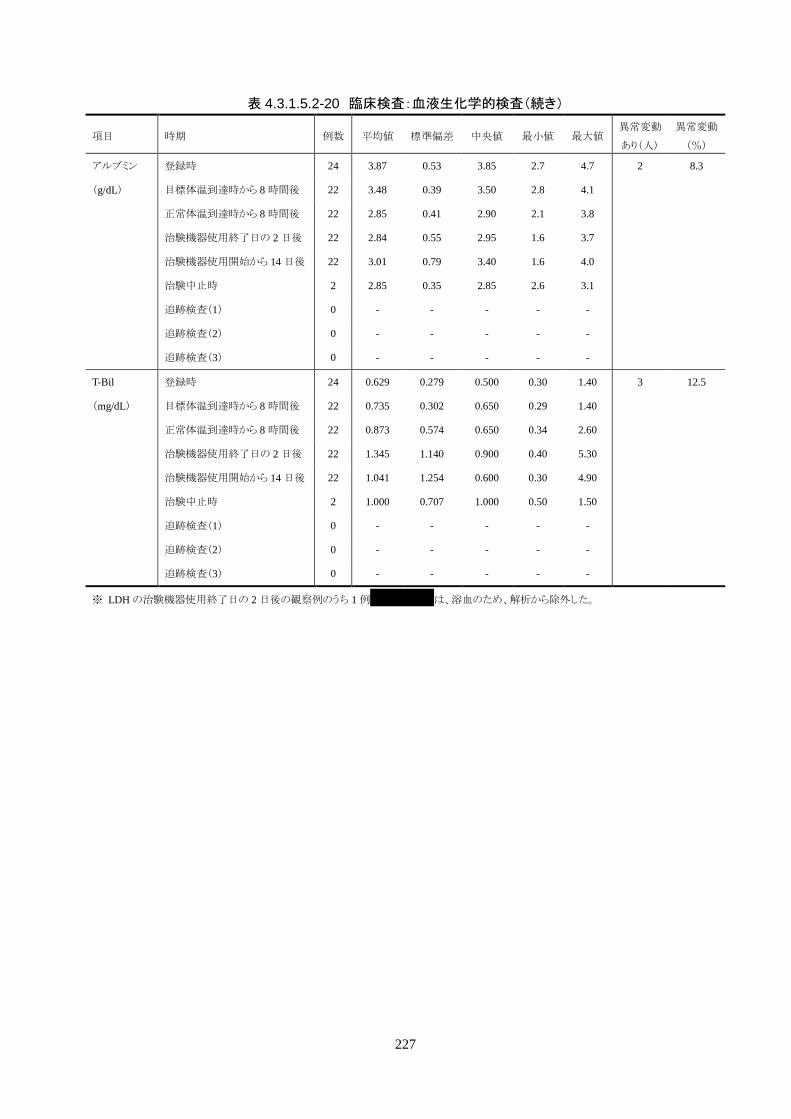
227
表 4.3.1.5.2-20 臨床検査:血液生化学的検査(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
アルブミン 登録時 24 3.87 0.53 3.85 2.7 4.7 2 8.3
(g/dL) 目標体温到達時から 8 時間後 22 3.48 0.39 3.50 2.8 4.1
正常体温到達時から 8 時間後 22 2.85 0.41 2.90 2.1 3.8
治験機器使用終了日の 2 日後 22 2.84 0.55 2.95 1.6 3.7
治験機器使用開始から 14 日後 22 3.01 0.79 3.40 1.6 4.0
治験中止時 2 2.85 0.35 2.85 2.6 3.1
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
T-Bil 登録時 24 0.629 0.279 0.500 0.30 1.40 3 12.5
(mg/dL) 目標体温到達時から 8 時間後 22 0.735 0.302 0.650 0.29 1.40
正常体温到達時から 8 時間後 22 0.873 0.574 0.650 0.34 2.60
治験機器使用終了日の 2 日後 22 1.345 1.140 0.900 0.40 5.30
治験機器使用開始から 14 日後 22 1.041 1.254 0.600 0.30 4.90
治験中止時 2 1.000 0.707 1.000 0.50 1.50
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
※ LDHの治験機器使用終了日の 2 日後の観察例のうち 1 例******は、溶血のため、解析から除外した。
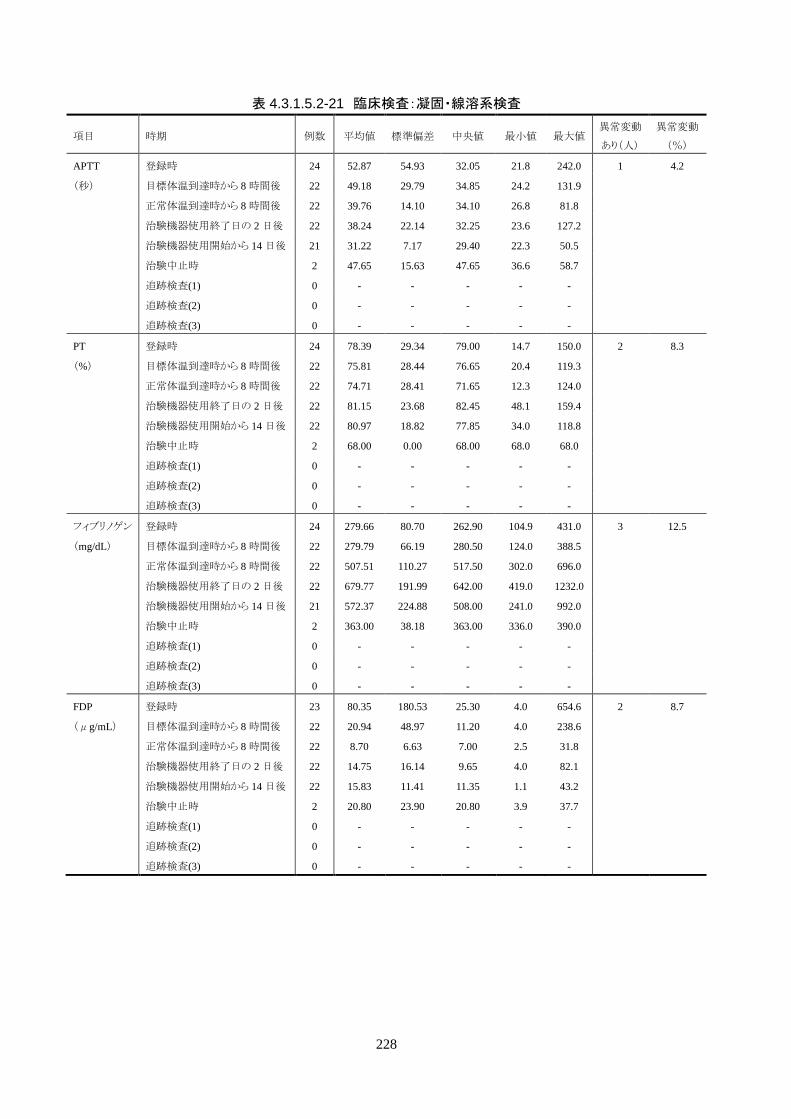
228
表 4.3.1.5.2-21 臨床検査:凝固・線溶系検査
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
APTT 登録時 24 52.87 54.93 32.05 21.8 242.0 1 4.2
(秒) 目標体温到達時から 8 時間後 22 49.18 29.79 34.85 24.2 131.9
正常体温到達時から 8 時間後 22 39.76 14.10 34.10 26.8 81.8
治験機器使用終了日の 2 日後 22 38.24 22.14 32.25 23.6 127.2
治験機器使用開始から 14 日後 21 31.22 7.17 29.40 22.3 50.5
治験中止時 2 47.65 15.63 47.65 36.6 58.7
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
PT 登録時 24 78.39 29.34 79.00 14.7 150.0 2 8.3
(%) 目標体温到達時から 8 時間後 22 75.81 28.44 76.65 20.4 119.3
正常体温到達時から 8 時間後 22 74.71 28.41 71.65 12.3 124.0
治験機器使用終了日の 2 日後 22 81.15 23.68 82.45 48.1 159.4
治験機器使用開始から 14 日後 22 80.97 18.82 77.85 34.0 118.8
治験中止時 2 68.00 0.00 68.00 68.0 68.0
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
フィブリノゲン 登録時 24 279.66 80.70 262.90 104.9 431.0 3 12.5
(mg/dL) 目標体温到達時から 8 時間後 22 279.79 66.19 280.50 124.0 388.5
正常体温到達時から 8 時間後 22 507.51 110.27 517.50 302.0 696.0
治験機器使用終了日の 2 日後 22 679.77 191.99 642.00 419.0 1232.0
治験機器使用開始から 14 日後 21 572.37 224.88 508.00 241.0 992.0
治験中止時 2 363.00 38.18 363.00 336.0 390.0
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
FDP 登録時 23 80.35 180.53 25.30 4.0 654.6 2 8.7
(μg/mL) 目標体温到達時から 8 時間後 22 20.94 48.97 11.20 4.0 238.6
正常体温到達時から 8 時間後 22 8.70 6.63 7.00 2.5 31.8
治験機器使用終了日の 2 日後 22 14.75 16.14 9.65 4.0 82.1
治験機器使用開始から 14 日後 22 15.83 11.41 11.35 1.1 43.2
治験中止時 2 20.80 23.90 20.80 3.9 37.7
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
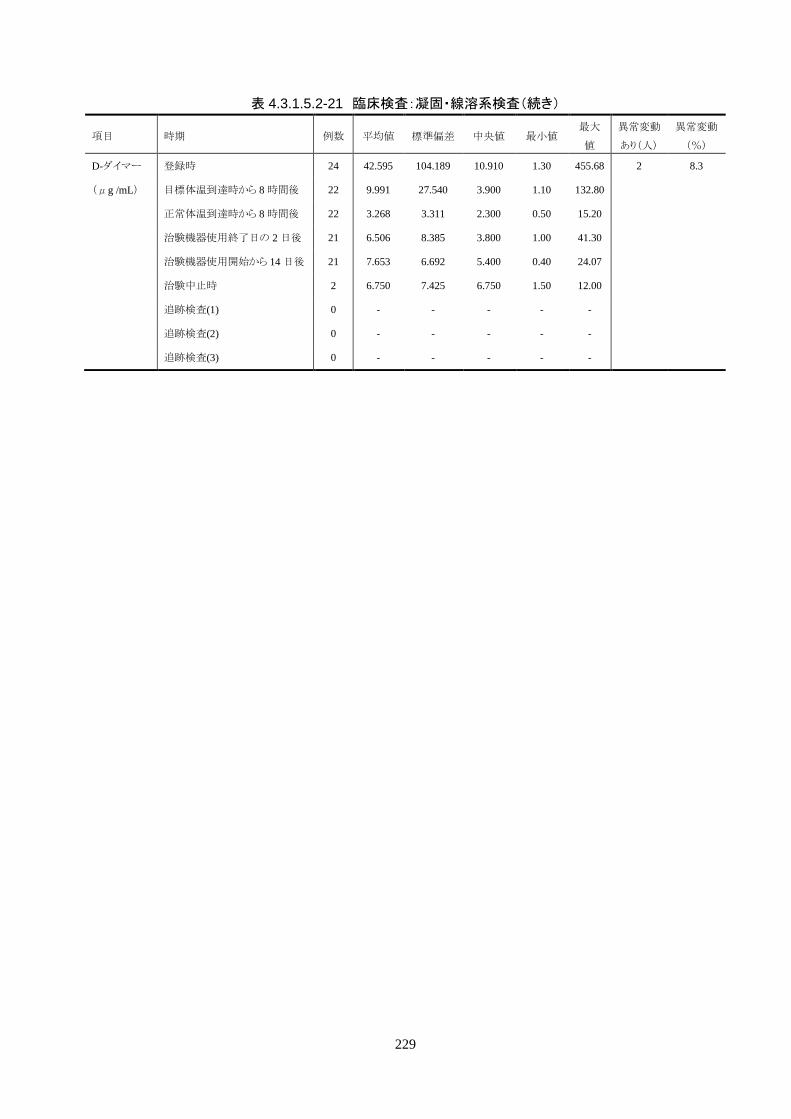
229
表 4.3.1.5.2-21 臨床検査:凝固・線溶系検査(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大
値
異常変動
あり(人)
異常変動
(%)
D-ダイマー 登録時 24 42.595 104.189 10.910 1.30 455.68 2 8.3
(μg /mL) 目標体温到達時から 8 時間後 22 9.991 27.540 3.900 1.10 132.80
正常体温到達時から 8 時間後 22 3.268 3.311 2.300 0.50 15.20
治験機器使用終了日の 2 日後 21 6.506 8.385 3.800 1.00 41.30
治験機器使用開始から 14 日後 21 7.653 6.692 5.400 0.40 24.07
治験中止時 2 6.750 7.425 6.750 1.50 12.00
追跡検査(1) 0 - - - - -
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
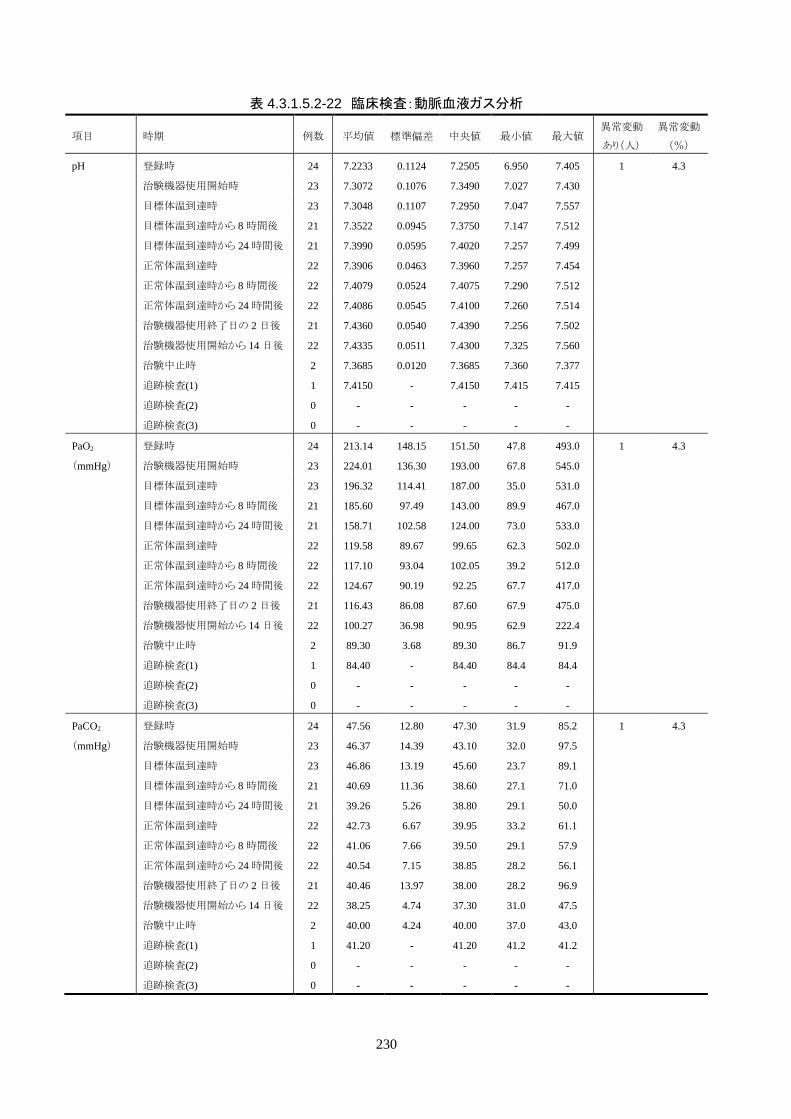
230
表 4.3.1.5.2-22 臨床検査:動脈血液ガス分析
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
pH 登録時 24 7.2233 0.1124 7.2505 6.950 7.405 1 4.3
治験機器使用開始時 23 7.3072 0.1076 7.3490 7.027 7.430
目標体温到達時 23 7.3048 0.1107 7.2950 7.047 7.557
目標体温到達時から 8 時間後 21 7.3522 0.0945 7.3750 7.147 7.512
目標体温到達時から 24時間後 21 7.3990 0.0595 7.4020 7.257 7.499
正常体温到達時 22 7.3906 0.0463 7.3960 7.257 7.454
正常体温到達時から 8 時間後 22 7.4079 0.0524 7.4075 7.290 7.512
正常体温到達時から 24時間後 22 7.4086 0.0545 7.4100 7.260 7.514
治験機器使用終了日の 2 日後 21 7.4360 0.0540 7.4390 7.256 7.502
治験機器使用開始から 14 日後 22 7.4335 0.0511 7.4300 7.325 7.560
治験中止時 2 7.3685 0.0120 7.3685 7.360 7.377
追跡検査(1) 1 7.4150 - 7.4150 7.415 7.415
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
PaO2 登録時 24 213.14 148.15 151.50 47.8 493.0 1 4.3
(mmHg) 治験機器使用開始時 23 224.01 136.30 193.00 67.8 545.0
目標体温到達時 23 196.32 114.41 187.00 35.0 531.0
目標体温到達時から 8 時間後 21 185.60 97.49 143.00 89.9 467.0
目標体温到達時から 24時間後 21 158.71 102.58 124.00 73.0 533.0
正常体温到達時 22 119.58 89.67 99.65 62.3 502.0
正常体温到達時から 8 時間後 22 117.10 93.04 102.05 39.2 512.0
正常体温到達時から 24時間後 22 124.67 90.19 92.25 67.7 417.0
治験機器使用終了日の 2 日後 21 116.43 86.08 87.60 67.9 475.0
治験機器使用開始から 14 日後 22 100.27 36.98 90.95 62.9 222.4
治験中止時 2 89.30 3.68 89.30 86.7 91.9
追跡検査(1) 1 84.40 - 84.40 84.4 84.4
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
PaCO2 登録時 24 47.56 12.80 47.30 31.9 85.2 1 4.3
(mmHg) 治験機器使用開始時 23 46.37 14.39 43.10 32.0 97.5
目標体温到達時 23 46.86 13.19 45.60 23.7 89.1
目標体温到達時から 8 時間後 21 40.69 11.36 38.60 27.1 71.0
