Embed Size (px)
Citation preview

Experimentelles 85
Rühren Rühren
Rühren Gelieren
Filtrieren Waschen
Kalzinierung
8. Experimentelles
8.1. Herstellung von Träger und Katalysatoren
Die eingesetzten Träger waren bis auf einen Sol-Gel-hergestellten handelsübliche Materialien.
Die Herstellung der Katalysatoren erfolgte durch das Aufbringen der Eisen- und
Natriumvorläufer auf die Silicagelträger, jeweils gefolgt von einer thermischen Behandlung.
8.1.1. Trägerherstellung
Die verwendeten Träger sind in der Tabelle 5.1 (S. 24) aufgelistet. Lediglich ein chemisch
reines Silicagel wurde selbst hergestellt und als Katalysatorträger verwendet.
Die Herstellung erfolgte nach der Vorschrift von [145] entsprechend dem Verfahrensschema
gemäß Abb. 8.1.a wie folgt: In einem Becherglas Nr. 1 (1000 ml) wird eine Mischung von
TEOS DDA
Ethanol i-Propanol Wasser
Filtrat
Rückstand (Kuchen)
Träger D (Sol-Gel Silicagel)
Abb. 8.1.a. Verfahrensschema zur Trägerherstellung.

Experimentelles 86
0.3 mol (67.2 ml) Tetraethylorthosilikat (TEOS), 1.95 mol (112.1 ml) Ethanol und 0.3 mol
(13.08 ml) Isopropanol hergestellt. In einem Becherglas Nr. 2 (500 ml) wird 0.09 mol (16.68
g) 1-Dodecylamin (DDA) in 10.8 mol (194.4 ml) Wasser gelöst, und danach unter Rühren in
das Becherglas Nr. 1 gegossen, 2 h weiter gerührt, und für 48 h im abgedeckten Becherglas
stehengelassen. Das darin entstandene Gel wird über ein Papierfilter abfiltriert und drei Mal
mit destilliertem Wasser gewaschen. Der Filterkuchen wird 24 h bei 40°C in einem Ofen
unter Belüftung getrocknet und anschließend entsprechend dem Temperaturprogramm in
Tabelle 8.1 kalziniert.
Tabelle 8.1. Temperaturprogramm bei der Kalzinierung der Träger und Katalysatoren
Aufheizen/Abkühlen Halten
in 1h auf 65°C 5h bei 65°C
in 0.5h auf 110°C 2h bei 110°C
in 5.5h auf 600°C 5h bei 600°C
in 6.5h auf 30°C
8.1.2. Katalysatorherstellung
Die Mehrzahl der Katalysatoren wurden durch Imprägnierung hergestellt. Bei dieser
Herstellung wurden die Träger durch die „incipient wetness“-Methode mit einer Fe(III)-
acetylacetonat-haltigen Toluol-Lösung bekannten Gehaltes imprägniert, so daß der Fe-Gehalt
der Probe nach der Kalzinierung (5h bei 600°C, Temperaturprogramm in Tabelle 8.1) den
gewünschten Wert erreichte. Einige käuflich erworbene Materialien, die schon einen
bestimmten Fe-Gehalt hatten, wurden lediglich 5h bei 600°C kalziniert.
Einige Katalysatoren wurden durch eine Sol-Gel Methode hergestellt. Die Herstellung
erfolgte ähnlich wie für den Träger D (Kap. 8.1.1). In dem Becherglas 1 wurde zusätzlich eine
Menge Fe(III)-acetylacetonat (Fe(acac)3) zugegeben, die dem gewünschten Fe-Gehalt des
Katalysators entspricht.
Einige Katalysatoren wurden von Firma Degussa durch Flammenhydrolyse hergestellt und für
die Untersuchungen bereitgestellt.
Die Mehrzahl der Eisenoxid-Katalysatoren wurden dann mit wäßrigen Natriumacetat-
Lösungen von unterschiedlichem Na-Gehalt imprägniert. Dazu befanden sich die
Katalysatoren 48 h bei Raumtemperatur in der Lösung, die einige Mal aufgerührt wurde (10 g
Katalysator in einem 200-300 ml Becher mit 100 ml Naac-Lösung). Die verwendeten

Experimentelles 87
Imprägnierung (incipient wetness)
Kalzinierung
Imprägnierung
Filtrieren Waschen
Kalzinierung
Natriumacetat(Naac)-Konzentrationen und die Bezeichnungen, die weiter in dem Namen der
Katalysatoren verwendet wurden, sind in Tabelle 8.2 zusammengestellt.
Tabelle 8.2. Verwendete Konzentrationen der Naac-Lösungen und die entsprechende
Bezeichnung
Konzentration Bezeichnung
0.01 mol/l (0.136 g Naac.3H2O und dest. Wasser bis 100 ml) N1
0.1 mol/l (1.36 g Naac.3H2O und dest. Wasser bis 100 ml) N2
1 mol/l (13.6 g Naac.3H2O und dest. Wasser bis 100 ml) N3
Nach Filtration und Waschen mit destilliertem Wasser wurden die mit Na-Ionen dotierten
Katalysatoren in der ersten Stufe 5h bei 600°C kalziniert. In einer zweiten Stufe wurden
Teilmengen von Katalysatoren zusätzlich 6h bei 700°C oder 800°C kalziniert.
Das Verfahrensschema zur Katalysatorherstellung durch Imprägnierung wird in Abb. 8.1.b
wiedergegeben.
Träger
Fe(acac)3
in Toluol
Naac in Wasser
Filtrat
Rückstand (Kuchen)
Katalysator
Abb. 8.1.b. Verfahrensschema zur Katalysatorherstellung.

