Embed Size (px)
Citation preview

NATURE CHEMISTRY | www.nature.com/naturechemistry 1
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1862
1
A bioinspired redox relay that mimics radical interactions of
the Tyr-His pairs of photosystem II
Jackson D. Megiatto, Jr.1#, Dalvin D. Méndez-Hernández1, Marely E. Tejeda-Ferrari1, Anne-
Lucie Teillout1,3, Manuel J. Llansola-Portolés1, Gerdenis Kodis1, Oleg G. Poluektov2*, Tijana
Rajh2*, Vladimiro Mujica1, Thomas L. Groy1, Devens Gust1*, Thomas A. Moore1* and Ana L.
Moore1*
1Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ 85287-1604, USA E-mail:
[email protected] ; [email protected] ; [email protected]
2 NanoBio Interface Group, Center for Nanoscale Materials and Chemistry Division, Argonne National Laboratory
Argonne, IL 60439, USA E-mail: [email protected]; [email protected]
3 Laboratoire de Chimie Physique, Groupe d'Electrochimie et de Photoélectrochimie, UMR 8000, CNRS, Université
Paris-Sud, Batiment 350, 91405 Orsay Cedex, France.
# Current address: Institute of Chemistry, Campinas State University (UNICAMP), P.O. Box 6154, Campinas, SP,
13084-861, Brazil.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 2
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1862
2
SUPPLEMENTARY INFORMATION
Table of contents :
1 – Materials 2
2 – Nuclear Magnetic Resonance 2
3 – Electron Paramagnetic Resonance (EPR) Section 3
4 – Theoretical Calculations 5
5 – Electrochemical Section 10
6 – Crystal Structure and X-Ray Data 15
7 – References 35
1 – Materials All chemicals were purchased from Aldrich, Alfa Aesar, and Acros and were used
without further purification. Solvents were obtained from EM Science and were used as received
unless otherwise noted.
2 – Nuclear Magnetic Resonance
1H-NMR spectra were recorded on a Varian spectrometer at 500 MHz. NMR samples
were prepared in deuterated solvents with tetramethylsilane as an internal reference using
Wilmad 528-PP 5 mm NMR tubes. Deuterated chloroform was distilled from CaH, whereas
deuterated acetone and acetonitrile were used as received. Chemical shifts (δ) are reported in
parts per million (ppm) and referenced to the residual solvent peak.
2
SUPPLEMENTARY INFORMATION
Table of contents :
1 – Materials 2
2 – Nuclear Magnetic Resonance 2
3 – Electron Paramagnetic Resonance (EPR) Section 3
4 – Theoretical Calculations 5
5 – Electrochemical Section 10
6 – Crystal Structure and X-Ray Data 15
7 – References 35
1 – Materials All chemicals were purchased from Aldrich, Alfa Aesar, and Acros and were used
without further purification. Solvents were obtained from EM Science and were used as received
unless otherwise noted.
2 – Nuclear Magnetic Resonance
1H-NMR spectra were recorded on a Varian spectrometer at 500 MHz. NMR samples
were prepared in deuterated solvents with tetramethylsilane as an internal reference using
Wilmad 528-PP 5 mm NMR tubes. Deuterated chloroform was distilled from CaH, whereas
deuterated acetone and acetonitrile were used as received. Chemical shifts (δ) are reported in
parts per million (ppm) and referenced to the residual solvent peak.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 3
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18623
3 – Electron Paramagnetic Resonance (EPR) Section
X-band EPR spectra were recorded on a Bruker Elexsys E580 spectrometer equipped
with standard Bruker resonator with optical access and a variable-temperature cryostat (Air
Products). Samples at 4.2 K were illuminated directly in the spectrometer cavity using a LX 300
UV xenon lamp (Atlas Specialty Lighting, Tampa, FL) with a 520 nm cut off filter. Temperature
was controlled using Lake Shore temperature controller model 321-Autotuning. The background
EPR signal was measured for each sample before illumination. The g-values were calibrated by
comparison to the Mn2+ standard in a SrO matrix (g = 2.0012 ± 0.0002)1 and coal sample No 4
from Argonne coal collection (g = 2.00285 ± 0.00005).2
Supplementary Figure S1. Photoinduced X-band (9.5 GHz) EPR spectrum, obtained after illumination of an acetonitrile suspension of triad-1 (BiP–PF10–TiO2) with light of wavelengths greater than 520 nm at 4.2 K, modulation amplitude 2 G, power 0.05 mW. Red: signal from BiP–PF10
•+; blue: signals from TiO2•– lattice.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 4
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18624
Supplementary Figure S2. Photoinduced X-band (9.5 GHz) EPR spectra, obtained after illumination of an acetonitrile suspension of triad-1 (red) and dyad-2 (black) with light of wavelengths greater than 520 nm at 4.2 K, modulation amplitude 5 G, power 2 mW. Note higher power and modulation amplitude that was used to enhance signal intensity and signal to noise ratio.
Supplementary Figure S3. Integrated (9.5 GHz) X-band spectrum of triad-1 (BiP–PF10–TiO2) at low power (0.02 mW) to avoid saturation and low modulation amplitude (2 G). The area under each radical species signal is proportional to the number of spins: BiP–PF10
•+, red, field 3270–3336 G and TiO2
•–, blue, field 3336–3450 G. The number of spins (radicals) was determined using calibrated weak pitch in a KCl sample under the same conditions (1.15 × 1013 spins cm-1, Bruker Instruments). It was determined to be 3.8 × 1013 spins cm-1 for BiP–PF10
•+ and 4.5 × 1013 spin cm–1 for TiO2
•–.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 5
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18625
4 – Theoretical Calculations
The theoretical structures were optimized with Gaussian 093 at the B3LYP/6-31G(d,p)4-9
level of theory using the Conductor-like Polarizable Continuum Model (CPCM)10,11 for the
acetonitrile environment. The g-values for all the structures were obtained with ORCA12 at the
B3LYP/EPR-II13 level of theory using the Conductor-Like Screening Model (COSMO)14 to
simulate the acetonitrile environment.
The spin-density population of the structure that corresponds to the carboxylic acid form
of dyad-2 depicted as a radical cation after the proton transfer from the phenol to the
benzimidazole is shown in Supplementary Fig. S4. In agreement with the experimental data, a
very strong intra-molecular hydrogen bond can be inferred from the small dihedral angle (less
than 2°) between the phenol and imidazole planes of the BiP moiety in the optimized structure.
The spin-density distribution reveals that the unpaired electron is delocalized to some extent
between both the porphyrin and the phenol rings. The ratio of the unpaired spin populations
(PF10:BiP) is approximately 35:65. This explains the observation that the gx-value of dyad-2 is
lower than those reported for other BiP systems.15,16
Supplementary Figure S4. Spin density (the orange and green colors show positive and negative spin densities, respectively) for a structure of the carboxylic acid form of dyad-2 as a radical cation after PCET geometry optimized using Gaussian 09 at the B3LYP/6-31G(d,p) level of theory and the Conductor-like Polarizable Continuum Model (CPCM) to include the
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 6
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18626
acetonitrile environment in the calculations. Carbon (grey), hydrogen (white), nitrogen (blue), fluorine (cyan) and oxygen (red).
Supplementary Figure S5. Structures of the carboxylic acid form of dyad-2, as a radical cation after PCET, geometry optimized with Gaussian 09 at the B3LYP/6-31G(d,p) level of theory and the Conductor-like Polarizable Continuum Model (CPCM) to include the acetonitrile environment in the calculations. Explicit water and/or acetonitrile molecules are included where indicated in calculated structures.