目標体温到達時から 24時間後 21 39.26 5.26 38.80 29.1 50.0
正常体温到達時 22 42.73 6.67 39.95 33.2 61.1
正常体温到達時から 8 時間後 22 41.06 7.66 39.50 29.1 57.9
正常体温到達時から 24時間後 22 40.54 7.15 38.85 28.2 56.1
治験機器使用終了日の 2 日後 21 40.46 13.97 38.00 28.2 96.9
治験機器使用開始から 14 日後 22 38.25 4.74 37.30 31.0 47.5
治験中止時 2 40.00 4.24 40.00 37.0 43.0
追跡検査(1) 1 41.20 - 41.20 41.2 41.2
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
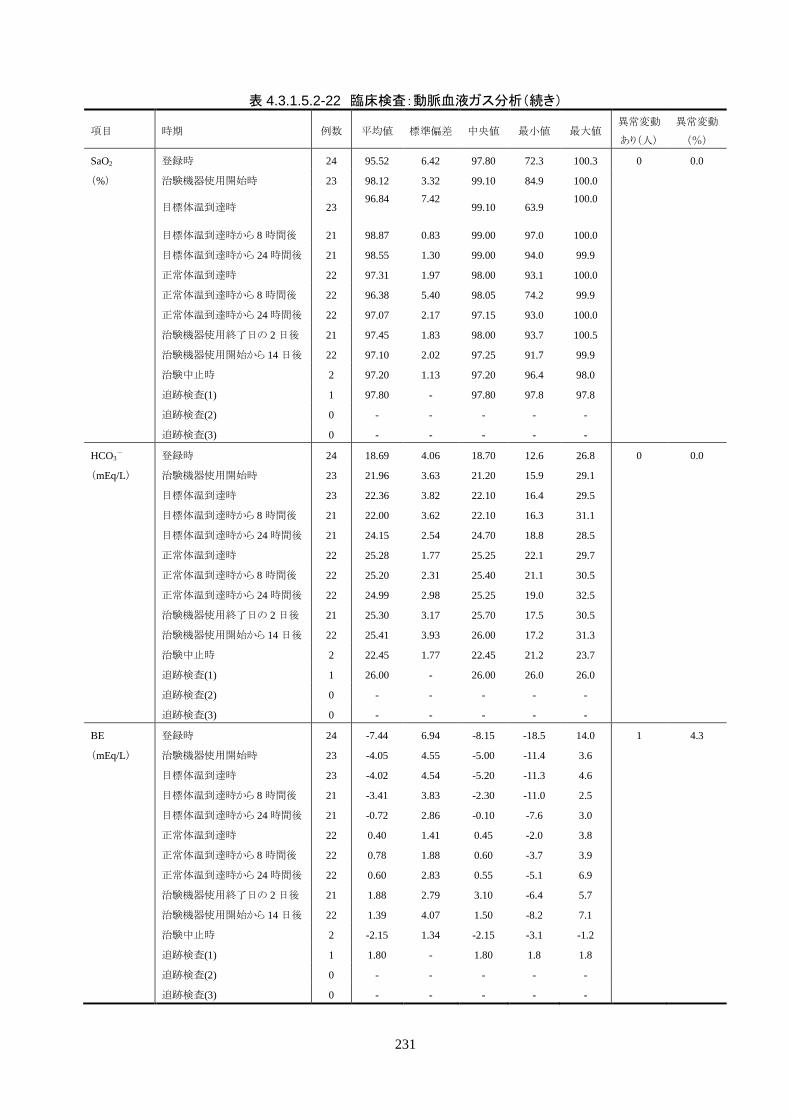
231
表 4.3.1.5.2-22 臨床検査:動脈血液ガス分析(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
SaO2 登録時 24 95.52 6.42 97.80 72.3 100.3 0 0.0
(%) 治験機器使用開始時 23 98.12 3.32 99.10 84.9 100.0
目標体温到達時 23 96.84
7.42
99.10 63.9
100.0
目標体温到達時から 8 時間後 21 98.87 0.83 99.00 97.0 100.0
目標体温到達時から 24時間後 21 98.55 1.30 99.00 94.0 99.9
正常体温到達時 22 97.31 1.97 98.00 93.1 100.0
正常体温到達時から 8 時間後 22 96.38 5.40 98.05 74.2 99.9
正常体温到達時から 24時間後 22 97.07 2.17 97.15 93.0 100.0
治験機器使用終了日の 2 日後 21 97.45 1.83 98.00 93.7 100.5
治験機器使用開始から 14 日後 22 97.10 2.02 97.25 91.7 99.9
治験中止時 2 97.20 1.13 97.20 96.4 98.0
追跡検査(1) 1 97.80 - 97.80 97.8 97.8
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
HCO3- 登録時 24 18.69 4.06 18.70 12.6 26.8 0 0.0
(mEq/L) 治験機器使用開始時 23 21.96 3.63 21.20 15.9 29.1
目標体温到達時 23 22.36 3.82 22.10 16.4 29.5
目標体温到達時から 8 時間後 21 22.00 3.62 22.10 16.3 31.1
目標体温到達時から 24時間後 21 24.15 2.54 24.70 18.8 28.5
正常体温到達時 22 25.28 1.77 25.25 22.1 29.7
正常体温到達時から 8 時間後 22 25.20 2.31 25.40 21.1 30.5
正常体温到達時から 24時間後 22 24.99 2.98 25.25 19.0 32.5
治験機器使用終了日の 2 日後 21 25.30 3.17 25.70 17.5 30.5
治験機器使用開始から 14 日後 22 25.41 3.93 26.00 17.2 31.3
治験中止時 2 22.45 1.77 22.45 21.2 23.7
追跡検査(1) 1 26.00 - 26.00 26.0 26.0
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
BE 登録時 24 -7.44 6.94 -8.15 -18.5 14.0 1 4.3
(mEq/L) 治験機器使用開始時 23 -4.05 4.55 -5.00 -11.4 3.6
目標体温到達時 23 -4.02 4.54 -5.20 -11.3 4.6
目標体温到達時から 8 時間後 21 -3.41 3.83 -2.30 -11.0 2.5
目標体温到達時から 24時間後 21 -0.72 2.86 -0.10 -7.6 3.0
正常体温到達時 22 0.40 1.41 0.45 -2.0 3.8
正常体温到達時から 8 時間後 22 0.78 1.88 0.60 -3.7 3.9
正常体温到達時から 24時間後 22 0.60 2.83 0.55 -5.1 6.9
治験機器使用終了日の 2 日後 21 1.88 2.79 3.10 -6.4 5.7
治験機器使用開始から 14 日後 22 1.39 4.07 1.50 -8.2 7.1
治験中止時 2 -2.15 1.34 -2.15 -3.1 -1.2
追跡検査(1) 1 1.80 - 1.80 1.8 1.8
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
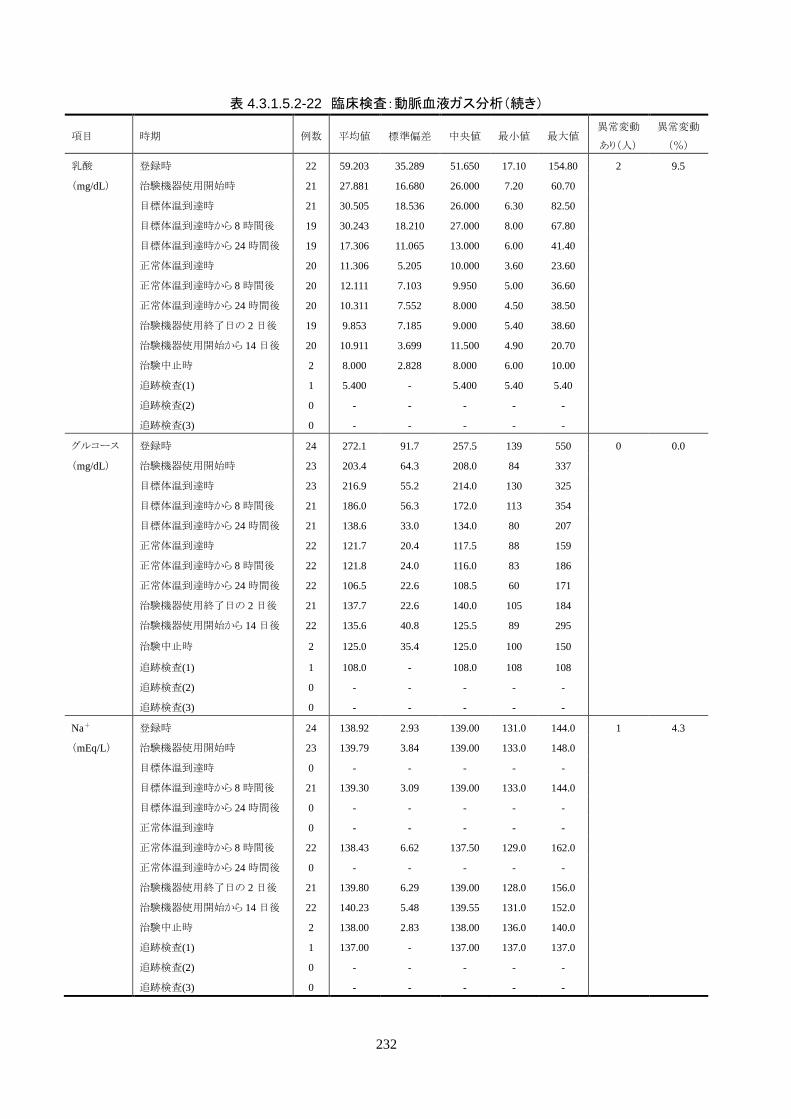
232
表 4.3.1.5.2-22 臨床検査:動脈血液ガス分析(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
乳酸 登録時 22 59.203 35.289 51.650 17.10 154.80 2 9.5
(mg/dL) 治験機器使用開始時 21 27.881 16.680 26.000 7.20 60.70
目標体温到達時 21 30.505 18.536 26.000 6.30 82.50
目標体温到達時から 8 時間後 19 30.243 18.210 27.000 8.00 67.80
目標体温到達時から 24時間後 19 17.306 11.065 13.000 6.00 41.40
正常体温到達時 20 11.306 5.205 10.000 3.60 23.60
正常体温到達時から 8 時間後 20 12.111 7.103 9.950 5.00 36.60
正常体温到達時から 24時間後 20 10.311 7.552 8.000 4.50 38.50
治験機器使用終了日の 2 日後 19 9.853 7.185 9.000 5.40 38.60
治験機器使用開始から 14 日後 20 10.911 3.699 11.500 4.90 20.70
治験中止時 2 8.000 2.828 8.000 6.00 10.00
追跡検査(1) 1 5.400 - 5.400 5.40 5.40
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
グルコース 登録時 24 272.1 91.7 257.5 139 550 0 0.0
(mg/dL) 治験機器使用開始時 23 203.4 64.3 208.0 84 337
目標体温到達時 23 216.9 55.2 214.0 130 325
目標体温到達時から 8 時間後 21 186.0 56.3 172.0 113 354
目標体温到達時から 24時間後 21 138.6 33.0 134.0 80 207
正常体温到達時 22 121.7 20.4 117.5 88 159
正常体温到達時から 8 時間後 22 121.8 24.0 116.0 83 186
正常体温到達時から 24時間後 22 106.5 22.6 108.5 60 171
治験機器使用終了日の 2 日後 21 137.7 22.6 140.0 105 184
治験機器使用開始から 14 日後 22 135.6 40.8 125.5 89 295
治験中止時 2 125.0 35.4 125.0 100 150
追跡検査(1) 1 108.0 - 108.0 108 108
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
Na+ 登録時 24 138.92 2.93 139.00 131.0 144.0 1 4.3
(mEq/L) 治験機器使用開始時 23 139.79 3.84 139.00 133.0 148.0
目標体温到達時 0 - - - - -
目標体温到達時から 8 時間後 21 139.30 3.09 139.00 133.0 144.0
目標体温到達時から 24時間後 0 - - - - -
正常体温到達時 0 - - - - -
正常体温到達時から 8 時間後 22 138.43 6.62 137.50 129.0 162.0
正常体温到達時から 24時間後 0 - - - - -
治験機器使用終了日の 2 日後 21 139.80 6.29 139.00 128.0 156.0
治験機器使用開始から 14 日後 22 140.23 5.48 139.55 131.0 152.0
治験中止時 2 138.00 2.83 138.00 136.0 140.0
追跡検査(1) 1 137.00 - 137.00 137.0 137.0
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
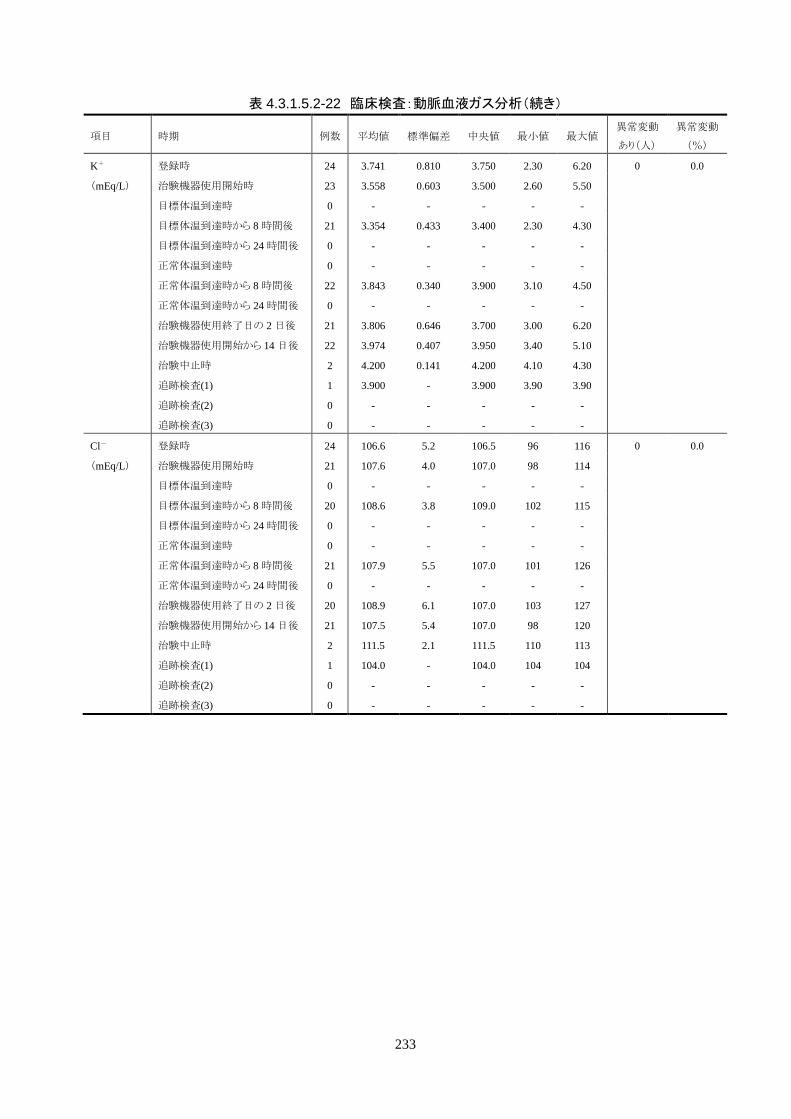
233
表 4.3.1.5.2-22 臨床検査:動脈血液ガス分析(続き)
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
K+ 登録時 24 3.741 0.810 3.750 2.30 6.20 0 0.0
(mEq/L) 治験機器使用開始時 23 3.558 0.603 3.500 2.60 5.50
目標体温到達時 0 - - - - -
目標体温到達時から 8 時間後 21 3.354 0.433 3.400 2.30 4.30
目標体温到達時から 24時間後 0 - - - - -
正常体温到達時 0 - - - - -
正常体温到達時から 8 時間後 22 3.843 0.340 3.900 3.10 4.50
