Embed Size (px)
Citation preview

62890 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
1 This draft guidance represents the agency’scurrent thinking on the selection of test proceduresand the setting and justification of acceptancecriteria for new chemical drug substances and newdrug products. It does not create or confer anyrights for or on any person and does not operate tobind FDA or the public. An alternative approachmay be used if such approach satisfies therequirements of the applicable statute, regulations,or both.
DEPARTMENT OF HEALTH ANDHUMAN SERVICES
Food and Drug Administration
[Docket No. 97D–0448]
International Conference onHarmonisation; Draft Guidance onSpecifications: Test Procedures andAcceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances
AGENCY: Food and Drug Administration,HHS.ACTION: Notice.
SUMMARY: The Food and DrugAdministration (FDA) is publishing adraft guidance entitled ‘‘Q6ASpecifications: Test Procedures andAcceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances.’’ The draftguidance was prepared under theauspices of the International Conferenceon Harmonisation of TechnicalRequirements for Registration ofPharmaceuticals for Human Use (ICH).The draft guidance provides guidanceon the selection of test procedures andthe setting and justification ofacceptance criteria for new chemicaldrug substances and new drug productsproduced from them. The draft guidanceis intended to assist in theestablishment of a single set of globalspecifications for new drug substancesand new drug products.DATES: Written comments by January 26,1998.ADDRESSES: Submit written commentson the draft guidance to the DocketsManagement Branch (HFA–305), Foodand Drug Administration, 12420Parklawn Dr., rm. 1–23, Rockville, MD20857. Copies of the draft guidance areavailable from the Drug InformationBranch (HFD–210), Center for DrugEvaluation and Research, Food andDrug Administration, 5600 FishersLane, Rockville, MD 20857, 301–827–4573.FOR FURTHER INFORMATION CONTACT:
Regarding the guidance: Eric B.Sheinin, Center for Drug Evaluationand Research (HFD–800), Food andDrug Administration, 5600 FishersLane, Rockville, MD 20857, 301–827–5918, or
Neil D. Goldman, Center for BiologicEvaluation and Research (HFM–416), Food and DrugAdministration, 8800 RockvillePike, Rockville, MD 20852, 301–827–0377.
Regarding the ICH: Janet J. Showalter,Office of Health Affairs (HFY–20),
Food and Drug Administration,5600 Fishers Lane, Rockville, MD20857, 301–827–0864.
SUPPLEMENTARY INFORMATION: In recentyears, many important initiatives havebeen undertaken by regulatoryauthorities and industry associations topromote international harmonization ofregulatory requirements. FDA hasparticipated in many meetings designedto enhance harmonization and iscommitted to seeking scientificallybased harmonized technical proceduresfor pharmaceutical development. One ofthe goals of harmonization is to identifyand then reduce differences in technicalrequirements for drug developmentamong regulatory agencies.
ICH was organized to provide anopportunity for tripartite harmonizationinitiatives to be developed with inputfrom both regulatory and industryrepresentatives. FDA also seeks inputfrom consumer representatives andothers. ICH is concerned withharmonization of technicalrequirements for the registration ofpharmaceutical products among threeregions: The European Union, Japan,and the United States. The six ICHsponsors are the European Commission,the European Federation ofPharmaceutical Industries Associations,the Japanese Ministry of Health andWelfare, the Japanese PharmaceuticalManufacturers Association, the Centersfor Drug Evaluation and Research andBiologics Evaluation and Research,FDA, and the Pharmaceutical Researchand Manufacturers of America. The ICHSecretariat, which coordinates thepreparation of documentation, isprovided by the InternationalFederation of PharmaceuticalManufacturers Associations (IFPMA).
The ICH Steering Committee includesrepresentatives from each of the ICHsponsors and the IFPMA, as well asobservers from the World HealthOrganization, the Canadian HealthProtection Branch, and the EuropeanFree Trade Area.
In July 1997, the ICH SteeringCommittee agreed that a draft guidanceentitled ‘‘Q6A Specifications: TestProcedures and Acceptance Criteria forNew Drug Substances and New DrugProducts: Chemical Substances’’ shouldbe made available for public comment.The draft guidance is the product of theQuality Expert Working Group of theICH. Comments about this draft will beconsidered by FDA and the QualityExpert Working Group. A relateddocument for biotechnology derivedproducts is the subject of a separateExpert Working Group.
In accordance with Good GuidancePractices (62 FR 8961, February 27,
1997), this document is now beingcalled a guidance, rather than aguideline.
The draft guidance provides guidanceon the selection of test procedures andthe setting and justification ofacceptance criteria for new drugsubstances of synthetic chemical origin,and new drug products produced fromthem, that have not been registeredpreviously in the United States, theEuropean Union, or Japan. The draftguidance is intended to assist in theestablishment of a single set of globalspecifications for new drug substancesand new drug products.
This draft guidance represents theagency’s current thinking on theselection of test procedures and thesetting and justification of acceptancecriteria for new chemical drugsubstances and new drug products. Itdoes not create or confer any rights foror on any person and does not operateto bind FDA or the public. Analternative approach may be used ifsuch approach satisfies therequirements of the applicable statute,regulations, or both.
Interested persons may, on or beforeJanuary 26, 1998, submit to the DocketsManagement Branch (address above)written comments on the draft guidance.Two copies of any comments are to besubmitted, except that individuals maysubmit one copy. Comments are to beidentified with the docket numberfound in brackets in the heading of thisdocument. The draft guidance andreceived comments may be seen in theoffice above between 9 a.m. and 4 p.m.,Monday through Friday. An electronicversion of this guidance is available onthe Internet at ‘‘http://www.fda.gov/cder/guidance.htm’’.
The text of the draft guidance follows:
Q6A Specifications: Test Procedures andAcceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances1
Table of Contents1. Introduction
1.1 Specifications1.2 Objective of the Guidance1.3 Scope of the Guidance
2. General Concepts2.1 Periodic/Skip Testing2.2 Release vs. Shelf-Life Acceptance
Criteria2.3 In-Process Tests

62891Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
2.4 Design and DevelopmentConsiderations
2.5 Limited Data Available at Filing2.6 Parametric Release2.7 Alternative Procedures2.8 Pharmacopoeial Tests and Acceptance
Criteria2.9 Evolving Technologies2.10 Impact of Drug Substance on Drug
Product Specifications2.11 Reference Standard
3. Guidelines3.1 Specifications: Definition and
Justification3.1.1 Definition of Specifications3.1.2 Justification of Specifications
3.2 Universal Tests/Criteria3.2.1 New Drug Substances3.2.2 New Drug Products
3.3 Specific Tests/Criteria3.3.1 New Drug Substances3.3.2 New Drug Products
4. Glossary5. References6. Attachments: Decision Trees #1 Through#8
1. Introduction
1.1 Specifications
A specification is defined as a list of tests,references to analytical procedures, andappropriate acceptance criteria that arenumerical limits, ranges, or other criteria forthe tests described. It establishes the set ofcriteria to which a drug substance or drugproduct should conform to be consideredacceptable for its intended use.‘‘Conformance to specifications’’ means thatthe drug substance and/or drug product,when tested according to the listed analyticalprocedures, will meet the listed acceptancecriteria. Specifications are binding qualitystandards that are agreed to between theappropriate governmental regulatory agencyand the applicant.
Specifications are one part of a totalcontrol strategy for the drug substance anddrug product designed to ensure productquality and consistency. Other parts of thisstrategy include thorough productcharacterization during development uponwhich specifications are based, adherence togood manufacturing practices (GMP’s), and avalidated manufacturing process, e.g., rawmaterial testing, in-process testing, stabilitytesting.
Specifications are chosen to confirm thequality of the drug substance and drugproduct rather than to establish fullcharacterization, and should focus on thosecharacteristics found to be useful in ensuringthe safety and efficacy of the drug substanceand drug product.
1.2 Objective of the Guidance
This guidance is intended to assist, to theextent possible, in the establishment of asingle set of global specifications for newdrug substances and new drug products. Itprovides guidance on the setting andjustification of acceptance criteria and theselection of test procedures for new drugsubstances of synthetic chemical origin, andnew drug products produced from them, thathave not been registered previously in theUnited States, the European Union, or Japan.
1.3 Scope of the Guidance
The quality of drug substances and drugproducts is determined by their design,development, in-process controls, GMPcontrols, and process validation, and byspecifications applied to them throughoutdevelopment and manufacture. Thisguidance addresses specifications, i.e., thosetests, procedures, and acceptance criteriaused to assure the quality of the new drugsubstance and new drug product at releaseand during shelf life. Specifications are animportant component of quality assurance,but are not its only component. All of theconsiderations listed above are necessary toensure consistent production of drugsubstances and drug products of high quality.
This guidance addresses only themarketing approval of new drug products(including combination products); it does notaddress drug substances or drug productsduring the clinical research stages of drugdevelopment. Biological/biotechnologicalproducts, peptides, oligonucleotides,radiopharmaceuticals, fermentation andsemisynthetic products derived therefrom,herbal products, and crude products ofanimal or plant origin are also not covered.A separate ICH guidance addressesspecifications, tests, and procedures forbiotechnological/biological products.
Guidance is provided with regard toacceptance criteria that should be establishedfor all new drug substances and new drugproducts, i.e., universal acceptance criteria,and those that are considered specific toindividual drug substances and/or dosageforms. This guidance reflects the current stateof the art at the time it has been written, andshould not be considered all-encompassing.New analytical technology, andmodifications to existing technology, arecontinuously being developed. Suchtechnologies should be used when justifiable.
Dosage forms addressed in this guidanceinclude solid oral dosage forms, liquid oraldosage forms, and parenterals (small andlarge volume). This is not meant to be an all-inclusive list, or to limit the number ofdosage forms to which this guidance applies.The dosage forms presented serve as modelsthat may be applicable to other dosage formsthat have not been discussed. The extendedapplication of the concepts in this guidanceto other dosage forms, e.g., inhalation dosageforms (powders, solutions, etc.), topicalformulations (creams, ointments, gels), andtransdermal systems, is encouraged.