Calculations of the anisotropic g-values for the series of radical cation structures
presented in Supplementary Fig. S5 and Supplementary Table S1 were performed following
methodology from the literature.16 Structures with solvent molecules involved in hydrogen bonds
with N–H sites were based on the fact that the distal N–H proton interacts strongly with solvent
molecules as revealed by 1H-NMR studies. Solvent molecules binding to the newly formed N–H
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 7
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18627
bond were chosen based on the expectation of that this new N–H site would behave similarly to
the distal N–H (See Fig. 4 and Fig. 5 in the manuscript and discussion therein).
Supplementary Table S1. Comparison between experimental g values for the carboxylic acid form of dyad-2, as a radical cation, initially formed at 13 K and after annealing at 100 K and returned to 13 K for the measurement in two states and the calculated g values for structures A•+–I•+ obtained with ORCA at the B3LYP/EPRII level of theory and using COSMO to simulate the acetonitrile environment.
gx gy gz
Exp. at 13 K 2.0056 2.0042 2.0022
Exp. after annealing at 100 K 2.0061 2.0042 2.0022
Theoretical Structure A•+ 2.0052 2.0041 2.0024
Theoretical Structure B•+ 2.0060 2.0044 2.0023
Theoretical Structure C•+ 2.0041 2.0032 2.0025
Theoretical Structure D•+ 2.0052 2.0040 2.0024
Theoretical Structure E•+ 2.0060 2.0045 2.0023
Theoretical Structure F•+ 2.0063 2.0044 2.0023
Theoretical Structure G•+ 2.0062 2.0044 2.0023
Theoretical Structure H• 2.0066 2.0047 2.0024
Theoretical Structure I•+ 2.0056 2.0042 2.0024
It is important to clarify that in our model we take into account explicitly two solvent
molecules and the solvent reorganization is defined in the following way. For example, in the
case of A•+ reorganizing to form B•+, we start with a structure in which the two explicit water
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 8
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18628
molecules are near the positions shown in A•+ and then run the geometry optimization program
to yield the actual structure shown as A•+. We then relocate the water molecule near the newly
formed N–H site and run the geometry optimization program again to yield structure B•+. The
change in solvent structure between the two geometry-optimized systems is the reorganization of
the explicit water (solvent). The remaining solvent effect is included using the conventional
dielectric model. Other reorganizations in the system include a change in the BiP dihedral angle
and concomitant changes in the distance between phenoxyl O and N–H site.
The calculated g-values for the radical cation of the carboxylic acid form of dyad-2
without taking into account hydrogen bond formation involving the new (proximal) N–H site and
an explicit solvent molecule yields results that are very close to the experimental g-values
observed at 13 K (Supplementary Table S1 and Supplementary Fig. S5, A•+, D•+ and I•+). This
agreement supports the interpretation that at 13 K the solvent molecules do not have enough
energy to reorganize to form a hydrogen bond with the newly formed (proximal) N–H site. Upon
warming of the solution to 100 K the energy available for solvent reorganization increases and
hydrogen bond formation involving the newly formed (proximal) N–H site is possible resulting
in an increase of the gx-value. The calculated g-values for radical cations including this new
hydrogen bond with solvent molecules are in excellent agreement with those observed at 100 K
(Supplementary Table S1 and Supplementary Fig. S5, B•+, E•+, F•+ and G•+). The calculated gx-
value for a structure before proton transfer (C•+) results in a value much lower than that observed
experimentally even at 13 K. For the case where the distal N–H proton is deprotonated by a base
yielding a neutral radical (H•) the calculated gx-value is much higher than that observed
experimentally even after annealing at 100 K.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 9
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.18629
Supplementary Table S2. Comparison between calculated structural parameters for structures A•+–I•+. The structures are geometry optimized with Gaussian 09 at the B3LYP/6-31G(d,p) level of theory and the Conductor-like Polarizable Continuum Model (CPCM) to include the acetonitrile environment in the calculations. (For structures see Supplementary Fig. S5).
Structure Charge, multiplicity
Number of explicit water
molecules
Number of explicit
acetonitrile molecules
BiP O–H distance
(Å)
BiP N–H distance
(Å)
BiP N–O distance
(Å)
BiP dihedral angle (°)
∆E relative to structure
C•+ (kcal/mol)a
A•+ 1,2 2 0 1.88 1.02 2.62 1.82 -19.05
B•+ 1,2 2 0 2.14 1.03 2.72 7.12 -21.93
C•+ 1,2 0 0 1.02 1.58 2.53 -0.05 Reference
D•+ 1,2 0 0 1.84 1.02 2.60 -1.69 -2.76
E•+ 1,2 1 0 2.34 1.04 2.80 24.4 -10.15
F•+ 1,2 1 1 2.01 1.02 2.67 3.87 -17.85
G•+ 1,2 0 1 1.99 1.02 2.66 2.80 -5.79
H• 0,2 0 0 1.94 1.02 2.65 -0.04 -
I•+ 1,2 1 0 1.86 1.02 2.62 1.55 -14.45
a) ∆E relative to structure C•+ (kcal/mol) was calculated by the following equation:
∆E = E(Structure) – E(C•+) – (E(explicit solvent) × number explicit solvent molecules).
The structure with the highest energy is C•+, corresponding to BiP–PF10•+ before the
proton is transferred from the phenol to the benzimidazole. The energy of this structure (C•+)
was used as the reference to calculate ∆E. Upon proton transfer to the benzimidazole (D•+), the
energy decreases. Adding a hydrogen bond acceptor to the distal N–H of the BiP further lowers
the energy (I•+). Addition of a water molecule hydrogen bonded to the nearby the N–H.....O bond
of the BiP stabilizes the system further (A•+ and B•+), but the gx-value increases only when a
bifurcated hydrogen bond involving the newly formed N–H site is formed (B•+). Structure E•+
shows the effect of water hydrogen bonded to the newly formed N–H site without the distal
water, and structures F•+ and G•+ show the effect of acetonitrile hydrogen bonded to the newly
formed N–H site with and without the distal water. Notice that acetonitrile is not hydrogen
bonded to the distal N–H in any of the structures. Due to steric considerations when an
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 10
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186210
acetonitrile molecule is included at this site the structures cannot be geometry optimized without
a serious distortion of the porphyrin macrocycle.
5 – Electrochemical Section
Cyclic voltammetry was performed with a CHI 650C potentiostat (CH Instrument) with
scan rates ranging from 0.1 V/s and 0.5V/s. For scan rates faster than 1 V/s, cyclic voltammetry
was performed with a potentiostat equipped with EG&G Parc Model 175 and Princeton applied
research Model 176 current follower. A custom glass cell with Teflon top accommodating a
three electrode setup and 1–2 mL of solution was used. The working electrode was either a
platinum (1.5 mm diameter) or a glassy carbon (1 mm diameter) disc electrode as indicated for
the particular experiment. The working electrode was cleaned between experiments by polishing
with alumina (50 mm diameter) slurry followed by solvent rinses. The counter electrode was a
platinum grid. The potential of the pseudoreference electrode was determined using the
ferrocenium-ferrocene redox couple as an internal standard (with Em taken as 0.45 V vs. SCE in
acetonitrile). The compound of interest was dissolved in acetonitrile (Aldrich, 99.99%) to yield a
solution with concentration between 4 × 10-4 and 8 × 10-4, to which was added
tetrabutylammonium hexafluorophosphate at a concentration of 100 mM as a supporting
electrolyte for all experiments. Prior to any experiment, the solution was purged with argon for 2
minutes and then run under an argon atmosphere.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 11
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186211
Supplementary Figure S6. Cyclic voltammetry of the porphyrin model compound S6 lacking the BIP moiety. The voltammogram was taken in acetonitrile with a platinum working electrode. and at a scan rate of 100 mV s-1. Sequential reductions of the porphyrin core occurred at -0.91 V and -1.36 V vs. SCE, and an oxidation was observed at 1.29 V vs. SCE.