正常体温到達時から 24時間後 0 - - - - -
治験機器使用終了日の 2 日後 21 3.806 0.646 3.700 3.00 6.20
治験機器使用開始から 14 日後 22 3.974 0.407 3.950 3.40 5.10
治験中止時 2 4.200 0.141 4.200 4.10 4.30
追跡検査(1) 1 3.900 - 3.900 3.90 3.90
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
Cl- 登録時 24 106.6 5.2 106.5 96 116 0 0.0
(mEq/L) 治験機器使用開始時 21 107.6 4.0 107.0 98 114
目標体温到達時 0 - - - - -
目標体温到達時から 8 時間後 20 108.6 3.8 109.0 102 115
目標体温到達時から 24時間後 0 - - - - -
正常体温到達時 0 - - - - -
正常体温到達時から 8 時間後 21 107.9 5.5 107.0 101 126
正常体温到達時から 24時間後 0 - - - - -
治験機器使用終了日の 2 日後 20 108.9 6.1 107.0 103 127
治験機器使用開始から 14 日後 21 107.5 5.4 107.0 98 120
治験中止時 2 111.5 2.1 111.5 110 113
追跡検査(1) 1 104.0 - 104.0 104 104
追跡検査(2) 0 - - - - -
追跡検査(3) 0 - - - - -
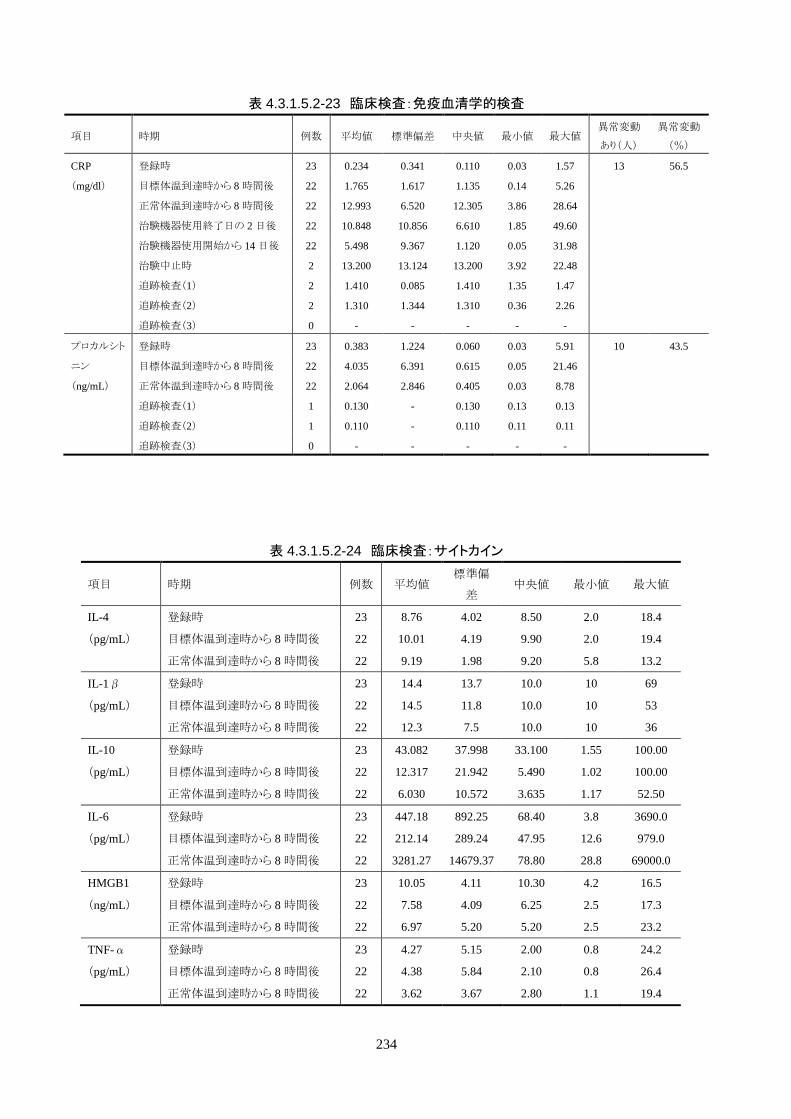
234
表 4.3.1.5.2-23 臨床検査:免疫血清学的検査
項目 時期 例数 平均値 標準偏差 中央値 最小値 最大値 異常変動
あり(人)
異常変動
(%)
CRP 登録時 23 0.234 0.341 0.110 0.03 1.57 13 56.5
(mg/dl) 目標体温到達時から 8 時間後 22 1.765 1.617 1.135 0.14 5.26
正常体温到達時から 8 時間後 22 12.993 6.520 12.305 3.86 28.64
治験機器使用終了日の 2 日後 22 10.848 10.856 6.610 1.85 49.60
治験機器使用開始から 14 日後 22 5.498 9.367 1.120 0.05 31.98
治験中止時 2 13.200 13.124 13.200 3.92 22.48
追跡検査(1) 2 1.410 0.085 1.410 1.35 1.47
追跡検査(2) 2 1.310 1.344 1.310 0.36 2.26
追跡検査(3) 0 - - - - -
プロカルシト 登録時 23 0.383 1.224 0.060 0.03 5.91 10 43.5
ニン 目標体温到達時から 8 時間後 22 4.035 6.391 0.615 0.05 21.46
(ng/mL) 正常体温到達時から 8 時間後 22 2.064 2.846 0.405 0.03 8.78
追跡検査(1) 1 0.130 - 0.130 0.13 0.13
追跡検査(2) 1 0.110 - 0.110 0.11 0.11
追跡検査(3) 0 - - - - -
表 4.3.1.5.2-24 臨床検査:サイトカイン
項目 時期 例数 平均値 標準偏
差 中央値 最小値 最大値
IL-4 登録時 23 8.76 4.02 8.50 2.0 18.4
(pg/mL) 目標体温到達時から 8 時間後 22 10.01 4.19 9.90 2.0 19.4
正常体温到達時から 8 時間後 22 9.19 1.98 9.20 5.8 13.2
IL-1β 登録時 23 14.4 13.7 10.0 10 69
(pg/mL) 目標体温到達時から 8 時間後 22 14.5 11.8 10.0 10 53
正常体温到達時から 8 時間後 22 12.3 7.5 10.0 10 36
IL-10 登録時 23 43.082 37.998 33.100 1.55 100.00
(pg/mL) 目標体温到達時から 8 時間後 22 12.317 21.942 5.490 1.02 100.00
正常体温到達時から 8 時間後 22 6.030 10.572 3.635 1.17 52.50
IL-6 登録時 23 447.18 892.25 68.40 3.8 3690.0
(pg/mL) 目標体温到達時から 8 時間後 22 212.14 289.24 47.95 12.6 979.0
正常体温到達時から 8 時間後 22 3281.27 14679.37 78.80 28.8 69000.0
HMGB1 登録時 23 10.05 4.11 10.30 4.2 16.5
(ng/mL) 目標体温到達時から 8 時間後 22 7.58 4.09 6.25 2.5 17.3
正常体温到達時から 8 時間後 22 6.97 5.20 5.20 2.5 23.2
TNF-α 登録時 23 4.27 5.15 2.00 0.8 24.2
(pg/mL) 目標体温到達時から 8 時間後 22 4.38 5.84 2.10 0.8 26.4
正常体温到達時から 8 時間後 22 3.62 3.67 2.80 1.1 19.4
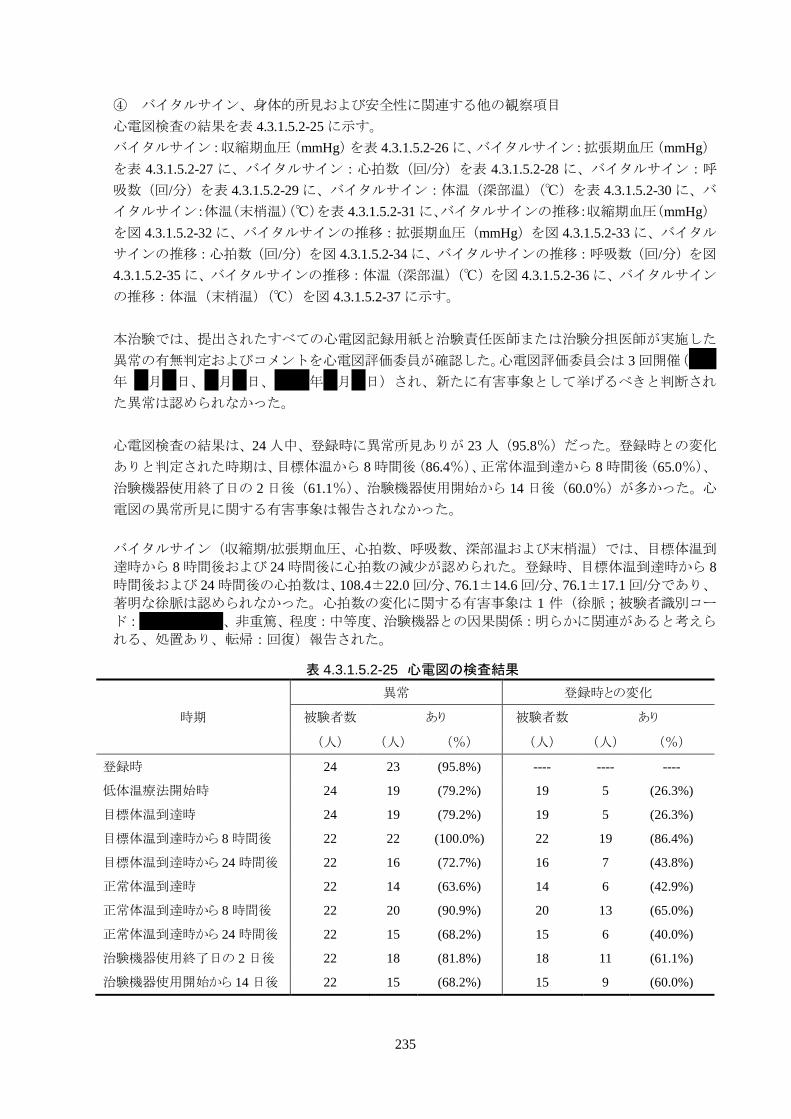
235
④ バイタルサイン、身体的所見および安全性に関連する他の観察項目
心電図検査の結果を表 4.3.1.5.2-25 に示す。
バイタルサイン:収縮期血圧(mmHg)を表 4.3.1.5.2-26 に、バイタルサイン:拡張期血圧(mmHg)
を表 4.3.1.5.2-27 に、バイタルサイン:心拍数(回/分)を表 4.3.1.5.2-28 に、バイタルサイン:呼
吸数(回/分)を表 4.3.1.5.2-29 に、バイタルサイン:体温(深部温)(℃)を表 4.3.1.5.2-30 に、バ
イタルサイン:体温(末梢温)(℃)を表 4.3.1.5.2-31 に、バイタルサインの推移:収縮期血圧(mmHg)
を図 4.3.1.5.2-32 に、バイタルサインの推移:拡張期血圧(mmHg)を図 4.3.1.5.2-33 に、バイタル
サインの推移:心拍数(回/分)を図 4.3.1.5.2-34 に、バイタルサインの推移:呼吸数(回/分)を図
4.3.1.5.2-35 に、バイタルサインの推移:体温(深部温)(℃)を図 4.3.1.5.2-36 に、バイタルサイン
の推移:体温(末梢温)(℃)を図 4.3.1.5.2-37 に示す。
本治験では、提出されたすべての心電図記録用紙と治験責任医師または治験分担医師が実施した
異常の有無判定およびコメントを心電図評価委員が確認した。心電図評価委員会は 3 回開催(****
年 *月*日、*月*日、*****年*月*日)され、新たに有害事象として挙げるべきと判断され
た異常は認められなかった。
心電図検査の結果は、24 人中、登録時に異常所見ありが 23 人(95.8%)だった。登録時との変化
ありと判定された時期は、目標体温から 8 時間後(86.4%)、正常体温到達から 8 時間後(65.0%)、
治験機器使用終了日の 2 日後(61.1%)、治験機器使用開始から 14 日後(60.0%)が多かった。心
電図の異常所見に関する有害事象は報告されなかった。
バイタルサイン(収縮期/拡張期血圧、心拍数、呼吸数、深部温および末梢温)では、目標体温到
達時から 8 時間後および 24 時間後に心拍数の減少が認められた。登録時、目標体温到達時から 8
時間後および 24 時間後の心拍数は、108.4±22.0 回/分、76.1±14.6 回/分、76.1±17.1 回/分であり、
著明な徐脈は認められなかった。心拍数の変化に関する有害事象は 1 件(徐脈;被験者識別コー
ド:******、非重篤、程度:中等度、治験機器との因果関係:明らかに関連があると考えら
れる、処置あり、転帰:回復)報告された。
表 4.3.1.5.2-25 心電図の検査結果
時期
異常 登録時との変化
被験者数 あり 被験者数 あり
(人) (人) (%) (人) (人) (%)
登録時 24 23 (95.8%) ---- ---- ----
低体温療法開始時 24 19 (79.2%) 19 5 (26.3%)
目標体温到達時 24 19 (79.2%) 19 5 (26.3%)
目標体温到達時から 8 時間後 22 22 (100.0%) 22 19 (86.4%)
目標体温到達時から 24 時間後 22 16 (72.7%) 16 7 (43.8%)
正常体温到達時 22 14 (63.6%) 14 6 (42.9%)
正常体温到達時から 8 時間後 22 20 (90.9%) 20 13 (65.0%)
正常体温到達時から 24 時間後 22 15 (68.2%) 15 6 (40.0%)
治験機器使用終了日の 2 日後 22 18 (81.8%) 18 11 (61.1%)
治験機器使用開始から 14 日後 22 15 (68.2%) 15 9 (60.0%)
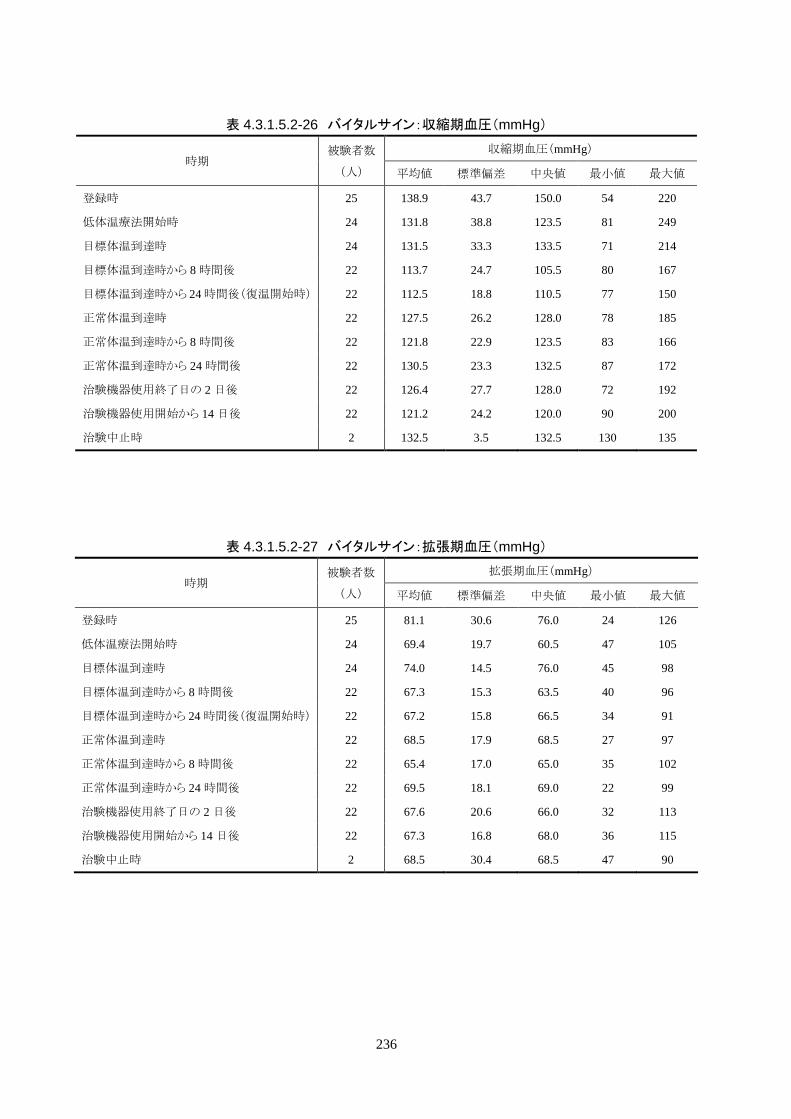
236
表 4.3.1.5.2-26 バイタルサイン:収縮期血圧(mmHg)
時期 被験者数
(人)
収縮期血圧(mmHg)
平均値 標準偏差 中央値 最小値 最大値
登録時 25 138.9 43.7 150.0 54 220
低体温療法開始時 24 131.8 38.8 123.5 81 249
目標体温到達時 24 131.5 33.3 133.5 71 214
目標体温到達時から 8 時間後 22 113.7 24.7 105.5 80 167
目標体温到達時から 24時間後(復温開始時) 22 112.5 18.8 110.5 77 150
正常体温到達時 22 127.5 26.2 128.0 78 185
正常体温到達時から 8 時間後 22 121.8 22.9 123.5 83 166