2. General Concepts
The following concepts are important inthe development and setting of harmonizedspecifications. They are not universallyapplicable, but each should be considered inparticular circumstances. This guidancepresents a brief definition of each conceptand an indication of the circumstances underwhich it may be applicable. Generally,proposals to implement these conceptsshould be justified by the applicant andapproved by the appropriate regulatoryauthority before being put into effect.2.1 Periodic/Skip Testing: Periodic or skiptesting is the performance of specified testsat release on preselected batches and/or atpredetermined intervals, rather than on a
batch-to-batch basis. This represents a lessthan full schedule of testing and shouldtherefore be justified and presented to theregulatory authority prior to implementation.This concept may be applicable to, forexample, dissolution (see section 2.4),residual solvents, and microbiologicaltesting, e.g., for solid oral dosage forms. It isrecognized that only limited data may beavailable at the time of submission of anapplication (see section 2.5). This conceptmay therefore sometimes be implementedpostapproval in accordance with GMP.2.2 Release Vs. Shelf-Life AcceptanceCriteria: The concept of different acceptancecriteria for release vs. shelf-life specificationsapplies to drug products only; it pertains tothe establishment of more restrictive criteriafor the release of a drug product than areapplied to the shelf-life. Examples where thismay be applicable include assay andimpurity (degradation product) levels. InJapan and the United States, this conceptmay only be applicable to inhouse criteria,and not to the regulatory release criteria. Inthe European Union, there is a regulatoryrequirement for distinct specifications forrelease and for shelf-life.2.3 In-Process Tests: In-process tests are teststhat may be performed during themanufacture of either the drug substance ordrug product, rather than as part of theformal battery of tests which are conductedprior to release. In-process tests that are usedfor the purpose of adjusting processparameters within an operating range, e.g.,hardness and friability of tablet cores thatwill be coated, are not included in thespecification. Certain tests conducted duringthe manufacturing process, where theacceptance criterion is identical to or tighterthan the release requirement (e.g., pH of asolution), may be acceptable to satisfyspecification requirements when the test isincluded in the specification.2.4 Design and Development Considerations:The experience and data accumulated duringthe development of a new drug substance orproduct should form the basis for the settingof specifications. It may be possible topropose excluding or replacing certain testson this basis. Some examples are:
• Microbiological testing for drugsubstances and solid dosage forms that havebeen shown during development not tosupport microbial viability or growth.
• Extractables from product containerswhere it has been reproducibly shown thateither no extractables are found in the drugproduct or the levels meet acceptedstandards for safety.
• Particle size testing may fall into thiscategory, may be performed as an in-processtest, or may be performed as a release test,depending on its relevance to productperformance.
• Dissolution testing for immediaterelease solid oral drug products made fromvery water soluble drug substances may bereplaced by disintegration testing, if theseproducts have been demonstrated duringdevelopment to have consistently rapid drugrelease characteristics. (See Decision trees#7(1) through #7(4)).2.5 Limited Data Available at Filing: It isrecognized that only a limited amount of data

62892 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
may be available at the time of filing, whichcan influence the process of settingacceptance criteria. As a result, it may benecessary to propose revised acceptancecriteria as additional experience is gainedwith the manufacture of a particular drugsubstance or drug product (example:acceptance limits for a specific impurity).The basis for the acceptance criteria at thetime of filing will focus necessarily on safetyand efficacy.2.6 Parametric Release: Parametric releasecan be used as an operational alternative toroutine release testing for the drug product.Sterility testing for terminally sterilized drugproducts is one example. In this case, therelease of a batch is based on results frommonitoring specific parameters, e.g.,temperature and pressure, during theterminal sterilization phase(s) of drugproduct manufacturing. These parameterscan generally be more accurately controlledand measured, so that they are more reliablein predicting sterility assurance than is end-product sterility testing. It is important tonote that the sterilization process should beadequately validated before parametricrelease is proposed. When parametric releaseis performed, the attribute which is indirectlycontrolled (e.g., sterility), together with areference to the associated test procedure,still should be included in the specifications.2.7 Alternative Procedures: Alternativeprocedures are those that may be used tomeasure an attribute when such procedurescontrol the quality of the drug substance ordrug product to an extent that is comparableor superior to the official procedure.Example: For tablets that have been shownnot to degrade during manufacture, it may bepermissible to use a spectrophotometricprocedure for release as opposed to theofficial procedure, which is chromatographic.However, the chromatographic procedureshould still be used to demonstratecompliance with the acceptance criteriaduring the shelf-life of the product.2.8 Pharmacopoeial Tests and AcceptanceCriteria: References to certain methods arefound in pharmacopoeias in each region.Wherever they are appropriate,pharmacopoeial methods should be utilized.Whereas differences in pharmacopoeialmethods and/or acceptance criteria haveexisted among the regions, a harmonizedspecification is possible only if the methodsand acceptance criteria defined areacceptable to regulatory authorities in allregions. This guidance is dependent on thesuccessful completion of harmonization ofpharmacopoeial methods for severalattributes commonly considered in thespecifications for new drug substances ornew drug products.
The following attributes are essentiallyharmonized with respect to analyticalmethod and acceptance criteria, exceptwhere noted, across the EuropeanPharmacopoeia (Ph. Eur.), JapanesePharmacopoeia (JP), and United StatesPharmacopeia (USP):
SterilityResidue on Ignition/Sulfated AshBacterial EndotoxinsColor/ClarityParticulate Matter
Dissolution (apparatus)Disintegration (apparatus)To signify the harmonized status of these
general methods, the pharmacopoeias willinclude a statement in the text that indicatesthat the methods and acceptance criteriafrom all three pharmacopoeias are consideredequivalent and are, therefore,interchangeable. An appropriate reference tothe harmonized method and acceptancecriteria is considered acceptable for aspecification in all three regions. Forexample, sterility data generated using the JPmethod, as well as the JP method itself andits acceptance criteria, are consideredacceptable for registration in all threeregions. An appropriate reference may beexpressed as JP/Ph. Eur./USP.
Harmonization of the following attributeswill be completed prior to approval of a step4 guidance:
Dissolution (media and acceptance criteria)Disintegration (media and acceptance
criteria)Uniformity of MassUniformity of ContentExtractable VolumePreservative Effectiveness (scope of test
and acceptance criteria)Microbial Contamination
2.9 Evolving Technologies: New analyticaltechnology and modifications to existingtechnology are continuously beingdeveloped. Such technologies should be usedwhen they are considered to offer additionalassurance of quality, or are otherwisejustifiable.2.10 Impact of Drug Substance on DrugProduct Specifications: In general, it shouldnot be necessary to test the drug product forquality attributes uniquely associated withthe drug substance. Example: It is normallynot necessary to test the drug product forsynthesis impurities that are controlled in thedrug substance and are not degradationproducts. Refer to the ICH guidance‘‘Impurities in New Drug Products’’ fordetailed information.2.11 Reference Standard: A referencestandard, or reference material, is a substanceprepared for use as the standard in an assay,identification, or purity test. The substancemay be either the new drug substance or aknown impurity. It has a quality appropriateto its use. For new drug substance referencestandards intended for use in assays, theimpurities should be adequately identifiedand/or controlled, and purity should bemeasured by a quantitative procedure.
3. Guidelines
3.1 Specifications: Definition andJustification
3.1.1 Definition of Specifications
A specification is defined as a list of tests,references to analytical procedures, andappropriate acceptance criteria that arenumerical limits, ranges, or other criteria forthe tests described. It establishes the set ofcriteria to which a new drug substance ornew drug product should conform to beconsidered acceptable for its intended use.‘‘Conformance to specifications’’ means thatthe drug substance and/or drug product,when tested according to the listed analytical
procedures, will meet the listed acceptancecriteria. Specifications are binding qualitystandards that are agreed to between theappropriate governmental regulatory agencyand the applicant.
It is possible that, in addition to releasetests, a specification may list in-process tests,periodic (skip) tests, and other tests whichare not always conducted on a batch-by-batchbasis. In such cases, the applicant shouldspecify which tests are routinely conductedbatch-by-batch, and which tests are not, withan indication and justification of the actualtesting frequency. In this situation, the drugsubstance and/or drug product should meetthe acceptance criteria if tested.
It should be noted that changes in thespecification after approval of the applicationmay need prior approval by the regulatoryauthority.
3.1.2 Justification of Specifications
When a specification is first proposed,justification should be presented for eachprocedure and each acceptance criterionincluded. The justification should refer torelevant development data, pharmacopoeialstandards, test data for drug substances anddrug products used in toxicology and clinicalstudies, and results from accelerated andlong term stability studies, as appropriate.Additionally, a reasonable range of expectedanalytical and manufacturing variabilityshould be considered. It is important toconsider all of this information.
Approaches other than those set forth inthis guidance may be applicable andacceptable. The applicant should justifyalternative approaches. Such justificationshould be based on data derived from thenew drug substance synthesis and/or the newdrug product manufacturing process. Thisjustification may consider theoreticaltolerances for a given procedure oracceptance criterion, but the actual resultsobtained should form the primary basis forwhatever approach is taken.
Test results from primary stability andscale-up/validation batches should beconsidered in setting and justifyingspecifications. If multiple manufacturingsites are planned, it may be valuable toconsider data from these sites in establishingthe initial tests and acceptance criteria. Thisis particularly true when there is limitedinitial experience with the manufacture ofthe drug substance or drug product at anyparticular site. If data from a singlerepresentative manufacturing site are used insetting tests and acceptance criteria, productmanufactured at all sites should still complywith these criteria.
Presentation of test results in graphicformat may be helpful in justifyingindividual acceptance criteria, particularlyfor assay values and impurity levels. Datafrom development work should be includedin such a presentation, along with stabilitydata available for new drug substance or newdrug product batches manufactured by theproposed commercial processes. Justificationfor exclusion of a test from the specificationshould be based on development data and onprocess validation data (where available).
When only limited data are available, theinitially approved tests and acceptancecriteria should be reviewed as more

62893Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
information is collected, with a view towardspossible modification. This could involveloosening, as well as tightening, acceptancecriteria as appropriate.
3.2 Universal Tests/Criteria
Implementation of the recommendations inthe following section should take intoaccount the ICH guidances ‘‘Text onValidation of Analytical Procedures’’ and‘‘Validation of Analytical Procedures:Methodology.’’
3.2.1 New Drug Substances
The following tests and acceptance criteriaare considered generally applicable to allnew drug substances.
(a) Description: A qualitative statementabout the state (e.g., solid, liquid) and colorof the new drug substance. If any of thesecharacteristics change during storage, thischange should be investigated andappropriate action taken.
(b) Identification: Identification testingshould optimally be able to discriminatebetween compounds of closely relatedstructure that are likely to be present.Identification tests should be specific for thenew drug substance, e.g., infraredspectroscopy (IR). Identification solely bychromatographic retention time, for example,is not regarded as being specific; however, acombination of tests into a single procedure,such as HPLC (high pressure/performanceliquid chromatography)/UV (ultraviolet)-diode array, HPLC/MS (mass spectroscopy),or GC (gas chromatography)/MS may beacceptable. If the new drug substance is asalt, identification testing should beperformed for the individual ions.
New drug substances which are opticallyactive may also need specific identificationtesting. Please refer to section 3.3.1.(d) in thisguidance for further discussion of this topic.
