Embed Size (px)
DESCRIPTION
Cell Biology once more. This is a series of ppt presentations made available to you guys.
Citation preview

Lysosomes: structure, function and storage diseases
Michael Kibe
Sept,2012

The discovery of lysosomes
In 1955, Belgian scientist Christian de Duve observed that the cells released an enzyme called acid phosphatase in much larger amounts when they were repeatedly frozen and thawed before centrifugation
de Duve C (1975) Exploring cells with a centrifuge. Science 189, 186-194
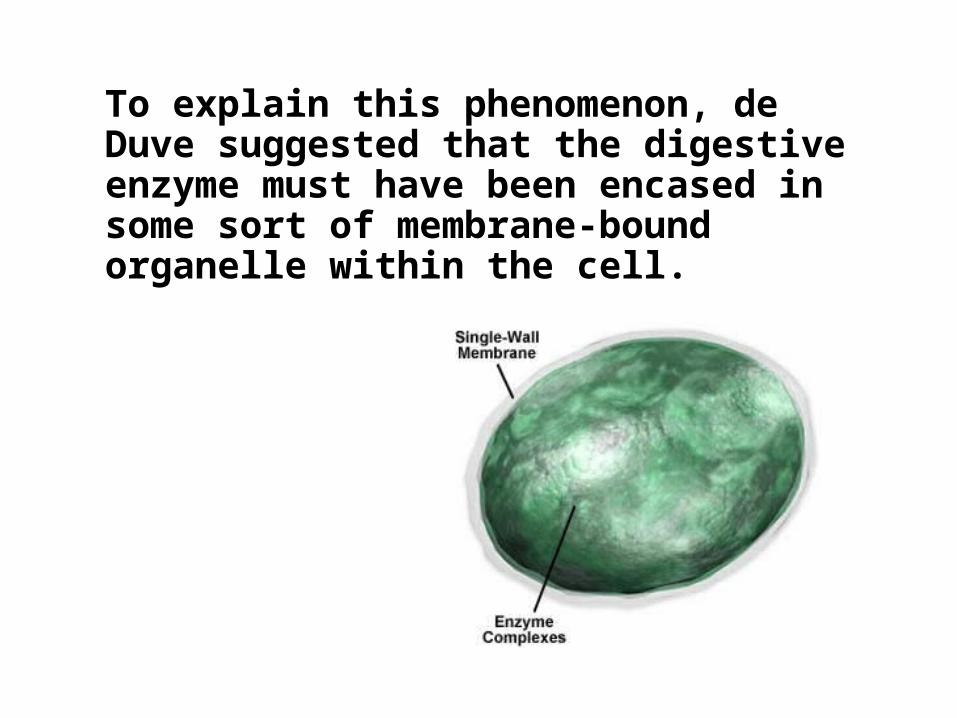
To explain this phenomenon, de Duve suggested that the digestive enzyme must have been encased in some sort of membrane-bound organelle within the cell.
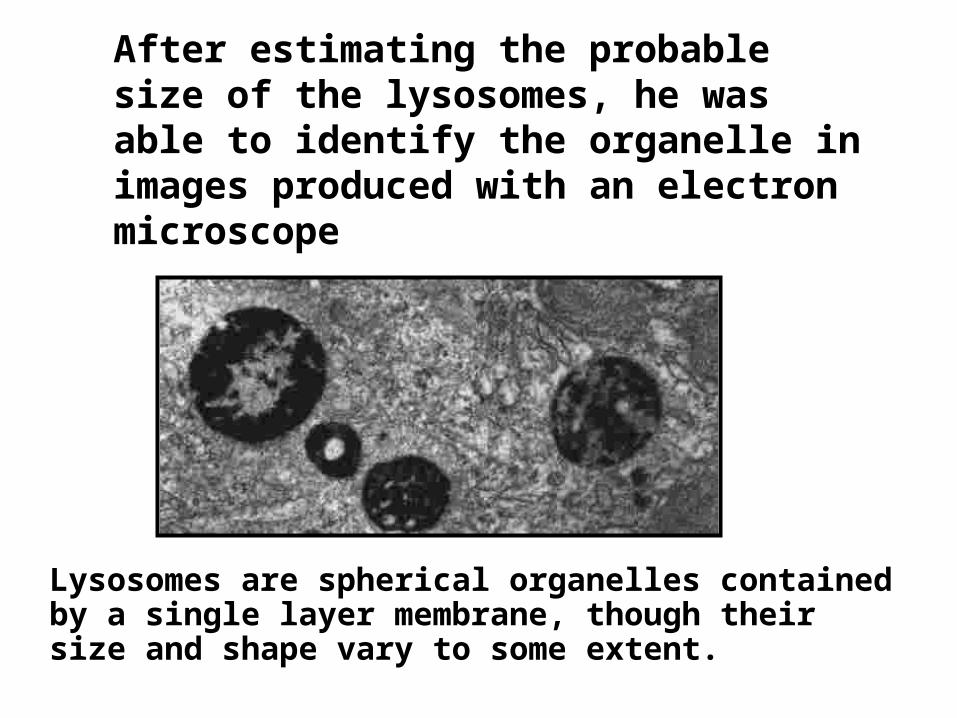
After estimating the probable size of the lysosomes, he was able to identify the organelle in images produced with an electron microscope
Lysosomes are spherical organelles contained by a single layer membrane, though their size and shape vary to some extent.
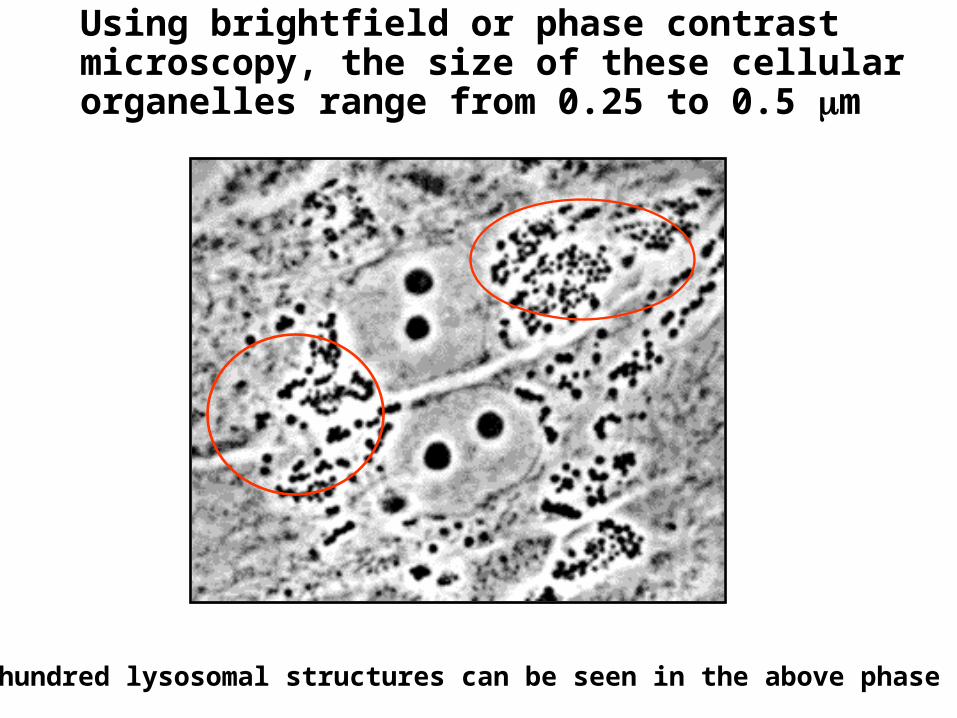
Using brightfield or phase contrast microscopy, the size of these cellular organelles range from 0.25 to 0.5 m
Several hundred lysosomal structures can be seen in the above phase micrograph

• "Lysosome" was the name given because of these enzymes' ability to "lyse" the cell
• Initially referred to as "suicide bags" since one of the functions of lysosomes is to rupture when the cell dies,
• Rupturing releases hydrolytic enzymes that digest all parts of the cell, including proteins, DNA, RNA, carbohydrates, lipids and cellulose.