Experimentelles 88
Die verwendeten Chemikalien bei der Herstellung von Trägern und Katalysatoren sind in der
Tabelle 8.3 zusammengestellt.
Tabelle 8.3. Verwendete Chemikalien
Stoff Spezifikation Hersteller/Lieferant
Tetraethylorthosilikat (TEOS) 98% Aldrich
Isopropanol >99,5% Fluka
Ethanol >99,8% Fluka
1-Dodecylamin (DDA) 98% Aldrich
Eisen(III)-acetylacetonat (Fe(acac)3) 97% Aldrich
Toluol 99% Fluka
Natriumacetat-trihydrat (Naac.3H2O) 99% Aldrich
8.2. Durchführung der Oxidationsexperimente
8.2.1. Beschreibung der Labor-Strömungsapparatur
Die für die Oxidation von Propen aufgebaute Labor-Strömungsapparatur ist in Abb. 8.2
schematisch dargestellt. Die Apparatur besteht aus drei Teilen, in denen die Funktionen
Gasdosierung, Reaktion und Produktgewinnung, und Gasanalyse realisiert wurden.
Zum Dosierteil gehört die Gasversorgung mit Propen, Distickstoffmonoxid, Luft und Inertgas
(Helium oder Stickstoff). Die Gase wurden aus Druckgasflaschen (Propen,
Distickstoffmonoxid, Helium) oder aus den zentralen Gasversorgung (Luft, Stickstoff)
entnommen. Die Spezifikationen der Gase sind in der Tabelle 8.4 aufgelistet.
Tabelle 8.4. Verwendete Gase
Gas Hersteller/Lieferant Spezifikation/Reinheit
Propen (C3H6) Messer-Griesheim 2.5/>99,5 Vol.%
Distickstoffmonoxid (N2O) Messer-Griesheim pro narcosi/>99,5 Vol.%
Helium (He) Messer-Griesheim 4.6/>99,996 Vol.%
Stickstoff (N2) Messer-Griesheim 5.0/>99,999 Vol.%
Luft zentrale Luftversorgung gefiltert, getrocknet
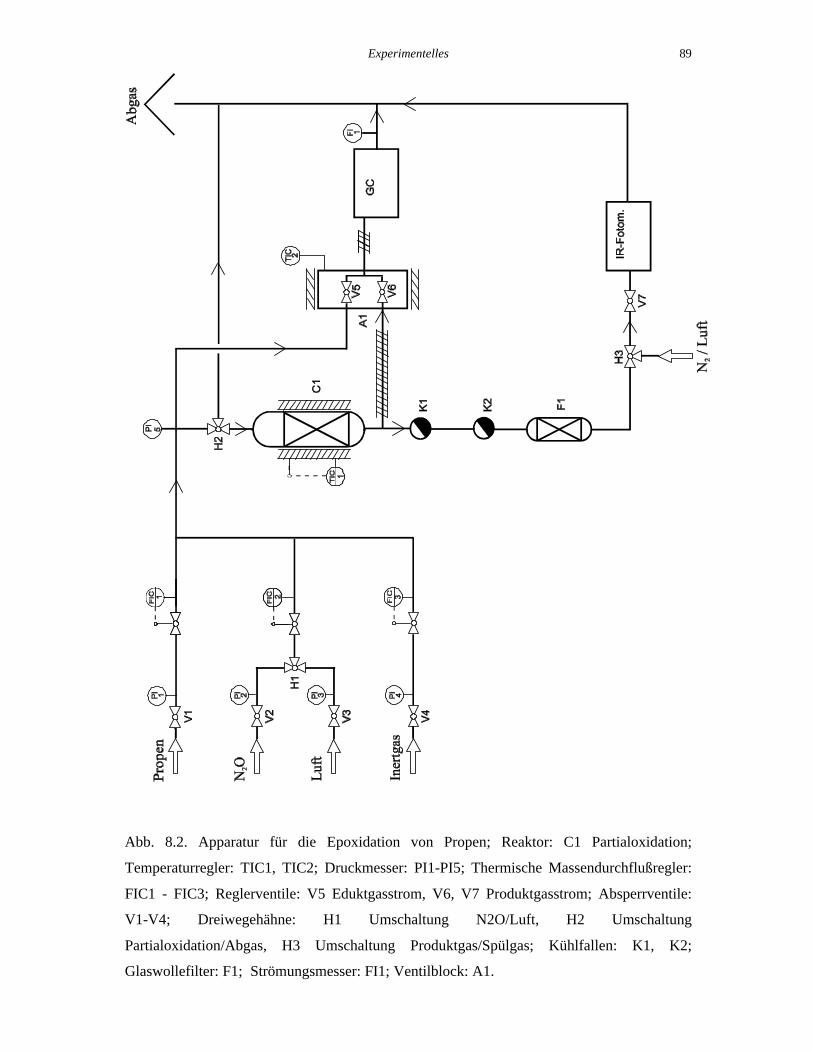
Experimentelles 89
Abb. 8.2. Apparatur für die Epoxidation von Propen; Reaktor: C1 Partialoxidation;
Temperaturregler: TIC1, TIC2; Druckmesser: PI1-PI5; Thermische Massendurchflußregler:
FIC1 - FIC3; Reglerventile: V5 Eduktgasstrom, V6, V7 Produktgasstrom; Absperrventile:
V1-V4; Dreiwegehähne: H1 Umschaltung N2O/Luft, H2 Umschaltung
Partialoxidation/Abgas, H3 Umschaltung Produktgas/Spülgas; Kühlfallen: K1, K2;
Glaswollefilter: F1; Strömungsmesser: FI1; Ventilblock: A1.

Experimentelles 90
Die Gase wurden über die Absperrventile V1-V4 der Anlage zugeführt. Über das
Dreiwegeventil H1 kann entweder Distickstoffmonoxid oder Luft in die Anlage eingespeist
werden. Die Gasströme wurden über die Massendurchflußreglere FIC1-FIC3 dosiert. Die
Massendurchflußregler wurden für die entsprechenden Gase kalibriert und zur Einstellung der
gewünschten Eduktzusammensetzung verwendet.
Zur Partialoxidation von Propen diente der Quarzreaktor C1 (s. Abb. 8.3 und [146]). Die
Katalysatorfüllung befand sich im unteren Teil des Reaktors direkt auf einer Quarzfritte. Der
Reaktor wurde von außen elektrisch beheizt und war thermisch isoliert. Die Temperatur
Φ1
Φ3
B
E
C
A
F
8
2
1
3
4
5
6
7
9
Φ2
Φ2
F
D
Abbildung 8.3. Schema des Oxidationsreaktors; 1 -Produktausgang zur Kondensation (Kern
19/9); 2 -Produktausgang zum GC (Schale 13/5); 3 - Quarzglasfritte; 4, 9 -Thermoelement-
führungsrohr (Quarzglas); 5 - Gaseingang (Schale 13/5); 6 - Gewinderohr GL 14; 7 -
Verschlußkappe GL 14; 8 - Silikon-Dichtung;
Maße (mm): A=260; B=35; C=195; D=30; E=15; F=40; Φ1=10x1,5; Φ2=5x1,5; Φ3=1,2x0,5.
Experimentelles 91
wurde mittels einem NiCr-Ni- Thermoelementes in der Mitte des Reaktors gemessen und mit
Hilfe des Temperaturreglers TIC1 eingestellt.
An den Reaktionsteil schließt sich der Gasanalytikteil an. Er besteht aus einem
Gaschromatographen (GC) und einem IR-Fotometer und diente der on-line-Bestimmung der
Reaktionsprodukte (Kap. 8.3.2). Vor dem IR-Fotometer befanden sich die Kühlfallen K1 und
K2 und das Glaswollefilter F1. Die Kühlfallen befanden sich in einem Dewargefäß mit
Kältemittel. Die Kühlfalle K1 befand sich in einem Wasser/Eis-Gemisch bei 0°C und die
Kühlfalle K2 in einem i-Propanol/Trockeneis-Gemisch bei –78°C.
Die Gasleitungen vor dem GC wurden elektrisch beheizt und thermisch isoliert, um das
Auskondensieren der Reaktionsprodukte zu vermeiden. Der Produktgasstrom wurde über V6
zum GC geleitet. Weitere Gasleitungen erlaubten die Führung des Eduktgasstromes zur GC-
Analyse über V5, die Führung des Eduktgasstromes zum Abzug über H2 und die Spülung und
Kalibrierung des IR-Fotometers über H3.
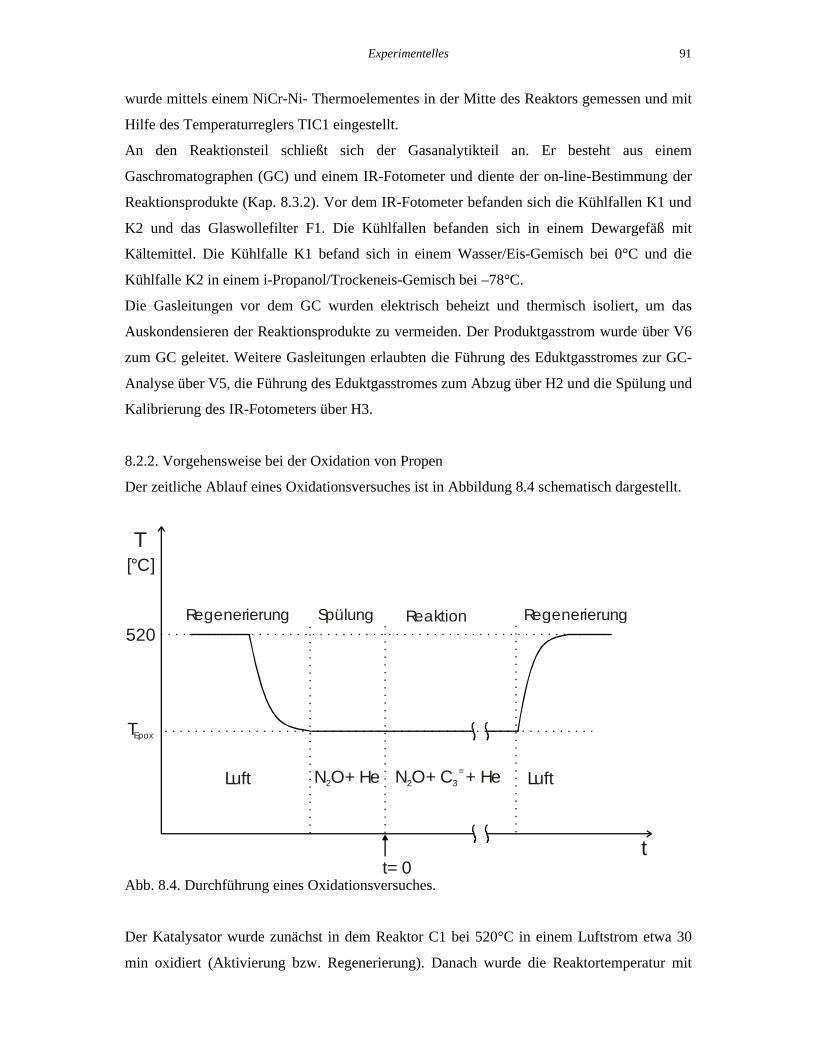
8.2.2. Vorgehensweise bei der Oxidation von Propen
Der zeitliche Ablauf eines Oxidationsversuches ist in Abbildung 8.4 schematisch dargestellt.
Abb. 8.4. Durchführung eines Oxidationsversuches.
Der Katalysator wurde zunächst in dem Reaktor C1 bei 520°C in einem Luftstrom etwa 30
min oxidiert (Aktivierung bzw. Regenerierung). Danach wurde die Reaktortemperatur mit
t
Luft N O+ He2 N O+ C + He2 3=
t= 0
T
520
TEpox
[°C]
Luft
Regenerierung Spülung Reaktion Regenerierung