(c) Assay: A specific, stability-indicatingprocedure should be included to determinethe content of the new drug substance. Inmany cases it is possible to employ the sameprocedure (e.g., HPLC) for both assay of thenew drug substance and quantitation ofimpurities.
In cases where use of a nonspecific assayis justified, other supporting analyticalprocedures should be used to achieve overallspecificity. For example, where titration isadopted to assay the drug substance, thecombination of the assay and a suitable testfor impurities can be used.
(d) Impurities: Organic and inorganicimpurities and residual solvents are includedin this category. Refer to the ICH guidances‘‘Impurities in New Drug Substances’’ and‘‘Residual Solvents in Pharmaceuticals’’ fordetailed information.
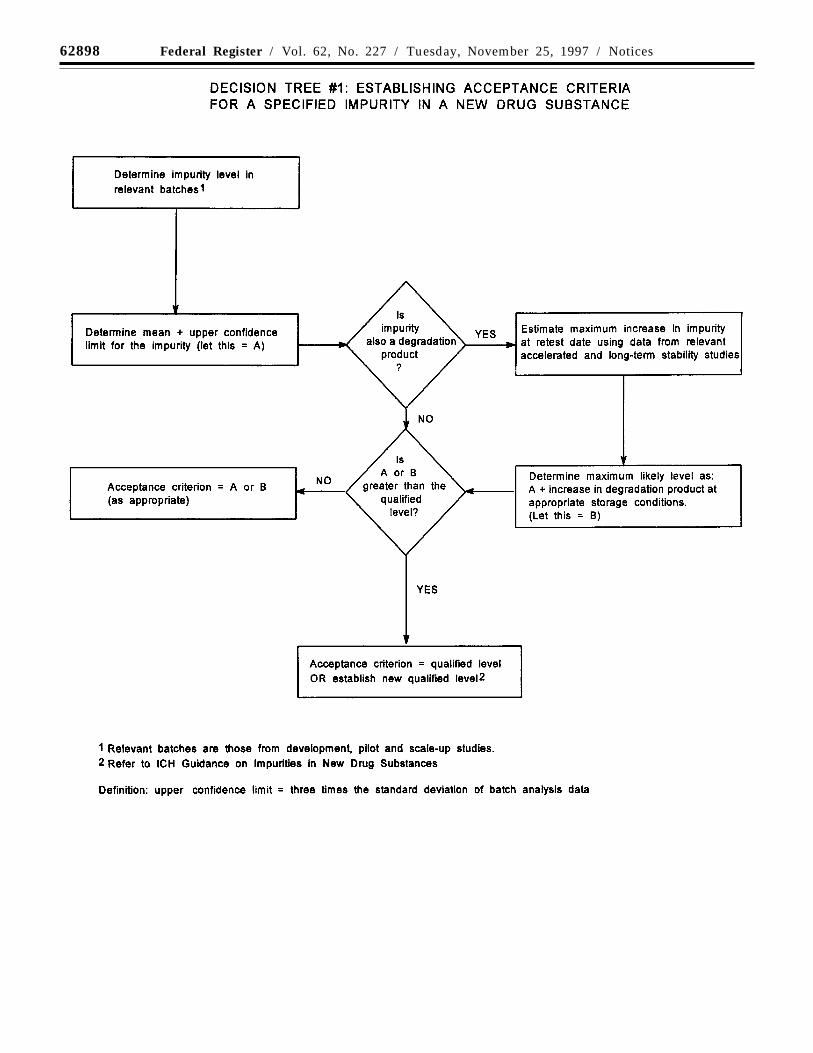
Decision tree #1 addresses theextrapolation of meaningful limits onimpurities from the body of data generatedduring development. At the time of filing, itis unlikely that sufficient data will beavailable to assess process consistency.Therefore, it is inappropriate to establishacceptance criteria that tightly encompass thebatch data at the time of filing. (See section2.5, limited data available at filing.)
3.2.2 New Drug Products
The following tests and acceptance criteriaare considered generally applicable to allnew drug products:
(a) Description: A qualitative description ofthe dosage form should be provided (e.g.,size, shape, color). If any of thesecharacteristics change during manufacture orstorage, this change should be investigatedand appropriate action taken. The acceptancecriteria should include the final acceptableappearance. If color changes during storage,a quantitative procedure may be appropriate.
(b) Identification: Identification testingshould establish the identity of the new drugsubstance(s) in the new drug product andshould be able to discriminate betweencompounds of closely related structurewhich are likely to be present. Identity testsshould be specific for the new drugsubstance, e.g., infrared spectroscopy.Identification solely by chromatographicretention time, for example, is not regardedas being specific; however, a combination oftests into a single procedure, such as HPLC/UV-diode array, may be acceptable.
(c) Assay: A specific, stability-indicatingassay to determine strength should beincluded for all new drug products. In manycases it is possible to employ the sameprocedure (e.g., HPLC) for both assay of thenew drug substance and quantitation ofimpurities. Results of content uniformitytesting for new drug products can be used forquantitation of drug product strength, if themethods used for content uniformity are alsoappropriate as assays.
In cases where use of a nonspecific assayis justified, other supporting analyticalprocedures should be used to achieve overallspecificity. For example, where titration isadopted to assay the drug substance, thecombination of the assay and a suitable testfor impurities can be used.
(d) Impurities: Organic and inorganicimpurities and residual solvents are includedin this category. Refer to the ICH guidances‘‘Impurities in New Drug Products’’ and‘‘Residual Solvents in Pharmaceuticals’’ fordetailed information.
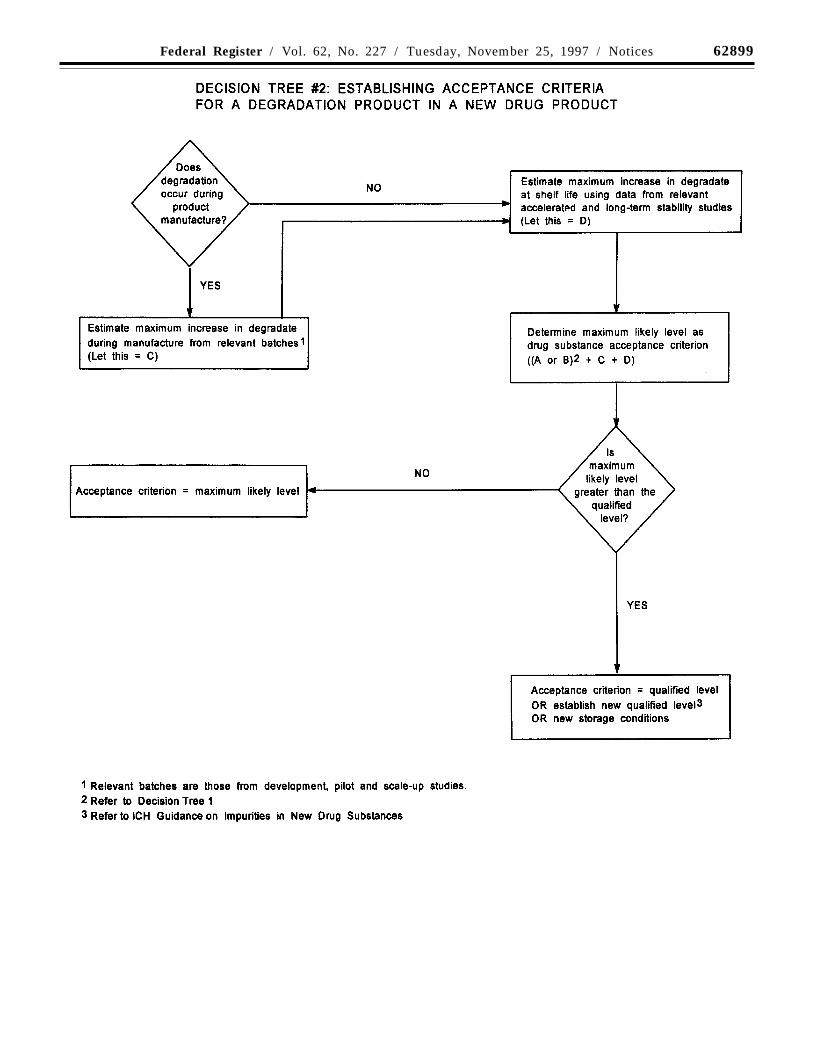
Organic impurities arising fromdegradation of the new drug substanceshould be monitored in the new drugproduct. Acceptance limits should be statedfor individual specified degradationproducts, which may include both identifiedand unidentified degradation products asappropriate, and total degradation products.Process impurities from the new drugsubstance synthesis are normally controlledduring drug substance testing, and thereforeare not included in the total impurities limit.When it has been conclusively demonstratedvia appropriate analytical methodology, witha significant body of data, that the drugsubstance does not degrade in the specificformulation, and under the specific storageconditions proposed in the new drugapplication, degradation product testing maybe reduced or eliminated upon approval bythe regulatory authorities.
Decision tree #2 addresses theextrapolation of meaningful limits ondegradation products from the body of datagenerated during development. At the time offiling, it is unlikely that sufficient data will
be available to assess process consistency.Therefore, it is inappropriate to establishacceptance criteria that tightly encompass thebatch data at the time of filing. (See section2.5, limited data available at filing).
3.3 Specific Tests/Criteria
In addition to the universal tests listedabove, the following tests may be consideredon a case by case basis for drug substancesand/or drug products. Individual tests/criteria should be included in thespecification when the tests have an impacton the quality of the drug substance and drugproduct for batch control. Tests other thanthose listed below may be needed inparticular situations or as new informationbecomes available.
3.3.1 New Drug Substances
(a) Physicochemical properties: These areproperties such as pH of an aqueous solution,melting point/range, and refractive index.The procedures used for the measurement ofthese properties are usually unique and donot need much elaboration, e.g., capillarymelting point, Abbe refractometry. The testsperformed in this category should bedetermined by the physical nature of the newdrug substance and by its intended use.