For their discoveries concerning
"the structural and functional organization of the cell"
Albert Claude, Christian de Duve and George Palade received the Nobel Prize in Physiology or Medicine in 1974

Lysosome structure• Single membrane bound organelles found in
animal cells• Derived from Golgi complex • Vary in shape and size and of two types:• Primary lysosomes:
– spherical and do not contain particulate or membrane debris,
– newly formed without substrates– Derived from coated vesicles that pinches off the
Golgi• Secondary lysosomes:
– Formed from primary lysosomes after fusion with other membrane orgenelles or vesicles
– They are larger and irregular shaped

Lysosomal enzymes
• Mostly acid hydrolases with an optimal pH of 5.0• Can degrade all major classes of biological
molecules eg– DNA and RNA degrade by nuclease to
mononucleotides– Proteins and peptides degraded by proteases to
amino acids– Phosphatases remove phosphate groups from
mononucleotides and phosholipids– Carbohydrates and glycolipids degraded by
hydrolases to smaller units

Lysosomal enzymes/degradation
• Lysosomal degradation is primarily directed to:– Extracellular molecules taken up by the cell– Aged, diffective and worn out organelles– Sometimes, denatured, misfolded and proteins taken up by the
cell are also degraded• Interior of lysosome maintained at pH 5.0 by two
ATPases on the lysosome membrane which work together to pump H+ and Cl- ions(HCl) from cytosol
• The acidic pH denatures the proteins so that hydrolases can act on them
• Lysosomal enzymes cause little or no degradation of cytosolic contents (if they leak) because they cannot act at pH 7.0-7.3, the pH of the cytosol

Lysosomal enzymes
• About 40 enzymes found in th lysosomes which include; Phosphatases (5), proteases (4), nucleases (2), lipases (6), glucosidases (12), aryl sulfatase (1), acid phosphatase (1) and others
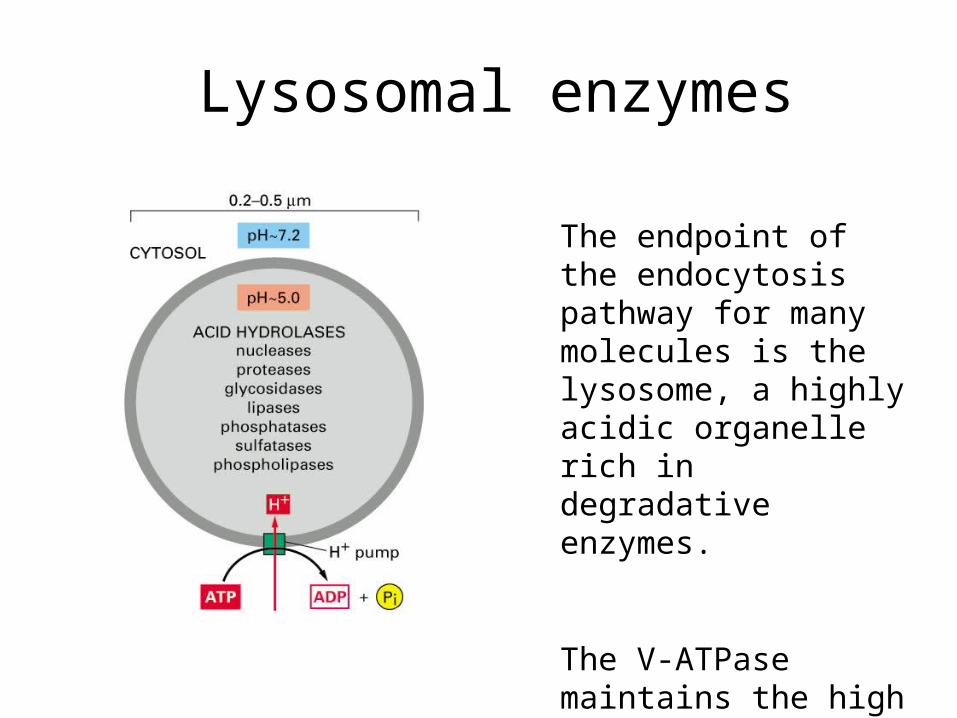
Lysosomal enzymes
The endpoint of the endocytosis pathway for many molecules is the lysosome, a highly acidic organelle rich in degradative enzymes.
The V-ATPase maintains the high acidity of the lumen by pumping protons across the lipid bilayer.

Lysosomal cellular digestion
• Material for digestion comes from within or outside the cell. If material is from outside, then it is taken in by the following procesess– Endocytosis: can be receptor mediated or by
pinocytosis– Phogocytosis
• Lysosomal enzymes are pinched off Golgi in coated vesicles which fuse to form a late endosome
• The late endosome then fuse with the lysosome
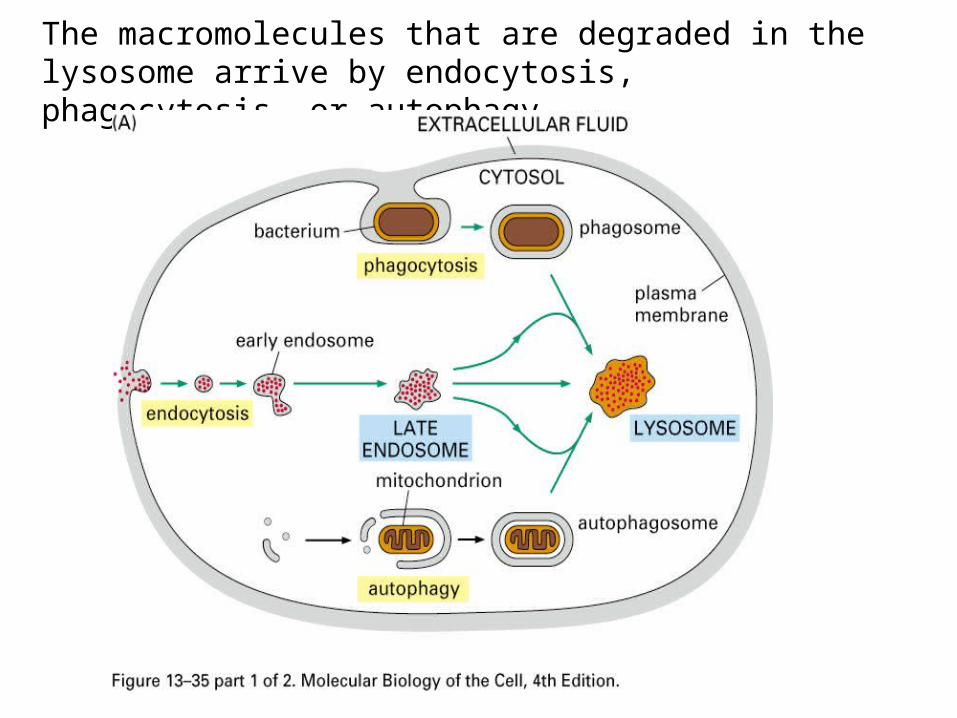
The macromolecules that are degraded in the lysosome arrive by endocytosis, phagocytosis, or autophagy.

Cellular processes of the lysosome
• Nutrition
• Defence
• Recycling of cellular components
• Differentiation
• Cell death

Autophagy
• Breakdown of cellular compartments and structures that are damaged or no longer needed
• The digestion of unwanted organelles and structures by the lysosome is called “self eating”
• The organelle is wrapped in ER membranes and the autophagic vacuole then fuses with the lysosome to give a secondary vacuole (autophagic lysosome)
• Autophagy is increased in cells stressed by starvation
• It occurs in certain developmental situations eg destruction of cell organelles such as mitochondria in maturing red blood cells
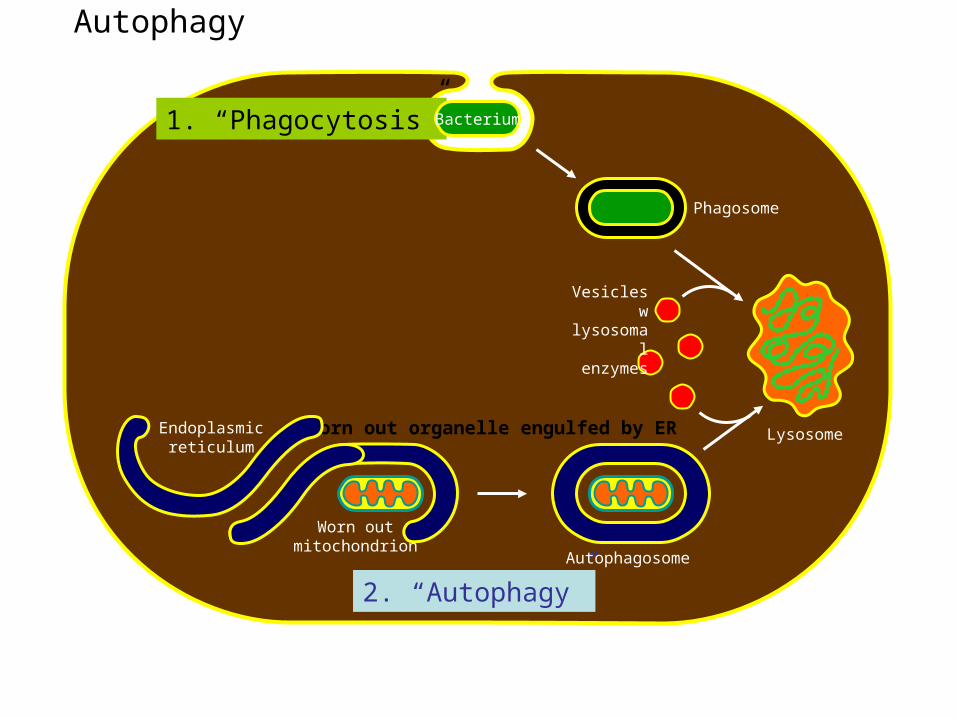
Autophagy (“Self-eating”); used to recycle worn-out organelles
Phagosome
Worn outmitochondrion
1. “Phagocytosis”
2. “Autophagy”Autophagosome
Bacterium
LysosomeWorn out organelle engulfed by EREndoplasmicreticulum
Vesicles wlysosomal enzymes