Experimentelles 92
Hilfe des Temperaturreglers TIC1 auf die gewünschte Reaktionstemperatur (TEpox) gestellt.
Wenn die Isttemperatur im Katalysatorbett den Sollwert für die Reaktion erreicht hatte, wurde
der Luftstrom abgestellt und ein Gasstrom aus N2O und He durch den Reaktor geleitet
(Spülung), wobei der N2O-Strom dem Sollwert für die Reaktion entspricht. Nachdem die
N2O-Konzentration am Ausgang des Reaktors konstant war, wurde ein bestimmter Propen-
Strom eingestellt und der He-Strom entsprechend verringert. Das war der Zeitpunkt Null für
die Katalysator-Laufzeit (time-on-stream, TOS) in dem jeweiligen Oxidationsversuch. Der
Druck im System, gemessen vor dem Oxidationsreaktor (PI5, Abb. 8.2), wurde im Laufe des
Versuches mit Hilfe der Ventile V6 und V7 konstant auf 1200 mbar gehalten.
Im Laufe des Oxidationsversuches wurden die Reaktionsprodukte gaschromatographisch und
IR-fotometrisch on-line analysiert (siehe 8.3.2.). Die organischen Reaktionsprodukte konnten
auch in den Kühlfallen K1 und K2 kondensiert und gesammelt oder in einer Gasmaus
gefangen und off-line analysiert werden.
Nach einer bestimmten Laufzeit wurde der Oxidationsversuch unterbrochen, indem die
Gasströme von Propen, N2O und He abgestellt wurden und erneut ein Luftstrom zur
Regenerierung durch den Reaktor bei 520°C geleitet wurde. Anschließend konnte ein neuer
Oxidationsversuch beginnen.
8.3. Analytische Methoden
8.3.1. Charakterisierung der Katalysatoren
Die Katalysatoren wurden mittels Stickstoffphysisorption, Röntgendiffraktometrie,
Transmissionselektronmikroskopie, Photoelektronspektroskopie, temperaturprogrammierter
Reduktion und Oxidation, Chemisorption und temperaturprogrammierter Desorption und IR-
Spektroskopie charakterisiert.
8.3.1.1. Durchführung der Stickstoffphysisorption
Die Stickstoffphysisorption wurde bei der Siedetemperatur von Stickstoff (77K) nach der
BET-Methode [36] durchgeführt und diente zur Ermittlung der spezifischen Oberflächen und
Porositäten.
Die Katalysatorprobe (etwa 0,1-0,2 g) wurde zunächst bei 130°C für 2h, dann bei
Raumtemperatur über Nacht evakuiert; der Restdruck der Evakuierung betrug ca. 10-6 mbar.
Anschließend wurden die Adsorptions-Desorptions-Isothermen mit dem Gerät Sorptomatic
1990 von Fisons Instruments unter Benutzung der Software Milestone 200 aufgenommen. Zur

Experimentelles 93
Berechnung der spezifischen Oberfläche wurde ein Platzbedarf von 0,162 nm2 für das
physisorbierte N2-Molekül angenommen. Die Porenverteilung wurde nach der Dollimore-
Heal Methode berechnet.
8.3.1.2. Durchführung der Röntgendiffraktometrie (XRD)
Die XRD diente zur Bestimmung der kristallographischen Struktur der Proben. Die XRD-
Messungen wurden am Lehrstuhl für Festkörperphysik der TU Chemnitz mit Hilfe eines
Diffraktometers HZG4 von Seifert-FPM durchgeführt.
Die Meßparameter waren:
Strahlung: Cu-Kα (α = 1,540598 ∆)
Filter: Ni
Spannung: 40 kV
Röhrenstrom: 35 mA
Winkelgeschwindigkeit: 1,5°/min
Meßbereich: 22 = 10-70°.
8.3.1.3. Durchführung der Transmissionselektronmikroskopie (TEM)
Die TEM-Messungen wurden zur Ermittlung der Eisenoxidpartikelgröße am Institut für
Angewandte Chemie (ACA) in Berlin-Adlershof an dem Gerät CM 20 von Fa. Philips
durchgeführt. Die Beschleunigungsspannung betrug 200kV. Die Proben wurden trocken auf
Kupfergrids mit Lacey-Kohlefilm aufgebracht.
8.3.1.4. Durchführung der Photoelektronspektroskopie (XPS)
Die XPS-Messungen dienten zur Bestimmung des Oxidationszustandes der Eisenionen in
einigen repräsentativen Katalysatorproben. Die Messungen wurden am Institut für
Angewandte Chemie (ACA) in Berlin-Adlershof an dem Gerät VG ESCALAB 220iXL
durchgeführt. In das Gerät war eine Hochdruck-Gaszelle integriert, die es erlaubte, vor den
Messungen bei 673K einen N2O-Strom über die Katalysatorproben zu leiten, um sie in den
Oxidationszustand unter Reaktionsbedingungen zu bringen. Die Korrektur der Peaklagen
erfolgte nach den bekannten Al 2p- und Si 2p-Signalen des Al2O3 bzw. SiO2.
8.3.1.5. Apparatur und Durchführung der temperaturprogrammierten Reduktion (TPR) und
Oxidation (TPO)
Die TPR und TPO dienten zur Ermittlung der Redoxeigenschaften der Katalysatoren.

Experimentelles 94
Die Versuchsapparatur wurde nach einer Beschreibung von [40] gebaut und ist in Abb. 8.5
schematisch dargestellt. In der verwendeten Versuchsapparatur wurden die Gase (Tabelle 8.5)
Ref. Meß.
2
Sauerstoff
Helium
Wasserstoff
Stickstoff
T1
T2 T4
T3
A1
A2
W1
V1 C1
WLD
PC
A3
K1
Abb. 8.5. TPR-TPO Apparatur; FIC1-FIC4 Massendurchflußregler; T1, T2 Trockentürme mit
Blaugel; T3, T4 Trockentürme mit P2O5; V1 Vierwegeventil; A1 WLD-Zelle; A2 Computer;
K1 Kühlfalle; C1 Reaktor; W1 Ofen; A3 Abzug.
mit Hilfe der thermischen Massendurchflußreglere FIC1-FIC4 dosiert und in den
Trockentürmen T1-T4 getrocknet. Durch das Vierwegeventil V1 wurde entweder das
Sauerstoff-Helium Gemisch oder das Wasserstoff-Stickstoff-Gemisch zur Referenzkammer
des Wärmeleitfähigkeitsdetektors geleitet. Von dort aus gelangte der Gasstrom zu dem
Quartzglasreaktor C1. Die Katalysatorprobe befand sich in dem Schenkel des U-förmigen
Reaktorrohres von 6 mm Inenndurchmesser. Der Reaktor wurde mit Hilfe des Ofens W1 der
Fa. Lenton Furnaces programmiert beheizt. Die Isttemperatur in der Katalysatorschüttung
wurde mit Hilfe eines NiCr-Ni Thermoelementes gemessen. Der Gasstrom gelangte nach
Tabelle 8.5. Verwendete Gase bei der TPR-TPO
Gas Hersteller/Lieferant Spezifikation/Reinheit
Sauerstoff Messer-Griesheim 5.0/>99,999 Vol.%
Helium Messer-Griesheim 4.6/>99,996 Vol.%
Wasserstoff Messer-Griesheim 5.0/>99,999 Vol.%
Stickstoff Messer-Griesheim 5.0/>99,999 Vol.%