-2.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0-1.5x10-5
-1.0x10-5
-5.0x10-6
0.0
5.0x10-6
1.0x10-5
1.5x10-5
Curr
ent i
(am
p.)
Potential E (Volts, vs. SCE)
Fc/Fc+
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 12
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186212
Supplementary Figure S7. Cyclic voltammetry of dyad-2. The voltammogram was taken in acetonitrile with a platinum working electrode and at a scan rate of 100 mV s-1. Sequential reductions of the porphyrin core occurred at -1.02 V and -1.52 V vs. SCE. An oxidation peak was observed at 1.00 V vs. SCE corresponding to the BIP group, whereas the porphyrin core oxidation took place at about 1.65 V vs. SCE.
-2.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0-1.0x10-5-8.0x10-6-6.0x10-6-4.0x10-6-2.0x10-6
0.02.0x10-64.0x10-66.0x10-68.0x10-61.0x10-51.2x10-51.4x10-5
Curr
ent i
(Am
p.)
Potential E (Volts vs. SCE)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 13
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186213
Supplementary Table S3: Redox potentials (V vs. SCE) for reduction and oxidation of the compounds shown on the left. The peak to peak separation (mV) is listed in parentheses for redox processes exhibiting either reversible, or quasi-reversible electrode kinetics.
a) E = Epeak irreversible process.
b) E = E1/2 reversible or quasi-reversible process; numbers between parentheses are given in mV
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 14
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186214
5.1 – Electrochemical Kinetic Study17-19 Variable scan rate cyclic voltammetry studies of dyad-2 on glassy carbon and gold
electrodes at scan rates varying from 0.1 to 200 V/s (Supplementary Fig. S8) in acetonitrile
reveal that the phenol oxidation is reversible with the apparent standard potentials independent of
the scan rate in the range investigated. These findings indicate an extremely fast phenolic proton
transfer reaction to the benzimidazole moiety upon electrochemical oxidation of dyad-2. The
reversibility of the phenoxyl/phenol couple in dyad-2 at high scan rates clearly reveals the ability
of the proton to easily shuttle between the phenolic oxygen and the nitrogen atom of the
benzimidazole in the corresponding Bi–PhOH reduced and BiH+–PhO• oxidized forms.
However, further variable scan rate cyclic voltammetry experiments with dyad-2 dissolved in
acetonitrile containing 2% (v/v) of either regular or deuterated methanol, followed by
comparison of the experimental data with those afforded by digital simulations using the
DigiSim software for estimation of the apparent rate constants in the two solvent systems (kappH
= 0.054 ± 0.009 and kappD = 0.056 ± 0.006 in regular and deuterated methanol, respectively),
disclose a negligible kinetic isotope effect (KIE) for the electrochemical oxidation of the phenol.
Supplementary Figure S8. Left: normalized cyclic voltammograms (i is the current and v is the scan rate) of the BIP group PCET of the dyad-2 at a glassy carbon electrode (1mm diameter) in ACN + 2% MeOD at 10 V/s (black), 50 V/s (red), 100 V/s (blue), 200 V/s (green). Right: Anodic (upper symbol) and cathodic (lower symbol) peak potentials as a function of the log of
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 15
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186215
scan rate. Simulation of the ‘‘trumpet plot’’ (full line) leads to the determination of the apparent standard rate constant ( from the peak current density:
, where is the electroactive species bulk concentration). In ACN + 2% MeOH, experimental peak potential (red circle) and corresponding simulation (red full line). In ACN + 2% MeOD, experimental peak potential (black square) and corresponding simulation (grey full line).
6 – Crystal Structure and X-Ray Data
A clear dark red needle-like specimen of C57H34F10N6O3, trimmed to approximate
dimensions 0.07 mm × 0.21 mm × 0.48 mm, was used for the X-ray crystallographic analysis.
The X-ray intensity data were measured as indicated in Supplementary Table S4 below with a
Bruker SMART APEX 2D CCD diffractometer.
Supplementary Table S4: Data collection details for dyad-2.
Axis dx/mm 2θ/° ω/° φ/° χ/° Width/° Frames Time/s Wavelength/Å Voltage/kV Current/mA Temperature/K
Omega 50.210 -34.00 -34.00 0.00 54.78 0.50 364 120.00 0.71073 50 30.0 123
Omega 50.210 -34.00 -34.00 120.00 54.78 0.50 364 120.00 0.71073 50 30.0 123
Omega 50.210 -34.00 -34.00 240.00 54.78 0.50 364 120.00 0.71073 50 30.0 123
A total of 1092 frames were collected. The total exposure time was 36.40 hours. The
frames were integrated with the Bruker SAINT20 software package using a narrow-frame
algorithm. The integration of the data using a monoclinic unit cell yielded a total of 47693
reflections to a maximum θ angle of 25.10° (0.84 Å resolution), of which 10916 were
independent (average redundancy 4.369, completeness = 99.4%, Rint = 9.06%, Rsig = 8.57%) and
5465 (50.06%) were greater than 2σ(F2). The final cell constants of a = 19.934(4) Å, b =
10.691(2) Å, c = 29.550(7) Å, β = 101.580(3)°, volume = 6169.(2) Å3, are based upon the
refinement of the XYZ-centroids of 3448 reflections above 20 σ(I) with 4.434° < 2θ < 40.09°.
Data were corrected for absorption effects using the multi-scan method (SADABS).21 The ratio
of minimum to maximum apparent transmission was 0.762. The calculated minimum and
maximum transmission coefficients (based on crystal size) are 0.9573 and 0.9941. CCDC
deposition number 949079.
126 .10.5 −−= scmDRTFDvCFjp
∞= 446.0/ ∞C
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 16
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186216
The structure was solved and refined using the Bruker SHELXTL22 Software Package,
using the space group P 1 2/c 1, with Z = 4 for the formula unit, C57H33F10N6O3. After fruitless
attempts at modeling the disordered solvent in the structure, the PLATON SQUEEZE23
algorithm was applied to the partially solved structure to produce a solvent-free data set upon
which the final refinements were performed. There were no significant changes in the structure
of the primary molecule between the original solution and the final solvent-free model. The final
anisotropic full-matrix least-squares refinement on F2 with 693 variables converged at R1 =
7.74%, for the observed data and wR2 = 22.76% for all data. The goodness-of-fit was 0.978. The
largest peak in the final difference electron density synthesis was 0.323 e-/Å3 and the largest hole
was -0.395 e-/Å3 with an RMS deviation of 0.063 e-/Å3. On the basis of the final model, the
calculated density was 1.120 g/cm3 and F(000), 2124 e-. The depictions of the crystallographic
structure were produced by the Bruker XShell24 display software, which is illustrated in
Supplementary Fig. S9. When run through IUCR's CheckCIF routine, the crystal structure .cif
files produced 2 Level B alerts. These were on account of minor rotational disorder about the
axis defined by C5 and C15, as indicated by the elongated thermal parameters perpendicular to
this axis.
Supplementary Figure S9. Thermal ellipsoid plot of dyad-2 shown at the 50% probability level. Hydrogen atoms have been omitted for clarity.
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 17
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186217
Supplementary Table S5. Sample and crystal data for dyad-2.