Autolysis
• A process in which unwanted cells are destrolyed by programmed cell death during development
• The loss of tail by the tadpole during metamorphosis is by lysosomal mediated autolysis

Extracellular digestion
• In rare cases, lysosomes discharge their enzymes outside the cell by exocytosis
• For example, during fertilization of the ovum the head of sperm releases lysosomal enzymes that degrade the chemical barriers and help the sperm penetrate the egg surface
• Inflamation of joints due to rheumatoid arthritis is as a result of release of lysosomal enzymes into the joints. Anti-inflamatory agents such as cortisone and hydrocortisone work by stabilizing lysosomal membranes

Endocytosis
• An important function of the lysosome is the degradation of foreign material brought in by endocytosis
• In endocytosis, there is invagination of the plasma membrane and budding of vesicles to the interior of the cell
• There are three different types of endocytosis– Phagocytosis – “cell eating”– Pinocytosis – “cell drinking”– Receptor mediated endocytosis
• The endocytic pathway is important for cells to take up nutrients in macromolecular form eg cholesterol in form of lipoprotein particles and iron complexed with trasferrin, a serum protein

Phagocytosis
• Macrophages, polymorphonuclear leukocytes and neutrophils can take up invading bacteria, viruses and other microorganisms by the process of phagocytosis
• This is non-selective actin mediated process in which the ingested material is enveloped by the plasma membrane forming a vesicle called phagosome
• The phagosome then fuses with primary lysosome formimg a secondary lysosome (heterophagic lysosome). Soluble products of digestion eg sugars, amino acids, nucleotides cross the lysosomal membrane and serve as source of nutrients. A residual body may accumulate in the cytoplasm after this digestion contributing to cellular aging
• Amoeba, cilliate protozoa, sponges etc use phagocytosis as a means of obtaining nutrients
Bacterium
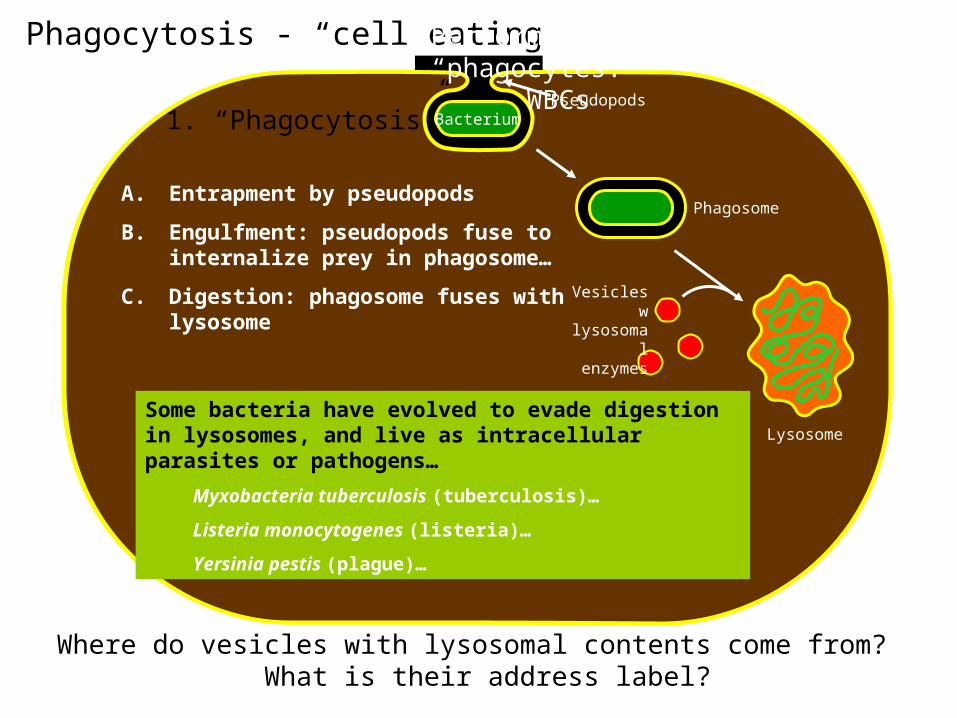
Phagocytosis - “cell eating”
Phagosome
Lysosome
1. “Phagocytosis”
A. Entrapment by pseudopods
B. Engulfment: pseudopods fuse to internalize prey in phagosome…
C. Digestion: phagosome fuses with lysosome
Pseudopods
Vesicles wlysosomal enzymes
Some bacteria have evolved to evade digestion in lysosomes, and live as intracellular parasites or pathogens…
Myxobacteria tuberculosis (tuberculosis)…
Listeria monocytogenes (listeria)…
Yersinia pestis (plague)…
Performed by specialized “phagocytes:”
WBCs
Where do vesicles with lysosomal contents come from? What is their address label?

Pinocytosis
• This occurs in many cell types such as macrophages, leukocytes, intestinal epithelial cells etc
• The plasma membrane invaginates forming a pinocytic vesicle, 0.05-0.1μm in diameter
• The pinocytic vesicle with small droplets of extracellular fluid and dissolved material is pinched off the plasma membrane to form a free vesicle
• The vesicle may fragment or coalesce into large ones which fuse with the primary lysosomes

Receptor mediated endocytosis
• Major mechanism of selective uptake of macromolecules or peptide hormones that bind to cell surface
• A specific receptor binds to the macromolecule or peptide ligand and the plasma membrane region containing the receptor ligand complex then buds off inward to form a transport vesicle
• The endocytosis process is initiated by clathrin coated pits leading to a clathrin coated vesicle
• This process is called receptor mediated endocytosis or adaptive pinocytosis
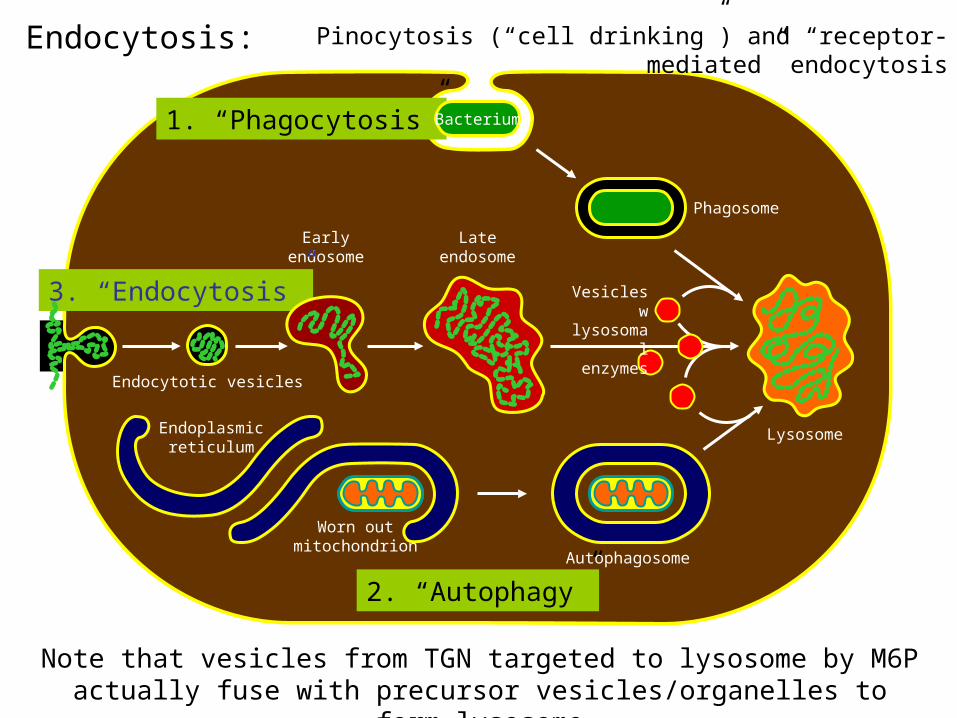
Endocytosis:
Phagosome
Lateendosome
Earlyendosome
Endoplasmicreticulum
1. “Phagocytosis”
3. “Endocytosis”
2. “Autophagy”Autophagosome
Bacterium
Worn outmitochondrion
Vesicles wlysosomal enzymes
Endocytotic vesicles
Lysosome
Pinocytosis (“cell drinking”) and “receptor-mediated” endocytosis
Note that vesicles from TGN targeted to lysosome by M6P actually fuse with precursor vesicles/organelles to form lysosome