Experimentelles 95
Verlassen des Reaktors durch eine mit CO2(s)/i-Propanol auf –78°C temperierte Kühlfalle K1
in die Meßkammer des WLD und trat aus der Apparatur aus.
Die Erfassung des WLD-Signales erfolgte mit Hilfe eines Computerprogrammes [147]. Die
Auswertung der erhaltenen Daten wurde mit dem Software Origin von Microcal durchgeführt.
Die Katalysatorprobe (ca. 0,3 g) wurde in den Quarzglasreaktor überführt. Die
Katalysatorschüttung war von beiden Seiten von Glaswolle umgeben. Der Reaktor wurde in
die Apparatur eingebaut und die Spitze des Thermoelementes in der Mitte der
Katalysatorschüttung positioniert.
Mit Hilfe der Durchflußreglere FIC1-FIC4 wurden die Gasströme eingestellt: 40 ml/min mit
12,5 vol.% O2 in He bei TPO und 30 ml/min mit 10 vol.% H2 in N2 bei TPR. Zunächst wurde
eine TPO durchgeführt um abzusichern, daß der Katalysator in dem oxidierten Zustand
vorlag, dann folgte die TPR und anschließend wurde erneut eine TPO durchgeführt.
Es wurde folgendes Temperaturprogramm (sowohl bei TPR als auch bei TPO) angewendet:
Starttemperatur: 30°C; Aufheizrate: 30°C/min; Endtemperatur: 700°C; Haltezeit: 30 min.
8.3.1.6. Apparatur und Durchführung der Chemisorption und temperaturprogrammierten
Desorption (TPD)
Die Chemisorption und TPD Messungen dienten zur Ermittlung der gesamten
chemisorbierten Menge eines Adsorbates auf einem Katalysator bzw. zur Bestimmung des
Desorptionsprofiles des Adsorbates mit der Temperatur.
Die verwendete Adsorptionsapparatur ist in Abb. 8.6 dargestellt. Die Vakuumpumpen P1 und
P2 dienten zur Evakuierung der Apparatur. Vor die Vakuumpumpen wurden die mit CO2(s)/i-
Propanol auf –78°C temperierten Kühlfallen K1-K3 geschaltet. Die Hähne H1 und H2
konnten die Verbindungen zwischen den Pumpen und den Rest der Apparatur abschließen.
Über die Verbindungen zu den Hähnen H4-H6 konnten die von der Katalysatorprobe zu
adsorbierenden Stoffe mit dem Behälter (B1) für flüssige oder feste (verdampfbare) Stoffe,
mit den Leitungen (A1) zu der zentralen Gasversorgung oder zu einer Gasflasche, oder eine
Bürette (B2) zugeführt werden. Die Bürette hatte einen eigenen Hahn H7.
Bei einigen Chemisorptionsmessungen wurde die Probe vorher reduziert. Das geschah durch
Anschließen der Bürette B2 mit der eingefüllten Probe an der TPR-TPO Apparatur und
Durchführung einer TPR nach der im Kap. 8.3.1.3 beschriebenen Vorgehensweise. Am Ende
der Reduktion wurde der Hahn H7 geschlossen.

Experimentelles 96
PI1
PI2
H1
H2
H3
H4 H5 H6
H7
K1
K2
K3
P1 P2
W1
A1B1
B2
A2
Abb. 8.6. Adsorptionsapparatur; P1 Vorvakuumpumpe; P2 Hochvakuumpumpe; K1 - K3
Kühlfallen; B1 Behälter; B2 Bürette; A1, A2 Verbindungsstück; PI1, PI2 Vakuummeßgeräte;
H1 - H7 Absperrhähne; W1 Ofen.
Danach wurden die Quarzglasbürette B2 mit der Katalysatorprobe (etwa 0,3 g) und der
Behälter B1 oder die Leitung A1 an die Adsorptionsapparatur angeschlossen. Die Hähne H2,
H4 und H5 wurden geschlossen, die Hähne H1, H3, H6 und H7 geöffnet und die
Vorvakuumpumpe P1 eingeschaltet. Damit begann die Evakuierung der Apparatur und der
Probe bei Raumtemperatur. Wenn der Druck in der Anlage, gemessen mit PI1, ca. 10-2 mbar
erreicht hatte, wurden der Hahn H2 geöffnet, die Hähne H1 und H3 geschlossen, die
Vorvakuumpumpe P1 ausgeschaltet und die Hochvakuumpumpe P2 eingeschaltet. Der Druck
in der Apparatur, gemessen mit PI2, erreichte nach einer gewisser Zeit ca. 10-6 mbar. Die
Evakuierung bei diesem Druck und Raumtemperatur wurde für ca. 30 min vorgenommen, und
danach wurde der Ofen W1 eingeschaltet und die Probe auf 700°C aufgeheizt. Die
Katalysatorprobe wurde bei dieser Temperatur etwa 60 min evakuiert.
Im Falle einer anschließenden TPD-Messung wurde dann die Bürette mit der Probe auf die
Adsorptionstemperatur von meist 400°C eingestellt. Der Hahn H2 wurde geschlossen, die
Pumpe P2 ausgeschaltet und der Hahn H4 oder H5 geöffnet. Der zu adsorbierende Stoff aus
dem Behälter B1 oder aus der Leitung A1 gelang damit zur Probe in B2. Nach 30 min
Adsorptionszeit wurde der Hahn H4 bzw. H5 geschlossen, die Probe auf Raumtemperatur
abkühlen gelassen und dann mit Hilfe der Vorvakuumpumpe P1 bis zu 10-2 mbar evakuiert.
Die Hähne H6 und H7 wurden geschlossen, die Bürette aus der Adsorptionsapparatur

Experimentelles 97
entnommen und an das Quadrupol-Massenspektrometer (QTMD) von Fisons Instruments
angeschlossen. Die Bürette wurde darin für zwei Stunden bei einem Druck von etwa 10-6
mbar evakuiert. Mit Hilfe eines Ofens der Fa. Netzsch wurde die Bürette auf 700°C mit
5°C/min aufgeheizt und 20 min bei 700°C gehalten. Die dabei desorbierenden Stoffe wurden
mit dem QTMD analysiert.
Während der einleitenden Evakuierung wurden durch Massenscans die Fragmente ermittelt,
die bei der Desorption eines bestimmten Stoffes auftreten. Bei der eigentlichen TPD wurde
dann die Desorption von bis zu sechs aufschlußreichen Fragmenten kontinuierlich verfolgt.
Bei den Experimenten mit Propen wurden die Molarmassen 12, 15, 16, 27, 41 und 42 benutzt;
im Fall von N2O waren es die Molarmassen 12, 14, 16, 28, 32 und 44.
Im Falle einer Chemisorptionsmessung, die nach einer statischen volumetrischen Methode
wie in [27b] beschrieben erfolgte, wurde die Bürette mit der Katalysatorprobe an einem Gerät
Sorptomatic 1990 von Fisons Instruments angeschlossen. Die Chemisorptionskurve wurde als
Differenz zwischen den Totaladsorptionskurve und die Physisorptionskurve berechnet.
8.3.1.7. Durchführung der IR-Spektroskopie
Für die Durchführung von Untersuchungen zur Adsorption von N2O und Propen auf der
Katalysatoroberfläche und die Aufnahme und Auswertung der IR-Spektren wurde eine
Adsorptionsapparatur in Verbindung mit einem FTIR-Spektrometer verwendet.
Die Adsorptionsapparatur wurde in Kap. 8.3.1.6.1. beschrieben.
Zur Aufnahme der IR-Spektren der adsorbierten Moleküle auf der Katalysatoroberfläche
wurde eine spezielle Küvette verwendet (Abb. 8.7). Die Küvette besteht aus zwei
Quarzglasteilen, dem eigentlichen Küvettenkörper, sowie aus einem oben geschlossenen
Rohr, an dem am unteren Ende ein Schliff angebracht ist. Am Küvettenkörper zweigt am
oberen und unteren Teil je ein Rohr ab, welche mit einem Hahn verbunden sind. Am oberen
Teil des Hahnes werden diese Rohre fortgeführt. An ihrem oberen Ende befindet sich jeweils
eine Schliffpfanne, eine ist mit einer Blindstopfen geschlossen und die andere wird für den
Anschluß der Küvette an den Anschluß A1 der Adsorptionsapparatur benutzt (vergl. Abb.
8.6). Der mittlere Teil des Küvettenkörpers ist mit einer isolierten Heizwicklung umgeben.
Mit Hilfe eines in die Isolierung eingebauten Ni/CrNi-Thermoelementes und eines
Temperaturreglers (Tempat 2000TS) wurde der mittlere Teil des Küvettenkörpers beheizt.
Am unteren Ende der Küvette befinden sich im Quarzglas zwei seitliche Öffnungen, auf die
die IR-durchlässigen NaCl-Scheiben mit Epoxidharz aufgekittet sind.