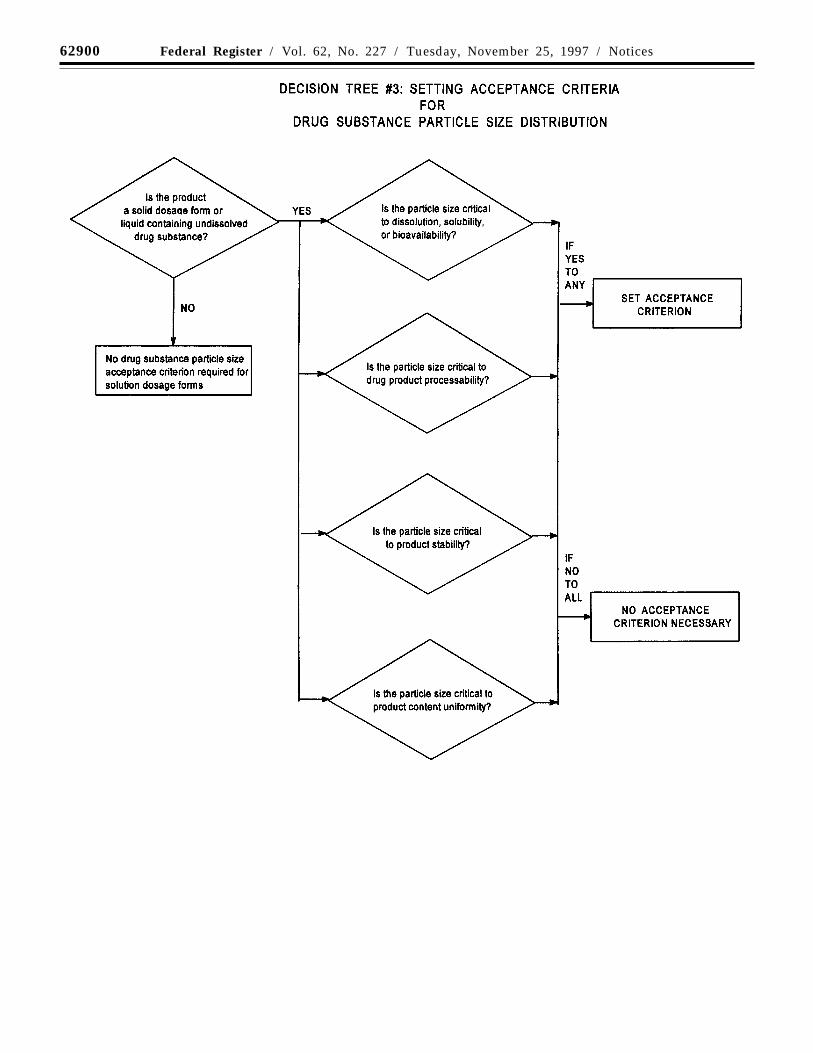
(b) Particle size: For some new drugsubstances intended for use in solid orsuspension drug products, particle size canhave a significant effect on dissolution rates,bioavailability, and/or stability. In suchinstances, testing for particle size distributionshould be carried out using an appropriateprocedure, and acceptance criteria should beprovided.
Decision tree #3 provides additionalguidance on when particle size testingshould be considered.
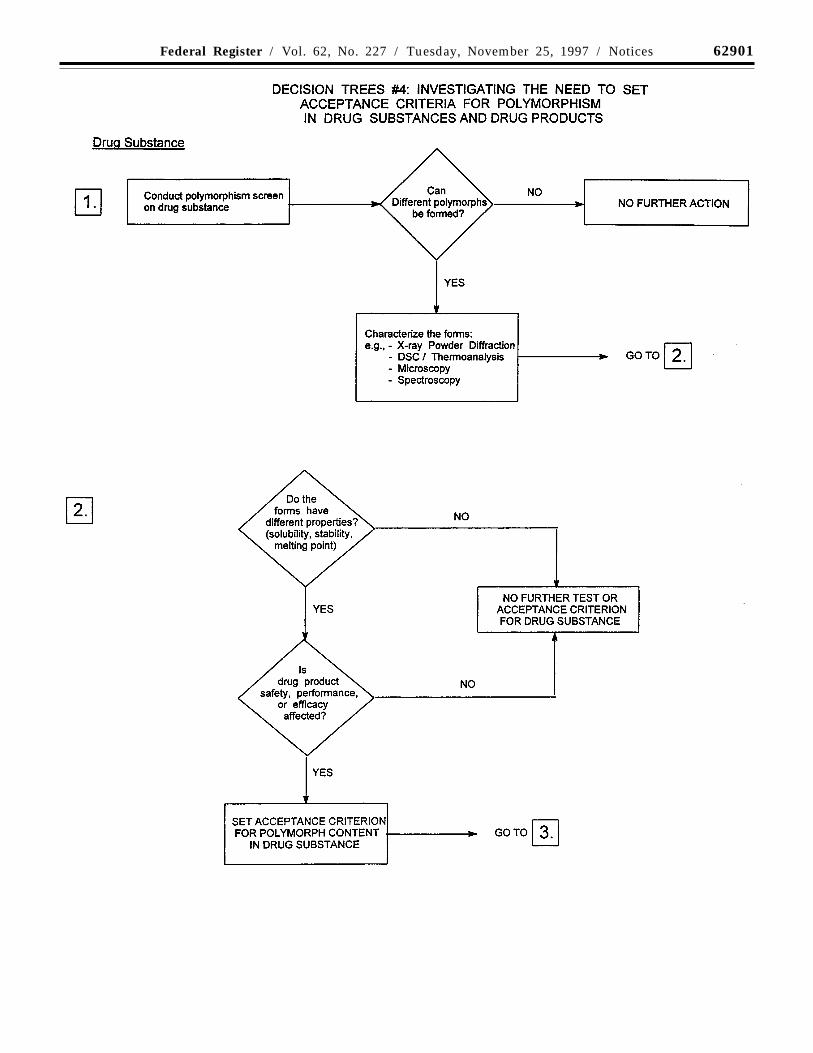
(c) Solid state forms: Some new drugsubstances exist in different solid state forms(polymorphs or solvates) that differ in theirphysical properties. Differences in theseforms could, in some cases, affect the qualityor performance of the new drug products. Incases where differences exist that have beenshown to affect drug product performance,bioavailability, or stability, then theappropriate solid state should be specified.
Physico-chemical measurements andtechniques are commonly used to determinewhether multiple forms exist. Examples ofthese procedures are: Melting point(including hot-stage microscopy), solid stateIR, X-ray powder diffraction, thermalanalysis procedures (like DSC (differentialscanning calorimetry), TGA(thermogravimetric analysis) and DTA(differential thermal analysis)), Ramanspectroscopy, scanning electron microscopy,and solid state NMR (nuclear magneticresonance spetroscopy).
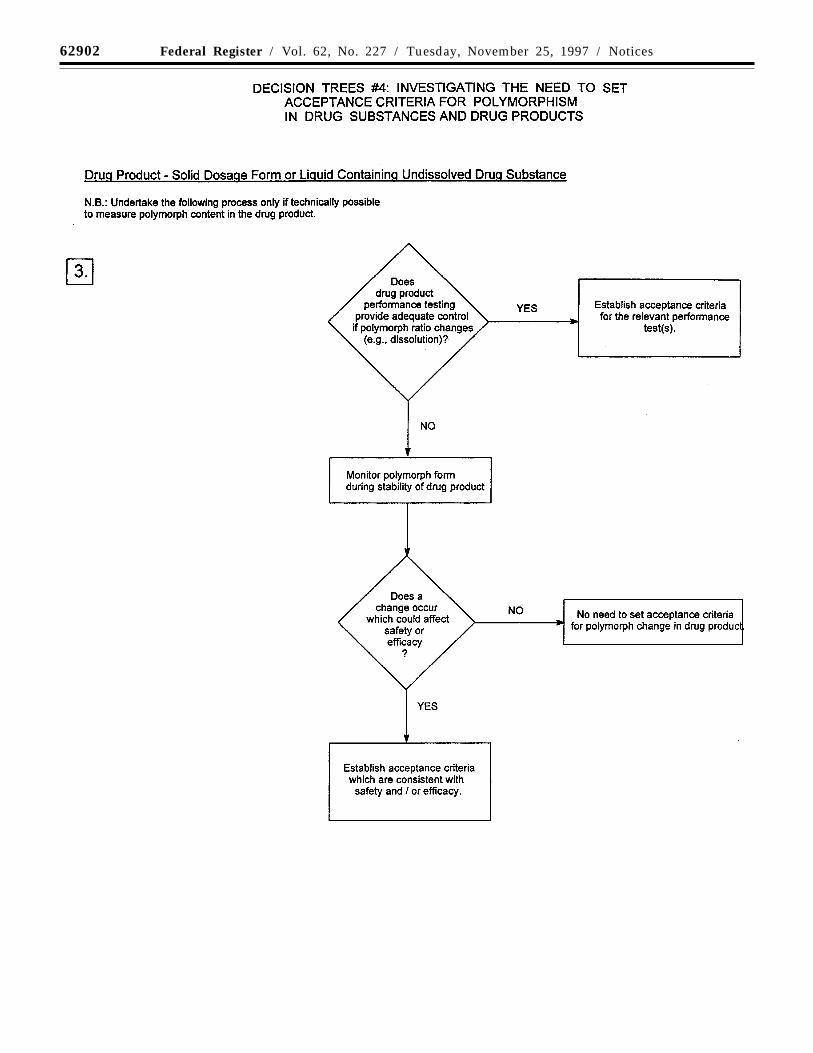
Decision trees #4(1) through #4(3) provideadditional guidance on when, and how, solidstate forms should be monitored andcontrolled.
Note: These decision trees should befollowed sequentially. Trees #1 and #2consider whether polymorphism is exhibitedby the drug substance and whether thedifferent polymorphic forms can affectperformance of the drug product. Tree #3should only be applied when polymorphismhas been demonstrated for the drug substance

62894 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
and has been shown to affect theseproperties. Tree #3 considers the potential forchange in polymorphic forms in the drugproduct and whether such a change has anyeffect on product performance.
It is generally technically very difficult tomeasure polymorphic changes in drugproducts. A surrogate test (e.g., dissolution)can generally be used to monitor productperformance, and polymorph content shouldonly be used as a test and acceptancecriterion of last resort.
The decision trees focus on polymorphism,but the same decision process can be appliedto other solid state criteria, such as hydrationand solvation, where appropriate.