正常体温到達時から 24 時間後 22 130.5 23.3 132.5 87 172
治験機器使用終了日の 2 日後 22 126.4 27.7 128.0 72 192
治験機器使用開始から 14 日後 22 121.2 24.2 120.0 90 200
治験中止時 2 132.5 3.5 132.5 130 135
表 4.3.1.5.2-27 バイタルサイン:拡張期血圧(mmHg)
時期 被験者数
(人)
拡張期血圧(mmHg)
平均値 標準偏差 中央値 最小値 最大値
登録時 25 81.1 30.6 76.0 24 126
低体温療法開始時 24 69.4 19.7 60.5 47 105
目標体温到達時 24 74.0 14.5 76.0 45 98
目標体温到達時から 8 時間後 22 67.3 15.3 63.5 40 96
目標体温到達時から 24時間後(復温開始時) 22 67.2 15.8 66.5 34 91
正常体温到達時 22 68.5 17.9 68.5 27 97
正常体温到達時から 8 時間後 22 65.4 17.0 65.0 35 102
正常体温到達時から 24 時間後 22 69.5 18.1 69.0 22 99
治験機器使用終了日の 2 日後 22 67.6 20.6 66.0 32 113
治験機器使用開始から 14 日後 22 67.3 16.8 68.0 36 115
治験中止時 2 68.5 30.4 68.5 47 90
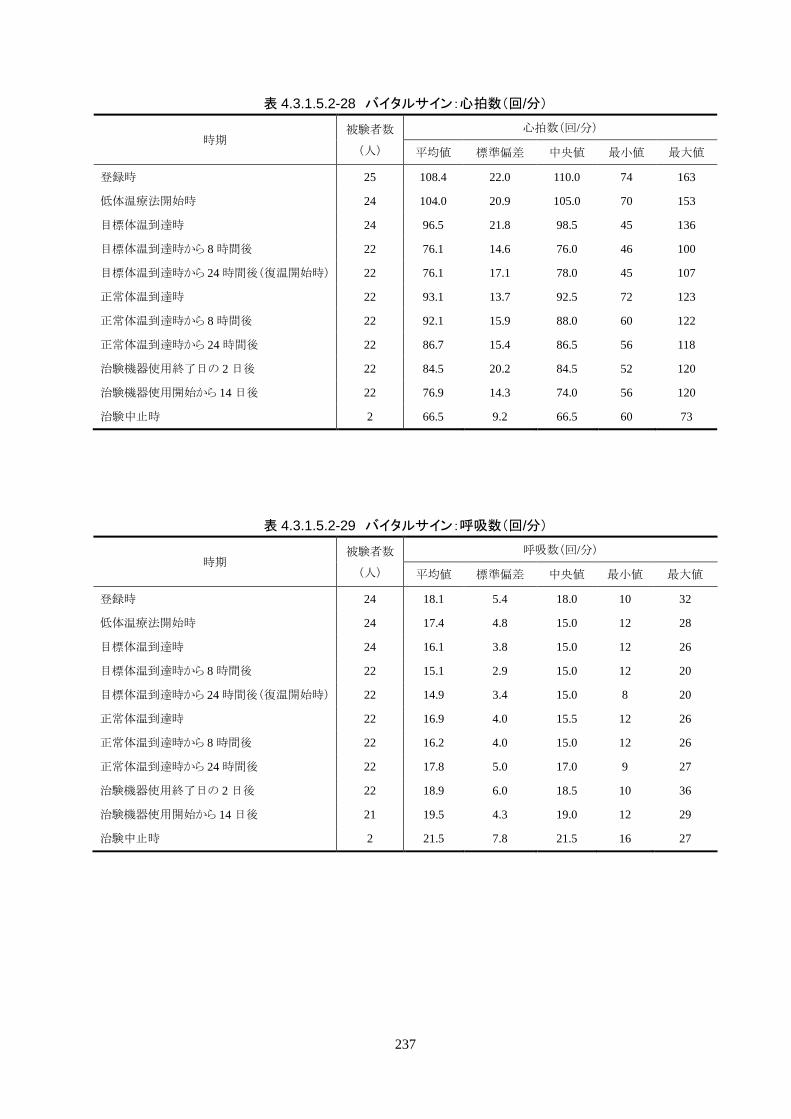
237
表 4.3.1.5.2-28 バイタルサイン:心拍数(回/分)
時期 被験者数
(人)
心拍数(回/分)
平均値 標準偏差 中央値 最小値 最大値
登録時 25 108.4 22.0 110.0 74 163
低体温療法開始時 24 104.0 20.9 105.0 70 153
目標体温到達時 24 96.5 21.8 98.5 45 136
目標体温到達時から 8 時間後 22 76.1 14.6 76.0 46 100
目標体温到達時から 24時間後(復温開始時) 22 76.1 17.1 78.0 45 107
正常体温到達時 22 93.1 13.7 92.5 72 123
正常体温到達時から 8 時間後 22 92.1 15.9 88.0 60 122
正常体温到達時から 24 時間後 22 86.7 15.4 86.5 56 118
治験機器使用終了日の 2 日後 22 84.5 20.2 84.5 52 120
治験機器使用開始から 14 日後 22 76.9 14.3 74.0 56 120
治験中止時 2 66.5 9.2 66.5 60 73
表 4.3.1.5.2-29 バイタルサイン:呼吸数(回/分)
時期 被験者数
(人)
呼吸数(回/分)
平均値 標準偏差 中央値 最小値 最大値
登録時 24 18.1 5.4 18.0 10 32
低体温療法開始時 24 17.4 4.8 15.0 12 28
目標体温到達時 24 16.1 3.8 15.0 12 26
目標体温到達時から 8 時間後 22 15.1 2.9 15.0 12 20
目標体温到達時から 24時間後(復温開始時) 22 14.9 3.4 15.0 8 20
正常体温到達時 22 16.9 4.0 15.5 12 26
正常体温到達時から 8 時間後 22 16.2 4.0 15.0 12 26
正常体温到達時から 24 時間後 22 17.8 5.0 17.0 9 27
治験機器使用終了日の 2 日後 22 18.9 6.0 18.5 10 36
治験機器使用開始から 14 日後 21 19.5 4.3 19.0 12 29
治験中止時 2 21.5 7.8 21.5 16 27
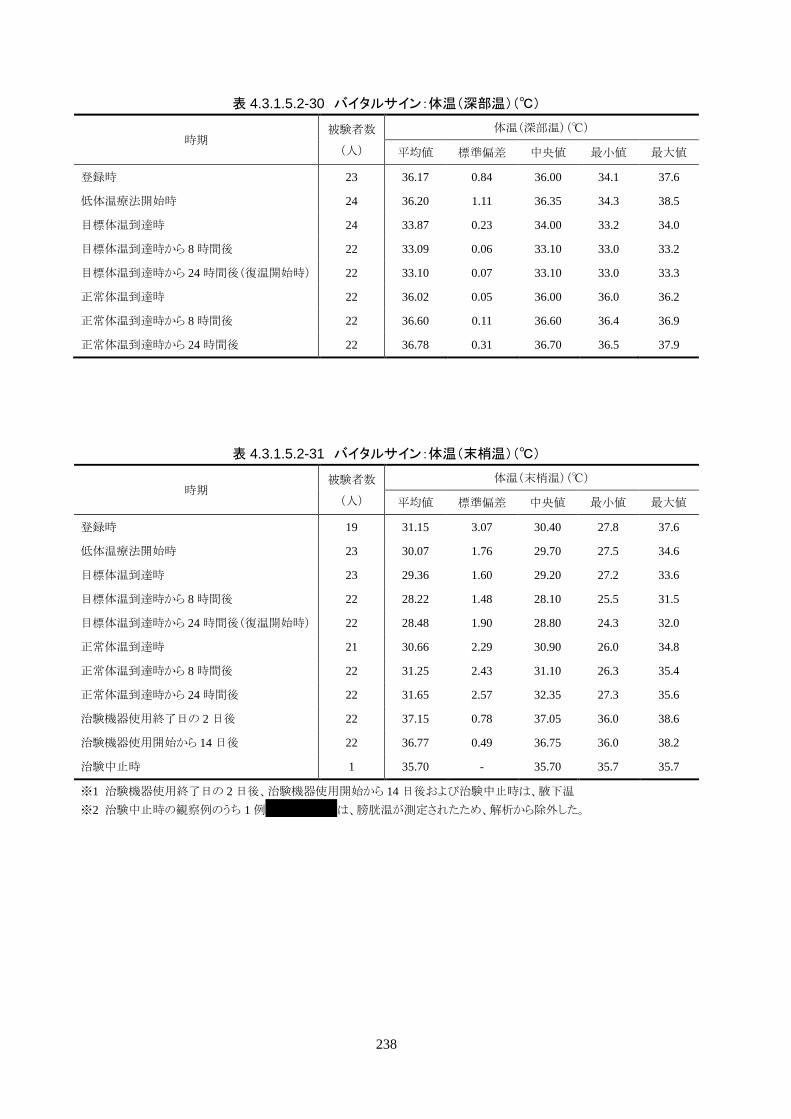
238
表 4.3.1.5.2-30 バイタルサイン:体温(深部温)(℃)
時期 被験者数
(人)
体温(深部温)(℃)
平均値 標準偏差 中央値 最小値 最大値
登録時 23 36.17 0.84 36.00 34.1 37.6
低体温療法開始時 24 36.20 1.11 36.35 34.3 38.5
目標体温到達時 24 33.87 0.23 34.00 33.2 34.0
目標体温到達時から 8 時間後 22 33.09 0.06 33.10 33.0 33.2
目標体温到達時から 24 時間後(復温開始時) 22 33.10 0.07 33.10 33.0 33.3
正常体温到達時 22 36.02 0.05 36.00 36.0 36.2
正常体温到達時から 8 時間後 22 36.60 0.11 36.60 36.4 36.9
正常体温到達時から 24 時間後 22 36.78 0.31 36.70 36.5 37.9
表 4.3.1.5.2-31 バイタルサイン:体温(末梢温)(℃)
時期 被験者数
(人)
体温(末梢温)(℃)
平均値 標準偏差 中央値 最小値 最大値
登録時 19 31.15 3.07 30.40 27.8 37.6
低体温療法開始時 23 30.07 1.76 29.70 27.5 34.6
目標体温到達時 23 29.36 1.60 29.20 27.2 33.6
目標体温到達時から 8 時間後 22 28.22 1.48 28.10 25.5 31.5
目標体温到達時から 24 時間後(復温開始時) 22 28.48 1.90 28.80 24.3 32.0
正常体温到達時 21 30.66 2.29 30.90 26.0 34.8
正常体温到達時から 8 時間後 22 31.25 2.43 31.10 26.3 35.4
正常体温到達時から 24 時間後 22 31.65 2.57 32.35 27.3 35.6
治験機器使用終了日の 2 日後 22 37.15 0.78 37.05 36.0 38.6
治験機器使用開始から 14 日後 22 36.77 0.49 36.75 36.0 38.2
治験中止時 1 35.70 - 35.70 35.7 35.7
※1 治験機器使用終了日の 2日後、治験機器使用開始から 14 日後および治験中止時は、腋下温
※2 治験中止時の観察例のうち 1 例******は、膀胱温が測定されたため、解析から除外した。
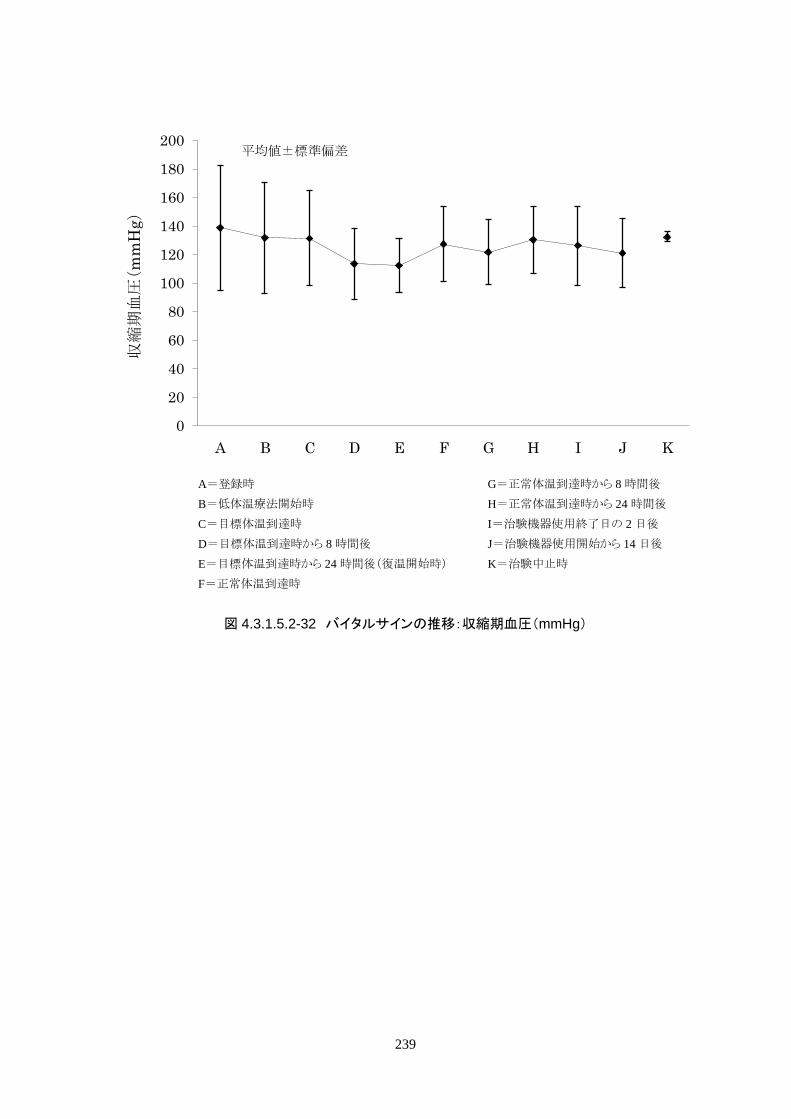
239
A=登録時 G=正常体温到達時から 8 時間後
B=低体温療法開始時 H=正常体温到達時から 24 時間後
C=目標体温到達時 I=治験機器使用終了日の 2 日後
D=目標体温到達時から 8 時間後 J=治験機器使用開始から 14 日後
E=目標体温到達時から 24 時間後(復温開始時) K=治験中止時
F=正常体温到達時
図 4.3.1.5.2-32 バイタルサインの推移:収縮期血圧(mmHg)
0
20
40
60
80
100
120
140
160
180
200
A B C D E F G H I J K
収縮期血圧(m
mH
g)
平均値±標準偏差
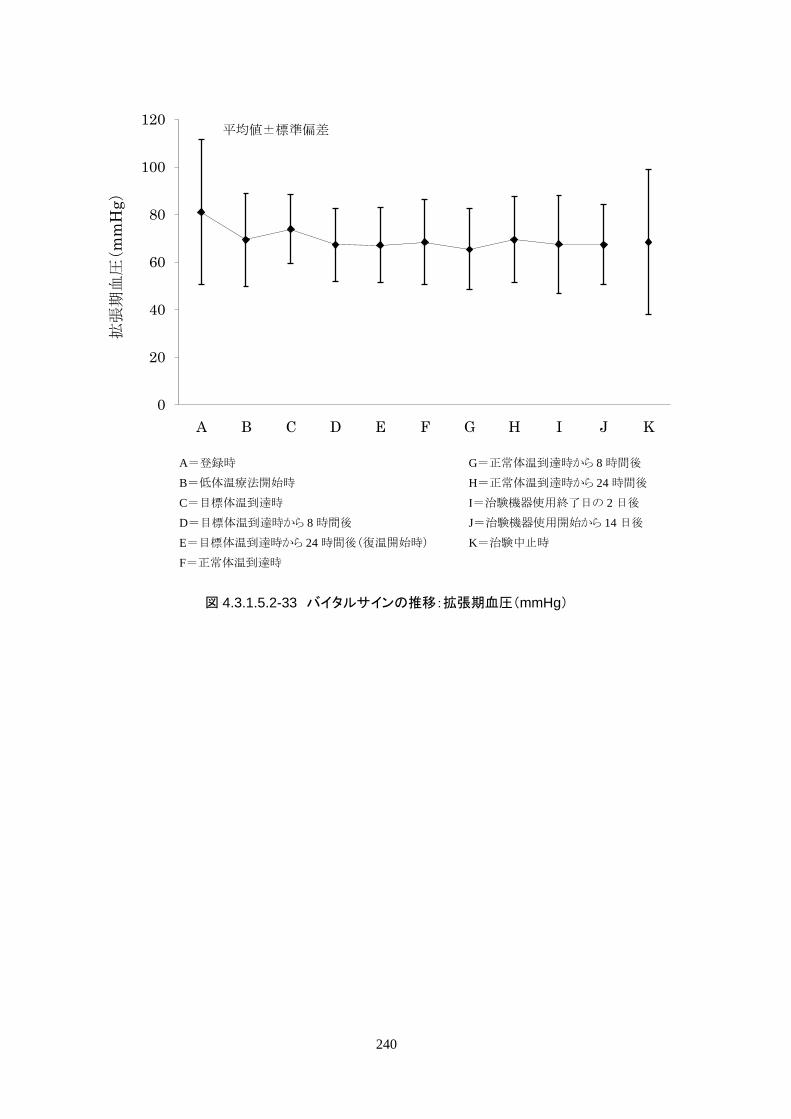
240
A=登録時 G=正常体温到達時から 8 時間後
B=低体温療法開始時 H=正常体温到達時から 24 時間後
C=目標体温到達時 I=治験機器使用終了日の 2 日後
D=目標体温到達時から 8 時間後 J=治験機器使用開始から 14 日後
E=目標体温到達時から 24 時間後(復温開始時) K=治験中止時
F=正常体温到達時
図 4.3.1.5.2-33 バイタルサインの推移:拡張期血圧(mmHg)
0
20
40
60
80
100
120
A B C D E F G H I J K
拡張期血圧(m
mH
g)
平均値±標準偏差
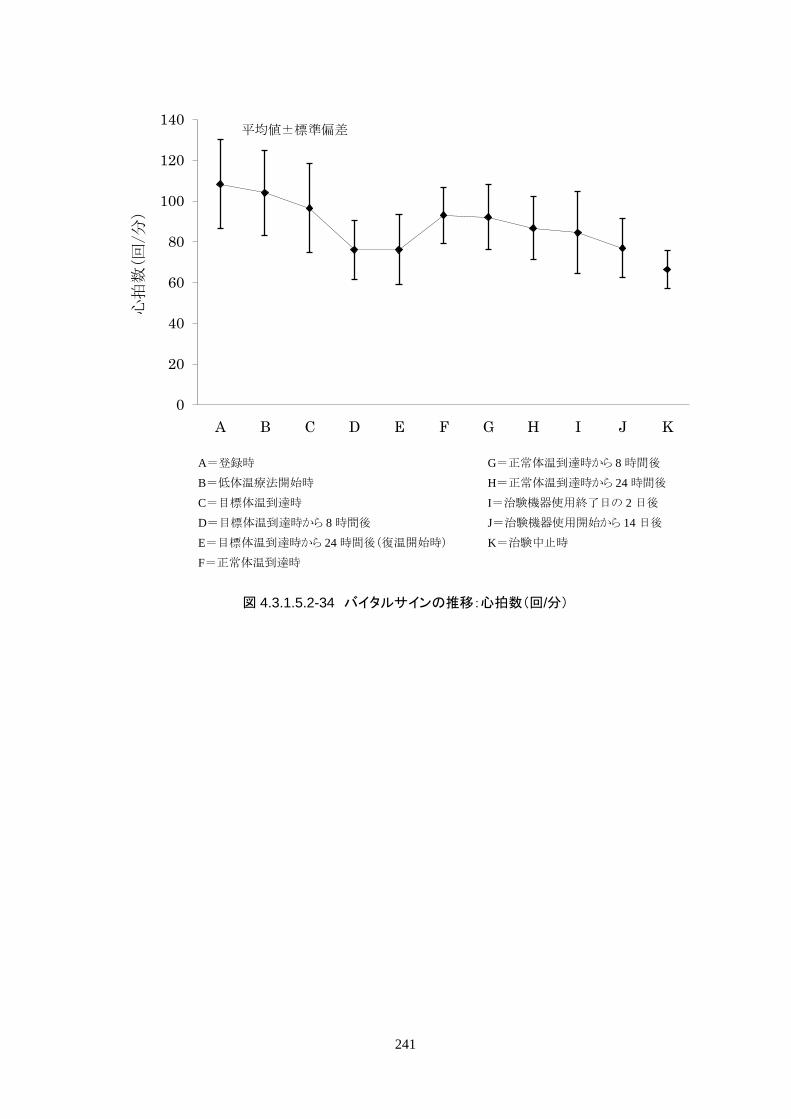
241
A=登録時 G=正常体温到達時から 8 時間後
B=低体温療法開始時 H=正常体温到達時から 24 時間後
C=目標体温到達時 I=治験機器使用終了日の 2 日後
D=目標体温到達時から 8 時間後 J=治験機器使用開始から 14 日後
E=目標体温到達時から 24 時間後(復温開始時) K=治験中止時
F=正常体温到達時
図 4.3.1.5.2-34 バイタルサインの推移:心拍数(回/分)
0
20
40
60
80
100
120
140
A B C D E F G H I J K
心拍数(回
/分)
平均値±標準偏差
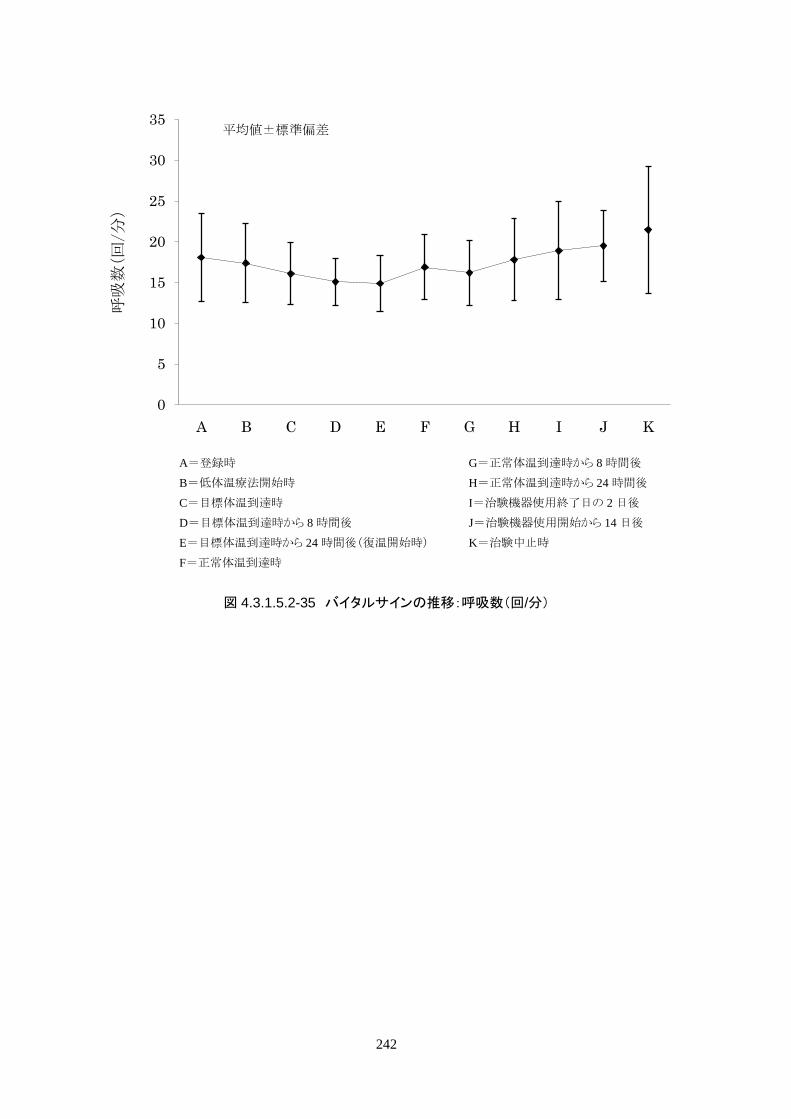
242
A=登録時 G=正常体温到達時から 8 時間後
B=低体温療法開始時 H=正常体温到達時から 24 時間後
C=目標体温到達時 I=治験機器使用終了日の 2 日後
D=目標体温到達時から 8 時間後 J=治験機器使用開始から 14 日後
E=目標体温到達時から 24 時間後(復温開始時) K=治験中止時
F=正常体温到達時
図 4.3.1.5.2-35 バイタルサインの推移:呼吸数(回/分)
0
5
10
15
20
25
30
35
A B C D E F G H I J K
呼吸数(回
/分)
平均値±標準偏差
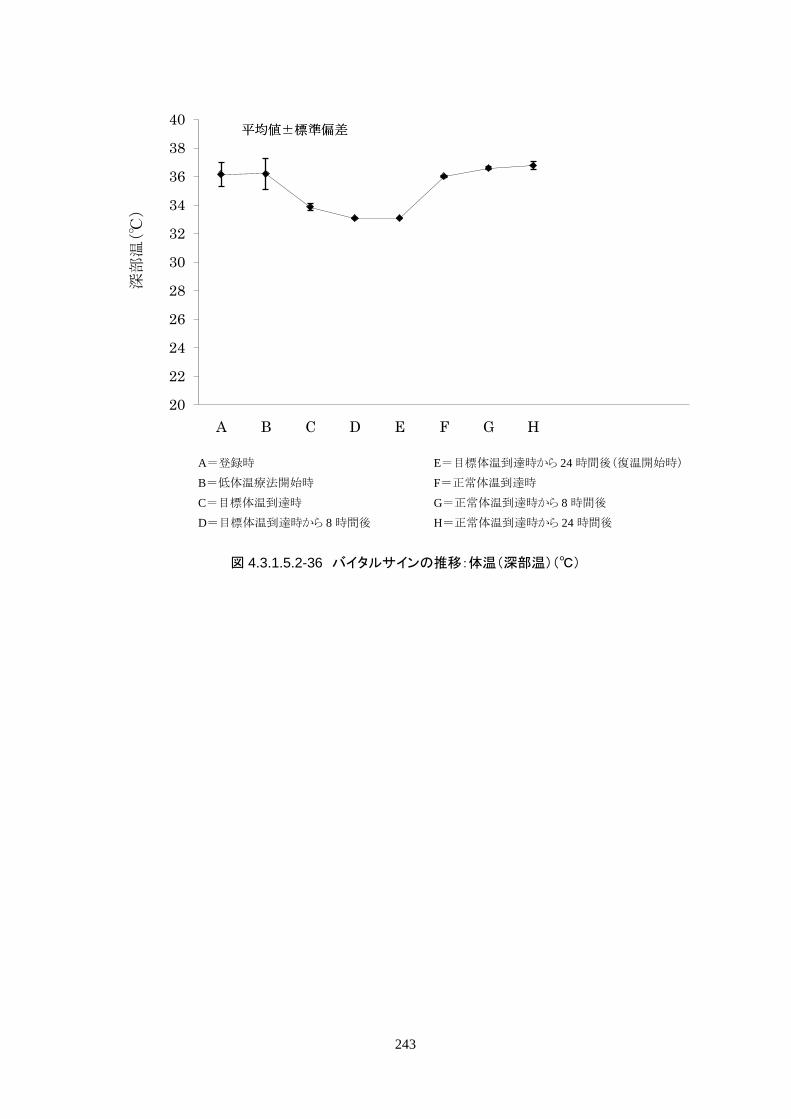
243
A=登録時 E=目標体温到達時から 24 時間後(復温開始時)
B=低体温療法開始時 F=正常体温到達時
C=目標体温到達時 G=正常体温到達時から 8 時間後
D=目標体温到達時から 8 時間後 H=正常体温到達時から 24 時間後
図 4.3.1.5.2-36 バイタルサインの推移:体温(深部温)(℃)
20
22
24
26
28
30
32
34
36
38
40
A B C D E F G H
深部温(℃)
平均値±標準偏差 平均値±標準偏差
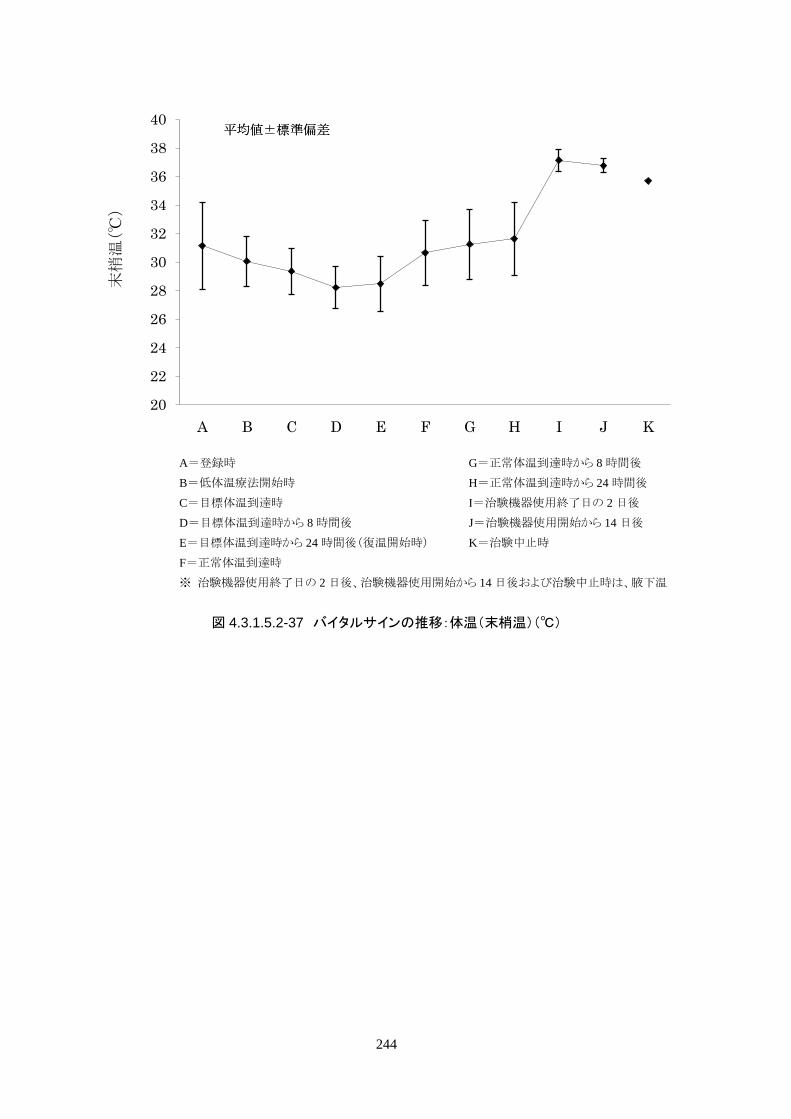
244
A=登録時 G=正常体温到達時から 8 時間後
B=低体温療法開始時 H=正常体温到達時から 24 時間後