Experimentelles 98
Abb. 8.7. IR-Küvette.
Der zu untersuchende Katalysator wurde zu einer Tablette gepreßt, in den unteren Teil des
Probenhalters eingebracht. Der Probenhalter ist in seinem oberen Teil als Glasstab
ausgebildet, in dessen Spitze ein Eisenkern eingeschmolzen war. Dieser Eisenkern
ermöglichte mit einem außerhalb der Küvette angebrachten Dauermagneten eine vertikale
Positionierung der Probenhalters. Somit konnte die Probe entweder im beheizten Teil der
Küvette oder im Bereich der NaCl-Fenster positioniert werden.
Zur Herstellung der Katalysator-Tablette wurde in einem Preßwerkzeug mit einem
Durchmesser von 16 mm 20 mg Katalysatorpulver eingefüllt und gleichmäßig auf dem
Zylinderboden des Unterstempels verteilt. Dabei wurde über und unter der Katalysatorpulver
jeweils ein Stück Papier, welches den Durchmesser des Preßwerkzeuges hatte, eingelegt. Mit

Experimentelles 99
Hilfe der Tablettenpresse (Gerät Graseby Specac) wurde nach Auflegen des Oberstempels
über einen am Gerät angebrachten Vakuumstutzen 5 min evakuiert und anschließend mit
einer Preßdauer von 5 min. bei einem konstanten Preßdruck von 6 bis 7 t eine selbsttragende
Katalysatortablette gepreßt. Anschließend wurde die Tablette dem Preßwerkzeug entnommen,
von dem Papier getrennt und in den Probenhalter der IR-Küvette überführt.
Der Probenhalter mit der Katalysatortablette wurde in die geöffnete Küvette eingebracht.
Anschließend wurde die Küvette verschlossen und an dem Anschluß A1 der Hochvakuum-
Apparatur (Abb. 8.6) angeschlossen. Dann wurden der Absperrhahn H5 und der Absperrhahn
der IR-Küvette geöffnet. Damit war die IR-Küvette mit dem Adsorptionsteil der
Hochvakuum-Apparatur verbunden. Anschließend wurden die Hähne H1 und H2
nacheinander geöffnet und mit Hilfe der Vorvakuumpumpe P1 und Hochvakuumpumpe P2
im Adsorptionsteil und in der Küvette ein Vakuum von ca. 10-3 mbar erzeugt. Die
Katalysatortablette wurde in der Mitte der Heizzone der IR-Küvette positioniert und zur
Entfernung des adsorbiertem Wassers ca. 3 h bei 400 °C unter dem genannten Vakuum
belassen.
Nachdem die vorher beschriebene Vorbehandlung abgeschlossen war, wurde die Adsorption
bei einer Temperatur von 400 °C durchgeführt. Der Absperrhahn H5 wurde geschlossen. Am
Adsorptionsapparatur wurde der Vorratsbehälter B1 mit der adsorbierenden Substanz
angebracht. Anschließend wurden der Absperrhahn H2 geschlossen, der Absperrhahn H4
geöffnet und Adsorptionsapparatur mit der adsorbierenden Substanz gefüllt. Danach wurde
dieser Absperrhahn geschlossen und der Absperrhahn H1 geöffnet. Nach dem Erreichen des
Vorvakuums von 10-2 mbar wurde durch Schließen des Absperrhahns H1 und anschließendem
Öffnen des Absperrhahns H2 wieder ein Vakuum von ca. 10-3 mbar erzeugt. Dieser Vorgang
wurde als Spülprozess insgesamt drei mal durchgeführt, so daß Fremdgase aus der
Adsorptionsapparatur entfernt wurden. Anschließend wurden der Absperrhahn H2
geschlossen und die Absperrhähne H4 und H5 geöffnet, so daß der Vorratsbehälter mit der
adsorbierenden Substanz mit der IR-Küvette verbunden war. Dabei stellte sich bei diesem
geschlossenen System ein für die betreffende adsorbierende Substanz entsprechender
Gleichgewichtsdruck ein. Das System wurde für eine Adsorptionszeit von 30 min in diesem
Gleichgewichtszustand belassen. Nach Ablauf der Adsorptionszeit wurden die Absperrhähne
H4 und H5 geschlossen und durch Öffnen des Absperrhahnes H1 die in der
Adsorptionsapparatur noch vorhandene nicht mehr benötigte adsorbierende Substanz

Experimentelles 100
evakuiert. Bei der Aufnahme der Infrarotspektren blieben die Absperrhähne H1, H2, H4 und
H5 geschlossen.
Die Aufnahme von Infrarotspektren erfolgte mit einem Fourier-Transformations-IR-
Spektrometer vom Typ IFS 66 der Firma Bruker. In der Tabelle 8.6. sind die Meßparameter
für die Aufnahme der Infrarotspektren zusammengefasst.
Tabelle 8.6. Meßparameter für die IR-Transmissionsmessung
Parameter
Meßbereich IR
max. Wellenzahl [cm-1] 4000
min. Wellenzahl [cm-1] 400
Scanzahl 64
Laser Wavenumber [cm-1] 15797,961
Acquisition mode Double Sided, Forward-Backward
Resolution [cm-1] 2,0
Measurement Channel Intern
Apodisation Blackmann Harris 3-Term
Scanner velocity 4 (5,0 kHz)
Apertur 6,0
Stabilisation Delay 2
Correlation Test Mode No
Strahlteiler KBr
Strahlquelle Globar
Detektor DTGS (Deuteriertes Triglycinsulfat)
Zur Aufnahme von Infrarotspektren wurde die IR-Küvette an die Hochvakuumapparatur
angeschlossen und deren NaCl-Fenster im Strahlengang des FTIR-Spektrometers positioniert.
Vor Beginn der Adsorption wurde ein Background-Spektrum der evakuierten Zelle
aufgenommen. Bei allen folgenden Spektrenaufnahmen erfolgte eine elektronische
Substraktion des Background-Spektrums, so dass Einflüsse durch den apparativen Aufbau
und die Strahlungscharakteristik der IR-Quelle eliminiert wurden.