Identification code Megiatto_Compound_2
Chemical formula C57H33F10N6O3
Formula weight 1039.89
Temperature 123(2) K
Wavelength 0.71073 Å
Crystal size 0.07 x 0.21 x 0.48 mm
Crystal habit clear dark red needle
Crystal system Monoclinic
Space group P 1 2/c 1
Unit cell dimensions a = 19.934(4) Å α = 90°
b = 10.691(2) Å β = 101.580(3)°
c = 29.550(7) Å γ = 90°
Volume 6169.(2) Å3
Z 4
Density (calculated) 1.120 Mg/cm3
Absorption coefficient 0.091 mm-1
F(000) 2124
Supplementary Table S6. Data collection and structure refinement for dyad-2.
Theta range for data collection 1.90 to 25.10°
Index ranges -23<=h<=23, -12<=k<=12, -35<=l<=34
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 18
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186218
Reflections collected 47693
Independent reflections 10916 [R(int) = 0.0906]
Coverage of independent reflections 99.4%
Absorption correction multi-scan
Max. and min. transmission 0.9941 and 0.9573
Structure solution technique direct methods
Structure solution program SHELXS-97 (Sheldrick, 2008)
Refinement method Full-matrix least-squares on F2
Refinement program SHELXL-97 (Sheldrick, 2008)
Function minimized Σ w(Fo2 - Fc
2)2
Data / restraints / parameters 10916 / 0 / 693
Goodness-of-fit on F2 0.978
Δ/σmax 0.001
Δ/σmean 0.000
Final R indices 5465 data; I>2σ(I) R1 = 0.0774, wR2 = 0.1946
all data R1 = 0.1501, wR2 = 0.2276
Weighting scheme w=1/[σ2(Fo2)+(0.1209P)2+0.0000P]
where P=(Fo2+2Fc
2)/3
Largest diff. peak and hole 0.323 and -0.395 eÅ-3
R.M.S. deviation from mean 0.063 eÅ-3
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 19
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186219
Supplementary Table S7. Atomic coordinates and equivalent isotropic atomic displacement parameters (Å2) for dyad-2.
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
x/a y/b z/c U(eq)
F1 0.73621(13) 0.5834(3) 0.91905(10) 0.0702(8)
F2 0.76968(15) 0.5159(3) 0.83868(11) 0.0934(10)
F3 0.76791(13) 0.6877(4) 0.77048(9) 0.0948(11)
F4 0.73458(12) 0.9256(3) 0.78366(9) 0.0833(10)
F5 0.70078(12) 0.9944(3) 0.86411(9) 0.0693(8)
F6 0.61751(14) 0.3111(3) 0.15356(10) 0.0752(8)
F7 0.59374(17) 0.4134(4) 0.23226(12) 0.1133(13)
F8 0.56954(13) 0.2663(4) 0.30166(9) 0.1165(14)
F9 0.57048(13) 0.0164(4) 0.29221(9) 0.1066(13)
F10 0.59588(13) 0.9121(3) 0.21475(9) 0.0754(8)
O1 0.10407(12) 0.1967(3) 0.13472(9) 0.0496(7)
O2 0.25113(16) 0.4226(3) 0.96729(17) 0.1049(16)
O3 0.18876(14) 0.5911(3) 0.96226(12) 0.0664(9)
N1 0.61115(13) 0.8308(3) 0.98553(10) 0.0360(8)
N2 0.57835(14) 0.9298(3) 0.06902(10) 0.0367(8)
N3 0.71975(14) 0.0171(3) 0.09885(10) 0.0370(8)
N4 0.74885(14) 0.9359(3) 0.01217(10) 0.0381(8)
N5 0.07442(15) 0.3964(3) 0.08522(12) 0.0501(9)
N6 0.96380(17) 0.4479(3) 0.05989(13) 0.0559(10)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 20
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186220
C1 0.63648(18) 0.7994(4) 0.94720(13) 0.0411(10)
C2 0.58396(18) 0.7292(4) 0.91677(14) 0.0449(10)
C3 0.53004(18) 0.7203(4) 0.93744(13) 0.0439(10)
C4 0.54524(17) 0.7834(3) 0.98070(12) 0.0360(9)
C5 0.50145(17) 0.8039(3) 0.01123(13) 0.0348(9)
C6 0.51695(17) 0.8771(4) 0.05146(12) 0.0354(9)
C7 0.46872(19) 0.9084(4) 0.08045(13) 0.0419(10)
C8 0.50212(19) 0.9782(4) 0.11512(13) 0.0446(10)
C9 0.57162(18) 0.9895(4) 0.10855(13) 0.0379(9)
C10 0.62423(18) 0.0499(4) 0.13967(13) 0.0414(10)
C11 0.69285(18) 0.0594(4) 0.13503(13) 0.0402(10)
C12 0.74705(19) 0.1169(4) 0.16806(14) 0.0491(11)
C13 0.80484(19) 0.1097(4) 0.15069(13) 0.0452(10)
C14 0.78811(17) 0.0501(4) 0.10661(13) 0.0371(9)
C15 0.83162(17) 0.0312(3) 0.07579(13) 0.0361(9)
C16 0.81285(17) 0.9806(3) 0.03143(13) 0.0368(9)
C17 0.85827(18) 0.9634(4) 0.99992(13) 0.0409(10)
C18 0.82205(18) 0.9111(4) 0.96123(14) 0.0431(10)
C19 0.75312(17) 0.8910(4) 0.96973(14) 0.0407(10)
C20 0.70114(18) 0.8291(4) 0.93913(13) 0.0447(10)
C21 0.71696(18) 0.7906(5) 0.89388(14) 0.0471(11)
C22 0.7349(2) 0.6710(5) 0.88628(16) 0.0580(13)
C23 0.7516(2) 0.6340(6) 0.8450(2) 0.0648(14)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 21
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186221