C=目標体温到達時 I=治験機器使用終了日の 2 日後
D=目標体温到達時から 8 時間後 J=治験機器使用開始から 14 日後
E=目標体温到達時から 24 時間後(復温開始時) K=治験中止時
F=正常体温到達時
※ 治験機器使用終了日の 2 日後、治験機器使用開始から 14日後および治験中止時は、腋下温
図 4.3.1.5.2-37 バイタルサインの推移:体温(末梢温)(℃)
20
22
24
26
28
30
32
34
36
38
40
A B C D E F G H I J K
末梢温(℃)
平均値±標準偏差 平均値±標準偏差

245
4.3.2 臨床試験成績のまとめ
4.3.2.1 試験結果及び考察
(1)有効性のまとめ
本治験は、心原性が疑われる内因性心停止・心拍再開後患者を対象に、IVTM-TG、IVTM-SK、IVTM-
CI または IVTM-CQ を用いた血管内冷却による低体温療法を実施した。
本治験の被験者の内訳は、同意取得被験者 25 人、治験を中止した被験者 3 人であった。
治験を中止した被験者 3 人の中止理由は、全員が「有害事象の発現により治験の継続が困難と治験責
任医師または治験分担医師が判断した場合」であった。3 人とも有害事象と治験機器との因果関係はない
と判断された。
被験者背景は、男性 22 人(88.0%)、女性 3 人(12.0%)、年齢 56.1±15.4 歳(27-78 歳)(平均値±
標準偏差、カッコ内は最小値-最大値;以下同様)、身長 168.5±8.8 cm(148-181 cm)、体重 69.0±
13.2 kg(39-100 kg)、BMI 24.2±3.5 kg/m2(16.8-31.1 kg/m2)、BSA 1.737±0.193 m2(1.24-2.15 m2)
であった。
心停止の原因疾患で多かったのは、急性心筋梗塞 5 人(20%)、異型狭心症 3 人(12.0%)、急性冠
症候群 3 人(12.0%)などであった。既往歴は、なし 20 人(80%)、あり 5 人(20%)だった。合
併症は、なし 5 人(20%)、あり 20 人(80%)だった。
本治験に参加した被験者の心停止場所は全員が院外で、うち 22 人(88.0%)が目撃ありだった。
初期波形は 25 人中 19 人(76.0%)が心室細動で、残り 6 人(24.0%)が無脈性電気活動だった。
心静止の被験者はいなかった。
心停止から ROSC までの時間は 24.6±14.5 分、ROSC から冷却(治験機器による治療)開始までの
時間は 262.3±73.5 分で、全員が治験機器による冷却開始前に冷電解質液が投与された。治療に使
用したカテーテルは IVTM-CI が 7 人(29.2%)、IVTM-CQ が 17 人(70.8%)だった。
本治験の主要評価項目は、「治験機器による冷却開始 3 時間以内に深部体温が目標体温に達した被
験者の割合」であった。治験機器による治療を開始した被験者全員(24 人)が 3 時間以内に目標
体温に到達し、目標達成率は 100%(95%CI 85.8-100%)であった。
冷却開始時の深部体温は 36.20±1.11℃で、冷却開始から目標体温到達までに要した時間は 54.63±
37.46 分(中央値 45.00 分、IQR 33.0-73.5 分)であった。
副次的評価項目は、治験機器使用開始から 14 日後の CPC および mRS、治験機器使用中の体温の
推移、単位時間あたりの体温変化量であった。
治験機器使用開始から 14 日後の CPC は、良好な神経学的転帰を示す CPC 1 または 2 が 12 人(50%)
だった。
治験機器使用開始から 14 日後の mRS は、良好な神経学的転帰を示す mRS 0~2 の被験者が 11 人
(45.9%)だった。
維持期(設定温度 33.0℃)の平均体温は 33.09±0.06℃、95%信頼区間は 33.08-33.10℃であった。
体温測定回数 242 回中、±0.2℃を超えたのは 2 回(0.8%)で±0.5℃および±1.0℃を超えた変動は

246
なかった。
正常体温維持期(設定温度 36.5℃)の平均体温は 36.64±0.17℃、95%信頼区間は 36.60-36.69℃で
あった。体温測定回数 188 回中、±0.2℃を超えたのは 30 回(16.0%)、±0.5℃を超えたのは 6 回
(3.2%)、±1.0℃を超えたのは 1 回(0.5%)であった。
維持期から正常体温維持期に移行する復温期は 0.1℃/hr で復温することとした。維持期から正常体
温到達時までの体温差は 3.0℃であり、復温時間は 28.69±4.03 時間(IQR 27.9-30.0)(FAS)だっ
た。
本治験に参加した被験者の体重の最小値は******の 39 kg、最大値は******の 100 kg
だった。2 人の被験者の冷却時間(冷却開始から目標体温到達までに要した時間)は、*****
が 50 分、******が 57 分だった。維持期および正常体温維持期の体温は、体重に関わりなく
安定して推移した。さらに、復温時間は******が 29 時間 15 分、******が 28 時間 29
分で、いずれの被験者も想定された復温時間に近い値であった。
導入期の体温冷却速度は、3.08±1.45℃/hr(中央値 2.70℃/hr、IQR 2.3-3.7℃/hr)だった。
以上の結果より、IVTM-TG、IVTM-SK、IVTM-CI または IVTM-CQ を使用した低体温療法は、被
験者の体温、体格等に関わらず速やかな低体温導入が可能だった。また、設定温度に関わらず、体
温の維持および変更にも優れた性能を発揮した。
(2)安全性のまとめ
本治験の同意を取得した全被験者を対象に安全性を評価した。
少なくとも 1 件以上の有害事象(重篤、非重篤を問わない、また因果関係も問わない)が報告され
た被験者数(有害事象発現率)(95%CI)は 25 人中 19 人(76.0%)(54.9-90.6%)だった。治験機
器との因果関係が否定できない有害事象の発現被験者数は 4 人(16.0%)(4.5-36.1%)であり、い
ずれの治験機器との因果関係が否定できない有害事象も非重篤だった。重篤な有害事象の発現被
験者数は 5 人(20%)(6.8-40.7%)であり、いずれも治験機器との因果関係はなかった。治験機器
との因果関係が否定できない重篤な有害事象は発現しなかった。
発現率の高かった重篤な有害事象は、心室細動 3 件(12.0%)だった。3 件とも重症度は高度で治
験機器との因果関係は「関連がない」だった。転帰は 2 件が軽快、1 件が死亡だった。
発現率の高かった非重篤な有害事象は、肺炎 6 件(24.0%)、高血圧 3 件(12.0%)だった。
重症度は肺炎(6 件)が軽度 3 件、中等度 3 件、高血圧(3 件)が軽度 3 件だった。
治験機器との因果関係が否定できない有害事象は、肺炎、徐脈、大静脈血栓症、血中クレアチンホ
スホキナーゼ増加、血圧低下がそれぞれ 1 件発現した。重症度はいずれも中等度だった。なお、徐
脈については、低体温療法を行う場合、心臓脈の減少は通常起こりえることであり、可逆的で予期
される効果であると考える。
発現時期は、大静脈血栓症のみ治療終了後に発現した。残りの治験機器との因果関係が否定できな
い有害事象は、治療期間中に発現したが、治験機器の使用状況は「変更なし(治療継続)」だった。
転帰は、いずれの治験機器との因果関係が否定できない有害事象も「回復」だった。治験機器との
因果関係が否定できない有害事象は、いずれも治験機器による治療継続が可能であったことや、処
置により回復していることから被験者の安全性には問題がないと考えられる。

247
本治験の登録被験者 25 人のうち、最終確認時の転帰が生存だったのは 23 人(92.0%)、死亡は 2
人(8.0%)であった。生存率の 95%信頼区間は 71.6-97.9%だった。死亡した被験者の死因は 2 人
とも原疾患の悪化で、死亡と治験機器との因果関係は関係ないと治験責任医師が判断した。いずれ
の被験者も治験中止後の死亡だった。
治験期間中の死亡はなかった。死亡以外の重篤な有害事象は 5 人 5 件(心室細動 3 件、心停止 1
件、急性呼吸窮迫症候群 1 件)が報告され、すべての重篤な有害事象は治験機器との因果関係がないと
判断された。重篤な有害事象のうち、心室細動 1 件と心停止 1 件は治験機器の使用前に発現した有
害事象だった。
臨床検査値に関する有害事象は、血中クレアチンホスホキナーゼ増加の 2 件だった。
臨床検査値で異常変動が多かったのは CRP(56.5%)、プロカルシトニン(43.5%)であった。いず
れの検査値も登録時から徐々に上昇し、治療中または中止時にピークを迎え、その後徐々に低下し
ていた。
心電図検査の結果は、24 人中、登録時に異常所見ありが 23 人(95.8%)だった。登録時との変化
ありと判定された時期は、目標体温から 8 時間後(86.4%)、正常体温到達から 8 時間後(65.0%)、
治験機器使用終了日の 2 日後(61.1%)、治験機器使用開始から 14 日後(60.0%)が多かった。心
電図の異常所見に関する有害事象は報告されなかった。
バイタルサイン(収縮期/拡張期血圧、心拍数、呼吸数、深部温および末梢温)では、目標体温到達
時から 8 時間後および 24 時間後に心拍数の減少が認められた。登録時、目標体温到達時から 8 時
間後および 24 時間後の心拍数は、108.4±22.0 回/分、76.1±14.6 回/分、76.1±17.1 回/分であり、被
験者全体では著明な徐脈は認められなかった。心拍数の変化に関する有害事象は 1 件(徐脈;被験
者識別コード:******、非重篤、程度:中等度、治験機器との因果関係:明らかに関連があ
ると考えられる、処置あり、転帰:回復)報告された。
治験機器の不具合は 2 件報告された。内訳は、IVTM-SK のチューブの亀裂による破損 1 件、IVTM-
CQ のキット内に同梱されているガイドワイヤの折れ曲がり 1 件だった。どちらの事象も、不具合