Experimentelles 101
Alle Infrarotspektren wurden in Transmission aufgenommen und graphisch als
Absorptionsspektren, welche dem negativen dekadischen Logarithmus der Transmission
entsprechen, dargestellt.
Nach der Aufnahme des Infrarotspektrums der Gasphase der adsorbierenden Substanz als
Referenzspektrum wurde die Katalysatortablette in den Strahlengang des FTIR-Spektrometers
abgesenkt. Nach einer Wartezeit von 15 min, wobei eine Abkühlung auf Raumtemperatur
erfolgte, wurde das Infrarotspektrum der Katalysatortablette mit der darauf adsorbierten
Substanz aufgenommen.
Für die Ermittlung der adsorbierten Katalysator-Adsorbat Wechselwirkungen auf der
Oberfläche des Katalysators wurden die Adsorbatspektren durch Differenzspektroskopie des
Infrarotspektrums der Katalysatortablette vor und nach der Adsorption mit dem IR-Spektrum
der adsorbierten Substanz in der Gasphase verglichen.
8.3.2. Bestimmung der Reaktionsprodukte
Die Reaktionsprodukte bei der Oxidation von Propen wurden on-line und off-line identifiziert
und quantifiziert. Die on-line Bestimmung der Reaktionsprodukte erfolgte mit Hilfe eines IR-
Fotometers und eines Gaschromatographen (Abb. 8.2). Das IR-Fotometer war ein Gerät Typ
Binos von Rosemount und war mit zwei Detektionskanälen zur Bestimmung von CO und
CO2 ausgestattet. Weil die IR-Absorptionsbanden von CO und N2O übereinander liegen, war
eine IR-fotometrische Bestimmung von CO nicht möglich. Das Signal vom CO-
Detektionskanal wurde lediglich dazu verwendet, das Erreichen einer konstanten N2O-
Konzentration in der Spülphase (Abb. 8.4) festzustellen. Das CO2 wurde IR-fotometrisch
bestimmt.
Der Gaschromatograph (GC) vom Typ 5890 II Plus der Firma Hewlett-Packard (HP) wurde
mit Hilfe der Software HP ChemStation computergesteuert. Der GC war mit einem Injektor,
zwei Trennsäulen – eine HP-Molsieb 5A und eine HP-FFAP – und zwei Detektoren – einen
Wärmeleitfähigkeitsdetektor (WLD) und einen Flammenionisationsdetektor (FID) –
ausgestattet. Die zwei Trennsäulen waren in Paralellschaltung über einen Splitter mit dem
Injektor verbunden. Die Molsieb-Säule zusammen mit dem WLD diente zur Bestimmung von
O2, N2, CO, N2O und weiterer Stickstoffoxide. Die FFAP-Säule mit dem FID diente zur
Bestimmung der organischen Produkte.
Experimentelles 102
Die off-line Analyse der organischen Produkte wurde mit Hilfe eines Gaschromatograph-
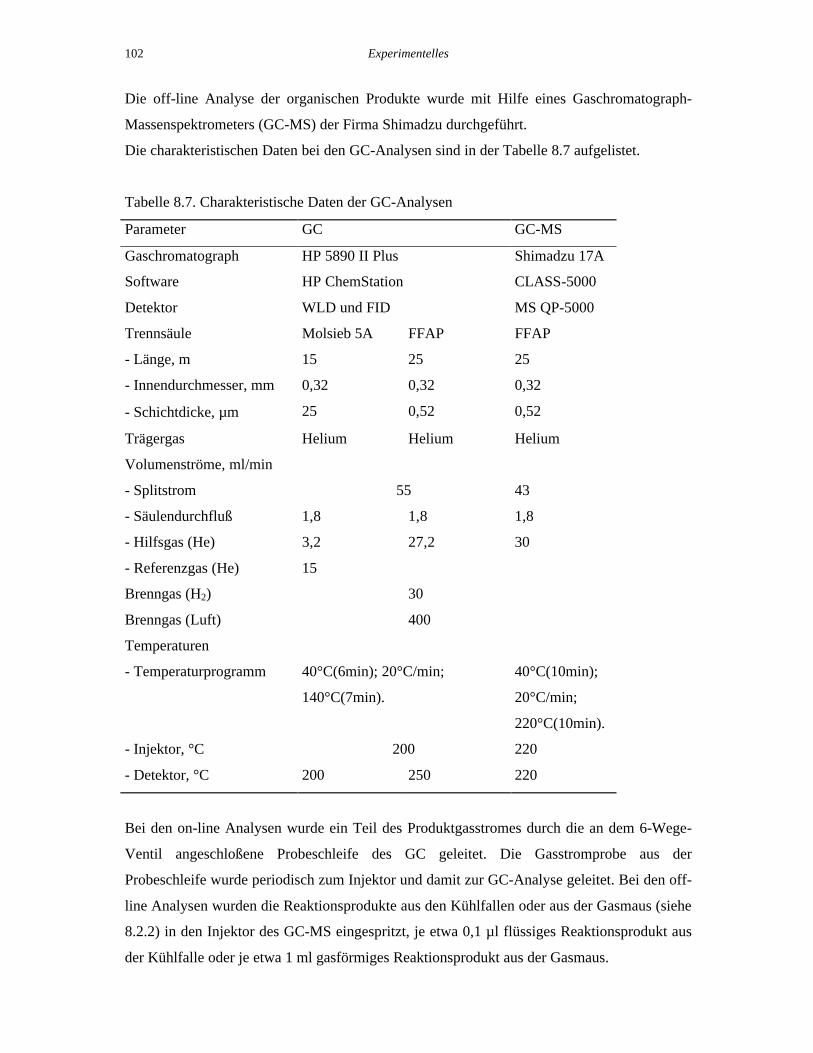
Massenspektrometers (GC-MS) der Firma Shimadzu durchgeführt.
Die charakteristischen Daten bei den GC-Analysen sind in der Tabelle 8.7 aufgelistet.
Tabelle 8.7. Charakteristische Daten der GC-Analysen
Parameter GC GC-MS
Gaschromatograph HP 5890 II Plus Shimadzu 17A
Software HP ChemStation CLASS-5000
Detektor WLD und FID MS QP-5000
Trennsäule Molsieb 5A FFAP FFAP
- Länge, m 15 25 25
- Innendurchmesser, mm 0,32 0,32 0,32
- Schichtdicke, µm 25 0,52 0,52
Trägergas Helium Helium Helium
Volumenströme, ml/min
- Splitstrom 55 43
- Säulendurchfluß 1,8 1,8 1,8
- Hilfsgas (He) 3,2 27,2 30
- Referenzgas (He) 15
Brenngas (H2) 30
Brenngas (Luft) 400
Temperaturen
- Temperaturprogramm 40°C(6min); 20°C/min;
140°C(7min).
40°C(10min);
20°C/min;
220°C(10min).
- Injektor, °C 200 220
- Detektor, °C 200 250 220
Bei den on-line Analysen wurde ein Teil des Produktgasstromes durch die an dem 6-Wege-
Ventil angeschloßene Probeschleife des GC geleitet. Die Gasstromprobe aus der
Probeschleife wurde periodisch zum Injektor und damit zur GC-Analyse geleitet. Bei den off-
line Analysen wurden die Reaktionsprodukte aus den Kühlfallen oder aus der Gasmaus (siehe
8.2.2) in den Injektor des GC-MS eingespritzt, je etwa 0,1 µl flüssiges Reaktionsprodukt aus
der Kühlfalle oder je etwa 1 ml gasförmiges Reaktionsprodukt aus der Gasmaus.

Experimentelles 103
8.3.3. Auswertung der Versuchsergebnisse
Die on-line Analysen lieferten die primären Meßdaten. Die Gaschromatogramme der
Reaktionsprodukte und die IR-fotometrisch bestimmte CO2-Konzentration wurden
aufgezeichnet. Die gaschromatographischen Signale wurden mit Hilfe der Software HP
ChemStation registriert und ausgewertet. Durch die Kalibrierung des IR-Fotometers wurde
die CO2-Konzentration im Produktgas bestimmt. Die Peakflächen und die CO2-Konzentration
wurden zur Berechnung des Umsatzgrades, der Selektivitäten und der Ausbeuten der
einzelnen Produkte verwendet.
8.3.3.1. Berechnung der reaktionsbezogenen Größen
Der Umsatzgrad von Propen wurde nach der Gleichung
V
NV
FFF
U−
= (8.1)
berechnet, wobei
U – Umsatzgrad
FV – Peakfläche Propen im Vorkanal (Eduktstrom)
FN - Peakfläche Propen im Nachkanal (Produktstrom)
Die FN wurde aus dem Gaschromatogramm des Produktstromes direkt ermittelt. Die
entsprechende FV wurde aus den Peakflächen der organischen Produkte und der
Kohlenoxidenkonzentrationen im Produktstrom berechnet.
Bei der Auswertung der FID-Peakflächen wurde berücksichtigt, daß die
Anzeigeempfindlichkeit des FID von der Natur der jeweiligen organischen Verbindung
abhängig ist. Die Anzeigeempfindlichkeit des FID wurde nach Ackman [148] durch
Berechnung des ECR („effective carbon response“) für die entsprechenden Verbindungen
ermittelt. Als Bezugsgruppe zur Berechnung diente die CH2-Gruppe mit einem ECR-Wert
von 100. Der ECR-Wert für eine Verbindung i wurde berechnet nach
2%
.%
CH
iZahli C
CCECR = (8.2)
wobei
ECRi = ECR-Wert der Verbindung i
CZahl = Zahl der C-Atome in Verbindung i
%Ci = prozentueller Kohlenstoffanteil in Verbindung i
%CCH2 = prozentueller Kohlenstoffanteil in Gruppe CH2