C24 0.7510(2) 0.7197(7) 0.81121(17) 0.0685(16)
C25 0.7348(2) 0.8406(7) 0.81780(16) 0.0664(15)
C26 0.71716(19) 0.8739(5) 0.85862(15) 0.0514(12)
C27 0.60716(18) 0.1078(5) 0.18134(14) 0.0464(11)
C28 0.6064(2) 0.2345(5) 0.18734(16) 0.0580(13)
C29 0.5936(2) 0.2882(7) 0.2277(2) 0.0776(17)
C30 0.5814(2) 0.2158(8) 0.26277(19) 0.079(2)
C31 0.5816(2) 0.0912(8) 0.25766(16) 0.078(2)
C32 0.5945(2) 0.0384(6) 0.21752(16) 0.0591(13)
C33 0.43409(17) 0.7365(4) 0.00109(12) 0.0374(9)
C34 0.3737(2) 0.7991(4) 0.98501(17) 0.0711(15)
C35 0.3125(2) 0.7333(5) 0.97526(18) 0.0707(15)
C36 0.30997(18) 0.6085(4) 0.98136(13) 0.0401(10)
C37 0.3702(2) 0.5460(5) 0.9983(2) 0.0772(17)
C38 0.4305(2) 0.6124(4) 0.00863(19) 0.0705(16)
C39 0.2437(2) 0.5414(4) 0.96952(14) 0.0460(11)
C40 0.1876(3) 0.3510(5) 0.9577(3) 0.145(4)
C41 0.90471(17) 0.0755(4) 0.09105(12) 0.0371(9)
C42 0.95144(18) 0.0037(4) 0.12105(13) 0.0414(10)
C43 0.01904(18) 0.0421(4) 0.13683(13) 0.0417(10)
C44 0.03811(17) 0.1571(4) 0.12131(13) 0.0385(10)
C45 0.99067(18) 0.2325(4) 0.09248(13) 0.0408(10)
C46 0.92391(18) 0.1890(4) 0.07797(13) 0.0425(10)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 22
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186222
C47 0.06948(19) 0.9617(4) 0.17212(14) 0.0522(11)
C48 0.0364(2) 0.8415(5) 0.18425(19) 0.0831(18)
C49 0.0926(2) 0.0386(5) 0.21686(15) 0.0667(14)
C50 0.1309(2) 0.9256(5) 0.15242(17) 0.0645(13)
C51 0.0096(2) 0.3572(4) 0.07912(14) 0.0470(11)
C52 0.0025(2) 0.5523(4) 0.05325(17) 0.0582(12)
C53 0.9842(2) 0.6704(5) 0.03606(19) 0.0752(15)
C54 0.0367(3) 0.7536(5) 0.03484(18) 0.0709(14)
C55 0.1048(2) 0.7210(5) 0.04931(17) 0.0639(13)
C56 0.1224(2) 0.6042(4) 0.06656(16) 0.0569(12)
C57 0.0707(2) 0.5196(4) 0.06943(15) 0.0506(11)
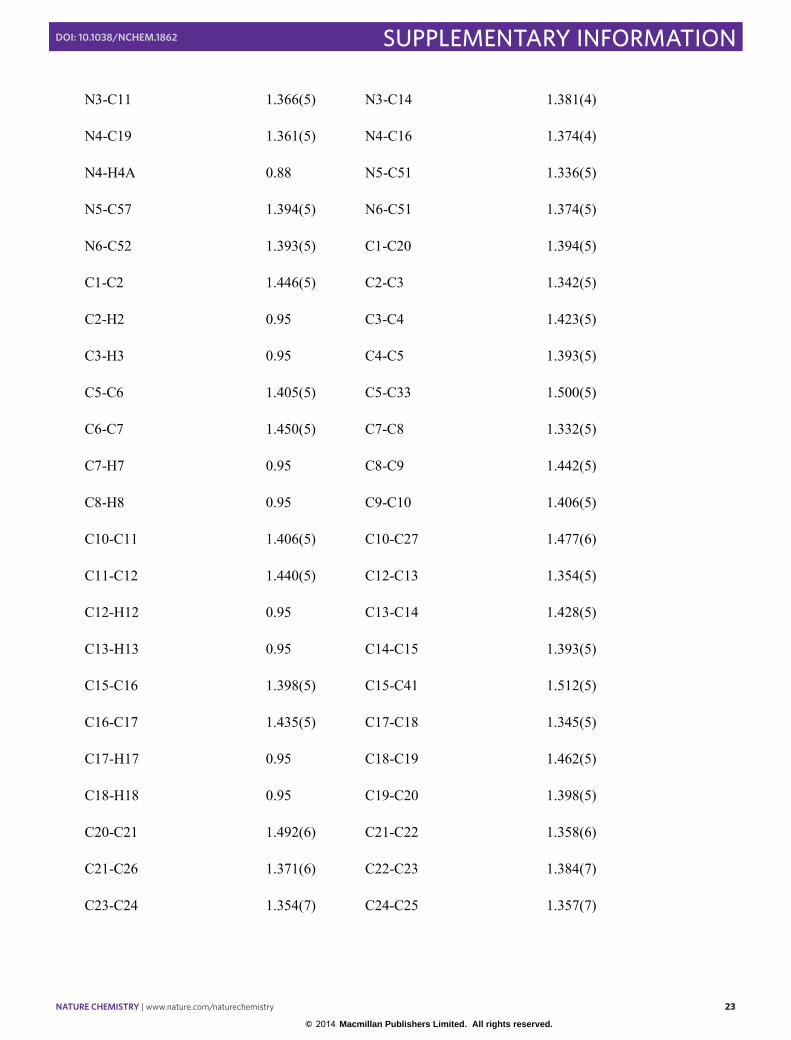
Supplementary Table S8. Bond lengths (Å) for dyad-2.
F1-C22 1.344(5) F2-C23 1.337(6)
F3-C24 1.357(5) F4-C25 1.357(6)
F5-C26 1.347(5) F6-C28 1.343(5)
F7-C29 1.344(7) F8-C30 1.333(6)
F9-C31 1.350(7) F10-C32 1.354(6)
O1-C44 1.362(4) O1-H1 0.84
O2-C39 1.281(5) O2-C40 1.459(5)
O3-C39 1.198(5) N1-C1 1.371(5)
N1-C4 1.388(4) N2-C6 1.353(4)
N2-C9 1.361(5) N2-H2A 0.88
© 2014 Macmillan Publishers Limited. All rights reserved.
NATURE CHEMISTRY | www.nature.com/naturechemistry 23
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186223
N3-C11 1.366(5) N3-C14 1.381(4)
N4-C19 1.361(5) N4-C16 1.374(4)
N4-H4A 0.88 N5-C51 1.336(5)
N5-C57 1.394(5) N6-C51 1.374(5)
N6-C52 1.393(5) C1-C20 1.394(5)
C1-C2 1.446(5) C2-C3 1.342(5)
C2-H2 0.95 C3-C4 1.423(5)
C3-H3 0.95 C4-C5 1.393(5)
C5-C6 1.405(5) C5-C33 1.500(5)
C6-C7 1.450(5) C7-C8 1.332(5)
C7-H7 0.95 C8-C9 1.442(5)
C8-H8 0.95 C9-C10 1.406(5)
C10-C11 1.406(5) C10-C27 1.477(6)
C11-C12 1.440(5) C12-C13 1.354(5)
C12-H12 0.95 C13-C14 1.428(5)
C13-H13 0.95 C14-C15 1.393(5)
C15-C16 1.398(5) C15-C41 1.512(5)
C16-C17 1.435(5) C17-C18 1.345(5)
C17-H17 0.95 C18-C19 1.462(5)
C18-H18 0.95 C19-C20 1.398(5)
C20-C21 1.492(6) C21-C22 1.358(6)
C21-C26 1.371(6) C22-C23 1.384(7)
C23-C24 1.354(7) C24-C25 1.357(7)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 24
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186224
C25-C26 1.369(6) C27-C32 1.366(6)
C27-C28 1.366(6) C28-C29 1.392(7)
C29-C30 1.355(8) C30-C31 1.342(8)
C31-C32 1.383(7) C33-C38 1.349(6)
C33-C34 1.376(5) C34-C35 1.388(6)
C34-H34 0.95 C35-C36 1.349(6)
C35-H35 0.95 C36-C37 1.378(5)
C36-C39 1.482(5) C37-C38 1.375(6)
C37-H37 0.95 C38-H38 0.95
C40-H40A 0.98 C40-H40B 0.98
C40-H40C 0.98 C41-C46 1.352(5)
C41-C42 1.382(5) C42-C43 1.396(5)
C42-H42 0.95 C43-C44 1.392(5)
C43-C47 1.554(5) C44-C45 1.395(5)
C45-C46 1.394(5) C45-C51 1.461(5)
C46-H46 0.95 C47-C50 1.507(6)
C47-C48 1.520(6) C47-C49 1.546(6)
C48-H48A 0.98 C48-H48B 0.98
C48-H48C 0.98 C49-H49A 0.98
C49-H49B 0.98 C49-H49C 0.98
C50-H50A 0.98 C50-H50B 0.98
C50-H50C 0.98 C52-C53 1.383(6)
C52-C57 1.392(6) C53-C54 1.379(6)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 25
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186225
C53-H53 0.95 C54-C55 1.384(6)
C54-H54 0.95 C55-C56 1.368(6)
C55-H55 0.95 C56-C57 1.386(6)
C56-H56 0.95
Supplementary Table S9. Bond angles (°) for dyad-2.