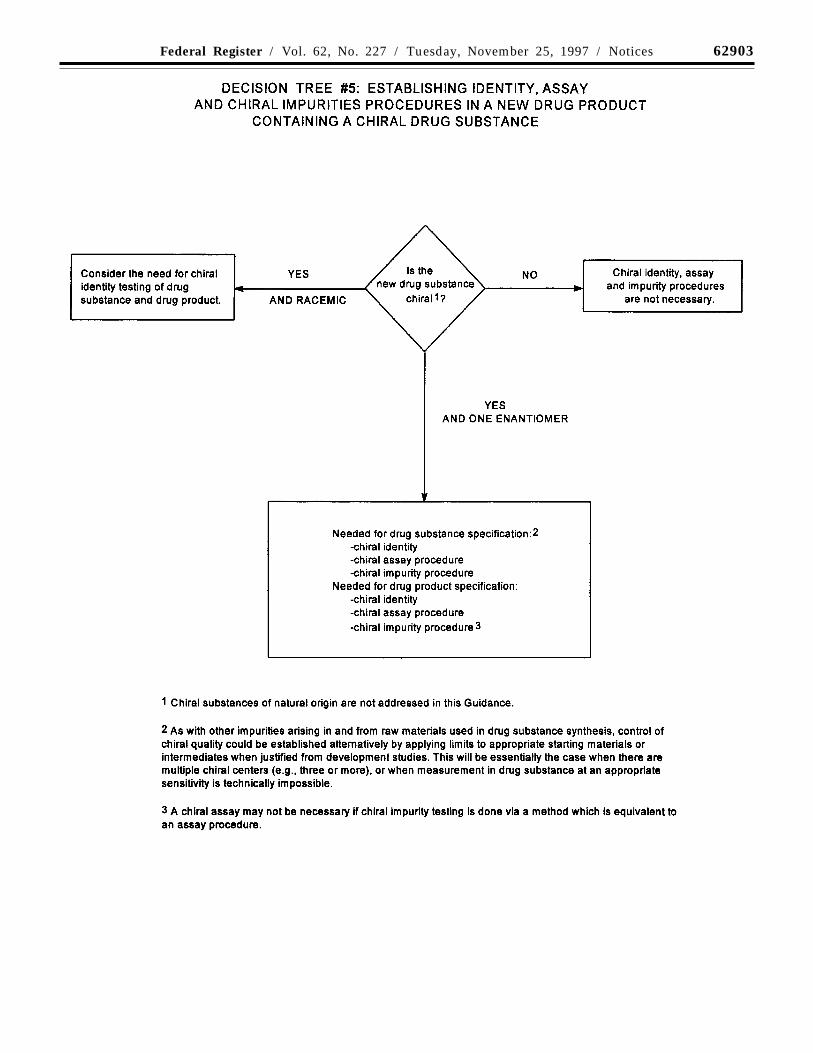
(d) Tests for new drug substances that areoptically active: Chiral impurities areexcluded from ICH guidances on ‘‘Impuritiesin New Drug Substances’’ and ‘‘Impurities inNew Drug Products’’ because of practicaldifficulties in quantifying them at thequalification and identification thresholdsgiven in those guidances. However, chiralimpurities in chiral new drug substances andthe resulting new drug products should betreated according to principles established inthose guidances.
Decision tree #5 summarizes when and ifchiral identity tests, impurity tests, andassays may be needed for both new drugsubstances and new drug products, accordingto the following concepts:
Drug Substance: Impurities. For chiral drugsubstances that are developed as a singleenantiomer, control of the other enantiomershould be considered in the same manner asfor other impurities. However, technicallimitations may preclude the same limits ofdetermination or qualification being applied.If it is technically difficult to effect controlin the drug substance itself, assurance ofcontrol could be given by appropriate testingof a starting material or intermediate, withsuitable justification.
Assay. An enantioselective determinationof the drug substance should be part of thespecification. It is considered acceptable forthis to be achieved either through use of achiral assay procedure or by the combinationof an achiral assay together with appropriatemethods of controlling the enantiomericimpurity.
Identity. The identity test(s) should becapable of distinguishing a single enantiomerfrom its opposite enantiomer. Where a drugsubstance is a racemate, the identity methodshould be capable of verifying the racemicnature and distinguishing it from eitherenantiomer.
Drug Product: Degradation products.Control of the other enantiomer in a drugproduct is necessary if that enantiomer hasbeen shown to be a degradation product.
Assay. Where development studies havedemonstrated that the enantiomer is not adegradation product, an achiral assay may besufficient. However, a chiral assay ispreferred or, alternatively, the combination ofan achiral assay plus a procedure to controlthe presence of the opposite enantiomer.
Identity. An identity test should beestablished that is capable of verifying thepresence of the correct enantiomer or theracemate, as appropriate.
(e) Water content: This test is important incases where the new drug substance is
known to be hygroscopic or degraded bymoisture or when the drug substance isknown to be a stoichiometric hydrate. Theacceptance criteria may be justified with dataon the effects of hydration or moistureabsorption. In some cases, a Loss on Dryingprocedure may be adequate; however, adetection procedure that is specific for water(e.g., Karl Fischer titration) is preferred.
(f) Inorganic impurities: The need forinclusion of tests and acceptance criteria forinorganic impurities should be studiedduring development and based on knowledgeof the manufacturing process. Wherejustified, procedures and acceptance criteriafor sulfated ash/residue on ignition shouldfollow pharmacopoeial precedents; otherinorganic impurities may be determined byother appropriate procedures, e.g., atomicabsorption spectroscopy.
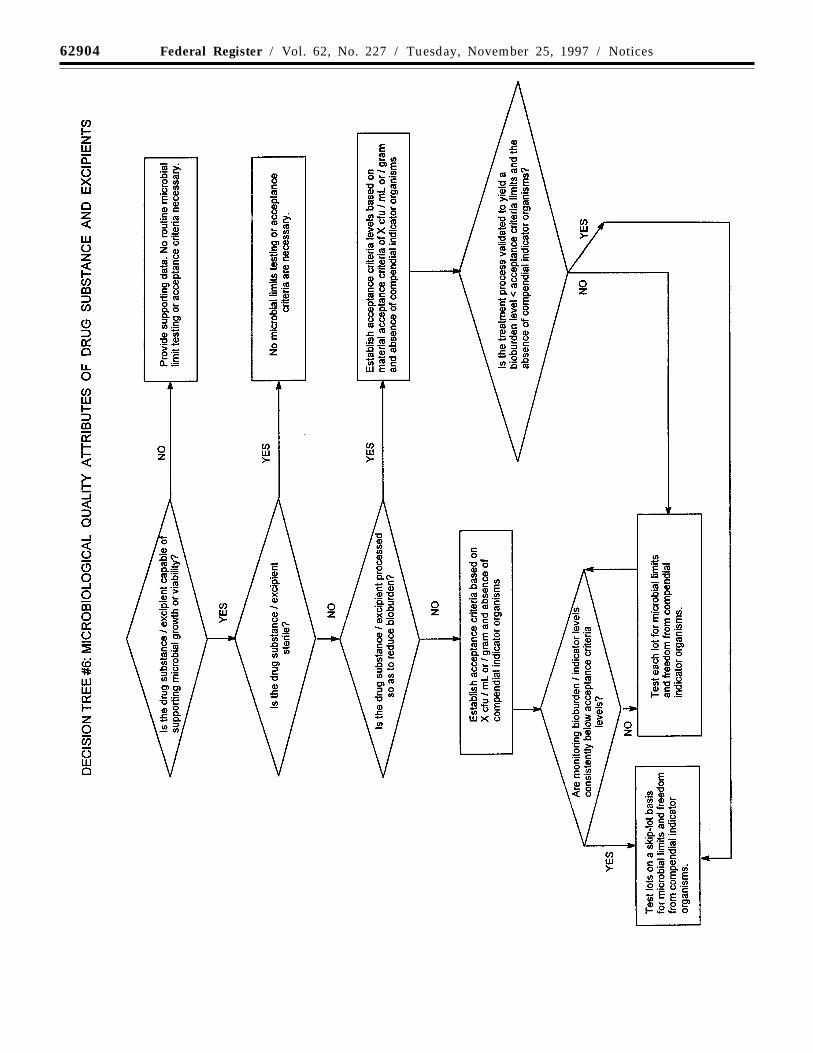
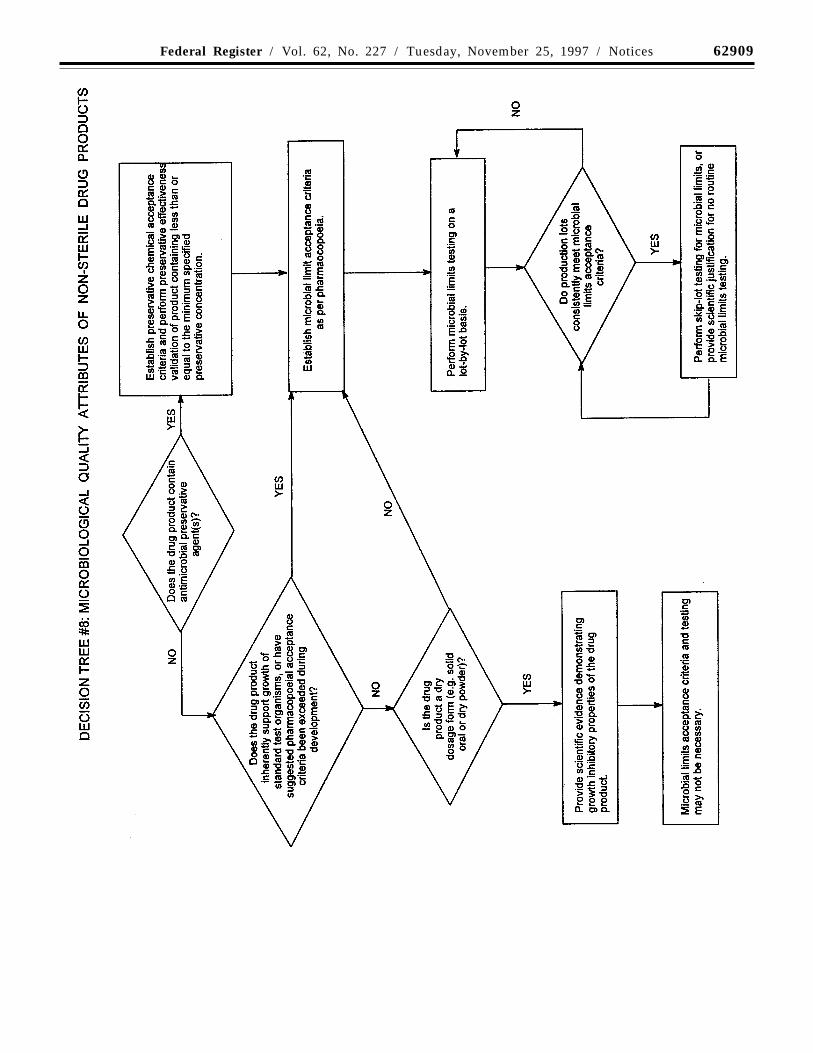
(g) Microbial limits: There may be a needto specify the total count of aerobicmicroorganisms, the total count of yeasts andmolds, and the absence of specificobjectionable bacteria (e.g., Staphylococcusaureus, Escherichia coli, Salmonella,Pseudomonas aeruginosa). These should besuitably determined using pharmacopoeialprocedures. In special cases, sterility testingor endotoxin testing may be appropriate. Forexample, the drug substance is manufacturedas sterile (sterility testing appropriate) or willbe used to formulate an injectable drugproduct (endotoxin testing appropriate).