Experimentelles 104
Die Korrekturfaktoren wurden unter Verwendung von Propen als Bezugsmolekül, mit einem
ECR-Wert von 300, berechnet:
i
openi ECR
ECRf Pr= (8.3)
fi = Korrekturfaktor für Verbindung i
Die ECR- und f-Werte sind in der Tabelle 5.3 aufgelistet.
Aus den WLD-Peakflächen für die anorganischen Produkte (O2, N2, CO und N2O) wurden
mittels Kalibrierung die entsprechenden Konzentrationen im Produktgas berechnet. Die CO2-
Konzentration wurde mit dem Schreiber des IR-Fotometers registriert. Aus den CO- und CO2-
Konzentrationen konnte dann die Ausbeute zu CO und CO2 berechnet werden:
KWZahl
COCO cC
cA x
x .= (8.4)
ACOx = Ausbeute zu COx
cCOx = Konzentration COx im Produktgas
CZahl = Zahl der C-Atome in dem organischen Edukt (=3 für Propen)
cKW = Konzentration des organischen Eduktes (Propen)
Die FV wird dann:
∑ +++= )(2 COCOViiNV AAFfFFF (8.5)
COCO
iiNV AA
fFFF
−−+
= ∑2
1 (8.6)
Fi = Peakfläche für Verbindung i
Die Selektivität Si zu einem organischen Produkt i wurde berechnet:
NV
iii FF
fFS
−=
. (8.7)
Die Ausbeute Ai zu einem Produkt i ist:
ii SUA .= (8.8)
Die Berechnung der oberflächenbezogenen Reaktionsgeschwindigkeit von Propen r erfolgte
nach den Gleichungen 8.9-8.11:
dtadc
rS .
−= (8.9)
c = Propenkonzentration
t = Reaktionszeit
aS = spezifische Oberfläche des Katalysators

Experimentelles 105
durch Einsetzen des Umsatzgrades:
KatS
E
S maUcV
aUc
r.
.... 0
.
0 ==τ
[l.m-2.h-1] (8.10)
c0 = Anfangskonzentration von Propen im Eduktstrom
oder
KatSm
Em maV
UcVr
.... 0
.
= [mol.m-2.h-1] (8.11)
rm = molare Reaktionsgeschwindigkeit; Umsetzungsrate
Die Bildungsgeschwindigkeit (Bildungsrate) rm,i für ein Produkt i ist:
KatSm
iEim maV
SUcVr
..... 0
.
, = [mol.m-2.h-1] (8.12)
oder
iKatSm
Eim A
maVcV
r .... 0
.
, = [mol.m-2.h-1] (8.13)
Die Temperaturabhängigkeit der Reaktionsgeschwindigkeit wird durch die Arrhenius’sche
Gleichung beschrieben. Für eine Reaktion n-ter Ordnung nckr .= (8.14)
ergibt sich
nTRE
cekrA
.. .0
−= (8.15)
Nach Einführung des Umsatzgrades erhält man
nnTRE
cUekrA
0.
0 .)1.(. −=−
(8.16)
Durch Gleichsetzen von (8.10) und (8.16) ergibt sich
nnTRE
S
cUekddU
ac A
0.
00 .)1.(. −=⋅
−
τ (8.17)
und durch Umformen
)1(0
.0 .)1.(.. −−
−= nnS
TRE
cUaekddU A
τ (8.18)
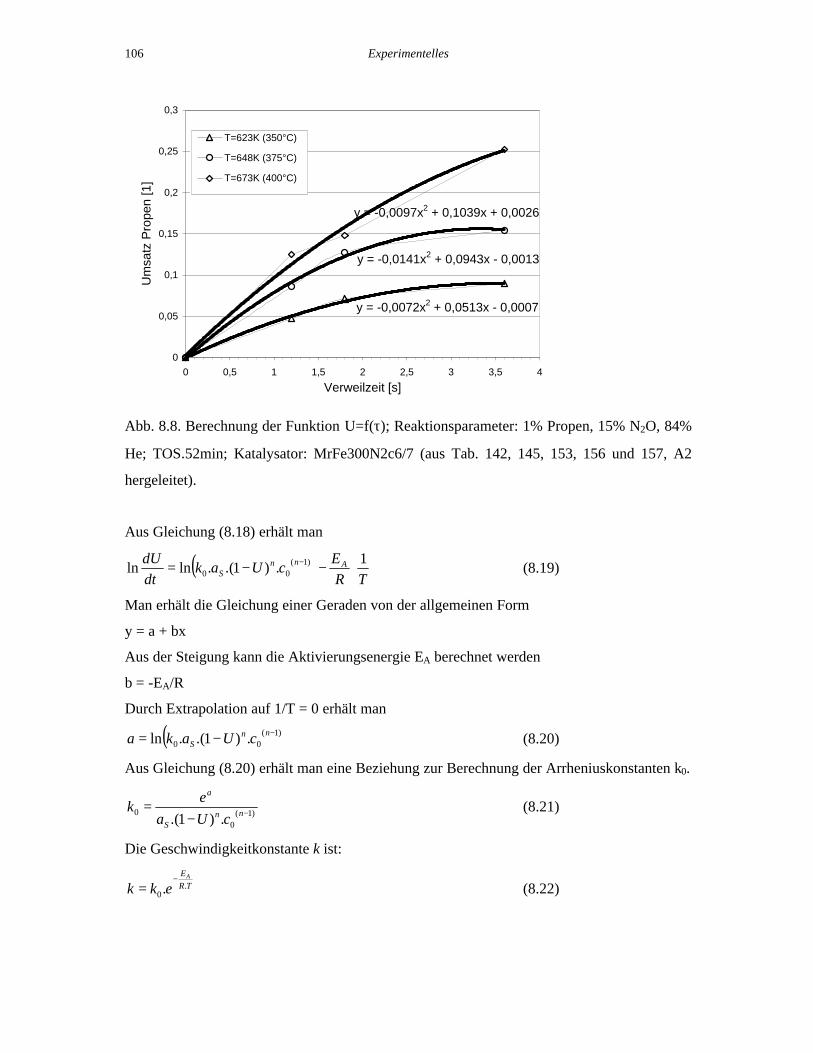
Die Funktion U=f(τ) läßt sich für einen bestimmten Katalysator bei einer vorgegebenen
Temperatur experimentell ermitteln. Der prinzipielle Verlauf ist in Abb. 8.8. für drei
verschiedene Temperaturen dargestellt.
Experimentelles 106
y = -0,0072x2 + 0,0513x - 0,0007
y = -0,0141x2 + 0,0943x - 0,0013
y = -0,0097x2 + 0,1039x + 0,0026
0
0,05
0,1
0,15
0,2
0,25
0,3
0 0,5 1 1,5 2 2,5 3 3,5 4
Verweilzeit [s]
Um
satz
Pro
pen
[1]
T=623K (350°C)
T=648K (375°C)
T=673K (400°C)
Polynomisch
Abb. 8.8. Berechnung der Funktion U=f(τ); Reaktionsparameter: 1% Propen, 15% N2O, 84%
He; TOS.52min; Katalysator: MrFe300N2c6/7 (aus Tab. 142, 145, 153, 156 und 157, A2
hergeleitet).
Aus Gleichung (8.18) erhält man
( )TR
EcUak
ddU Ann
S1
.)1.(.lnln )1(00 ⋅−−= −
τ (8.19)
Man erhält die Gleichung einer Geraden von der allgemeinen Form
y = a + bx
Aus der Steigung kann die Aktivierungsenergie EA berechnet werden
b = -EA/R
Durch Extrapolation auf 1/T = 0 erhält man
( ))1(00 .)1.(.ln −−= nn
S cUaka (8.20)
Aus Gleichung (8.20) erhält man eine Beziehung zur Berechnung der Arrheniuskonstanten k0.
)1(0
0 .)1.( −−= nn
S
a
cUae
k (8.21)
Die Geschwindigkeitkonstante k ist:
TREA
ekk .0.
−= (8.22)