Clathrin coated pits
• Cell surface receptors mediating receptor mediated endocytosis are transmembrane glycoproteins
• The receptors are located in specialized regions of the plasma membrane called coated pits where they cogregate whether they have a bound ligand or not
• Receptor mediated endocytosis begins with invagination of a coated pit
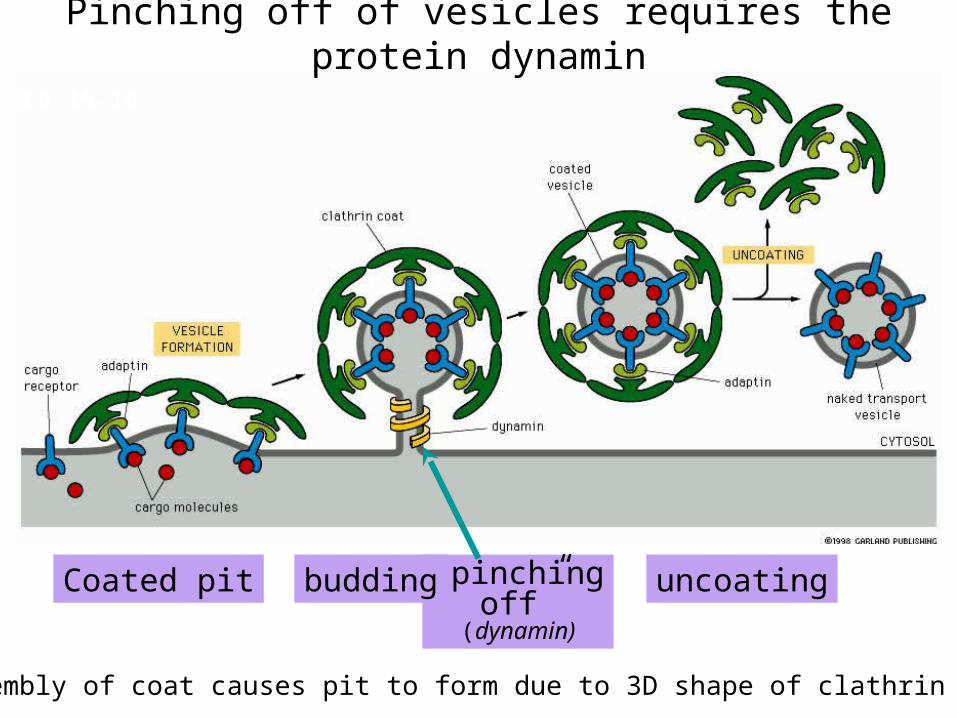
• Clathrin forms a lattice around a coated pit excising it from the plasma membrane to form a coated vesicle (80μm in diameter)