C44-O1-H1 109.5 C39-O2-C40 115.2(4)
C1-N1-C4 108.9(3) C6-N2-C9 106.7(3)
C6-N2-H2A 126.7 C9-N2-H2A 126.7
C11-N3-C14 108.4(3) C19-N4-C16 106.2(3)
C19-N4-H4A 126.9 C16-N4-H4A 126.9
C51-N5-C57 105.4(3) C51-N6-C52 106.4(3)
N1-C1-C20 126.3(3) N1-C1-C2 107.4(3)
C20-C1-C2 126.2(4) C3-C2-C1 107.5(4)
C3-C2-H2 126.3 C1-C2-H2 126.3
C2-C3-C4 109.2(3) C2-C3-H3 125.4
C4-C3-H3 125.4 N1-C4-C5 125.4(3)
N1-C4-C3 107.0(3) C5-C4-C3 127.5(3)
C4-C5-C6 125.2(3) C4-C5-C33 116.5(3)
C6-C5-C33 118.2(3) N2-C6-C5 125.8(3)
N2-C6-C7 109.5(3) C5-C6-C7 124.7(3)
C8-C7-C6 107.2(3) C8-C7-H7 126.4
C6-C7-H7 126.4 C7-C8-C9 106.8(4)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 26
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186226
C7-C8-H8 126.6 C9-C8-H8 126.6
N2-C9-C10 126.0(3) N2-C9-C8 109.8(3)
C10-C9-C8 124.1(4) C11-C10-C9 125.6(4)
C11-C10-C27 116.2(3) C9-C10-C27 118.1(3)
N3-C11-C10 126.5(3) N3-C11-C12 108.2(3)
C10-C11-C12 125.3(4) C13-C12-C11 107.4(4)
C13-C12-H12 126.3 C11-C12-H12 126.3
C12-C13-C14 108.1(3) C12-C13-H13 126.0
C14-C13-H13 126.0 N3-C14-C15 125.3(3)
N3-C14-C13 107.9(3) C15-C14-C13 126.8(3)
C14-C15-C16 125.8(3) C14-C15-C41 116.7(3)
C16-C15-C41 117.5(3) N4-C16-C15 125.1(3)
N4-C16-C17 109.9(3) C15-C16-C17 125.0(3)
C18-C17-C16 107.7(3) C18-C17-H17 126.2
C16-C17-H17 126.2 C17-C18-C19 106.1(3)
C17-C18-H18 126.9 C19-C18-H18 126.9
N4-C19-C20 126.0(3) N4-C19-C18 109.9(3)
C20-C19-C18 124.0(4) C1-C20-C19 126.0(4)
C1-C20-C21 117.4(3) C19-C20-C21 116.6(3)
C22-C21-C26 116.4(4) C22-C21-C20 121.2(4)
C26-C21-C20 122.4(4) F1-C22-C21 120.3(4)
F1-C22-C23 117.5(5) C21-C22-C23 122.2(5)
F2-C23-C24 119.8(5) F2-C23-C22 120.9(5)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 27
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186227
C24-C23-C22 119.2(5) C23-C24-C25 120.4(5)
C23-C24-F3 121.2(6) C25-C24-F3 118.4(6)
C24-C25-F4 119.8(5) C24-C25-C26 119.0(5)
F4-C25-C26 121.2(6) F5-C26-C25 117.8(5)
F5-C26-C21 119.4(4) C25-C26-C21 122.8(5)
C32-C27-C28 115.4(5) C32-C27-C10 122.3(4)
C28-C27-C10 122.2(4) F6-C28-C27 120.0(4)
F6-C28-C29 118.1(5) C27-C28-C29 121.9(6)
F7-C29-C30 119.2(6) F7-C29-C28 120.0(6)
C30-C29-C28 120.8(6) F8-C30-C31 120.3(7)
F8-C30-C29 121.3(7) C31-C30-C29 118.4(6)
C30-C31-F9 119.9(6) C30-C31-C32 120.5(6)
F9-C31-C32 119.6(7) F10-C32-C27 119.1(5)
F10-C32-C31 117.9(5) C27-C32-C31 123.0(6)
C38-C33-C34 117.6(4) C38-C33-C5 121.0(3)
C34-C33-C5 121.4(4) C33-C34-C35 119.8(4)
C33-C34-H34 120.1 C35-C34-H34 120.1
C36-C35-C34 121.8(4) C36-C35-H35 119.1
C34-C35-H35 119.1 C35-C36-C37 118.4(4)
C35-C36-C39 120.2(4) C37-C36-C39 121.4(4)
C38-C37-C36 119.3(4) C38-C37-H37 120.4
C36-C37-H37 120.4 C33-C38-C37 123.0(4)
C33-C38-H38 118.5 C37-C38-H38 118.5
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 28
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186228
O3-C39-O2 122.7(4) O3-C39-C36 124.6(4)
O2-C39-C36 112.7(4) O2-C40-H40A 109.5
O2-C40-H40B 109.5 H40A-C40-H40B 109.5
O2-C40-H40C 109.5 H40A-C40-H40C 109.5
H40B-C40-H40C 109.5 C46-C41-C42 119.3(3)
C46-C41-C15 120.5(3) C42-C41-C15 120.1(3)
C41-C42-C43 122.2(4) C41-C42-H42 118.9
C43-C42-H42 118.9 C44-C43-C42 117.3(3)
C44-C43-C47 121.8(3) C42-C43-C47 120.9(4)
O1-C44-C43 119.3(3) O1-C44-C45 119.6(3)
C43-C44-C45 121.0(3) C46-C45-C44 118.9(4)
C46-C45-C51 120.2(4) C44-C45-C51 120.8(3)
C41-C46-C45 121.3(4) C41-C46-H46 119.4
C45-C46-H46 119.4 C50-C47-C48 107.4(4)
C50-C47-C49 109.9(3) C48-C47-C49 108.5(4)
C50-C47-C43 110.4(4) C48-C47-C43 111.9(3)
C49-C47-C43 108.7(4) C47-C48-H48A 109.5
C47-C48-H48B 109.5 H48A-C48-H48B 109.5
C47-C48-H48C 109.5 H48A-C48-H48C 109.5
H48B-C48-H48C 109.5 C47-C49-H49A 109.5
C47-C49-H49B 109.5 H49A-C49-H49B 109.5
C47-C49-H49C 109.5 H49A-C49-H49C 109.5
H49B-C49-H49C 109.5 C47-C50-H50A 109.5
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 29
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186229
C47-C50-H50B 109.5 H50A-C50-H50B 109.5
C47-C50-H50C 109.5 H50A-C50-H50C 109.5
H50B-C50-H50C 109.5 N5-C51-N6 112.3(4)
N5-C51-C45 123.0(4) N6-C51-C45 124.8(3)
C53-C52-C57 121.5(4) C53-C52-N6 132.1(4)
C57-C52-N6 106.3(4) C54-C53-C52 116.9(5)
C54-C53-H53 121.5 C52-C53-H53 121.5
C53-C54-C55 122.2(5) C53-C54-H54 118.9
C55-C54-H54 118.9 C56-C55-C54 120.5(4)
C56-C55-H55 119.8 C54-C55-H55 119.8
C55-C56-C57 118.7(4) C55-C56-H56 120.6
C57-C56-H56 120.6 C56-C57-C52 120.1(4)
C56-C57-N5 130.3(4) C52-C57-N5 109.5(3)
Supplementary Table S10. Anisotropic atomic displacement parameters (Å2) for dyad-2.