Decision tree #6 provides additionalguidance on when microbial limits should beincluded.
3.3.2 New Drug Products
Additional tests and acceptance criteriagenerally should be included for particularnew drug products. The following selectionpresents a representative sample of both thedrug products and the types of tests andacceptance criteria which may beappropriate. The specific dosage formsaddressed include solid oral drug products,liquid oral drug products, and parenterals(small and large volume). Application of theconcepts in this guidance to other dosageforms is encouraged. Note that issues relatedto optically active drug substances and tosolid state considerations for drug productsare discussed in section 3.3.1 of thisguidance.
3.3.2.1 The following tests are applicable totablets (coated and uncoated) and hardcapsules. One or more of these tests may alsobe applicable to soft capsules and granules.
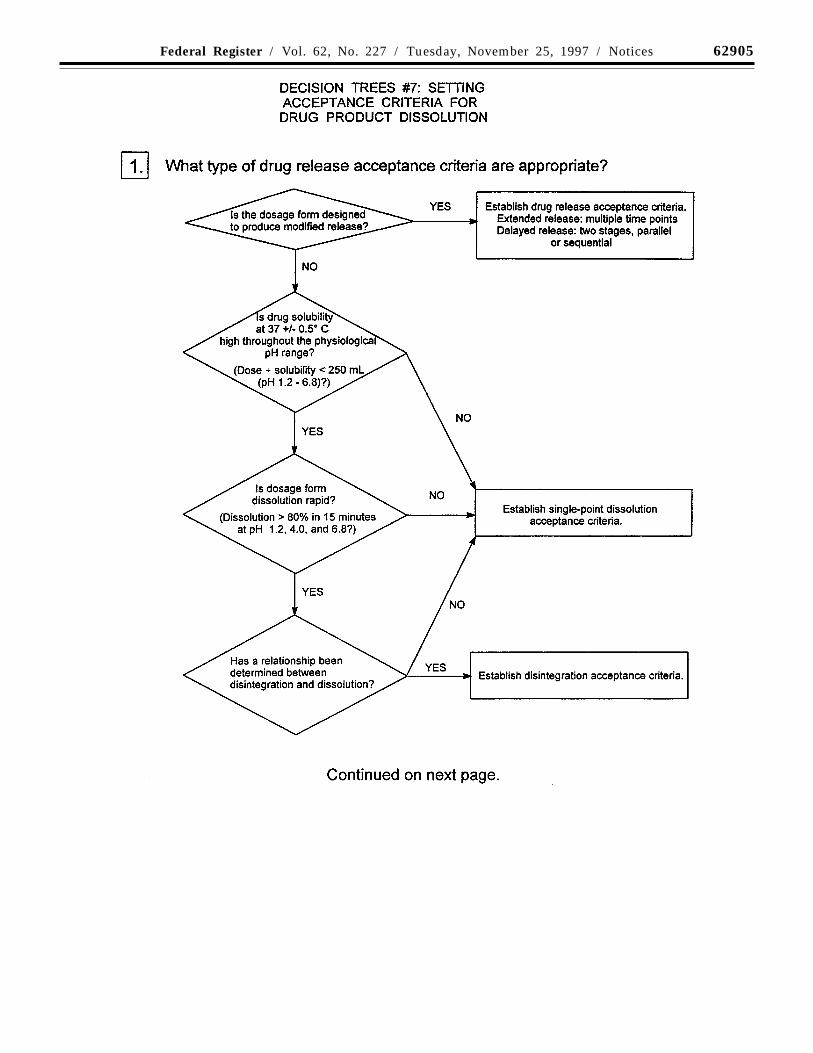
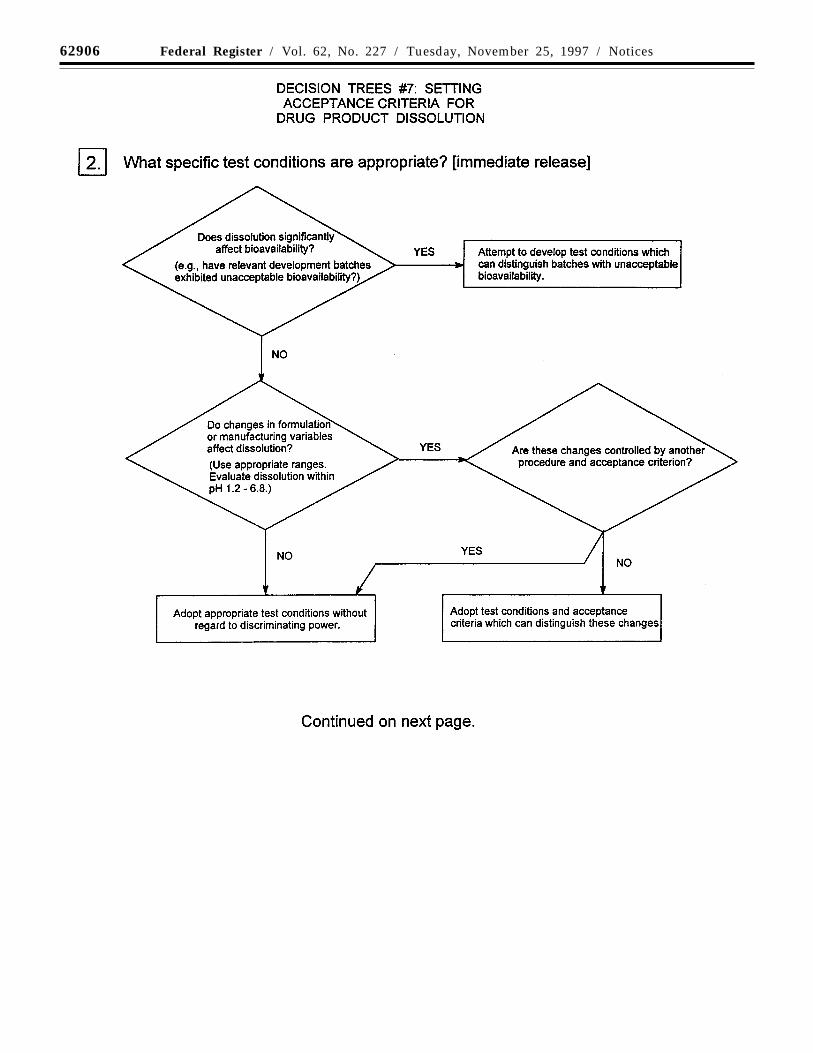
(a) Dissolution/disintegration: For rapidlydissolving products containing drugs that arehighly soluble throughout the physiologicalpH range, disintegration testing maysometimes be sufficient. Disintegrationtesting is most appropriate when arelationship to dissolution has beenestablished or when disintegration is shownto be more discriminating than dissolution.In such cases, dissolution testing may notalways be necessary, or may be proposed asa skip test. It is expected that developmentinformation will be provided to support therobustness of the formulation andmanufacturing process with respect to theselection of dissolution vs. disintegrationtesting.
Single-point measurements are normallyconsidered to be suitable for immediaterelease dosage forms. For modified releasedosage forms, appropriate test conditions andsampling procedures should be established.For example, multiple-time-point samplingshould be performed for extended releasedosage forms, and two-stage testing (usingdifferent media in succession or in parallel,as appropriate) may be appropriate fordelayed release dosage forms. In these casesit is important to consider the populations ofindividuals who will be taking the drugproduct (e.g., achlorhydric elderly) whendesigning the tests and acceptance criteria.
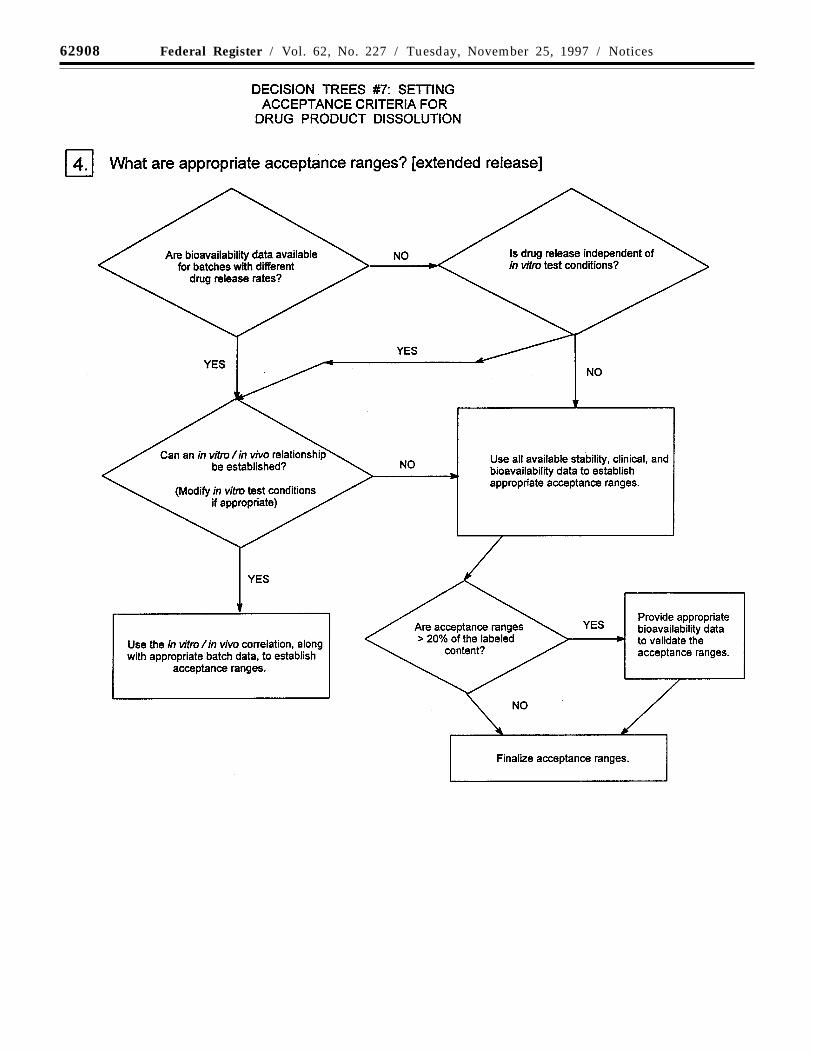
Where multiple-point acceptance criteriaare necessary, in vitro/in vivo correlationmay be used to establish these criteria whenhuman bioavailability data are available forformulations exhibiting different releaserates. Where such data are not available, anddrug release cannot be shown to beindependent of in vitro test conditions, thenacceptance criteria should be established onthe basis of available batch data. Normally,the permitted variability in release rate at anygiven time point should not exceed a totalnumerical difference of +/-10 percent of thelabeled content of drug substance (i.e., a totalvariability of 20 percent: a requirement of 50+/-10 percent thus means an acceptable rangefrom 40 to 60 percent) unless a wider rangeis supported by a bioequivalency study.