に伴う有害事象はなかった。
体温のオーバーシュートは 1 件認められた。被験者識別コード******の正常体温到達から
22 時間後(設定体温 36.5℃)の体温が 37.6℃まで上昇した。オーバーシュート時に使用していた
治験機器に不具合はなかった。
以上のことから、心原性が疑われる内因性心停止・心拍再開後患者に対する本治験機器の安全性は
十分許容できることが確認できた。

248
4.3.2.2 考察と全般的結論
(1)性能、安全性およびその他のエンドポイントの試験結果に関する考察
治療開始時体温と登録時体温の差に及ぼす冷電解質液/冷血漿増量剤の投与量の影響を図 4.3.2.2.1-
1 に、治療開始時体温と登録時体温の差に及ぼす冷電解質液/冷血漿増量剤の投与開始から治療開
始までの時間の影響を図 4.3.2.2.1-2 に、治療開始時体温と冷却時間の検討を図 4.3.2.2.1-3 に、BMI
と冷却時間の検討を図 4.3.2.2.1-4 に、BSA と冷却時間の検討を図 4.3.2.2.1-5 に示す。
本治験では、冷電解質液または冷血漿増量剤の輸液(30 mL/kg を上限とする)を、ROSC から治療
開始までの間の必須療法とした。冷却輸液の投与量や投与のタイミングが被験者体温にどの程度
影響を与えたか検討した。
冷却輸液の投与量が治療開始時と登録時の深部体温の差に及ぼす影響を検討するため回帰分析を
行い、回帰係数、R2 乗値および p 値を算出し、さらに両者の関係を散布図として図示した(図
4.3.2.2.1-1)。また、冷却輸液の投与から治療開始までの時間が治療開始時と登録時の深部体温の差
に及ぼす影響を検討するため、同様に回帰分析で検討した(図 4.3.2.2.1-2)。
冷却輸液の投与量は個々の被験者ごとに異なり、最小投与量 10 mL/kg、最大投与量 30 mL/kg だっ
た。また、冷却輸液の投与から治療開始までの時間は最短 28 分、最長 335 分だった。治療開始時
と登録時の深部体温の差がマイナス(登録時より治療開始時の体温が低い)だったのは 10 人、変
化なしが 3 人、プラス(登録時より治療開始時の体温が高い)が 9 人(以上、登録時体温データが
欠側を除く 22 人のデータ)で、体温差は-0.9℃~+2.5℃だった。
冷却輸液の投与量、冷却輸液の投与から治療開始までの時間ともに R2値は小さく(それぞれ 0.0012、
0.0000)、これら 2 つと治療開始時と登録時の深部体温の差には関係性は認められなかった。
治療開始時の体温が冷却時間に及ぼす影響を検討するため、回帰分析(線形回帰)を行って回帰式
を求め図示した(図 4.3.2.2.1-3)。また、BMI および BSA が冷却時間に及ぼす影響についても、同
様に回帰分析(線形回帰)で検討した(図 4.3.2.2.1-4、図 4.3.2.2.1-5)。
治療開始時の体温は、最も低い被験者が 34.3℃、最も高い被験者が 38.5℃だった。治療開始時の体
温は、目標体温(34.0℃)との差を説明変数として検討した。線形回帰で R2=0.6393 の直線性が認
められた。
BMI は最小値 16.8 kg/m2、最大値 31.1 kg/m2だった。BSA は最小値 1.24 m2、最大値 2.15 m2だった。
冷却時間は最短 7 分、最長 180 分だった。BMI および BSA は種々の変数変換を施して検討したが、
冷却時間に対する関係性は見いだせなかった。個々の被験者の検討でも、最小体重および最大体重
のいずれの場合も速やかに目標体温に到達し、その後の体温維持・管理も安定していたことから、
本品は被験者の体格に関係なく高い性能を発揮することが示唆された。
本治験のデータでは、少ない冷却輸液投与量でも体温差がマイナスの被験者がいた一方、冷却輸液
を多く投与しても体温差がプラスの被験者や、輸液による体温変化がない被験者がいた。また、冷
却輸液の投与から治療開始までの時間と体温差も大きくばらついた。この結果、本治験では冷却輸
液の投与量および投与のタイミングと、治療開始時と登録時の深部体温の差は相関しなかったが、
臨床では、低体温導入時に冷却輸液を投与して冷却を開始している報告も多い[8-11]。
一方、治療開始時の体温は本治験機器の冷却時間と相関していることが確認された。これは、一般
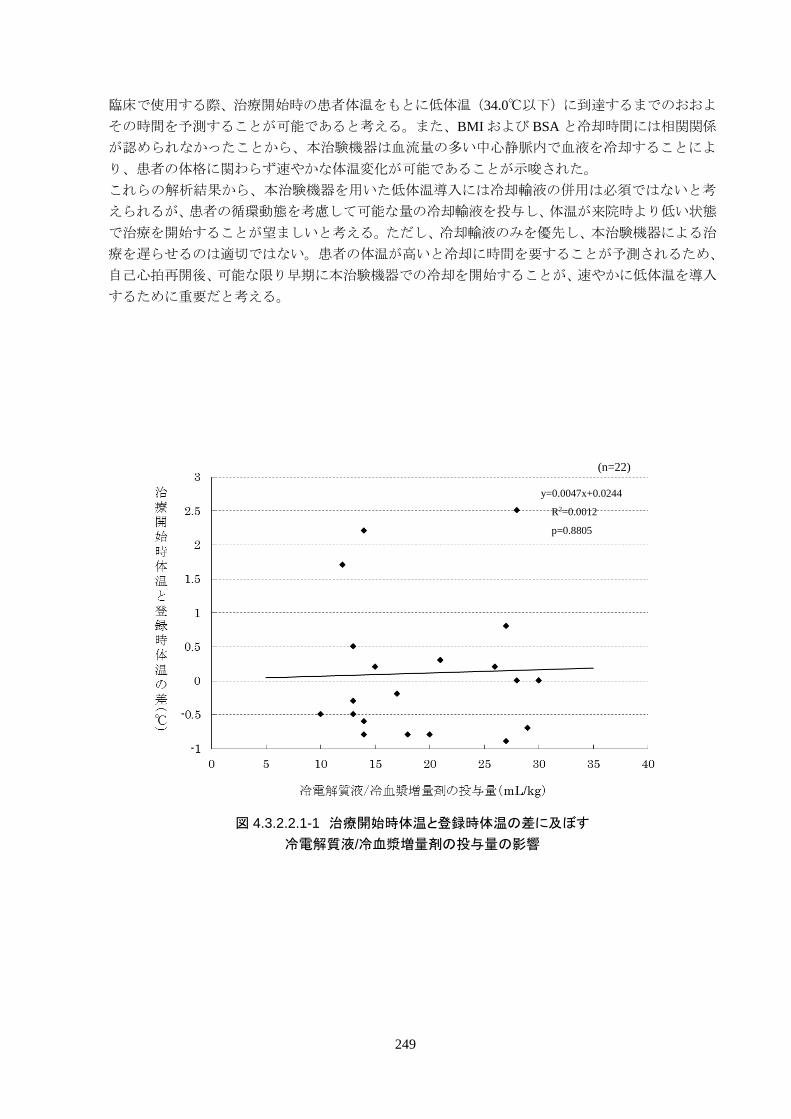
249
臨床で使用する際、治療開始時の患者体温をもとに低体温(34.0℃以下)に到達するまでのおおよ
その時間を予測することが可能であると考える。また、BMI および BSA と冷却時間には相関関係
が認められなかったことから、本治験機器は血流量の多い中心静脈内で血液を冷却することによ
り、患者の体格に関わらず速やかな体温変化が可能であることが示唆された。
これらの解析結果から、本治験機器を用いた低体温導入には冷却輸液の併用は必須ではないと考
えられるが、患者の循環動態を考慮して可能な量の冷却輸液を投与し、体温が来院時より低い状態
で治療を開始することが望ましいと考える。ただし、冷却輸液のみを優先し、本治験機器による治
療を遅らせるのは適切ではない。患者の体温が高いと冷却に時間を要することが予測されるため、
自己心拍再開後、可能な限り早期に本治験機器での冷却を開始することが、速やかに低体温を導入
するために重要だと考える。
図 4.3.2.2.1-1 治療開始時体温と登録時体温の差に及ぼす
冷電解質液/冷血漿増量剤の投与量の影響
y=0.0047x+0.0244
R2=0.0012
p=0.8805
(n=22)
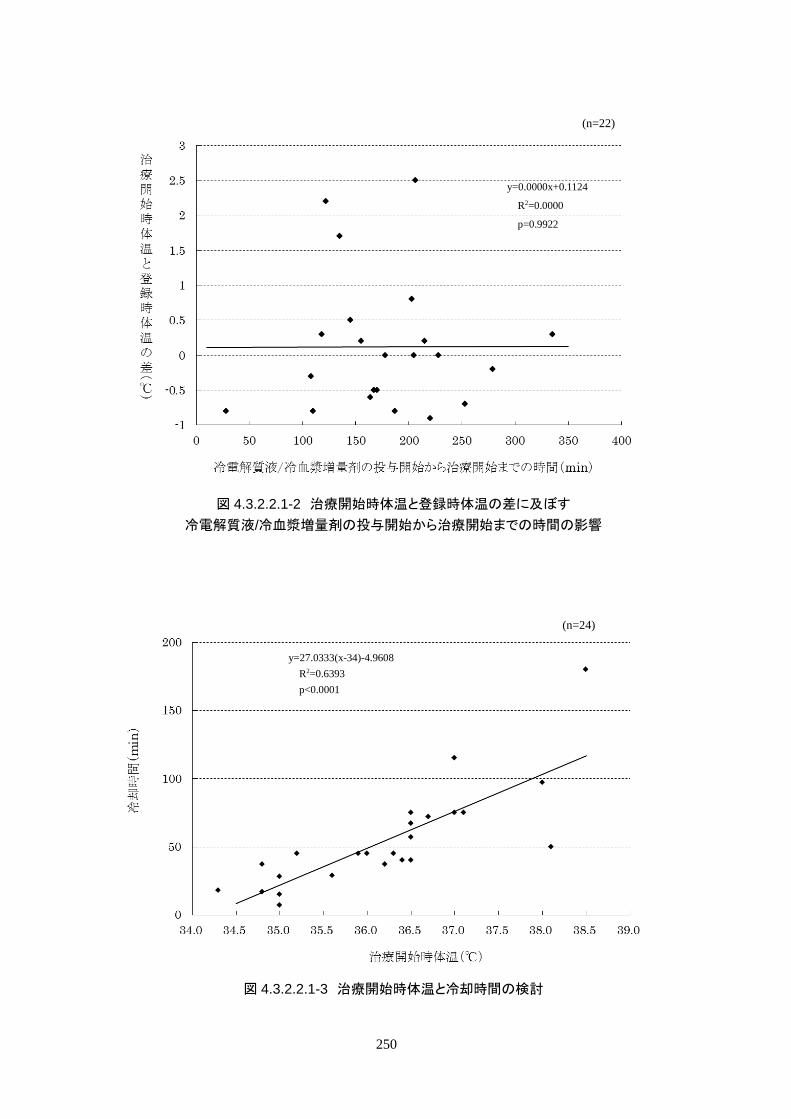
250
図 4.3.2.2.1-2 治療開始時体温と登録時体温の差に及ぼす
冷電解質液/冷血漿増量剤の投与開始から治療開始までの時間の影響
図 4.3.2.2.1-3 治療開始時体温と冷却時間の検討
y=0.0000x+0.1124
R2=0.0000
p=0.9922
y=27.0333(x-34)-4.9608
R2=0.6393
p<0.0001
(n=24)
(n=22)
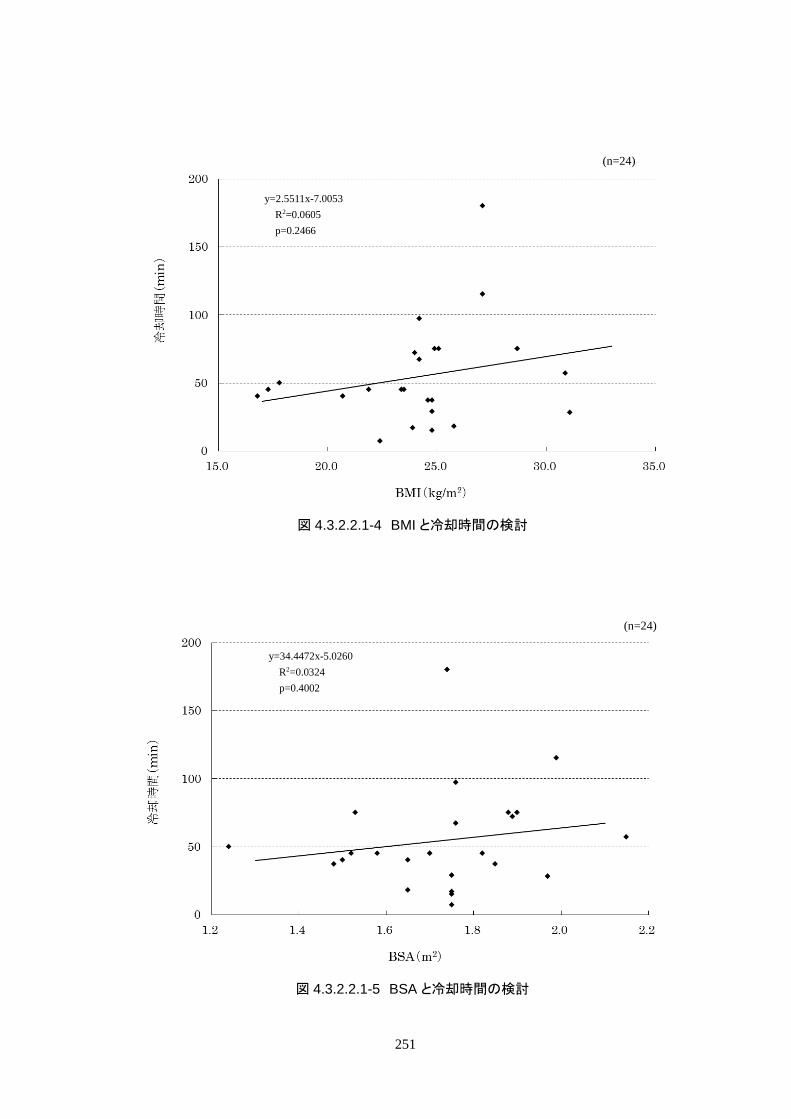
251
図 4.3.2.2.1-4 BMI と冷却時間の検討
図 4.3.2.2.1-5 BSA と冷却時間の検討
y=2.5511x-7.0053
R2=0.0605
p=0.2466
(n=24)
y=34.4472x-5.0260
R2=0.0324
p=0.4002
(n=24)

252
(2)他の既存データを考慮した上での治験結果の臨床的意義および重要性に関する検討
心停止から治験機器による治療終了時までの各時間の要約統計量を表 4.3.2.2.2-1 に、本治験と類似
医療機器を使用した代表的な先行論文[12-14]との比較を表 4.3.2.2.2-2 に示す。
本治験と先行する代表的な試験では、治療開始から目標到達に要した時間、冷却速度に違いがあっ
た。また、本治験では設定温度から±0.2℃を超えた回数で、維持期と正常体温維持期の割合に違
いがあった。
治療開始から目標到達に要した時間は、本治験が 54.63±37.46 分、先行する 3 試験は 219 分、83±
85 分、記載なしだった。冷却速度は、本治験が 3.08±1.45℃/hr、先行する 3 試験は 0.8±0.3℃/hr、
記載なし、1.46±0.42℃/hr だった。これらは、目標到達の定義が各試験で異なること、先行する 3
試験のうち 2 試験では薬剤の使用状況が不明であること、試験実施時期が異なること、対象患者に
違いがみられることなどにより単純な比較はできないと考えられるが、本治験結果は先行する試
験と同等以上の機器性能が発揮されていると考える。
被験者の体温が設定温度から±0.2℃を超えた割合は、本治験では 33.0℃(維持期)が 0.8%、36.5℃
(正常体温維持期)が 16.0%だった。一方、先行論文[14]では、33.0℃(低体温群)が 3.2±4.8%、
37.0℃(通常体温群)が 4.2±5.1%だった。本治験では、維持期に比べ正常体温維持期で設定温度
から±0.2℃を超えた割合が高かった。先行論文では、設定温度から±0.2℃を超えた割合は低体温、
通常体温で同程度だった。
しかしながら本治験では、正常体温維持期(36.5℃)の平均体温値は 36.64℃、平均値の 95%信頼
区間は 36.60~36.69℃であり、本治験機器の装置本体の品目仕様で設定した設定温度±0.2℃の範
囲に収まっていることから、治験機器を用いた体温管理の正確性を示すことができた。
これは、治療中の投薬に関する違いが考えられる。本治験では、低体温の導入および維持期は鎮痛、
鎮静、シバリング抑制プロトコルに示した薬剤を必ず投与していたが、復温期および正常体温維持
期は、治験責任(分担)医師が被験者の状況に合わせ必要に応じた投与が可能だった。そのため、
正常体温維持期のドロペリドールとパンクロニウム(ベクロニウム、ロクロニウムを含む)の使用
率は維持期に比べて低下していた(それぞれ、23 人→13 人、24 人→16 人へ減少)。一方、先行す
る試験では低体温群、通常体温群ともに、鎮静薬、神経筋遮断薬、鎮痛薬が全員に投与されていた。
本治験では、正常体温維持期に鎮静薬と筋弛緩薬が使用されないことにより、患者の体温が上昇し
±0.2℃を超えた割合が多くなったと思われる。しかしながら、正常体温維持期(36.5℃)の実測体
温は 36.64±0.17℃であり、臨床使用には十分な精度でコントロールできていると考える。
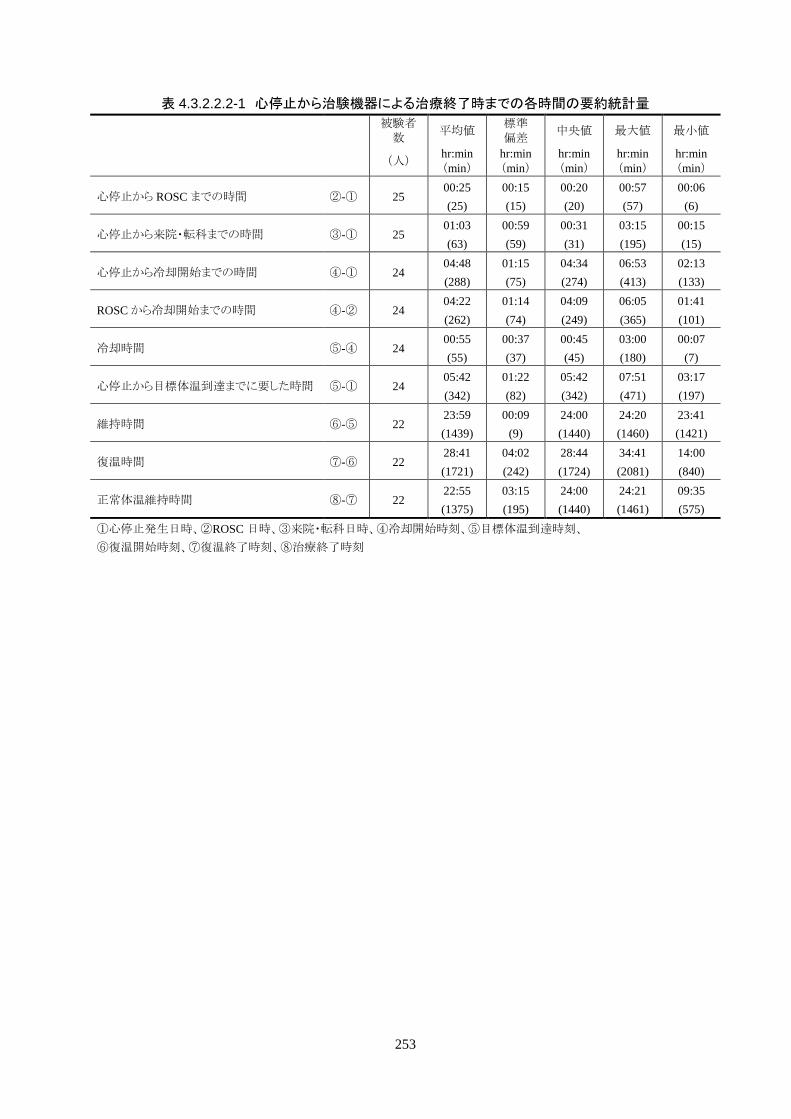
253
表 4.3.2.2.2-1 心停止から治験機器による治療終了時までの各時間の要約統計量
被験者数
平均値 標準 偏差
中央値 最大値 最小値
(人) hr:min
(min)
hr:min
(min)
hr:min
(min)
hr:min
(min)
hr:min
(min)
心停止から ROSC までの時間 ②-① 25 00:25