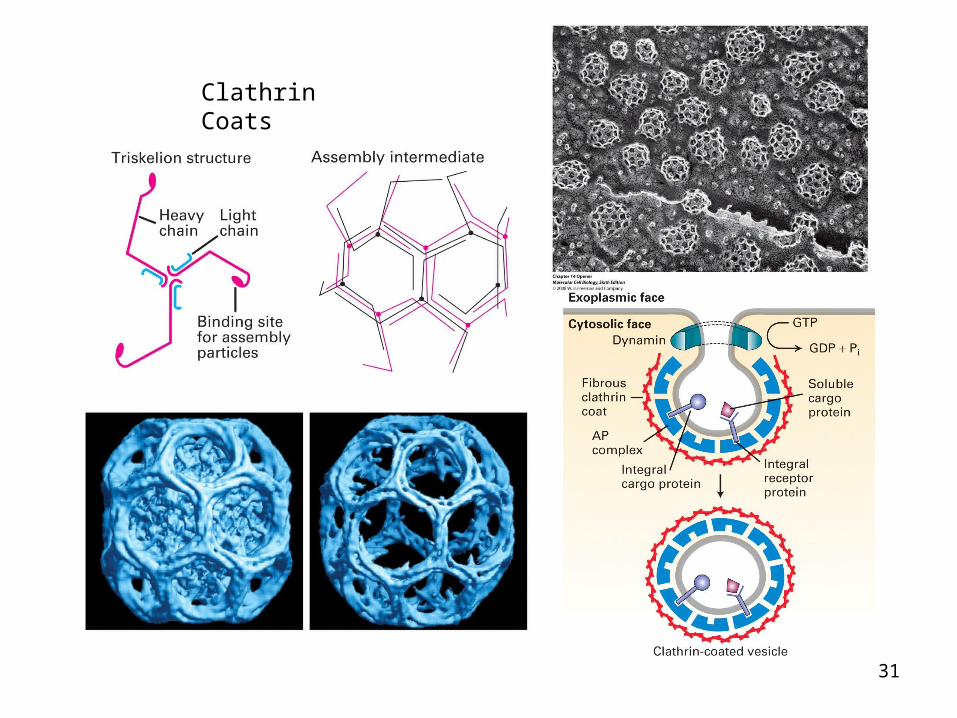
Clathrin and coated pits formation• Clathrin can form closed polyhedral lattices• It consists of 3 heavy (H-180 kDa) and 3 light (L-35 kDa)
polypeptide chains• In EM it looks like a three legged – triskelion. The
flexibility of the triskelion enables it to fit into a pentagon or a hexagon
• Triskelions polymerize into a polyhedral network in the cytosolic side of the membrane. They interact with assembly particles (adapter proteins) that select the cargo to be carried in the clathrin cage
• There are four types of assembly particles (AP1, AP2, AP3 and GGA). AP1 and AP2 are specific for proteins that buds off the Golgi and plasma membrane, respectively
• AP1 bind to cytosolic domains that buds off trans Golgi (eg mannose 6P receptor). AP2 bind to cytosolic domains of plasma membrane receptors inetrnalized by receptor mediated endocytosis eg LDL receptor
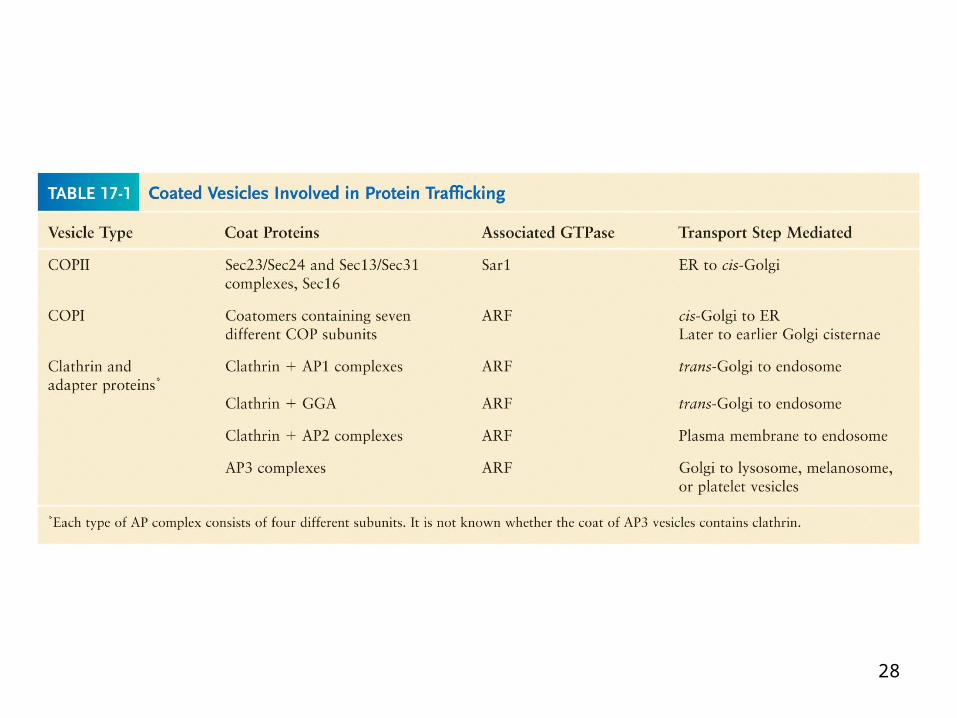
28
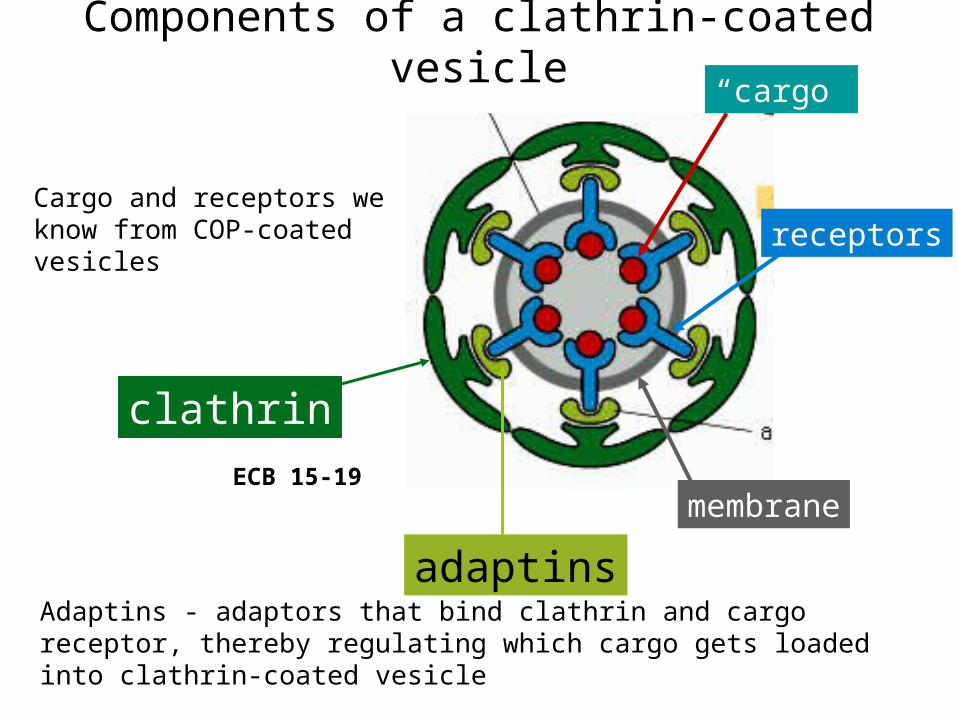
ECB 15-19
clathrin
adaptins
membrane
receptors
“cargo”
Components of a clathrin-coated vesicle
Cargo and receptors we know from COP-coated vesicles
Adaptins - adaptors that bind clathrin and cargo receptor, thereby regulating which cargo gets loaded into clathrin-coated vesicle
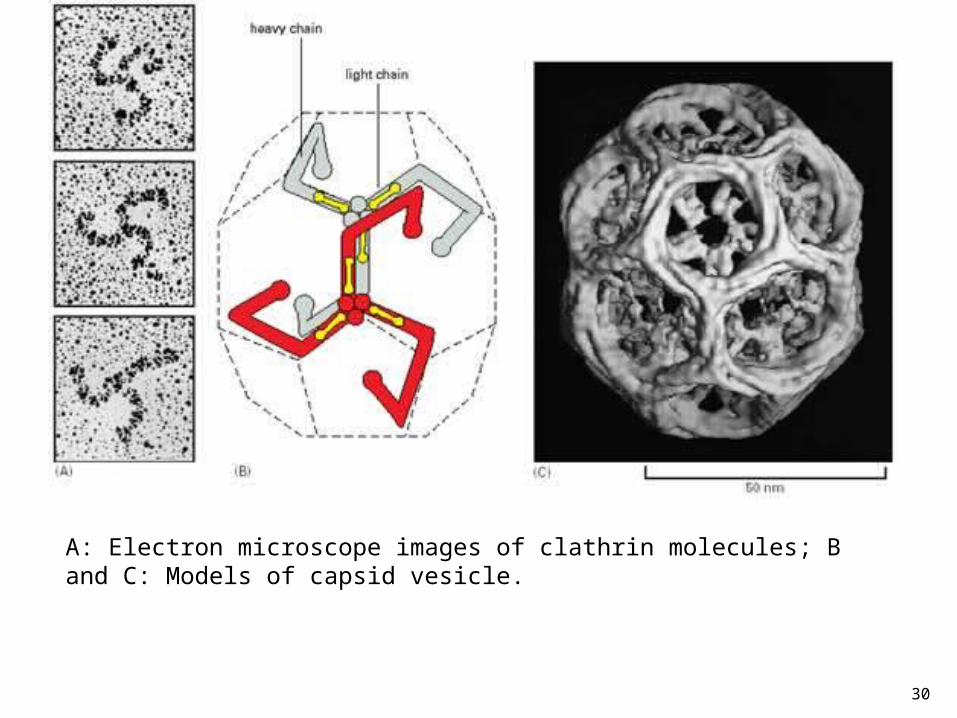
30
A: Electron microscope images of clathrin molecules; B and C: Models of capsid vesicle.
31
Clathrin Coats
ECB 15-19
Coated pit “pinchingoff”
(dynamin)
budding uncoating
Pinching off of vesicles requires the protein dynamin
Assembly of coat causes pit to form due to 3D shape of clathrin coat
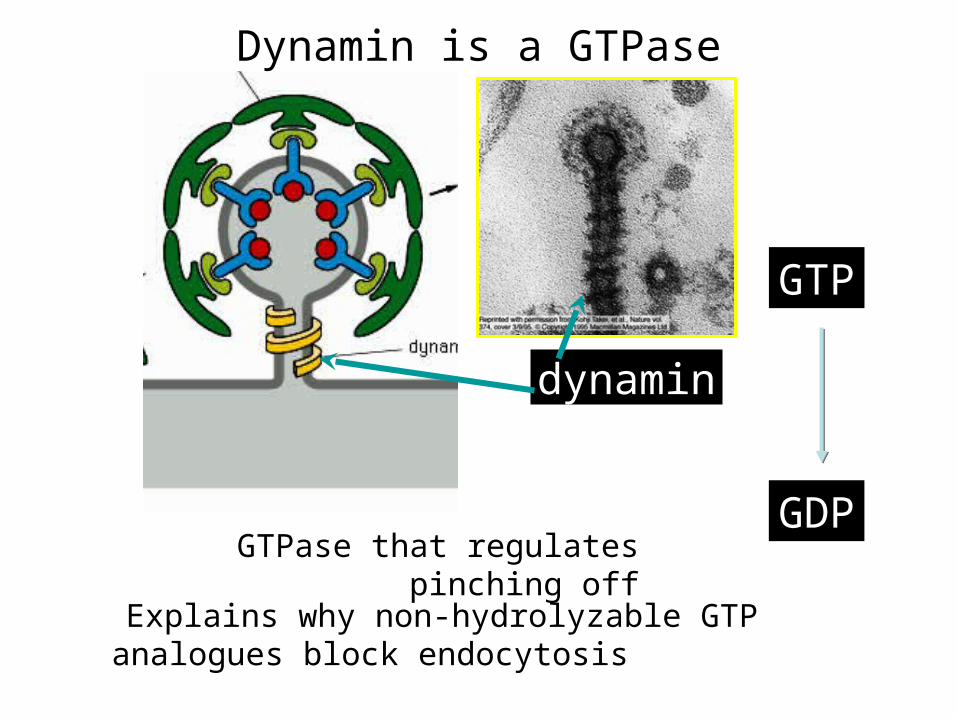
dynamin
GTP
GDPGTPase that regulates pinching off
Dynamin is a GTPase