The anisotropic atomic displacement factor exponent takes the form: -2π2[ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
U11 U22 U33 U23 U13 U12
F1 0.0603(18) 0.0771(19) 0.0736(19) -0.0158(17) 0.0146(14) 0.0057(14)
F2 0.075(2) 0.121(3) 0.085(2) -0.035(2) 0.0167(17) 0.0020(19)
F3 0.0479(17) 0.183(3) 0.0509(17) -0.0312(19) 0.0045(13) -0.0095(18)
F4 0.0366(15) 0.164(3) 0.0444(15) 0.0104(19) -0.0046(11) -0.0140(16)
F5 0.0440(15) 0.101(2) 0.0570(17) 0.0143(16) -0.0033(12) -0.0008(15)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 30
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186230
U11 U22 U33 U23 U13 U12
F6 0.0703(19) 0.0764(19) 0.0712(19) -0.0104(16) -0.0045(15) -0.0005(15)
F7 0.086(2) 0.137(3) 0.101(3) -0.060(2) -0.0182(19) 0.029(2)
F8 0.0394(16) 0.253(4) 0.0488(17) -0.059(2) -0.0099(13) 0.034(2)
F9 0.0397(16) 0.239(4) 0.0403(16) 0.014(2) 0.0050(12) 0.000(2)
F10 0.0508(17) 0.123(3) 0.0499(16) 0.0180(17) 0.0028(12) -0.0166(16)
O1 0.0232(14) 0.072(2) 0.0478(17) 0.0025(16) -0.0063(12) -0.0137(13)
O2 0.039(2) 0.042(2) 0.210(5) 0.026(2) -0.030(2) -0.0142(15)
O3 0.0216(16) 0.063(2) 0.104(3) 0.0109(18) -0.0112(15) -0.0039(14)
N1 0.0161(15) 0.053(2) 0.0373(18) 0.0037(16) 0.0003(13) -0.0006(14)
N2 0.0174(16) 0.050(2) 0.0408(19) 0.0037(16) 0.0014(13) -0.0043(14)
N3 0.0203(16) 0.055(2) 0.0331(18) 0.0083(16) -0.0010(13) -0.0044(14)
N4 0.0140(15) 0.061(2) 0.0368(19) 0.0065(17) -0.0020(13) -0.0035(14)
N5 0.0258(19) 0.065(3) 0.056(2) 0.0045(19) -0.0019(15) -0.0152(16)
N6 0.0310(19) 0.056(2) 0.076(3) 0.015(2) -0.0021(17) -0.0092(17)
C1 0.019(2) 0.060(3) 0.040(2) -0.001(2) -0.0032(16) -0.0048(18)
C2 0.024(2) 0.065(3) 0.042(2) -0.005(2) -0.0034(17) -0.0044(19)
C3 0.018(2) 0.062(3) 0.045(2) -0.003(2) -0.0081(17) -0.0090(18)
C4 0.023(2) 0.046(2) 0.035(2) 0.0101(19) -0.0045(16) -0.0048(17)
C5 0.0189(19) 0.042(2) 0.040(2) 0.0129(19) -0.0024(16) -0.0033(16)
C6 0.0213(19) 0.047(2) 0.035(2) 0.0079(19) -0.0024(16) -0.0071(17)
C7 0.022(2) 0.059(3) 0.042(2) 0.009(2) -0.0001(17) -0.0047(18)
C8 0.028(2) 0.069(3) 0.034(2) 0.005(2) -0.0011(17) -0.004(2)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 31
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186231
U11 U22 U33 U23 U13 U12
C9 0.023(2) 0.054(3) 0.034(2) 0.008(2) -0.0011(16) -0.0035(17)
C10 0.024(2) 0.061(3) 0.036(2) 0.003(2) -0.0033(17) -0.0036(18)
C11 0.024(2) 0.057(3) 0.035(2) 0.006(2) -0.0055(17) -0.0067(18)
C12 0.025(2) 0.081(3) 0.037(2) -0.006(2) -0.0042(17) -0.009(2)
C13 0.022(2) 0.067(3) 0.042(2) 0.001(2) -0.0048(17) -0.0062(19)
C14 0.0153(19) 0.051(2) 0.042(2) 0.006(2) -0.0028(16) -0.0004(16)
C15 0.0189(19) 0.043(2) 0.041(2) 0.0081(19) -0.0067(16) -0.0026(16)
C16 0.0183(19) 0.045(2) 0.043(2) 0.0098(19) -0.0035(16) -0.0046(16)
C17 0.0201(19) 0.052(3) 0.047(2) 0.005(2) -0.0009(17) -0.0093(17)
C18 0.023(2) 0.063(3) 0.043(2) 0.002(2) 0.0043(17) -0.0028(19)
C19 0.0180(19) 0.055(3) 0.045(2) 0.006(2) -0.0031(16) -0.0062(17)
C20 0.021(2) 0.069(3) 0.041(2) 0.000(2) -0.0016(17) 0.0027(19)
C21 0.015(2) 0.079(3) 0.043(3) -0.005(3) -0.0048(17) -0.002(2)
C22 0.038(3) 0.094(4) 0.040(3) -0.004(3) 0.001(2) -0.004(3)
C23 0.034(3) 0.085(4) 0.072(4) -0.019(3) 0.002(2) -0.002(2)
C24 0.032(3) 0.130(5) 0.040(3) -0.030(4) -0.002(2) -0.014(3)
C25 0.025(2) 0.127(5) 0.040(3) 0.003(3) -0.010(2) -0.016(3)
C26 0.021(2) 0.087(4) 0.042(3) -0.006(3) -0.0004(18) -0.002(2)
C27 0.021(2) 0.077(3) 0.037(2) -0.003(2) -0.0027(17) -0.004(2)
C28 0.032(2) 0.088(4) 0.049(3) -0.017(3) -0.003(2) 0.002(2)
C29 0.038(3) 0.114(5) 0.069(4) -0.035(4) -0.016(3) 0.020(3)
C30 0.023(3) 0.166(7) 0.041(3) -0.029(4) -0.010(2) 0.017(3)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 32
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186232
U11 U22 U33 U23 U13 U12
C31 0.018(2) 0.185(7) 0.029(3) -0.006(4) -0.0020(19) 0.004(3)
C32 0.022(2) 0.098(4) 0.053(3) 0.005(3) -0.0046(19) -0.007(2)
C33 0.0198(19) 0.051(3) 0.038(2) 0.0025(19) -0.0026(16) -0.0049(17)
C34 0.033(3) 0.063(3) 0.102(4) 0.034(3) -0.024(2) -0.011(2)
C35 0.026(2) 0.070(3) 0.103(4) 0.031(3) -0.019(2) -0.008(2)
C36 0.027(2) 0.044(3) 0.047(2) 0.011(2) 0.0001(17) -0.0016(18)
C37 0.030(3) 0.053(3) 0.136(5) 0.027(3) -0.012(3) -0.004(2)
C38 0.025(2) 0.047(3) 0.125(4) 0.022(3) -0.019(2) -0.006(2)
C39 0.027(2) 0.054(3) 0.051(3) 0.015(2) -0.0045(18) -0.012(2)
C40 0.045(3) 0.059(4) 0.292(10) 0.049(5) -0.058(4) -0.030(3)
C41 0.0188(19) 0.051(3) 0.038(2) 0.006(2) -0.0023(16) -0.0058(17)
C42 0.024(2) 0.055(3) 0.043(2) 0.005(2) -0.0006(17) -0.0055(18)
C43 0.019(2) 0.061(3) 0.041(2) -0.001(2) -0.0035(16) -0.0027(18)
C44 0.0180(19) 0.058(3) 0.038(2) -0.006(2) 0.0020(16) -0.0106(18)
C45 0.027(2) 0.049(3) 0.043(2) 0.001(2) 0.0004(17) -0.0108(18)
C46 0.021(2) 0.055(3) 0.047(2) 0.010(2) -0.0029(17) -0.0032(18)
C47 0.027(2) 0.073(3) 0.049(3) 0.013(2) -0.0103(18) -0.005(2)
C48 0.044(3) 0.093(4) 0.093(4) 0.043(3) -0.034(3) -0.009(3)
C49 0.039(3) 0.105(4) 0.049(3) 0.009(3) -0.009(2) -0.010(3)
C50 0.030(2) 0.080(3) 0.075(3) 0.001(3) -0.010(2) 0.007(2)
C51 0.031(2) 0.052(3) 0.056(3) 0.002(2) 0.0031(19) -0.008(2)
C52 0.041(3) 0.053(3) 0.078(3) 0.012(3) 0.006(2) -0.015(2)
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 33
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186233
U11 U22 U33 U23 U13 U12
C53 0.044(3) 0.075(4) 0.105(4) 0.010(3) 0.013(3) -0.010(3)
C54 0.059(3) 0.063(3) 0.091(4) 0.018(3) 0.013(3) -0.013(3)
C55 0.050(3) 0.066(3) 0.077(3) -0.001(3) 0.014(3) -0.019(2)
C56 0.039(3) 0.065(3) 0.065(3) -0.005(3) 0.005(2) -0.016(2)
C57 0.033(2) 0.060(3) 0.058(3) -0.004(2) 0.007(2) -0.015(2)
Supplementary Table S11. Hydrogen atomic coordinates and isotropic atomic displacement parameters (Å2) for dyad-2.