(25)
00:15
(15)
00:20
(20)
00:57
(57)
00:06
(6)
心停止から来院・転科までの時間 ③-① 25 01:03
(63)
00:59
(59)
00:31
(31)
03:15
(195)
00:15
(15)
心停止から冷却開始までの時間 ④-① 24 04:48
(288)
01:15
(75)
04:34
(274)
06:53
(413)
02:13
(133)
ROSCから冷却開始までの時間 ④-② 24 04:22
(262)
01:14
(74)
04:09
(249)
06:05
(365)
01:41
(101)
冷却時間 ⑤-④ 24 00:55
(55)
00:37
(37)
00:45
(45)
03:00
(180)
00:07
(7)
心停止から目標体温到達までに要した時間 ⑤-① 24 05:42
(342)
01:22
(82)
05:42
(342)
07:51
(471)
03:17
(197)
維持時間 ⑥-⑤ 22 23:59
(1439)
00:09
(9)
24:00
(1440)
24:20
(1460)
23:41
(1421)
復温時間 ⑦-⑥ 22 28:41
(1721)
04:02
(242)
28:44
(1724)
34:41
(2081)
14:00
(840)
正常体温維持時間 ⑧-⑦ 22 22:55
(1375)
03:15
(195)
24:00
(1440)
24:21
(1461)
09:35
(575)
①心停止発生日時、②ROSC 日時、③来院・転科日時、④冷却開始時刻、⑤目標体温到達時刻、
⑥復温開始時刻、⑦復温終了時刻、⑧治療終了時刻
254
表 4.3.2.2.2-2 本治験と類似医療機器を使用した代表的な先行論文との比較
本治験 Al-Senani FM ら[12] Kliegel A ら[13] Hoedemaekers CW ら[14]
被験者数 登録: 25 人
治療開始: 24 人
(男 22/女 3)
登録: 13 人
治療開始: 13 人
(男 6/女 7)
26 人
(男 19/女 7)
類似医療機器群:10 人(低
体温 5/通常体温 5)
(低体温:男 4/女 1)
(通常体温: 男 5)
年齢(歳) 56.1±15.4 60.2±18.7 50(中央値)
(IQR: 42-62)
低体温: 60.4±14.6
通常体温: 48.8±12.8
BMI (kg/m2) 24.2 記載なし 記載なし 低体温: 24.4±3.2
通常体温: 24.9±1.8
対象 心原性が疑われる内
因性心停止・自己心
拍再開後患者
心停止・心拍再開後
患者
心原性心停止 23 人、
肺疾患 1 人、溺水 1
人、不明 1 人
低体温: 心原性と推定さ
れる院外心停止/院内心
停止/外傷性脳損傷後の
頭蓋内圧コントロール不
良
通常体温: くも膜下出血
/外傷性脳損傷
蘇生時間
(分)1)
24.6±14.5
(range: 6-57)
14.3
(range: 5-32.5)
20(中央値)
(IQR: 13-29)
記載なし
使用した
カテーテル
IVTM-CI ま た は
IVTM-CQ( 熱交換用
バルーン数 3 または
4)
Icy TM カテーテル(熱
交換用バルーン数 3)
Icy TM カテーテル(熱
交換用バルーン数 3)
Icy TM カテーテル(熱交換
用バルーン数 3)
冷却輸液 あり
20.2±6.6 mL/kg
なし あり
24±7 mL/kg
類似医療機器群は投与
なし
深部温測定
部位
食道 膀胱 膀胱 直腸
治療中の投薬
(薬剤名の後に
記載した数字
は、使用した
患者数)
33.0℃ : ミダゾラム
24/ドロペリドール
23/フェンタニル 23
/パンクロニウム 2)
24
36.5℃ : ミダゾラム
21/ドロペリドール
13/フェンタニル 19
/パンクロニウム 2)
16
冷却速度を最大にす
るため、シバリング
は鎮静剤と麻酔で抑
制した。鎮静剤と筋
弛緩薬は復温終了ま
で継続した。
鎮静および鎮痛にミ
ダゾラム、フェンタ
ニルを使用、筋弛緩
薬はロクロニウムを
使用
低体温: 鎮静剤 5/神経筋
遮断薬 5/鎮痛薬 5/アセ
トアミノフェン 2
通常体温: 鎮静剤 5/神経
筋遮断薬 5/鎮痛薬 5/ア
セトアミノフェン 5
治療開始時
体温(℃)
36.20±1.11
(range: 34.3-38.5)
35.9
(range: 34.1-37.7)
入院時: 35.6 記載なし
治療開始から
目標到達に要
した時間(分)
34.0℃以下まで 54.63
±37.46
33.0℃まで
219
(IQR 80-315)
33±1℃まで
83±85
記載なし
冷却速度
(℃/hr)
3.08±1.45 0.8±0.3 記載なし 低体温: 1.46±0.42
通常体温:
1.02±0.55
復温時間(hr) 36.0℃以上まで 28.69
±4.03
36.5℃まで
18.3±5.9
24 時間低体温維持
後、 8 時間以内に
36.0℃以上まで復温
記載なし
設定温度との
体温差
33.0℃: 33.09±0.06
36.5℃: 36.64±0.17
設定温度から±0.2℃
を超えた割合
33.0℃: 0.8%
36.5℃: 16.0%
33.0℃: 32.7±0.5 記載なし 設定温度から±0.2℃を超
えた割合
33.0℃: 3.2±4.8%
37.0℃: 4.2±5.1%
1):心停止から ROSC までの時間 2):ベクロニウム、ロクロニウムへの代替えも可能

255
【有効性と安全性の根拠】
低体温療法は、心停止後患者の治療法として 10 年以上の臨床試験および臨床診療によって安全性
および有効性が確立された。表面冷却、冷却された輸液、血管内冷却等を含む様々な体温療法の有
効性および安全性を検証するため、それぞれの方法で調査等を行い、系統的レビューが実施された。
2012 年、Arrich らは心停止後の患者に対する低体温療法の安全性および有効性を評価するために
系統的レビューを行った[15]。このレビューは、世界中の複数のデータベースを検索した結果であ
り、2011 年 7 月までに公表されたすべての文献が含まれている。4 つの臨床試験および 1 つの要約
が分析され、合計 481 名の患者が評価された。3 つの臨床試験(383 名の患者)において、表面冷
却や冷却された輸液、血管内冷却を含む一般的な冷却方法が評価された。さらに、心停止後低体温
療法の有効性(特に退院までの生存および良好な神経学的転帰)が評価された。
Arrich は退院までの生存率について、心停止後患者のうち低体温療法を受けた患者は、平温のまま
の患者に比較して、生存率が顕著に優位であると結論した。また、Arrich は退院時の良好な神経学
的転帰について、心停止後患者のうち低体温療法を受けた患者は、平温のままの患者に比較して優
位であると結論した。
また、Arrich らは系統的レビューを更新し、IVTM システム全体を使用した試験を含む、新たに公
表された試験を追加した。未公開の更新版でも、低体温療法を受けた患者は、34℃以上のままの患
者より良好な神経学的転帰が優位となる、更新前と同様な有効性を示す結果となった。さらに、低
体温療法を受けた患者と平温のままの患者の間に有害事象発現率の差異は確認されなかった。2012
年の Arrich らの系統的レビューに含まれた臨床試験の死亡率、有害事象発現率、良好な神経学的
転帰率は、本申請品を含む IVTM システム全体を調査した国内治験と同等であった。
2013 年、Xiao らは、Arrich らの結果を心停止後の別のレビューおよびメタ分析により確認した[16]。
このレビューは、2011 年 6 月までに公表された 1742 件の要約と 63 件の臨床試験を解析した。63
件の公表された研究文献を調査し、様々な方法で冷却された 1 万名以上の心停止後患者の安全性
および有効性を総合的に評価した。本レビューは、低体温療法の安全面および短期・長期の転帰を
精査し、メタ分析を行うようにデザインされた。
Xiao は、低体温療法を受けた患者が平温のままの患者と比較して、低体温療法と関連が考えられ
る合併症のほとんどの発生率は差異がないと結論づけた。また、このメタ分析は、表面冷却された
患者の有害事象発現率が、血管内冷却された患者の有害事象発現率(特に出血、院内死亡、肺炎、
徐脈)と差異がないと結論した。
本申請品を含む IVTM システム全体を調査した今回の国内治験の結果のうち、不整脈の有害事象
発現率はメタ分析の不整脈(37 件)の有害事象発現率と同等であった。さらに、本申請品を用い
た国内治験の肺炎の有害事象発現率は、メタ分析の肺炎(23 件)より大幅に下回った。
また、Xiao のメタ分析は、低体温療法を受けた患者が冷却されなかった患者(平温のままの患者)よ
り全死亡率が顕著に優位であることを示した。これは最初の心停止時から 6 か月後のフォローア
ップまですべての時点で観察された。本申請品を含む IVTM システム全体を調査した今回の国内
治験での死亡率は Xiao らのメタ分析より顕著に下回った。
Xiao のレビューでは「低体温療法は心停止後の患者に対して一般的に安全である」と結論し、Xiao
は既に広く認められている治療法の全体的な安全性を記述した。本申請品を含む IVTM システム
全体を用いたこの国内治験は、一般的に認められている治療法と比較して、同等あるいは優位な有

256
害事象発現率および死亡率を確認した。
IVTM システムを調査した国内治験の結果は、Arrich および Xiao の系統レビューの結果[15-16]と同
等あるいは優位になった。さらに、多数の臨床試験と数百名の患者を評価した系統的レビューは、
心停止後患者が低体温療法を受ける場合、平温のままの患者と比較して、同等な有害事象発現率、
優位な生存率および良好な神経学的転帰率を示す。
(3)リスク集団に対する特定のベネフィットまたは注意事項(リスクおよびベネフィットの評価)
特殊な状況や特別な集団に対する本治験機器のリスクを検討する。本治験は、選択および除外基準