Explains why non-hydrolyzable GTP analogues block endocytosis
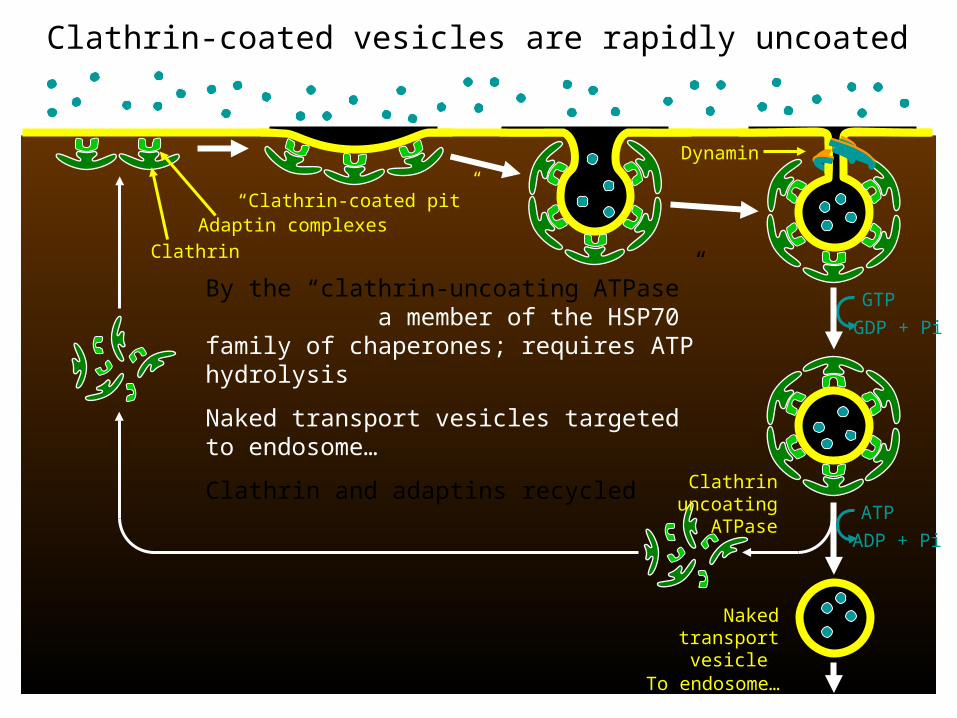
GTP
GDP + Pi
Clathrin uncoating ATPase
Naked transport
vesicle
Dynamin
ATP
ADP + Pi
Clathrin-coated vesicles are rapidly uncoated
By the “clathrin-uncoating ATPase” a member of the HSP70 family of chaperones; requires ATP hydrolysis
Naked transport vesicles targeted to endosome…
Clathrin and adaptins recycled
“Clathrin-coated pit”
To endosome…
Clathrin
Adaptin complexes
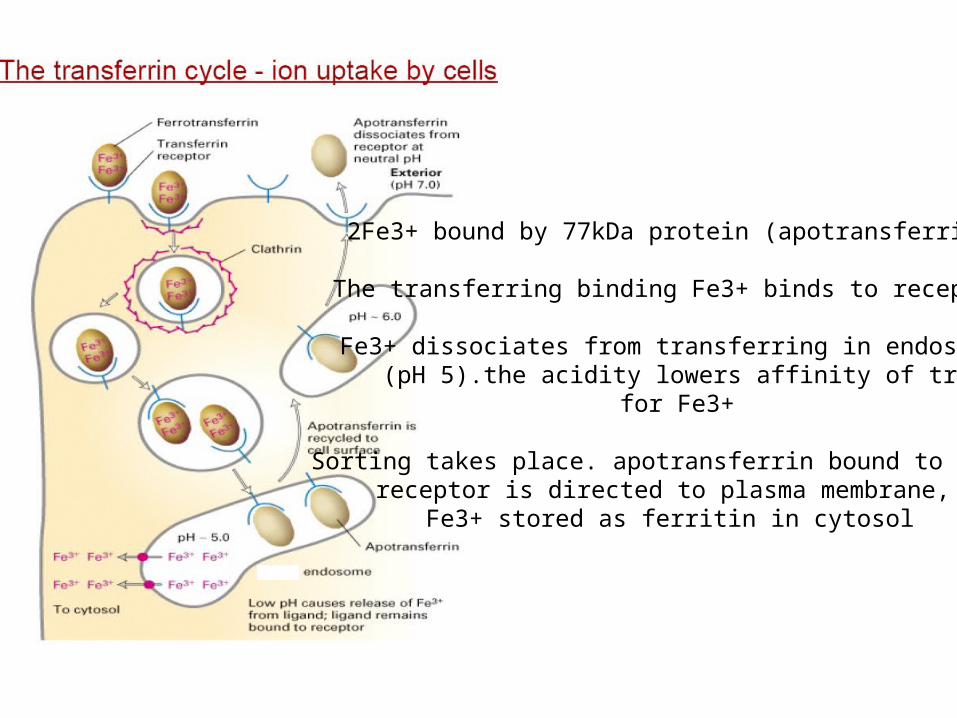
2Fe3+ bound by 77kDa protein (apotransferrin)
The transferring binding Fe3+ binds to receptor
Fe3+ dissociates from transferring in endosome (pH 5).the acidity lowers affinity of transferring
for Fe3+
Sorting takes place. apotransferrin bound to the receptor is directed to plasma membrane,
Fe3+ stored as ferritin in cytosol
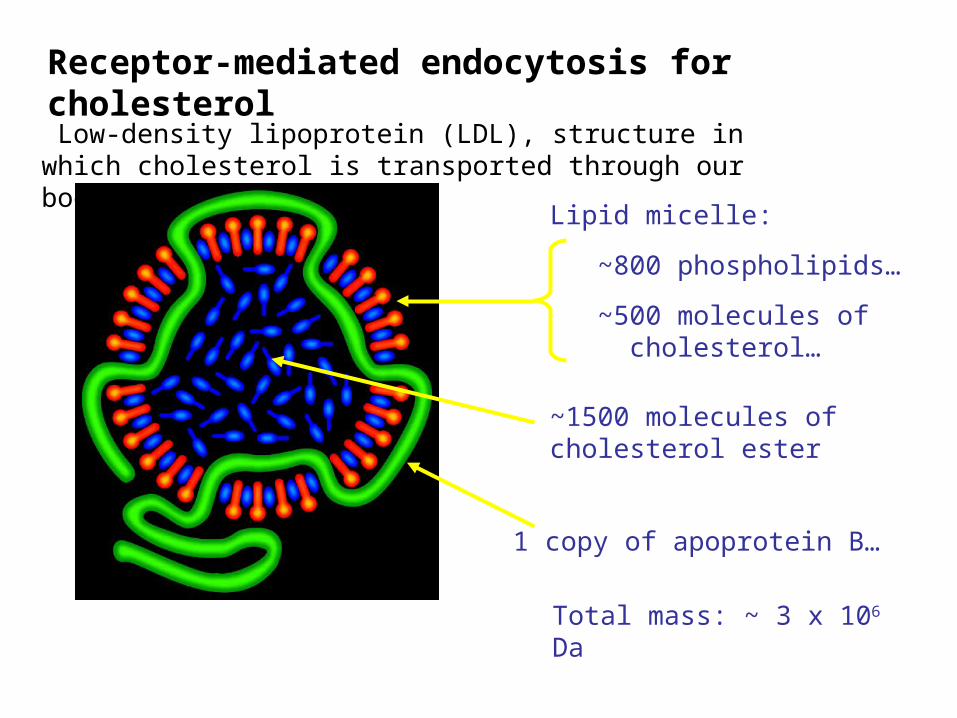
Lipid micelle:
~800 phospholipids…
~500 molecules of cholesterol…
Low-density lipoprotein (LDL), structure in which cholesterol is transported through our bodies
Receptor-mediated endocytosis for cholesterol
Total mass: ~ 3 x 106 Da
~1500 molecules of cholesterol ester
1 copy of apoprotein B…
37
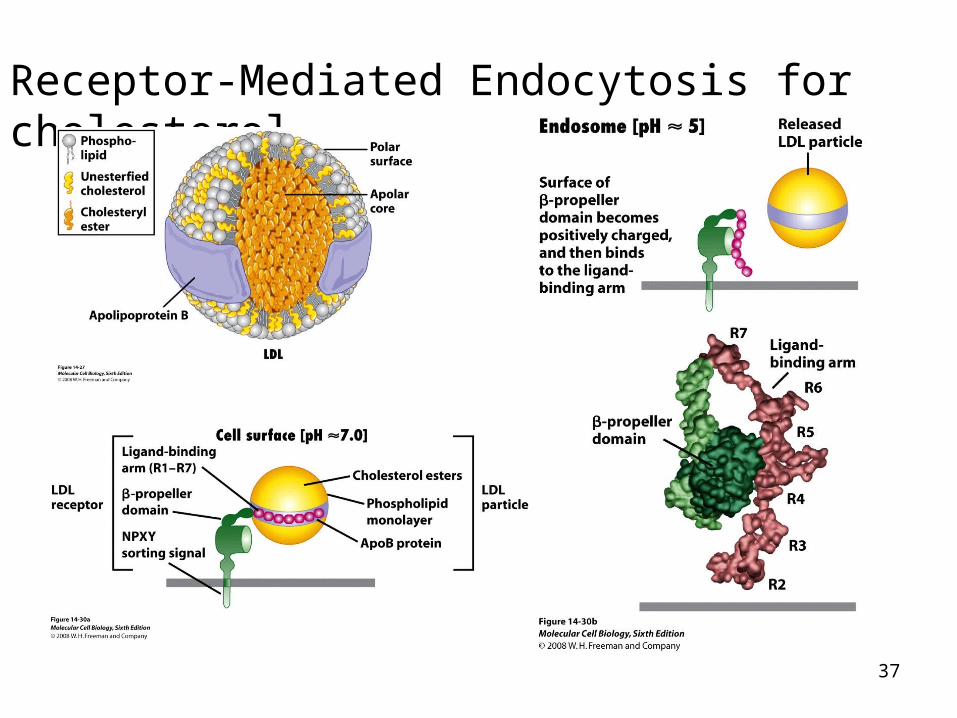
Receptor-Mediated Endocytosis for cholesterol
38
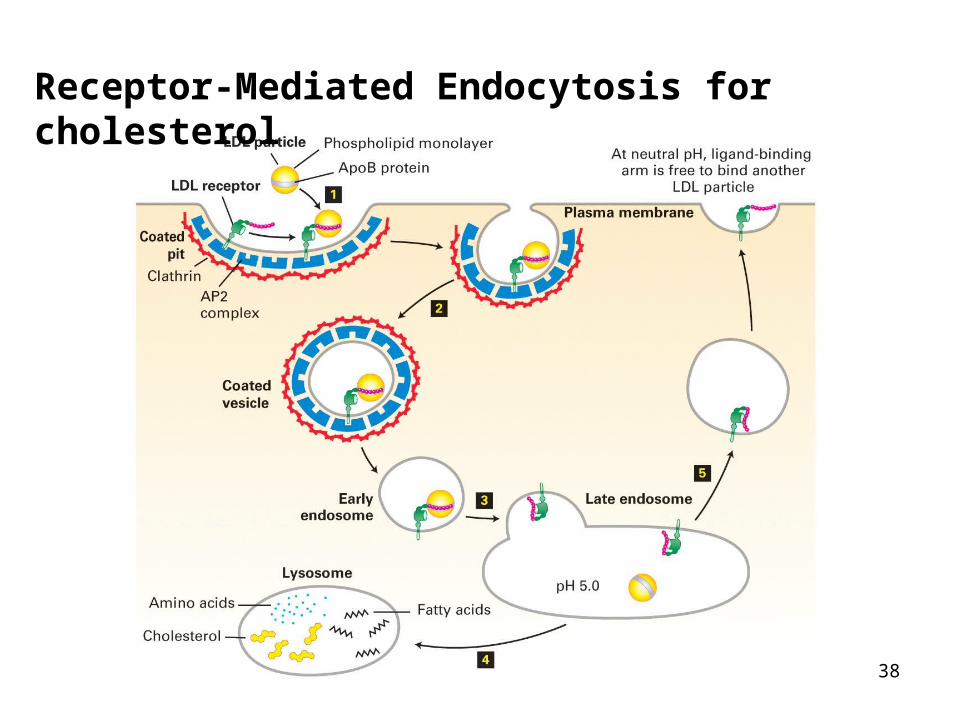
Receptor-Mediated Endocytosis for cholesterol
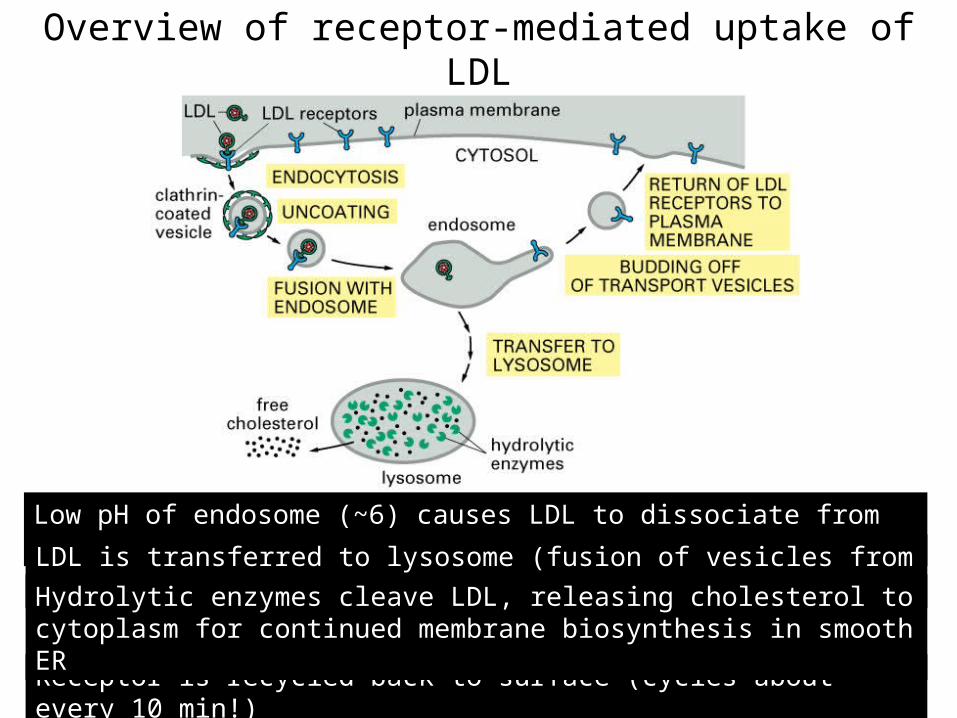
Overview of receptor-mediated uptake of LDL
Low pH of endosome (~6) causes LDL to dissociate from receptor
Receptor is recycled back to surface (cycles about every 10 min!)
LDL is transferred to lysosome (fusion of vesicles from TGN)
Hydrolytic enzymes cleave LDL, releasing cholesterol to cytoplasm for continued membrane biosynthesis in smooth ER
ECB 15-32