Experimentelles 107
8.3.3.2. Berechnung der hydrodynamischen Größen
Die Raumgeschwindigkeit RG ergibt sich zu:
Kat
E
VV
RG
.
= [h-1] (8.23)
.
EV = Eduktvolumenstrom [l/h]
VKat = Katalysatorschüttvolumen [l]
Die Verweilzeit τ ist:
.
1
E
Kat
V
VRG
==τ [h] (8.24)
Die modifizierte Raumgeschwindigkeit RG* wurde berechnet:
Kat
E
mV
RG
.
* = [l.g-1.h-1] (8.25)
mKat = Katalysatormasse [g]
Zur Berechnung der Reynoldszahl für die Strömung durch das Katalysatorbett wurde eine
Gleichung für die Strömung in Schüttschichten [149] verwendet.
νuL
=(Re) (8.26)
wobei
)1(64
ε−= Pd
L [m], mit Partikeldurchmesser dP [m] und Porösität der Schüttung ε [1]
u = mittlere Geschwindigkeit im leeren Rohr [m.s-1]
ν = kinematischer Viskositätskoeffizient des Fluids [m2.s-1]
Die kritische Reynolds-Zahl beträgt in diesem Fall (Re)k ≈ 20.
Die mittlere Geschwindigkeit im leeren Reaktor ist
0
02
.
02
0
0.
0.
4
4PT
TPdV
dPT
TPV
AV
uRRR ππ
=== (8.27)
.
V = Gasvolumenstrom durch Reaktor [m3.s-1]
AR = Reaktorkreisfläche [m2]

Experimentelles 108
Der kinematische Viskositätskoeffizient eines Gasgemisches wird approximiert:
∑=i i
ixνν
1 (8.28)
wobei
xi = molarer Anteil der Komponente i im Gasgemisch
0
0
0, PTTP
i
i
i
ii ρ
ηρη
ν == (8.29)
ηi = dynamischer Viskositätskoeffizient der Komponente i [Pa.s]
ρi = Gasdichte der Komponente i [kg.m-3]
m
ii V
M=0,ρ (8.30)
Mi = molekulare Masse der Komponente i [kg.mol-1]
Vm = Molvolumen [m3.mol-1]
Für ein Gas A wird der dynamische Viskositätskoeffizient errechnet:
VAA
ArA
TM
Ω= 2
,026693,0σ
η (8.31)
Mr,A = relative molekulare Masse von A [1]
T = Temperatur [K]
σAA = Stoßdurchmesser [pm]
ΩV = Stoßintegral [1]
Der kinematische Viskositätskoeffizient des Reaktandengasgemisches ist
TPPTxxx
Inert
InertInert
ON
ONON
C
CC
0
00,0,0,
2
22
3
331
++=
=
==
ηρ
ηρ
η
ρ
ν (8.32)
damit wird die Reynolds-Zahl
++
−=
=
++
−=
=
==
=
==
Inert
InertInert
ON
ONON
C
CC
MR
P
Inert
InertInert
ON
ONON
C
CC
R
P
MxMxMx
VddV
xxx
ddV
ηηηεπ
ηρ
ηρ
η
ρ
επ
2
22
3
33
2
22
3
33
)1(38
)1(616
(Re)
2
.
0
0,0,0,2
.
0
(8.33)
Für die Katalysatorkornfraktion von 0,12-0,25 mm wird für den Partikeldurchmesser ein
Mittelwert von dP = 1,85 x 10-4 m für die Berechnungen verwendet.
Der Reaktordurchmesser dR betrug 0,01 m.
Die Porösität der Katalysatorschüttung betrug ε ≈ 0,55.
Experimentelles 109
Die dynamischen Viskositätskoeffizienten wurden für eine Temperatur von T=648K
berechnet und sind in der Tabelle 8.8 aufgelistet.
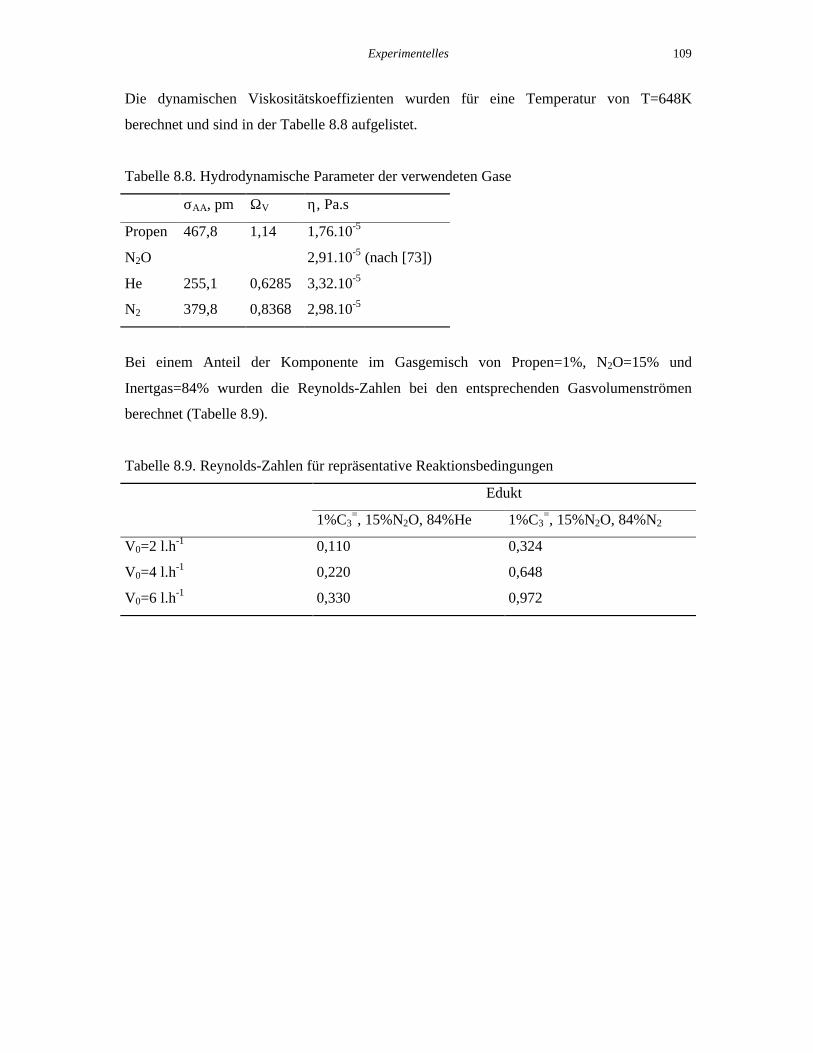
Tabelle 8.8. Hydrodynamische Parameter der verwendeten Gase
σAA, pm ΩV η, Pa.s
Propen 467,8 1,14 1,76.10-5
N2O 2,91.10-5 (nach [73])
He 255,1 0,6285 3,32.10-5
N2 379,8 0,8368 2,98.10-5
Bei einem Anteil der Komponente im Gasgemisch von Propen=1%, N2O=15% und
Inertgas=84% wurden die Reynolds-Zahlen bei den entsprechenden Gasvolumenströmen
berechnet (Tabelle 8.9).
Tabelle 8.9. Reynolds-Zahlen für repräsentative Reaktionsbedingungen
Edukt
1%C3=, 15%N2O, 84%He 1%C3
=, 15%N2O, 84%N2
V0=2 l.h-1 0,110 0,324
V0=4 l.h-1 0,220 0,648
V0=6 l.h-1 0,330 0,972
















![Wetterstation ENGINEER - lambrecht.net · Wetterstation ENGINEER Ultraschall-Sensor u[sonic] für Windgeschwindigkeit und Windrichtung · seewasserfester Sensor, optimal beheizt ·](https://img.pdfslide.net/doc/110x75/5e10c1c819466036b63d07b3/wetterstation-engineer-wetterstation-engineer-ultraschall-sensor-usonic-fr.jpg)