で妊婦、20 歳未満および 80 歳以上の患者、出血のリスクが高い患者、重篤な肝機能または腎機能
障害のある患者の登録を不可とした。これらの逸脱はなかったため、治験実施計画書で除外した特
別な集団は、本治験に参加していない。しかし、本治験の対象は心原性が疑われる心停止・心拍再
開後の患者であり、集学的治療が必要な重症の患者集団という観点では被験者全員がリスク集団
とも考えられる。
本治験では重篤な有害事象は 5 件発現したが、いずれも治験機器との因果関係は関連がないもの
だった。発現率の高かった有害事象は心室細動(3 件)で、いずれも原疾患によるもので 1 件は治
験機器使用前に発現したものだった。治験機器との因果関係が否定できない有害事象は肺炎、徐脈、
大静脈血栓症、血中クレアチンホスホキナーゼ増加、血圧低下がそれぞれ 1 件認められた。なお、
徐脈については、低体温療法を行う場合、心臓脈の減少は通常起こりえることであり、可逆的で予期さ
れる効果であると考える。バイタルサインでは、治療中に心拍数の減少があったが著明な徐脈は認
められなかった。有効性は、治験機器を使用した 24 人全員が 3 時間以内に目標体温に到達し、冷
却速度は 3.08±1.45℃/hr、設定温度に対するコントロールも良好であることが確認できた。
以上のことから、患者に対する十分な観察は必要だが、本治験機器は ICU 等の医療体制が整った
環境で十分な安全配慮の下に使用されており、同様の環境で使用される場合は集学的治療が必要
な重症度の高い患者集団に対しても特別なリスクはないと考えられる。
(4)本治験の限界と将来的な試験実施の可能性
本治験は限られた医療機関で、患者条件を厳密に規定した少数例での検討である。しかしながら、
本治験の対象となった患者集団に対する本品の性能および安全性は確認できたことから、性能や
安全性を評価する追加試験は不要と考える。今後、一般臨床で広く使用し、有用性および安全性の
データを蓄積していくことが必要だと考える。
(5)全般的結論
治験機器(IVTM-TG、IVTM-SK、IVTM-CI または IVTM-CQ)を用いた血管内体温管理による低体
温療法は、心原性が疑われる内因性心停止・心拍再開後患者に対して安全に実施できた。また、本
治験機器は目標体温への到達、低体温の管理・維持、および復温と正常体温の管理・維持が治験実
施計画書どおり有効に実施できた。本治験機器による低体温療法が安全および有効に実施できる
ことが示された本治験結果から、中心静脈カテーテルが留置可能な心停止・心拍再開後患者に対し
て、本治験機器は対象集団の体温管理に用いる医療機器として適切であると考える。

257
4.3.3 治験依頼者と規制当局間の合意/協議結果
本治験は、**年 *月 *日に医療機器治験相談(番号:****号)を実施した。相談および助言
の概要を以下に示す。
<相談者からの相談事項の概要>
<独立行政法人医薬品医療機器総合機構の助言の概要>

258

259
4.3.4 参考文献
1. 日本蘇生協議会・日本救急医療財団, JRC 蘇生ガイドライン 2010. へるす出版, 東京, 2011;91-
93.
2. Yokoyama H, Nagao K, et al., Impact of Therapeutic Hypothermia in the Treatment of Patients With Out-
of-Hospital Cardiac Arrest From the J-PULSE-HYPO Study Registry. Circulation J 2011 May; 75:1063-
1070.
3. Wolff B, Machill K, et al., Early achievement of mild therapeutic hypothermia and the neurologic outcome
after cardiac arrest. Int J Cardiol. 2009 Apr 3; 133(2):223-8.
4. Tømte Ø, Drægni T, et al., A comparison of intravascular and surface cooling techniques in comatose
cardiac arrest survivors. Crit Care Med. 2011 Mar; 39(3):443-9.
5. The Hypothermia after Cardiac Arrest Study Group, Mild therapeutic hypothermia to improve the
neurologic outcome after cardiac arrest. N Engl J Med. 2002 Feb 21; 346(8):549-56. Erratum in: N Engl J
Med. 2002 May 30; 346(22):1756.
6. Kokubu N, Hase M, et al., Abstract 135: Impacts of Rewarming Speed Differences on Outcomes of
Therapeutic Hypothermia in Out-of-Hospital Cardiac Arrest: an Analysis in J-Pulse Hypo-Registry, a
Multicenter Hypothermia Registry in Japan. Circulation. 2010; 122:A135.
7. 藤本 薫喜,渡邊 孟,他,日本人の体表面積に関する研究,第 18 篇 三期にまとめた算出式.
日本衛生学会誌.1968; 23(5):443-450.
8. Gaieski DF, Band RA, et al., Early goal-directed hemodynamic optimization combined with therapeutic
hypothermia in comatose survivors of out-of-hospital cardiac arrest. Resuscitation. 2009 Apr; 80(4): 418-
424.
9. Sunde K, Pytte M, et al., Implementation of a standardised treatment protocol for post resuscitation care
after out-of-hospital cardiac arrest. Resuscitation. 2007 Apr; 73(1): 29-39.
10. Jeppesen JB, Kjaergaard J, et al., The impact of therapeutic hypothermia on neurological function and
quality of life after cardiac arrest. Resuscitation. 2009 Feb; 80(2): 171-176.
11. Knafelj R, Radsel P, et al., Primary percutaneous coronary intervention and mild induced hypothermia in
comatose survivors of ventricular fibrillation with ST-elevation acute myocardial infarction. Resuscitation.
2007 Aug; 74(2): 227-234.
12. Al-Senani FM, Graffagnino C, et al., A prospective, multicenter pilot study to evaluate the feasibility and
safety of using the CoolGardTM System and IcyTM catheter following cardiac arrest. Resuscitation. 2004
Aug; 62(2):143-50.
13. Kliegel A, Losert H, et al., Cold simple intravenous infusions preceding special endovascular cooling for
faster induction of mild hypothermia after cardiac arrest- a feasibility study. Resuscitation. 2005;
64(3):347-351.
14. Hoedemaekers CW, Ezzahti M, et al., Comparison of cooling methods to induce and maintain normo and
hypothermia in intensive care unit patients: a prospective intervention study. Crit Care. 2007; 11(4):R91
15. Arrich J, et al., Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation (Review).
The Cochrane Library 2012, Issue 9.
16. Guoguang Xiao, et al., Safety profile and outcome of mild therapeutic hypothermia in patients following
cardiac arrest: systematic review and meta-analysis. Emerg Med J 2013;30:91–100. doi:10.1136/emermed-
2012-201120





