Defects in LDL endocytosis are associated with “familial hypercholesterolemia”…
–Severe atherosclerosis at early age (strokes and heart attacks in pre-teens)–Excess LDL in circulating blood–LDL not properly internalized by cells–Recessive/single gene… encoding plasma membrane receptor for LDL (LDL-receptor or LDL-R)
•Disease provided insight into mechanism of receptor-mediated endocytosis and identification/function of LDL-receptor
–Mutations in N-terminal domain: LDL-R doesn’t bind LDL…–Mutations in C-terminal domain: LDL-R is not internalized…
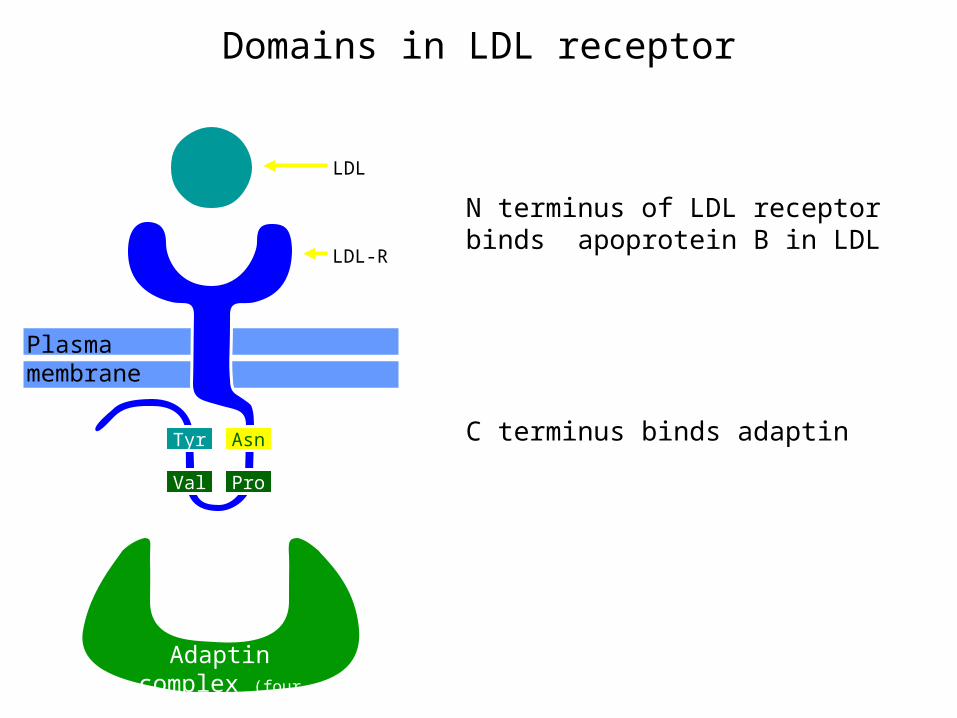
Domains in LDL receptor
Adaptin complex (four polypeptides)
Plasma membrane
Val
Tyr
Pro
Asn
LDL-R
LDL
HOOC
N terminus of LDL receptor binds apoprotein B in LDL
C terminus binds adaptin
NH2
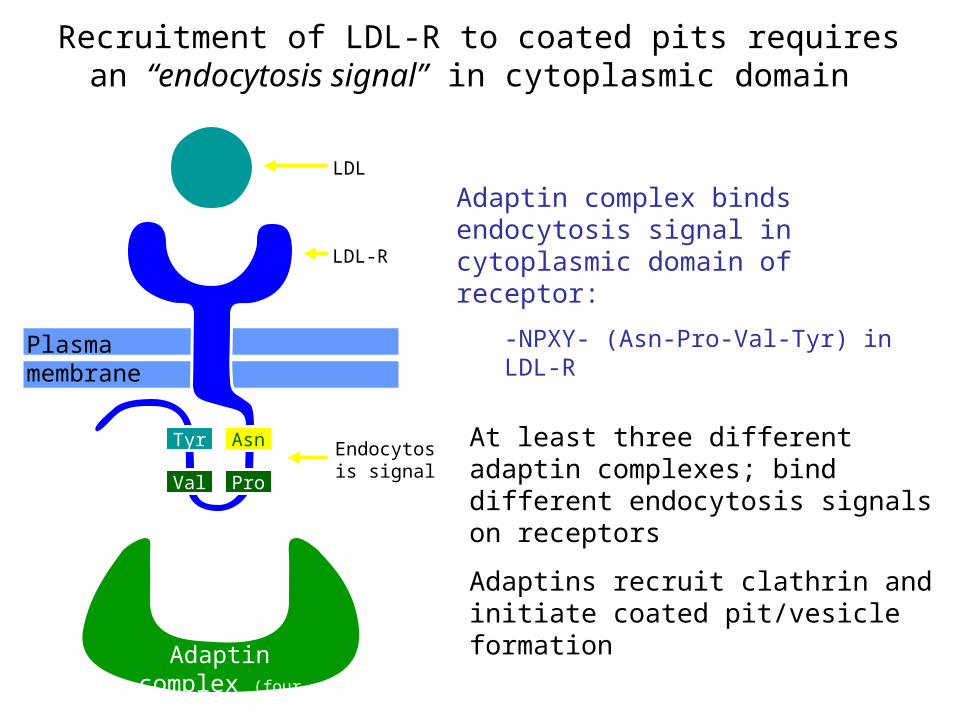
Recruitment of LDL-R to coated pits requires an “endocytosis signal” in cytoplasmic domain
Adaptin complex (four polypeptides)
Plasma membrane
Adaptin complex binds endocytosis signal in cytoplasmic domain of receptor:
-NPXY- (Asn-Pro-Val-Tyr) in LDL-R
At least three different adaptin complexes; bind different endocytosis signals on receptors
Adaptins recruit clathrin and initiate coated pit/vesicle formation
Val
Tyr
Pro
Asn
LDL-R
Endocytosis signal
LDL
Based on MBoC (3) figure 13-53
HOOC
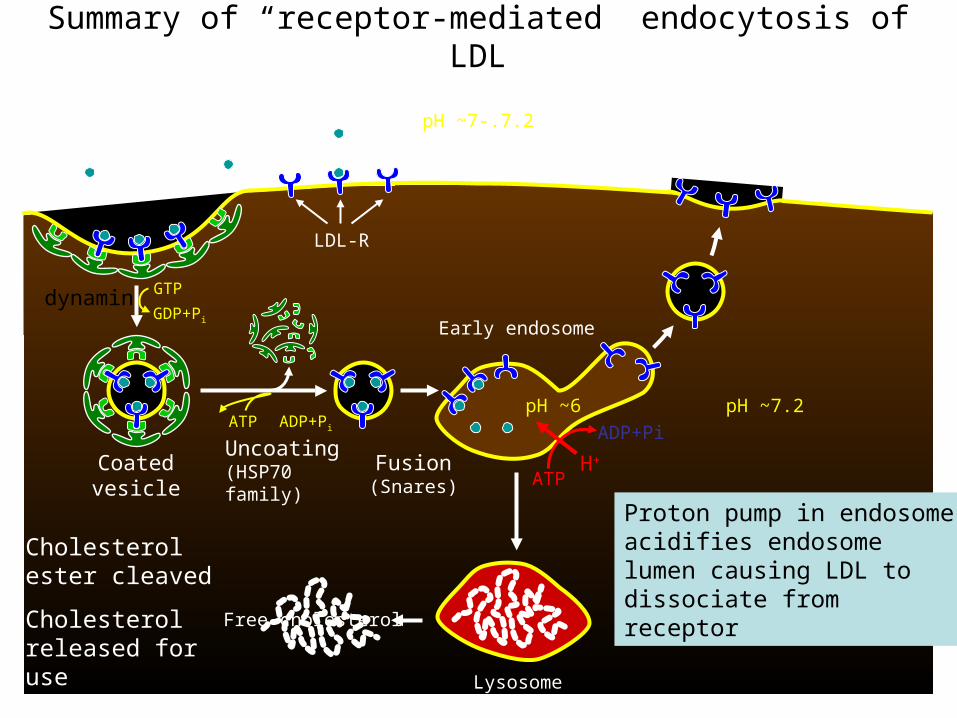
Summary of “receptor-mediated” endocytosis of LDL
ATP
ADP+Pi
H+
Lysosome
Early endosome
ATP ADP+Pi
Uncoating(HSP70 family)
GTP
GDP+Pi
Coatedvesicle
Fusion(Snares)
Cholesterol ester cleaved
Cholesterol released for use
A single receptor makes hundreds of trips (~10 min/cycle)
Free cholesterol
pH ~7.2pH ~6
LDL-R
pH ~7-.7.2Low density lipoprotein (LDL)
Proton pump in endosome acidifies endosome lumen causing LDL to dissociate from receptor
dynamin