x/a y/b z/c U(eq)
H1 1.1085 1.2672 0.1231 0.074
H2A 0.6154 0.9262 0.0572 0.044
H4A 0.7124 0.9363 0.0247 0.046
H2 0.5870 0.6956 -0.1125 0.054
H3 0.4883 0.6784 -0.0749 0.053
H7 0.4220 0.8839 0.0756 0.05
H8 0.4837 1.0136 0.1395 0.054
H12 0.7430 1.1531 0.1967 0.059
H13 0.8488 1.1391 0.1653 0.054
H17 0.9054 0.9851 0.0054 0.049
H18 0.8380 0.8913 -0.0661 0.052
H34 0.3739 0.8871 -0.0194 0.085
H35 0.2711 0.7775 -0.0360 0.085
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 34
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186234
x/a y/b z/c U(eq)
H37 0.3702 0.4581 0.0028 0.093
H38 0.4714 0.5690 0.0217 0.085
H40A 0.1679 0.3478 -0.0145 0.217
H40B 0.1970 0.2658 -0.0515 0.217
H40C 0.1550 0.3913 -0.0673 0.217
H42 0.9371 0.9257 0.1312 0.05
H46 0.8913 1.2400 0.0585 0.051
H48A 1.0206 0.7924 0.1561 0.125
H48B 0.9973 0.8618 0.1984 0.125
H48C 1.0699 0.7928 0.2060 0.125
H49A 1.1248 0.9892 0.2393 0.1
H49B 1.0525 1.0598 0.2299 0.1
H49C 1.1150 1.1157 0.2097 0.1
H50A 1.1632 0.8784 0.1755 0.097
H50B 1.1533 1.0013 0.1440 0.097
H50C 1.1161 0.8738 0.1249 0.097
H53 0.9376 1.6932 0.0256 0.09
H54 1.0257 1.8360 0.0237 0.085
H55 1.1396 1.7802 0.0473 0.077
H56 1.1691 1.5814 0.0764 0.068
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 35
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186235
7 – References:
1. Rosenthal, J. & Yarmus, L. SrO:Mn as EPR marker and intensity standard. Rev. Sci. Instrum. 37,
381–381 (1966).
2. Brezgunov, A. Y., Dubinskii, A. A., Poluektov, O. G., Vorob’eva, G. A., Lebedev, Y. S.
Electron paramagnetic resonance of coals. New approaches to an old problem with
multifrequency electron paramagnetic resonance and spin echo. J. Chem. Soc., Faraday Trans.
86, 3185–3189 (1990).
3. Frisch, M. J. et al. Gaussian 09, Revision A.02 (Gaussian Inc., Wallingford, 2009).
4. Lee, C., Yang, W., Parr, R. G. Development of the Colle-Salvetti correlation-energy formula
into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
5. Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic
behavior. Phys. Rev. A 38, 3098–3100 (1988).
6. Becke, A. D. Density–functional thermochemistry. IV. A new dynamical correlation
functional and implications for exact–exchange mixing. J. Chem. Phys. 104, 1040–1046 (1996).
7. Francl, M. M. et. al. Self–consistent molecular orbital methods. XXIII. A polarization–type
basis set for second–row elements. J. Chem. Phys. 77, 3654–3665 (1982).
8. Harihan, P. C., Pople, J. A. The influence of polarization functions on molecular orbital
hydrogenation energies. Theor. Chim. Acta 28, 213–222 (1973).
9. Rassolov, V. A., Pople, J. A., Ratner, M. A., Windus, T. L. 6-31G* basis set for atoms K
through Zn. J. Chem. Phys. 109, 1223–1229 (1998).
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 36
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186236
10. Barone, V., Cossi, M. Quantum calculation of molecular energies and energy gradients in
solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998).
11. Cossi, M., Rega, N., Scalmani, G., Barone, V. Energies, structures, and electronic properties
of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681
(2003).
12. Neese, F. ORCA, An ab initio, density functional and semi-empirical program package,
Version 2.4.13, Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der Ruhr,
Germany (2004).
13. V. Barone in Recent Advances in Density Functional Methods (Ed.: Chong, D. P.) (World
Scientific Publishing, Singapore, 1996).
14. A. Klamt, G. Schüürmann. COSMO: a new approach to dielectric screening in solvents with
explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 25,
799–805 (1993).
15. Benisvy, L. et. al. Phenoxyl radicals hydrogen-bonded to imidazolium: Analogues of tyrosyl
D● of Photosystem II: High-Field EPR and DFT studies. Angew. Chem. Int. Ed. 44, 5314–5317
(2005).
16. Orio, M. et. al. Geometric and electronic structures of phenoxyl radicals hydrogen bonded to
neutral and cationic partners. Chem. Eur. J. 18, 5416–5429 (2012).
© 2014 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 37
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.186237
17. Costentin, C., Robert, M., Saveant, J.-M. Electrochemical concerted proton and electron
transfers. Potential-dependent rate constant, reorganization factors, proton tunneling and isotope
effects J. Electroanal. Chem. 588, 197–206 (2006).
18. Savéant, J.-M. Elements of Molecular and Biomolecular Electrochemistry Ch. 1 (Wiley-
Interscience, New York, 2006).
19. Using DigiElch software, see: Rudolph, M. J. Electroanal. Chem. 543, 23–39 (2003).
20. Bruker SAINT-Plus (Bruker AXS Inc., Madison, Wisconsin, USA, 2009).
21. Bruker APEX2, SADABS, (Bruker AXS Inc., Madison, Wisconsin, USA, 2010).
22. Sheldrick, G. M., SHELXS and SHELXL, Acta Cryst. A, 2008, 64, 112–122.
23. Speck, A. L., "PLATON, a multipurpose crystallographic tool." Utrecht University, Utrecht,
The Netherlands, 2001, http://www.cryst.chem.uu.nl/platon/.
24. Bruker XShell ver. 6.3.1, (Bruker AXS Inc., Madison, Wisconsin, USA, 2004).
© 2014 Macmillan Publishers Limited. All rights reserved.