Decision trees #7(1) through #7(4) provideadditional guidance on the use of dissolutionand disintegration testing.
(b) Hardness/friability: It is normallyappropriate to perform hardness and/orfriability testing as an in-process control (seesection 2.3). Under these circumstances, it isnormally not necessary to include theseattributes in the specification. If thecharacteristics of hardness and friability havea critical impact on drug product quality(e.g., chewable tablets), acceptance criteriashould be included in the specification.
(c) Uniformity of dosage units: This termincludes both uniformity of content anduniformity of mass; a pharmacopoeialprocedure should be used. If appropriate,these tests may be performed as in-processcontrols; the acceptance criteria should beincluded in the specification.
(d) Water content: A test for water contentshould be included when appropriate. Theacceptance criteria may be justified with dataon the effects of hydration or waterabsorption on the drug product. In somecases, a Loss on Drying procedure may beadequate; however, a detection procedurewhich is specific for water (e.g., Karl Fischertitration) is preferred.
(e) Microbial limits: Microbial limit testingis seen as an attribute of GMP, as well as ofquality assurance. In general, it is advisableto test the drug product unless itscomponents are tested before manufactureand the manufacturing process is known,through validation studies, not to carry asignificant risk of microbial contamination. Itshould be noted that, whereas this guidancedoes not directly address excipientselsewhere, the principles discussed here maybe applicable to excipients as well as to newdrug products. Skip testing may be anappropriate approach in both cases.

62895Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
Acceptance criteria should be set for thetotal count of aerobic microorganisms, thetotal count of yeasts and molds, and theabsence of specific objectionable bacteria(e.g., Staphylococcus aureus, Escherichiacoli, Salmonella, Pseudomonas). Theseshould be determined by suitable procedures,using pharmacopoeial procedures, and at asampling frequency or time point inmanufacture that is justified by data andexperience. With acceptable scientificjustification, it may be possible to propose nomicrobial limit testing for solid oral dosageforms.
Decision tree #8 provides additionalguidance on the use of microbial limittesting.
3.3.2.2 Oral liquids: One or more of thefollowing specific tests will normally beapplicable to oral liquids and to powdersintended for reconstitution as oral liquids.
(a) Uniformity of dosage units: This termincludes both uniformity of content anduniformity of mass. Generally, acceptancecriteria should be set for weight variation, fillvolume, and/or uniformity of fill.Pharmacopoeial procedures should be used.
If appropriate, tests may be performed asin-process controls; however, the acceptancecriteria should be included in thespecification. This concept may be applied toboth single-dose and multiple-dose packages.
The dosage unit is considered to be thetypical dose taken by the patient. If the actualunit dose, as taken by the patient, iscontrolled, it may either be measured directlyor calculated based on the total measuredweight or volume of drug divided by the totalnumber of doses expected. If dispensingequipment (such as medicine droppers ordropper tips for bottles) is an integral part ofthe packaging, this equipment should beused to measure the dose. Otherwise, astandard volume measure should be used.The dispensing equipment to be used isnormally determined during development.
For powders for reconstitution, uniformityof mass testing is generally consideredacceptable.
(b) pH: Acceptance criteria for pH shouldbe provided where applicable and theproposed range justified.
(c) Microbial limits: Microbial limit testingis seen as an attribute of GMP, as well as ofquality assurance. In general, it is advisableto test the drug product unless itscomponents are tested before manufactureand the manufacturing process is known,through validation studies, not to carry asignificant risk of microbial contamination. Itshould be noted that, whereas this guidancedoes not directly address excipientselsewhere, the principles discussed here maybe applicable to excipients as well as to newdrug products. Skip testing may be anappropriate approach in both cases. Withacceptable scientific justification, it may bepossible to propose no microbial limit testingfor powders intended for reconstitution asoral liquids.
Acceptance criteria should be set for thetotal count of aerobic microorganisms, totalcount of yeasts and molds, and the absenceof specific objectionable bacteria (e.g.,Staphylococcus aureus, Escherichia coli,Salmonella, Pseudomonas). These should be
determined by suitable procedures, usingpharmacopoeial procedures, and at asampling frequency or time point inmanufacture which is justified by data andexperience.
Decision tree #8 provides additionalguidance on the use of microbial limittesting.
(d) Antimicrobial preservative content: Fororal liquids needing an antimicrobialpreservative, acceptance criteria forpreservative content may be appropriate.These criteria should be based on the levelsnecessary to maintain microbiologicalproduct quality throughout the shelf-life. Thelowest specified concentration ofantimicrobial preservative should bedemonstrated to be effective in controllingmicroorganisms by using a pharmacopoeialantimicrobial preservative effectiveness test.
Release testing for antimicrobialpreservative content should normally beperformed. Under certain circumstances, in-process testing may suffice in lieu of releasetesting. When antimicrobial preservativecontent testing is performed as an in-processtest, the acceptance criteria should remainpart of the specification.
Antimicrobial preservative effectivenessshould be demonstrated during development,during scaleup, and throughout the shelf-life(e.g., in stability testing, see the ICH guidance‘‘Stability Testing of New Drug Substancesand Products’’), although chemical testing forpreservative content is the attribute normallyincluded in the specification.
(e) Antioxidant preservative content:Release testing for antioxidant contentshould normally be performed. Under certaincircumstances, where justified bydevelopmental and stability data, shelf-lifetesting may be unnecessary, and in-processtesting may suffice in lieu of release testing.When antioxidant content testing isperformed as an in-process test, theacceptance criteria should remain part of thespecification. If only release testing isperformed, this decision should bereinvestigated whenever either themanufacturing procedure or the container/closure system changes.
(f) Extractables: Generally, wheredevelopment and stability data show nosignificant evidence of extractables,elimination of this test may be proposed.This should be reinvestigated if thecontainer/closure system changes.
Where data demonstrate the need, tests andacceptance criteria for extractables from thecontainer/closure system components (e.g.,rubber stopper, cap liner, plastic bottle) areconsidered appropriate for oral solutionspackaged in nonglass systems, or in glasscontainers with nonglass closures. Thecontainer/closure components should belisted, and data collected for thesecomponents as early in the developmentprocess as possible.
(g) Alcohol content: Where it is declaredquantitatively on the label in accordancewith pertinent regulations, the alcoholcontent should be specified. It may beassayed or calculated.
(h) Dissolution: In addition to the attributesrecommended immediately above, it may beappropriate (e.g., insoluble drug substance) to
include dissolution testing and acceptancecriteria for oral suspensions and dry powderproducts for resuspension. The testingapparatus, media, and conditions should bepharmacopoeial, if possible, or otherwisejustified. Dissolution procedures using eitherpharmacopoeial or non-pharmacopoeialapparatus and conditions should bevalidated.
Single-point measurements are normallyconsidered suitable for immediate releasedosage forms. Multiple-point sampling, atappropriate intervals, should be performedfor modified release dosage forms.Acceptance criteria should be set based onthe observed range of variation, and shouldtake into account the dissolution profiles ofthe batches that showed acceptableperformance in vivo. Developmental datashould be considered when determining theneed for either a dissolution procedure or aparticle size distribution procedure.
Dissolution testing may be performed as anin-process test, or as a release test, dependingon its relevance to product performance. Thediscussion of dissolution for solid oraldosage forms (above), and of particle sizedistribution (immediately following), shouldalso be considered here.
(i) Particle size distribution: Quantitativeacceptance criteria and a procedure fordetermination of particle size distributionmay be appropriate for oral suspensions.Developmental data should be consideredwhen determining the need for either adissolution procedure or a particle sizedistribution procedure for theseformulations.
Particle size distribution testing may beperformed as an in-process test or as a releasetest, depending on its relevance to productperformance. If these products have beendemonstrated during development to haveconsistently rapid drug releasecharacteristics, exclusion of a particle sizedistribution test from the specification maybe proposed.
Particle size distribution testing may alsobe proposed in place of dissolution testing;justification should be provided. Theacceptance criteria should include acceptableparticle size distribution in terms of thepercent of total particles in given size ranges.The mean, upper, and/or lower particle sizelimits should be well defined.
Acceptance criteria should be set based onthe observed range of variation, and shouldtake into account the dissolution profiles ofthe batches that showed acceptableperformance in vivo, as well as the intendeduse of the product. The potential for particlegrowth should be investigated duringproduct development; the acceptance criteriashould take the results of these studies intoaccount.
(j) Redispersibility: For oral suspensionswhich settle on storage (produce sediment),acceptance criteria for redispersibility may beappropriate. Shaking may be an appropriatetest. The procedure (mechanical or manual)should be indicated. Time required toachieve resuspension by the indicatedprocedure should be clearly defined. Datagenerated during product development maybe sufficient to justify skip lot testing orelimination of this attribute from thespecification.

62896 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
(k) Rheological properties: For relativelyviscous solutions or suspensions, it may beappropriate to include rheological properties(viscosity) in the specification. The test andacceptance criteria should be stated. Datagenerated during product development maybe sufficient to justify skip lot testing orelimination of this attribute from thespecification.
(l) Specific gravity: For oral suspensions orrelatively viscous or nonaqueous solutions,acceptance criteria for specific gravity may beappropriate. Testing may be performed as anin-process control.
(m) Reconstitution time: Acceptancecriteria for reconstitution time should beprovided for dry powder products whichrequire reconstitution. The choice of diluentshould be justified. Data generated duringproduct development may be sufficient tojustify skip lot testing or elimination of thisattribute from the specification.
(n) Water content: For oral productsrequiring reconstitution, a test andacceptance criterion for water content shouldbe proposed when appropriate. Loss ondrying is generally considered sufficient ifthe effect of absorbed moisture vs. water ofhydration has been adequately characterizedduring the development of the product. Incertain cases, a more specific procedure (e.g.,Karl Fischer titration) may be preferable.
3.3.2.3 Parenteral Drug Products: Thefollowing tests may be applicable toparenteral drug products.
(a) Uniformity of dosage units: This termincludes both uniformity of content anduniformity of mass; a pharmacopoeialprocedure should be used. Generally,acceptance criteria should be set for weightvariation, fill volume, or uniformity of fill.
If appropriate, these tests may beperformed as in-process controls; theacceptance criteria should be included in thespecification. This test may be applied toboth single-dose and multiple-dose packages.
For powders for reconstitution, uniformityof mass testing is generally consideredacceptable.
(b) pH: Acceptance criteria for pH shouldbe provided where applicable and theproposed range justified.