Lysosomal storage disease • A group of disorders known as lysosomal storage
diseases are cause by the absence (defficiency) of one or more lysosomal enzymes
• This defficiency leads to several lysosomal storage diseases characterized by accumulation and storage of excessive amounts of substances such as– Polysaccharides– Iipids– Glycosaminoglycans– Glycoproteins and glycolipids– Glycogen etc
• There is impairment of function or cell destruction leading to symptoms such as muscle weakness, skeletal deformities, mental retardation etc

I cell disease
• This is a severe form of lysosomal storage disease in which many enzymes are missing from the lysosome
• Affected individuals lack N- acetyl glucosamine phosphotransferase, the enzyme required for phosphorylating a mannose residue in all lysosomal enzymes in the cis – Golgi so that a receptor can recognize this phosphorylated mannose and direct the enzymes to the lysosomes
• Lack of this phosphorylation results in mistargeting of lysosomal enzymes

I cell disease
• The disease is inherited as an autosomal recessive trait
• Patients have a high level of enzymes in the blood and urine since the enzymes are mislocated ie they are synthesized but are exported instead of being sequestered in the lysosome
• The lysosomes of these individuals contain large inclusions (I cell disease) of undigested glycosaminoglycans and glycolipids due to the fact that eight acid hydrolases required for their degradation are missing from the lysosome
• Patients have severe psycomotor retardation and skeletal deformities

Type II glycogen storage (Pompe’s) disease
• This is a condition which is due to lack of the enzyme α 1 – 4 glucosidase, a hydrolytic enzyme found in lysosomes hat catalyse glycogen hydrolysis into glucose units
• Lysosomes become engorged with glycogen due to lack of this enzyme
• The disease is inherited as an autosomal recessive trait
• Children accumulate excessive amounts of glycogen in the liver, heart and muscles and they have cardiorespiratory failure before 2 years leading to death

Hurlers/Hunter syndrome (mucopolysaccharidosis)
• These syndromes are due to lack of the enzyme α – L – Iduronidase in the lysosome
• Due to lack of this enzyme, there is defects in the degradation of glycosaminoglycans. These are anionic polysaccharide chains which include chondroitin sulfate, keratan sulfate, heparin hyaluronate and darmatan sulfate
• The condition is characterised by bone deformaities

Tay Sachs disease
• This disease is due to lack of one of the enzymes involved in lysosomal breakdown of gangliosides
• The condition is common in Jews and is inherited as an autosomal recessive trait
• The enzyme lacking is β- N-acetyl hexosaminidase A which cleaves off N-acetylgalactosamine residues from carbohydrate portion of gangliosides
• There is accumulation of the ganglioside GM2 in the brain and nerve cells of children affected by this condition (nerve cells are enlarged with swollen GM2 filled lysosomes)
• Affected children normally show symptoms of dementia (mental retardation) and are blind by year 2 and die within 3 years

Fabrys disease• The condition was first described by Fabrys and
Anderson in 1898• In 1963, Seeley and Klonsky described the structure of
Gal-Gal-Glc-Ceramide as the major lipid that accumulate in Fabrys affected kidneys
• In 1967, Roscoe Brady showed that the disease was due to the lack of the enzyme α- galactosidase A, a lysosomal enzyme that degrades the above trihexosyl ceramide
• The characteristic symptoms are skin rash, pain in extremities and renal impairment.
• Patients are normal until into their 40’s when the kidneys fail.
• There is no effective treatment apart from a kidney transplant. However, the disease is rare – only a few hundred cases worldwide