(c) Sterility: All parenteral products shouldhave a test procedure and acceptancecriterion for evaluation of sterility. Wheredata generated during development andvalidation justify parametric release, thisapproach may be proposed for terminallysterilized drug products.
(d) Endotoxins: A test procedure andacceptance criterion for endotoxins, using aprocedure such as the limulus amoebocytelysate test, should be included in thespecification.
(e) Pyrogens: Pyrogenicity testing may beproposed as an alternative to endotoxintesting where justified.
(f) Particulate matter: Parenteral productsshould have appropriate acceptance criteriafor particulate matter. This will normallyinclude limits for visible particulates (alsodesignated ‘‘foreign matter’’) and/or clarity ofsolution, as well as for subvisibleparticulates.
(g) Water content: For nonaqueousparenterals, and for parenteral products for
reconstitution, a test procedure andacceptance criterion for water content shouldbe proposed when appropriate. Loss ondrying is generally considered sufficient forparenteral products if the effect of absorbedmoisture vs. water of hydration has beenadequately characterized duringdevelopment. In certain cases, a morespecific procedure (e.g., Karl Fischertitration) may be preferred.
(h) Antimicrobial preservative content: Forparenteral products needing an antimicrobialpreservative, acceptance criteria forpreservative content may be appropriate.These criteria should be based on the levelsnecessary to maintain microbiologicalproduct quality throughout the shelf-life. Thelowest specified concentration ofantimicrobial preservative should bedemonstrated to be effective in controllingmicroorganisms by using a pharmacopoeialantimicrobial preservative effectiveness test.
Release testing for antimicrobialpreservative content should normally beperformed. Under certain circumstances, in-process testing may suffice in lieu of releasetesting. When antimicrobial preservativecontent testing is performed as an in-processtest, the acceptance criteria should remainpart of the specification.
Antimicrobial preservative effectivenessshould be demonstrated during development,during scaleup, and throughout the shelf-life(e.g., in stability testing, see the ICH guidance‘‘Stability Testing of New Drug Substancesand Products’’), although chemical testing forpreservative content is the attribute normallyincluded in the specification.
(i) Antioxidant preservative content:Release testing for antioxidant contentshould normally be performed. Under certaincircumstances, where justified bydevelopmental and stability data, shelf-lifetesting may be unnecessary and in-processtesting may suffice in lieu of release testing.When antioxidant content testing isperformed as an in-process test, theacceptance criteria should remain part of thespecification. If only release testing isperformed, this decision should bereinvestigated whenever either themanufacturing procedure or the container/closure system changes.
(j) Extractables: Control of extractables isconsidered significantly more important forparenteral products than for oral liquids.However, where development and stabilitydata show no significant evidence ofextractables, elimination of this test may beproposed. This should be reinvestigated ifthe container/closure system changes.
Where data demonstrate the need,acceptance criteria for extractables from thecontainer/closure components are consideredappropriate for parenteral products packagedin nonglass systems or in glass containerswith elastomeric closures. This testing maybe performed at release only, where justifiedby data obtained during development. Thecontainer/closure system components (e.g.,rubber stopper) should be listed, and datacollected for these components as early in thedevelopment process as possible.
(k) Functionality testing of deliverysystems: Parenteral formulations packaged inprefilled syringes, autoinjector cartridges, or
the equivalent, should have test proceduresand acceptance criteria related to thefunctionality of the delivery system. Thesemay include control of syringeability,pressure, and seal integrity (leakage), and/orparameters such as tip cap removal force,piston release force, piston travel force, andpower injector function force. Data generatedduring product development may besufficient to justify skip lot testing orelimination of some attributes from thespecification.
(l) Osmolality: When the tonicity of aproduct is declared in its labeling,appropriate control of its osmolality shouldbe performed. Data generated duringdevelopment and validation may besufficient to justify performance of thisprocedure as an in-process control, skip lottesting, or direct calculation of this attribute.
(m) Particle size distribution: Quantitativeacceptance criteria and a procedure fordetermination of particle size distributionmay be appropriate for injectablesuspensions. Developmental data should beconsidered when determining the need foreither a dissolution procedure or a particlesize distribution procedure.
Particle size distribution testing may beperformed as an in-process test or as a releasetest, depending on its relevance to productperformance. If the product has beendemonstrated during development to haveconsistently rapid drug releasecharacteristics, exclusion of particle sizecontrols from the specification may beproposed.
Particle size distribution testing may alsobe proposed in place of dissolution testingwhen development studies demonstrate thatparticle size is the primary factor influencingdissolution; justification should be provided.The acceptance criteria should includeacceptable particle size distribution in termsof the percent of total particles in given sizeranges. The mean, upper, and/or lowerparticle size limits should be well defined.
Acceptance criteria should be set based onthe observed range of variation, and shouldtake into account the dissolution profiles ofthe batches that showed acceptableperformance in vivo and the intended use ofthe product. The potential for particle growthshould be investigated during productdevelopment; the acceptance criteria shouldtake the results of these studies into account.
(n) Redispersibility: For injectablesuspensions which settle on storage (producesediment), acceptance criteria forredispersibility may be appropriate. Shakingmay be an appropriate test. The procedure(mechanical or manual) should be indicated.Time required to achieve resuspension by theindicated procedure should be clearlydefined. Data generated during productdevelopment may be sufficient to justify skiplot testing or elimination of this attributefrom the specification.
(o) Reconstitution time: Acceptance criteriafor reconstitution time should be providedfor all parenteral products which requirereconstitution. The choice of diluent shouldbe justified. Data generated during productdevelopment may be sufficient to justify skiplot testing or elimination of this attributefrom the specification.

62897Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
4. GlossaryAcceptance criteria: Numerical limits,
ranges, or other suitable measures foracceptance of the results of analyticalprocedures.
Chiral: Not superposable with its mirrorimage, as applied to molecules,conformations, and macroscopic objects,such as crystals. The term has been extendedto samples of substances whose moleculesare chiral, even if the macroscopic assemblyof such molecules is racemic.
Combination product: A drug product thatcontains more than one drug substance.
Degradation product: A molecule resultingfrom a chemical change in the drug moleculebrought about over time and/or by the actionof e.g., light, temperature, pH, water, or byreaction with an excipient and/or theimmediate container/closure system. Alsocalled decomposition product.
Enantiomers: Compounds with the samemolecular formula as the drug substance, thatdiffer in the spatial arrangement of atomswithin the molecule and arenonsuperimposable mirror images.
Impurity: (1) Any component of the newdrug substance that is not the chemical entitydefined as the new drug substance. (2) Anycomponent of the drug product that is not thechemical entity defined as the drug substanceor an excipient in the drug product.
Identified impurity: An impurity for whicha structural characterization has beenachieved.
New drug product: A pharmaceuticalproduct type, for example, tablet, capsule,solution, cream, that has not previously beenregistered in a region or Member State, andwhich contains a drug ingredient generally,but not necessarily, in association withexcipients.
New drug substance: The designatedtherapeutic moiety, that has not previously
been registered in a region or Member State(also referred to as a new molecular entity ornew chemical entity). It may be a complex,simple ester, or salt of a previously approveddrug substance.
Polymorphism: The occurrence of differentcrystalline forms of the same drug substance.This may include solvation or hydrationproducts (also known as pseudopolymorphs)and amorphous forms.
Quality: The suitability of either a drugsubstance or drug product for its intendeduse. This term includes such attributes as theidentity, strength, and purity of the article.
Racemate: A composite (solid, liquid,gaseous, or in solution) of equimolarquantities of two enantiomeric species. It isdevoid of optical activity.
Reagent: A substance, other than a startingmaterial or solvent, that is used in themanufacture of a new drug substance.
Solvent: An inorganic or an organic liquidused as a vehicle for the preparation ofsolutions or suspensions in the synthesis ofa new drug substance or the manufacture ofa new drug product.
Specification: A list of tests, references toanalytical procedures, and appropriateacceptance criteria that are numerical limits,ranges, or other criteria for the testsdescribed. It establishes the set of criteria towhich a drug substance or drug productshould conform to be considered acceptablefor its intended use. ‘‘Conformance tospecifications’’ means that the drugsubstance and/or drug product, when testedaccording to the listed analytical procedures,will meet the listed acceptance criteria.Specifications are binding quality standardsthat are agreed to between the appropriategovernmental regulatory agency and theapplicant.
Specific test: A test that is considered to beapplicable to particular new drug substancesor particular new drug products depending
on their specific properties and/or intendeduse.
Specified impurity: An identified orunidentified impurity that is selected forinclusion in the new drug substance or newdrug product specification and isindividually listed and limited in order toassure the quality of the new drug substanceor new drug product.
Unidentified impurity: An impurity that isdefined solely by qualitative analyticalproperties (e.g., chromatographic retentiontime).
Universal test: A test that is considered tobe potentially applicable to all new drugsubstances, or all new drug products (e.g.,appearance, identification, assay, andimpurity tests).
5. References
International Conference onHarmonisation, ‘‘Impurities in New DrugSubstances,’’ 1995.
International Conference onHarmonisation, ‘‘Impurities in New DrugProducts,’’ 1996.
International Conference onHarmonisation, ‘‘Stability Testing of NewDrug Substances and Products,’’ 1994.
International Conference onHarmonisation, ‘‘Text on Validation ofAnalytical Procedures,’’ 1994.
International Conference onHarmonisation, ‘‘Validation of AnalyticalProcedures: Methodology,’’ 1996.
International Conference onHarmonisation, ‘‘Residual Solvents inPharmaceuticals,’’ 1996.
6. Attachments: Decision Trees #1 through #8
For the decision trees referenced in thisguidance, see the following pages.
BILLING CODE 4160–01–F
62898 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62899Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62900 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62901Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62902 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62903Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62904 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62905Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62906 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices

62907Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62908 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
62909Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices

62910 Federal Register / Vol. 62, No. 227 / Tuesday, November 25, 1997 / Notices
Dated: November 18, 1997.William K. Hubbard,Associate Commissioner for PolicyCoordination.[FR Doc. 97–30916 Filed 11–24–97; 8:45 am]BILLING CODE 4160–01–C







![colombophile-du-calaisis.e-monsite.comcolombophile-du-calaisis.e-monsite.com/medias/files/siege-unique.pdf · de de Financemenf Gr STBC S. TAR..] : 24 route Nationale 62890 Zouafqu](https://img.pdfslide.net/doc/110x75/5b82d9dc7f8b9a7d3a8b83ec/colombophile-du-calaisise-de-de-financemenf-gr-stbc-s-tar-24-route-nationale.jpg)





















